إطار فثالوسيانين معدني مرتبط بالديثيين مع طبقات متموجة لإنتاج H2O2 الكهربائي بكفاءة عالية واستقرار Dithiine-linked metalphthalocyanine framework with undulated layers for highly efficient and stable H2O2 electroproduction
تحقيق مستوى مستقر وصناعيلا يزال الإنتاج الكهربائي يواجه تحديات كبيرة جزئيًا بسبب سهولة التحلل.. هنا، تم الحصول على إطار عضوي تساهمي (COF) قائم على فثالوسيانين الكوبالت (CoPc) مرتبط بالثيوين ثنائي الأبعاد (CoPc-S-COF) من تفاعل فثالوسيانين الكوبالت (II) الهكسادكافلوري مع 1،2،4،5-بيزنتيترا ثيول. يؤدي إدخال ذرات الكبريت ذات نصف القطر الذري الكبير وزوجين من الإلكترونات الحرة في وحدة الربط C-S-C إلى هيكل طبقي متموج وزيادة كثافة الإلكترون في مركز الكوبالت لـ CoPc-S-COF وفقًا لسلسلة من التجارب بالتزامن مع الحسابات النظرية. يسمح التأثير الهيكلي السابق بفتح المزيد من مواقع الكوبالت لتعزيز أداء COF التحفيزي، بينما ينشط التأثير الإلكتروني الأخير تفاعل اختزال الأكسجينORR) ولكن يقوم بإلغاء تفعيلقدرة التحلل لنفس مركز الكوبالت، مما يمكّن CoPc-S-COF من إظهار كفاءة كهربائية جيدة.أداء الإنتاج بشكل ملحوظانتقائيةوإسطبلالإنتاج مع تركيز علىتحت كثافة تيار عالية منعند جهد مطبق يبلغ حوالي 0.67 فولت مقابل RHE لمدة 20 ساعة في خلية تدفق، مما يمثل الأفضل المبلغ عنه حتى الآنتحفيز كهربائي لمركبات الإطار العضوي.
بيروكسيد الهيدروجين ) هو مادة كيميائية غير عضوية مهمة ومواد مؤكسدة صديقة للبيئة مع تطبيقات واسعة في التبييض، والتعقيم، ومعالجة مياه الصرف الصحي، والتركيب العضوي في الصناعة، يتم استخدام طريقة الأنثراكوينون لإنتاج أكثر منمنومع ذلك، فإنها تتطلب طاقة كبيرة وتنتج كمية كبيرة من المنتجات الثانوية السامةمن أجل مستقبل مستدام، من الضروري تطوير استراتيجية فعالة من حيث الطاقة وصديقة للبيئة لعملية التخليقيجب أن تعمل في الموقع سواء على نطاق واسع أو صغير. ونتيجة لذلك، التحفيز الكهربائيتفاعل اختزال الأكسجينتم اعتبار ORR كأكثر الطرق البديلة الواعدة حيث يمكن أن تحقق الطلب الأخضر والموزع عند الطلب.توليد تحت الظروف المحيطة الشروط. حتى الآن، تشمل المحفزات الكهربائية المختلفة الكربون المعدلالمعادن النبيلة والسبائكالمعادن غير النبيلةإطارات معدنية عضوية (MOFs)وأطر عضوية تساهمية (COFs)تم تطويرها لتعزيزORR لـالتحليل الكهربائي. ومع ذلك، فإن التحفيز الكهربائي على نطاق واسعلا يزال من الصعب تحقيق الإنتاج بسبب الذوبانية المحدودة للأكسجين في محاليل الإلكتروليت وسهولة تحلله.خاصة في وجود مراكز نشطة معدنية، والتي عادة ما تؤدي إلى تيارات عمل صغيرة ) ومنخفض تركيز. بالإضافة إلى ذلك، الديناميكا الحرارية المواتية لتوليد جزيئات الماء عبر المسار يقلل حتمًا منقدرة التوليد خلال.
تتكون COFs من وحدات بناء عضوية مرتبطة بروابط تساهمية.التي جذبت اهتمامًا كبيرًا في البحث لتطبيقاتها الواسعة في تخزين الغاز والفصلالأجهزة البصرية الإلكترونيةتحفيزوتخزين الطاقةنظرًا لتفوقها في المسامية العالية، والاستقرار القوي، والكثافة المنخفضة. نتيجة للثقوب المرتبة التي تعزز المزيد من المواقع النشطة المكشوفة للتفاعل مع جزيئات الركيزة، أظهرت COFs إمكانيات تطبيق كبيرة كعوامل تحفيز كهربائية واعدة لمختلف التفاعلات بما في ذلك اختزال الأكسجين.تفاعل تطور الأكسجينتفاعل تطور الهيدروجين، و تفاعل الاختزالعلى وجه الخصوص، تم الكشف عن أن الأطر العضوية المتشابكة ثنائية الأبعاد (2D) ذات الروابط العطرية المدمجة الفائقة القوة تظهر موصلية عالية وثبات حراري/كيميائي جوهري، مما يعزز الأداء الكهروكيميائي.. ومع ذلك، فإن تصميم الروابط المناسبة وتحسين ظروف التفاعل لبناء COFs لا يزال مهمة صعبة للكيميائيين الصناعيين. الديوكسينفينازين وبيبيرازين تم إنشاء تشكيل الروابط في COF مترافق ثنائي الأبعاد من خلال الاستبدال الأروماتي النوكليوفيلي كروابط عضوية غير متجانسة ملتحمة لبناء COFs بلورية ومستقرة. وقد تم تطبيق COFs المقابلة على هذه الروابط في التحفيز وأجهزة تخزين الطاقة. ومع ذلك، فإن هذه الروابط عادة ما تؤدي إلى تباعد طبقات قريب نسبيًا مرتبط بخصائصها المسطحة النموذجية بين الطبقات.-ترتيب التكديس، مما يؤدي إلى دفن المواقع النشطة الداخلية إلى حد ما. مؤخرًا، كاسكل وزملاؤه قاموا ببناء إطار عضوي مرتبط بالديثيين مع طبقات متموجة بسبب الانحناء على طول الجسر ولكن تم الحفاظ على العطرية والبلورية في الهيكل العام لـ COF، مما يوفر دليلاً لاستخدام أكثر كفاءة للمواقع النشطة المدفونة الداخلية. بالإضافة إلى اختيار الربط في COFs المترافقة ثنائية الأبعاد، تشمل المواد الأولية المترافقة المسطحة البورفيرين.الفثالوسيانين (Pc)وهكسانزينكورونينعادة ما تم اختيارها ككتل بناء بسبب استقرارها القوي وموصليتها الكهربائية العالية الجوهرية.
وحدات بناء الفثالوسيانين المعدنية (MPc) معلقد تم إثبات أن تكوين التنسيق يعمل كمواقع نشطة عالية الكفاءة لتحفيز سلسلة من التفاعلات كما يتضح من الكفاءةنشاط ORR لفيتالوسيانين الكوبالت (CoPc).
في هذا السياق، تم الحصول على COF قائم على CoPc مرتبط بـ dithiine، CoPc-S-COF، من تفاعل كوبالت (II) هكسادكافلوروفتالوسيانيناتو. ) مع 1،2،4،5-بنزينتترايثيول (BTT). لغرض المقارنة، تم أيضًا تحضير COF قائم على CoPc مرتبط بالديكسين التقليدي ثنائي الأبعاد، CoPc-O-COF، من خلال التفاعل بين و 5-تتراهيدروكسي بنزين (THB). تكشف نتائج تحليل حيود الأشعة السينية بالمسحوق (PXRD) وتحليل المجهر الإلكتروني عن الإطار البلوري المسامي لـ CoPc-S-COF مع هيكل طبقي متموج بسبب الانحناء على طول جسر C-S-C المرتبط بنصف قطر الذرة الكبير واثنين من أزواج الإلكترونات الوحيدة لذرات الكبريت في وحدة الربط، مما يؤدي إلى تعرض شبه مزدوج لمواقع الكوبالت النشطة.ORR مقارنةً بـ CoPc-O-COF مع وضعية متداخلة-نموذج التكديس وفقًا للتحليل الكهروكيميائي. هذا، بالاشتراك مع المنشطORR ولكن معطلقدرة التحلل لنفس مركز الكوبالت بسبب تأثير التبرع بالإلكترونات من ذرات الكبريت، تمكن CoPc-S-COF من عرض نشاط كهربائي تحفيزي متفوق.أداء ORR بشكل ملحوظانتقائيةوإسطبلالإنتاج تحت كثافة تيار عالية من عند جهد مطبق يبلغ حوالي 0.67 فولت مقابل RHE لمدة 20 ساعة في خلية تدفق، مما ينتج محلول بتركيز.
النتائج
تحضير المواد وخصائصها
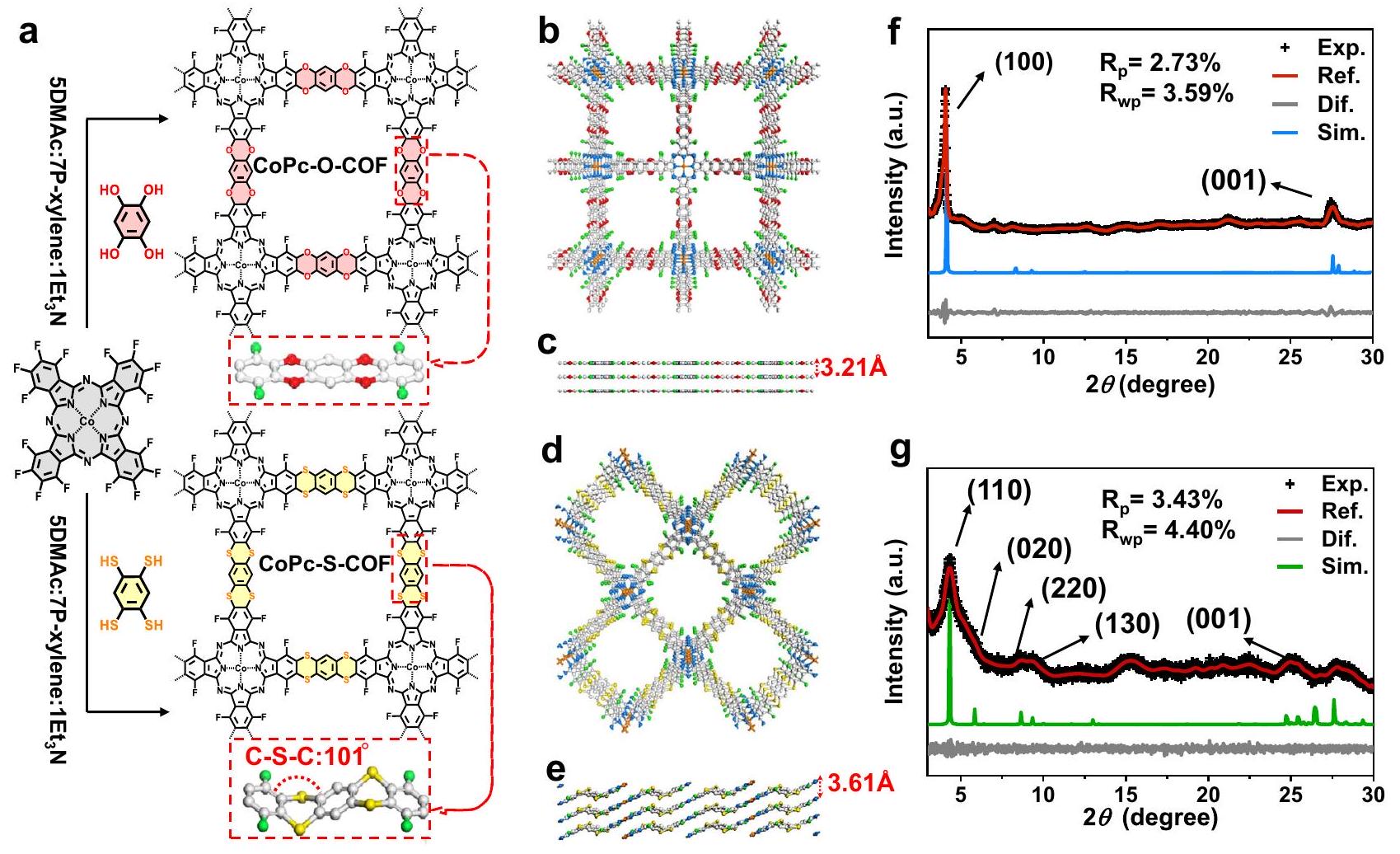
يتم توضيح تخليق CoPc-O-COF و CoPc-S-COF في الشكل 1a وتظهر نماذجها الهيكلية المحاكاة في الشكل 1b-e. تفاعل الاستبدال النوكليوفيلي لـمع THB و BTT، على التوالي، في ثنائي ميثيل أسيتاميد (DMAC) و p-xylene مع
الشكل 1 | مخطط للـ COFs الاصطناعية وتمثيلات هيكلية للـ COFs. أ مسار تخليق CoPc-O-COF و CoPc-S-COF. التكديس المحاكى AA لـ ب، ج CoPc-O-COF و د، هـ CoPc-S-COF (Co: برتقالي؛ C: رمادي فاتح؛ N: أزرق؛ O: أحمر؛ S: أصفر؛ F: أخضر). PXRD من CoPc-O-COF و CoPc-S-COF: PXRD التجريبي الملف (أسود)، الملف المصقول (أحمر)، الفرق بين PXRD التجريبي والمصقول (رمادي)، ونمط المحاكاة بناءً على طريقة تكديس AA (أزرق وأخضر). ثلاثي إيثيل أمين ( ) كعامل مساعد يوفر و في العائد 75 ومراقبة الفرقة فيبسببسنداتفي طيف الأشعة تحت الحمراء بواسطة تحويل فورييه (FT-IR) يظهر التكوين الناجح لجسر الديوكسين فيCOF، الشكل التوضيحي التكميلي 1. الشريط المميز لرابطة C-S-Cيظهر فيفي طيف FT-IR، الشكل التوضيحي 2، يؤكد على النجاح في تشكيل جسر الديثيين في CoPc-S-COF. الحالة الصلبة تكشف مطيافية الرنين المغناطيسي النووي (NMR) بتقنية الاستقطاب المتقاطع/الدوران بزاوية سحرية (CP/MAS) عن إشارات الكربون العطرية المميزة عند 149 و143 جزء في المليون لـ CoPc-O-COF وCoPc-S-COF، على التوالي، مما يدعم بشكل أكبر توليد COFs المرتبطة بالديوكسين/الديثين، الأشكال التكميلية 3 و4. كلا من CoPc-O-COF وCoPc-S-COF يظهران إشارة عند حوالي -124 جزء في المليون في حالتهما الصلبة.طيف NMR لـ CP/MAS، مما يدل على طبيعة مجموعة C-F المماثلة، الشكل التوضيحي الإضافي 5. تم الكشف عن درجة حرارة التحلل لتكون أعلى منلكلا COFs وفقًا للتحليل الحراري الوزني، مما يدل على استقرارهما الحراري الكبير، الشكل التوضيحي الإضافي 6. علاوة على ذلك، تم إعادة جمع أنماط PXRD لكلا COFs بعد نقعهما في محاليل مختلفة بما في ذلكماء نقي، THF، DMF، أسيتون، إيثانول ولثلاثة أيام تبقى دون تغيير، مما يكشف عن الاستقرار الكيميائي الجيد لكل من COFs، الأشكال التكميلية 7 و 8. على وجه الخصوص، تعرض كل من COFs طيف FTIR وصور TEM متشابهة جداً قبل وبعد معالجة النقع في، مما يثبت أكثر متانة هذين المركبين العضويين الإطارين فيالحل، الأشكال التكميلية 9 و 10.
تم تقدير الهياكل البلورية لهذين COFs من خلال قياس PXRD المدمج مع المحاكاة الحاسوبية. كما هو موضح في الشكل 1f، يظهر CoPc-O-COF ذروتين قويتين عند 4.13 و، التي تتوافق مع الوجوه (100) و (001) على التوالي. علاوة على ذلك، فإن النمط التجريبي لـ يتفق مع القيم المحسوبة مع تكدسات طبقات AA استنادًا إلى طريقة محاكاة فورسايت الهندسية، الشكل 1b، c. علاوة على ذلك، نمط PXRD المصقول بواسطة باولي لـ CoPc-O-COF باستخدام فراغ المنحنى التجريبي الملحوظ كما أثبتت عوامل التوافق الجيدة و الشكل 1f. يظهر نمط PXRD لـ CoPc-S-COF قمة قوية واحدة عندوأربع انعكاسات متوسطة الشدة عند، و “، والتي تتوافق مع الوجوه (110)، (020)، (220)، (130)، و(001). تشير مجموعة من المحاكاة النظرية وتحسين باولي إلى أن CoPc-S-COF يتبنى هيكلًا مكدسًا متموجًا بسبب التكوين غير المستوي لوحدات C-S-C بزاوية ثنائية.مما يوفر معلمات الشبكة لـ، و فيمجموعة الفضاء مع و الشكل 1c، f، g. يوفر الهيكل البلوري الأحادي لمركبات النموذج التي تحتوي على وحدات ديثيين دعماً إضافياً للتكوين غير المستوي لـ CoPc-S-COF، الشكل التكميلي 11 والجدول التكميلي 1. تم تسجيل طيف رامان لكلا COFs لاستكشاف انقساماتها الاهتزازية بناءً على الروابط الوظيفية المقابلة، الشكل التكميلي 12. تشير القمم المتماثلة الواضحة الناتجة عن روابط الكربون العطرية وكربون-أكسجين إلى طبيعة الاهتزاز في المستوى.مراقبة سلسلة من نطاقات التمدد غير المتماثل الناتجة عن اهتزازات الكربون العطري وكبريت الكربون لـتشير إلى الانقسامات الاهتزازية المختلفة لحالات الطاقة لرابطة الكربون – الكبريت المرتبطة بالانحناء أو الالتواء خارج المستوى للرابط.في الواقع، على عكس التركيب المرتبط بالديكسين لـ CoPc-O-COF، فإن وحدات C-S-C في CoPc-S-COF مستقرّة في تكوين غير مستوٍ لتقليل تنافر أزواج الإلكترونات الوحيدة لذرات الكبريت ذات نصف القطر الذري الكبير في الطبقات المجاورة.مما أدى إلى هيكله المكدس ذو الطبقات المتموجة.
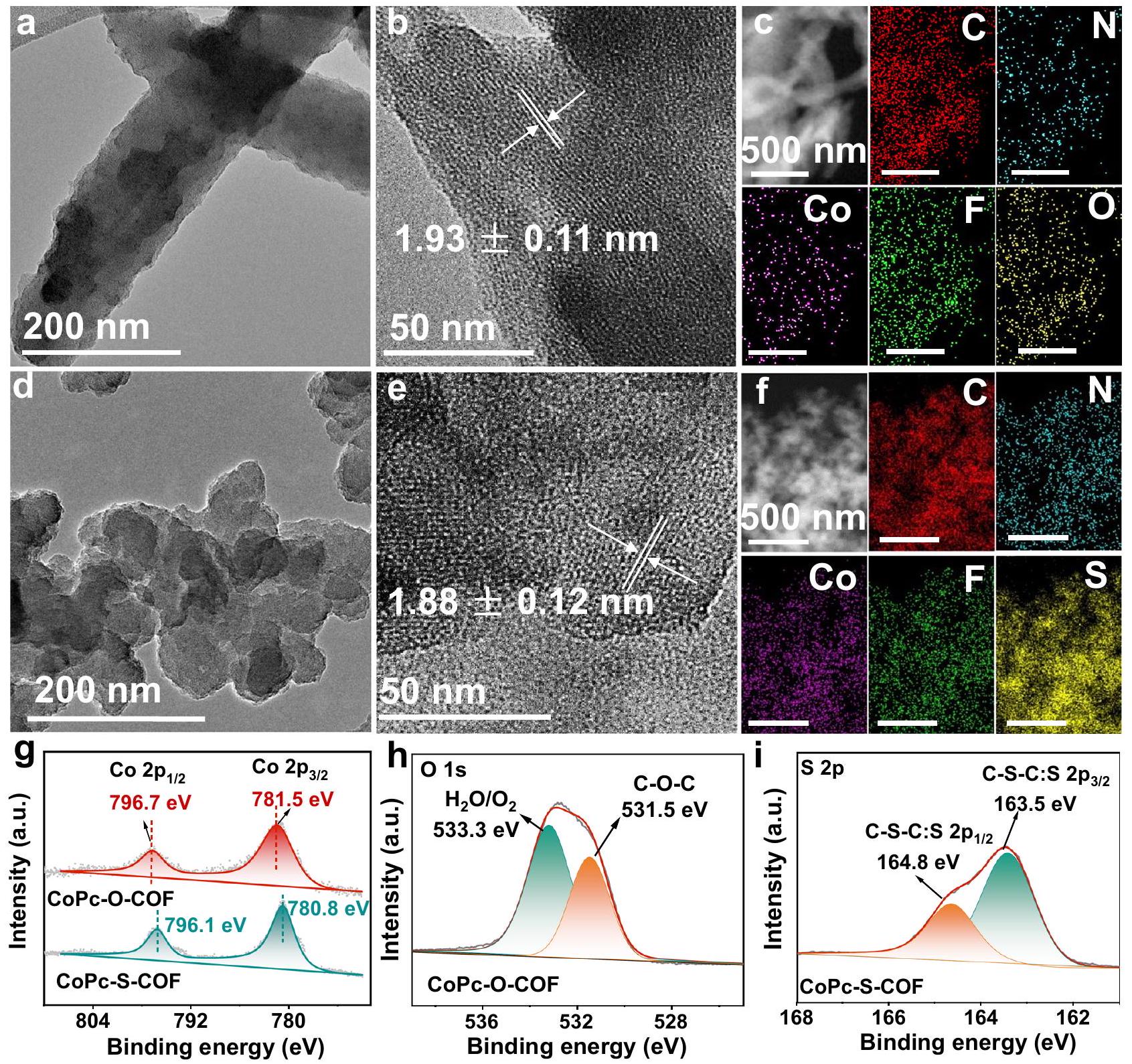
تمت دراسة مورفولوجيا CoPc-O-COF و CoPc-S-COF بواسطة المجهر الإلكتروني الماسح (SEM) وصور المجهر الإلكتروني الناقل (TEM)، الشكل 2a-f والشكل التكميلي 13. كما يمكن ملاحظته، يظهر CoPc-O-COF مورفولوجيا غير منتظمة على مقياس الميكرومتر، تختلف عن مورفولوجيا الكتل غير المنتظمة لـ CoPc-S-COF بسبب نمط الطبقات المكدسة المتموجة. كلا من CoPc-O-COF ويوضح CoPc-S-COF حواف شبكة مميزة مع تباعد قدره و في صورهم عالية الدقة بتقنية TEM (HR-TEM)، والتي تُنسب إلى المستوى (100) من CoPc-O-COF والمستوى (110) من CoPc-S-COF، على التوالي، وتؤكد بدورها على بلوريتها العالية، الشكل 2b، e. بالإضافة إلى ذلك، تظهر حواف شبكية واضحة تنتمي إلى المستوى (001) من هذين COFs عند حوالي.لـ CoPc-O-COF وحوالي.لـ CoPc-S-COF. ومع ذلك، فإن تحليل تحويل فورييه السريع (FFT) المقابل يظهر البقعة البلورية، الشكل التكميلية 14، مما يوضح مزيدًا من جودتها البلورية. يكشف تحليل رسم الخرائط بالأشعة السينية المشتتة للطاقة (EDX) عن التركيب العنصري لـ، و F في كلا COFs بالإضافة إلى عنصر O فيوعنصر S فيمع نسب الذرات المقابلة القريبة من القيم النظرية، الشكل 2c، f، الشكل التكميلي 15، والجدول التكميلي 2. كما هو موضح في الأشكال التكميلية 16 و17، تكشف قياسات امتصاص-إزالة N2 عن مساميتها الدائمة مع مساحة سطح بروناوير-إيميت-تيلر (BET) تبلغلـ CoPc-O-COF ولـ CoPc-S-COF مع حجم المسام المحسوب الخاص بهم و 0.14 ، على التوالي. تم تحديد توزيع حجم المسام لـ CoPc-O-COF و CoPc-S-COF على أنه و على التوالي، بمتوسط حجم مسام يبلغ 1.7 و 1.5 نانومتر.
تم إجراء مطيافية الإلكترون الضوئي بالأشعة السينية (XPS) لاستكشاف التركيب العنصري وحالات الفلزات في كل من COFs. تكشف طيف XPS لكل من COFs عن القمم الناتجة عنوعناصر F، الشكل التوضيحي التكميلي 18 والجدول التكميلي 3، بما يتماشى مع نتائج رسم الخرائط EDX المقابلة. الكوبالتطيف XPS لـ CoPc-O-COF يظهر ذروتين عند 781.5 و 796.7 إلكترون فولت، تُنسب إلى و من Co (II)، الشكل 2 ج. ومع ذلك، فإن Co وشركاه تتحول قمم CoPc-S-COF إلى طاقة ارتباط أقل تبلغ 780.8 و 796.1 إلكترون فولت مقارنةً بـ، بسبب التأثير الكبير المانح للإلكترونات لذرات الكبريت فيالقمم المميزة الناتجة عنوالممتص كيميائيًاتظهر عند 531.5 و 533.3 إلكترون فولت، على التوالي، في طيف XPS عالي الدقة لـ CoPc-O-COF، وذروتان مركزتان عند 164.8 و 163.5 إلكترون فولت بسببتُلاحظ في الدقة العاليةطيف XPS لـ CoPc-S-COF، الشكل 2h، i، يؤكد بشكل إضافي النجاح في توليد COFs المرتبطة بالديوكسين/ثنائي الكبريت. كلا من CoPc-O-COF و CoPc-S-COF يظهران قمة F 1s عند 687.2 eV في Fطيف XPS، على التوالي، مما يشير إلى طبيعة مجموعة C-F المماثلة، الشكل التكميلي 19. بالإضافة إلى ذلك، تم جمع هيكل الامتصاص القريب من الحافة بالأشعة السينية (XANES)، وطيف هيكل الامتصاص الدقيق الممتد بالأشعة السينية (EXAFS) لتحديد الحالة الكيميائية والبيئة التنسيقية المحلية لأنواع الكوبالت. كما هو موضح في الشكل التكميلي 20، فإن متوسط حالة الأكسدة لمراكز الكوبالت في كلا COFs قريب من +2 وفقًا لحافة الامتصاص المماثلة لتلك الخاصة بـ CoPc في Co.-طيف XANES عند حافة الطاقة. من الجدير بالذكر أن CoPc-S-COF يظهر قمة ما قبل الحافة عند 7710 إلكترون فولت، وهي أقل من تلك الخاصة بـ، مما يشير إلى أن حالة الأكسدة للكوبيالت تتحول إلى قيمة أصغر بعد إدخال الكبريت. علاوة على ذلك،طيف XANES ذو الحواف الثلاثة لـتم جمع COF و CoPc-S-COF أيضًا، الشكل التوضيحي التكميلي 21. كما هو موضح، فإن طاقات الفوتون المقابلة لـقمم الخط الأبيض الحاد لـ CoPc-S-COF أقل بمقدار 0.5 إلكترون فولت من تلك الخاصة بـ CoPc-O-COF، مما يتماشى مع نتائج تحليل XPS و K-edge XAFS، مؤكداً أن حالة الأكسدة لـ Co تنتقل إلى قيمة أصغر بعد إدخال الكبريت. تشير هذه النتائج أيضاً إلى أن ذرات الكبريت قادرة على تعديل الهيكل الإلكتروني لمواقع الكوبالت النشطة الذرية، بما يتماشى مع نتائج XPS. علاوة على ذلك، تظهر طيف EXAFS المحول فورييه (FT) والنتائج الملائمة المقابلة لكل من COFs و CoPc قمم عند حوالي 1.53 و 2.49 و 3.10 Å تُنسب إلى مسارات تشتت Co-N (الطبقة الأولى) و Co-C (الطبقة الثانية) و Co-N (الطبقة الثالثة) مع عدد تنسيق يبلغ 4 و 8 و 4، الأشكال التكميلية 22 و 23 والجدول التكميلية 4. بشكل خاص، محتوى الكوبالت في CoPc-O-COF ويبلغ 5.91 وعلى التوالي، وفقًا لقياس الطيف الضوئي بالتحليل الطيفي للموصلية المحفزة بالحث (ICP-OES)، قريب جدًا من القيم النظرية، الجدول التكميلي 5.
الشكل 2 | الشكل والمميزات لـ COFs. صور TEM لـ CoPc-O-COF وصور HR-TEM لـ CoPc-S-COF. CoPc-O-COF و CoPc-S-COF. تحليل EDX منشركةلـ CoPc-O-COF و CoPc-S-COF، أو لـ CoPc-O-COF و iSلـ CoPc-S-COF. تحليل الخرائط لـ CoPc-O-COF و CoPc-S-COF. طيف XPS عالي الدقة
أداء التحفيز الكهربائي لتفاعل اختزال الأكسجين
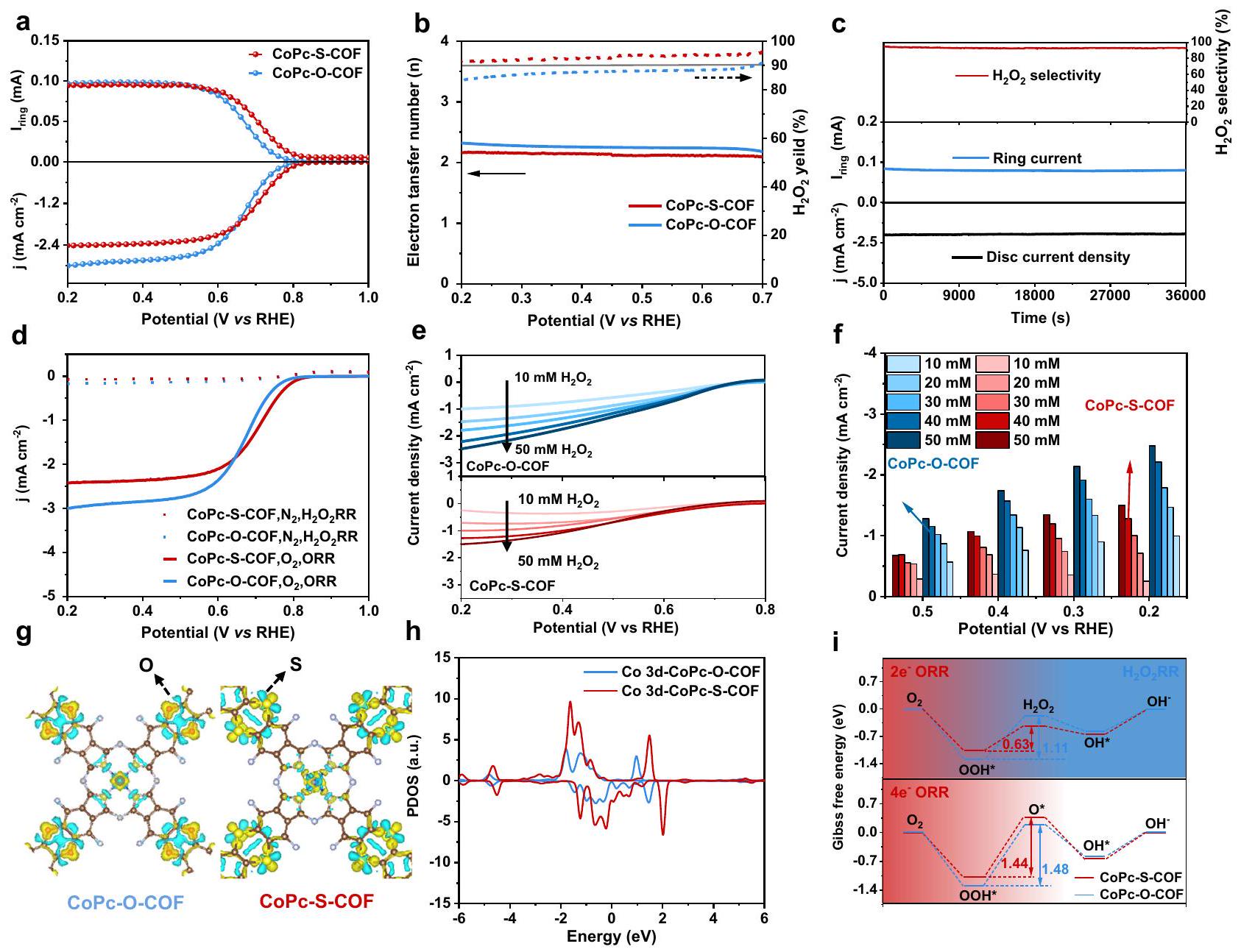
تم إجراء قياسات ORR على نظام الثلاثة أقطاب باستخدام القطب الدوار ذو الحلقة والقرص (RRDE) كقطب عمل في ظروف وسط قلوي. كما هو موضح في الشكل 3a، منحنيات استقطاب ORR وتم جمع تيار الكشف لكل من COFs على RRDE عند 1600 دورة في الدقيقة في-مشبع 0.1 م KOH. توفر أقطاب CoPc-SCOF نشاطًا كهربائيًا عاليًا لتقليل الأكسجين مما يؤدي إلى جهد بدء قدره 0.81 فولت مقابل القطب الهيدروجيني القابل للعكس (RHE) (المحدد عندمنتيار جزئيأعلى من ذلك منالقطب الكهربائي (0.78 فولت). علاوة على ذلك، تم حساب انحدارات تافل لـ CoPc-O-COF و CoPc-S-COF لتكون 62 وعلى التوالي، أصغر من ذلك الخاص بالفردالشكل التوضيحي 24. هذا يشير إلى الأسرعديناميكية تفاعل الأكسدة والاختزال لكل من COFs، والتي قد تُعزى إلى كفاءتها العالية في نقل الإلكترونات الأولية وسطحها النشط الكبير خلال عملية التحفيز. الشكل 3ب يعرض الـالانتقائية وعدد نقل الإلكترونخلال ORR لكلا COFs.انتقائية CoPc-S-
COF يعادل أكبر منفي النطاق المحتمل لـ مقابل RHE مع قيمة لـمما يشير إلى أنه واعدأداء ORR. في هذه الأثناء،قيمة الانتقائيةأقل قليلاً من تلك الخاصة بـ CoPc-S-COF في نفس نطاق الجهد. وفقًا لرسوم الاستقطاب لتفاعل الأكسدة الاختزالية للأكسجين (ORR) عند سرعات دوران مختلفة ومعادلة كوتيكي-ليفش (K-L)، فإن عدد نقل الإلكتروناتتم تحديد النشاط الكتلي للكوبالت (CoPc-O-COF و CoPc-S-COF) ليكون حوالي 2.3 و 2.0، على التوالي، الشكل التوضيحي 25، وهو متسق مع نتيجة RRDE. تم أيضًا حساب النشاط الكتلي (MA) لكلا COFs، الشكل التوضيحي 26. يظهر CoPc-S-COF MA قدرهعند 0.7 فولت مقابل RHE، أفضل منعند 0.7 فولت مقابل RHE. بالإضافة إلى ذلك، تم تحليل موصلية كلا القطبين COF من خلال قياسات طيف الامتياز الكهربائي (EIS). كما هو موضح في الشكل التوضيحي 27، تظهر أقطاب CoPc-O-COF و CoPc-S-COF قطر نصف دائرة EIS صغير يبلغ 112 و“، مما يشير إلى نقل الشحن المحسن لهذين الهيكلين من COFs بفضل بنيتهما المترافقة. ومع ذلك، فإن سعات الطبقة المزدوجة ( ) من كلا
الشكل 3 |الإنتاج الكهربائي وحساب DFT. منحنيات LSVs لـ CoPc-O-COF و CoPc-S-COF عند 1600 دورة في الدقيقة في-مشبعالانتقائية وعدد نقل الإلكترون من CoPc-O-COF و CoPc-S-COF. ج قياس الكرونوأمبيرومترية لـ CoPc-S-COF لمدة 36000 ثانية عند 0.52 فولت مقابل RHE. د منحنيات استقطاب ORR في -مشبع 0.1 م كOH ومنحنيات الاستقطاب في محلول KOH بتركيز 0.1 م مشبع بالأرجون. e منحنيات الاستقطاب والحالي
كثافات CoPc-O-COF و CoPc-S-COF في محلول KOH بتركيز 0.1 م مشبع بالأرجون يحتوي على تركيزات مختلفة منتحميل كتلة المحفز: : ). توزيع الشحنة التفاضلية على كل من الشظايا الدورية المحاكية لكل من COFs (مستوى السطح المتساوي=0.01). h الكثافة الجزئية لحالات (PDOS) مدارات Co 3d في نماذج مختلفة. i تغيير الطاقة الحرة للتفاعل من أجلأو آر آر،ORR وعملية كل من COFs.
تم اشتقاق COFs، التي تتناسب مع مساحتها السطحية الكهروكيميائية، من منحنيات CV عند معدلات مسح مختلفة، الشكل التوضيحي 28.تم حساب CoPc-S-COF كـأكبر بكثير من تلك الخاصة بـ CoPc-O-COF،، مما يشير إلى المواقع النشطة الأكثر توفرًا داخل CoPc-S-COF الناتجة عن هيكله الطبقي الملتوي، مما يؤدي بدوره إلى ارتفاعهأداء ORR. وفقًا لهذه النقطة، يتم حساب المواقع النشطة كهربائيًا على سطح قطب CoPc-SCOF لتكونوفقًا للتيار الذروي لمنحنيات CV كدالة لمعدل المسحالشكل التوضيحي التكميلي 29، يكشف عن 11.0% من إجمالي وحدات الكوبالت-فثالوسيانين تعمل كمواقع نشطة. هذه القيمة تقريبًا ضعف تلك الخاصة بقطب CoPc-O-COF،، مما يؤكد المواقع النشطة الأكثر تعرضًا في CoPc-SCOF، الشكل التوضيحي 30. وهذا بدوره يصبح مسؤولًا عن نشاط ORR لـ CoPc-S-COF إلى CoPc-O-COF.
تم تقييم الاستقرار الحفزي لكل من COFs على RRDE من خلال قياسات الكرونوأمبيرومترية، الشكل 3c والشكل التكميلي 31. يُظهر قطب Co-S-COF إشارات تيار شبه ثابتة تقريبًا وحافظ علىانتقائيةلمدة 10 ساعات عند 0.52 فولت مقابل RHE، مما يثبت متانتها القوية كعامل حفاز لـORR في الوسط القلوي. كما هو موضح في الشكل التكميلي 31، يظهر Co-O-COF أيضًا استقرارًا تحفيزيًا جيدًا على RRDE. علاوة على ذلك، فإن كل من CoPc-O-COF و CoPc-S-COF تظهر أنماط PXRD مشابهة قبل وبعد اختبارات الاستقرار، التي تثبت طبيعة إطارها القوي، الشكل التوضيحي التكميلي 32. تم أيضًا إجراء قياسات المجهر الإلكتروني الناقل الماسح وXANES لكل من COFs قبل وبعد اختبارات الاستقرار. كما هو موضح في الشكل التوضيحي التكميلي 33، لا يمكن ملاحظة أي جزيئات نانوية من الكوبالت في صور STEM لكل من COFs قبل وبعد اختبارات الاستقرار، مما يستبعد تسرب الكوبالت وتجمعه خلال الاختبار الكهروكيميائي. بشكل خاص، تظهر طيف EXAFS عند حافة كاكي للكوبالت لكل من COFs بعد اختبارات الاستقرار أيضًا فقط القمة عندبسبب مسار تشتت Co-N دون إعطاء الذروة عندبسبب رابطة الكوبالت-الكوبالت، مما يؤكد غياب جزيئات الكوبالت النانوية الناتجة عن تسرب الكوبالت خلال الاختبار الكهروكيميائي وبالتالي المتانة العالية للـ COFs الاثنين، الشكل التكميلية 34. ثم تم إجراء تجربة السم لتحديد الموقع الحفاز باستخدامالأيونات، التي تميل إلى ربط ذرات المعدن من CoPc وتعيق امتصاص وسائط التفاعل، مما يؤثر سلبًا على الأداء التحفيزي. كما هو موضح في الشكل التكميلي 35، يحدث تدهور كبير في النشاط التحفيزي بعد الإضافةإلى الإلكتروليت، مما يوضح الطبيعة الفعلية لموقع النشاط لمركز معدن الكوبالت لـORR في الاثنين. بالإضافة إلى ذلك، تم إجراء تجارب تسمم مركز الكوبالت في الموقع باستخدام النيتريت، كما هو موضح في الشكل التكميلية 36. كما يمكن ملاحظته، هناك تدهور كبير في تحدث النشاط التحفيزي لـ CoPc-S-COF بعد إضافة النيتريت إلى الإلكتروليت، مما يؤكد بشكل أكبر طبيعة مواقع النشاط لذرات الكوبالت تجاهORR في CoPc-S-COF. على وجه الخصوص، منحنيات الاستقطاب لـتفاعل اختزال ) لكلا COFs تم تسجيلها في محلول 0.1 M KOH المشبع بالآر كما هو موضح في الشكل 3d، كلا العينتين تظهران تأثيرًا ضئيلًاالنشاط. لمقارنة تثبيط بشكل أوضحتحلل كل من COFs،منحنيات الاستقطاب و فيمحاليل KOH بتركيز 0.1 م المشبعة التي تحتوي على تركيزات مختلفة منتم قياسها، الشكل 3e، f. يظهر CoPc-O-COF أكبر التيارات أكثر من تلك الخاصة بـ CoPc-S-COF خاصة في الارتفاع تركيز ). تشير هذه النتيجة إلى التثبيط المتفوق لـ CoPc -S-COF إلى تحلل خلالعملية ORR بسبب الطبيعة الغنية بالإلكترونات لمركز الكوبالت المرتبطة بذرات الكبريت المانحة للإلكترونات في وحدة الربط، مما يفضل إنتاج تركيز عالٍ.حل.
لاستكشاف تأثير ذرة الفلور على النشاط التحفيزي والانتقائية للكونترات في CoPc-O-COF و CoPc-S-COF، تم دراسة النشاط التحفيزي لجزيئات الفثالات الكوبالت الصغيرة بما في ذلكوالفثالوسيانين الخالي من المعادنتم إعداد واختبار الأقطاب الكهربائية أيضًا على RRDE بسرعة 1600 دورة في الدقيقة في-المشبعة 0.1 م KOH ، الشكل التوضيحي 37. من الجدير بالذكر أن CoPc وتظهر الأقطاب الكهربائية نفس جهد البداية البالغ 0.78 فولت مقابل RHE والانتقائية ) في النطاق المحتمل لـ مقابل RHE مع قيمة تتراوح بين 2.5-2.7، مما يدل على تفاعليتهما الواضحة المتشابهة لـتقليل الأكسجين مع تأثير ضئيل لذرات الفلور. ومع ذلك، فإن المادة الخالية من المعادنيظهر القطب الكهربائي أداءً كهربائيًا تحفيزيًا أقل بكثير مع جهد بدء أقل يبلغ 0.68 فولت مقابل RHE والانتقائية )، مؤكداً طبيعة المراكز النشطة. بهدف توضيح تأثير ذرات الأكسجين والكبريت على النشاط التحفيزي والانتقائية لمراكز الكوبالت في CoPc-O-COF وCoPc-S-COF، تم تخليق جزيئين صغيرين من فثالوسيانين الكوبالت بما في ذلك CoPc-S وCoPc-O اللذين يحتويان على مجموعات C-S-C وC-O-C، على التوالي، مع تم تقييم أداء ORR، الأشكال التكميلية 38 و39. كما يتضح، يظهر CoPc-S أداءً أفضلنشاط ORR والأسوأالنشاط بالمقارنة مع، مما يثبت تأثير S على تعزيزأداء ORR لـ. بالإضافة إلى ذلك،مخدرتم تحضير COF (المسمى CoPc-O-COF-S 20%) وتم اختبار أدائه الكهروكيميائي. كما هو متوقع، يظهر CoPc-O-COF-S 20% نشاطًا عاليًا في اختزال الأكسجين مع جهد بدء يبلغ 0.80 فولت مقابل RHE وعدد نقل الإلكترونات يتراوح بين 2.2-2.3، الأشكال التكميلية 40-42. ومع ذلك، يظهر CoPc-O-COF-S 20% أصغرتيارات أكثر من تلك التيفي محلول KOH بتركيز 0.1 م المشبع بالأرجون، كاشفًا تأثير إضافة الكبريت على تقليل النشاط التحفيزي تجاهتحلل.
للحصول على مزيد من الفهم حولتمت دراسة نشاط ORR للـ COFs، وتم التحقيق في الهيكل الإلكتروني لـ CoPc-O-COF و CoPc-S-COF من خلال حسابات نظرية الكثافة الوظيفية (DFT). طاقة الامتزاز ( ) من على ذرات مختلفة من COFs الاثنين بما في ذلك، و تم حسابه أولاً لاستكشاف الموقع النشط لـالإنتاج. كما يتضح في الشكل التكميلي 43 والجدول التكميلي 6، القيمة الأصغر بشكل ملحوظ لـلذرة الكوبالت،، من تلك الخاصة بذرات أخرى (إلى -0.05 إلكترون فولت لـ. لـ يكشف طبيعة موقع النشاط لذرة الكوبالت في COFs تجاه تفاعل اختزال الأكسجين. بالإضافة إلى ذلك، فإن كثافة الإلكترون على جزء CoPc بما في ذلك حول موقع الكوبالت في أعلى من ذلك لـ CoPc-O-COF بسبب تأثير التبرع بالإلكترونات من ذرات الكبريت، مما يسهل نقل الشحنة بين مواقع الكوبالت النشطة والوسطاء، وبالتالي يوفر نشاطًا تحفيزيًا معززًا لـالشكل 3g والشكل التكميلي 44. علاوة على ذلك، تحليل السكان الطبيعي (NPA)تم أيضًا إجراء حساب توزيع الشحنات لكل من COFs لتقييم عملية نقل الإلكترونات في كلا COFs. كما هو موضح في الجدول التكميلي 7، فإن تغيير الشحنات الذرية على السلسلة المترافقة يتم بطريقة موجية. (NCo)خلال عملية نقل الإلكترونات من ذرات S/O إلى ذرات Co المركزية، مما يظهر تأثير استقطاب بديل على طولمسار نقل الإلكترون في كلاهما. بشكل خاص، يحصل ذرة الكوبالت المركزية على شحنة إضافية من عندما يتم استبدال ذرات الأكسجين في CoPc-O-COF بذرات الكبريت في CoPc-S-COF، مما يؤكد بشكل أكبر انتقال الشحنة الأكثر وضوحًا من الكبريت إلى الكوبالت مقارنةً بتلك التي من الأكسجين إلى الكوبالت. علاوة على ذلك، تكشف كثافة الحالات المتوقعة المحسوبة (pDOS) عن انخفاض فيموضع مركز النطاق -1.33 إلكترون فولت لـ CoPc-S-COF مع كثافة أعلى من القمم بالقرب من مستوى فيرمي ) مقارنةً بـ CoPc-O-COF الذي يمتلك موضع مركز نطاق d بقيمة -1.17 eV، الشكل 3h. وهذا يشير إلى أن كثافة أكبر من الإلكترونات النشطة حول مراكز الكوبالت في CoPc-S-COF تشارك في تفاعل الاختزال الكهربائي للأكسجين، مما يؤكد النشاط التحفيزي الأعلى لـ CoPc-S-COF بسبب تأثير التبرع بالإلكترونات من ذرات الكبريت. ومع ذلك، يقدم الشكل 3i الفروقات المحسوبة في الطاقة الحرة لجيبس ( ) مخططات الـ ORR وعمليات ORR على و . كما يمكن العثور على، تحويل إلىهو الخطوة المحددة لمعدلORR على كلاهما و مع حاجز طاقة قدره 0.63 و 1.11 إلكترون فولت، على التوالي. هذه القيم أصغر من حاجز الطاقة لـإلىعملية التحويل (الخطوة المحددة للتقييم)ORR) على كلا COFs، 1.44 و 1.48 إلكترون فولت، مما يدل على سرعة حركية التفاعلأو أكثر منORR على كل من COFs وبالتالي انتقائيتها العالية تجاهأو.ر.ر. بشكل خاص، الأدنىإلىحاجز طاقة التحويلCOF بالمقارنة مع ذلك لـيشير إلى المعززنشاط ORR للنوع السابق على النوع اللاحق، بينما الأعلىإلىحاجز طاقة التحويل لـ CoPc-S-COF مقارنةً بـيظهر النشاط المتناقص تجاهتحللإلىتفسير التحفيز الكهربائي الأكثر استقرارًاإنتاج.
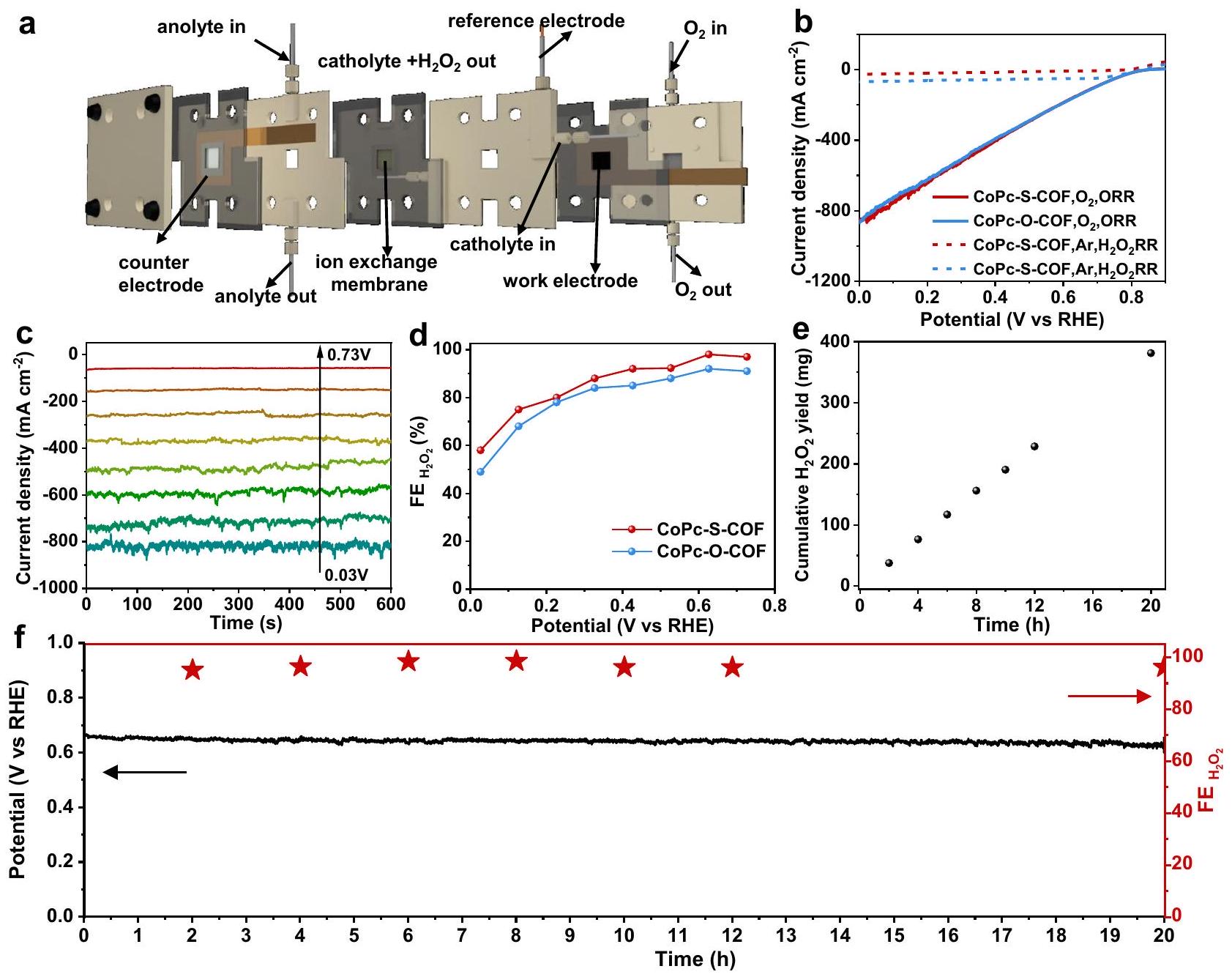
تم استخدام أجهزة أقطاب الغاز (GDE) لاستكشاف المزيد من إمكانيات التطبيق العملي لـ COFs المعتمدة على CoPc التي تم تحضيرها.ORR. خلية تدفق ثلاثية الطور، الشكل 4a، حيث يتم ترسيب المحفز على طبقة انتشار الغاز (GDL) كقطب عمل، يُعتبر أنها قادرة على توفير كثافات تيار اختزال أعلى من خلال زيادة تركيز الأكسجين على GDL وتحسينمعدل الإنتاج. تم إجراء القياسات المقابلة لكل من COFs في 1 M KOH. كما هو موضح في الشكل 4b، يظهر كلا COFs نشاطًا عاليًا في التحفيز الكهربائي مع كثافة تيار أعلى بكثير في خلية التدفق مقارنةً بقياسات RRDE. تم إجراء قياسات الكرونوأمبيرومترية عند فولتية مطبقة مختلفة مع الجهد الناتج.محدد بواسطةطريقة المعايرة، الشكل 4c، والأشكال التكميلية 45 و46. يوضح الشكل 4d الكفاءة الفارادية المحددة لـلكلا COFs. يمكن أن يظهر CoPc-O-COF و CoPc-S-COF أكثر منفي نطاق من 0.73 إلى 0.33 فولت مقابل RHE، وهو أعلى حتى من قياسات RRDE في النطاق من 0.73 إلى 0.53 فولت. من المRemarkably،من CoPc-S-COF يصل إلىعند 0.63 فولت مقابل RHE (ما يعادل جهد زائد قدره 130 مللي فولت) معمنفي 1 م كوه عند درجة حرارة الغرفة، والتي تزداد أكثر إلىعند 0.33 فولت مقابل RHE (ما يعادل جهد زائد قدره 430 مللي فولت) معلا يزال أعلى من، متفوق على الشكل 4 ج، د، والشكل التكميلي 45.تم استكشاف أداء كل من COFs أيضًا في خلية التدفق. كما هو موضح في الشكل 4b، يظهر CoPc-S-COF حجمًا أصغر بكثيركثافات التيار المخفضة مقارنة بـ، متسق مع النتيجة في نظام RRDE. هذا في الواقع يشير إلى أنيمكن أن يتم تثبيطه في CoPc-S-COF وبالتالي يؤدي إلى زيادة و التركيز. علاوة على ذلك، فإن الاستقرار الطويل الأمدتم تسجيل إنتاج CoPc-S-COF عند كثافة تيار ثابتة منالشكل 4e، f. يحافظ CoPc-S-COF علىفي المستمرالإنتاج الكهربائي لمدة 20 ساعة. ومع ذلك، فإنتزداد الكمية المنتجة بشكل خطي مع زيادة وقت التشغيل بمعدل إنتاج شبه ثابت قدره. وجهد تشغيل شبه ثابت 0.67 فولت مقابل RHE، الشكل 4f، مما يؤكد المزيد من الاستقرار العالي لـ
الشكل 4 |الإنتاج الكهربائي في خلية التدفق. رسم تخطيطي لخلية التدفق.LSVs لـ CoPc-O-COF و CoPc-S-COF في خلية التدفق.قياسات الكرونوأمبيرومترية عند فولتية مطبقة متنوعة. د من و CoPc-S-COF عند فولتية مطبقة متنوعة. عائدات CoPc-S-COF.
منحنى الكرونوپوتنشيومتر عند كثافة تياروال correspondenteفي خلية التدفق لـ CoPc-S-COF (حمولة كتلة المحفز: ).
CoPc-S-COF وكذلك تعطيله تجاهالتحلل. والأهم من ذلك،العائد يصل إلى 377 ملغ بعد 20 ساعة من التحليل الكهربائي مما يتوافق معتركيزمن الجدير بالذكر أن ميزات COFs مثل القابلية لتعديل الهيكل الجزيئي والمسامية الدائمة المرغوبة تمكّن هذه الفئة من المواد من أن تكون محفزات متقدمة واعدة ذات هيكل محدد جيدًا ومواقع نشطة لمختلف التطبيقات الكهروكيميائية.مؤخراً، تم تطوير هياكل الإطار العضوي ثنائي الأبعاد مثل PYTA-TPEDH-COFTP-TDCOFو Py-TD-COFبالإضافة إلى COFs ثلاثية الأبعاد بما في ذلك BUCT-COF-1 و BUCT-COF- تم توظيفها كعوامل تحفيز كهربائية خالية من المعادن لتعزيزORR معانتقائية تصل إلىمقدمة لمواقع نشطة عالية الكفاءة لـORR إلىيوفر تحسينهالانتقائية ) في الحالة الحالية. ومع ذلك، فإن الهيكل المكدس ذو الطبقات المتموجة لـ CoPc-S-COF يعزز من تعرض المواقع النشطة، مما يؤدي إلى زيادة معدل الإنتاج بالمقارنة مع جميع المحفزات الكهروكيميائية المعتمدة على COF التي تم الإبلاغ عنها حتى الآن، الجدول التكميلي 8. في الواقع، فإن التحفيز الكهروكيميائيأداء إنتاج CoPc-S-COF تنافسي أيضًا مع المواد الحفازة غير العضوية المتطورة، الجدول التكميلي 8، مما يوضح إمكانيته الكبيرة في التطبيقات العملية.الإنتاج. علاوة على ذلك، بعد اختبار الاستقرار، يظهر CoPc-S-COF هيكلًا ثابتًا وفقًا لتحليل FTIR وXPS، مما يثبت استقراره القوي، الأشكال التكميلية 47 و48.
لكشف المزيد عن الإمكانيات التطبيقية لـ CoPc-S-COF، التحفيز الكهربائيتم تقييم نشاط إنتاج CoPc -S-COF أيضًا فيفي جهاز GDE. كما هو موضح في الشكل التكميلي 49، يظهر CoPc-S-COF أداءً عاليًانشاط ORR في الإلكتروليت المحايد مع مستوى عالٍالانتقائية (حوالي ) في النطاق المحتمل لـ مقابل RHE. والأهم من ذلك، COF يعرض استقرارًاإنتاج عاليمنتحت كثافة تيارعند جهد مطبق قدره. 0.30 فولت مقابل RHE لمدة 20 ساعة، مما يولد، مقارنةً بالنتيجة التي تم الحصول عليها في الإلكتروليت القلوي. تؤكد هذه النتائج التجريبية الإضافية أيضًا على جودة التحفيز الكهربائيأداء إنتاج CoPc-S-COF، والذي يفيد في تصميم وتحضير المحفزات الكهربائية عالية الأداء ومنخفضة التكلفة على مستوى الصناعةالإنتاج الكهربائي
نقاش
باختصار، تم تصنيع إطار عضوي شبكي قائم على CoPc مرتبط بديثيين مسامي. يتميز CoPc-S-COF بهيكل مكدس ذو طبقات متموجة بسبب الانحناء على طول جسر C-S-C للسماح بمزيد من مراكز الكوبالت المكشوفة.ORR. هذا، بالاشتراك مع المفعلORR ولكن معطلقدرة التحلل لمركز الكوبالت بسبب تأثير التبرع بالإلكترونات من ذرات الكبريت، يمكّنلإظهار مستوى عالٍالانتقائية وتحقيق النطاق الواسعالإنتاج في خلية التدفق. يجب أن تكون هذه العمل مفيدًا في تصميم وتحضير المحفزات الكهربية عالية الأداء ومنخفضة التكلفة لـالتحليل الكهربائي
طرق
تركيب CoPc-O-COF
و THB ( ) تم إضافتها إلى المذيب المختلط لـ -زيلين و0.5 مل من DMAc في أنبوب بايركس سعة 16 مل. تم استخدام الموجات فوق الصوتية للمزيج لمدة 5 دقائق لتكوين تعليق متجانس. ثمتم إضافة ثلاثي إيثيل أمين إلى الخليط. بعد ثلاث دورات من التجميد والضخ والتسخين، تم إغلاق أنبوب بايركس وتسخينه في فرن عند لمدة 7 أيام. تم جمع الراسب الأسود-الأخضر عن طريق الطرد المركزي وغسله بأسيتون، وثنائي كلورو الميثان، وTHF في جهاز سوكسهليت لمدة يوم واحد. أخيرًا، تم الحصول على CoPc-O-COF كمسحوق أسود ب yield من .
تركيب CoPc-S-COF
و BTT ( ) تم إضافتها إلى المذيب المختلط من -زيلين و0.5 مل من DMAc في أنبوب بايركس سعة 16 مل. تم استخدام الموجات فوق الصوتية للمزيج لمدة 5 دقائق لتكوين تعليق متجانس. ثمتم إضافة ثلاثي إيثيل الأمين إلى الخليط. بعد ثلاث دورات من التجميد والضخ والذوبان، تم إغلاق أنبوب البيركس وتسخينه في فرن عند لمدة 7 أيام. تم تشكيل مادة صلبة داكنة خضراء داخل أنبوب بايركس خلال عملية التفاعل. تم جمع المنتج عن طريق الطرد المركزي ثم تم استبدال المذيب بإيثانول جاف لمدة أسبوع تلاه تجفيف فائق الحرج. أخيرًا، تم الحصول على CoPc-S-COF كمنتج داكن أخضر رقيق ب yield من .
توفر البيانات
جميع البيانات ذات الصلة التي تدعم نتائج هذه الدراسة موجودة في المخطوطة وملف المعلومات التكميلية. تم توفير بيانات المصدر مع هذه الورقة.
References
Yang, X. et al. Tuning two-electron oxygen-reduction pathways for electrosynthesis via engineering atomically dispersed single metal site catalysts. Adv. Mater. 34, e2107954 (2022).
Chen, J. et al. Kinetically restrained oxygen reduction to hydrogen peroxide with nearly 100% selectivity. Nat. Commun. 13, 2808 (2022).
Wang, N. et al. Recent progress of electrochemical production of hydrogen peroxide by two-electron oxygen reduction reaction. Adv. Sci. 8, e2100076 (2021).
Wang, Y., Waterhouse, G. I. N., Shang, L. & Zhang, T. Electrocatalytic oxygen reduction to hydrogen peroxide: from homogeneous to heterogeneous electrocatalysis. Adv. Energy Mater. 11, 2003323 (2020).
Fei, H. et al. Atomic cobalt on nitrogen-doped graphene for hydrogen generation. Nat. Commun. 6, 8668 (2015).
Bu, Y. et al. Carbon-based electrocatalysts for efficient hydrogen peroxide production. Adv. Mater. 33, e2103266 (2021).
Jiang, K. et al. Catalyst design for electrochemical oxygen reduction toward hydrogen peroxide. Adv. Funct. Mater. 30, 2003321 (2020).
Kim, H. W. et al. Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts. Nat. Catal. 1, 282-290 (2018).
Tian, Y. et al. Edge-hosted atomic sites on hierarchical porous carbon for highly selective two-electron oxygen reduction reaction. Angew. Chem. Int. Ed. 61, e202213296 (2022).
Li, B. Q. et al. Electrosynthesis of hydrogen peroxide synergistically catalyzed by atomic Co-Nx-C sites and oxygen functional groups in noble-metal-free electrocatalysts. Adv. Mater. 31, e1808173 (2019).
Sun, Y. et al. A comparative perspective of electrochemical and photochemical approaches for catalytic production. Chem. Soc. Rev. 49, 6605-6631 (2020).
Li, D. et al. Metal-free thiophene-sulfur covalent organic frameworks: precise and controllable synthesis of catalytic active sites for oxygen reduction. J. Am. Chem. Soc. 142, 8104-8108 (2020).
Melchionna, M. et al. The rise of hydrogen peroxide as the main product by metal-free catalysis in oxygen reductions. Adv. Mater. 31, e1802920 (2019).
Lobyntseva, E. et al. Electrochemical synthesis of hydrogen peroxide: rotating disk electrode and fuel cell studies. Electrochim. Acta. 52, 7262-7269 (2007).
Lee, K. et al. Structure-controlled graphene electrocatalysts for high-performance production. Energ. Environ. Sci. 15, 2858-2866 (2022).
Tammeveski, K. et al. Surface redox catalysis for reduction on quinone-modified glassy carbon electrodes. J. Electroanal. Chem. 515, 101-112 (2001).
Sarapuu, A. et al. Electrochemical reduction of oxygen on anthraquinone-modified glassy carbon electrodes in alkaline solution. J. Electroanal. Chem. 541, 23-29 (2003).
Vaik, K. et al. Oxygen reduction on phenanthrenequinone-modified glassy carbon electrodes in 0.1 M KOH. J. Electroanal. Chem. 564, 159-166 (2004).
Sarapuu, A. et al. Kinetics of oxygen reduction on quinone-modified HOPG and BDD electrodes in alkaline solution. Electrochem. SolidState Lett. 8, E30 (2005).
Vaik, K. et al. Electrocatalytic oxygen reduction on glassy carbon grafted with anthraquinone by anodic oxidation of a carboxylate substituent. Electrochim. Acta 50, 5126-5131 (2005).
Siahrostami, S. et al. Enabling direct production through rational electrocatalyst design. Nat. Mater. 12, 1137-1143 (2013).
Cao, P. et al. Metal single-site catalyst design for electrocatalytic production of hydrogen peroxide at industrial-relevant currents. Nat. Commun. 14, 172 (2023).
Zhang, C. et al. Crystal engineering enables cobalt-based metalorganic frameworks as high-performance electrocatalysts for production. J. Am. Chem. Soc. 145, 7791-7799 (2023).
Guo, Y. et al. Precise design of covalent organic frameworks for electrocatalytic hydrogen peroxide production. CHEM-ASIAN. J. 16, 498-502 (2021).
Lee, B.-H. et al. Supramolecular tuning of supported metal phthalocyanine catalysts for hydrogen peroxide electrosynthesis. Nat. Catal. 6, 234-243 (2023).
Lin, R. et al. Approaching theoretical performances of electrocatalytic hydrogen peroxide generation by cobalt-nitrogen moieties. Angew. Chem. Int. Ed. 62, e202301433 (2023).
Zhao, X. et al. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc 143, 9423-9428 (2021).
Diercks, C. S. et al. The atom, the molecule, and the covalent organic framework. Science 355, eaal1585 (2017).
Yu, B. et al. Linkage conversions in single-crystalline covalent organic frameworks. Nat. Chem. 16, 114-121 (2024).
Pramudya, Y. et al. Design principles for high storage using chelation of abundant transition metals in covalent organic frameworks for 0-700 bar at 298 K. J. Am. Chem. Soc. 138, 15204-15213 (2016).
Wang, Z. et al. Covalent organic frameworks for separation applications. Chem. Soc. Rev. 49, 708-735 (2020).
Keller, N. et al. Optoelectronic processes in covalent organic frameworks. Chem. Soc. Rev. 50, 1813-1845 (2021).
Huang, S. et al. Porphyrin and phthalocyanine based covalent organic frameworks for electrocatalysis. Coord. Chem. Rev. 464, 214563 (2022).
Yang, X. et al. lonothermal synthesis of fully conjugated covalent organic frameworks for high-capacity and ultrastable potassiumion batteries. Adv. Mater. 34, e2207245 (2022).
Bhunia, S. et al. 2,1,3]-Benzothiadiazole-spaced co-porphyrin-based covalent organic frameworks for O(2) reduction. ACS Nano 17, 3492-3505 (2023).
Liu, M. et al. Construction of catalytic covalent organic frameworks with redox-active sites for the oxygen reduction and the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202213522 (2022).
Mondal, S. et al. A thiadiazole-based covalent organic framework: a metal-free electrocatalyst toward oxygen evolution reaction. ACS Catal 10, 5623-5630 (2020).
Lin, C. Y. et al. Covalent organic framework electrocatalysts for clean energy conversion. Adv. Mater. 30, 1703646 (2018).
Han, B. et al. Two-dimensional covalent organic frameworks with cobalt(II)-phthalocyanine sites for efficient electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 143, 7104-7113 (2021).
Zhong, H. et al. Boosting the electrocatalytic conversion of nitrogen to ammonia on metal-phthalocyanine-based two-dimensional conjugated covalent organic frameworks. J. Am. Chem. Soc. 143, 19992-20000 (2021).
Yang, C. et al. Theory-driven design and targeting synthesis of a highly-conjugated basal-plane 2D covalent organic framework for metal-free electrocatalytic OER. ACS Energy Lett 4, 2251-2258 (2019).
Lu, M. et al. Stable dioxin-linked metallophthalocyanine covalent organic frameworks (COFs) as photo-coupled electrocatalysts for reduction. Angew. Chem. Int. Ed. 60, 4864-4871 (2021).
Yue, Y. et al. Stable bimetallic polyphthalocyanine covalent organic frameworks as superior electrocatalysts. J. Am. Chem. Soc. 143, 18052-18060 (2021).
Huang, N. et al. A stable and conductive metallophthalocyanine framework for electrocatalytic carbon dioxide reduction in water. Angew. Chem. Int. Ed. 59, 16587-16593 (2020).
Zhi, Q. et al. Piperazine-LInked Metalphthalocyanine Frameworks for Highly Efficient Visible-light-driven photosynthesis. J. Am. Chem. Soc. 144, 21328-21336 (2022).
Haldar, S. et al. Porous dithiine-linked covalent organic framework as a dynamic platform for covalent polysulfide anchoring in lithiumsulfur battery cathodes. J. Am. Chem. Soc. 144, 9101-9112 (2022).
Wang, K. et al. Tetrapyrrole macrocycle based conjugated twodimensional mesoporous polymers and covalent organic frameworks: from synthesis to material applications. Coord. Chem. Rev. 378, 188-206 (2019).
Lin, S. et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 349, 1208-1213 (2015).
Qiu, X. F. et al. A stable and conductive covalent organic framework with isolated active sites for highly selective electroreduction of carbon dioxide to acetate. Angew. Chem. Int. Ed. 61, e202206470 (2022).
Yu, H.-Y. et al. A stack-guiding unit constructed 2D COF with improved charge carrier transport and versatile photocatalytic functions. Chem. Eng. J. 445, 136713 (2022).
Yuan, Y. et al. Deciphering the selectivity descriptors of heterogeneous metal phthalocyanine electrocatalysts for hydrogen peroxide production. Chem. Sci. 13, 11260-11265 (2022).
Tiekink, E. R. T. & Zukerman-Schpector, J. Stereochemical activity of lone pairs of electrons and supramolecular aggregation patterns based on secondary interactions involving tellurium in its 1,1dithiolate structures. Coord. Chem. Rev. 254, 46-76 (2010).
Li, N. et al. Polyarylether-Based 2D Covalent-Organic Frameworks with In-Plane D-A Structures and Tunable Energy Levels for Energy Storage. Adv. Sci. 9, e2104898 (2022).
Shen, H. et al. Synergistic effects between atomically dispersed Fe-N-C and C-S-C for the oxygen reduction reaction in acidic media. Angew. Chem. Int. Ed. 56, 13800-13804 (2017).
Xia, Y. et al. Highly active and selective oxygen reduction to on boron-doped carbon for high production rates. Nat. Commun. 12, 4225 (2021).
Han, B. et al. Maximizing electroactive sites in a three-dimensional covalent organic framework for significantly improved carbon dioxide reduction electrocatalysis. Angew. Chem. Int. Ed. 61, e202114244 (2022).
Chen, S . et al. Identification of the highly active coordination motif for selective oxygen reduction to hydrogen peroxide. J. Am. Chem. Soc. 144, 14505-14516 (2022).
Lin, L. et al. Atomic-level modulation-induced electron redistribution in Co coordination polymers elucidates the oxygen reduction mechanism. ACS Catal 12, 7531-7540 (2022).
Carpenter, J. E. Extension of Lewis Structure Concepts to Open-shell and Excited-state Molecular Species. (PhD thesis. University of Wisconsin, Madison, WI, 1987).
Wu, C. et al. Polarization Engineering of Covalent Triazine Frameworks for Highly Efficient Photosynthesis of Hydrogen Peroxide from Molecular Oxygen and Water. Adv. Mater. 34, 2110266 (2022).
Zhao, X. et al. Covalent organic frameworks (COFs) for electrochemical applications. Chem. Soc. Rev. 50, 6871-6913 (2021).
An, S. et al. One-dimensional covalent organic frameworks for the oxygen reduction reaction. Angew. Chem. Int. Ed. 62, e202218742 (2023).
Huang, S. et al. Covalent organic frameworks with molecular electronic modulation as metal-free electrocatalysts for efficient hydrogen peroxide production. Small Struct. 4, 2200387 (2023).
Huang, S. et al. Linkage engineering in covalent organic frameworks as metal-free oxygen reduction electrocatalysts for hydrogen peroxide production. Appl. Catal. B 340, 123216 (2024).
Bao, R. et al. Designing thiophene-enriched fully conjugated 3D covalent organic framework as metal-free oxygen reduction catalyst for hydrogen fuel cells. Angew. Chem. Int. Ed. 62, e202216751 (2023).
Zhang, Y. et al. Multicomponent synthesis of imidazole-linked fully conjugated 3D covalent organic framework for efficient electrochemical hydrogen peroxide production. Angew. Chem. Int. Ed. 62, e202314539 (2023).
شكر وتقدير
الدعم المالي من مؤسسة العلوم الطبيعية في الصين، رقم المنح 22235001 (J.J.)، 22175020 (J.J.) و12305372 (Y.L.). يُعرب عن الشكر لجامعة العلوم والتكنولوجيا في بكين. كما يرغب المؤلفون في شكر الدعم المقدم من منشأة خط الشعاع 4B9A في منشأة الإشعاع السنكروتروني في بكين (BSRF).
مساهمات المؤلفين
تصور: ق.ز.، ك.و.، وج.ج. المنهجية: ق.ز.، ر.ج.، وك.و. التحقيق: ق.ز.، ر.ج.، وي.ل. التصور: ق.ز. وX.ي. حساب DFT: ي.ج. و د.ق. الكتابة – المسودة الأصلية: ق.ز. الكتابة – المراجعة والتحرير: ق.ز.، ر.ج.، X.ي.، ي.ج.، د.ق.، ك.و.، وج.ج.
مركز بكين المتقدم للابتكار في هندسة جينوم المواد، مختبر بكين الرئيسي لعلوم وتطبيق المواد الجزيئية والبلورية الوظيفية، قسم الكيمياء والهندسة الكيميائية، كلية الكيمياء والهندسة البيولوجية، جامعة العلوم والتكنولوجيا في بكين، بكين 100083، الصين.مرفق إشعاع السنكروترون في بكين، معهد الفيزياء عالية الطاقة، الأكاديمية الصينية للعلوم، بكين 100049، الصين. البريد الإلكتروني: كانغوانغ@ustb.edu.cn; liuyunpeng@ihep.ac.cn; jianzhuang@ustb.edu.cn
Dithiine-linked metalphthalocyanine framework with undulated layers for highly efficient and stable electroproduction
Received: 26 July 2023
Accepted: 9 January 2024
Published online: 23 January 2024
Check for updates
Qianjun Zhi , Rong Jiang , Xiya Yang , Yucheng Jin , Dongdong Qi , Kang Wang , Yunpeng Liu & Jianzhuang Jiang
Realization of stable and industrial-level electroproduction still faces great challenge due large partly to the easy decomposition of . Herein, a two-dimensional dithiine-linked phthalocyaninato cobalt (CoPc)-based covalent organic framework (COF), CoPc-S-COF, was afforded from the reaction of hexadecafluorophthalocyaninato cobalt (II) with 1,2,4,5-benzenetetrathiol. Introduction of the sulfur atoms with large atomic radius and two lone-pairs of electrons in the C-S-C linking unit leads to an undulated layered structure and an increased electron density of the Co center for CoPc-S-COF according to a series of experiments in combination with theoretical calculations. The former structural effect allows the exposition of more Co sites to enhance the COF catalytic performance, while the latter electronic effect activates the oxygen reduction reaction ( ORR) but deactivates the decomposition capability of the same Co center, as a total result enabling CoPc-S-COF to display good electrocatalytic production performance with a remarkable selectivity of and a stable production with a concentration of under a high current density of at an applied potential of ca. 0.67 V versus RHE for 20 h in a flow cell, representing the thus far reported best synthesis COFs electrocatalysts.
Hydrogen peroxide ( ) is an important inorganic chemical and environmentally friendly oxidant with extensive applications in bleaching, disinfection, wastewater treatment, and organic synthesis . In industry, anthraquinone method is employed to generate more than of , which however is energy-intensive and produces a large amount of toxic by-products . For a sustainable future, it is essential to develop an energy efficient and ecofriendly strategy for the synthesis of that should operate onsite even on large or small scales. As a consequence, electrocatalytic oxygen reduction reaction ( ORR) has been considered as the most promising alternative approach since it can realize the green and distributed on-demand generation under ambient
conditions . Thus far, various electrocatalysts including modified carbon , noble-metal and alloys , non-noble metals , metalorganic frameworks (MOFs) , and covalent organic frameworks (COFs) have been developed to promote ORR for electrosynthesis. However, large-scale electrocatalytic production is still hard to be realized because of the limited solubility of oxygen in electrolyte solutions and easy decomposition of especially in the presence of metal active centers, which usually result in small working currents ( ) and low concentration ( . In addition, the favorable thermodynamics to generate water molecules via the pathway inevitably reduces the generation capability during .
COFs consist of organic building blocks linked by covalent bonds , which have drawn great research attention for extensive applications in gas storage and separation , optoelectronic devices , catalysis , and energy storage owing to their superiority of high porosity, robust stability, and low density. As a consequence of the ordered pores that favor more exposed active sites to contact with substrate molecules, COFs have shown a great application potentials as promising electrocatalysts for various reactions including ORR , oxygen evolution reaction , hydrogen evolution reaction , and reduction reaction . In particular, two-dimensional (2D) conjugated COFs with ultrastrong fused aromatic linkage have been revealed to exhibit intrinsically high conductivity and thermal/chemical stability, promoting enhanced electrocatalytic performance . However, the design of suitable linkers and optimization of reaction conditions for COFs construction remain a demanding task for synthetic chemists. The dioxin , phenazine , and piperazine linkage formation in a 2D conjugated COF by nucleophilic aromatic substitution have been established as fused heterocyclic organic linkage to build up crystalline and stable COFs. Corresponding COFs upon these linkages have been applied for catalysis and energy storage devices. However, these linkages usually result in relatively close layers spacing associated with their typical planar interlayer -stacking arrangement, leading to the inner active-sites being buried to some degree. Recently, Kaskel and co-workers constructed a dithiine-linked COF with undulated layers due to the bending along the bridge but the aromaticity and crystallinity of the overall COF structure still maintained, providing a heuristic for more efficient utilization of buried inner active-sites. In addition to the choice of linkage in 2D conjugated COFs, planar conjugated precursors including porphyrin , phthalocyanine (Pc) , and hexabenzocoronene have usually been selected as building blocks owing to their robust stability and intrinsic high electrical conductivity.
Particularly, metal phthalocyanine (MPc) building units with coordination configuration have been demonstrated to act as highefficiency active sites for catalyzing a series of reactions as exemplified by the efficient ORR activity of cobalt phthalocyanine (CoPc) .
Herein, a dithiine-linked 2D CoPc-based COF, CoPc-S-COF, was afforded from the reaction of hexadecafluorophthalocyaninato cobalt (II) ( ) with 1,2,4,5-benzenetetrathiol (BTT). For the purpose of comparison, a conventional dioxin-linked 2D CoPc-based COF, CoPc-O-COF, was also prepared by reaction between and , 5-tetrahydroxybenzene (THB). Powder X-ray diffraction (PXRD) and electron microscopy analysis results reveal the crystalline porous framework of CoPc-S-COF with an undulated layered structure due to the bending along the C-S-C bridge associated with the large atomic radius and two lone-pairs of electrons of the sulfur atoms in the linking unit, resulting in almost double exposed active Co sites for ORR compared to CoPc-O-COF with an eclipsed -stacking model according to the electrochemical analysis. This, in combination with the activated ORR but deactivated decomposition capability of the same Co center due to the electron-donating effect of S atoms, enables CoPc-S-COF to display a superior electrocatalytic ORR performance with a remarkable selectivity of and a stable production under a high current density of at an applied potential of ca. 0.67 V versus RHE for 20 h in a flow cell, generating solution with a concentration of .
Results
Materials synthesis and characterization
The synthesis of CoPc-O-COF and CoPc-S-COF is illustrated in Fig. 1a and their simulated structural models are displayed in Fig. 1b-e. Nucleophilic substitution reaction of with THB and BTT, respectively, in dimethylacetamide (DMAC) and p-xylene with
Fig. 1 | Schematic of synthetic COFs and structural representations of COFs. a The synthesis route of CoPc-O-COF and CoPc-S-COF. The simulated AA stacking of b, c CoPc-O-COF and d, e CoPc-S-COF (Co: orange; C: light gray; N: blue; O: red; S: yellow; F: green). PXRD of CoPc-O-COF and CoPc-S-COF: experimental PXRD
profile (black), refined profile (red), the difference between the experimental and refined PXRD (gray), and simulation pattern based on the AA stacking manner (blue and green).
triethylamine ( ) as catalyst affords and in the yield of 75 and . Observation of the band at due to the bonds in the Fourier-transform infrared (FT-IR) spectrum demonstrates the successful formation of dioxin bridge in COF, Supplementary Fig. 1. The characteristic band of the C-S-C linkage gets appeared at in the FT-IR spectrum, Supplementary Fig. 2, verifying the successful formation of dithiine bridge in CoPc-S-COF. The solid-state cross-polarization/magic-angle-spinning (CP/MAS) NMR spectroscopy reveals the characteristic aromatic carbon signals at 149 and 143 ppm for CoPc-O-COF and CoPc-S-COF, respectively, further supporting the generation of the dioxin/dithiinelinked COFs, Supplementary Figs 3 and 4. Both CoPc-O-COF and CoPc-S-COF exhibit a signal at ca. -124 ppm in their solid-state CP/MAS NMR spectra, indicating their same C-F group nature, Supplementary Fig. 5. The decomposition temperature was revealed to be above for both COFs according to thermogravimetric analysis, indicating their great thermal stability, Supplementary Fig. 6. Moreover, the PXRD patterns of both COFs recollected after soaking in different solutions including , pure water, THF, DMF, acetone, ethanol and for three days remain unchanged, unveiling the good chemical stability of both COFs, Supplementary Figs 7 and 8. In particular, both COFs display very similar FTIR spectra and TEM images before and after the soaking treatment in , further proving the durability of these two COFs in solution, Supplementary Figs. 9 and 10.
The crystalline structures of these two COFs were estimated by PXRD measurement combined with computational simulation. As displayed in Fig. 1f, CoPc-O-COF shows two strong peaks at 4.13 and , corresponding to (100) and (001) facets, respectively. Moreover, the experimental pattern of agrees with the calculated one with AA layer stackings on the basis of Forcite geometrical simulation method, Fig. 1b, c. Furthermore, the Pawley-refined PXRD pattern of CoPc-O-COF using the space the observed experimental curve as proved by the good agreement factors of and , Fig. 1f. The PXRD pattern of CoPc-S-COF exhibits one strong peak at and four medium intensity reflections at , and , corresponding to (110), (020), (220), (130), and (001) facets. Combination of the theoretical simulation and Pawley refinement indicates that CoPc-S-COF adopts undulated layerstacked structure owing to the nonplanar configuration of the C-S-C units with a dihedral angle of , affording the lattice parameters of , and in space group with and , Fig. 1c, f, g. The single crystal structure of the model compounds with dithiine moieties provides additional support for the nonplanar configuration of CoPc-S-COF, Supplementary Fig. 11 and Supplementary Table 1. The Raman spectra of both COFs were recorded to explore their vibrational splittings based on corresponding functional bonds, Supplementary Fig. 12. The obvious symmetric peaks due to the aromatic carbon and carbonoxygen bonds indicate the in-plane vibration nature for . Observation of the series of asymmetric stretching bands due to the aromatic carbon and carbon-sulfur vibrations for indicates the different vibrational splittings of the energy states of the C – S bond associated with the bending or out-of-plane twisting of the bonds . Actually unlike the dioxin-linked structure for CoPc-O-COF, the C-S-C units in CoPc-S-COF are stabilized in a nonplanar configuration to minimize the lone pair electron repulsion of sulfur atoms with large atomic radius in neighboring layers , resulting in its undulated layer-stacked structure.
The morphology of CoPc-O-COF and CoPc-S-COF was investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images, Fig. 2a-f and Supplementary Fig. 13. As can be found, CoPc-O-COF exhibits a micrometer scale irregular sheet morphology, different from the irregular cluster morphology of CoPc -S-COF due to the undulated layer-stacked mode. Both CoPc-O-COF
and CoPc-S-COF exhibit distinct lattice fringes with a spacing of and in their high-resolution TEM (HR-TEM) images, which are attributed to the (100) plane of CoPc-O-COF and (110) plane of CoPc-S-COF, respectively, and in turn confirm their high crystallinity, Fig. 2b, e. In addition, clear lattice fringes belonging to the (001) plane of these two COFs get appeared at ca. for CoPc-O-COF and ca. for CoPc-S-COF. Nevertheless, the corresponding fast Fourier transform (FFT) analysis displays the crystalline spot, Supplementary Fig. 14, further demonstrating their good crystallinity. Energy dispersive X-ray (EDX) mapping analysis reveals the elemental composition of , and F in both COFs as well as O element in and S element in with corresponding atom ratios close to the theoretical values, Fig. 2c, f, Supplementary Fig. 15, and Supplementary Table 2. As displayed in Supplementary Figs 16 and 17, N2 adsorption-desorption measurements reveal their permanent porosity with a Brunauer-Emmett-Teller (BET) surface area of for CoPc-O-COF and for CoPc-S-COF with their calculated pore volume of and 0.14 , respectively. The pore size distribution of CoPc-O-COF and CoPc-S-COF is determined to be and , respectively, with an average pore size of 1.7 and 1.5 nm .
X-ray photoelectron spectroscopy (XPS) was also performed to explore the elemental composition and metal valence states in both COFs. The XPS spectra of both COFs disclose the peaks due to , and F elements, Supplementary Fig. 18 and Supplementary Table 3, in agreement with corresponding EDX mapping results. The Co XPS spectrum of CoPc-O-COF displays two peaks at 781.5 and 796.7 eV , attributed to and of Co (II), Fig. 2 g . Nevertheless, the Co and Co peaks of CoPc-S-COF shift to a lower binding energy of 780.8 and 796.1 eV compared to , due to the significant electron-donating effect of S atoms in . The characteristic peaks due to the and chemisorbed appear at 531.5 and 533.3 eV , respectively, in the high-resolution O 1s XPS spectrum of CoPc-O-COF, and two peaks centered at 164.8 and 163.5 eV due to are observed in the high-resolution XPS spectrum of CoPc-S-COF, Fig. 2h, i, further confirming the successful generation of dioxin/dithiine-connected CoPc-based COFs. Both CoPc-O-COF and CoPc-S-COF exhibit a F 1s peak at 687.2 eV in their F XPS spectra, respectively, indicating their same C-F group nature, Supplementary Fig. 19. Additionally, the X-ray absorption near-edge structure (XANES), and extended Xray absorption fine structure (EXAFS) spectra were collected to determine the chemical state and local coordination environment of the Co species. As displayed in Supplementary Fig. 20, the average oxidation state of Co centers in both COFs is close to +2 according to their similar absorption edge to that for CoPc in Co -edge XANES spectra. It is noteworthy that CoPc-S-COF displays the pre-edge peak at 7710 eV , smaller than that of , indicating the oxidization state of Co shifts to a smaller value after S introduction. Morever, the , 3-edge XANES spectra of COF and CoPc-S-COF were also collected, Supplementary Fig. 21. As shown, the corresponding photon energies of the edge white line peaks for CoPc-S-COF are 0.5 eV smaller than those of CoPc-O-COF, consisting with the results of XPS and K-edge XAFS analysis, confirming the oxidization state of Co shifts to a smaller value after S introduction. These results further suggest the S atoms are able to modulate the electron structure of the atomic Co active sites, in line with the XPS results. Moreover, the Fourier transform (FT) EXAFS spectra and corresponding fitting results of both COFs and CoPc show peaks at ca. 1.53, 2.49, and 3.10 Å are attributed to the Co-N (first shell), Co-C (second shell), and Co-N (third shell) scattering paths with a coordination number of 4, 8, and 4, Supplementary Figs 22, 23 and Supplementary Table 4. In particular, the Co content of CoPc-O-COF and amounts to 5.91 and , respectively, according to inductively coupled plasma-optical emission spectrometry (ICP-OES), very close to the theoretical values, Supplementary Table 5.
Fig. 2 | Morphology and characterization of COFs. TEM images of a CoPc-O-COF and CoPc-S-COF. HR-TEM images of CoPc-O-COF and CoPc-S-COF. The EDX
of Co for CoPc-O-COF and CoPc-S-COF, O for CoPc-O-COF and iS for CoPc-S-COF.
mapping analysis of CoPc-O-COF and CoPc-S-COF. High-resolution XPS spectra
Electrocatalytic ORR performance
The ORR measurements were performed on the three-electrode system with the rotating ring-disk electrode (RRDE) used as the working electrode in the condition of alkaline media. As shown in Fig. 3a, the ORR polarization curves and detection current of both COFs are collected on RRDE at 1600 rpm in -saturated 0.1 M KOH . CoPc-SCOF electrodes offer high electrocatalytic activity for oxygen reduction to render an onset potential of 0.81 V versus reversible hydrogen electrode (RHE) (defined at of partial current) , higher than that of electrode ( 0.78 V ). Moreover, the Tafel slopes of CoPc-O-COF and CoPc-S-COF are calculated to be 62 and , respectively, smaller than that of individual , Supplementary Fig. 24. This indicates the faster ORR kinetics of both COFs, which might be attributed to their high initial electron transfer efficiency and large active surface during the catalytic process. Figure 3b presents the selectivity and electron transfer number during ORR for both COFs. The selectivity of CoPc-S-
COF amounts to larger than in the potential range of versus RHE with an value of , suggesting its promising ORR performance. Meanwhile, the selectivity value of is slightly lower than that of CoPc-S-COF in the same potential range. According to the ORR polarization curves at different rotation rates and Koutecky-Levich (K-L) equation, the electron transfer number of CoPc-O-COF and CoPc-S-COF is determined to be ca. 2.3 and 2.0, respectively, Supplementary Fig. 25, consistent with the RRDE result. The Co mass activity (MA) of both COFs was also calculated, Supplementary Fig. 26. CoPc-S-COF exhibits a MA of at 0.7 V vs RHE, superior to that of at 0.7 V vs RHE. In addition, the conductivity of both COF electrodes was analyzed by electrochemical impedance spectroscopy (EIS) measurements. As shown in Supplementary Fig. 27, CoPc-O-COF and CoPc-S-COF electrodes exhibit a small EIS semicircle diameter of 112 and , indicating the optimized charge transfer of these two COFs owing to their conjugated structure. Nevertheless, the double-layer capacitances ( ) of both
Fig. 3 | electroproduction and DFT calculation. a LSVs of CoPc-O-COF and CoPc-S-COF at 1600 rpm in -saturated selectivity and electron transfer number of CoPc-O-COF and CoPc-S-COF. c Chronoamperometry measurement of CoPc-S-COF for 36000 s at 0.52 V versus RHE. d ORR polarization curves in -saturated 0.1 M KOH and polarization curves in Ar-saturated 0.1 M KOH containing . e polarization curves and current
densities of CoPc-O-COF and CoPc-S-COF in Ar-saturated 0.1 M KOH containing different concentrations of (Catalyst mass loading: : ). Differential charge distribution on both simulated periodic fragment of both COFs (isosurface Level=0.01). h Partial density of states (PDOS) of Co 3dorbital in different models. i Reaction free energy change for ORR, ORR and process of both COFs.
COFs, which is proportional to their electrochemical surface area, were derived from the CV curves at different sweep rates, Supplementary Fig. 28. The of CoPc-S-COF is calculated as , much larger than that of CoPc-O-COF, , indicating the more available active sites within CoPc-S-COF originated from its twisted layered structure, in turn leading to its higher ORR performance. In line with this point, the surface electrochemical active sites on the CoPc-SCOF electrode are calculated to be according to the peak current of CV curves as a function of scan rate , Supplementary Fig. 29, revealing 11.0% of the total cobalt-phthalocyanine units acting as active sites. This value is almost twice of that for the CoPc-O-COF electrode, , confirming the more exposed active sites in CoPc-SCOF, Supplementary Fig. 30. This in turn becomes responsible for the much superior ORR activity of CoPc-S-COF to CoPc-O-COF.
The catalytic stability of both COFs on RRDE was assessed by the chronoamperometry measurements, Fig. 3c and Supplementary Fig. 31. The Co-S-COF electrode shows almost unchanged current signals and maintained selectivity of for 10 h at 0.52 V versus RHE, verifying its robust catalytic durability for ORR in alkaline media. As displayed in Supplementary Fig. 31, Co-O-COF also shows good catalytic stability on RRDE. Moreover, both CoPc-O-COF and CoPc-S-COF exhibit similar PXRD patterns before and after the
stability tests, proving their robust framework nature, Supplementary Fig. 32. The scanning transmission election microscope and XANES measurements were also carried out for both COFs before and after the stability tests. As exhibited in Supplementary Fig. 33, no Co nanoparticles can be observed in the STEM images of the two COFs before and after the stability tests, excluding the Co leaching and aggregation during the electrochemical test. Particularly, the Co K-edge EXAFS spectra of both COFs after the stability tests also show only the peak at A due to the Co-N scattering path without giving the peak at due to the Co-Co bond, further confirming the absence of Co nanoparticles formed from the Co leaching during the electrochemical test and in turn the high durability of the two COFs, Supplementary Fig. 34. The poison experiment was then carried out to identify the catalytic site by using ions, which tend to bind the metal atoms of CoPc and block the adsorption of reaction intermediates, thus negatively affecting the catalytic performance. As shown in Supplementary Fig. 35, significant degradation of the catalytic activity occurs after adding into the electrolyte, demonstrating the actual active site nature of the Co metal center for ORR in the two . Additionally, in-situ Co center poisoning experiments with nitrite has also been carried out and shown in Supplementary Fig. 36. As can be found, significant degradation of the
catalytic activity for CoPc-S-COF occurs after adding nitrite into the electrolyte, further confirming the active site nature of the Co atoms towards ORR in CoPc-S-COF. Particularly, the polarization curves of reduction reaction ( ) of both COFs were recorded in the Ar-saturated 0.1 M KOH solution containing . As revealed in Fig. 3d, both samples display negligible activity. To more clearly compare the inhibition of decomposition for both COFs, the polarization curves of and in saturated 0.1 M KOH solutions containing different concentrations of were measured, Fig. 3e, f. CoPc-O-COF exhibits larger currents than those of CoPc-S-COF especially in high concentration ( ). This result indicates the superior inhibition of CoPc -S-COF to decomposition during the ORR process due to the enriched electron nature of the Co center associated with the elelctron-donating sulfur atoms in the linking unit, favouring the production of high concentration solution.
To explore the impact of F atom on the catalytic activity and selectivity of the Conters in CoPc-O-COF and CoPc-S-COF, the catalytic activity of the small molecule Co phthalocyanines including , and the metal-free phthalocyanine electrodes have also been prepared and tested on RRDE at 1600 rpm in -saturated 0.1 M KOH , Supplementary Fig. 37. It is worth noting that the CoPc and electrodes exhibit the same onset potential of 0.78 V versus RHE and similar selectivity ( ) in the potential range of versus RHE with an value of 2.5-2.7, demonstrating their obvious similar reactivity for oxygen reduction with negligible effect of F atoms. However, the metal-free electrode displays much inferior electrocatalytic performance with lower onset potential of 0.68 V versus RHE and selectivity ( ), confirming the nature of Co active centers. For the purpose of further clarifying the impact of O and S atoms on the catalytic activity and selectivity of the Co centers in CoPc-O-COF and CoPc-S-COF, two small molecule Co phthalocyanines including CoPc-S and CoPc-O containing C-S-C and C-O-C groups, respectively, were synthesized with their ORR performance assessed, Supplementary Figs 38 and 39. As can be seen, CoPc-S shows better ORR activity and worse activity in comparison with , proving the effect of S on enhancing the ORR performance of . In addition, the doped COF (named CoPc-O-COF-S 20%) was prepared with its electrocatalytic performance tested. As expected, CoPc-O-COF-S 20% shows high ORR activity with an onset potential of 0.80 V versus RHE and an electron transfer number of 2.2-2.3, Supplementary Figs. 40-42. Nevertheless, CoPc-O-COF-S 20% displays smaller currents than those of in the Ar-saturated 0.1 M KOH solution containing , revealing the effect of S -doping on diminishing the catalytic activity towards decomposition.
To gain further insight into the ORR activity of the COFs, the electronic structure of CoPc-O-COF and CoPc-S-COF was investigated by density functional theory (DFT) calculation. The adsorption energy ( ) of on various atoms of the two COFs including , and has been firstly calculated to explore the active site of production. As can be seen in Supplementary Fig. 43 and Supplementary Table 6, the significantly smaller value of for Co atom, , than those for other atoms ( to -0.05 eV for . for ) reveals the active site nature of Co atom in the two COFs towards ORR. In addition, the electron density on the CoPc moiety including around Co site in is higher than that for CoPc-O-COF due to the electron-donating effect of S atoms, which facilitates the charge transfer between Co active sites and intermediates and in turn affords enhanced catalytic activity for , Fig. 3g and Supplementary Fig. 44. Moreover, the natural population analysis (NPA) charge distribution calculation of both COFs was also carried out to assess the electron transferring process in both COFs. As displayed in Supplementary Table 7, the change of atomic charges on the conjugated chain is in a wave manner of
(NCo) during the electron transferring process from S/O atoms to central Co atoms, exhibiting an alternative polarization effect along the electron transferring pathway in both . In particular, the central Co atom gains an additional charge of when the O atoms in CoPc-O-COF are replaced by S atoms in CoPc-S-COF, further confirming a more obvious charge transfer from S to Co compared to that from O to Co . Furthermore, the calculated projected density of states (pDOS) discloses a lower band center position of -1.33 eV for CoPc-S-COF with higher intensity of peaks near the Fermi level ( ) compared to CoPc-O-COF with a d band center position of -1.17 eV , Fig. 3h. This indicates that larger density of active electrons around Co centers in CoPc-S-COF participates in the electrochemical ORR reaction, confirming the higher catalytic activity of CoPc-S-COF due to the electron-donating effect of S atoms. Nevertheless, Fig. 3i presents the calculated Gibbs free energy differences ( ) diagrams of the ORR and ORR processes on and . As can be found, the conversion of to is the rate-determining step of ORR on both and with an energy barrier of 0.63 and 1.11 eV , respectively. These values are smaller than the energy barrier of the to conversion process (the ratedetermining step of ORR) on both COFs, 1.44 and 1.48 eV , demonstrating the faster reaction kinetics of ORR than ORR on both COFs and in turn their high selectivity towards ORR. In particular, the lower to conversion energy barrier of COF in comparison with that for indicates the enhanced ORR activity of the former species over the latter one, while the higher to conversion energy barrier of CoPc-S-COF than that for illustrates the diminished activity towards decomposition of to , rationalizing the more stable electrocatalytic production of .
Gas diffusion electrode (GDE) devices were used to further explore the practical application potential of the as-prepared CoPcbased COFs towards ORR. A three-phase flow cell, Fig. 4a, in which the catalyst is deposited on a gas diffusion layer (GDL) as the work electrode, is deemed to be able to afford higher reduction current densities by increasing oxygen concentration on GDL and improve the production rate. Corresponding measurements of both COFs were carried out in 1 M KOH . As exhibited in Fig. 4b, both COFs show high electrocatalystic activity with much higher current density in flow cell compared to RRDE measurements. The chronoamperometry measurements at varied applied voltages were conducted with the generated determined by the titration method , Fig. 4c, and Supplementary Figs. 45 and 46. Figure 4d shows the determined Faradaic efficiency of for both COFs. CoPc-O-COF and CoPc-S-COF could exhibit over in the range of 0.73 to 0.33 V versus RHE, even higher than the RRDE measurements in the range from 0.73 to 0.53 V . Remarkably, the of CoPc-S-COF reaches at 0.63 V versus RHE (equivalent to an overpotential of 130 mV ) with a of in 1 M KOH at room temperature, which gets further increased to at 0.33 V versus RHE (equivalent to an overpotential of 430 mV ) with still higher than , superior to , Fig. 4c, d, and Supplementary Fig. 45. The performance of both COFs was also explored in the flow cell. As displayed in Fig. 4b, CoPc-S-COF shows much smaller reduction current densities compared to , consistent with the result in the RRDE system. This actually suggests that could be inhibited in CoPc-S-COF and therefore leads to higher and concentration. Furthermore, the long standing and stable production of CoPc-S-COF has been recorded at a fixed current density of , Fig. 4e, f. CoPc-S-COF maintains in the continuous electroproduction for 20 h . Nevertheless, the amount produced gets linearly increased along with increasing the operating time with an almost constant production rate of . and an almost unchanged operating voltage of . 0.67 V versus RHE, Fig. 4f, further confirming the high stability of
Fig. 4 | electroproduction in the flow cell. a Schematic diagram of the flow cell. LSVs of CoPc-O-COF and CoPc-S-COF in flow cell. The chronoamperometry measurements at varied applied voltages of . d of and CoPc-S-COF at varied applied voltages. e yields of CoPc-S-COF.
f Chronopotentiometry curve at a current density of and the corresponding in the flow cell for CoPc-S-COF (Catalyst mass loading: ).
CoPc-S-COF as well as its inactivation towards decomposition. More importantly, the yield reaches up to 377 mg after 20 h electrolysis corresponding to a concentration of , Fig. 4e. It is worth noting that the features of COFs such as the molecularly structural tunability and desired permanent porosity enable this class of materials to be promising advanced catalysts with welldefined structure and active sites for various electrochemical applications . Recently, 2D COFs such as PYTA-TPEDH-COF , TP-TDCOF , and Py-TD-COF as well as 3D COFs including BUCT-COF-1 and BUCT-COF- have been employed as metal-free electrocatalysts to promote ORR with selectivity up to . Introduction of highly efficient Co active sites for ORR into affords its enhanced selectivity ( ) in the present case. Nevertheless, the undulated layer-stacked structure of CoPc-S-COF favours the exposure of active sites, resulting in the higher production rate in comparison with all the thus far reported COF-based electrocatalysts, Supplementary Table 8. Actually, the electrocatalytic production performance of CoPc-S-COF is also competitive with that of the state-of-the-art inorganic catalytic materials, Supplementary Table 8, demonstrating its great application potential towards practical production. Moreover, after the stability test, CoPc-S-COF shows constant structure according to the FTIR and XPS analysis, further proving its robust stability, Supplementary Figs. 47 and 48.
To further reveal the application potential of CoPc-S-COF, the electrocatalytic production activity of CoPc -S-COF has also been evaluated in in the GDE device. As shown in Supplementary Fig. 49, CoPc-S-COF displays high ORR activity in the neutral electrolyte with a high selectivity (ca. ) in the potential range of versus RHE. More importantly, COF displays a stable production with a high of under a current density of at an applied potential of . 0.30 V vs RHE for 20 h , generating , comparable to the result obtained in alkaline electrolyte. These additional experimental results further confirm the good electrocatalytic production performance of CoPc-S-COF, which is beneficial to the design and preparation of high-performance and low-cost electrocatalysts towards industrial-level electroproduction.
Discussion
In summary, a porous dithiine-linked CoPc-based COF was fabricated. CoPc-S-COF possesses an undulated layer-stacked structure due to the bending along the C-S-C bridge to allow more exposed Co centers for ORR. This, in combination with the activated ORR but deactivated decomposition capability of the Co center because of the electron-donating effect of S atoms, enables to display a high selectivity and realize the large-scale production in the
flow cell. This work should be beneficial to the design and preparation of high-performance and low-cost electrocatalysts for electrosynthesis.
Methods
Synthesis of CoPc-O-COF
and THB ( ) were added into the mixed solvent of -xylene and 0.5 mL DMAc in a 16 mL Pyrex tube. The mixture was sonicated for 5 min to form a homogeneous suspension. Then triethylamine was added into the mixture. After three freeze-pump-thaw cycles, the Pyrex tube was sealed and heated in an oven at for 7 days. The black-green precipitate was collected by centrifugation and rinsed with acetone, dichloromethane, and THF in a Soxhlet extractor for one day. Finally, CoPc-O-COF was then obtained as black powder in a yield of .
Synthesis of CoPc-S-COF
and BTT ( ) were added into the mixed solvent of -xylene and 0.5 mL DMAc in a 16 mL Pyrex tube. The mixture was sonicated for 5 min to form a homogeneous suspension. Then triethylamine was added into the mixture. After three freeze-pump-thaw cycles, the Pyrex tube was sealed and heated in an oven at for 7 days. A dark-green solid was formed inside the Pyrex tube during the reaction process. The product was collected by centrifugation and then solvent exchanged with dry ethanol for a week followed by supercritical drying. Finally, CoPc-S-COF was obtained as fluffy dark-green product in a yield of .
Data availability
All relevant data that support the findings of this study are presented in the manuscript and supplementary information file. Source data are provided with this paper.
References
Yang, X. et al. Tuning two-electron oxygen-reduction pathways for electrosynthesis via engineering atomically dispersed single metal site catalysts. Adv. Mater. 34, e2107954 (2022).
Chen, J. et al. Kinetically restrained oxygen reduction to hydrogen peroxide with nearly 100% selectivity. Nat. Commun. 13, 2808 (2022).
Wang, N. et al. Recent progress of electrochemical production of hydrogen peroxide by two-electron oxygen reduction reaction. Adv. Sci. 8, e2100076 (2021).
Wang, Y., Waterhouse, G. I. N., Shang, L. & Zhang, T. Electrocatalytic oxygen reduction to hydrogen peroxide: from homogeneous to heterogeneous electrocatalysis. Adv. Energy Mater. 11, 2003323 (2020).
Fei, H. et al. Atomic cobalt on nitrogen-doped graphene for hydrogen generation. Nat. Commun. 6, 8668 (2015).
Bu, Y. et al. Carbon-based electrocatalysts for efficient hydrogen peroxide production. Adv. Mater. 33, e2103266 (2021).
Jiang, K. et al. Catalyst design for electrochemical oxygen reduction toward hydrogen peroxide. Adv. Funct. Mater. 30, 2003321 (2020).
Kim, H. W. et al. Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts. Nat. Catal. 1, 282-290 (2018).
Tian, Y. et al. Edge-hosted atomic sites on hierarchical porous carbon for highly selective two-electron oxygen reduction reaction. Angew. Chem. Int. Ed. 61, e202213296 (2022).
Li, B. Q. et al. Electrosynthesis of hydrogen peroxide synergistically catalyzed by atomic Co-Nx-C sites and oxygen functional groups in noble-metal-free electrocatalysts. Adv. Mater. 31, e1808173 (2019).
Sun, Y. et al. A comparative perspective of electrochemical and photochemical approaches for catalytic production. Chem. Soc. Rev. 49, 6605-6631 (2020).
Li, D. et al. Metal-free thiophene-sulfur covalent organic frameworks: precise and controllable synthesis of catalytic active sites for oxygen reduction. J. Am. Chem. Soc. 142, 8104-8108 (2020).
Melchionna, M. et al. The rise of hydrogen peroxide as the main product by metal-free catalysis in oxygen reductions. Adv. Mater. 31, e1802920 (2019).
Lobyntseva, E. et al. Electrochemical synthesis of hydrogen peroxide: rotating disk electrode and fuel cell studies. Electrochim. Acta. 52, 7262-7269 (2007).
Lee, K. et al. Structure-controlled graphene electrocatalysts for high-performance production. Energ. Environ. Sci. 15, 2858-2866 (2022).
Tammeveski, K. et al. Surface redox catalysis for reduction on quinone-modified glassy carbon electrodes. J. Electroanal. Chem. 515, 101-112 (2001).
Sarapuu, A. et al. Electrochemical reduction of oxygen on anthraquinone-modified glassy carbon electrodes in alkaline solution. J. Electroanal. Chem. 541, 23-29 (2003).
Vaik, K. et al. Oxygen reduction on phenanthrenequinone-modified glassy carbon electrodes in 0.1 M KOH. J. Electroanal. Chem. 564, 159-166 (2004).
Sarapuu, A. et al. Kinetics of oxygen reduction on quinone-modified HOPG and BDD electrodes in alkaline solution. Electrochem. SolidState Lett. 8, E30 (2005).
Vaik, K. et al. Electrocatalytic oxygen reduction on glassy carbon grafted with anthraquinone by anodic oxidation of a carboxylate substituent. Electrochim. Acta 50, 5126-5131 (2005).
Siahrostami, S. et al. Enabling direct production through rational electrocatalyst design. Nat. Mater. 12, 1137-1143 (2013).
Cao, P. et al. Metal single-site catalyst design for electrocatalytic production of hydrogen peroxide at industrial-relevant currents. Nat. Commun. 14, 172 (2023).
Zhang, C. et al. Crystal engineering enables cobalt-based metalorganic frameworks as high-performance electrocatalysts for production. J. Am. Chem. Soc. 145, 7791-7799 (2023).
Guo, Y. et al. Precise design of covalent organic frameworks for electrocatalytic hydrogen peroxide production. CHEM-ASIAN. J. 16, 498-502 (2021).
Lee, B.-H. et al. Supramolecular tuning of supported metal phthalocyanine catalysts for hydrogen peroxide electrosynthesis. Nat. Catal. 6, 234-243 (2023).
Lin, R. et al. Approaching theoretical performances of electrocatalytic hydrogen peroxide generation by cobalt-nitrogen moieties. Angew. Chem. Int. Ed. 62, e202301433 (2023).
Zhao, X. et al. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc 143, 9423-9428 (2021).
Diercks, C. S. et al. The atom, the molecule, and the covalent organic framework. Science 355, eaal1585 (2017).
Yu, B. et al. Linkage conversions in single-crystalline covalent organic frameworks. Nat. Chem. 16, 114-121 (2024).
Pramudya, Y. et al. Design principles for high storage using chelation of abundant transition metals in covalent organic frameworks for 0-700 bar at 298 K. J. Am. Chem. Soc. 138, 15204-15213 (2016).
Wang, Z. et al. Covalent organic frameworks for separation applications. Chem. Soc. Rev. 49, 708-735 (2020).
Keller, N. et al. Optoelectronic processes in covalent organic frameworks. Chem. Soc. Rev. 50, 1813-1845 (2021).
Huang, S. et al. Porphyrin and phthalocyanine based covalent organic frameworks for electrocatalysis. Coord. Chem. Rev. 464, 214563 (2022).
Yang, X. et al. lonothermal synthesis of fully conjugated covalent organic frameworks for high-capacity and ultrastable potassiumion batteries. Adv. Mater. 34, e2207245 (2022).
Bhunia, S. et al. 2,1,3]-Benzothiadiazole-spaced co-porphyrin-based covalent organic frameworks for O(2) reduction. ACS Nano 17, 3492-3505 (2023).
Liu, M. et al. Construction of catalytic covalent organic frameworks with redox-active sites for the oxygen reduction and the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202213522 (2022).
Mondal, S. et al. A thiadiazole-based covalent organic framework: a metal-free electrocatalyst toward oxygen evolution reaction. ACS Catal 10, 5623-5630 (2020).
Lin, C. Y. et al. Covalent organic framework electrocatalysts for clean energy conversion. Adv. Mater. 30, 1703646 (2018).
Han, B. et al. Two-dimensional covalent organic frameworks with cobalt(II)-phthalocyanine sites for efficient electrocatalytic carbon dioxide reduction. J. Am. Chem. Soc. 143, 7104-7113 (2021).
Zhong, H. et al. Boosting the electrocatalytic conversion of nitrogen to ammonia on metal-phthalocyanine-based two-dimensional conjugated covalent organic frameworks. J. Am. Chem. Soc. 143, 19992-20000 (2021).
Yang, C. et al. Theory-driven design and targeting synthesis of a highly-conjugated basal-plane 2D covalent organic framework for metal-free electrocatalytic OER. ACS Energy Lett 4, 2251-2258 (2019).
Lu, M. et al. Stable dioxin-linked metallophthalocyanine covalent organic frameworks (COFs) as photo-coupled electrocatalysts for reduction. Angew. Chem. Int. Ed. 60, 4864-4871 (2021).
Yue, Y. et al. Stable bimetallic polyphthalocyanine covalent organic frameworks as superior electrocatalysts. J. Am. Chem. Soc. 143, 18052-18060 (2021).
Huang, N. et al. A stable and conductive metallophthalocyanine framework for electrocatalytic carbon dioxide reduction in water. Angew. Chem. Int. Ed. 59, 16587-16593 (2020).
Zhi, Q. et al. Piperazine-LInked Metalphthalocyanine Frameworks for Highly Efficient Visible-light-driven photosynthesis. J. Am. Chem. Soc. 144, 21328-21336 (2022).
Haldar, S. et al. Porous dithiine-linked covalent organic framework as a dynamic platform for covalent polysulfide anchoring in lithiumsulfur battery cathodes. J. Am. Chem. Soc. 144, 9101-9112 (2022).
Wang, K. et al. Tetrapyrrole macrocycle based conjugated twodimensional mesoporous polymers and covalent organic frameworks: from synthesis to material applications. Coord. Chem. Rev. 378, 188-206 (2019).
Lin, S. et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 349, 1208-1213 (2015).
Qiu, X. F. et al. A stable and conductive covalent organic framework with isolated active sites for highly selective electroreduction of carbon dioxide to acetate. Angew. Chem. Int. Ed. 61, e202206470 (2022).
Yu, H.-Y. et al. A stack-guiding unit constructed 2D COF with improved charge carrier transport and versatile photocatalytic functions. Chem. Eng. J. 445, 136713 (2022).
Yuan, Y. et al. Deciphering the selectivity descriptors of heterogeneous metal phthalocyanine electrocatalysts for hydrogen peroxide production. Chem. Sci. 13, 11260-11265 (2022).
Tiekink, E. R. T. & Zukerman-Schpector, J. Stereochemical activity of lone pairs of electrons and supramolecular aggregation patterns based on secondary interactions involving tellurium in its 1,1dithiolate structures. Coord. Chem. Rev. 254, 46-76 (2010).
Li, N. et al. Polyarylether-Based 2D Covalent-Organic Frameworks with In-Plane D-A Structures and Tunable Energy Levels for Energy Storage. Adv. Sci. 9, e2104898 (2022).
Shen, H. et al. Synergistic effects between atomically dispersed Fe-N-C and C-S-C for the oxygen reduction reaction in acidic media. Angew. Chem. Int. Ed. 56, 13800-13804 (2017).
Xia, Y. et al. Highly active and selective oxygen reduction to on boron-doped carbon for high production rates. Nat. Commun. 12, 4225 (2021).
Han, B. et al. Maximizing electroactive sites in a three-dimensional covalent organic framework for significantly improved carbon dioxide reduction electrocatalysis. Angew. Chem. Int. Ed. 61, e202114244 (2022).
Chen, S . et al. Identification of the highly active coordination motif for selective oxygen reduction to hydrogen peroxide. J. Am. Chem. Soc. 144, 14505-14516 (2022).
Lin, L. et al. Atomic-level modulation-induced electron redistribution in Co coordination polymers elucidates the oxygen reduction mechanism. ACS Catal 12, 7531-7540 (2022).
Carpenter, J. E. Extension of Lewis Structure Concepts to Open-shell and Excited-state Molecular Species. (PhD thesis. University of Wisconsin, Madison, WI, 1987).
Wu, C. et al. Polarization Engineering of Covalent Triazine Frameworks for Highly Efficient Photosynthesis of Hydrogen Peroxide from Molecular Oxygen and Water. Adv. Mater. 34, 2110266 (2022).
Zhao, X. et al. Covalent organic frameworks (COFs) for electrochemical applications. Chem. Soc. Rev. 50, 6871-6913 (2021).
An, S. et al. One-dimensional covalent organic frameworks for the oxygen reduction reaction. Angew. Chem. Int. Ed. 62, e202218742 (2023).
Huang, S. et al. Covalent organic frameworks with molecular electronic modulation as metal-free electrocatalysts for efficient hydrogen peroxide production. Small Struct. 4, 2200387 (2023).
Huang, S. et al. Linkage engineering in covalent organic frameworks as metal-free oxygen reduction electrocatalysts for hydrogen peroxide production. Appl. Catal. B 340, 123216 (2024).
Bao, R. et al. Designing thiophene-enriched fully conjugated 3D covalent organic framework as metal-free oxygen reduction catalyst for hydrogen fuel cells. Angew. Chem. Int. Ed. 62, e202216751 (2023).
Zhang, Y. et al. Multicomponent synthesis of imidazole-linked fully conjugated 3D covalent organic framework for efficient electrochemical hydrogen peroxide production. Angew. Chem. Int. Ed. 62, e202314539 (2023).
Acknowledgements
Financial support from the Natural Science Foundation of China, grant Nos. 22235001 (J.J.), 22175020 (J.J.) and 12305372 (Y.L.). University of Science and Technology Beijing is gratefully acknowledged. The authors also wish to thank the facility support of the 4B9A beamline of Beijing Synchrotron Radiation Facility (BSRF).
Author contributions
Conceptualization: Q.Z., K.W., and J.J. Methodology: Q.Z., R.J., and K.W. Investigation: Q.Z., R.J., and Y.L. Visualization: Q.Z. and X.Y. DFT calculation: Y.J. and D.Q. Writing—original draft: Q.Z. Writing—review and editing: Q.Z., R.J., X.Y., Y.J., D.Q., K.W., and J.J.
Correspondence and requests for materials should be addressed to Kang Wang, Yunpeng Liu or Jianzhuang Jiang.
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Beijing Advanced Innovation Center for Materials Genome Engineering, Beijing Key Laboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry and Chemical Engineering, School of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing 100083, China. Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Science, Beijing 100049, China. e-mail: kangwang@ustb.edu.cn; liuyunpeng@ihep.ac.cn; jianzhuang@ustb.edu.cn