أزواج Fe1−Ti المستقطبة للدوران من أجل اختزال النترات بكفاءة عالية إلى الأمونيا Spin polarized Fe1−Ti pairs for highly efficient electroreduction nitrate to ammonia
يقدم تقليل النترات الكهربائي إلى الأمونيا حلاً جذابًا للاستدامة البيئية وإنتاج الطاقة النظيفة، ولكنه يعاني من بطء هدرجة *NO مع انتقالات حالة الدوران. هنا، نبلغ أن التلاعب في فراغات الأكسجين يمكن أن يحقق توازنًا مغناطيسيًا.أزواج على إلكترود تيتانيوم أحادي الكتلة يظهر جاذبيةمعدل العائدوارتفاعالكفاءة الفارادية لـعند -0.4 فولت مقابل RHE، أفضل بكثير من النظير الذي يعاني من انخفاض الدورانأزواج ( ) والعوامل المحفزة الكهربائية الأكثر شيوعًا. يمكن للإلكترونات ذات الدوران غير المتزاوج في ذرات الحديد والتيتانيوم التفاعل بفعالية مع الوسائط الرئيسية، مما يسهل عملية هدرجة *NO. ربط جهاز التحليل الكهربائي المتدفق مع نظام قائم على الغشاءوحدة الاسترداد، تقليل النترات المتزامن وتم تحقيق الاسترداد. يقدم هذا العمل استراتيجية رائدة للتلاعب بالاستقطاب المغزلي للمواد المحفزة الكهروكيميائية ضمن مواقع الأزواج لمعالجة مياه الصرف الصحي المحتوية على النترات.
الأمونيا ) كمصادر أساسية لإنتاج الأسمدة الصناعية وحامل للطاقة خالٍ من الكربون له أهمية كبيرة للمجتمع الحديث في الوقت الحاضر، العالميةالطلب يتجاوز 150 مليون طن سنويًا، مما يعتمد بشكل كبير على عملية هابر-بوش التي تتطلب طاقة عالية المرتبطة بالاستهلاك لـالطاقة العالمية، مع المساهمة في الوقت نفسهانبعاثات الكربون العالمية. مؤخرًا، تم تقليل النيتروجين كهربائيًا ( ) إلى مستوحاة من الميكروبات الطبيعيةالتركيز، قد جذب اهتمامًا هائلًا. ومع ذلك، فإن القويوانخفاض الذوبانية في وسط الماء لـيؤدي إلى انخفاضمعدل العائد (أقل من ) وكثافات التيار الجزئي (أقل من “، مما يعيق تجارته الواسعة النطاق. بدلاً من ذلك، النترات (يمكن أن يتم فصلها إلى أنواع غير مؤكسجة بشكل أكبر طاقة أقلمما يجعله مصدراً أكثر ملاءمة للنيتروجين لـالتحليل الكهربائي، خاصة فيما يتعلق بـ يتوزع على نطاق واسع في مياه الصرف الصناعي والمياه الجوفية الملوثة، مما يؤدي إلى الإثراء الغذائي واضطراب النظم البيئية.. لذلك، فإن التخفيض الانتقائي لـإلى(NITRR) يقدم مسارًا واعدًا لتخفيف أزمة الطاقة والبيئة بشكل متزامن.
بشكل عام، يبدأ NITRR من امتصاص النترات ) على الأقطاب الكهربائية تليها إزالة الأكسجين بشكل متسلسل إلى النيتريت الممتص (* ) وأكسيد النيتريك (*NO) . بعد ذلك، *NO يتحول إلى *NHO المهدرج/أنواع NOH مع انتقال حالة الدوران. أخيرًا، يمكن أن تتفاعل الأنواع المهدرجة بشكل تدريجي مع الهيدروجين لتكوين الهيدروكسي أمين ) والمنتج المستهدف
n من * . وقد تم الإبلاغ عن أن كل من امتصاصوأن الهدرجة اللاحقة لـ *NO ضرورية للتفاعل السريع مع NITRR، وبالتالي تعزيز يمكن أن يكون الامتزاز و/أو تسريع هدرجة *NO فعالين لتحسين نشاط NITRR. حاليًا، تم تعزيزتم الإبلاغ عن الامتزاز على نطاق واسع من خلال تعديل السطحتطعيم الذرات غير العضويةوخلط المعادنللأقطاب الكهربائية. بينما نادراً ما يتم التحقيق في تسريع الهيدروجين *NO المرتبط بانتقال الدوران من خلال التلاعب المناسب في حالات الدوران للمواد المحفزة الكهربية، على الرغم من أن هذه العملية مشابهة للتفاعلات المحفزة الكهربية المرتبطة بالأكسجين المبلغ عنها مثل تفاعل تطور الأكسجين (OER) وتفاعل اختزال الأكسجين (ORR).، وعادة ما يتطلب طاقة هائلة ويكون بطيئًا حركيًا، مما يعيق بشكل كبير تحول -إلى-لذلك، من الضروري تصميم محفزات كهربائية متقدمة مستقطبة للدوران لتعزيز نقل الإلكترونات المعتمد على الدوران من أجل تحسين NITRR.
تم الإبلاغ عن أن استقطاب الدوران لمواقع المعادن النشطة هو وسيلة فعالة لتسريع الانتقال بين حالات الدوران للوسطاء المتفاعلين، لأن مواقع المعادن النشطة المستقطبة قادرة على دفع تفاعل تبادل الدوران الكمي وتوفير قناة لنقل الإلكترونات الدورانية نحو التفاعل الكهروكيميائي المعتمد على الدوران.في ضوء هذا المبدأ، تم التحقيق بشكل مكثف في الحديد البارامغناطيسي (Fe)، خاصةً لتفاعلات التحفيز الكهربائي المتعلقة بالأكسجين (مثل OER و ORR)، وذلك بفضل درجة حرية الدوران الإلكترونية القابلة للتعديل بشكل جيد.. على سبيل المثال، دوران منخفض ( ) وحالات الدوران المتوسطة ( ) من الحديد (Fe) أكثر ملاءمة لاختراق المدار المضاد للرابطة لـ مقارنةً بحالة الدوران العالي للحديد )، مما يسهل الانتقال الدوراني من الحالة الفردية إلى حالة الثلاثي. والأكثر إثارة للإعجاب، وُجد أن ذرات الحديد الفردية على مصفوفة الكربون كانت فعالة في تقليل النيتروجين في تفاعل النيتروجين.، تظهر قدرة جيدة على اختزال النترات كهربائيًا مع الكفاءة فارادائية لـومعدل عائد قدرهمن الواضح أن التحكم بدقة في تأثير استقطاب الدوران للمواد القائمة على الحديد يمكن أن يوفر حلاً لتحسين NITRR.
في هذه الدراسة، تم تثبيت ذرات الحديد الفردية على طبقة الأكسيد السطحية المتأصلة لرغوة التيتانيوم، حيث يمكن أن تؤدي الفراغات الأكسجينية في أكسيد التيتانيوم إلى تحفيز الاستقطاب المغزلي المتزامن لذرات الحديد والتيتانيوم المجاورة. تم تصميم الاستقطاب المغزليالأزواج التي تم تسليمها عاليةالكفاءة الفارادية لـوانطباع مثير للإعجابمعدل العائدعند -0.4 فولت مقابل RHE لـ NITRR، متفوقًا بكثير على نظيره من الإلكترود الأحادي مع انخفاض الدوران-أزواج تي ( ) والمحولات الكهربية NITRR التي تم الإبلاغ عنها بشكل رئيسي. الإلكترونات المغزولة الموجودة في المدارات 3d لذرات الحديد والتيتانيوم في حالة الاستقطاب المغزلي تم العثور على مواقع الزوج التي تسهم بشكل رئيسي في هذا الأداء الممتاز من خلال التفاعل مع الوسائط الرئيسية، مما يعزز إزالة الأكسجين منوهدرجة *NO. تم تطوير القطب الأحادي مع الاستقطاب الدورانيتم استخدام أزواج أيضًا لبناء إلكتروليزر يتدفق عبره NITRR مرتبط بوحدة فصل غشائية لتحقيق تحويل نترات عالي الكفاءة عند شدة تيار على مستوى صناعي واسترداد الأمونيا عالية النقاء في الموقع من مياه الصرف الصحي المحتوية على النترات.
النتائج
التلاعب في استقطاب الدوران لـ – تي بيرز
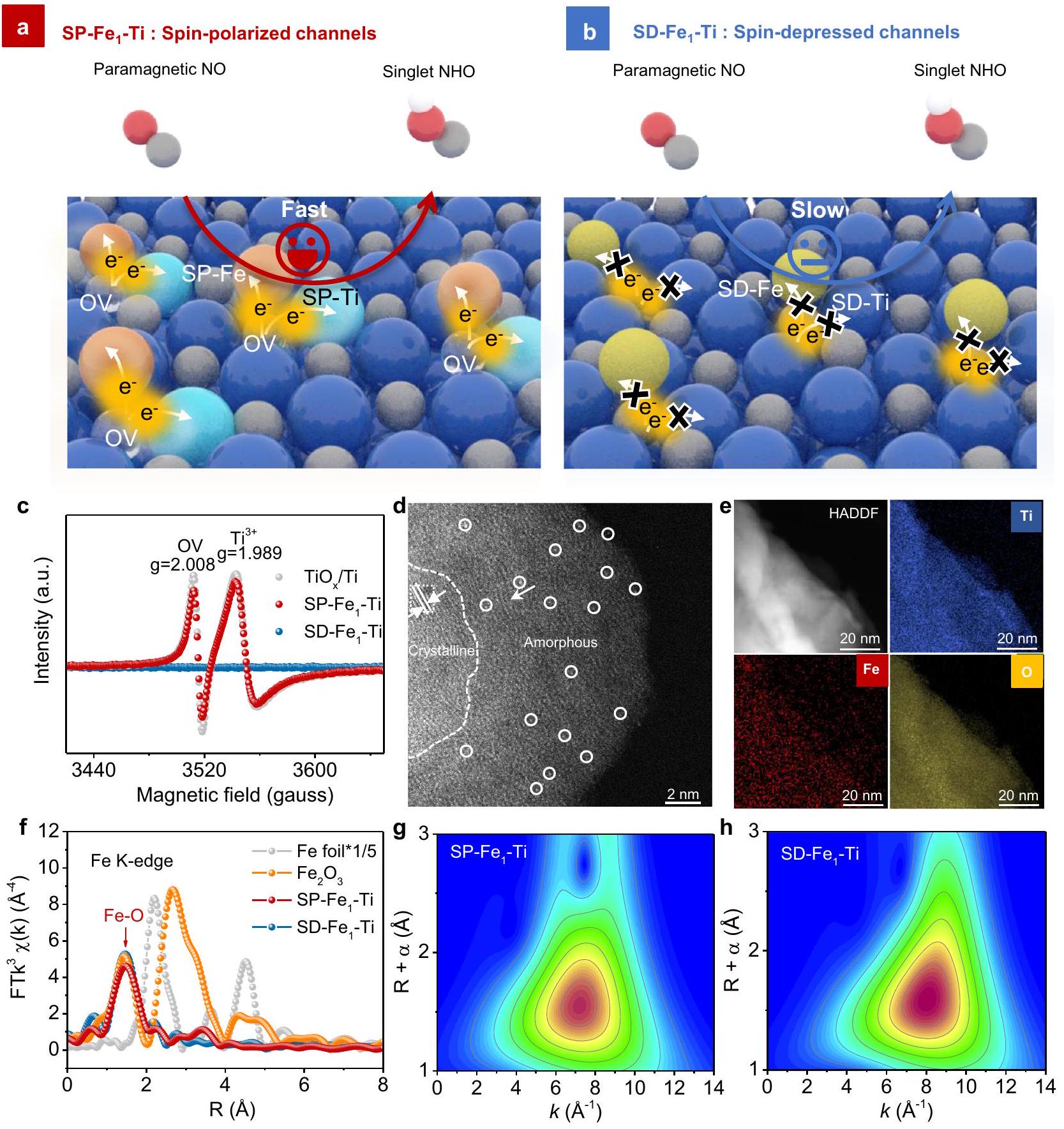
تم تحضير ذرات الحديد المفردة المثبتة على قطب تيتانيوم أحادي الكتلة بدرجات مختلفة من استقطاب الدوران من خلال تنظيم العيوب السطحية لطبقة الأكسيد المتأصلة من رغوة التيتانيوم، كما هو موضح في الشكل 1a و b. يكمن جوهر عملية التخليق في التحكم في البيئة الغازية الاختزالية/الأكسدية أثناء المعالجة الحرارية (الشكل التكميلي 1، انظر التفاصيل في الطرق). بشكل محدد، تم استخدام جو اختزالي.تم تنفيذ ( ) لإنشاء فراغات أكسجين (OVs) في طبقة الأكسيد المتأصلة لقطب التيتانيوم لتحفيز استقطاب الدوران لذرات الحديد الفردية وذرات التيتانيوم المجاورة (الشكل التكميلي 2)، مما ينتج عنه استقطاب الدوران أزواج ( ) بينما جو مؤكسد ( تم استخدام ) لتجنب توليد
OVs للحصول على الانخفاض في الدورانأزواج (للمقارنة. خلال معالجة الهيدروجين لطبقة الأكسيد المتأصلة في رغوة التيتانيوم، تم نقل الإلكترونات أولاً من ذرات الهيدروجين (H) إلى ذرات الأكسجين (O) في شبكة طبقة الأكسيد المتأصلة. ثم، تترك ذرات الأكسجين في الشبكة مع ذرة الهيدروجين لتكوينكما يتضح من خلال ذروة التطور عند حواليخلال قياس التفاعل المبرمج بالحرارة (TPR) لرغوة التيتانيوم النقية في 5% (الشكل التكميلي 3)، وتتكون الفيروسات المعوية على سطح الرغوة. تم استخدام طيف رنين دوران الإلكترون (ESR) لتحديد الحالات الإلكترونية المحبوسة في العيوب الأكسجينية (OVs) (الشكل 1c)، والذي أظهر بوضوح النجاح في إنشاء العيوب الأكسجينية في SP-إلكترود التيتانيوم، وغياب العيوب في SD-تم تأكيد ذلك أيضًا من خلال إشارات ESR الصامتة.كما يتضح من صور المجهر الإلكتروني الناقل عالي الدقة (HRTEM) لـ SP-و SD- (الأشكال التكميلية 4، 5)، لوحظت الحدود المميزة بين التيتانيوم البلوري الداخلي وطبقة أكسيد التيتانيوم غير المتبلور السطحية ( ) ، متسق مع عملنا السابق . أظهرت صورة المجهر الإلكتروني الناقل الماسح ذو الزاوية العالية والحقل المظلم المصحح للانحراف (HAADF-STEM) أن ذرات الحديد الفردية (المعلمة بدوائر بيضاء) كانت موزعة بشكل جيد في طبقة الأكسيد لرغوة التيتانيوم لكل من SPو SD- (الشكل 1d والشكل التكميلي 6)، ولم تُلاحظ تجمعات أو جزيئات الحديد، مما يتماشى مع غياب قمم حيود الحديد المعدني في نمط حيود الأشعة السينية (الشكل التكميلي 7). وقد أكد ملف شدة السطح ثلاثي الأبعاد على طول السهم الأصفر مزيدًا من التأكيد على توزيع الحديد على المستوى الذري على الـالرغوة (الشكل التوضيحي الإضافي 8). كشفت صور رسم الخرائط الطيفية العنصرية (EDS) عن التوزيع المتجانس لذرات الحديد فيو (الشكل 1e والشكل التوضيحي 9).
استخدمنا أيضًا مطيافية امتصاص الأشعة السينية الدقيقة (XAFS) لتأكيد التشتت الذري لذرات الحديد على. كما يتضح في الشكل 1f، فإن طيف الامتصاص الدقيق للأشعة السينية الممتد عند حافة الحديد K المحول فورييه (FT-EXAFS) لـ SP-أظهر ذروة سائدة عند، الذي يتوافق مع التنسيق في القشرة الأولىغيابمسار التشتت فياستبعدت تجمع الحديد المعدني في SP-كما استخدمنا تحليل تحويل الموجات (WT)-EXAFS لتقديم دقة في كل من المسافة الشعاعية وفضاء k، ولتكملة تحليل FT-EXAFS. كما هو موضح في الشكل 1g، فإن طيف WT-EXAFS لـ SP-عرضت الذروة بأقصى شدة عندعلى عكس رقائق الحديد (الشكل التكميلي 10)، التي تم تخصيصها لـمسار التشتت. هيكل محلي مشابه دقيق لذرات الحديد على SD-تمت ملاحظة القطب الكهربائي أيضًا (الشكل 1f-h). علاوة على ذلك، فإن الموصلية الكهربائية لـكان قريبًا جدًا من SD- (الجدول التكميلي 1)، مستفيدًا من الموصلية الكهربائية الممتازة لركيزة التيتانيوم.
أدلة تجريبية على استقطاب الدوران لـ-أزواج Ti الناتجة عن OVs
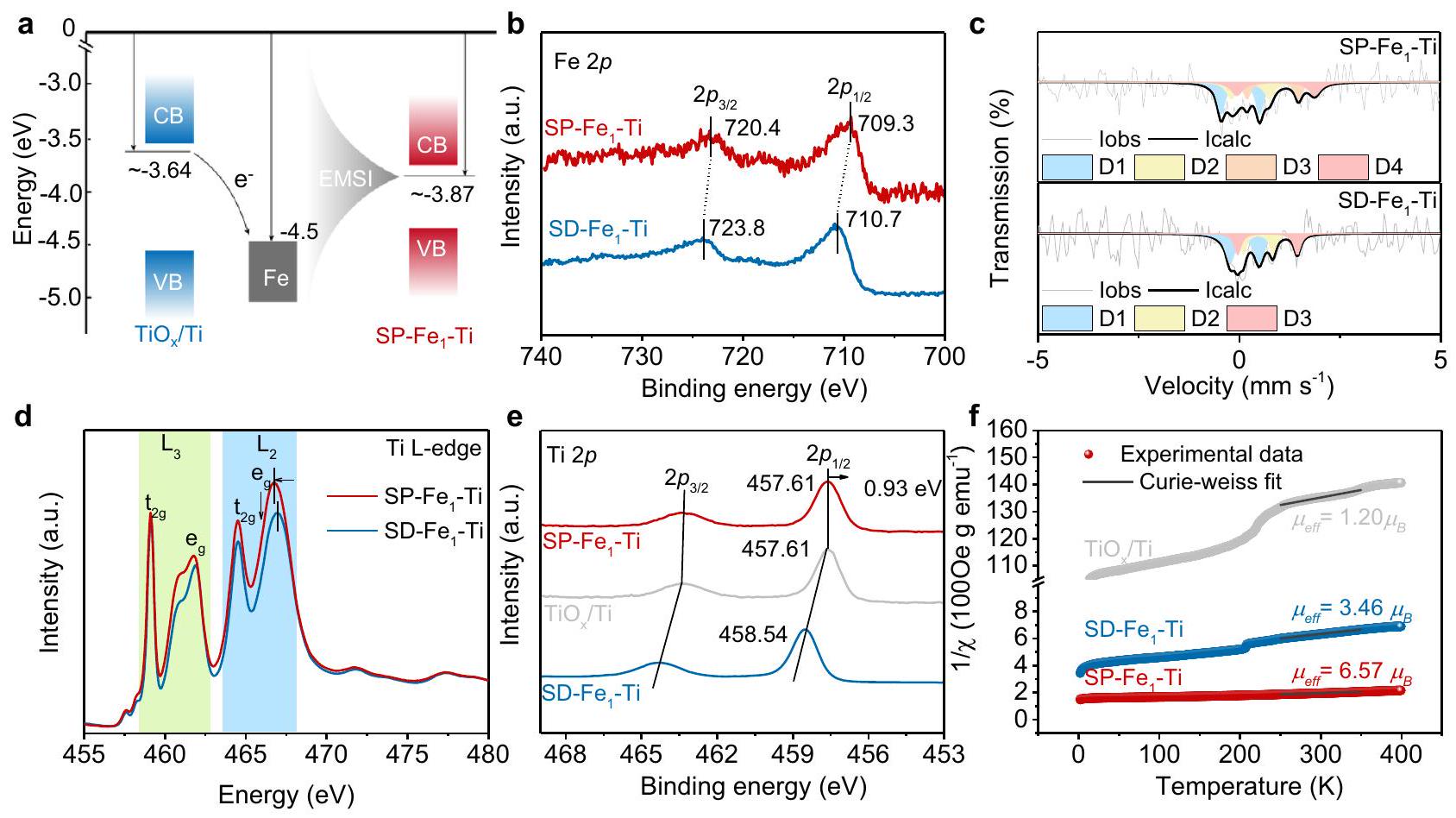
من المتوقع أن تنتقل الإلكترونات غير المتزاوجة الموجودة في العيوب إلى الفراغالمستويات التابعة لذرات التيتانيوم والحديد المجاورة لـ، مما يؤدي إلى نتيجتين محتملتين بما في ذلك تحول سلبي في طاقات الربط الأساسية لذرات التيتانيوم أو الحديد المخفضة، ووجود إلكترون غير مزدوج (دوران) في غلاف ذرات التيتانيوم أو الحديد. بينما الخصائص الإلكترونية للحديد والتيتانيوم في SD-يجب أن يكون القطب الكهربائي بدون OVs مختلفًا عن تلك الخاصة بـ SP-القطب الكهربائي، الحفاظ على دوران منخفضوالدول. لتأكيد الهياكل الإلكترونية وحالات الدوران لـ SP-و SD-تم إجراء قياسات ESR، وطيف الإلكترون الضوئي بالأشعة السينية (XPS)، وطيف امتصاص الأشعة السينية الناعمة (XAS)، وطيف موسباور، وقياسات المغنطة المعتمدة على درجة الحرارة (M-T). قمنا أولاً بالتحقيق في محاذاة طاقة مستويات فيرمي.، انظر تفاصيل الحساب في الشكل التوضيحي 11)و Fe (الشكل 2أ). وُجد أن الحساباتمنكان فوق ذلك منلذا فإن الإلكترونات المحاصرة عند العيوب في المادة غير المتبلورةيمكن أن تنتقل السطح إلى ذرات الحديد المدعومة، مما يؤدي إلىزيادة السرعة. كما يُرى من الأعلى -دقة Feتمت ملاحظة قمتين عند 710.7 و 723.8 إلكترون فولت في طيف المستوى الأساسي في الشكل 2ب.الذي يمكن تخصيصه لـومدارات ثلاثية التكافؤنوع. بالمقارنة مع طيف SD- ، وقممأظهر تحولًا واضحًا في طاقة الربط منخفضة الطاقة، مما يشير إلى تقليل حالة الفالنس لعنصر الحديد الناتجة عن العيوب في الأكسجين وظهور حالة غنية بالإلكترونات.الأنواع. على هذا النحو، الفيروسات الغير متبلورةسطحيمكن أن ينقل الإلكترون إلى ذرات الحديد الفردية ويؤدي إلى زيادة كثافة الإلكترون في ذرات الحديد الفردية. يحدث مثل هذا النقل للإلكترون من العيوب الأكسجينية إلى ذرات الحديد حتى داخل دون معالجة حرارية، كما يتضح من ظهورالأنواع في حديدهطيف XPS (الشكل التوضيحي 12). قمنا أيضًا بتطبيق مطيافية موسباور للكشف عن حالة الدوران لذرات الحديد الفردية على القطب الأحادي من التيتانيوم (الشكل 2c)، ولم نجد أي مجموعات أو مفردات ولكن تم اشتقاق ثنائيات من فك تشفير طيف موسباور لـ SP-و SD-، مما يدل على غياب سنداتكما هو متوقع،امتلك محتوى أعلى بكثير منأنواع (40.2%) بتكوين استقطاب دوران عالي ) من SD- وفقًا للنتائج الملائمة (الجدول التكميلي 2).
الشكل 1 | التلاعب في استقطاب الدوران لـ-أزواج Ti. توضيح تخطيطي لاستراتيجية التلاعب في استقطاب الدوران لـأزواج على سطح أكسيد متأصل لرغوة التيتانيوممستقطب الدورانومكتئب الدورانتمثل الكرات البرتقالية والصفراء والسماوية والزرقاء والرمادية والشفافة الحديد المقطب الدوراني، والحديد المنخفض الدوران، والتيتانيوم المقطب الدوراني، والتيتانيوم المنخفض الدوران، والأكسجين. وفراغ الأكسجين (OV) على التوالي. طيف ESR لـو SP –-تي. صورة HAADF-STEM لـ SP- حيث تم الإشارة إلى ذرات الحديد الفردية بواسطة دائرة بيضاء. صورة HADDF ورسم الخرائط العنصرية STEM لـ SP--تي. طيف FT-EXAFS لـرقائقعند حافة ك لعنصر الحديد.-طيف WT -EXAFS الموزونوعند حافة الحديد (Fe K).
الشكل 2 | الأدلة التجريبية لاستقطاب الدوران لـ-أزواج Ti الناتجة عن OVs. أ محاذاة الطاقة لـلـ، ورقة الحديد.طيف XPS لـ SD- و SP- . طيف ميسباور لـ SP-و SD طيف XAS الناعم لحدود Ti. d Ti L لـ SD-تي و SP-تي. إي تيطيف XPS لـ SD- و SP- .مخططات مقابل درجة الحرارة والحساباتمن و SP- .
علاوة على ذلك، وجدنا أن الفيروسات الغدية على كلا الجانبينورافق ذلك ظهور استقطاب الدورانأنواع تحتوي على إشارة EPR فيالذي كان غير مرئي في طيف EPR لـبدون OVs (الشكل 1c). تشير هذه الفجوة إلى أن إدخال OVs يمكن أن يحفز الاستقطاب المغزلي لمواقع التيتانيوم. لذلك استخدمنا قياس الأشعة السينية الناعمة عند حافة L للتيتانيوم، والذي يتمتع بحساسية عالية لحالة الشحنة، واحتلال المدارات، واتجاه دوران الإلكترونات.لكشف تشكيل الاستقطاب الدورانيالأنواع على SP-Fe1-Ti (الشكل 2d). الميزات الطيفية المتعددة لـ SD-كانت مشابهة جدًا لـتشير إلىحالة التكافؤ. بالمقارنة مع التحول إلى طاقة أقل وذروة شدة SP-تحقق من تشكيل الاستقطاب الدورانيالأنواع المستحثة بواسطة، والذي تم تأكيده بشكل أكبر من خلال الانتقال إلى طاقة ربط أقل في طيف XPS لكلاو SP (الشكل 2e).
لذلك قمنا بإجراء قياس القابلية المغناطيسية المعتمدة على درجة الحرارةقياس لفك اللف المغناطيسي الفعال الكلي )، والتي يمكن تحديدها من خلال قانون كوري-وايس . وفقًا لـمخططات مقابل درجة الحرارةو SD- (الشكل 2f)، ترتيب الحساباتكان في تسلسل SP-Fe ( )، مما يشير إلى SP- امتلكت عددًا أكبر بكثير من الإلكترونات الدوارة منوبشكل عام، أظهرت النتائج المذكورة أعلاه بوضوح أن استقطاب الدوران للذرات الموزعة على المستوى الذرييمكن بسهولة التلاعب بالأزواج بواسطة OVs، مما يوفر فرصة لتطوير محفزات ذرات مفردة قائمة على الحديد ذات استقطاب مغزلي فريد تجاه NITRR وتعميق الفهم حول تأثير الاستقطاب المغزلي على NITRR.
تقييم أداء NITRR تجاه أقطاب مختلفة
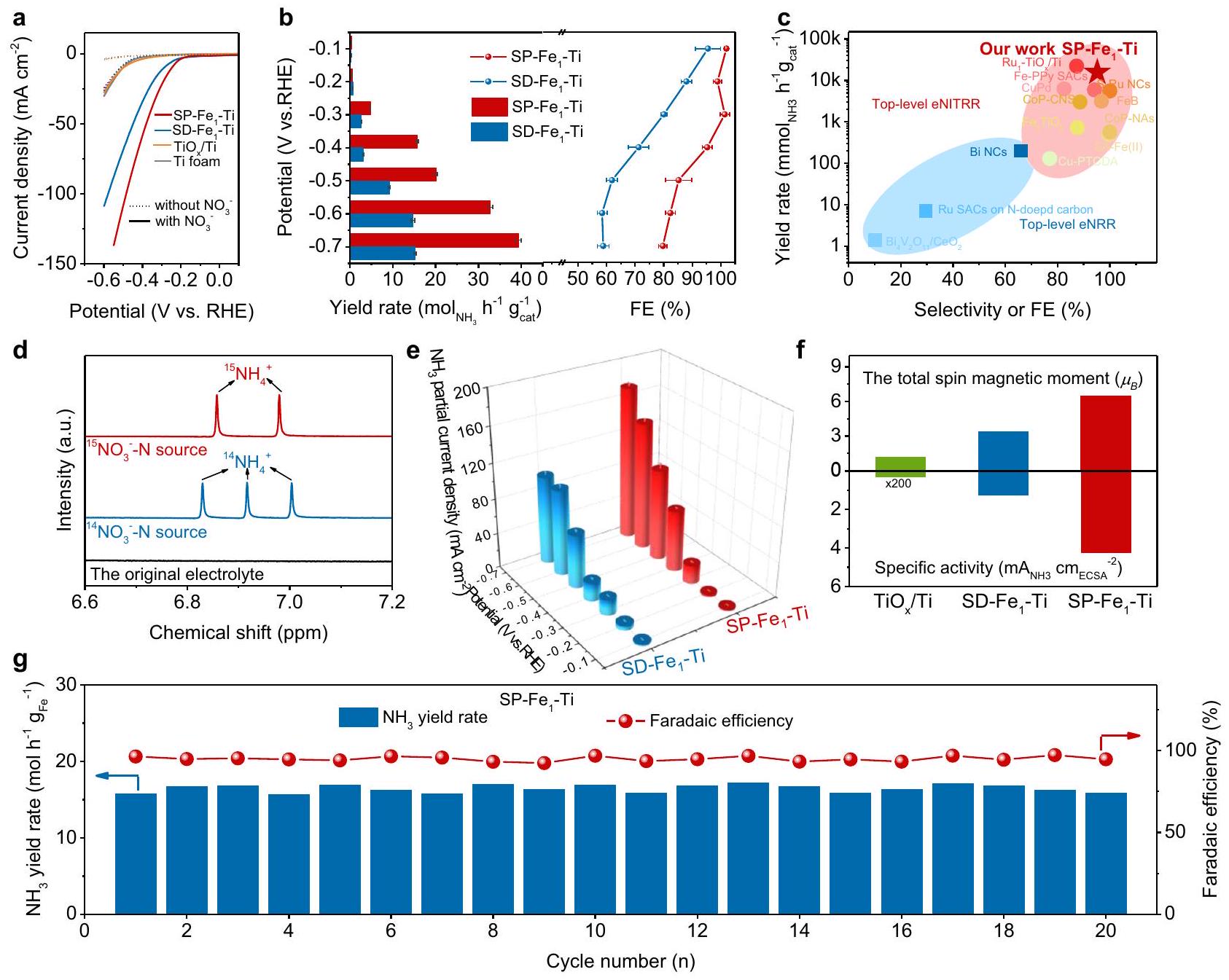
تمت دراسة أداء NITRR على أقطاب مختلفة في خلية إلكتروليتية من نوع H تحت ظروف محيطية. بالنسبة لتقنية الفولتميترية ذات المسح الخطي (LSV) في 1 M KOH (الشكل 3a)، زاد إضافة النترات بشكل كبير من كثافة التيار.القطب الكهربائي أكثر من تلك رغوة التيتانيوم،والأقطاب الكهربائية. قمنا أيضًا بتحديد كمية الـالكفاءة فارادايكية ) و معدل العائد عند إمكانيّة معينة بعد ساعتين من التحليل الكهربائي عبر مطيافية الأشعة فوق البنفسجية والمرئية (الأشكال التكميلية 13 و14). كما هو متوقع، فإن SPأظهر القطب الكهربائي مستوى أعلىومعدل العائد منالقطب الكهربائي (الشكل 3ب). بشكل مثير للإعجاب، أظهر القطب الكهربائي SP-Fe1-Ti جاذبيةمعدل العائد وكفاءة فاراداي عالية عند -0.4 فولت مقابل RHE، والتي تم التحقق منها أيضًا من النتائج التي تم الحصول عليها منقياس الرنين المغناطيسي النووي (NMR، الشكل التوضيحي 15). مثل هذا النشاط الممتاز لـ NITRR لـ SP-كان القطب الكهربائي متفوقًا بكثير على تلك المحفزات الكهربائية الحديثة NITRR ومحفزات هابر-بوشمحفزات الاختزال الكهروكيميائية (الشكل 3c والجدول التكميلي 3). بالإضافة إلى ذلك،يمتلك القطب الكهربائي إمكانيات تطبيق واعدة لتعظيمإمكانات البداية وكثافة التيار عند جهد معين (الجدول التكميلي 4) على معظم أقطاب NITRR من المستوى العالي التي تتكون من مكونات منخفضة السعر (على سبيل المثال،، و Fe في الجدول التكميلي 5). كما أننا نضع مسحوق محفز SP-Fe1-Ti على ورق الألياف الكربونية بتحميل كتلة المحفز لتقييم الأداء. كما هو موضح في الشكل التكميلي 16،يعرض المحفز كثافة تيار كاثودي عالية منعند -0.4 فولت مقابل RHE، متفوقًا على معظم المحفزات NITRR من المستوى الأعلى.معدل العائد المحدد بواسطة مساحة القطب الكهربائي لـ SP-تم تحديد المحفز على أنهالذي يتفوق على معظم المحفزات المعدنية منخفضة السعر وحتى العديد من المحفزات المعدنية عالية السعر (الجدول التكميلي 6). والأهم من ذلك، الطبيعة الأحادية لـالقطب الكهربائي أكثر جدوى بكثير من شكل المسحوق من المحفزات الكهربائية NITRR عالية المستوى السابقة للاستخدام العملي.
تم تقييم مساهمات المنتجات المحتملة الأخرى في التيار الفارادي أيضًا باستخدام كروماتوغرافيا الغاز المزودة بكاشف الموصلية الحرارية (TCD) وطيفية الأشعة فوق البنفسجية والمرئية.وتم اكتشافها (الشكل التكميلي 17 والشكل التكميلي 18)، وتم حساب قيمتها من حيث FEs لتكونوعلى التوالي،
الشكل 3 | أداء NITRR لأقطاب مختلفة. أ منحنيات LSV لرغوة التيتانيوم، و SP- أقطاب كهربائية.معدل العائد ومن SD و SP- أقطاب كهربائية عند إمكانيات مختلفة.مقارنة أداء التحفيز الكهربائي لتفاعل اختزال النيتروجين (NITRR) لـ SP-الكاثود مع محفزات كهربائية أخرى تم الإبلاغ عنها بشكل واسع.طيف الرنين المغناطيسي النووي للمنتجات الناتجة خلال التحفيز الكهربائي لتقليل النيتروجين على SP-القطب الأحادي عند -0.4 فولت مقابل RHE. جزئيكثافات التيار الحالية لـ SD-و SP-أقطاب التيتانيوم تم تطبيعها على المساحة الهندسية عند إمكانيات مختلفة.النشاط المحدد المعدل إلى ECSA و SP- أقطاب التيتانيوم عند -0.4 فولت مقابل RHE.معدل العائد وكفاءة فاراداي لـ SP-القطب تحت الجهد المطبق -0.4 فولت مقابل RHE خلال 20 دورة تحليل كهربائي متتالية. عند -0.4 فولت مقابل RHE. بعد أخذ التفاعلات الجانبية في الاعتبار، كانت الكفاءة الكلية (الشكل التكميلي 19) هيكما هو متوقع. علاوة على ذلك،انتقائية SP-تم تحديد قطب “Ti” على أنهعند -0.4 فولت مقابل RHE، مما يؤكد انتقائيته العالية تجاهالإنتاج من NITRR.
لذا استخدمنا الرنين المغناطيسي النووي لتأكيد مصدر النيتروجين الناتجعبر تجارب وسم النظائر مع-موسومكعامل كيميائي. مختلف عنمع ثلاثة قمم فيطيف الرنين المغناطيسي النووي، فقط قمتانظهر فيطيف الرنين المغناطيسي النووي عندماتم استخدامه ككاشف (الشكل 3د)، مما يوضح أن المنتجكان من المواد الخام النيتروجينية بدلاً من التلوثات. بالإضافة إلى ذلك، قممغائبون فيطيف NMR للإلكتروليت الأصلي الذي يحتوي على، مما يؤكد أكثر أن المنتج نشأ ذلك من تقليل المواد الخام النيتروجينية بدلاً من التلوثات. ومن الجدير بالذكر أن SP-أظهر القطب الكهربائي أيضًا حجمًا أكبركثافة التيار الجزئي لـعند -0.7 فولت مقابل RHE منالقطب الكهربائي (الشكل 3e)، مما يشير إلى إمكانيات كبيرة في التطبيقات العملية. كما قمنا بتقييم النشاط الجوهري لـ SPالقطب الكهربائي من خلال حساب النشاط النوعي (SA) عن طريق تطبيع نشاط القطب الكهربائي إلى المساحة السطحية الكهروكيميائية (ECSA، الشكل التوضيحي التكميلي 20)، ووجدنا أن SA لـ SP- كان الجهد الكهربائي على الأقطاب الكهربائية أعلى على ما يبدو منوالأقطاب عند -0.4 فولت مقابل RHE (الشكل 3f)، تكشف عن النشاط التحفيزي العالي intrinsically لتفاعل اختزال النيتروجين في وجود دوران مغزليالأزواج والمساهمة الكبيرة لاستقطاب الدوران لمواقع المعادن النشطة في NITRR.
نظرًا لاختلاف تركيز النترات في المصادر المختلفة، قمنا أيضًا بفحص نشاط NITRR لـ SP-القطب الكهربائي في الإلكتروليت مع اختلافتركيزات (، و 1 م ) عند -0.4 فولت مقابل RHE. كما هو موضح في الشكل التوضيحي 21، فإن كفاءات -إلى-التحويل كانوفي المحلول الكهربائي مع، و 1 م، على التوالي. تشير هذه النتائج إلى أن لا توجد تأثيرات واضحة على التركيز فيمن SP--إلكترود Ti، مما يدل على الواسعتوافق التركيز. بالإضافة إلى ذلك، لاحظنا أنتم تحسين معدل العائد من خلال زيادةتركيزات من 0.1 إلى 1 م بسبب تسريع نقل الكتلة. قمنا أيضًا بتقييم نشاط NITRR لـ SP-القطب في الحالة المحايدة. بشكل مثير للإعجاب، SP-عرضت مقارنةمعدل العائد ) و FE (96.1%) إلى تلك الموجودة في الحالة القلوية (الشكل التكميلي 22)، مما يوضح بشكل أكبر إمكانيته العالية للتطبيق البيئي.
الأهم من ذلك،ومعدل العائدالقطب الكهربائي ظل مستقرًا جدًا خلال 20 دورة من التحليل الكهربائي المتتالي
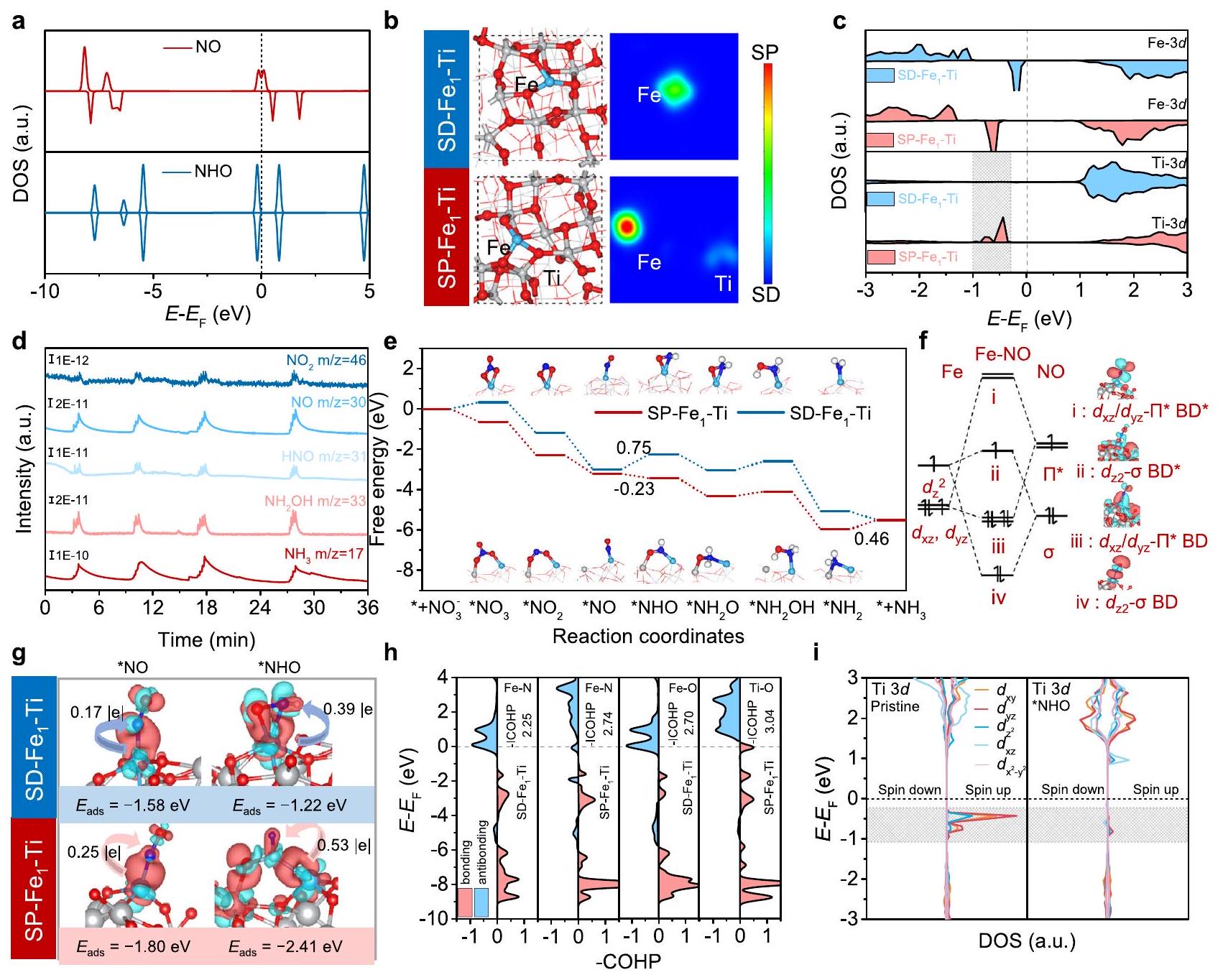
الشكل 4 | رؤى حول آلية NITRR. أ DOSs من أنواع NO و NHO. الهياكل المحسّنة والرسوم البيانية لكثافة الدوران ثنائية الأبعاد لـ SD-و. c DOSs لذرات الحديد والتيتانيوم علىو. قياسات DEMS لـ NITRR فوق SP-القطب الكهربائي. e تم حساب مخططات الطاقة الحرة لـ NITRR على SD- و SP- . تفاعلات مدارية رئيسية بين ذرة الحديد وجزيء NO بالإضافة إلى مخططات المدارات الجزيئية المقابلة (BD)
وتمثل المدارات الرابطة والمدارات المضادة للرابطة، على التوالي).اختلافات كثافة الشحنة لـ NO و NHO الممتصة علىو (يمثل اللون الأحمر تراكم الإلكترونات واللون الساين استنفادها، على التوالي). COHPs من NHO الممتصة على SD- و SP- . DOSs لذرة التيتانيوم على SP-قبل وبعد امتصاص NHO. يتم الإشارة إلى ذرات التيتانيوم والأكسجين والحديد والنيتروجين والهيدروجين باللون الرمادي والأحمر والسماوي والأزرق والأبيض، على التوالي. في خلية إلكتروليتية من نوع H عند -0.4 فولت مقابل RHE (الشكل 3 ج والشكل التكميلي 23)، مما يدل على متانتها على المدى الطويل وإمكاناتها العالية للتطبيق العملي. كما كشفت تحليلات XRD وXPS وTEM عن الاستقرار العالي لـ SP--Ti الكاثود خلال NITRR (الأشكال التكميلية 24-26)، كما تأكد من خلال الذوبان الضئيل للحديد في SP-القطب الكهربائي خلال 20 دورة من التحليل الكهربائي المتتالي (الشكل التوضيحي 27).
رؤى حول آلية NITRR تجاه SP-Fe-أزواج تي
تم استخدام حسابات نظرية الكثافة الوظيفية (DFT) للحصول على رؤى حول آلية NITRR على SP-الأزواج. استكشفنا أولاً كثافة الحالات (DOSs) للأنواع الرئيسية NO وNHO المعنية في NITRR (الشكل 4a)، وأكدنا الحالة المغزلية غير المتناظرة لـ NO في DOS لحالات الدوران لأعلى ولأسفل، والتي كانت مختلفة عن تلك الخاصة بـ NHO، مما يشير إلى أن تحويل NO إلى NHO كان مصحوبًا بانتقال في حالة الدوران. كما كشفت النماذج التي تم بناؤها لـ SPو SD- (الشكل 4ب)، كانت ذرات الحديد الخاصة بهم منسقة على التوالي مع ثلاث وأربع ذرات أكسجين لـويتماشى ذلك بشكل جيد مع نتائج EXAFS الملائمة (الشكل التوضيحي 28 والجدول 7). أظهرت مخططات كثافة الدوران أن ذرة الحديد فقط هي التي كانت مستقطبة الدوران لـبينما كل من ذرة الحديد وذرة التيتانيوم متجاورتان تم استقطاب دوران ذرة الحديد في حالة (الشكل 4ب). هذه الفروق المغناطيسية أكدت بشكل أكبر زيادة الـتم قياس العزم المغناطيسي مع وجود OV (الشكل 2f). كانت الحالات المغناطيسية واضحة أيضًا من دوال الكثافة لحالات الإلكترون (DOSs) لذرات الحديد والتيتانيوم. كانت مكونات الدوران لأعلى ولأسفل لذرة الحديد حول مستوى فيرمي.كانت مختلفة تمامًا (الشكل 4ج)، مما يدل على وجود إلكترونات غير متزاوجة فيالمدارات. بالإضافة إلى ذلك، مكونات الدوران لأعلى ولأسفل لذرة التيتانيوم في SDكانت تقريبًا متشابهة، لكن حالات إلكترونية غير متزاوجة ظهرت بالقرب منبعد إدخال OV (الشكل 4c). ساهمت هذه الذرات المغناطيسية من التيتانيوم والحديد في تحقيق NITRR بكفاءة عالية.
نظرًا لأن مصدر الهيدروجين لـ NITRR تحت الظروف القلوية يأتي منتفكك الماء بما في ذلكإن انكسار الروابط وقدرة نقل البروتون ستؤثر بشكل كبير على النشاط التحفيزي لـ NITRR. وقد وُجد أنكان الانفصال طفيفًا ماصًا للحرارة بمقدار 0.08 و 0.03 إلكترون فولت، مع حواجز قدرها 0.28 و 0.29 إلكترون فولت على SD- و SP- على التوالي، مما يشير إلى أنكان الانفصال على كلا القطبين مفضلاً للغاية وقابلًا للمقارنة (الشكل التكميلي 29). كما استخدمنا تقنية الرنين المغناطيسي الإلكتروني (ESR) باستخدام 5,5-ثنائي ميثيل-1-بيرولين-N-أكسيد (DMPO) كعامل لاحتجاز الجذور للتحقيق في توليد ودور الجذور الهيدروجينية الناتجة عن انفصال الماء. لفهم آلية اختزال النترات. تم ملاحظة تسعة قمم ESR بنسبة شدة 1:1:2:1:2:1:2:1:1 في الشكل التكميلية 30 لعملية التحفيز الكهربائي على الـإلكترود في النقاء، الذي يمكن أن يُنسب إلى الناتج الدوراني لـ DMPO-H، مؤكدًا توليد . عند الإضافة في KOH ، شدة الإشارة لـاختفى. مثل هذه الظاهرة كشفت أن الناتجتم استهلاك من خلال تفكك الماء، مما يشير إلى المشاركة العميقة لـفي عملية الهيدروجين الناتج عن NITRR. قمنا أيضًا بإجراء تجارب تأثير النظائر الحركية (KIE) لتقدير قدرة نقل البروتونات لـ SP.وخلال NITRR من خلال مقارنة نسب كثافة التيار. قيمة KIE لـ NITRR (1.40) لـ SP-كان قريبًا من ذلك (1.53) من SD-عند -0.4 فولت مقابل RHE (الشكل التكميلي 31)، مما يشير إلى قدرة نقل الهيدروجين المماثلة على القطبين. لذلك، فإن قدرات تفكك الماء ونقل الهيدروجين لها تأثير ضئيل على الأنشطة المميزة لـ NITRR لـ SP-و SD-.
لفهم الدور الفريد للسبين المستقطبالأزواج على NITRR، مسارات تفاعل NITRR على SD-وتم التحقيق فيها. لبناء وصف شامل لآلية التفاعل، تم استخدام مطيافية الكتلة الكهربائية التفاضلية على الإنترنت (DEMS) للكشف عن الوسائط والمنتجات الرئيسية على الإنترنت (الشكل 4d). لقد لاحظناإشارات، و 17 من ، و “، على التوالي. استنادًا إلى نتائج DEMS، قمنا بحساب الطاقة الحرة للوسط الفردي علىو (الشكل 4e، الشكل التوضيحي 32) تحت جهد 0 فولت مقابل.. في الواقع، إلى *لا خضعت لـخطوة الامتزاز وخطوتان لإزالة الأكسجين، وهما، * * * * لا. في حالة النظام،خضعت الامتزاز لتغير في الطاقة الحرة لجيبس بمقدار 0.34 إلكترون فولت. بعد ذلك،أولاً إزالة الأكسجين إلىمع تغيير في الطاقة الحرة نحو الأسفل قدره -1.52 إلكترون فولت. بعد ذلك،استمر في إزالة أكسجين واحد لتكوين *NO مع إطلاق طاقة قدرها 1.81 إلكترون فولت. أما بالنسبة لـ SP-النظام، الخطوات الثلاثة أعلاه تطلق طاقة0.93 إلكترون فولت، على التوالي. أخيرًا،تم إزالة الأكسجين إلى *NO مع تغيير إجمالي في طاقة غيبس الحرة بمقدار -3.00 إلكترون فولت على SDبينما كانت التغير الكلي في الطاقة الحرة للانحدار للخطوات المذكورة أعلاه -3.22 إلكترون فولت. لذلك، إزالة الأكسجين إلى *NO على SP-كان أكثر ملاءمة من الناحية الديناميكية الحرارية. بالنسبة لـ NITRR علىكانت الخطوة المحددة للإمكانات (PDS) هي *NO إلى *NHO مع حاجز حراري قدره 0.75 إلكترون فولت، بينما تم تحديد PDS ليكون *إلىمع حاجز حراري ديناميكي قدره 0.46 إلكترون فولت على، مما يؤدي بالتالي إلى زيادة نشاط NITRR مقارنةً بـ SD. جاء هذا التغيير في PDS نتيجة لتأثير كل من مواقع الحديد والتيتانيوم المستقطبة بالدوران على، مما أدى إلى انخفاض ملحوظ في الطاقة الحرة لـ *NO إلى *NHO من 0.98 إلكترون فولت إلى -0.23 إلكترون فولت. في الوقت نفسه، كانت نسبة شدة الذروة أصغر بكثير (~3.92) لـ *NO إلى *NHO لـمن ذلك ( تمت ملاحظته، مما يؤكد بشكل إضافي تسريع عملية *NO الهيدروجينية علىالقطب الكهربائي. لتسليط الضوء بشكل أكبر على التأثير التآزري لـأزواج الدوران، قمنا بحساب الطاقة الحرة للوسط *NHO الممتص علىالأزواج التي تتضمن ذرة تيتانيوم بدون استقطاب مغناطيسي للمقارنة (الشكل التكميلي 34)، وكانت القيمة المحسوبة 0.37 إلكترون فولت أعلى بكثير من تلك على، مما يشير إلى أن الاستقطاب الدوراني لذرة التيتانيوم ساهم بشكل كبير في زيادة نشاط تفاعل اختزال النيتروجين. بعد ذلك، قمنا بدراسة تفاعل تطور الهيدروجين التنافسي على الاستقطاب الدورانيالأزواج (الشكل التوضيحي التكميلي 35)، والطاقة الحرة المحسوبة لـالامتزاز على مواقع الحديد والتيتانيومكانت 1.13 و 0.78 إلكترون فولت، على التوالي، أضعف بكثير من ذلك لـممتص على مغناطيسية الدورانأزواج ( -0.64 eV ). لذلك، الـتوليد على الاستقطاب الدورانيتم تثبيط الأزواج، مما ساعد على تعزيز الكفاءة الفارادية لتقنية NITRR.
لفهم تأثير الاستقطاب الدوراني بشكل أفضلتم تحليل التفاعلات المدارية بين *NO/*NHO وزوج Fe-Ti بالتفصيل، حيث تم التركيز على تعزيز نشاط NITRR. تم اقتراح أن الهدرجة تتحكم عمومًا في النشاط. لعدم امتصاص NO على، وُجد أن NO يمتص بشكل مفضل علىأقوى من ذلك على. كانت المساهمة الرئيسية في التفاعل بين الحديد و *NO من مدارات ذرة الحديد ومدارات NO (الشكل 4f)، وفقًا لتكوين الاستقطاب العالي الدوران ) من الحديد، كما ذُكر أعلاه. لاحظنا أن ولم تتمكن مدارات الحديد من التهجين مع المدار الخاص بأكسيد النيتريك بسبب الحفاظ على التماثل، وبالتالي لم يتم اعتبار تفاعل مداراتها.. وبالتالي، في مثل هذا النموذج للترابط، فإن يمكن لذرات الحديد أن تقبل الإلكترونات من المستويات المملوءةمدار NO، وبالمقابل إلكترونات Feيمكن أن تقوم المدارات بحقن في الحالة المشغولة جزئيًامدارات لتقليل ترتيب الرابطة لـ. هذه “التبرع-التبرع العكسي” للإلكترونات أدى عمومًا إلى قبول NO لـ 0.25 |e| من الركيزة (الشكل 4 ج والشكل التكميلية 36)، مما نتج عنه طاقة ربط قدرها -1.80 eV لامتصاص NO على موقع الحديد في SP-أقوى من -1.58 إلكترون فولت في موقع الحديد لـالذي نقل أقل من 0.17 إلكترون إلى *NO. علاوة على ذلك، يمكن أن تكون الأزواج المستقطبة مغناطيسياً مفيدة أيضاً في هدرجة *NO من خلال استقرار وسائط *NHO المهدرجة بشكل كبير. الطاقة المحسوبة لعملية الامتزاز البالغة -2.41 إلكترون فولت لـ *NHO التي تفضل الامتزاز على SP-من خلال تشكيلوكان أقوى بكثير من -1.22 eV علىعبر التشكيلو الروابط، لأن المزيد من الإلكترونات تم نقلها إلى *NHO من SP-Fe أكثر من ذلك من SDكما يتضح من تحليل شحنة بادر (الشكل 4 ج)، الذي تم دعمه من خلال تحليل كثافات هاملتونيان البلورات (COHP) (الشكل 4 ح). القيم السلبية المدمجة لـ COHP (-COHP) لـوالتفاعلات علىكانت أكثر إيجابية من تلك لـوالتفاعلات على SD-، مما يشير إلى أن الاستقطاب الدورانيأظهرت الأزواج تفاعلًا أقوى مع *NHO. كما ذُكر سابقًا، فإن ذرة التيتانيوم المجاورة لذرة الحديد منمرت بانتقال حالة الدوران من عدم استقطاب الدوران إلى استقطاب الدوران بعد إدخال العيوب، كما تدعمه ظهور غير المتزاوجين، و الدول القريبة من(الشكل 5ي). علاوة على ذلك، الـحالة الدوران بالقرب منذرة التيتانيوم علىاختفى بعد امتصاص *NHO، مما يعني أن الإلكترون غير المتزاوج من ذرة التيتانيوم تم حقنه في *. بالمثل، الإلكترون ذو الدوران غير المتزاوج فييمكن لحالة Fe أيضًا أن تضخ جزئيًا في *NHO (الشكل التوضيحي التكميلي 37)، مما يعزز نقل الإلكترون منزوج الدوران لتثبيت *NHO بشكل أكبر. وبشكل قاطع، كلاهما موجهان بالدورانيمكن لمواقع الزوج أن تسهل بشكل فعال إزالة الأكسجينلتحقيق *NO واستقراره و intermediates *NHO لتعزيز *NO الهدرجة اللاحقة، بالإضافة إلى تثبيط تفاعل تطور الهيدروجين، مما يؤدي إلى نشاط عالي وانتقائية.القطب الكهربائي نحو NITRR.
معدات متكاملة لخلية التحليل الكهربائي NITRR ذات التدفق المستمر واسترداد الأمونيا في الموقع
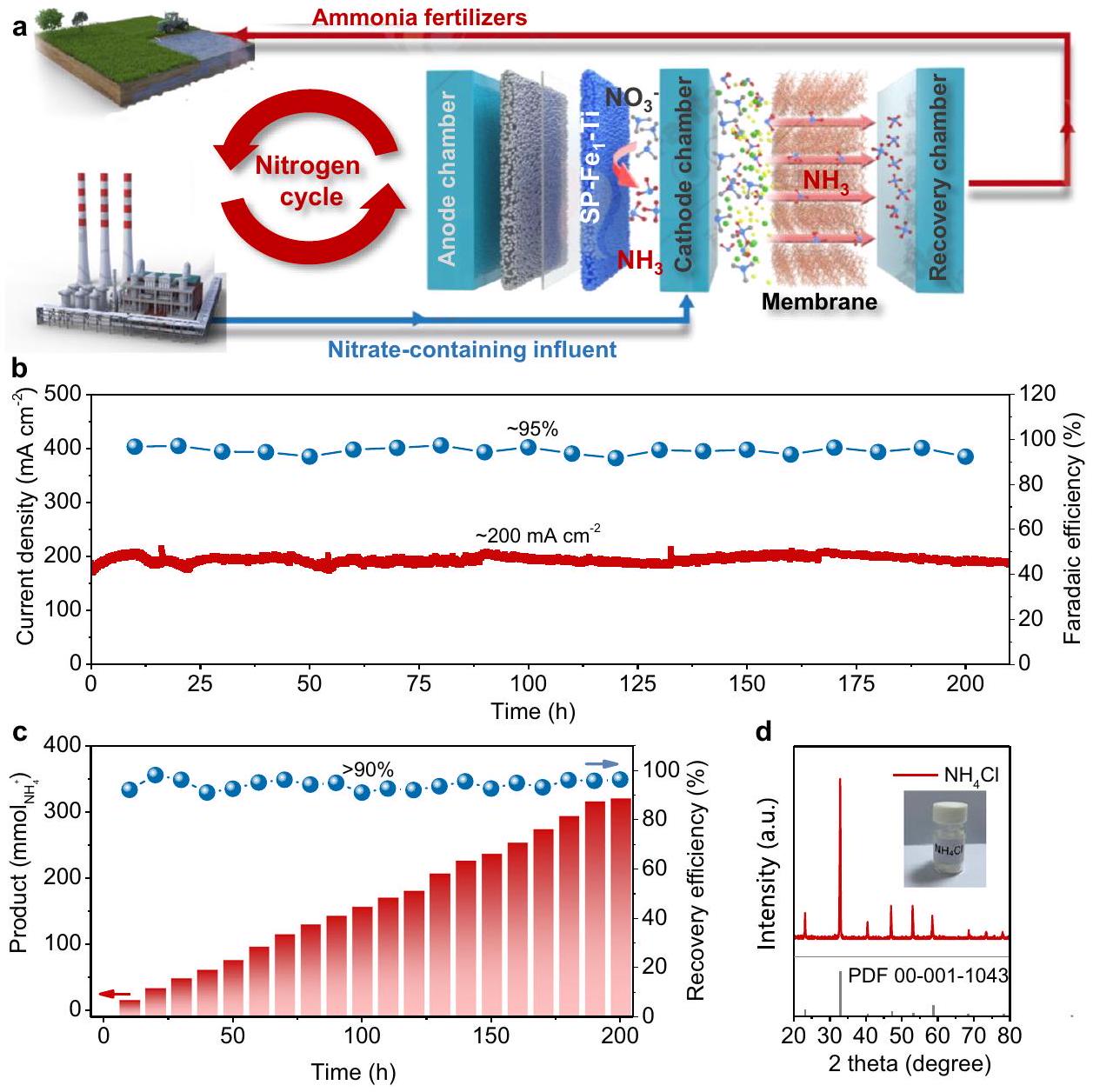
بجانب أداء القطب الكهربائي،فصل المنتج واستعادته أمران حاسمان أيضًا للتطبيق العملي لـ NITRR. من هذا الجانب، صممنا جهازًا متكاملًا يتكون من إلكتروليزر NITRR يعمل بتدفق مستمر ووحدة استرداد الأمونيا قائمة على الغشاء لتحقيق NITRR الفعال وفي الموقعالاسترداد، كما هو موضح بشكل تخطيطي في الشكل 5a والشكل التكميلي 38. قمنا بتصميم إلكتروليزر مدمج يعمل بتدفق مستمر يتكون من SP-الكاثود والأنود المستقر من نوع الشبكة التجارية (DSA،أقطاب كهربائية بسمك طلاء ) كـ NITRR الكهربائي. أثناء التشغيل، -محلول يحتوي علىتم معالجة المياه العادمة الاصطناعية من خلال الاختزال الكهروكيميائي علىالكاثود، ويتم تدويره بواسطة مضخة بيرستالتية بمعدل تدفق. ثم -تم توجيه المياه الملوثة المحتوية على المواد إلى مصفوفات الألياف البولي بروبيلين (PP) المجوفة المغمورة في محلول 1 م HCl (800 مل) لاستعادةمفتاح NITRR المتزامن واستعادة المنتج هي التدفق المستمر لـمن خلال الغشاء الهيدروفوبي القابل لاختراق الغاز ثم
الشكل 5 | جهاز متكامل لخلية التحليل الكهربائي NITRR ذات التدفق المستمر وفي الموقعاستعادة. توضيح تخطيطي لمعدات متكاملة تتكون من مفاعل خلية تدفق وفاصل غشائي من خلال استخدامكاثود أحادي الكتلة لـ NITRR وغشاء هيدروفوبي قابل لاختراق الغاز لـالشفاء. اختبار الاستقرار على المدى الطويل لـ SP-القطب عند -0.4 فولت مقابل RHE باستخدام المعدات المتكاملة. جكفاءة الإنتاج والاسترداد للمعدات المتكاملة خلال اختبار الاستقرار على المدى الطويل.نمط حيود الأشعة السينية (XRD) الناتجمسحوق. العودة إلى غرفة الكاثود. خلال هذه العملية، يتم إنتاجيمكن أن يتخلل من خلال الغشاء بين غرفة الكاثود وغرفة الاسترداد ويتم امتصاصه بواسطة محلول الحمض في غرفة الاسترداد. من خلال استخدام الكاثود الأحادي، يمكن للجهاز المتزاوج أن يعمل بشكل قوي عند كثافة تيار على مستوى صناعي منلمدة تصل إلى 200 ساعة مع مستوى عالٍمن (الشكل 5ب). ونتيجة لذلك، حقق هذا النظام المستدام واللامركزي معدل استرداد النيتروجين العالي في الوقت نفسهالشفاء مع ما يقرب منالانتقائية وحولكفاءة الاسترداد (الشكل 5c). بعد التحليل الكهربائي على المدى الطويل، تم تحديد زاوية الاتصال المقاسة للغشاء (الشكل التوضيحي 39) لتكون، مشابه لذلك من الغشاء النقي ( )، مما يشير إلى أن الغشاء بعد التفاعل لا يزال يحتفظ بخاصية الكارهية للماء. بعد المزيد من التبخر الدوراني، يمكن تحويل الأمونيا المعاد تدويرها في المحلول إلى نقاء عالٍ مسحوق، كما أكد نمط حيود الأشعة السينية (الشكل 5d). أظهرت هذه النتائج إمكانية التحويل الانتقائي لمياه الصرف المحتوية على النترات إلى أسمدة الأمونيا المحسّنة من خلال دمج الأداء العالي لـ SP- – الكاثود الأحادي مع الجهاز المدمج، مما يوفر طريقة مستدامة لدورة النيتروجين.
نقاش
في الختام، الاستقطاب الدورانيتم تصميم الأزواج من خلال التلاعب بفجوات الأكسجين في طبقة الأكسيد السطحية المتأصلة على قطب التيتانيوم الأحادي، وكان القطب الناتج قادرًا على تقديم نشاط استثنائي في تقليل النيتروجين مع أداء مثير للإعجاب.معدل العائد n( ) ومرتفع الكفاءة الفارادية لـعند -0.4 فولت مقابل RHE، مما يثبط نظير الدوران المنخفضالقطب الكهربائي ومعظم الأقطاب الكهربائية المعروفة. وقد تم إثبات أن التدوير المستقطبيمكن أن توفر الأزواج إلكترونات الدوران للتفاعل مع الوسائط التفاعلية، وبالتالي تسهيل إزالة الأكسجين منإلى *NO وهدرجة *NO من خلال تعزيز امتصاصها. من خلال ربط جهاز التحليل الكهربائي المتدفق NITRR بتقنية فصل الأغشية، نجحنا في تحقيق استعادة الأمونيا في موقع إنتاج الأسمدة من الإلكتروليت المحتوي على النترات بطريقة مستدامة ومركزية، من خلال الاستفادة من الدوران الناتج.أزواج الأقطاب. دراستنا لا تعرض فقط بناء الأقطاب الموجهة بالدورانأزواج لـ NITRR وتطبيقات محتملة لمعالجة مياه الصرف أو إنتاج الأمونيا، ولكنها تقدم أيضًا استراتيجية مبتكرة لتطوير الأقطاب المتقدمة مع تأثير استقطاب الدوران تجاه التفاعلات المتعلقة بالدوران.
طرق
المواد الكيميائية
تم شراء القطب الأحادي المصنوع من التيتانيوم من شركة كونشان جوانغجيابوان للمواد الجديدة المحدودة، الصين. كلوريد الحديد ( ) ، تارترات البوتاسيوم والصوديوم ( )، DMSO-d6، حمض الماليك نترات الصوديومهيدروكسيد البوتاسيومنترات البوتاسيوموتم شراء الإيثانول من
شركة سينوفارم للكيماويات. تم استخدام جميع المواد الكيميائية دون مزيد من التنقية.
تحضير الأقطاب الكهربائية
مسار التخليق لـتم توضيح القطب في الشكل التوضيحي 1a، كما تم الإبلاغ عنه سابقًا. عادةً، الـإلكترود وحيد الكتلة معتمت معالجة الحجم أولاً في جو مختزل ) في لمدة 3 ساعات بمعدل تسخينلإنشاء فراغات الأكسجين (OVs) على سطح القطب. ثم، يتم تحضير محلول متجانس من سلفيد الحديد بتركيز كتلة من الحديد قدرهتم ترسيبها بشكل موحد في الإيثانول على العينة المحضرةالقطب الأحادي. في هذه الأثناء، كانت عملية الترسيب تحت مصباح الأشعة تحت الحمراء الذي يضيء لتسريع تبخر المذيب وتجنب تجمع المذيب الناتج عن توتر السطح. أخيرًا، الـ SP-تم الحصول على إلكترود -Ti عن طريق معالجة الإلكترود المودع عليه الحديد في جو مختزل. ) في لمدة 3 ساعات بمعدل تسخينمرة أخرى. الـ SD-تبع القطب الكهربائي نفس مسار التخليق ولكن في جو مؤكسدالهواء) لتجنب توليد OVs للمقارنة (الشكل التوضيحي التكميلي 1b).
توصيف
تم تسجيل أنماط حيود الأشعة السينية (XRD) باستخدام جهاز Rigaku Miniflex-600.و 15 مللي أمبير). تم الحصول على صور الميكروسكوب الإلكتروني الناقل بتقنية الحقل الداكن الزاوي العالي (HAADF-STEM) على جهاز JEOL JEM-ARM200F TEM/STEM مع مصحح الانحراف الكروي يعمل عند 200 كيلو فولت. تم إجراء مطيافية الأشعة السينية للألكترونات (XPS) على مجهر الأشعة السينية الماسح (PHI 5000 Verasa، ULAC-PHI، Inc.) باستخدام إشعاع Al Ka. تعتبر قمة C 1 s عند 284.6 eV معيارًا داخليًا لمعايرة الطاقة. تم إجراء مطيافية الأشعة السينية المشتتة للطاقة (EDS) على جهاز FEI Talos F200X. تم إجراء رنين دوران الإلكترون (ESR) على جهاز EMX micro-6/1 تحت 100 كلفن. تم الحصول على طيف بنية الامتصاص الدقيقة للأشعة السينية عند حافة Fe K في مركز مصدر الضوء المتزامن في سنغافورة (SSLS، يعمل عند 2.5 جيجا فولت مع تيار أقصى قدره 200 مللي أمبير). تم تحديد أطياف EXAFS عن طريق طرح الخلفية بعد الحافة من الامتصاص الكلي ثم تم تطبيعها إلى خطوة قفزة الحافة. بعد ذلك، تم تحويل البيانات باستخدام تحويل فورييه إلى الشكل الحقيقيالفضاء باستخدام نوافذ هانين ) لفصل مساهمات EXAFS من قذائف التنسيق المختلفة. للحصول على المعلمات الهيكلية الكمية حول الذرات المركزية، تم إجراء ملاءمة معلمات منحنى بأقل المربعات باستخدام وحدة ARTEMIS من حزم برامج IFEFFIT. تم جمع طيف XAS لحافة Ti-L في خط الشعاع BL 11 A من المركز الوطني لأبحاث الإشعاع السنكروتروني (NSRRC) في تايوان. قمنا بقياس الموصلية الكهربائية لـ SP-و SD-القطب عند درجة حرارة الغرفة من خلال تقنية الأربعة مجسات للتيار المستمر.
القياسات الكهروكيميائية
تم إجراء اختبار NITRR في خلية من نوع H نموذجية، مفصولة بغشاء نافيوني 117.أقطاب كهربائية بدرجة استقطاب دوران محددةو SD- )، لوحة بلاتينية و تم استخدام الأقطاب الكهربائية كقطب عمل وقطب مضاد وقطب مرجعي، على التوالي. إذا لم يُذكر خلاف ذلك، فإن جميع الجهود المبلغ عنها في هذا العمل كانت مرجعة إلى قطب هيدروجين عكسي (RHE). قبل كل قياس، تم تطهير المحلول الكهربائي بالغاز الأرجون. ) لمدة 30 دقيقة. تم تسجيل منحنيات الفولتمترية المسح الخطي (LSV) بمعدل مسح تحت التحريك بمعدل دوران 500 دورة في الدقيقة. تم إجراء اختبارات جهد ثابت عند إمكانيات معينة ضمن نافذة NITRR لمدة ساعتين. تم استخدام طريقة كاشف نيسلر لت quantifying الناتج.في المحلول الكهربائي لتحديد الكفاءة الفارادية بشكل أكبر ) و معدل العائد. على وجه الخصوص، تم سحب كمية معينة من الإلكتروليت من الخلية وتخفيفها إلى نطاق الكشف. ثم، تم إعداد محلول مائي من تارترات البوتاسيوم والصوديوم.ومحلول نيسلر تمت إضافتها إلى الإلكتروليت المخففللحصول على الخليط المتجانس. اتركه لمدة 10 دقائق. تم جمع شدة الامتصاص في الأشعة فوق البنفسجية والمرئية عند ذروة الامتصاص عند 420 نانومتر لتحديدتركيز سلسلة من محاليل كلوريد الأمونيوم القياسية والمحلول المختلط. تم رسم منحنى التركيز-الامتصاص لحساب الـتركيز المحلول المختلط (الشكل التوضيحي 14).تم حساب معدل العائد من خلال العلاقة التالية:
التم حسابه من خلال العلاقة التالية:
أينهو المقاس التركيز، V هو حجم الإلكتروليت، t هو زمن التفاعل، هو كتلة الحديد على القطب الأحادي، F هو ثابت فاراداي ( ) و Q هو الشحنة الكلية. الـ تم تحديد الكمية بشكل إضافي بواسطة 1 H NMR (600 ميغاهرتز، بروكير أفانس III) معدي إم إس أو-d6 كحقل قفل الدوران وحمض الماليك ) كمعيار داخلي، على التوالي. قبل كل قياس 1H NMR، تم ضبط الرقم الهيدروجيني للمحلول الناتج إلى 2.0 باستخدام HCl. تركيز تم رسمه مقابل نسبة مساحة الذروة لـوللحصول على المنحنى القياسي. تم توليديمكن تحديد التركيز من المنحنى القياسي. تم إجراء تجربة وسم النظائر في الإلكتروليت الذي يحتوي علىولمدة ساعتين عند -0.4 فولت مقابل RHE لتحديد مصدر النيتروجين لـ. تم الحصول علىتمت ملاحظته في طيف الرنين المغناطيسي النووي ^1H NMR. تم الكشف عن المنتجات الغازية المحتملة بواسطة كروماتوغرافيا الغاز (GC، Thermal Trace-1300) المزودة بكاشف الموصلية الحرارية (TCD). تم استخدام اختبار غريس لتحديدتركيز الإلكتروليتات بعد الاختبار البوتنشيومتري. بشكل عام،نفتيل إيثيلينديامين ثنائي الهيدروكلوريد (0.04 جرام)، p-أمينوبنزول سلفوناميد، و “تم إذابتها في ماء ديونيزيد (10 مل) باستخدام كاشف غريس. الإلكتروليت المخففتم خلطه مع عامل جريس (100 ميكرولتر) وترك لمدة 10 دقائق في درجة حرارة الغرفة لإجراء اختبارات الأشعة فوق البنفسجية والمرئية.تم تحديد تركيزات الإلكتروليت من خلال الامتصاص عند 540 نانومتر. في نفس العملية، تم استخدام أنواع مختلفةتم استخدام المحاليل المائية كعينات قياسية للحصول على منحنى المعايرة (الشكل التكميلي 17). كانت الكفاءة من المنتج الثانويتم حسابه من خلال العلاقة التالية:
انتقائيةتم حسابه من خلال العلاقة التالية:
أينهو عدد الإلكترونات المنقولة إلى المنتج الثانويهو الناتج الثانوي المقاس التركيز، V هو حجم الإلكتروليت أو المساحة العليا، F هو ثابت فاراداي و Q هو الشحنة الكلية.
المكثف الكهربائي ذو الطبقة المزدوجةتم تحديدها من منحنيات شحن الطبقة المزدوجة باستخدام الفولتموجرامات الدورية (CVs) ضمن منطقة الجهد غير الفارادي بين 0.4 و 0.5 فولت مقابل RHE عند معدلات مسح مختلفة منإلىنصف فرق كثافة التيار الحاليتم رسم الجهد المركزي مقابل معدل المسح (v) لحساب ميل الخط المستقيم الملائم والحصول على القيمة. تم حساب المساحة السطحية النشطة كهربائياً (ECSA) عن طريق قسمة سعة الطبقة المزدوجة ( بقدرة سعة محددةتم إجراء قياسات مطيافية الكتلة الكهروكيميائية التفاضلية (DEMS) على خلية كهروكيميائية مصنوعة خصيصًا مع تدفق مستمر للإلكتروليت من خلال مضخة بيرستالتية. خلال الاختبار، تم الحفاظ على غاز الأرجون يتصاعد. إلى الإلكتروليت. تم جمع إشارات الكتلة باستمرار خلال قياسات LSV من 0.1 إلى -0.6 فولت بمعدل مسحعندما عاد إشارة الكتلة إلى المستوى الأساسي، تم إجراء ثلاث دورات أخرى تحت نفس الظروف لتجنب الخطأ العرضي.
توفر البيانات
جميع البيانات التي تدعم نتائج هذه الدراسة موجودة في الورقة والمعلومات التكميلية. يمكن الحصول على مزيد من المعلومات من المؤلفين المراسلين عند الطلب المعقول.
References
Ashida, Y., Arashiba, K., Nakajima, K. & Nishibayashi, Y. Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water. Nature 568, 536-540 (2019).
Service, R. F. New recipe produces ammonia from air, water, and sunlight. Science 345, 610-610 (2014).
Suryanto, B. H. R. et al. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nat. Catal. 2, 290-296 (2019).
Tang, C. & Qiao, S. -Z. How to explore ambient electrocatalytic nitrogen reduction reliably and insightfully. Chem. Soc. Rev. 48, 3166-3180 (2019).
Hao, Y. -C. et al. Promoting nitrogen electroreduction to ammonia with bismuth nanocrystals and potassium cations in water. Nat. Catal. 2, 448-456 (2019).
Geng, Z. et al. Achieving a record-high yield rate of 120.9 for electrochemical reduction over Ru single-atom catalysts. Adv. Mater. 30, 1803498 (2018).
Lv, C. et al. An amorphous noble-metal-free electrocatalyst that enables nitrogen fixation under ambient conditions. Angew. Chem. Int. Ed. 57, 6073-6076 (2018).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036-7046 (2020).
Wu, Z. -Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Chen, G. -F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper-molecular solid catalyst. Nat. Energy 5, 605-613 (2020).
Chen, F. -Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotech. 17, 759-767 (2022).
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290-294 (2021).
Duca, M. & Koper, M. T. M. Powering denitrification: the perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726-9742 (2012).
Wang, Y. et al. Nitrate electroreduction: mechanism insight, in situ characterization, performance evaluation, and challenges. Chem. Soc. Rev. 50, 6720-6733 (2021).
Xu, H. et al. Electrocatalytic reduction of nitrate-a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710-2758 (2022).
Shafirovich, V. & Lymar, S. V. Nitroxyl and its anion in aqueous solutions: spin states, protic equilibria, and reactivities toward oxygen and nitric oxide. Proc. Nat. Acad. Sci. 99, 7340-7345 (2002).
Ford, P. C. & Miranda, K. M. The solution chemistry of nitric oxide and other reactive nitrogen species. Nitric Oxide 103, 31-46 (2020).
Janaway, G. A. & Brauman, J. I. Direct observation of spin forbidden proton-transfer reactions: . J. Phys. Chem. A 104, 1795-1798 (2000).
Gao, Q. et al. Breaking adsorption-energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine-learned insights. Nat. Commun. 13, 2338 (2022).
Zhang, S. et al. High-ammonia selective metal-organic framework-derived Co -doped catalysts for
electrochemical nitrate reduction. Proc. Nat. Acad. Sci. 119, e2115504119 (2022).
Wang, Y. et al. Enhanced nitrate-to-ammonia sctivity on copper-nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702-5708 (2020).
Cheng, X. -F. et al. Coordination symmetry breaking of single-atom catalysts for robust and efficient nitrate electroreduction to ammonia. Adv. Mater. 34, 2205767 (2022).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper-supported rhodium cluster and single-atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Clayborne, A., Chun, H. -J., Rankin, R. B. & Greeley, J. Elucidation of pathways for NO electroreduction on Pt(111) from first principles. Angew. Chem. Int. Ed. 54, 8255-8258 (2015).
Mtangi, W. et al. Control of electrons’ spin eliminates hydrogen peroxide formation during water splitting. J. Am. Chem. Soc. 139, 2794-2798 (2017).
Do, V. -H. & Lee, J. -M. Orbital occupancy and spin polarization: from mechanistic study to rational design of transition metal-based electrocatalysts toward energy applications. ACS Nano 16, 17847-17890 (2022).
Chen, R. R. et al. Antiferromagnetic inverse spinel oxide with spin-polarized channels for water oxidation. Adv. Mater. 32, 1907976 (2020).
Liang, Y. et al. Enhancement of electrocatalytic oxygen evolution by chiral molecular functionalization of hybrid 2D electrodes. Nat. Commun. 13, 3356 (2022).
Ren, X. et al. Spin-polarized oxygen evolution reaction under magnetic field. Nat. Commun. 12, 2608 (2021).
Wu, T. et al. Spin pinning effect to reconstructed oxyhydroxide layer on ferromagnetic oxides for enhanced water oxidation. Nat. Commun. 12, 3634 (2021).
Li, X. et al. Identification of the electronic and structural dynamics of catalytic centers in single-Fe-atom material. Chem 6, 3440-3454 (2020).
Li, Z. et al. Spin engineering of single-site metal catalysts. Innovation 3, 100268 (2022).
Dai, J. et al. Single-phase perovskite oxide with super-exchange induced atomic-scale synergistic active centers enables ultrafast hydrogen evolution. Nat. Commun. 11, 5657 (2020).
Yao, Y. et al. Single atom Ru monolithic electrode for efficient chlorine evolution and nitrate reduction. Angew. Chem. Int. Ed. 61, e202208215 (2022).
Zhang, S. et al. Electrocatalytically active single-atom sites for efficient reduction of nitrogen to ammonia. Angew. Chem. Int. Ed. 59, 13423-13429 (2020).
Muller, D. A. et al. Atomic-scale imaging of nanoengineered oxygen vacancy profiles in . Nature 430, 657-661 (2004).
Chakraborty, T., Meneghini, C., Aquilanti, G. & Ray, S. Microscopic distribution of metal dopants and anion vacancies in Fe-doped single crystals. J. Phys. Condens. Matter 25, 236002 (2013).
Sala, G. et al. Vacancy defects and monopole dynamics in oxygendeficient pyrochlores. Nat. Mater. 13, 488-493 (2014).
Yang, T.-Y. et al. A new hematite photoanode doping strategy for solar water splitting: oxygen vacancy generation. Phys. Chem. Chem. Phys. 15, 2117-2124 (2013).
Wei, K. et al. Strained zero-valent iron for highly efficient heavy metal removal. Adv. Funct. Mater. 32, 2200498 (2022).
Mao, C. et al. Energy-confined solar thermal ammonia synthesis with . Appl. Catal. B Environ. 224, 612-620 (2018).
Xie, J. et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 1, 889-896 (2018).
Mondal, P. et al. Behavior of with system and the reduction to As(O). J. Phys. Chem. C. 118, 21614-21621 (2014).
Liu, W. et al. Discriminating catalytically active species of atomically dispersed catalyst for selective oxidation of the C-H bond. J. Am. Chem. Soc. 139, 10790-10798 (2017).
Zitolo, A. et al. Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials. Nat. Mater. 14, 937-942 (2015).
Ohtomo, A., Muller, D. A., Grazul, J. L. & Hwang, H. Y. Artificial charge-modulationin atomic-scale perovskite titanate superlattices. Nature 419, 378-380 (2002).
Pan, L. et al. Manipulating spin polarization of titanium dioxide for efficient photocatalysis. Nat. Commun. 11, 418 (2020).
Chang, C. F. et al. c-axis dimer and its electronic breakup: the insulator-to-metal transition in . Phys. Rev. X 8, 021004 (2018).
Schlappa, J. et al. Resonant soft x-ray scattering from stepped surfaces of . J. Phys. Condens. Matter 24, 035501 (2012).
Guo, Y. et al. Engineering the electronic state of a perovskite electrocatalyst for synergistically enhanced oxygen evolution reaction. Adv. Mater. 27, 5989-5994 (2015).
Miao, J. et al. Spin-state-dependent peroxymonosulfate activation of single-atom M-N moieties via a radical-free pathway. ACS Catal. 11, 9569-9577 (2021).
Dai, J. et al. Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis. Nat. Commun. 13, 1189 (2022).
Wang, Y. et al. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350-5354 (2020).
Gao Q., et al. Synthesis of core/shell nanocrystals with ordered intermetallic single-atom alloy layers for nitrate electroreduction to ammonia. Nat. Synth. https://doi.org/10.1038/s44160-023-00258x (2023).
Liu, H. et al. Low-coordination Rhodium catalysts for an efficient electrochemical nitrate reduction to ammonia. ACS Catal. 13, 1513-1521 (2023).
Long, J. et al. Direct electrochemical ammonia synthesis from nitric oxide. Angew. Chem. Int. Ed. 59, 9711-9718 (2020).
Sun, Y. et al. Spin-related electron transfer and orbital interactions in oxygen electrocatalysis. Adv. Mater. 32, 2003297 (2020).
Wei, X. et al. Tuning the spin state of Fe single atoms by Pd nanoclusters enables robust oxygen reduction with dissociative pathway. Chem 9, 181-197 (2023).
Li, J. et al. solar-to-ammonia efficiency from nitrate using Fe single atomic catalyst supported on nanosheets. Adv. Funct. Mater. 32, 2108316 (2022).
Niu, H. et al. A feasible strategy for identifying single-atom catalysts toward electrochemical NO-to- conversion. Small 17, 2102396 (2021).
Gao, R. et al. Pt/ with Pt-Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy 6, 614-623 (2021).
Liu, K. et al. Insights into the activity of single-atom Fe-N-C catalysts for oxygen reduction reaction. Nat. Commun. 13, 2075 (2022).
Gao, J. et al. Electrocatalytic upcycling of nitrate wastewater into an ammonia fertilizer via an electrified membrane. Environ. Sci. Technol. 56, 11602-11613 (2022).
Gao, J. et al. Electrochemically selective ammonia extraction from nitrate by coupling electron- and phase-transfer reactions at a three-phase interface. Environ. Sci. Technol. 55, 10684-10694 (2021).
الشكر والتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2021YFA1201701، 2022YFA150470، L.Z.)، الوطني
مؤسسة العلوم الطبيعية في الصين (U22A204O2، U21A20286، L.Z.)، (22102100، Y.Y.)، (22206121، J.D.)، (22273068، U22A20394، X.G.)، برنامج العلوم والتكنولوجيا في شنتشن (JCYJ2O220818095601002، L.Z.)، مؤسسة العلوم الطبيعية في شنغهاي (22ZR1431700، Y.Y.)، مؤسسة ما بعد الدكتوراه في الصين (2021M702117، 2022M722080، J.D.)، البرنامج الوطني لما بعد الدكتوراه للمواهب المبتكرة (BX2O220197، J.D.) وبرنامج التميز لما بعد الدكتوراه في شنغهاي (2021182، J.D.)، (2022381، G.Z.). يقر المؤلفون بالدعم من مركز ماكس بلانك-بوستيك-هسينتشو للمواد المعقدة، ومركز التحليل الآلي في جامعة شنغهاي جياو تونغ، ومركز التحليل الآلي في كلية علوم البيئة والهندسة، ومختبر شيا يانجيا وشركة بروكير للمساعدة في التوصيفات والقياسات التجريبية، ومركز الحوسبة الفائقة في جامعة ووهان.
مساهمات المؤلفين
قام Y. Y. و X. G. و L.Z. Z. بتصور الفكرة. نفذ J. D. و L. Z. التجارب. قام Y. T. بإجراء حسابات DFT. كتب J. D. و Y. T. و Y. Y. و X. G. و L.Z. Z. الورقة. ساعد Z. H. و C. C. و C. K. و G. Z. و J. W. و X. Z. و Q. Z. و W. H. و R. W. و K. W. و R. Z. في تحليل البيانات وتحرير المخطوطة. ناقش جميع المؤلفين النتائج وقدموا تعليقات خلال إعداد المخطوطة.
كلية علوم البيئة والهندسة، جامعة جياو تونغ بشنغهاي، شنغهاي 200240، الصين.كلية الطاقة والهندسة الميكانيكية، جامعة ووهان، ووهان 430072، الصين.معهد ماكس بلانك لفيزياء الكيمياء للمواد الصلبة، شارع نوتنيتزر 40، 01187 دريسدن، ألمانيا.مركز أبحاث الإشعاع السنكروتروني الوطني، 101 طريق هسين-آن، هسينتشو 300092 تايوان، الصين.قسم الفيزياء الكهربائية، جامعة يانغ مينغ تشياو تونغ الوطنية، هسينتشو، تايوان، الصين.ساهم هؤلاء المؤلفون بالتساوي: جي داي، يوان تونغ، لونغ تشاو.البريد الإلكتروني:xiangkuigu@whu.edu.cn؛yyancai@sjtu.edu.cn؛zhanglz@ccnu.edu.cn
Electrochemical nitrate reduction to ammonia offers an attractive solution to environmental sustainability and clean energy production but suffers from the sluggish *NO hydrogenation with the spin-state transitions. Herein, we report that the manipulation of oxygen vacancies can contrive spin-polarized pairs on monolithic titanium electrode that exhibits an attractive yield rate of and a high Faradic efficiency of at -0.4 V vs. RHE, far superior to the counterpart with spin-depressed pairs ( ) and the mostly reported electrocatalysts. The unpaired spin electrons of Fe and Ti atoms can effectively interact with the key intermediates, facilitating the *NO hydrogenation. Coupling a flow-through electrolyzer with a membrane-based recovery unit, the simultaneous nitrate reduction and recovery was realized. This work offers a pioneering strategy for manipulating spin polarization of electrocatalysts within pair sites for nitrate wastewater treatment.
Ammonia ( ) as the critical feedstocks for artificial fertilizers production and a carbon-free energy carrier is of great significance to the modern society . Nowadays, global demand exceeds 150 million tons per year, which heavily relies on the energy-intensive Haber -Bosch process associated with the consumption of global energy, concurrently contributing to of global carbon emissions . Recently, electrochemical reduction of nitrogen ( ) to , inspired by the natural microbial fixation, has attracted tremendous interest . However, the robust and poor solubility in water medium of lead to a low yield rate (less than ) and partial current densities (less than , hindering its widespread commercialization. Alternatively, nitrate ( ) can be dissociated into deoxygenated species with a much
lower energy of , making it as a more suitable nitrogen source for electrosynthesis , especially regarding that is widely distributed in industrial wastewaters and polluted groundwater, resulting in the eutrophication and the disturbance of ecosystems . Therefore, selective reduction of to (NITRR) offers a promising route to synchronously relieve energy and environmental crisis.
Generally, NITRR initiates from the adsorption of nitrate ( ) on electrodes followed by the sequential deoxygenation of to adsorbed nitrite (* ) and nitric oxide (*NO) . Subsequently, *NO hydrogenates to the hydrogenated *NHO/NOH species with a spin state transition . Finally, the hydrogenated species can stepwise hydrogenate to the hydroxylamine ( ) and the targeted product
of * . It has been reported that both the adsorption of and the subsequent hydrogenation of *NO are crucial for the rapid NITRR , and thus the enhancement of adsorption and/or the acceleration of *NO hydrogenation would be effective to improve the activity of NITRR. Currently, the enhanced adsorption has been widely reported by the surface modification , heteroatom doping , and alloying of electrodes. While the acceleration of spin-transition related *NO hydrogenation via properly manipulation the spin states of electrocatalysts is seldom investigated, although this process is similar with reported oxygen-related electrocatalytic reactions such as oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) processes , and generally demands tremendous energy and is kinetically sluggish, thus strongly retarding the transformation of -to- . Therefore, it is urgent to design advanced spin-polarized electrocatalysts to boost the spin-dependent electron transfer for superior NITRR.
Spin-polarization of metal active sites has been reported to be an effective way to accelerate the spin-state transition between reactant intermediates, because spin-polarized metal active sites are able to drive quantum spin exchange interaction and offer a spin electron transfer channel towards spin-dependent electrocatalytic reaction . In light of this principle, paramagnetic iron ( Fe ) has been extensively investigated, especially for oxygen-related electrocatalytic reactions (e.g., OER and ORR), owing to its well-tunable spin degree of electronic freedom . For instance, low spin ( ) and intermediate spin states ( ) of Fe are more favorable to penetrate the antibonding orbital of as compared with the high spin state of Fe ( ), thus facilitating the spin transition from the single-state to triplet-state . More impressively, Fe single atoms on carbon matrix were found to be effective for NITRR , exhibiting decent nitrate electroreduction capacity with an Faradaic efficiency of and a yield rate of . Obviously, finely manipulating the spin polarization effect of Fe -based materials can provide a solution to superior NITRR.
In this study, Fe single atoms were anchored on the inherent surface oxide layer of titanium foam, where the oxygen vacancies (OVs) of titanium oxide could trigger the synchronous spin polarization of Fe and the adjacent Ti atoms. The designed spin-polarized pairs delivered a high Faradic efficiency of and an impressive yield rate of at -0.4 V vs. RHE for NITRR, far superior to the counterpart monolithic electrode with spin-depressed -Ti pairs ( ) and the mostly reported NITRR electrocatalysts. The spin electrons located in the 3d orbitals of Fe and Ti atoms in the spin-polarized pair sites were found to dominantly contribute this excellent performance via interacting with the key intermediates, boosting the deoxygenation of and the hydrogenation of *NO. The developed monolithic electrode with spin-polarized pairs was also employed to construct a NITRR flow-through electrolyzer coupled with a membrane separation unit for a high-efficient nitrate conversion at an industrial-level current intensity and in-situ high-purity ammonia recovery from nitrate-containing wastewater.
Results
Manipulating spin polarization of – Ti pairs
Fe single atoms anchored on a monolithic Ti electrode with different spin polarization degrees were prepared by regulating the OVs of the inherent surface oxide layer of titanium foam, as illustrated in Fig.1a, b. The core of the synthesis process lies in the control of the reductive/ oxidative gaseous environment during the thermal treatment (Supplementary Fig. 1, See details in Methods). Specifically, a reductive atmosphere ( ) was performed to create oxygen vacancies (OVs) in the inherent oxide layer of Ti electrode to evoke the spin polarization of Fe single atoms and adjacent Ti atoms (Supplementary Fig. 2), thus producing spin-polarized pairs ( ), while an oxidative atmosphere ( ) was employed to avoid the generation of
OVs to obtain the spin-depressed pairs ( ) for comparison. During hydrogen treatment of inherent oxide layer of Ti foam, electrons were first transferred from hydrogen (H) atoms to the oxygen ( O ) atoms in the lattice of inherent oxide layer. Then, the lattice O leaves with the H atom to form , as evidenced by an obvious evolution peak at about during the temperature programmed reaction (TPR) measurement of pristine Ti foam in 5% (Supplementary Fig. 3), and the OVs form on the surface of foam. Electron spin resonance (ESR) spectra was utilized to identify the electronic states trapped in OVs (Fig. 1c), which clearly showed the successful creation of OVs in the SP- Ti electrode, and the absence of OVs in SD- was also confirmed by its silent ESR signals . As evidenced by high-resolution transmission electron microscopy (HRTEM) images of SP- and SD- (Supplementary Figs. 4, 5), the distinct boundaries were observed between the interior crystalline Ti and surface amorphous titanium oxide layer ( ), consistent with our previous work . The aberration-corrected high-angle annular darkfield scanning transmission electron microscopy (HAADF -STEM) image demonstrated that Fe single atoms (marked by white circles) were well dispersed in the oxide layer of Ti foam for both SP and SD- (Fig. 1d and Supplementary Fig. 6), and the Fe clusters or nanoparticles were not observed, consistent with the absence of metallic Fe diffraction peaks in the X-ray diffraction (XRD) pattern (Supplementary Fig. 7). The 3D surface intensity profile along the yellow arrow further confirmed the atomically dispersed Fe on the foam (Supplementary Fig. 8). The elemental EDS mapping images revealed the uniform dispersion of Fe atoms in and (Fig. 1e and Supplementary Fig. 9).
We further utilized X-ray absorption fine structure spectrometry (XAFS) to confirm the atomic dispersion of Fe atoms on . As evidenced in Fig. 1f, the Fourier-transformed Fe K-edge extended X -ray adsorption fine structure (FT-EXAFS) spectroscopy of SP- manifested a dominant peak at , corresponding to coordination in the first shell . The absence of scattering path at ruled out the metallic Fe aggregation in SP- . We also employed the wavelet-transform (WT)-EXAFS analysis to offer resolution in both radial distance and k -space and supplement the FT-EXAFS analysis. As shown in Fig. 1g, the WT-EXAFS spectra of SP- exhibited the peak with a maximum intensity at in contrast to Fe foil (Supplementary Fig. 10), which was assigned to the scattering path. Similar locally fine structure of Fe atoms on the SD- electrode was also observed (Fig. 1f-h). Moreover, the electrical conductivity of was very close to that of SD- (Supplementary Table 1), benefiting from the excellent electrical conductivity of Ti substrate.
Experimental evidences for the spin polarization of -Ti pairs induced by OVs
The unpaired electrons located in OVs are expected to transfer to the empty levels belonging to Ti and Fe atoms adjacent to , resulting in two possible consequences including a negative shift in the core level binding energies of the reduced Ti or Fe atoms, and the presence of an unpaired electron (spin) in the shell of the Ti or Fe atoms . While the electronic properties of Fe and Ti in SD- electrode without OVs should be different from those of SP- electrode, maintaining spin-depressed and states. To confirm the electronic structures and spin states of SP- and SD- , ESR, X-ray photoelectron spectroscopy (XPS), soft X-ray absorption spectroscopy (XAS), Mössbauer spectroscopy, and temperaturedependent magnetizations (M-T) measurements were carried out. We first investigated the energy alignment of the Fermi levels ( , see the calculation details in Supplementary Fig. 11) for , and Fe (Fig. 2a). It was found that the calculated of was above that of , so the trapped electrons at the OVs on the amorphous surface can transfer to the supported Fe atoms, resulting in the upshift of . As seen from the high
-resolution Fe core level spectra in Fig. 2b, two peaks at 710.7 and 723.8 eV were observed for , which could be assigned to the and orbitals of trivalent species . Compared with the spectra of SD- , the and peaks of showed an obviously lower-energy shift in binding energy, suggesting the reduction of Fe valence state induced by OVs and the appearance of electron-rich species. As such, OVs on the amorphous surface of can transfer electron to the Fe single atoms and lead to an increased electron density of Fe single atoms. Such an electron transfer from OVs to Fe atoms even occurs within without thermal treatment, as evidenced by the appearance of species in its Fe XPS spectra (Supplementary Fig. 12). We further applied the Mössbauer spectroscopy to reveal the spin state of Fe single atoms on the Ti monolithic electrode (Fig. 2c), and found no sextets or singlets but doublets were derived from the deconvolution of the Mössbauer spectra for SP- and SD- , indicating the absence of bonds . As expected, possessed a much higher content of species (40.2%) with high spin polarization configuration ( ) than SD- according to the fitted results (Supplementary Table 2).
Fig. 1 | Manipulating spin polarization of -Ti pairs. Schematic illustration of the manipulation strategy of spin polarization for pairs on inherent oxide surface of Ti foam for spin-polarized and spin-depressed . Orange, yellow, cyan, blue, grey and transparent balls represent spin-polarized Fe, spin-depressed Fe, spin-polarized Ti, spin-depressed Ti, oxygen
and oxygen vacancy (OV), respectively. c ESR spectra of and SP – -Ti. d HAADF-STEM image of SP- , where Fe single atoms were indicated by a white circle. e HADDF image and STEM elemental mapping of SP- -Ti. f FT-EXAFS spectra of foil and at the Fe K-edge. k -weighted WT -EXAFS spectra of and at the Fe K-edge.
Fig. 2 | Experimental evidences for the spin polarization of -Ti pairs induced by OVs. a Energy alignment of the for , and Fe foil. XPS spectra of SD- and SP- . c Mössbauer spectra for SP- and SD Ti. d Ti L-edge soft-XAS spectra of SD- Ti and SP- Ti. e Ti XPS spectra of SD- and SP- . versus temperature plots and the calculated of and SP- .
Furthermore, we found that OVs on both and were accompanied by the appearance of spin-polarized species with an EPR signal at , which was invisible in the EPR spectra of without OVs (Fig. 1c). This difference indicated that the introduction of OVs could trigger the spin-polarization of Ti sites. We thus employed the Ti L-edge soft-XAS measurement that is highly sensitive to charge state, orbitals occupation and the spin orientation of electrons to reveal the formation of spin-polarized species on SP-Fe1-Ti (Fig. 2d). The multiple spectral features of SD- were very similar to that of indicating valence state . In comparison with , the lower-energy shift and higher peak intensity of SP- verified the formation of spin-polarized species induced by , which was further confirmed by the shift to lower binding energy in XPS spectra for both and SP (Fig. 2e).
We therefore carried out the temperature-dependent magnetic susceptibility ( ) measurement to unravel the total effective magnetic moment ( ), which can be determined via the Curie-Weiss law . According to the versus temperature plots of and SD- (Fig. 2f), the order of calculated was in the sequence of SP-Fe ( ), suggesting that SP- possessed much more spin electrons than and . Taken together, these above results evidently showed that the spin polarization of atomically dispersed pairs can be easily manipulated by OVs, providing an opportunity for developing unique spin-polarized Fe -based single atom catalysts towards NITRR and deepening insights into spin effect on NITRR.
Evaluation of NITRR performance towards different electrodes
NITRR performance was investigated on different electrodes in a H -type electrolytic cell under ambient conditions. For the linear sweep voltammetry (LSV) in 1 M KOH (Fig. 3a), the addition of nitrate much more increased the current density of electrode than those
of Ti foam, and electrodes. We further quantified the Faradaic efficiency ( ) and yield rate at a certain potential after 2 h of electrolysis via ultraviolet-visible (UV-Vis) spectrophotometry (Supplementary Figs. 13 and 14). As expected, the SP electrode exhibited higher and yield rate than the electrode (Fig. 3b). Impressively, the SP-Fe1-Ti electrode displayed an attractive yield rate of ) and a high Faradaic efficiency of at -0.4 V vs. RHE, which were also verified by the results obtained from nuclear magnetic resonance (NMR, Supplementary Fig. 15) measurement. Such an excellent NITRR activity of SP- electrode was much superior to those of state-of-the-art NITRR electrocatalysts, Haber-Bosch catalysts and reduction electrocatalysts (Fig. 3c and Supplementary Table 3). Additionally, electrode possesses promising application potential for maximizing , onset potential and current density at certain potential (Supplementary Table 4) over almost all of top-level NITRR electrodes consisting of the low-price components (e.g., , and Fe in Supplementary Table 5). We also drop -casted SP-Fe1-Ti catalyst powders on the carbon fiber paper with the catalyst mass loading of for performance assessment. As shown in Supplementary Fig. 16, catalyst displays a high cathodic current density of at -0.4 V vs. RHE, superior to most of the top-level NITRR catalysts. The yield rate defined by the electrode area of SP- catalyst was determined to be , which outperforms most of low-price metal -based catalysts and even several high-price metal-based catalysts (Supplementary Table 6). More importantly, the monolithic nature of electrode is much more feasible than the powder form of previous top-level NITRR electrocatalysts for practical application.
The contributions of other possible products to the faradaic current were also evaluated using gas chromatography equipped with thermal conductivity detector (TCD) and UV-Vis spectrometry. Only and were detected (Supplementary Fig. 17 and Supplementary Fig. 18), and their FEs were calculated to be and , respectively,
Fig. 3 | NITRR performances of different electrodes. a LSV curves of Ti foam, and SP- electrodes. yield rate and of SD and SP- electrodes at various potentials. Comparison of the electrocatalytic NITRR performance of SP- electrode with other extensively reported electrocatalysts. NMR spectrum of the products generated during the electrocatalytic NITRR on the SP- monolithic electrode at -0.4 V vs. RHE.
e Partial current densities of SD- and SP- Ti electrodes normalized to the geometric area at various potentials. Specific activity normalized to ECSA of and SP- Ti electrodes at -0.4 V vs. RHE. g yield rate and Faradaic efficiency of SP- electrode under the applied potential of -0.4 V vs. RHE during 20 consecutive electrolysis cycles.
at -0.4 V vs. RHE. After taking the two side-reactions into consideration, the total FE (Supplementary Fig. 19) was as expected. Moreover, the selectivity of SP- -Ti electrode was determined to be at -0.4 V vs. RHE, further confirming its high selectivity towards production from NITRR.
We thus employed NMR to confirm the N source of generated via the isotope labeling experiments with -labeled as the reagent. Different from with three peaks in the NMR spectra, only two peaks of appeared in NMR spectra when was used as the reagent (Fig. 3d), demonstrating that the produced was from nitrate feedstock instead of contaminations. In addition, peaks of are absent in NMR spectra of the original electrolyte containing , further confirming that the produced was originated from the reduction of nitrate feedstock instead of contaminations. Remarkably, the SP- electrode also showed a larger partial current density of at -0.7 V vs. RHE than the electrode (Fig. 3e), suggesting a great potential in practical applications. We also assessed the intrinsic activity of SP electrode through calculating the specific activity (SA) by normalizing the electrode activity to the electrochemical surface area (ECSA, Supplementary Fig. 20), and found that the SA of SP-
electrode was apparently higher than and electrodes at -0.4 V vs. RHE (Fig. 3f), revealing the intrinsically high catalytic NITRR activity of spin-polarized pairs and the great contribution of spin-polarization of active metal sites to NITRR.
Since the nitrate concentration varies in different sources, we also checked the NITRR activity of SP- electrode in the electrolyte with different concentrations ( , and 1 M ) at -0.4 V vs. RHE. As shown in Supplementary Fig. 21, the FEs of -to- conversion were and in the electrolyte with , and 1 M , respectively. These results suggest that the concentration has no obvious impacts on of SP- -Ti electrode, indicating the wide concentration compatibility. In addition, we observed that the yield rate was enhanced by increasing the concentrations from 0.1 to 1 M because of the accelerated mass transfer . We further evaluated the NITRR activity of the SP- electrode at the neutral condition. Impressively, the SP- exhibited comparable yield rate ( ) and FE (96.1%) to that of at the alkaline condition (Supplementary Fig. 22), further demonstrating its high potential of environmental application.
More importantly, the and yield rate of electrode kept very stable during 20 cycles of consecutive electrolysis
Fig. 4 | Insights into the NITRR mechanism. a DOSs of NO and NHO species.
b Optimized structures and calculated 2D spin density diagrams of SD- and . c DOSs of Fe and Ti atoms on and . d DEMS measurements of NITRR over SP- electrode. e Calculated free energy diagrams for NITRR on SD- and SP- . f Major orbital interactions between Fe atom and NO molecule as well as the corresponding molecular orbital diagrams (BD
and represent bonding and antibonding orbital, respectively). Charge density differences of NO and NHO adsorbed on and (red and cyan represent electron accumulation and depletion, respectively). COHPs of NHO adsorbed on SD- and SP- . i DOSs of Ti atom on SP- before and after NHO adsorption. Ti, O, Fe, N, and H atoms are denoted by grey, red, cyan, blue, and white, respectively.
in a H -type electrolytic cell at -0.4 V vs. RHE (Fig. 3 g and Supplementary Fig. 23), indicating its long-term durability and high potential for practical application. XRD, XPS, and TEM analyses further revealed the high stability of SP- -Ti electrode during NITRR (Supplementary Figs. 24-26), as confirmed by the negligible Fe dissolution of SP- electrode within 20 cycles consecutive electrolysis (Supplementary Fig. 27).
Insights into NITRR mechanism towards SP-Fe -Ti pairs
Density functional theory (DFT) calculations were employed to get insights into NITRR mechanism over SP- pairs. We first explored the density of states (DOSs) of the key NO and NHO species involved in NITRR (Fig. 4a), and confirmed the asymmetric spin state of NO in DOS of spin-up and spin-down states, which was different from that of NHO, indicating that the transformation of NO to NHO was accompanied by a spin-state transition. As revealed by the constructed models of SP and SD- (Fig. 4b), their Fe atoms were respectively coordinated with three and four O atoms for and , consistent well with the fitting EXAFS results (Supplementary Fig. 28 and Table 7). The spin density diagrams displayed that only Fe atom was spin-polarized for , while both Fe atom and Ti atom adjacent
to Fe atom were spin-polarized in the case of (Fig. 4b). This magnetic difference further verified the increase of the measured magnetic moment with the existence of OV (Fig. 2f). Magnetic states were also evident from the DOSs of Fe and Ti atoms. The spin up and spin down components of Fe atom around Fermi level ( ) were quite different (Fig. 4c), indicative of unpaired electrons in the orbitals. Besides, the spin-up and spin-down components of Ti atom in SD were roughly the same, but unpaired electronic states appeared near after the introduction of OV (Fig. 4c). These magnetic Ti and Fe atoms contributed to the highly efficient NITRR.
Given that the hydrogen source for NITRR under alkaline condition comes from , water dissociation including the bond cleavage and the proton transfer ability would strongly influence the catalytic activity of NITRR. It was found that dissociation was slightly endothermic by 0.08 and 0.03 eV , with barriers of 0.28 and 0.29 eV on SD- and SP- , respectively, suggesting that dissociation on the both electrodes were much favorable and comparable (Supplementary Fig. 29). We further employed the electron spin resonance (ESR) technique using 5,5-dimethyl-1-pyrroline-N -oxide (DMPO) as the radical trapping reagent to investigate the generation and the role of hydrogen radicals from water dissociation
to understand the mechanism of nitrate reduction. Nine ESR peaks with an intensity ratio of 1:1:2:1:2:1:2:1:1 were observed in Supplementary Fig. 30 for the electrocatalysis on the electrode in pure , which could be assigned to the spin adduct of DMPO-H, confirming the generation of . When adding in KOH , the signal intensity of disappeared. Such a phenomenon revealed that the generated through water dissociation was consumed, suggesting the deep participation of in the hydrogeneration process of NITRR. We further conducted kinetic isotope effect (KIE) experiments to estimate the proton transfer ability of SP and during NITRR by comparing the current density ratios. The NITRR KIE value (1.40) of SP- was close to that (1.53) of SD- at -0.4 V vs. RHE (Supplementary Fig. 31), suggesting a similar hydrogen transfer ability on the two electrodes. Therefore, the water dissociation and hydrogen transfer abilities have tiny impact on the distinct NITRR activities of SP- and SD- .
To understand the unique role of spin-polarized pairs on NITRR, the NITRR reaction pathways on SD- and were investigated. To construct a comprehensive description of the reaction mechanism, online differential electrochemical mass spectrometry (DEMS) was utilized to on-line detect the key intermediates and products (Fig. 4d). We observed the signals of , and 17 from , and , respectively. On the basis of DEMS results, we calculated the free energy of individual intermediate on and (Fig. 4e, Supplementary Fig. 32) under a potential of 0 V vs. . Actually, to *NO underwent an adsorption step and two deoxygenation steps, namely, * * * * NO . In the case of the system, the adsorption underwent an uphill Gibbs free energy change of 0.34 eV . Afterward, first deoxygenated to with a downhill free energy change of -1.52 eV . Subsequently, continued to remove one oxygen to form *NO with an energy release of 1.81 eV . As for the SP- system, the above three steps release the energy of , and 0.93 eV , respectively. Finally, deoxygenated to *NO with a total downhill Gibbs free energy change of -3.00 eV on SD , while the total downhill free energy change for the above steps was -3.22 eV on . Therefore, deoxygenation to *NO on SP- was more favorable thermodynamically. For NITRR on , the potential-determining step (PDS) was *NO to *NHO with an thermodynamic barrier of 0.75 eV , while the PDS was determined to be * to with a thermodynamic barrier of 0.46 eV on , consequently resulting in a higher NITRR activity than SD . This PDS change stemmed from the effect of both spin-polarized Fe and Ti sites on , resulting in the remarkably decreased free energy of *NO to *NHO from 0.98 eV to -0.23 eV . Meanwhile, a much smaller peak intensity ratio (~3.92) of *NO to *NHO for than that of ( , Supplementary Fig. 33) was observed, further confirming an accelerated *NO hydrogenation process on the electrode. To further highlight the synergistic effect of spin pairs, we calculated the free energy of *NHO intermediate adsorbed on the pairs involving a Ti atom without spin-polarization for comparison (Supplementary Fig. 34), and the calculated value of 0.37 eV was much higher than that on , suggesting that the spin-polarization of Ti atom was also significantly contributed the higher activity of NITRR. Subsequently, we investigated the competitive hydrogen evolution reaction on spin-polarized pairs (Supplementary Fig. 35), and the calculated free energy for adsorption on Fe and Ti sites of were 1.13 and 0.78 eV , respectively, much weaker than that of adsorbed on spin -polarized pairs ( -0.64 eV ). Therefore, the generation on the spin-polarized pairs were inhibited, facilitating to enhance the Faradaic efficiency for NITRR.
To further understand the spin-polarization effect of pairs on promoting the activity of NITRR, the orbital interactions between *NO/*NHO and the Fe-Ti pair were analyzed in detail, since *NO
hydrogenation was generally suggested to govern the activity . For NO adsorption on , it was found that NO preferably adsorbed on , stronger than that on . The main contribution to the interaction between Fe and *NO was from the orbitals of Fe atom and the orbitals of NO (Fig. 4f), according to the high spin polarization configuration ( ) of Fe , as mentioned above. We noted that the and orbitals of Fe could not hybridize with the NO orbital owing to symmetry conservation, their orbital interaction was thus not considered . Thus, in such a bonding model, the semioccupied of Fe can accept electrons from the occupied orbital of NO, and in turn the electrons of Fe orbitals can inject into the partially occupied orbitals to weaken the bond order of . This “donation-backdonation” of electrons generally led to the NO’s acceptance of 0.25 |e| from the substrate (Fig. 4 g and Supplementary Fig. 36), resulting in a binding energy of -1.80 eV for NO adsorption on the Fe site of SP- , stronger than that of -1.58 eV on the Fe site of that transferred less electron of 0.17 le| to *NO. Moreover, the spin polarized pairs can also be advantageous of the hydrogenation of *NO through significantly stabilizing hydrogenated *NHO intermediates. The calculated adsorption energy of -2.41 eV for *NHO that preferably adsorbed on SP- via the formation of and was much stronger than that of -1.22 eV on via forming and bonds, because more electrons transferred to *NHO from SP-Fe than that from SD , as evidenced by the Bader charge analysis (Fig. 4 g ), which was supported by the Crystal Orbital Hamiltonian Populations (COHP) analysis (Fig. 4h). The negatively integrated COHP (-COHP) values for and interactions on were more positive than those for and interactions on SD- , suggesting that the spin-polarized pairs exhibited a stronger interaction with *NHO. As stated before, Ti atom adjacent to Fe atom of underwent a spin state transition from unspin polarization to spin polarization after the introduction of OVs, as supported by the appearance of unpaired , and states near the (Fig. 5i). Furthermore, the spin state near the of Ti atom on disappeared after the adsorption of *NHO, implying that the unpaired spin electron of Ti atom injected into * . Similarly, the unpaired spin electron in state of Fe can also partially inject into *NHO (Supplementary Fig. 37), enhancing the electron transfer from spin pair to further stabilize *NHO. Conclusively, both spin-polarized pair sites can effectively facilitate the deoxygenation of to *NO and stabilize *NO and *NHO intermediates to boost the subsequent *NO hydrogenation, as well as inhibit the hydrogen evolution reaction, leading to high activity and selectivity of electrode towards NITRR.
An integrated equipment for flow-through NITRR electrolyzer and in-situ ammonia recovery
Besides the electrode performance, product separation and recovery are also crucial for the practical application of NITRR . From this aspect, we designed an integrated device composed of a flow -through NITRR electrolyzer and a membrane-based ammonia recovery unit to realize the efficient NITRR and in-situ recovery, as schematically illustrated in Fig. 5a and Supplementary Fig. 38. We designed a compact flow-through electrolyzer consisting of SP- cathode and the mesh-type commercial dimension stable anode (DSA, the electrodes with a coating thickness of ) as the NITRR electrolyzer. During operation, the -containing solution as synthetic effluent was treated through electrochemical reduction on cathode, and circulated by a peristaltic pump with a flow rate of . Then the -containing effluent was directly guided into the hollow polypropylene (PP) fiber arrays immersed into 1 M HCl solution ( 800 mL ) to recover . The key of simultaneous NITRR and product recovery is the continuous flow of through the gas-permeable hydrophobic membrane and then
Fig. 5 | An integrated equipment for flow-through NITRR electrolyzer and in -situ recovery. a Schematic illustration of integrated equipment composed of flow-cell reactor and membrane separator by employing the monolithic cathode for NITRR and a gas-permeable hydrophobic membrane for recovery.
b Long-term stability test of SP- electrode at -0.4 V vs. RHE using the integrated equipment. c production and recovery efficiency of the integrated equipment during long-term stability test. XRD pattern of the obtained powder.
back to the cathode chamber. During this process, the produced could permeate through membrane between the cathode chamber and the recovery chamber and be absorbed by acid solution in the recovery chamber . By using the monolithic cathode, the coupled device could robustly work at an industrial-level current density of for up to 200 h with a high of (Fig. 5b). As a result, this sustainable and decentralized system achieved high NITRR and simultaneous recovery with nearly selectivity and about recovery efficiency (Fig. 5c). After long-term electrolysis, the measured contact angle of membrane (Supplementary Fig. 39) was determined to be , similar with that of pristine membrane ( ), suggesting that the post-reacted membrane still kept its hydrophobicity. After further rotary evaporation, the recycled ammonia in solution could be converted into high-purity powder , as confirmed by the XRD pattern (Fig. 5d). These findings demonstrated the feasibility of selective conversion of nitrate-containing wastewater into upgraded ammonia fertilizers through combining the high-performance SP- – Ti monolithic cathode with the integrated device, providing a sustainable way for nitrogen cycle.
Discussion
In conclusion, the spin-polarized pairs were designed by manipulating oxygen vacancies of the inherent surface oxide layer on monolithic Ti electrode, and the resultant electrode could deliver extraordinary NITRR activity with an impressive yield rate
( ) and a high Faradic efficiency of at -0.4 V vs. RHE, suppressing the counterpart of spin-depressed electrode and most of the well-known electrodes. It was demonstrated that spin-polarized pairs could provide spin electrons to interact with the reaction intermediates, and consequently facilitate to the deoxygenation of to *NO and the hydrogenation of *NO via strengthening their adsorption. By coupling the NITRR flow-through electrolyzer with the membrane separation technology, we successfully realized the on-site ammonia recovery in the fertilizer’s formation from the nitrate-containing electrolyte in a sustainable and decentralized manner, by taking advantage of the resultant spin-polarized pairs electrode. Our study not only showcases the construction of spin-polarized pairs for NITRR and potential wastewater treatment or ammonia production applications, but also offers an innovative strategy to develop the advanced electrodes with spin polarization effect towards spin-related reactions.
Methods
Chemicals
The titanium monolithic electrode was purchased from Kunshan Guangjiayuan New Material Co., Ltd., China. Iron chloride ( ), potassium sodium tartrate ( ), DMSO-d6, maleic acid , sodium nitrate , potassium hydroxide , potassium nitrate and ethanol were purchased from
Sinopharm Chemical Regent Company. All chemicals were used without further purification.
Electrode preparation
The synthesis route of electrode was illustrated in Supplementary Fig. 1a, as previously reported . Typically, the monolithic electrode with in size was first pretreated in reductive atmosphere ( ) at for 3 h with a heating rate of to create the oxygen vacancies (OVs) on the electrode surface. Then, the homogeneous Fe precursor solution with a Fe mass concentration of in ethanol was uniformly deposited onto the as-prepared monolithic electrode. Meanwhile, the deposition process was under the infrared lamp illuminating to fasten the solvent evaporation and avoid the solvent aggregation arising from surface tension. Finally, the SP- -Ti electrode was obtained by treating the Fe-deposited electrode in reductive atmosphere ( ) at for 3 h with a heating rate of once more. The SD- electrode followed the similar synthesis route but in an oxidative atmosphere ( Air ) to avoid the generation of OVs for comparison (Supplementary Fig. 1b).
Characterization
X-ray diffraction (XRD) patterns were recorded by employing a Rigaku Miniflex-600 ( and 15 mA ). The high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) images were obtained on a JEOL JEM-ARM200F TEM/STEM with a spherical aberration corrector working at 200 kV . X-ray photoelectron spectroscopy (XPS) was carried out on scanning X-ray microprobe (PHI 5000 Verasa, ULAC-PHI, Inc.) with Al Ka radiation. The C 1 s peak at 284.6 eV is as internal standard for energy calibration. The energy dispersive X-ray spectroscopy (EDS) was performed on FEI Talos F200X. Electron spin resonance (ESR) was conducted on the EMX micro-6/1 under 100 K . The X-ray absorption fine structure spectra of Fe K-edge was obtained at Singapore Synchrotron Light Source center (SSLS, operating at 2.5 GeV with a maximum current of 200 mA ). The EXAFS spectra were determined via subtracting the post-edge background from the overall absorption and then normalized to the edge-jump step. Subsequently, the data was Fourier transformed to real space by using a hanning windows ( ) to separate the EXAFS contributions from different coordination shells. To obtain the quantitative structural parameters around central atoms, least-squares curve parameter fitting was performed with using the ARTEMIS module of IFEFFIT software packages. XAS spectra of Ti-L edge were collected at the BL 11 A beamline of the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. We measured electrical conductivities of SP- and SD- electrode at room temperature through the four probe DC technique.
Electrochemical measurements
NITRR test was conducted in a typical H-type cell, separated by a Nafion 117 membrane. The electrodes with specific spin polarization degree ( and SD- ), platinum plate and electrode were used as working electrode, counter electrode and reference electrode, respectively. If not specified otherwise, all potentials reported in this work were referenced to a reversible hydrogen electrode (RHE). Before each measurement, the electrolyte was purged with Ar ( ) for 30 min . The linear sweep voltammetry (LSV) curves were recorded at a scan rate of under stirring with rotation rate of 500 rpm . Potentiostatic tests were conducted at certain applied potentials within NITRR window for 2 h . Nessler’s reagent method was employed to quantify the generated in electrolyte to further determine the faradaic efficiency ( ) and yield rate. In particular, a certain amount of electrolyte was taken out from the cell and diluted to the detection range. Then, an aqueous solution of potassium sodium tartrate and Nessler’s reagent were added into the diluted electrolyte to obtain the uniform mixture. Let stand for 10 min . The UV-Vis absorption intensity at the absorbance peak at 420 nm was collected to determine the concentration of a series of standard ammonium chloride solutions and the mixed solution. The concentration-absorbance curve was plotted for the calculation of the concentration of the mixed solution (Supplementary Fig. 14). The yield rate was calculated via the following relation:
The was calculated via the following relation:
where is the measured concentration, V is the volume of the electrolyte, t is the reaction time, is the Fe mass on the monolithic electrode, F is the Faraday constant ( ) and Q is the total charge. The amount was further determined by 1 H NMR ( 600 MHz , Bruker Avance III) with DMSO-d6 as the spin-lock field and maleic acid ( ) as the internal standard, respectively. Before each 1H NMR measurement, the pH of obtained solution was adjusted to 2.0 with HCl . The concentration of was plotted versus the peak area ratio of and to obtain the standard curve. The generated concentration can be determined from standard curve. An isotope-labeling experiment was performed in the electrolyte containing and for 2 h at -0.4 V vs. RHE to specify the nitrogen source of . The obtained was observed in 1 H NMR spectra. The possible gas products were detected by a gas chromatography (GC, Thermal Trace-1300) equipped with thermal conductivity detector (TCD). The Griess test was used to determine the concentration in electrolytes after the potentiostatic test. Generally, naphthyl ethylenediamine dihydrochloride ( 0.04 g ), p -aminobenzene sulfonamide , and were dissolved in Dl water ( 10 mL ) using the Griess reagent. The diluted electrolyte was mixed with the Griess agent ( 100 uL ) and rested for 10 min at room temperature to conduct UV-vis tests. concentrations of the electrolyte were determined by the absorbance at 540 nm . In the same operation, various aqueous solutions were used as the standard samples to obtain the calibration curve (Supplementary Fig. 17). The FE of byproduct was calculated via the following relation:
The selectivity of was calculated via the following relation:
where is the number of electrons transferred to byproduct is the measured byproduct concentration, V is the volume of the electrolyte or the upper space, F is the Faraday constant and Q is the total charge.
The electrical double layer capacitor ( ) was determined from double-layer charging curves using cyclic voltammograms (CVs) within non-faradaic potential region between 0.4 and 0.5 V vs. RHE at different scan rates from to . Half of the current density difference ( ) at the centered potential was plotted against the scan rate (v) to calculate the slope of fitted straight line and obtained the value. The electrochemical active surface area (ECSA) was calculated by dividing the double-layer capacitance ( ) by specific capacitance ( ). Differential Electrochemical Mass Spectrometry (DEMS) measurements were carried out on a specially-made electrochemical cell with electrolyte constantly flowing in through a peristaltic pump. During the test, Ar was kept bubbling
into the electrolyte. The mass signals were constantly collected during the LSV measurements from 0.1 to -0.6 V at a scan rate of . When the mass signal returned to baseline, another three cycles were conducted under identical condition to avoid the accidental error.
Data availability
All data that support the findings of this study are present in the paper and the Supplementary Information. Further information can be acquired from the corresponding authors upon reasonable request.
References
Ashida, Y., Arashiba, K., Nakajima, K. & Nishibayashi, Y. Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water. Nature 568, 536-540 (2019).
Service, R. F. New recipe produces ammonia from air, water, and sunlight. Science 345, 610-610 (2014).
Suryanto, B. H. R. et al. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nat. Catal. 2, 290-296 (2019).
Tang, C. & Qiao, S. -Z. How to explore ambient electrocatalytic nitrogen reduction reliably and insightfully. Chem. Soc. Rev. 48, 3166-3180 (2019).
Hao, Y. -C. et al. Promoting nitrogen electroreduction to ammonia with bismuth nanocrystals and potassium cations in water. Nat. Catal. 2, 448-456 (2019).
Geng, Z. et al. Achieving a record-high yield rate of 120.9 for electrochemical reduction over Ru single-atom catalysts. Adv. Mater. 30, 1803498 (2018).
Lv, C. et al. An amorphous noble-metal-free electrocatalyst that enables nitrogen fixation under ambient conditions. Angew. Chem. Int. Ed. 57, 6073-6076 (2018).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036-7046 (2020).
Wu, Z. -Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Chen, G. -F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper-molecular solid catalyst. Nat. Energy 5, 605-613 (2020).
Chen, F. -Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotech. 17, 759-767 (2022).
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290-294 (2021).
Duca, M. & Koper, M. T. M. Powering denitrification: the perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726-9742 (2012).
Wang, Y. et al. Nitrate electroreduction: mechanism insight, in situ characterization, performance evaluation, and challenges. Chem. Soc. Rev. 50, 6720-6733 (2021).
Xu, H. et al. Electrocatalytic reduction of nitrate-a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710-2758 (2022).
Shafirovich, V. & Lymar, S. V. Nitroxyl and its anion in aqueous solutions: spin states, protic equilibria, and reactivities toward oxygen and nitric oxide. Proc. Nat. Acad. Sci. 99, 7340-7345 (2002).
Ford, P. C. & Miranda, K. M. The solution chemistry of nitric oxide and other reactive nitrogen species. Nitric Oxide 103, 31-46 (2020).
Janaway, G. A. & Brauman, J. I. Direct observation of spin forbidden proton-transfer reactions: . J. Phys. Chem. A 104, 1795-1798 (2000).
Gao, Q. et al. Breaking adsorption-energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine-learned insights. Nat. Commun. 13, 2338 (2022).
Zhang, S. et al. High-ammonia selective metal-organic framework-derived Co -doped catalysts for
electrochemical nitrate reduction. Proc. Nat. Acad. Sci. 119, e2115504119 (2022).
Wang, Y. et al. Enhanced nitrate-to-ammonia sctivity on copper-nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702-5708 (2020).
Cheng, X. -F. et al. Coordination symmetry breaking of single-atom catalysts for robust and efficient nitrate electroreduction to ammonia. Adv. Mater. 34, 2205767 (2022).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper-supported rhodium cluster and single-atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Clayborne, A., Chun, H. -J., Rankin, R. B. & Greeley, J. Elucidation of pathways for NO electroreduction on Pt(111) from first principles. Angew. Chem. Int. Ed. 54, 8255-8258 (2015).
Mtangi, W. et al. Control of electrons’ spin eliminates hydrogen peroxide formation during water splitting. J. Am. Chem. Soc. 139, 2794-2798 (2017).
Do, V. -H. & Lee, J. -M. Orbital occupancy and spin polarization: from mechanistic study to rational design of transition metal-based electrocatalysts toward energy applications. ACS Nano 16, 17847-17890 (2022).
Chen, R. R. et al. Antiferromagnetic inverse spinel oxide with spin-polarized channels for water oxidation. Adv. Mater. 32, 1907976 (2020).
Liang, Y. et al. Enhancement of electrocatalytic oxygen evolution by chiral molecular functionalization of hybrid 2D electrodes. Nat. Commun. 13, 3356 (2022).
Ren, X. et al. Spin-polarized oxygen evolution reaction under magnetic field. Nat. Commun. 12, 2608 (2021).
Wu, T. et al. Spin pinning effect to reconstructed oxyhydroxide layer on ferromagnetic oxides for enhanced water oxidation. Nat. Commun. 12, 3634 (2021).
Li, X. et al. Identification of the electronic and structural dynamics of catalytic centers in single-Fe-atom material. Chem 6, 3440-3454 (2020).
Li, Z. et al. Spin engineering of single-site metal catalysts. Innovation 3, 100268 (2022).
Dai, J. et al. Single-phase perovskite oxide with super-exchange induced atomic-scale synergistic active centers enables ultrafast hydrogen evolution. Nat. Commun. 11, 5657 (2020).
Yao, Y. et al. Single atom Ru monolithic electrode for efficient chlorine evolution and nitrate reduction. Angew. Chem. Int. Ed. 61, e202208215 (2022).
Zhang, S. et al. Electrocatalytically active single-atom sites for efficient reduction of nitrogen to ammonia. Angew. Chem. Int. Ed. 59, 13423-13429 (2020).
Muller, D. A. et al. Atomic-scale imaging of nanoengineered oxygen vacancy profiles in . Nature 430, 657-661 (2004).
Chakraborty, T., Meneghini, C., Aquilanti, G. & Ray, S. Microscopic distribution of metal dopants and anion vacancies in Fe-doped single crystals. J. Phys. Condens. Matter 25, 236002 (2013).
Sala, G. et al. Vacancy defects and monopole dynamics in oxygendeficient pyrochlores. Nat. Mater. 13, 488-493 (2014).
Yang, T.-Y. et al. A new hematite photoanode doping strategy for solar water splitting: oxygen vacancy generation. Phys. Chem. Chem. Phys. 15, 2117-2124 (2013).
Wei, K. et al. Strained zero-valent iron for highly efficient heavy metal removal. Adv. Funct. Mater. 32, 2200498 (2022).
Mao, C. et al. Energy-confined solar thermal ammonia synthesis with . Appl. Catal. B Environ. 224, 612-620 (2018).
Xie, J. et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 1, 889-896 (2018).
Mondal, P. et al. Behavior of with system and the reduction to As(O). J. Phys. Chem. C. 118, 21614-21621 (2014).
Liu, W. et al. Discriminating catalytically active species of atomically dispersed catalyst for selective oxidation of the C-H bond. J. Am. Chem. Soc. 139, 10790-10798 (2017).
Zitolo, A. et al. Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials. Nat. Mater. 14, 937-942 (2015).
Ohtomo, A., Muller, D. A., Grazul, J. L. & Hwang, H. Y. Artificial charge-modulationin atomic-scale perovskite titanate superlattices. Nature 419, 378-380 (2002).
Pan, L. et al. Manipulating spin polarization of titanium dioxide for efficient photocatalysis. Nat. Commun. 11, 418 (2020).
Chang, C. F. et al. c-axis dimer and its electronic breakup: the insulator-to-metal transition in . Phys. Rev. X 8, 021004 (2018).
Schlappa, J. et al. Resonant soft x-ray scattering from stepped surfaces of . J. Phys. Condens. Matter 24, 035501 (2012).
Guo, Y. et al. Engineering the electronic state of a perovskite electrocatalyst for synergistically enhanced oxygen evolution reaction. Adv. Mater. 27, 5989-5994 (2015).
Miao, J. et al. Spin-state-dependent peroxymonosulfate activation of single-atom M-N moieties via a radical-free pathway. ACS Catal. 11, 9569-9577 (2021).
Dai, J. et al. Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis. Nat. Commun. 13, 1189 (2022).
Wang, Y. et al. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350-5354 (2020).
Gao Q., et al. Synthesis of core/shell nanocrystals with ordered intermetallic single-atom alloy layers for nitrate electroreduction to ammonia. Nat. Synth. https://doi.org/10.1038/s44160-023-00258x (2023).
Liu, H. et al. Low-coordination Rhodium catalysts for an efficient electrochemical nitrate reduction to ammonia. ACS Catal. 13, 1513-1521 (2023).
Long, J. et al. Direct electrochemical ammonia synthesis from nitric oxide. Angew. Chem. Int. Ed. 59, 9711-9718 (2020).
Sun, Y. et al. Spin-related electron transfer and orbital interactions in oxygen electrocatalysis. Adv. Mater. 32, 2003297 (2020).
Wei, X. et al. Tuning the spin state of Fe single atoms by Pd nanoclusters enables robust oxygen reduction with dissociative pathway. Chem 9, 181-197 (2023).
Li, J. et al. solar-to-ammonia efficiency from nitrate using Fe single atomic catalyst supported on nanosheets. Adv. Funct. Mater. 32, 2108316 (2022).
Niu, H. et al. A feasible strategy for identifying single-atom catalysts toward electrochemical NO-to- conversion. Small 17, 2102396 (2021).
Gao, R. et al. Pt/ with Pt-Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy 6, 614-623 (2021).
Liu, K. et al. Insights into the activity of single-atom Fe-N-C catalysts for oxygen reduction reaction. Nat. Commun. 13, 2075 (2022).
Gao, J. et al. Electrocatalytic upcycling of nitrate wastewater into an ammonia fertilizer via an electrified membrane. Environ. Sci. Technol. 56, 11602-11613 (2022).
Gao, J. et al. Electrochemically selective ammonia extraction from nitrate by coupling electron- and phase-transfer reactions at a three-phase interface. Environ. Sci. Technol. 55, 10684-10694 (2021).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA1201701, 2022YFA150470, L.Z.), the National
Natural Science Foundation of China (U22A204O2, U21A20286, L.Z.), (22102100, Y.Y.), (22206121, J.D.), (22273068, U22A20394, X.G.), Shenzhen Science and Technology Program (JCYJ2O220818095601002, L.Z.), the Natural Science Foundation of Shanghai (22ZR1431700, Y.Y.), the China Postdoctoral Science Foundation (2021M702117, 2022M722080, J.D.), the National Postdoctoral Program for Innovative Talents (BX2O220197, J.D.) and the Shanghai Postdoctoral Excellence Program (2021182, J.D.), (2022381, G.Z.). The authors acknowledge the support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials, the Instrumental analysis center of Shanghai Jiao Tong University, Instrumental analysis center of School of Environmental science and engineering, Shiyanjia Lab and Bruker Corporation for the help in characterizations and experimental measurements, and Supercomputing Center of Wuhan University.
Author contributions
Y. Y., X. G., and L.Z. Z. conceived the idea. J. D. and L. Z. carried out the experiments. Y. T. carried out the DFT calculations. J. D., Y. T., Y. Y., X. G., and L.Z. Z. wrote the paper. Z. H., C. C., C. K., G. Z., J. W., X. Z., Q. Z., W. H., R. W., K. W., and R. Z. helped with data analysis and manuscript polishing. All the authors discussed results and provided comments during the manuscript preparation.
Correspondence and requests for materials should be addressed to Xiang-Kui Gu, Yancai Yao or Lizhi Zhang.
Peer review information Nature Communications thanks Wenhui He, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
School of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. School of Power and Mechanical Engineering, Wuhan University, Wuhan 430072, China. Max Planck Institute for Chemical Physics of Solids, Nothnitzer Strasse 40, 01187 Dresden, Germany. National Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu 300092 Taiwan, China. Department of Electrophysics, National Yang Ming Chiao Tung University, Hsinchu, Taiwan, China. These authors contributed equally: Jie Dai, Yawen Tong, Long Zhao. e-mail: xiangkuigu@whu.edu.cn; yyancai@sjtu.edu.cn; zhanglz@ccnu.edu.cn