DOI: https://doi.org/10.1038/s41467-024-46708-8
PMID: https://pubmed.ncbi.nlm.nih.gov/38509067
تاريخ النشر: 2024-03-20
إعادة بناء ذاتية ديناميكية محدودة نحو أداء عالي في أكسدة مياه البحر المباشرة
تم القبول: 7 مارس 2024
نُشر على الإنترنت: 20 مارس 2024
(أ) التحقق من التحديثات
الملخص
تطوير محفزات كهربائية عالية الكفاءة لتفكيك مياه البحر مباشرة مع وظائف ثنائية لمنع إعادة بناء الأكسدة الأنودية وتفاعلات تطور الأكسجين الانتقائية هو تحدٍ كبير. هنا، نبلغ عن محفز كهربائي لتأكسد مياه البحر مباشرة يحقق استقرارًا طويل الأمد لأكثر من 1000 ساعة عند
إعادة تشكيلها لتكوين أكاسيد المعادن الهيدروكسيلية في النطاق المحتمل، والذي يعتبر “المحفز الحقيقي” لـ
تعتمد طبقة CoMo-LDH المستقرة المعاد بناؤها على التنافر الكهروستاتيكي لمزيد من الكلور الكاره للماء، مما يحقق أكسدة انتقائية لمياه البحر. تتكون الخلية الكهروكيميائية الجارية من
النتائج
التركيب والتوصيف
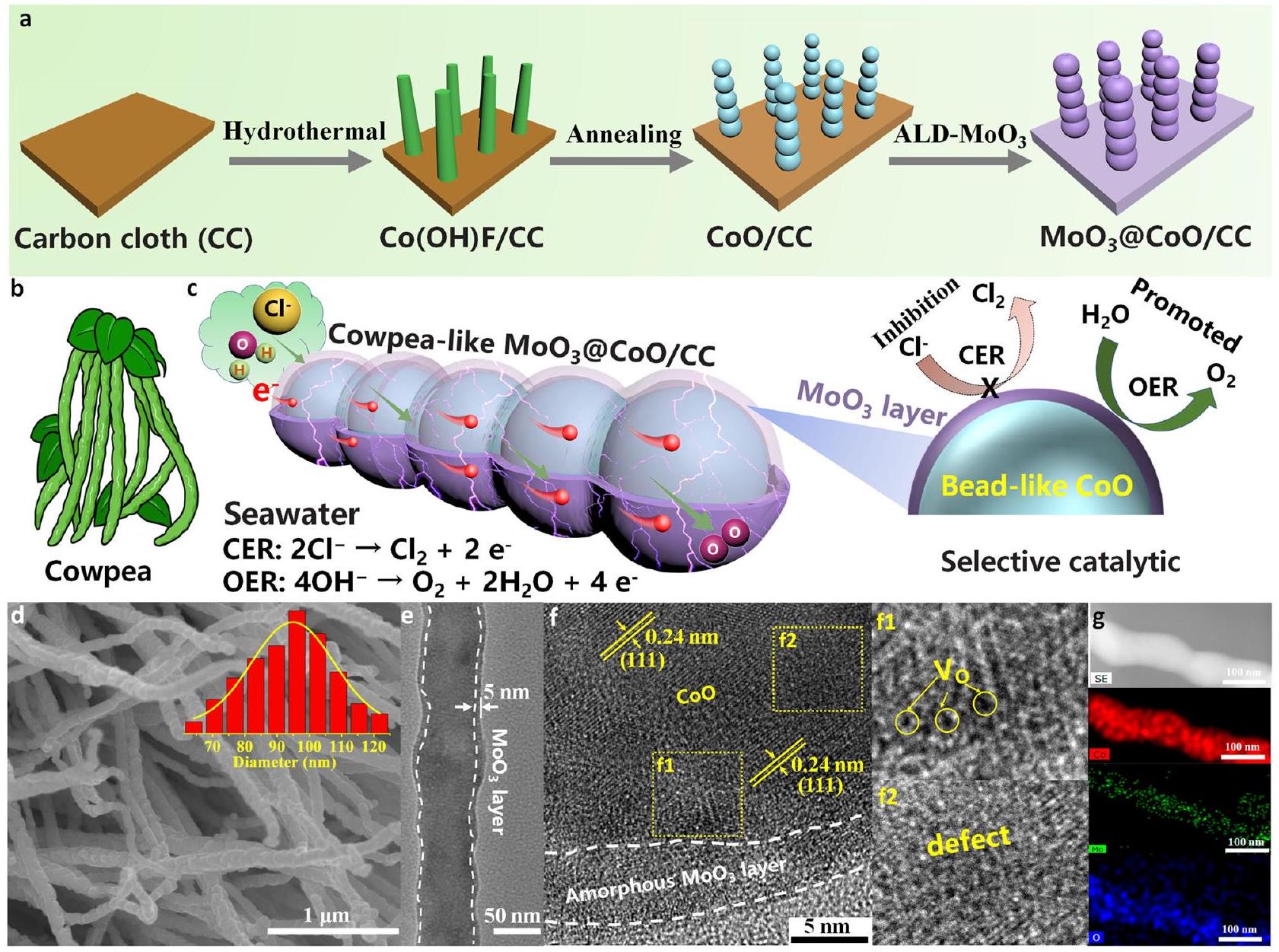
عملية التحويل للامتزاز الانتقائي لـ
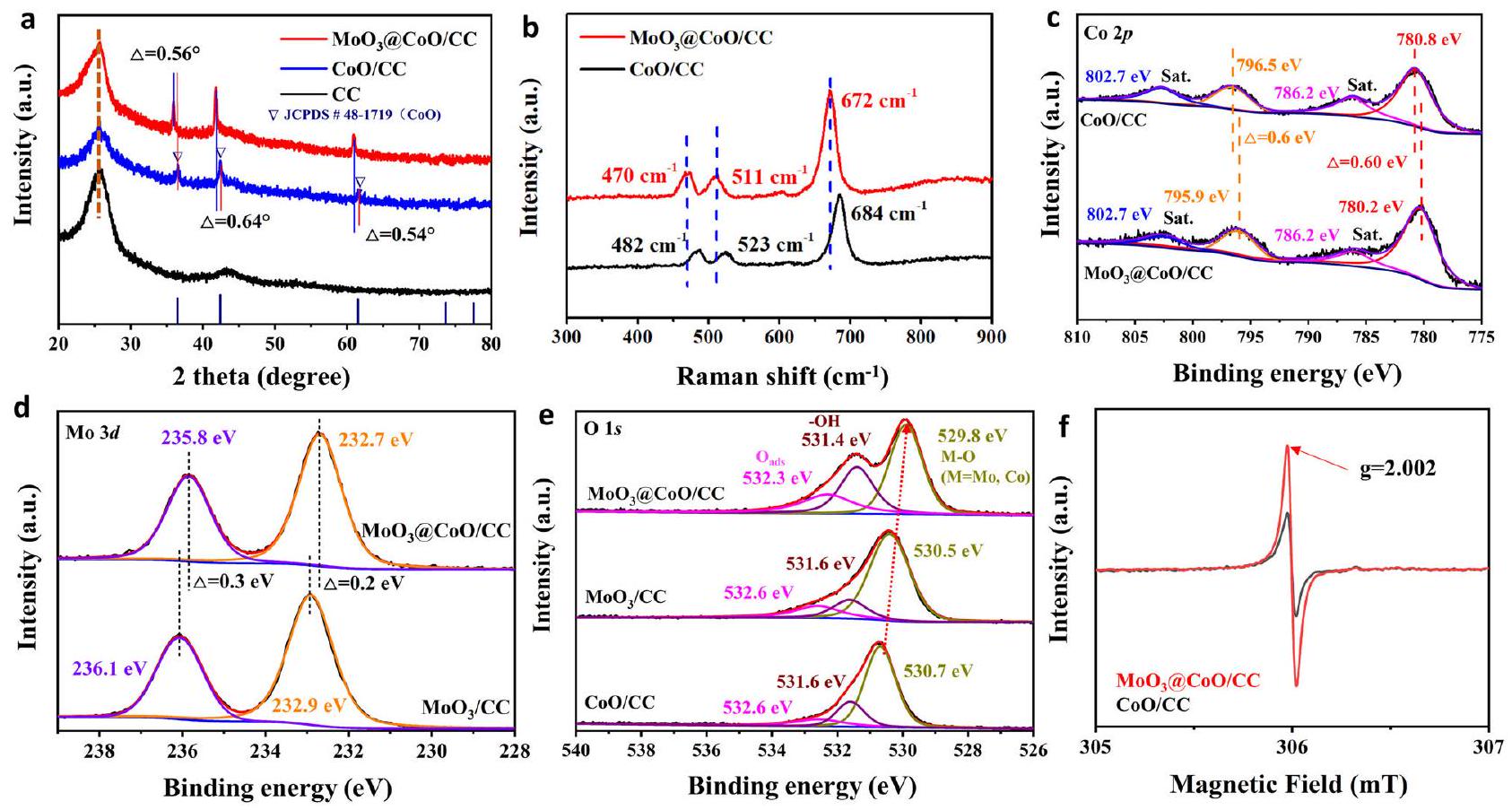
التحليل الهيكلي
و
اختبار كيميائي كهربائي
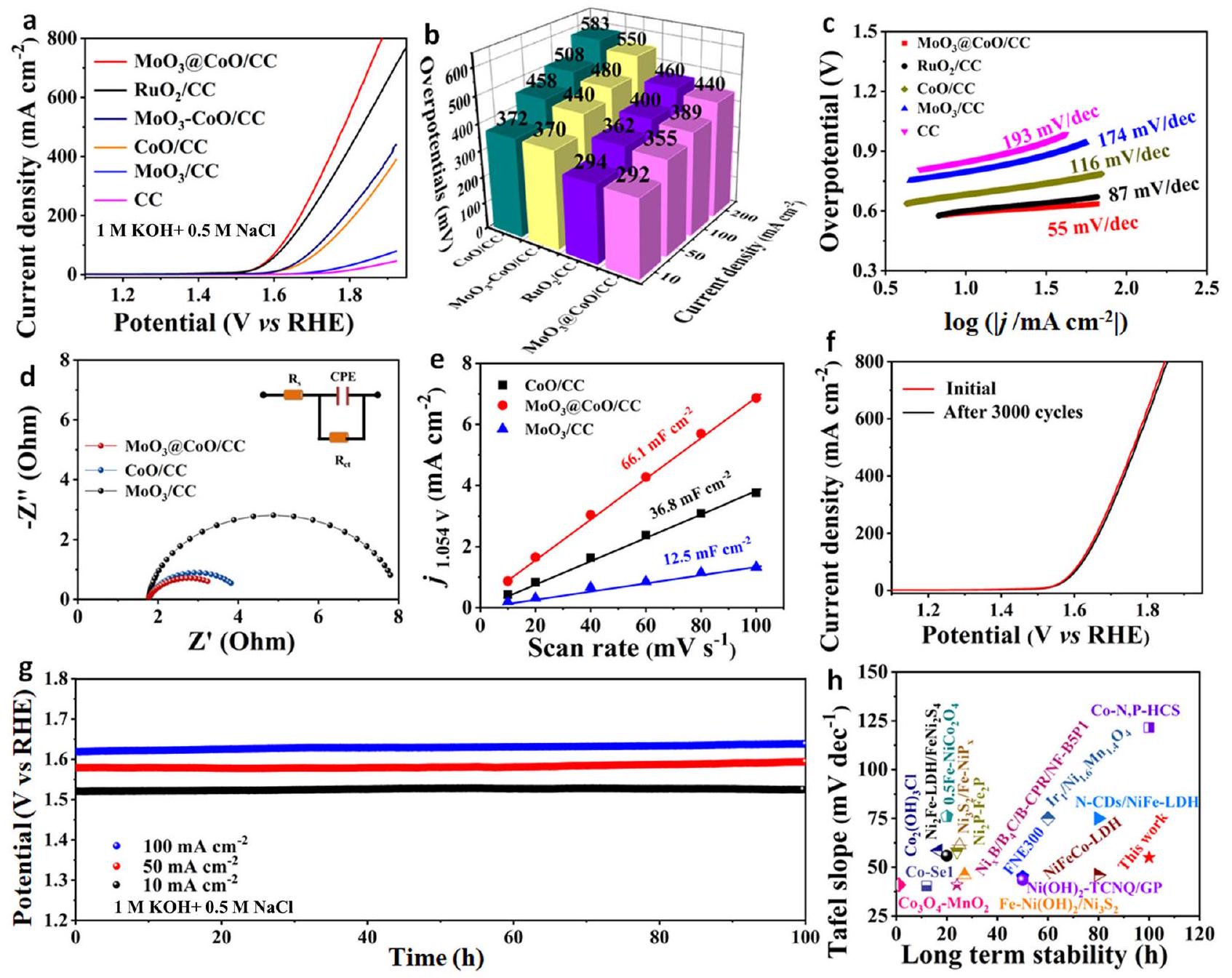
الدورات. يمكن أن يُعزى هذا الانخفاض إلى طبقة الحماية السميكة بشكل مفرط، والتي تقلل من المساحة النشطة للمحفز (الشكل التكميلي 6). تشير هذه النتائج إلى أن واجهة المحفز تعمل كمركز نشط للتفاعل. من الشكل 3c، ميل تافل (
تحليل آلية تثبيط CER
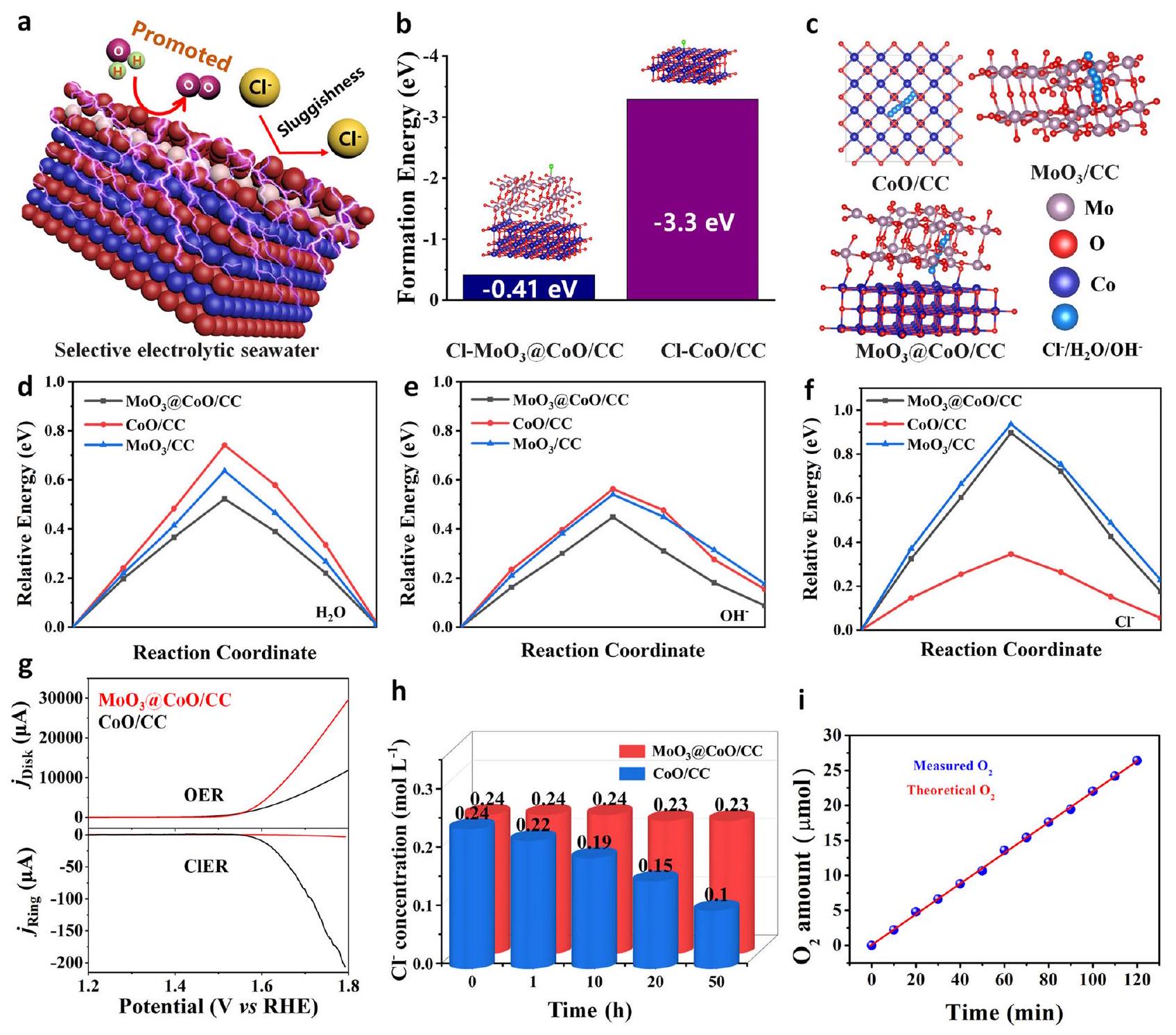
نموذج هيكلي لمسار الهجرة
كاشف (الشكل 4h).
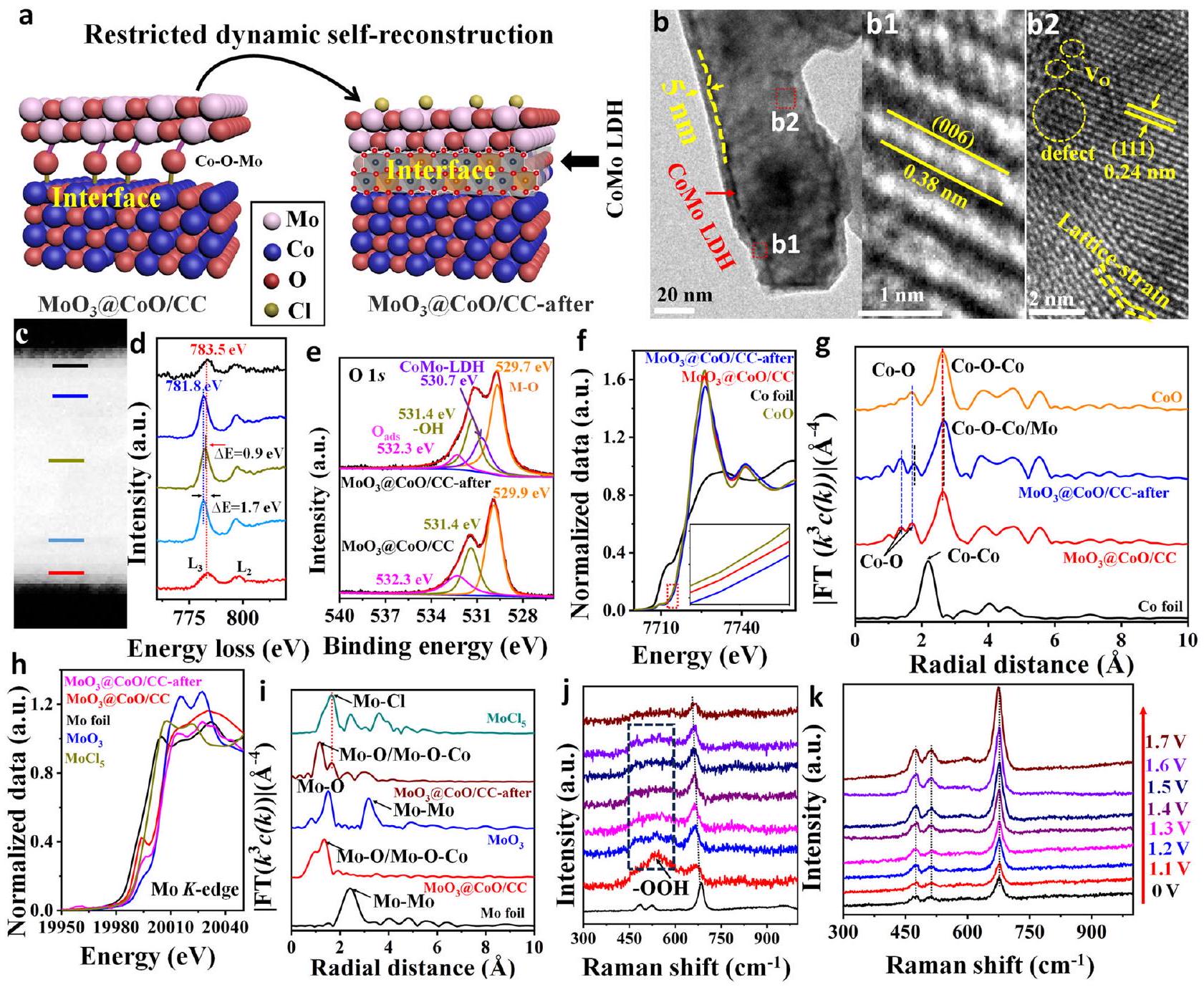
إعادة بناء السطح الديناميكي الذاتي المقيد
أقوى من القمة الرئيسية (
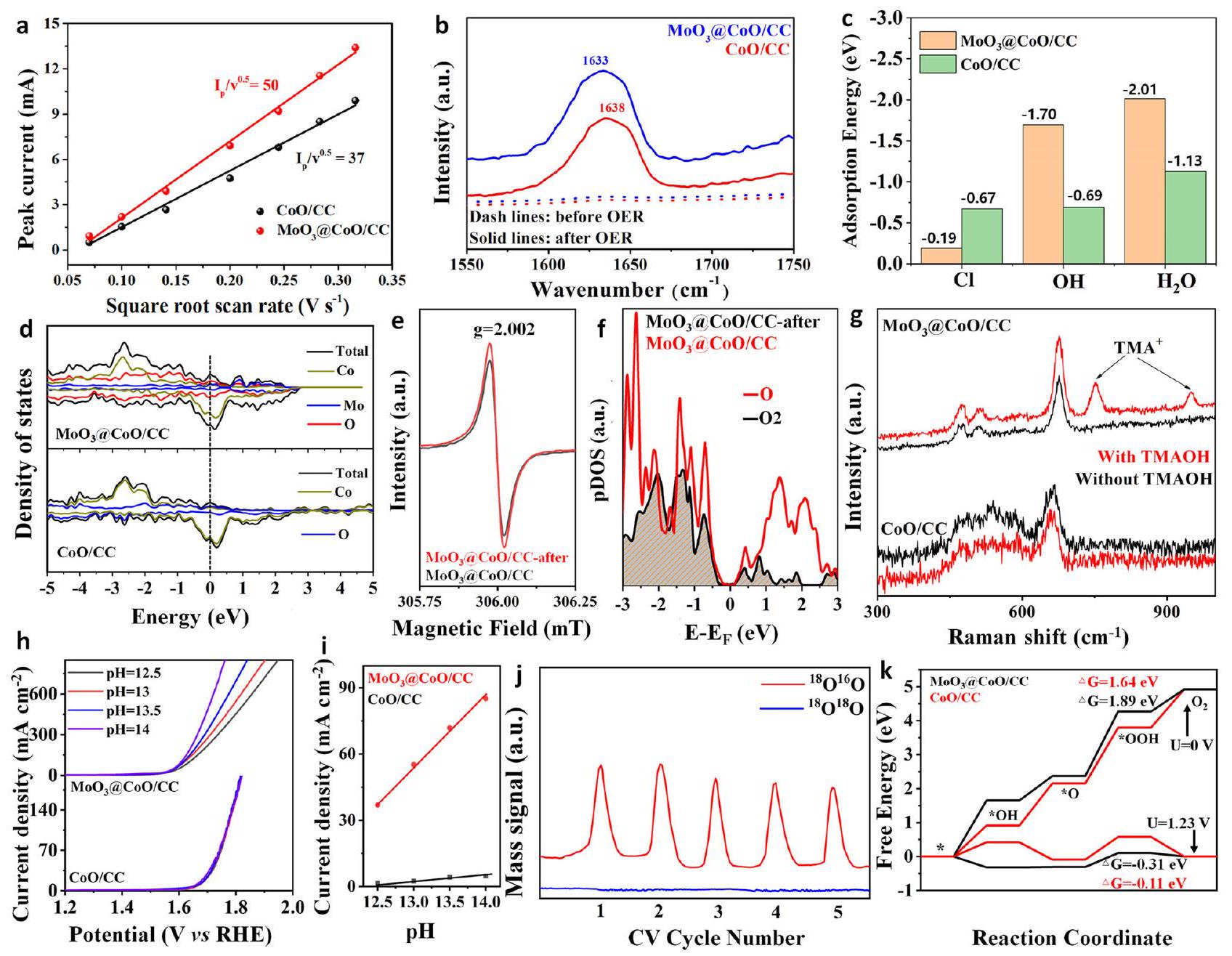
تحليل آلية أكسدة مياه البحر
مما يشير إلى وجود نقل شحنات كبير يفضل التفاعل الحفزي (الشكل التكميلية 26).
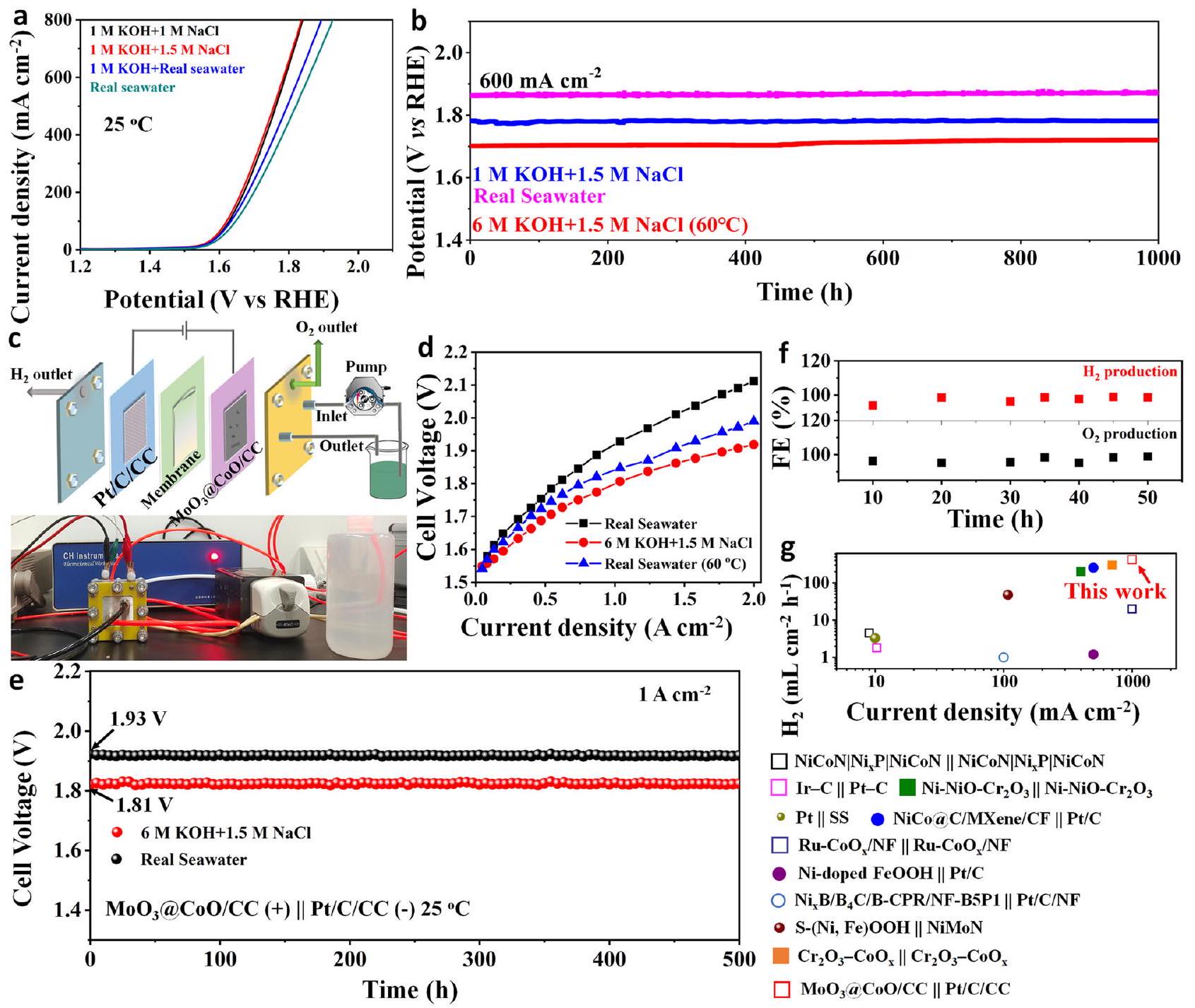
تطبيقات التحليل الكهربائي العملية
على التوالي. طيف رامان لمختلف المحفزات التي تم اختبارها بعد غسلها بالماء بعد الاختبار مع أو بدون TMAOH. منحنيات LSV لمختلف العينات عند قيم pH مختلفة.
أن
نقاش
بيئات متنوعة. الاستقرار الكهربائي المستمر. كتلة الـ
واجهة نشطة من المحفز، مما يحقق التحفيز الانتقائي؛ (2) الرقيق للغاية
طرق
المواد
غاز النيتروجين هو
تحضير CoO/CC
تحضير
بلازما. تتكون دورة ALD واحدة من
توفر البيانات
References
- Turner, J. A. Sustainable hydrogen production. Science 305, 972-974 (2004).
- Dresp, S. et al. Efficient direct seawater electrolysers using selective alkaline NiFe-LDH as OER catalyst in asymmetric electrolyte feeds. Energy Environ. Sci. 13, 1725-1729 (2020).
- Sun, F. et al. Energy-saving hydrogen production by chlorine-free hybrid seawater splitting coupling hydrazine degradation. Nat. Commun. 12, 4182 (2021).
- Schmidtko, S., Heywood, K. J., Thompson, A. F. & Aoki, S. Multidecadal warming of Antarctic waters. Science 346, 1227-1231 (2014).
- Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: toward boosted and sustained water oxidation catalysis. Joule 5, 2164-2176 (2021).
- Huang, Y. et al. Plasma-induced Mo-doped
with enriched oxygen vacancies for electrocatalytic oxygen evolution in water splitting. Carbon Energy 5, 279 (2022). - Yu, L. et al. Non-noble metal-nitride based electrocatalysts for highperformance alkaline seawater electrolysis. Nat. Commun. 10, 5106-5115 (2019).
- Yu, L. et al. Ultrafast room-temperature synthesis of porous S-doped Ni/Fe (oxy)hydroxide electrodes for oxygen evolution catalysis in seawater splitting. Energy Environ. Sci. 13, 3439-3446 (2020).
- Grimaud, A. et al. Activation of surface oxygen sites on an iridiumbased model catalyst for the oxygen evolution reaction. Nat. Energy 2, 16189 (2016).
- Qiu, Z. et al. Direct observation of active catalyst surface phases and the effect of dynamic self-optimization in NiFe-layered double hydroxides for alkaline water splitting. Energy Environ. Sci. 12, 572-581 (2019).
- Chung, D. Y. et al. Dynamic stability of active sites in hydr(oxy) oxides for the oxygen evolution reaction. Nat. Energy 5, 222-230 (2020).
- Zhao, S. et al. Structural transformation of highly active metal-organic framework electrocatalysts during the oxygen evolution reaction. Nat. Energy 5, 881 (2020).
- Lin, Q. et al. Tuning the interface of
by atomic replacement strategy toward high-performance electrocatalytic oxygen evolution. ACS Nano 16, 15460-15470 (2022). - Chen, F.-Y. et al. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704-1731 (2021).
- Zhang, Y. et al. Rapid synthesis of cobalt nitride nanowires: highly efficient and low-cost catalysts for oxygen evolution. Angew. Chem. Int. Ed. 55, 8670-8674 (2016).
- Minguzzi, A. How to improve the lifetime of an electrocatalyst. Nat. Catal. 3, 687-689 (2020).
- Ning, M. et al. Boosting efficient alkaline fresh water and seawater electrolysis via electrochemical reconstruction. Energy Environ. Sci. 15, 3945-3957 (2022).
- Chala, S. A. et al. Tuning dynamically formed active phases and catalytic mechanisms of in situ electrochemically activated layered
double hydroxide for oxygen evolution reaction. ACS Nano 15, 14996-15006 (2021). - Peng, L. et al. Atomic cation-vacancy engineering of NiFe-layered double hydroxides for improved activity and stability towards the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 24612-24619 (2021).
- Ye, S.H. et al. Deeply self-reconstructing
to lowcrystalline with motifs for oxygen evolution reaction. Appl. Catal. B 304, 120986 (2022). - Kang, J. X. et al. Valence oscillation and dynamic active sites in monolayer NiCo hydroxides for water oxidation. Nat. Catal. 4, 1050-1058 (2021).
- Duan, Y. et al. Anodic oxidation enabled cation leaching for promoting surface reconstruction in water oxidation. Angew. Chem. Int. Ed. 60, 7418-7425 (2021).
- Wang, H. Y. et al. In operando identification of geometrical-sitedependent water oxidation activity of spinel
. J. Am. Chem. Soc. 138, 36-39 (2016). - Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212-222 (2021).
- Wang, W. et al. Structural reconstruction of catalysts in electroreduction reaction: identifying, understanding, and manipulating. Adv. Mater. 34, 2110699 (2022).
- Karlsson, R. K. B. et al. Selectivity between oxygen and chlorine evolution in the chlor-alkali and chlorate processes. Chem. Rev. 116, 2982-3028 (2016).
- Kuai, C. et al. Phase segregation reversibility in mixed-metal hydroxide water oxidation catalysts. Nat. Catal. 3, 743-753 (2020).
- Yang, L. et al. Transition metal-based electrocatalysts for seawater oxidation. Adv. Mater. Interfaces 9, 2201486 (2022).
- Wu, Q. et al. A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater. Nat. Commun. 12, 483 (2021).
- Dresp, S. et al. Direct electrolytic splitting of seawater: opportunities and challenges. ACS Energy Lett. 4, 933-942 (2019).
- Song, H. J. et al. Electrocatalytic selective oxygen evolution of carbon-coated
nanoparticles for alkaline seawater electrolysis. ACS Catal. 10, 702-709 (2019). - Guo, D. et al. A tandem interfaced
composite fabricated by atomic layer deposition as efficient HER electrocatalyst. Small 18, 2201896 (2022). - Cao, L. et al. Atomically dispersed iron hydroxide anchored on Pt for preferential oxidation of CO in
. Nature 565, 631-635 (2019). - Meng, L. et al. Atomic layer deposition triggered Fe-In-S cluster and gradient energy band in ZnInS photoanode for improved oxygen evolution reaction. Nat. Commun. 12, 5247 (2021).
- Guo, D. et al. Strategic atomic layer deposition and electrospinning of cobalt sulfide/nitride composite as efficient bifunctional electrocatalysts for overall water splitting. Small 16, 2002432 (2020).
- Mao, S. et al. High-performance bi-functional electrocatalysts of 3D crumpled graphene-cobalt oxide nanohybrids for oxygen reduction and evolution reactions. Energy Environ. Sci. 7, 609-616 (2014).
- Tian, Y. et al. Engineering crystallinity and oxygen vacancies of Co(II) oxide nanosheets for high performance and robust rechargeable Zn-Air batteries. Adv. Funct. Mater. 31, 2101239 (2021).
- Ling, T. et al. Engineering surface atomic structure of single-crystal cobalt (II) oxide nanorods for superior electrocatalysis. Nat. Commun. 7, 12876 (2016).
- Wu, L. et al. Boron-modified cobalt iron layered double hydroxides for high efficiency seawater oxidation. Nano Energy 83, 105838 (2021).
- Yang, G. et al. Interfacial engineering of
heterojunction for highly efficient hydrogen evolution coupled with biomass electrooxidation. Adv. Mater. 32, 2000455 (2020). - Huang, C. et al. The debut and spreading the landscape for excellent vacancies-promoted electrochemical energy storage of nanoarchitected molybdenum oxides. Mater. Today Energy 30, 101154 (2022).
- Vos, J. G. & Koper, M. T. M. Measurement of competition between oxygen evolution and chlorine evolution using rotating ring-disk electrode voltammetry. J. Electroanal. Chem. 819, 260-268 (2018).
- Shen, W. et al. Defect engineering of layered double hydroxide nanosheets as inorganic photosensitizers for NIR-III photodynamic cancer therapy. Nat. Commun. 13, 3384 (2022).
- Cheng, W. et al. Lattice-strained metal-organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 4, 115-122 (2019).
- Guo, D. et al. TiN@Co5.47N composite material constructed by atomic layer deposition as reliable electrocatalyst for oxygen evolution reaction. Adv. Funct. Mater. 31, 2008511 (2021).
- Yan, H. et al. Holey reduced graphene oxide coupled with an
heterojunction for efficient hydrogen evolution. Adv. Mater. 30, 1704156 (2018). - Lu, M. et al. Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci. Adv. 8, eabq3563 (2022).
- Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in Co-Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329-338 (2019).
- Tan, X. H. et al. Electrochemical etching switches electrocatalytic oxygen evolution pathway of IrOx/
from adsorbate evolution mechanism to lattice-oxygen-mediated mechanism. Small 19, 2303249 (2023). - Wang, F. et al. Activating lattice oxygen in high-entropy LDH for robust and durable water oxidation. Nat. Commun. 14, 6019 (2023).
- Zhang, N. & Yang, C. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647-4671 (2021).
- Niu, Z. et al. Robust Ru-
bifunctional catalysts for all-pH overall water splitting. Adv. Mater. 36, 2310690 (2023). - Zhai, P. et al. Regulating electronic states of nitride/hydroxide to accelerate kinetics for oxygen evolution at large current density. Nat. Commun. 14, 1873 (2023).
- Kuang, Y. et al. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl Acad. Sci. USA 116, 6624-6629 (2019).
- Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264-272 (2023).
- Hausmann, J. N. et al. Is direct seawater splitting economically meaningful? Energy Environ. Sci. 14, 3679-3685 (2021).
- Chen, Z.-J. et al. Acidic enol electrooxidation-coupled hydrogen production with ampere-level current density. Nat. Commun. 14, 4210 (2023).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-46708-8.
http://www.nature.com/reprints
© المؤلف(ون) 2024
المختبر الرئيسي لمواد الكربون في مقاطعة تشجيانغ، كلية الكيمياء وهندسة المواد، جامعة ونتشو، ونتشو 325035، الصين. - البريد الإلكتروني: guody@wzu.edu.cn; xianchen@wzu.edu.cn; شونوانغ@wzu.edu.cn
DOI: https://doi.org/10.1038/s41467-024-46708-8
PMID: https://pubmed.ncbi.nlm.nih.gov/38509067
Publication Date: 2024-03-20
A restricted dynamic surface selfreconstruction toward high-performance of direct seawater oxidation
Accepted: 7 March 2024
Published online: 20 March 2024
(A) Check for updates
Abstract
The development of highly efficient electrocatalysts for direct seawater splitting with bifunctionality for inhibiting anodic oxidation reconstruction and selective oxygen evolution reactions is a major challenge. Herein, we report a direct seawater oxidation electrocatalyst that achieves long-term stability for more than 1000 h at
reconstructed to form hydroxyl metal oxides in the potential range, which is considered the “real catalyst” of
reconstructed stable CoMo-LDH layer relies on electrostatic repulsion to further hydrophobic chlorine, thus achieving selective seawater oxidation. The flow electrolytic cell composed of
Results
Synthesis and characterization
conversion process of selective adsorption of
Structural analysis
and
Electrochemical test
cycles. This decrease can be attributed to the excessively thick shielding layer, which reduces the active area of the catalyst (Supplementary Fig. 6). These results imply that the catalyst interface serves as the catalytic active center. From Fig. 3c, the Tafel slope (
Mechanism analysis of inhibiting CER
structural model of the migration path of
detector (Fig. 4h). The
Restricted dynamic surface self-reconstruction
stronger than the main peak (
Mechanism analysis of seawater oxidation
indicating that there is significant charge transfer favoring the catalytic reaction (Supplementary Fig. 26).
Practical electrolysis applications
respectively. g Raman spectrum of various catalysts tested after washing with water after testing with or without TMAOH. h LSV curves of various samples at different pH values.
that the
Discussion
various environments. e Continuous electrolytic stability. The mass of the
active interface of the catalyst, thus realizing selective catalysis; (2) the ultra-thin
Methods
Materials
nitrogen gas is
Preparation of CoO/CC
Preparation of
plasma. One ALD cycle consists of a
Data availability
References
- Turner, J. A. Sustainable hydrogen production. Science 305, 972-974 (2004).
- Dresp, S. et al. Efficient direct seawater electrolysers using selective alkaline NiFe-LDH as OER catalyst in asymmetric electrolyte feeds. Energy Environ. Sci. 13, 1725-1729 (2020).
- Sun, F. et al. Energy-saving hydrogen production by chlorine-free hybrid seawater splitting coupling hydrazine degradation. Nat. Commun. 12, 4182 (2021).
- Schmidtko, S., Heywood, K. J., Thompson, A. F. & Aoki, S. Multidecadal warming of Antarctic waters. Science 346, 1227-1231 (2014).
- Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: toward boosted and sustained water oxidation catalysis. Joule 5, 2164-2176 (2021).
- Huang, Y. et al. Plasma-induced Mo-doped
with enriched oxygen vacancies for electrocatalytic oxygen evolution in water splitting. Carbon Energy 5, 279 (2022). - Yu, L. et al. Non-noble metal-nitride based electrocatalysts for highperformance alkaline seawater electrolysis. Nat. Commun. 10, 5106-5115 (2019).
- Yu, L. et al. Ultrafast room-temperature synthesis of porous S-doped Ni/Fe (oxy)hydroxide electrodes for oxygen evolution catalysis in seawater splitting. Energy Environ. Sci. 13, 3439-3446 (2020).
- Grimaud, A. et al. Activation of surface oxygen sites on an iridiumbased model catalyst for the oxygen evolution reaction. Nat. Energy 2, 16189 (2016).
- Qiu, Z. et al. Direct observation of active catalyst surface phases and the effect of dynamic self-optimization in NiFe-layered double hydroxides for alkaline water splitting. Energy Environ. Sci. 12, 572-581 (2019).
- Chung, D. Y. et al. Dynamic stability of active sites in hydr(oxy) oxides for the oxygen evolution reaction. Nat. Energy 5, 222-230 (2020).
- Zhao, S. et al. Structural transformation of highly active metal-organic framework electrocatalysts during the oxygen evolution reaction. Nat. Energy 5, 881 (2020).
- Lin, Q. et al. Tuning the interface of
by atomic replacement strategy toward high-performance electrocatalytic oxygen evolution. ACS Nano 16, 15460-15470 (2022). - Chen, F.-Y. et al. Stability challenges of electrocatalytic oxygen evolution reaction: from mechanistic understanding to reactor design. Joule 5, 1704-1731 (2021).
- Zhang, Y. et al. Rapid synthesis of cobalt nitride nanowires: highly efficient and low-cost catalysts for oxygen evolution. Angew. Chem. Int. Ed. 55, 8670-8674 (2016).
- Minguzzi, A. How to improve the lifetime of an electrocatalyst. Nat. Catal. 3, 687-689 (2020).
- Ning, M. et al. Boosting efficient alkaline fresh water and seawater electrolysis via electrochemical reconstruction. Energy Environ. Sci. 15, 3945-3957 (2022).
- Chala, S. A. et al. Tuning dynamically formed active phases and catalytic mechanisms of in situ electrochemically activated layered
double hydroxide for oxygen evolution reaction. ACS Nano 15, 14996-15006 (2021). - Peng, L. et al. Atomic cation-vacancy engineering of NiFe-layered double hydroxides for improved activity and stability towards the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 24612-24619 (2021).
- Ye, S.H. et al. Deeply self-reconstructing
to lowcrystalline with motifs for oxygen evolution reaction. Appl. Catal. B 304, 120986 (2022). - Kang, J. X. et al. Valence oscillation and dynamic active sites in monolayer NiCo hydroxides for water oxidation. Nat. Catal. 4, 1050-1058 (2021).
- Duan, Y. et al. Anodic oxidation enabled cation leaching for promoting surface reconstruction in water oxidation. Angew. Chem. Int. Ed. 60, 7418-7425 (2021).
- Wang, H. Y. et al. In operando identification of geometrical-sitedependent water oxidation activity of spinel
. J. Am. Chem. Soc. 138, 36-39 (2016). - Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212-222 (2021).
- Wang, W. et al. Structural reconstruction of catalysts in electroreduction reaction: identifying, understanding, and manipulating. Adv. Mater. 34, 2110699 (2022).
- Karlsson, R. K. B. et al. Selectivity between oxygen and chlorine evolution in the chlor-alkali and chlorate processes. Chem. Rev. 116, 2982-3028 (2016).
- Kuai, C. et al. Phase segregation reversibility in mixed-metal hydroxide water oxidation catalysts. Nat. Catal. 3, 743-753 (2020).
- Yang, L. et al. Transition metal-based electrocatalysts for seawater oxidation. Adv. Mater. Interfaces 9, 2201486 (2022).
- Wu, Q. et al. A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater. Nat. Commun. 12, 483 (2021).
- Dresp, S. et al. Direct electrolytic splitting of seawater: opportunities and challenges. ACS Energy Lett. 4, 933-942 (2019).
- Song, H. J. et al. Electrocatalytic selective oxygen evolution of carbon-coated
nanoparticles for alkaline seawater electrolysis. ACS Catal. 10, 702-709 (2019). - Guo, D. et al. A tandem interfaced
composite fabricated by atomic layer deposition as efficient HER electrocatalyst. Small 18, 2201896 (2022). - Cao, L. et al. Atomically dispersed iron hydroxide anchored on Pt for preferential oxidation of CO in
. Nature 565, 631-635 (2019). - Meng, L. et al. Atomic layer deposition triggered Fe-In-S cluster and gradient energy band in ZnInS photoanode for improved oxygen evolution reaction. Nat. Commun. 12, 5247 (2021).
- Guo, D. et al. Strategic atomic layer deposition and electrospinning of cobalt sulfide/nitride composite as efficient bifunctional electrocatalysts for overall water splitting. Small 16, 2002432 (2020).
- Mao, S. et al. High-performance bi-functional electrocatalysts of 3D crumpled graphene-cobalt oxide nanohybrids for oxygen reduction and evolution reactions. Energy Environ. Sci. 7, 609-616 (2014).
- Tian, Y. et al. Engineering crystallinity and oxygen vacancies of Co(II) oxide nanosheets for high performance and robust rechargeable Zn-Air batteries. Adv. Funct. Mater. 31, 2101239 (2021).
- Ling, T. et al. Engineering surface atomic structure of single-crystal cobalt (II) oxide nanorods for superior electrocatalysis. Nat. Commun. 7, 12876 (2016).
- Wu, L. et al. Boron-modified cobalt iron layered double hydroxides for high efficiency seawater oxidation. Nano Energy 83, 105838 (2021).
- Yang, G. et al. Interfacial engineering of
heterojunction for highly efficient hydrogen evolution coupled with biomass electrooxidation. Adv. Mater. 32, 2000455 (2020). - Huang, C. et al. The debut and spreading the landscape for excellent vacancies-promoted electrochemical energy storage of nanoarchitected molybdenum oxides. Mater. Today Energy 30, 101154 (2022).
- Vos, J. G. & Koper, M. T. M. Measurement of competition between oxygen evolution and chlorine evolution using rotating ring-disk electrode voltammetry. J. Electroanal. Chem. 819, 260-268 (2018).
- Shen, W. et al. Defect engineering of layered double hydroxide nanosheets as inorganic photosensitizers for NIR-III photodynamic cancer therapy. Nat. Commun. 13, 3384 (2022).
- Cheng, W. et al. Lattice-strained metal-organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 4, 115-122 (2019).
- Guo, D. et al. TiN@Co5.47N composite material constructed by atomic layer deposition as reliable electrocatalyst for oxygen evolution reaction. Adv. Funct. Mater. 31, 2008511 (2021).
- Yan, H. et al. Holey reduced graphene oxide coupled with an
heterojunction for efficient hydrogen evolution. Adv. Mater. 30, 1704156 (2018). - Lu, M. et al. Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci. Adv. 8, eabq3563 (2022).
- Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in Co-Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329-338 (2019).
- Tan, X. H. et al. Electrochemical etching switches electrocatalytic oxygen evolution pathway of IrOx/
from adsorbate evolution mechanism to lattice-oxygen-mediated mechanism. Small 19, 2303249 (2023). - Wang, F. et al. Activating lattice oxygen in high-entropy LDH for robust and durable water oxidation. Nat. Commun. 14, 6019 (2023).
- Zhang, N. & Yang, C. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647-4671 (2021).
- Niu, Z. et al. Robust Ru-
bifunctional catalysts for all-pH overall water splitting. Adv. Mater. 36, 2310690 (2023). - Zhai, P. et al. Regulating electronic states of nitride/hydroxide to accelerate kinetics for oxygen evolution at large current density. Nat. Commun. 14, 1873 (2023).
- Kuang, Y. et al. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl Acad. Sci. USA 116, 6624-6629 (2019).
- Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264-272 (2023).
- Hausmann, J. N. et al. Is direct seawater splitting economically meaningful? Energy Environ. Sci. 14, 3679-3685 (2021).
- Chen, Z.-J. et al. Acidic enol electrooxidation-coupled hydrogen production with ampere-level current density. Nat. Commun. 14, 4210 (2023).
Acknowledgements
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-46708-8.
http://www.nature.com/reprints
© The Author(s) 2024
Key Laboratory of Carbon Materials of Zhejiang Province, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, China.