DOI: https://doi.org/10.1038/s41467-024-47023-y
PMID: https://pubmed.ncbi.nlm.nih.gov/38531862
تاريخ النشر: 2024-03-26
إطار عضوي تساهمي كيتو-أنتراكينون لعملية التمثيل الضوئي لـ H2O2 مع الأكسجين والماء القلوي
تم القبول: 18 مارس 2024
نُشر على الإنترنت: 26 مارس 2024
(أ) التحقق من التحديثات
الملخص
تعاني عملية التمثيل الضوئي لبيروكسيد الهيدروجين من نشاط تحفيزي غير كافٍ بسبب الحاجز الطاقي العالي لاستخراج الهيدروجين من
النتائج والمناقشة
تخليق وتوصيف الهيكل
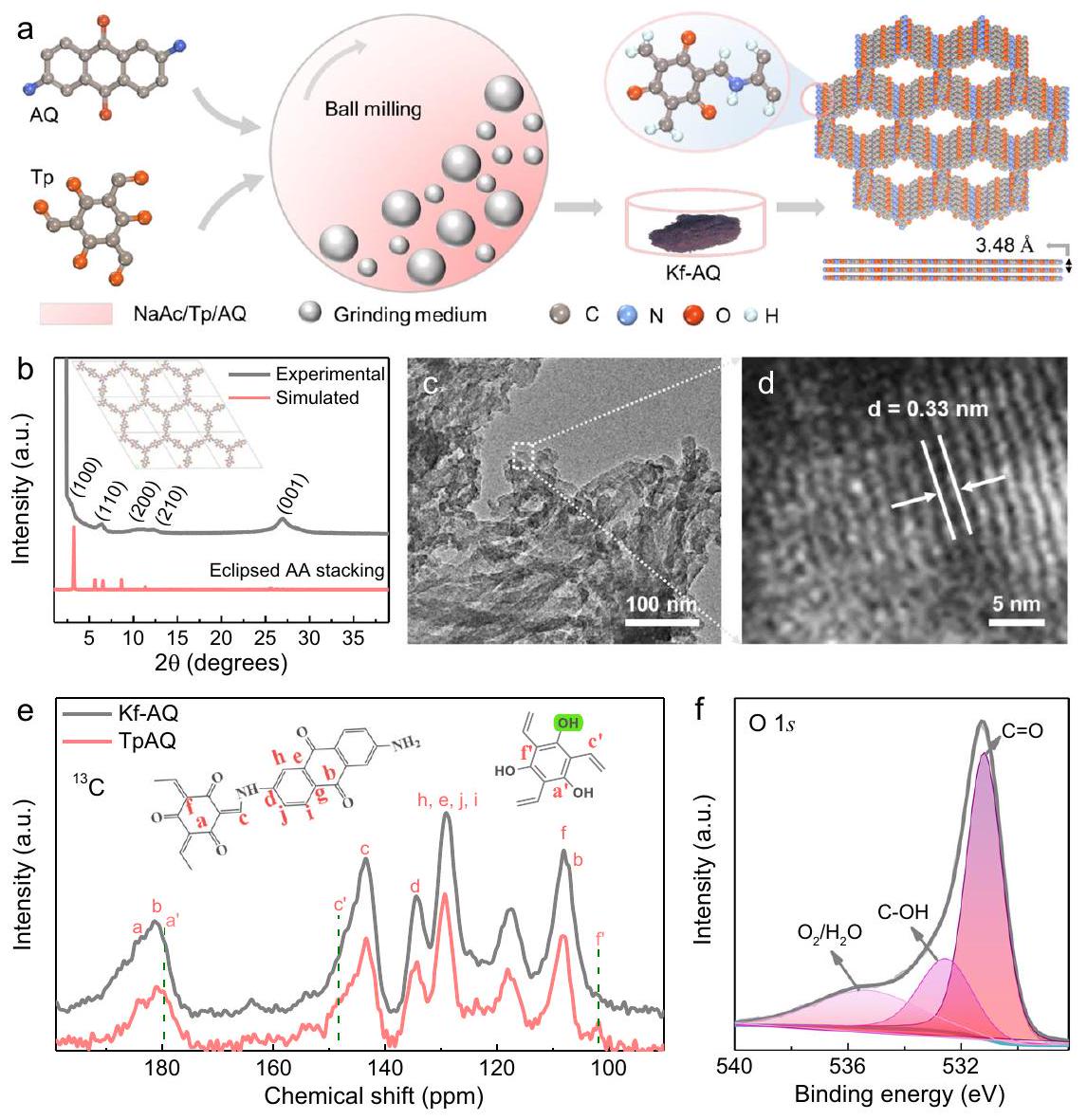
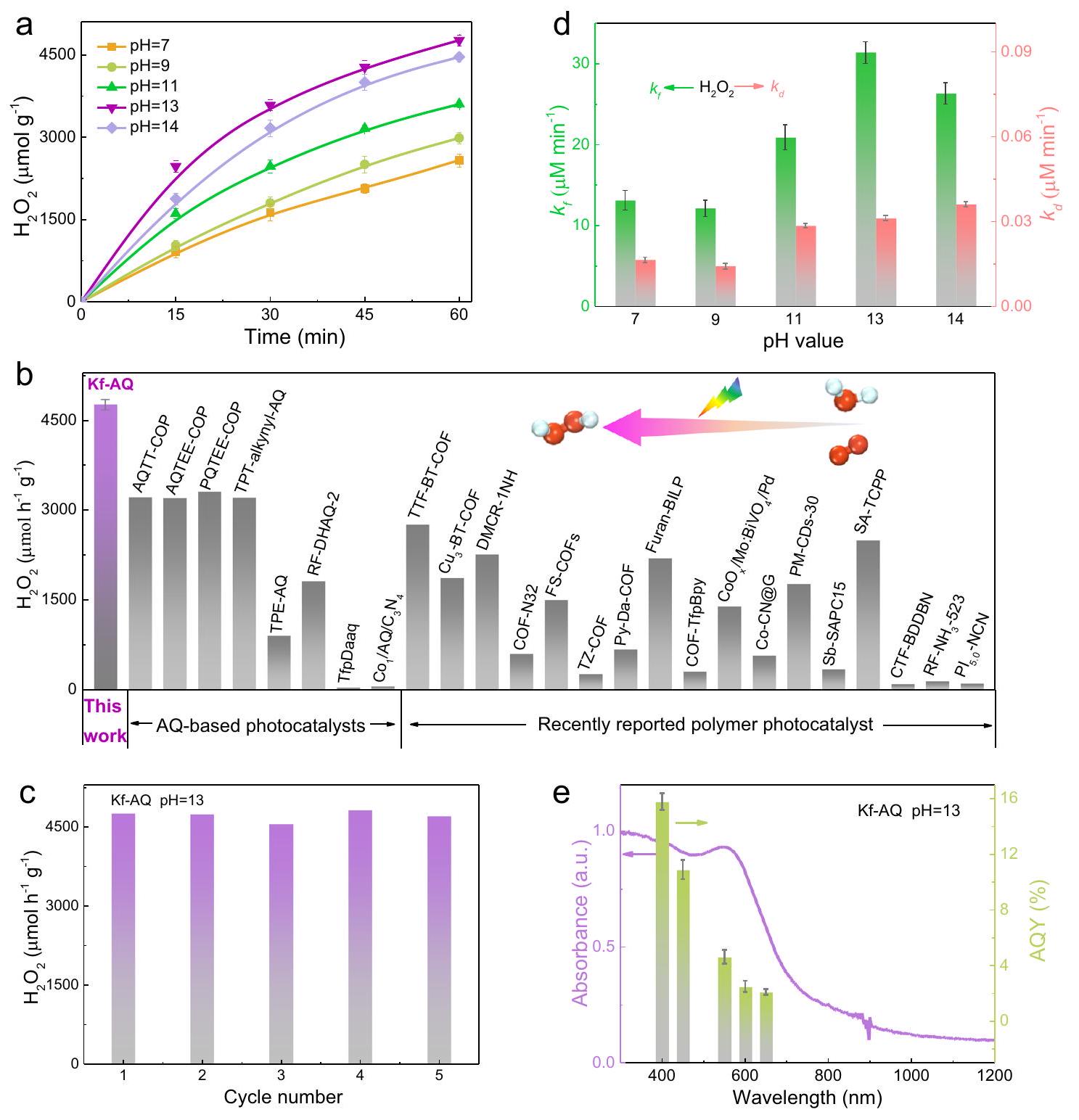
فعال
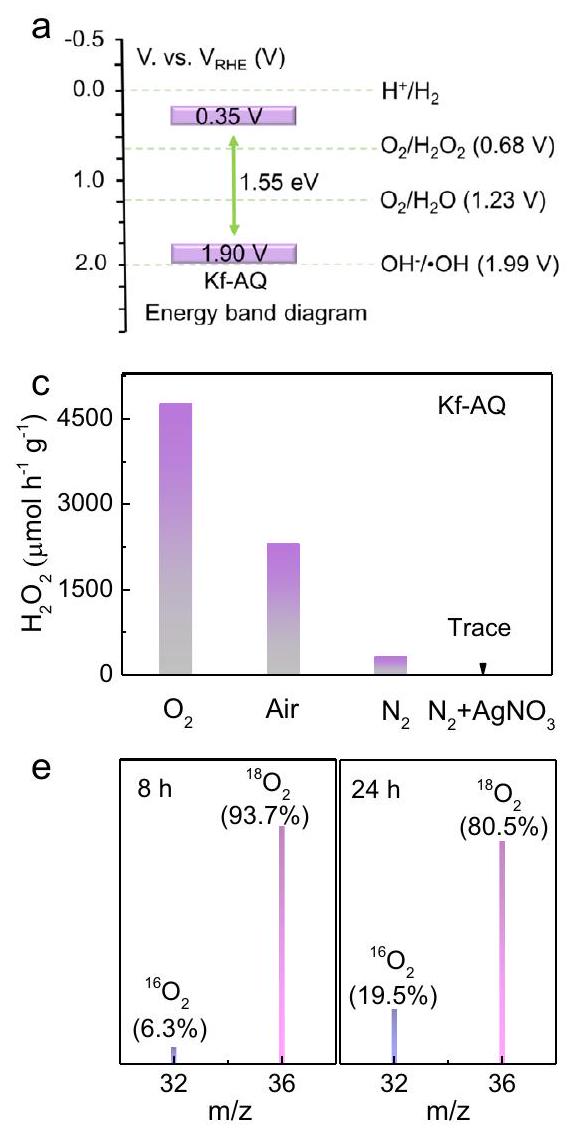
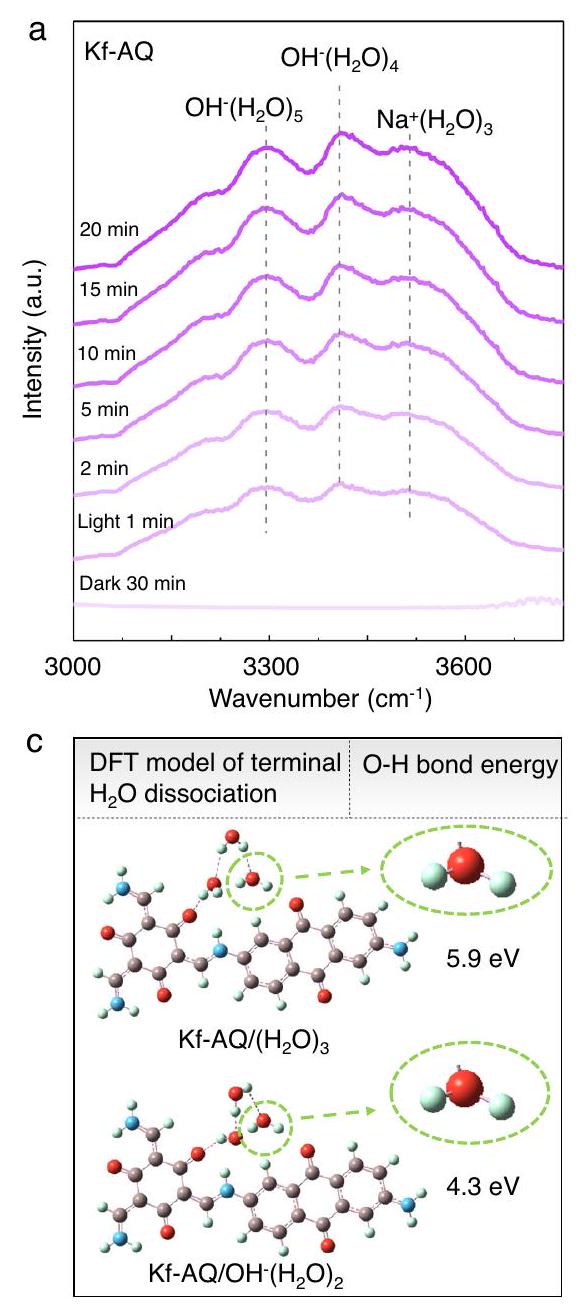
تحقيق في الآلية
شبكة المياه (
ال
الأكسدة واستخراج الهيدروجين من
نقاش
طرق
تركيب Kf-AQ
قطر و عشرة كرات من العقيق بقطر 5 مم. ثم تم طحن الخليط في درجة حرارة الغرفة بسرعة دوران 400 دورة في الدقيقة لمدة 6 ساعات. بعد ذلك، تم غسل المواد الأولية الناتجة بمادة N، N -ثنائي ميثيل الفورماميد والأسيتون، ثم تم تجفيفها في فرن مفرغ.
تركيب TpAQ
قياسات FTIR في الموقع
التجارب النظيرية
حسابات DFT
توفر البيانات
References
- Sun, X . et al. Molecular oxygen enhances
utilization for the photocatalytic conversion of methane to liquid-phase oxygenates. Nat. Commun. 13, 6677 (2022). - Shi, X. et al. Electrochemical synthesis of
by two-electron water oxidation reaction. Chem 7, 38-63 (2021). - Wang, X., Du, J. & Xu, C. Reactions in activated peroxide systems and their influences on bleaching performance. Mini-Rev. Org. Chem. 18, 836-840 (2021).
- Xu, J. et al. Organic wastewater treatment by a single-atom catalyst and electrolytically produced
. Nat. Sustain. 4, 233-241 (2020). - Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962-6984 (2006).
- Chen, Z., Yao, D., Chu, C. & Mao, S. Photocatalytic
production systems: design strategies and environmental applications. Chem. Eng. J. 451, 138489 (2023). - Kondo, Y., Kuwahara, Y., Mori, K. & Yamashita, H. Design of metalorganic framework catalysts for photocatalytic hydrogen peroxide production. Chem 8, 2924-2938 (2022).
- Wu, S. & Quan, X. Design principles and strategies of photocatalytic
production from reduction. ACS ES&T Eng. 2, 1068-1079 (2022). - Zeng, X., Liu, Y., Hu, X. & Zhang, X. Photoredox catalysis over semiconductors for light-driven hydrogen peroxide production. Green Chem 23, 1466-1494 (2021).
- Zhao, Y. et al. Efficient exciton dissociation in ionically interacted methyl viologen and polymeric carbon nitride for superior
photoproduction. ACS Catal 5, 2790-2801 (2023). - Agarwal, R. G. et al. Free energies of proton-coupled electron transfer reagents and their applications. Chem. Rev. 122, 1-49 (2022).
- Zhang, J., Muck-Lichtenfeld, C. & Studer, A. Photocatalytic phosphine-mediated water activation for radical hydrogenation. Nature 619, 506-513 (2023).
- Ma, X. et al. Hydrogen-bond network promotes water splitting on the
surface. J. Am. Chem. Soc. 144, 13565-13573 (2022). - Du, Y. et al. Two pathways for water interaction with oxygen adatoms on
(110). Phys. Rev. Lett. 102, 096102 (2009). - Tan, S. et al. Interfacial hydrogen-bonding dynamics in surfacefacilitated dehydrogenation of water on
. J. Am. Chem. Soc. 142, 826-834 (2020). - Chu, W. et al. Ultrafast charge transfer coupled to quantum proton motion at molecule/metal oxide interface. Sci. Adv. 8, eabo2675 (2022).
- Wang, Y. et al. Enabling high-energy-density aqueous batteries with hydrogen bond-anchored electrolytes. Matter 5, 162-179 (2022).
- Wang, X., Xu, C., Jaroniec, M., Zheng, Y. & Qiao, S. Z. Anomalous hydrogen evolution behavior in high-pH environment induced by locally generated hydronium ions. Nat. Commun. 10, 4876 (2019).
- Wang, H., Yang, C., Chen, F., Zheng, G. & Han, Q. A Crystalline partially fluorinated triazine covalent organic framework for efficient photosynthesis of hydrogen peroxide. Angew. Chem. Int. Ed. 61, e202202328 (2022).
- Yang, C., Wan, S., Zhu, B., Yu, J. & Cao, S. Calcination-regulated microstructures of donor-acceptor polymers towards enhanced and stable photocatalytic
production in pure water. Angew. Chem. Int. Ed. 61, e202208438 (2022). -
. et al. Anthraquinone-based conjugated organic polymers containing dual oxidation centers for photocatalytic production from and under visible-light irradiation. ACS Appl. Poly. Mater. 9, 7571-7580 (2023). -
. et al. Conjugated organic polymers with anthraquinone redox centers for efficient photocatalytic hydrogen peroxide production from water and oxygen under visible light irradiation without any additives. ACS Catal 12, 12954-12963 (2022). - Ye, Y. X. et al. A solar-to-chemical conversion efficiency up to
achieved in ambient conditions. Proc. Natl. Acad. Sci. USA 118, e2115666118 (2021). - Zhang, X. et al. Keto-enamine-based covalent organic framework with controllable anthraquinone moieties for superior
photosynthesis from and water. Chem. Eng. J. 466, 143085 (2023). - Kandambeth, S. et al. Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc. 134, 19524-19527 (2012).
- Wu, S. et al. The Keto-switched photocatalysis of reconstructed covalent organic frameworks for efficient hydrogen evolution. Angew. Chem. Int. Ed. 135, e202309026 (2023).
- Li, Q. et al. Visible-light-responsive anthraquinone functionalized covalent organic frameworks for metal-free selective oxidation of sulfides: effects of morphology and structure. ACS Catal 10, 6664-6675 (2020).
- DeBlase, C. R., Silberstein, K. E., Truong, T. T., Abruna, H. D. & Dichtel, W. R. beta-Ketoenamine-linked covalent organic frameworks capable of pseudocapacitive energy storage. J. Am. Chem. Soc. 135, 16821-16824 (2013).
- Khayum, M. A. et al. Chemically delaminated free-standing ultrathin covalent organic nanosheets. Angew. Chem. Int. Ed. 55, 15604-15608 (2016).
- Dubois, M. et al. Solid state NMR study of nanodiamond surface chemistry. Solid State Nucl. Magn. Reson. 40, 144-154 (2011).
- Wu, Z. et al. Covalent-organic frameworks with keto-enol tautomerism for efficient photocatalytic oxidative coupling of amines to imines under visible light. Sci. Chi. Chem. 64, 169-2179 (2021).
- Chen, S. et al. Chemical identification of catalytically active sites on oxygen-doped carbon nanosheet to decipher the high activity for electro-synthesis hydrogen peroxide. Angew. Chem. Int. Ed. 133, 16743-16750 (2021).
- Zhang, L. et al. A facile solution-phase synthetic approach for constructing phenol-based porous organic cages and covalent organic frameworks. Green Chem 22, 2498-2504 (2020).
- Zhang, M. et al. Construction of flexible amine-linked covalent organic frameworks by catalysis and reduction of formic acid via the eschweiler-clarke reaction. Angew. Chem. Int. Ed. 60, 12396-12405 (2021).
- Chiang, Y., Kresge, A. J., Santaballa, J. A. & Wirz, J. Ketonization of acetophenone enol in aqueous buffer solutions. Rate-equilibrium relations and mechanism of the uncatalyzed reaction. J. Am. Chem. Soc. 110, 5506-5510 (1988).
- Wu, Q. et al. A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater. Nat. Commun. 12, 483 (2021).
- Shiraishi, Y. et al. Resorcinol-formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion. Nat. Mater. 18, 985-993 (2019).
- Xu, X. et al. The construction of conjugated organic polymers containing phenanthrenequinone redox centers for visible-lightdriven
production from and without any additives. Chem. Eng. J. 454, 139929 (2023). - Yan, H. et al. Spontaneous exciton dissociation in organic photocatalyst under ambient conditions for highly efficient synthesis of hydrogen peroxide. Proc. Natl. Acad. Sci. USA 119, e2202913119 (2022).
- Zhao, C. et al. Molecular level modulation of anthraquinonecontaining resorcinol-formaldehyde resin photocatalysts for
production with exceeding 1.2% efficiency. Angew. Chem. Int. Ed. 62, e202218318 (2023). - Kou, M. et al. Molecularly engineered covalent organic frameworks for hydrogen peroxide photosynthesis. Angew. Chem. Int. Ed. 61, 202200413 (2022).
- Chu, C. et al. Spatially separating redox centers on 2D carbon nitride with cobalt single atom for photocatalytic
production. Proc. Natl. Acad. Sci. USA 117, 6376-6382 (2020). - Chang, J. N. et al. Oxidation-reduction molecular junction covalent organic frameworks for full reaction photosynthesis of
. Angew. Chem. Int. Ed. 62, e202218868 (2023). - Chang, J. N. et al. Regulation of redox molecular junctions in covalent organic frameworks for
photosynthesis coupled with biomass valorization. Angew. Chem. Int. Ed. 62, e202303606 (2023). - Das, P. et al. Integrating bifunctionality and chemical stability in covalent organic frameworks via one-pot multicomponent reactions for solar-driven
Production. J. Am. Chem. Soc. 145, 2975-2984 (2023). - Liu, F. et al. Covalent organic frameworks for direct photosynthesis of hydrogen peroxide from water, air and sunlight. Nat. Commun. 14, 4344 (2023).
- Luo, Y. et al. Sulfone-modified covalent organic frameworks enabling efficient photocatalytic hydrogen peroxide generation via one-step two-electron
reduction. Angew. Chem. Int. Ed. 62, e202305355 (2023). - Mou, Y. et al. Linkage microenvironment of azoles-related covalent organic frameworks precisely regulates photocatalytic generation of hydrogen peroxide. Angew. Chem. Int. Ed. 62, 202309480 (2023).
- Sun, J. et al. Pyrene-based covalent organic frameworks for photocatalytic hydrogen peroxide production. Angew. Chem. Int. Ed. 19, e202216719 (2023).
- Yang, T. et al. Covalent furan-benzimidazole-linked polymer hollow fiber membrane for clean and efficient photosynthesis of hydrogen peroxide. Adv. Func. Mater. 34, 202300714 (2023).
- Liu, T. et al. Overall photosynthesis of
by an inorganic semiconductor. Nat. Commun. 13, 1034 (2022). - Wang, W. et al. Photothermal-enabled single-atom catalysts for high-efficiency hydrogen peroxide photosynthesis from natural seawater. Nat. Commun. 14, 2493 (2023).
- Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374-384 (2021).
- Zhang, Y. et al.
generation from and on a near-infrared absorbing porphyrin supramolecular photocatalyst. Nat. Energy 8, 361-371 (2023). - Chen, L. et al. Acetylene and diacetylene functionalized covalent triazine frameworks as metal-free photocatalysts for hydrogen peroxide production: a new two-electron water oxidation pathway. Adv. Mater. 32, e1904433 (2020).
- Yang, L. et al. Two-channel photocatalytic production of
over nanosheets modified with perylene imides. J. Catal. 352, 274-281 (2017). - Li, L., Xu, L., Hu, Z. & Yu, J. C. Enhanced mass transfer of oxygen through a gas-liquid-solid interface for photocatalytic hydrogen peroxide production. Adv. Funct. Mater. 22, 2106120 (2021).
- Chu, C. et al. Photocatalytic
production driven by cyclodextrin-pyrimidine polymer in a wide pH range without electron donor or oxygen aeration. Appl. Catal. B: Environ. 314, 121485 (2022). - Yu, F. Y., Zhou, Y. J., Tan, H. Q., Li, Y. G. & Kang, Z. H. Versatile photoelectrocatalysis strategy raising up the green production of hydrogen peroxide. Adv. Energy Mater. 13, 202300119 (2023).
- Mitsui, T., Rose, M. K., Fomin, E., Ogletree, D. F. & Salmeron, M. Water diffusion and clustering on Pd(111). Science 297, 1850-1852 (2002).
- Peng, J. et al. The effect of hydration number on the interfacial transport of sodium ions. Nature 557, 701-705 (2018).
- Nilsson, A. & Pettersson, L. G. The structural origin of anomalous properties of liquid water. Nat. Commun. 6, 8998 (2015).
- Shen, R. et al. Coupling oxygen vacancy and hetero-phase junction for boosting catalytic activity of Pd toward hydrogen generation. Appl. Catal. B: Environ. 328, 122484 (2023).
- Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81-85 (2021).
- Chen, S. et al. Aqueous rechargeable zinc air batteries operated at
. Chem 9, 497-510 (2022). - Zeng, H. et al. pH-independent production of hydroxyl radical from atomic
-Mediated electrocatalytic reduction: a green fenton process without byproducts. Environ. Sci. Technol. 54, 14725-14731 (2020). - Liu, R. et al. Defect sites in ultrathin Pd nanowires facilitate the highly efficient electrochemical hydrodechlorination of pollutants by
. Environ. Sci. Technol. 52, 9992-10002 (2018). - Strmcnik, D. et al. The role of non-covalent interactions in electrocatalytic fuel-cell reactions on platinum. Nat. Chem. 1, 466-472 (2009).
- Tan, H. et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 13, 2024 (2022).
- Yu, W. et al. High-density frustrated lewis pair for high-performance hydrogen evolution. Adv. Energy Mater. 13, 2203136 (2023).
- Wei, Y. et al. Quantification of photocatalytically-generated hydrogen peroxide in the presence of organic electron donors:
72. Gu, S. et al. Tunable redox chemistry and stability of radical intermediates in 2D covalent organic frameworks for high performance sodium ion batteries. J. Am. Chem. Soc. 141, 9623-9628 (2019).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-47023-y.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى مينغسي لونغ أو ليزhi تشانغ.
http://www.nature.com/reprints
(ج) المؤلف(ون) 2024
مدرسة علوم البيئة والهندسة، جامعة شنغهاي جياو تونغ، شنغهاي 200240، الصين. مدرسة العلوم البيئية والإيكولوجية، المختبر الرئيسي لعمليات الإيكولوجيا الحضرية واستعادة البيئة، جامعة شرق الصين العادية، شنغهاي 200241، الصين. - البريد الإلكتروني: long_mc@sjtu.edu.cn; zhanglizhi@sjtu.edu.cn
DOI: https://doi.org/10.1038/s41467-024-47023-y
PMID: https://pubmed.ncbi.nlm.nih.gov/38531862
Publication Date: 2024-03-26
Keto-anthraquinone covalent organic framework for
Accepted: 18 March 2024
Published online: 26 March 2024
(A) Check for updates
Abstract
Hydrogen peroxide photosynthesis suffers from insufficient catalytic activity due to the high energy barrier of hydrogen extraction from
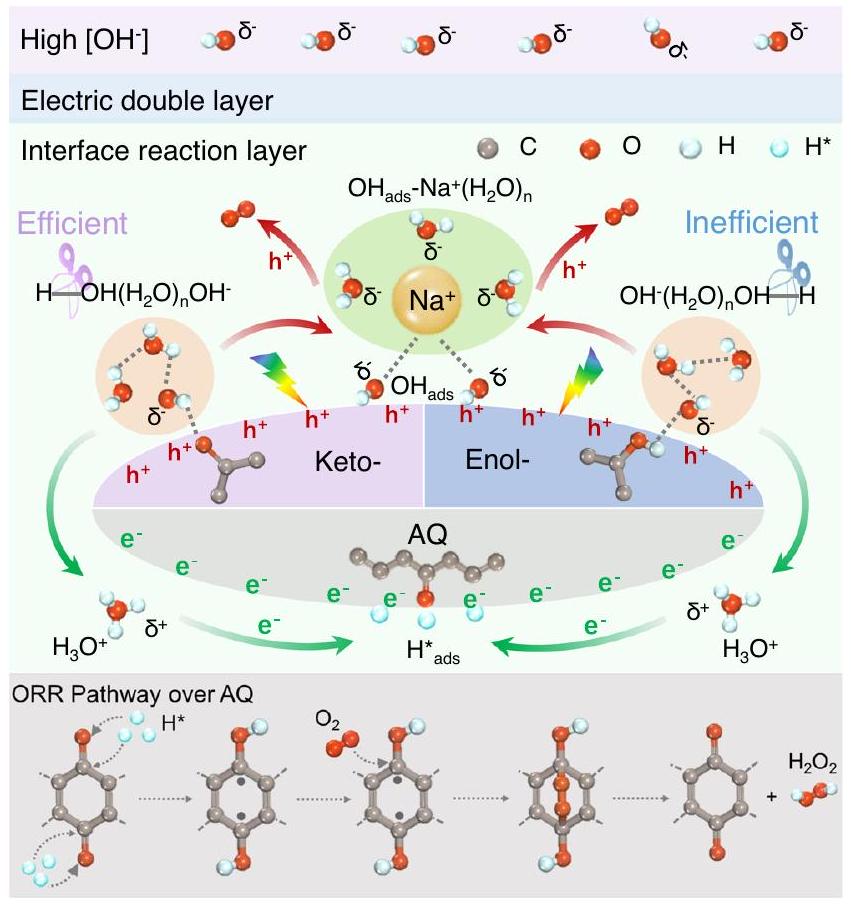
Results and discussion
Synthesis and structure characterization
Efficient
Mechanism investigation
water network (
the
oxidation and hydrogen extraction from
Discussion
Methods
Synthesis of Kf-AQ
diameter and ten 5 mm diameter agate balls. Then, the mixture was ground at room temperature with a rotation speed of 400 rpm for 6 h . After that, the obtained precursors were washed with N , N -dimethylformamide and acetone, and then dried in a vacuum oven at
Synthesis of TpAQ
In-situ FTIR measurements
Isotopic experiments
DFT calculations
Data availability
References
- Sun, X . et al. Molecular oxygen enhances
utilization for the photocatalytic conversion of methane to liquid-phase oxygenates. Nat. Commun. 13, 6677 (2022). - Shi, X. et al. Electrochemical synthesis of
by two-electron water oxidation reaction. Chem 7, 38-63 (2021). - Wang, X., Du, J. & Xu, C. Reactions in activated peroxide systems and their influences on bleaching performance. Mini-Rev. Org. Chem. 18, 836-840 (2021).
- Xu, J. et al. Organic wastewater treatment by a single-atom catalyst and electrolytically produced
. Nat. Sustain. 4, 233-241 (2020). - Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962-6984 (2006).
- Chen, Z., Yao, D., Chu, C. & Mao, S. Photocatalytic
production systems: design strategies and environmental applications. Chem. Eng. J. 451, 138489 (2023). - Kondo, Y., Kuwahara, Y., Mori, K. & Yamashita, H. Design of metalorganic framework catalysts for photocatalytic hydrogen peroxide production. Chem 8, 2924-2938 (2022).
- Wu, S. & Quan, X. Design principles and strategies of photocatalytic
production from reduction. ACS ES&T Eng. 2, 1068-1079 (2022). - Zeng, X., Liu, Y., Hu, X. & Zhang, X. Photoredox catalysis over semiconductors for light-driven hydrogen peroxide production. Green Chem 23, 1466-1494 (2021).
- Zhao, Y. et al. Efficient exciton dissociation in ionically interacted methyl viologen and polymeric carbon nitride for superior
photoproduction. ACS Catal 5, 2790-2801 (2023). - Agarwal, R. G. et al. Free energies of proton-coupled electron transfer reagents and their applications. Chem. Rev. 122, 1-49 (2022).
- Zhang, J., Muck-Lichtenfeld, C. & Studer, A. Photocatalytic phosphine-mediated water activation for radical hydrogenation. Nature 619, 506-513 (2023).
- Ma, X. et al. Hydrogen-bond network promotes water splitting on the
surface. J. Am. Chem. Soc. 144, 13565-13573 (2022). - Du, Y. et al. Two pathways for water interaction with oxygen adatoms on
(110). Phys. Rev. Lett. 102, 096102 (2009). - Tan, S. et al. Interfacial hydrogen-bonding dynamics in surfacefacilitated dehydrogenation of water on
. J. Am. Chem. Soc. 142, 826-834 (2020). - Chu, W. et al. Ultrafast charge transfer coupled to quantum proton motion at molecule/metal oxide interface. Sci. Adv. 8, eabo2675 (2022).
- Wang, Y. et al. Enabling high-energy-density aqueous batteries with hydrogen bond-anchored electrolytes. Matter 5, 162-179 (2022).
- Wang, X., Xu, C., Jaroniec, M., Zheng, Y. & Qiao, S. Z. Anomalous hydrogen evolution behavior in high-pH environment induced by locally generated hydronium ions. Nat. Commun. 10, 4876 (2019).
- Wang, H., Yang, C., Chen, F., Zheng, G. & Han, Q. A Crystalline partially fluorinated triazine covalent organic framework for efficient photosynthesis of hydrogen peroxide. Angew. Chem. Int. Ed. 61, e202202328 (2022).
- Yang, C., Wan, S., Zhu, B., Yu, J. & Cao, S. Calcination-regulated microstructures of donor-acceptor polymers towards enhanced and stable photocatalytic
production in pure water. Angew. Chem. Int. Ed. 61, e202208438 (2022). -
. et al. Anthraquinone-based conjugated organic polymers containing dual oxidation centers for photocatalytic production from and under visible-light irradiation. ACS Appl. Poly. Mater. 9, 7571-7580 (2023). -
. et al. Conjugated organic polymers with anthraquinone redox centers for efficient photocatalytic hydrogen peroxide production from water and oxygen under visible light irradiation without any additives. ACS Catal 12, 12954-12963 (2022). - Ye, Y. X. et al. A solar-to-chemical conversion efficiency up to
achieved in ambient conditions. Proc. Natl. Acad. Sci. USA 118, e2115666118 (2021). - Zhang, X. et al. Keto-enamine-based covalent organic framework with controllable anthraquinone moieties for superior
photosynthesis from and water. Chem. Eng. J. 466, 143085 (2023). - Kandambeth, S. et al. Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc. 134, 19524-19527 (2012).
- Wu, S. et al. The Keto-switched photocatalysis of reconstructed covalent organic frameworks for efficient hydrogen evolution. Angew. Chem. Int. Ed. 135, e202309026 (2023).
- Li, Q. et al. Visible-light-responsive anthraquinone functionalized covalent organic frameworks for metal-free selective oxidation of sulfides: effects of morphology and structure. ACS Catal 10, 6664-6675 (2020).
- DeBlase, C. R., Silberstein, K. E., Truong, T. T., Abruna, H. D. & Dichtel, W. R. beta-Ketoenamine-linked covalent organic frameworks capable of pseudocapacitive energy storage. J. Am. Chem. Soc. 135, 16821-16824 (2013).
- Khayum, M. A. et al. Chemically delaminated free-standing ultrathin covalent organic nanosheets. Angew. Chem. Int. Ed. 55, 15604-15608 (2016).
- Dubois, M. et al. Solid state NMR study of nanodiamond surface chemistry. Solid State Nucl. Magn. Reson. 40, 144-154 (2011).
- Wu, Z. et al. Covalent-organic frameworks with keto-enol tautomerism for efficient photocatalytic oxidative coupling of amines to imines under visible light. Sci. Chi. Chem. 64, 169-2179 (2021).
- Chen, S. et al. Chemical identification of catalytically active sites on oxygen-doped carbon nanosheet to decipher the high activity for electro-synthesis hydrogen peroxide. Angew. Chem. Int. Ed. 133, 16743-16750 (2021).
- Zhang, L. et al. A facile solution-phase synthetic approach for constructing phenol-based porous organic cages and covalent organic frameworks. Green Chem 22, 2498-2504 (2020).
- Zhang, M. et al. Construction of flexible amine-linked covalent organic frameworks by catalysis and reduction of formic acid via the eschweiler-clarke reaction. Angew. Chem. Int. Ed. 60, 12396-12405 (2021).
- Chiang, Y., Kresge, A. J., Santaballa, J. A. & Wirz, J. Ketonization of acetophenone enol in aqueous buffer solutions. Rate-equilibrium relations and mechanism of the uncatalyzed reaction. J. Am. Chem. Soc. 110, 5506-5510 (1988).
- Wu, Q. et al. A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater. Nat. Commun. 12, 483 (2021).
- Shiraishi, Y. et al. Resorcinol-formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion. Nat. Mater. 18, 985-993 (2019).
- Xu, X. et al. The construction of conjugated organic polymers containing phenanthrenequinone redox centers for visible-lightdriven
production from and without any additives. Chem. Eng. J. 454, 139929 (2023). - Yan, H. et al. Spontaneous exciton dissociation in organic photocatalyst under ambient conditions for highly efficient synthesis of hydrogen peroxide. Proc. Natl. Acad. Sci. USA 119, e2202913119 (2022).
- Zhao, C. et al. Molecular level modulation of anthraquinonecontaining resorcinol-formaldehyde resin photocatalysts for
production with exceeding 1.2% efficiency. Angew. Chem. Int. Ed. 62, e202218318 (2023). - Kou, M. et al. Molecularly engineered covalent organic frameworks for hydrogen peroxide photosynthesis. Angew. Chem. Int. Ed. 61, 202200413 (2022).
- Chu, C. et al. Spatially separating redox centers on 2D carbon nitride with cobalt single atom for photocatalytic
production. Proc. Natl. Acad. Sci. USA 117, 6376-6382 (2020). - Chang, J. N. et al. Oxidation-reduction molecular junction covalent organic frameworks for full reaction photosynthesis of
. Angew. Chem. Int. Ed. 62, e202218868 (2023). - Chang, J. N. et al. Regulation of redox molecular junctions in covalent organic frameworks for
photosynthesis coupled with biomass valorization. Angew. Chem. Int. Ed. 62, e202303606 (2023). - Das, P. et al. Integrating bifunctionality and chemical stability in covalent organic frameworks via one-pot multicomponent reactions for solar-driven
Production. J. Am. Chem. Soc. 145, 2975-2984 (2023). - Liu, F. et al. Covalent organic frameworks for direct photosynthesis of hydrogen peroxide from water, air and sunlight. Nat. Commun. 14, 4344 (2023).
- Luo, Y. et al. Sulfone-modified covalent organic frameworks enabling efficient photocatalytic hydrogen peroxide generation via one-step two-electron
reduction. Angew. Chem. Int. Ed. 62, e202305355 (2023). - Mou, Y. et al. Linkage microenvironment of azoles-related covalent organic frameworks precisely regulates photocatalytic generation of hydrogen peroxide. Angew. Chem. Int. Ed. 62, 202309480 (2023).
- Sun, J. et al. Pyrene-based covalent organic frameworks for photocatalytic hydrogen peroxide production. Angew. Chem. Int. Ed. 19, e202216719 (2023).
- Yang, T. et al. Covalent furan-benzimidazole-linked polymer hollow fiber membrane for clean and efficient photosynthesis of hydrogen peroxide. Adv. Func. Mater. 34, 202300714 (2023).
- Liu, T. et al. Overall photosynthesis of
by an inorganic semiconductor. Nat. Commun. 13, 1034 (2022). - Wang, W. et al. Photothermal-enabled single-atom catalysts for high-efficiency hydrogen peroxide photosynthesis from natural seawater. Nat. Commun. 14, 2493 (2023).
- Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374-384 (2021).
- Zhang, Y. et al.
generation from and on a near-infrared absorbing porphyrin supramolecular photocatalyst. Nat. Energy 8, 361-371 (2023). - Chen, L. et al. Acetylene and diacetylene functionalized covalent triazine frameworks as metal-free photocatalysts for hydrogen peroxide production: a new two-electron water oxidation pathway. Adv. Mater. 32, e1904433 (2020).
- Yang, L. et al. Two-channel photocatalytic production of
over nanosheets modified with perylene imides. J. Catal. 352, 274-281 (2017). - Li, L., Xu, L., Hu, Z. & Yu, J. C. Enhanced mass transfer of oxygen through a gas-liquid-solid interface for photocatalytic hydrogen peroxide production. Adv. Funct. Mater. 22, 2106120 (2021).
- Chu, C. et al. Photocatalytic
production driven by cyclodextrin-pyrimidine polymer in a wide pH range without electron donor or oxygen aeration. Appl. Catal. B: Environ. 314, 121485 (2022). - Yu, F. Y., Zhou, Y. J., Tan, H. Q., Li, Y. G. & Kang, Z. H. Versatile photoelectrocatalysis strategy raising up the green production of hydrogen peroxide. Adv. Energy Mater. 13, 202300119 (2023).
- Mitsui, T., Rose, M. K., Fomin, E., Ogletree, D. F. & Salmeron, M. Water diffusion and clustering on Pd(111). Science 297, 1850-1852 (2002).
- Peng, J. et al. The effect of hydration number on the interfacial transport of sodium ions. Nature 557, 701-705 (2018).
- Nilsson, A. & Pettersson, L. G. The structural origin of anomalous properties of liquid water. Nat. Commun. 6, 8998 (2015).
- Shen, R. et al. Coupling oxygen vacancy and hetero-phase junction for boosting catalytic activity of Pd toward hydrogen generation. Appl. Catal. B: Environ. 328, 122484 (2023).
- Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81-85 (2021).
- Chen, S. et al. Aqueous rechargeable zinc air batteries operated at
. Chem 9, 497-510 (2022). - Zeng, H. et al. pH-independent production of hydroxyl radical from atomic
-Mediated electrocatalytic reduction: a green fenton process without byproducts. Environ. Sci. Technol. 54, 14725-14731 (2020). - Liu, R. et al. Defect sites in ultrathin Pd nanowires facilitate the highly efficient electrochemical hydrodechlorination of pollutants by
. Environ. Sci. Technol. 52, 9992-10002 (2018). - Strmcnik, D. et al. The role of non-covalent interactions in electrocatalytic fuel-cell reactions on platinum. Nat. Chem. 1, 466-472 (2009).
- Tan, H. et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 13, 2024 (2022).
- Yu, W. et al. High-density frustrated lewis pair for high-performance hydrogen evolution. Adv. Energy Mater. 13, 2203136 (2023).
- Wei, Y. et al. Quantification of photocatalytically-generated hydrogen peroxide in the presence of organic electron donors:
72. Gu, S. et al. Tunable redox chemistry and stability of radical intermediates in 2D covalent organic frameworks for high performance sodium ion batteries. J. Am. Chem. Soc. 141, 9623-9628 (2019).
Acknowledgements
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-47023-y.
Correspondence and requests for materials should be addressed to Mingce Long or Lizhi Zhang.
http://www.nature.com/reprints
(c) The Author(s) 2024
School of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. School of Ecological and Environmental Science, Key Laboratory for Urban Ecological Processes and Eco-Restoration, East China Normal University, Shanghai 200241, China.