تعتبر المحفزات الكهروكيميائية الفعالة لتفاعل تطور الأكسجين ضرورية لتحويل الطاقة النظيفة المستدامة. ومع ذلك، فإن المواد الحفازة تتبع آلية تطور الامتصاص التقليدية (AEM) مع العلاقة المتوازنة الموروثة بين الوسائط الرئيسية للأكسجين *OOH و*OH، أو آلية الوساطة بالأكسجين الشبكي (LOM) مع إمكانية هجرة الأكسجين الشبكي وإعادة بناء الهيكل، والتي لا تتناسب مع التوازن بين النشاط العالي والاستقرار. هنا، نقترح آلية غير تقليدية للتآزر الجزئي لموقعين مزدوجين من الكوبالت والحديد (DSSM) لمحفز مغناطيسي أحادي النطاق.النانوكلاسترز على الأنابيب النانوية الكربونية (CNT) (CFS )، والتي يمكن أن تكسر العلاقة التناسبية بفعالية دون التضحية بالاستقرار. (ل.س، تقدم أقوىطاقة الامتزاز، بينما (ماجستير، ) يكشف عن قوة الامتزاز. تنتج هذه المواقع المزدوجة بشكل متآزرالمتوسطات، مما يسرع من إطلاق الأكسجين في حالة الثلاثي (كما تم التنبؤ، فإن المحفزات المحضرة من CFSACs/CNT تظهر جهدًا زائدًا أقل من ذلك الخاص بـ IrO التجارية.بالإضافة إلى حوالي 633 ساعة من الاستقرار دون فقدان محتمل كبير.
لا يزال تطوير المحفزات الكهربائية الممتازة ذات النشاط العالي والمتانة على المدى الطويل يمثل تحديًا بسبب تدمير سطح المحفز من قبل البيئة المؤكسدة القوية خلال عملية تفاعل تطور الأكسجين (OER) وتفاعلها المعقد متعدد الخطوات المرتبط بالبروتون.. المبادئ التوجيهية لتعزيز الأداء الكهروكيميائي تعتمد بشكل أساسي على الاستراتيجيات التالية: (1) وفقًا لآلية تطور الامتصاص (AEM)، فإن نشاط تفاعل الأكسدة (OER) يتأثر بشكل رئيسي بالطاقة الامتصاصية للوسائط الأكسجينية. و وفقًا لمبدأ ساباتيه. ومع ذلك، هناك علاقة خطية متأصلة بين طاقات الامتزاز للوسطين التفاعليين *OOH و *OH، مما يجعل الحد الأدنى من الجهد الزائد النظري مرتفعًا كما هوحتى لأفضل المواد الممكنة. (2) بالمقارنة مع AEM التقليدي، فإن آلية الوساطة بالأكسجين الشبكي (LOM) يتغلب على قيود LSR من خلال تفعيل اقتران O-O، مما يمكن أن يقلل من حاجز الطاقة إلى. ومع ذلك، في هذه الآلية، غالبًا ما يتطلب الأمر أن تشارك الأكسجين الشبكي في اقتران الأكسجين-الأكسجين، وتسريع إطلاق الأكسجين في حالة الثلاثي. بالإضافة إلى ذلك، تؤدي العديد من الفراغات الأكسجينية إلى هجرة الأكسجين الشبكي على السطح، وإعادة بناء هيكلية شديدة، وعدم استقرار كافٍ.لذلك، لا تعتبر المحفزات المعتمدة على LOM تنافسية مع تلك المعتمدة على AEM من حيث الاستقرار والجدوى للتطبيقات العملية، على الرغم من كفاءتها التحفيزية الأعلى..
بدلاً من ذلك، فإن آلية مسار الأكسيد (OPM) للمواد الحفازة المتجانسة تستحق المزيد من تحسينات أداء تفاعل الأكسدة (OER)، حيث إنها تسمح بربط الجذور الحرة O-O مباشرةً دون عيوب فراغات الأكسجين أو وسائط تفاعل إضافية (*OOH). ربط O-O
تتطلب الآلية وجود فقط و الوسطاء خلال عملية OER، والتي تشمل نوعين: (1) اقتران O-O الناتج عن الأكسجين الشبكي من المحفز الجوهري وأكسجين المذيب معًا (LOM)؛ (2) اقتران O-O المقدم من المذيب فقط (OPM). ومع ذلك، فإن آلية LOM لها شروط تفعيل معقدة، مثل تنشيط الأكسجين الشبكي: طاقة Oيجب أن يتم رفع المدار بالقرب من مستوى فيرمي لتمكين التداخل مع المركز-مدار المعدن (أي، تعزيز التساهمية لـالرابطة)، وبالتالي تغطي الطاقة المختزلة للأكسجين في الشبكة. في هذه الأثناء، من الضروري إنشاء العديد من الثقوب لزعزعة ذرات الأكسجين في الشبكة وتمكين إطلاق بالمقارنة مع LOM، تعتبر عملية OPM، التي تُعتبر مسارًا مثاليًا، تسمح بوجود مباشر الاقتران. في هذه العملية، يتم فصلها من خلال العمل التآزري لمواقع المعادن النشطة، مما يؤدي إلىالاقتران الجذري واقتران O-O المدفوع. ومع ذلك، تتطلب هذه الآلية مواقع ثنائية معدنية متناظرة مع مسافات ذرية مناسبة للسماح باقتران الجذري O-O مع حواجز طاقة منخفضة.ومع ذلك، فإن آلية OPM أقل تقاريرًا في البيئات القلوية، لذا فإن استكشاف المحفزات ذات المواقع المتعددة المعادن المناسبة التي تتبع آلية OPM في البيئات القلوية لا يزال يمثل تحديًا.
مع تطوير المحفزات الممتازة من المواد الوفيرةالمعادن، أثرت الاضطرابات في المجال المغناطيسي الخارجي على حركيات التفاعل المحفز بالدوران وتنوع الدوران الداخلي الجوهري، مما سمح بمسارات التفاعل من الأنواع المحظورة بالدوران (*OH، *OOH و *O إلى ) إلى الأنواع المسموح بها للدوران وتسريع إطلاق ترتيب الدوران المتوازي للأكسجين في حالة الثلاثي . لذلك، فإن بناء المحفزات الفيرومغناطيسية هو شرط أساسي لتحقيق حركيات تفاعل سريعة محفزة بالدوران. من بين تفاعل OER، كمواد مغناطيسية حديدية نموذجية، تظهر حافة رائدة مع نشاط عالي في تفاعل الأكسدة في البيئات القلوية.. بالنسبة للتفاعلات الحفزية التي تتضمن نقل الإلكترون المرتبط بالبروتون (مثل OER)، فإن الطاقات السطحية العالية لمحفزات الذرات المزدوجة غير المتجانسة وانخفاض استخدام الذرات في المحفزات البلورية تجعل من الصعب تحقيق توازن بين نشاط OER والاستقرار. من المعقول أن نستنتج أنه إذا تم التحكم في حجم الذرات للمحفز الفيرومغناطيسي القائم على الكوبالت والحديد ليكون في منطقة ذات مجال واحد (حتى الكتل)، سيحدث تفاعل الأكسدة الانتقائية للأكسجين الموجه (OER) في غياب مجال مغناطيسيلذلك، فإن تسريع إطلاق حالة الأكسجين الثلاثي المتوازي المتراص بواسطة الحقل الكهربائي المدمج الذي يسبب اقتران مغناطيسي موضعي هو طريق فعال لتحسين محفزات تفاعل أكسدة الأكسجين.. فيما يتعلق بذلك، الوسطاء الرئيسيون (تبعها اقتران مغناطيسي حديدي لفهم شامل للاقتباس المباشر O-O تحت آلية OPM يوفر منظورًا جديدًا، وقد يُعتبر في النهاية حلاً لعائق الاستقرار والنشاط الأساسي للمحفزات المعتمدة على LOM أو AEM.
من أجل تحليل آلية اقتران الأكسجين-الأكسجين التي يمكن أن تتغلب على قيود لاندو-سيربرغ وتوازن بين النشاط العالي والثبات القوي، اخترنا المواد المغناطيسية الحديدية.موقعين مزدوجينالعناقيد المدعومة على مادة أنابيب الكربون النانوية CNT كمنصة لتحفيز تفاعل الأكسدة (OER) وتوضيح العلاقة الجوهرية بين نشاط OER وحالة الدوران لكل موقع معدني. في الوقت نفسه، كنا نشك في أن التركيز على الامتصاص التفضيلي للوسائط الأكسجينية الرئيسية على المعادن يقدم منظورًا بحثيًا واعدًا لتعميق الفهم حول الربط المباشر بين ذرتي الأكسجين. كما يتم النظر في مناقشة تطور حالات الدوران للحديد (II) والحديد (III) خلال عملية OER.
النتائج
التصميم والتركيب العقلاني
التم تحضير النانوكلاسترات المدعومة على الأنابيب النانوية (CFS-ACs/CNT) من خلال طريقة سهلة للاختزال بالامتصاص-الهيدروحرارية. أولاً، تم الحصول على الشكل غير المتبلورتم تحضير النانوصفائح ودعمها على أنابيب الكربون النانوية المنظفة مسبقًا (CFO-p/CNT) من خلال اختزال NaBH4. عملية، وتفاعلات قوية بين سوائل المعادن وخصائص الأكسجين الغنية على أنابيب الكربون النانوية. بعد ذلك، تم تصنيع النانوكلاسترز والحفاظ عليها من خلال البيئة الحمضية المرغوبة القابلة للتشغيل الناتجة عن تحلل الثيوأسيتيمايد.خلال العملية الهيدروحرارية (الشكل 1أ).
تم فحص التركيب الكيميائي، الشكل، والحالة الكيميائية للمنتج النهائي بواسطة حيود الأشعة السينية (XRD) وميكروسكوب الإلكترون الناقل (TEM). أظهرت أنماط حيود الأشعة السينية لـ CFSACs/CNT و CFO-p/CNT (الشكل S1) عدم وجود قمم حيود واضحة تنتمي إلىالهجينة القائمة على -، والتي تشير إلى هيكل غير متبلور لأن تحليل حيود الأشعة السينية حساس فقط للمواد البلورية.تمتاز المواد غير المتبلورة ذات المدى الطويل والمشوشة التي تحتوي على عدد قليل من الذرات التي تظهر ترتيبًا قصير المدى بوجود مواقع نشطة أكثر. كما أن لديها نطاقًا أوسع من التركيبات الكيميائية ومرونة هيكلية أكبر مقارنة بالمواد البلورية، مما يؤدي إلى نشاط كهربائي تحفيزي متفوق.. عادةً ما تكون صور TEM للمواد النقيةتظهر شكل الأغشية النانوية المتجانسة (الشكل S2)، وأنابيب الكربون النانوية المتميزة بقطرتُلاحظ أيضًا في الشكل S3 مع خطوط شبكية واضحة وكاملة. بعدالخفض، أظهرت صور TEM (الشكل S4) أن CFO-p/CNT احتفظت بالشكل أحادي الأبعاد لـ CNTs، التي عليها صغيرةتم تزيين النانوشرائح بشكل موحد. بالمقارنة مع صورة TEM لأسطوانات الكربون النانوية النقية، تظهر خطوط الشبكة في أسطوانات الكربون النانوية في محفز CFO-p/CNT تشوهات واضحة وكسور بسبب تضمين الـالأغشية النانوية (الموضحة بواسطة الدوائر الصفراء)، والتي أكدت النجاح في إدخالالأنواع. مع إدخال مصدر الكبريت،تم تفكيك النانوصفائح وإعادة تكوينها إلى CFS-ACs أصغر على أسطح CNTs (الشكل 1ب، ج). بالإضافة إلى ذلك، تم إجراء تصوير مجهر إلكتروني بالمسح المظلم عالي الزاوية (HAADF-STEM) بدقة ذرية للتحقيق في توزيع CFS-ACs على المقياس الذري. كما هو موضح في الشكل 1د، كانت هناك العديد من CFS-ACs الموزعة بشكل جيد (المعلمة بدوائر حمراء) على دعم CNTs. كما أظهرت الخرائط العنصرية توزيعًا موحدًا لعناصر مختلفة. ) إلى (الشكل 1e). الكوبالت ( ) و تم تحديد محتويات CFS-ACs/CNT باستخدام مطيافية الانبعاث الضوئي بالتحليل الطيفي البلازمي المقترن بالحث (ICP-OES)، والتي تم استخدامها كمعيار للدراسات اللاحقة.
تم استخدام مطيافية الإلكترونات الضوئية بالأشعة السينية (XPS) للتحقق بشكل أكبر من بيئة التنسيق والحالات الكيميائية لجميع العينات المحضرة (الأشكال S5-S9). في الشكل S5a، الكوبالتأظهرت طيفيات XPS لـ CFO-p/CNT و CFS-ACs/CNT وجود نوعين رئيسيين من الكوبالت وشركاه انقسام الدوران-المدار. الكوبالتقمم لـ CFS-ACs/CNT تقع عند 779.9 و تم تعيينهم إلى و على التوالي. في الوقت نفسه، مقارنةً بالسابقة،أنواع CFS-ACs/CNT أظهرت تحولات قمة إيجابية، مما يشير إلى حالة تكافؤ أعلى. بالنسبة للمنتج الموزع بشكل جيد، وجودونقل الإلكترون المحيطي لكل من الكوبالت والحديد بسبب إدخال الكبريت، زاد من نشاط تفاعل أكسدة الأكسجين.. بعد عملية الكبريت، تغيرت حالة أكسدة المعدن، بينما كانت الكل تم تحويل الطيف بشكل إيجابي (الشكل S6). الـ Sطيف XPS (الشكل S7a) يحتوي على ثلاثة قمم: أو التنسيق عند 162.5 إلكترون فولت، الحالة المؤكسدةفي و كما هو موضح في الشكل S7b، القمم عند 322 و340 ونُسِبَت إلى و أنماط من و أنواع الروابط، على التوالي، مقارنةً مع المدير الماليطيف مطياف الأشعة تحت الحمراء لتحويل فورييه (FT-IR) لمحفز CFS-ACs/CNT (الشكل S8) أظهر اهتزازًا مميزًا لرباط C-S. وجود C-S-C أكد مرة أخرى إدخال ذرات الكبريت في الأنابيب النانوية الكربونية (CNTs). في الوقت نفسه، كانت القمم المميزة لـ (الشكل S9) تم تحويلها مقارنة بتلك الخاصة بأنابيب الكربون النانوية النقية، مما يشير إلى أن التفاعل القوي بين المعدن والداعم الناتج عن إدخال ذرات الكبريت أدى إلى الاستقرار الهيكلي المتفوق، وموصلية أعلى، وانتقال أسرع للإلكترونات أثناء عملية الأكسدة. تم استخدام مطيافية رامان لفحص نسب الشدة ( ) من نطاق D إلى نطاق G في العينات، مما يشير إلى العيوب السطحية في المادة الكربونية (الشكل S10). أظهرت النتائج أن نسبة CFS-ACs/CNT كانت
الشكل 1 | الخصائص الشكلية والهياكل لـ CFS-ACs/CNT. أ تخليق والخصائص الشكلية لـ CFS-ACs/CNT. ب، ج صور مجهر إلكتروني ناقل (TEM). د تصحيح الانحراف الزاوي عالي الزاوية.
صور مجهر الإلكترون الناقل (STEM) بتقنية الحقل المظلم (HAADF) مع صورة مكبرة تظهرعنقود (دائرة حمراء)، ورسم خرائط طيف الأشعة السينية المشتتة للطاقة (EDS) للعناصر الفردية، وشركاه). أصغر من تلك الخاصة بـ CFO-p/CNT. مستويات أقل من العيوب والاضطراب المنخفض في المواد الكربونية تشير إلى ارتفاع نسبة الإشغال في المواقع وتوزيع جيد لمواقع المعادن النشطة.
الإلكترون وحالة الدوران للعامل المساعد
هيكل امتصاص الأشعة السينية بالقرب من الحافة (XANES) لأنواع الحديد في كل من CFS-ACs/CNT و FeS أظهرت CNT (الشكل 2أ) حالات التكافؤ و . في هذه الأثناء، أظهر محفز CFS-ACs/CNT تحولًا طفيفًا نحو طاقة فوتون أقل، مما يدل على حالة كيميائية تعادل تقريبًا +2 في تلك العينة، وهو ما يتماشى مع تحليل XPS. وبالمثل، كانت هناك قمة مميزة قبل الحافة عندتم تعيين حافة ك-edge XANES لـ CFS-ACs/CNT إلى عينة Co-O (الشكل S11a)، بينما كانت حافة الامتصاص لكتالوج CFS-ACs/CNT تقع بين رقائق الكوبالت وCo-O، بالقرب من تلك الخاصة بالفثالوسيانين.كما هو موضح في الشكل 2ب، كشف طيف الامتصاص الدقيق للأشعة السينية الممتد (EXAFS) لـ CFO-p/CNT عن قمة بارزة مميزة عندفيالفراغ، الذي تزامن مع تنسيق الحديد والأكسجين. في هذه الأثناء، هناك قمة سائدة في طيف CFS-ACs/CNT ومحمل فيتمت ملاحظته، والذي كان يقع بين Fe-O و Fe-S. وبالمثل، فإن أعلى قمة لحديد-حديد عندكان غياب رقائق الحديد ل CFS-ACs/CNT.
وبالمثل، أظهر طيف EXAFS عند حافة كربون Co K لـ CFS-ACs/CNT (الشكل S11b) انزياحًا سلبيًا مع وجود قمة بارزة عندأ ، الذي كان يقع بين و . علاوة على ذلك، فإن إدخال المواقع المزدوجة زاد من تقصير رابطة M-S مقارنةً بمحفز الموقع المعدني الفردي، مما أدى إلى تلبية شرط التحفيز لآلية اقتران O-O المباشر إلى حد ما. كما هو موضح في الشكل S12، تم استخدام ملاءمة FT-EXAFS الكمية لتحديد المعلمات الهيكلية لـالبيئة الكيميائية المحلية. كانت أعداد تنسيق Co-S و Fe-S 5.5 و 5.3 (الجدول S1) على التوالي، مما يشير إلى تشابه البيئات الكيميائية لـ Co و Fe. للتحقق من الهيكل بشكل أكبر، تم حساب طيف XANES عند حافة الحديد K بناءً علىتم الحصول على الهيكل، الذي تزامن بشكل جيد مع طيف XANES عند حافة الحديد K لـ CFS-ACs/CNT (الشكل S13). علاوة على ذلك، أظهر تحليل EXAFS باستخدام تحويل الموجات (الشكل 2c والشكل S14) عند حافة الحديد K قمة مميزة في الشدة عندالذي نُسب إلى تنسيق الحديد والكبريت.
الإلكترون غير المتزاوج في المدارات الذرية للحديد في CFS-ACs/CNT وتم تحديد العينات بشكل إضافي باستخدام طيف الرنين المغناطيسي الإلكتروني (EPR). كما هو موضح في الشكل S15، كان إشارة ذروة EPR أكثر وضوحًا بعد إدخالالذي
الشكل 2 | تحليل XASطيفية ميسباور وقابلية المغناطيسية. XANES عند حافة الحديد K طيف EXAFS المحول فورييه و CFS-ACs/CNT، و . تحويلات الموجات المتقطعة للتجربة-طيف EXAFS الموزون لـ CFS-ACs/CNT. دالاستجابة X والمعكوسمن CFS-
ACs/CNT ودرجة حرارة الغرفةطيف موسباور لـ CFSACs/CNT. طيف XANES لحدود كوبالت لـ CFS-ACs/CNT و FeS CNT . g توضيح تخطيطي لنقل حالة الدوران للنشاط العالي لتفاعل تطور الأكسجين (OER) لقطب CFS-ACs/CNT (اليسار هو شغل المدار d لـ، الصحيح هو CFS-ACs/CNT). يوحي بوجود المزيد من الإلكترونات غير المتزاوجة في Fe-3d. بشكل عام، تترافق التغيرات في شغل المدارات وإعادة توزيع الشحنات مع تحول تكوين دوران الإلكترونات في Fe-3d. لذلك، لتحديد التغير المحتمل في حالة دوران الإلكترون، تم فحص السلوك المغناطيسي (الشكل S16). بعد تشكيل الكبريتيدات في العملية الهيدروحرارية، كانت هناك اعتماد على الحقول المغناطيسية في CFSACs/CNT وأظهرت العينات ظاهرة تذبذب ملحوظة. بالمقارنة معكان لدى محفز CFS-ACs/CNT مع مغنطة تشبع أكبر (Ms) المزيد من الإلكترونات غير المتزاوجة وغيرت التكوين الإلكتروني الداخلي للمعادن، مما صحح حالة الدوران المحلية.الألكترونات غير المتزاوجة المعتدلة مكنت من تحقيق الأمثلالتفاعلات المدارية بين مدارات M-3d ومدارات O-2p، مما يسهل امتصاص/إطلاق الوسائط الأكسجينية من المحفز الفيرومغناطيسي CFS-ACs خلال خطوة تحديد معدل تفاعل الأكسدة، وبالتالي تسريع حركية عملية الأكسدة.. بعد ذلك، تم تحديد تكوين دوران الإلكترون لـ CFS-ACs/CNT من خلال قياس القابلية المغناطيسية المعتمدة على درجة الحرارة (MT). توضح الشكل 2d والشكل S17 أن انتقالات الطور من المغناطيسية البارامغناطيسية إلى المغناطيسية الحديدية حدثت عند درجات حرارة كوري ( ) لجميع المركبات. مقارنةً مع ذلك من ، أدى جيل CFS-ACs/CNT إلى تقليل حالة البارامغناطيسية لأنواع الحديد، ونقلإلى درجات حرارة أعلى مع توليد كمية كبيرة من استجابة مغناطيسية“، مما يوحي بزيادة الاضطراب وزيادة حركة الإلكترونات الحرة حول أنواع الحديد. من خلال ملاءمة قانون كوري-وايس، تم حساب العزم المغناطيسي الفعال ( ) تم حساب CFS-ACs/CNT كـ . ثم، عدد غير المتزاوجينإلكترونتم تحديد ( ) لموقع Fe (II) وفقًا للمعادلة:حيثكانلـ CFS-ACs/CNT وأعلى من ذلكتم إجراء مطيافية ميسباور (الشكل 2e والجدول S2) للتحقق من حالات الدوران المحلية لمواقع الحديد الفردية في المحفزات الفيرومغناطيسية متعددة المعادن. تم ضبط القمة الرئيسية مع ثنائي واحد من D1 (الأزرق)، المعين لـ M.S Fe(II).. لذلك، لم يكن هناك إلكترونات غير متزاوجة زائدة مخصصة لموقع الكوبالت. من المعقول افتراض وجود الكوبالت في حالة دوران منخفضة. )، التي تحتوي على فراغات غير مملوءة المدارات والروابط القوية مع المادة الممتصةالمتوسطات. علاوة على ذلك، تم استخدام طيف الامتصاص الدقيق للأشعة السينية عند حافة L لكوبالت (NEXAFS) للتحقيق في شدة كوبالت.حالة إلكترونية فارغة/مملوءة جزئيًا لـ CFS-ACs/CNT وكما هو موضح في الشكل 2f، هناك قمتان (A1 و A2) لـكانت الحواف مهيمنة عليها ميزات “الخط الأبيض” المعززة التي تشير إلى عدم وجود شغل و المدارات، على التوالي. تم تخصيص القمة A1 لانتقال من الكوبالتإلىمداري، بينما أشارت القمم A2 إلى انتقال إلىمداري. كو غير المأهولةمدارات CFS-ACs/CNT و تم عكس العينات مباشرة من خلال نسبة شدة الامتصاص لـالحافة، التي تعكس إلى حد كبير حالة الدوران الأساسية الناتجة عن تأثير مجال البلورةعرضت أكبرنسبة الشدة 3.5، بينما كانت نسبة CFS-ACs/CNT 2.5. لذلك، تم تقليل طاقة الانقسام لـاقترح تأثير حقل بلوري ضعيف يفضل الأعلىالإشغال المداري لمركز الكوبالت. مقترن معمنحنياتالمحفز كشف عن غير ممتلئشغل المدارات، يسمى حالة الدوران المنخفض ). ليس من المستغرب أن هذا كان متسقًا مع افتراضنا الأولي المعقول بأن ذرات الكوبالت في CFS-ACs/CNT كانت غير مملوءة -مدارات. يانغ شاو هورن وآخرون. اقترح أن الأمثل كان شغل المدارات -orbital لربط وسائط OER تقريبًا 1.2. وبالتالي، مقارنة بموقع الحديد الفردي معالتكوين، CFS-ACs/CNT المعرضة المثلى-شغل المدارات (الشكل 2g). كنا نتوقع أن تأثير التآزر للتكوين المحلي لـولاية ميسيسيبي ) و دولة ل.س (سوف يؤدي ذلك إلى هيكل إلكتروني مع طاقة امتصاص مثالية لوسائط تفاعل الأكسدة، مما ينتج عنه نشاط تحفيزي أعلى لتفاعل الأكسدة.
أداء التحفيز الكهربائي لـ CFS-ACs في تفاعل الأكسدة الكهربية للأكسجين
تم فحص أداء OER لـ CFS-ACs/CNT فيمحاليل KOH المشبعة 1.0 م بنظام ثلاثي الأقطاب نموذجي. للمقارنة،جملةأنابيب الكربون النانوية النظيفة والتجاريةتم قياسها أيضًا. تم تكوين جميع المحفزات المحضرة في حبر متجانس وأضيفت إلى سطح ركيزة رغوة النيكل (NF) بشكل قطرة متجانسة. ) لتشكيل القطب العامل (الشكل S18). تم إعداد العينات القياسية (CFS-ACs/CNT) من خلال سلسلة من ضوابط المحتوى للحصول على النسبة المثلى من و S (الشكل. ). تظهر الفولتمترية الخطية المستقرة (LSV) في الشكل 3a. أظهرت CFS-ACs/CNT نشاطًا ممتازًا في تفاعل الأكسدة (OER) مع جهد زائد أقل يبلغ 270 مللي فولت عند ، والتي كانت أقل بمقدار 100 مللي فولت و80 مللي فولت من تلك الخاصة بالتجاري و ، وقدم أصغر قيمة لميل تافل (الشكل S20 والشكل 3b) من. بالإضافة إلى ذلك، منحنيات LSV معتعويض CFS-ACs/CNT،وتم توفير CNT النقي في الشكل S21a-e. وبالمثل، أظهرت CFS-ACs/CNT أيضًا نشاط OER متفوق مقارنةً بالعناصر الأخرى المعدة. لاستبعاد
الشكل 3 | قياسات أداء OER. أ منحنيات LSV لـ CFS-ACs/CNT، FeS CNT، ، CNT نقي، تجاريوركيزة رغوة النيكل بدونتعويض، يقاس بتغطية المحفز على رغوة النيكلمع تحميل الكتلة منمقارنة النشاط لجهد البداية عندوميل تافل لـ CFS-ACs/CNT (أحمر)، (أزرق)، (أخضر)، (بني) و CNT نقي (رمادي). ج طيف EIS لـ CFS-ACs/CNT مع شبه دائرة صغيرة حول ) ، مع شبه دائرةحول ) ، و التيار السعوي المزدوج النقي. ) من CFS-ACs/CNT، FeS CNT و CNT النقي. منحنى الكرونو بوتنشيومتر لـ و CFS-ACs/ CNT في . مقارنة بين نشاط واستقرار مختلف المحفزات المعدنية الانتقالية باستخدام رموز مختلفة لتمثيلها. تمثل المناطق الحمراء المحفزات التي تتمتع بتوازن جيد بين النشاط والاستقرار، بينما تمثل المناطق الصفراء مستوى معتدل، والمناطق الخضراء ضعيفة. تم توفير المراجع لنقاط البيانات في الجدول التكميلي 4. تأثير ركيزة NF، تم تقييم عينة NF النقية التي خضعت لإجراء مشابه، وأظهرت نشاطًا ضئيلًا في OER (الشكل S22a). في الوقت نفسه، تم تقييم نشاط OER لـ CFO-p/CNT أيضًا (الشكل S22b). وبالمثل، تم تقييم أداء OER للكتلةتم تقديره من خلال منحنى LSV ورسم كثافة التيار مع CFS-ACs/CNT وفي (الشكل S23) لتسليط الضوء على الفعالية والرؤية المستقبلية للتصميم العقلاني لهيكل الكتلة. لتقييم نقل الشحنة بشكل أكبر، تم قياس مخططات نايكويست بواسطة طيف الامتياز الكهربائي (EIS) لـ CFS-ACs/ CNT، وتم عرض CNT في الشكل 3c. أظهر عينة النتيجة مقاومة نقل شحن أصغر ( )، حركية أسرع، والتي تزامنت مع قيمة منحدر تافل الأقل تم تقييم المساحات السطحية الكهروكيميائية (ECSAs) للمحفزات التي تم تحضيرها من خلال السعات الكهربائية الثنائية الطبقة (ال. ) في 1 م كوه التي تم الحصول عليها بواسطة الفولتمترية الدورية (CV) بمعدلات مسح مختلفة (التجارب التفصيلية في الشكل التكميلي 24 والشكل 3d). الأمثل أشارت CFS-ACs/CNT إلى وجود مواقع نشطة سطحية يمكن الوصول إليها كهربائيًا بشكل أكبر، مما يتوافق مع الهيكل العنقودي الأمثل. في الوقت نفسه، أظهر منحنى LSV المصحح بـ ECSA (الشكل S25) الذي تم الحصول عليه بعد تطبيع كثافة التيار باستخدام ECSA أن CFS-ACs/CNT كانت لديها أعلى نشاط داخلي. عدم كفاية الاستقرار هو مشكلة رئيسية أخرى لمعظم المحفزات النشطة للغاية والموزعة بشكل كبير في تفاعل الأكسدة. أولاً، تم تقييم مقاومة التآكل من خلال دورة CV في الشكل S26. لم يكن هناك تدهور ملحوظ قبل وبعد 2000 دورة CV ضمن نطاق الجهد. مقابل RHE حول الفائض الجهد، مما يدل على المقاومة العالية للتآكل لـ CFS-ACs/CNT. كما هو موضح في الشكل 3e والشكل S27، قياس الجهد الزمني لـ CFS-ACs/CNT وتم إجراء ذلك في 1 م KOH. أظهرت CFS-ACs/CNT استقرارًا كيميائيًا كهربائيًا قويًا مع استمرار اختبار OER لمدة 633 ساعة دون انخفاض ملحوظ في الجهد، بينمالقد أظهر زيادة ملحوظة في الجهد الزائد بعدقياس الكرونو بوتنشيومتر المستمر مع انخفاض في الجهد. في هذه الأثناء، المنحنيات بعدتعويض CFS-ACs/CNT، و عند كثافة تيار عالية ( ) تم توفيره أيضًا لتقدير استقرارها الكهروكيميائي بشكل أكبر. كما هو موضح في الشكل S21f، بعد قياس مستمر لمدة 70 ساعة عند كثافة تيار عالية، انخفضت كثافة التيار لعنصر CFS-ACs/CNT فقط بمقدار 4 مللي فولت. علاوة على ذلك، للتحقق من الاستقرار الهيكلي الذي توفره ذرات الكبريت، تم إجراء قياسات الكرونوپوتنشيومترية أيضًا لعنصر CFO-p/CNT في 1 م KOH. كما هو موضح في الشكل S28، أظهر عنصر CFO-p/CNT استقرارًا ضعيفًا، مع انخفاض في كثافة التيار بعد 70 ساعة من اختبار OER الثابت، مما أظهر بشكل أكبر أن إدخال الكبريت قد حسّن الهيكل الإلكتروني للمحفز ونجح في تقليل حالة التجمع.العناقيد خلال العملية التحفيزية. متسقة مع التقارير السابقةكانت حالة الأكسدة لمركبات الكوبالت والحديد عرضة للتغيرات خلال عملية تطوير الأكسجين. لذلك، تم إجراء تحليل باستخدام طيف الأشعة السينية للأشعة السطحية (XPS) وطيف رامان لدراسة التركيب أو الأنواع التي تطورت بعد عملية تطوير الأكسجين. بعد تطبيق الجهد لفترة من الزمن، أعيد تشكيل السطح على المحفزات لإنتاج هيدروكسيدات الحديد غير المتبلورة (الشكل S29، 30 والملاحظات التكميلية المفصلة)، وتم أكسدة أنواع الحديد إلى حالة الفالنسية +3. في الوقت نفسه، بعد الاستقرار، زاد محتوى ذرات الأكسجين مع انخفاض الأنواع النشطة.بينما شدة الـذروة CFS-ACs/CNT أظهرت فقط تدهورًا طفيفًا في محتوى الكبريت من 19.01-14.8 عند النسبة المئوية (الجدول S3)، مما أظهر المزيد من الاستقرار الهيكلي بعد إدخال ذرات الكوبالت والكبريت. علاوة على ذلك، تفوقت CFS-ACs/CNT على أفضل المحفزات التي تم الإبلاغ عنها سابقًا في الأدبيات (الجدول S4)، من حيث النشاط الكهروكيميائي والاستقرار (الشكل 3f). مقارنة نشاط OER واستقرار CFS-ACs/CNT مع تلك المتعلقة بالذرات الفردية.تشير الأكاسيد وغيرها من المحفزات القائمة على المعادن الانتقالية إلى تأثير هيكل ACs وآلية الاقتران.
استكشاف الآلية الحفازة من منظور حالة الدوران
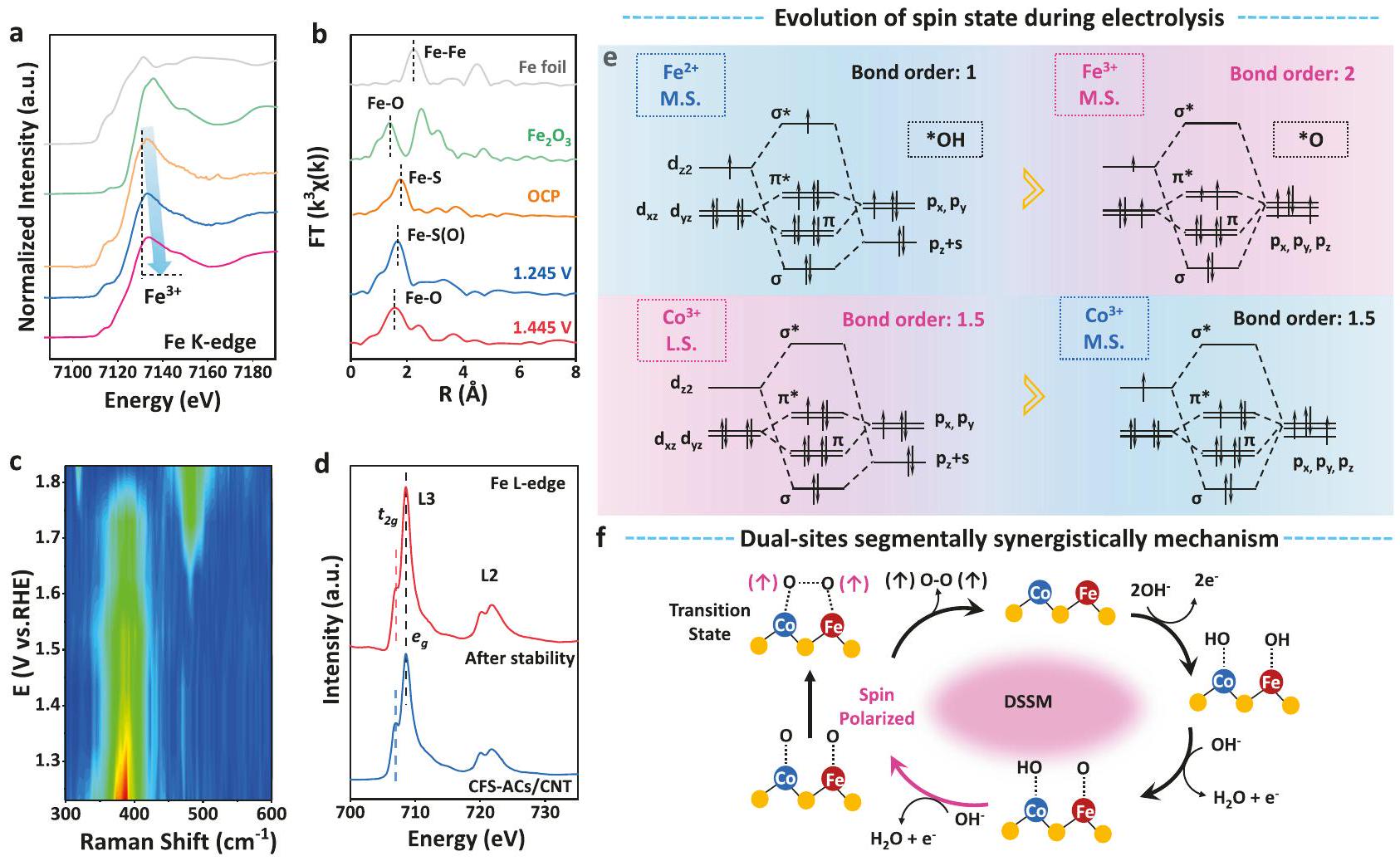
للمزيد من التحقيق في العلاقة بين حالة الدوران لتحويل الأنواع النشطة من وجهة نظر الانتقال الإلكتروني خلال عملية OER، تم أيضًا إجراء قياسات XAS وRaman في الموقع. أظهرت طيف XANES الموضح في الشكل 4a، أن هناك قمة امتصاص بارزة تقع عند 7715 eV قد انتقلت تدريجياً نحو طاقة أعلى مع زيادة الجهد المطبق، مما يدل على تغيير في حالة الأكسدة خلال عملية OER. خلال عملية الأكسدة، اختفاءتم أكسدة المواقع تدريجياً إلىالذي تزامن مع طيف XPS بعد قياس طويل الأمد (الشكل S30). في الشكل 4b، مع زيادة الجهد المطبق من OCP،، انتقل القمة الرئيسية تدريجياً في اتجاه الطاقة المنخفضة، مما اقترح تحولاً في بيئة تنسيق الحديد والكبريت وتغيراً في هيكل الانتقال الإلكتروني للحديد خلال عملية تطوير الأكسجين.عندما وصل الجهد إلى 1.445 فولت، كان الذروة موجودة فيقريب من ذلك لـبيئة التنسيقأثبت تنسيق Fe-O بشكل غير مباشر أن CFS-ACs انتقلت تدريجياً إلى المرحلة النشطة الحقيقية من أكاسيد الهيدروكسيد المعدنية خلال عملية OER، وكان ذلك مصحوباً بالاختفاء التدريجي لـسنداتأكدت مطيافية رامان في الموقع تطور الأنواع النشطة خلال القياس الكهروكيميائي (الشكل 4c والشكل S31). أظهر طيف رامان عند الجهد الابتدائي 1.23 فولت وجود قمتين رئيسيتين فقط عند 344 والتي نُسِبَت إلى و أنماط تمدد الحديد والكبريت في CFSACs/CNT، على التوالي. مع زيادة الإمكانية، زادت شدة الـانخفض الذروة بشكل كبير، مما يشير إلى حدوث انتقال في الطور. ظهرت شريحة عندبجهد محتمل قدره 1.43 فولت واختفى فوق 1.73 فولت، بينما توجد شريحة عندظهرت عند 1.73 فولت (الشكل S31). تم تعيين كلا النطاقين لأكسيد الحديد وأوكسي هيدروكسيد الحديد، على الرغم من غياب العديد من النطاقات المصاحبة. وبالتالي، من المحتمل أن تكون هذين النطاقين مرتبطتين بأنماط اهتزاز Fe-O للطور غير المتبلور بشكل جيد الذي تشكل خلال الأكسدة السطحية. علاوة على ذلك،المرحلة النشطة تنتمي إلىلم يتم العثور عليه.
بشكل عام، يتم مرافقة إعادة توزيع الشحنات ونقلها بقفزة فيتكوين دوران الإلكترون. كما هو موضح في الشكل 4d، أظهرت CFSACs/CNT قيمة أكبرنسبة الشدة 1.6، بينما كانت نسبة CFS-ACs/CNT بعد الاستقرار 1.3. لذلك، فإن قمع هيكل XANES المنقسم لـ CFS-ACs/CNT يشير إلى وجود مجال بلوري ضعيف مما يتيح مستوى أعلىالإشغال المداري لمركز الحديد.موقع المحفز CFS-ACs/CNT أظهر نصف ممتلئشغل المدارات بحالة دوران متوسطةلذلك، لأن CFSACs/CNT قبل وبعد استقرار OER أظهرت نسبة كثافة تقريبية،يجب أن يكون مشابهًاشغل المدارات. بالمثل، كما هو موضح في XANES حافة Co L (الشكل S32)، أظهرت CFS-ACs/CNT قيمة أصغرنسبة الشدة مقارنةً بـ CFS-ACs/CNT بعد الاستقرار.موقع المحفزات CFSACs/CNT كان غير ممتلئالأوربيتالز ذات حالة الدوران المنخفضة ). لذلك، الموقع بعد الاستقرار قد يكون قد امتلأ جزئيًاالأوربيتالز بحالة الدوران المتوسطةبسبب طاقة الانقسام الأصغروفقًا لنظرية ترتيب الروابط والتفاعلات المدارية بين الكاتيونات والوسائط الأكسجينية في الشكل 4e،ولاية ميسيسيبي ) أظهرت تفاعلات ربط الدوران-المدار أضعف من ولاية L.S. ( )، مما يوحي بضعف سعة الامتزاز. في السنوات الأخيرة، تم تكريس العديد من التحقيقات لكشف العلاقة بين النشاط التحفيزي وتكوينات الدوران للعوامل الحفازة، ومن بين هذه العناصر، كان العنصر الفيرومغناطيسي الحديد (Fe) هو الأكثر دراسة (الجدول S5). هيونغ-سوك أوه وآخرون. اقترح تطور حالة الدوران لـموقع من L.S.إلى م.س. ( ) عند الجهد المطبق 1.4 فولت. وبالمثل، بعد عملية الأكسدة، تم أكسدته إلىبسبب توليد أكسيد الحديد الهيدروكسي، لكن حالة M.S. تحافظ، في هذه النقطة، على حالة الدوران لـتغير من L.S. إلى M.S.، لكن حالة التكافؤ ظلت دون تغيير. الـأظهر M.S. 1 من-شغل المدارات، يمتلك قوة قليلاً قويةطاقة الامتزاز لتفاعل الأكسدة. دعم هذا في
الشكل 4 | تحليل حالة الدوران خلال عملية OER. أ طيف XANES عند حافة الحديد K، ب FT-EXAFS عند حافة الحديد K خلال عملية OER مع الجهود المختلفة (مقابل RHE) من OCV، 1.245 فولت، و 1.445 فولت في 1 م KOH، على التوالي.طيف رامان في الموقع لمحفز CFS فيطيف XANES لحدود الحديد قبل وبعد استقرار CFSACs/CNT. التفاعلات المدارية بين الكاتيونات والوسائط الوسيطة لتفاعل الأكسدة.
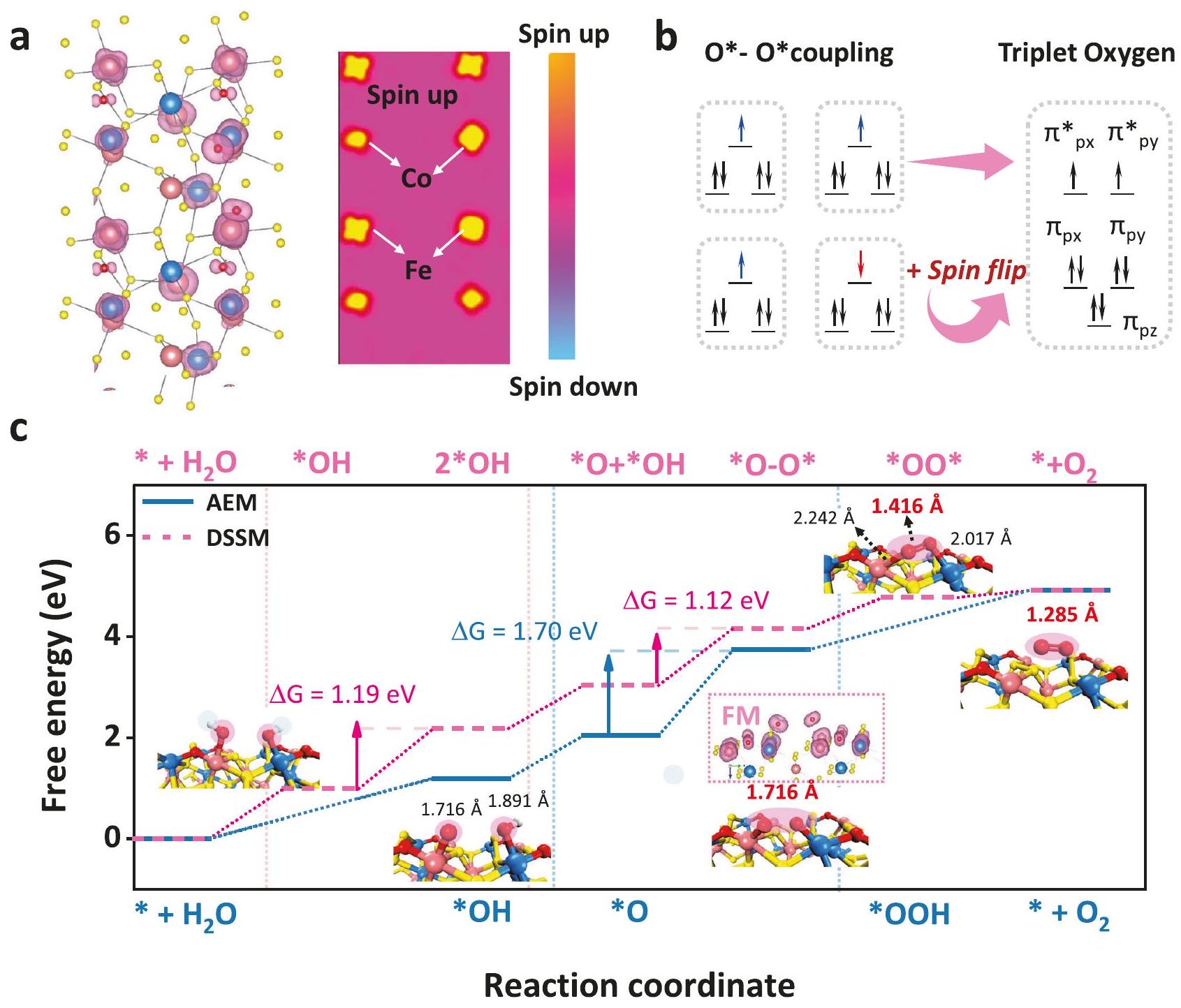
الحديد والفاناديوم خلال عملية OER وفقًا لنظرية ترتيب الروابط. تمثل المنطقة الحمراء تفاعلات اقتران الدوران المداري المعدنية الأكثر ملاءمة.المخطط التخطيطي لآليات تطور الممتزات في موقعين مزدوجين لارتباط O-O أثناء عملية OER (الأزرق لذرات الكوبالت، والأحمر لذرات الحديد، والأصفر لذرة الكبريت). أنظمة مختلفة، نظم زانغ وآخرون L.S. إلى M.S.تحويل حالة الدورانمع تقديملتعديل احتلال الحالة المضادة للرابطة-مدار الأكسجين. وبالمثل، قام وانغ وآخرون بتحسين نشاط تفاعل اختزال الأكسجين (ORR) من خلال تحسينامتصاصموقع بواسطة أكسيالرابطة. هيرين، المواقع (م.س. ) أظهر الأمثل الامتزاز، عندما المواقع (L.S., ) أظهرت امتصاصًا مهيمنًا لـ *OH خلال عملية OER. لذلك، فإن التفاعل الكهربائيتم ربط وسائط قاعدة لويس OH بشكل تفضيلي في مواقع الحديد وفقدت بذلك الإلكترونات لإنتاجالوسطاء. من التغيرات في حالة التكافؤ أو الدوران في مواقع مختلفة أثناء الأكسدة، توقعنا أن ذلك يمكّن من التمييز الوظيفي الفعال للأنواع النشطة في خطوات نقل الإلكترون المرتبط بالبروتون والتجزئة المتزامنة لتعزيز حركية تفاعل الأكسدة. من خلال دمج تمثيل تخطيطي لتفاعل الدوران-المدار للمعادن مع الوسطاء الأكسجينيين وتغير التكافؤ لموقع المعدن عند الجهد المطبق، يتم اقتراح آلية OPM مزدوجة الموقع من CoFe (أي، DSSM المذكورة أعلاه): (الشكل 4f): آلية تحفيز تآزري مزدوج الموقع بشكل جزئي تمكّن من القوة.سعة الامتزاز عند موقع الكوبالت والقوةسعة الامتزاز لموقع الحديد، مما سمح بالوجود المتزامن لـأنواع معوأدى ذلك إلى تفعيل مفتاح آلية اقتران O-O، مما حفز تشكيل الوسط الممتص Co-O-O-Fe.
محفز فيرومغناطيسي ذو نطاق واحدحفز وجود حقل كهربائي داخل المحفز وأدى إلى أكسدة الماء ذات الاستقطاب المغزلي إلى وسائط الأكسجين الرئيسية، مما ساهم في الاستقطاب الموازي لتقليل حاجز التكوين لـالمتوسطات المرتبطة وبالتالي سهلت إزالة الأكسجين الثلاثي بشكل مباشرلذلك، العلاقة التناسبية بين و الذي يقيد تطور امتصاص OER، كان تكسرت أثناء تجنب الاستقرار الهيكلي الضعيف الناتج عن آلية الربط التقليدية O-O.
حساب DFT
علاوة على ذلك، لتقييم ما إذا كانستوفر آلية الاقتران أفضل أداء لتفاعل تطور الأكسجين، وتم إجراء حسابات نظرية الكثافة. كمرجع، تم أيضًا النظر في آليات AEM النموذجية ونماذج كبريتيد الحديد (موقع واحد).كان المحلول الكهربائي الغني مصحوبًا بعملية استبدال طويلة للطور النشط، لذا اخترنا الطور الوسيط المستقر مؤقتًا.تم إنتاجه من خلال الاستبدال الجزئي لتحليل آلية OER. لذلك، تم محاكاة عملية OER باستخدام حسابات DFT الموجهة بالدوران من خلال تطبيق طريقة DFT +U لدراسة أنشطة OER لموقع CoFe الثنائي مع آليات مختلفة. توضح الأشكال S33-37 مسار 4e لآلية AEM النموذجية وآلية OPM المثلى مع CoFe كنوع نشط في الموقع الثنائي. علاوة على ذلك، توزيع التpolarization الدوراني المكاني لـتم حساب الموقع مع وسائط الأكسجين لتقييم تفاعلات تبادل الدوران للإلكترونات المتنقلة بالقرب منالمنطقة (الشكل S 38 والشكل 5a). النقل الانتقائي للإلكترونات من الحالة الخطية الفرديةأوكان الانتقال إلى حالة الأكسجين الثلاثي ضروريًا لضمان التشغيل السلس لعملية نقل 4 إلكترونات OER. ومع ذلك، فإن هذه عملية محظورة من حيث الدوران. بالنسبة للمحفزات الكهربية الفيرومغناطيسية، فإن تنافر الإلكترونات يكون صغيرًا عندما يتزاوج دوران الإلكترون القصير المدى مع الأكسجين الممتص (المتفاعل)، مما يؤدي إلى توصيل يعتمد على الدوران ويقلل من طاقة الرابطة المحددة للسرعة..
في هذه الأثناء، لتقييم تأثير حالات الدوران السطحية على طاقة امتصاص المواد الماصة في حسابات نظرية الكثافة، وخاصة فيما يتعلق بالمواد المغناطيسية، تم تقديم سطحي كوبالت-حديد للمقارنة: أحدهما مع لحظة دوران مميزة (لحظة دوران الحالة الأساسية) والآخر غير مستقطب للدوران، تم تحقيقه عن طريق ضبط الدوران على الصفر في
الشكل 5 | تحليل آلية OER استنادًا إلى حسابات DFT. أ خريطة كثافة الدوران وتوزيع مستوي للدوران القطبي للوسائط الرئيسية للأكسجينتمثل الأسطح العازلة ذات اللون الأرجواني الوردي حالات الدوران للأعلى. الترتيب المتوازي للإلكترونات المغزلية في اقتران الأكسجين-الأكسجين لمواقع المعادن المجاورة يسهل إنتاج الأكسجين الثلاثي. ج مخطط الطاقة الحرة من
آلية AEM النموذجية ومسار DSSM لتفاعل أكسدة الماء (OER) بما في ذلك جميع الوسائط الأكسجينية و تظهر الإضافات مخطط كثافة الدوران لـعلى نموذج CFS الفيرومغناطيسي نحو الانتقال من *O إلىكرات زرقاء لذرات الكوبالت، كرات وردية لذرات الحديد، كرات صفراء لذرات الكبريت، كرات حمراء لذرات الأكسجين وكرات بيضاء لذرات الهيدروجين. نظام وإعادة حساب ثابت الشبكة. كما هو موضح في الشكل S39، أظهرت الأسطح غير المقطبة للدوران طاقة امتصاص أقوى من تلك الخاصة بالأسطح المقطبة للدوران في الحالة الأساسية، خاصة في خطوة RDS. في الشكل 5a، الحالة الوسيطة الرئيسيةكشف عن الاقتران الفيرومغناطيسي بين هذين الموقعين المزدوجين من الكوبالت والحديد، مما يوحي بأن المحاذاة المتوازية للدوران للوسط الأكسجيني يمكن أن تسرع بشكل فعال خطوة الانفصال (إطلاق الحالة الثلاثية). كما هو موضح في الشكل 5ب، بسبب الاقتران المضاد للمغناطيسية، كانت هناك حاجة لتطبيق طاقة إضافية لتجاوز حاجز الطاقة الثنائي وتحفيز المحاذاة غير المتوازنة للدوران، مما يمكن أن يتسبب في تقلبات الدوران وتحقيق محاذاة نهائية متوازية للدوران لإطلاق حالة الأكسجين الثلاثية. في الوقت نفسه، فإن اتجاه الاقتران المغناطيسي.أظهرت المزيد من التفضيل لـالترابط بدلاً من القيود الخطية علىالأنواع وكفاءة إطلاق الأكسجين، وهي ميزة من ميزات آلية OPM. علاوة على ذلك، تُظهر الشكل 5c مخططات الطاقة الحرة لجيبس لطريقتين مختلفتين: آلية AEM النموذجية وآلية DSSM. كما هو موضح في الشكل 5c و S37، تم حساب الخطوة المحددة لمعدل التفاعل (RDS) لتكون تشكيل *OOH أو أوه-أوهالمتوسطات من خطوة * O لكل من CFS-ACs/CNT و، مما يدل على ارتباط أقوى لـ *O على موقع الحديد، والذي كان متسقًا مع العلاقة المعروفة. كانت حاجز RDS لطريق DSSM هو 1.12 eV، مما يظهر ميزة كبيرة تقريبًا مقارنةً بتلك الخاصة بطريق AEM. كما هو متوقع، أظهرت الذرتان O ترتيبًا متوازيًا للدوران في المواقع النشطة المرتبطة بالمغناطيسية، مما كان أكثر ملاءمة للتخلص لتشكيل حالة الثلاثي الدوران..
نقاش
باختصار، قمنا بتصنيع محفزات CFS-ACs صغيرة الحجم (1.5 ~3 نانومتر) مرتبطة بـ CNT بواسطة تقنية الهيدروحرارية منخفضة الحرارة، والتي أظهرت نشاطًا ممتازًا في تفاعل الأكسدة (OER) مع جهد زائد قدره 270 مللي فولت عندومتانة ملحوظة (633 ساعة دون فقدان محتمل) في 1 م KOH. وفقًا للتوصيف المغناطيسي وطيف ميسباور، طيف NEXAFS، حلقة هيسترسيس كبيرة، والتصلب، ومنحنى الفيرومغناطيسية المملوك يعود إلى CFS-ACs/CNT. لقد ميزنا للمرة الأولى الوظائف وحالة الدوران لمواقع النشاط المختلفة مع (ماجستير، ) بالإضافة إلى (ل.س، استنادًا إلى قياسات الامتصاص بالأشعة السينية في الموقع/أثناء التشغيل وطيف رامان، اقترحنا آلية تحفيز متآزرة جزئيًا تعتمد على تطور المعدن-الأكسجين. تفاعلات مدارية متوسطة بسبب التغيرات فيشغل الإلكترون المداري لموقع المعدن النشط خلال عملية OER، حيثآلية الاقتران تجنبت بنجاح قيود العلاقة التناسبية بين آلية امتصاص الأكسجين و . في هذه الأثناء، الأحجام الصغيرة ( يمكن اعتبار محفز CFS-ACs الفيرومغناطيسي بمثابة محفز فيرومغناطيسي أحادي المجال، مما حفز وجود مجال كهربائي داخل المحفز، وأدى إلى أكسدة الماء الموجهة بالسبين للأنواع الوسيطة الرئيسية من الأكسجين وولد محاذاة متوازية للسبين لتقليل حاجز التكوين.المتوسطات المرتبطة وبالتالي تسهيل إزالة الأكسجين الثلاثي مباشرة ). كشفت حسابات DFT أيضًا أن آلية اقتران O-O ممكنة وتتفوق على آلية الامتصاص ذات الموقع الواحد النموذجية. لم يحقق عملنا فقط تعزيزًا متزامنًا لتوليد وسائط اقتران O-O وإصدار، ولكنه يقدم أيضًا استراتيجية قابلة للتوجيه ورؤى فريدة حول تأثير تغييرات حالة الدوران على وسائط امتصاص الأكسجين أثناء عملية OER.
طرق
المواد الكيميائية
نترات الحديدنترات الكوبالتوزتم شراء ثياكetamيد (TAA) جميعها من شركة بكين إينوكاي للتكنولوجيا المحدودة. كبريتات البوتاسيوم ( ) وهيدروكسيد الصوديوم ( KOH ) تم شراؤها من شركة سينوفارم للكيماويات. أنبوب الكربون متعدد الجدران (CNT، في القطر وتم الحصول على (بالطول) من شركة بكين ديكي داوجين للتكنولوجيا المحدودة. بوروهيدريد الصوديومتم الاستحواذ عليها من قبل شركة تيانجين فنغتشوان لتكنولوجيا الكواشف الكيميائية المحدودة. تم تزويد رقائق النيكل (NF، 99.8%) من قبل شركة سوتشو شينجرنو للتكنولوجيا المحدودة، ونافيو.تم الحصول على ) من سيغما-ألدريتش. تم استخدام جميع المواد الكيميائية كما هي دون أي تنقية إضافية. الـتم الحصول على المحفز من شركة جونسون ماثي. كانت المياه المنزوعة الأيونات المستخدمة في جميع التجارب فائقة النقاء.
تركيب CNT (CFO-p/CNT)
أولاً، تم تفريق 70 ملغ من CNT في 50 مل من الإيثانول مع الموجات فوق الصوتية المستمرة لمدة 30 دقيقة لتكوين المحلول A. ثانياً، 2 مليمول و تم إذابتها في 20 مل من الإيثانول تحت تحريك مغناطيسي لمدة 10 دقائق لتكوين المحلول B. تم إضافة المحلول B ببطء قطرة قطرة إلى المحلول A مع التحريك المستمر لمدة 10 ساعات، والذي يسمى المحلول C. بعد ذلك، تم إضافة 10 مل من 1.25 مليمول منتمت الإضافة ببطء قطرة قطرة إلى المحلول C في بيئة من الماء المثلج. أخيرًا، تم غسل المحلول بواسطة الطرد المركزي باستخدام الإيثانول البارد والماء النقي، ثم تم تجفيفه بالتجميد للحصول على سلفيد CFO-p/ CNT. بالمثل، و تم تحضير السلف باستخدام نفس العملية دون إضافة مصدر الكوبالت ومصدر الحديد، على التوالي.
تركيب CFS-ACs/CNT
تم تفريق 80 ملغ من سلفيد الكربون المحضر مسبقًا CFO-p/CNT في 40 مل من الماء المقطر تحت التحريك بقوة لمدة 30 دقيقة. ثم، تم إضافة 25 مل من 40 مليمول من ثيوأسيتيمايد بالتنقيط مع التحريك لمدة 30 دقيقة. تم نقل المحلول الناتج إلى وعاء أوتوكلاف من الفولاذ المقاوم للصدأ مبطن بتفلون سعة 100 مل، تلاه تسخين عندلمدة 6 ساعات. أخيرًا، تم الحصول على CFS-ACs/CNT النهائي عن طريق الغسل والتجفيف بالتجميد. بالمثل، و تم تصنيعها بنفس كمية الثيوأسيتيمايد. في هذه الأثناء، CNT، و تم إعدادها بمقادير مختلفة منو 1 مليمول و ; 10 مليمول ثيوأسيتيمايد و 50 مليمول ثيوأسيتيمايد.
توصيفات
تمت دراسة مكون جميع العينات المعدة بواسطة أنماط حيود الأشعة السينية (XRD)، التي تم جمعها بواسطة جهاز Y-2000 للأشعة السينية.
مقياس الانكسار باستخدام النحاسإشعاع ) عند 40 كيلوفولت، 40 مللي أمبير. تم إثبات هيكل العينات بواسطة مطيافية الأشعة السينية للأشعة الكهروضوئية (XPS)، التي تم إجراؤها باستخدام مطياف ثيرمو ساينتيفيك K-Alpha باستخدام شعاع أشعة سينية أحادي اللون مركّز من نوع Al Ka (1486.6 eV) بقطر تم جمع أطياف رامان باستخدام جهاز HORIBA Scientific LabRAM HR Evolution مع مصدر ليزر بقدرة 532 نانومتر. تم توصيف شكل العينات باستخدام مجاهر الإلكترون الناقل (TEM، FEI Tecnai G220) بجهد تسريع يبلغ 200 كيلو فولت ومجهر إلكتروني مسح ميداني (FE-SEM، JEOL JSM-6700F). تم الحصول على صور HAADF-STEM من جهاز JEOL JEM-ARM200F بجهد تسريع يبلغ 200 كيلو فولت. تم دراسة هيكل الامتصاص القريب من حافة الأشعة السينية (XANES) لحافة الحديد (Fe K-edge) وحافة الكوبالت (Co K-edge) وهيكل الامتصاص الدقيق الممتد (EXAFS) في خط شعاع BL14W1 في منشأة الإشعاع المتزامن في شنغهاي (SSRF) في وضع الفلورة باستخدام مقياس الطيف الثنائي البلوري الثابت Si (111). تم معالجة بيانات XAFS التي تم الحصول عليها في برنامج Athena (الإصدار 0.9.26) لتصحيح الخلفية، وخط ما قبل الحافة، وخط ما بعد الحافة. ثم تم إجراء تحويل فورييه في برنامج Artemis (الإصدار 0.9.26). تم استخدام وزن k3،-نطاق و نطاق منتم استخدامها لتركيب الألواح؛ نطاق k لـونطاق R منتم استخدامها لتجهيز عينات الحديد والكوبالت. المعلمات الأربعة، عدد التنسيق، طول الرابطة، عامل ديباي-والر، وانزياح EO (CN، R، ) تم تركيبها دون أن يتم تثبيت أو تقييد أو ربط أي شخص. بالنسبة لتحليل تحويل المويجات، تم استيراد البيانات المصدرة من أثينا إلى كود فورتران في حما. تم سرد المعلمات كما يلي: نطاق R،نطاق kلعينات الحديد والكوبالت؛ وزن k، 2؛ ودالة مورلي معتم استخدامه كالموجة الأم لتوفير التوزيع العام.تم الحصول على طيف ميسباور للمواد الحفازة باستخدام مقياس طيف Topologic 500 A مدفوعًا بعداد متناسب عند درجة حرارة الغرفة. تم مراقبة شعاع الأشعة السينية الساقط بواسطة غرفة تأين مملوءة بـ، وتم استخدام كاشف من نوع ليتل مملوء بالآر في كشف الفلورسنس بالأشعة السينية. ثم تم طرح الخلفية من بيانات EXAFS الخام، وتطبيعها وتحويلها فورييه باستخدام إجراءات قياسية مع حزمة IFEFFIT.
قياسات XAS المعتمدة على السنكروترون
تم قياس طيف امتصاص الأشعة السينية عند حواف الحديد والكوبالت (XAS) في خطوط الأشعة MCD-A وMCD-B (خط الأشعة سو تشو للطاقة المواد) في المختبر الوطني للإشعاع المتزامن (NSRL، الصين). خلال القياس، تم وضع العينات في درجة حرارة الغرفة. تم قياس كل من حواف الحديد والكوبالت باستخدام عائد الإلكترون الكلي (TEY)، حيث تم قياس تيار التصريف كدالة لطاقة الفوتون. تم قياس عدة مسحات ومتوسطها.
هيكل الامتصاص الدقيق للأشعة السينية في الموقع (XAFS)
أولاً، المعالجة المسبقة كما يلي: تم معالجة الأقطاب الكهربائية العاملة مسبقًا بواسطة دورات فولتامترية بين 0.2 و 0.8 فولت بمعدلفي-محلول مائي مشبع 1 م من هيدروكسيد البوتاسيوم. تم تطبيق المحفز بواسطة فرشاة يدوية على ورق الكربون (تم اختيار 1.245 فولت مقابل RHE و1.445 فولت مقابل RHE كحالتين تمثيليتين، وتم جمع إشارات الامتصاص بالأشعة السينية في الموقع عندما وصل القطب العامل إلى حالة مستقرة. تم الحفاظ على القطب تحت كل حالة لمدة 30 دقيقة قبل قياس الطيف. تم جمع طيف XANES وEXAFS لحدود الحديد K في مصدر الفوتون في تايوان 44 A وBW14 من SSRF. تم استخدام رقائق الحديد لمعايرة طاقة خط الشعاع ومقارنة العينات. تم تنفيذ الكشف في وضع الفلورية باستخدام مقياس الطيف البلوري المزدوج Si (111). تم استخدام كاشف غرفة الأيونات للعينات والعائد الكلي للإلكترونات لقياس العينات ذات التركيزات العالية، مثل المراجع.
رامان في الموقع
تم استخدام خلية كيميائية كهربائية مخصصة ذات حجرة مزدوجة وثلاثة أقطاب مع نافذة من الكوارتز لقياسات رامان في الموقع.. تم إسقاط تعليق CFS على القطب الكربوني الزجاجي كقطب عمل. تم استخدام قضيب جرافيت كقطب مضاد.إلكترود ) تم استخدامه كإلكترود مرجعي. تم استخدام جهاز قياس الجهد Gamry Reference 600+ للتحكم في جهد الإلكترود. قبل قياس رامان، تم تلميع الإلكترود الكربوني الزجاجي بواسطة تعليق الألومينا بقطر 0.05 ميكرومتر. تم إجراء قياسات طيف رامان باستخدام مجهر LabRAM HR Evolution (هوربا جوبين يفون) المزود بالليزر ب wavelength 532 نانومتر،هدف ( )، وكاشف CCD. تم معايرة تردد رامان باستخدام شريحة سيليكون قبل كل تجربة. تم ضبط الفلتر ليكون للحفاظ على شدة الليزر منخفضة لتجنب أي تعديلات ناتجة عن الإشعاع على سطح المضاف العضوي المودع. تم ضبط وقت الاكتساب على 40 ثانية لكل طيف.
تفاصيل حساب DFT
تم إجراء جميع حسابات DFT باستخدام حزمة المحاكاة الأولية فيينا (VASP). تم إجراء حسابات DFT الموجهة نحو الدوران باستخدام تقريب التدرج العام ودالة بيردو-بورك-إرنزرهوف (PBE).كما هو مُنفذ في حزمة المحاكاة فيينا من البدايةطريقة الموجة المعززة بواسطة البروجيكتورتم استخدامه لعلاج تفاعلات الأيونات والإلكترونات. تم تحديد طاقة القطع عند 450 إلكترون فولت وكانت درجة تحمل المجال الذاتي المتسق (SCF) تم تصحيح لحظة ثنائي القطب وفقًا لذلك فيالاتجاه، ونظام التصحيح التجريبي لجريميتم أيضًا إجراء ذلك، حيث تم تضمين تأثير تفاعلات فان دير فال بشكل صريح. لوصف التفاعل المحلي القوي من و تم استخدام دالة تبادل-ترابط PBEsol مع معامل هوبارد الفعال ضمن نهج دوداريف بقيمة 3 إلكترون فولت للحديد و3.3 إلكترون فولت للكوبالت، مأخوذة من المرجع.تم اختيار السطح مع التكرار فيخلية وحدة وعرض فراغتم استخدامه. تم تثبيت الطبقتين السفليتين، بينما تم السماح للطبقة العليا والمواد الممتصة بالاسترخاء. 3٣تم أخذ عينة من نقاط k لشبكة Monkhorst-Pack بمقدار 1 لجميع حسابات السطح. ولا تتجاوز جميع فروق الطاقة لجميع المستويات 0.02 إلكترون فولت. تم استخدام حزمة VASPKIT لمعالجة نتائج الحسابات بعد الانتهاء..
تم حساب الطاقة الحرة لجيبس بواسطة DFT استنادًا إلى نموذج القطب الهيدروجيني الحاسوبي (CHE) المقدم من نørسكوف.، حيث تكون الطاقة الكلية لـيساوي في الظروف القياسية ( ). الطاقة الحرة لـ يتم حسابه بواسطة
فيما يتعلق بكل خطوة أساسية، يتم حساب تغير الطاقة الحرة لجيبس من خلال المعادلة (2):
أين هو إجمالي الطاقة المستخرجة في نظرية الكثافة، ZPE هو طاقة النقطة الصفرية، هي درجة الحرارة ( 298.15 كيلفن )، و هو الإنتروبيا. بالتفصيل، يتم حساب الطاقة الحركية الصفرية للمواد الممتصة بواسطة المعادلة (3):
قمنا بتحديد إنتروبيا جزيئات الطور الغازي من دليل CRCيتم حساب إنتروبيا أنواع الامتصاص بواسطة المعادلة (4):
أين و هي ثابت بلانك، سرعة الضوء وتردد الاهتزاز على التوالي. يتم حساب المساهمة من الإنتروبيا TS بواسطة المعادلة (5) باستخدام تقريب المذبذب التوافقي كالتالي:
أين، و ثابت بلانك وتردد الاهتزاز على التوالي.
وبناءً عليه، تم حساب تغييرات الطاقة الحرة لجيبس لخطوات أكسدة الماء باستخدام آلية AEM باستخدام المعادلات التالية:
طاقة امتصاص الوسائطيمكن استخدام طاقة الحالة الأساسية المحسوبة بواسطة DFT لـ M-OH و M-O و M-OOH و M-OO كالتالي:
علاوة على ذلك، تم حساب تغييرات الطاقة الحرة لجيبس لخطوات أكسدة الماء باستخدام آلية OPM باستخدام المعادلات التالية:
طاقة امتصاص الوسائطيمكن استخدام طاقة الحالة الأساسية المحسوبة بواسطة DFT لـ M-OH و M-O و M-OO-M كالتالي:
القياسات الكهروكيميائية
عادةً، تم إجراء جميع الاختبارات الكهروكيميائية في نظام ثلاثي الأقطاب في محطة كهروكيميائية (محطة CHI الكهروكيميائية (النموذج 760 E)) في درجة حرارة الغرفة. (في 3.0 م كCl) تم استخدامه كإلكترود مرجعي، وقضيب كربوني كإلكترود مضاد. أولاً، تم تحضير حبر المحفز عن طريق تشتت 5 ملغ من المحفز في 1 مل من الماء/الإيزوبروبانول (مذيب يحتوي علىنافيوين وتمت الموجات فوق الصوتية لمدة 30 دقيقة لتشكيل محلول متجانس. ثم تم تحميل 600 ميكرولتر من المحلول المتجانس على NF نظيف. )، تم تجفيفه بالتفريغ واستخدامه كقطب عمل. كانت الحمولة النهائية لجميع المحفزات على NF حوالي خلال عملية قياس تفاعل تطور الأكسجين (OER)، تم اختبار الفولتمترية المسحية الخطية (LSV) في 1.0 M KOH بمعدل مسحيتم تحضير المحلول الكهربائي حديثًا لكل قياس كيميائي كهربائي. في هذه الأثناء، يتم إجراء جميع الاختبارات الكيميائية الكهربائية في خلية تيفلون كهربائية من نوع H. جميع الجهود مرجعية إلى القطب الهيدروجيني القابل للعكس (RHE) بواسطة معادلة نيرنست:
وتم حساب الجهد الزائد لتفاعل الأكسدة (OER) وفقًا للمعادلة:
تم الحصول على طيف الامتزاز الكهربائي الكيميائي (EIS) عند جهد الدائرة المفتوحة منتم إجراء استقرار العينات على المدى الطويل باستخدام طريقة الكرونو بوتنشيومترية مع كثافة تيار مستقرة منلتحديد المساحة السطحية الكهروكيميائية (ECSA) يمكن الحصول عليها من المعادلة التالية: ECSAهو السعة النوعية في 1 م كوه ) و هو سعة الطبقة المزدوجة الكهروكيميائية المقاسة باستخدام منحنيات الفولتمترية الدورية (CV، تم اختبارها فييتراوح بينتم تفعيل السيرة الذاتية قبل كل اختبار كيميائي كهربائي.
توفر البيانات
البيانات التي تدعم الرسوم البيانية متاحة ضمن هذه الورقة ومعلوماتها التكميلية. جميع البيانات الأخرى ذات الصلة التي تدعم نتائج هذه الدراسة متاحة من المؤلفين المقابلين عند الطلب المعقول. البيانات الخاصة بالأشكال 2-5، والأشكال التكميلية 1، 5-7، 8-13، 15-32 و37 التي تم توليدها في هذه الدراسة متوفرة في ملفات البيانات المصدرية. يتم توفير بيانات المصدر مع هذه الورقة.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294-303 (2012).
Montoya, J. H. et al. Materials for solar fuels and chemicals. Nat. Mater. 16, 70-81 (2017).
Görlin, M. et al. Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of oxide water splitting electrocatalysts. J. Am. Chem. Soc. 138, 5603-5614 (2016).
Suntivich, J. et al. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science. 334, 1383-1385 (2011).
Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: toward boosted and sustained water oxidation catalysis. Joule. 5, 2164-2176 (2021).
Xiao, K., Wang, Y., Wu, P., Hou, L. & Liu, Z.-Q. Activating lattice oxygen in spinel through filling oxygen vacancies with fluorine for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 62, e202301408 (2023).
Xu , J. et al. with lattice water-assisted oxygen exchange for high-performance proton exchange membrane water electrolyzers. Sci. Adv. 9, eadh1718 (2023).
Grimaud, A. et al. Activation of surface oxygen sites on an iridiumbased model catalyst for the oxygen evolution reaction. Nat. Energy. 2, 17002 (2017).
Chen, F.-Y. et al. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule. 5, 1704-1731 (2021).
Huang, Z.-F. et al. Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat. Commun. 12, 3992-4000 (2021).
Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy. 4, 329-338 (2019).
Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829-7836 (2023).
. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021).
Ren, X. et al. Spin-polarized oxygen evolution reaction under magnetic field. Nat. Commun. 12, 2608-2620 (2021).
Garcés-Pineda, F. A. et al. Direct magnetic enhancement of electrocatalytic water oxidation in alkaline media. Nat. Energy. 4, 519-525 (2019).
Yan, J. et al. Direct magnetic reinforcement of electrocatalytic ORR/ OER with electromagnetic induction of magnetic catalysts. Adv. Mater. 33, 2007525 (2021).
Zhuang, L. et al. Sulfur-modified oxygen vacancies in iron-cobalt oxide nanosheets: enabling extremely high activity of the oxygen evolution reaction to achieve the industrial water splitting benchmark. Angew. Chem. Int. Ed. 59, 14664-14670 (2020).
Li, X. et al. Adaptive bifunctional electrocatalyst of amorphous CoFe Oxide @ 2D black phosphorus for overall water splitting. Angew. Chem. Int. Ed. 59, 21106-21113 (2020).
Ge, J. et al. Multi-domain versus single-domain: a magnetic field is not a must for promoting spin-polarized water oxidation. Angew. Chem. Int. Ed. 62, e202301721 (2023).
Sun, T. et al. Ferromagnetic single-atom spin catalyst for boosting water splitting. Nat. Nanotechnol. 18, 763-771 (2023).
Deng, J. et al. Multiscale structural and electronic control of molybdenum disulfide foam for highly efficient hydrogen production. Nat. Commun. 8, 14430-14437 (2017).
Jiang, S. et al. Insight into the catalytic aactivity of amorphous multimetallic catalysts under a magnetic field toward the oxygen evolution reaction. ACS Appl. Mater. Inter. 14, 10227-10236 (2022).
Wu, T. et al. Iron-facilitated dynamic active-site generation on spinel with self-termination of surface reconstruction for water oxidation. Nat. Catal. 2, 763-772 (2019).
Zhang, S. et al. In situ embedding into nitrogen and sulfur codoped hollow porous carbon as a bifunctional electrocatalyst for oxygen reduction and hydrogen evolution reactions. Appl. Catal. B: Environ. 254, 186-193 (2019).
Zhang, J. et al. Tuning the coordination environment in single-atom catalysts to achieve highly efficient oxygen reduction reactions. J. Am. Chem. Soc. 141, 20118-20126 (2019).
Peng, W. et al. Deciphering the dynamic structure evolution of Feand Ni-Codoped CoS for enhanced water oxidation. ACS Catal. 12, 3743-3751 (2022).
Li, C. et al. Tailor-made open porous 2D CoFe/SN-carbon with slightly weakened adsorption strength of ORR/OER intermediates as remarkable electrocatalysts toward zinc-air batteries. Appl. Catal. B: Environ. 269, 118771 (2020).
Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped single-atom catalyst for enhanced electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022).
Zhuang, Z. et al. Reversely trapping atoms from a perovskite surface for high-performance and durable fuel cell cathodes. Nat. Catal. 5, 300-310 (2022).
Wan, X. & Shui, J. Exploring durable single-atom catalysts for proton exchange membrane fuel cells. ACS Energy Lett. 7, 1696-1705 (2022).
Li, Y. et al. Local spin-state tuning of iron single-atom electrocatalyst by S-coordinated doping for kinetics-boosted ammonia synthesis. Adv. Mater. 34, 2202240 (2022).
Wu, T. et al. Spin pinning effect to reconstructed oxyhydroxide layer on ferromagnetic oxides for enhanced water oxidation. Nat. Commun. 12, 3634-3645 (2021).
Zhuang, Z. et al. Atomically dispersed nonmagnetic electron traps improve oxygen reduction activity of perovskite oxides. Energy Environ. Sci. 14, 1016-1028 (2021).
Wang, X. et al. Engineering 3d-2p-4f gradient orbital coupling to enhance electrocatalytic oxygen reduction. Adv. Mater. 34, 2206540 (2022).
Mehmood, A. et al. High loading of single atomic iron sites in Fe-N-C oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 5, 311-323 (2022).
Kroll, T. et al. Transition metal phthalocyanines: insight into the electronic structure from soft X-ray spectroscopy. J. Chem. Phys. 137, 054306 (2012).
Lee, W. H. et al. Electrode reconstruction strategy for oxygen evolution reaction: maintaining phase with intermediate-spin state during electrolysis. Nat. Commun. 13, 605 (2022).
Ding, J. et al. Atomic high-spin cobalt(II) center for highly selective electrochemical CO reduction to . Nat. Commun. 14, 6550 (2023).
Liu, D. et al. A template editing strategy to create interlayerconfined active species for efficient and durable oxygen evolution reaction. Adv. Mater. 35, 2203420 (2023).
Xiao, Z. et al. Operando identification of the dynamic behavior of oxygen vacancy-rich for oxygen evolution reaction. J. Am. Chem. Soc. 142, 12087-12095 (2020).
Lei, Z. et al. Coordination modulation of iridium single-atom catalyst maximizing water oxidation activity. Nat. Commun. 13, 24-34 (2022).
Wang, C. et al. Identification of the origin for reconstructed active sites on oxyhydroxide for oxygen evolution reaction. Adv. Mater. 35, 2209307 (2022).
Chen, X., Shi, T., Zhong, K., Wu, G. & Lu, Y. Capacitive behavior of decorated with @carbon nanospheres. Chem. Eng. J. 379, 122240 (2020).
Hu, Y. et al. Interfacial evolution on Co-based oxygen evolution reaction electrocatalysts probed by using in situ surface-enhanced raman spectroscopy. Anal. Chem. 95, 1703-1709 (2023).
Zhang, Z. et al. Regulating spin states in oxygen electrocatalysis. Angew. Chem. Int. Ed. 62, e202216837 (2023).
Yang, G. et al. Regulating Fe-spin state by atomically dispersed MnN in Fe-N-C catalysts with high oxygen reduction activity. Nat. Commun. 12, 1734-1744 (2021).
Liu, Y. et al. Tuning the spin state of the iron center by bridgebonded ligands for enhanced oxygen reduction. Angew. Chem. Int. Ed. 61, e202117617 (2022).
Yang, H. et al. Intramolecular hydroxyl nucleophilic attack pathway by a polymeric water oxidation catalyst with single cobalt sites. Nat. Catal. 5, 414-429 (2022).
Gao, W. et al. Electron spin polarization-enhanced photoinduced charge separation in ferromagnetic . ACS Energy Lett. 6, 2129-2137 (2021).
Cao, A. & Nørskov, J. K. Spin effects in chemisorption and catalysis. ACS Catal. 13, 3456-3462 (2023).
Zhao, K. et al. Enhancing hydrogen oxidation and evolution kinetics by tuning the interfacial hydrogen-bonding environment on functionalized platinum surfaces. Angew. Chem. Int. Ed. 61, e202207197 (2022).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558-561 (1993).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787-1799 (2006).
Csonka, G. I. et al. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 79, 155107 (2009).
Wang, V. et al. VASPKIT: A user-friendly interface facilitating highthroughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886-17892 (2004).
Lide, D. R. CRC Handbook of Chemistry and Physics 89th edn (CRC Press, 2009).
Campbell, C. T. & Sellers, J. R. V. The entropies of adsorbed. Mol. J. Am. Chem. Soc. 134, 18109-18115 (2012).
شكر وتقدير
تم دعم هذا العمل ماليًا من قبل المشاريع الرئيسية لمؤسسة العلوم الطبيعية الوطنية في الصين (U22A20107، J.N.Z.)، والمشاريع الرئيسية لصندوق البحث والتطوير التكنولوجي المشترك لبرنامج العلوم والتكنولوجيا في مقاطعة هنان (222301420001، J.N.Z.)، وفريق الابتكار للعلماء الشباب المتميزين في جامعة تشنغتشو (32320275، J.N.Z.)، ومشروع البحث والممارسة لإصلاح التعليم العالي في مقاطعة هنان (2021SJGLX093Y، J.N.Z.). نشكر مرفق الإشعاع المتزامن في بكين (BSRF) محطة 1W1B لقياسات XAS. لي-رونغ تشنغ ممتن للدعم المقدم لقياسات XAS. نشكر مجموعة ب.-ج. شو من كلية الكيمياء والهندسة الجزيئية، جامعة بكين، على اختبار الطيف رامان في الموقع. نشكر خطوط الشعاع MCD-A وMCD-B (خط شعاع سوتشو لمواد الطاقة) في المختبر الوطني للإشعاع المتزامن (NSRL، الصين).
مساهمات المؤلفين
قام ج.ن.ز. بالإشراف على هذا البحث. وصمم س.ر.إكس. المشروع وأعد العمل، وقام س.ر.إكس. و م.ل.ل. بتنفيذ التجارب، بما في ذلك تخليق المواد، والتوصيفات، والقياسات الكهروكيميائية. وقام ك.ي.ز. و ب.ج.إكس. بتنفيذ التوصيفات الرامانية في الموقع. وقدمت س.هـ.ف. و ج.و. قياسات XAS المعتمدة على السنكروترون. قدم ي.ي. الدعم الحسابي النظري، وقام د.ب.إكس. و ج.ن.ز. بمراجعة وتحرير المخطوطة. وقد وافق جميع المؤلفين على النسخة النهائية من المخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى جيا-نان تشانغ.
معلومات مراجعة الأقران تشكر مجلة Nature Communications يانغ هو، هيونغ-سوك أوه والمراجعين المجهولين الآخرين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
معلومات إعادة الطباعة والتصاريح متاحة على http://www.nature.com/reprints ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
المختبر الرئيسي لتحضير المواد الوظيفية والعوامل الحفازة للطاقة المتقدمة، كلية علوم المواد والهندسة، جامعة تشنغتشو، تشنغتشو 450001، الصين.مختبر الإشعاع السنكروتروني الوطني، جامعة العلوم والتكنولوجيا في الصين، هيفي 230026 آنهوي، الصين.كلية الكيمياء والهندسة الجزيئية، جامعة بكين، بكين، الصين.المختبر الوطني الرئيسي لموارد الفحم الكوك الأخضر، تشنغتشو 450001، الصين.ساهم هؤلاء المؤلفون بالتساوي: سيران شو وسهوا فنغ.
Efficient oxygen evolution reaction electrocatalysts are essential for sustainable clean energy conversion. However, catalytic materials followed the conventional adsorbate evolution mechanism (AEM) with the inherent scaling relationship between key oxygen intermediates *OOH and *OH, or the lattice-oxygen-mediated mechanism (LOM) with the possible lattice oxygen migration and structural reconstruction, which are not favorable to the balance between high activity and stability. Herein, we propose an unconventional CoFe dual-site segmentally synergistic mechanism (DSSM) for single-domain ferromagnetic catalyst nanoclusters on carbon nanotubes (CNT) (CFS ), which can effectively break the scaling relationship without sacrificing stability. (L.S, ) supplies the strongest adsorption energy, while (M.S, ) exposes strong adsorption. These dual-sites synergistically produce of intermediates, thereby accelerating the release of triplet-state oxygen ( ). As predicted, the prepared CFSACs/CNT catalyst exhibits less overpotential than that of commercial IrO , as well as approximately 633 h of stability without significant potential loss.
The development of excellent electrocatalysts with high activity and long-term durability remains challenging due to destruction of the catalyst surface by the strong oxidizing environment during the oxygen evolution reaction (OER) process and its complex multi-step proton-coupling reaction . The guiding principles for enhancing electrochemical performance are mainly based on the following strategies: (1) According to the adsorption evolution mechanism (AEM), OER activity is mainly influenced by the adsorption energetics of the oxygen intermediates ( and ) according to the Sabatier principle . However, there is an inherent linear scaling relation (LSR) between the adsorption energies of reaction intermediates *OOH and *OH, thus rendering a minimum theoretical overpotential as high as even for the best possible material. (2) Compared with the conventional AEM, the lattice-oxygen-mediated mechanism (LOM)
overcomes the LSR constraint by triggering O-O coupling, which can decrease the energy barrier to the . Nevertheless, in this mechanism, lattice oxygen is often required to be involved in O-O coupling, and accelerating the release of triplet-state oxygen. Additionally, the many oxygen vacancies lead to surface migration of the lattice oxygen, severe structural reconstruction and insufficient stability . Therefore, catalysts based on LOM are not considered competitive with those based on AEM in terms of stability and feasibility for practical applications, despite their higher catalytic efficiency .
Alternatively, the oxide path mechanism (OPM) for homogeneous catalysts is more worthy of further OER performance improvements, since it allows direct O-O radical coupling without oxygen vacancy defects or additional reaction intermediates (*OOH). The O-O coupling
mechanism require the presence of only and intermediates during the OER process, which includes two types: (1) The O-O coupling generated by the lattice oxygen of the intrinsic catalyst and the solvent oxygen together (LOM); (2) The O-O coupling supplied by the solvent only (OPM). However, the LOM mechanism has complex trigger conditions, such as activation of lattice oxygen: The energy of the O orbital must be moved up near the Fermi level to enable overlap with the central -orbital of the metal (i.e., enhancing the covalency of the bond), thus spanning the redox energy of the lattice oxygen . Meanwhile, it is necessary to construct numerous holes to destabilize lattice oxygen atoms and enable the release of . Compared with LOM, OPM process, regarded as an ideal pathway, allows a direct coupling. In this process, is dissociated through the synergistic action of active metal sites, triggering radical coupling and driving O-O coupling. However, this mechanism requires symmetric bimetallic positions with appropriate atomic distances to allow O-O radical coupling with low energy potential barriers . However, OPM mechanism is less reported in alkaline environments, so exploring catalysts with appropriate polymetallic sites following the OPM mechanism in alkaline environments remains challenging. However, OPM mechanism is less reported in alkaline environments, so exploring catalysts with appropriate polymetallic sites following the OPM mechanism in alkaline environments remains challenging.
With the development of excellent catalysts from abundant metals, external magnetic field disturbances affected the spincatalyzed reaction kinetics and internal intrinsic spin diversity, allowing reaction pathways from spin-forbidden species (*OH, *OOH and *O to ) to spin-allowed species and accelerating the release of spinparallel arrangement of triplet-state oxygen . Therefore, the construction of ferromagnetic catalysts is a prerequisite for achieving fast spin-catalyzed reaction kinetics. Among OER reaction, as typical ferromagnetic materials, show a leading edge with high OER activity in alkaline environments . For catalytic reactions involving proton-coupled electron transfer (such as OER), the high surface energies of heterogeneous dual-atom catalysts and the low atomic utilization of crystalline bulk catalysts make it difficult to balance OER activity and stability. It is reasonable to infer that if the atomic size of the Co-Fe-based ferromagnetic catalyst was controlled to a single-domain region ( even clusters), spin-polarized OER would occur in the absence of a magnetic field . Therefore, accelerating the release of parallel spin alignment triplet state oxygen by built-in electric field-induced localized ferromagnetic coupling is an effective pathway for superior OER catalysts . Regarding this, the key intermediates ( ) followed by ferromagnetic coupling for a comprehensive understanding of the O-O direct coupling under the OPM mechanism provides a new perspective, and might be regarded as ultimately solving the primary stability and activity bottleneck of catalysts based on LOM or AEM.
In order to analyze the O-O coupling mechanism that can overcome the LSR and balance the high activity and strong stability, we chose ferromagnetic dual-site clusters supported on carbon nanotube CNT material as a platform to catalyze OER and elucidate the intrinsic relationship between the OER activity and the spin state of each metal site. Meanwhile, we suspected that focusing on the preferential adsorption of key oxygen intermediates on metals offers a promising research perspective for deepening understanding direct O-O coupling. The evolution of Fe (II) and Fe (III) spin states during the OER process is also considered and discussed.
Results
Rational design and synthesis
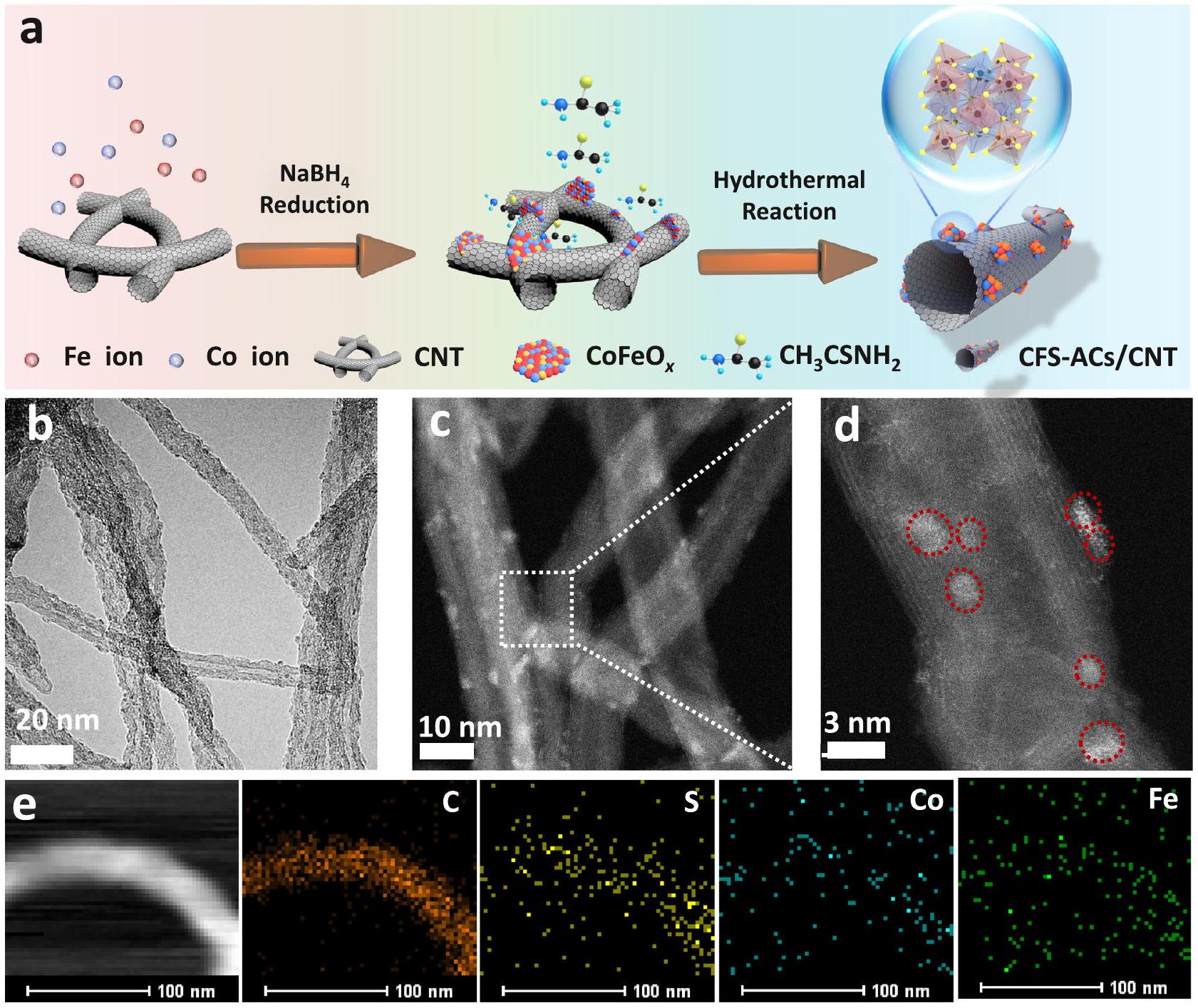
The nanoclusters supported on nanotubes (CFS-ACs/CNT) were synthesized via a facile adsorption reduction-hydrothermal method. First, amorphous nanosheets were prepared and supported onto pre-cleaned CNTs (CFO-p/CNT) via a NaBH4 reduction
process , and strong interactions between metal precursors and rich oxygen functionalities on CNTs. Subsequently, nanoclusters were fabricated and maintained through the operable desired acidic environment generated by the decomposition of thioacetamide during the hydrothermal process (Fig. 1a).
The chemical composition, morphology and chemical state of the final product were examined by X-ray powder diffraction (XRD) and transmission electron microscopy (TEM). The XRD patterns of CFSACs/CNT and CFO-p/CNT (Fig. S1) showed no obvious diffraction peaks belonging to -based hybrids, which implied an amorphous structure because XRD is only sensitive to the crystalline materials . Long-range disordered amorphous materials with a few atom exhibiting short-range order atoms tend to be endowed with more active sites. A broader range of chemical compositions, and greater structural flexibility than those of crystalline materials, and this leads to superior electrocatalytic activity . Typically, TEM images of pure show the homogeneous nanosheets morphology (Fig. S2), and distinct CNTs with diameters of are also observed in Fig. S3 with clear and complete lattice stripes. After reduction, the TEM images (Fig. S4) showed that CFO-p/CNT retained the one-dimensional morphology of the CNTs, on which small nanosheets were uniformly decorated. Compared with the TEM image of the pure CNTs, the lattice stripes of the CNTs in the CFO-p/CNT catalyst show obvious distortions and fracture with due to embedding of the nanosheets (shown by yellow circles), which confirmed the successful introduction of species. With the introduction of sulfur source, the nanosheets were broken up and reconstituted into smaller CFS-ACs on the surfaces of the CNTs (Fig. 1b, c). In addition, atomic resolution high-angle annular dark-field scanning TEM (HAADF-STEM) was performed to investigate the distribution of CFS-ACs at the atomic scale. As shown in Fig. 1d, numerous welldispersed CFS-ACs (marked by red circles) were present on CNTs support. The elemental maps further exhibited a uniform distributions of different atoms ( ) into (Fig. 1e). The Co ( ) and contents of CFS-ACs/CNT were determined with inductively coupled plasma-optical emission spectroscopy (ICP-OES), which was used as a standard for subsequent studies.
The X-ray photoelectron spectroscopy (XPS) was provided to further validate the coordination environment and chemical states of the all-prepared samples (Figs. S5-S9). In Fig. S5a, the Co XPS spectra of CFO-p/CNT and CFS-ACs/CNT showed two major Co and Co spin-orbit splitting. The Co peaks for CFS-ACs/CNT located at 779.9 and were assigned to and respectively. Meanwhile, compared with the precursor, species of CFS-ACs/CNT exhibited positive peak shifts, suggesting a higher valence state. For the well-dispersed product, the presence of and the peripheral electron transfer of Co and Fe due to the introduction of S , further enhanced the OER activity . After sulfidation, the metal oxidation state changed, while the whole spectrum was positively shifted (Fig. S6). The S XPS spectrum (Fig. S7a) contained three peaks: or coordination at 162.5 eV , oxidized state at and . As shown in Fig. S7b, the peaks at 322,340 and were attributed to and modes of and bonds species, respectively, compared with the CFO. Fourier Transform Infrared Spectrometer (FT-IR) spectrum of the CFS-ACs/CNT catalyst (Fig. S8) exposed a distinct stretching vibration of C-S bond. The presence of C-S-C further verified the introduction of S atoms into the CNTs. Meanwhile, the feature peaks of (Fig. S9) were shifted compared to those of the pure CNTs, which suggested that the strong metal-support interaction arising from the introduction of sulfur atoms led to the superior structural stability, higher conductivity and faster electron transfer during the OER. Raman spectroscopy was used to examine the intensity ratios ( ) of the D -band to the G -band in the samples, indicating the surface defects in the carbon material (Fig. S10). The results showed that the ratio of CFS-ACs/CNT was
Fig. 1 | Morphology and structures characterizations of CFS-ACs/CNT. a Synthesis and morphological characterizations of CFS-ACs/CNT. b, c Transmission electron microscopy (TEM) images. d Aberration corrected high angle annular
dark-field (HAADF)-scanning transmission electron microscopy (STEM) images with zoom-in image showing a cluster (red circle), and e energy dispersive X-ray spectroscopy (EDS) mappings of individual elements ( , and Co ).
smaller than that of CFO-p/CNT. Fewer levels of defects and the reduced disorder of the carbon materials suggested high site occupancy and well-dispersed metal active sites.
Electron and spin state of the catalyst
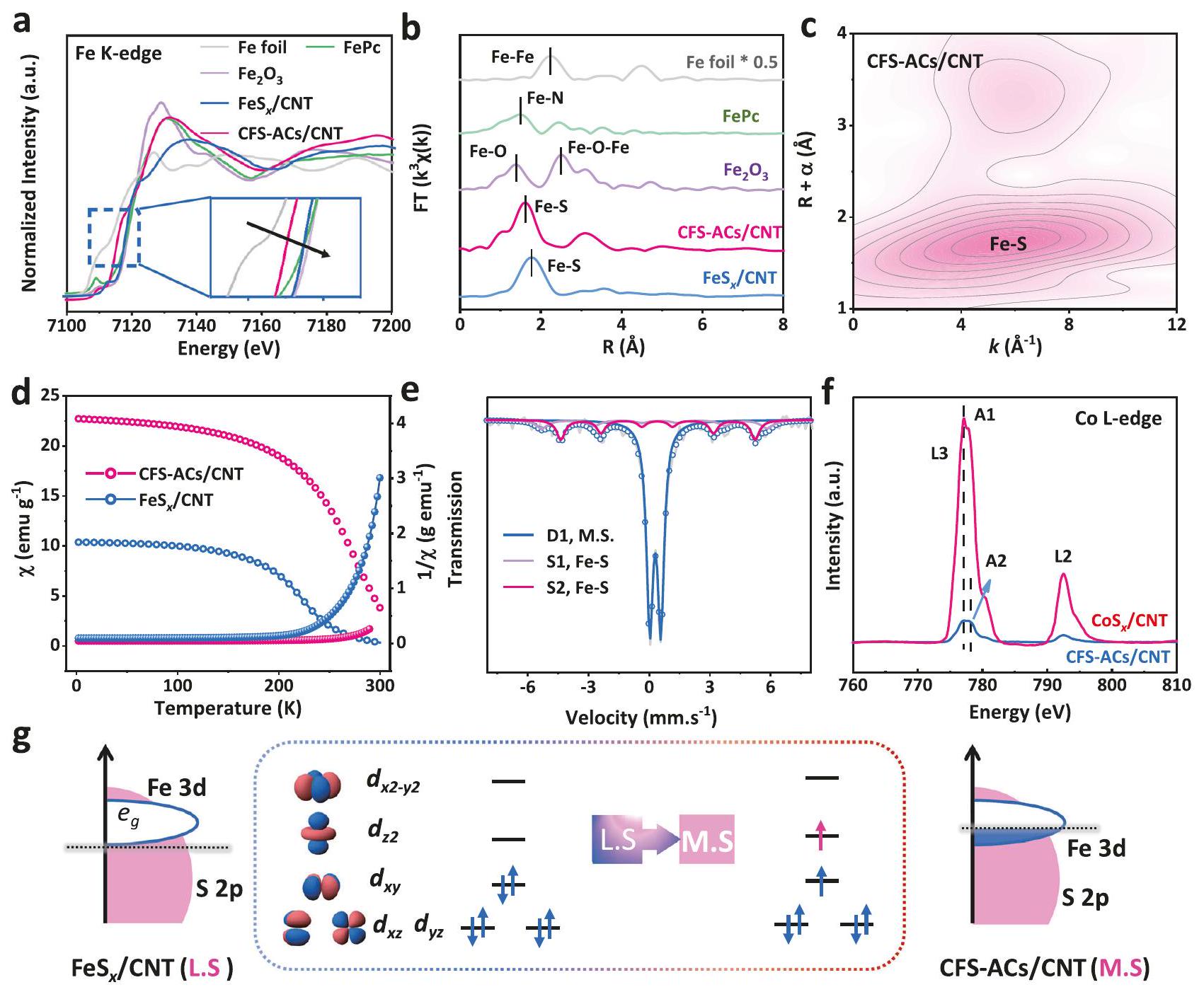
The X-ray absorption near-edge structure (XANES) of the Fe species in both CFS-ACs/CNT and FeS CNT (Fig. 2a) showed the valence states and . Meanwhile, CFS-ACs/CNT catalyst exhibited a slight shift to a lower photon energy, which indicated a chemical valence of approximately +2 in that sample, which was consistent with the XPS analysis. Similarly, the distinct pre-edge peak at of Co K-edge XANES of CFS-ACs/CNT was assigned to the Co-O sample (Fig. S11a), while the absorption edge for the CFS-ACs/CNT catalyst was located between Co foil and Co-O, close to that of the phthalocyanine . As depicted in Fig. 2b, the extended X-ray absorption fine structure (EXAFS) spectrum of CFO-p/CNT exposed a distinct predominant peak at in space, which coincided with the Fe-O coordination of . Meanwhile, a dominant peak in the spectrum of CFS-ACs/CNT and loaded at was observed, which was situated between Fe-O and Fe-S . Correspondingly, the highest Fe-Fe peak at of Fe foil was absent for CFS-ACs/CNT.
Similarly, Co K-edge EXAFS spectrum of CFS-ACs/CNT (Fig. S11b) showed a negative shift with a predominant peak at a , which was located between and . Moreover, the introduction of dual sites further aggravated the shortening of the M-S bond compared with the single metal site catalyst, which satisfied the trigger condition of the direct O-O coupling mechanism to a certain extent. As shown in Fig. S12, quantitative FT-EXAFS fitting was used to determine the structural parameters for the local chemical environment. Co-S and Fe-S coordination numbers were 5.5 and 5.3 (Table S1), respectively, which implied the similar chemical environments of Co and Fe . For further verifying the structure, Fe K-edge XANES spectrum calculated based on structure was obtained, which coincided well with the Fe K-edge XANES spectra of CFS-ACs/CNT (Fig. S13). Furthermore, the wavelet transform EXAFS analysis (Fig. 2c and Fig. S14) of the Fe K-edge showed an intensity characteristic peak at , which was attributed to Fe-S coordination.
The unpaired electron in the Fe atomic orbitals in CFS-ACs/CNT and samples were further identified with electron paramagnetic resonance (EPR) spectra. As shown in Fig. S15, the EPR peak signal was more pronounced after the introduction of , which
Fig. 2 | XAS analysis, Mössbauer spectroscopy and magnetic susceptibility. Fe K-edge XANES and Fourier-transform EXAFS spectra and of CFS-ACs/CNT, and . c Wavelet transforms of the experimental -weighted EXAFS spectra of CFS-ACs/CNT. d susceptibility X and reciprocal of CFS-
ACs/CNT and . e Room-temperature Mossbauer spectrum of CFSACs/CNT. f Co L-edge XANES spectra of CFS-ACs/CNT and FeS CNT . g Schematic illustration of the transfer of spin-state of the high OER activity for CFS-ACs/CNT electrode (Left is d-orbital occupancy of , right is CFS-ACs/CNT).
implied more Fe-3d unpaired electrons. Generally, the changes in orbital occupation and the charge redistribution are accompanied by the transformation of the Fe-3d electron spin configuration. Therefore, to identify the possible variation of the electron spin state, magnetic behavior (Fig. S16) was probed. After the formation of sulfides in the hydrothermal process, the dependence on the magnetic fields in CFSACs/CNT and samples showed a significant hysteresis phenomenon. Compared with , CFS-ACs/CNT catalyst with greater saturation magnetization (Ms) had more unpaired electrons and changed the internal electronic configuration of the metal, thus correcting the local spin state . The moderate unpaired electrons enabled optimal orbital interactions between M-3d and the O-2p orbitals, facilitating adsorption/desorption of the oxygen intermediates from the ferromagnetic catalyst CFS-ACs catalyst during the OER rate-limiting step, thus accelerating the OER process kinetics . Subsequently, the electron spin configuration of CFS-ACs/CNT was determined with a temperature-dependent magnetic susceptibility (MT) measurement. Figure 2d and Fig. S17 show that paramagneticferromagnetic phase transitions occured at the Curie temperatures ( ) for all compounds. Compared with that of , the generation of CFS-ACs/CNT decreased the paramagnetism state of Fe species, and shifted to higher temperatures while generating a significant
magnetic response , which implied the enhanced disorder and more free electron travel around the Fe species. From the Curie-Weiss law fitting, the effective magnetic moment ( ) for CFS-ACs/CNT was calculated as . Then, the number of unpaired electron ( ) was determined for the Fe (II) site according to the equation: , whereby was for CFS-ACs/CNT and higher than that of Mössbauer spectroscopy (Fig. 2e and Table S2) was conducted to verify the localized spin states of a single the Fe sites in multi-metallic ferromagnetic catalysts. The main peak was fitted with one doublet of D1 (blue), assigned to M.S Fe(II) . Therefore, there were no excess unpaired electrons assigned to the Co site. It is reasonable to assume the existence of Co in a low-spin state ( ), which has unfilled orbitals and strong bonding with the adsorbed intermediates. Furthermore, Co L-edge near edge X-ray absorption fine structure (NEXAFS) spectroscopy was used to investigate the intensity of Co empty/partially filled electronic state of CFS-ACs/CNT and . As shown in Fig. 2f, two peaks (A1 and A2) for the edges were dominated by enhanced “white line” features that implied unoccupied and orbitals, respectively. The peak A1 was assigned to a transition from the Co to the orbital, while peaks A2 indicated a transition to the orbital . The unoccupied Co orbitals of the CFS-ACs/CNT and
samples were directly reflected by the absorption intensity ratio of the edge, which largely reflects the spin ground state generated by the crystal field effect displayed a larger intensity ratio of 3.5 , while that of CFS-ACs/CNT was 2.5 . Therefore, suppression of the splitting energy of suggested a weak crystal field effect that favored higher orbtial occupancy of the Co center. Combined with curves, catalyst exposed a unfilled orbitals occupancy, called low spin state ( ). Not surprisingly, This was consistent with our initial reasonable assumption that Co atoms of CFS-ACs/CNT had unfilled -orbitals. Yang ShaoHorn et al . proposed that the optimal -orbitals occupancy for binding the OER intermediates was almost 1.2. Thus, compared with single Fe site with configuration, CFS-ACs/CNT exposed optimal -orbitals occupancy (Fig. 2g). We expected that the synergism effect of local configuration of M.S state ( ) and L.S state ( ) would lead to an electronic structure with optimal adsorption energy for OER intermediates, resulting in higher OER catalytic activity.
OER electrocatalytic performance of CFS-ACs
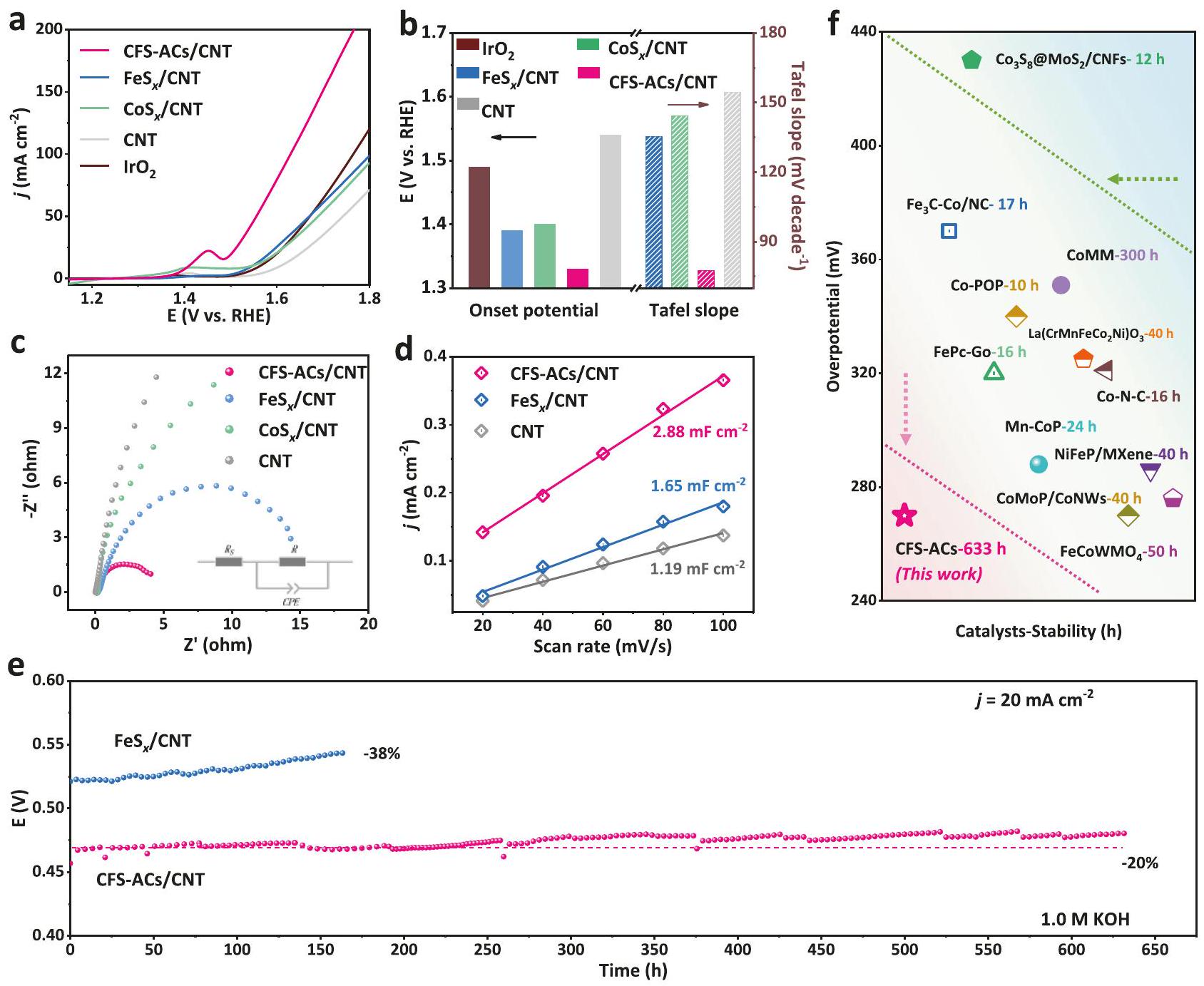
The OER performance of CFS-ACs/CNT was examined in -saturated 1.0 M KOH solutions with a typical three-electrode system. For comparison, , bulk , clean CNTs and commercial were also measured. All prepared catalysts were configured into a homogeneous ink and added to the surface of Ni foam (NF) substrate in a uniform drop ( ) to form working electrode (Fig. S18). The standard samples (CFS-ACs/CNT) were prepared by a series of content controls to obtain the optimal ratio of and S (Fig. ). The steady-state linear sweep voltammetry (LSV) shown in Fig. 3a. CFS-ACs/CNT exhibited excellent OER activity with a lower overpotential of 270 mV at , which was 100 mV and 80 mV less than those of commercial and , and presented the smallest Tafel slope (Fig. S20 and Fig. 3b) value of . Additionally, LSV curves with compensation of CFS-ACs/CNT, and pure CNT are provided in Fig. S21a-e. Similarly, CFS-ACs/CNT also showed superior OER activity than others prepared samples. To rule out the
Fig. 3 | OER performance measurements. a LSV curves of CFS-ACs/CNT, FeS CNT, , pure CNT, commercial and Ni foam substrate without compensation, measured by coating the catalyst on Ni foam with the mass loading of Activity comparison of onset potential at and Tafel slope for of CFS-ACs/CNT (red), (blue), (green), (brown) and pure CNT (gray). c EIS spectra of CFS-ACs/CNT with a smaller quasisemicircle ( about ), with a quasi-semicircle ( about ), and pure CNT. d double capacitive current ( ) of CFS-ACs/CNT, FeS CNT and pure CNT. e The chronopotentiometry curve of and CFS-ACs/ CNT in . Comparison of the activity and stability of different transition metal catalysts using different symbols to represent them. The red areas represent catalysts with a good balance of activity and stability, the yellow areas are moderate, and the green areas are weak. The references to the data points are supplied in Supplementary Table 4.
influence of NF substrate, the pure NF sample that underwent a similar procedure was also evaluated, and exposed a negligible OER activity (Fig. S22a). Meanwhile, the OER acitivity of CFO-p/CNT was also evaluated (Fig. S22b). Similarly, the OER performance of bulk was estimated via LSV curve and current density plot with CFS-ACs/CNT and at (Fig. S23) to highlight the effectiveness and foresight for the rational design of cluster structure. To further assess the charge transfer, the Nyquist plots measured by electrochemical impedance spectroscopy (EIS) of CFS-ACs/ CNT, and CNT were shown in Fig. 3c. The result sample exhibited a smaller charge transfer resistance ( ), a faster kinetics, which coincided with the lower Tafel slope value . The electrochemical surface areas (ECSAs) of as-prepared catalysts were evaluated through the electrochemical double-layer capacitances ( ) in 1 M KOH which were acquired by cyclic voltammetry (CV) at different scan rates (the detailed experiments in Supplementary Fig. 24 and Fig. 3d). The optimal of CFS-ACs/CNT indicated more electrochemically accessible surface active sites, which coincided with the optimal cluster structure. Meanwhile, the ECSA-corrected LSV curve (Fig. S25) obtained after normalizing the current density using ECSA showed that CFS-ACs/CNT had the highest intrinsic activity. Insufficient stability is another major problem for most highly active and highly dispersed OER catalysts. First, corrosion resistance was characterized by CV cycling in Fig. S26. There was no distinct decay before and after 2000 CV cycles over the potential range vs. RHE about the overpotentials, which implied the high corrosion resistance for CFS-ACs/CNT. As shown in Fig. 3e and Fig. S27, chronopotentiometry measurement of CFS-ACs/CNT and in 1 M KOH were carried out. CFS-ACs/CNT exposed strong electrochemical stability with continuous OER test run for 633 h without a distinct potential drop, while has shown a significantly increasing overpotential after continuous chronopotentiometry measure with a potential drop of . Meanwhile, curves after compensation of CFS-ACs/CNT, and at high current density ( ) also provided to further estimate their electrochemical stability. As shown in Fig. S21f, after the 70 h continuous measure at high current density, the current density of CFS-ACs/CNT catalyst only decreased by 4 mV . Furthermore, to validate the structural stability provided by sulfur atoms, chronopotentiometry measurements was also carried out for CFO-p/CNT in 1 M KOH. As shown in Fig. S28, the CFO-p/CNT catalyst exhibited weak stability, with a drop in current density after constant 70 h of constant OER testing, which further demonstrated that the introduction of S optimized the electronic structure of the catalyst and successfully attenuated the aggregation state of clusters during the catalytic process. Consistent with previous reports , oxidation state of Co and Fe species were susceptible to changes during the OER. Therefore, XPS and Raman spectra were accomplished to analyze the structure or species evolved after the OER process. After the potential was applied for a period of time, the surface on the catalysts was reconstituted to produce amorphous Fe oxyhydroxides (Fig. S29, 30 and detailed Supplementary Notes), and Fe species were oxidized to +3 valence state. Meanwhile, after stabilization, the O atom content increased with the decreases of active species , while intensity of the peak for CFS-ACs/CNT only showed slight degradation of S content from 19.01-14.8 at % (Table S3), which further demonstrated the structural stability after the introduction Co, S atom. Furthermore, CFS-ACs/CNT outperformed those of the best catalysts previously reported in literature (Table S4), in terms of both electrochemical activity and stability (Fig. 3f). The comparison of OER activity and stability of CFS-ACs/CNT with those of related single atom , oxides and other transition metal-based catalysts denoted the effect of ACs structure and coupling mechanism.
Exploration of the catalytic mechanism from spin state perspective
To further investigate the relationship between spin state of active species transformation from the viewpoint of electronic transition during OER process, in-situ XAS and Raman measurements is also performed. The XANES spectra shown in Fig. 4a, revealed that a conspicuous absorption peak located at 7715 eV gradually shifted toward higher energy with the promotion of the applied potential, which signified an oxidation state change during the OER process. During the oxidation process, the disappearance of sites were gradually oxidized to , which coincided with XPS spectra after long-term measurement (Fig. S30). In Fig. 4b, with the increase in applied potential from the OCP, , the main peak progressively shifted in the low-energy direction, which suggested a transformation in the Fe-S coordination environment and changed in the electronic structure transition of Fe during the OER process . When the potential reached to 1.445 V , the peak was located at , close to that of coordination environment . Fe-O coordination indirectly proved that CFS-ACs gradually transfered to the real active phase metal oxyhydroxides during the OER process, and this was accompanied by gradual disappearance of bonds . In-situ Raman spectroscopy further verified the evolution of the active species during electrochemical measurement (Fig. 4c and Fig. S31). The Raman spectrum at the initial potential of 1.23 V showed only two main peaks at 344 and , which were attributed to and modes of Fe-S stretching in CFSACs/CNT, respectively . As the potential increased, the intensity of the peak decreased significantly, indicating that a phase transition occured. A band at appeared at with a potential of 1.43 V and disappeared above 1.73 V , while a band at emerged at 1.73 V (Fig. S31). Both bands were assigned to iron oxide and oxyhydroxide, although many accompanying bands were absent. Thus, these two bands were likely associated with the Fe-O vibration modes of poorly crystalline phases formed during the surface oxidation. Moreover, active phase belong to was not found .
In general, charge redistribution and transfer are accompanied by a leap in the electron spin configuration. As shown in Fig. 4d, CFSACs/CNT displayed a larger intensity ratio of 1.6 , while that of CFS-ACs/CNT after stability was 1.3. Therefore, the suppression of the splitting XANES structure of CFS-ACs/CNT suggested a weak crystal field enableing a higher orbtial occupancy of the Fe center. The site of the CFS-ACs/CNT catalyst exhibited a half-filled orbital occupancy with a medium spin state ( ). Therefore, because CFSACs/CNT before and after OER stability showed approximate intensity ratio, should have a similar orbital occupancy of . Similarly, as shown in Co L-edge XANES (Fig. S32), CFS-ACs/CNT displayed a smaller intensity ratio than CFS-ACs/CNT after stability. site of CFSACs/CNT catalyst had unfilled orbitals with the low spin state ( ). Therefore, site after stability may have partially filled orbitals with the medium spin state due to smaller splitting energy . According to the bond order theorem and orbital interactions between cations and the oxygen intermediates in Fig. 4e, M.S. state ( ) exhibited poorer spin-orbit coupling interactions than the L.S. state ( ), implying a weaker adsorption capacity. In recent years, numerous investigations have been devoted to revealing the relationship between catalytic activity and the spin configurations of catalysts, among these, the ferromagnetic element Fe has been the most studied (Table S5). Hyung-Suk Oh et al . proposed the evolution of the spin state of site from L.S. to M.S. ( ) at the applied potential of 1.4 V . Similarly, after oxidation process, was oxidized to due to the generation of Fe oxyhydroxides but the M.S. state maintains, at which point the spin state of changed from L.S. to M.S., but the valence state remained unchanged. The M.S. showed 1 of -orbital occupancy, possessed slightly strong adsorption energy for the OER. Supporting this in
Fig. 4 | Spin state analysis during OER process. a Fe K-edge XANES spectra, b Fe K-edge FT-EXAFS during OER process with the different potentials (vs RHE) of OCV, 1.245 V , and 1.445 V in 1 M KOH , respectively. In-situ Raman spectroscopy of CFS catalyst in Fe L-edge XANES spectra before and after stability of CFSACs/CNT. e The orbital interactions between cations and the OER intermediates of
Fe and Co during OER process according to the bond order theorem. The red region represents more favorable metal spin-orbit coupling interactions. The schematic diagram of dual-site adsorbate evolution mechanisms for coupled O-O bonding during the OER (blue for Co atoms, red for Fe atoms, yellow for S atom).
different systems, Zhang et al. regulated the L.S. to M.S. ( ) spin state transformation of with the introduction of to modulate the occupy of antibonding -orbital of oxygen. Similarly, Wang et al. enhanced the activity of oxygen reduction reaction (ORR) through optimizing adsorption of site by axial ligand . Herien, sites (M.S., ) showed the optimal adsorption, when sites (L.S., ) demonstrated dominant *OH adsorption during OER process. Therefore, electrophilic intermediates of Lewis basic OH were preferentially bound at the Fe sites and lost thereby losing electrons to produce intermediates. From the valence or spin state changes at different sites during oxidation, we expected that it enables effective functional differentiation of active species in the proton-coupled electron transfer steps and simultaneous segmentation to promote OER kinetics. Combining a schematic representation of the spin-orbit interaction of the metal with the oxygen intermediate and the valence change of the metal site at the applied potential, a feasible CoFe dual-site OPM mechanism (i.e., the DSSM mentioned above) is proposed: (Fig. 4f): a dual-site segmentally synergistic catalysis mechanism enabled the strong adsorption capacity at the Co site and the strong adsorption capacity of the Fe site, this permited the simultaneous presence of species with and triggered the switch of the O-O coupling mechanism, therefore stimulating the formation of the adsorbed Co-O-O-Fe intermediate.
A single-domain ferromagnetic catalyst stimulated the presence of an electric field inside the catalyst and induced spinpolarized water oxidation to key oxygen intermediates, which was contributing to the parallel spin to reduce the formation barrier of coupled intermediates and thus facilitated the direct desorption of triplet oxygen ( . Therefore, scaling relationship between and , which constrains the OER adsorption evolution, was
broken while avoiding the poor structural stability caused by the traditional O-O coupling mechanism .
DFT calculation
Furthermore, to evaluate whether coupling mechanism would afford the best OER performance, DFT calculations were performed. As a comparison, typical AEM mechanisms and iron sulfide (single site) models were also considered. The rich electrolyte was accompanied by a long substitution process of the active phase, so we choose the transiently stable intermediate phase produced by partial substitution for the analysis of the OER mechanism. Therefore, OER process was simulated with spin-polarized DFT calculations by applying DFT +U method to study the OER activities of the CoFe dual-site with different mechanisms. Figure S33-37 shows 4e pathway for typical AEM mechanism and optimal OPM mechanism with CoFe as dual-site active species. Furthermore, the spin polarization spatial distribution of site with oxygen intermediates was calculated to evaluate the spin exchange interactions of the itinerant electrons near region (Fig. S 38 and Fig. 5a). The spin-selective electron transfer from the single-linear state or to triplet-state oxygen was necessary to guarantee the smooth operation of the OER 4 e transfer process. However, this is a spin-forbidden process. For ferromagnetic electrocatalysts, electron-electron repulsion are small when the short-range quantum spin-electron couples with the adsorbed oxygen (reactant), thus inducing spin-dependent conductivity and lowering the rate-limiting bond energy .
Meanwhile, to assess the impact of surface spin states on adsorbate adsorption energy in DFT calculations, particularly concerning magnetic materials, two Co-Fe surfaces were introduced for comparison: one with a distinct spin moment (ground state spin moment) and the other non-spin polarized, achieved by setting the spin to zero in the
Fig. 5 | OER mechanism analysis based on DFT calculations. a Spin density and planar distribution maps of the spin polarization of the key oxygen intermediates . The pinkish purple isosurfaces represent the spin-up states. The parallel arrangement of spin electrons in the oxygen-oxygen coupling of adjacent metal sites facilitates triplet oxygen production. c The free energy diagram of
typical AEM mechanism and DSSM pathway of OER including all oxygen intermediates and . Insets show the spin density plot of over the ferromagnetic CFS model towards the transition from *O to . Blue balls for Co atoms, pink balls for Fe atoms, yellow balls for S atoms, red balls for O atoms and white balls for H atoms.
system and recalculating the lattice constant . As shown in Fig. S39, for a range of adsorbates, non-spin polarized surfaces showed stronger the adsorption energy than that of spin-polarized ground state surfaces, especially in RDS step. In Fig. 5a, the key intermediate state revealed the ferromagnetic coupling between these CoFe dual-site, which implied that the spin-parallel alignment of the oxygen intermediate can effectively accelerate the desorption step (release of the triplet-state ). As illustrated in Fig. 5b, due to antiferromagnetic coupling, the application of additional energy was required to overcome the pairwise energy barrier and induce the spinunaligned alignment, which can cause spin flips and achieve a final spin-parallel alignment to release the triplet state oxygen. Meanwhile, the trend of ferromagnetic coupling of further demonstrated the preference of coupling rather than linear constraints on species and the efficiency of oxygen release, which is a feature of the OPM mechanism . Moreover, Fig. 5c shows the Gibbs free energy diagrams of two different pathways: typical AEM and DSSM mechanism. As plotted in Fig. 5c and S37, the rate determining step (RDS) was calculated to be the formation of *OOH or O-O intermediates from the step of * O for both CFS-ACs/CNT and , indicating the
stronger binding of *O on Fe site, which was consistent with the established scaling relationship. The RDS barrier for DSSM pathway was 1.12 eV , showing almost great advantage compared with that of AEM pathway. As excepted, the two O atoms exhibited parallel spin arrangements at the ferromagnetic-coupled active sites, which was more favorable for desorption to form the spin triplet state .
Discussion
In summary, we fabricated a small-sized (1.5 ~3 nm) CFS-ACs catalyst tied to CNT by a low-temperature hydrothermal technology, which exhibited excellent OER activity with the overpotential of 270 mV at and remarkable durability ( 633 h without potential loss) in 1 M KOH . According to magnetic characterization and Mössbauer spectroscopy, NEXAFS spectrum, large hysteresis loop and coercivity and proprietary ferromagnetic curve belongs to CFS-ACs/CNT. We distinguished for the first time the functionality and spin-state of different active sites with (M.S, ) as well as (L.S, ). Based on in-situ/operando X-ray adsorption measurements and Raman spectroscopy, we proposed a dual-site segmentally synergistic catalytic mechanism based on the evolution of the metal-oxygen
intermediate orbital interactions due to changes in the orbital electron occupation of the metal active site during the OER process, in which coupling mechanism successfully avoided the limitations of the scaling relationship between the adsorption oxygen mechanism and . Meanwhile, small-sized ( ) ferromagnetic CFS-ACs catalyst can be regarded as a single-domain ferromagnetic catalyst, which stimulated the presence of an electric field inside the catalyst, induced spin-polarized water oxidation of key intermediate oxygen species and generated a spin parallel alignment to reduce the formation barrier of coupled intermediates and thus facilitate the direct desorption of triplet oxygen ( ). DFT calculations further revealed that O-O coupling mechanism is feasible and outperformed to typical single-site adsorption mechanism. Our work has not only achieved simultaneous promotion of O-O coupling intermediates generation and release, but also offers a steerable strategy and unique insights into the impact of spin state changes on oxygen adsorption intermediates during OER.
Methods
Chemicals
Ferric nitrate , cobaltous nitrate , thiacetamide (TAA) all were purchased from Beijing Enokai Technology Co.,LTD. Potassium sulfate ( ) and sodium hydroxide ( KOH ) were purchased from Sinopharm Chem. Reagent Co., Ltd. Multi-walled carbon nanotube (CNT, in diameter and in length) was obtained from Beijing Deke Daojin Technology Co., LTD. Sodium borohydride was acquired by Tianjin Fengchuan Chemical Reagent Technology Co., Ltd. Nickle foil (NF, 99.8%) was supplied by Suzhou Shengerno Technology Co., Ltd, and Nafion ( ) was obtained from Sigma-Aldrich. All chemicals were used as received without any further purification. The catalyst was obtained from Johnson Matthey. Deionized water used in all experiments was ultrapure.
Synthesis of CNT (CFO-p/CNT)
Firstly, 70 mg CNT were dispersed in 50 mL ethanol with continuous sonicating for 30 min to form solution A. Secondly, 2 mmol and were dissolved in 20 mL ethanol under 10 min magnetic stirring to form solution B. Solution B was slowly added dropwise to solution A with continuous stirring for 10 h , called solution C. Subsequently, 10 mL of 1.25 mmol of was slowly added dropwise to solution C in an ice-water environment. Finally, the solution was washed by centrifugation with ice-cold ethanol and ultrapure water, followed by freeze-dried to obtain the CFO-p/ CNT precursor. Similarly, and precursor were prepared with the same process without the addition of Co source and Fe source, respectively.
Synthesis of CFS-ACs/CNT
80 mg as-prepared CFO-p/CNT precursor was dispersed in 40 mL deionized water under stirring vigorously for 30 min . Then, 25 mL 40 mmol thioacetamide was added dropwise while stirring for 30 min . The resulting solution was transferred into a 100 mL Teflon-lined stainless-steel autoclave, followed by heating at for 6 h . Finally, the final CFS-ACs/CNT was obtained by washing and freeze-drying. Similarly, and were fabricated with the same amount of thioacetamide. Meanwhile, CNT, and were prepared with the differernt amount of and 1 mmol and ; 10 mmol thioacetamide and 50 mmol thioacetamide.
Characterizations
The component of all-prepared samples was characterized by X-ray diffraction (XRD) patterns, which were collected in by a Y-2000 X-ray
Diffractometer using copper radiation ( ) at 40 kV , 40 mA . The structure of samples was proved by X-ray photoelectron spectroscopy (XPS), which were performed with thermo Scientific K-Alpha spectrometer using a focused monochromatic Al Ka ( 1486.6 eV ) X-ray beam with a diameter . Raman spectra were collected on a HORIBA Scientific LabRAM HR Evolution with a 532 nm laser source. The morphology of the samples was characterized by transmission electron microscopes (TEM, FEI Tecnai G220) with an accelerating voltage of 200 kV and field-emission scanning electron microscope (FE-SEM, JEORJSM-6700F). HAADF-STEM images were acquired from JEOL JEM-ARM200F at an accelerating voltage of 200 kV . The Fe K-edge and Co K-edge X-ray absorption near edge structure (XANES) and the extended X-ray absorption fine structure (EXAFS) were investigated at the BL14W1 beamline of Shanghai Synchrotron Radiation Facility (SSRF) in the fluorescence mode using a fixed-exit Si (111) double crystal monochromator. The obtained XAFS data was processed in Athena (version 0.9.26) for background, preedge line and post-edge line calibrations. Then Fourier transformed fitting was carried out in Artemis (version 0.9.26). The k3 weighting, -range of and range of were used for the fitting of foils; k-range of and R range of were used for the fitting of Fe and Co samples. The four parameters, coordination number, bond length, Debye-Waller factor and EO shift (CN, R, ) were fitted without anyone was fixed, constrained, or correlated. For Wavelet Transform analysis, the exported from Athena was imported into the Hama Fortran code. The parameters were listed as follow: R range, , k range, for Fe and Co samples; k weight, 2 ; and Morlet function with was used as the mother wavelet to provide the overall distribution. Mössbauer spectra of the catalysts were obtained on a Topologic 500 A spectrometer driven with a proportional counter at room temperature. The incident X-ray beam was monitored by an ionization chamber filled with , and the X-ray fluorescence detection used a Lytle-type detector filled with Ar. The EXAFS raw data were then background-subtracted, normalized and Fourier transformed by standard procedures with the IFEFFIT package.
Synchrotron-based XAS measurements
The Fe and CoL-edge X-ray absorption (XAS) spectra were measured at the Beamlines MCD-A and MCD-B (Soochow Beamline for Energy Materials) at National Synchrotron Radiation Laboratory (NSRL, China). During the measurement, samples were placed at room temperature. Both the Fe and Co L-edge were measured by means of total electron yield (TEY), measuring the drain current as a function of the photon energy. Multiple scans were measured and averaged.
In-Situ X-ray Absorption Fine Structure (XAFS)
Firstly, the pretreatment as follows: the working electrodes were pretreated by voltammetric cycles between 0.2 and 0.8 V at a rate of in -saturated 1 M KOH aqueous solution. The catalyst was hand-brushed onto carbon paper ( ). OCV, 1.245 V vs. RHE and 1.445 V vs. RHE were chosen as three representative conditions, the in situ X-ray absorption signals were collected when the working electrode reached a steady state. The electrode was maintained under each condition for 30 min before measuring the spectrum. The Fe K-edge XANES and EXAFS spectra were collected at the Taiwan Photon Source 44 A beamline and BW14 of SSRF. Fe foils was used to calibrate the beamline energy and compare samples. Fluorescence mode detection was performed using a Si (111) double crystal monochromator. An ion chamber detector for samples and the total electron yield was used to measure samples with high concentrations, such as references.
In-situ Raman
A custom-made two-compartment three-electrode electrochemical cell with a quartz window was used for in situ Raman measurements . of CFS suspension was dropcast onto the the glassy carbon electrode as a working electrode. A graphite rod was used as the counter electrode. electrode ( ) was used as the reference electrode. A Gamry Reference 600+ Potentiostat was used to control the electrode potential. Prior to Raman measurement, the glassy carbon electrode was polished by 0.05 um alumina suspension. The Raman spectroscopy measurements were performed using a LabRAM HR Evolution microscope (Horiba Jobin Yvon) equipped with a 532 nm laser, a objective ( ), and a CCD detector. Raman frequency was calibrated using a Si wafer before each experiment. The filter was set to be to keep a low laser intensity to avoid any irradiation-induced modifications of the organic additive deposited surface. The acquisition time was set to 40 s for each spectrum.
DFT calculation details
All DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP). The spin-polarized DFT calculations were performed with generalized gradient approximation and Perdew-Burke-Ernzerhof (PBE) functional as implemented in the Vienna ab initio Simulation Package . The projector augmentedwave method was used to treat the ion-electron interactions. The cutoff energy of 450 eV was set and the self-consistent field (SCF) tolerance was . Dipole moment corrected was made accordingly in the direction, and the empirical correction scheme of Grimme was also performed, where the effect of vdW interactions was included explicitly. To describe the strongly-localized interaction from and electrons, the PBEsol exchange-correlation functional was used together with an effective Hubbard parameter within the Dudarev approach with 3 eV for Fe and 3.3 eV for Co , taken from Ref. surface was selected with repeated in unit cell and a vacuum width of was used. The bottom two layers were fixed, while the top one layers and the adsorbates was allowed to relax. The 331 Monkhorst-Pack mesh k-points was sampled for all surface calculation. And all energy difference of all planes does not exceed 0.02 eV . The VASPKIT package was used for post-processing of the calculation results .
The Gibbs free energy calculated by DFT is based on computational hydrogen electrode (CHE) model provided by Nørskov , in which the total energy of is equal to at standard condition ( ). The free energy of is calculated by
In terms of each elementary step, the Gibbs free energy change is calculated via Eq. (2):
where is the total energy obtained in DFT, ZPE is the zero-point energy, is the temperature ( 298.15 K ), and is the entropy. In detail, the ZPE of the adsorbate is calculated by Eq. (3):
We determined the entropy of gas-phase molecules from the CRC Handbook , the entropy of the adsorbate species are calculated by Eq. (4):
where and are Plank constant, the speed of light and vibrational frequency respectively.
the contribution from entropy TS is calculated by Eq. (5) with a Harmonic oscillator approximation as:
where , and are Plank constant and vibrational frequency respectively.
Accordingly, the Gibbs free energy changes for the water oxidation steps using AEM mechanism was calculated using the following Equations:
The intermediates adsorption energy for M-OH, M-O, M-OOH and M-OO can be used DFT ground state energy calculated as:
Moreover, the Gibbs free energy changes for the water oxidation steps using OPM mechanism was calculated using the following Equations:
The intermediates adsorption energy for M-OH, M-O and M-OO-M can be used DFT ground state energy calculated as:
Electrochemical measurements
Typically, all the electrochemical tests were performed in a threeelectrode system at an electrochemical station (CHI Electrochemical Station (Model 760 E )) at a room temperature. (in 3.0 M KCl ) was used as the reference electrode, and a carbon rod as the counter electrode. Firstly, catalyst ink was prepared by dispersing 5 mg of catalyst into 1 mL of water/isopropanol ( ) solvent containing Nafion and sonicated for 30 min to form a homogeneous solution. Then, 600 uL homogeneous solution was loaded on a clean NF ( ), vacuum dried and used as working electrode. The final loading for all catalysts on the NF was about . During oxygen evolution reaction (OER) measurement process, linear sweep voltammetry (LSV) was tested in 1.0 M KOH with a scan rate of . The electrolyte is freshly prepared for each electrochemical measurement. Meanwhile, all the electrochemical test is performed in an H-type electrolytic Teflon cell. All potentials are referenced to the reversible hydrogen electrode (RHE) by the Nernst equation:
and the overpotential of OER was calculated according to the equation:
The electrochemical impedance spectroscopy (EIS) was obtained at the open-circuit voltage from . The longterm stability of the samples was carried out by the chronopotentiometry method with a stable current density of . To determine the electrochemical surface area (ECSA) can be obtained by the following equation: ECSA is the specific capacitance in 1 M KOH ( ) and is the electrochemical double layer capacitance measured using cyclic voltammetry curves ( CV , tested in ranging from ). CV activation was performed prior to each electrochemical test.
Data availability
The data that support the plots are available within this paper and its Supplementary Information. All other relevant data that support the findings of this study are available from the corresponding authors on reasonable request. Figures 2-5, Supplementary Figs. 1, 5-7, 8-13, 15-32 and 37 data generated in this study are provided in the Source data files. Source data are provided with this paper.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294-303 (2012).
Montoya, J. H. et al. Materials for solar fuels and chemicals. Nat. Mater. 16, 70-81 (2017).
Görlin, M. et al. Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of oxide water splitting electrocatalysts. J. Am. Chem. Soc. 138, 5603-5614 (2016).
Suntivich, J. et al. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science. 334, 1383-1385 (2011).
Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: toward boosted and sustained water oxidation catalysis. Joule. 5, 2164-2176 (2021).
Xiao, K., Wang, Y., Wu, P., Hou, L. & Liu, Z.-Q. Activating lattice oxygen in spinel through filling oxygen vacancies with fluorine for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 62, e202301408 (2023).
Xu , J. et al. with lattice water-assisted oxygen exchange for high-performance proton exchange membrane water electrolyzers. Sci. Adv. 9, eadh1718 (2023).
Grimaud, A. et al. Activation of surface oxygen sites on an iridiumbased model catalyst for the oxygen evolution reaction. Nat. Energy. 2, 17002 (2017).
Chen, F.-Y. et al. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule. 5, 1704-1731 (2021).
Huang, Z.-F. et al. Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat. Commun. 12, 3992-4000 (2021).
Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy. 4, 329-338 (2019).
Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829-7836 (2023).
. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021).
Ren, X. et al. Spin-polarized oxygen evolution reaction under magnetic field. Nat. Commun. 12, 2608-2620 (2021).
Garcés-Pineda, F. A. et al. Direct magnetic enhancement of electrocatalytic water oxidation in alkaline media. Nat. Energy. 4, 519-525 (2019).
Yan, J. et al. Direct magnetic reinforcement of electrocatalytic ORR/ OER with electromagnetic induction of magnetic catalysts. Adv. Mater. 33, 2007525 (2021).
Zhuang, L. et al. Sulfur-modified oxygen vacancies in iron-cobalt oxide nanosheets: enabling extremely high activity of the oxygen evolution reaction to achieve the industrial water splitting benchmark. Angew. Chem. Int. Ed. 59, 14664-14670 (2020).
Li, X. et al. Adaptive bifunctional electrocatalyst of amorphous CoFe Oxide @ 2D black phosphorus for overall water splitting. Angew. Chem. Int. Ed. 59, 21106-21113 (2020).
Ge, J. et al. Multi-domain versus single-domain: a magnetic field is not a must for promoting spin-polarized water oxidation. Angew. Chem. Int. Ed. 62, e202301721 (2023).
Sun, T. et al. Ferromagnetic single-atom spin catalyst for boosting water splitting. Nat. Nanotechnol. 18, 763-771 (2023).
Deng, J. et al. Multiscale structural and electronic control of molybdenum disulfide foam for highly efficient hydrogen production. Nat. Commun. 8, 14430-14437 (2017).
Jiang, S. et al. Insight into the catalytic aactivity of amorphous multimetallic catalysts under a magnetic field toward the oxygen evolution reaction. ACS Appl. Mater. Inter. 14, 10227-10236 (2022).
Wu, T. et al. Iron-facilitated dynamic active-site generation on spinel with self-termination of surface reconstruction for water oxidation. Nat. Catal. 2, 763-772 (2019).
Zhang, S. et al. In situ embedding into nitrogen and sulfur codoped hollow porous carbon as a bifunctional electrocatalyst for oxygen reduction and hydrogen evolution reactions. Appl. Catal. B: Environ. 254, 186-193 (2019).
Zhang, J. et al. Tuning the coordination environment in single-atom catalysts to achieve highly efficient oxygen reduction reactions. J. Am. Chem. Soc. 141, 20118-20126 (2019).
Peng, W. et al. Deciphering the dynamic structure evolution of Feand Ni-Codoped CoS for enhanced water oxidation. ACS Catal. 12, 3743-3751 (2022).
Li, C. et al. Tailor-made open porous 2D CoFe/SN-carbon with slightly weakened adsorption strength of ORR/OER intermediates as remarkable electrocatalysts toward zinc-air batteries. Appl. Catal. B: Environ. 269, 118771 (2020).
Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped single-atom catalyst for enhanced electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022).
Zhuang, Z. et al. Reversely trapping atoms from a perovskite surface for high-performance and durable fuel cell cathodes. Nat. Catal. 5, 300-310 (2022).
Wan, X. & Shui, J. Exploring durable single-atom catalysts for proton exchange membrane fuel cells. ACS Energy Lett. 7, 1696-1705 (2022).
Li, Y. et al. Local spin-state tuning of iron single-atom electrocatalyst by S-coordinated doping for kinetics-boosted ammonia synthesis. Adv. Mater. 34, 2202240 (2022).
Wu, T. et al. Spin pinning effect to reconstructed oxyhydroxide layer on ferromagnetic oxides for enhanced water oxidation. Nat. Commun. 12, 3634-3645 (2021).
Zhuang, Z. et al. Atomically dispersed nonmagnetic electron traps improve oxygen reduction activity of perovskite oxides. Energy Environ. Sci. 14, 1016-1028 (2021).
Wang, X. et al. Engineering 3d-2p-4f gradient orbital coupling to enhance electrocatalytic oxygen reduction. Adv. Mater. 34, 2206540 (2022).
Mehmood, A. et al. High loading of single atomic iron sites in Fe-N-C oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 5, 311-323 (2022).
Kroll, T. et al. Transition metal phthalocyanines: insight into the electronic structure from soft X-ray spectroscopy. J. Chem. Phys. 137, 054306 (2012).
Lee, W. H. et al. Electrode reconstruction strategy for oxygen evolution reaction: maintaining phase with intermediate-spin state during electrolysis. Nat. Commun. 13, 605 (2022).
Ding, J. et al. Atomic high-spin cobalt(II) center for highly selective electrochemical CO reduction to . Nat. Commun. 14, 6550 (2023).
Liu, D. et al. A template editing strategy to create interlayerconfined active species for efficient and durable oxygen evolution reaction. Adv. Mater. 35, 2203420 (2023).
Xiao, Z. et al. Operando identification of the dynamic behavior of oxygen vacancy-rich for oxygen evolution reaction. J. Am. Chem. Soc. 142, 12087-12095 (2020).
Lei, Z. et al. Coordination modulation of iridium single-atom catalyst maximizing water oxidation activity. Nat. Commun. 13, 24-34 (2022).
Wang, C. et al. Identification of the origin for reconstructed active sites on oxyhydroxide for oxygen evolution reaction. Adv. Mater. 35, 2209307 (2022).
Chen, X., Shi, T., Zhong, K., Wu, G. & Lu, Y. Capacitive behavior of decorated with @carbon nanospheres. Chem. Eng. J. 379, 122240 (2020).
Hu, Y. et al. Interfacial evolution on Co-based oxygen evolution reaction electrocatalysts probed by using in situ surface-enhanced raman spectroscopy. Anal. Chem. 95, 1703-1709 (2023).
Zhang, Z. et al. Regulating spin states in oxygen electrocatalysis. Angew. Chem. Int. Ed. 62, e202216837 (2023).
Yang, G. et al. Regulating Fe-spin state by atomically dispersed MnN in Fe-N-C catalysts with high oxygen reduction activity. Nat. Commun. 12, 1734-1744 (2021).
Liu, Y. et al. Tuning the spin state of the iron center by bridgebonded ligands for enhanced oxygen reduction. Angew. Chem. Int. Ed. 61, e202117617 (2022).
Yang, H. et al. Intramolecular hydroxyl nucleophilic attack pathway by a polymeric water oxidation catalyst with single cobalt sites. Nat. Catal. 5, 414-429 (2022).
Gao, W. et al. Electron spin polarization-enhanced photoinduced charge separation in ferromagnetic . ACS Energy Lett. 6, 2129-2137 (2021).
Cao, A. & Nørskov, J. K. Spin effects in chemisorption and catalysis. ACS Catal. 13, 3456-3462 (2023).
Zhao, K. et al. Enhancing hydrogen oxidation and evolution kinetics by tuning the interfacial hydrogen-bonding environment on functionalized platinum surfaces. Angew. Chem. Int. Ed. 61, e202207197 (2022).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558-561 (1993).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787-1799 (2006).
Csonka, G. I. et al. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 79, 155107 (2009).
Wang, V. et al. VASPKIT: A user-friendly interface facilitating highthroughput computing and analysis using VASP code. Comput. Phys. Commun. 267, 108033 (2021).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886-17892 (2004).
Lide, D. R. CRC Handbook of Chemistry and Physics 89th edn (CRC Press, 2009).
Campbell, C. T. & Sellers, J. R. V. The entropies of adsorbed. Mol. J. Am. Chem. Soc. 134, 18109-18115 (2012).
Acknowledgements
This work was financially supported by the key projects of the National Natural Science Foundation of China (U22A20107, J.N.Z.), and the key projects of the Henan Provincial Science and Technology R&D Program Joint Fund (222301420001, J.N.Z.), the Distinguished Young Scholars Innovation Team of Zhengzhou University (32320275, J.N.Z.), Higher Education Teaching Reform Research and Practice Project of Henan Province (2021SJGLX093Y, J.N.Z.). Acknowledge the Beijing Synchrotron Radiation Facility (BSRF) 1W1B station for XAS measurements. Li-Rong Zheng is grateful to the support of XAS. Acknowledge the B.-J.Xu’s group of College of Chemistry and Molecular Engineering, Peking University Beijing for In-situ Raman spectroscopic testing. Acknowledge the Beamlines MCD-A and MCD-B (Soochow Beamline for Energy Materials) at National Synchrotron Radiation Laboratory (NSRL, China).
Author contributions
J.-N.Z. supervised this research. and S.-R.X. conceived the project and designed the work and S.-R.X., M.-L.L. performed the experiments, including materials synthesis, characterizations, and electrochemical measurements. And K.-Y.Z., B.-J.X. performed In-situ Raman characterizations. S.-H.F., C.W. provided Synchrotron-based XAS measurement. Y.Y. provided theoretical computational support and D.-P.X., J.-N.Z. reviewed and edited the manuscript. All authors have approved the final version of the manuscript.
Correspondence and requests for materials should be addressed to Jia-Nan Zhang.
Peer review information Nature Communications thanks Yang Hou, Hyung-Suk Oh and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Reprints and permissions information is available at http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key Laboratory of Advanced Energy Catalytic and Functional Materials Preparation, College of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, China. National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230026 Anhui, China. College of Chemistry and Molecular Engineering, Peking University, Beijing, China. State Key Laboratory of Coking Coal Resources Green Exploitation, Zhengzhou 450001, China. These authors contributed equally: Siran Xu and Sihua Feng.