DOI: https://doi.org/10.1038/s41422-023-00918-9
PMID: https://pubmed.ncbi.nlm.nih.gov/38267638
تاريخ النشر: 2024-01-24
أدوار وآليات تنظيم إشارات TGF-β و BMP في تطوير العظام والغضاريف، التوازن والمرض
الملخص
عامل تحويل النمو-
مقدمة
نظرة عامة على TGF-
الليغاندات والمستقبلات: الهيكل، التنوع، والانتقائية
| تصنيف | جين | KO/CKO/Tg/ضرب في | الظاهرة | المراجع |
| TGF-
|
Tgfb1 | إلى | وفاة مبكرة (شهر واحد) | ٨٨ |
| Tgfb1
|
انخفاض كثافة العظام؛ OB
|
152 | ||
| العمود 1
|
تثخن في الديافز، حجم العظام المتقلب، زيادة في إعادة تشكيل العظام، عرضة للكسر؛
|
152 | ||
| العمود 1
|
التهاب المفاصل العظمي في الركبة ومفصل الفك الصدغي
|
٢٢١،٢٢٢ | ||
| Tgfb2 | ضربة قاضية | وفيات ما حول الولادة؛ عيب القوس العصبي؛ عظمة صدرية متشعبة؛ قصر في عظمة الكعبرة وعظمة الزند | 85 | |
| Tgfb3 | إلى | يموتون خلال 20 ساعة من الولادة؛ فشل الأرفف الحنكية في الاندماج مما يؤدي إلى الشق الحنكي | ٨٦، ٨٧ | |
| ليغاندات BMP | Bmp2 | العمود 2
|
تشوهات غضروفية شديدة؛ قامة وأطراف قصيرة؛ تكاثر الخلايا الغضروفية
|
50 |
| Prx1-Cre | تشكيل الأطراف الطبيعي | 51 | ||
| Bmp7 | ضربة قاضية | الموت عند الولادة؛ عيوب قاعدة الجمجمة؛ تشوهات الأضلاع وعظمة الصدر؛ تعدد الأصابع في الأطراف الخلفية | ٣٢٦ | |
| Bmp7، Alk6 | دي كي أو | عظام زائدة مشوهة ومختصرة مقارنة بـ Bmp
|
٢٥ | |
| Bmp2، 7 | Prx1-Cre;Bmp2
|
هيكل عظمي طرفي متناقص قليلاً؛ مفقود الفلانكس الأخير في الإصبع الثالث؛ شظايا مشوهة | 51 | |
| Bmp4 | العمود 2
|
تشوه غضروفي خفيف | 50 | |
| Prx1-Cre | تعدد الأصابع | ٥١، ٥٢ | ||
| بي إم بي 2، 4 | العمود 2
|
تشوه غضروفي شديد؛ عظام طويلة مشوهة أو مفقودة بشكل شديد ومفاصل ملتحمة؛ تكاثر الخلايا الغضروفية
|
50 | |
| Prx 1 -Cre | تعدد الأصابع؛ التصاق الأصابع الكامل؛ تأخر التمعدن؛ تكوين الغضاريف
|
51 | ||
| بي إم بي 3 / جي دي إف 10 | كول1 بروم-بمب3 تي جي | تمايز الغضاريف المتضخمة المتأخر
|
١١٥ | |
| ضربة قاضية | زيادة كثافة العظام | ٢٨ | ||
| Bmp14/GDF5 | بي بي
|
عظام محورية طبيعية؛ أطراف وأصابع قصيرة؛ مفاصل مفقودة في الأطراف؛ عناصر سلاميات مفقودة | ٢٥,٣٢٧ | |
| لا تأخير في شفاء الكسور | ٣٢٨ | |||
| زيادة تلف المفاصل في التهاب المفاصل الناتج عن الكولاجين؛ انخفاض كثافة العظام | ٣٢٩ | |||
| GDF5، Alk6 |
|
نفس الشيء مثل bp
|
٢٥ | |
| مستقبلات النوع الأول | Alk2/Acvr1 | العمود 2
|
قاعدة جمجمة قصيرة؛ فقرات عنقية ناقصة التنسج | ٤٩ |
| أوسك-كري | كثافة عظم الفك السفلي
|
113 | ||
| Q207D Tg | التنسج الليفي العظمي التقدمي | 127,130,132 | ||
| أكفر1
|
التنسج الليفي العظمي التقدمي | 128 | ||
| إدخال R206H | التنسج الليفي العظمي التقدمي | ١٢٩ | ||
| Alk3/Bmpr1A | العمود 2
|
أقواس ظهرية مقسمة؛ أطراف قصيرة؛ لوح كتف ناقص التنسج؛ خلل التنسج الغضروفي، تكاثر الخلايا الغضروفية
|
٤٨، ٤٩، ٦٥ | |
| جيدف5-كري | تطور التهاب المفاصل العظمي تلقائيًا | ٣٣٠ | ||
| العمود 1
|
كتلة العظام في العظام الطويلة والأضلاع
|
148,149 | ||
| سب7-كري | كتلة العظم الإسفنجي
|
147 | ||
| Dmp-Cre | كتلة العظم الإسفنجي
|
١٤٧,١٧٦ | ||
| Alk6/Bmpr1B | ضربة قاضية | براكيوبوديزم؛ تقليل العناصر السلامية؛ اندماج المفاصل الزائدة، مشابه لطفرات GDF5 (bp
|
٢٥، ٤٨، ٤٩، ٦٥ |
| تصنيف | جين | KO/CKO/Tg/ضرب في | الظاهرة | المراجع |
| كُو | هشاشة العظام العابرة والمحددة حسب الجنس الناتجة عن انخفاض تكوين العظام من الخلايا الجذعية المولدة للعظام | ٣٣١ | ||
| عمود1
|
انخفاض كثافة العظام وحجم العظام؛ انخفاض عدد الخلايا البانية للعظام والخلايا الآكلة للعظام | ٣٣٢ | ||
| الك3، الك6 | كول2ا1-كري؛ ألك3
|
الظاهرة الشبيهة والأكثر شدة من فئران Alk3 CKO | ٤٨,٦٥ | |
| Col2-Cre;Alk3
|
عيوب شديدة في تشكيل الغضاريف وتكوين الهيكل العظمي | ٤٨ | ||
| الك2، الك3 | كول2-كري | هيكل محور مشوه (فقرات، مناطق عنقية وصدرية)؛ عيوب طرفية أكثر شدة من فئران Alk3 CKO | ٤٩ | |
| الك2، الك6 | Col2-Cre;Alk2
|
عيوب أكثر شدة في المحور والأطراف مقارنة بكل عيب فردي | ٤٩ | |
| الك5/تي جي إف بي آر 1 | ديرمو-كري | عظام طويلة قصيرة وعريضة، بروز غضروفي خارج المكان، أحجام عظام مخفضة | 83 | |
| العمود 2
|
أطراف ممدودة؛ تكاثر الخلايا الغضروفية
|
68 | ||
| العمود 2
|
التهاب المفاصل التنكسي التلقائي | ٢٠٨,٣٣٣ | ||
| مستقبلات النوع الثاني | تي جي إف بي آر 2 | نيستين-كريER | التهاب المفاصل العظمي في الركبة
|
221 |
| كول إكس-كري | تأخر التمايز النهائي للخلايا الغضروفية؛ عرقلة التمعدن | 84 | ||
| بروكس-كري | الموت عند الولادة؛ انخفاض في عظام اللثة والعظام الجبهية؛ تقصير الأطراف؛ انقسام عظمة الصدر؛ اندماج مفاصل الأطراف؛ انخفاض في التمعدن؛ تكاثر الخلايا الغضروفية
|
78,81 | ||
| كول
|
البقاء على قيد الحياة؛ عيب في القوس العصبي؛ الأقراص الفقرية المفقودة/غير المكتملة؛ انخفاض تدريجي في طول العظام الطويلة | 79,80 | ||
| سب7-كري (دوكس) | وفاة مبكرة (1 م)؛ حجم جسم مخفض؛ حجم عظام مخفض؛ زيادة في نسيج الدهون في نخاع العظام؛ اضطراب في تشكيل الأسنان الضرسية؛ OB
|
157,334 | ||
| أوكن-كري | كثافة العظام
|
172 | ||
| Bmpr2 | Prx1-Cre | حجم العظم الإسفنجي
|
335 | |
| العمود 1
|
قصر القامة؛ تأخر التمعدن؛ حجم العظام
|
114 | ||
| أكتريه/أكفر2أ | أوكن-كري | حجم العظم الإسفنجي
|
٣٣٦ | |
| أكتري آي بي / أكفر 2 بي | ضربة قاضية | تمايز الغضاريف المتضخمة المتأخر
|
١١٥ | |
| أوكن-كري | عادي | ٣٣٦ | ||
| أكتريه A، أكتريه B | أوكن-كري | حجم العظم الإسفنجي
|
٣٣٦ | |
| المسار القياسي | سماد1 | العمود 1
|
نقص كثافة العظام؛ تكاثر وتمايز خلايا العظام
|
٣٣٧ |
| العمود 2
|
لوحة النمو المختصرة؛ تضخم الخلايا الغضروفية
|
68 | ||
| سماد1/5 | كول2ا1-كري؛سماد1
|
ظاهرة مشابهة وأكثر شدة من Smad1 CKO | 68 | |
| سماد1/5 | العمود 2
|
تشوه الغضاريف؛ أطراف قصيرة؛ زيادة سمك الغلاف الغضروفي؛ إنتاج المصفوفة
|
69 | |
| سماد8 | ضربة قاضية | عادي | 69 | |
| سماد1/5/8 | كول2أ1-كري؛سماد1
|
غياب الهيكل العظمي المحوري؛ عظام طرفية غير منظمة بشدة | 69 | |
| سماد2 | كول
|
ظاهرة مشابهة وأكثر شدة من فئران Smad3 KO | 89 | |
| سماد2/3 | كول2أ1-كري؛سماد2
|
ظاهرة مشابهة وأكثر شدة من فئران Smad3 KO | 89 | |
| سماد3 | كُو | قزامة ما بعد الولادة؛ منطقة عمودية موسعة ومنطقة تضخم؛ تكاثر الخلايا الغضروفية
|
89 | |
| ضربة قاضية | التهاب المفاصل العظمي في الركبة ومفصل الفك الصدغي | ٢١٤، ٢١٥ |
| الجدول 1. متابعة | ||||
| تصنيف | جين | KO/CKO/Tg/ضرب في | الظاهرة | المراجع |
| إلى | نقص كثافة العظام؛ موت الخلايا العظمية والخلية العظمية
|
156 | ||
| سماد4 | تي بي إكس 18-كري | أطراف قصيرة، تكوين الغضاريف
|
40 | |
| سب7-كري (دوكس) | زيادة كتلة العظم الإسفنجي | 147 | ||
| سب7-كري | نمو متوقف؛ كسور عفوية؛ زيادة في حجم العظم التربيقي؛ انخفاض في كثافة العظام المعدنية؛ مجموعة من الميزات التي تُرى في هشاشة العظام الخلقية، وعسر التنسج القحفي الترقوي، ومتلازمات نقص Wnt. | ١١٨ | ||
| Dmp-Cre | زيادة كتلة العظم الإسفنجي
|
147 | ||
| عمود1
|
زيادة كتلة العظم الإسفنجي؛ الحماية من فقدان العظم الناتج عن تعليق الذيل؛ عدد خلايا العظم البانية وخلايا العظم المحبوسة
|
178 | ||
| أوكن-كري | كتلة عظمية أقل من 6 أشهر، كتلة عظمية أكثر من 7 أشهر | ١٧٧ | ||
| سي تي إس كيه – كري | انخفاض كتلة العظام؛ OC
|
186 | ||
| مسار غير تقليدي | تاك1 | العمود 2
|
تأخر النمو؛ علامات التهاب المفاصل العظمي
|
71 |
| أوسك-كري | ظاهرة مشابهة لخلل التنسج القحفي الترقوي (CCD) (نقص تنسج الترقوة وتأخر التحام اليافوخ)؛ OB
|
١١٦ | ||
| العمود 2
|
أطراف أقصر؛ تكاثر الخلايا الغضروفية
|
٣٣٨ | ||
| Prx1-Cre | اندماجات مفصلية واسعة الانتشار؛ تضخم وتكاثر الخلايا الغضروفية | ٣٣٨ | ||
| p38 | أوكن-كري | نشاط OB و BFR
|
١١٧ | |
| العمود 2
|
أطراف قصيرة؛ التهاب المفاصل العظمي في الركبة
|
٢١٠ | ||
|
|
انخفاض كبير في تكلس العظام الطويلة وتأثير أكثر اعتدالًا على القحف | ١١٦ | ||
| MKKs |
|
ظاهرة مشابهة لـ Tak1
|
١١٦ | |
| تنظيم I-SMAD واليوبيكويتين | سماد6 | عمود11
|
القزامة وهشاشة العظام؛ تضخم الخلايا الغضروفية
|
٢٧٩ |
| ضربة قاضية | قصر القامة؛ عيوب في العظام المحورية والأطراف؛ تأخر ظهور التضخم | ٢٨٠ | ||
| سماد6;سمرف1 | عمود11
|
تأخر أكثر حدة في التكلس الغضروفي مقارنة بـ Smad6 Tg | ٢٧٩ | |
| سماد7 |
|
هشاشة العظام؛ BFR
|
٢٨٢ | |
| ضربة قاضية | تكاثر الخلايا الغضروفية وتضخمها
|
٣٣٩ | ||
| Prx1 Prom-Tg؛ Col11 Enh-Tg؛ Col11 Prom-Tg | تشوه الغضروف؛ تكاثف الميزانشيم
|
٢٨٣ | ||
| سمرف1 | ضربة قاضية | كتلة العظام
|
286 | |
| العمود 1
|
هشاشة العظام؛ BFR
|
٢٨٥ | ||
| سمرف 2 | كول
|
التهاب المفاصل العظمي؛ تنكس القرص الفقري | ٢٩١، ٢٩٢ | |
| ضربة قاضية | الحماية من التهاب المفاصل التنكسي المرتبط بالعمر والمسبب بواسطة DMM | ٢٩٣ | ||
| ضربة قاضية | هشاشة العظام
|
٢٩٦ | ||
| ضربة قاضية | تعزيز تشكيل العظام خارج الموقع الناتج عن BMP | 294 | ||
| PLEKHO1 | أوستيريكس-كري | الحماية من فقدان العظام المرتبط بالعمر | ٢٨٩ | |
| أوستيريكس بروم-تي جي | فقدان العظام المرتبط بالعمر | ٢٨٩ | ||
| NEDD4 | العمود 1
|
كتلة العظام
|
298 | |
| كول
|
كتلة العظام
|
298 | ||
| جاب1 | أوسك-كري | قصر القامة؛ كتلة العظام الشبكية
|
٣٠٣ | |
| تصنيف | جين | KO/CKO/Tg/ضرب في | الظاهرة | المراجع |
| خصوم | نوجين | أوكن بروم-تي جي | هشاشة العظام؛ BFR
|
٢٥٠,٢٥٣ |
| أوكن-كري | هشاشة العظام | 254 | ||
| إلى | فرط تنسج الغضروف؛ فشل في تطوير المفاصل؛ عيوب هيكلية متعددة مرتبطة بتشكيل الأنبوب العصبي والسمات (فشل إغلاق الأنبوب العصبي، أطراف عريضة على شكل نادي، فقدان الفقرات الذيلية، محور جسم قصير، واحتفاظ بذيل ضامر صغير) | 66,67 | ||
| جرم1 | أوكن-كري | كتلة العظام
|
258 | |
| أوكن بروم-تي جي | كسور العظام؛ كتلة العظام
|
259 | ||
| FS | تي جي | كتلة العظام
|
262 | |
| المستقبلات المساعدة |
|
ضربة قاضية | تطور غير طبيعي في الحنك مع OB
|
267 |
| نربس | ضربة قاضية | كتلة العظام
|
٢٦٨ | |
| نيوجينين | ضربة قاضية | لوحة نمو ممدودة؛ تكاثر الخلايا الغضروفية وموتها المبرمج
|
٢٧٦ | |
| جهات تنظيمية أخرى | تم53 | ضربة قاضية | العظم المتصلب | ٣٠٨ |
| إندوفين | كول1 بروم-إندوفين F872A Tg | كتلة العظام
|
304 |
(1) العمود 1
(2)
| جين | مرض | MIM# | اضطرابات العظام | المراجع |
| نوجين، جي دي إف 5 | التصاق الأصابع | 185800، 186500، 184460، 615298 | التصاق أو التحام المفاصل بين السلاميات | ٥٩، ٦٠ |
| نوجين | متلازمة الائتلاف بين العظام القاربية والعظام الرسغية | 186570 | اندماج العظام الرسغية والعظام الكاحلية والعظام السلامية؛ قصر العظام المشطية الأولى مما يسبب قصر الأصابع؛ اندماج عظم العضد والعظم الكعبرة | 61 |
| نوجين، BMP2، BMPR1B، GDF5 | قصر الأصابع | 611377، 112600، 113100 | قصر الأصابع | 60،62-64 |
| TGFBR1، TGFBR2، TGFB2، TGFB3، SMAD2، SMAD3 | متلازمة لويز-ديتس | 609192، 610168، 613795، 614816، 615582، 619656 | تشوهات هيكلية متغيرة (بما في ذلك فرط نمو الهيكل العظمي، تشوه الصدر، التهاب المفاصل، الفتق، إلخ) | 72-76 |
| ACVR1 | التنسج الليفي العظمي المتقدم | 135100 | تكوين العظام غير الطبيعية التقدمية في العضلات والأوتار والأربطة والمفاصل | ١٢٢,١٢٣ |
| TGFB1 | مرض كاموراتي-إنجلمان | 131300 | آفات عظمية متصلبة في العظام الطويلة والجمجمة مع زيادة في إعادة التشكيل؛ التهاب المفاصل العظمي | 158 |
| SMAD3، MAP2K1، LEMD3 | ميلوريوستوز | 155950 | ميلوريوستوز (مرض العظام المتصلب الخاص) | 159-161، 306، 307 |
| LEMD3 | أوستيوبويكولوزيس؛ متلازمة بوشكي أولندورف | 166700 | العظم المتصلب | ٣٠٦، ٣٠٧ |
| TMEM53 | خلل التنسج القحفي الأنبوب، نوع إكيغاوا | 619727 | فرط التكلس؛ قصر القامة المرتبط بزيادة حجم الرأس، أو طول الرأس، أو جبهة بارزة | ٣٠٨ |
| FBN-1 | متلازمة مارفان | 154700 | تشوهات هيكلية متغيرة بما في ذلك زيادة طول العظام الطويلة | 77 |
| FBN-2 | تشوهات خلقية في الأوتار العنكبوتية | 121050 | أطراف طويلة (دوليكستينومليا) وأصابع وأصابع قدم طويلة ونحيلة (أراخنوداكتيلية)، ومفاصل مثنية بشكل دائم (انقباضات) | ٢٣٠ |
| أدامتس إل 2 | خلل التنسج الجيولوجي | 231050 | قصر القامة، قصر الأطراف، والعيوب الهيكلية | 243 |
| أدامتس10، أدامتس17 | متلازمة وايل-مارشيساني | 277600,608328 | قصر القامة، قصر الأصابع، وانزياح العدسة | 245 |
| COL1A1، COL1A2 | تكوّن العظام الناقص | 259420 | خلل تنسج العظام يتميز بتشوهات عظمية، كسور، ومعدل مرتفع من عدم التئام العظام بسبب انخفاض كتلة العظام وضعف جودة العظام | 235 |
| EXT1، EXT2 | التشوهات العظمية المتعددة الوراثية | ٣٣٧٠٠، ١٣٣٧٠١ | تكوين نمو عظمي مغطى بالغضروف (أوستيوكوندروما) في نهايات العظام | 238 |
| تزلج | متلازمة شبرينتزن-غولدبرغ | 182212 | مجموعة واسعة من التشوهات الهيكلية بما في ذلك التحام الجمجمة المبكر، ميزات وجه مميزة، أصابع طويلة، أطراف طويلة، انخماص الصدر أو بروز الصدر، والجنف | 313 |
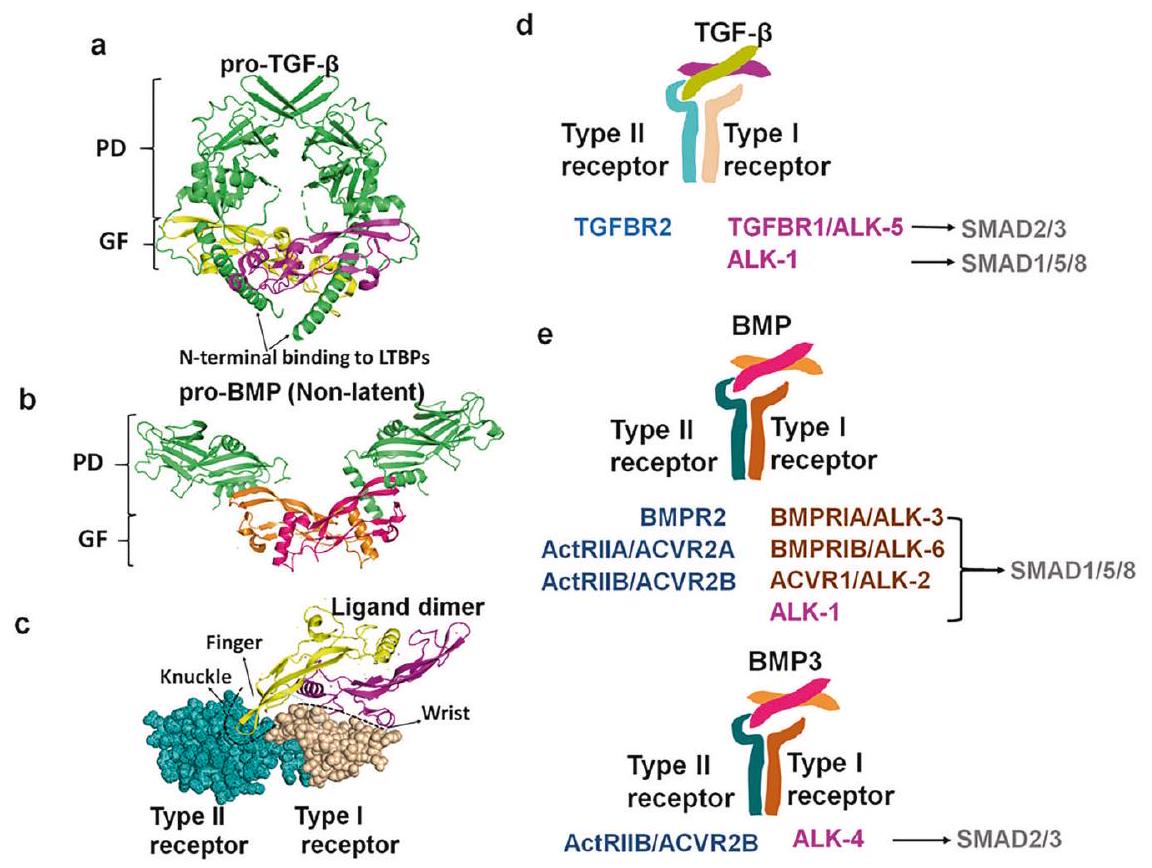
بروتين (LTBP) لتشكيل المركب الكامن الكبير (LLC)، الذي يرتبط ببروتينات مصفوفة خارج الخلية مثل الفيبريلين (FBN).
مستقبل (BMPRIA)/ALK3، مستقبل نوع BMP IB (BMPRIB)/ALK6، مستقبل نوع Activin I (ACVR1)/ALK2، وALK1.
الإشارات الكانونية وغير الكانونية
ت antagonize نشاطه النسخي لقمع تكوين العظام المستحث بواسطة BMP2.
المصفوفة المستهدفة
(مادة مستحثة بشكل خاص من الخلايا الدبقية القديمة)، وKLF4 (عامل كروبل الشبيه 4). قد تعمل هذه العوامل النسخية معًا أو في مجرى SMAD لتنظيم النتائج البيولوجية التي يسببها BMP2. باستخدام تقنيات RNA-seq وChIP-seq، قام يان وآخرون.
تGF-
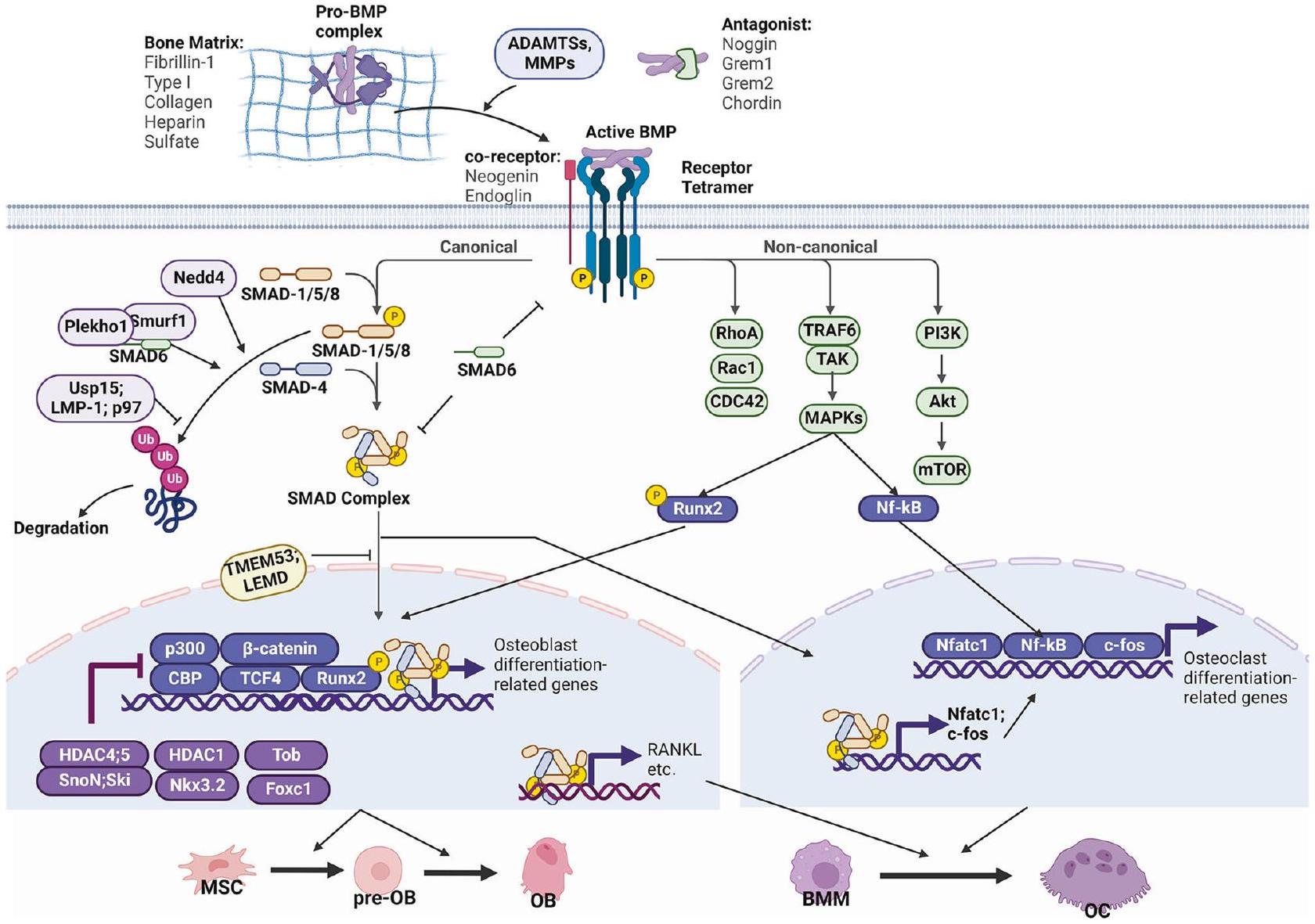
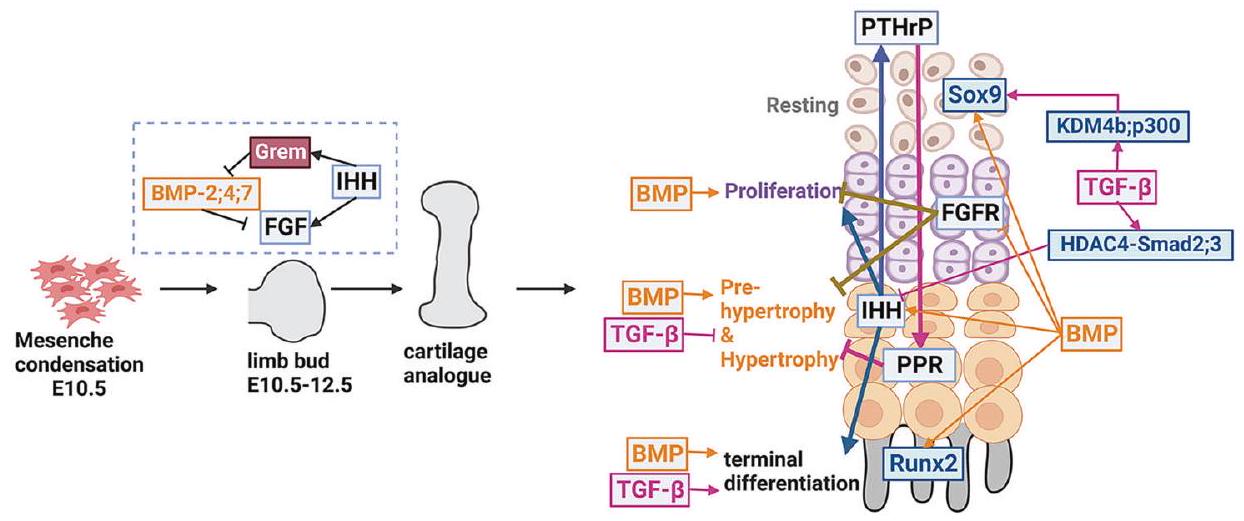
إشارة BMP في تطوير الهيكل العظمي
الأولي، وتشكيل الهيكل العظمي، وتطوير صفائح النمو (الشكل 4). كما ذُكر سابقاً، تتكون إشارة BMP من مجموعة متنوعة من الروابط والمستقبلات ذات affinities وأنماط ربط غير متجانسة، مما ينتج عنه نتائج فسيولوجية متغيرة. تمتلك روابط BMP أنماط تعبير مختلفة خلال تطوير الهيكل العظمي، مما يحدد وظائفها الفسيولوجية المتنوعة. على سبيل المثال، يتمتع Bmp14 ومستقبله Alk6 بنمط تعبير مقيد في العظام الزائدة.
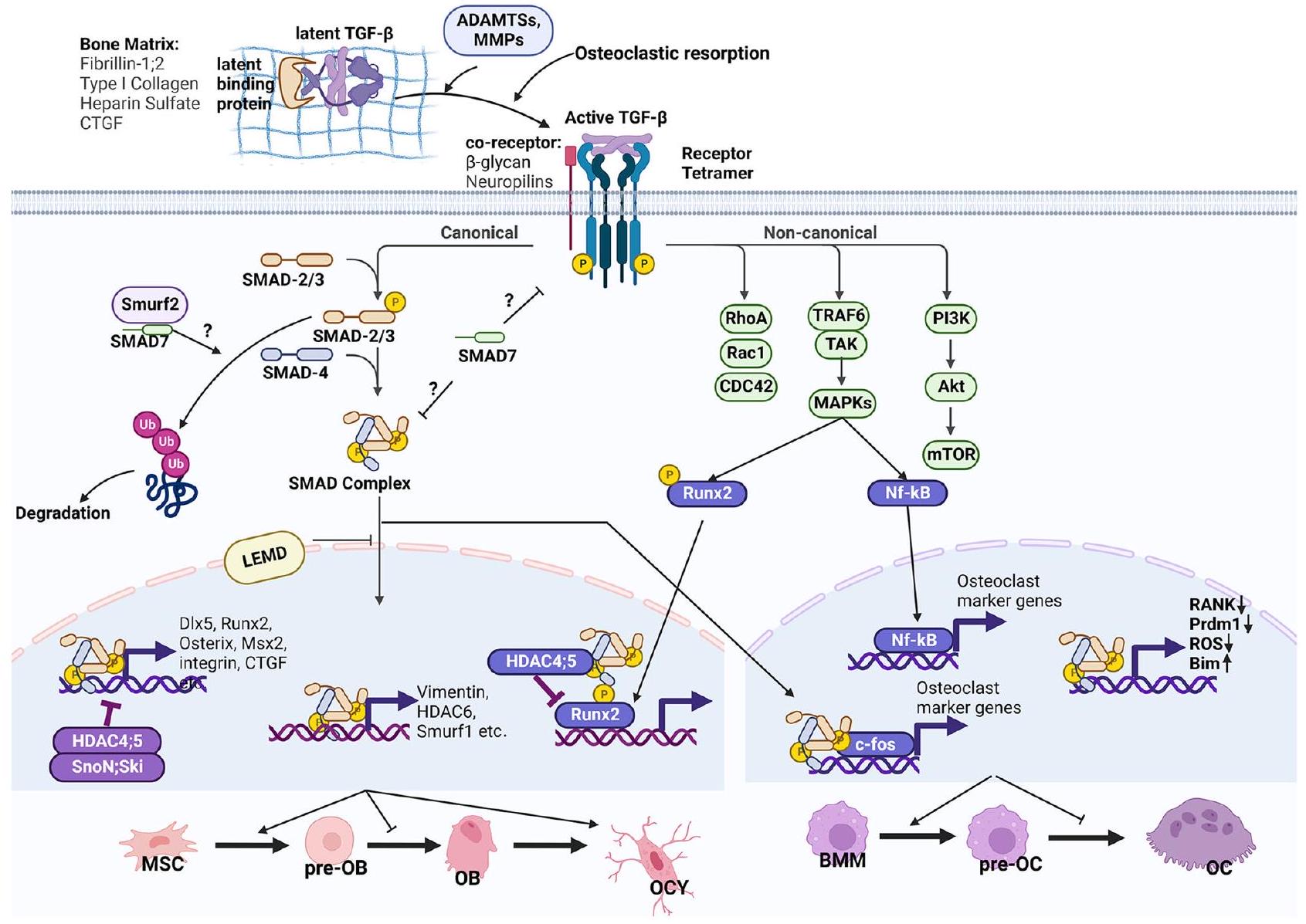
إشارة TGF-
M. Wu et al.
شدة، مما يشير إلى أن Smad2 يلعب دوراً أكثر أهمية من Smad3 في تطوير العظام الغضروفية.
إشارة TGF-
إشارات BMP في تكوين العظام وتمايز الخلايا العظمية
إشارة.
تGF-
BMP و TGF-
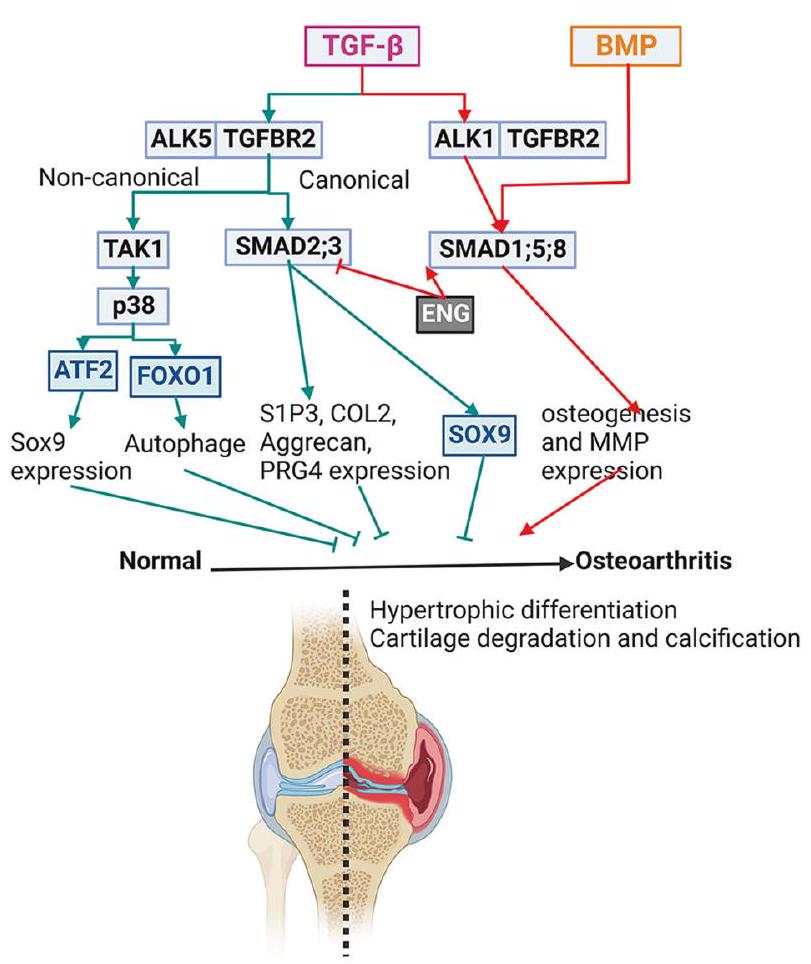
دور TGF-
م. وو وآخرون
لتحكم الخلايا العظمية في جودة العظام.
تGF-
من مستقبلات النوع I الخاصة بها، ALK1 و ALK5، لنقل الإشارات إلى SMAD1/5/8 و SMAD2/3، على التوالي، في الخلايا الغضروفية.
تنظيم TGF-
زمن الاستجابة والتحكم في إطلاق الروابط
تنشيط غير صحيح لـ TGF-
مضادات خارج الخلية
المستقبلات المساعدة
م. وو وآخرون
إنتاج.
آلية التنظيم في السيتوبلازم
تنظيم في النواة
إزالة الأسيتيل HDAC4 وHDAC5 كمثبطات مشتركة.
c-Fos، وهو عامل نسخ رئيسي للخلايا العظمية، يتفاعل مباشرة مع SMAD-2 و-3 لتعزيز تمايز الخلايا العظمية.
الاستنتاج ووجهات النظر
عدة جينات في TGF-
- لماذا BMP و TGF-
هل للإشارات وظائف ديناميكية؟ كما تم استعراضه هنا، يمكن أن يُجاب جزئيًا عن هذا السؤال من خلال تنوع تركيبات الليغاند-المستقبلات والشبكة التنظيمية المعقدة داخل الخلايا التي تسبب القراءة الديناميكية لـ TGF- وإشارات BMP. على وجه الخصوص، يتكون مسار إشارات BMP من عدة ليغاندات ومستقبلات تتفاعل بشكل عشوائي مع بعضها البعض. أظهرت سلسلة من الأعمال من مجموعة الدكتور مايكل ب. إلوويتز أن أنظمة تفاعل الليغاند-المستقبل العشوائية في إشارات BMP حاسمة لتنظيماتها الديناميكية. عملهم أوضح كيف تعالج مسارات BMP المدخلات متعددة الروابط باستخدام مجموعة من الآليات الحاسوبية، بما في ذلك الاستشعار النسبي، واكتشاف التوازن، واكتشاف عدم التوازن. نظرًا لأن الخلايا لديها أنماط تعبير مختلفة من المستقبلات والروابط، فإن نظام التفاعل المتنوع يسمح لعدد قليل من الروابط، التي تعمل في مجموعات، بمعالجة مسألة عدد أكبر من أنواع الخلايا الفردية. - ما هي الآلية النسخية التي تعمل على تحقيق تنوع النتائج النسخية التي تنشأ في أنواع الخلايا المختلفة استجابةً لنفس الليغاند؟ يتطلب الإجابة على هذا السؤال استخدام تقنيات متطورة مثل ChIP-seq و co-IP/MS و ATAC-seq و CUT&Tag-seq. حالة تعديل الحمض النووي والهستون تختلف في أنواع الخلايا المختلفة وقد تؤثر على ألفة ارتباط مركب SMAD مع الكروموسومات. لذلك، فإن تحليل العلامات الوراثية على تسلسلات ارتباط عوامل النسخ سيساعد في الإجابة على هذا السؤال. قد يفسر توصيف نمط تفاعل المستقبل-الليغاند وحالة الكروماتين في سياقات خلوية محددة أيضًا سبب TGF-
الإشارات لها وظائف تعتمد على المرحلة في معظم خلايا الهيكل العظمي. - كيفية تجاوز الآثار الجانبية لبروتينات BMP وTGF-
متى يتم تطبيقها في البيئات السريرية؟ زيادة BMP و TGF- يرتبط الإشارات بعدة شذوذات في أنسجة العظام. لذلك، هناك حاجة إلى مزيد من الدراسة والتدخل لمنع تلك الآثار الجانبية عند تطبيق BMPs و TGF- في البيئات السريرية. على الرغم من أن TGF- الإشارات تحافظ على تنكس الغضروف، TGF مفرط النشاط الإشارات تزيد من تفاقم التهاب المفاصل. على الرغم من وظائفه المزدوجة، TGF-
لا يزال يُقترح الإشارات كعلاج محتمل لتخفيف التهاب المفاصل العظمي، على الرغم من أنه يحتاج إلى مزيد من الدراسة لتصميم التوقيت والجرعة المناسبة للعلاج. - كيفية تعديل BMP و TGF- بأمان وفعالية
الإشارات في الاضطرابات الهيكلية الناتجة عن خللها؟ استهداف BMP و TGF- يُقترح الإشارات كاستراتيجية علاجية لعلاج أو اضطرابات التصلب العظمي بينما لا يزال العلاج الفعال قيد التطوير.
REFERENCES
- Moses, H. L., Roberts, A. B. & Derynck, R. The discovery and early days of TGF-
: A historical perspective. Cold Spring Harb. Perspect. Biol. 8, a021865 (2016). - Katagiri, T. & Watabe, T. Bone morphogenetic proteins. Cold Spring Harb. Perspect. Biol. 8, a021899 (2016).
- Derynck, R. & Budi, E. H. Specificity, versatility, and control of TGF-
family signaling. Sci. Signal. 12, eaav5183 (2019). - Mahmood, A., Harkness, L., Schrøder, H. D., Abdallah, B. M. & Kassem, M. Enhanced differentiation of human embryonic stem cells to mesenchymal progenitors by inhibition of TGF-beta/activin/nodal signaling using SB-431542. J. Bone Miner. Res. 25, 1216-1233 (2010).
- Cianciolo, G. et al. The role of activin: the other side of chronic kidney diseasemineral bone disorder? Nephrol. Dial. Transplant. 36, 966-974 (2021).
- Lee, S. J. et al. Targeting myostatin/activin A protects against skeletal muscle and bone loss during spaceflight. Proc. Natl. Acad. Sci. USA 117, 23942-23951 (2020).
- Maridas, D. E., et al. Chapter 48 – Bone morphogenetic proteins. In: Bilezikian J. P., Martin T. J., Clemens T. L., Rosen C. J. eds. Principles of Bone Biology (Fourth Edition): Academic Press: 1189-1197 (2020).
- Xu, X. & Cao, X. Chapter 47 – Transforming growth factor-
and skeletal homeostasis1. In: Bilezikian J. P., Martin T. J., Clemens T. L., Rosen C. J. eds. Principles of Bone Biology (Fourth Edition): Academic Press: 1153-1187 (2020). - Gipson, G. R. et al. Structural perspective of BMP ligands and signaling. Bone 140, 115549-115549 (2020).
- Shi, M. et al. Latent TGF-
structure and activation. Nature 474, 343-349 (2011). - Karsdal, M. A. et al. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J. Biol. Chem. 277, 44061-44067 (2002).
- Dallas, S. L., Rosser, J. L., Mundy, G. R. & Bonewald, L. F. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 277, 21352-21360 (2002).
- D’Angelo, M., Billings, P. C., Pacifici, M., Leboy, P. S. & Kirsch, T. Authentic matrix vesicles contain active metalloproteases (MMP). a role for matrix vesicleassociated MMP-13 in activation of transforming growth factor-beta. J. Biol. Chem. 276, 11347-11353 (2001).
- Wang, J. et al. Atp6i deficient mouse model uncovers transforming growth factor-
Smad as a key signaling pathway regulating odontoblast differentiation and tooth root formation. Int. J. Oral Sci. 15, 35 (2023). - Salmon, R. M. et al. Molecular basis of ALK1-mediated signalling by BMP9/ BMP10 and their prodomain-bound forms. Nat. Commun. 11, 1621 (2020).
- Neugebauer, J. M. et al. The prodomain of BMP4 is necessary and sufficient to generate stable BMP4/7 heterodimers with enhanced bioactivity in vivo. Proc. Natl. Acad. Sci. USA 112, E2307-E2316 (2015).
- Sengle, G., Ono, R. N., Sasaki, T. & Sakai, L. Y. Prodomains of transforming growth factor beta (TGFbeta) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 286, 5087-5099 (2011).
- Gregory, K. E. et al. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J. Biol. Chem. 280, 27970-27980 (2005).
- Martinez-Hackert, E., Sundan, A. & Holien, T. Receptor binding competition: A paradigm for regulating TGF-
family action. Cytokine Growth Factor Rev. 57, 39-54 (2021). - Finnson, K. W., Parker, W. L., ten Dijke, P., Thorikay, M. & Philip, A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res. 23, 896-906 (2008).
- Goumans, M. J. et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 12, 817-828 (2003).
- Mang, T. et al. BMPR1A is necessary for chondrogenesis and osteogenesis, whereas BMPR1B prevents hypertrophic differentiation. J. Cell Sci. 133, jcs246934 (2020).
- Zhu, D. et al. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1 mediated pathway. J. Cell Mol. Med. 19, 165-174 (2015).
- Kokabu, S. et al. BMP3 suppresses osteoblast differentiation of bone marrow stromal cells via interaction with Acvr2b. Mol. Endocrinol. 26, 87-94 (2012).
- Yi, S. E., Daluiski, A., Pederson, R., Rosen, V. & Lyons, K. M. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development 127, 621-630 (2000).
- van Caam, A. et al. The high affinity ALK1-ligand BMP9 induces a hypertrophylike state in chondrocytes that is antagonized by TGF
1. Osteoarthritis Cartilage 23, 985-995 (2015). - Zhang, D. et al. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J. Bone Miner. Res. 18, 1593-1604 (2003).
- Daluiski, A. et al. Bone morphogenetic protein- 3 is a negative regulator of bone density. Nat. Genet. 27, 84-88 (2001).
- Macias, M. J., Martin-Malpartida, P. & Massagué, J. Structural determinants of Smad function in TGF-
signaling. Trends Biochem. Sci. 40, 296-308 (2015). - Gámez, B., Rodríguez-Carballo, E., Graupera, M., Rosa, J. L. & Ventura, F. Class I PI-3-kinase signaling is critical for bone formation through regulation of SMAD1 activity in osteoblasts. J. Bone Miner. Res. 31, 1617-1630 (2016).
- Zhu, Y. et al. Crosstalk between Smad
and specific isoforms of ERK in TGF- induced TIMP-3 expression in rat chondrocytes. PLoS Genet. 21, 1781-1790 (2017). - Baron, R. et al. Balancing BMP signaling through integrated inputs into the Smad1 linker. Nat. Commun. 25, 441-454 (2007).
- Urata, M. et al. A peptide that blocks the interaction of NF-kB p65 subunit with Smad4 enhances BMP2-induced osteogenesis. J. Cell. Physiol. 233, 7356-7366 (2018).
- Sun, X. et al. TGF-
inhibits osteogenesis by upregulating the expression of ubiquitin ligase SMURF1 via MAPK-ERK signaling. J. Cell. Physiol. 233, 596-606 (2018). - Kua, H. Y. et al. c-Abl promotes osteoblast expansion by differentially regulating canonical and non-canonical BMP pathways and p16INK4a expression. Nat. Cell Biol. 14, 727-737 (2012).
- Martin-Malpartida, P. et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nat. Commun. 8, 2070 (2017).
- Miyazono, K., Maeda, S. & Imamura, T. BMP receptor signaling: Transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 16, 251-263 (2005).
- Omata, Y. et al. Genomewide comprehensive analysis reveals critical cooperation between Smad and c-Fos in RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 30, 869-877 (2015).
- Yu, S. et al. BMP2-dependent gene regulatory network analysis reveals Klf4 as a novel transcription factor of osteoblast differentiation. Cell Death Dis. 12, 197 (2021).
- Yan, J. et al. Smad4 deficiency impairs chondrocyte hypertrophy via the Runx2 transcription factor in mouse skeletal development. J. Biol. Chem. 293, 9162-9175 (2018).
- Berendsen, A. D. & Olsen, B. R. Bone development. Bone 80, 14-18 (2015).
- Long, F. & Ornitz, D. M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 5, a008334 (2013).
- Tang, C. Y. et al. Run
up-regulates chondrocyte to osteoblast lineage commitment and promotes bone formation by enhancing both chondrogenesis and osteogenesis. Biochem. J. 477, 2421-2438 (2020). - Tang, J. et al. Runt-related transcription factor 1 is required for murine osteoblast differentiation and bone formation. J. Biol. Chem. 295, 11669-11681 (2020).
- Tian, F. et al. Core binding factor beta (
) controls the balance of chondrocyte proliferation and differentiation by upregulating Indian hedgehog (Ihh) expression and inhibiting parathyroid hormone-related protein receptor (PPR) expression in postnatal cartilage and bone formation. J. Bone Miner. Res. 29, 1564-1574 (2014). - Wu, M. et al. Deletion of core-binding factor
in mesenchymal progenitor cells provides new insights into Runxs complex function in cartilage and bone development. Bone 65, 49-59 (2014). - Wu, M. et al. Chondrocyte-specific knockout of
reveals the indispensable function of in chondrocyte maturation, growth plate development and trabecular bone formation in mice. Int. J. Biol. Sci. 10, 861-872 (2014). - Yoon, B. S. et al. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. USA 102, 5062-5067 (2005).
- Rigueur, D. et al. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J. Bone Miner. Res. 30, 733-741 (2015).
- Shu, B. et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 124, 3428-3440 (2011).
- Bandyopadhyay, A. et al. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2, e216 (2006).
- Selever, J., Liu, W., Lu, M. F., Behringer, R. R. & Martin, J. F. Bmp4 in limb bud mesoderm regulates digit pattern by controlling AER development. Dev. Biol. 276, 268-279 (2004).
- Nissim, S., Hasso, S. M., Fallon, J. F. & Tabin, C. J. Regulation of Gremlin expression in the posterior limb bud. Dev. Biol. 299, 12-21 (2006).
- Maatouk, D. M., Choi, K. S., Bouldin, C. M. & Harfe, B. D. In the limb AER Bmp2 and Bmp4 are required for dorsal-ventral patterning and interdigital cell death but not limb outgrowth. Dev. Biol. 327, 516-523 (2009).
- Choi, K. S., Lee, C., Maatouk, D. M. & Harfe, B. D. Bmp2, Bmp4 and Bmp7 are corequired in the mouse AER for normal digit patterning but not limb outgrowth. PLoS One 7, e37826 (2012).
- Pajni-Underwood, S., Wilson, C. P., Elder, C., Mishina, Y. & Lewandoski, M. BMP signals control limb bud interdigital programmed cell death by regulating FGF signaling. Development 134, 2359-2368 (2007).
- Benazet, J. D. & Zeller, R. Dual requirement of ectodermal Smad4 during AER formation and termination of feedback signaling in mouse limb buds. Genesis 51, 660-666 (2013).
- Pignatti, E., Zeller, R. & Zuniga, A. To BMP or not to BMP during vertebrate limb bud development. Semin. Cell Dev. Biol. 32, 119-127 (2014).
- Takano, K. et al. A novel nonsense mutation in the NOG gene causes familial NOGrelated symphalangism spectrum disorder. Hum. Genome Var. 3, 16023 (2016).
- Seemann, P. et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J. Clin. Invest. 115, 2373-2381 (2005).
- Dixon, M. E., Armstrong, P., Stevens, D. B. & Bamshad, M. Identical mutations in NOG can cause either tarsal/carpal coalition syndrome or proximal symphalangism. Genet. Med. 3, 349-353 (2001).
- Lehmann, K. et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am. J. Hum. Genet. 81, 88-396 (2007).
- Dathe, K. et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am. J. Hum. Genet. 84, 483-492 (2009).
- Lehmann, K. et al. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc. Natl. Acad. Sci. USA 100, 12277-12282 (2003).
- Yoon, B. S. et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 133, 4667-4678 (2006).
- Brunet, L. J., McMahon, J. A., McMahon, A. P. & Harland, R. M. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science 280, 1455-1457 (1998).
- McMahon, J. A. et al. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 12, 1438-1452 (1998).
- Keller, B. et al. Interaction of TGF
and BMP signaling pathways during chondrogenesis. PLoS One 6, e16421 (2011). - Retting, K. N., Song, B., Yoon, B. S. & Lyons, K. M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 136, 1093-1104 (2009).
- Tsumaki, N. et al. Role of CDMP-1 in skeletal morphogenesis: promotion of mesenchymal cell recruitment and chondrocyte differentiation. J. Cell Biol. 144, 161-173 (1999).
- Gao, L. et al. TAK1 regulates SOX9 expression in chondrocytes and is essential for postnatal development of the growth plate and articular cartilages. J. Cell Sci. 126, 5704-5713 (2013).
- Bertoli-Avella, A. M. et al. Mutations in a TGF-
ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 65, 1324-1336 (2015). - Lindsay, M. E. et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 44, 922-927 (2012).
- van de Laar, I. M. et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 43, 121-126 (2011).
- Loeys, B. L. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 37, 275-281 (2005).
- Vandeloo, B. et al. Spontaneous coronary artery dissection in a man with a novel missense mutation in SMAD2 treated by optical coherence tomography-guided percutaneous coronary intervention. JACC Cardiovasc. Interv. 12, e45-e47 (2019).
- Milewicz, D. M. et al. Marfan syndrome. Nat. Rev. Dis. Primers 7, 64 (2021).
- Seo, H. S. & Serra, R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev. Biol. 310, 304-316 (2007).
- Baffi, M. O. et al. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev. Biol. 276, 124-142 (2004).
- Sohn, P., Cox, M., Chen, D. & Serra, R. Molecular profiling of the developing mouse axial skeleton: a role for Tgfbr2 in the development of the intervertebral disc. BMC Dev. Biol. 10, 29 (2010).
- Spagnoli, A. et al. TGF-beta signaling is essential for joint morphogenesis. J. Cell Biol. 177, 1105-1117 (2007).
- Longobardi, L. et al. TGF-
type II receptor/MCP-5 axis: at the crossroad between joint and growth plate development. Cancers 23, 71-81 (2012). - Matsunobu, T. et al. Critical roles of the TGF-beta type I receptor ALK5 in perichondrial formation and function, cartilage integrity, and osteoblast differentiation during growth plate development. Dev. Biol. 332, 325-338 (2009).
- Sueyoshi, T., Yamamoto, K. & Akiyama, H. Conditional deletion of Tgfbr2 in hypertrophic chondrocytes delays terminal chondrocyte differentiation. Matrix Biol. 31, 352-359 (2012).
- Sanford, L. P. et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124, 2659-2670 (1997).
- Proetzel, G. et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 11, 409-414 (1995).
- Kaartinen, V. et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 11, 415-421 (1995).
- Kulkarni, A. B. et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 90, 770-774 (1993).
- Wang, W., Song, B., Anbarchian, T., Shirazyan, A. & Sadik, J. E. Smad2 and Smad3 regulate chondrocyte proliferation and differentiation in the growth plate. PLOS Genet. 12, e1006352 (2016).
- Edwards, J. R. & Mundy, G. R. Advances in osteoclast biology: old findings and new insights from mouse models. Nat. Rev. Rheumatol. 7, 235-243 (2011).
- Long, F. Building strong bones: molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 13, 27-38 (2011).
- Chen, W. et al.
deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of required for skeletal development. Proc. Natl. Acad. Sci. USA 111, 8482-8487 (2014). - Wu, M. et al. Cbf
governs osteoblast-adipocyte lineage commitment through enhancing -catenin signaling and suppressing adipogenesis gene expression. Proc. Natl. Acad. Sci. USA 114, 10119-10124 (2017). - Tang, C. Y. et al. Runx1 is a central regulator of osteogenesis for bone homeostasis by orchestrating BMP and WNT signaling pathways. PLoS Genet. 17, e1009233 (2021).
- Delgado-Calle, J. & Bellido, T. The osteocyte as a signaling cell. Physiol. Rev. 102, 379-410 (2022).
- Langdahl, B. L., Carstens, M., Stenkjaer, L. & Eriksen, E. F. Polymorphisms in the transforming growth factor beta 1 gene and osteoporosis. Bone 32, 297-310 (2003).
- Panach, L. et al. Comparative transcriptome analysis identifies CARM1 and DNMT3A as genes associated with osteoporosis. Sci. Rep. 10, 16298 (2020).
- Gregson, C. L. et al. Genome-wide association study of extreme high bone mass: Contribution of common genetic variation to extreme BMD phenotypes and potential novel BMD-associated genes. Bone 114, 62-71 (2018).
- Pei, Y. F. et al. Genome-wide association meta-analyses identified 1 q 43 and 2q32.2 for hip Ward’s triangle areal bone mineral density. Bone 91, 1-10 (2016).
- He, J. W., Yue, H., Hu, W. W., Hu, Y. Q. & Zhang, Z. L. Contribution of the sclerostin domain-containing protein 1 (SOSTDC1) gene to normal variation of peak bone mineral density in Chinese women and men. J. Bone Miner. Metab. 29, 571-581 (2011).
- Moffett, S. P. et al. Identification and association analysis of single nucleotide polymorphisms in the human noggin (NOG) gene and osteoporosis phenotypes. Bone 44, 999-1002 (2009).
- Wang, H. et al. Association of bone morphogenetic protein-2 gene polymorphisms with susceptibility to ossification of the posterior longitudinal ligament of the spine and its severity in Chinese patients. Eur. Spine J. 17, 956-964 (2008).
- Lin, G. T. et al. SNP combinations in chromosome-wide genes are associated with bone mineral density in Taiwanese women. Chin. J. Physiol. 51, 32-41 (2008).
- Medici, M. et al. BMP-2 gene polymorphisms and osteoporosis: the Rotterdam Study. J. Bone Miner. Res. 21, 845-854 (2006).
- Gregson, C. L. et al. A rare mutation in SMAD9 associated with high bone mass identifies the SMAD-dependent BMP signaling pathway as a potential anabolic target for osteoporosis. J. Bone Miner. Res. 35, 92-105 (2020).
- Kim, B. J. et al. Association of SMAD2 polymorphisms with bone mineral density in postmenopausal Korean women. Osteoporos. Int. 22, 2273-2282 (2011).
- Lowery, J. W. & Rosen, V. Bone morphogenetic protein-based therapeutic approaches. Cold Spring Harb. Perspect. Biol. 10, a022327 (2018).
- Begam, H., Nandi, S. K., Kundu, B. & Chanda, A. Strategies for delivering bone morphogenetic protein for bone healing. Mater. Sci. Eng. C Mater. Biol. Appl. 70, 856-869 (2017).
- Bharadwaz, A. & Jayasuriya, A. C. Osteogenic differentiation cues of the bone morphogenetic protein-9 (BMP-9) and its recent advances in bone tissue regeneration. Mater. Sci. Eng. C Mater. Biol. Appl. 120, 111748 (2021).
- Eiraku, N. et al. BMP9 directly induces rapid GSK3-
phosphorylation in a Wntindependent manner through class I PI3K-Akt axis in osteoblasts. FASEB J. 33, 12124-12134 (2019). - Tsuji, K. et al. BMP4 is dispensable for skeletogenesis and fracture-healing in the limb. J. Bone Joint Surg. Am. 90, 14-18 (2008).
- Tsuji, K. et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet. 38, 1424-1429 (2006).
- Hu, Y. Acvr1 deletion in osteoblasts impaired mandibular bone mass through compromised osteoblast differentiation and enhanced sRANKL-induced osteoclastogenesis. J. Cell. Physiol. 236, 4580-4591 (2021).
- Yang, C., Yang, L., Wan, M. & Cao, X. Generation of a mouse model with expression of bone morphogenetic protein type II receptor lacking the cytoplasmic domain in osteoblasts. Ann. N. Y. Acad. Sci. 1192, 286-291 (2010).
- Gamer, L. W., Cox, K., Carlo, J. M. & Rosen, V. Overexpression of BMP3 in the developing skeleton alters endochondral bone formation resulting in spontaneous rib fractures. Dev. Dyn. 238, 2374-2381 (2009).
- Greenblatt, M. B. et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Invest. 120, 2457-2473 (2010).
- Thouverey, C. & Caverzasio, J. The p38a MAPK positively regulates osteoblast function and postnatal bone acquisition. Cell Mol. Life Sci. 69, 3115-3125 (2012).
- Salazar, V. S. et al. Embryonic ablation of osteoblast Smad4 interrupts matrix synthesis in response to canonical Wnt signaling and causes an osteogenesis-imperfecta-like phenotype. J. Cell Sci. 126, 4974-4984 (2013).
- Salazar, V. S. et al. Postnatal ablation of osteoblast Smad4 enhances proliferative responses to canonical Wnt signaling through interactions with
-catenin. J. Cell Mol. Med. 126, 5598-5609 (2013). - Zhang, J. et al. The inhibition effects of insulin on BMP2-induced muscle heterotopic ossification. Biomaterials 35, 9322-9331 (2014).
- Agarwal, S. et al. Strategic targeting of multiple BMP receptors prevents traumainduced heterotopic ossification. Mol. Ther. 25, 1974-1987 (2017).
- van Dinther, M. et al. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 25, 1208-1215 (2010).
- Fukuda, T. et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem. Biophys. Res. Commun. 377, 905-909 (2008).
- Hatsell, S. J. et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 7, 303ra137 (2015).
- Hino, K. et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 112, 15438-15443 (2015).
- Billings, P. C. et al. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 23, 305-313 (2008).
- Yu, P. B. et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat. Med. 14, 1363-1369 (2008).
- Lees-Shepard, J. B. et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 9, 471 (2018).
- Chakkalakal, S. A. et al. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 27, 1746-1756 (2012).
- Fukuda, T. et al. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 44, 159-167 (2016).
- Lodberg, A. Principles of the activin receptor signaling pathway and its inhibition. Cytokine Growth Factor Rev. 60, 1-17 (2021).
- Shimono, K. et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-
agonists. Nat. Med. 17, 454-460 (2011). - Pignolo, R. J. et al. Reduction of new heterotopic ossification (HO) in the openlabel, phase 3 MOVE trial of palovarotene for fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 38, 381-394 (2023).
- Meng, X., Wang, H. & Hao, J. Recent progress in drug development for fibrodysplasia ossificans progressiva. Mol. Cell. Biochem. 477, 2327-2334 (2022).
- Williams, E. et al. Saracatinib is an efficacious clinical candidate for fibrodysplasia ossificans progressiva. JCI Insight 6, e95042 (2021).
- Wang, Q. et al. Bone morphogenetic protein 2 activates Smad6 gene transcription through bone-specific transcription factor Runx2. J. Biol. Chem. 282, 10742-10748 (2007).
- Jeon, E. J. et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J. Biol. Chem. 281, 16502-16511 (2006).
- Jun, J. H. et al. BMP2-activated Erk/MAP kinase stabilizes Runx2 by increasing p300 levels and histone acetyltransferase activity. J. Biol. Chem. 285, 36410-36419 (2010).
- Rodríguez-Carballo, E. et al. Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways. J. Bone Miner. Res. 26, 718-729 (2011).
- Pawaputanon Na Mahasarakham, C. et al. BMP-2 enhances Lgr4 gene expression in osteoblastic cells. J. Cell. Physiol. 231, 887-895 (2016).
- Ko, F. C. et al. Acute phosphate restriction impairs bone formation and increases marrow adipose tissue in growing mice. J. Bone Miner. Res. 31, 2204-2214 (2016).
- Yang, G. et al. BMP-2 induction of Dlx3 expression is mediated by p38/ Smad5 signaling pathway in osteoblastic MC3T3-E1 cells. J. Cell. Physiol. 229, 943-954 (2014).
- Hopkins, A., Mirzayans, F. & Berry, F. Foxc1 expression in early osteogenic differentiation is regulated by BMP4-SMAD activity. J. Cell. Biochem. 117, 1707-1717 (2016).
- Ramazzotti, G. et al. BMP-2 induced expression of PLC
that is a positive regulator of osteoblast differentiation. J. Cell. Physiol. 231, 623-629 (2016). - Guo, Y. et al. BMP-IHH-mediated interplay between mesenchymal stem cells and osteoclasts supports calvarial bone homeostasis and repair. Bone Res. 6, 30 (2018).
- Liu, Z. et al. Molecules mimicking Smad1 interacting with Hox stimulate bone formation. J. Biol. Chem. 279, 11313-11319 (2004).
- Lim, J. et al. Dual function of Bmpr1a signaling in restricting preosteoblast proliferation and stimulating osteoblast activity in mouse. Development 143, 339-347 (2016).
- Zhang, H. et al. Loss of BMP signaling mediated by BMPR1A in osteoblasts leads to differential bone phenotypes in mice depending on anatomical location of the bones. Bone 137, 115402 (2020).
- Zhang, Y. et al. Loss of BMP signaling through BMPR1A in osteoblasts leads to greater collagen cross-link maturation and material-level mechanical properties in mouse femoral trabecular compartments. Bone 88, 74-84 (2016).
- Kamiya, N. et al. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J. Bone Miner. Res. 25, 200-210 (2010).
- Liu, Z., Tang, Y., Qiu, T., Cao, X. & Clemens, T. L. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J. Biol. Chem. 281, 17156-17163 (2006).
- Tang, Y. et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 15, 757-765 (2009).
- Seo, H. S. & Serra, R. Tgfbr2 is required for development of the skull vault. Dev. Biol. 334, 481-490 (2009).
- Peters, S. B., Wang, Y. & Serra, R. Tgfbr2 is required in osterix expressing cells for postnatal skeletal development. Bone 97, 54-64 (2017).
- Corps, K., Stanwick, M., Rectenwald, J., Kruggel, A. & Peters, S. B. Skeletal deformities in Osterix-Cre;Tgfbr2(f/f) mice may cause postnatal death. Genes 12, 975 (2021).
- Borton, A. J., Frederick, J. P., Datto, M. B., Wang, X. F. & Weinstein, R. S. The loss of Smad3 results in a lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J. Bone Miner. Res. 16, 1754-1764 (2001).
- Wang, Y., Cox, M. K., Coricor, G., MacDougall, M. & Serra, R. Inactivation of Tgfbr2 in Osterix-Cre expressing dental mesenchyme disrupts molar root formation. Dev. Biol. 382, 27-37 (2013).
- Kinoshita, A. et al. Domain-specific mutations in TGFB1 result in CamuratiEngelmann disease. Nat. Genet. 26, 19-20 (2000).
- Velchev, J. D., Verstraeten, A. & Loeys, B. Hide and seek: Somatic SMAD3 mutations in melorheostosis. J. Exp. Med. 217, e20200185 (2020).
- Kang, H. et al. Somatic SMAD3-activating mutations cause melorheostosis by up-regulating the TGF-
SMAD. Pathway. J. Exp. Med. 217, e20191499 (2020). - Kang, H. et al. Somatic activating mutations in MAP2K1 cause melorheostosis. Nat. Commun. 9, 1390 (2018).
- Nesti, L. J. et al. TGF-beta1-stimulated osteoblasts require intracellular calcium signaling for enhanced alpha5 integrin expression. Ann. N. Y. Acad. Sci. 961, 178-182 (2002).
- Arnott, J. A. et al. Molecular requirements for induction of CTGF expression by TGF-beta1 in primary osteoblasts. Bone 42, 871-885 (2008).
- Li, J. et al. Smad2 overexpression enhances Smad4 gene expression and suppresses CBFA1 gene expression in osteoblastic osteosarcoma ROS17/2.8 cells and primary rat calvaria cells. J. Biol. Chem. 273, 31009-31015 (1998).
- Lin, H. T. et al. Dynamic expression of SMAD3 is critical in osteoblast differentiation of PDMCs. Int. J. Mol. Med. 43, 1085-1093 (2019).
- Kang, J. S., Alliston, T., Delston, R. & Derynck, R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 24, 2543-2555 (2005).
- Hjelmeland, A. B., Schilling, S. H., Guo, X., Quarles, D. & Wang, X. F. Loss of Smad3-mediated negative regulation of Runx2 activity leads to an alteration in cell fate determination. Mol. Cell. Biol. 25, 9460-9468 (2005).
- Lian, N. et al. Transforming growth factor
suppresses osteoblast differentiation via the vimentin activating transcription factor 4 (ATF4) axis. J. Biol. Chem. 287, 35975-35984 (2012). - Ehnert, S. et al. TGF-
impairs mechanosensation of human osteoblasts via HDAC6-mediated shortening and distortion of primary cilia. J. Mol. Med. 95, 653-663 (2017). - Nam, B. & Park, H. TGF
suppressed matrix mineralization of osteoblasts differentiation by regulating SMURF1-C/EBP -DKK1 axis. Int. J. Mol. Sci. 21, 9771 (2020). - Ochiai, H. et al. Inhibition of insulin-like growth factor-1 (IGF-1) expression by prolonged transforming growth factor-
(TGF- ) administration suppresses osteoblast differentiation. J. Biol. Chem. 287, 22654-22661 (2012). - Qiu, T. et al. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat. Cell Biol. 12, 224-234 (2010).
- Kwok, S., Qin, L., Partridge, N. C. & Selvamurugan, N. Parathyroid hormone stimulation and PKA signaling of latent transforming growth factor-beta binding protein-1 (LTBP-1) mRNA expression in osteoblastic cells. J. Cell. Biochem. 95, 1002-1011 (2005).
- Sowa, H. et al. Parathyroid hormone-Smad3 axis exerts anti-apoptotic action and augments anabolic action of transforming growth factor beta in osteoblasts. J. Biol. Chem. 278, 52240-52252 (2003).
- Kamiya, N. et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J. Bone Miner. Res. 23, 2007-2017 (2008).
- Kamiya, N. et al. Targeted disruption of BMP signaling through type IA receptor (BMPR1A) in osteocyte suppresses SOST and RANKL, leading to dramatic increase in bone mass, bone mineral density and mechanical strength. Bone 91, 53-63 (2016).
- Tan, X. et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J. Cell Sci. 120, 2162-2170 (2007).
- Moon, Y. J. et al. Smad4 controls bone homeostasis through regulation of osteoblast/osteocyte viability. Exp. Mol. Med. 48, e256 (2016).
- Wu, H. et al. Inhibitory effects of combined bone morphogenetic protein 2, vascular endothelial growth factor, and basic fibroblast growth factor on osteoclast differentiation and activity. Tissue Eng. Part A 27, 1387-1398 (2021).
- Tasca, A. et al. Smad1/5 and Smad4 expression are important for osteoclast differentiation. J. Cell. Biochem. 116, 1350-1360 (2015).
- Miao, X. et al. Bone morphogenetic protein-2 promotes osteoclasts-mediated osteolysis via Smad1 and p65 signaling pathways. Spine 46, E234-E242 (2021).
- Omi, M., Kaartinen, V. & Mishina, Y. Activin A receptor type 1-mediated BMP signaling regulates RANKL-induced osteoclastogenesis via canonical SMADsignaling pathway. J. Biol. Chem. 294, 17818-17836 (2019).
- Okamoto, M. et al. Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J. Bone Miner. Res. 26, 2511-2522 (2011).
- Crane, J. L. & Cao, X. Bone marrow mesenchymal stem cells and TGF-
signaling in bone remodeling. J. Clin. Invest. 124, 466-472 (2014). - Karsdal, M. A. et al. Transforming growth factor-beta controls human osteoclastogenesis through the p38 MAPK and regulation of RANK expression. J. Biol. Chem. 278, 44975-44987 (2003).
- Morita, M. et al. Smad4 is required to inhibit osteoclastogenesis and maintain bone mass. Sci. Rep. 6, 35221 (2016).
- Pan, W. et al. SIS3 suppresses osteoclastogenesis and ameliorates bone loss in ovariectomized mice by modulating Nox4-dependent reactive oxygen species. Biochem. Pharmacol. 195, 114846 (2022).
- Houde, N., Chamoux, E., Bisson, M. & Roux, S. Transforming growth factor-beta1 (TGF-beta1) induces human osteoclast apoptosis by up-regulating Bim. J. Biol. Chem. 284, 23397-23404 (2009).
- Quinn, J. M. et al. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J. Bone Miner. Res. 16, 1787-1794 (2001).
- Dole, N. S. et al. Osteocyte-intrinsic TGF-
signaling regulates bone quality through perilacunar/canalicular remodeling. Cell Rep. 21, 2585-2596 (2017). - Dole, N. S., Yee, C. S., Mazur, C. M., Acevedo, C. & Alliston, T. TGF
regulation of perilacunar/canalicular remodeling is sexually dimorphic. J. Bone Miner. Res. 35, 1549-1561 (2020). - Schurman, C. A., Verbruggen, S. W. & Alliston, T. Disrupted osteocyte connectivity and pericellular fluid flow in bone with aging and defective TGF-
signaling. Proc. Natl. Acad. Sci. USA 118, e2023999118 (2021). - Liu, W. et al. TGF-
facilitates cell-cell communication in osteocytes via con-nexin43- and pannexin1-dependent gap junctions. Cell Death Discov. 5, 141 (2019). - Jähn, K. et al. Phenotype and viability of MLO-Y4 cells is maintained by
in a serum-dependent manner within a 3D-co-culture with MG-63 cells. Int. J. Mol. Sci. 19, 1932 (2018). - Bailey, K. N. et al. Mechanosensitive control of articular cartilage and subchondral bone homeostasis in mice requires osteocytic transforming growth factor
signaling. Arthritis Rheumatol. 73, 414-425 (2021). - Grol, M. W. & Lee, B. H. Gene therapy for repair and regeneration of bone and cartilage. Curr. Opin. Pharmacol. 40, 59-66 (2018).
- Patil, A. S., Sable, R. B. & Kothari, R. M. An update on transforming growth factor
(TGF- ): sources, types, functions and clinical applicability for cartilage/bone healing. J. Cell. Physiol. 226, 3094-3103 (2011). - He, Y. et al. Reduction of Smad2 caused by oxidative stress leads to necrotic death of hypertrophic chondrocytes associated with an endemic osteoarthritis. Rheumatology 61, 440-451 (2021).
- Li, T. F. et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J. Bone Miner. Res. 21, 4-16 (2006).
- Motaung, S. C. K. M., Di Cesare, P. E. & Hari Reddi, A. Differential response of cartilage oligomeric matrix protein (COMP) to morphogens of bone morphogenetic protein/transforming growth factor-
family in the surface, middle and deep zones of articular cartilage. J. Tissue Eng. Regen. Med. 5, e87-e96 (2011). - Niikura, T. & Reddi, A. H. Differential regulation of lubricin/superficial zone protein by transforming growth factor
bone morphogenetic protein superfamily members in articular chondrocytes and synoviocytes. Arthritis Rheum. 56, 2312-2321 (2007). - Malemud, C. J., Killeen, W., Hering, T. M. & Purchio, A. F. Enhanced sulfatedproteoglycan core protein synthesis by incubation of rabbit chondrocytes with recombinant transforming growth factor-
. J. Cell. Physiol. 149, 152-159 (1991). - Takahashi, N. et al. Elucidation of IL-1/TGF-
interactions in mouse chondrocyte cell line by genome-wide gene expression. Osteoarthritis Cartilage 13, 426-438 (2005). - Wiegertjes, R. et al. TGF-
dampens IL- 6 signaling in articular chondrocytes by decreasing IL-6 receptor expression. Osteoarthritis Cartilage 27, 1197-1207 (2019). - Cherian, J. J. et al. Preliminary results of a phase II randomized study to determine the efficacy and safety of genetically engineered allogeneic human chondrocytes expressing TGF-
in patients with grade 3 chronic degenerative joint disease of the knee. Osteoarthritis Cartilage 23, 2109-2118 (2015). - Ramaswamy, G., Sohn, P., Eberhardt, A. & Serra, R. Altered responsiveness to TGF-
results in reduced Papss2 expression and alterations in the biomechanical properties of mouse articular cartilage. Arthritis Res. Ther. 14, R49 (2012). - van Caam, A. et al. TGF
-induced SMAD2 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthritis Res. Ther. 19, 112 (2017). - Wang, Q. & Tan, Q. Postnatal deletion of Alk5 gene in meniscal cartilage accelerates age-dependent meniscal degeneration in mice. J. Cell. Physiol. 234, 595-605 (2018).
- Shen, J. et al. Deletion of the transforming growth factor
receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 65, 3107-3119 (2013). - Namdari, S., Wei, L., Moore, D. & Chen, Q. Reduced limb length and worsened osteoarthritis in adult mice after genetic inhibition of p38 MAP kinase activity in cartilage. Arthritis Rheum. 58, 3520-3529 (2008).
- Frazier, K. et al. Inhibition of ALK5 signaling induces physeal dysplasia in rats. Toxicol. Pathol. 35, 284-295 (2007).
- Prasadam, I. et al. Inhibition of p38 pathway leads to OA-like changes in a rat animal model. Rheumatology 51, 813-823 (2012).
- Wang, C., Shen, J., Ying, J., Xiao, D. & O’Keefe, R. J. FoxO1 is a crucial mediator of TGF-
TAK1 signaling and protects against osteoarthritis by maintaining articular cartilage homeostasis. Proc. Natl. Acad. Sci. USA 117, 30488-30497 (2020). - Mori, H., Izawa, T. & Tanaka, E. Smad3 deficiency leads to mandibular condyle degradation via the sphingosine 1-phosphate (S1P)/S1P3 signaling axis. Am. J. Pathol. 185, 2742-2756 (2015).
- Wang, H., Zhang, J., Sun, Q. & Yang, X. Altered gene expression in articular chondrocytes of Smad3(ex8/ex8) mice, revealed by gene profiling using microarrays. J. Genet. Genomics 34, 698-708 (2007).
- Zhang, Y. et al. Runx1 is a key regulator of articular cartilage homeostasis by orchestrating YAP, TGF
, and Wnt signaling in articular cartilage formation and osteoarthritis. Bone Res. 10, 63 (2022). - Ueland, T. et al. Increased serum and bone matrix levels of transforming growth factor {beta}1 in patients with GH deficiency in response to GH treatment. Eur. J. Endocrinol. 165, 393-400 (2011).
- Pombo-Suarez, M., Castaño-Oreja, M. T., Calaza, M., Gomez-Reino, J. & Gonzalez, A. Differential upregulation of the three transforming growth factor beta isoforms in human osteoarthritic cartilage. Ann. Rheum. Dis. 68, 568-571 (2009).
- Blaney Davidson, E. N., Vitters, E. L., van der Kraan, P. M. & van den Berg, W. B. Expression of transforming growth factor-beta (TGFbeta) and the TGFbeta signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: role in cartilage degradation, chondrogenesis and osteophyte formation. Ann. Rheum. Dis. 65, 1414-1421 (2006).
- François, R. J., Neure, L., Sieper, J. & Braun, J. Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumour necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Ann. Rheum. Dis. 65, 713-720 (2006).
- Zhen, G. et al. Inhibition of TGF-
signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 19, 704-712 (2013). - Zheng, L. et al. Aberrant activation of latent transforming growth factor-
initiates the onset of temporomandibular joint osteoarthritis. Bone Res. 6, 26 (2018). - Blaney Davidson, E. N. et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 182, 7937-7945 (2009).
- Occhetta, P. et al. Developmentally inspired programming of adult human mesenchymal stromal cells toward stable chondrogenesis. Proc. Natl. Acad. Sci. USA 115, 4625-4630 (2018).
- Janssens, K., ten Dijke, P., Janssens, S. & Van Hul, W. Transforming growth factor
to the bone. Endocr. Rev. 26, 743-774 (2005). - Dabovic, B. et al. Osteopetrosis-like phenotype in latent TGF-beta binding protein 3 deficient mice. Bone 37, 25-31 (2005).
- Dabovic, B. et al. Bone defects in latent TGF-beta binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-beta presentation. J. Endocrinol. 175, 129-141 (2002).
- Koli, K., Ryynänen, M. J. & Keski-Oja, J. Latent TGF-beta binding proteins (LTBPs)1 and -3 coordinate proliferation and osteogenic differentiation of human mesenchymal stem cells. Bone 43, 679-688 (2008).
- Kitahama, S. et al. Expression of fibrillins and other microfibril-associated proteins in human bone and osteoblast-like cells. Bone 27, 61-67 (2000).
- Putnam, E. A., Zhang, H., Ramirez, F. & Milewicz, D. M. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat. Genet. 11, 456-458 (1995).
- Nistala, H. et al. Fibrillin-1 and -2 differentially modulate endogenous TGF-
and BMP bioavailability during bone formation. J. Cell Biol. 190, 1107-1121 (2010). - Nistala, H., Lee-Arteaga, S., Smaldone, S., Siciliano, G. & Ramirez, F. Extracellular microfibrils control osteoblast-supported osteoclastogenesis by restricting TGF{beta} stimulation of RANKL production. J. Biol. Chem. 285, 34126-34133 (2010).
- Craft, C. S. et al. Oophorectomy-induced bone loss is attenuated in MAGP1deficient mice. J. Cell. Biochem. 113, 93-99 (2012).
- Craft, C. S. et al. Microfibril-associated glycoprotein-1, an extracellular matrix regulator of bone remodeling. J. Biol. Chem. 285, 23858-23867 (2010).
- Marini, J. C. et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers 3, 17052 (2017).
- Zieba, J. et al. Fracture healing in collagen-related preclinical models of osteogenesis imperfecta. J. Bone Miner. Res. 35, 1132-1148 (2020).
- Grafe, I. et al. Excessive transforming growth factor-
signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 20, 670-675 (2014). - Pacifici, M. The pathogenic roles of heparan sulfate deficiency in hereditary multiple exostoses. Matrix Biol. 71-72, 28-39 (2018).
- Kawashima, K. et al. Heparan sulfate deficiency leads to hypertrophic chondrocytes by increasing bone morphogenetic protein signaling. Osteoarthritis Cartilage 28, 1459-1470 (2020).
- Inubushi, T., Nozawa, S., Matsumoto, K., Irie, F. & Yamaguchi, Y. Aberrant perichondrial BMP signaling mediates multiple osteochondromagenesis in mice. JCI Insight 2, e90049 (2017).
- Inubushi, T., Lemire, I., Irie, F. & Yamaguchi, Y. Palovarotene inhibits osteochondroma formation in a mouse model of multiple hereditary exostoses. J. Bone Miner. Res. 33, 658-666 (2018).
- Tang, X. et al. Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGF
. Ann. Rheum. Dis. 77, 1372-1380 (2018). - Le Goff, C. et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet. 40, 1119-1123 (2008).
- Delhon, L. et al. Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency. FASEB J. 33, 2707-2718 (2019).
- Morales, J. et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet. 85, 558-568 (2009).
- Oichi, T. et al. Adamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway. Cell Mol. Life Sci. 76, 4795-4809 (2019).
- Groppe, J. et al. Structural basis of BMP signaling inhibition by Noggin, a novel twelve-membered cystine knot protein. J. Bone Joint Surg. Am. 85-A, 52-58 (2003).
- Pregizer, S. K. & Mortlock, D. P. Dynamics and cellular localization of Bmp2, Bmp4, and Noggin transcription in the postnatal mouse skeleton. J. Bone Miner. Res. 30, 64-70 (2015).
- Yoshimura, Y. et al. Colocalization of noggin and bone morphogenetic protein-4 during fracture healing. J. Bone Miner. Res. 16, 876-884 (2001).
- Wu, X. B. et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J. Clin. Invest. 112, 924-934 (2003).
- Iwata, T. et al. Noggin blocks osteoinductive activity of porcine enamel extracts. J. Dent. Res. 81, 387-391 (2002).
- Wan, D. C. et al. Noggin suppression enhances in vitro osteogenesis and accelerates in vivo bone formation. J. Biol. Chem. 282, 26450-26459 (2007).
- Devlin, R. D. et al. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology 144, 1972-1978 (2003).
- Canalis, E., Brunet, L. J., Parker, K. & Zanotti, S. Conditional inactivation of noggin in the postnatal skeleton causes osteopenia. Endocrinology 153, 1616-1626 (2012).
- Xie, Z. et al. Imbalance between bone morphogenetic protein 2 and noggin induces abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. Arthritis Rheumatol. 68, 430-440 (2016).
- Warren, S. M., Brunet, L. J., Harland, R. M., Economides, A. N. & Longaker, M. T. The BMP antagonist noggin regulates cranial suture fusion. Nature 422, 625-629 (2003).
- Nolan, K. et al. Structure of Gremlin-2 in complex with GDF5 gives insight into DAN-family-mediated BMP antagonism. Cell Rep. 16, 2077-2086 (2016).
- Gazzerro, E. et al. Conditional deletion of gremlin causes a transient increase in bone formation and bone mass. J. Biol. Chem. 282, 31549-31557 (2007).
- Gazzerro, E. et al. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology 146, 655-665 (2005).
- Worthley, D. L. et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269-284 (2015).
- Eijken, M. et al. The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. FASEB J. 21, 2949-2960 (2007).
- Suh, J. et al. GDF11 promotes osteogenesis as opposed to MSTN, and follistatin, a MSTN/GDF11 inhibitor, increases muscle mass but weakens bone. Proc. Natl. Acad. Sci. USA 117, 4910-4920 (2020).
- Abe, Y., Abe, T., Aida, Y., Hara, Y. & Maeda, K. Follistatin restricts bone morphogenetic protein (BMP)-2 action on the differentiation of osteoblasts in fetal rat mandibular cells. Proc. Natl. Acad. Sci. USA 19, 1302-1307 (2004).
- Zhang, D. et al. A role for the BMP antagonist chordin in endochondral ossification. J. Bone Miner. Res. 17, 293-300 (2002).
- Petryk, A. et al. Twisted gastrulation and chordin inhibit differentiation and mineralization in MC3T3-E1 osteoblast-like cells. Bone 36, 617-626 (2005).
- Cook, L. M. et al. Betaglycan drives the mesenchymal stromal cell osteogenic program and prostate cancer-induced osteogenesis. Oncogene 38, 6959-6969 (2019).
- Hill, C. R., Jacobs, B. H., Brown, C. B., Barnett, J. V. & Goudy, S. L. Type III transforming growth factor beta receptor regulates vascular and osteoblast development during palatogenesis. Dev. Dyn. 244, 122-133 (2015).
- Verlinden, L. et al. Nrp2 deficiency leads to trabecular bone loss and is accompanied by enhanced osteoclast and reduced osteoblast numbers. Bone 55, 465-475 (2013).
- Ishibashi, O. et al. Endoglin is involved in BMP-2-induced osteogenic differentiation of periodontal ligament cells through a pathway independent of Smad-1/5/8 phosphorylation. J. Cell. Physiol. 222, 465-473 (2010).
- Lawera, A. et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 116, 17800-17808 (2019).
- Finnson, K. W. et al. Endoglin differentially regulates TGF-
-induced Smad and Smad1/5 signalling and its expression correlates with extracellular matrix production and cellular differentiation state in human chondrocytes. Osteoarthritis Cartilage 18, 1518-1527 (2010). - Parker, W. L., Goldring, M. B. & Philip, A. Endoglin is expressed on human chondrocytes and forms a heteromeric complex with betaglycan in a ligand and
type II TGFbeta receptor independent manner. J. Bone Miner. Res. 18, 289-302 (2003). - Alzahrani, A. et al. Endoglin haploinsufficiency is associated with differential regulation of extracellular matrix production during skin fibrosis and cartilage repair in mice. J. Cell Commun. Signal. 12, 379-388 (2018).
- Bianchi, V. J., Parsons, M., Backstein, D. & Kandel, R. A. Endoglin level is critical for cartilage tissue formation in vitro by passaged human chondrocytes. Tissue Eng. Part A 27, 1140-1150 (2021).
- Hagihara, M. et al. Neogenin, a receptor for bone morphogenetic proteins. J. Biol. Chem. 286, 5157-5165 (2011).
- Zhou, Z. et al. Neogenin regulation of BMP-induced canonical Smad signaling and endochondral bone formation. Dev. Cell 19, 90-102 (2010).
- Miyazawa, K. & Miyazono, K. Regulation of TGF-
family signaling by inhibitory Smads. Cold Spring Harb. Perspect. Biol. 9, a022095 (2017). - Timberlake, A. T. et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife 5, e20125 (2016).
- Horiki, M. et al. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J. Cell Biol. 165, 433-445 (2004).
- Estrada, K. D., Retting, K. N., Chin, A. M. & Lyons, K. M. Smad6 is essential to limit BMP signaling during cartilage development. J. Bone Miner. Res. 26, 2498-2510 (2011).
- Shen, R. et al. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J. Biol. Chem. 281, 3569-3576 (2006).
- Li, N. et al. Partial loss of Smad7 function impairs bone remodeling, osteogenesis and enhances osteoclastogenesis in mice. Bone 67, 46-55 (2014).
- Iwai, T., Murai, J., Yoshikawa, H. & Tsumaki, N. Smad7 inhibits chondrocyte differentiation at multiple steps during endochondral bone formation and down-regulates p38 MAPK pathways. J. Biol. Chem. 283, 27154-27164 (2008).
- Zhao, M., Qiao, M., Oyajobi, B. O., Mundy, G. R. & Chen, D. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J. Biol. Chem. 278, 27939-27944 (2003).
- Zhao, M. et al. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. Int. J. Mol. Sci. 279, 12854-12859 (2004).
- Yamashita, M. et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 121, 101-113 (2005).
- Sapkota, G. et al. signaling through integrated inputs into the Smad1 linker. Mol. Cell 25, 441-454 (2007).
- Liang, C. et al. Inhibition of osteoblastic Smurf1 promotes bone formation in mouse models of distinctive age-related osteoporosis. Nat. Commun. 9, 3428 (2018).
- Liu, J. et al. Increased PLEKHO1 within osteoblasts suppresses Smad-dependent BMP signaling to inhibit bone formation during aging. Aging Cell 16, 360-376 (2017).
- Wu, Q. et al. Regulation of embryonic endochondral ossification by Smurf2. J. Orthop. Res. 26, 704-712 (2008).
- Wu, Q. et al. Induction of an osteoarthritis-like phenotype and degradation of phosphorylated Smad3 by Smurf2 in transgenic mice. Arthritis Rheum. 58, 3132-3144 (2008).
- Wu, Q. & Huang, J. H. Ectopic expression of Smurf2 and acceleration of agerelated intervertebral disc degeneration in a mouse model. J. Neurosurg. Spine 27, 116-126 (2017).
- Huang, H., Veien, E. S., Zhang, H., Ayers, D. C. & Song, J. Skeletal characterization of Smurf2-deficient mice and in vitro analysis of Smurf2-deficient chondrocytes. PLoS One 11, e0148088 (2016).
- Kushioka, J. & Kaito, T. A novel negative regulatory mechanism of Smurf2 in BMP/Smad signaling in bone. Bone Res. 8, 41 (2020).
- Tang, L. Y. et al. Ablation of Smurf2 reveals an inhibition in TGF-
signalling through multiple mono-ubiquitination of Smad3. EMBO J. 30, 4777-4789 (2011). - Xu, Z. et al. SMURF2 regulates bone homeostasis by disrupting SMAD3 interaction with vitamin D receptor in osteoblasts. Nat. Commun. 8, 14570 (2017).
- Kim, B. G., Lee, J. H., Yasuda, J., Ryoo, H. M. & Cho, J. Y. Phospho-Smad1 modulation by nedd4 E3 ligase in BMP/TGF-
signaling. J. Bone Miner. Res. 26, 1411-1424 (2011). - Jeon, S. A., Lee, J. H., Kim, D. W. & Cho, J. Y. E3-ubiquitin ligase NEDD4 enhances bone formation by removing TGF
-induced pSMAD1 in immature osteoblast. Bone 116, 248-258 (2018). - Herhaus, L. et al. USP15 targets ALK3/BMPR1A for deubiquitylation to enhance bone morphogenetic protein signalling. Open Biol. 4, 140065 (2014).
- Wang, W., Zhu, Y., Sun, Z., Jin, C. & Wang, X. Positive feedback regulation between USP15 and ERK2 inhibits osteoarthritis progression through TGF-
/ SMAD2 signaling. Arthritis Res. Ther. 23, 84 (2021). - Sangadala, S. et al. Characterization of a unique motif in LIM mineralization protein-1 that interacts with jun activation-domain-binding protein 1. Mol. Cell. Biochem. 385, 145-157 (2014).
- Li, H. et al. VCP/p97 increases BMP signaling by accelerating ubiquitin ligase Smurf1 degradation. FASEB J. 33, 2928-2943 (2019).
- Samsa, W. E. et al. The master developmental regulator Jab1/Cops5/Csn5 is essential for proper bone growth and survival in mice. Bone 143, 115733 (2021).
- Zhang, F. et al. Sustained BMP signaling in osteoblasts stimulates bone formation by promoting angiogenesis and osteoblast differentiation. J. Bone Miner. Res. 24, 1224-1233 (2009).
- Durbano, H. W. et al. Aberrant BMP2 signaling in patients diagnosed with osteoporosis. Int. J. Mol. Sci. 21, 6909 (2020).
- Mumm, S. et al. Deactivating germline mutations in LEMD3 cause osteopoikilosis and Buschke-Ollendorff Syndrome, but not sporadic melorheostosis. J. Bone Miner. Res. 22, 243-250 (2007).
- Hellemans, J. et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat. Genet. 36, 1213-1218 (2004).
- Guo, L. et al. Deficiency of TMEM53 causes a previously unknown sclerosing bone disorder by dysregulation of BMP-SMAD signaling. Nat. Commun. 12, 2046 (2021).
- Mullin, B. H. et al. Genome-wide association study meta-analysis for quantitative ultrasound parameters of bone identifies five novel loci for broadband ultrasound attenuation. Hum. Mol. Genet. 26, 2791-2802 (2017).
- Moayyeri, A. et al. Genetic determinants of heel bone properties: genome-wide association meta-analysis and replication in the GEFOS/GENOMOS consortium. Hum. Mol. Genet. 23, 3054-3068 (2014).
- Luo, K. Negative regulation of BMP signaling by the ski oncoprotein. J. Bone Joint Surg. Am. 85-A, 39-43 (2003).
- Kim, K. O. et al. Ski inhibits TGF-
phospho-Smad3 signaling and accelerates hypertrophic differentiation in chondrocytes. J. Cell. Biochem. 113, 2156-2166 (2012). - Doyle, A. J. et al. Mutations in the TGF-
repressor SKI cause ShprintzenGoldberg syndrome with aortic aneurysm. Nat. Genet. 44, 1249-1254 (2012). - Kawamura, I. et al. SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-
and bone morphogenetic protein pathways. J. Biol. Chem. 287, 29101-29113 (2012). - Ehnert, S. et al. Transforming growth factor
inhibits bone morphogenic protein (BMP)-2 and BMP-7 signaling via upregulation of Ski-related novel protein N (SnoN): possible mechanism for the failure of BMP therapy? BMC Med. 10, 101 (2012). - Kim, D. W. & Lassar, A. B. Smad-dependent recruitment of a histone deacetylase/ Sin3A complex modulates the bone morphogenetic protein-dependent transcriptional repressor activity of Nkx3.2. Mol. Cell. Biol. 23, 8704-8717 (2003).
- Yoshida, Y. et al. Negative regulation of BMP/Smad signaling by Tob in osteoblasts. Cell 103, 1085-1097 (2000).
- Caddy, J. C., Luoma, L. M. & Berry, F. B. FOXC1 negatively regulates BMP-SMAD activity and Id1 expression during osteoblast differentiation. J. Cell. Biochem. 121, 3266-3277 (2020).
- Leboy, P. et al. Smad-Runx interactions during chondrocyte maturation. J. Bone Joint Surg. Am. 83-A, S15-S22 (2001).
- Javed, A. et al. Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J. Biol. Chem. 283, 8412-8422 (2008).
- Furumatsu, T., Tsuda, M., Taniguchi, N., Tajima, Y. & Asahara, H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 280, 8343-8350 (2005).
- Lee, H. L., Yu, B., Deng, P., Wang, C. Y. & Hong, C. Transforming growth factor-
induced KDM4B promotes chondrogenic differentiation of human mesenchymal stem cells. Stem Cells 34, 711-719 (2016). - Su, C. J. et al. Ligand-receptor promiscuity enables cellular addressing. Cell Syst. 13, 408-425.e12 (2022).
- Klumpe, H. E. et al. The context-dependent, combinatorial logic of BMP signaling. Cell Syst. 13, 388-407.e10 (2022).
- Antebi, Y. E. et al. Combinatorial signal perception in the BMP pathway. Cell 170, 1184-1196.e24 (2017).
- Luo, G. et al. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 9, 2808-2820 (1995).
- Storm, E. E. et al. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 368, 639-643 (1994).
- Coleman, C. M., Scheremeta, B. H., Boyce, A. T., Mauck, R. L. & Tuan, R. S. Delayed fracture healing in growth differentiation factor 5-deficient mice: a pilot study. Clin. Orthop. Relat. Res. 469, 2915-2924 (2011).
- Daans, M., Luyten, F. P. & Lories, R. J. GDF5 deficiency in mice is associated with instability-driven joint damage, gait and subchondral bone changes. Ann. Rheum. Dis. 70, 208-213 (2011).
- Rountree, R. B. et al. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2, e355 (2004).
- Shi, C. et al. Deletion of BMP receptor type IB decreased bone mass in association with compromised osteoblastic differentiation of bone marrow mesenchymal progenitors. Sci. Rep. 6, 24256 (2016).
- Zhao, M. et al. Bone morphogenetic protein receptor signaling is necessary for normal murine postnatal bone formation. J. Cell Biol. 157, 1049-1060 (2002).
- Wang, Q. et al. Cartilage-specific deletion of Alk5 gene results in a progressive osteoarthritis-like phenotype in mice. Osteoarthritis Cartilage 25, 1868-1879 (2017).
- Abou-Ezzi, G. et al. TGF-
signaling plays an essential role in the lineage specification of mesenchymal stem/progenitor cells in fetal bone marrow. Stem Cell Rep. 13, 48-60 (2019). - Lowery, J. W. et al. Loss of BMPR2 leads to high bone mass due to increased osteoblast activity. J. Cell Sci. 128, 1308-1315 (2015).
- Goh, B. C. et al. Activin receptor type 2A (ACVR2A) functions directly in osteoblasts as a negative regulator of bone mass. J. Biol. Chem. 292, 13809-13822 (2017).
- Wang, M. et al. Smad1 plays an essential role in bone development and postnatal bone formation. Osteoarthritis Cartilage 19, 751-762 (2011).
- Gunnell, L. M. et al. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J. Bone Miner. Res. 25, 1784-1797 (2010).
- Estrada, K. D. et al. Smad7 regulates terminal maturation of chondrocytes in the growth plate. Dev. Biol. 382, 375-384 (2013).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
قسم علم الخلايا وعلم الأحياء التطوري، كلية علوم الحياة، جامعة تشجيانغ، هانغتشو، تشجيانغ، الصين. قسم الطب الخلوي والجزيئي، قسم علم الأمراض وطب المختبرات، كلية الطب بجامعة تولين، جامعة تولين، نيو أورلينز، لويزيانا، الولايات المتحدة الأمريكية. البريد الإلكتروني:mengruiwu@zju.edu.cn; yli81@tulane.edu
DOI: https://doi.org/10.1038/s41422-023-00918-9
PMID: https://pubmed.ncbi.nlm.nih.gov/38267638
Publication Date: 2024-01-24
The roles and regulatory mechanisms of TGF-
Abstract
Transforming growth factor-
INTRODUCTION
OVERVIEW OF TGF-
Ligands and receptors: structure, diversity, and selectivity
| Classification | Gene | KO/CKO/Tg/knock-in | Phenotype | References |
| TGF-
|
Tgfb1 | ко | Early death (1 month) | 88 |
| Tgfb1
|
Reduced bone density; OB
|
152 | ||
| Col1
|
Diaphyseal thickening, fluctuating bone volume, increased bone remodeling, prone to fracture;
|
152 | ||
| Col1
|
Knee and temporomandibular joint osteoarthritis
|
221,222 | ||
| Tgfb2 | KO | Perinatal mortality; neural arch defect; bifurcated sternum; shortened radius and ulna | 85 | |
| Tgfb3 | ко | Die within 20 h of birth; failure of the palatal shelves to fuse leading to cleft palate | 86,87 | |
| BMP ligands | Bmp2 | Col2
|
Severe chondrodysplasia; shortened stature and limbs; chondrocyte proliferation
|
50 |
| Prx1-Cre | Normal limb patterning | 51 | ||
| Bmp7 | KO | Die at birth; skull base defects; rib & sternum malformation; hindlimb polydactyly | 326 | |
| Bmp7, Alk6 | DKO | Malformed and shortened appendicular bones compared to Bmp
|
25 | |
| Bmp2, 7 | Prx1-Cre;Bmp2
|
Slightly diminished appendicular skeleton; missing the last phalanx in digit III; malformed fibulae | 51 | |
| Bmp4 | Col2
|
Mild chondrodysplasia | 50 | |
| Prx1-Cre | Polydactyly | 51,52 | ||
| Bmp2, 4 | Col2
|
Severe chondrodysplasia; severely shortened and malformed or missing long bone skeleton element & fused joints; chondrocyte proliferation
|
50 | |
| Prx 1 -Cre | Polydactyly; complete syndactyly; delayed mineralization; chondrogenesis
|
51 | ||
| BMP3/GDF10 | Col1 Prom-Bmp3 Tg | Late hypertrophic chondrocyte differentiation
|
115 | |
| KO | Increased bone density | 28 | ||
| Bmp14/GDF5 | bp
|
Normal axial bones; shortened limbs and digits; missing joints of autopods; missing phalange elements | 25,327 | |
| No delay in fracture healing | 328 | |||
| Increased joint damage in collagen-induced arthritis; reduced bone density | 329 | |||
| GDF5, Alk6 |
|
Same as bp
|
25 | |
| Type I receptors | Alk2/Acvr1 | Col2
|
Shortened cranial base; hypoplastic cervical vertebrae | 49 |
| Osx-Cre | Mandibular bone density
|
113 | ||
| Q207D Tg | Fibrodysplasia ossificans progressive | 127,130,132 | ||
| Acvr1
|
Fibrodysplasia ossificans progressive | 128 | ||
| R206H knock-in | Fibrodysplasia ossificans progressive | 129 | ||
| Alk3/Bmpr1A | Col2
|
Split dorsal arches; shortened limbs; hypoplastic scapula; chondrodysplasia, chondrocyte proliferation
|
48,49,65 | |
| Gdf5-Cre | Automatically develop osteoarthritis | 330 | ||
| Col1
|
bone mass in long bones and ribs
|
148,149 | ||
| Sp7-Cre | Trabecular bone mass
|
147 | ||
| Dmp-Cre | Trabecular bone mass
|
147,176 | ||
| Alk6/Bmpr1B | KO | Brachypodism; reduced phalangeal elements; the fusion of appendicular joints, similar to GDF5 mutant (bp
|
25,48,49,65 |
| Classification | Gene | KO/CKO/Tg/knock-in | Phenotype | References |
| кO | Transient and gender-specific osteopenia caused by reduced osteogenesis from MSCs | 331 | ||
| Col1
|
Reduced BMD and bone volume; reduced osteoblast and osteoclast number | 332 | ||
| Alk3, Alk6 | Col2a1-Cre;Alk3
|
Phenotype resembling and more severe than Alk3 CKO mice | 48,65 | |
| Col2-Cre;Alk3
|
Severe defects in cartilage formation and skeletogenesis | 48 | ||
| Alk2, Alk3 | Col2-Cre | Malformed axis skeleton (vertebra, cervical and thoracic regions); more severe appendicular defects than Alk3 CKO mice | 49 | |
| Alk2, Alk6 | Col2-Cre;Alk2
|
More severe axis and appendicular defects than each single KO | 49 | |
| Alk5/TGFBR1 | Dermo-Cre | Short and wide long bones, ectopic cartilaginous protrusions, reduced bone volumes | 83 | |
| Col2
|
Elongated limbs; chondrocyte proliferation
|
68 | ||
| Col2
|
Automatic osteoarthritis | 208,333 | ||
| Type II receptors | Tgfbr2 | Nestin-CreER | Knee osteoarthritis
|
221 |
| ColX-Cre | Delayed chondrocyte terminal differentiation; impeded mineralization | 84 | ||
| Prx-Cre | Die at birth; reduced periodontal and frontal bone; shortened limbs; split sternum; autopod joint fusion; reduced mineralization; chondrocyte proliferation
|
78,81 | ||
| Col
|
Survive; neural arch defect; missing/incomplete intervetebral discs; progressive reduction in long bone length | 79,80 | ||
| Sp7-Cre (Dox) | Early death (1 M); reduced body size; reduced bone volume; increased bone marrow adipose tissue; disrupted molar tooth formation; OB
|
157,334 | ||
| Ocn-Cre | bone density
|
172 | ||
| Bmpr2 | Prx1-Cre | Trabecular bone volume
|
335 | |
| Col1
|
Dwarfism; delayed mineralization; bone volume
|
114 | ||
| ActRIIA/Acvr2A | Ocn-Cre | Trabecular bone volume
|
336 | |
| ActRIIB/Acvr2B | KO | Late hypertrophic chondrocyte differentiation
|
115 | |
| Ocn-Cre | Normal | 336 | ||
| ActRIIA, ActRIIB | Ocn-Cre | Trabecular bone volume
|
336 | |
| Canonical pathway | Smad1 | Col1
|
Osteopenia; OB proliferation and differentiation
|
337 |
| Col2
|
Shortened growth plate; chondrocyte hypertrophy
|
68 | ||
| Smad1/5 | Col2a1-Cre;Smad1
|
Similar and more severe phenotype than Smad1 CKO | 68 | |
| Smad1/5 | Col2
|
Chondrodysplasia; shortened limbs; thicker perichondrium; matrix production
|
69 | |
| Smad8 | KO | Normal | 69 | |
| Smad1/5/8 | Col2a1-Cre;Smad1
|
Absence of an axial skeleton; severely disorganized appendicular bones | 69 | |
| Smad2 | Col
|
Similar and more severe phenotype than Smad3 KO mice | 89 | |
| Smad2/3 | Col2a1-Cre;Smad2
|
Similar and more severe phenotype than Smad3 KO mice | 89 | |
| Smad3 | кO | Postnatal dwarfism; expanded columnar and hypertrophic zone; chondrocyte proliferation
|
89 | |
| KO | Knee and temporomandibular joint osteoarthritis | 214,215 |
| Table 1. continued | ||||
| Classification | Gene | KO/CKO/Tg/knock-in | Phenotype | References |
| ко | Osteopenia; OB and OCY apoptosis
|
156 | ||
| Smad4 | Tbx18-Cre | Short limbs, chondrogenesis
|
40 | |
| Sp7-Cre (Dox) | Increased trabecular bone mass | 147 | ||
| Sp7-Cre | Stunted growth; spontaneous fractures; increased trabecular bone volume; decreased BMD; a combination of features seen in osteogenesis imperfecta, cleidocranial dysplasia, and Wnt-deficiency syndromes | 118 | ||
| Dmp-Cre | Increased trabecular bone mass (
|
147 | ||
| Col1
|
Increased trabecular bone mass; protection from tail suspension-induced bone loss; OB & OCY number
|
178 | ||
| Ocn-Cre | Lower bone mass < 6-month, more bone mass > 7-month | 177 | ||
| Ctsk-Cre | Reduced bone mass; OC
|
186 | ||
| Non-canonical pathway | TAK1 | Col2
|
Growth retardation; osteoarthritis markers
|
71 |
| Osx-Cre | Cleidocranial dysplasia (CCD)-like phenotype (clavicular hypoplasia and delayed fontanelle fusion); OB
|
116 | ||
| Col2
|
Shorter limbs; chondrocyte proliferation
|
338 | ||
| Prx1-Cre | Widespread joint fusions; chondrocyte hypertrophy and proliferation | 338 | ||
| p38 | Ocn-Cre | OB activity and BFR
|
117 | |
| Col2
|
Shortened limbs; knee joint osteoarthritis
|
210 | ||
|
|
A substantial decrease in long bone mineralization and a more modest effect on the calvarium | 116 | ||
| MKKs |
|
Similar phenotype to Tak1
|
116 | |
| I-SMAD and ubiquitin-related regulation | Smad6 | Col11
|
Dwarfism and osteopenia; chondrocyte hypertrophy
|
279 |
| KO | Dwarfism; defects in axial and appendicular bones; delayed onset of hypertrophy | 280 | ||
| Smad6;Smurf1 | Col11
|
More severely delayed endochondral ossification than Smad6 Tg | 279 | |
| Smad7 |
|
Osteopenia; BFR
|
282 | |
| KO | Chondrocyte proliferation and hypertrophy
|
339 | ||
| Prx1 Prom-Tg; Col11 Enh-Tg; Col11 Prom-Tg | Chondrodysplasia; mesenchymal condensation
|
283 | ||
| Smurf1 | KO | Bone mass
|
286 | |
| Col1
|
Osteopenia; BFR
|
285 | ||
| Smurf2 | Col
|
Osteoarthritis; intervertebral disc degeneration | 291,292 | |
| KO | Protection from age-related and DMM-induced osteoarthritis | 293 | ||
| KO | Osteopenia
|
296 | ||
| KO | Enhanced BMP-induced ectopic bone formation | 294 | ||
| PLEKHO1 | Osterix-Cre | Protection from age-related bone loss | 289 | |
| Osterix Prom-Tg | Age-related bone loss | 289 | ||
| NEDD4 | Col1
|
Bone mass
|
298 | |
| Coll
|
Bone mass
|
298 | ||
| Jab1 | Osx-Cre | Dwarfism; trabecular bone mass
|
303 | |
| Classification | Gene | KO/CKO/Tg/knock-in | Phenotype | References |
| Antagonists | Noggin | Ocn Prom-Tg | Osteopenia; BFR
|
250,253 |
| Ocn-Cre | Osteopenia | 254 | ||
| ко | Hyperplasia of cartilage; joint development failure; multiple skeletal defects related to neural tube and somite patterning (failure of neural tube closure, broad club-shaped limbs, loss of caudal vertebrae, a shortened body axis, and retention of a small vestigial tail) | 66,67 | ||
| Grem1 | Ocn-Cre | Bone mass
|
258 | |
| Ocn Prom-Tg | Bone fractures; bone mass
|
259 | ||
| FS | Tg | Bone mass
|
262 | |
| Co-receptors |
|
KO | Defective palate development with OB
|
267 |
| Nrps | KO | Bone mass
|
268 | |
| Neogenin | KO | Elongated growth plate; chondrocyte proliferation & apoptosis
|
276 | |
| Other regulators | Tmem53 | KO | Sclerosing bone | 308 |
| Endofin | Col1 Prom-Endofin F872A Tg | Bone mass
|
304 |
(1) Col1
(2)
| Gene | Disease | MIM# | Bone disorders | References |
| NOGGIN, GDF5 | Symphalangism | 185800, 186500, 184460, 615298 | Ankylosis or synostosis of the interphalangeal joints | 59,60 |
| NOGGIN | Tarsal-carpal coalition syndrome | 186570 | Fusion of the carpals, tarsals, and phalanges; short first metacarpals causing brachydactyly; humeroradial fusion | 61 |
| NOGGIN, BMP2, BMPR1B, GDF5 | Brachydactyly | 611377, 112600, 113100 | Brachydactyly | 60,62-64 |
| TGFBR1, TGFBR2, TGFB2, TGFB3, SMAD2, SMAD3 | Loeys-Dietz syndrome | 609192, 610168, 613795, 614816, 615582, 619656 | Variable skeletal anomalies (including skeletal overgrowth, pectus deformity, osteoarthritis, hernias, etc.) | 72-76 |
| ACVR1 | Fibrodysplasia ossificans progressiva | 135100 | Progressive heterotopic bone formation in muscles, tendons, ligaments, and joints | 122,123 |
| TGFB1 | Camurati-Engelmann disease | 131300 | Osteosclerotic lesions in the long bones and skull with increased remodeling; osteoarthritis | 158 |
| SMAD3, MAP2K1, LEMD3 | Melorheostosis | 155950 | Melorheostosis (special sclerosing bone disease) | 159-161,306,307 |
| LEMD3 | Osteopoikilosis; BuschkeOllendorff syndrome | 166700 | Sclerosing bone | 306,307 |
| TMEM53 | Craniotubular dysplasia, Ikegawa type | 619727 | Hyperostosis; short stature in association with macrocephaly, dolichocephaly, or a prominent forehead | 308 |
| FBN-1 | Marfan syndrome | 154700 | Variable skeletal anomalies including long bone overgrowth | 77 |
| FBN-2 | Congenital contractural arachnodactyly | 121050 | Long limbs (dolichostenomelia) and long, slender fingers and toes (arachnodactyly), permanently bent joints (contractures) | 230 |
| ADAMTSL2 | Geleophysic dysplasia | 231050 | Short stature, short extremities, and skeletal abnormalities | 243 |
| ADAMTS10, ADAMTS17 | Weill-Marchesani syndrome | 277600,608328 | Short stature brachydactyly, and ectopia lentis | 245 |
| COL1A1, COL1A2 | Osteogenesis imperfecta | 259420 | A bone dysplasia characterized by bone deformities, fractures, and a high un-union rate caused by low bone mass and impaired bone quality | 235 |
| EXT1, EXT2 | hereditary multiple exostoses | 33700, 133701 | Formation of cartilage-capped bony growths (osteochondroma) at the ends of the bones | 238 |
| SKI | Shprintzen-Goldberg syndrome | 182212 | A wide range of skeletal abnormalities including craniosynostosis, distinctive facial features, arachnodactyly, long limbs, pectus excavatum or carinatum, and scoliosis | 313 |
protein (LTBP) to form the large latent complex (LLC), which binds to ECM proteins such as fibrillin (FBN).
receptor (BMPRIA)/ALK3, BMP type IB receptor (BMPRIB)/ALK6, Activin type I receptor (ACVR1)/ALK2, and ALK1.
Canonical and non-canonical signaling
antagonize its transcriptional activity to suppress BMP2-induced bone formation.
Target transcriptome
(old astrocyte specifically induced substance), and KLF4 (Krüppellike factor 4). These transcriptional factors may function together with or downstream of SMAD proteins to regulate the biological outcomes induced by BMP2. With RNA-seq and ChIP-seq techniques, Yan et al.
TGF-
BMP signaling in skeleton development
primordium formation, skeleton patterning, and growth plate development (Fig. 4). As mentioned earlier, BMP signaling consists of a variety of ligands and receptors with heterogeneous binding affinities and patterns, which produce variable physiological outcomes. BMP ligands have different expression patterns during skeleton development, delineating their diverse physiological functions. For example, Bmp14 and its receptor Alk6 have a restricted expression pattern in appendicular bones.
TGF-
M. Wu et al.
severe phenotype, indicating that Smad2 plays a more critical role than Smad3 in endochondral bone development.
TGF-
BMP signaling in bone formation and osteoblast differentiation
signaling.
TGF-
BMP and TGF-
The role of TGF-
M. Wu et al.
osteocyte to control bone quality.
TGF-
of its type I receptors, ALK1 and ALK5, to transduce signals to SMAD1/5/8 and SMAD2/3, respectively, in chondrocytes.
REGULATION OF TGF-
Latency and release control of the ligands
improper activation of TGF-
Extracellular antagonists
Co-receptors
M. Wu et al.
production.
Regulation machinery in the cytoplasm
Regulation in the nucleus
deacetylases HDAC4 and HDAC5 as co-repressors.
c-Fos, a key osteoclastic transcription factor, interacts directly with SMAD-2 and -3 to promote osteoclast diffrentiation.
CONCLUSION AND PERSPECTIVES
studies have identified several genes in TGF-
- Why do BMP and TGF-
signalings have dynamic functions? As reviewed here, this question could be partially answered by the diversity of ligand-receptor combinations and the complex intracellular regulatory network that causes the dynamic readout of the TGF- and BMP signaling. In particular, the BMP signaling pathway comprises multiple ligands and receptors that interact promiscuously with one another. A series of works from Dr. Michael B. Elowitz’s group demonstrated that the promiscuous ligand-receptor interaction systems of BMP signaling are critical for its dynamic regulations. Their work elucidated how the BMP pathway processes multi-ligand inputs using a repertoire of computational mechanisms, including ratiometric sensing, balance detection, and imbalance detection. Since cells have different expression patterns of receptors and ligands, the promiscuous interaction system allows a small number of ligands, acting in combinations, to address the issue of a larger number of individual cell types. - What transcriptional mechanism operates to bring about the diversity of transcriptional outcomes that arise in different cell types in response to the same ligand? Answering this question would require using state-of-theart techniques such as ChIP-seq, co-IP/MS, ATAC-seq, and CUT&Tag-seq. DNA and histone modification status varies in different cell types and might alter the affinity of SMAD complex binding with the chromosomes. Therefore, analyzing the epigenetic marks on the transcription factor binding sequences would help answer this question. Characterization of the receptor-ligand interaction mode and chromatin status in specific cell contexts might also explain why TGF-
signaling has stage-dependent functions in most skeletal cells. - How to circumvent the side effects of BMPs and TGF-
s when applying them in clinical settings? Excessive BMP and TGF- signaling is associated with multiple anomalies in bone tissues. Thus, further study and intervention are needed to prevent those side effects when applying BMPs and TGF- s in clinical settings. Although TGF- signaling maintains cartilage degeneration, hyperactivated TGF- signaling aggravates OA. Despite its dual functions, TGF-
signaling is still proposed as a potential treatment to alleviate OA, although it needs more study to design the proper timing and dose for the treatment. - How to safely and effectively modulate BMP and TGF-
signaling in skeletal disorders caused by their dysfunctions? Targeting BMP and TGF- signaling is proposed as the therapeutic strategy to treat , or osteosclerosis disorders while effective treatment is still under development.
REFERENCES
- Moses, H. L., Roberts, A. B. & Derynck, R. The discovery and early days of TGF-
: A historical perspective. Cold Spring Harb. Perspect. Biol. 8, a021865 (2016). - Katagiri, T. & Watabe, T. Bone morphogenetic proteins. Cold Spring Harb. Perspect. Biol. 8, a021899 (2016).
- Derynck, R. & Budi, E. H. Specificity, versatility, and control of TGF-
family signaling. Sci. Signal. 12, eaav5183 (2019). - Mahmood, A., Harkness, L., Schrøder, H. D., Abdallah, B. M. & Kassem, M. Enhanced differentiation of human embryonic stem cells to mesenchymal progenitors by inhibition of TGF-beta/activin/nodal signaling using SB-431542. J. Bone Miner. Res. 25, 1216-1233 (2010).
- Cianciolo, G. et al. The role of activin: the other side of chronic kidney diseasemineral bone disorder? Nephrol. Dial. Transplant. 36, 966-974 (2021).
- Lee, S. J. et al. Targeting myostatin/activin A protects against skeletal muscle and bone loss during spaceflight. Proc. Natl. Acad. Sci. USA 117, 23942-23951 (2020).
- Maridas, D. E., et al. Chapter 48 – Bone morphogenetic proteins. In: Bilezikian J. P., Martin T. J., Clemens T. L., Rosen C. J. eds. Principles of Bone Biology (Fourth Edition): Academic Press: 1189-1197 (2020).
- Xu, X. & Cao, X. Chapter 47 – Transforming growth factor-
and skeletal homeostasis1. In: Bilezikian J. P., Martin T. J., Clemens T. L., Rosen C. J. eds. Principles of Bone Biology (Fourth Edition): Academic Press: 1153-1187 (2020). - Gipson, G. R. et al. Structural perspective of BMP ligands and signaling. Bone 140, 115549-115549 (2020).
- Shi, M. et al. Latent TGF-
structure and activation. Nature 474, 343-349 (2011). - Karsdal, M. A. et al. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J. Biol. Chem. 277, 44061-44067 (2002).
- Dallas, S. L., Rosser, J. L., Mundy, G. R. & Bonewald, L. F. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 277, 21352-21360 (2002).
- D’Angelo, M., Billings, P. C., Pacifici, M., Leboy, P. S. & Kirsch, T. Authentic matrix vesicles contain active metalloproteases (MMP). a role for matrix vesicleassociated MMP-13 in activation of transforming growth factor-beta. J. Biol. Chem. 276, 11347-11353 (2001).
- Wang, J. et al. Atp6i deficient mouse model uncovers transforming growth factor-
Smad as a key signaling pathway regulating odontoblast differentiation and tooth root formation. Int. J. Oral Sci. 15, 35 (2023). - Salmon, R. M. et al. Molecular basis of ALK1-mediated signalling by BMP9/ BMP10 and their prodomain-bound forms. Nat. Commun. 11, 1621 (2020).
- Neugebauer, J. M. et al. The prodomain of BMP4 is necessary and sufficient to generate stable BMP4/7 heterodimers with enhanced bioactivity in vivo. Proc. Natl. Acad. Sci. USA 112, E2307-E2316 (2015).
- Sengle, G., Ono, R. N., Sasaki, T. & Sakai, L. Y. Prodomains of transforming growth factor beta (TGFbeta) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 286, 5087-5099 (2011).
- Gregory, K. E. et al. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J. Biol. Chem. 280, 27970-27980 (2005).
- Martinez-Hackert, E., Sundan, A. & Holien, T. Receptor binding competition: A paradigm for regulating TGF-
family action. Cytokine Growth Factor Rev. 57, 39-54 (2021). - Finnson, K. W., Parker, W. L., ten Dijke, P., Thorikay, M. & Philip, A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res. 23, 896-906 (2008).
- Goumans, M. J. et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 12, 817-828 (2003).
- Mang, T. et al. BMPR1A is necessary for chondrogenesis and osteogenesis, whereas BMPR1B prevents hypertrophic differentiation. J. Cell Sci. 133, jcs246934 (2020).
- Zhu, D. et al. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1 mediated pathway. J. Cell Mol. Med. 19, 165-174 (2015).
- Kokabu, S. et al. BMP3 suppresses osteoblast differentiation of bone marrow stromal cells via interaction with Acvr2b. Mol. Endocrinol. 26, 87-94 (2012).
- Yi, S. E., Daluiski, A., Pederson, R., Rosen, V. & Lyons, K. M. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development 127, 621-630 (2000).
- van Caam, A. et al. The high affinity ALK1-ligand BMP9 induces a hypertrophylike state in chondrocytes that is antagonized by TGF
1. Osteoarthritis Cartilage 23, 985-995 (2015). - Zhang, D. et al. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J. Bone Miner. Res. 18, 1593-1604 (2003).
- Daluiski, A. et al. Bone morphogenetic protein- 3 is a negative regulator of bone density. Nat. Genet. 27, 84-88 (2001).
- Macias, M. J., Martin-Malpartida, P. & Massagué, J. Structural determinants of Smad function in TGF-
signaling. Trends Biochem. Sci. 40, 296-308 (2015). - Gámez, B., Rodríguez-Carballo, E., Graupera, M., Rosa, J. L. & Ventura, F. Class I PI-3-kinase signaling is critical for bone formation through regulation of SMAD1 activity in osteoblasts. J. Bone Miner. Res. 31, 1617-1630 (2016).
- Zhu, Y. et al. Crosstalk between Smad
and specific isoforms of ERK in TGF- induced TIMP-3 expression in rat chondrocytes. PLoS Genet. 21, 1781-1790 (2017). - Baron, R. et al. Balancing BMP signaling through integrated inputs into the Smad1 linker. Nat. Commun. 25, 441-454 (2007).
- Urata, M. et al. A peptide that blocks the interaction of NF-kB p65 subunit with Smad4 enhances BMP2-induced osteogenesis. J. Cell. Physiol. 233, 7356-7366 (2018).
- Sun, X. et al. TGF-
inhibits osteogenesis by upregulating the expression of ubiquitin ligase SMURF1 via MAPK-ERK signaling. J. Cell. Physiol. 233, 596-606 (2018). - Kua, H. Y. et al. c-Abl promotes osteoblast expansion by differentially regulating canonical and non-canonical BMP pathways and p16INK4a expression. Nat. Cell Biol. 14, 727-737 (2012).
- Martin-Malpartida, P. et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nat. Commun. 8, 2070 (2017).
- Miyazono, K., Maeda, S. & Imamura, T. BMP receptor signaling: Transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 16, 251-263 (2005).
- Omata, Y. et al. Genomewide comprehensive analysis reveals critical cooperation between Smad and c-Fos in RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 30, 869-877 (2015).
- Yu, S. et al. BMP2-dependent gene regulatory network analysis reveals Klf4 as a novel transcription factor of osteoblast differentiation. Cell Death Dis. 12, 197 (2021).
- Yan, J. et al. Smad4 deficiency impairs chondrocyte hypertrophy via the Runx2 transcription factor in mouse skeletal development. J. Biol. Chem. 293, 9162-9175 (2018).
- Berendsen, A. D. & Olsen, B. R. Bone development. Bone 80, 14-18 (2015).
- Long, F. & Ornitz, D. M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 5, a008334 (2013).
- Tang, C. Y. et al. Run
up-regulates chondrocyte to osteoblast lineage commitment and promotes bone formation by enhancing both chondrogenesis and osteogenesis. Biochem. J. 477, 2421-2438 (2020). - Tang, J. et al. Runt-related transcription factor 1 is required for murine osteoblast differentiation and bone formation. J. Biol. Chem. 295, 11669-11681 (2020).
- Tian, F. et al. Core binding factor beta (
) controls the balance of chondrocyte proliferation and differentiation by upregulating Indian hedgehog (Ihh) expression and inhibiting parathyroid hormone-related protein receptor (PPR) expression in postnatal cartilage and bone formation. J. Bone Miner. Res. 29, 1564-1574 (2014). - Wu, M. et al. Deletion of core-binding factor
in mesenchymal progenitor cells provides new insights into Runxs complex function in cartilage and bone development. Bone 65, 49-59 (2014). - Wu, M. et al. Chondrocyte-specific knockout of
reveals the indispensable function of in chondrocyte maturation, growth plate development and trabecular bone formation in mice. Int. J. Biol. Sci. 10, 861-872 (2014). - Yoon, B. S. et al. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. USA 102, 5062-5067 (2005).
- Rigueur, D. et al. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J. Bone Miner. Res. 30, 733-741 (2015).
- Shu, B. et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 124, 3428-3440 (2011).
- Bandyopadhyay, A. et al. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2, e216 (2006).
- Selever, J., Liu, W., Lu, M. F., Behringer, R. R. & Martin, J. F. Bmp4 in limb bud mesoderm regulates digit pattern by controlling AER development. Dev. Biol. 276, 268-279 (2004).
- Nissim, S., Hasso, S. M., Fallon, J. F. & Tabin, C. J. Regulation of Gremlin expression in the posterior limb bud. Dev. Biol. 299, 12-21 (2006).
- Maatouk, D. M., Choi, K. S., Bouldin, C. M. & Harfe, B. D. In the limb AER Bmp2 and Bmp4 are required for dorsal-ventral patterning and interdigital cell death but not limb outgrowth. Dev. Biol. 327, 516-523 (2009).
- Choi, K. S., Lee, C., Maatouk, D. M. & Harfe, B. D. Bmp2, Bmp4 and Bmp7 are corequired in the mouse AER for normal digit patterning but not limb outgrowth. PLoS One 7, e37826 (2012).
- Pajni-Underwood, S., Wilson, C. P., Elder, C., Mishina, Y. & Lewandoski, M. BMP signals control limb bud interdigital programmed cell death by regulating FGF signaling. Development 134, 2359-2368 (2007).
- Benazet, J. D. & Zeller, R. Dual requirement of ectodermal Smad4 during AER formation and termination of feedback signaling in mouse limb buds. Genesis 51, 660-666 (2013).
- Pignatti, E., Zeller, R. & Zuniga, A. To BMP or not to BMP during vertebrate limb bud development. Semin. Cell Dev. Biol. 32, 119-127 (2014).
- Takano, K. et al. A novel nonsense mutation in the NOG gene causes familial NOGrelated symphalangism spectrum disorder. Hum. Genome Var. 3, 16023 (2016).
- Seemann, P. et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J. Clin. Invest. 115, 2373-2381 (2005).
- Dixon, M. E., Armstrong, P., Stevens, D. B. & Bamshad, M. Identical mutations in NOG can cause either tarsal/carpal coalition syndrome or proximal symphalangism. Genet. Med. 3, 349-353 (2001).
- Lehmann, K. et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am. J. Hum. Genet. 81, 88-396 (2007).
- Dathe, K. et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am. J. Hum. Genet. 84, 483-492 (2009).
- Lehmann, K. et al. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc. Natl. Acad. Sci. USA 100, 12277-12282 (2003).
- Yoon, B. S. et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 133, 4667-4678 (2006).
- Brunet, L. J., McMahon, J. A., McMahon, A. P. & Harland, R. M. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science 280, 1455-1457 (1998).
- McMahon, J. A. et al. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 12, 1438-1452 (1998).
- Keller, B. et al. Interaction of TGF
and BMP signaling pathways during chondrogenesis. PLoS One 6, e16421 (2011). - Retting, K. N., Song, B., Yoon, B. S. & Lyons, K. M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 136, 1093-1104 (2009).
- Tsumaki, N. et al. Role of CDMP-1 in skeletal morphogenesis: promotion of mesenchymal cell recruitment and chondrocyte differentiation. J. Cell Biol. 144, 161-173 (1999).
- Gao, L. et al. TAK1 regulates SOX9 expression in chondrocytes and is essential for postnatal development of the growth plate and articular cartilages. J. Cell Sci. 126, 5704-5713 (2013).
- Bertoli-Avella, A. M. et al. Mutations in a TGF-
ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 65, 1324-1336 (2015). - Lindsay, M. E. et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 44, 922-927 (2012).
- van de Laar, I. M. et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 43, 121-126 (2011).
- Loeys, B. L. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 37, 275-281 (2005).
- Vandeloo, B. et al. Spontaneous coronary artery dissection in a man with a novel missense mutation in SMAD2 treated by optical coherence tomography-guided percutaneous coronary intervention. JACC Cardiovasc. Interv. 12, e45-e47 (2019).
- Milewicz, D. M. et al. Marfan syndrome. Nat. Rev. Dis. Primers 7, 64 (2021).
- Seo, H. S. & Serra, R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev. Biol. 310, 304-316 (2007).
- Baffi, M. O. et al. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev. Biol. 276, 124-142 (2004).
- Sohn, P., Cox, M., Chen, D. & Serra, R. Molecular profiling of the developing mouse axial skeleton: a role for Tgfbr2 in the development of the intervertebral disc. BMC Dev. Biol. 10, 29 (2010).
- Spagnoli, A. et al. TGF-beta signaling is essential for joint morphogenesis. J. Cell Biol. 177, 1105-1117 (2007).
- Longobardi, L. et al. TGF-
type II receptor/MCP-5 axis: at the crossroad between joint and growth plate development. Cancers 23, 71-81 (2012). - Matsunobu, T. et al. Critical roles of the TGF-beta type I receptor ALK5 in perichondrial formation and function, cartilage integrity, and osteoblast differentiation during growth plate development. Dev. Biol. 332, 325-338 (2009).
- Sueyoshi, T., Yamamoto, K. & Akiyama, H. Conditional deletion of Tgfbr2 in hypertrophic chondrocytes delays terminal chondrocyte differentiation. Matrix Biol. 31, 352-359 (2012).
- Sanford, L. P. et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124, 2659-2670 (1997).
- Proetzel, G. et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 11, 409-414 (1995).
- Kaartinen, V. et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 11, 415-421 (1995).
- Kulkarni, A. B. et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 90, 770-774 (1993).
- Wang, W., Song, B., Anbarchian, T., Shirazyan, A. & Sadik, J. E. Smad2 and Smad3 regulate chondrocyte proliferation and differentiation in the growth plate. PLOS Genet. 12, e1006352 (2016).
- Edwards, J. R. & Mundy, G. R. Advances in osteoclast biology: old findings and new insights from mouse models. Nat. Rev. Rheumatol. 7, 235-243 (2011).
- Long, F. Building strong bones: molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 13, 27-38 (2011).
- Chen, W. et al.
deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of required for skeletal development. Proc. Natl. Acad. Sci. USA 111, 8482-8487 (2014). - Wu, M. et al. Cbf
governs osteoblast-adipocyte lineage commitment through enhancing -catenin signaling and suppressing adipogenesis gene expression. Proc. Natl. Acad. Sci. USA 114, 10119-10124 (2017). - Tang, C. Y. et al. Runx1 is a central regulator of osteogenesis for bone homeostasis by orchestrating BMP and WNT signaling pathways. PLoS Genet. 17, e1009233 (2021).
- Delgado-Calle, J. & Bellido, T. The osteocyte as a signaling cell. Physiol. Rev. 102, 379-410 (2022).
- Langdahl, B. L., Carstens, M., Stenkjaer, L. & Eriksen, E. F. Polymorphisms in the transforming growth factor beta 1 gene and osteoporosis. Bone 32, 297-310 (2003).
- Panach, L. et al. Comparative transcriptome analysis identifies CARM1 and DNMT3A as genes associated with osteoporosis. Sci. Rep. 10, 16298 (2020).
- Gregson, C. L. et al. Genome-wide association study of extreme high bone mass: Contribution of common genetic variation to extreme BMD phenotypes and potential novel BMD-associated genes. Bone 114, 62-71 (2018).
- Pei, Y. F. et al. Genome-wide association meta-analyses identified 1 q 43 and 2q32.2 for hip Ward’s triangle areal bone mineral density. Bone 91, 1-10 (2016).
- He, J. W., Yue, H., Hu, W. W., Hu, Y. Q. & Zhang, Z. L. Contribution of the sclerostin domain-containing protein 1 (SOSTDC1) gene to normal variation of peak bone mineral density in Chinese women and men. J. Bone Miner. Metab. 29, 571-581 (2011).
- Moffett, S. P. et al. Identification and association analysis of single nucleotide polymorphisms in the human noggin (NOG) gene and osteoporosis phenotypes. Bone 44, 999-1002 (2009).
- Wang, H. et al. Association of bone morphogenetic protein-2 gene polymorphisms with susceptibility to ossification of the posterior longitudinal ligament of the spine and its severity in Chinese patients. Eur. Spine J. 17, 956-964 (2008).
- Lin, G. T. et al. SNP combinations in chromosome-wide genes are associated with bone mineral density in Taiwanese women. Chin. J. Physiol. 51, 32-41 (2008).
- Medici, M. et al. BMP-2 gene polymorphisms and osteoporosis: the Rotterdam Study. J. Bone Miner. Res. 21, 845-854 (2006).
- Gregson, C. L. et al. A rare mutation in SMAD9 associated with high bone mass identifies the SMAD-dependent BMP signaling pathway as a potential anabolic target for osteoporosis. J. Bone Miner. Res. 35, 92-105 (2020).
- Kim, B. J. et al. Association of SMAD2 polymorphisms with bone mineral density in postmenopausal Korean women. Osteoporos. Int. 22, 2273-2282 (2011).
- Lowery, J. W. & Rosen, V. Bone morphogenetic protein-based therapeutic approaches. Cold Spring Harb. Perspect. Biol. 10, a022327 (2018).
- Begam, H., Nandi, S. K., Kundu, B. & Chanda, A. Strategies for delivering bone morphogenetic protein for bone healing. Mater. Sci. Eng. C Mater. Biol. Appl. 70, 856-869 (2017).
- Bharadwaz, A. & Jayasuriya, A. C. Osteogenic differentiation cues of the bone morphogenetic protein-9 (BMP-9) and its recent advances in bone tissue regeneration. Mater. Sci. Eng. C Mater. Biol. Appl. 120, 111748 (2021).
- Eiraku, N. et al. BMP9 directly induces rapid GSK3-
phosphorylation in a Wntindependent manner through class I PI3K-Akt axis in osteoblasts. FASEB J. 33, 12124-12134 (2019). - Tsuji, K. et al. BMP4 is dispensable for skeletogenesis and fracture-healing in the limb. J. Bone Joint Surg. Am. 90, 14-18 (2008).
- Tsuji, K. et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet. 38, 1424-1429 (2006).
- Hu, Y. Acvr1 deletion in osteoblasts impaired mandibular bone mass through compromised osteoblast differentiation and enhanced sRANKL-induced osteoclastogenesis. J. Cell. Physiol. 236, 4580-4591 (2021).
- Yang, C., Yang, L., Wan, M. & Cao, X. Generation of a mouse model with expression of bone morphogenetic protein type II receptor lacking the cytoplasmic domain in osteoblasts. Ann. N. Y. Acad. Sci. 1192, 286-291 (2010).
- Gamer, L. W., Cox, K., Carlo, J. M. & Rosen, V. Overexpression of BMP3 in the developing skeleton alters endochondral bone formation resulting in spontaneous rib fractures. Dev. Dyn. 238, 2374-2381 (2009).
- Greenblatt, M. B. et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Invest. 120, 2457-2473 (2010).
- Thouverey, C. & Caverzasio, J. The p38a MAPK positively regulates osteoblast function and postnatal bone acquisition. Cell Mol. Life Sci. 69, 3115-3125 (2012).
- Salazar, V. S. et al. Embryonic ablation of osteoblast Smad4 interrupts matrix synthesis in response to canonical Wnt signaling and causes an osteogenesis-imperfecta-like phenotype. J. Cell Sci. 126, 4974-4984 (2013).
- Salazar, V. S. et al. Postnatal ablation of osteoblast Smad4 enhances proliferative responses to canonical Wnt signaling through interactions with
-catenin. J. Cell Mol. Med. 126, 5598-5609 (2013). - Zhang, J. et al. The inhibition effects of insulin on BMP2-induced muscle heterotopic ossification. Biomaterials 35, 9322-9331 (2014).
- Agarwal, S. et al. Strategic targeting of multiple BMP receptors prevents traumainduced heterotopic ossification. Mol. Ther. 25, 1974-1987 (2017).
- van Dinther, M. et al. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 25, 1208-1215 (2010).
- Fukuda, T. et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem. Biophys. Res. Commun. 377, 905-909 (2008).
- Hatsell, S. J. et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 7, 303ra137 (2015).
- Hino, K. et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 112, 15438-15443 (2015).
- Billings, P. C. et al. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 23, 305-313 (2008).
- Yu, P. B. et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat. Med. 14, 1363-1369 (2008).
- Lees-Shepard, J. B. et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 9, 471 (2018).
- Chakkalakal, S. A. et al. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 27, 1746-1756 (2012).
- Fukuda, T. et al. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis 44, 159-167 (2016).
- Lodberg, A. Principles of the activin receptor signaling pathway and its inhibition. Cytokine Growth Factor Rev. 60, 1-17 (2021).
- Shimono, K. et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-
agonists. Nat. Med. 17, 454-460 (2011). - Pignolo, R. J. et al. Reduction of new heterotopic ossification (HO) in the openlabel, phase 3 MOVE trial of palovarotene for fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 38, 381-394 (2023).
- Meng, X., Wang, H. & Hao, J. Recent progress in drug development for fibrodysplasia ossificans progressiva. Mol. Cell. Biochem. 477, 2327-2334 (2022).
- Williams, E. et al. Saracatinib is an efficacious clinical candidate for fibrodysplasia ossificans progressiva. JCI Insight 6, e95042 (2021).
- Wang, Q. et al. Bone morphogenetic protein 2 activates Smad6 gene transcription through bone-specific transcription factor Runx2. J. Biol. Chem. 282, 10742-10748 (2007).
- Jeon, E. J. et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J. Biol. Chem. 281, 16502-16511 (2006).
- Jun, J. H. et al. BMP2-activated Erk/MAP kinase stabilizes Runx2 by increasing p300 levels and histone acetyltransferase activity. J. Biol. Chem. 285, 36410-36419 (2010).
- Rodríguez-Carballo, E. et al. Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways. J. Bone Miner. Res. 26, 718-729 (2011).
- Pawaputanon Na Mahasarakham, C. et al. BMP-2 enhances Lgr4 gene expression in osteoblastic cells. J. Cell. Physiol. 231, 887-895 (2016).
- Ko, F. C. et al. Acute phosphate restriction impairs bone formation and increases marrow adipose tissue in growing mice. J. Bone Miner. Res. 31, 2204-2214 (2016).
- Yang, G. et al. BMP-2 induction of Dlx3 expression is mediated by p38/ Smad5 signaling pathway in osteoblastic MC3T3-E1 cells. J. Cell. Physiol. 229, 943-954 (2014).
- Hopkins, A., Mirzayans, F. & Berry, F. Foxc1 expression in early osteogenic differentiation is regulated by BMP4-SMAD activity. J. Cell. Biochem. 117, 1707-1717 (2016).
- Ramazzotti, G. et al. BMP-2 induced expression of PLC
that is a positive regulator of osteoblast differentiation. J. Cell. Physiol. 231, 623-629 (2016). - Guo, Y. et al. BMP-IHH-mediated interplay between mesenchymal stem cells and osteoclasts supports calvarial bone homeostasis and repair. Bone Res. 6, 30 (2018).
- Liu, Z. et al. Molecules mimicking Smad1 interacting with Hox stimulate bone formation. J. Biol. Chem. 279, 11313-11319 (2004).
- Lim, J. et al. Dual function of Bmpr1a signaling in restricting preosteoblast proliferation and stimulating osteoblast activity in mouse. Development 143, 339-347 (2016).
- Zhang, H. et al. Loss of BMP signaling mediated by BMPR1A in osteoblasts leads to differential bone phenotypes in mice depending on anatomical location of the bones. Bone 137, 115402 (2020).
- Zhang, Y. et al. Loss of BMP signaling through BMPR1A in osteoblasts leads to greater collagen cross-link maturation and material-level mechanical properties in mouse femoral trabecular compartments. Bone 88, 74-84 (2016).
- Kamiya, N. et al. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J. Bone Miner. Res. 25, 200-210 (2010).
- Liu, Z., Tang, Y., Qiu, T., Cao, X. & Clemens, T. L. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J. Biol. Chem. 281, 17156-17163 (2006).
- Tang, Y. et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 15, 757-765 (2009).
- Seo, H. S. & Serra, R. Tgfbr2 is required for development of the skull vault. Dev. Biol. 334, 481-490 (2009).
- Peters, S. B., Wang, Y. & Serra, R. Tgfbr2 is required in osterix expressing cells for postnatal skeletal development. Bone 97, 54-64 (2017).
- Corps, K., Stanwick, M., Rectenwald, J., Kruggel, A. & Peters, S. B. Skeletal deformities in Osterix-Cre;Tgfbr2(f/f) mice may cause postnatal death. Genes 12, 975 (2021).
- Borton, A. J., Frederick, J. P., Datto, M. B., Wang, X. F. & Weinstein, R. S. The loss of Smad3 results in a lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J. Bone Miner. Res. 16, 1754-1764 (2001).
- Wang, Y., Cox, M. K., Coricor, G., MacDougall, M. & Serra, R. Inactivation of Tgfbr2 in Osterix-Cre expressing dental mesenchyme disrupts molar root formation. Dev. Biol. 382, 27-37 (2013).
- Kinoshita, A. et al. Domain-specific mutations in TGFB1 result in CamuratiEngelmann disease. Nat. Genet. 26, 19-20 (2000).
- Velchev, J. D., Verstraeten, A. & Loeys, B. Hide and seek: Somatic SMAD3 mutations in melorheostosis. J. Exp. Med. 217, e20200185 (2020).
- Kang, H. et al. Somatic SMAD3-activating mutations cause melorheostosis by up-regulating the TGF-
SMAD. Pathway. J. Exp. Med. 217, e20191499 (2020). - Kang, H. et al. Somatic activating mutations in MAP2K1 cause melorheostosis. Nat. Commun. 9, 1390 (2018).
- Nesti, L. J. et al. TGF-beta1-stimulated osteoblasts require intracellular calcium signaling for enhanced alpha5 integrin expression. Ann. N. Y. Acad. Sci. 961, 178-182 (2002).
- Arnott, J. A. et al. Molecular requirements for induction of CTGF expression by TGF-beta1 in primary osteoblasts. Bone 42, 871-885 (2008).
- Li, J. et al. Smad2 overexpression enhances Smad4 gene expression and suppresses CBFA1 gene expression in osteoblastic osteosarcoma ROS17/2.8 cells and primary rat calvaria cells. J. Biol. Chem. 273, 31009-31015 (1998).
- Lin, H. T. et al. Dynamic expression of SMAD3 is critical in osteoblast differentiation of PDMCs. Int. J. Mol. Med. 43, 1085-1093 (2019).
- Kang, J. S., Alliston, T., Delston, R. & Derynck, R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 24, 2543-2555 (2005).
- Hjelmeland, A. B., Schilling, S. H., Guo, X., Quarles, D. & Wang, X. F. Loss of Smad3-mediated negative regulation of Runx2 activity leads to an alteration in cell fate determination. Mol. Cell. Biol. 25, 9460-9468 (2005).
- Lian, N. et al. Transforming growth factor
suppresses osteoblast differentiation via the vimentin activating transcription factor 4 (ATF4) axis. J. Biol. Chem. 287, 35975-35984 (2012). - Ehnert, S. et al. TGF-
impairs mechanosensation of human osteoblasts via HDAC6-mediated shortening and distortion of primary cilia. J. Mol. Med. 95, 653-663 (2017). - Nam, B. & Park, H. TGF
suppressed matrix mineralization of osteoblasts differentiation by regulating SMURF1-C/EBP -DKK1 axis. Int. J. Mol. Sci. 21, 9771 (2020). - Ochiai, H. et al. Inhibition of insulin-like growth factor-1 (IGF-1) expression by prolonged transforming growth factor-
(TGF- ) administration suppresses osteoblast differentiation. J. Biol. Chem. 287, 22654-22661 (2012). - Qiu, T. et al. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat. Cell Biol. 12, 224-234 (2010).
- Kwok, S., Qin, L., Partridge, N. C. & Selvamurugan, N. Parathyroid hormone stimulation and PKA signaling of latent transforming growth factor-beta binding protein-1 (LTBP-1) mRNA expression in osteoblastic cells. J. Cell. Biochem. 95, 1002-1011 (2005).
- Sowa, H. et al. Parathyroid hormone-Smad3 axis exerts anti-apoptotic action and augments anabolic action of transforming growth factor beta in osteoblasts. J. Biol. Chem. 278, 52240-52252 (2003).
- Kamiya, N. et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J. Bone Miner. Res. 23, 2007-2017 (2008).
- Kamiya, N. et al. Targeted disruption of BMP signaling through type IA receptor (BMPR1A) in osteocyte suppresses SOST and RANKL, leading to dramatic increase in bone mass, bone mineral density and mechanical strength. Bone 91, 53-63 (2016).
- Tan, X. et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J. Cell Sci. 120, 2162-2170 (2007).
- Moon, Y. J. et al. Smad4 controls bone homeostasis through regulation of osteoblast/osteocyte viability. Exp. Mol. Med. 48, e256 (2016).
- Wu, H. et al. Inhibitory effects of combined bone morphogenetic protein 2, vascular endothelial growth factor, and basic fibroblast growth factor on osteoclast differentiation and activity. Tissue Eng. Part A 27, 1387-1398 (2021).
- Tasca, A. et al. Smad1/5 and Smad4 expression are important for osteoclast differentiation. J. Cell. Biochem. 116, 1350-1360 (2015).
- Miao, X. et al. Bone morphogenetic protein-2 promotes osteoclasts-mediated osteolysis via Smad1 and p65 signaling pathways. Spine 46, E234-E242 (2021).
- Omi, M., Kaartinen, V. & Mishina, Y. Activin A receptor type 1-mediated BMP signaling regulates RANKL-induced osteoclastogenesis via canonical SMADsignaling pathway. J. Biol. Chem. 294, 17818-17836 (2019).
- Okamoto, M. et al. Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J. Bone Miner. Res. 26, 2511-2522 (2011).
- Crane, J. L. & Cao, X. Bone marrow mesenchymal stem cells and TGF-
signaling in bone remodeling. J. Clin. Invest. 124, 466-472 (2014). - Karsdal, M. A. et al. Transforming growth factor-beta controls human osteoclastogenesis through the p38 MAPK and regulation of RANK expression. J. Biol. Chem. 278, 44975-44987 (2003).
- Morita, M. et al. Smad4 is required to inhibit osteoclastogenesis and maintain bone mass. Sci. Rep. 6, 35221 (2016).
- Pan, W. et al. SIS3 suppresses osteoclastogenesis and ameliorates bone loss in ovariectomized mice by modulating Nox4-dependent reactive oxygen species. Biochem. Pharmacol. 195, 114846 (2022).
- Houde, N., Chamoux, E., Bisson, M. & Roux, S. Transforming growth factor-beta1 (TGF-beta1) induces human osteoclast apoptosis by up-regulating Bim. J. Biol. Chem. 284, 23397-23404 (2009).
- Quinn, J. M. et al. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J. Bone Miner. Res. 16, 1787-1794 (2001).
- Dole, N. S. et al. Osteocyte-intrinsic TGF-
signaling regulates bone quality through perilacunar/canalicular remodeling. Cell Rep. 21, 2585-2596 (2017). - Dole, N. S., Yee, C. S., Mazur, C. M., Acevedo, C. & Alliston, T. TGF
regulation of perilacunar/canalicular remodeling is sexually dimorphic. J. Bone Miner. Res. 35, 1549-1561 (2020). - Schurman, C. A., Verbruggen, S. W. & Alliston, T. Disrupted osteocyte connectivity and pericellular fluid flow in bone with aging and defective TGF-
signaling. Proc. Natl. Acad. Sci. USA 118, e2023999118 (2021). - Liu, W. et al. TGF-
facilitates cell-cell communication in osteocytes via con-nexin43- and pannexin1-dependent gap junctions. Cell Death Discov. 5, 141 (2019). - Jähn, K. et al. Phenotype and viability of MLO-Y4 cells is maintained by
in a serum-dependent manner within a 3D-co-culture with MG-63 cells. Int. J. Mol. Sci. 19, 1932 (2018). - Bailey, K. N. et al. Mechanosensitive control of articular cartilage and subchondral bone homeostasis in mice requires osteocytic transforming growth factor
signaling. Arthritis Rheumatol. 73, 414-425 (2021). - Grol, M. W. & Lee, B. H. Gene therapy for repair and regeneration of bone and cartilage. Curr. Opin. Pharmacol. 40, 59-66 (2018).
- Patil, A. S., Sable, R. B. & Kothari, R. M. An update on transforming growth factor
(TGF- ): sources, types, functions and clinical applicability for cartilage/bone healing. J. Cell. Physiol. 226, 3094-3103 (2011). - He, Y. et al. Reduction of Smad2 caused by oxidative stress leads to necrotic death of hypertrophic chondrocytes associated with an endemic osteoarthritis. Rheumatology 61, 440-451 (2021).
- Li, T. F. et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J. Bone Miner. Res. 21, 4-16 (2006).
- Motaung, S. C. K. M., Di Cesare, P. E. & Hari Reddi, A. Differential response of cartilage oligomeric matrix protein (COMP) to morphogens of bone morphogenetic protein/transforming growth factor-
family in the surface, middle and deep zones of articular cartilage. J. Tissue Eng. Regen. Med. 5, e87-e96 (2011). - Niikura, T. & Reddi, A. H. Differential regulation of lubricin/superficial zone protein by transforming growth factor
bone morphogenetic protein superfamily members in articular chondrocytes and synoviocytes. Arthritis Rheum. 56, 2312-2321 (2007). - Malemud, C. J., Killeen, W., Hering, T. M. & Purchio, A. F. Enhanced sulfatedproteoglycan core protein synthesis by incubation of rabbit chondrocytes with recombinant transforming growth factor-
. J. Cell. Physiol. 149, 152-159 (1991). - Takahashi, N. et al. Elucidation of IL-1/TGF-
interactions in mouse chondrocyte cell line by genome-wide gene expression. Osteoarthritis Cartilage 13, 426-438 (2005). - Wiegertjes, R. et al. TGF-
dampens IL- 6 signaling in articular chondrocytes by decreasing IL-6 receptor expression. Osteoarthritis Cartilage 27, 1197-1207 (2019). - Cherian, J. J. et al. Preliminary results of a phase II randomized study to determine the efficacy and safety of genetically engineered allogeneic human chondrocytes expressing TGF-
in patients with grade 3 chronic degenerative joint disease of the knee. Osteoarthritis Cartilage 23, 2109-2118 (2015). - Ramaswamy, G., Sohn, P., Eberhardt, A. & Serra, R. Altered responsiveness to TGF-
results in reduced Papss2 expression and alterations in the biomechanical properties of mouse articular cartilage. Arthritis Res. Ther. 14, R49 (2012). - van Caam, A. et al. TGF
-induced SMAD2 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthritis Res. Ther. 19, 112 (2017). - Wang, Q. & Tan, Q. Postnatal deletion of Alk5 gene in meniscal cartilage accelerates age-dependent meniscal degeneration in mice. J. Cell. Physiol. 234, 595-605 (2018).
- Shen, J. et al. Deletion of the transforming growth factor
receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 65, 3107-3119 (2013). - Namdari, S., Wei, L., Moore, D. & Chen, Q. Reduced limb length and worsened osteoarthritis in adult mice after genetic inhibition of p38 MAP kinase activity in cartilage. Arthritis Rheum. 58, 3520-3529 (2008).
- Frazier, K. et al. Inhibition of ALK5 signaling induces physeal dysplasia in rats. Toxicol. Pathol. 35, 284-295 (2007).
- Prasadam, I. et al. Inhibition of p38 pathway leads to OA-like changes in a rat animal model. Rheumatology 51, 813-823 (2012).
- Wang, C., Shen, J., Ying, J., Xiao, D. & O’Keefe, R. J. FoxO1 is a crucial mediator of TGF-
TAK1 signaling and protects against osteoarthritis by maintaining articular cartilage homeostasis. Proc. Natl. Acad. Sci. USA 117, 30488-30497 (2020). - Mori, H., Izawa, T. & Tanaka, E. Smad3 deficiency leads to mandibular condyle degradation via the sphingosine 1-phosphate (S1P)/S1P3 signaling axis. Am. J. Pathol. 185, 2742-2756 (2015).
- Wang, H., Zhang, J., Sun, Q. & Yang, X. Altered gene expression in articular chondrocytes of Smad3(ex8/ex8) mice, revealed by gene profiling using microarrays. J. Genet. Genomics 34, 698-708 (2007).
- Zhang, Y. et al. Runx1 is a key regulator of articular cartilage homeostasis by orchestrating YAP, TGF
, and Wnt signaling in articular cartilage formation and osteoarthritis. Bone Res. 10, 63 (2022). - Ueland, T. et al. Increased serum and bone matrix levels of transforming growth factor {beta}1 in patients with GH deficiency in response to GH treatment. Eur. J. Endocrinol. 165, 393-400 (2011).
- Pombo-Suarez, M., Castaño-Oreja, M. T., Calaza, M., Gomez-Reino, J. & Gonzalez, A. Differential upregulation of the three transforming growth factor beta isoforms in human osteoarthritic cartilage. Ann. Rheum. Dis. 68, 568-571 (2009).
- Blaney Davidson, E. N., Vitters, E. L., van der Kraan, P. M. & van den Berg, W. B. Expression of transforming growth factor-beta (TGFbeta) and the TGFbeta signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: role in cartilage degradation, chondrogenesis and osteophyte formation. Ann. Rheum. Dis. 65, 1414-1421 (2006).
- François, R. J., Neure, L., Sieper, J. & Braun, J. Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumour necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Ann. Rheum. Dis. 65, 713-720 (2006).
- Zhen, G. et al. Inhibition of TGF-
signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 19, 704-712 (2013). - Zheng, L. et al. Aberrant activation of latent transforming growth factor-
initiates the onset of temporomandibular joint osteoarthritis. Bone Res. 6, 26 (2018). - Blaney Davidson, E. N. et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 182, 7937-7945 (2009).
- Occhetta, P. et al. Developmentally inspired programming of adult human mesenchymal stromal cells toward stable chondrogenesis. Proc. Natl. Acad. Sci. USA 115, 4625-4630 (2018).
- Janssens, K., ten Dijke, P., Janssens, S. & Van Hul, W. Transforming growth factor
to the bone. Endocr. Rev. 26, 743-774 (2005). - Dabovic, B. et al. Osteopetrosis-like phenotype in latent TGF-beta binding protein 3 deficient mice. Bone 37, 25-31 (2005).
- Dabovic, B. et al. Bone defects in latent TGF-beta binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-beta presentation. J. Endocrinol. 175, 129-141 (2002).
- Koli, K., Ryynänen, M. J. & Keski-Oja, J. Latent TGF-beta binding proteins (LTBPs)1 and -3 coordinate proliferation and osteogenic differentiation of human mesenchymal stem cells. Bone 43, 679-688 (2008).
- Kitahama, S. et al. Expression of fibrillins and other microfibril-associated proteins in human bone and osteoblast-like cells. Bone 27, 61-67 (2000).
- Putnam, E. A., Zhang, H., Ramirez, F. & Milewicz, D. M. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat. Genet. 11, 456-458 (1995).
- Nistala, H. et al. Fibrillin-1 and -2 differentially modulate endogenous TGF-
and BMP bioavailability during bone formation. J. Cell Biol. 190, 1107-1121 (2010). - Nistala, H., Lee-Arteaga, S., Smaldone, S., Siciliano, G. & Ramirez, F. Extracellular microfibrils control osteoblast-supported osteoclastogenesis by restricting TGF{beta} stimulation of RANKL production. J. Biol. Chem. 285, 34126-34133 (2010).
- Craft, C. S. et al. Oophorectomy-induced bone loss is attenuated in MAGP1deficient mice. J. Cell. Biochem. 113, 93-99 (2012).
- Craft, C. S. et al. Microfibril-associated glycoprotein-1, an extracellular matrix regulator of bone remodeling. J. Biol. Chem. 285, 23858-23867 (2010).
- Marini, J. C. et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers 3, 17052 (2017).
- Zieba, J. et al. Fracture healing in collagen-related preclinical models of osteogenesis imperfecta. J. Bone Miner. Res. 35, 1132-1148 (2020).
- Grafe, I. et al. Excessive transforming growth factor-
signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 20, 670-675 (2014). - Pacifici, M. The pathogenic roles of heparan sulfate deficiency in hereditary multiple exostoses. Matrix Biol. 71-72, 28-39 (2018).
- Kawashima, K. et al. Heparan sulfate deficiency leads to hypertrophic chondrocytes by increasing bone morphogenetic protein signaling. Osteoarthritis Cartilage 28, 1459-1470 (2020).
- Inubushi, T., Nozawa, S., Matsumoto, K., Irie, F. & Yamaguchi, Y. Aberrant perichondrial BMP signaling mediates multiple osteochondromagenesis in mice. JCI Insight 2, e90049 (2017).
- Inubushi, T., Lemire, I., Irie, F. & Yamaguchi, Y. Palovarotene inhibits osteochondroma formation in a mouse model of multiple hereditary exostoses. J. Bone Miner. Res. 33, 658-666 (2018).
- Tang, X. et al. Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGF
. Ann. Rheum. Dis. 77, 1372-1380 (2018). - Le Goff, C. et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet. 40, 1119-1123 (2008).
- Delhon, L. et al. Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency. FASEB J. 33, 2707-2718 (2019).
- Morales, J. et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet. 85, 558-568 (2009).
- Oichi, T. et al. Adamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway. Cell Mol. Life Sci. 76, 4795-4809 (2019).
- Groppe, J. et al. Structural basis of BMP signaling inhibition by Noggin, a novel twelve-membered cystine knot protein. J. Bone Joint Surg. Am. 85-A, 52-58 (2003).
- Pregizer, S. K. & Mortlock, D. P. Dynamics and cellular localization of Bmp2, Bmp4, and Noggin transcription in the postnatal mouse skeleton. J. Bone Miner. Res. 30, 64-70 (2015).
- Yoshimura, Y. et al. Colocalization of noggin and bone morphogenetic protein-4 during fracture healing. J. Bone Miner. Res. 16, 876-884 (2001).
- Wu, X. B. et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J. Clin. Invest. 112, 924-934 (2003).
- Iwata, T. et al. Noggin blocks osteoinductive activity of porcine enamel extracts. J. Dent. Res. 81, 387-391 (2002).
- Wan, D. C. et al. Noggin suppression enhances in vitro osteogenesis and accelerates in vivo bone formation. J. Biol. Chem. 282, 26450-26459 (2007).
- Devlin, R. D. et al. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology 144, 1972-1978 (2003).
- Canalis, E., Brunet, L. J., Parker, K. & Zanotti, S. Conditional inactivation of noggin in the postnatal skeleton causes osteopenia. Endocrinology 153, 1616-1626 (2012).
- Xie, Z. et al. Imbalance between bone morphogenetic protein 2 and noggin induces abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. Arthritis Rheumatol. 68, 430-440 (2016).
- Warren, S. M., Brunet, L. J., Harland, R. M., Economides, A. N. & Longaker, M. T. The BMP antagonist noggin regulates cranial suture fusion. Nature 422, 625-629 (2003).
- Nolan, K. et al. Structure of Gremlin-2 in complex with GDF5 gives insight into DAN-family-mediated BMP antagonism. Cell Rep. 16, 2077-2086 (2016).
- Gazzerro, E. et al. Conditional deletion of gremlin causes a transient increase in bone formation and bone mass. J. Biol. Chem. 282, 31549-31557 (2007).
- Gazzerro, E. et al. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology 146, 655-665 (2005).
- Worthley, D. L. et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269-284 (2015).
- Eijken, M. et al. The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. FASEB J. 21, 2949-2960 (2007).
- Suh, J. et al. GDF11 promotes osteogenesis as opposed to MSTN, and follistatin, a MSTN/GDF11 inhibitor, increases muscle mass but weakens bone. Proc. Natl. Acad. Sci. USA 117, 4910-4920 (2020).
- Abe, Y., Abe, T., Aida, Y., Hara, Y. & Maeda, K. Follistatin restricts bone morphogenetic protein (BMP)-2 action on the differentiation of osteoblasts in fetal rat mandibular cells. Proc. Natl. Acad. Sci. USA 19, 1302-1307 (2004).
- Zhang, D. et al. A role for the BMP antagonist chordin in endochondral ossification. J. Bone Miner. Res. 17, 293-300 (2002).
- Petryk, A. et al. Twisted gastrulation and chordin inhibit differentiation and mineralization in MC3T3-E1 osteoblast-like cells. Bone 36, 617-626 (2005).
- Cook, L. M. et al. Betaglycan drives the mesenchymal stromal cell osteogenic program and prostate cancer-induced osteogenesis. Oncogene 38, 6959-6969 (2019).
- Hill, C. R., Jacobs, B. H., Brown, C. B., Barnett, J. V. & Goudy, S. L. Type III transforming growth factor beta receptor regulates vascular and osteoblast development during palatogenesis. Dev. Dyn. 244, 122-133 (2015).
- Verlinden, L. et al. Nrp2 deficiency leads to trabecular bone loss and is accompanied by enhanced osteoclast and reduced osteoblast numbers. Bone 55, 465-475 (2013).
- Ishibashi, O. et al. Endoglin is involved in BMP-2-induced osteogenic differentiation of periodontal ligament cells through a pathway independent of Smad-1/5/8 phosphorylation. J. Cell. Physiol. 222, 465-473 (2010).
- Lawera, A. et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 116, 17800-17808 (2019).
- Finnson, K. W. et al. Endoglin differentially regulates TGF-
-induced Smad and Smad1/5 signalling and its expression correlates with extracellular matrix production and cellular differentiation state in human chondrocytes. Osteoarthritis Cartilage 18, 1518-1527 (2010). - Parker, W. L., Goldring, M. B. & Philip, A. Endoglin is expressed on human chondrocytes and forms a heteromeric complex with betaglycan in a ligand and
type II TGFbeta receptor independent manner. J. Bone Miner. Res. 18, 289-302 (2003). - Alzahrani, A. et al. Endoglin haploinsufficiency is associated with differential regulation of extracellular matrix production during skin fibrosis and cartilage repair in mice. J. Cell Commun. Signal. 12, 379-388 (2018).
- Bianchi, V. J., Parsons, M., Backstein, D. & Kandel, R. A. Endoglin level is critical for cartilage tissue formation in vitro by passaged human chondrocytes. Tissue Eng. Part A 27, 1140-1150 (2021).
- Hagihara, M. et al. Neogenin, a receptor for bone morphogenetic proteins. J. Biol. Chem. 286, 5157-5165 (2011).
- Zhou, Z. et al. Neogenin regulation of BMP-induced canonical Smad signaling and endochondral bone formation. Dev. Cell 19, 90-102 (2010).
- Miyazawa, K. & Miyazono, K. Regulation of TGF-
family signaling by inhibitory Smads. Cold Spring Harb. Perspect. Biol. 9, a022095 (2017). - Timberlake, A. T. et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife 5, e20125 (2016).
- Horiki, M. et al. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J. Cell Biol. 165, 433-445 (2004).
- Estrada, K. D., Retting, K. N., Chin, A. M. & Lyons, K. M. Smad6 is essential to limit BMP signaling during cartilage development. J. Bone Miner. Res. 26, 2498-2510 (2011).
- Shen, R. et al. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J. Biol. Chem. 281, 3569-3576 (2006).
- Li, N. et al. Partial loss of Smad7 function impairs bone remodeling, osteogenesis and enhances osteoclastogenesis in mice. Bone 67, 46-55 (2014).
- Iwai, T., Murai, J., Yoshikawa, H. & Tsumaki, N. Smad7 inhibits chondrocyte differentiation at multiple steps during endochondral bone formation and down-regulates p38 MAPK pathways. J. Biol. Chem. 283, 27154-27164 (2008).
- Zhao, M., Qiao, M., Oyajobi, B. O., Mundy, G. R. & Chen, D. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J. Biol. Chem. 278, 27939-27944 (2003).
- Zhao, M. et al. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. Int. J. Mol. Sci. 279, 12854-12859 (2004).
- Yamashita, M. et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 121, 101-113 (2005).
- Sapkota, G. et al. signaling through integrated inputs into the Smad1 linker. Mol. Cell 25, 441-454 (2007).
- Liang, C. et al. Inhibition of osteoblastic Smurf1 promotes bone formation in mouse models of distinctive age-related osteoporosis. Nat. Commun. 9, 3428 (2018).
- Liu, J. et al. Increased PLEKHO1 within osteoblasts suppresses Smad-dependent BMP signaling to inhibit bone formation during aging. Aging Cell 16, 360-376 (2017).
- Wu, Q. et al. Regulation of embryonic endochondral ossification by Smurf2. J. Orthop. Res. 26, 704-712 (2008).
- Wu, Q. et al. Induction of an osteoarthritis-like phenotype and degradation of phosphorylated Smad3 by Smurf2 in transgenic mice. Arthritis Rheum. 58, 3132-3144 (2008).
- Wu, Q. & Huang, J. H. Ectopic expression of Smurf2 and acceleration of agerelated intervertebral disc degeneration in a mouse model. J. Neurosurg. Spine 27, 116-126 (2017).
- Huang, H., Veien, E. S., Zhang, H., Ayers, D. C. & Song, J. Skeletal characterization of Smurf2-deficient mice and in vitro analysis of Smurf2-deficient chondrocytes. PLoS One 11, e0148088 (2016).
- Kushioka, J. & Kaito, T. A novel negative regulatory mechanism of Smurf2 in BMP/Smad signaling in bone. Bone Res. 8, 41 (2020).
- Tang, L. Y. et al. Ablation of Smurf2 reveals an inhibition in TGF-
signalling through multiple mono-ubiquitination of Smad3. EMBO J. 30, 4777-4789 (2011). - Xu, Z. et al. SMURF2 regulates bone homeostasis by disrupting SMAD3 interaction with vitamin D receptor in osteoblasts. Nat. Commun. 8, 14570 (2017).
- Kim, B. G., Lee, J. H., Yasuda, J., Ryoo, H. M. & Cho, J. Y. Phospho-Smad1 modulation by nedd4 E3 ligase in BMP/TGF-
signaling. J. Bone Miner. Res. 26, 1411-1424 (2011). - Jeon, S. A., Lee, J. H., Kim, D. W. & Cho, J. Y. E3-ubiquitin ligase NEDD4 enhances bone formation by removing TGF
-induced pSMAD1 in immature osteoblast. Bone 116, 248-258 (2018). - Herhaus, L. et al. USP15 targets ALK3/BMPR1A for deubiquitylation to enhance bone morphogenetic protein signalling. Open Biol. 4, 140065 (2014).
- Wang, W., Zhu, Y., Sun, Z., Jin, C. & Wang, X. Positive feedback regulation between USP15 and ERK2 inhibits osteoarthritis progression through TGF-
/ SMAD2 signaling. Arthritis Res. Ther. 23, 84 (2021). - Sangadala, S. et al. Characterization of a unique motif in LIM mineralization protein-1 that interacts with jun activation-domain-binding protein 1. Mol. Cell. Biochem. 385, 145-157 (2014).
- Li, H. et al. VCP/p97 increases BMP signaling by accelerating ubiquitin ligase Smurf1 degradation. FASEB J. 33, 2928-2943 (2019).
- Samsa, W. E. et al. The master developmental regulator Jab1/Cops5/Csn5 is essential for proper bone growth and survival in mice. Bone 143, 115733 (2021).
- Zhang, F. et al. Sustained BMP signaling in osteoblasts stimulates bone formation by promoting angiogenesis and osteoblast differentiation. J. Bone Miner. Res. 24, 1224-1233 (2009).
- Durbano, H. W. et al. Aberrant BMP2 signaling in patients diagnosed with osteoporosis. Int. J. Mol. Sci. 21, 6909 (2020).
- Mumm, S. et al. Deactivating germline mutations in LEMD3 cause osteopoikilosis and Buschke-Ollendorff Syndrome, but not sporadic melorheostosis. J. Bone Miner. Res. 22, 243-250 (2007).
- Hellemans, J. et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat. Genet. 36, 1213-1218 (2004).
- Guo, L. et al. Deficiency of TMEM53 causes a previously unknown sclerosing bone disorder by dysregulation of BMP-SMAD signaling. Nat. Commun. 12, 2046 (2021).
- Mullin, B. H. et al. Genome-wide association study meta-analysis for quantitative ultrasound parameters of bone identifies five novel loci for broadband ultrasound attenuation. Hum. Mol. Genet. 26, 2791-2802 (2017).
- Moayyeri, A. et al. Genetic determinants of heel bone properties: genome-wide association meta-analysis and replication in the GEFOS/GENOMOS consortium. Hum. Mol. Genet. 23, 3054-3068 (2014).
- Luo, K. Negative regulation of BMP signaling by the ski oncoprotein. J. Bone Joint Surg. Am. 85-A, 39-43 (2003).
- Kim, K. O. et al. Ski inhibits TGF-
phospho-Smad3 signaling and accelerates hypertrophic differentiation in chondrocytes. J. Cell. Biochem. 113, 2156-2166 (2012). - Doyle, A. J. et al. Mutations in the TGF-
repressor SKI cause ShprintzenGoldberg syndrome with aortic aneurysm. Nat. Genet. 44, 1249-1254 (2012). - Kawamura, I. et al. SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-
and bone morphogenetic protein pathways. J. Biol. Chem. 287, 29101-29113 (2012). - Ehnert, S. et al. Transforming growth factor
inhibits bone morphogenic protein (BMP)-2 and BMP-7 signaling via upregulation of Ski-related novel protein N (SnoN): possible mechanism for the failure of BMP therapy? BMC Med. 10, 101 (2012). - Kim, D. W. & Lassar, A. B. Smad-dependent recruitment of a histone deacetylase/ Sin3A complex modulates the bone morphogenetic protein-dependent transcriptional repressor activity of Nkx3.2. Mol. Cell. Biol. 23, 8704-8717 (2003).
- Yoshida, Y. et al. Negative regulation of BMP/Smad signaling by Tob in osteoblasts. Cell 103, 1085-1097 (2000).
- Caddy, J. C., Luoma, L. M. & Berry, F. B. FOXC1 negatively regulates BMP-SMAD activity and Id1 expression during osteoblast differentiation. J. Cell. Biochem. 121, 3266-3277 (2020).
- Leboy, P. et al. Smad-Runx interactions during chondrocyte maturation. J. Bone Joint Surg. Am. 83-A, S15-S22 (2001).
- Javed, A. et al. Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J. Biol. Chem. 283, 8412-8422 (2008).
- Furumatsu, T., Tsuda, M., Taniguchi, N., Tajima, Y. & Asahara, H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 280, 8343-8350 (2005).
- Lee, H. L., Yu, B., Deng, P., Wang, C. Y. & Hong, C. Transforming growth factor-
induced KDM4B promotes chondrogenic differentiation of human mesenchymal stem cells. Stem Cells 34, 711-719 (2016). - Su, C. J. et al. Ligand-receptor promiscuity enables cellular addressing. Cell Syst. 13, 408-425.e12 (2022).
- Klumpe, H. E. et al. The context-dependent, combinatorial logic of BMP signaling. Cell Syst. 13, 388-407.e10 (2022).
- Antebi, Y. E. et al. Combinatorial signal perception in the BMP pathway. Cell 170, 1184-1196.e24 (2017).
- Luo, G. et al. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 9, 2808-2820 (1995).
- Storm, E. E. et al. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 368, 639-643 (1994).
- Coleman, C. M., Scheremeta, B. H., Boyce, A. T., Mauck, R. L. & Tuan, R. S. Delayed fracture healing in growth differentiation factor 5-deficient mice: a pilot study. Clin. Orthop. Relat. Res. 469, 2915-2924 (2011).
- Daans, M., Luyten, F. P. & Lories, R. J. GDF5 deficiency in mice is associated with instability-driven joint damage, gait and subchondral bone changes. Ann. Rheum. Dis. 70, 208-213 (2011).
- Rountree, R. B. et al. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2, e355 (2004).
- Shi, C. et al. Deletion of BMP receptor type IB decreased bone mass in association with compromised osteoblastic differentiation of bone marrow mesenchymal progenitors. Sci. Rep. 6, 24256 (2016).
- Zhao, M. et al. Bone morphogenetic protein receptor signaling is necessary for normal murine postnatal bone formation. J. Cell Biol. 157, 1049-1060 (2002).
- Wang, Q. et al. Cartilage-specific deletion of Alk5 gene results in a progressive osteoarthritis-like phenotype in mice. Osteoarthritis Cartilage 25, 1868-1879 (2017).
- Abou-Ezzi, G. et al. TGF-
signaling plays an essential role in the lineage specification of mesenchymal stem/progenitor cells in fetal bone marrow. Stem Cell Rep. 13, 48-60 (2019). - Lowery, J. W. et al. Loss of BMPR2 leads to high bone mass due to increased osteoblast activity. J. Cell Sci. 128, 1308-1315 (2015).
- Goh, B. C. et al. Activin receptor type 2A (ACVR2A) functions directly in osteoblasts as a negative regulator of bone mass. J. Biol. Chem. 292, 13809-13822 (2017).
- Wang, M. et al. Smad1 plays an essential role in bone development and postnatal bone formation. Osteoarthritis Cartilage 19, 751-762 (2011).
- Gunnell, L. M. et al. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J. Bone Miner. Res. 25, 1784-1797 (2010).
- Estrada, K. D. et al. Smad7 regulates terminal maturation of chondrocytes in the growth plate. Dev. Biol. 382, 375-384 (2013).
ACKNOWLEDGEMENTS
AUTHOR CONTRIBUTIONS
COMPETING INTERESTS
ADDITIONAL INFORMATION
Department of Cell and Developmental Biology, College of Life Sciences, Zhejiang University, Hangzhou, Zhejiang, China. Division in Cellular and Molecular Medicine, Department of Pathology and Laboratory Medicine, Tulane University School of Medicine, Tulane University, New Orleans, LA, USA. email: mengruiwu@zju.edu.cn; yli81@tulane.edu