DOI: https://doi.org/10.1038/s41392-023-01688-x
PMID: https://pubmed.ncbi.nlm.nih.gov/38185705
تاريخ النشر: 2024-01-08
إصابة نقص التروية-إعادة التروية: الآليات الجزيئية والأهداف العلاجية
الملخص
تحدث إصابة نقص التروية-إعادة التروية (I/R) بشكل متناقض أثناء إعادة التروية بعد نقص التروية، مما يؤدي إلى تفاقم الضرر الأولي في الأنسجة. إن الفهم المحدود للآليات المعقدة التي تكمن وراء إصابة I/R يعيق تطوير تدخلات علاجية فعالة. يظهر مسار إشارة Wnt تداخلًا واسعًا مع مسارات أخرى متعددة، مما يشكل نظام شبكة من مسارات الإشارة المعنية في إصابة I/R. توضح هذه المقالة الاستعراضية الآليات الأساسية المعنية في إشارة Wnt، بالإضافة إلى التفاعل المعقد بين Wnt ومسارات أخرى، بما في ذلك Notch، كيناز الفوسفاتيديلينوزيتول 3/كيناز البروتين B، عامل النمو المحول-
المقدمة
المحتوية على محول تحفيز الإنترفيرون-
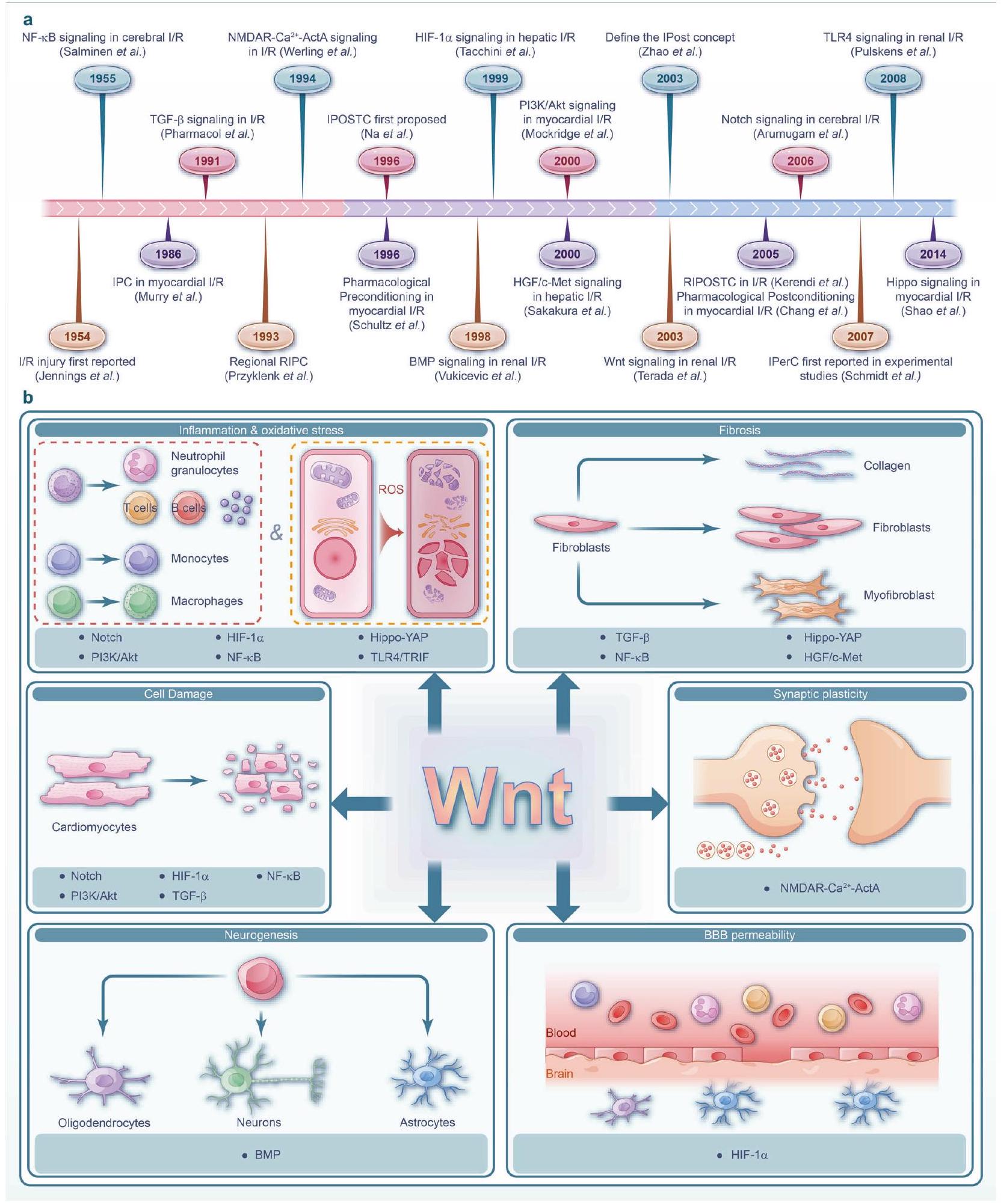
التداخلات بين مسارات الإشارة المختلفة، لا يزال يمثل تحديًا كبيرًا. لذلك، لا يزال هناك نقص في الإجماع في الأبحاث الحالية،
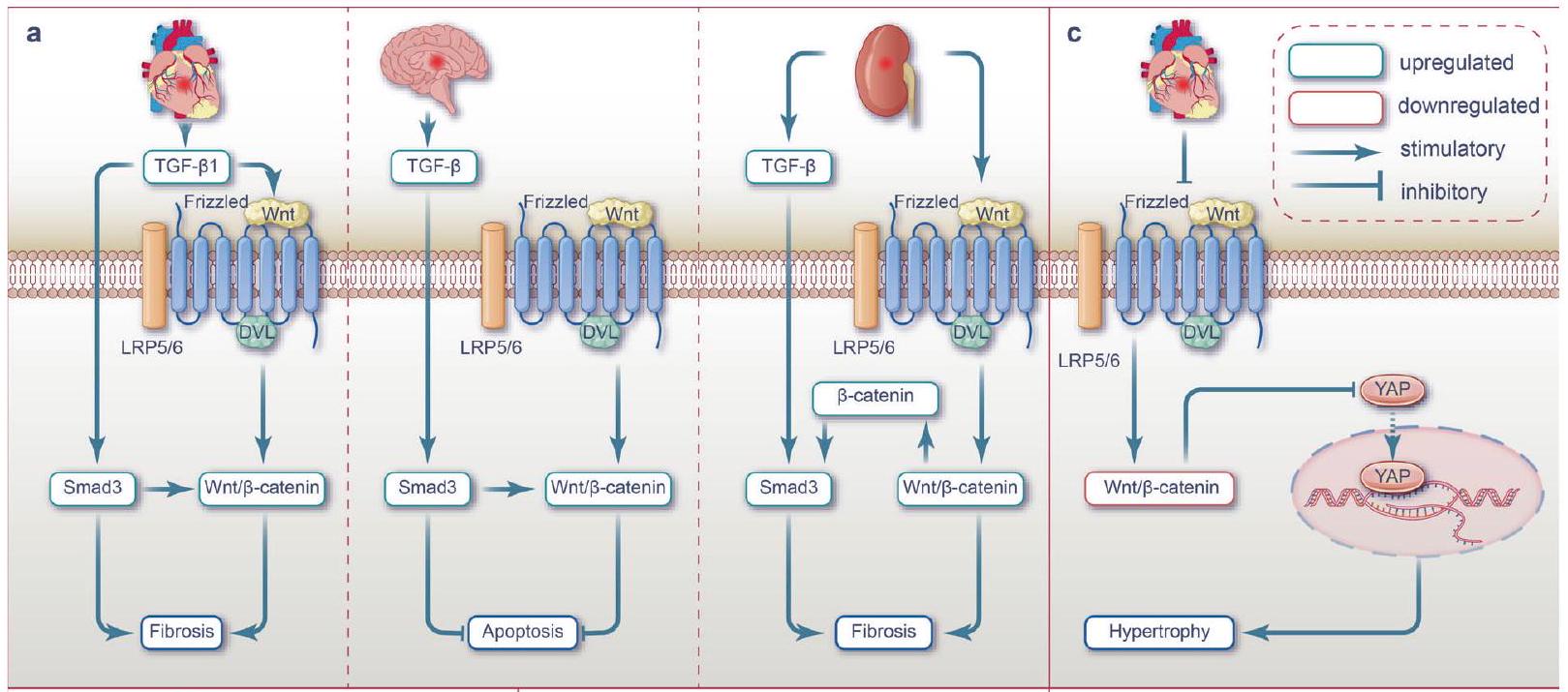
نماذج الخلايا. بالإضافة إلى ذلك، من خلال توضيح الآليات الأساسية، نحدد الاستراتيجيات العلاجية السريرية وما قبل السريرية الحالية التي تستهدف مسار Wnt والشبكات المرتبطة بالإشارات (الشكل 1أ). بالنظر إلى الطبيعة المعقدة لتلف الأعضاء في إصابة نقص التروية/إعادة التروية، فإن استهداف مسارات الإشارات الشبكية أمر حاسم للتدخلات الفعالة. يجب أن تركز الدراسات المستقبلية على تطوير استراتيجيات تعدل هذه الإشارات المترابطة بفعالية للتخفيف من الآثار الضارة لتلف نقص التروية/إعادة التروية.
مسارات WNT وإصابة نقص التروية في الأعضاء
مسارات Wnt
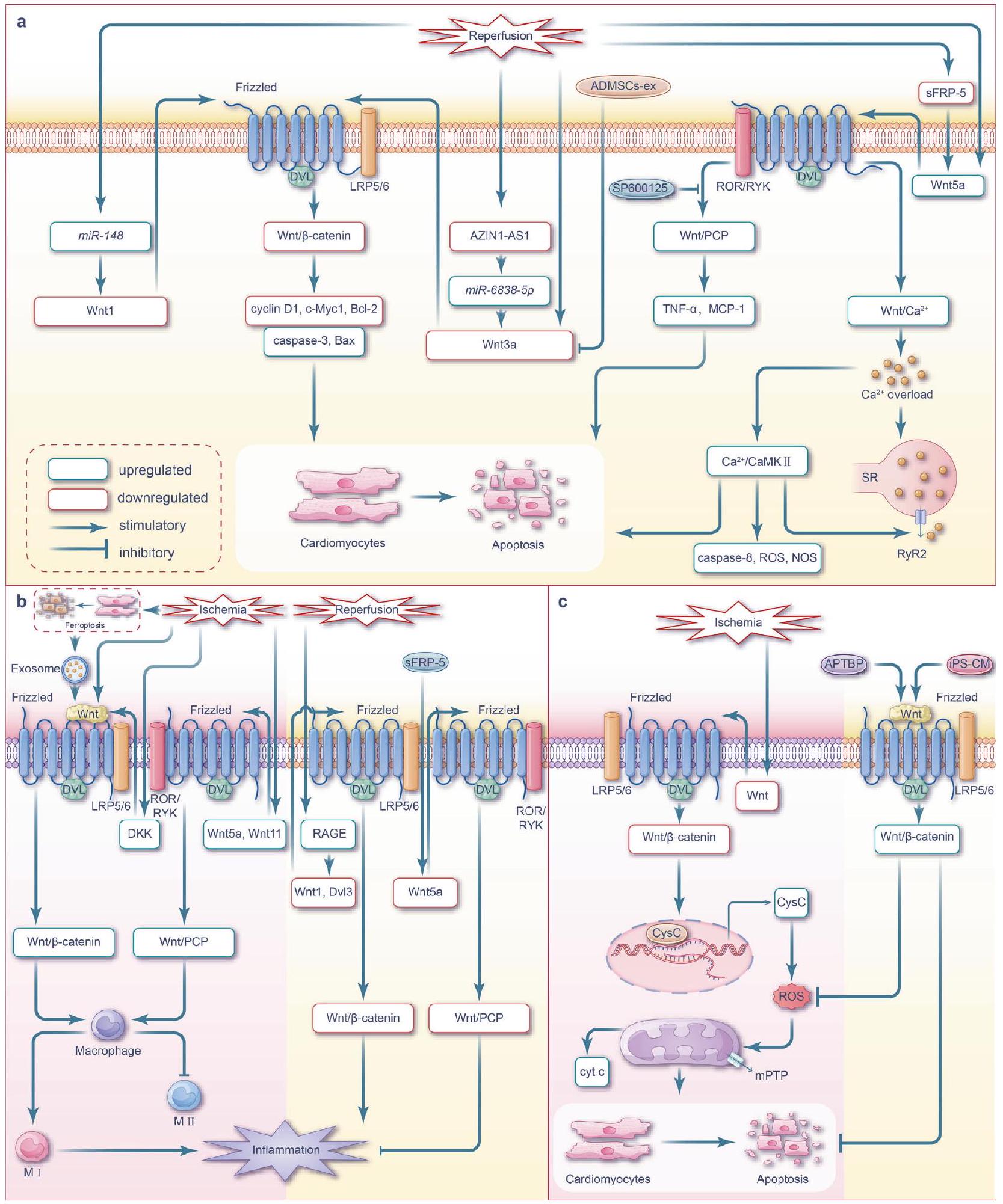
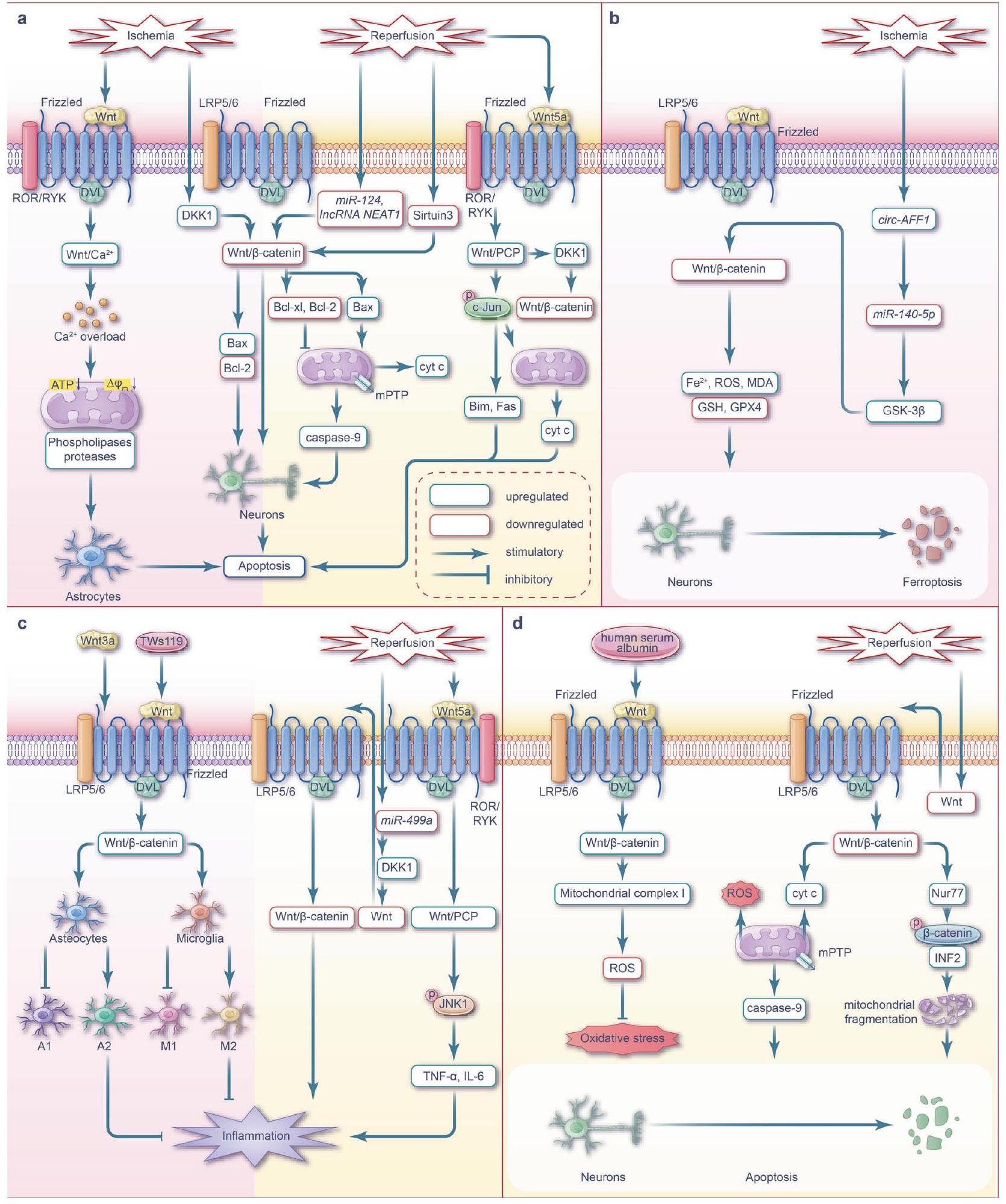
تنشيط كيناز جين c-Jun N-terminal (JNK)، الذي يلعب بدوره دورًا حاسمًا في إعادة ترتيب الهيكل الخلوي للأكتين وتنظيم قطبية الخلايا وتعزيز الهجرة.
تم تنظيمه بشكل سلبي، بينما نسبة الكاسبيز 3 المقسوم و p-GSK
تثبيط Wnt/
فئران نشطت البلعميات في القلب المصاب. هذه التنشيط زاد بعد ذلك من تعبير Wnt5a، وزاد من فسفرة JNK، ورفع مستويات تعبير السيتوكينات الالتهابية IL-1.
لقد أظهرت الدراسات أن نقص تروية عضلة القلب ينشط مسار Wnt/
في البلعميات القلبية، خاصة في الأنماط الفرعية المؤيدة للالتهاب.
في استقطاب البلعميات.
داخل المنطقة المتعفنة بعد أسبوع من احتشاء القلب؛ بالإضافة إلى ذلك، تعبير البروتين الموجود في أعلى مسار Wnt/
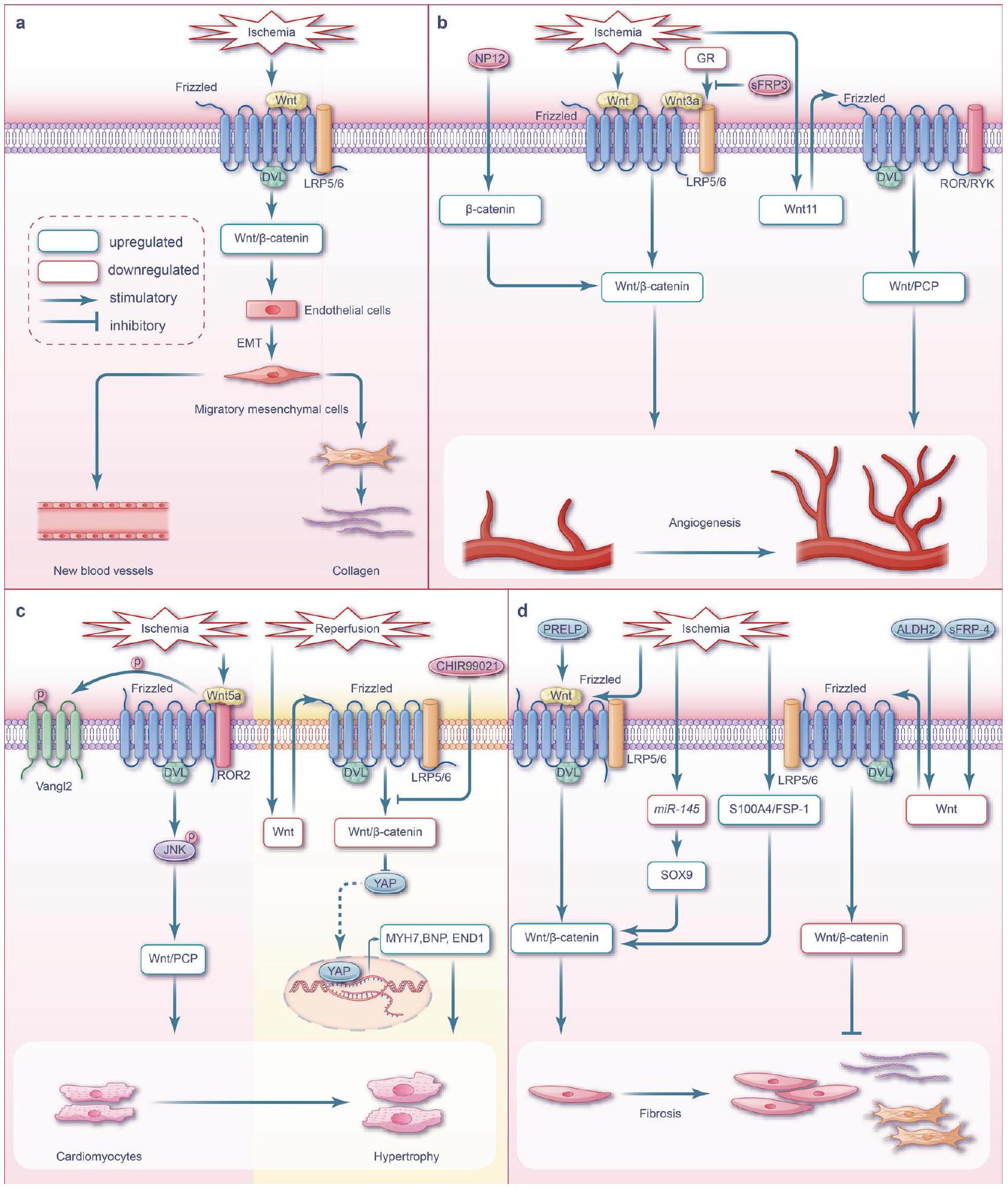
إصابة عضلة القلب الإقفارية، مما يؤدي إلى بدء إعادة تشكيل المصفوفة خارج الخلوية المفرطة والتليف. ومن المثير للاهتمام أن مسار Wnt/
إن التعرف والتدخل في الوقت المناسب للسكتة الدماغية الإقفارية أمر في غاية الأهمية. العلاج الموصى به حاليًا
تشمل استراتيجية السكتة الدماغية الإقفارية التحلل الخثاري، من خلال حقن منشط البلازمينوجين النسيجي المؤتلف (rtPA) خلال 4.5 ساعات من بداية الأعراض،
يتحول مسار إشارة Wnt من حالة نشطة إلى حالة مثبطة مع زيادة وقت الإقفار، ويتم تنظيمه بواسطة العديد من العمليات مثل البلعمة الذاتية.
أثناء نقص التروية.
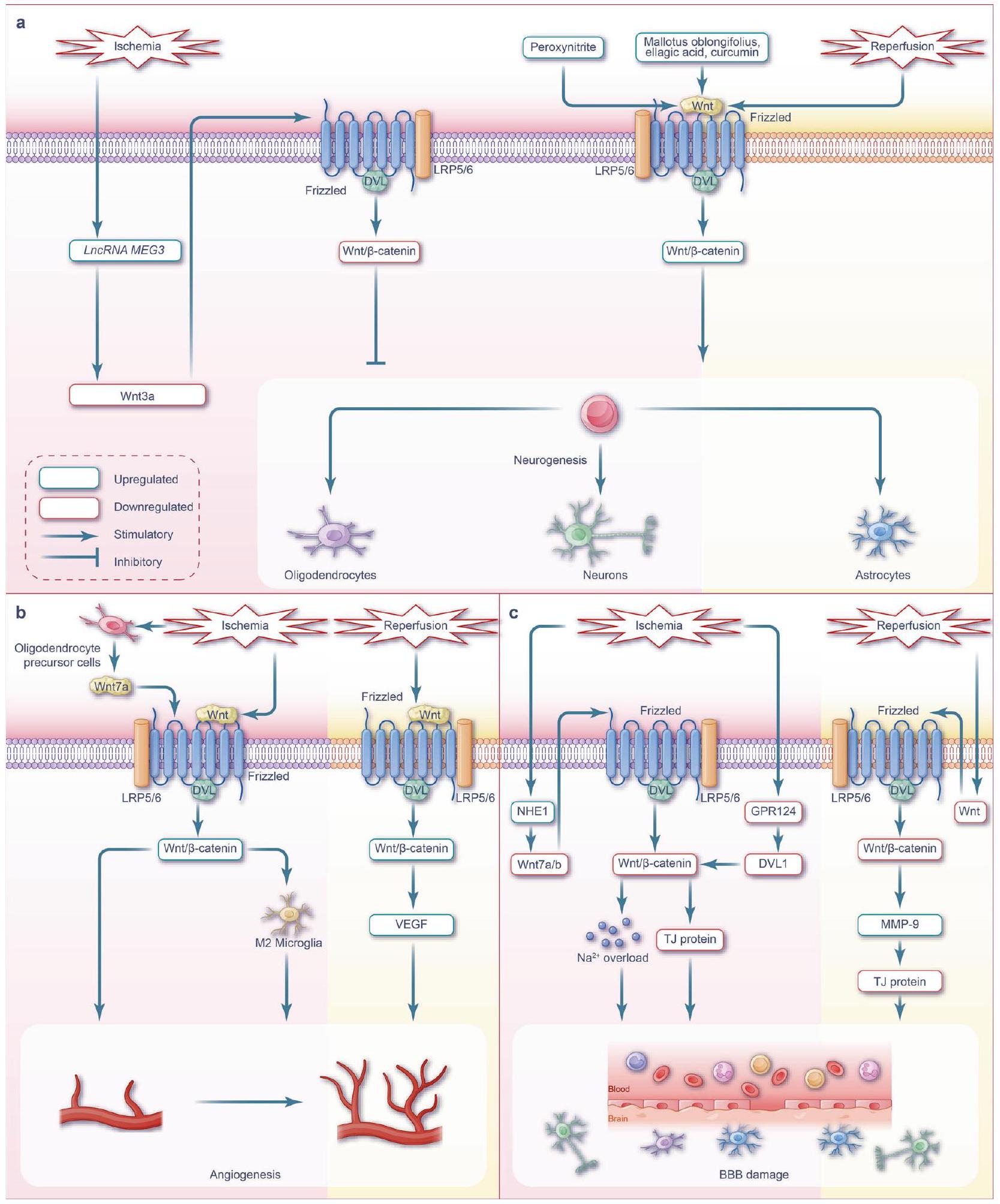
مسام الانتقال النفاذية والموت الخلوي اللاحق.
يتم تحفيز مستقبل الهرمون نور77، مما يعزز
زيادة تنظيم جزيئات مختلفة في مجرى العمل. يعزز تكوين الأعصاب من شفاء إصابات الدماغ من خلال مكافحة موت الخلايا المبرمج.
الإشارات تحفز استقطاب الميكروغليا التفاعلية نحو النمط الظاهري M2.
مسارات Wnt خلال إصابة الكلى الناتجة عن نقص التروية
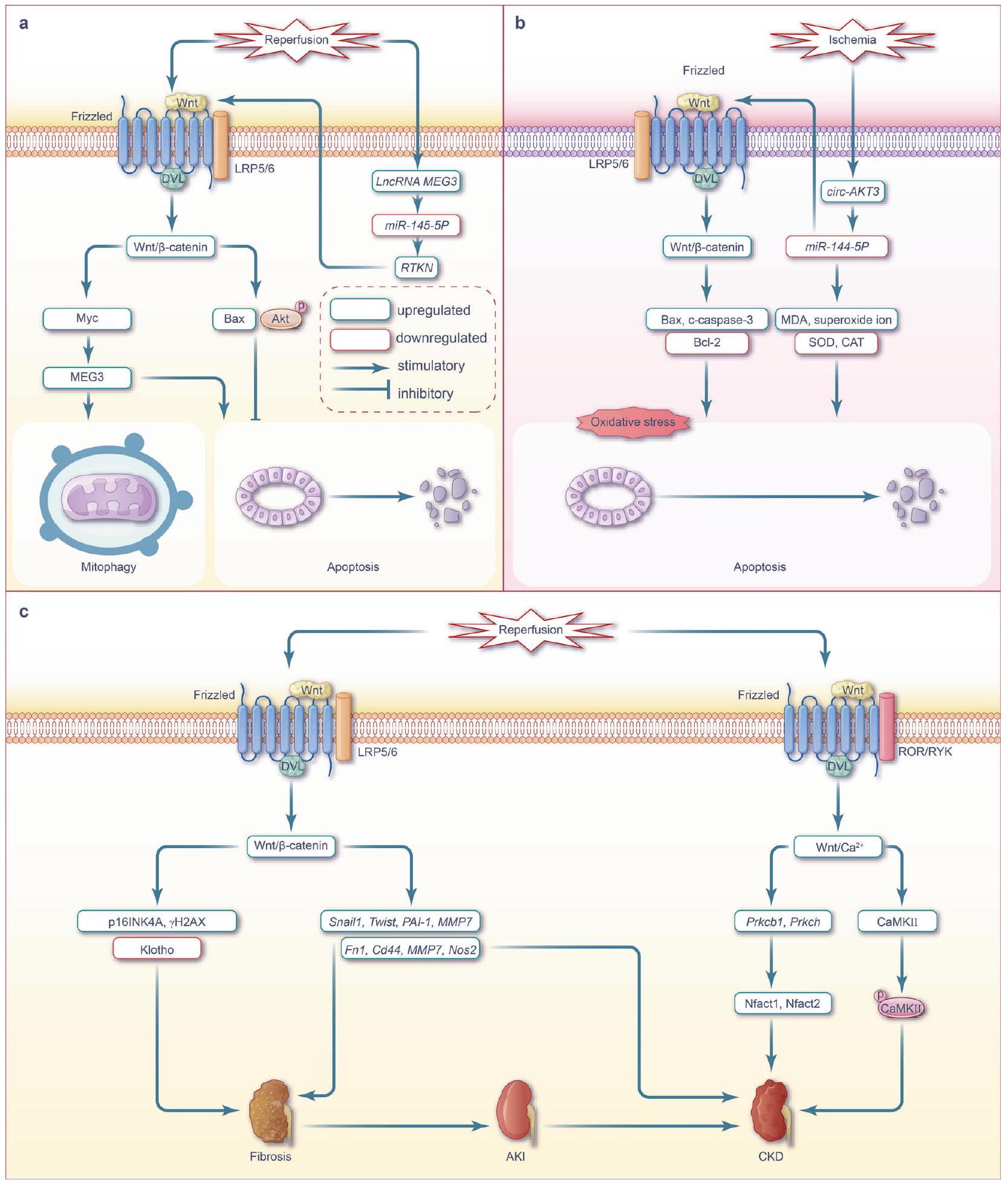
مسارات Wnt خلال إصابة الكبد الناتجة عن نقص التروية/إعادة التروية
تشخيص وظيفة الكبد. يتأثر مدى إصابة الكبد الناتجة عن نقص التروية بالحرارة ومدة نقص التروية والنطاق. تكون إصابة خلايا الكبد الناتجة عن نقص التروية الدافئ أكثر شدة من تلك الناتجة عن نقص التروية الباردة، بينما تكون إصابة خلايا الأوعية الشعرية الكبدية على العكس من ذلك.
زيادة ملحوظة في تكاثر الخلايا الكبدية مع تقليل الموت الخلوي والنخر في الوقت نفسه.
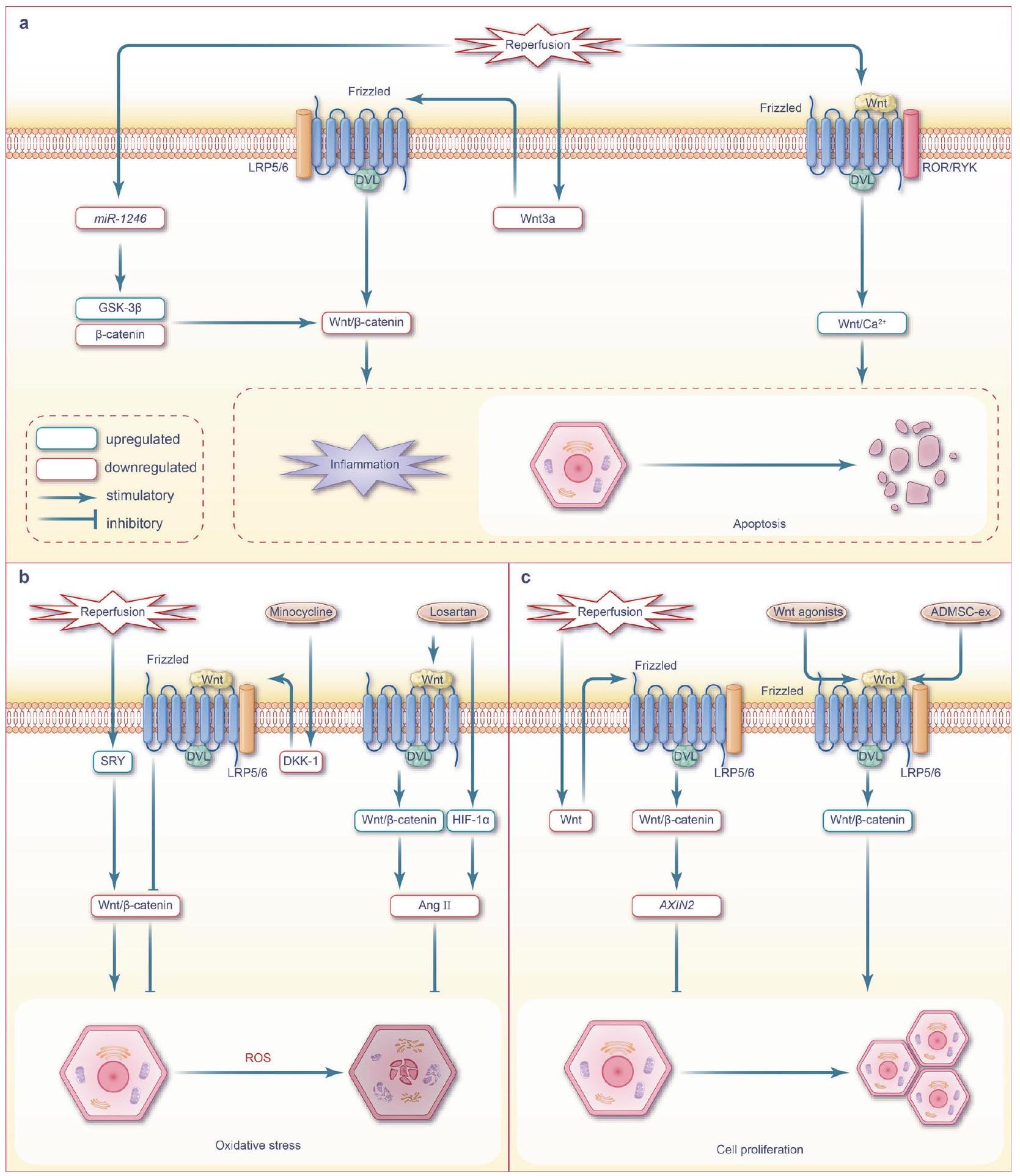
التداخل بين إشارة WNT ومسارات أخرى مرتبطة بنقص التروية/إعادة التروية
يمثل مسار إشارة Notch، وهو مسار محفوظ تطوريًا بشكل كبير ويشارك في تطوير الأجنة وإصلاح الأنسجة،
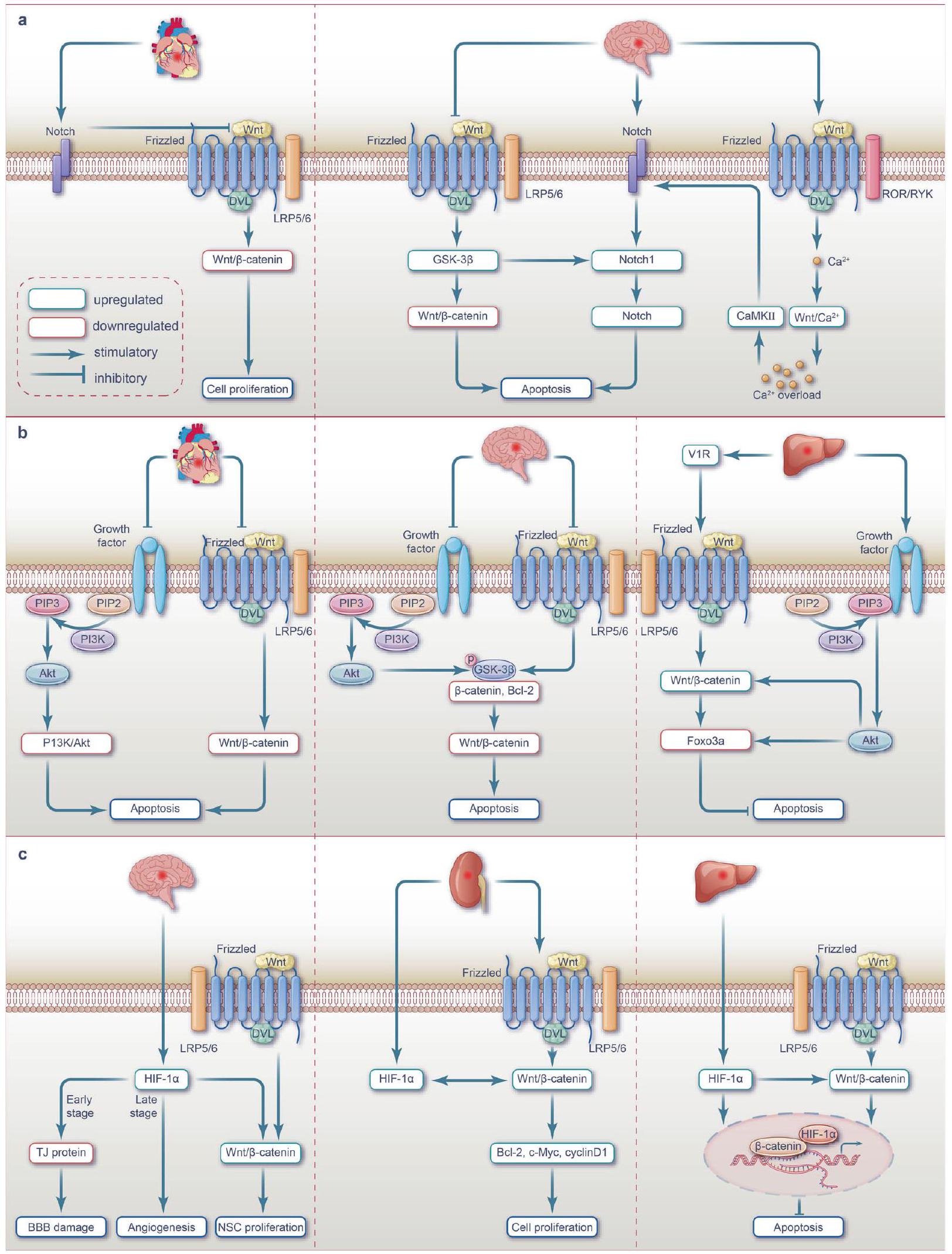
. تؤدي هذه التعديلات المزدوجة إلى بروتينات مشبكية وتقليل الموت الخلوي في الجرذان حديثة الولادة المصابة بنقص التروية الدماغية.
إلى تثبيط مسارات إشارة Wnt3a/
ضمور.
يمثل مسار إشارة PI3K/Akt، الذي سمي على اسم مكوناته الرئيسية، كيناز الفوسفاتيديلينوسيتول 3 (PI3K) و AKT (المعروف أيضًا باسم كيناز البروتين
مضاد للموت الخلوي المبرمج.
يمثل مسار إشارة عامل نقص الأكسجة-1a (HIF-1a) المسؤول عن منح التكيف مع الظروف منخفضة الأكسجين ويلعب دورًا حاسمًا في تكوين الأوعية الدموية والإجهاد التأكسدي وعمليات التمثيل الغذائي الخلوي في ظل ظروف نقص الأكسجة.
ويظهر عمر نصف قصير في ظل ظروف الأكسجين العالي ويتم تكسيره بسرعة بواسطة البروتيازومات. ومع ذلك، فإنه مستقر في ظل ظروف نقص الأكسجة؛ في ظل هذه الظروف، ينتقل HIF-1 إلى النواة، ويتعاون مع HIF-1
تي جي إف-
يشكل معقدًا مع
التأثيرات الليفية الناتجة عن التفاعل المتبادل بين TGF-
تم تأسيس NF-кB في البداية كعامل نسخي مهم في تحفيز استجابات المناعة والالتهابات المختلفة.
بينما تنشط مسار إشارة Wnt2/
إشارة Hippo-YAP
إشارات BMP
مسار الإشارة.
NMDAR-
إشارات TLR4/TRIF
اضطراب وضعف إدراكي ناتج عن حرمان من نوم حركة العين السريعة.
إشارات HGF/c-Met
استراتيجيات علاجية
لقد أظهرت عدة أساليب علاجية تركز على إشارات Wnt، بما في ذلك العلاج بالخلايا والعلاج بالإكسوزومات، والعلاج الجيني، والعلاج البروتيني، والعلاج الدوائي، آفاقًا واعدة للتطبيق السريري. تلخص الجدول 1 الاستراتيجيات العلاجية المستهدفة لإشارات Wnt لعلاج إصابة نقص التروية/إعادة التروية.
في سعيهم للتغلب على إصابات نقص التروية وإعادة التروية، يقوم الأطباء حاليًا باستخدام أساليب ما قبل التكييف وما بعد التكييف. لقد أظهرت هذه الاستراتيجيات تأثيرات علاجية واعدة من خلال استهداف الشبكة المعقدة من مسارات الإشارات المرتبطة بمرض نقص التروية وإعادة التروية.
إصابة الكلى الناتجة عن نقص التروية عندما يتم تطبيق استراتيجية علاج نقص التروية باستخدام العلاج الوقائي.
النقاش والآفاق
| الجدول 1. استراتيجيات علاجية تستهدف إشارات Wnt لعلاج إصابة نقص التروية/إعادة التروية | |||||
| استراتيجية علاجية | مسارات الهدف | استراتيجية/جزيئية/أدوية | عضو | آثار | المراجع |
| علاج الخلايا | وينت النشط/
|
الإكسوزومات المعزولة من خلايا جذعية ميزانشيمية مشتقة من الدهون، ADMSCs-EX | قلب | تنظيم Wnt3a بشكل إيجابي؛ تثبيط الموت الخلوي المبرمج | 95 |
| زراعة خلايا سلف الأليغودندروسايت | دماغ | تعزيز تكوين الأوعية الدموية؛ إصلاح سلامة الحاجز الدموي الدماغي | 239 | ||
| إكسوزومات خلايا الدهون الجذعية المزروعة (ADSCsExo) | كبد | تثبيط مسار NF-кB؛ تقليل البيروبتوزيس في الكبد التالف | ٢٩٦ | ||
| العلاج الجيني | وينت النشط/
|
الميكرو RNA-148b المنخفض | قلب | تنظيم Wnt1 بشكل إيجابي؛ تثبيط الموت الخلوي المبرمج والأضرار التأكسدية | 93 |
| تنظيم مفرط لـ LncRNA AZIN1-AS1 | قلب | الـ LncRNA AZIN1-AS1/miR-6838-5p ينشط Wnt3a
|
94 | ||
| إزالة جين نور77 | دماغ | تثبيط تجزئة الميتوكوندريا | ١٧٦ | ||
| زيادة التعبير عن LncRNA NEAT1 | دماغ | Wnt3a المستقر؛ تثبيط الموت الخلوي | 174 | ||
| تنظيم تنازلي لـ IncRNA MEG | دماغ | تعزيز تكوين الأعصاب | ٢٣٢,٢٥٢ | ||
| العلاج الجزيئي | تثبيط مسارات Wnt غير التقليدية | زيادة تنظيم Sfrp5 | قلب | تثبيط Wnt5a/JNK وWnt/PCP؛ تثبيط الموت الخلوي والالتهاب | ٩٦-٩٩، ١٠٢ |
| دواء | وينت النشط/
|
بولي ببتيد من بروتين ساق التونة، APTBP | قلب | تثبيط موت الخلايا المبرمج | 127,128 |
| فيلانثوس إمبليكا (P. إمبليكا) | قلب | نشاط PI3K/Akt/GSK3
|
٣٣٤ | ||
| CHIR99021 (GSK3
|
قلب | مسار الهيبو النشط؛ تثبيط الموت الخلوي وتضخم الخلايا | ١٣٧ | ||
| إيزوفلوران | دماغ | تثبيط موت الخلايا المبرمج | 165 | ||
| XQ-1H؛ غاسترودين | دماغ | تثبيط الموت الخلوي؛ تعزيز تكوين الأعصاب | ١٧٥,٢٥٢ | ||
| بيروكسينيترايت؛ مالوتوس أوبلونغيفوليوس؛ حمض الإيلاجيك | دماغ | تعزيز تكوين الأعصاب | ٢٢١، ٢٢٩، ٢٣٠ | ||
| الكركمين | دماغ | تعزيز تكوين الأعصاب؛ تثبيط موت الخلايا المبرمج؛ تخفيف الالتهاب | ٢٠٨، ٢١١، ٢٣١ | ||
| TWS119 | دماغ | إصلاح الحاجز الدموي الدماغي؛ تقليل الالتهاب العصبي | ٢٠٩ | ||
| TWS119 | دماغ | تثبيط نوتش؛ تثبيط الموت الخلوي المبرمج | 318 | ||
| كويرسيتين | دماغ | إصلاح BBB | ٢٤٦,٢٥٤ | ||
| ألبومين مصل الإنسان | دماغ | تقليل الإجهاد التأكسدي | ٢١٢ | ||
| غالانجين | دماغ | تثبيط HIF-1
|
343 | ||
| ديكسمديتوميدين | دماغ | تنشيط PI3K/AKT؛ تقليل حجم السكتة الدماغية؛ تعزيز بقاء الخلايا العصبية | ٣٣٣,٣٣٤ | ||
| منشط Wnt (بيريميدين صناعي) | كلى | تثبيط الاستجابة الالتهابية والإجهاد التأكسدي | ٢٩٣ | ||
| مينوسكلين | كبد | تقليل الإجهاد التأكسدي؛ تثبيط إفراز السيتوكينات المؤيدة للالتهابات | 287 | ||
| أغماطين | كبد | تعزيز تكاثر الخلايا؛ تقليل الالتهاب والموت الخلوي | ٢٨٢ | ||
| لوسارتان | كبد | زيادة تنظيم HIF-1
|
٢٨٥ | ||
| تثبيط Wnt/
|
ديكساميثازون، ديكس | قلب | مي آر-208ب-3ب/ميد13/وينت/
|
٣٣٣,٣٣٤ | |
| بايكاين | قلب | تقليل الضرر التأكسدي لعضلات القلب | 414 | ||
| حبوب هوكسين | قلب | تثبيط NF-кB؛ تثبيط الالتهاب | ١٠٧٣٦٦ | ||
| الميلاتونين | كلى | تحسين تليف الكلى | 385 | ||
| تثبيط مسارات Wnt غير التقليدية | الكركمين | دماغ | تثبيط Wnt/PCP؛ تعزيز تكوين الأعصاب؛ تثبيط موت الخلايا المبرمج؛ تخفيف الالتهاب | ٢١١,٢٣١ | |
| مرحلة | مسار إشارة Wnt | نشاط | عضو | أثر | المراجع |
| نقص التروية | Wnt/
|
مفعل | قلب | تعزيز الالتهاب، إعادة تشكيل المصفوفة خارج الخلوية، تكوين الأوعية الدموية، التليف | ١١٥-١١٧، ١٢٩، ١٣٠، ١٣٨-١٤١ |
| دماغ | تعزيز تكوين الأعصاب (في المختبر) | 221 | |||
| كلى | تعزيز موت الخلايا المبرمج والإجهاد التأكسدي | 256 | |||
| مُثبَط | قلب | تعزيز الإجهاد التأكسدي | 78,126 | ||
| دماغ | تعزيز الموت الخلوي المبرمج، الموت الحديدي، الالتهاب، تثبيط تكوين الأعصاب، تثبيط تكوين الأوعية الدموية، تدمير الحاجز الدموي الدماغي | 169-171، 173، 193، 209، 210، 228، 239، 242، 248، 343، 395 | |||
| Wnt/PCP | مفعل | قلب | تعزيز الالتهاب، تضخم الخلايا | 146 | |
| Wnt/
|
مفعل | دماغ | تعزيز الاستماتة (في المختبر) | 167 | |
| إعادة التروية | Wnt/
|
مُثبَط | قلب | تعزيز الالتهاب المسبب للموت الخلوي، تضخم الخلايا | 93-95,120,137 |
| دماغ | تعزيز موت الخلايا المبرمج، الالتهاب، الإجهاد التأكسدي، تثبيط تكوين الأعصاب، تثبيط تكوين الأوعية الدموية، تدمير الحاجز الدموي الدماغي | ١٠١٦٤١٦٥١٧٤١٧٥٢٠٧٢٣٢٢٤٤٢٤٦ | |||
| كبد | تعزيز موت الخلايا المبرمج، الإجهاد التأكسدي، الالتهاب، تثبيط تكاثر الخلايا | 267-271، 283، 285، 293، 296 | |||
| مفعل | دماغ | تثبيط موت الخلايا المبرمج (في المختبر) | 166 | ||
| كلى | تثبيط موت الخلايا المبرمج، تعزيز موت الخلايا المبرمج (في المختبر)، الميتوفاجي، الالتهام الذاتي للخلايا، شيخوخة الخلايا وتليف الكلى | ٢٥،٢٥٢،٢٥٣،٢٥٨-٢٦٣ | |||
| Wnt/PCP | مفعل | قلب | تعزيز موت الخلايا المبرمج، الالتهاب، تكوين الأوعية الدموية | ٥٠,١٠٢ | |
| دماغ | تعزيز موت الخلايا المبرمج، الالتهاب | ٢٩,٢٠٨ | |||
| Wnt/
|
مفعل | قلب | تعزيز موت الخلايا المبرمج | ١٠٣ | |
| كلى | تعزيز تليف الكلى | 264 | |||
| كبد | تعزيز الاستماتة | ١٠٣,٢٧٢,٢٧٣ |
فهم شامل لدور Wnt/
تفعيل مسار إشارة PI3K/Akt وتخفيف إصابة الدماغ الناتجة عن نقص التروية. بالإضافة إلى ذلك، تم العثور على أن هذه العلاجات التقليدية الصينية تعدل أيضًا نشاط مسار Wnt/
شكر وتقدير
مساهمات المؤلفين
تمويل
معلومات إضافية
REFERENCES
- Frangogiannis, N. G. Pathophysiology of myocardial infarction. Compr. Physiol. 5, 1841-1875 (2015).
- Zhao, Y., Zhang, X., Chen, X. & Wei, Y. Neuronal injuries in cerebral infarction and ischemic stroke: from mechanisms to treatment (Review). Int. J. Mol. Med. 49, 15 (2022).
- Yapca, O. E., Borekci, B. & Suleyman, H. Ischemia-reperfusion damage. Eurasia. J. Med. 45, 126-127 (2013).
- Eltzschig, H. K. & Eckle, T. Ischemia and reperfusion-from mechanism to translation. Nat. Med. 17, 1391-1401 (2011).
- Wu, M. Y. et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell. Physiol. Biochem. 46, 1650-1667 (2018).
- Hosohata, K., Harnsirikarn, T. & Chokesuwattanaskul, S. Ferroptosis: a potential therapeutic target in acute kidney injury. Int. J. Mol. Sci. 23, 6583 (2022).
- Tan, H., Chen, L. & Ma, J. Penehyclidine hydrochloride post-conditioning reduces ischemia/reperfusion-induced cardiomyocyte apoptosis in rats. Exp. Ther. Med 14, 4272-4278 (2017).
- Liu, H. et al. Inhibition of Brd4 alleviates renal ischemia/reperfusion injuryinduced apoptosis and endoplasmic reticulum stress by blocking FoxO4mediated oxidative stress. Redox Biol. 24, 101195 (2019).
- Guo, Z. et al. NLRP3 is involved in ischemia/reperfusion injury. CNS Neurol. Disord. Drug Targets 15, 699-712 (2016).
- Ji, Y. B. et al. Lithium alleviates blood-brain barrier breakdown after cerebral ischemia and reperfusion by upregulating endothelial Wnt/
-catenin signaling in mice. Neuropharmacology 186, 108474 (2021). - Burke, R. M., Burgos Villar, K. N. & Small, E. M. Fibroblast contributions to ischemic cardiac remodeling. Cell Signal 77, 109824 (2021).
- Wu, X., Reboll, M. R., Korf-Klingebiel, M. & Wollert, K. C. Angiogenesis after acute myocardial infarction. Cardiovasc. Res. 117, 1257-1273 (2021).
- Smiley, D. et al. Increased fibrosis and progression to heart failure in MRL mice following ischemia/reperfusion injury. Cardiovasc. Pathol. 23, 327-334 (2014).
- Salminen, A., Liu, P. K. & Hsu, C. Y. Alteration of transcription factor binding activities in the ischemic rat brain. Biochem. Biophys. Res. Commun. 212, 939-944 (1995).
- Werling, L. L. et al. Increased activation of L-type voltage-dependent calcium channels is associated with glycine enhancement of N-methyl-D-aspartate-stimulated dopamine release in global cerebral ischemia/reperfusion. J. Neurochem. 63, 215-221 (1994).
- Lefer, A. M. Mechanisms of the protective effects of transforming growth factorbeta in reperfusion injury. Biochem. Pharm. 42, 1323-1327 (1991).
- Tacchini, L., Radice, L. & Bernelli-Zazzera, A. Differential activation of some transcription factors during rat liver ischemia, reperfusion, and heat shock. J. Cell Physiol. 180, 255-262 (1999).
- Vukicevic, S. et al. Osteogenic protein-1 (bone morphogenetic protein-7) reduces the severity of injury after ischemic acute renal failure in rat. J. Clin. Investig. 102, 202-214 (1998).
- Mockridge, J. W., Marber, M. S. & Heads, R. J. Activation of Akt during simulated ischemia/reperfusion in cardiac myocytes. Biochem. Biophys. Res. Commun. 270, 947-952 (2000).
- Sakakura, Y. et al. Recombinant human hepatocyte growth factor protects the liver against hepatic ischemia and reperfusion injury in rats. J. Surg. Res. 92, 261-266 (2000).
21. Arumugam, T. V. et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcomes in ischemic stroke. Nat. Med. 12, 621-623 (2006).
22. Terada, Y. et al. Expression and function of the developmental gene Wnt-4 during experimental acute renal failure in rats. J. Am. Soc. Nephrol. 14, 1223-1233 (2003).
23. Shao, D. et al. A functional interaction between Hippo-YAP signaling and FoxO1 mediates the oxidative stress response. Nat. Commun. 5, 3315 (2014).
24. Pulskens, W. P. et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One 3, e3596 (2008).
25. Dong, Q. et al. Wnt/
26. Meyer, I. S. et al. Blockade of Wnt Secretion Attenuates Myocardial IschemiaReperfusion Injury by Modulating the Inflammatory Response. Int. J. Mol. Sci. 23, 12252 (2022).
27. Liu, J., Zheng, X., Zhang, C., Zhang, C. & Bu, P. Lcz696 alleviates myocardial fibrosis after myocardial infarction through the sFRP-1/Wnt/
28. Fuping, Z. et al. Tao-Hong-Si-Wu decoction reduces ischemia reperfusion rat myoblast cells calcium overloading and inflammation through the Wnt/IP3R/ CAMKII pathway. J. Cell. Biochem. 120, 13095-13106 (2019).
29. Wei, X. et al. Targeting the Dvl-1/
30. Gao, C. & Chen, Y. G. Dishevelled: the hub of Wnt signaling. Cell. Signal 22, 717-727 (2010).
31. Ben-Ghedalia-Peled, N. & Vago, R. Wnt Signaling in the Development of Bone Metastasis. Cells. 11, 3934 (2022).
32. Carmon, K. S., Gong, X., Lin, Q., Thomas, A. & Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl Acad. Sci. USA 108, 11452-11457 (2011).
33. de Lau, W. et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293-297 (2011).
34. Kasai, M., Satoh, K. & Akiyama, T. Wnt signaling regulates the sequential onset of neurogenesis and gliogenesis via induction of BMPs. Genes Cells 10, 777-783 (2005).
35. Trifan, G., Biller, J. & Testai, F. D. Mechanical thrombectomy vs bridging therapy for anterior circulation large vessel occlusion stroke: systematic review and meta-analysis. Neurology 98, e1361-e1373 (2022).
36. Kalogeris, T., Baines, C. P., Krenz, M. & Korthuis, R. J. Ischemia/reperfusion. Compr. Physiol. 7, 113-170 (2016).
37. Peng, T. I. & Jou, M. J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201, 183-188 (2010).
38. Salvadori, M., Rosso, G. & Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: pathogenesis and treatment. World J. Transpl. 5, 52-67 (2015).
39. Malis, C. D. & Bonventre, J. V. Mechanism of calcium potentiation of oxygen free radical injury to renal mitochondria. a model for post-ischemic and toxic mitochondrial damage. J. Biol. Chem. 261, 14201-14208 (1986).
40. Nieuwenhuijs-Moeke, G. J. et al. Ischemia and reperfusion injury in kidney transplantation: relevant mechanisms in injury and repair. J Clin Med. 9, 253 (2020).
41. Gujral, J. S., Bucci, T. J., Farhood, A. & Jaeschke, H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology 33, 397-405 (2001).
42. Glinka, A. et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/
43. de Lau, W., Peng, W. C., Gros, P. & Clevers, H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 28, 305-316 (2014).
44. Molenaar, M. et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 86, 391-399 (1996).
45. Pell, V. R. et al. Ischemic preconditioning protects against cardiac ischemia reperfusion injury without affecting succinate accumulation or oxidation. J. Mol. Cell Cardiol. 123, 88-91 (2018).
46. Lee, S. M., Hutchinson, M., Staikopoulos, V. & Saint, D. A. Amitriptyline pharmacologically preconditions rat hearts against cardiac ischemic-reperfusion injury. Int. J. Cardiol. 190, 353-359 (2015).
47. Li, Y., Cai, M., Xu, Y., Swartz, H. M. & He, G. Late phase ischemic preconditioning preserves mitochondrial oxygen metabolism and attenuates post-ischemic myocardial tissue hyper oxygenation. Life Sci. 88, 57-64 (2011).
48. Sárközy, M. et al. Ischemic preconditioning protects the heart against ischemiareperfusion injury in chronic kidney disease in both males and females. Biol. Sex. Differ. 12, 49 (2021).
49. Nusse, R. & Clevers, H. Wnt/
50. Wang, J. et al. WNT11-conditioned medium promotes angiogenesis through the activation of non-canonical WNT-PKC-JNK signaling pathway. Genes 11, 1277 (2020).
51. Gajos-Michniewicz, A. & Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 21, 4852 (2020).
52. Rim, E. Y., Clevers, H. & Nusse, R. The Wnt pathway: from signaling mechanisms to synthetic modulators. Annu. Rev. Biochem 91, 571-598 (2022).
53. Wang, H. et al. The Wnt signaling pathway in diabetic nephropathy. Front. Cell. Dev. Biol. 9, 701547 (2021).
54. Malik, S. A., Modarage, K. & Goggolidou, P. The Role of Wnt Signalling in Chronic Kidney Disease (CKD). Genes 11, 496 (2020).
55. Wang, H. Y., Liu, T. & Malbon, C. C. Structure-function analysis of Frizzleds. Cell. Signal 18, 934-941 (2006).
56. Joiner, D. M., Ke, J., Zhong, Z., Xu, H. E. & Williams, B. O. LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. 24, 31-39 (2013).
57. Menck, K., Heinrichs, S., Baden, C. & Bleckmann, A. The WNT/ROR Pathway in Cancer: From Signaling to Therapeutic Intervention. Cells. 10, 142 (2021).
58. Jung, Y. S. & Park, J. I. Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond
59. Chae, W. J. & Bothwell, A. L. M. Canonical and non-canonical Wnt signaling in immune cells. Trends Immunol. 39, 830-847 (2018).
60. Tran, F. H. & Zheng, J. J. Modulating the wnt signaling pathway with small molecules. Protein Sci. 26, 650-661 (2017).
61. Akoumianakis, I., Polkinghorne, M. & Antoniades, C. Non-canonical WNT signaling in cardiovascular disease: mechanisms and therapeutic implications. Nat. Rev. Cardiol. 19, 783-797 (2022).
62. Jin, Z. et al. Neuroprotective effects of irisin against cerebral ischemia/ reperfusion injury via Notch signaling pathway. Biomed. Pharmacother. 120, 109452 (2019).
63. Gordon, M. D. & Nusse, R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281, 22429-22433 (2006).
64. Veeman, M. T., Axelrod, J. D. & Moon, R. T. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev. Cell. 5, 367-377 (2003).
65. Kikuchi, A. & Yamamoto, H. Tumor formation due to abnormalities in the beta-catenin-independent pathway of Wnt signaling. Cancer Sci. 99, 202-208 (2008).
66. Shi, D. L. Decoding dishevelled-mediated Wnt signaling in vertebrate early development. Front. Cell Dev. Biol. 8, 588370 (2020).
67. Lerner, U. H. & Ohlsson, C. The WNT system: background and its role in bone. J. Intern. Med. 277, 630-649 (2015).
68. VanderVorst, K. et al. Wnt/PCP signaling contribution to carcinoma collective cell migration and metastasis. Cancer Res. 79, 1719-1729 (2019).
69. Frenquelli, M. & Tonon, G. WNT signaling in hematological malignancies. Front. Oncol. 10, 615190 (2020).
70. Cho, S. J. et al. Wip1 directly dephosphorylates NLK and increases Wnt activity during germ cell development. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1013-1022 (2017).
71. Xiao, Q., Chen, Z., Jin, X., Mao, R. & Chen, Z. The many postures of noncanonical Wnt signaling in development and diseases. Biomed. Pharmacother. 93, 359-369 (2017).
72. Ma, L. & Wang, H. Y. Mitogen-activated protein kinase p38 regulates the Wnt/ cyclic GMP/Ca2+ non-canonical pathway. J. Biol. Chem. 282, 28980-28990 (2007).
73. Hausenloy, D. J. & Yellon, D. M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J. Clin. Investig. 123, 92-100 (2013).
74. Vaage, J. & Valen, G. Pathophysiology and mediators of ischemia-reperfusion injury with special reference to cardiac surgery. A review. Scand. J. Thorac. Cardiovasc Surg. Suppl. 41, 1-18 (1993).
75. Tanaka, M. et al. Cardiomyocyte-specific Bcl-2 overexpression attenuates ischemia-reperfusion injury, immune response during acute rejection, and graft coronary artery disease. Blood 104, 3789-3796 (2004).
76. Kleinbongard, P., Heusch, G. & Schulz, R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharm. Ther. 127, 295-314 (2010).
77. Frohlich, G. M., Meier, P., White, S. K., Yellon, D. M. & Hausenloy, D. J. Myocardial reperfusion injury: looking beyond primary PCl . Eur. Heart J. 34, 1714-1722 (2013).
78. Shen, J. et al. Wnt 3a protects myocardial injury in elderly acute myocardial infarction by inhibiting serum cystatin C/ROS-induced mitochondrial damage. Front. Physiol. 13, 950960 (2022).
79. Piper, H. M., García-Dorado, D. & Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 38, 291-300 (1998).
80. Yellon, D. M. & Hausenloy, D. J. Myocardial reperfusion injury. N. Engl. J. Med 357, 1121-1135 (2007).
81. Logue, S. E., Gustafsson, A. B., Samali, A. & Gottlieb, R. A. Ischemia/reperfusion injury at the intersection with cell death. J. Mol. Cell Cardiol. 38, 21-33 (2005).
82. Gottlieb, R. A. Cell death pathways in acute ischemia/reperfusion injury. J. Cardiovasc. Pharm. Ther. 16, 233-238 (2011).
83. Chen, Y. et al. Ferroptosis: a novel therapeutic target for ischemia-reperfusion injury. Front. Cell Dev. Biol. 9, 688605 (2021).
84. Hamacher-Brady, A., Brady, N. R. & Gottlieb, R. A. The interplay between prodeath and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther. 20, 445-462 (2006).
85. Deb, A. Cell-cell interaction in the heart via Wnt/beta-catenin pathway after cardiac injury. Cardiovasc. Res. 102, 214-223 (2014).
86. Lorenzon, A. et al. Wnt/beta-catenin pathway in arrhythmogenic cardiomyopathy. Oncotarget 8, 60640-60655 (2017).
87. Bergmann, M. W. WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ. Res. 107, 1198-1208 (2010).
88. Oerlemans, M. I. et al. Active Wnt signaling in response to cardiac injury. Basic Res. Cardiol. 105, 631-641 (2010).
89. Haybar, H., Khodadi, E. & Shahrabi, S. Wnt/
90. Litvinukova, M. et al. Cells of the adult human heart. Nature 588, 466-472 (2020).
91. Bastakoty, D. et al. Temporary, Systemic inhibition of the WNT/beta-catenin pathway promotes regenerative cardiac repair following myocardial infarct. Cell Stem Cells Regen. Med. 2, 16966 (2016).
92. Haybar, H., Khodadi, E. & Shahrabi, S. Wnt/beta-catenin in ischemic myocardium: interactions and signaling pathways as a therapeutic target. Heart Fail Rev. 24, 411-419 (2019).
93. Yang, M., Kong, D. Y. & Chen, J. C. Inhibition of miR-148b ameliorates myocardial ischemia/reperfusion injury via regulation of Wnt/beta-catenin signaling pathway. J. Cell Physiol. 234, 17757-17766 (2019).
94. Zhang, G. et al. LncRNA AZIN1-AS1 ameliorates myocardial ischemia-reperfusion injury by targeting miR-6838-5p/WNT3A axis to activate Wnt-beta/catenin signaling pathway. Vitr. Cell Dev. Biol. Anim. 58, 54-68 (2022).
95. Cui, X. et al. Exosomes from adipose-derived mesenchymal stem cells protect the myocardium against ischemia/reperfusion injury through Wnt/beta-catenin signaling pathway. J. Cardiovasc. Pharm. 70, 225-231 (2017).
96. Finch, P. W. et al. Purification and molecular cloning of a secreted, Frizzledrelated antagonist of Wnt action. Proc. Natl Acad. Sci. USA 94, 6770-6775 (1997).
97. Bovolenta, P., Esteve, P., Ruiz, J. M., Cisneros, E. & Lopez-Rios, J. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J. Cell Sci. 121, 737-746 (2008).
98. Ouchi, N. et al. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 329, 454-457 (2010).
99. Li, Y. et al. Sfrp5 coordinates foregut specification and morphogenesis by antagonizing both canonical and noncanonical Wnt11 signaling. Genes Dev. 22, 3050-3063 (2008).
100. Kikuchi, R. et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med 20, 1464-1471 (2014).
101. Fuster, J. J. et al. Noncanonical Wnt signaling promotes obesity-induced adipose tissue inflammation and metabolic dysfunction independent of adipose tissue expansion. Diabetes 64, 1235-1248 (2015).
102. Nakamura, K. et al. Secreted Frizzled-related protein 5 diminishes cardiac inflammation and protects the heart from ischemia/reperfusion injury. J. Biol. Chem. 291, 2566-2575 (2016).
103. Zhou, S. S., He, F., Chen, A. H., Hao, P. Y. & Song, X. D. Suppression of rat Frizzled2 attenuates hypoxia/reoxygenation-induced
104. Zhang, L. et al. Inhibition of Rac1 reduces store overload-induced calcium release and protects against ventricular arrhythmia. J. Cell Mol. Med. 20, 1513-1522 (2016).
105. Belevych, A. E. et al. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res 84, 387-395 (2009).
106. Fauconnier, J. et al. Ryanodine receptor leak mediated by caspase- 8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc. Natl Acad. Sci. USA 108, 13258-13263 (2011).
107. He, J. et al. Huoxin pill prevents excessive inflammation and cardiac dysfunction following myocardial infarction by inhibiting adverse Wnt/betacatenin signaling activation. Phytomedicine 104, 154293 (2022).
108. Hu, Y. et al. Class A scavenger receptor attenuates myocardial infarction-induced cardiomyocyte necrosis through suppressing M1 macrophage subset polarization. Basic Res. Cardiol. 106, 1311-1328 (2011).
109. Cutolo, M., Campitiello, R., Gotelli, E. & Soldano, S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front. Immunol. 13, 867260 (2022).
110. Zhang, R. Y. K., Cochran, B. J., Thomas, S. R. & Rye, K. A. Impact of reperfusion on temporal immune cell dynamics after myocardial infarction. J. Am. Heart Assoc. 12, e027600 (2023).
111. Prabhu, S. D. & Frangogiannis, N. G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 119, 91-112 (2016).
112. Nahrendorf, M. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037-3047 (2007).
113. Cheng, B., Chen, H. C., Chou, I. W., Tang, T. W. & Hsieh, P. C. Harnessing the early post-injury inflammatory responses for cardiac regeneration. J. Biomed. Sci. 24, 7 (2017).
114. Huang, C. K. et al. Lgr4 governs a pro-inflammatory program in macrophages to antagonize post-infarction cardiac repair. Circ. Res. 127, 953-973 (2020).
115. Zhao, G. et al. CXCR6 deficiency ameliorated myocardial ischemia/reperfusion injury by inhibiting infiltration of monocytes and IFN-
116. Gombozhapova, A. et al. Macrophage activation and polarization in postinfarction cardiac remodeling. J. Biomed. Sci. 24, 13 (2017).
117. Yuan, C. et al. Modulation of Wnt/
118. Palevski, D. et al. Loss of macrophage Wnt secretion improves remodeling and function after myocardial infarction in mice. J. Am. Heart Assoc. 6, e004387 (2017).
119. Frangogiannis, N. G. The inflammatory response in myocardial injury, repair, and remodeling. Nat. Rev. Cardiol. 11, 255-265 (2014).
120. Sun, S., Wu, Y., Maimaitijiang, A., Huang, Q. & Chen, Q. Ferroptotic cardiomyocyte-derived exosomes promote cardiac macrophage M1 polarization during myocardial infarction. PeerJ. 10, e13717 (2022).
121. Huang, L., Xiang, M., Ye, P., Zhou, W. & Chen, M. Beta-catenin promotes macrophage-mediated acute inflammatory response after myocardial infarction. Immunol. Cell. Biol. 96, 100-113 (2018).
122. Aisagbonhi, O. et al. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model Mech. 4, 469-483 (2011).
123. Aleshin, A. et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol. 294, H1823-H1832 (2008).
124. Bucciarelli, L. G. et al. Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury. Circulation 113, 1226-1234 (2006).
125. Park, H. et al. RAGE siRNA-mediated gene silencing provides cardioprotection against ventricular arrhythmias in acute ischemia and reperfusion. J. Control Release 217, 315-326 (2015).
126. Rauner, M. et al. WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Min. Res. 27, 575-585 (2012).
127. Meyer, I. S. et al. The cardiac microenvironment uses non-canonical WNT signaling to activate monocytes after myocardial infarction. EMBO Mol. Med. 9, 1279-1293 (2017).
128. Moon, J. et al. Blockade to pathological remodeling of infarcted heart tissue using a porcupine antagonist. Proc. Natl Acad. Sci. USA 114, 1649-1654 (2017).
129. Pereira, C., Schaer, D. J., Bachli, E. B., Kurrer, M. O. & Schoedon, G. Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler. Thromb. Vasc. Biol. 28, 504-510 (2008).
130. Port, F. et al. Wingless secretion promotes and requires retromer-dependent cycling of Wntless. Nat. Cell Biol. 10, 178-185 (2008).
131. Guo, X. et al. Induced pluripotent stem cell-conditional medium inhibits H9C2 cardiomyocytes apoptosis via autophagy flux and Wnt/beta-catenin pathway. J. Cell. Mol. Med 23, 4358-4374 (2019).
132. Je, J. Y., Qian, Z. J., Byun, H. G. & Kim, S. K. Purification and characterization of an antioxidant peptide obtained from tuna backbone protein by enzymatic hydrolysis. Process Biochem. 42, 840-846 (2007).
133. Zhang, L. et al. The restoration of Wnt/
134. Blankesteijn, W. M., van Gijn, M. E., Essers-Janssen, Y. P., Daemen, M. J. & Smits, J. F. Beta-catenin, an inducer of uncontrolled cell proliferation and migration in malignancies, is localized in the cytoplasm of vascular endothelium during neovascularization after myocardial infarction. Am. J. Pathol. 157, 877-883 (2000).
32
135. Barandon, L. et al. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation 108, 2282-2289 (2003).
136. Baruah, J. et al. The allosteric glycogen synthase kinase-3 inhibitor NP12 limits myocardial remodeling and promotes angiogenesis in an acute myocardial infarction model. J. Biol. Chem. 292, 20785-20798 (2017).
137. Liu, B. et al. Loss of endothelial glucocorticoid receptor promotes angiogenesis via upregulation of Wnt/beta-catenin pathway. Angiogenesis 24, 631-645 (2021).
138. MacLellan, W. R. & Schneider, M. D. Genetic dissection of cardiac growth control pathways. Annu. Rev. Physiol. 62, 289-319 (2000).
139. Nakamura, M. & Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387-407 (2018).
140. Shimizu, I. & Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 97, 245-262 (2016).
141. Hagenmueller, M. et al. Dapper-1 is essential for Wnt5a induced cardiomyocyte hypertrophy by regulating the Wnt/PCP pathway. FEBS Lett. 588, 2230-2237 (2014).
142. Khan, K., Makhoul, G., Yu, B., Schwertani, A. & Cecere, R. The cytoprotective impact of yes-associated protein 1 after ischemia-reperfusion injury in AC16 human cardiomyocytes. Exp. Biol. Med. 244, 802-812 (2019).
143. Zhao, X. et al. Aldehyde dehydrogenase-2 protects against myocardial infarction-related cardiac fibrosis through modulation of the Wnt/beta-catenin signaling pathway. Ther. Clin. Risk Manag. 11, 1371-1381 (2015).
144. Qian, L. et al. Downregulation of S100A4 Alleviates Cardiac Fibrosis via Wnt/beta -Catenin Pathway in Mice. Cell Physiol. Biochem 46, 2551-2560 (2018).
145. Cui, S. et al. miR-145 attenuates cardiac fibrosis through the AKT/GSK-3beta/ beta-catenin signaling pathway by directly targeting SOX9 in fibroblasts. J. Cell Biochem. 122, 209-221 (2021).
146. Matsushima, K. et al. Secreted frizzled related protein 4 reduces fibrosis scar size and ameliorates cardiac function after ischemic injury. Tissue Eng. Part A 16, 3329-3341 (2010).
147. Duan, J. et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 31, 429-442 (2012).
148. Zhang, Y. et al. PRELP promotes myocardial fibrosis and ventricular remodeling after acute myocardial infarction by the wnt/
149. Jean LeBlanc, N. et al. Canonical Wnt pathway maintains blood-brain barrier integrity upon ischemic stroke and its activation ameliorates tissue plasminogen activator therapy. Mol. Neurobiol. 56, 6521-6538 (2019).
150. Abuelazm, M. et al. The efficacy and safety of tenecteplase versus alteplase for acute ischemic stroke: an updated systematic review, pairwise, and network meta-analysis of randomized controlled trials. J. Thromb. Thrombolysis 55, 322-338 (2023).
151. Berge, E. et al. European Stroke Organisation (ESO) guidelines on intravenous thrombolysis for acute ischaemic stroke. Eur Stroke J. 6, I-Ixii (2021).
152. Emberson, J. et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomized trials. Lancet 384, 1929-1935 (2014).
153. Xiong, Y., Wakhloo, A. K. & Fisher, M. Advances in Acute Ischemic Stroke Therapy. Circ. Res. 130, 1230-1251 (2022).
154. Katsanos, A. H. et al. Intravenous thrombolysis prior to mechanical thrombectomy in large vessel occlusions. Ann. Neurol. 86, 395-406 (2019).
155. Fischer, U. et al. Primary thrombectomy in tPA (Tissue-Type Plasminogen Activator) eligible stroke patients with proximal intracranial occlusions. Stroke 49, 265-269 (2018).
156. Rai, A. T. et al. Intravenous thrombolysis before endovascular therapy for large vessel strokes can lead to significantly higher hospital costs without improving outcomes. J. Neurointerv. Surg. 10, 17-21 (2018).
157. Goyal, N. et al. Impact of pretreatment with intravenous thrombolysis on reperfusion status in acute strokes treated with mechanical thrombectomy. J. Neurointerv Surg. 11, 1073-1079 (2019).
158. Rossi, R. et al. Does prior administration of rtPA influence acute ischemic stroke clot composition? Findings from the analysis of clots retrieved with mechanical thrombectomy from the RESTORE registry. J. Neurol. 269, 1913-1920 (2022).
159. Muroyama, Y., Kondoh, H. & Takada, S. Wnt proteins promote neuronal differentiation in neural stem cell culture. Biochem. Biophys. Res. Commun. 313, 915-921 (2004).
160. Maretto, S. et al. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl Acad. Sci. USA 100, 3299-3304 (2003).
161. McKenzie, M. G. et al. Non-canonical Wnt signaling through Ryk regulates the generation of somatostatin- and parvalbumin-expressing cortical interneurons. Neuron 103, 853-864.e854 (2019).
162. Lewis, J. L. et al. Reiterated Wnt signaling during zebrafish neural crest development. Development 131, 1299-1308 (2004).
163. Hutchins, B. I., Li, L. & Kalil, K. Wnt-induced calcium signaling mediates axon growth and guidance in the developing corpus callosum. Sci. Signal. 5, pt1 (2012).
164. Rosso, S. B., Sussman, D., Wynshaw-Boris, A. & Salinas, P. C. Wnt signaling through Dishevelled, Rac, and JNK regulates dendritic development. Nat. Neurosci. 8, 34-42 (2005).
165. Liebner, S. et al. Wnt/beta-catenin signaling controls development of the bloodbrain barrier. J. Cell Biol. 183, 409-417 (2008).
166. Benz, F. et al. Low wnt/
167. Shi, Z. Y. et al. Protective effect of autophagy in neural ischemia and hypoxia: negative regulation of the Wnt/
168. Ji, Y. B., Wang, T. X., Gao, Q., Huang, X. W. & Chang, J. Normalization of noncanonical Wnt signalings does not compromise blood-brain barrier protection conferred by upregulating endothelial Wnt/
169. Zhao, H. et al. Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt
170. Zhang, G. et al. Wnt/
171. Li, T. et al. DIXDC1 prevents oxygen-glucose deprivation/reoxygenation-induced injury in hippocampal neurons in vitro by promoting Wnt/
172. Niu, L. J., Xu, R. X., Zhang, P., Du, M. X. & Jiang, X. D. Suppression of Frizzled-2mediated Wnt/
173. Kunz, A., Dirnagl, U. & Mergenthaler, P. Acute pathophysiological processes after ischaemic and traumatic brain injury. Best. Pr. Res. Clin. Anaesthesiol. 24, 495-509 (2010).
174. Seifert-Held, T. et al. Circulating Dickkopf-1 in acute ischemic stroke and clinically stable cerebrovascular disease. Atherosclerosis 218, 233-237 (2011).
175. Cappuccio, I. et al. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J. Neurosci. 25, 2647-2657 (2005).
176. Mastroiacovo, F. et al. Induction of the Wnt antagonist, Dickkopf-1, contributes to the development of neuronal death in models of brain focal ischemia. J. Cereb. Blood Flow. Metab. 29, 264-276 (2009).
177. Scali, C. et al. Inhibition of Wnt signaling, modulation of Tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol. Dis. 24, 254-265 (2006).
178. Che, Q. Q., Huang, T., Zhang, Y. D. & Qian, X. J. Effect of miR-124 on neuronal apoptosis in rats with cerebral infarction through Wnt/
179. Zhou, Z., Ren, X., Zheng, L., Li, A. & Zhou, W. LncRNA NEAT1 stabilized Wnt3a via U2AF2 and activated Wnt/
180. Xu, D. et al. XQ-1H alleviates cerebral ischemia in mice through inhibition of apoptosis and promotion of neurogenesis in a Wnt/
181. Zhao, H., Pan, W., Chen, L., Luo, Y. & Xu, R. Nur77 promotes cerebral ischemiareperfusion injury via activating INF2-mediated mitochondrial fragmentation. J. Mol. Histol. 49, 599-613 (2018).
182. Chong, Z. Z. & Maiese, K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol. Histopathol. 19, 495-504 (2004).
183. Guo, C. & Whitmarsh, A. J. The beta-arrestin-2 scaffold protein promotes c-Jun N -terminal kinase- 3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 283, 15903-15911 (2008).
184. Kuan, C. Y. et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl Acad. Sci. USA 100, 15184-15189 (2003).
185. Zhang, Q. G., Wang, R., Khan, M., Mahesh, V. & Brann, D. W. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J. Neurosci. 28, 8430-8441 (2008).
186. Cheng, Y. L. et al. Evidence that collaboration between HIF-1a and Notch-1 promotes neuronal cell death in ischemic stroke. Neurobiol. Dis. 62, 286-295 (2014).
187. Burchell, S. R., Dixon, B. J., Tang, J. & Zhang, J. H. Isoflurane provides neuroprotection in neonatal hypoxic ischemic brain injury. J. Investig. Med. 61, 1078-1083 (2013).
188. Lan, X. B. et al. Neuroprotective effects of oxymatrine on hypoxic-ischemic brain damage in neonatal rats by activating the Wnt/
189. Yan, H. F., Tuo, Q. Z., Yin, Q. Z. & Lei, P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool. Res. 41, 220-230 (2020).
190. Li, D. & Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target Ther. 5, 108 (2020).
191. Li, L., Li, Y. W., Zhao, J. Y., Liu, Y. Z. & Holscher, C. Quantitative analysis of iron concentration and expression of ferroportin 1 in the cortex and hippocampus of rats induced by cerebral ischemia. J. Clin. Neurosci. 16, 1466-1472 (2009).
192. Won, S. M. et al. Iron mediates endothelial cell damage and blood-brain barrier opening in the hippocampus after transient forebrain ischemia in rats. Exp. Mol. Med. 43, 121-128 (2011).
193. Hällgren, R., Terent, A., Wide, L., Bergström, K. & Birgegård, G. Cerebrospinal fluid ferritin in patients with cerebral infarction or bleeding. Acta Neurol. Scand. 61, 384-392 (1980).
194. Shi, Y. et al. Selenium alleviates cerebral ischemia/reperfusion injury by regulating oxidative stress, mitochondrial fusion and ferroptosis. Neurochem. Res. 47, 2992-3002 (2022).
195. Groenendaal, F., Shadid, M., McGowan, J. E., Mishra, O. P. & van Bel, F. Effects of deferoxamine, a chelator of free iron, on NA(+), K(+)-ATPase activity of cortical brain cell membrane during early reperfusion after hypoxia-ischemia in newborn lambs. Pediatr. Res. 48, 560-564 (2000).
196. Shadid, M. et al. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neurosci. Lett. 248, 5-8 (1998).
197. Zhao, Y. et al. Nano-liposomes of lycopene reduces ischemic brain damage in rodents by regulating iron metabolism. Free Radic. Biol. Med. 124, 1-11 (2018).
198. Yin, M. et al. circAFF1 enhances intracerebral hemorrhage induced neuronal ferroptosis by targeting miR-140-5p to regulate GSK-3
199. Wu, X. et al. Regulation of GSK3
200. Armagan, G. et al. Regulation of the Nrf2 Pathway by Glycogen Synthase Kinase
201. Wang, L., Ouyang, S., Li, B., Wu, H. & Wang, F. GSK-3
202. Candelario-Jalil, E., Dijkhuizen, R. M. & Magnus, T. Neuroinflammation, stroke, blood-brain barrier dysfunction, and imaging modalities. Stroke 53, 1473-1486 (2022).
203. Lengfeld, J. E. et al. Endothelial Wnt/
204. Liu, Z. & Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 144, 103-120 (2016).
205. Ma, Y., Wang, J., Wang, Y. & Yang, G. Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 157, 247-272 (2017).
206. Kanazawa, M., Ninomiya, I., Hatakeyama, M., Takahashi, T. & Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 18, 2135 (2017).
207. Wang, Y. et al. Antioxidants & Redox Signaling. Antioxid. Redox Signal. 32, 213-214 (2020).
208. Zolezzi, J. M. & Inestrosa, N. C. Wnt/TLR Dialog in Neuroinflammation, Relevance in Alzheimer’s Disease. Front. Immunol. 8, 187 (2017).
209. Yeh, H., Woodbury, M. E., Ingraham Dixie, K. L., Ikezu, T. & Ikezu, S. Microglial WNT5A supports dendritic spines maturation and neuronal firing. Brain Behav. Immun. 107, 403-413 (2023).
210. Mecha, M. et al. Involvement of Wnt7a in the role of M2c microglia in neural stem cell oligodendrogenesis. J. Neuroinflamm 17, 88 (2020).
211. Xie, K., Cai, Y., Yang, P., Du, F. & Wu, K. Upregulating microRNA-874-3p inhibits CXCL12 expression to promote angiogenesis and suppress inflammatory response in ischemic stroke. Am. J. Physiol. Cell Physiol. 319, C579-c588 (2020).
212. Zhao, J., Li, L. & Fang, G. Salvianolic acid A attenuates cerebral ischemia/ reperfusion injury induced rat brain damage, inflammation, and apoptosis by regulating miR-499a/DDK1. Am. J. Transl. Res. 12, 3288-3301 (2020).
213. Zhou, J., Wu, N. & Lin, L. Curcumin suppresses apoptosis and inflammation in hypoxia/reperfusion-exposed neurons via Wnt Signaling pathway. Med Sci. Monit. 26, e920445 (2020).
214. Song, D. et al. Wnt canonical pathway activator TWS119 drives microglial antiinflammatory activation and facilitates neurological recovery following experimental stroke. J. Neuroinflamm 16, 256 (2019).
215. Zhao, B., Wang, P., Yu, J. & Zhang, Y. MicroRNA-376b-5p targets SOX7 to alleviate ischemic brain injury in a mouse model through activating Wnt/
216. Kalogeris, T., Bao, Y. & Korthuis, R. J. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2, 702-714 (2014).
217. Tang, Y., Shen, J., Zhang, F., Yang, F. Y. & Liu, M. Human serum albumin attenuates global cerebral ischemia/reperfusion-induced brain injury in a Wnt/
218. Ten, V. S. & Starkov, A. Hypoxic-ischemic injury in the developing brain: the role of reactive oxygen species originating in mitochondria. Neurol. Res. Int. 2012, 542976 (2012).
219. Korobova, F., Ramabhadran, V. & Higgs, H. N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339, 464-467 (2013).
220. Alvarez-Buylla, A. & Garcia-Verdugo, J. M. Neurogenesis in adult subventricular zone. J. Neurosci. 22, 629-634 (2002).
221. Alvarez-Buylla, A. & Lim, D. A. For the long run: maintaining germinal niches in the adult brain. Neuron 41, 683-686 (2004).
222. Adachi, K. et al. Beta-catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells 25, 2827-2836 (2007).
223. Lie, D. C. et al. Wnt signaling regulates adult hippocampal neurogenesis. Nature 437, 1370-1375 (2005).
224. Jin, K. et al. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl Acad. Sci. USA 103, 13198-13202 (2006).
225. Martí-Fàbregas, J. et al. Proliferation in the human ipsilateral subventricular zone after ischemic stroke. Neurology 74, 357-365 (2010).
226. Chen, X. et al. Peroxynitrite enhances self-renewal, proliferation, and neuronal differentiation of neural stem/progenitor cells through activating HIF-1a and Wnt/
227. Tiwari, S. K. et al. Inhibitory effects of Bisphenol-A on neural stem cells proliferation and differentiation in the rat brain are dependent on Wnt/
228. Gan, Q. et al. Pax6 mediates ß-catenin signaling for self-renewal and neurogenesis by neocortical radial glial stem cells. Stem Cells 32, 45-58 (2014).
229. Kuwabara, T. et al. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 12, 1097-1105 (2009).
230. Gao, Z. et al. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat. Neurosci. 12, 1090-1092 (2009).
231. Joksimovic, M. & Awatramani, R. Wnt/
232. Yi, H., Hu, J., Qian, J. & Hackam, A. S. Expression of brain-derived neurotrophic factor is regulated by the Wnt signaling pathway. Neuroreport 23, 189-194 (2012).
233. Wei, Z. Z. et al. Neuroprotective and regenerative roles of intranasal Wnt-3a administration after focal ischemic stroke in mice. J. Cereb. Blood Flow. Metab. 38, 404-421 (2018).
234. Li, S. R. et al. Mallotus oblongifolius extracts ameliorate ischemic nerve damage by increasing endogenous neural stem cell proliferation through the Wnt/
235. Liu, Q. et al. Ellagic acid improves endogenous neural stem cells proliferation and neurorestoration through Wnt/
236. Yang, X. et al. Curcumin promotes neurogenesis of hippocampal dentate gyrus via Wnt/
237. You, D. & You, H. Repression of long non-coding RNA MEG3 restores nerve growth and alleviates neurological impairment after cerebral ischemiareperfusion injury in a rat model. Biomed. Pharmacother. 111, 1447-1457 (2019).
238. Stenman, J. M. et al. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247-1250 (2008).
239. Zhou, Y. & Nathans, J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev. Cell 31, 248-256 (2014).
240. Hu, Y., Zheng, Y., Wang, T., Jiao, L. & Luo, Y. VEGF, a Key Factor for Blood Brain Barrier Injury After Cerebral Ischemic Stroke. Aging Dis. 13, 647-654 (2022).
241. Green, D. R. Caspases and Their Substrates. Cold Spring Harb. Perspect. Biol. 14, a041012 (2022).
242. Tian, Y. et al. IL-4-polarized BV2 microglia cells promote angiogenesis by secreting exosomes. Adv. Clin. Exp. Med. 28, 421-430 (2019).
243. Krupinski, J., Kaluza, J., Kumar, P., Kumar, S. & Wang, J. M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 25, 1794-1798 (1994).
244. Wang, L. P. et al. Oligodendrocyte precursor cell transplantation promotes angiogenesis and remyelination via Wnt/
245. Jiang, X. et al. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 163-164, 144-171 (2018).
246. Ta, S. et al. Variants of WNT7A and GPR124 are associated with hemorrhagic transformation following intravenous thrombolysis in ischemic stroke. CNS Neurosci. Ther. 27, 71-81 (2021).
247. Chang, J. et al. Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat. Med. 23, 450-460 (2017).
248. Hussain, B. et al. Endothelial
249. Chen, X. Y. et al. Inhibition of the immunoproteasome LMP2 ameliorates ischemia/hypoxia-induced blood-brain barrier injury through the Wnt/
250. Langen, U. H., Ayloo, S. & Gu, C. Development and cell biology of the bloodbrain barrier. Annu. Rev. Cell Dev. Biol. 35, 591-613 (2019).
251. Jin, Z., Ke, J., Guo, P., Wang, Y. & Wu, H. Quercetin improves blood-brain barrier dysfunction in rats with cerebral ischemia reperfusion via Wnt signaling pathway. Am. J. Transl. Res. 11, 4683-4695 (2019).
252. Kintner, D. B. et al. Increased tolerance to oxygen and glucose deprivation in astrocytes from
253. Song, S. et al. Activation of endothelial Wnt/
254. Zhao, H., Alam, A., Soo, A. P., George, A. J. T. & Ma, D. Ischemia-reperfusion injury reduces long term renal graft survival: mechanism and beyond. EBioMedicine 28, 31-42 (2018).
255. Wang, W., Sai, W. L. & Yang, B. [The role of macrophage polarization and interaction with renal tubular epithelial cells in ischemia-reperfusion induced acute kidney injury]. Sheng Li Xue Bao 74, 28-38 (2022).
256. He, W. et al. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 20, 765-776 (2009).
257. Liu, D., Liu, Y., Zheng, X. & Liu, N. c-MYC-induced long noncoding RNA MEG3 aggravates kidney ischemia-reperfusion injury through activating mitophagy by upregulation of RTKN to trigger the Wnt/
258. Zhou, D. et al. Tubule-specific ablation of endogenous
259. Wang, Y. et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 28, 231-243 (2021).
260. Wei, X. et al. Identification of subtypes and a delayed graft function predictive signature based on ferroptosis in renal ischemia-reperfusion injury. Front. Cell Dev. Biol. 10, 800650 (2022).
261. Xu, Y. et al. circ-AKT3 aggravates renal ischaemia-reperfusion injury via regulating miR-144-5p /Wnt/
262. Sturmlechner, I., Durik, M., Sieben, C. J., Baker, D. J. & van Deursen, J. M. Cellular senescence in renal aging and disease. Nat. Rev. Nephrol. 13, 77-89 (2017).
263. Xiao, L. et al. Sustained Activation of Wnt/
264. Zhou, L. et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/
265. Zhou, D. et al. Matrix metalloproteinase-7 is an urinary biomarker and pathogenic mediator of kidney fibrosis. J. Am. Soc. Nephrol. 28, 598-611 (2017).
266. He, W. et al. Plasminogen activator inhibitor-1 is a transcriptional target of the canonical pathway of Wnt/beta-catenin signaling. J. Biol. Chem. 285, 24665-24675 (2010).
267. Simon-Tillaux, N. & Hertig, A. Snail and kidney fibrosis. Nephrol. Dial. Transpl. 32, 224-233 (2017).
268. Luo, C. et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J. Am. Soc. Nephrol. 29, 1238-1256 (2018).
269. von Toerne, C. et al. Wnt pathway regulation in chronic renal allograft damage. Am. J. Transpl. 9, 2223-2239 (2009).
270. Sun, Q. et al. Allogeneic mesenchymal stem cells as induction therapy are safe and feasible in renal allografts: pilot results of a multicenter randomized controlled trial. J. Transl. Med. 16, 52 (2018).
271. Peralta, C., Jiménez-Castro, M. B. & Gracia-Sancho, J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J. Hepatol. 59, 1094-1106 (2013).
272. Russell, J. O. & Monga, S. P. Wnt/
273. Dar, W. A., Sullivan, E., Bynon, J. S., Eltzschig, H. & Ju, C. Ischaemia reperfusion injury in liver transplantation: cellular and molecular mechanisms. Liver Int. 39, 788-801 (2019).
274. Lehwald, N. et al. Wnt-
275. Liu, X. et al. Signaling through hepatocyte vasopressin receptor 1 protects mouse liver from ischemia-reperfusion injury. Oncotarget 7, 69276-69290 (2016).
276. Xie, K., Liu, L., Chen, J. & Liu, F. Exosomes derived from human umbilical cord blood mesenchymal stem cells improve hepatic ischemia reperfusion injury via delivering miR-1246. Cell Cycle 18, 3491-3501 (2019).
277. Sakon, M., Ariyoshi, H., Umeshita, K. & Monden, M. Ischemia-reperfusion injury of the liver with special reference to calcium-dependent mechanisms. Surg. Today 32, 1-12 (2002).
278. Hu, X. et al. Inhibition of Frizzled-2 by small interfering RNA protects rat hepatic BRL-3A cells against cytotoxicity and apoptosis induced by Hypoxia/Reoxygenation. Gastroenterol. Hepatol. 43, 107-116 (2020).
279. Yim, S. Y. et al. Risk factors for developing hyponatremia during terlipressin treatment: a retrospective analyses in variceal bleeding. J. Clin. Gastroenterol. 49, 607-612 (2015).
280. Koshimizu, T. A. et al. Vasopressin V1a and V1b receptors: from molecules to physiological systems. Physiol. Rev. 92, 1813-1864 (2012).
281. Kohler, A., Perrodin, S., De Gottardi, A., Candinas, D. & Beldi, G. Effectiveness of terlipressin for prevention of complications after major liver resection-A randomized placebo-controlled trial. HPB 22, 884-891 (2020).
282. Hong, S. H. et al. Perioperative assessment of terlipressin infusion during living donor liver transplantation. J. Int. Med. Res. 40, 225-236 (2012).
283. Reis, D. J. & Regunathan, S. Is agmatine a novel neurotransmitter in brain? Trends Pharm. Sci. 21, 187-193 (2000).
284. Kim, D. J. et al. Protective effect of agmatine on a reperfusion model after transient cerebral ischemia: Temporal evolution on perfusion MR imaging and histopathologic findings. AJNR Am. J. Neuroradiol. 27, 780-785 (2006).
285. Sugiura, T. et al. Protective effect of agmatine on ischemia/reperfusion-induced renal injury in rats. J. Cardiovasc. Pharm. 51, 223-230 (2008).
286. Greenberg, S. et al. The effect of agmatine administration on ischemicreperfused isolated rat heart. J. Cardiovasc. Pharm. Ther. 6, 37-45 (2001).
287. Han, Z. et al. Agmatine attenuates liver ischemia reperfusion injury by activating Wnt/
288. Dong, J. et al. SRY is a Key Mediator of Sexual Dimorphism in Hepatic Ischemia/ Reperfusion Injury. Ann. Surg. 276, 345-356 (2022).
289. O’Neill, M. J. & O’Neill, R. J. Whatever happened to SRY? Cell Mol. Life Sci. 56, 883-893 (1999).
290. Yang, Y. Y. et al. Involvement of the HIF-1a and Wnt/
291. Griffin, M. O., Ceballos, G. & Villarreal, F. J. Tetracycline compounds with nonantimicrobial organ protective properties: possible mechanisms of action. Pharm. Res. 63, 102-107 (2011).
292. Li, Y., Li, T., Qi, H. & Yuan, F. Minocycline protects against hepatic ischemia/ reperfusion injury in a rat model. Biomed. Rep. 3, 19-24 (2015).
293. Bataller, R. et al. Prolonged infusion of angiotensin II into normal rats induces stellate cell activation and proinflammatory events in liver. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G642-G651 (2003).
294. Kanno, K., Tazuma, S., Nishioka, T., Hyogo, H. & Chayama, K. Angiotensin II participates in hepatic inflammation and fibrosis through MCP-1 expression. Dig. Dis. Sci. 50, 942-948 (2005).
295. Bataller, R. et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Investig. 112, 1383-1394 (2003).
296. Harrison, D. G., Cai, H., Landmesser, U. & Griendling, K. K. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 4, 51-61 (2003).
297. Guo, L. et al. Role of the renin-angiotensin system in hepatic ischemia reperfusion injury in rats. Hepatology 40, 583-589 (2004).
298. Kuncewitch, M. et al. Wnt agonist attenuates liver injury and improves survival after hepatic ischemia/reperfusion. Shock 39, 3-10 (2013).
299. Sun, T. et al. AXIN2(+) Pericentral hepatocytes have limited contributions to liver homeostasis and regeneration. Cell Stem Cell 26, 97-107.e106 (2020).
300. Katoh, M. Multi-layered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/
301. Piao, C. et al. Effects of Exosomes Derived from Adipose-Derived Mesenchymal Stem Cells on Pyroptosis and Regeneration of Injured Liver. Int. J. Mol. Sci. 23, 12065 (2022).
302. Zhou, B. et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 7, 95 (2022).
303. Kopan, R. & llagan, M. X. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216-233 (2009).
304. Chen, G. et al. The canonical Notch signaling was involved in the regulation of intestinal epithelial cells apoptosis after intestinal ischemia/reperfusion injury. Int. J. Mol. Sci. 15, 7883-7896 (2014).
305. Guo, P. et al. Dexmedetomidine alleviates myocardial ischemia-reperfusion injury by down-regulating miR-34b-3p to activate the Jagged1/Notch signaling pathway. Int. Immunopharmacol. 116, 109766 (2023).
306. Li, H. et al. Botch protects neurons from ischemic insult by antagonizing Notchmediated neuroinflammation. Exp. Neurol. 321, 113028 (2019).
307. Pei, H. et al. Notch1 cardioprotection in myocardial ischemia/reperfusion involves reduction of oxidative/nitrative stress. Basic Res. Cardiol. 108, 373 (2013).
308. Yu, H. C. et al. Canonical notch pathway protects hepatocytes from ischemia/ reperfusion injury in mice by repressing reactive oxygen species production through JAK2/STAT3 signaling. Hepatology 54, 979-988 (2011).
309. Chatterjee, S. & Sil, P. C. Targeting the crosstalks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharm. Res. 142, 251-261 (2019).
310. Patni, A. P. et al. Comprehending the crosstalk between Notch, Wnt, and Hedgehog signaling pathways in oral squamous cell carcinoma – clinical implications. Cell Oncol. 44, 473-494 (2021).
311. Kim, H. A. et al. Notch1 counteracts WNT/
312. Kim, W. et al. Hippo signaling interactions with Wnt/
313. Sprinzak, D. & Blacklow, S. C. Biophysics of Notch Signaling. Annu. Rev. Biophys. 50, 157-189 (2021).
314. Gude, N. A. et al. Activation of Notch-mediated protective signaling in the myocardium. Circ. Res. 102, 1025-1035 (2008).
315. Ashton, K. J., Willems, L., Holmgren, K., Ferreira, L. & Headrick, J. P. Ageassociated shifts in cardiac gene transcription and transcriptional responses to ischemic stress. Exp. Gerontol. 41, 189-204 (2006).
316. Zhao, L. et al. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl Acad. Sci. USA 111, 1403-1408 (2014).
317. Zhao, L., Ben-Yair, R., Burns, C. E. & Burns, C. G. Endocardial Notch signaling promotes cardiomyocyte proliferation in the regenerating zebrafish heart through wnt pathway antagonism. Cell Rep. 26, 546-554.e545 (2019).
318. Zhang, H. P. et al. The neuroprotective effects of isoflurane preconditioning in a murine transient global cerebral ischemia-reperfusion model: the role of the Notch signaling pathway. Neuromolecular Med. 16, 191-204 (2014).
319. Yang, Q. et al. Activation of canonical notch signaling pathway is involved in the ischemic tolerance induced by sevoflurane preconditioning in mice. Anesthesiology 117, 996-1005 (2012).
320. Zhang, H. et al. [Expressions of Notch3, Notch4, Frizzled2, and Tead1 in rats with focal cerebral ischemia-reperfusion]. Zhonghua Yi Xue Za Zhi 95, 3766-3769 (2015).
321. Arboleda-Velasquez, J. F. et al. Linking Notch signaling to ischemic stroke. Proc. Natl Acad. Sci. USA 105, 4856-4861 (2008).
322. Arumugam, T. V. et al. Notch signaling and neuronal death in stroke. Prog. Neurobiol. 165-167, 103-116 (2018).
323. Gao, L., Yang, L. & Cui, H. GSK-3
324. Ma, R. et al. l-Borneol and d-Borneol promote transdifferentiation of astrocytes into neurons in rats by regulating Wnt/Notch pathway to exert neuroprotective effect during recovery from cerebral ischemia. Phytomedicine 109, 154583 (2023).
325. Huang, S. et al. Zhongfenggao protects brain microvascular endothelial cells from oxygen-glucose deprivation/reoxygenation-induced injury by angiogenesis. Biol. Pharm. Bull. 42, 222-230 (2019).
326. Zhang, Z., Yao, L., Yang, J., Wang, Z. & Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 18, 3547-3554 (2018).
327. Dong, J., Xu, X., Zhang, Q., Yuan, Z. & Tan, B. The PI3K/AKT pathway promotes fracture healing through its crosstalk with Wnt/
328. Deng, S. et al. PI3K/AKT signaling tips the balance of cytoskeletal forces for cancer progression. Cancers 14, 1652 (2022).
329. Papadimitrakopoulou, V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J. Thorac. Oncol. 7, 1315-1326 (2012).
330. Tewari, D., Patni, P., Bishayee, A., Sah, A. N. & Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 80, 1-17 (2022).
331. Xiao, C. L. et al. The role of PI3K/Akt signaling pathway in spinal cord injury. Biomed. Pharmacother. 156, 113881 (2022).
332. Potz, B. A. et al. Calpain inhibition modulates glycogen synthase kinase
333. Chen, B. et al. Co-expression of Akt1 and Wnt11 promotes the proliferation and cardiac differentiation of mesenchymal stem cells and attenuates hypoxia/ reoxygenation-induced cardiomyocyte apoptosis. Biomed. Pharmacother. 108, 508-514 (2018).
334. Zhuang, Q. et al. Stimulated CB1 cannabinoid receptor inducing ischemic tolerance and protecting neurons from cerebral ischemia. Cent. Nerv. Syst. Agents Med. Chem. 17, 141-150 (2017).
335. Blankesteijn, W. M., van de Schans, V. A., ter Horst, P. & Smits, J. F. The Wnt/ frizzled/GSK-3 beta pathway: a novel therapeutic target for cardiac hypertrophy. Trends Pharm. Sci. 29, 175-180 (2008).
336. Hur, E. M. & Zhou, F. Q. GSK3 signaling in neural development. Nat. Rev. Neurosci. 11, 539-551 (2010).
337. Xing, X. S., Liu, F. & He, Z. Y. Akt regulates
338. Li, P., Zhang, Y. & Liu, H. The role of Wnt/ß-catenin pathway in the protection process by dexmedetomidine against cerebral ischemia/reperfusion injury in rats. Life Sci. 236, 116921 (2019).
339. Thirunavukkarasu, M. et al. Protective effects of Phyllanthus emblica against myocardial ischemia-reperfusion injury: the role of PI3-kinase/glycogen synthase kinase
340. Fei, Y., Zhao, B., Zhu, J., Fang, W. & Li, Y. XQ-1H promotes cerebral angiogenesis via activating PI3K/Akt/GSK3
341. Martínez-Sánchez, G. & Giuliani, A. Cellular redox status regulates hypoxia inducible factor-1 activity. Role in tumor development. J. Exp. Clin. Cancer Res. 26, 39-50 (2007).
342. Semenza, G. L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 76, 39-56 (2014).
343. Tan, Z. et al. Lithium and copper induce the osteogenesis-angiogenesis coupling of bone marrow mesenchymal stem cells via crosstalk between canonical Wnt and HIF-1a signaling pathways. Stem Cells Int. 2021, 6662164 (2021).
344. Tang, K. et al. HIF-1a stimulates the progression of oesophageal squamous cell carcinoma by activating the Wnt/
345. Zhang, Q. et al. Wnt/
346. DeFrates, K. G., Franco, D., Heber-Katz, E. & Messersmith, P. B. Unlocking mammalian regeneration through hypoxia inducible factor one alpha signaling. Biomaterials 269, 120646 (2021).
347. Engelhardt, S., Al-Ahmad, A. J., Gassmann, M. & Ogunshola, O. O. Hypoxia selectively disrupts brain microvascular endothelial tight junction complexes through a hypoxia-inducible factor-1 (HIF-1) dependent mechanism. J. Cell Physiol. 229, 1096-1105 (2014).
348. Wu, C. et al. Wnt/
349. Kaidi, A., Williams, A. C. & Paraskeva, C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 9, 210-217 (2007).
350. Xu, Z. H. et al. Hypoxia-inducible factor protects against acute kidney injury via the Wnt/
351. Peng, D., Fu, M., Wang, M., Wei, Y. & Wei, X. Targeting TGF-
352. Li, T. F. et al. Transforming growth factor-beta stimulates cyclin D1 expression through activation of beta-catenin signaling in chondrocytes. J. Biol. Chem. 281, 21296-21304 (2006).
353. Liu, J., Jin, J., Liang, T. & Feng, X. H. To Ub or not to Ub: a regulatory question in TGF-
354. Działo, E., Tkacz, K. & Błyszczuk, P. Crosstalk between the TGF-
355. Eid, R. A. et al. Exendin-4 Attenuates Remodeling in the Remote Myocardium of Rats After an Acute Myocardial Infarction by Activating
356. Wang, S. et al. Transforming growth-beta 1 contributes to isoflurane postconditioning against cerebral ischemia-reperfusion injury by regulating the c-Jun N-terminal kinase signaling pathway. Biomed. Pharmacother. 78, 280-290 (2016).
357. Chen, D. Q. et al. Combined melatonin and poricoic acid A inhibits renal fibrosis through modulating the interaction of Smad3 and
358. Tian, X. et al. Association of
359. Vallée, A. & Lecarpentier, Y. TGF-
360. Huber, N. et al. Age-related decrease in proteasome expression contributes to defective nuclear factor-kappaB activation during hepatic ischemia/reperfusion. Hepatology 49, 1718-1728 (2009).
361. Liang, W. et al. Preactivation of Notch1 in remote ischemic preconditioning reduces cerebral ischemia-reperfusion injury through crosstalk with the NF-кB pathway. J. Neuroinflamm. 16, 181 (2019).
362. Ling, H. et al.
363. Sakai, N. et al. Receptor activator of nuclear factor-
364. Oeckinghaus, A., Hayden, M. S. & Ghosh, S. Crosstalk in NF-kB signaling pathways. Nat. Immunol. 12, 695-708 (2011).
365. Mitchell, S., Vargas, J. & Hoffmann, A. Signaling via the NFкB system. Wiley Interdiscip. Rev. Syst. Biol. Med 8, 227-241 (2016).
366. Yu, H., Lin, L., Zhang, Z., Zhang, H. & Hu, H. Targeting NF-кВ pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct. Target Ther. 5, 209 (2020).
367. Yin, C. et al. Elevated Wnt2 and Wnt4 activate NF-кB signaling to promote cardiac fibrosis by cooperation of Fzd4/2 and LRP6 following myocardial infarction. EBioMedicine 74, 103745 (2021).
368. Lin, J. C. et al. Enhancement of beta-catenin in cardiomyocytes suppresses survival protein expression but promotes apoptosis and fibrosis. Cardiol. J. 24, 195-205 (2017).
369. Lin, J. C. et al.
370. Spiegelman, V. S. et al. Wnt/beta-catenin signaling induces the expression and activity of betaTrCP ubiquitin ligase receptor. Mol. Cell 5, 877-882 (2000).
371. He, J. et al. Huoxin pill prevents excessive inflammation and cardiac dysfunction following myocardial infarction by inhibiting adverse Wnt/
372. Winston, J. T. et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and betacatenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 13, 270-283 (1999).
373. Noubissi, F. K. et al. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signaling. Nature 441, 898-901 (2006).
374. Hoeflich, K. P. et al. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86-90 (2000).
375. Zhuang, X. et al. Differential effects on lung and bone metastasis of breast cancer by Wnt signaling inhibitor DKK1. Nat. Cell Biol. 19, 1274-1285 (2017).
376. El-Sayyad, S. M., Soubh, A. A., Awad, A. S. & El-Abhar, H. S. Mangiferin protects against intestinal ischemia/reperfusion-induced liver injury: Involvement of PPAR-
377. Jiang, S., Huang, L., Zhang, W. & Zhang, H. Vitamin D/VDR in acute kidney injury: a potential therapeutic target. Curr. Med. Chem. 28, 3865-3876 (2021).
378. Ali, R. M., Al-Shorbagy, M. Y., Helmy, M. W. & El-Abhar, H. S. Role of Wnt4/
379. Wang, J., Liu, S., Heallen, T. & Martin, J. F. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 15, 672-684 (2018).
380. Zhou, Q., Li, L., Zhao, B. & Guan, K. L. The hippo pathway in heart development, regeneration, and diseases. Circ. Res. 116, 1431-1447 (2015).
381. Heallen, T. et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458-461 (2011).
382. Gong, P. et al. Hippo/YAP signaling pathway mitigates blood-brain barrier disruption after cerebral ischemia/reperfusion injury. Behav. Brain Res. 356, 8-17 (2019).
383. Yu, H. et al. RRM2 improves cardiomyocyte proliferation after myocardial ischemia reperfusion injury through the hippo-YAP pathway. Dis. Mark. 2021, 5089872 (2021).
384. Zheng, Z. et al. Hippo-YAP/MCP-1 mediated tubular maladaptive repair promote inflammation in renal failed recovery after ischemic AKI. Cell Death Dis. 12, 754 (2021).
385. Zhou, J. et al. TNFAIP3 interacting protein 3 is an activator of Hippo-YAP signaling protecting against hepatic ischemia/reperfusion injury. Hepatology 74, 2133-2153 (2021).
386. Zheng, A., Chen, Q. & Zhang, L. The Hippo-YAP pathway in various cardiovascular diseases: focusing on the inflammatory response. Front. Immunol. 13, 971416 (2022).
387. Nishina, H. Physiological and pathological roles of the Hippo-YAP/TAZ signaling pathway in liver formation, homeostasis, and tumorigenesis. Cancer Sci. 113, 1900-1908 (2022).
388. Liu, S. et al. Yap promotes noncanonical Wnt signals from cardiomyocytes for heart regeneration. Circ. Res. 129, 782-797 (2021).
389. Azzolin, L. et al. YAP/TAZ incorporation in the
390. Ma, W. Y. et al. Melatonin promotes cardiomyocyte proliferation and heart repair in mice with myocardial infarction via miR-143-3p/Yap/Ctnnd1 signaling pathway. Acta Pharm. Sin. 42, 921-931 (2021).
391. Amani, H. et al. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci. Rep. 9, 6044 (2019).
392. Kanzler, B., Foreman, R. K., Labosky, P. A. & Mallo, M. BMP signaling is essential for development of skeletogenic and neurogenic cranial neural crest. Development 127, 1095-1104 (2000).
393. Mabie, P. C., Mehler, M. F. & Kessler, J. A. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J. Neurosci. 19, 7077-7088 (1999).
394. Baker, J. C., Beddington, R. S. & Harland, R. M. Wnt signaling in Xenopus embryos inhibits bmp4 expression and activates neural development. Genes Dev. 13, 3149-3159 (1999).
395. Antebi, Y. E. et al. Combinatorial signal perception in the BMP pathway. Cell 170, 1184-1196.e1124 (2017).
396. Zhang, Y. & Que, J. BMP signaling in development, stem cells, and diseases of the gastrointestinal tract. Annu. Rev. Physiol. 82, 251-273 (2020).
397. Fujita, K., Ogawa, R., Kawawaki, S. & Ito, K. Roles of chromatin remodelers in maintenance mechanisms of multipotency of mouse trunk neural crest cells in the formation of neural crest-derived stem cells. Mech. Dev. 133, 126-145 (2014).
398. Dizon, M. L., Maa, T. & Kessler, J. A. The bone morphogenetic protein antagonist noggin protects white matter after perinatal hypoxia-ischemia. Neurobiol. Dis. 42, 318-326 (2011).
399. Guan, J. et al. Bone morphogenetic protein-7 (BMP-7) mediates ischemic preconditioning-induced ischemic tolerance via attenuating apoptosis in rat brain. Biochem. Biophys. Res. Commun. 441, 560-566 (2013).
400. Chen, C., Yang, Y. & Yao, Y. HBO promotes the differentiation of neural stem cells via interactions between the Wnt3/
401. Lei, Z. N., Liu, F., Zhang, L. M., Huang, Y. L. & Sun, F. Y. Bcl-2 increases strokeinduced striatal neurogenesis in adult brains by inhibiting BMP-4 function via activation of
402. Baik, J., Borges, L., Magli, A., Thatava, T. & Perlingeiro, R. C. Effect of endoglin overexpression during embryoid body development. Exp. Hematol. 40, 837-846 (2012).
403. Zhang, L. et al. Modulation of TGF-
404. Borges, L. et al. A critical role for endoglin in the emergence of blood during embryonic development. Blood 119, 5417-5428 (2012).
405. Baik, J. et al. Endoglin integrates BMP and Wnt signalling to induce haematopoiesis through JDP2. Nat. Commun. 7, 13101 (2016).
406. Ahmadi, A. et al. Recent advances on small molecules in osteogenic differentiation of stem cells and the underlying signaling pathways. Stem Cell Res. Ther. 13, 518 (2022).
407. Zhang, X., Shi, X., Wang, J., Xu, Z. & He, J. Enriched environment remedies cognitive dysfunctions and synaptic plasticity through NMDAR-
408. Liu, S. et al. Icaritin alleviates cerebral ischemia-reperfusion injury by regulating NMDA receptors through ERK signaling. Eur. J. Pharm. 941, 175492 (2023).
409. Abe, K. & Takeichi, M. NMDA-receptor activation induces calpain-mediated betacatenin cleavages for triggering gene expression. Neuron 53, 387-397 (2007).
410. Villmann, C. & Becker, C. M. On the hypes and falls in neuroprotection: targeting the NMDA receptor. Neuroscientist 13, 594-615 (2007).
411. Jolly, S. et al. G protein-coupled receptor 37-like 1 modulates astrocyte glutamate transporters and neuronal NMDA receptors and is neuroprotective in ischemia. Glia 66, 47-61 (2018).
412. Luo, Y. et al. Focal cerebral ischemia and reperfusion induce brain injury through a28-1-Bound NMDA receptors. Stroke 49, 2464-2472 (2018).
413. Kawai, T. & Akira, S. TLR signaling. Semin. Immunol. 19, 24-32 (2007).
414. Tong, Y. et al. WISP1 mediates hepatic warm ischemia reperfusion injury via TLR4 signaling in mice. Sci. Rep. 6, 20141 (2016).
415. Undi, R. B., Sarvothaman, S., Narasaiah, K., Gutti, U. & Gutti, R. K. Toll-like receptor 2 signalings: significance in megakaryocyte development through wnt signalling cross-talk and cytokine induction. Cytokine 83, 245-249 (2016).
416. Martín-Medina, A. et al. TLR/WNT: A Novel Relationship in Immunomodulation of Lung Cancer. Int. J Mol Sci. 23, 6539 (2022).
417. Christman, M. A. 2nd et al. Wnt5a is expressed in murine and human atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 294, H2864-H2870 (2008).
418. He, W. et al. Lipopolysaccharide enhances Wnt5a expression through toll-like receptor 4, myeloid differentiating factor 88, phosphatidylinositol 3-OH kinase/ AKT and nuclear factor kappa B pathways in human dental pulp stem cells. J. Endod. 40, 69-75 (2014).
419. El-Ela, S. R. A., Zaghloul, R. A. & Eissa, L. A. Promising cardioprotective effect of baicalin in doxorubicin-induced cardiotoxicity through targeting toll-like receptor
420. Liu, B., Li, F., Xu, Y., Wu, Q. & Shi, J. Gastrodin improves cognitive dysfunction in REM Sleep-deprived rats by regulating TLR4/NF-кB and Wnt/
421. Tanaka, R., Terai, M., Londin, E. & Sato, T. The role of HGF/MET signaling in metastatic uveal melanoma. Cancers 13, 5457 (2021).
422. Demkova, L. & Kucerova, L. Role of the HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol. Cancer 17, 26 (2018).
423. Zhang, Y. et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 17, 45 (2018).
424. Liu, S. et al. A self-assembling peptide hydrogel-based drug co-delivery platform to improve tissue repair after ischemia-reperfusion injury. Acta Biomater. 103, 102-114 (2020).
425. Humphreys, B. D. et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2, 284-291 (2008).
426. Koraishy, F. M., Silva, C., Mason, S., Wu, D. & Cantley, L. G. Hepatocyte growth factor (Hgf) stimulates low density lipoprotein receptor-related protein (Lrp) 5/6 phosphorylation and promotes canonical Wnt signaling. J. Biol. Chem. 289, 14341-14350 (2014).
427. Maarouf, O. H. et al. Paracrine Wnt1 drives interstitial fibrosis without inflammation by tubulointerstitial cross-talk. J. Am. Soc. Nephrol. 27, 781-790 (2016).
428. Zhou, D. et al. Fibroblast-specific
429. Doeppner, T. R. et al. Acute hepatocyte growth factor treatment induces longterm neuroprotection and stroke recovery via mechanisms involving neural precursor cell proliferation and differentiation. J. Cereb. Blood Flow. Metab. 31, 1251-1262 (2011).
430. Shang, J. et al. Antiapoptotic and anti autophagic effects of glial cell line-derived neurotrophic factor and hepatocyte growth factor after transient middle cerebral artery occlusion in rats. J. Neurosci. Res. 88, 2197-2206 (2010).
431. Nakaguchi, K. et al. Growth factors released from gelatin hydrogel microspheres increase new neurons in the adult mouse brain. Stem Cells Int. 2012, 915160 (2012).
432. Chaparro, R. E. et al. Sustained functional improvement by hepatocyte growth factor-like small molecule BB3 after focal cerebral ischemia in rats and mice. J. Cereb. Blood Flow. Metab. 35, 1044-1053 (2015).
433. Matsunaga, S., Fujishiro, H. & Takechi, H. Efficacy and Safety of Glycogen Synthase Kinase 3 Inhibitors for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 69, 1031-1039 (2019).
434. del Ser, T. et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J. Alzheimers Dis. 33, 205-215 (2013).
435. O’Leary, O. & Nolan, Y. Glycogen synthase kinase-3 as a therapeutic target for cognitive dysfunction in neuropsychiatric disorders. CNS Drugs 29, 1-15 (2015).
436. Singh, A. P. et al. Inhibition of GSK-3 to induce cardiomyocyte proliferation: a recipe for in situ cardiac regeneration. Cardiovasc. Res. 115, 20-30 (2019).
437. Fu, W. B., Wang, W. E. & Zeng, C. Y. Wnt signaling pathways in myocardial infarction and the therapeutic effects of Wnt pathway inhibitors. Acta Pharm. Sin. 40, 9-12 (2019).
438. Saraswati, S. et al. Pyrvinium, a potent small molecule Wnt inhibitor, promotes wound repair and post-MI cardiac remodeling. PLoS One 5, e15521 (2010).
439. Laeremans, H. et al. Blocking of frizzled signaling with a homologous peptide fragment of wnt3a/wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation 124, 1626-1635 (2011).
440. Sasaki, T., Hwang, H., Nguyen, C., Kloner, R. A. & Kahn, M. The small molecule Wnt signaling modulator ICG-001 improves contractile function in chronically infarcted rat myocardium. PLoS One 8, e75010 (2013).
441. Jiang, J. et al. A novel porcupine inhibitor blocks WNT pathways and attenuates cardiac hypertrophy. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3459-3467 (2018).
442. Xie, S. et al. Discovering small molecules as Wnt inhibitors that promote heart regeneration and injury repair. J. Mol. Cell Biol. 12, 42-54 (2020).
443. Ni, T. T. et al. Discovering small molecules that promote cardiomyocyte generation by modulating Wnt signaling. Chem. Biol. 18, 1658-1668 (2011).
444. Kumar, K., Singh, N., Jaggi, A. S. & Maslov, L. Clinical applicability of conditioning techniques in ischemia-reperfusion injury: a review of the literature. Curr. Cardiol. Rev. 17, 306-318 (2021).
445. Liu, G. S. et al. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 84, 350-356 (1991).
446. Goto, M. et al. Role of bradykinin in protection of ischemic preconditioning in rabbit hearts. Circ. Res. 77, 611-621 (1995).
447. Cohen, M. V. et al. Preconditioning-mimetics bradykinin and DADLE activate PI3kinase through divergent pathways. J. Mol. Cell Cardiol. 42, 842-851 (2007).
448. Schultz, J. E., Rose, E., Yao, Z. & Gross, G. J. Evidence for involvement of opioid receptors in ischemic preconditioning in rat hearts. Am. J. Physiol. 268, H2157-H2161 (1995).
449. Banerjee, A. et al. Preconditioning against myocardial dysfunction after ischemia and reperfusion by an alpha 1-adrenergic mechanism. Circ. Res. 73, 656-670 (1993).
450. Yao, Z. & Gross, G. J. Role of nitric oxide, muscarinic receptors, and the ATPsensitive
451. Kim, J. et al. Adenosine and Cordycepin Accelerate Tissue Remodeling Process through Adenosine Receptor Mediated Wnt/
452. Kim, J., Shin, J. Y., Choi, Y. H., Kang, N. G. & Lee, S. Anti-Hair Loss Effect of Adenosine Is Exerted by cAMP Mediated Wnt/
453. Borhani, S., Corciulo, C., Larranaga-Vera, A. & Cronstein, B. N. Adenosine A(2A) receptor (A2AR) activation triggers Akt signaling and enhances nuclear localization of
454. Yan, L., Yao, X., Bachvarov, D., Saifudeen, Z. & El-Dahr, S. S. Genome-wide analysis of gestational gene-environment interactions in the developing kidney. Physiol. Genom. 46, 655-670 (2014).
455. Liu, Y. et al. Wnt/
456. Li, Y. et al. Propoxyphene mediates oxyhemoglobin-induced injury in rat cortical neurons through up-regulation of active-
457. Wang, J. et al. Pentazocine Protects SN4741 Cells Against MPP(+)-Induced Cell Damage via Up-Regulation of the Canonical Wnt/
458. Guan, M., Huang, Y. & Lin, X. Sufentanil inhibits the proliferation and epithelial mesenchymal transition of lung cancer cells through Wnt/beta-catenin signaling pathway. Bioengineered 13, 10857-10865 (2022).
459. Mocanu, M. M., Bell, R. M. & Yellon, D. M. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J. Mol. Cell Cardiol. 34, 661-668 (2002).
460. Jonassen, A. K., Mjøs, O. D. & Sack, M. N. p70s6 kinase is a functional target of insulin activated Akt cell-survival signaling. Biochem. Biophys. Res. Commun. 315, 160-165 (2004).
461. Tong, H., Chen, W., Steenbergen, C. & Murphy, E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ. Res. 87, 309-315 (2000).
462. Juhaszova, M. et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 113, 1535-1549 (2004).
463. Barandon, L. et al. Involvement of FrzA/sFRP-1 and the Wnt/frizzled pathway in ischemic preconditioning. Circ. Res. 96, 1299-1306 (2005).
464. Vigneron, F. et al. GSK-3
465. Correa-Costa, M. et al. Transcriptome analysis of renal ischemia/reperfusion injury and its modulation by ischemic pre-conditioning or hemin treatment. PLoS One 7, e49569 (2012).
466. Przyklenk, K., Bauer, B., Ovize, M., Kloner, R. A. & Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87, 893-899 (1993).
467. Kambakamba, P. et al. Novel benefits of remote ischemic preconditioning through VEGF-dependent protection from resection-induced liver failure in the mouse. Ann. Surg. 268, 885-893 (2018).
468. Sawashita, Y. et al. Remote ischemic preconditioning reduces myocardial ischemia-reperfusion injury through unacylated ghrelin-induced activation of the JAK/STAT pathway. Basic Res. Cardiol. 115, 50 (2020).
469. Sörensson, P. et al. Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart 96, 1710-1715 (2010).
470. Woo, J. S. et al. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler. Thromb. Vasc. Biol. 33, 2252-2260 (2013).
471. Koyama, T. et al. Impact of postconditioning with lactate-enriched blood on inhospital outcomes of patients with ST-segment elevation myocardial infarction. Int. J. Cardiol. 220, 146-148 (2016).
472. Zhu, M. et al. Ischemic postconditioning protects remodeled myocardium via the PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc. Res. 72, 152-162 (2006).
473. Wagner, C., Tillack, D., Simonis, G., Strasser, R. H. & Weinbrenner, C. Ischemic post-conditioning reduces infarct size of the in vivo rat heart: role of PI3-K, mTOR, GSK-3beta, and apoptosis. Mol. Cell. Biochem. 339, 135-147 (2010).
474. Guo, J. Y. et al. Ischemic postconditioning attenuates liver warm ischemiareperfusion injury through Akt-eNOS-NO-HIF pathway. J. Biomed. Sci. 18, 79 (2011).
475. Darling, C. E. et al. Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2. Am. J. Physiol. Heart Circ. Physiol. 289, H1618-H1626 (2005).
476. Tsang, A., Hausenloy, D. J., Mocanu, M. M. & Yellon, D. M. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ. Res. 95, 230-232 (2004).
477. Hausenloy, D. J., Tsang, A. & Yellon, D. M. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc. Med. 15, 69-75 (2005).
478. Díaz-Ruíz, J. L. et al. Redox signaling in ischemic postconditioning protection involves PKCɛ and Erk1/2 pathways and converges indirectly in Nrf2 activation. Cell Signal 64, 109417 (2019).
479. He, N. et al. Remote ischemic perconditioning prevents liver transplantationinduced ischemia/reperfusion injury in rats: role of ROS/RNS and eNOS. World J. Gastroenterol. 23, 830-841 (2017).
480. Qiu, Y. et al. Hyperglycemia-Induced Overexpression of PH Domain Leucine-Rich Repeat Protein Phosphatase 1 (PHLPP1) Compromises the Cardioprotective Effect of Ischemic Postconditioning Via Modulation of the Akt/Mst1 Pathway Signaling. Cardiovasc. Drugs Ther. https://doi.org/10.1007/s10557-022-07349-5 (2022).
481. Chen, H., Shen, J. & Zhao, H. Ischemic postconditioning for stroke treatment: current experimental advances and future directions. Cond. Med. 3, 104-115 (2020).
482. Xue, R. et al. Selective inhibition of PTEN preserves ischaemic post-conditioning cardioprotection in STZ-induced Type 1 diabetic rats: role of the PI3K/Akt and JAK2/STAT3 pathways. Clin. Sci. 130, 377-392 (2016).
483. Kerendi, F. et al. Remote postconditioning. Brief renal ischemia and reperfusion applied before coronary artery reperfusion reduces myocardial infarct size via endogenous activation of adenosine receptors. Basic Res. Cardiol. 100, 404-412 (2005).
484. Sun, J. et al. Protective effect of delayed remote limb ischemic postconditioning: role of mitochondrial K(ATP) channels in a rat model of focal cerebral ischemic reperfusion injury. J. Cereb. Blood Flow. Metab. 32, 851-859 (2012).
485. Hu, X., Lv, T., Yang, S. F., Zhang, X. H. & Miao, Y. F. Limb remote ischemic post-conditioning reduces injury and improves long-term behavioral recovery in rats following subarachnoid hemorrhage: possible involvement of the autophagic process. Mol. Med. Rep. 17, 21-30 (2018).
486. Peng, B. et al. Remote ischemic postconditioning protects the brain from global cerebral ischemia/reperfusion injury by up-regulating endothelial nitric oxide synthase through the PI3K/Akt pathway. Brain Res. 1445, 92-102 (2012).
487. Qi, Z. F. et al. AKT/GSK3
488. Gao, S. et al. Remote ischemic postconditioning protects against renal ischemia/ reperfusion injury by activation of T-LAK-cell-originated protein kinase (TOPK)/ PTEN/Akt signaling pathway mediated anti-oxidation and anti-inflammation. Int. Immunopharmacol. 38, 395-401 (2016).
489. Danielisová, V., Némethová, M., Gottlieb, M. & Burda, J. The changes in endogenous antioxidant enzyme activity after postconditioning. Cell. Mol. Neurobiol. 26, 1181-1191 (2006).
490. Wang, Q. et al. Limb remote postconditioning alleviates cerebral reperfusion injury through reactive oxygen species-mediated inhibition of delta protein kinase C in rats. Anesth. Analg. 113, 1180-1187 (2011).
491. Niu, D. G. et al. Morphine promotes cancer stem cell properties, contributing to chemoresistance in breast cancer. Oncotarget 6, 3963-3976 (2015).
492. Wang, K. P., Bai, Y., Wang, J. & Zhang, J. Z. Morphine protects SH-SY5Y human neuroblastoma cells against Dickkopf1-induced apoptosis. Mol. Med. Rep. 11, 1174-1180 (2015).
493. Zhou, Z., Liu, T. & Zhang, J. Morphine activates blast-phase chronic myeloid leukemia cells and alleviates the effects of tyrosine kinase inhibitors. Biochem. Biophys. Res. Commun. 520, 560-565 (2019).
494. Xue, J. J. et al. Protective effect of propofol on hydrogen peroxide-induced human esophageal carcinoma via blocking the Wnt/
495. Zhan, K., Song, X., Zhang, Q., Yang, J. & Lu, S. Propofol-Induced miR-493-3p Inhibits Growth and Invasion of Gastric Cancer through Suppression of DKK1Mediated Wnt/
496. Gong, T. et al. Propofol-induced miR-219-5p inhibits growth and invasion of hepatocellular carcinoma through suppression of GPC3-mediated Wnt/
497. Zhang, Y. F., Li, C. S., Zhou, Y. & Lu, X. H. Effects of propofol on colon cancer metastasis through STAT3/HOTAIR axis by activating WIF-1 and suppressing Wnt pathway. Cancer Med. 9, 1842-1854 (2020).
498. Lin, X. H. et al. Norepinephrine-stimulated HSCs secrete sFRP1 to promote HCC progression following chronic stress via augmentation of a Wnt16B/
499. Zhao, X. et al. Aldehyde dehydrogenase-2 protects against myocardial infarction-related cardiac fibrosis through modulation of the Wnt/
500. Qian, L. et al. Downregulation of S100A4 alleviates cardiac fibrosis via Wnt/
501. Cui, S. et al. miR-145 attenuates cardiac fibrosis through the AKT/GSK-3
502. Guo, X. et al. Induced pluripotent stem cell-conditional medium inhibits H9C2 cardiomyocytes apoptosis via autophagy flux and Wnt/
503. Liu, C. & Li, Y. Propofol relieves inflammation in MIRI rats by inhibiting Rho/Rock signaling pathway. Eur. Rev. Med. Pharm. Sci. 25, 976-984 (2021).
504. Chen, F. et al. Activation of EphA4 induced by EphrinA1 exacerbates disruption of the blood-brain barrier following cerebral ischemia-reperfusion via the Rho/ ROCK signaling pathway. Exp. Ther. Med. 16, 2651-2658 (2018).
505. Zhou, D., Zhang, M., Min, L., Jiang, K. & Jiang, Y. Cerebral ischemia-reperfusion is modulated by macrophage-stimulating 1 through the MAPK-ERK signaling pathway. J. Cell Physiol. 235, 7067-7080 (2020).
506. Zhang, F., Cao, X., Zhao, C., Chen, L. & Chen, X. Empagliflozin activates JAK2/ STAT3 signaling and protects cardiomyocytes from hypoxia/reoxygenation injury under high glucose conditions. J. Thromb. Thrombolysis 55, 116-125 (2023).
507. Li, Z. et al. Theaflavin ameliorates renal ischemia/reperfusion injury by activating the Nrf2 signaling pathway in vivo and in vitro. Biomed. Pharmacother. 134, 111097 (2021).
508. Wu, J. W., Hu, H., Hua, J. S. & Ma, L. K. ATPase inhibitory factor 1 protects the heart from acute myocardial ischemia/reperfusion injury through activating AMPK signaling pathway. Int J. Biol. Sci. 18, 731-741 (2022).
509. Zhao, N., Gao, Y., Jia, H. & Jiang, X. Anti-apoptosis effect of traditional Chinese medicine in the treatment of cerebral ischemia-reperfusion injury. Apoptosis. 28, 702-729 (2023).
510. Dong, L. et al. Research Progress of Chinese Medicine in the Treatment of Myocardial Ischemia-Reperfusion Injury. Am. J. Chin. Med. 51, 1-17 (2023).
511. Yin, B., Hou, X. W. & Lu, M. L. Astragaloside IV attenuates myocardial ischemia/ reperfusion injury in rats via inhibition of calcium-sensing receptor-mediated apoptotic signaling pathways. Acta Pharm. Sin. 40, 599-607 (2019).
512. Jiang, M. et al. Astragaloside IV Attenuates Myocardial Ischemia-Reperfusion Injury from Oxidative Stress by Regulating Succinate, Lysophospholipid Metabolism, and ROS Scavenging System. Oxid. Med. Cell. Longev. 2019, 9137654 (2019).
513. Song, M., Huang, L., Zhao, G. & Song, Y. Beneficial effects of a polysaccharide from Salvia miltiorrhiza on myocardial ischemia-reperfusion injury in rats. Carbohydr. Polym. 98, 1631-1636 (2013).
514. Zeng, H. et al. Activated PKB/GSK-
activity: Therapeutic efficacy of dihydrotanshinone-I. Acta Pharm. Sin. B 11, 71-88 (2021).
515. Huang, C. Y. et al. Protective effect of Danggui (Radix Angelicae Sinensis) on angiotensin II-induced apoptosis in H9c2 cardiomyoblast cells. BMC Complement Alter. Med. 14, 358 (2014).
516. Zhang, S. et al. Extraction, chemical analysis of Angelica sinensis polysaccharides and antioxidant activity of the polysaccharides in ischemia-reperfusion rats. Int. J. Biol. Macromol. 47, 546-550 (2010).
517. Wang, K., Lou, Y., Xu, H., Zhong, X. & Huang, Z. Harpagide from Scrophularia protects rat cortical neurons from oxygen-glucose deprivation and reoxygenation-induced injury by decreasing endoplasmic reticulum stress. J. Ethnopharmacol. 253, 112614 (2020).
518. Mo, Z. T., Liao, Y. L., Zheng, J. & Li, W. N. Icariin protects neurons from endoplasmic reticulum stress-induced apoptosis after OGD/R injury via suppressing IRE1a-XBP1 signaling pathway. Life Sci. 255, 117847 (2020).
519. Pang, Y., Zhu, S. & Pei, H. Pachymic acid protects against cerebral ischemia/ reperfusion injury by the PI3K/Akt signaling pathway. Metab. Brain Dis. 35, 673-680 (2020).
520. Tong, C. et al. Intravenous administration of lycopene, a tomato extract, protects against myocardial ischemia-reperfusion injury. Nutrients 8, 138 (2016).
521. Wu, S. et al. Effects of lycopene attenuating injuries in ischemia and reperfusion. Oxid. Med. Cell. Longev. 2022, 9309327 (2022).
522. Liu, Y., Qu, X., Yan, M., Li, D. & Zou, R. Tricin attenuates cerebral ischemia/ reperfusion injury through inhibiting nerve cell autophagy, apoptosis and inflammation by regulating the PI3K/Akt pathway. Hum. Exp. Toxicol. 41, 9603271221125928 (2022).
523. Wang, G., Guo, H. & Wang, X. Platycodin D protects cortical neurons against oxygen-glucose deprivation/reperfusion in neonatal hypoxic-ischemic encephalopathy. J. Cell. Biochem 120, 14028-14034 (2019).
524. Yang, S. et al. Baicalein administered in the subacute phase ameliorates ischemia-reperfusion-induced brain injury by reducing neuroinflammation and neuronal damage. Biomed. Pharmacother. 117, 109102 (2019).
525. Wang, Z. et al. Lupeol alleviates cerebral ischemia-reperfusion injury in correlation with modulation of PI3K/Akt pathway. Neuropsychiatr. Dis. Treat. 16, 1381-1390 (2020).
526. Wang, P. C. et al. Combination of paeoniflorin and calycosin-7-glucoside alleviates ischaemic stroke injury via the PI3K/AKT signaling pathway. Pharm. Biol. 60, 1469-1477 (2022).
527. Jian, J., Xuan, F., Qin, F. & Huang, R. Bauhinia championii flavone inhibits apoptosis and autophagy via the PI3K/Akt pathway in myocardial ischemia/ reperfusion injury in rats. Drug Des. Dev. Ther. 9, 5933-5945 (2015).
528. Zhang, H. & Li, H. Tricin enhances osteoblastogenesis through the regulation of Wnt/
529. Lee, H., Bae, S., Kim, Y. S. & Yoon, Y. WNT/ß-catenin pathway mediates the antiadipogenic effect of platycodin D , a natural compound found in Platycodon grandiflorum. Life Sci. 89, 388-394 (2011).
530. Xia, X. et al. Baicalein blocked cervical carcinoma cell proliferation by targeting CCND1 via Wnt/
531. Tarapore, R. S., Siddiqui, I. A., Adhami, V. M., Spiegelman, V. S. & Mukhtar, H. The dietary terpene lupeol targets colorectal cancer cells with constitutively active Wnt/
532. Wang, Y. et al. Construing the biochemical and molecular mechanism underlying the in vivo and in vitro chemotherapeutic efficacy of ruthenium-baicalein complex in colon cancer. Int. J. Biol. Sci. 15, 1052-1071 (2019).
533. Tarapore, R. S. et al. Specific targeting of Wnt/
534. Zhang, L., Tu, Y., He, W., Peng, Y. & Qiu, Z. A novel mechanism of hepatocellular carcinoma cell apoptosis induced by lupeol via Brain-derived neurotrophic factor inhibition and glycogen synthase kinase 3 beta reactivation. Eur. J. Pharm. 762, 55-62 (2015).
535. Wu, X. T. et al. The enhanced effect of lupeol on the destruction of gastric cancer cells by NK cells. Int. Immunopharmacol. 16, 332-340 (2013).
536. Zhou, Y. et al. Paeoniflorin Affects Hepatocellular Carcinoma Progression by Inhibiting Wnt/
537. Li, H. et al. Bauhinia championi (Benth.) Benth. polysaccharides upregulate Wnt/
538. Zhang, C. et al. Asiaticoside alleviates cerebral ischemia-reperfusion injury via NOD2/Mitogen-Activated Protein Kinase (MAPK)/Nuclear Factor kappa B (NF-кB) Signaling Pathway. Med. Sci. Monit. 26, e920325 (2020).
539. Ye, B. et al. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des. Dev. Ther. 13, 975-990 (2019).
540. Pan, J. et al. Ginkgetin attenuates cerebral ischemia-reperfusion induced autophagy and cell death via modulation of the NF-кB/p53 signaling pathway. Biosci. Rep. 39, BSR20191452 (2019).
541. Cao, W., Feng, S. J. & Kan, M. C. Naringin Targets NFKB1 to Alleviate OxygenGlucose Deprivation/Reoxygenation-Induced Injury in PC12 Cells Via Modulating HIF-1a/AKT/mTOR-Signaling Pathway. J. Mol. Neurosci. 71, 101-111 (2021).
542. An, B. et al. Crocin regulates the proliferation and migration of neural stem cells after cerebral ischemia by activating the Notch1 pathway. Folia Neuropathol. 58, 201-212 (2020).
543. Dibben, G. et al. Exercise-based cardiac rehabilitation for coronary heart disease. Cochrane Database Syst. Rev. 11, Cd001800 (2021).
544. Wang, H. et al. Programmed exercise attenuates familial hypertrophic cardiomyopathy in transgenic E22K mice via inhibition of PKC-a/NFAT pathway. Front. Cardiovasc. Med. 9, 808163 (2022).
545. Cheedipudi, S. M. et al. Exercise restores dysregulated gene expression in a mouse model of arrhythmogenic cardiomyopathy. Cardiovasc. Res. 116, 1199-1213 (2020).
© The Author(s) 2023
The Collaborative Innovation Center, Jining Medical University, Jining, Shandong 272067, China; Clinical Medical College, Jining Medical University, Jining, Shandong 272067, China; Second Clinical Medical College, Jining Medical University, Jining, Shandong 272067, China; Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital and Institutes of Biomedical Sciences, Fudan University, Shanghai, China; Institute of Zoology, Chinese Academy of Sciences, Beijing, China and Department of Physiology, Basic medical school, Xuzhou Medical University, Xuzhou 221004, China
Correspondence: Shijun Wang (shijun_w@126.com) or Rubin Tan (tanrubin11@126.com) or Jinxiang Yuan (yuanjinxiang18@163.com)
These authors contributed equally: Meng Zhang, Qian Liu, Hui Meng
DOI: https://doi.org/10.1038/s41392-023-01688-x
PMID: https://pubmed.ncbi.nlm.nih.gov/38185705
Publication Date: 2024-01-08
Ischemia-reperfusion injury: molecular mechanisms and therapeutic targets
Abstract
Ischemia-reperfusion (I/R) injury paradoxically occurs during reperfusion following ischemia, exacerbating the initial tissue damage. The limited understanding of the intricate mechanisms underlying I/R injury hinders the development of effective therapeutic interventions. The Wnt signaling pathway exhibits extensive crosstalk with various other pathways, forming a network system of signaling pathways involved in I/R injury. This review article elucidates the underlying mechanisms involved in Wnt signaling, as well as the complex interplay between Wnt and other pathways, including Notch, phosphatidylinositol 3-kinase/protein kinase B, transforming growth factor-
INTRODUCTION
receptor domain-containing adapter-inducing interferon-
the interconnections among various signaling pathways, remains significantly challenging. Therefore, there remains a lack of consensus in current research,
cell models. Additionally, by elucidating the underlying mechanisms, we outline current clinical and preclinical therapeutic strategies that target the Wnt pathway and interconnected signaling pathway networks (Fig. 1a). Considering the complex nature of organ damage in I/R injury, targeting network signaling pathways is crucial for effective interventions. Future studies should focus on developing strategies that effectively modulate these interconnected signals to mitigate the detrimental effects of I/R damage.
WNT PATHWAYS AND ORGAN I/R INJURY
Wnt pathways
the activation of c-Jun N-terminal kinase (JNK), which in turn plays a critical role in the rearrangement of the actin cytoskeleton and regulating cell polarity and promoting migration.
downregulated, while the ratio of cleaved caspase 3 and p-GSK
inhibition of Wnt/
mice activated macrophage in the injured heart. This activation subsequently upregulated Wnt5a expression, increased JNK phosphorylation, and elevated the expression levels of inflammatory cytokines IL-1
studies have shown that myocardial I/R activates Wnt/
in cardiac macrophages, especially in pro-inflammatory subsets.
in macrophage polarization.
within the infarcted area one-week post Ml; additionally, expression of the protein upstream of the Wnt/
ischemic myocardial injury, thereby initiating excessive ECM remodeling and fibrosis. Interestingly, the Wnt/
The timely identification and intervention of ischemic stroke are of utmost importance. The current recommended therapeutic
strategy for ischemic stroke involves thrombolysis, via recombinant tissue plasminogen activator (rtPA) injection within 4.5 h post onset,
the Wnt signaling pathway transitions from an activated state to an inhibited state as ischemia time increases, and is regulated by many processes such as autophagy.
during ischemia.
permeability transition pores and subsequent cell death.
hormone receptor Nur77 is stimulated, promoting
the upregulation of various downstream molecules. Neurogenesis alleviates brain injury by combating apoptosis.
signaling induces the polarization of reactive microglia toward the M2 phenotype.
Wnt pathways during renal I/R injury
Wnt pathways during hepatic I/R injury
the prognosis of liver function. The degree of liver I/R injury is influenced by temperature, ischemic time, and range. The injury of liver cells caused by warm ischemia is more severe than that caused by cold ischemia, whereas the injury to liver sinusoidal ECs is the opposite.
remarkable increase in hepatocyte proliferation while concurrently reducing apoptosis and necrosis.
CROSSTALK BETWEEN WNT SIGNALING AND OTHER I/RASSOCIATED PATHWAYS
The Notch signaling pathway, a highly evolutionarily conserved pathway involved in embryonic development and tissue injury repair,
pathway. This dual modulation results in synaptic proteins and reduced apoptosis in neonatal rats with cerebral ischemia.
inhibited the Wnt3a/
atrophy.
The PI3K/Akt signaling pathway, named after its key components, phosphatidylinositol 3-kinase (PI3K) and AKT (also known as protein kinase
anti-apoptotic effect.
The hypoxia-inducible factor-1a (HIF-1a) signaling pathway is responsible for conferring adaptation to hypoxic conditions and plays a crucial role in angiogenesis, oxidative stress, and cell metabolism under hypoxic conditions.
exhibits a short half-life under high oxygen conditions and is rapidly degraded by proteasomes. It is, however, stable under hypoxic conditions; under such conditions, HIF-1 translocates to the nucleus, co-polymerizes with HIF-1
TGF-
forms a complex with
fibrotic effects resulting from the crosstalk between TGF-
NF-кB was initially established as an important transcription factor in the induction of various immune and inflammatory responses.
phosphorylation while activating the Wnt2/
Hippo-YAP signaling
BMP signaling
signaling pathway.
NMDAR-
TLR4/TRIF signaling
disturbance and cognitive dysfunction induced by REM sleep deprivation.
HGF/c-Met signaling
THERAPEUTIC STRATEGIES
Several therapeutic approaches focusing on Wnt signaling, including cell and exosome therapy, gene therapy, protein therapy, and drug therapy, have shown promising prospects for clinical application. Table 1 summarizes the therapeutic strategies targeting Wnt signaling for the treatment of I/R injury.
In the pursuit of overcoming I/R injuries, clinicians are currently utilizing preconditioning and postconditioning approaches. These strategies have shown promising therapeutic effects by specifically targeting the intricate network of signaling pathways implicated in I/R pathology.
renal I/R injury when the ischemic IPC treatment strategy is applied.
DISCUSSION AND PERSPECTIVES
| Table 1. Therapeutic strategies targeting Wnt signaling for the treatment of I/R injury | |||||
| Therapeutic strategy | Target pathways | Strategy/Molecular/ Drugs | Organ | Effects | References |
| Cell therapy | Active Wnt/
|
Exosomes isolated from adipose-derived mesenchymal stem cells, ADMSCs-EX | Heart | Up-regulate Wnt3a; Inhibiting apoptosis | 95 |
| Oligodendrocyte precursor cell transplantation | Brain | Promoting angiogenesis; Repair BBB integrity | 239 | ||
| Transplanted adipose stem cell exosomes (ADSCsExo) | Liver | Inhibit NF-кB pathway; Reducing pyroptosis of damaged liver | 296 | ||
| Gene therapy | Active Wnt/
|
Down-regulated miR-148b | Heart | Up-regulate Wnt1; Inhibiting apoptosis and oxidative damage | 93 |
| Up-regulation of LncRNA AZIN1-AS1 | Heart | LncRNA AZIN1-AS1/miR-6838-5p active Wnt3a /
|
94 | ||
| Nur77 gene ablation | Brain | Inhibiting mitochondrial fragmentation | 176 | ||
| Overexpression of LncRNA NEAT1 | Brain | Stable Wnt3a; Inhibiting apoptosis | 174 | ||
| Down-regulation of IncRNA MEG | Brain | Promoting neurogenesis | 232,252 | ||
| Molecular therapy | Inhibit Wnt noncanonical pathways | Up-regulation of Sfrp5 | Heart | Inhibit Wnt5a/JNK and Wnt/PCP; Inhibiting apoptosis and inflammation | 96-99,102 |
| Medication | Active Wnt/
|
A polypeptide of tuna stem protein, APTBP | Heart | Inhibiting apoptosis | 127,128 |
| Phyllanthus emblica (P. emblica) | Heart | Active PI3K/Akt/GSK3
|
334 | ||
| CHIR99021 (GSK3
|
Heart | Active Hippo pathway; Inhibiting apoptosis and cell hypertrophy | 137 | ||
| Isoflurane | Brain | Inhibiting apoptosis | 165 | ||
| XQ-1H; gastrodin | Brain | Inhibiting apoptosis; Promoting neurogenesis | 175,252 | ||
| Peroxynitrite; Mallotus oblongifolius;ellagic acid | Brain | Promoting neurogenesis | 221,229,230 | ||
| Curcumin | Brain | Promoting neurogenesis; Inhibiting apoptosis; Relieving inflammation | 208,211,231 | ||
| TWS119 | Brain | Repairing BBB; Reducing neuroinflammation | 209 | ||
| TWS119 | Brain | Inhibit Notch; Inhibiting apoptosis | 318 | ||
| Quercetin | Brain | Repairing BBB | 246,254 | ||
| Human serum albumin | Brain | Reducing oxidative stress | 212 | ||
| Galangin | Brain | nhibit HIF-1
|
343 | ||
| Dexmedetomidine | Brain | Active PI3K/AKT; Reducing cerebral infarct volume; Promoting neuronal survival | 333,334 | ||
| Wnt agonist (a synthetic pyrimidine) | Kidney | Inhibiting inflammatory response and oxidative stress | 293 | ||
| Minocycline | Liver | Reducing oxidative stress; Inhibiting the release of proinflammatory cytokines | 287 | ||
| Agmatine | Liver | Promoting cell proliferation; Reducing inflammation and apoptosis | 282 | ||
| Losartan | Liver | Up-regulate HIF-1
|
285 | ||
| Inhibit Wnt/
|
Dexamethasone, Dex | Heart | MiR-208b-3p/Med13/Wnt/
|
333,334 | |
| Baicalin | Heart | Reducing oxidative damage to cardiomyocytes | 414 | ||
| Huoxin pill | Heart | Inhibit NF-кB; Inhibiting inflammation | 107,366 | ||
| Melatonin | Kidney | Improving renal fibrosis | 385 | ||
| Inhibit Wnt noncanonical pathways | Curcumin | Brain | Inhibit Wnt/PCP; Promoting neurogenesis; Inhibiting apoptosis; Relieving inflammation | 211,231 | |
| Phase | Wnt signaling pathway | Activity | Organ | Effect | References |
| Ischemia | Wnt/
|
Activated | Heart | Promoting inflammation, ECM remodeling, angiogenesis, fibrosis | 115-117,129,130,138-141 |
| Brain | Promoting neurogenesis (in vitro) | 221 | |||
| Kidney | Promoting apoptosis and oxidative stress | 256 | |||
| Inhibited | Heart | Promoting oxidative stress | 78,126 | ||
| Brain | Promoting apoptosis, ferroptosis, inflammation, inhibiting neurogenesis, inhibiting angiogenesis, destroying BBB | 169-171,173,193,209,210,228,239,242,248,343,395 | |||
| Wnt/PCP | Activated | Heart | Promoting inflammation, cell hypertrophy | 146 | |
| Wnt/
|
Activated | Brain | Promoting apoptosis (in vitro) | 167 | |
| Reperfusion | Wnt/
|
Inhibited | Heart | Promoting apoptosis inflammation, cell hypertrophy | 93-95,120,137 |
| Brain | Promoting apoptosis, inflammation, oxidative stress, inhibiting neurogenesis, inhibiting angiogenesis, destroying BBB | 10,164,165,174,175,207,232,244,246 | |||
| Liver | Promoting apoptosis, oxidative stress, inflammation, inhibiting cell proliferation | 267-271,283,285,293,296 | |||
| Activated | Brain | Inhibiting apoptosis (in vitro) | 166 | ||
| Kidney | Inhibiting apoptosis, promoting apoptosis (in vitro), mitophagy, cell autophagy, cell aging and renal fibrosis | 25,252,253,258-263 | |||
| Wnt/PCP | Activated | Heart | Promoting apoptosis, inflammation, angiogenesis | 50,102 | |
| Brain | Promote apoptosis, inflammation | 29,208 | |||
| Wnt/
|
Activated | Heart | Promoting apoptosis | 103 | |
| Kidney | Promoting renal fibrosis | 264 | |||
| Liver | Promoting apoptosis | 103,272,273 |
comprehensive understanding of the role of Wnt/
activate the PI3K/Akt signaling pathway and mitigate brain I/R injury. Additionally, these TCM treatments have also been found to modulate the activity of the Wnt/
ACKNOWLEDGEMENTS
AUTHOR CONTRIBUTIONS
FUNDING
ADDITIONAL INFORMATION
REFERENCES
- Frangogiannis, N. G. Pathophysiology of myocardial infarction. Compr. Physiol. 5, 1841-1875 (2015).
- Zhao, Y., Zhang, X., Chen, X. & Wei, Y. Neuronal injuries in cerebral infarction and ischemic stroke: from mechanisms to treatment (Review). Int. J. Mol. Med. 49, 15 (2022).
- Yapca, O. E., Borekci, B. & Suleyman, H. Ischemia-reperfusion damage. Eurasia. J. Med. 45, 126-127 (2013).
- Eltzschig, H. K. & Eckle, T. Ischemia and reperfusion-from mechanism to translation. Nat. Med. 17, 1391-1401 (2011).
- Wu, M. Y. et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell. Physiol. Biochem. 46, 1650-1667 (2018).
- Hosohata, K., Harnsirikarn, T. & Chokesuwattanaskul, S. Ferroptosis: a potential therapeutic target in acute kidney injury. Int. J. Mol. Sci. 23, 6583 (2022).
- Tan, H., Chen, L. & Ma, J. Penehyclidine hydrochloride post-conditioning reduces ischemia/reperfusion-induced cardiomyocyte apoptosis in rats. Exp. Ther. Med 14, 4272-4278 (2017).
- Liu, H. et al. Inhibition of Brd4 alleviates renal ischemia/reperfusion injuryinduced apoptosis and endoplasmic reticulum stress by blocking FoxO4mediated oxidative stress. Redox Biol. 24, 101195 (2019).
- Guo, Z. et al. NLRP3 is involved in ischemia/reperfusion injury. CNS Neurol. Disord. Drug Targets 15, 699-712 (2016).
- Ji, Y. B. et al. Lithium alleviates blood-brain barrier breakdown after cerebral ischemia and reperfusion by upregulating endothelial Wnt/
-catenin signaling in mice. Neuropharmacology 186, 108474 (2021). - Burke, R. M., Burgos Villar, K. N. & Small, E. M. Fibroblast contributions to ischemic cardiac remodeling. Cell Signal 77, 109824 (2021).
- Wu, X., Reboll, M. R., Korf-Klingebiel, M. & Wollert, K. C. Angiogenesis after acute myocardial infarction. Cardiovasc. Res. 117, 1257-1273 (2021).
- Smiley, D. et al. Increased fibrosis and progression to heart failure in MRL mice following ischemia/reperfusion injury. Cardiovasc. Pathol. 23, 327-334 (2014).
- Salminen, A., Liu, P. K. & Hsu, C. Y. Alteration of transcription factor binding activities in the ischemic rat brain. Biochem. Biophys. Res. Commun. 212, 939-944 (1995).
- Werling, L. L. et al. Increased activation of L-type voltage-dependent calcium channels is associated with glycine enhancement of N-methyl-D-aspartate-stimulated dopamine release in global cerebral ischemia/reperfusion. J. Neurochem. 63, 215-221 (1994).
- Lefer, A. M. Mechanisms of the protective effects of transforming growth factorbeta in reperfusion injury. Biochem. Pharm. 42, 1323-1327 (1991).
- Tacchini, L., Radice, L. & Bernelli-Zazzera, A. Differential activation of some transcription factors during rat liver ischemia, reperfusion, and heat shock. J. Cell Physiol. 180, 255-262 (1999).
- Vukicevic, S. et al. Osteogenic protein-1 (bone morphogenetic protein-7) reduces the severity of injury after ischemic acute renal failure in rat. J. Clin. Investig. 102, 202-214 (1998).
- Mockridge, J. W., Marber, M. S. & Heads, R. J. Activation of Akt during simulated ischemia/reperfusion in cardiac myocytes. Biochem. Biophys. Res. Commun. 270, 947-952 (2000).
- Sakakura, Y. et al. Recombinant human hepatocyte growth factor protects the liver against hepatic ischemia and reperfusion injury in rats. J. Surg. Res. 92, 261-266 (2000).
21. Arumugam, T. V. et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcomes in ischemic stroke. Nat. Med. 12, 621-623 (2006).
22. Terada, Y. et al. Expression and function of the developmental gene Wnt-4 during experimental acute renal failure in rats. J. Am. Soc. Nephrol. 14, 1223-1233 (2003).
23. Shao, D. et al. A functional interaction between Hippo-YAP signaling and FoxO1 mediates the oxidative stress response. Nat. Commun. 5, 3315 (2014).
24. Pulskens, W. P. et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One 3, e3596 (2008).
25. Dong, Q. et al. Wnt/
26. Meyer, I. S. et al. Blockade of Wnt Secretion Attenuates Myocardial IschemiaReperfusion Injury by Modulating the Inflammatory Response. Int. J. Mol. Sci. 23, 12252 (2022).
27. Liu, J., Zheng, X., Zhang, C., Zhang, C. & Bu, P. Lcz696 alleviates myocardial fibrosis after myocardial infarction through the sFRP-1/Wnt/
28. Fuping, Z. et al. Tao-Hong-Si-Wu decoction reduces ischemia reperfusion rat myoblast cells calcium overloading and inflammation through the Wnt/IP3R/ CAMKII pathway. J. Cell. Biochem. 120, 13095-13106 (2019).
29. Wei, X. et al. Targeting the Dvl-1/
30. Gao, C. & Chen, Y. G. Dishevelled: the hub of Wnt signaling. Cell. Signal 22, 717-727 (2010).
31. Ben-Ghedalia-Peled, N. & Vago, R. Wnt Signaling in the Development of Bone Metastasis. Cells. 11, 3934 (2022).
32. Carmon, K. S., Gong, X., Lin, Q., Thomas, A. & Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl Acad. Sci. USA 108, 11452-11457 (2011).
33. de Lau, W. et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293-297 (2011).
34. Kasai, M., Satoh, K. & Akiyama, T. Wnt signaling regulates the sequential onset of neurogenesis and gliogenesis via induction of BMPs. Genes Cells 10, 777-783 (2005).
35. Trifan, G., Biller, J. & Testai, F. D. Mechanical thrombectomy vs bridging therapy for anterior circulation large vessel occlusion stroke: systematic review and meta-analysis. Neurology 98, e1361-e1373 (2022).
36. Kalogeris, T., Baines, C. P., Krenz, M. & Korthuis, R. J. Ischemia/reperfusion. Compr. Physiol. 7, 113-170 (2016).
37. Peng, T. I. & Jou, M. J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201, 183-188 (2010).
38. Salvadori, M., Rosso, G. & Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: pathogenesis and treatment. World J. Transpl. 5, 52-67 (2015).
39. Malis, C. D. & Bonventre, J. V. Mechanism of calcium potentiation of oxygen free radical injury to renal mitochondria. a model for post-ischemic and toxic mitochondrial damage. J. Biol. Chem. 261, 14201-14208 (1986).
40. Nieuwenhuijs-Moeke, G. J. et al. Ischemia and reperfusion injury in kidney transplantation: relevant mechanisms in injury and repair. J Clin Med. 9, 253 (2020).
41. Gujral, J. S., Bucci, T. J., Farhood, A. & Jaeschke, H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology 33, 397-405 (2001).
42. Glinka, A. et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/
43. de Lau, W., Peng, W. C., Gros, P. & Clevers, H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 28, 305-316 (2014).
44. Molenaar, M. et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 86, 391-399 (1996).
45. Pell, V. R. et al. Ischemic preconditioning protects against cardiac ischemia reperfusion injury without affecting succinate accumulation or oxidation. J. Mol. Cell Cardiol. 123, 88-91 (2018).
46. Lee, S. M., Hutchinson, M., Staikopoulos, V. & Saint, D. A. Amitriptyline pharmacologically preconditions rat hearts against cardiac ischemic-reperfusion injury. Int. J. Cardiol. 190, 353-359 (2015).
47. Li, Y., Cai, M., Xu, Y., Swartz, H. M. & He, G. Late phase ischemic preconditioning preserves mitochondrial oxygen metabolism and attenuates post-ischemic myocardial tissue hyper oxygenation. Life Sci. 88, 57-64 (2011).
48. Sárközy, M. et al. Ischemic preconditioning protects the heart against ischemiareperfusion injury in chronic kidney disease in both males and females. Biol. Sex. Differ. 12, 49 (2021).
49. Nusse, R. & Clevers, H. Wnt/
50. Wang, J. et al. WNT11-conditioned medium promotes angiogenesis through the activation of non-canonical WNT-PKC-JNK signaling pathway. Genes 11, 1277 (2020).
51. Gajos-Michniewicz, A. & Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 21, 4852 (2020).
52. Rim, E. Y., Clevers, H. & Nusse, R. The Wnt pathway: from signaling mechanisms to synthetic modulators. Annu. Rev. Biochem 91, 571-598 (2022).
53. Wang, H. et al. The Wnt signaling pathway in diabetic nephropathy. Front. Cell. Dev. Biol. 9, 701547 (2021).
54. Malik, S. A., Modarage, K. & Goggolidou, P. The Role of Wnt Signalling in Chronic Kidney Disease (CKD). Genes 11, 496 (2020).
55. Wang, H. Y., Liu, T. & Malbon, C. C. Structure-function analysis of Frizzleds. Cell. Signal 18, 934-941 (2006).
56. Joiner, D. M., Ke, J., Zhong, Z., Xu, H. E. & Williams, B. O. LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. 24, 31-39 (2013).
57. Menck, K., Heinrichs, S., Baden, C. & Bleckmann, A. The WNT/ROR Pathway in Cancer: From Signaling to Therapeutic Intervention. Cells. 10, 142 (2021).
58. Jung, Y. S. & Park, J. I. Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond
59. Chae, W. J. & Bothwell, A. L. M. Canonical and non-canonical Wnt signaling in immune cells. Trends Immunol. 39, 830-847 (2018).
60. Tran, F. H. & Zheng, J. J. Modulating the wnt signaling pathway with small molecules. Protein Sci. 26, 650-661 (2017).
61. Akoumianakis, I., Polkinghorne, M. & Antoniades, C. Non-canonical WNT signaling in cardiovascular disease: mechanisms and therapeutic implications. Nat. Rev. Cardiol. 19, 783-797 (2022).
62. Jin, Z. et al. Neuroprotective effects of irisin against cerebral ischemia/ reperfusion injury via Notch signaling pathway. Biomed. Pharmacother. 120, 109452 (2019).
63. Gordon, M. D. & Nusse, R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281, 22429-22433 (2006).
64. Veeman, M. T., Axelrod, J. D. & Moon, R. T. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev. Cell. 5, 367-377 (2003).
65. Kikuchi, A. & Yamamoto, H. Tumor formation due to abnormalities in the beta-catenin-independent pathway of Wnt signaling. Cancer Sci. 99, 202-208 (2008).
66. Shi, D. L. Decoding dishevelled-mediated Wnt signaling in vertebrate early development. Front. Cell Dev. Biol. 8, 588370 (2020).
67. Lerner, U. H. & Ohlsson, C. The WNT system: background and its role in bone. J. Intern. Med. 277, 630-649 (2015).
68. VanderVorst, K. et al. Wnt/PCP signaling contribution to carcinoma collective cell migration and metastasis. Cancer Res. 79, 1719-1729 (2019).
69. Frenquelli, M. & Tonon, G. WNT signaling in hematological malignancies. Front. Oncol. 10, 615190 (2020).
70. Cho, S. J. et al. Wip1 directly dephosphorylates NLK and increases Wnt activity during germ cell development. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1013-1022 (2017).
71. Xiao, Q., Chen, Z., Jin, X., Mao, R. & Chen, Z. The many postures of noncanonical Wnt signaling in development and diseases. Biomed. Pharmacother. 93, 359-369 (2017).
72. Ma, L. & Wang, H. Y. Mitogen-activated protein kinase p38 regulates the Wnt/ cyclic GMP/Ca2+ non-canonical pathway. J. Biol. Chem. 282, 28980-28990 (2007).
73. Hausenloy, D. J. & Yellon, D. M. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J. Clin. Investig. 123, 92-100 (2013).
74. Vaage, J. & Valen, G. Pathophysiology and mediators of ischemia-reperfusion injury with special reference to cardiac surgery. A review. Scand. J. Thorac. Cardiovasc Surg. Suppl. 41, 1-18 (1993).
75. Tanaka, M. et al. Cardiomyocyte-specific Bcl-2 overexpression attenuates ischemia-reperfusion injury, immune response during acute rejection, and graft coronary artery disease. Blood 104, 3789-3796 (2004).
76. Kleinbongard, P., Heusch, G. & Schulz, R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharm. Ther. 127, 295-314 (2010).
77. Frohlich, G. M., Meier, P., White, S. K., Yellon, D. M. & Hausenloy, D. J. Myocardial reperfusion injury: looking beyond primary PCl . Eur. Heart J. 34, 1714-1722 (2013).
78. Shen, J. et al. Wnt 3a protects myocardial injury in elderly acute myocardial infarction by inhibiting serum cystatin C/ROS-induced mitochondrial damage. Front. Physiol. 13, 950960 (2022).
79. Piper, H. M., García-Dorado, D. & Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 38, 291-300 (1998).
80. Yellon, D. M. & Hausenloy, D. J. Myocardial reperfusion injury. N. Engl. J. Med 357, 1121-1135 (2007).
81. Logue, S. E., Gustafsson, A. B., Samali, A. & Gottlieb, R. A. Ischemia/reperfusion injury at the intersection with cell death. J. Mol. Cell Cardiol. 38, 21-33 (2005).
82. Gottlieb, R. A. Cell death pathways in acute ischemia/reperfusion injury. J. Cardiovasc. Pharm. Ther. 16, 233-238 (2011).
83. Chen, Y. et al. Ferroptosis: a novel therapeutic target for ischemia-reperfusion injury. Front. Cell Dev. Biol. 9, 688605 (2021).
84. Hamacher-Brady, A., Brady, N. R. & Gottlieb, R. A. The interplay between prodeath and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther. 20, 445-462 (2006).
85. Deb, A. Cell-cell interaction in the heart via Wnt/beta-catenin pathway after cardiac injury. Cardiovasc. Res. 102, 214-223 (2014).
86. Lorenzon, A. et al. Wnt/beta-catenin pathway in arrhythmogenic cardiomyopathy. Oncotarget 8, 60640-60655 (2017).
87. Bergmann, M. W. WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ. Res. 107, 1198-1208 (2010).
88. Oerlemans, M. I. et al. Active Wnt signaling in response to cardiac injury. Basic Res. Cardiol. 105, 631-641 (2010).
89. Haybar, H., Khodadi, E. & Shahrabi, S. Wnt/
90. Litvinukova, M. et al. Cells of the adult human heart. Nature 588, 466-472 (2020).
91. Bastakoty, D. et al. Temporary, Systemic inhibition of the WNT/beta-catenin pathway promotes regenerative cardiac repair following myocardial infarct. Cell Stem Cells Regen. Med. 2, 16966 (2016).
92. Haybar, H., Khodadi, E. & Shahrabi, S. Wnt/beta-catenin in ischemic myocardium: interactions and signaling pathways as a therapeutic target. Heart Fail Rev. 24, 411-419 (2019).
93. Yang, M., Kong, D. Y. & Chen, J. C. Inhibition of miR-148b ameliorates myocardial ischemia/reperfusion injury via regulation of Wnt/beta-catenin signaling pathway. J. Cell Physiol. 234, 17757-17766 (2019).
94. Zhang, G. et al. LncRNA AZIN1-AS1 ameliorates myocardial ischemia-reperfusion injury by targeting miR-6838-5p/WNT3A axis to activate Wnt-beta/catenin signaling pathway. Vitr. Cell Dev. Biol. Anim. 58, 54-68 (2022).
95. Cui, X. et al. Exosomes from adipose-derived mesenchymal stem cells protect the myocardium against ischemia/reperfusion injury through Wnt/beta-catenin signaling pathway. J. Cardiovasc. Pharm. 70, 225-231 (2017).
96. Finch, P. W. et al. Purification and molecular cloning of a secreted, Frizzledrelated antagonist of Wnt action. Proc. Natl Acad. Sci. USA 94, 6770-6775 (1997).
97. Bovolenta, P., Esteve, P., Ruiz, J. M., Cisneros, E. & Lopez-Rios, J. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J. Cell Sci. 121, 737-746 (2008).
98. Ouchi, N. et al. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 329, 454-457 (2010).
99. Li, Y. et al. Sfrp5 coordinates foregut specification and morphogenesis by antagonizing both canonical and noncanonical Wnt11 signaling. Genes Dev. 22, 3050-3063 (2008).
100. Kikuchi, R. et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med 20, 1464-1471 (2014).
101. Fuster, J. J. et al. Noncanonical Wnt signaling promotes obesity-induced adipose tissue inflammation and metabolic dysfunction independent of adipose tissue expansion. Diabetes 64, 1235-1248 (2015).
102. Nakamura, K. et al. Secreted Frizzled-related protein 5 diminishes cardiac inflammation and protects the heart from ischemia/reperfusion injury. J. Biol. Chem. 291, 2566-2575 (2016).
103. Zhou, S. S., He, F., Chen, A. H., Hao, P. Y. & Song, X. D. Suppression of rat Frizzled2 attenuates hypoxia/reoxygenation-induced
104. Zhang, L. et al. Inhibition of Rac1 reduces store overload-induced calcium release and protects against ventricular arrhythmia. J. Cell Mol. Med. 20, 1513-1522 (2016).
105. Belevych, A. E. et al. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res 84, 387-395 (2009).
106. Fauconnier, J. et al. Ryanodine receptor leak mediated by caspase- 8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc. Natl Acad. Sci. USA 108, 13258-13263 (2011).
107. He, J. et al. Huoxin pill prevents excessive inflammation and cardiac dysfunction following myocardial infarction by inhibiting adverse Wnt/betacatenin signaling activation. Phytomedicine 104, 154293 (2022).
108. Hu, Y. et al. Class A scavenger receptor attenuates myocardial infarction-induced cardiomyocyte necrosis through suppressing M1 macrophage subset polarization. Basic Res. Cardiol. 106, 1311-1328 (2011).
109. Cutolo, M., Campitiello, R., Gotelli, E. & Soldano, S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front. Immunol. 13, 867260 (2022).
110. Zhang, R. Y. K., Cochran, B. J., Thomas, S. R. & Rye, K. A. Impact of reperfusion on temporal immune cell dynamics after myocardial infarction. J. Am. Heart Assoc. 12, e027600 (2023).
111. Prabhu, S. D. & Frangogiannis, N. G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 119, 91-112 (2016).
112. Nahrendorf, M. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037-3047 (2007).
113. Cheng, B., Chen, H. C., Chou, I. W., Tang, T. W. & Hsieh, P. C. Harnessing the early post-injury inflammatory responses for cardiac regeneration. J. Biomed. Sci. 24, 7 (2017).
114. Huang, C. K. et al. Lgr4 governs a pro-inflammatory program in macrophages to antagonize post-infarction cardiac repair. Circ. Res. 127, 953-973 (2020).
115. Zhao, G. et al. CXCR6 deficiency ameliorated myocardial ischemia/reperfusion injury by inhibiting infiltration of monocytes and IFN-
116. Gombozhapova, A. et al. Macrophage activation and polarization in postinfarction cardiac remodeling. J. Biomed. Sci. 24, 13 (2017).
117. Yuan, C. et al. Modulation of Wnt/
118. Palevski, D. et al. Loss of macrophage Wnt secretion improves remodeling and function after myocardial infarction in mice. J. Am. Heart Assoc. 6, e004387 (2017).
119. Frangogiannis, N. G. The inflammatory response in myocardial injury, repair, and remodeling. Nat. Rev. Cardiol. 11, 255-265 (2014).
120. Sun, S., Wu, Y., Maimaitijiang, A., Huang, Q. & Chen, Q. Ferroptotic cardiomyocyte-derived exosomes promote cardiac macrophage M1 polarization during myocardial infarction. PeerJ. 10, e13717 (2022).
121. Huang, L., Xiang, M., Ye, P., Zhou, W. & Chen, M. Beta-catenin promotes macrophage-mediated acute inflammatory response after myocardial infarction. Immunol. Cell. Biol. 96, 100-113 (2018).
122. Aisagbonhi, O. et al. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model Mech. 4, 469-483 (2011).
123. Aleshin, A. et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol. 294, H1823-H1832 (2008).
124. Bucciarelli, L. G. et al. Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury. Circulation 113, 1226-1234 (2006).
125. Park, H. et al. RAGE siRNA-mediated gene silencing provides cardioprotection against ventricular arrhythmias in acute ischemia and reperfusion. J. Control Release 217, 315-326 (2015).
126. Rauner, M. et al. WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Min. Res. 27, 575-585 (2012).
127. Meyer, I. S. et al. The cardiac microenvironment uses non-canonical WNT signaling to activate monocytes after myocardial infarction. EMBO Mol. Med. 9, 1279-1293 (2017).
128. Moon, J. et al. Blockade to pathological remodeling of infarcted heart tissue using a porcupine antagonist. Proc. Natl Acad. Sci. USA 114, 1649-1654 (2017).
129. Pereira, C., Schaer, D. J., Bachli, E. B., Kurrer, M. O. & Schoedon, G. Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler. Thromb. Vasc. Biol. 28, 504-510 (2008).
130. Port, F. et al. Wingless secretion promotes and requires retromer-dependent cycling of Wntless. Nat. Cell Biol. 10, 178-185 (2008).
131. Guo, X. et al. Induced pluripotent stem cell-conditional medium inhibits H9C2 cardiomyocytes apoptosis via autophagy flux and Wnt/beta-catenin pathway. J. Cell. Mol. Med 23, 4358-4374 (2019).
132. Je, J. Y., Qian, Z. J., Byun, H. G. & Kim, S. K. Purification and characterization of an antioxidant peptide obtained from tuna backbone protein by enzymatic hydrolysis. Process Biochem. 42, 840-846 (2007).
133. Zhang, L. et al. The restoration of Wnt/
134. Blankesteijn, W. M., van Gijn, M. E., Essers-Janssen, Y. P., Daemen, M. J. & Smits, J. F. Beta-catenin, an inducer of uncontrolled cell proliferation and migration in malignancies, is localized in the cytoplasm of vascular endothelium during neovascularization after myocardial infarction. Am. J. Pathol. 157, 877-883 (2000).
32
135. Barandon, L. et al. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation 108, 2282-2289 (2003).
136. Baruah, J. et al. The allosteric glycogen synthase kinase-3 inhibitor NP12 limits myocardial remodeling and promotes angiogenesis in an acute myocardial infarction model. J. Biol. Chem. 292, 20785-20798 (2017).
137. Liu, B. et al. Loss of endothelial glucocorticoid receptor promotes angiogenesis via upregulation of Wnt/beta-catenin pathway. Angiogenesis 24, 631-645 (2021).
138. MacLellan, W. R. & Schneider, M. D. Genetic dissection of cardiac growth control pathways. Annu. Rev. Physiol. 62, 289-319 (2000).
139. Nakamura, M. & Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387-407 (2018).
140. Shimizu, I. & Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 97, 245-262 (2016).
141. Hagenmueller, M. et al. Dapper-1 is essential for Wnt5a induced cardiomyocyte hypertrophy by regulating the Wnt/PCP pathway. FEBS Lett. 588, 2230-2237 (2014).
142. Khan, K., Makhoul, G., Yu, B., Schwertani, A. & Cecere, R. The cytoprotective impact of yes-associated protein 1 after ischemia-reperfusion injury in AC16 human cardiomyocytes. Exp. Biol. Med. 244, 802-812 (2019).
143. Zhao, X. et al. Aldehyde dehydrogenase-2 protects against myocardial infarction-related cardiac fibrosis through modulation of the Wnt/beta-catenin signaling pathway. Ther. Clin. Risk Manag. 11, 1371-1381 (2015).
144. Qian, L. et al. Downregulation of S100A4 Alleviates Cardiac Fibrosis via Wnt/beta -Catenin Pathway in Mice. Cell Physiol. Biochem 46, 2551-2560 (2018).
145. Cui, S. et al. miR-145 attenuates cardiac fibrosis through the AKT/GSK-3beta/ beta-catenin signaling pathway by directly targeting SOX9 in fibroblasts. J. Cell Biochem. 122, 209-221 (2021).
146. Matsushima, K. et al. Secreted frizzled related protein 4 reduces fibrosis scar size and ameliorates cardiac function after ischemic injury. Tissue Eng. Part A 16, 3329-3341 (2010).
147. Duan, J. et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 31, 429-442 (2012).
148. Zhang, Y. et al. PRELP promotes myocardial fibrosis and ventricular remodeling after acute myocardial infarction by the wnt/
149. Jean LeBlanc, N. et al. Canonical Wnt pathway maintains blood-brain barrier integrity upon ischemic stroke and its activation ameliorates tissue plasminogen activator therapy. Mol. Neurobiol. 56, 6521-6538 (2019).
150. Abuelazm, M. et al. The efficacy and safety of tenecteplase versus alteplase for acute ischemic stroke: an updated systematic review, pairwise, and network meta-analysis of randomized controlled trials. J. Thromb. Thrombolysis 55, 322-338 (2023).
151. Berge, E. et al. European Stroke Organisation (ESO) guidelines on intravenous thrombolysis for acute ischaemic stroke. Eur Stroke J. 6, I-Ixii (2021).
152. Emberson, J. et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomized trials. Lancet 384, 1929-1935 (2014).
153. Xiong, Y., Wakhloo, A. K. & Fisher, M. Advances in Acute Ischemic Stroke Therapy. Circ. Res. 130, 1230-1251 (2022).
154. Katsanos, A. H. et al. Intravenous thrombolysis prior to mechanical thrombectomy in large vessel occlusions. Ann. Neurol. 86, 395-406 (2019).
155. Fischer, U. et al. Primary thrombectomy in tPA (Tissue-Type Plasminogen Activator) eligible stroke patients with proximal intracranial occlusions. Stroke 49, 265-269 (2018).
156. Rai, A. T. et al. Intravenous thrombolysis before endovascular therapy for large vessel strokes can lead to significantly higher hospital costs without improving outcomes. J. Neurointerv. Surg. 10, 17-21 (2018).
157. Goyal, N. et al. Impact of pretreatment with intravenous thrombolysis on reperfusion status in acute strokes treated with mechanical thrombectomy. J. Neurointerv Surg. 11, 1073-1079 (2019).
158. Rossi, R. et al. Does prior administration of rtPA influence acute ischemic stroke clot composition? Findings from the analysis of clots retrieved with mechanical thrombectomy from the RESTORE registry. J. Neurol. 269, 1913-1920 (2022).
159. Muroyama, Y., Kondoh, H. & Takada, S. Wnt proteins promote neuronal differentiation in neural stem cell culture. Biochem. Biophys. Res. Commun. 313, 915-921 (2004).
160. Maretto, S. et al. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl Acad. Sci. USA 100, 3299-3304 (2003).
161. McKenzie, M. G. et al. Non-canonical Wnt signaling through Ryk regulates the generation of somatostatin- and parvalbumin-expressing cortical interneurons. Neuron 103, 853-864.e854 (2019).
162. Lewis, J. L. et al. Reiterated Wnt signaling during zebrafish neural crest development. Development 131, 1299-1308 (2004).
163. Hutchins, B. I., Li, L. & Kalil, K. Wnt-induced calcium signaling mediates axon growth and guidance in the developing corpus callosum. Sci. Signal. 5, pt1 (2012).
164. Rosso, S. B., Sussman, D., Wynshaw-Boris, A. & Salinas, P. C. Wnt signaling through Dishevelled, Rac, and JNK regulates dendritic development. Nat. Neurosci. 8, 34-42 (2005).
165. Liebner, S. et al. Wnt/beta-catenin signaling controls development of the bloodbrain barrier. J. Cell Biol. 183, 409-417 (2008).
166. Benz, F. et al. Low wnt/
167. Shi, Z. Y. et al. Protective effect of autophagy in neural ischemia and hypoxia: negative regulation of the Wnt/
168. Ji, Y. B., Wang, T. X., Gao, Q., Huang, X. W. & Chang, J. Normalization of noncanonical Wnt signalings does not compromise blood-brain barrier protection conferred by upregulating endothelial Wnt/
169. Zhao, H. et al. Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt
170. Zhang, G. et al. Wnt/
171. Li, T. et al. DIXDC1 prevents oxygen-glucose deprivation/reoxygenation-induced injury in hippocampal neurons in vitro by promoting Wnt/
172. Niu, L. J., Xu, R. X., Zhang, P., Du, M. X. & Jiang, X. D. Suppression of Frizzled-2mediated Wnt/
173. Kunz, A., Dirnagl, U. & Mergenthaler, P. Acute pathophysiological processes after ischaemic and traumatic brain injury. Best. Pr. Res. Clin. Anaesthesiol. 24, 495-509 (2010).
174. Seifert-Held, T. et al. Circulating Dickkopf-1 in acute ischemic stroke and clinically stable cerebrovascular disease. Atherosclerosis 218, 233-237 (2011).
175. Cappuccio, I. et al. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is required for the development of ischemic neuronal death. J. Neurosci. 25, 2647-2657 (2005).
176. Mastroiacovo, F. et al. Induction of the Wnt antagonist, Dickkopf-1, contributes to the development of neuronal death in models of brain focal ischemia. J. Cereb. Blood Flow. Metab. 29, 264-276 (2009).
177. Scali, C. et al. Inhibition of Wnt signaling, modulation of Tau phosphorylation and induction of neuronal cell death by DKK1. Neurobiol. Dis. 24, 254-265 (2006).
178. Che, Q. Q., Huang, T., Zhang, Y. D. & Qian, X. J. Effect of miR-124 on neuronal apoptosis in rats with cerebral infarction through Wnt/
179. Zhou, Z., Ren, X., Zheng, L., Li, A. & Zhou, W. LncRNA NEAT1 stabilized Wnt3a via U2AF2 and activated Wnt/
180. Xu, D. et al. XQ-1H alleviates cerebral ischemia in mice through inhibition of apoptosis and promotion of neurogenesis in a Wnt/
181. Zhao, H., Pan, W., Chen, L., Luo, Y. & Xu, R. Nur77 promotes cerebral ischemiareperfusion injury via activating INF2-mediated mitochondrial fragmentation. J. Mol. Histol. 49, 599-613 (2018).
182. Chong, Z. Z. & Maiese, K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol. Histopathol. 19, 495-504 (2004).
183. Guo, C. & Whitmarsh, A. J. The beta-arrestin-2 scaffold protein promotes c-Jun N -terminal kinase- 3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 283, 15903-15911 (2008).
184. Kuan, C. Y. et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl Acad. Sci. USA 100, 15184-15189 (2003).
185. Zhang, Q. G., Wang, R., Khan, M., Mahesh, V. & Brann, D. W. Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J. Neurosci. 28, 8430-8441 (2008).
186. Cheng, Y. L. et al. Evidence that collaboration between HIF-1a and Notch-1 promotes neuronal cell death in ischemic stroke. Neurobiol. Dis. 62, 286-295 (2014).
187. Burchell, S. R., Dixon, B. J., Tang, J. & Zhang, J. H. Isoflurane provides neuroprotection in neonatal hypoxic ischemic brain injury. J. Investig. Med. 61, 1078-1083 (2013).
188. Lan, X. B. et al. Neuroprotective effects of oxymatrine on hypoxic-ischemic brain damage in neonatal rats by activating the Wnt/
189. Yan, H. F., Tuo, Q. Z., Yin, Q. Z. & Lei, P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool. Res. 41, 220-230 (2020).
190. Li, D. & Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target Ther. 5, 108 (2020).
191. Li, L., Li, Y. W., Zhao, J. Y., Liu, Y. Z. & Holscher, C. Quantitative analysis of iron concentration and expression of ferroportin 1 in the cortex and hippocampus of rats induced by cerebral ischemia. J. Clin. Neurosci. 16, 1466-1472 (2009).
192. Won, S. M. et al. Iron mediates endothelial cell damage and blood-brain barrier opening in the hippocampus after transient forebrain ischemia in rats. Exp. Mol. Med. 43, 121-128 (2011).
193. Hällgren, R., Terent, A., Wide, L., Bergström, K. & Birgegård, G. Cerebrospinal fluid ferritin in patients with cerebral infarction or bleeding. Acta Neurol. Scand. 61, 384-392 (1980).
194. Shi, Y. et al. Selenium alleviates cerebral ischemia/reperfusion injury by regulating oxidative stress, mitochondrial fusion and ferroptosis. Neurochem. Res. 47, 2992-3002 (2022).
195. Groenendaal, F., Shadid, M., McGowan, J. E., Mishra, O. P. & van Bel, F. Effects of deferoxamine, a chelator of free iron, on NA(+), K(+)-ATPase activity of cortical brain cell membrane during early reperfusion after hypoxia-ischemia in newborn lambs. Pediatr. Res. 48, 560-564 (2000).
196. Shadid, M. et al. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neurosci. Lett. 248, 5-8 (1998).
197. Zhao, Y. et al. Nano-liposomes of lycopene reduces ischemic brain damage in rodents by regulating iron metabolism. Free Radic. Biol. Med. 124, 1-11 (2018).
198. Yin, M. et al. circAFF1 enhances intracerebral hemorrhage induced neuronal ferroptosis by targeting miR-140-5p to regulate GSK-3
199. Wu, X. et al. Regulation of GSK3
200. Armagan, G. et al. Regulation of the Nrf2 Pathway by Glycogen Synthase Kinase
201. Wang, L., Ouyang, S., Li, B., Wu, H. & Wang, F. GSK-3
202. Candelario-Jalil, E., Dijkhuizen, R. M. & Magnus, T. Neuroinflammation, stroke, blood-brain barrier dysfunction, and imaging modalities. Stroke 53, 1473-1486 (2022).
203. Lengfeld, J. E. et al. Endothelial Wnt/
204. Liu, Z. & Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 144, 103-120 (2016).
205. Ma, Y., Wang, J., Wang, Y. & Yang, G. Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 157, 247-272 (2017).
206. Kanazawa, M., Ninomiya, I., Hatakeyama, M., Takahashi, T. & Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 18, 2135 (2017).
207. Wang, Y. et al. Antioxidants & Redox Signaling. Antioxid. Redox Signal. 32, 213-214 (2020).
208. Zolezzi, J. M. & Inestrosa, N. C. Wnt/TLR Dialog in Neuroinflammation, Relevance in Alzheimer’s Disease. Front. Immunol. 8, 187 (2017).
209. Yeh, H., Woodbury, M. E., Ingraham Dixie, K. L., Ikezu, T. & Ikezu, S. Microglial WNT5A supports dendritic spines maturation and neuronal firing. Brain Behav. Immun. 107, 403-413 (2023).
210. Mecha, M. et al. Involvement of Wnt7a in the role of M2c microglia in neural stem cell oligodendrogenesis. J. Neuroinflamm 17, 88 (2020).
211. Xie, K., Cai, Y., Yang, P., Du, F. & Wu, K. Upregulating microRNA-874-3p inhibits CXCL12 expression to promote angiogenesis and suppress inflammatory response in ischemic stroke. Am. J. Physiol. Cell Physiol. 319, C579-c588 (2020).
212. Zhao, J., Li, L. & Fang, G. Salvianolic acid A attenuates cerebral ischemia/ reperfusion injury induced rat brain damage, inflammation, and apoptosis by regulating miR-499a/DDK1. Am. J. Transl. Res. 12, 3288-3301 (2020).
213. Zhou, J., Wu, N. & Lin, L. Curcumin suppresses apoptosis and inflammation in hypoxia/reperfusion-exposed neurons via Wnt Signaling pathway. Med Sci. Monit. 26, e920445 (2020).
214. Song, D. et al. Wnt canonical pathway activator TWS119 drives microglial antiinflammatory activation and facilitates neurological recovery following experimental stroke. J. Neuroinflamm 16, 256 (2019).
215. Zhao, B., Wang, P., Yu, J. & Zhang, Y. MicroRNA-376b-5p targets SOX7 to alleviate ischemic brain injury in a mouse model through activating Wnt/
216. Kalogeris, T., Bao, Y. & Korthuis, R. J. Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2, 702-714 (2014).
217. Tang, Y., Shen, J., Zhang, F., Yang, F. Y. & Liu, M. Human serum albumin attenuates global cerebral ischemia/reperfusion-induced brain injury in a Wnt/
218. Ten, V. S. & Starkov, A. Hypoxic-ischemic injury in the developing brain: the role of reactive oxygen species originating in mitochondria. Neurol. Res. Int. 2012, 542976 (2012).
219. Korobova, F., Ramabhadran, V. & Higgs, H. N. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339, 464-467 (2013).
220. Alvarez-Buylla, A. & Garcia-Verdugo, J. M. Neurogenesis in adult subventricular zone. J. Neurosci. 22, 629-634 (2002).
221. Alvarez-Buylla, A. & Lim, D. A. For the long run: maintaining germinal niches in the adult brain. Neuron 41, 683-686 (2004).
222. Adachi, K. et al. Beta-catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells 25, 2827-2836 (2007).
223. Lie, D. C. et al. Wnt signaling regulates adult hippocampal neurogenesis. Nature 437, 1370-1375 (2005).
224. Jin, K. et al. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl Acad. Sci. USA 103, 13198-13202 (2006).
225. Martí-Fàbregas, J. et al. Proliferation in the human ipsilateral subventricular zone after ischemic stroke. Neurology 74, 357-365 (2010).
226. Chen, X. et al. Peroxynitrite enhances self-renewal, proliferation, and neuronal differentiation of neural stem/progenitor cells through activating HIF-1a and Wnt/
227. Tiwari, S. K. et al. Inhibitory effects of Bisphenol-A on neural stem cells proliferation and differentiation in the rat brain are dependent on Wnt/
228. Gan, Q. et al. Pax6 mediates ß-catenin signaling for self-renewal and neurogenesis by neocortical radial glial stem cells. Stem Cells 32, 45-58 (2014).
229. Kuwabara, T. et al. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 12, 1097-1105 (2009).
230. Gao, Z. et al. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat. Neurosci. 12, 1090-1092 (2009).
231. Joksimovic, M. & Awatramani, R. Wnt/
232. Yi, H., Hu, J., Qian, J. & Hackam, A. S. Expression of brain-derived neurotrophic factor is regulated by the Wnt signaling pathway. Neuroreport 23, 189-194 (2012).
233. Wei, Z. Z. et al. Neuroprotective and regenerative roles of intranasal Wnt-3a administration after focal ischemic stroke in mice. J. Cereb. Blood Flow. Metab. 38, 404-421 (2018).
234. Li, S. R. et al. Mallotus oblongifolius extracts ameliorate ischemic nerve damage by increasing endogenous neural stem cell proliferation through the Wnt/
235. Liu, Q. et al. Ellagic acid improves endogenous neural stem cells proliferation and neurorestoration through Wnt/
236. Yang, X. et al. Curcumin promotes neurogenesis of hippocampal dentate gyrus via Wnt/
237. You, D. & You, H. Repression of long non-coding RNA MEG3 restores nerve growth and alleviates neurological impairment after cerebral ischemiareperfusion injury in a rat model. Biomed. Pharmacother. 111, 1447-1457 (2019).
238. Stenman, J. M. et al. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247-1250 (2008).
239. Zhou, Y. & Nathans, J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev. Cell 31, 248-256 (2014).
240. Hu, Y., Zheng, Y., Wang, T., Jiao, L. & Luo, Y. VEGF, a Key Factor for Blood Brain Barrier Injury After Cerebral Ischemic Stroke. Aging Dis. 13, 647-654 (2022).
241. Green, D. R. Caspases and Their Substrates. Cold Spring Harb. Perspect. Biol. 14, a041012 (2022).
242. Tian, Y. et al. IL-4-polarized BV2 microglia cells promote angiogenesis by secreting exosomes. Adv. Clin. Exp. Med. 28, 421-430 (2019).
243. Krupinski, J., Kaluza, J., Kumar, P., Kumar, S. & Wang, J. M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 25, 1794-1798 (1994).
244. Wang, L. P. et al. Oligodendrocyte precursor cell transplantation promotes angiogenesis and remyelination via Wnt/
245. Jiang, X. et al. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 163-164, 144-171 (2018).
246. Ta, S. et al. Variants of WNT7A and GPR124 are associated with hemorrhagic transformation following intravenous thrombolysis in ischemic stroke. CNS Neurosci. Ther. 27, 71-81 (2021).
247. Chang, J. et al. Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat. Med. 23, 450-460 (2017).
248. Hussain, B. et al. Endothelial
249. Chen, X. Y. et al. Inhibition of the immunoproteasome LMP2 ameliorates ischemia/hypoxia-induced blood-brain barrier injury through the Wnt/
250. Langen, U. H., Ayloo, S. & Gu, C. Development and cell biology of the bloodbrain barrier. Annu. Rev. Cell Dev. Biol. 35, 591-613 (2019).
251. Jin, Z., Ke, J., Guo, P., Wang, Y. & Wu, H. Quercetin improves blood-brain barrier dysfunction in rats with cerebral ischemia reperfusion via Wnt signaling pathway. Am. J. Transl. Res. 11, 4683-4695 (2019).
252. Kintner, D. B. et al. Increased tolerance to oxygen and glucose deprivation in astrocytes from
253. Song, S. et al. Activation of endothelial Wnt/
254. Zhao, H., Alam, A., Soo, A. P., George, A. J. T. & Ma, D. Ischemia-reperfusion injury reduces long term renal graft survival: mechanism and beyond. EBioMedicine 28, 31-42 (2018).
255. Wang, W., Sai, W. L. & Yang, B. [The role of macrophage polarization and interaction with renal tubular epithelial cells in ischemia-reperfusion induced acute kidney injury]. Sheng Li Xue Bao 74, 28-38 (2022).
256. He, W. et al. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 20, 765-776 (2009).
257. Liu, D., Liu, Y., Zheng, X. & Liu, N. c-MYC-induced long noncoding RNA MEG3 aggravates kidney ischemia-reperfusion injury through activating mitophagy by upregulation of RTKN to trigger the Wnt/
258. Zhou, D. et al. Tubule-specific ablation of endogenous
259. Wang, Y. et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 28, 231-243 (2021).
260. Wei, X. et al. Identification of subtypes and a delayed graft function predictive signature based on ferroptosis in renal ischemia-reperfusion injury. Front. Cell Dev. Biol. 10, 800650 (2022).
261. Xu, Y. et al. circ-AKT3 aggravates renal ischaemia-reperfusion injury via regulating miR-144-5p /Wnt/
262. Sturmlechner, I., Durik, M., Sieben, C. J., Baker, D. J. & van Deursen, J. M. Cellular senescence in renal aging and disease. Nat. Rev. Nephrol. 13, 77-89 (2017).
263. Xiao, L. et al. Sustained Activation of Wnt/
264. Zhou, L. et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/
265. Zhou, D. et al. Matrix metalloproteinase-7 is an urinary biomarker and pathogenic mediator of kidney fibrosis. J. Am. Soc. Nephrol. 28, 598-611 (2017).
266. He, W. et al. Plasminogen activator inhibitor-1 is a transcriptional target of the canonical pathway of Wnt/beta-catenin signaling. J. Biol. Chem. 285, 24665-24675 (2010).
267. Simon-Tillaux, N. & Hertig, A. Snail and kidney fibrosis. Nephrol. Dial. Transpl. 32, 224-233 (2017).
268. Luo, C. et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J. Am. Soc. Nephrol. 29, 1238-1256 (2018).
269. von Toerne, C. et al. Wnt pathway regulation in chronic renal allograft damage. Am. J. Transpl. 9, 2223-2239 (2009).
270. Sun, Q. et al. Allogeneic mesenchymal stem cells as induction therapy are safe and feasible in renal allografts: pilot results of a multicenter randomized controlled trial. J. Transl. Med. 16, 52 (2018).
271. Peralta, C., Jiménez-Castro, M. B. & Gracia-Sancho, J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J. Hepatol. 59, 1094-1106 (2013).
272. Russell, J. O. & Monga, S. P. Wnt/
273. Dar, W. A., Sullivan, E., Bynon, J. S., Eltzschig, H. & Ju, C. Ischaemia reperfusion injury in liver transplantation: cellular and molecular mechanisms. Liver Int. 39, 788-801 (2019).
274. Lehwald, N. et al. Wnt-
275. Liu, X. et al. Signaling through hepatocyte vasopressin receptor 1 protects mouse liver from ischemia-reperfusion injury. Oncotarget 7, 69276-69290 (2016).
276. Xie, K., Liu, L., Chen, J. & Liu, F. Exosomes derived from human umbilical cord blood mesenchymal stem cells improve hepatic ischemia reperfusion injury via delivering miR-1246. Cell Cycle 18, 3491-3501 (2019).
277. Sakon, M., Ariyoshi, H., Umeshita, K. & Monden, M. Ischemia-reperfusion injury of the liver with special reference to calcium-dependent mechanisms. Surg. Today 32, 1-12 (2002).
278. Hu, X. et al. Inhibition of Frizzled-2 by small interfering RNA protects rat hepatic BRL-3A cells against cytotoxicity and apoptosis induced by Hypoxia/Reoxygenation. Gastroenterol. Hepatol. 43, 107-116 (2020).
279. Yim, S. Y. et al. Risk factors for developing hyponatremia during terlipressin treatment: a retrospective analyses in variceal bleeding. J. Clin. Gastroenterol. 49, 607-612 (2015).
280. Koshimizu, T. A. et al. Vasopressin V1a and V1b receptors: from molecules to physiological systems. Physiol. Rev. 92, 1813-1864 (2012).
281. Kohler, A., Perrodin, S., De Gottardi, A., Candinas, D. & Beldi, G. Effectiveness of terlipressin for prevention of complications after major liver resection-A randomized placebo-controlled trial. HPB 22, 884-891 (2020).
282. Hong, S. H. et al. Perioperative assessment of terlipressin infusion during living donor liver transplantation. J. Int. Med. Res. 40, 225-236 (2012).
283. Reis, D. J. & Regunathan, S. Is agmatine a novel neurotransmitter in brain? Trends Pharm. Sci. 21, 187-193 (2000).
284. Kim, D. J. et al. Protective effect of agmatine on a reperfusion model after transient cerebral ischemia: Temporal evolution on perfusion MR imaging and histopathologic findings. AJNR Am. J. Neuroradiol. 27, 780-785 (2006).
285. Sugiura, T. et al. Protective effect of agmatine on ischemia/reperfusion-induced renal injury in rats. J. Cardiovasc. Pharm. 51, 223-230 (2008).
286. Greenberg, S. et al. The effect of agmatine administration on ischemicreperfused isolated rat heart. J. Cardiovasc. Pharm. Ther. 6, 37-45 (2001).
287. Han, Z. et al. Agmatine attenuates liver ischemia reperfusion injury by activating Wnt/
288. Dong, J. et al. SRY is a Key Mediator of Sexual Dimorphism in Hepatic Ischemia/ Reperfusion Injury. Ann. Surg. 276, 345-356 (2022).
289. O’Neill, M. J. & O’Neill, R. J. Whatever happened to SRY? Cell Mol. Life Sci. 56, 883-893 (1999).
290. Yang, Y. Y. et al. Involvement of the HIF-1a and Wnt/
291. Griffin, M. O., Ceballos, G. & Villarreal, F. J. Tetracycline compounds with nonantimicrobial organ protective properties: possible mechanisms of action. Pharm. Res. 63, 102-107 (2011).
292. Li, Y., Li, T., Qi, H. & Yuan, F. Minocycline protects against hepatic ischemia/ reperfusion injury in a rat model. Biomed. Rep. 3, 19-24 (2015).
293. Bataller, R. et al. Prolonged infusion of angiotensin II into normal rats induces stellate cell activation and proinflammatory events in liver. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G642-G651 (2003).
294. Kanno, K., Tazuma, S., Nishioka, T., Hyogo, H. & Chayama, K. Angiotensin II participates in hepatic inflammation and fibrosis through MCP-1 expression. Dig. Dis. Sci. 50, 942-948 (2005).
295. Bataller, R. et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Investig. 112, 1383-1394 (2003).
296. Harrison, D. G., Cai, H., Landmesser, U. & Griendling, K. K. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 4, 51-61 (2003).
297. Guo, L. et al. Role of the renin-angiotensin system in hepatic ischemia reperfusion injury in rats. Hepatology 40, 583-589 (2004).
298. Kuncewitch, M. et al. Wnt agonist attenuates liver injury and improves survival after hepatic ischemia/reperfusion. Shock 39, 3-10 (2013).
299. Sun, T. et al. AXIN2(+) Pericentral hepatocytes have limited contributions to liver homeostasis and regeneration. Cell Stem Cell 26, 97-107.e106 (2020).
300. Katoh, M. Multi-layered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/
301. Piao, C. et al. Effects of Exosomes Derived from Adipose-Derived Mesenchymal Stem Cells on Pyroptosis and Regeneration of Injured Liver. Int. J. Mol. Sci. 23, 12065 (2022).
302. Zhou, B. et al. Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 7, 95 (2022).
303. Kopan, R. & llagan, M. X. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216-233 (2009).
304. Chen, G. et al. The canonical Notch signaling was involved in the regulation of intestinal epithelial cells apoptosis after intestinal ischemia/reperfusion injury. Int. J. Mol. Sci. 15, 7883-7896 (2014).
305. Guo, P. et al. Dexmedetomidine alleviates myocardial ischemia-reperfusion injury by down-regulating miR-34b-3p to activate the Jagged1/Notch signaling pathway. Int. Immunopharmacol. 116, 109766 (2023).
306. Li, H. et al. Botch protects neurons from ischemic insult by antagonizing Notchmediated neuroinflammation. Exp. Neurol. 321, 113028 (2019).
307. Pei, H. et al. Notch1 cardioprotection in myocardial ischemia/reperfusion involves reduction of oxidative/nitrative stress. Basic Res. Cardiol. 108, 373 (2013).
308. Yu, H. C. et al. Canonical notch pathway protects hepatocytes from ischemia/ reperfusion injury in mice by repressing reactive oxygen species production through JAK2/STAT3 signaling. Hepatology 54, 979-988 (2011).
309. Chatterjee, S. & Sil, P. C. Targeting the crosstalks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharm. Res. 142, 251-261 (2019).
310. Patni, A. P. et al. Comprehending the crosstalk between Notch, Wnt, and Hedgehog signaling pathways in oral squamous cell carcinoma – clinical implications. Cell Oncol. 44, 473-494 (2021).
311. Kim, H. A. et al. Notch1 counteracts WNT/
312. Kim, W. et al. Hippo signaling interactions with Wnt/
313. Sprinzak, D. & Blacklow, S. C. Biophysics of Notch Signaling. Annu. Rev. Biophys. 50, 157-189 (2021).
314. Gude, N. A. et al. Activation of Notch-mediated protective signaling in the myocardium. Circ. Res. 102, 1025-1035 (2008).
315. Ashton, K. J., Willems, L., Holmgren, K., Ferreira, L. & Headrick, J. P. Ageassociated shifts in cardiac gene transcription and transcriptional responses to ischemic stress. Exp. Gerontol. 41, 189-204 (2006).
316. Zhao, L. et al. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl Acad. Sci. USA 111, 1403-1408 (2014).
317. Zhao, L., Ben-Yair, R., Burns, C. E. & Burns, C. G. Endocardial Notch signaling promotes cardiomyocyte proliferation in the regenerating zebrafish heart through wnt pathway antagonism. Cell Rep. 26, 546-554.e545 (2019).
318. Zhang, H. P. et al. The neuroprotective effects of isoflurane preconditioning in a murine transient global cerebral ischemia-reperfusion model: the role of the Notch signaling pathway. Neuromolecular Med. 16, 191-204 (2014).
319. Yang, Q. et al. Activation of canonical notch signaling pathway is involved in the ischemic tolerance induced by sevoflurane preconditioning in mice. Anesthesiology 117, 996-1005 (2012).
320. Zhang, H. et al. [Expressions of Notch3, Notch4, Frizzled2, and Tead1 in rats with focal cerebral ischemia-reperfusion]. Zhonghua Yi Xue Za Zhi 95, 3766-3769 (2015).
321. Arboleda-Velasquez, J. F. et al. Linking Notch signaling to ischemic stroke. Proc. Natl Acad. Sci. USA 105, 4856-4861 (2008).
322. Arumugam, T. V. et al. Notch signaling and neuronal death in stroke. Prog. Neurobiol. 165-167, 103-116 (2018).
323. Gao, L., Yang, L. & Cui, H. GSK-3
324. Ma, R. et al. l-Borneol and d-Borneol promote transdifferentiation of astrocytes into neurons in rats by regulating Wnt/Notch pathway to exert neuroprotective effect during recovery from cerebral ischemia. Phytomedicine 109, 154583 (2023).
325. Huang, S. et al. Zhongfenggao protects brain microvascular endothelial cells from oxygen-glucose deprivation/reoxygenation-induced injury by angiogenesis. Biol. Pharm. Bull. 42, 222-230 (2019).
326. Zhang, Z., Yao, L., Yang, J., Wang, Z. & Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 18, 3547-3554 (2018).
327. Dong, J., Xu, X., Zhang, Q., Yuan, Z. & Tan, B. The PI3K/AKT pathway promotes fracture healing through its crosstalk with Wnt/
328. Deng, S. et al. PI3K/AKT signaling tips the balance of cytoskeletal forces for cancer progression. Cancers 14, 1652 (2022).
329. Papadimitrakopoulou, V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J. Thorac. Oncol. 7, 1315-1326 (2012).
330. Tewari, D., Patni, P., Bishayee, A., Sah, A. N. & Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 80, 1-17 (2022).
331. Xiao, C. L. et al. The role of PI3K/Akt signaling pathway in spinal cord injury. Biomed. Pharmacother. 156, 113881 (2022).
332. Potz, B. A. et al. Calpain inhibition modulates glycogen synthase kinase
333. Chen, B. et al. Co-expression of Akt1 and Wnt11 promotes the proliferation and cardiac differentiation of mesenchymal stem cells and attenuates hypoxia/ reoxygenation-induced cardiomyocyte apoptosis. Biomed. Pharmacother. 108, 508-514 (2018).
334. Zhuang, Q. et al. Stimulated CB1 cannabinoid receptor inducing ischemic tolerance and protecting neurons from cerebral ischemia. Cent. Nerv. Syst. Agents Med. Chem. 17, 141-150 (2017).
335. Blankesteijn, W. M., van de Schans, V. A., ter Horst, P. & Smits, J. F. The Wnt/ frizzled/GSK-3 beta pathway: a novel therapeutic target for cardiac hypertrophy. Trends Pharm. Sci. 29, 175-180 (2008).
336. Hur, E. M. & Zhou, F. Q. GSK3 signaling in neural development. Nat. Rev. Neurosci. 11, 539-551 (2010).
337. Xing, X. S., Liu, F. & He, Z. Y. Akt regulates
338. Li, P., Zhang, Y. & Liu, H. The role of Wnt/ß-catenin pathway in the protection process by dexmedetomidine against cerebral ischemia/reperfusion injury in rats. Life Sci. 236, 116921 (2019).
339. Thirunavukkarasu, M. et al. Protective effects of Phyllanthus emblica against myocardial ischemia-reperfusion injury: the role of PI3-kinase/glycogen synthase kinase
340. Fei, Y., Zhao, B., Zhu, J., Fang, W. & Li, Y. XQ-1H promotes cerebral angiogenesis via activating PI3K/Akt/GSK3
341. Martínez-Sánchez, G. & Giuliani, A. Cellular redox status regulates hypoxia inducible factor-1 activity. Role in tumor development. J. Exp. Clin. Cancer Res. 26, 39-50 (2007).
342. Semenza, G. L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 76, 39-56 (2014).
343. Tan, Z. et al. Lithium and copper induce the osteogenesis-angiogenesis coupling of bone marrow mesenchymal stem cells via crosstalk between canonical Wnt and HIF-1a signaling pathways. Stem Cells Int. 2021, 6662164 (2021).
344. Tang, K. et al. HIF-1a stimulates the progression of oesophageal squamous cell carcinoma by activating the Wnt/
345. Zhang, Q. et al. Wnt/
346. DeFrates, K. G., Franco, D., Heber-Katz, E. & Messersmith, P. B. Unlocking mammalian regeneration through hypoxia inducible factor one alpha signaling. Biomaterials 269, 120646 (2021).
347. Engelhardt, S., Al-Ahmad, A. J., Gassmann, M. & Ogunshola, O. O. Hypoxia selectively disrupts brain microvascular endothelial tight junction complexes through a hypoxia-inducible factor-1 (HIF-1) dependent mechanism. J. Cell Physiol. 229, 1096-1105 (2014).
348. Wu, C. et al. Wnt/
349. Kaidi, A., Williams, A. C. & Paraskeva, C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 9, 210-217 (2007).
350. Xu, Z. H. et al. Hypoxia-inducible factor protects against acute kidney injury via the Wnt/
351. Peng, D., Fu, M., Wang, M., Wei, Y. & Wei, X. Targeting TGF-
352. Li, T. F. et al. Transforming growth factor-beta stimulates cyclin D1 expression through activation of beta-catenin signaling in chondrocytes. J. Biol. Chem. 281, 21296-21304 (2006).
353. Liu, J., Jin, J., Liang, T. & Feng, X. H. To Ub or not to Ub: a regulatory question in TGF-
354. Działo, E., Tkacz, K. & Błyszczuk, P. Crosstalk between the TGF-
355. Eid, R. A. et al. Exendin-4 Attenuates Remodeling in the Remote Myocardium of Rats After an Acute Myocardial Infarction by Activating
356. Wang, S. et al. Transforming growth-beta 1 contributes to isoflurane postconditioning against cerebral ischemia-reperfusion injury by regulating the c-Jun N-terminal kinase signaling pathway. Biomed. Pharmacother. 78, 280-290 (2016).
357. Chen, D. Q. et al. Combined melatonin and poricoic acid A inhibits renal fibrosis through modulating the interaction of Smad3 and
358. Tian, X. et al. Association of
359. Vallée, A. & Lecarpentier, Y. TGF-
360. Huber, N. et al. Age-related decrease in proteasome expression contributes to defective nuclear factor-kappaB activation during hepatic ischemia/reperfusion. Hepatology 49, 1718-1728 (2009).
361. Liang, W. et al. Preactivation of Notch1 in remote ischemic preconditioning reduces cerebral ischemia-reperfusion injury through crosstalk with the NF-кB pathway. J. Neuroinflamm. 16, 181 (2019).
362. Ling, H. et al.
363. Sakai, N. et al. Receptor activator of nuclear factor-
364. Oeckinghaus, A., Hayden, M. S. & Ghosh, S. Crosstalk in NF-kB signaling pathways. Nat. Immunol. 12, 695-708 (2011).
365. Mitchell, S., Vargas, J. & Hoffmann, A. Signaling via the NFкB system. Wiley Interdiscip. Rev. Syst. Biol. Med 8, 227-241 (2016).
366. Yu, H., Lin, L., Zhang, Z., Zhang, H. & Hu, H. Targeting NF-кВ pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct. Target Ther. 5, 209 (2020).
367. Yin, C. et al. Elevated Wnt2 and Wnt4 activate NF-кB signaling to promote cardiac fibrosis by cooperation of Fzd4/2 and LRP6 following myocardial infarction. EBioMedicine 74, 103745 (2021).
368. Lin, J. C. et al. Enhancement of beta-catenin in cardiomyocytes suppresses survival protein expression but promotes apoptosis and fibrosis. Cardiol. J. 24, 195-205 (2017).
369. Lin, J. C. et al.
370. Spiegelman, V. S. et al. Wnt/beta-catenin signaling induces the expression and activity of betaTrCP ubiquitin ligase receptor. Mol. Cell 5, 877-882 (2000).
371. He, J. et al. Huoxin pill prevents excessive inflammation and cardiac dysfunction following myocardial infarction by inhibiting adverse Wnt/
372. Winston, J. T. et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and betacatenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 13, 270-283 (1999).
373. Noubissi, F. K. et al. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signaling. Nature 441, 898-901 (2006).
374. Hoeflich, K. P. et al. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86-90 (2000).
375. Zhuang, X. et al. Differential effects on lung and bone metastasis of breast cancer by Wnt signaling inhibitor DKK1. Nat. Cell Biol. 19, 1274-1285 (2017).
376. El-Sayyad, S. M., Soubh, A. A., Awad, A. S. & El-Abhar, H. S. Mangiferin protects against intestinal ischemia/reperfusion-induced liver injury: Involvement of PPAR-
377. Jiang, S., Huang, L., Zhang, W. & Zhang, H. Vitamin D/VDR in acute kidney injury: a potential therapeutic target. Curr. Med. Chem. 28, 3865-3876 (2021).
378. Ali, R. M., Al-Shorbagy, M. Y., Helmy, M. W. & El-Abhar, H. S. Role of Wnt4/
379. Wang, J., Liu, S., Heallen, T. & Martin, J. F. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 15, 672-684 (2018).
380. Zhou, Q., Li, L., Zhao, B. & Guan, K. L. The hippo pathway in heart development, regeneration, and diseases. Circ. Res. 116, 1431-1447 (2015).
381. Heallen, T. et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458-461 (2011).
382. Gong, P. et al. Hippo/YAP signaling pathway mitigates blood-brain barrier disruption after cerebral ischemia/reperfusion injury. Behav. Brain Res. 356, 8-17 (2019).
383. Yu, H. et al. RRM2 improves cardiomyocyte proliferation after myocardial ischemia reperfusion injury through the hippo-YAP pathway. Dis. Mark. 2021, 5089872 (2021).
384. Zheng, Z. et al. Hippo-YAP/MCP-1 mediated tubular maladaptive repair promote inflammation in renal failed recovery after ischemic AKI. Cell Death Dis. 12, 754 (2021).
385. Zhou, J. et al. TNFAIP3 interacting protein 3 is an activator of Hippo-YAP signaling protecting against hepatic ischemia/reperfusion injury. Hepatology 74, 2133-2153 (2021).
386. Zheng, A., Chen, Q. & Zhang, L. The Hippo-YAP pathway in various cardiovascular diseases: focusing on the inflammatory response. Front. Immunol. 13, 971416 (2022).
387. Nishina, H. Physiological and pathological roles of the Hippo-YAP/TAZ signaling pathway in liver formation, homeostasis, and tumorigenesis. Cancer Sci. 113, 1900-1908 (2022).
388. Liu, S. et al. Yap promotes noncanonical Wnt signals from cardiomyocytes for heart regeneration. Circ. Res. 129, 782-797 (2021).
389. Azzolin, L. et al. YAP/TAZ incorporation in the
390. Ma, W. Y. et al. Melatonin promotes cardiomyocyte proliferation and heart repair in mice with myocardial infarction via miR-143-3p/Yap/Ctnnd1 signaling pathway. Acta Pharm. Sin. 42, 921-931 (2021).
391. Amani, H. et al. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci. Rep. 9, 6044 (2019).
392. Kanzler, B., Foreman, R. K., Labosky, P. A. & Mallo, M. BMP signaling is essential for development of skeletogenic and neurogenic cranial neural crest. Development 127, 1095-1104 (2000).
393. Mabie, P. C., Mehler, M. F. & Kessler, J. A. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J. Neurosci. 19, 7077-7088 (1999).
394. Baker, J. C., Beddington, R. S. & Harland, R. M. Wnt signaling in Xenopus embryos inhibits bmp4 expression and activates neural development. Genes Dev. 13, 3149-3159 (1999).
395. Antebi, Y. E. et al. Combinatorial signal perception in the BMP pathway. Cell 170, 1184-1196.e1124 (2017).
396. Zhang, Y. & Que, J. BMP signaling in development, stem cells, and diseases of the gastrointestinal tract. Annu. Rev. Physiol. 82, 251-273 (2020).
397. Fujita, K., Ogawa, R., Kawawaki, S. & Ito, K. Roles of chromatin remodelers in maintenance mechanisms of multipotency of mouse trunk neural crest cells in the formation of neural crest-derived stem cells. Mech. Dev. 133, 126-145 (2014).
398. Dizon, M. L., Maa, T. & Kessler, J. A. The bone morphogenetic protein antagonist noggin protects white matter after perinatal hypoxia-ischemia. Neurobiol. Dis. 42, 318-326 (2011).
399. Guan, J. et al. Bone morphogenetic protein-7 (BMP-7) mediates ischemic preconditioning-induced ischemic tolerance via attenuating apoptosis in rat brain. Biochem. Biophys. Res. Commun. 441, 560-566 (2013).
400. Chen, C., Yang, Y. & Yao, Y. HBO promotes the differentiation of neural stem cells via interactions between the Wnt3/
401. Lei, Z. N., Liu, F., Zhang, L. M., Huang, Y. L. & Sun, F. Y. Bcl-2 increases strokeinduced striatal neurogenesis in adult brains by inhibiting BMP-4 function via activation of
402. Baik, J., Borges, L., Magli, A., Thatava, T. & Perlingeiro, R. C. Effect of endoglin overexpression during embryoid body development. Exp. Hematol. 40, 837-846 (2012).
403. Zhang, L. et al. Modulation of TGF-
404. Borges, L. et al. A critical role for endoglin in the emergence of blood during embryonic development. Blood 119, 5417-5428 (2012).
405. Baik, J. et al. Endoglin integrates BMP and Wnt signalling to induce haematopoiesis through JDP2. Nat. Commun. 7, 13101 (2016).
406. Ahmadi, A. et al. Recent advances on small molecules in osteogenic differentiation of stem cells and the underlying signaling pathways. Stem Cell Res. Ther. 13, 518 (2022).
407. Zhang, X., Shi, X., Wang, J., Xu, Z. & He, J. Enriched environment remedies cognitive dysfunctions and synaptic plasticity through NMDAR-
408. Liu, S. et al. Icaritin alleviates cerebral ischemia-reperfusion injury by regulating NMDA receptors through ERK signaling. Eur. J. Pharm. 941, 175492 (2023).
409. Abe, K. & Takeichi, M. NMDA-receptor activation induces calpain-mediated betacatenin cleavages for triggering gene expression. Neuron 53, 387-397 (2007).
410. Villmann, C. & Becker, C. M. On the hypes and falls in neuroprotection: targeting the NMDA receptor. Neuroscientist 13, 594-615 (2007).
411. Jolly, S. et al. G protein-coupled receptor 37-like 1 modulates astrocyte glutamate transporters and neuronal NMDA receptors and is neuroprotective in ischemia. Glia 66, 47-61 (2018).
412. Luo, Y. et al. Focal cerebral ischemia and reperfusion induce brain injury through a28-1-Bound NMDA receptors. Stroke 49, 2464-2472 (2018).
413. Kawai, T. & Akira, S. TLR signaling. Semin. Immunol. 19, 24-32 (2007).
414. Tong, Y. et al. WISP1 mediates hepatic warm ischemia reperfusion injury via TLR4 signaling in mice. Sci. Rep. 6, 20141 (2016).
415. Undi, R. B., Sarvothaman, S., Narasaiah, K., Gutti, U. & Gutti, R. K. Toll-like receptor 2 signalings: significance in megakaryocyte development through wnt signalling cross-talk and cytokine induction. Cytokine 83, 245-249 (2016).
416. Martín-Medina, A. et al. TLR/WNT: A Novel Relationship in Immunomodulation of Lung Cancer. Int. J Mol Sci. 23, 6539 (2022).
417. Christman, M. A. 2nd et al. Wnt5a is expressed in murine and human atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 294, H2864-H2870 (2008).
418. He, W. et al. Lipopolysaccharide enhances Wnt5a expression through toll-like receptor 4, myeloid differentiating factor 88, phosphatidylinositol 3-OH kinase/ AKT and nuclear factor kappa B pathways in human dental pulp stem cells. J. Endod. 40, 69-75 (2014).
419. El-Ela, S. R. A., Zaghloul, R. A. & Eissa, L. A. Promising cardioprotective effect of baicalin in doxorubicin-induced cardiotoxicity through targeting toll-like receptor
420. Liu, B., Li, F., Xu, Y., Wu, Q. & Shi, J. Gastrodin improves cognitive dysfunction in REM Sleep-deprived rats by regulating TLR4/NF-кB and Wnt/
421. Tanaka, R., Terai, M., Londin, E. & Sato, T. The role of HGF/MET signaling in metastatic uveal melanoma. Cancers 13, 5457 (2021).
422. Demkova, L. & Kucerova, L. Role of the HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol. Cancer 17, 26 (2018).
423. Zhang, Y. et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 17, 45 (2018).
424. Liu, S. et al. A self-assembling peptide hydrogel-based drug co-delivery platform to improve tissue repair after ischemia-reperfusion injury. Acta Biomater. 103, 102-114 (2020).
425. Humphreys, B. D. et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2, 284-291 (2008).
426. Koraishy, F. M., Silva, C., Mason, S., Wu, D. & Cantley, L. G. Hepatocyte growth factor (Hgf) stimulates low density lipoprotein receptor-related protein (Lrp) 5/6 phosphorylation and promotes canonical Wnt signaling. J. Biol. Chem. 289, 14341-14350 (2014).
427. Maarouf, O. H. et al. Paracrine Wnt1 drives interstitial fibrosis without inflammation by tubulointerstitial cross-talk. J. Am. Soc. Nephrol. 27, 781-790 (2016).
428. Zhou, D. et al. Fibroblast-specific
429. Doeppner, T. R. et al. Acute hepatocyte growth factor treatment induces longterm neuroprotection and stroke recovery via mechanisms involving neural precursor cell proliferation and differentiation. J. Cereb. Blood Flow. Metab. 31, 1251-1262 (2011).
430. Shang, J. et al. Antiapoptotic and anti autophagic effects of glial cell line-derived neurotrophic factor and hepatocyte growth factor after transient middle cerebral artery occlusion in rats. J. Neurosci. Res. 88, 2197-2206 (2010).
431. Nakaguchi, K. et al. Growth factors released from gelatin hydrogel microspheres increase new neurons in the adult mouse brain. Stem Cells Int. 2012, 915160 (2012).
432. Chaparro, R. E. et al. Sustained functional improvement by hepatocyte growth factor-like small molecule BB3 after focal cerebral ischemia in rats and mice. J. Cereb. Blood Flow. Metab. 35, 1044-1053 (2015).
433. Matsunaga, S., Fujishiro, H. & Takechi, H. Efficacy and Safety of Glycogen Synthase Kinase 3 Inhibitors for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 69, 1031-1039 (2019).
434. del Ser, T. et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J. Alzheimers Dis. 33, 205-215 (2013).
435. O’Leary, O. & Nolan, Y. Glycogen synthase kinase-3 as a therapeutic target for cognitive dysfunction in neuropsychiatric disorders. CNS Drugs 29, 1-15 (2015).
436. Singh, A. P. et al. Inhibition of GSK-3 to induce cardiomyocyte proliferation: a recipe for in situ cardiac regeneration. Cardiovasc. Res. 115, 20-30 (2019).
437. Fu, W. B., Wang, W. E. & Zeng, C. Y. Wnt signaling pathways in myocardial infarction and the therapeutic effects of Wnt pathway inhibitors. Acta Pharm. Sin. 40, 9-12 (2019).
438. Saraswati, S. et al. Pyrvinium, a potent small molecule Wnt inhibitor, promotes wound repair and post-MI cardiac remodeling. PLoS One 5, e15521 (2010).
439. Laeremans, H. et al. Blocking of frizzled signaling with a homologous peptide fragment of wnt3a/wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation 124, 1626-1635 (2011).
440. Sasaki, T., Hwang, H., Nguyen, C., Kloner, R. A. & Kahn, M. The small molecule Wnt signaling modulator ICG-001 improves contractile function in chronically infarcted rat myocardium. PLoS One 8, e75010 (2013).
441. Jiang, J. et al. A novel porcupine inhibitor blocks WNT pathways and attenuates cardiac hypertrophy. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3459-3467 (2018).
442. Xie, S. et al. Discovering small molecules as Wnt inhibitors that promote heart regeneration and injury repair. J. Mol. Cell Biol. 12, 42-54 (2020).
443. Ni, T. T. et al. Discovering small molecules that promote cardiomyocyte generation by modulating Wnt signaling. Chem. Biol. 18, 1658-1668 (2011).
444. Kumar, K., Singh, N., Jaggi, A. S. & Maslov, L. Clinical applicability of conditioning techniques in ischemia-reperfusion injury: a review of the literature. Curr. Cardiol. Rev. 17, 306-318 (2021).
445. Liu, G. S. et al. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation 84, 350-356 (1991).
446. Goto, M. et al. Role of bradykinin in protection of ischemic preconditioning in rabbit hearts. Circ. Res. 77, 611-621 (1995).
447. Cohen, M. V. et al. Preconditioning-mimetics bradykinin and DADLE activate PI3kinase through divergent pathways. J. Mol. Cell Cardiol. 42, 842-851 (2007).
448. Schultz, J. E., Rose, E., Yao, Z. & Gross, G. J. Evidence for involvement of opioid receptors in ischemic preconditioning in rat hearts. Am. J. Physiol. 268, H2157-H2161 (1995).
449. Banerjee, A. et al. Preconditioning against myocardial dysfunction after ischemia and reperfusion by an alpha 1-adrenergic mechanism. Circ. Res. 73, 656-670 (1993).
450. Yao, Z. & Gross, G. J. Role of nitric oxide, muscarinic receptors, and the ATPsensitive
451. Kim, J. et al. Adenosine and Cordycepin Accelerate Tissue Remodeling Process through Adenosine Receptor Mediated Wnt/
452. Kim, J., Shin, J. Y., Choi, Y. H., Kang, N. G. & Lee, S. Anti-Hair Loss Effect of Adenosine Is Exerted by cAMP Mediated Wnt/
453. Borhani, S., Corciulo, C., Larranaga-Vera, A. & Cronstein, B. N. Adenosine A(2A) receptor (A2AR) activation triggers Akt signaling and enhances nuclear localization of
454. Yan, L., Yao, X., Bachvarov, D., Saifudeen, Z. & El-Dahr, S. S. Genome-wide analysis of gestational gene-environment interactions in the developing kidney. Physiol. Genom. 46, 655-670 (2014).
455. Liu, Y. et al. Wnt/
456. Li, Y. et al. Propoxyphene mediates oxyhemoglobin-induced injury in rat cortical neurons through up-regulation of active-
457. Wang, J. et al. Pentazocine Protects SN4741 Cells Against MPP(+)-Induced Cell Damage via Up-Regulation of the Canonical Wnt/
458. Guan, M., Huang, Y. & Lin, X. Sufentanil inhibits the proliferation and epithelial mesenchymal transition of lung cancer cells through Wnt/beta-catenin signaling pathway. Bioengineered 13, 10857-10865 (2022).
459. Mocanu, M. M., Bell, R. M. & Yellon, D. M. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J. Mol. Cell Cardiol. 34, 661-668 (2002).
460. Jonassen, A. K., Mjøs, O. D. & Sack, M. N. p70s6 kinase is a functional target of insulin activated Akt cell-survival signaling. Biochem. Biophys. Res. Commun. 315, 160-165 (2004).
461. Tong, H., Chen, W., Steenbergen, C. & Murphy, E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ. Res. 87, 309-315 (2000).
462. Juhaszova, M. et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 113, 1535-1549 (2004).
463. Barandon, L. et al. Involvement of FrzA/sFRP-1 and the Wnt/frizzled pathway in ischemic preconditioning. Circ. Res. 96, 1299-1306 (2005).
464. Vigneron, F. et al. GSK-3
465. Correa-Costa, M. et al. Transcriptome analysis of renal ischemia/reperfusion injury and its modulation by ischemic pre-conditioning or hemin treatment. PLoS One 7, e49569 (2012).
466. Przyklenk, K., Bauer, B., Ovize, M., Kloner, R. A. & Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87, 893-899 (1993).
467. Kambakamba, P. et al. Novel benefits of remote ischemic preconditioning through VEGF-dependent protection from resection-induced liver failure in the mouse. Ann. Surg. 268, 885-893 (2018).
468. Sawashita, Y. et al. Remote ischemic preconditioning reduces myocardial ischemia-reperfusion injury through unacylated ghrelin-induced activation of the JAK/STAT pathway. Basic Res. Cardiol. 115, 50 (2020).
469. Sörensson, P. et al. Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart 96, 1710-1715 (2010).
470. Woo, J. S. et al. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler. Thromb. Vasc. Biol. 33, 2252-2260 (2013).
471. Koyama, T. et al. Impact of postconditioning with lactate-enriched blood on inhospital outcomes of patients with ST-segment elevation myocardial infarction. Int. J. Cardiol. 220, 146-148 (2016).
472. Zhu, M. et al. Ischemic postconditioning protects remodeled myocardium via the PI3K-PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc. Res. 72, 152-162 (2006).
473. Wagner, C., Tillack, D., Simonis, G., Strasser, R. H. & Weinbrenner, C. Ischemic post-conditioning reduces infarct size of the in vivo rat heart: role of PI3-K, mTOR, GSK-3beta, and apoptosis. Mol. Cell. Biochem. 339, 135-147 (2010).
474. Guo, J. Y. et al. Ischemic postconditioning attenuates liver warm ischemiareperfusion injury through Akt-eNOS-NO-HIF pathway. J. Biomed. Sci. 18, 79 (2011).
475. Darling, C. E. et al. Postconditioning via stuttering reperfusion limits myocardial infarct size in rabbit hearts: role of ERK1/2. Am. J. Physiol. Heart Circ. Physiol. 289, H1618-H1626 (2005).
476. Tsang, A., Hausenloy, D. J., Mocanu, M. M. & Yellon, D. M. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ. Res. 95, 230-232 (2004).
477. Hausenloy, D. J., Tsang, A. & Yellon, D. M. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc. Med. 15, 69-75 (2005).
478. Díaz-Ruíz, J. L. et al. Redox signaling in ischemic postconditioning protection involves PKCɛ and Erk1/2 pathways and converges indirectly in Nrf2 activation. Cell Signal 64, 109417 (2019).
479. He, N. et al. Remote ischemic perconditioning prevents liver transplantationinduced ischemia/reperfusion injury in rats: role of ROS/RNS and eNOS. World J. Gastroenterol. 23, 830-841 (2017).
480. Qiu, Y. et al. Hyperglycemia-Induced Overexpression of PH Domain Leucine-Rich Repeat Protein Phosphatase 1 (PHLPP1) Compromises the Cardioprotective Effect of Ischemic Postconditioning Via Modulation of the Akt/Mst1 Pathway Signaling. Cardiovasc. Drugs Ther. https://doi.org/10.1007/s10557-022-07349-5 (2022).
481. Chen, H., Shen, J. & Zhao, H. Ischemic postconditioning for stroke treatment: current experimental advances and future directions. Cond. Med. 3, 104-115 (2020).
482. Xue, R. et al. Selective inhibition of PTEN preserves ischaemic post-conditioning cardioprotection in STZ-induced Type 1 diabetic rats: role of the PI3K/Akt and JAK2/STAT3 pathways. Clin. Sci. 130, 377-392 (2016).
483. Kerendi, F. et al. Remote postconditioning. Brief renal ischemia and reperfusion applied before coronary artery reperfusion reduces myocardial infarct size via endogenous activation of adenosine receptors. Basic Res. Cardiol. 100, 404-412 (2005).
484. Sun, J. et al. Protective effect of delayed remote limb ischemic postconditioning: role of mitochondrial K(ATP) channels in a rat model of focal cerebral ischemic reperfusion injury. J. Cereb. Blood Flow. Metab. 32, 851-859 (2012).
485. Hu, X., Lv, T., Yang, S. F., Zhang, X. H. & Miao, Y. F. Limb remote ischemic post-conditioning reduces injury and improves long-term behavioral recovery in rats following subarachnoid hemorrhage: possible involvement of the autophagic process. Mol. Med. Rep. 17, 21-30 (2018).
486. Peng, B. et al. Remote ischemic postconditioning protects the brain from global cerebral ischemia/reperfusion injury by up-regulating endothelial nitric oxide synthase through the PI3K/Akt pathway. Brain Res. 1445, 92-102 (2012).
487. Qi, Z. F. et al. AKT/GSK3
488. Gao, S. et al. Remote ischemic postconditioning protects against renal ischemia/ reperfusion injury by activation of T-LAK-cell-originated protein kinase (TOPK)/ PTEN/Akt signaling pathway mediated anti-oxidation and anti-inflammation. Int. Immunopharmacol. 38, 395-401 (2016).
489. Danielisová, V., Némethová, M., Gottlieb, M. & Burda, J. The changes in endogenous antioxidant enzyme activity after postconditioning. Cell. Mol. Neurobiol. 26, 1181-1191 (2006).
490. Wang, Q. et al. Limb remote postconditioning alleviates cerebral reperfusion injury through reactive oxygen species-mediated inhibition of delta protein kinase C in rats. Anesth. Analg. 113, 1180-1187 (2011).
491. Niu, D. G. et al. Morphine promotes cancer stem cell properties, contributing to chemoresistance in breast cancer. Oncotarget 6, 3963-3976 (2015).
492. Wang, K. P., Bai, Y., Wang, J. & Zhang, J. Z. Morphine protects SH-SY5Y human neuroblastoma cells against Dickkopf1-induced apoptosis. Mol. Med. Rep. 11, 1174-1180 (2015).
493. Zhou, Z., Liu, T. & Zhang, J. Morphine activates blast-phase chronic myeloid leukemia cells and alleviates the effects of tyrosine kinase inhibitors. Biochem. Biophys. Res. Commun. 520, 560-565 (2019).
494. Xue, J. J. et al. Protective effect of propofol on hydrogen peroxide-induced human esophageal carcinoma via blocking the Wnt/
495. Zhan, K., Song, X., Zhang, Q., Yang, J. & Lu, S. Propofol-Induced miR-493-3p Inhibits Growth and Invasion of Gastric Cancer through Suppression of DKK1Mediated Wnt/
496. Gong, T. et al. Propofol-induced miR-219-5p inhibits growth and invasion of hepatocellular carcinoma through suppression of GPC3-mediated Wnt/
497. Zhang, Y. F., Li, C. S., Zhou, Y. & Lu, X. H. Effects of propofol on colon cancer metastasis through STAT3/HOTAIR axis by activating WIF-1 and suppressing Wnt pathway. Cancer Med. 9, 1842-1854 (2020).
498. Lin, X. H. et al. Norepinephrine-stimulated HSCs secrete sFRP1 to promote HCC progression following chronic stress via augmentation of a Wnt16B/
499. Zhao, X. et al. Aldehyde dehydrogenase-2 protects against myocardial infarction-related cardiac fibrosis through modulation of the Wnt/
500. Qian, L. et al. Downregulation of S100A4 alleviates cardiac fibrosis via Wnt/
501. Cui, S. et al. miR-145 attenuates cardiac fibrosis through the AKT/GSK-3
502. Guo, X. et al. Induced pluripotent stem cell-conditional medium inhibits H9C2 cardiomyocytes apoptosis via autophagy flux and Wnt/
503. Liu, C. & Li, Y. Propofol relieves inflammation in MIRI rats by inhibiting Rho/Rock signaling pathway. Eur. Rev. Med. Pharm. Sci. 25, 976-984 (2021).
504. Chen, F. et al. Activation of EphA4 induced by EphrinA1 exacerbates disruption of the blood-brain barrier following cerebral ischemia-reperfusion via the Rho/ ROCK signaling pathway. Exp. Ther. Med. 16, 2651-2658 (2018).
505. Zhou, D., Zhang, M., Min, L., Jiang, K. & Jiang, Y. Cerebral ischemia-reperfusion is modulated by macrophage-stimulating 1 through the MAPK-ERK signaling pathway. J. Cell Physiol. 235, 7067-7080 (2020).
506. Zhang, F., Cao, X., Zhao, C., Chen, L. & Chen, X. Empagliflozin activates JAK2/ STAT3 signaling and protects cardiomyocytes from hypoxia/reoxygenation injury under high glucose conditions. J. Thromb. Thrombolysis 55, 116-125 (2023).
507. Li, Z. et al. Theaflavin ameliorates renal ischemia/reperfusion injury by activating the Nrf2 signaling pathway in vivo and in vitro. Biomed. Pharmacother. 134, 111097 (2021).
508. Wu, J. W., Hu, H., Hua, J. S. & Ma, L. K. ATPase inhibitory factor 1 protects the heart from acute myocardial ischemia/reperfusion injury through activating AMPK signaling pathway. Int J. Biol. Sci. 18, 731-741 (2022).
509. Zhao, N., Gao, Y., Jia, H. & Jiang, X. Anti-apoptosis effect of traditional Chinese medicine in the treatment of cerebral ischemia-reperfusion injury. Apoptosis. 28, 702-729 (2023).
510. Dong, L. et al. Research Progress of Chinese Medicine in the Treatment of Myocardial Ischemia-Reperfusion Injury. Am. J. Chin. Med. 51, 1-17 (2023).
511. Yin, B., Hou, X. W. & Lu, M. L. Astragaloside IV attenuates myocardial ischemia/ reperfusion injury in rats via inhibition of calcium-sensing receptor-mediated apoptotic signaling pathways. Acta Pharm. Sin. 40, 599-607 (2019).
512. Jiang, M. et al. Astragaloside IV Attenuates Myocardial Ischemia-Reperfusion Injury from Oxidative Stress by Regulating Succinate, Lysophospholipid Metabolism, and ROS Scavenging System. Oxid. Med. Cell. Longev. 2019, 9137654 (2019).
513. Song, M., Huang, L., Zhao, G. & Song, Y. Beneficial effects of a polysaccharide from Salvia miltiorrhiza on myocardial ischemia-reperfusion injury in rats. Carbohydr. Polym. 98, 1631-1636 (2013).
514. Zeng, H. et al. Activated PKB/GSK-
activity: Therapeutic efficacy of dihydrotanshinone-I. Acta Pharm. Sin. B 11, 71-88 (2021).
515. Huang, C. Y. et al. Protective effect of Danggui (Radix Angelicae Sinensis) on angiotensin II-induced apoptosis in H9c2 cardiomyoblast cells. BMC Complement Alter. Med. 14, 358 (2014).
516. Zhang, S. et al. Extraction, chemical analysis of Angelica sinensis polysaccharides and antioxidant activity of the polysaccharides in ischemia-reperfusion rats. Int. J. Biol. Macromol. 47, 546-550 (2010).
517. Wang, K., Lou, Y., Xu, H., Zhong, X. & Huang, Z. Harpagide from Scrophularia protects rat cortical neurons from oxygen-glucose deprivation and reoxygenation-induced injury by decreasing endoplasmic reticulum stress. J. Ethnopharmacol. 253, 112614 (2020).
518. Mo, Z. T., Liao, Y. L., Zheng, J. & Li, W. N. Icariin protects neurons from endoplasmic reticulum stress-induced apoptosis after OGD/R injury via suppressing IRE1a-XBP1 signaling pathway. Life Sci. 255, 117847 (2020).
519. Pang, Y., Zhu, S. & Pei, H. Pachymic acid protects against cerebral ischemia/ reperfusion injury by the PI3K/Akt signaling pathway. Metab. Brain Dis. 35, 673-680 (2020).
520. Tong, C. et al. Intravenous administration of lycopene, a tomato extract, protects against myocardial ischemia-reperfusion injury. Nutrients 8, 138 (2016).
521. Wu, S. et al. Effects of lycopene attenuating injuries in ischemia and reperfusion. Oxid. Med. Cell. Longev. 2022, 9309327 (2022).
522. Liu, Y., Qu, X., Yan, M., Li, D. & Zou, R. Tricin attenuates cerebral ischemia/ reperfusion injury through inhibiting nerve cell autophagy, apoptosis and inflammation by regulating the PI3K/Akt pathway. Hum. Exp. Toxicol. 41, 9603271221125928 (2022).
523. Wang, G., Guo, H. & Wang, X. Platycodin D protects cortical neurons against oxygen-glucose deprivation/reperfusion in neonatal hypoxic-ischemic encephalopathy. J. Cell. Biochem 120, 14028-14034 (2019).
524. Yang, S. et al. Baicalein administered in the subacute phase ameliorates ischemia-reperfusion-induced brain injury by reducing neuroinflammation and neuronal damage. Biomed. Pharmacother. 117, 109102 (2019).
525. Wang, Z. et al. Lupeol alleviates cerebral ischemia-reperfusion injury in correlation with modulation of PI3K/Akt pathway. Neuropsychiatr. Dis. Treat. 16, 1381-1390 (2020).
526. Wang, P. C. et al. Combination of paeoniflorin and calycosin-7-glucoside alleviates ischaemic stroke injury via the PI3K/AKT signaling pathway. Pharm. Biol. 60, 1469-1477 (2022).
527. Jian, J., Xuan, F., Qin, F. & Huang, R. Bauhinia championii flavone inhibits apoptosis and autophagy via the PI3K/Akt pathway in myocardial ischemia/ reperfusion injury in rats. Drug Des. Dev. Ther. 9, 5933-5945 (2015).
528. Zhang, H. & Li, H. Tricin enhances osteoblastogenesis through the regulation of Wnt/
529. Lee, H., Bae, S., Kim, Y. S. & Yoon, Y. WNT/ß-catenin pathway mediates the antiadipogenic effect of platycodin D , a natural compound found in Platycodon grandiflorum. Life Sci. 89, 388-394 (2011).
530. Xia, X. et al. Baicalein blocked cervical carcinoma cell proliferation by targeting CCND1 via Wnt/
531. Tarapore, R. S., Siddiqui, I. A., Adhami, V. M., Spiegelman, V. S. & Mukhtar, H. The dietary terpene lupeol targets colorectal cancer cells with constitutively active Wnt/
532. Wang, Y. et al. Construing the biochemical and molecular mechanism underlying the in vivo and in vitro chemotherapeutic efficacy of ruthenium-baicalein complex in colon cancer. Int. J. Biol. Sci. 15, 1052-1071 (2019).
533. Tarapore, R. S. et al. Specific targeting of Wnt/
534. Zhang, L., Tu, Y., He, W., Peng, Y. & Qiu, Z. A novel mechanism of hepatocellular carcinoma cell apoptosis induced by lupeol via Brain-derived neurotrophic factor inhibition and glycogen synthase kinase 3 beta reactivation. Eur. J. Pharm. 762, 55-62 (2015).
535. Wu, X. T. et al. The enhanced effect of lupeol on the destruction of gastric cancer cells by NK cells. Int. Immunopharmacol. 16, 332-340 (2013).
536. Zhou, Y. et al. Paeoniflorin Affects Hepatocellular Carcinoma Progression by Inhibiting Wnt/
537. Li, H. et al. Bauhinia championi (Benth.) Benth. polysaccharides upregulate Wnt/
538. Zhang, C. et al. Asiaticoside alleviates cerebral ischemia-reperfusion injury via NOD2/Mitogen-Activated Protein Kinase (MAPK)/Nuclear Factor kappa B (NF-кB) Signaling Pathway. Med. Sci. Monit. 26, e920325 (2020).
539. Ye, B. et al. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des. Dev. Ther. 13, 975-990 (2019).
540. Pan, J. et al. Ginkgetin attenuates cerebral ischemia-reperfusion induced autophagy and cell death via modulation of the NF-кB/p53 signaling pathway. Biosci. Rep. 39, BSR20191452 (2019).
541. Cao, W., Feng, S. J. & Kan, M. C. Naringin Targets NFKB1 to Alleviate OxygenGlucose Deprivation/Reoxygenation-Induced Injury in PC12 Cells Via Modulating HIF-1a/AKT/mTOR-Signaling Pathway. J. Mol. Neurosci. 71, 101-111 (2021).
542. An, B. et al. Crocin regulates the proliferation and migration of neural stem cells after cerebral ischemia by activating the Notch1 pathway. Folia Neuropathol. 58, 201-212 (2020).
543. Dibben, G. et al. Exercise-based cardiac rehabilitation for coronary heart disease. Cochrane Database Syst. Rev. 11, Cd001800 (2021).
544. Wang, H. et al. Programmed exercise attenuates familial hypertrophic cardiomyopathy in transgenic E22K mice via inhibition of PKC-a/NFAT pathway. Front. Cardiovasc. Med. 9, 808163 (2022).
545. Cheedipudi, S. M. et al. Exercise restores dysregulated gene expression in a mouse model of arrhythmogenic cardiomyopathy. Cardiovasc. Res. 116, 1199-1213 (2020).
© The Author(s) 2023
The Collaborative Innovation Center, Jining Medical University, Jining, Shandong 272067, China; Clinical Medical College, Jining Medical University, Jining, Shandong 272067, China; Second Clinical Medical College, Jining Medical University, Jining, Shandong 272067, China; Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital and Institutes of Biomedical Sciences, Fudan University, Shanghai, China; Institute of Zoology, Chinese Academy of Sciences, Beijing, China and Department of Physiology, Basic medical school, Xuzhou Medical University, Xuzhou 221004, China
Correspondence: Shijun Wang (shijun_w@126.com) or Rubin Tan (tanrubin11@126.com) or Jinxiang Yuan (yuanjinxiang18@163.com)
These authors contributed equally: Meng Zhang, Qian Liu, Hui Meng