إعادة تصميم حاسوبية لإنزيم هيدرولاز لتفكيك PET بشكل شبه كامل عند تحميل عالي من المواد الصلبة ذو صلة صناعية Computational redesign of a hydrolase for nearly complete PET depolymerization at industrially relevant high-solids loading
لقد ظهرت إعادة تدوير البلاستيك باستخدام التكنولوجيا الحيوية كخيار مناسب لمعالجة أزمة التلوث. تم تحقيق تقدم كبير في التحلل البيولوجي للبولي (إيثيلين تيريفثاليت) (PET) من خلال استخدام نوع LCC، مما يسمح بـالتحويل على مستوى صناعي. على الرغم من الإنجازات، فإن تطبيقاته قد تأثرت بـ 10% المتبقية من PET غير القابل للتحلل. هنا، نتناول التحديات الحالية من خلال استخدام استراتيجية حسابية لتصميم هيدراز من البكتيريا HR29. النسخة المعاد تصميمها، TurboPETase، تتفوق على غيرها من الهيدرازات المعروفة لـ PET. يتم تحقيق تحلل بوليمري شبه كامل في 8 ساعات عند تحميل المواد الصلبة بـتشير التحليلات الحركية والهيكلية إلى أن الأداء المحسن قد يُعزى إلى وجود تجويف ربط PET أكثر مرونة، مما يسهل استهداف مواقع هجوم أكثر تحديدًا. بشكل جماعي، تشكل نتائجنا تقدمًا كبيرًا في فهم وهندسة الهيدرازات البوليستر القابلة للتطبيق صناعيًا، وتوفر إرشادات للجهود المستقبلية على أنواع البوليمرات الأخرى.
تم تكريس جهود كبيرة لاكتشاف وهندسة إنزيمات تحلل PET على مدى العقدين الماضيين، مما ساهم في تطوير تحلل PET الحيوي من الكشف عن كميات ضئيلة من المنتجات المنبعثة إلى تحقيق تحويل عالي.عند مواجهة التحديات التي تطرحها إعادة تدوير PET باستخدام الإنزيمات، يجب أخذ المعايير الرئيسية بعين الاعتبار لتحقيق عملية صناعية قابلة للتطبيق اقتصاديًا، وخاصة تحميل المواد الصلبة. ) وعائد المنتج ( . ومع ذلك، فإن معظم الدراسات تحقق عوائد تحلل كبيرة عند تحميل المواد الصلبة أقل من وهو ما يقل بكثير عن المستوى ذي الصلة بالصناعةعند زيادة تحميل المواد الصلبة إلى مستوى عالٍ، يُلاحظ عادةً انخفاض في معدل التحلل المائي والتحويل (يشار إليه باسم “أثر المواد الصلبة”)، كما هو موضح في تفاعلات غير متجانسة أخرى مثل عملية تحويل الكتلة الحيوية..
تم تحقيق تقدم مع نوع معدل من LCC (LCC ) التي عرضت تحلل البوليمر للمواد البلاستيكية المعالجة مسبقًا من PET عند تحميل المواد الصلبة بشكل ذي صلة صناعيًا. ومع ذلك، تبقى 10% من PET غير القابلة للتحلل ووصلت إلى مستوى عالٍ من البلوريةبسبب الشيخوخة الفيزيائية، مما يعيق إعادة الاستخدام الفوري لتحلل PET. أشار تقرير صادر عن الجمعية الوطنية لموارد حاويات PET إلى أن زجاجات PET التي تم جمعها من المستهلكين لإعادة التدوير بلغت 800 كيلوطن في الولايات المتحدة.، وسيؤدي ذلك إلى توليد أكثر من 80 كيلوطن من نفايات PET غير القابلة للتحلل سنويًا إذا تم استخدام استراتيجية التحلل الحيوي الحالية، مما يشكل تهديدًا كبيرًا لاقتصاد إعادة تدوير PET. زيادة مدى التحويل من إلىسيقوم بتخفيض الحد الأدنى لسعر البيع (MSP) لـ TPA بمقدار (الولايات المتحدة وفقًا لنموذج TEA المقترح من قبل مختبر الطاقة المتجددة الوطني في الولايات المتحدة؛ وبالتالي، فإن مستوى التحويل هو العامل الأكبر الذي يؤثر على MSP في عملية التحلل البوليمري بعد تحميل المواد الصلبة.لقد أثارت كل من القضايا البيئية والاجتماعية الاقتصادية اهتمامًا كبيرًا في تحسين مستوى التدهور لتسريع الانتقال نحو الاقتصاد الدائري.
وفقًا للنتائج الحركية لتبلور PET (الشكل التكميلي 1)، يمكن تقليل الشيخوخة الفيزيائية بشكل كبير عن طريق خفض درجة حرارة التفاعل، ولكن كفاءة التحفيز لإنزيمات هيدرولاز PET الحرارية تتعرض للتضحية في الوقت نفسه.، مما يزيد من الطلب على المحفزات الحيوية ذات كفاءة التحلل العالي عند درجات حرارة منخفضة لتشوه سلاسل PET غير المتبلورة.
شهدت السنوات القليلة الماضية تقدمًا ملحوظًا في تخصيص الإنزيمات الطبيعية من خلال استراتيجيات إعادة التصميم الحاسوبية.استلهمنا من الإنجازات في الذكاء الاصطناعي لمعالجة مشهد ملاءمة البروتين لاستكشاف المعلومات التطورية المخفية، حيث استخدمنا استراتيجية حسابية تتضمن نموذج لغة البروتين وخوارزميات قائمة على مجالات القوة لتصميم إنزيمات هيدراز PET تتمتع بتوازن بين الثبات الحراري والقدرة التحليلية. وقد تفوق المتغير المعاد تصميمه (TurboPETase) الناتج عن هذه الحملة على أكثر إنزيمات هيدراز PET كفاءة المعترف بها حاليًا في هذا المجال.بهرPETaseفاست بيتيزهوتبيتيزديبوبيتيزCaPETase، و PES-H1 ) على مدى مجموعة من درجات الحرارة ( ). الأداء الاستثنائي في التحلل الذي توفره TurboPETase سمح بتحلل شبه كامل لزجاجات PET بعد الاستهلاك في 8 ساعات عند تحميل ركيزة عالي ذي صلة صناعيًا من بمعدل إنتاج أقصى يبلغ 61.3 جرام من PET المحلل، معالجة التحدي المتعلق بالنفايات المتبقية من PET غير القابلة للتحلل.
النتائج
إعادة تصميم حسابي لإنزيم هيدراز البولي إيثيلين تيريفثاليت الفعال
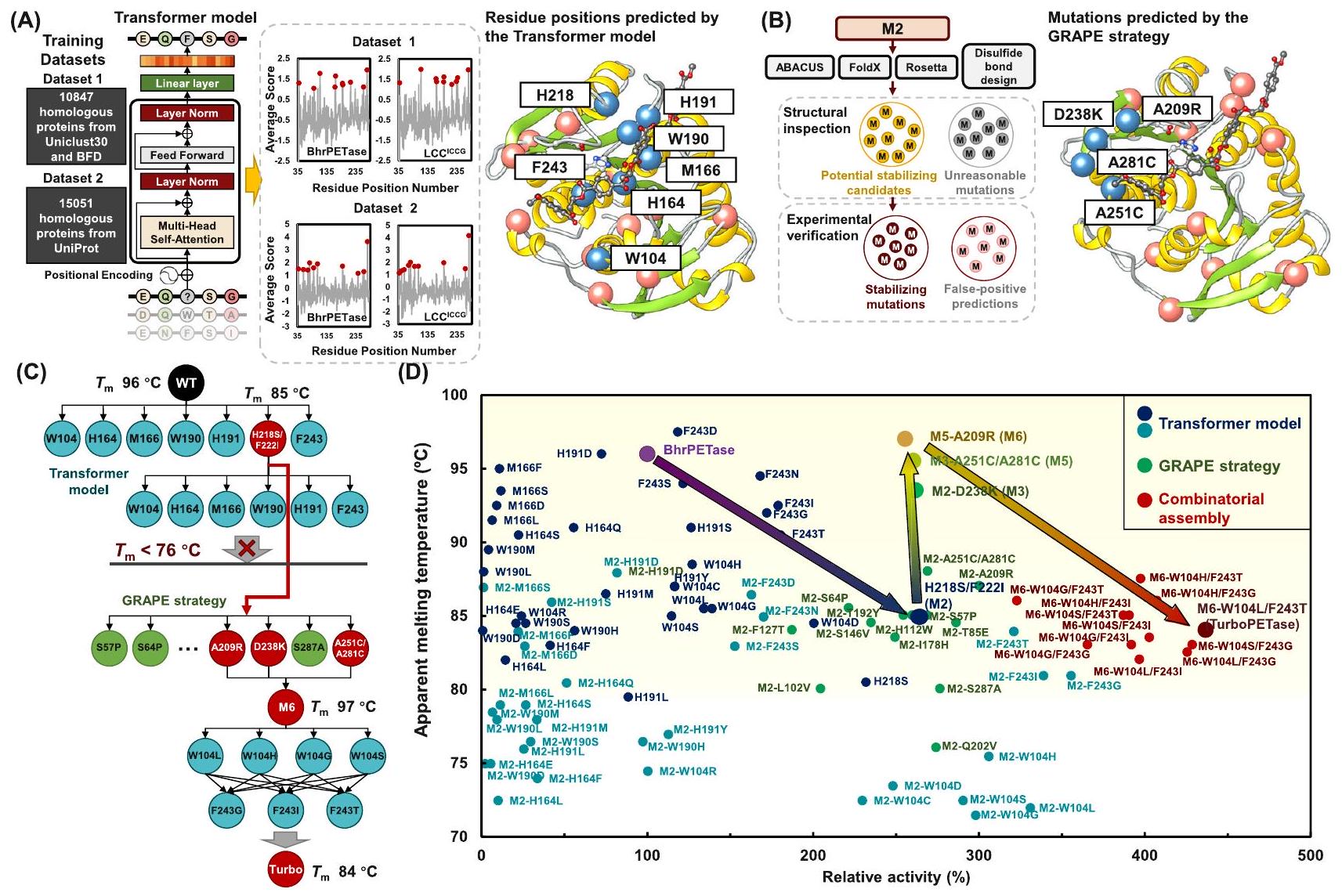
تنتمي إنزيمات التحلل المائي للبولي إيثيلين تيرفثالات (PET) إلى عائلة إنزيمات التحلل المائي السيرينية، وهي مجموعة منتشرة على نطاق واسع معروفة بخصوصيتها المنخفضة نسبيًا تجاه الركائز. يُعتقد أن وظيفة التحلل لإنزيمات PET كانت موجودة مسبقًا كوظيفة غير محددة، ثم تطورت لتصبح وظيفة أساسية.. نظرًا لمعرفتنا المحدودة بكيفية ترميز تسلسل الوظائف التحفيزية في إنزيمات تحلل البوليمر معالجة التحدي من خلال استغلال إعادة التصميم الحسابية المعتمدة على الفيزياء وطرق التصميم العقلاني هي مهمة صعبة. بدلاً من ذلك، تتضمن الطريقة الناجحة استخدام نماذج التعلم العميق لرسم العملية من تسلسل البروتين إلى الوظيفة، كما تم إثباته في العديد من الحالات.. يمكن أن تلتقط هذه النماذج المعلومات المخفية التي تشير إلى تحسين تحلل البوليمر على طول مسار التطور من الملاءمة النسبية لمتغيرات البروتين. لهذا الغرض، استخدمنا نموذج لغة تم تدريبه على مجموعتين من البيانات تضمان حوالي 26,000 تسلسل متجانس من إنزيمات هيدراز PET عبر التطور، للتنبؤ باحتمالية تغير الأحماض الأمينية من المشهد التطوري (الشكل 1A). لاختيار إنزيم النموذج، قمنا بمقارنة نشاط LCC، LCCبروتين BhrPETase وFastPETase وHotPETase باستخدام أفلام PET غير المتبلورة (Gf-PET، من المورد Goodfellow) عبر مجموعة من درجات الحرارة (الشكل التوضيحي التكميلي 2). يشارك BhrPETase هوية تسلسلية عالية منمع LCC، في حين أن LCCيمثل نوعًا من LCC يتميز بأربعة بدائل من الأحماض الأمينية (F243I / D238C / S283C / Y127G). كل من FastPETase و HotPETase هما مشتقات تم هندستها من IsPETase (الشكل التكميلي 3). فيبهرPETase و LCCأظهرت أعلى أداء تحفيزي من بين جميع إنزيمات هيدرولاز PET ودرجات الحرارة المختبرة وتم اختيارها كمدخلات. تم استخدام مشفر Transformer لمعالجة تسلسلات الأحماض الأمينية المدخلة مع تضمين الموقع المطلق. تم فرز مواقع البقايا حسب متوسط لوغاريتمات 19 طفرة تم تعيينها للأحماض الأمينية من النوع البري في كل موقع. تم اختيار أعلى عشرة مواقع للأحماض الأمينية ذات أعلى درجات متوسطة من كل نموذج. بعد إزالة مواقع الأحماض الأمينية المكررة، تم الحصول على 18 موقعًا للأحماض الأمينية حيث كانت بقايا النوع البري تتناسب بشكل أقل من البدائل المحتملة (الجدول التكميلي 1).
وفقًا للهياكل البلورية لبروتين BhrPETase (رمز PDB: 7EOA) و LCC S165A في مركب مع MHET (رمز PDB: 7VVE )، تم اقتراح أن 7 من 18 موضعًا للأحماض الأمينية المتولدة (W104، H164، M166، W190، H191، H218، وF/I243) تكون مدمجة في تجويف ربط PET. نظرًا لدرجة الاستقرار الحراري الأعلى نسبيًا لـ
قمنا بتعريض BhrPETase لسبع مواضع على هذا الإنزيم لتوليد 34 متغيرًا. أفاد تشين وآخرون أن الطفرات المزدوجة Ser214/Ile218 لعدة إنزيمات متجانسة لـ IsPETase أظهرت نشاطًا معززًا في تحلل PET (بزيادة لا تقل عن 1.3 مرة) ولكنها انخفضت بشكل كبير.القيم (بنحو. نظرًا لأن His 218 (المقابل لـ Ser214 في IsPETase) كان متورطًا في مرشحينا المتوقعين، تم استكشاف الأداء التحللي لمتغير H218S/F222I أيضًا (الجدول التكميلي 2). من بين المتغيرات، BhrPETase (المشار إليه باسم BhrPETase M2) أدى إلى أعلى تحسينات مع زيادة بمقدار 1.7 مرة في النشاط الهيدروليتيكي لـ PET عند ، مما يجعل هذه السلالة القالب الجديد لتراكم الطفرات في الجولات الثانية في المواقع الستة المتبقية. خلال مرحلة التراكم الثانية، أظهرت الطفرات فقط في مواقع W104 و F243 (W104L، W104S، W104H، W104G، F243I، F243T، و F243G) على M2 أنشطة تحليل مائي محسّنة (عن طريق إلى )، على الرغم من الانخفاض الكبير في استقرار درجة الحرارة (الجدول التكميلي 3).
تشير البيانات التجريبية إلى أنأعلى منضروري لطول عمر المحفز. ومن ثم، فإن إنزيم الهيدراز PET مع على الأقل أكثر منيفضل من أجل تحلل PET الفعال. ومن الجدير بالذكر أن المتغير M2 أظهر درجة انصهار تبلغالذي كانأقل من ذلك من إنزيم النوع البري. أدت الطفرات النشطة في مواضع W104 على M2 إلى تقليل الاستقرار بشكل أكبر، معقيم تتراوح منإلىإن الانخفاض غير القابل للتجاهل في الاستقرار حد من المزيد من التوليف، وكان من الضروري إدخال طفرات تعويضية أولاً لتقليل الآثار الضارة. لذلك، طبقنا استراتيجيتنا السابقة التي وضعناها والمعروفة باسم GRAPE.التي تستخدم أربعة خوارزميات تكاملية، وهي، Fold (دالة طاقة حقل القوة)، روزيتا_كارتيسيان_ddg (دالة طاقة حقل القوة)، ABACUS (دالة الطاقة الإحصائية) و DDD (دالة طاقة حقل القوة) ، لتصميم طفرات مستقرة لتعويض وتخفيف الطفرات غير المستقرة (الشكل 1B). بعد التحقق التجريبي، أسفرت 3 طفرات مفيدة (A209R، D238K، وA251C-A281C) عن تحسين الاستقرار الحراري دون التأثير على النشاط (الجدول التكميلي 4). أضفنا الطفرات المستقرة إلى متغير M2 باستخدام استراتيجية دمج تدريجية وأسفرت عن متغير BhrPETase M6 (BhrPETase )، الذي أظهر درجة انصهار مستعادة من دون التضحية بالنشاط. بعد ذلك، تم تجميع الطفرات النشطة في مواضع W104 و F243 بشكل تركيبي وتراكمها على المتغير M6 المستقر حرارياً، مما أدى إلى توليد 12 متغيراً جديداً (الشكل 1C والجدول التكميلي 5). بعد النظر في كل من أداء التحلل الحراري والثبات الحراري، كان أفضل متغير تركيبي هو BhrPETase. A209R/D238K/A251C/A281C/W104L/F243T (المشار إليه باسم TurboPETase) تم اختياره مع من وتحسين بمقدار 3.4 مرة في النشاط المحدد لـ PET تجاه أفلام GF-PET مقارنة بـ BhrPETase من النوع البري (الشكل 1D). في السيناريوهات الصناعية، يتم عادةً إجراء التحلل المائي الإنزيمي في الماء بدلاً من نظام مخفف مخصص لتبسيط المعالجة اللاحقة. تماشيًا مع التفضيلات الصناعية، قمنا أيضًا بتقييم 12 متغيرًا تحت تركيز منخفض من المخفف. كشفت النتائج أنه باستثناء احتفظت جميع المتغيرات إلى حد كبير بنشاطها التحللي في محلول فوسفات البوتاسيوم بتركيز 100 مللي مول (الشكل التوضيحي 4 والجدول التوضيحي 6). كان TurboPETase يتفوق باستمرار على المتغيرات الأخرى تحت تركيزات منخفضة وعالية من المحلول، مما يجعله المتغير الأكثر فعالية لمزيد من التحقيق.
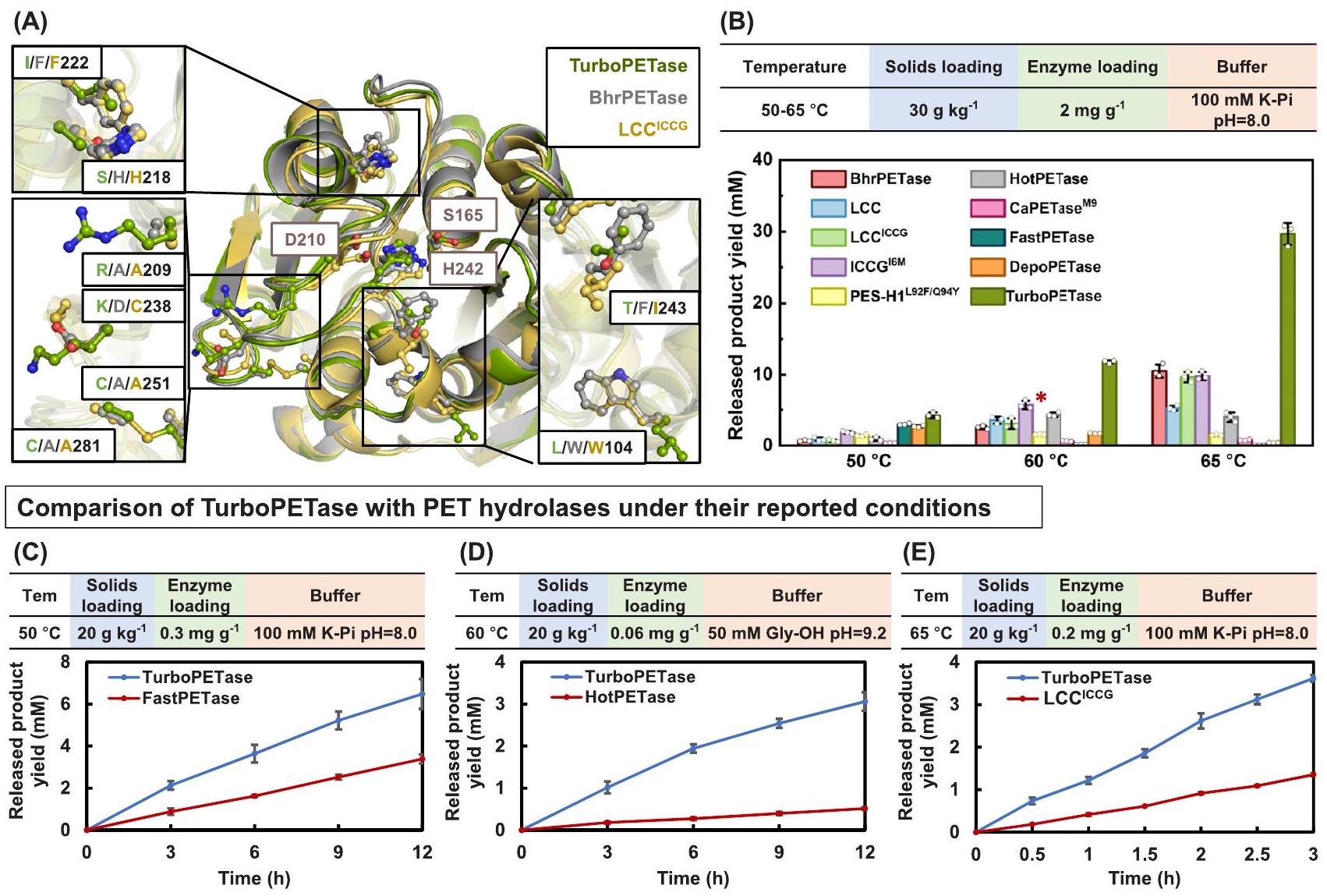
تم تقييم أداء التحلل البوليمري لـ TurboPETase بعد ذلك عبر نطاق درجات حرارة فيما يتعلق بالهيدرازات الأخرى للبولي إيثيلين تيريفثاليت (الشكل 2A وB، والشكل التوضيحي 5). عند أظهرت TurboPETase أعلى معدل تحلل إجمالي بين جميع إنزيمات تحلل PET ودرجات الحرارة المختبرة، حيث أطلقت 29.66 مللي مول من المنتجات (مجموع BHET وMHET وTPA) في 3 ساعات. نظراء مثل BhrPETase وLCC وLCCICCG، و PES-H1 التي تظهر أداءً عاليًا في التحلل عند درجات حرارة مرتفعة، جعلت النشاط المائي أقل بمقدار 1.8 و 4.7 و 2.1 و 2.0 و 19 مرة مقارنةً بـ TurboPETase، على التوالي. عند درجات الحرارة غير المثلى، TurboPETase
الشكل 1 | تمثيل تخطيطي لإعادة تصميم إنزيمات هيدروكسي PET باستخدام الطريقة الحاسوبية الهجينة. تم التنبؤ بالطفرات المحتملة المفيدة باستخدام نموذج المحول المدرب على مجموعتين من البيانات (اللوحة اليسرى). تم اختيار أفضل عشرة مواقع للأحماض الأمينية المتوقعة (دوائر حمراء). أدى إزالة المواقع المكررة إلى 18 موقعًا من البقايا، من بينها تم اقتراح 7 مواقع لتكون موجودة في موقع ارتباط PET (اللوحة اليمنى).تظهر ذرات هذه المواقع من الأحماض الأمينية ككرات زرقاء، بينما تظهر المواقع الـ 11 المتبقية ككرات مرجانية. يتم تمثيل PET والثلاثي الحفاز كتمثيلات كروية وعصوية. ب تمثيل تخطيطي لاستراتيجية GRAPE. تم استخدام أربعة خوارزميات للتنبؤ بالطفرات المثبتة (اللوحة اليسرى). بعد التصفية الهيكلية، تم التحقق من المرشحين المحتملين المثبتين تجريبيًا (اللوحة اليمنى).تظهر ذرات الطفرات ذات الاستقرار الحراري المحسن ككرات زرقاء، بينما تظهر الطفرات الأخرى التي تم التحقق منها تجريبياً ككرات مرجانية. ج تمثيل تخطيطي لخطوة التراكم. تم إضافة الطفرات في 7 مواقع من البقايا الموجودة في موقع ارتباط PET إلى النوع البري. إنزيم (BhrPETase)، مما أدى إلى أفضل نتيجة BhrPETase (M2). إضافة الطفرات في المواقع الستة المتبقية إلى M2 أدت إلى متغيرات نشطة ذات استقرار منخفض بشكل ملحوظ، وبالتالي تم استخدام استراتيجية GRAPE. تم دمج الطفرات المثبتة في M2 وأسفرت عن المتغير المستقر حرارياً M6، الذي يمكنه تعويض الطفرات غير المستقرة في خطوات التراكم اللاحقة. تم تجميع الطفرات النشطة في مواقع W104 وF243 بشكل تركيبي على M6، مما أدى إلى الطفرة النهائية TurboPETase. د الاستقرار الحراري والنشاط النسبي لتفكيك PET لـ BhrPETase والمتغيرات. تم توحيد الأنشطة وفقًا لـ BhrPETase. البيانات من تجربة مستقلة واحدة. الطفرات التي تم التنبؤ بها بواسطة نموذج Transformer واستراتيجية GRAPE ملونة باللون الأزرق والأخضر، على التوالي. الطفرات المزدوجة في مواقع W104 وF243 على M6 موضحة باللون الأحمر. الأسهم تمثل مسار التراكم. المنطقة الصفراء تمثل نطاق درجة حرارة الانصهار المثالي لإنزيمات تحلل PET لتمكين التحلل الفعال.. تجاوزت باستمرار إنزيمات الهيدراز الأخرى للبولي إيثيلين تيريفثاليت، على الرغم من انخفاض النشاط التحللي. عند درجة الحرارة المثلى لـ HotPETase و CaPETaseمنأنتج TurboPETase 11.73 مللي مول من المنتجات الأحادية في 3 ساعات، بينما أنتج HotPETase و CaPETase أنتجت 4.32 مللي مول و 0.60 مللي مول من المونومرات، والتي كانت أقل بمقدار 1.7 و 18 مرة مقارنة بـ TurboPETase. عند التعرض لـ سجل TurboPETase كفاءات تحليلية تفوق تلك الخاصة بـ FastPETase و DepoPETase بـ و ، على التوالي. في ضوء تركيزات الإنزيمات المنخفضة المستخدمة في بعض التقارير، قمنا بإعادة معايرة تفاعلاتنا لتعكس هذه الظروف لضمان مقارنة أكثر عدلاً (الشكل 2C-E). تحت ظروف التفاعل المبلغ عنها، كانت معدلات تفاعل LCCكانت قيم HotPETase و FastPETase أقل بمقدار 1.8 و 4.9 و 1.0 مرة، على التوالي، مقارنةً بـ TurboPETase. مؤخرًا، تم إجراء تجارب مماثلة لتقييم أداء التحلل لـ FastPETase و HotPETase و PES-H1. و LCC تحت الظروف الصناعيةأظهرت نتائجهم أن HotPETase أظهر نشاطًا نوعيًا أعلى مقارنةً بـ PES-H1.عند تحميل منخفض للركيزة. ومع ذلك، كما أن تركيزات الركيزة زيادة إلى و كان تحويل PET لـ HotPETase أقل بكثير من تلك الخاصة بـ PES-H1. يُقترح أن تكون هذه الكفاءة المنخفضة عند تحميل الركيزة العالي ناتجة عن الاستقرار الحراري المحدود وخصائص التحفيز الأخرى لـ HotPETase، مثل تثبيط المنتج. على الرغم من الأداء الجيد لـ PES-H1تحت ظروف ذات صلة صناعية، كانت عملية التحلل النهائي لا تزال أقل من تلك التي حققتها LCC.نظرًا للتحسينات المبلغ عنها في PES-H1في محلول فوسفات البوتاسيوم 1 م، قمنا أيضًا بمقارنة TurboPETase مع PES-H1 تحت تركيز عازل مرتفع، مع استمرار TurboPETase في التفوق، مما ينتج ما يصل إلى 2.5 مرة من منتجات التحلل لـ PES-H1 / فيج (الشكل التوضيحي التكميلي 6). لتقييم استقرار TurboPETase على المدى الطويل، قمنا بتمديد وقت التفاعل. أظهرت النتائج أداءً مستمرًا للإنزيم TurboPETase عند درجات حرارة مرتفعة، حيث أنتج 130 مللي مول من المنتجات الأحادية القابلة للذوبان على مدار 12 ساعة عندبينما المنتجات المفرج عنها مع BhrPETase و LCC و LCCكانت أقل بكثير (الشكل التوضيحي 7). تسلط نتائج التحلل الضوء على التفوق الكبير في التحلل المائي
الشكل 2 | مقارنة أداء التحلل البوليمري لـ TurboPETase مع إنزيمات الهيدرولاز المعروفة الأخرى لـ PET. A موقع الطفرات المفيدة في TurboPETase ونظائرها. TurboPETase وBhrPETase وLCCتظهر بالأخضر والرمادي والأصفر، على التوالي. يتم عرض البقايا في تمثيلات الكرة والعصا. ب مقارنة النشاط الهيدروليتيكي لـ TurboPETase ومواد هيدرولاز PET الأخرى تجاه أفلام Gf-PET عند درجات حرارة تتراوح من 50 إلىيستخدمتحميل المواد الصلبةتحميل الإنزيم في محلول فوسفات البوتاسيوم 100 مللي مول، pH 8.0. يوضح الرسم البياني الشريطي متوسط التحلل بعد 3 ساعات من التفاعل. تُعرض البيانات كمتوسط. س.د. ( التجارب المستقلة بيولوجيًا). تمثل الدوائر الأرقام الفردية. *في التحقيق الذي أجراه أرنال وآخرون.كفاءة التحلل البوليمري لـ PESتجاوز بشكل ملحوظ أداء HotPETase، ومع ذلك ظل أداؤه أقل من LCC.، تحت أحمال الركيزة ذات الصلة الصناعية من و . وقت C
تفاعلات دورة TurboPETase و FastPETase تجاه أفلام Gf-PET عندتحت ظروف التفاعل المبلغ عنها ج تحميل الإنزيمتحميل المواد الصلبة، 100 مللي مول من محلول فوسفات البوتاسيوم، pH 8.0). يتم تقديم البيانات كمتوسط س.د. ( تجارب مستقلة بيولوجيًا).تفاعلات زمنية لـ TurboPETase و HotPETase تجاه أفلام Gf-PET عندتحت ظروف التفاعل المبلغ عنهاتحميل الإنزيمتحميل المواد الصلبة، 50 مللي مول من محلول غليسين-NaOH، pH 9.2). يتم تقديم البيانات كمتوسط س.د. ( تجارب مستقلة بيولوجيًا). تفاعلات زمنية لإنزيم TurboPETase وLCCنحو أفلام Gf-PET فيباستخدام تحميل الإنزيم المبلغ عنه في محلول فوسفات البوتاسيوم بتركيز 100 مللي مولار ). كانت نسبة المواد الصلبة تُعرض البيانات كمتوسط س.د. ( تجارب مستقلة بيولوجيًا). يتم توفير بيانات المصدر كملف بيانات مصدر. أداء TurboPETase عبر درجات حرارة مختلفة وظروف تفاعل أخرى تم اختبارها.
نهج مايكلس-منتن والتحليل الهيكلي
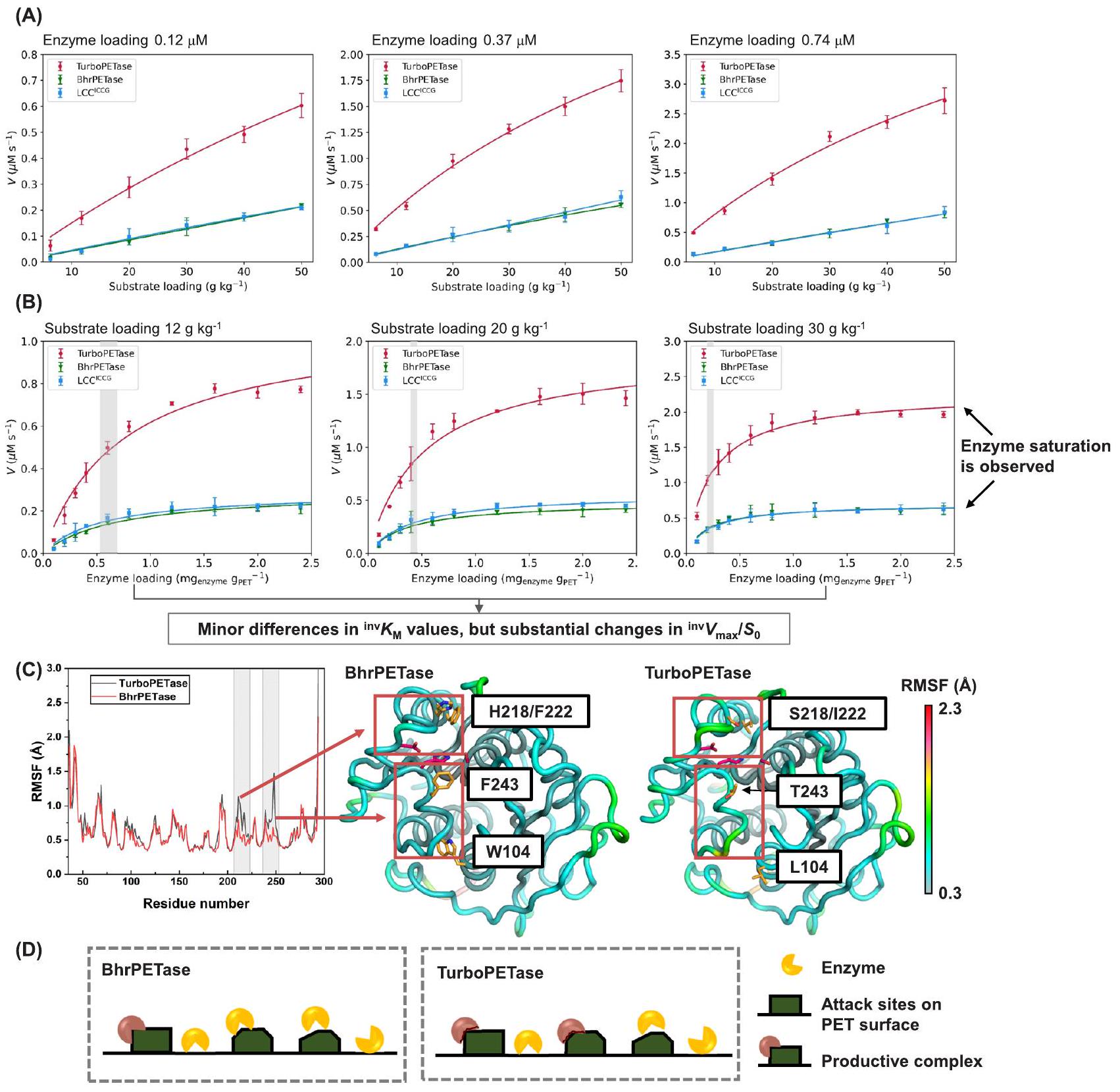
لتفسير أداء التحلل المائي المحسن، تم تحليل الحركيات من خلال نموذج مايكلس-منتن التقليدي ونموذج مايكلس-منتن العكسي. كما هو موضح في الشكل 3A، أظهرت المنحنيات علاقات قريبة من الخطية لمعدل البداية وتحميل الركيزة تحت جميع الظروف لـ TurboPETase وBhrPETase وLCC.لم يتم ملاحظة سلوك التشبع التقليدي لأنه حتى أدنى تركيزات الإنزيم المستخدمة هنا ) كانت مرتفعة جدًا بحيث لا يكون النهج التقليدي صالحًا، مما يشير إلى معدلات التفكك السريعة جدًا للإنزيمات من سطح PET. بالإضافة إلى ذلك، أجرينا مقارنة حركية للركائز القابلة للذوبان (MHET و ) كما هو موضح في الشكل التوضيحي التكميلي 8 والجدول التكميلي 7. أظهر الكفاءة التحفيزية لـ TurboPETase تجاه MHET زيادة متواضعة فقط مقارنةً بـ PET (مع زيادة بنسبة 32%)، مما يشير إلى أنه قد تكون هناك عوامل أخرى، ربما زيادة الامتصاص على السطح، تساهم في زيادة كفاءة تحلل PET. وعلى العكس، فإن الانخفاض الطفيف في “TurboPETaseلـNPB مصحوبًا بانخفاض في قوة الارتباط، مما يشير إلى تغييرات محتملة في مجال ارتباط الركيزة، مما يجعله أقل ملاءمة لتفاعلات الجزيئات الصغيرة الأخرى.
على الرغم من أن التحلل المائي للبولي إيثيلين تيريفثاليت لم يستطع تلبية المعايير للنهج التقليدي، إلا أن نموذج مايكلس-منتن العكسي كان أكثر ملاءمة. لقد تم استخدام معادلة مايكلس-منتن العكسية بنجاح لدراسة حركيات الإنزيمات غير المتجانسة مثل السليلوزات، والتي يمكن من خلالها تقدير الفعالية التحفيزية ضد مواقع الهجوم المتاحة على سطح البوليمر.كعملية تآكل سطحية نموذجية، فإن التحلل المائي الإنزيمي للبولي إيثيلين تيريفثاليت (PET) يكاد يكون غير قادر على اختراق النواة الداخلية للبوليmer، مما يؤدي إلى الوصول إلى عدد محدود من الروابط الاسترية السطحية (المعروفة أيضًا بمواقع الهجوم) حتى لو كانت الإنزيمات بكمية كبيرة. وبالتالي يحدث التشبع عندما تصبح جميع المواقع على السطح مشغولة، وتتكاثر جزيئات الإنزيم الزائدة في المذيب.يجب ملاحظة أن ليس جميع مواقع الامتزاز قادرة على التحويل الحفزي، كما أن الامتزاز غير المحدد يمثل أيضًا نسبة كبيرة.قمنا بقياس تركيزات الإنزيمات الحرة،وقام بتحويله إلى تغطية الركيزة،لحساب إجمالي امتصاص الإنزيمات على سطح PET (الشكل التكميلي 9 و
الشكل 3 | المعلمات الحركية والتحليل الهيكلي لـ TurboPETase. مخططات مايكلس-منتن التقليدية والعكسية لـ TurboPETase و BhrPETase وفيفي محلول فوسفات البوتاسيوم 100 مللي مولار، pH 8.0. تُعرض البيانات كمتوسط س.د. ( تجارب مستقلة بيولوجيًا). يتم توفير بيانات المصدر كملف بيانات مصدر. قيم C RMSF (في ) للجميع ذرات BhrPETase و TurboPETase خلال محاكاة الديناميكا الجزيئية، مما يشير إلى تغييرات عالمية في مرونة البروتين، خاصةً في تجويف ارتباط PET. تم تمثيل الطفرات H218S/F222I وW104L وF243T كذرات برتقالية، بينما تم تمثيل الثلاثي الحفاز كذرات وردية ساخنة. D مخطط تخطيطي لعمليات تحلل PET. مقارنةً بالإنزيم البري، قد يكون TurboPETase أكثر تنوعًا بسبب زيادة المرونة على طول تجويف ارتباط البروتين PET، مما يسمح له بالهجوم على هياكل سطحية مختلفة.
الجدول التكميلي 8).يمثل الإنزيمات المرتبطة المشبعة (الظاهرة)، بينماهو ثابت التفكك. عند تركيزات إنزيم منخفضة، أظهر TurboPETase امتصاصًا أعلى على سطح PET مقارنةً بنظرائه. ومع ذلك، عند الوصول إلى التشبع، كانت سعات الامتصاص القصوى متشابهة ( ) لوحظت، مما يبرز إمكانيات الربط العامة المتسقة عبر الإنزيمات. يمكن توسيع مقارنات هذه الإنزيمات من خلال النظر في التغيرات المحددة في معلمات مايكلس-منتن العكسية، والتي يمكن أن تعكس القدرة على الربط بمواقع الهجوم على سطح PET. كما هو موضح في الشكل 3B والجدول 1، أظهرت القيم اختلافات طفيفة بين TurboPETase وBhrPETase وLCC، خاصة للقيم عندتحميل الركيزة، مما يشير إلى أن السعة الامتصاصية المحددة لـ TurboPETase عند مواقع الهجوم على لم يتعرض سطح PET للتلف. من الجدير بالذكر أن TurboPETase أظهر زيادة بمقدار 2.1 مرة في مقارنةً بـ BhrPETase و LCC المعامل العكسي، أقصى سرعة تفاعل لكل موقع تفاعلي متاح )، يحدد المعدل عندما تكون جميع مواقع الهجوم مغطاة بالإنزيم . نظرًا لعدم وجود اختلافات جوهرية في و تمت ملاحظة قيم مرتفعة بين الإنزيمات،قد تشير القيم إلى استهداف موسع لـ TurboPETase نحو مواقع هجوم محددة عندما حافظت الإنزيمات على مستوى امتصاص عام مستقر. وبالتالي، افترضنا أن الأداء المحسن في التحلل البوليمري لـ TurboPETase قد يعتمد، على الأقل جزئيًا، على القدرة المعززة على مهاجمة طيف أوسع من مواقع الهجوم المحددة التي يمكن أن تتحلل لتشكيل مركب منتج.
الجدول 1 | المعلمات الحركية لـ TurboPETase و BhrPETase و LCCمستمدة من تجارب مايكاليز-مينتن العكسية
المعلمات
تحميل الركيزة
تيربوبيتيز
0.67
0.46
0.24
0.081
0.082
0.071
بهرPETase
0.69
0.40
0.20
0.020
0.022
0.022
شركة الطيران منخفضة التكلفة
0.54
0.42
0.23
0.021
0.027
0.023
تحليل هيكلي متعمق يساعدنا أيضًا على فهم الأسس الجزيئية لتحسينات الأداء. وفقًا لنموذج TurboPETase الذي توقعته AlphaFold2وتمت محاكاة الديناميكا الجزيئية (MD) اللاحقة، قد يُعزى الأداء المحسن لـ TurboPETase إلى الجوانب الرئيسية التالية: تحسين مرونة شق ربط الركيزة (H218S/F222I، W104L وF243T)، تحسين التفاعلات الشحنية على سطح البروتين (A209R وD238K)، وإدخال رابطة ثنائية الكبريت (A251C-A281C). يُقترح أن تساهم A209R وD238K ورابطة الكبريت الثنائية A251C-A281C بشكل أساسي في تحسين الاستقرار الحراري مع الحفاظ على النشاط (الأشكال التكميلية 10 و11)، بينما قد يُعزى تحسين فعالية التحلل المائي إلى الاستبدالات القريبة من المواقع النشطة. يُقترح أن H218 تشكل تعبئة وثيقة مع W190 المحفوظة في الإنزيمات المماثلة. وجد تشين وآخرون أن النشاط التحللي لـ PET يمكن أن يستفيد من موقع نشط أكثر مرونة في الطفرة المزدوجة H214S/F218I (التي تتوافق مع H218S/F222I في BhrPETase).في الدراسة الحالية، كشفت محاكاة الديناميكا الجزيئية لشكل الأبو من TurboPETase عن حرية دوران موسعة لـ W190 التي منحها الطفرة H218S/F222I (الشكل التكميلي 12)، وهو ما يتماشى مع ملاحظة التشكيلات المتنوعة لـ W156 المقابل في IsPETase.عند الارتباط بـ PET، يتم تقليل اهتزاز W190 وتثبيته بواسطةالتفاعلات مع ركيزة PET. تم تعزيز هذه المرونة في شق ارتباط PET بشكل أكبر من خلال التفاعلات التآزرية التي تم الحصول عليها من إضافة W104L و F243T، كما تم الكشف عنه بواسطة Cنتائج تقلب الجذر التربيعي المتوسط (RMSF) (الشكل 3C). تم الإبلاغ سابقًا عن أن W104 يتجمع ضد P248 المجاورة لاستقرار P248.. في TurboPETase المعاد تصميمه، قد يؤدي التخلي عن هذا التفاعل من خلال استبدال الليوسين إلى زيادة المرونة الشكلية داخل منطقة الحلقة (N246-A250)، كما يتضح من الانخفاض الكبير في الارتباط المتبادل لهذه المناطق (الشكل التكميلي 13). بالنسبة لاستبدال آخر F243T في تجويف ارتباط PET، يبدو أن الملف الاستيراتيكي لـ F243 يحدد موقع ارتباط أكثر محيطية لـ PET. ومع ذلك، قد يؤدي تحوله إلى الثريونين، الذي يتمتع بميزة استيراتيكية أقل وضوحًا، إلى تحرير المساحة لارتباط PET بحالة أكثر مرونة. والأهم من ذلك، أنه بدون الملف الاستيراتيكي للحلقة العطرية، قد يجذب T243 PET أعمق في التجويف، مما يقرب الكربونيل القابل للتغيير من السيرين الحفاز، مع تقليص المسافات البينية من إلى (الشكل التوضيحي 14). في الوقت نفسه، قد تؤثر المرونة المعززة سلبًا على استقرار البروتين، وهو ما يتماشى مع الانخفاض الملحوظ في درجات حرارة الانصهار للطفرات النقطية الفردية. من خلال دمج تحليلنا الهيكلي مع البيانات الحركية، افترضنا أن المرونة المتزايدة بشكل كبير على طول تجويف ربط PET قد توفر مساحة أكبر لاستيعاب مجموعة متنوعة من أشكال الهجوم من خلال الربط الديناميكي، وهو ما قد يكون حاسمًا لتكوين مجمعات كاتاليتيكية قادرة على العمل على هياكل سطحية مختلفة (الشكل 3D). بناءً على النتائج المذكورة أعلاه، استنتجنا أن TurboPETase ربما يكون أكثر تنوعًا فيما يتعلق بتكوين خيط PET الذي يهاجمه. ومع ذلك، فإن التفاصيل يتطلب تحليل الآلية مزيدًا من الجهود من خلال أبحاث أكثر عمقًا.
أداء TurboPETase على الركائز البديلة وفي أنظمة الإنزيم المزدوجة
أظهرت الدراسات السابقة كفاءة محدودة في التحلل البيولوجي لإنزيم ISPETase ونسخه تجاه البوليستر شبه العطري الآخر، وبالتحديد PBT، عند. مقارنةً بـ PET، يتمتع PBT بدرجة حرارة انتقال زجاجي أقل ( )، الذي يتراوح بين 37 و. في هذه الدراسة، على الرغم من أنيتجاوزمن PBT، مما يعزز بشكل كبير من حركة سلاسل بوليمر PBT، أظهرت جميع الإنزيمات التي تم فحصها كفاءة تدهور مخفضة بشكل ملحوظ تجاه أفلام PBT عند فيما يتعلق بتدهور PET (الشكل التكميلي 15). على وجه التحديد، أنتج TurboPETase كميات أكبر من المنتجات التحليلية ( ) من كل من BhrPETase و LCC تشير هذه النتائج إلى أن المواقع النشطة للإنزيمات الحالية التي تقوم بتفكيك PET كانت أقل كفاءة في الارتباط بالسلاسل الأليفاتية الممتدة في PBT مقارنةً بـ PET. وبالتالي، لا تزال هناك حاجة إلى جهود مخصصة في اكتشاف الإنزيمات أو الهندسة المخصصة لتحسين عملية التحلل البوليمري لفئات جديدة من البوليستر شبه العطرية.
تمت بذل جهود متنوعة لتطوير أنظمة إنزيمية مزدوجة لإزالة المنتجات الوسيطة BHET و MHET.، والتي تُعرف بأنها مثبطات إنزيمات هيدراز PET. أفاد هاوغويتز وآخرون بنظام إنزيم مزدوج يجمع بين طفرات TfCa المهندسة وإنزيم PETase PM الذي حقق زيادة بمقدار 4 أضعاف في المنتجات الكلية تجاه أفلام GfPET مقارنةً بـ PETase PM بمفرده عند فوققمنا بمزيد من استكشاف إمكانيات تطبيق TurboPETase من خلال ربطه بالإنزيم المقاوم للحرارة الذي تم الإبلاغ عنه مؤخرًا والذي يقوم بتحلل BHET، وهو BHETase.، في نظام إنزيمي مزدوج. عند تحميل منخفض للركيزة ( )، نظام الإنزيم المزدوج زاد بشكل فعال من العائد الإجمالي للمنتجات، مقارنةً بالاستخدام الفردي لـ TurboPETase (الشكل التكميلي 16). ومع ذلك، ظهرت ملاحظة مثيرة عند تحميل PET بمستوى مرتفع من تجاوزت TurboPETase وحدها العوائد من معظم نسب الإنزيمات في نظام الإنزيم المزدوج عندالاستثناء الوحيد هونسبة. ومع ذلك، في التحقيق الحالي، تم فقط تحليل تدهور المواد البولي إيثيلين تيريفثاليت غير المتبلور عندتم تقييمها. يمكن أن تتوسع الأبحاث المستقبلية لفحص فعالية نظام الإنزيم المزدوج على أنواع مختلفة من الهيدرازات MHET وركائز PET ذات الخصائص الفيزيائية المتنوعة تحت ظروف تفاعل مختلفة، بما في ذلك درجات حرارة متنوعة.
تحلل شبه كامل لمنتجات PET بعد الاستهلاك عند تحميل عالي من المواد الصلبة ذو صلة صناعية
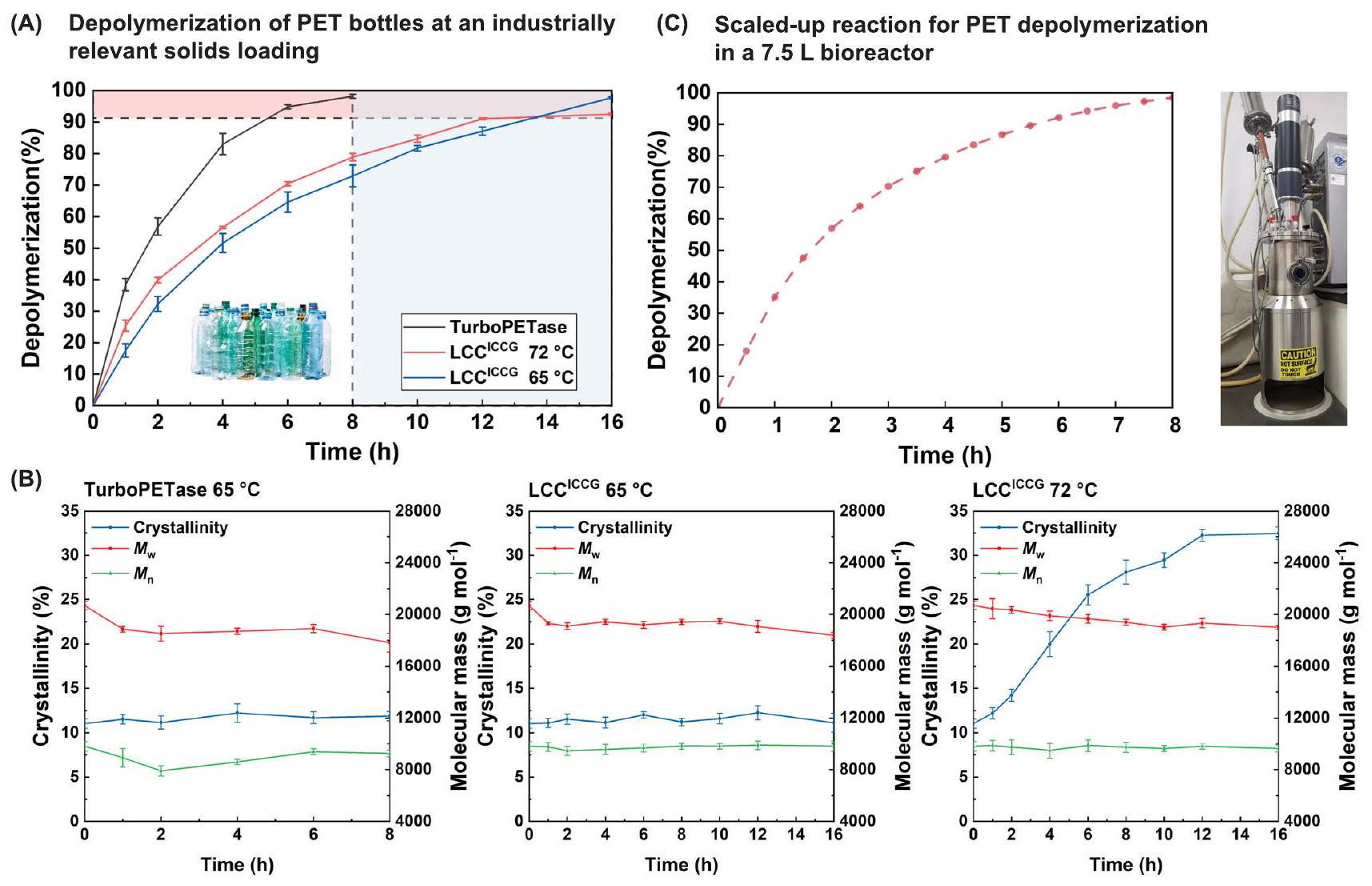
لتقييم أداء التحلل البوليمري لـ TurboPETase عند مستويات تحميل المواد الصلبة ذات الصلة بالصناعة بشكل أفضل، قمنا بمقارنة التحلل البوليمري لنفايات PET الملونة المعالجة مسبقًا (PcPET) باستخدام TurboPETase و LCC.عند تحميل الركيزة بـحقق TurboPETase تحللًا شبه كامل للبوليمر.، تم حسابها من بيانات HPLC) لنفايات PcPET في 8 ساعات (الشكل 4A)، مع معدل إنتاج أقصى قدره 61.3 جرام من PET المحلل (الشكل التكميلي 17 والجداول التكملية 9 و 10). لوحظ تغيير طفيف في بلورية PcPET (وصلت إلىبعد 8 ساعات)، مما يسمح بإعادة استخدام نفايات PcPET المتبقية على الفور لعملية التحلل البوليمري. على النقيض من ذلك، LCCمطلوب 16 ساعة للوصولتحلل البوليمر (محسوب من بيانات HPLC) عند، وأظهر PcPET المتبقي بلورية مشابهة لـ، مما يدل على أن عملية “الشيخوخة الفيزيائية” تم قمعها عند درجات حرارة أقل. عند درجة الحرارة المثلى للتفاعل التي تم الإبلاغ عنها سابقًا شركة ذات مسؤولية محدودةوصلت إلى أقصى تحويل لـ (محسوب من بيانات HPLC) على مدى 12 ساعة، ولم يتم الحصول على أي زيادة أخرى بعد فترة تفاعل مطولة بسبب قابلية تشوه سلاسل PET العالية. أظهر PcPET المتبقي مستوى عالٍ من
الشكل 4 | التحلل البوليمري لـ PcPET باستخدام TurboPETase و LCCفي المفاعلات الحيوية. مقارنة بين حركيات تحلل PcPET لـ TurboPETae عند و في و تمت التفاعلات معتحميل الركيزة وتحميل الإنزيم عند pH 8.0. تم حساب نسب تحلل PET بناءً على المنتجات المُحررة التي تم قياسها بواسطة HPLC. تُعرض البيانات كمتوسط. س.د. ( تجارب مستقلة بيولوجيًا). ب مسارات زمنية للبلورية، و التغييرات خلال
التحلل المائي الإنزيمي. تُعرض البيانات كمتوسط س.د. ( تجارب مستقلة بيولوجيًا). ج تقريبًا تدهور كامل لزجاجات PcPET المعالجة مسبقًا بواسطة TurboPETase عندمعتحميل الركيزة وتحميل الإنزيم في محلول مائي في مفاعل حيوي سعة 7.5 لتر. تم حساب نسب تحلل PET بناءً على استهلاك NaOH (تم تنظيم درجة حموضة خليط التفاعل عند 8.0). البيانات من تجربة مستقلة واحدة. تم توفير بيانات المصدر كملف بيانات مصدر. البلورية، المقدرة بـوهو يتماشى مع تقرير سابق.
يمكن أن تؤثر العديد من العوامل على الهجوم الإنزيمي ضد البلاستيك: مثل البلورية، حركة السلسلة، حجم الجزيء، الطبوغرافيا السطحية والخصائص الكارهة للماء.. من بين هذه، تم دراسة دور انخفاض البلورية في تحلل PET بشكل موسع. بلورية تتجاوزتم اقتراحه سابقًا بشكل كبير لعرقلة عملية التحلل الإنزيمي. في التحلل البوليمري لمساحيق PET غير المعالجة بالذوبان (البلورية)، وجدنا انخفاضًا كبيرًا في أداء التحلل (الشكل التكميلي 18 والجدول التكميلي 11). ومع ذلك، خلال التحلل المائي لـ PcPET المعالج مسبقًا، حتى عندما وصلت البلورية إلىبعد 4 ساعات من التحلل بواسطةفيلم نلاحظ تراجعًا سريعًا في معدل التدهور. أظهر بفاف وآخرون أن LCCأكثر كفاءة في تحليل البوليمرات الأقصرأظهرت الدراسات الحديثة أيضًا أن PET المعالج بشكل مكثف، مع درجة بوليمرية (DP) أقل من 20 وبلورية عالية، يمكن أن يظل قابلًا للتحلل بدرجة عالية.. هذه النتائج أكدت على أهمية عوامل أخرى غير البلورية في التحلل الإنزيمي، وشجعت على مزيد من التحقيق من خلال تحليل كروماتوغرافيا النفاذ الهلامي (GPC)، الذي كشف عن متوسط وزن منخفض بشكل ملحوظ ( ) ومتوسط العدد ( الكتل الجزيئية في مساحيق PcPET مقارنة بمساحيق PET غير المعالجة بالذوبان (الجدول التكميلي 12). من نتائج GPC، نفترض أن الوزن الجزيئي المنخفض لـ PcPET المعالج مسبقًا قد يساهم في الحفاظ على معدل التحلل بواسطةفيخلال الساعات الأولى من التفاعل، ولكن زيادة إضافية في البلورية إلىأدى إلى تقليل كبير في غير المتبلور المناطق، مما يعيق المزيد من التحفيز الإنزيمي. علاوة على ذلك، خلال عملية التحلل البوليمري، لاحظنا أيضًا انخفاضًا طفيفًا فيوتغييرات معتدلة فيعلى الرغم من تحقيق أكثر منتحلل البوليمر (الشكل 4B). هذا يتماشى مع الدراسات السابقة حول تحلل PET المائي بواسطة Cut190.لقد تم اقتراح أن التحلل المائي الإنزيمي للبولي إيثيلين تيريفثاليت (PET) يبدأ عادةً بقطع سلسلة من النوع الداخلي بعد ارتباط الإنزيم بسطح PET. يحدث مزيد من التحلل المائي للروابط الاستر المجاورة داخل المناطق غير المتبلورة من خلال قطع سلسلة من النوع الخارجي، مما يؤدي إلى إطلاق منتجات قابلة للذوبان.بالنسبة للبوليمرات التي تخضع للتقطيع العشوائي، فإن احتمال التقطيع الخارجي يرتبط عكسيًا بطول السلسلة.بالنسبة للبولي إيثيلين تيريفثاليت (PET) مع درجة بوليمرية (DP) حوالي 200، فإن احتمال الانقسام الخارجي أقل بكثير من الانقسام الداخلي. في هذه الدراسة، كانت درجة البوليمرية للبولي إيثيلين تيريفثاليت المعالج مسبقًا (PcPET)، والتي تبلغ حوالي 50، أقل بكثير من تلك الخاصة بمواد PET الخام النموذجية. وقد أدى ذلك إلى زيادة كبيرة في عدد نهايات السلاسل المتاحة للانقسامات الخارجية. وبالتالي، لم يعد الانقسام من النوع الداخلي، الذي كان سيقلل من درجة البوليمرية لـ PET، يحتل نسبة مهيمنة، مما قد يفسر التغيرات الطفيفة في الحجم الجزيئي التي لوحظت خلال عملية التحلل المائي لـ PET.
لاستكشاف الجدوى الصناعية، قمنا بتكبير التفاعل وأجريناه في مفاعل حيوي سعة 7.5 لتر مع تحميل PET منفي محلول مائي عند درجة حموضة 8.0. يعتبر تحميل الإنزيم مصدر قلق خاص للصناعة حيث أن تكلفة الإنزيم من بين العوامل الأساسية في عملية تحلل الكتلة الحيوية. على عكس استخدام تركيزات عالية من الإنزيم تصل إلىفي عملية تحويل الكتلة الحيويةتحميل الإنزيم مشبع في تحلل PET. استنادًا إلى نموذج TEA، تغيير من إلىمستويات تحميل الإنزيمات تؤدي فقط إلىزيادة في MSP (الولايات المتحدة) )، في حين أن تكاليف رأس المال والتشغيل ستزداد بمقدار لا يقل عن للحفاظ على مدة التفاعل المطولة عند استخدام مستويات تحميل إنزيم منخفضة. والأهم من ذلك، في التفاعلات على نطاق واسع مععند تحميل الركيزة، كان الانخفاض في نشاط الإنزيم وكفاءة التحلل البوليمري أكثر وضوحًا عند مستويات تحميل إنزيم أقل.. في عملية تحلل PET التي أبلغ عنها تورنييه وآخرون، كانت نسبة التحويل النهائية تم تحقيقه بواسطة LCCيستخدمتحميل الإنزيم مقارنة بـتحويل معتحميل الإنزيم. ومن ثم، تم اختيار تحميل الإنزيم لتحقيق توازن بين الإنتاجية وتكلفة الإنزيم. كما هو موضح في الشكل 4C، فإن تقدم التفاعل المقاس تقريبًا خطي خلال الساعتين الأوليين مع تحقيق حوالي 57% من التحلل البوليمري، يليه مرحلة أبطأ من 2 إلى 8 ساعات. وفقًا للمعلمات الحركية المستخلصة من الشكل 3، اقترحنا أن معدل التفاعل المنخفض قد يُعزى إلى الانخفاض الكبير في تحميل PET عند مستويات التحويل العالية. على الرغم من انخفاض الكفاءة التحليلية، فإن حواليتحلل البوليمرمحسوب من بيانات HPLC،محسوب من NaOH المستهلكة وتم حسابه من فقدان الوزن، كما هو مدرج في الجدول التكميلي 10) الذي تحقق خلال 8 ساعات أثناء التفاعل الموسع يجعل الإنتاج على نطاق تجريبي ممكنًا. يمكن أن يؤدي المزيد من تحسين العملية إلى تحسين كفاءة التحلل البوليمري لتركيزات أقل من نفايات PET.
نقاش
تقدم التحلل الحيوي للبولي إيثيلين تيريفثاليت (PET) نهجًا مستدامًا وفعالًا من حيث الطاقة لإعادة تدوير PET، مما يمثل بديلاً أكثر صداقة للبيئة لطرق التخلص الحالية مثل المدافن والحرق. على الرغم من تحقيق تقدم كبير في هذا الصدد، فإن الهدف النهائي هو تطوير إنزيمات وعمليات مناسبة للتطبيقات على نطاق صناعي. كل من محتوى المواد الصلبة العالي وتحويلها يفيدان اقتصاديات التحلل الحيوي لـ PET، حيث يقلل من النفقات الرأسمالية والتشغيلية. ومع ذلك، فإن معلق المادة يظهر لزوجة ظاهرة عالية بسبب تحميل المواد الصلبة العالي، مما يؤدي إلى محدودية نقل الكتلة والحرارة، مما يقلل من كفاءة الإنزيمات في المراحل المبكرة من التحلل المائي. والأهم من ذلك، أن زيادة تحميل المواد الصلبة إلى مستويات ذات صلة صناعية ستقلل من عائد التحلل بسبب التثبيط الناتج عن تركيزات المنتجات العالية.. وبالتالي، قد لا تكون الاستقرار الحراري للإنزيم كافية لتحلل PET في الصناعة. تتداخل عوامل متعددة، تؤثر على فعالية الإنزيم في السيناريوهات الواقعية. أحد الجوانب التي غالبًا ما يتم تجاهلها من قبل المجتمع العلمي هو التباين في التحلل المائي الإنزيمي غير المتجانس بين التجارب المخبرية والإنتاج الصناعي. على سبيل المثال، يمكن تعزيز نشاط التحلل للبولي إيثيلين تيريفثاليت (PET) في المختبر من خلال الدمج مع وحدات ربط غير تحفيزية لزيادة تركيز الإنزيم على سطح PET. ومع ذلك، فإن هذه الميزة تضيع تمامًا عند مستوى تحميل المواد الصلبة في الصناعة.على الرغم من التقدمات الأخيرة في هندسة إنزيمات الهيدراز PET لتحسين أدائها في تحلل PET، لا تزال الجهود مطلوبة في السعي نحو إنزيمات هيدراز PET جديدة لمعالجة فقدان التحويل تحت الظروف الصناعية ذات الصلة.
هنا، استخدمنا استراتيجية حسابية هجينة لإعادة تصميم إنزيم هيدراز PET الذي يتفوق بشكل كبير على إنزيمات هيدراز PET المعروفة الأخرى. تم تحقيق تدهور شبه كامل لزجاجات PET بعد الاستهلاك بمستوى تحميل صلب ذي صلة صناعيًا، مما يجعل هذا الإنزيم المحسن عالي الكفاءة مرشحًا جيدًا للتطبيقات المستقبلية في عمليات إعادة تدوير البلاستيك الصناعية. تم إثبات الآلية المسؤولة عن تعزيز أداء الإنزيم من خلال تحليلات حركية مستمدة من نظام تفاعل مائي عكسي من نوع مايكلس-منتن بالإضافة إلى التحليل الهيكلي، مما يبرز أهمية تحسين التفاعلات البوليمرية المحددة على مواقع الهجوم المحددة بدلاً من الامتصاص السطحي غير المحدد العام. قد تساعد النتائج في تعزيز المعرفة حول تحفيز التفاعلات غير المتجانسة وستكون مفيدة في تصميم عمليات تدهور البلاستيك القابلة للتطبيق صناعيًا. الإنزيمات لمعالجة التحديات المرتبطة بالبلاستيكات الأخرى الأكثر وفرة، مثل البولي يوريثانات ذات الهياكل القابلة للتحلل المائي. بينما يدفع إمكان تصميم الإنزيمات المزيد من التطورات في تحسين أداء الإنزيمات، من الضروري تقييم كفاءتها في التحلل البوليمري تحت ظروف ذات صلة صناعية لإظهار الجدوى العملية.
طرق
تدريب نموذج المحول
مجموعة بيانات التدريب غير المراقب. تسلسلات متجانسة من BhrPETase و LCCتم البحث عنهم من Uniclust30 (الإصدار 2018_08) وقاعدة بيانات BFD باستخدام HHblits (تم تعيين عدد التكرارات إلى 4، وتم ترك المعلمات الأخرى كقيم افتراضية) باستخدام 15 تسلسلًا أساسيًا في عائلة Pfam PF01083 كاستعلامات. تم تجميع جميع التسلسلات التي تم البحث عنها في الهوية باستخدام CD-HIT للحصول على 10847 تسلسل. نظرًا لأن BFD و Uniclust30 تم تجميعهما عند تشابه منخفض جدًا، فقد يكونان قد تم أخذ عينات منهما بشكل غير كافٍ لنمذجة اللياقة في المناطق القريبة جدًا. العمل السابق الذي قام به فريزر وآخرون.تم أخذ عينة من MSA المبنية على بروتينات أكثر تشابهًا للتنبؤ بمتغيرات الأمراض. كما استرجعنا 15051 تسلسلًا ينتمي إلى PF01083 من قاعدة بيانات UniProt.
تفاصيل التدريب وتوقع المرشحين “الأقل ملاءمة”. لنمذجة مسافة الملاءمة للتسلسلات، قمنا بتدريب شبكة عصبية مع مشفر-فك مشفر من الصفر. استخدمنا مشفر Transformer لمعالجة تسلسلات الأحماض الأمينية المدخلة مع تضمين الموضع المطلق. على عكس النماذج الأخرى مثل الشبكات المتكررة والشبكات التلافيفية، لم يقم Transformer بفرض أي افتراضات حول ترتيب التسلسل وكان أكثر قوة في التقاط العلاقات بعيدة المدى في التسلسل بسبب آلية الانتباه (المعادلة 1):
قمنا بتطبيق الانتباه الذاتي متعدد الرؤوس كما هو موصوف في المرجع 60. يتكون المشفر من 3 طبقات من المحولات مع 8 رؤوس باستخدام حجم تضمين قدره 512. استنادًا إلى تضمينات المشفر، يقوم المفكك بتوليد احتمالات لكل رمز. تم تدريب النموذج بهدف نمذجة اللغة المقنعة للتنبؤ بالحمض الأميني الحقيقي في الموضع المقنع. في هذه الدراسة، تم استبدال عدد من الرموز برموز القناع أثناء التدريب. استخدمنا مُحسِّن آدم مع معدل تعلم محدد إلىتم تدريب النماذج لمدة 20 دورة باستخدام حجم دفعة قدره 32. تم تصفية المتبقيات عن طريق الفرز حسب القيم اللوجستية المعينة للأحماض الأمينية WT. تم اختيار أعلى عشرة مواقع متبقية ذات أعلى درجات متوسطة من توقع كل نموذج. تم حساب الدرجة المتوسطة باستخدام المعادلة التالية:
أين هو متوسط درجة موقع البقايا المتوقعة، و هي القيم المتوقعة للوجيتات للطفرات النقطية الفردية في موضع البقايا والأحماض الأمينية من النوع البري، على التوالي. باستثناء المواضع المكررة، تم توليد ما مجموعه 18 موضع بقايا، من بينها تم اقتراح أن تكون W104 وH164 وM166 وW190 وH191 وH218 وF/I243 تقع على الأخدود المرتبط بـ PET.
تصميم الطفرات المستقرة بواسطة استراتيجية GRAPE
استراتيجية GRAPE المبلغ عنها في دراستنا السابقةتم استخدامه لتحسين استقرار البروتين. تسلسل BhrPETaseتم تقديمه إلى خادم GRAPE-WEB عبر الإنترنت (https://nmdc.cn/grape-web/“). استنادًا إلى معلومات تسلسل BhrPETaseألفا فولد 2تم استخدامه للتنبؤ بنموذج الهيكل. بعد ذلك، تم إجراء حسابات الطاقة باستخدام FoldXروزيتا_كارتيسيان_دي دي جي، و ABACUSتم استخدامه للتنبؤ بالطفرات المستقرة. خوارزمية DDDتم استخدامه للتنبؤ بـ روابط ثنائي الكبريتيد المناسبة. تم تعيين العتبات لـ ABACUS و FoldX و Rosetta إلىو -1.5 وحدة حرارية مكافئة، على التوالي.
الربط الجزيئي ومحاكاة الديناميكا الجزيئية
تم نمذجة TurboPETase بواسطة AlphaFold2. ثم تم محاكاة البروتين لمدة 20 نانوثانية. تم استخدام الهيكل التمثيلي الذي تم الحصول عليه من محاكاة الديناميكا الجزيئية وهيكل بلورة BhrPETase (معرف PDB: 7EOA) لمزيد من التوصيل. تم إنشاء ركيزة نموذجية تتكون من ثلاث وحدات PET متتالية، تحاكي المجموعات الطرفية N وC لتغطية سلسلة البوليمر من كلا الطرفين. تم استخدام YASARA لإجراء توصيل جزيئي لـ PET إلى TurboPETase وBhrPETase. تم اختيار نموذج أعلى طاقة ربط. تم إخضاع النموذج المختار لتوصيل محلي لـ 999 جولة. تم محاكاة النموذج المحسن لمجمعات TurboPETase-PET وBhrPETase-PET في AMBER.باستخدام حقل القوة ff16SB. تم بروتنة بقايا His242 في حالة HID.تم إضافة الأيونات للحفاظ على حيادية النظام. ثم تم حل النظام في صندوق ثماني الأوجه مقطوع باستخدام نموذج الماء TIP3P معالمسافة حول المذاب. بعد عملية الذوبان، خضعت الأنظمة لتقليل من 12,000 خطوة وتم تسخينها تدريجياً من 0 كلفن إلى 338 كلفن بتردد تصادمبعد التوازن، تم إجراء محاكاة الديناميكا الجزيئية لمدة 100 نانوثانية لكل مركب، باستخدام خطوة زمنية قدرها 2 فيمتوثانية.
استنساخ
الجينات التي تشفر BhrPETase (رقم الوصول في GenBank: GBD22443)، LCC (رقم الوصول في GenBank: AEV21261)، LCC (LCC S283C/1127G)، ICCG (ICCG فاستبيتيز (ISPETaseرقم الوصول إلى GenBank لـ ISPETase: BBYR01000074)، HotPETase (IsPETase S214Y/O119K/S213E/R90T/O182M/N212K/R224L/S58A/S61V/K95N/M154G/N241/K252M/T270Q)، DepoPET ase (ISPETase ), PES-H1 و CaPETase (CaPETase، رقم الوصول إلى GenBank لـ PETase: SHM40309) تم تصنيعه وتحسينه للتعبير في الإشريكية القولونية (General Biosystems، آنهوي، الصين). ببتيد الإشارة لـ BhrPETase، LCC، LCCICCG و بيتايزتمت إزالتها من الحمض النووي الاصطناعي. تم تصنيع الجينات لـ BhrPETase و LCC و LCC PES-H1 وتم استنساخ FastPETase في مواقع Nhel و Xhol من متجه pBAD (الذي يحتوي على علامة هيستيدين في الطرف N)، بينما تم استنساخ جين ICCGديبوبيتيز، كابيتازتم استنساخ HotPETase في مواقع Ndel و Xhol من متجه pET-21a(+) (الذي يحتوي على علامة هيستيدين في الطرف C). تم تقديم قائمة بتسلسلات النوكليوتيدات في الجدول التكميلي 13.
تحوير موجه للموقع
تم إنتاج طفرات BhrPETase باستخدام مجموعة التحوير الموجه QuickChange (Agilent Technologies، سانتا كلارا، كاليفورنيا، الولايات المتحدة الأمريكية). تم معالجة منتجات PCR بعد ذلك باستخدام DpnI (New England Biolabs، إبسويتش، ماساتشوستس، الولايات المتحدة الأمريكية) لهضم قالب الحمض النووي الأصلي، ثم تم إدخالها في خلايا E. coli TOP10. تم التحقق من الطفرات المدخلة من خلال تسلسل الحمض النووي (Tianyi Huiyuan، بكين، الصين).
تنقية البروتين
البلازميدات التي تحتوي على جينات BhrPETase،تم تحويل FastPETase ونسخها إلى E. coli BW25113. بلازميدات ICCGديبوبيتيز وكابيتيزتم تحويلها إلى E. coli C41(DE3)، بينما تم تحويل البلازميد الخاص بـ HotPETase إلىخلايا إيشيريشيا كولاي روزيتا غامي-ب (تم شراء خلايا التعبير القابلة للتنافس من زومان بيوتيك، بكين، الصين). تم زراعة الخلايا فيوسيط فيإلىمنثم تم تحفيز تعبير البروتين عن طريق إضافة L-أرابينوز (BhrPETase، LCC، LCC PES-H1 ، FastPETase ونسخها) أو 1 مللي مول من الإيزوبروبيل-D-thيوقالكوبيرانويد (ICCGديبوبيتيز، كابيتازو HotPETase). تم زراعة الخلايا لمدة 20 ساعة فيتم حصاده بواسطة الطرد المركزي ) ومعلقة في محلول التحلل ( و 20 مللي مول من الإيميدازول، pH 7.5 ). تم إجراء تكسير الخلايا من خلال الموجات فوق الصوتية على الثلج. تم الحصول على مستخلصات الخلايا الناتجة بعد الطرد المركزي عند لمدة 60 دقيقة عند، تليها عملية الترشيح من خلال فلتر ميلكس لإزالة الرواسب. تم غسل البروتينات غير المرتبطة في عمود HisTrap HP سعة 5 مل باستخدام محلول الغسيل.، و 60 مللي مول من الإيميدازول، pH 7.5)، تم استرجاع البروتين المستهدف باستخدام محلول الإخراج ( و 300 مليمول من الإيميدازول، pH 7.5 ). بعد ذلك، تم استبدال المحلول العازل بمحلول تخزين و 100 مليمول من NaCl، pH 7.5) باستخدام HiPrepعمود إزالة الملح. تم تخزين الإنزيم المنقى فيتم تحديد تركيزات البروتين باستخدام طريقة BCA مع ألبومين مصل البقر كمرجع.
اختبار تحلل PET باستخدام أفلام Gf-PET
تقييم نشاط الطفرات خلال هندسة BhrPETase. فيلم Gf-PET غير المتبلور (Goodfellow،سمك، رقم المنتج ES301445،حوالي 15 ملغ) تم نقعها فيمن محلول 1 م من فوسفات البوتاسيومتحميل المواد الصلبة) معمن الإنزيم المنقى (“تحميل الإنزيمات” في لمدة 3 ساعات. في تحليل HPLC، كانت المرحلة المتنقلة تتكون من مخزن و مخزن (المخزن A: حمض الفورميك في الماء المقطر؛ العازل ب: أسيتونتريل). كانت سرعة التدفق وتم تنفيذ الفصل في مع إجراء الكشف عند 260 نانومتر.
مقارنة TurboPETase مع إنزيمات التحلل المائي الأخرى للبولي إيثيلين تيريفثاليت تحت ظروف تفاعل متطابقة. تم تقدير الأنشطة الأولية لإنزيمات التحلل المائي للبولي إيثيلين تيريفثاليت عند درجات حرارة مختلفة. فيلم Gf-PET (حوالي 15 ملغ) تم نقعها فيمحلول عازل فوسفات البوتاسيوم بتركيز 100 مللي مولار (تحميل المواد الصلبة) مع ( 0.3 تحميل الإنزيمات) ( تحميل الإنزيم) للإنزيم المنقى عند 50، 60 أولمدة 3 ساعات، ثم تم تحليل السائل العلوي بواسطة HPLC لتحديد تركيز المونومرات المحررة من PET.
نظرًا للدرجة العالية من التحويل بعد التفاعلات المطولة، تم إجراء تحليل مسار الزمن في محلول فوسفات البوتاسيوم 1 م (pH 8.0) لتوفير سعة تخزين كافية. فيلم Gf-PET (حوالي 15 ملغ) تم نقعها فيمن محلول 1 م من فوسفات البوتاسيومتحميل المواد الصلبة) مع ( تحميل الإنزيم) للإنزيم المنقى عند 50، 60 أولمدة 12 ساعة.
مقارنة TurboPETase مع إنزيمات التحلل الأخرى للبولي إيثيلين تيريفثاليت تحت ظروف التفاعل المبلغ عنها. فيلم Gf-PET (حوالي 15 ملغ) تم نقعها فيمخزن ( تحميل المواد الصلبة) لمدة 12 ساعة. للمقارنة مع FastPETase، تم نقع فيلم Gf-PET في محلول فوسفات البوتاسيوم 100 مللي مول (pH 8.0) فيمعمن الإنزيم المنقى ( ج تحميل الإنزيم). للمقارنة مع HotPETase، تم نقع فيلم Gf-PET في محلول 50 مللي مول من غليسين-NaOH (رقم الهيدروجيني 9.2) فيمعمن الإنزيم المنقى (تحميل الإنزيم). للمقارنة مع LCCتم نقع فيلم Gf-PET في محلول فوسفات البوتاسيوم 100 مللي مولار (pH 8.0) فيمعمن الإنزيم المنقى (تحميل الإنزيم) لمدة 3 ساعات.
تحلل البوليمر للبلاستيكات PET المعالجة مسبقًا
معالجة مسبقة لزجاجات PET. تم جمع زجاجات PET المستعملة من محطة جمع القمامة في بكين، الصين. تم غسل الزجاجات بالماء المنزوع الأيونات وتجفيفها، ثم تم طحنها إلى رقائق صغيرة باستخدام كسارة JZ-T-005 (أدوات شيايان الدقيقة، دونغقوان، الصين). بعد ذلك، تم تحويل الرقائق إلى حالة غير متبلورة باستخدام جهاز بثق مزدوج اللولب SY-6219-20/32 (أدوات شيايان الدقيقة، دونغقوان، الصين). كانت درجات الحرارة المحددة هيفي مناطق البثق، في مضخة الانصهار، وفي مناطق تغيير الشاشة. تم ميكرونيزه الألياف غير المتبلورة من PET بواسطة مطحنة DFY-1000D (دينغلي، ونتشو، الصين) في درجة حرارة الغرفة. بعد الغربلة، تم الحصول على مساحيق PET بحجم جزيئات أقل منتم الحصول عليها (11.1% بلورية في المتوسط).
مقارنة أداء التحلل البوليمري لـ TurboPETase و LCCتم خلط عشرين جرامًا من مسحوق PET المعالج مسبقًا و80 مل من محلول فوسفات البوتاسيوم 100 مليمول (pH 8.0) الذي يحتوي على 40 ملغ من الإنزيم المنقى في مفاعل حيوي سعة 200 مل (Kusnc، شنغهاي، الصين). تم استخدام خلاط مغناطيسي للحفاظ على التحريك المستمر بسرعة 300 دورة في الدقيقة. تم تنظيم درجة الحرارة عند (أو لـ ) عن طريق غمر الحمام المائي، وتم تنظيم الرقم الهيدروجيني عند 8.0 عن طريق إضافة محلول NaOH بتركيز 4 م. تم إجراء التجارب ثلاث مرات وتم تقييمها وفقًا لذلك.
خلال التفاعل، تم تقييم نسبة تحلل PET من خلال تحليل السائل العلوي باستخدام HPLC. تم التحقق من العائد النهائي للتحلل من خلال فحص وزن PET المتبقي. تم تصفية خليط التفاعل من خلال أوراق فلترة نوعية من Whatman الدرجة 1 (GE Healthcare، الولايات المتحدة الأمريكية). تم غسل الرواسب مرتين بالماء المنزوع الأيونات ثم تم تجفيفها عندبين عشية وضحاها. لتقييم نسبة PET في المتبقي، تم تحضين 200 ملغ من المتبقي المجفف في 20 مل من محلول NaOH بتركيز 4 م.لمدة 3 ساعات. بعد ذلك، تم قياس كمية TPA المحررة بواسطة تحليل HPLC. الوزن الإجمالي للمواد المتبقية ونسبة PET موضحة في الجدول التكميلي 9.
تقدير أداء التحلل البوليمري لـ TurboPETase على نطاق أوسع. تم دمج 0.5 كجم من مسحوق PET المعالج مسبقًا، و1.9 لتر من الماء المقطر و0.1 لتر من محلول TurboPETase (الذي يحتوي على 1 جرام من الإنزيم المنقى) في مفاعل حيوي سعة 7.5 لتر (New Brunswick Scientific، إديسون، نيو جيرسي، الولايات المتحدة الأمريكية). تم الحفاظ على معدل التحريك عند 300 دورة في الدقيقة، وتم تنظيم درجة الحرارة عند، وتم تنظيم الرقم الهيدروجيني عند 8.0 عن طريق إضافة محلول NaOH بتركيز 4 م. تم تقييم نسبة تحلل PET خلال التفاعل وفقًا لاستهلاك القاعدة. بالإضافة إلى ذلك، تم التحقق من العائد النهائي للتحلل عن طريق تحليل المتبقي (كانت طرق التحلل القلوي وتحليل HPLC متطابقة مع البروتوكول أعلاه)، والذي تم جمعه عن طريق الطرد المركزي ( )، تم غسله مرتين بالماء منزوع الأيونات وتجفيفه في بين عشية وضحاها.
تحديد درجات حرارة الانصهار الظاهرة، بلورية PET والأوزان الجزيئية
لتحديد درجات حرارة الانصهار الظاهرة، تم استخدام اختبار استقرار حراري قائم على الفلورية.تم إضافة صبغة SYPRO Orange المخففة بمقدار -fold (Molecular Probes، Life Technologies، الولايات المتحدة الأمريكية) فيمحلول البروتين. تم وضع الخليط في لوحة PCR ذات 96 بئر ذات جدران رقيقة، وتم إغلاقها بشريط لاصق بجودة بصرية. تم تسخين الخليط في نظام تفاعل البوليميراز المتسلسل (PCR) CFX 96 (BioRad، هيركوليس، كاليفورنيا، الولايات المتحدة الأمريكية) من 25 إلىبمعدل تسخين قدرهتم تحليل بلورية PET و PBT بواسطة المسح الحراري التفاضلي (DSC). تم ضبط المعايير وفقًا لإجراء تم الإبلاغ عنه سابقًا.لتحليل الوزن الجزيئي للبولي إيثيلين تيرفثالات (PET)، تم تطبيق تحليل كروماتوغرافيا النفاذ الهلامي (GPC). تم إجراء قياسات GPC باستخدام نظام Agilent 1260 (Agilent 1260، شركة Agilent Technologies Inc.، الولايات المتحدة الأمريكية) مع كاشف مؤشر الانكسار (RI). كانت المرحلة المتنقلة تتكون من 1،1،1،3،3،3-هيكسافلورو-2-بروبانول (HFIP)، وتم إجراء التحليل عندبمعدل تدفق قدره.
تحليل الحركيات
لإعادة بوليمرة PET، تم جمع قياسات معدل البداية لـ و مجموعات البيانات. بالنسبة لـمجموعة البيانات، أفلام Gf-PET على مدىتم علاجهم فيمن محلول فوسفات البوتاسيوم بتركيز 100 مللي مولار (pH 8.0) مع تحميلات إنزيمية من، و . من أجل مجموعة البيانات،، ، و تم معالجة أفلام Gf-PET فيمن محلول فوسفات البوتاسيوم بتركيز 100 مللي مولار (pH 8.0) مع مستويات تحميل الإنزيم تتراوح من 0 إلىتم تحضين خليط التفاعل فيلمدة ساعة واحدة. تم إجراء جميع التفاعلات ثلاث مرات وانتهت بالتسخين إلى لمدة 10 دقائق. السائل الطافي الذي تم الحصول عليه عن طريق الطرد المركزي (ثم تم تحليلها بواسطة HPLC. تم ملاءمة البيانات باستخدام Matplotlib في بايثون.
بالنسبة لبيوتيرات 4-نيتروفينول (pNPB)، تم إجراء جميع التفاعلات في ألواح 96 بئر في حجم تفاعل إجمالي منتركيزات النهائية لـتتراوح من 0.2 إلى 1.4 مللي مول، وكانت التركيز النهائي للإنزيماتحلNPB (في الإيثانول اللامائي)من محلول فوسفات البوتاسيوم بتركيز 10 مللي مولار (pH 8.0)، ومن الإنزيمات تم حضنها عند لمدة 3 دقائق. ثم، تم إضافة الإيثانول اللامائي لإيقاف التفاعلات الإنزيمية. تم إجراء كل تفاعل ثلاث مرات.تم قياس منتجات النيتروفينول عند 405 نانومتر باستخدام جهاز إنفينيتقارئ الميكرو بلايت PRO (تيكان، سويسرا). تم تعريف وحدة واحدة من نشاط الإنزيم على أنها الكمية المطلوبة من الإنزيم لتحويلمنNPB لكل دقيقة. تم تعديل البيانات باستخدام Matplotlib في بايثون.
بالنسبة لـ MHET، تم إجراء التفاعلات في حجم إجمالي منتتراوح التركيزات النهائية لـ MHET من 6 إلى 60 مللي مولار (في محلول فوسفات البوتاسيوم 100 مللي مولار، pH 8.0)، وكان التركيز النهائي للإنزيمات هوتمت التفاعلات فيفي ثلاث نسخ لمدة 15 دقيقة، ثم تم تبريدها بإضافةمن ثنائي ميثيل سلفوكسيد. تم قياس مدى التحلل المائي باستخدام HPLC. تم تعديل البيانات باستخدام Matplotlib في بايثون.
المنحنيات المرتبطة
قياسات الامتزاز لـ TurboPETase و BhrPETase و LCCتمت التجارب باستخدام ألبومين مصل البقر (BSA) في محلول فوسفات البوتاسيوم بتركيز 100 مللي مولار (pH 8.0) باستخدام أنابيب إيبندورف ذات الالتصاق المنخفض.. فيلم GfPET (حوالي 15 ملغ) تم نقعها فيمن المحاليل العازلة التي تحتوي على تركيزات بروتين تتراوح من 0 إلى، maintained at لفترة توازن مدتها ساعة واحدة. بعد ذلك،تم خلط السائل العلوي معمحلول العمل BCA الطازج المحضر. خضعت هذه الخلطة للحضانة فيلمدة 30 دقيقة، تلتها قياسات الامتصاص عند 562 نانومتر باستخدام جهاز قراءة الألواح لتحديد تركيز البروتينات الحرة داخل المحلول. ثم تم استنتاج كميات البروتينات المرتبطة من خلال حساب الفرق بين تركيزات البروتين الكلي وتركيزات البروتينات الحرة.
تحديد بقايا السيستين الحرة
لتحديد بقايا السيستين الحرة في محلول TurboPETase، تم استخدام 5,5′-Dithiobis-(2-nitrobenzoic acid) (DTNB)محلول الإنزيممن BhrPETase أو TurboPETase) أو محلول L-cysteine ( و ) ، من محلول DTNB الطازج المحضرDTNB في محلول فوسفات البوتاسيوم بتركيز 100 مللي مولار، pH 8.0)تم خلط محلول EDTA (100 مللي مولار من حمض الإيثيلين diamine tetraacetic في 100 مللي مولار من محلول فوسفات البوتاسيوم، pH 8.0) بشكل جيد وتم حضنه لمدة 15 دقيقة. بعد ذلك، تم قياس امتصاص العينات عند 412 نانومتر باستخدام جهاز الطيف الضوئي (MAPADA V-1100D، شنغهاي، الصين).
تحلل البوليمر لبولي بيوتيلين تيريفثاليت
تم نقع أفلام PBT فيمحلول عازل فوسفات البوتاسيوم بتركيز 100 مللي مولار (تحميل المواد الصلبة) معمن الإنزيم المنقى (“تحميل الإنزيمات” فيلمدة 3 ساعات.
تحلل البوليمر باستخدام نظام إنزيمي مزدوج
فيلم Gf-PET (، حوالي 2 ملغ ) تم نقعه في 1 مل من محلول فوسفات البوتاسيوم 100 مللي مولار (تحميل المواد الصلبة) يحتوي على نظام إنزيمي مزدوج (تيربوبيتيز تحميل BHETase يتراوح بين 0 و ) وتم حضنه في لمدة 9 ساعات. لحمولة الركيزة منفيلم GfPET (حوالي 15 ملغ) تم نقعها فيمحلول عازل فوسفات البوتاسيوم بتركيز 100 مللي مولار (تحميل المواد الصلبة) التي تحتوي على نظام الإنزيمين. تم تحضين المزيج فيلمدة ساعة واحدة مع تركيزات TurboPETase و BHETase تتراوح من 0 إلىتم تحليل السائل العلوي بواسطة HPLC.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
جميع البيانات التي تدعم نتائج هذه الدراسة موجودة في المقالة وملفاتها التكميلية. لأي استفسارات أو طلبات لمزيد من المعلومات، يرجى الاتصال بالمؤلفين المعنيين. يتم توفير بيانات المصدر مع هذه الورقة.
Jambeck, J. R. et al. Plastic waste inputs from land into the ocean. Science 347, 768-771 (2015).
MacLeod, M. et al. The global threat from plastic pollution. Science 373, 61-65 (2021).
Wei, R. & Zimmermann, W. Microbial enzymes for the recycling of recalcitrant petroleumbased plastics: how far are we? Microb. Biotechnol. 10, 1308-1322 (2017).
Bornscheuer, U. T. Feeding on plastic. Science 351, 1154-1155 (2016).
Ellis, L. D. et al. Chemical and biological catalysis for plastics recycling and upcycling. Nat. Catal. 4, 539-556 (2021).
Kakadellis, S. & Rosetto, G. Achieving a circular bioeconomy for plastics. Science 373, 49-50 (2021).
Wei, R. et al. Possibilities and limitations of biotechnological plastic degradation and recycling. Nat. Catal. 3, 867-871 (2020).
Wei, R. et al. Mechanism-based design of efficient PET hydrolases. ACS Catal. 12, 3382-3396 (2022).
Taniguchi, I. et al. Biodegradation of PET: current status and application aspects. ACS Catal. 9, 4089-4105 (2019).
Kawai, F., Kawabata, T. & Oda, M. Current knowledge on enzymatic PET degradation and its possible application to waste stream management and other fields. Appl. Microbiol. Biotechnol. 103, 4253-4268 (2019).
Modenbach, A. A. & Nokes, S. E. Enzymatic hydrolysis of biomass at high-solids loadings-a review. Biomass-. Bioenergy 56, 526-544 (2013).
Singh, A. et al. Techno-economic, life-cycle, and socioeconomic impact analysis of enzymatic recycling of poly(ethylene terephthalate). Joule 5, 2479-2503 (2021).
Graham, R. et al. The role of binding modules in enzymatic poly (ethylene terephthalate) hydrolysis at high-solids loadings. Chem. Catal. 2, 2644-2657 (2022).
Ayla, S. D. S. et al. Constraints and advances in high-solids enzymatic hydrolysis of lignocellulosic biomass: a critical review. Biotechnol. Biofuels 13, 58 (2020).
Tournier, V. et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 580, 216-219 (2020).
Bell, E. L. et al. Directed evolution of an efficient and thermostable PET depolymerase. Nat. Catal. 5, 673-681 (2022).
Wei, R. et al. Biocatalytic degradation efficiency of postconsumer polyethylene terephthalate packaging determined by their polymer microstructures. Adv. Sci. 6, 1900491 (2019).
Cui, Y., Sun, J. & Wu, B. Computational enzyme redesign: large jumps in function. Trends Chem. 4, 409-419 (2022).
Sulaiman, S. et al. Isolation of a novel cutinase homolog with polyethylene terephthalate-degrading activity from leaf-branch compost by using a metagenomic approach. Appl. Environ. Microbiol. 78, 1556-1562 (2012).
Ding, Z. et al. Rational redesign of thermophilic PET hydrolase LCCICCG to enhance hydrolysis of high crystallinity polyethylene terephthalates. J. Hazard. Mater. 453, 131386 (2023).
Xi, X. et al. Secretory expression in Bacillus subtilis and biochemical characterization of a highly thermostable polyethylene terephthalate hydrolase from bacterium HR29. Enzym. Microb. Technol. 143, 109715 (2021).
Lu, H. et al. Machine learning-aided engineering of hydrolases for PET depolymerization. Nature 604, 662-667 (2022).
Shi, L. et al. Complete depolymerization of PET wastes by an evolved PET hydrolase from directed evolution. Angew. Chem. Int. Ed. Engl. 62, e202218390 (2023).
Hong, H. et al. Discovery and rational engineering of PET hydrolase with both mesophilic and thermophilic PET hydrolase properties. Nat. Commun. 14, 4556 (2023).
Pfaff, L. et al. Multiple substrate binding mode-guided engineering of a thermophilic PET hydrolase. ACS Catal. 12, 9790-9800 (2022).
Joho, Y. et al. Ancestral sequence reconstruction identifies structural changes underlying the evolution of Ideonella Sakaiensis PETase and variants with improved stability and activity. Biochemistry 62, 437-450 (2023).
Erickson, E. et al. Sourcing thermotolerant poly (ethylene terephthalate) hydrolase scaffolds from natural diversity. Nat. Commun. 13, 7850 (2022).
Wei, R. et al. Conformational fitting of a flexible oligomeric substrate does not explain the enzymatic PET degradation. Nat. Commun. 10, 5581 (2019).
Ding, X. & Zou, Z. & Brooks lii, C. L. Deciphering protein evolution and fitness landscapes with latent space models. Nat. Commun. 10, 5644 (2019).
Alley, E. C. et al. Unified rational protein engineering with sequencebased deep representation learning. Nat. Methods 16, 1315-1322 (2019).
Biswas, S. et al. Low-N protein engineering with data-efficient deep learning. Nat. Methods 18, 389-396 (2021).
Zeng, W. et al. Substrate-binding mode of a thermophilic PET hydrolase and engineering the enzyme to enhance the hydrolytic efficacy. ACS Catal. 12, 3033-3040 (2022).
Chen, C.-C. et al. General features to enhance enzymatic activity of poly(ethylene terephthalate) hydrolysis. Nat. Catal. 4, 425-430 (2021).
Cui, Y. et al. Computational redesign of a PETase for plastic biodegradation under ambient condition by the GRAPE strategy. ACS Catal. 11, 1340-1350 (2021).
Delgado, J., Radusky, L. G., Cianferoni, D. & Serrano, L. FoldX 5.0: working with RNA, small molecules and a new graphical interface. Bioinformatics 35, 4168-4169 (2019).
Park, H. et al. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J. Chem. Theory Comput. 12, 6201-6212 (2016).
Xiong, P. et al. Protein design with a comprehensive statistical energy function and boosted by experimental selection for foldability. Nat. Commun. 5, 5330 (2014).
Arabnejad, H. et al. A robust cosolvent-compatible halohydrin dehalogenase by computational library design. Protein Eng. Des. Sel. 30, 173-187 (2017).
Arnal, G. et al. Assessment of four engineered PET degrading enzymes considering large-scale industrial applications. ACS Catal. 13, 13156-13166 (2023).
Kari, J., Andersen, M., Borch, K. & Westh, P. An inverse Michaelis-Menten approach for interfacial enzyme kinetics. ACS Catal. 7, 4904-4914 (2017).
Schiano-di-Cola, C. et al. Systematic deletions in the cellobiohydrolase (CBH) Cel7A from the fungus Trichoderma reesei reveal flexible loops critical for CBH activity. J. Biol. Chem. 294, 1807-1815 (2019).
Baath, J. A. et al. Comparative biochemistry of four polyester (PET) hydrolases. Chem. Bio. Chem. 22, 1627-1637 (2021).
Erickson, E. et al. Comparative performance of PETase as a function of reaction conditions, substrate properties, and product accumulation. Chem. Sus. Chem. 15, e202101932 (2022).
Badino, S. et al. Adsorption of enzymes with hydrolytic activity on polyethylene terephthalate. Enzym. Microb. Technol. 152, 109937 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583-589 (2021).
Heidrich, D. & Gehde, M. The 3-phase structure of polyesters (PBT, PET) after isothermal and non-isothermal crystallization. Polymers 14, 793 (2022).
Barth, M. et al. A dual enzyme system composed of a polyester hydrolase and a carboxylesterase enhances the biocatalytic degradation of polyethylene terephthalate films. Biotechnol. J. 11, 1082-1087 (2016).
Knott, B. C. et al. Characterization and engineering of a two-enzyme system for plastics depolymerization. Proc. Natl Acad. Sci. USA. 117, 25476-25485 (2020).
Mrigwani, A. et al. Conversion of polyethylene terephthalate into pure terephthalic acid through synergy between a solid-degrading cutinase and a reaction intermediate-hydrolysing carboxylesterase. Green. Chem. 24, 6707-6719 (2022).
Haugwitz, G. et al. Structural insights into (tere) phthalate-ester hydrolysis by a carboxylesterase and its role in promoting PET depolymerization. ACS Catal. 12, 15259-15270 (2022).
Tarazona, N. A. et al. Rapid depolymerization of poly (ethylene terephthalate) thin films by a dual-enzyme system and its impact on material properties. Chem. Catal. 2, 3573-3589 (2022).
Li, A. et al. Discovery and mechanism-guided engineering of BHET hydrolases for improved PET recycling and upcycling. Nat. Commun. 14, 4169 (2023).
Guo, B. et al. Conformational selection in biocatalytic plastic degradation by PETase. ACS Catal. 12, 3397-3409 (2022).
Guo, B. et al. Fast depolymerization of PET bottle mediated by microwave pre-treatment and an engineered PETase. Chem. Sus. Chem. 16, e202300742 (2023).
Kawai, F. et al. Efficient depolymerization of polyethylene terephthalate (PET) and polyethylene furanoate by engineered PET hydrolase Cut190. AMB Express 12, 134 (2022).
Resch, M. G. et al. Clean fractionation pretreatment reduces enzyme loadings for biomass saccharification and reveals the mechanism of free and cellulosomal enzyme synergy. ACS Sustain. Chem. Eng. 2, 1377-1387 (2014).
Frazer, J. et al. Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91-95 (2021).
Vaswani, A. et al. Attention is all you need. In 31st Conference on Neural Information Processing Systems. 5998-6008 (Association of Computational Machinery, 2017).
Case, D. et al. AMBER 14, University of California, San Francisco. (2014).
شكر وتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (رقم المنحة 2021YFC2103600 إلى C.L.L.)، ومؤسسة العلوم الطبيعية الوطنية في الصين (32225002 إلى B.W.، 31822002 إلى B.W. و32170033 إلى Y.L.C.)، والبرنامج البحثي الرئيسي للعلوم الحدودية (ZDBS-LY-SM014 إلى B.W.)، وبرنامج الموارد البيولوجية (KFJ-BRP009 إلى B.W. وKFJ-BRP-017-58 إلى Y.L.C.) من الأكاديمية الصينية للعلوم، وخطة المعلوماتية من الأكاديمية الصينية للعلوم (CAS-WX2021SF-0111 إلى B.W.)، وجمعية تعزيز الابتكار للشباب في الأكاديمية الصينية للعلوم (2022086 إلى Y.L.C.)، وبرنامج زمالة ما بعد الدكتوراه من CPSF (GZC2O232929 إلى T.Z.).
مساهمات المؤلفين
بدأ ب.و. المشروع. قام ي.ل.س. وج.ي.س. بأداء العمل الحاسوبي، وقام ي.س.س. و.ه.ب. و.س.ل. بإجراء التجارب البيوكيميائية والبيوكاتاليتيكية، وقام ت.ز. و.و.س.ج. بإجراء تجارب تحلل البوليمر، وقام ي.ل.س. و ب.و. بصياغة المخطوطة، التي تم مراجعتها والموافقة عليها من قبل جميع المؤلفين.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى ينغلو كوي أو بيان وو.
معلومات مراجعة الأقران تشكر مجلة ناتشر كوميونيكيشنز بيدرو ألكسندرينو فيرنانديز والمراجع(ين) المجهول(ين) على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
مركز AIM، معهد الميكروبيولوجيا، الأكاديمية الصينية للعلوم، بكين، الصين.جامعة الأكاديمية الصينية للعلوم، بكين، الصين.كلية الكيمياء، جامعة نانكاي، تيانجين، الصين.ساهم هؤلاء المؤلفون بالتساوي: يينغلو كوي، يانشون تشين، جينيون صن، تونغ زو. البريد الإلكتروني: cuiyinglu@im.ac.cn; wub@im.ac.cn
Biotechnological plastic recycling has emerged as a suitable option for addressing the pollution crisis. A major breakthrough in the biodegradation of poly(ethylene terephthalate) (PET) is achieved by using a LCC variant, which permits conversion at an industrial level. Despite the achievements, its applications have been hampered by the remaining 10% of nonbiodegradable PET. Herein, we address current challenges by employing a computational strategy to engineer a hydrolase from the bacterium HR29. The redesigned variant, TurboPETase, outperforms other well-known PET hydrolases. Nearly complete depolymerization is accomplished in 8 h at a solids loading of . Kinetic and structural analysis suggest that the improved performance may be attributed to a more flexible PET-binding groove that facilitates the targeting of more specific attack sites. Collectively, our results constitute a significant advance in understanding and engineering of industrially applicable polyester hydrolases, and provide guidance for further efforts on other polymer types.
Substantial efforts have been dedicated to discovering and engineering PET hydrolases over the past two decades, contributing to the development of PET biodegradation from the detection of trace amounts of released products to the attainment of high conversion When combating the challenges posed by enzymatic PET recycling, key parameters should be taken into consideration to attain an economically viable industrial process, in particular solids loading ( ) and product yield ( . However, almost all studies obtain appreciable depolymerization yields at solids loadings lower than , which is considerably below an industrially relevant level . When increasing the solids loading to a high level, a reduction in the hydrolysis rate and conversion is commonly observed (referred to as “solids effect”), as demonstrated in other heterogeneous reactions such as the biomass conversion process .
A breakthrough was achieved with an engineered LCC variant (LCC ) that exhibited depolymerization of pretreated PET waste
at an industrially relevant solids loading . However, the 10% nonbiodegradable PET remained and reached a high crystallinity level of because of physical aging, hindering immediate reuse for PET depolymerization. A report issued by the National Association for PET Container Resources indicated that the postconsumer PET bottles collected for recycling reached 800 kilotons in the US , and this would generate over 80 kilotons of nonbiodegradable PET waste per year if the current biodegradation strategy was employed, posing a significant threat to the PET recycling economy. Increasing the extent of conversion from to would cut the minimum selling price (MSP) of TPA by (US ) according to the TEA model proposed by the National Renewable Energy Laboratory in the US; thus, second to solids loading, the conversion level is the largest factor affecting MSP in the depolymerization process . Both environmental and socioeconomic concerns have fueled intense interest in improving the degradation level to accelerate the transition towards the circular
economy. According to the kinetic results of PET crystallization (Supplementary Fig. 1), physical aging can be vastly suppressed by decreasing the reaction temperature, but the catalytic efficiency of thermophilic PET hydrolases is concomitantly sacrificed , thus raising demands for biocatalysts with high depolymerization efficiency at low deformability temperatures of amorphous PET chains.
The last few years have witnessed impressive progress in tailoring natural enzymes by computational redesign strategies . Inspired by the achievements in artificial intelligence for addressing the protein fitness landscape to probe hidden evolutionary information, we employed a computational strategy that incorporates a protein language model and force-field-based algorithms to engineer PET hydrolases with balanced thermostability and hydrolytic capacity. The redesigned variant (TurboPETase) derived from this campaign outperformed the most efficient PET hydrolases currently recognized in the field ( , BhrPETase , FastPETase , HotPETase , DepoPETase , CaPETase , and PES-H1 ) over a range of temperatures ( ). The extraordinary degradation performance afforded by TurboPETase allowed nearly complete depolymerization of post-consumer PET bottles in 8 h at a high industrially relevant substrate loading of , with a maximum production rate of 61.3 ghydrolyzed PET , addressing the challenge regarding residual nonbiodegradable PET waste.
Results
Computational redesign of an efficient PET hydrolase
PET hydrolases belong to serine-hydrolase family, a widely distributed group known for their relatively low substrate specificity. The degradation function of PET hydrolases is thought to preexist as a promiscuous function, which then evolves into a primary function . Given our limited knowledge of how a sequence encodes catalytic functions in polymer-degrading enzymes , addressing the challenge by exploiting physics-based computational redesign and rational design approaches is a difficult task. Alternatively, a successful approach involves utilizing deep learning models to map the process from protein sequence to function, as demonstrated in many cases . These models can capture hidden information indicating the improvement in polymer degradation along the evolution trajectory from the relative fitness of protein variants. To this end, we employed a language model trained on two datasets that involved approximately 26,000 homologous sequences of PET hydrolases across evolution, to predict the probability of amino acid variation from the evolutionary landscape (Fig. 1A). To select the template enzyme, we compared the activity of LCC, LCC , BhrPETase, FastPETase, and HotPETase using amorphous PET films (Gf-PET, from the supplier Goodfellow) across a range of temperatures (Supplementary Fig. 2). BhrPETase shares a high sequence identity of with LCC, whereas LCC represents a variant of LCC characterized by four amino acid substituents (F243I/ D238C/S283C/Y127G). Both FastPETase and HotPETase are derivatives engineered from IsPETase (Supplementary Fig. 3). At , BhrPETase and LCC exhibited the highest catalytic performance of all PET hydrolases and temperatures tested and were chosen as the inputs. A Transformer encoder was used to process input amino acid sequences with absolute position embedding. Residue positions were sorted by the mean of the logits of 19 mutations assigned to the wild type amino acid at each position. The top ten amino acid positions with the highest average scores of each model were selected. After duplicated amino acid positions were removed, 18 amino acid positions at which the wild-type residues fit less well than potential substitutions were obtained (Supplementary Table 1).
According to the crystal structures of BhrPETase (PDB code: 7EOA) and LCC S165A in complex with MHET (PDB code: 7VVE ), 7 of the 18 generated amino acid positions (W104, H164, M166, W190, H191, H218, and F/I243) were suggested to be embedded in a PETbinding groove. Due to the relatively higher thermostability of
BhrPETase, we subjected the 7 positions on this enzyme to generate 34 variants. Chen et al. reported that the Ser214/Ile218 double mutants of several IsPETase-homologous enzymes showed enhanced PET hydrolysis activity (by at least 1.3 -fold) but vastly decreased values (by approximately . Since His 218 (corresponding to Ser214 in IsPETase) was involved in our predicted candidates, the hydrolytic performance of the H218S/F222I variant was also explored (Supplementary Table 2). Among the variants, BhrPETase (referred to as BhrPETase M2) resulted in the highest improvements with a 1.7-fold increase in PET-hydrolytic activity at , rendering this variant the new template for the second-round accumulation of the mutations at the remaining 6 positions. During the 2nd accumulation stage, only mutations at W104 and F243 positions (W104L, W104S, W104H, W104G, F243I, F243T, and F243G) on M2 exhibited improved hydrolytic activities (by to ), albeit with significant decrease in thermostability (Supplementary Table 3).
Empirical data have suggested that a higher than the is necessary for catalyst longevity . Hence, a PET hydrolase with a at least over is preferred for efficient PET degradation. Notably, the M2 variant exhibited a melting temperature of , which was lower than that of the wild-type enzyme. The active mutations at the W104 positions on M2 reduced the stability even further, with values ranging from to . The nonnegligible decrease in stability limited further combination, and compensatory mutations needed to be introduced first to suppress the deleterious effects. Therefore, we applied our previously devised GRAPE strategy , which employs four complementary algorithms, namely, Fold (force field energy function), Rosetta_cartesian_ddg (force field energy function), ABACUS (statistical energy function) and DDD (force field energy function), to design stabilizing mutations to compensate and buffer the destabilizing mutations (Fig. 1B). Upon experimental validation, 3 beneficial variants (A209R, D238K, and A251C-A281C) resulted in improved thermostability without compromising the activity (Supplementary Table 4). We added the stabilizing variants to the M2 variant using a stepwise combining strategy and resulted in a BhrPETase M6 variant (BhrPETase ), which exhibited a restored melting temperature of without sacrificing activity. Subsequently, active mutations at the W104 and F243 positions were combinatorically assembled and accumulated onto the thermostable M6 variant, generating 12 new variants (Fig. 1C and Supplementary Table 5). After a consideration of both depolymerization performance and thermostability, the best combination variant, BhrPETase A209R/D238K/A251C/A281C/W104L/F243T (referred to as TurboPETase), was selected with a of and a 3.4 -fold improvement in PET-specific activity towards GF-PET films compared to wild-type BhrPETase (Fig. 1D). In industrial scenarios, enzymatic hydrolysis is typically conducted in water rather than in a dedicated buffered system to simplify downstream processing. Aligned with the industrial preferences, we also evaluated the 12 variants under a low buffer concentration. The results revealed that with the exception , all variants largely retained their degrading activity in 100 mM potassium phosphate buffer (Supplementary Fig. 4 and Supplementary Table 6). TurboPETase consistently outperformed the other variants under both low and high buffer concentrations, making it the most effective variant for further investigation.
The depolymerization performance of TurboPETase was subsequently evaluated across a temperature range of with respect to other PET hydrolases (Fig. 2A, B, and Supplementary Fig. 5). At , TurboPETase exhibited the highest overall degradation of all PET hydrolases and temperatures tested, releasing 29.66 mM of the products (sum of BHET, MHET, and TPA) in 3 h . Counterparts like BhrPETase, LCC, LCC , ICCG , and PES-H1 which exhibit high degradation performance at elevated temperatures, rendered hydrolytic activity 1.8-, 4.7-, 2.1-, 2.0-, and 19-fold lower than that of TurboPETase, respectively. At suboptimal temperatures, TurboPETase
Fig. 1 | Schematic representation of the redesign of PET hydrolases using the hybrid computational method. A Potentially beneficial mutations are predicted with the Transformer model trained on two datasets (left panel). The top ten predicted amino acid positions (red circles) were selected. Removal of duplicate positions resulted in 18 residue positions, of which 7 positions were suggested to be located at the PET binding site (right panel). The atoms of these amino acid positions are shown as blue spheres, whereas the 11 remaining positions are shown as coral spheres. PET and the catalytic triad are shown as ball-and-stick representations. B Schematic representation of the GRAPE strategy. Four algorithms were employed to predict the stabilizing mutations (left panel). After structural filtering, the potential stabilizing candidates were verified experimentally (right panel). The atoms of the mutations with improved thermostability are shown as blue spheres, whereas other experimentally verified mutations are shown as coral spheres. C Schematic representation of the accumulation step. Mutations at the 7 residue positions located at the PET binding site were added to the wild type
enzyme (BhrPETase), resulting in the best hit BhrPETase (M2). Adding the mutations at the remaining 6 positions to M 2 led to active variants with dramatically decreased stability, and therefore, the GRAPE strategy was employed. The stabilizing mutations were combined into M2 and resulted in the thermostable variant M6, which can compensate for the destabilizing mutations in further accumulation steps. The active mutations at the W104 and F243 positions were combinatorically assembled on M6, leading to the final mutant TurboPETase. D Thermostability and relative PET-degrading activity of BhrPETase and the variants. The activities were homogenized according to BhrPETase. Data are from one independent experiment. Mutations predicted by the Transformer model and the GRAPE strategy are colored blue and green, respectively. The double mutants at the W104 and F243 positions on M6 are shown in red. Arrows represent the accumulation path. The yellow area represents the optimal melting temperature range for PET hydrolases to enable efficient depolymerization at .
consistently outperformed other PET hydrolases, albeit the decreased hydrolytic activity. At the optimal temperature of HotPETase and CaPETase of , TurboPETase generated 11.73 mM monomer products in 3 h , whereas HotPETase and CaPETase produced 4.32 mM and 0.60 mM monomers, which were 1.7 -, and 18 -fold lower than that of TurboPETase. When subjected to , TurboPETase registered hydrolytic efficiencies exceeding those of FastPETase and DepoPETase by and , respectively. In light of the lower enzyme concentrations employed in certain reports, we recalibrated our reactions to mirror these conditions to ensure a fairer comparison (Fig. 2C-E). Under their reported reaction conditions, the reaction rates of LCC , HotPETase, and FastPETase were 1.8-, 4.9-, and 1.0-fold lower, respectively, than that of TurboPETase. Recently, similar experiments were conducted to evaluate the degradation performance of FastPETase, HotPETase, PES-H1 and LCC under industrial conditions . Their results demonstrated that HotPETase exhibited a higher specific activity compared to PES-H1 at a low substrate loading of . However, as the substrate concentrations
increased to and , the PET conversion of HotPETase were significantly lower than those of PES-H1 . This reduced efficiency at higher substrate loadings is suggested to be attributed to the limited thermostability and other catalytic properties of HotPETase, such as product inhibition. Despite the good performance of PES-H1 under industrially relevant conditions, the final depolymerization was still lower than that achieved by LCC . Given the reported enhancements of PES-H1 in 1 M potassium phosphate buffer, we also compared TurboPETase with PES-H1 under elevated buffer concentration, with TurboPETase still outpacing, yielding up to 2.5 times the degradation products of PES-H1 / at C (Supplementary Fig. 6). To evaluate the long-time stability of TurboPETase, we extended the reaction time. The results demonstrated continuous enzyme performance of TurboPETase at elevated temperatures, as it produced 130 mM of soluble monomer products over 12 h at , whereas the released products with BhrPETase, LCC, LCC were substantially lower (Supplementary Fig. 7). The degradation results highlight the substantially superior hydrolytic
Fig. 2 | Comparison of depolymerization performance of TurboPETase with other known PET hydrolases. A Location of the beneficial mutations in TurboPETase and its counterparts. TurboPETase, BhrPETase and LCC are shown in green, grey and yellow, respectively. Residues are shown in ball and stick representations. B Comparison of the PET-hydrolytic activity of TurboPETase and other PET hydrolases towards Gf-PET films at temperatures ranging from 50 to using solids loading and enzyme loading in 100 mM potassium phosphate buffer, pH 8.0 . The bar chart shows the mean depolymerization after 3 h of reaction. Data are presented as mean s.d. ( biologically independent experiments). The circles represent the individual numbers. *In the investigation conducted by Arnal et al. , the depolymerization efficiency of PES notably surpassed that of HotPETase, yet it remained inferior to LCC , under industrially relevant substrate loadings of and . C Time
course reactions of TurboPETase and FastPETase towards Gf-PET films at under its reported reaction conditions ( g enzyme loading, solids loading, 100 mM potassium phosphate buffer, pH 8.0 ). Data are presented as mean s.d. ( biologically independent experiments). Time course reactions of TurboPETase and HotPETase towards Gf-PET films at under its reported reaction conditions ( enzyme loading, solids loading, 50 mM Glycine-NaOH buffer, pH 9.2 ). Data are presented as mean s.d. ( biologically independent experiments). E Time course reactions of TurboPETase and LCC towards Gf-PET films at using its reported enzyme loading of in 100 mM potassium phosphate buffer ( ). The solids loading was . Data are presented as mean s.d. ( biologically independent experiments). Source data are provided as a Source Data file.
performance of TurboPETase across various temperatures and other reaction conditions tested.
Michaelis-Menten approach and structural analysis
To interpret the enhanced hydrolysis performance, the kinetics were analysed through conventional Michaelis-Menten model and inverse Michaelis-Menten model. As shown in Fig. 3A, the curves exhibited near-linear relationships for the initial rate and substrate loading under all conditions for TurboPETase, BhrPETase and LCC . Conventional saturation behaviour was not observed because even the lowest enzyme concentrations used here ( ) were too high for the conventional approach to be valid, which indicates the very fast rates of dissociation of the enzymes from the PET surface. Additionally, we conducted a kinetic comparison for soluble substrates (MHET and ) as detailed in Supplementary Fig. 8 and Supplementary Table 7. The catalytic efficiency of TurboPETase towards MHET showed only a modest enhancement relative to PET (with a 32% increase), suggesting that there may be other factors, potentially increased adsorption to the surface, contributing to the amplified degradation efficiency of PET. Conversely, the slight decrement in TurboPETase’s for NPB,
accompanied by a reduced binding affinity, inferred potential changes in the substrate binding domain, rendering it less conducive for other small molecule interactions.
Although the hydrolysis of PET could not meet the criteria for the conventional approach, the inverse Michaelis-Menten model was more applicable. The inverse Michaelis-Menten equation has been successfully employed to study the kinetics of heterogeneous enzymes such as cellulases, by which the catalytic efficacy against accessible attack sites on the polymer surface can be estimated . As a typical surface erosion process, enzymatic hydrolysis of PET can hardly permeate the inner core of the polymer, resulting in a limited number of superficial ester bonds (also termed attack sites) being accessed even if the enzymes are in great excess. Saturation thus occurs when all sites on the surface become occupied, and the excess enzyme molecules accumulate in the solvent . It should be noted that not all adsorption sites are competent for catalytic conversion, nonspecific adsorption also accounts for a considerable proportion . We measured free enzyme concentrations, , and converted it to substrate coverage, to calculated the total adsorption of the enzymes to PET surface (Supplementary Fig. 9 and
Fig. 3 | Kinetic parameters and structural analysis of TurboPETase.
A Conventional and B inverse Michaelis-Menten plots of TurboPETase, BhrPETase, and at in 100 mM potassium phosphate buffer, pH 8.0 . Data are presented as mean s.d. ( biologically independent experiments). Source data are provided as a Source Data file. C RMSF values (in ) for all atoms of BhrPETase and TurboPETase during the MD simulations, indicating global changes
in protein flexibility, especially for the PET-binding groove. The H218S/F222I, W104L, and F243T mutations are shown as orange atoms, whereas the catalytic triad is shown as hot pink atoms. D Schematic diagram of PET depolymerization processes. Compared to the wild-type enzyme, TurboPETase may be more promiscuous due to the increased flexibility along the protein PET-binding groove, which allows it to attack different surface structures.
Supplementary Table 8). represents the (apparent) saturated bound enzymes, whereas is a dissociation constant . At lower enzyme concentrations, TurboPETase exhibited a higher adsorption to the PET surface compared to its counterparts. However, upon reaching saturation, similar maximum adsorption capacities ( ) were observed, underscoring a consistent overall binding potential across the enzymes. Comparisons of these enzymes could be expanded by considering specific changes in inverse Michaelis-Menten parameters, which can reflect the binding capability to the attack sites of the PET surface. As shown in Fig. 3B and Table 1, the values revealed marginal differences between TurboPETase, BhrPETase, and LCC , especially for the values at substrate loading, indicating that the specific adsorption capacity of TurboPETase at the attack sites on
the PET surface was not impaired. It’s noteworthy that TurboPETase exhibited a 2.1-fold increase in compared with BhrPETase and LCC . The inverse parameter, maximal reaction velocity per available reactive site ( ), specifies the rate when all attack sites are covered with the enzyme . Since no substantial differences in and values were observed among the enzymes, the elevated values may imply a broadened targeting of TurboPETase towards specific attack sites when the enzymes maintained a stable overall adsorption level. Consequently, we presumed that the enhanced depolymerization performance of TurboPETase may rely, at least in part, on the enhanced ability to attack a broader spectrum of specific attack sites that can be hydrolysed to form a productive complex.
Table 1 | Kinetic parameters of TurboPETase, BhrPETase, and LCC derived from the inverse Michaelis-Menten experiments
Parameters
Substrate loading
TurboPETase
0.67
0.46
0.24
0.081
0.082
0.071
BhrPETase
0.69
0.40
0.20
0.020
0.022
0.022
LCC
0.54
0.42
0.23
0.021
0.027
0.023
An in-depth structural analysis also helps us to understand the molecular underpinnings of the performance improvements. According to the model of TurboPETase predicted by AlphaFold2 and subsequent molecular dynamics (MD) simulations, the improved performance of TurboPETase may be attributed to the following key aspects: improved flexibility of the substrate binding cleft (H218S/ F222I, W104L and F243T), optimized charge-charge interactions at the protein surface (A209R and D238K), and introduction of a disulfide bond (A251C-A281C). A209R, D238K, and the disulfide bond A251CA281C are suggested to primarily contribute to improving thermostability while maintaining activity (Supplementary Figs. 10 and 11), whereas the enhanced hydrolysis efficacy may be attributed to substitutions in proximity to the active sites. H 218 is suggested to form an intimate packing with the conserved W190 in analogous enzymes. Chen et al. found that PET hydrolytic activity could benefit from a more flexible active site in the H214S/F218I double mutant (corresponds to H218S/F222I in BhrPETase) . In the present study, MD simulations of the apo form of TurboPETase revealed an expanded rotational freedom of W190 endowed by the H218S/F222I mutation (Supplementary Fig. 12), which is consistent with the observation of diverse conformations of the corresponding W156 of IsPETase . When binding to the PET, the wobbling of W190 is curtailed and anchored by the interactions with the PET substrate. This flexibility of the PET binding cleft was further enhanced by the synergistic interactions conferred by the addition of W104L and F243T, as revealed by the C root-meansquare fluctuation (RMSF) results (Fig. 3C). W104 is previously reported to pack against the adjacent P248 to stabilize the P248-situated . In the redesigned TurboPETase, the relinquishment of this interaction by leucine substitution may engender increased conformational malleability within the loop region (N246-A250), as demonstrated by the largely reduced cross-correlation of these regions (Supplementary Fig. 13). For another substitution F243T in the PET binding cleft, the steric profile of F243 appears to dictate a more peripheral binding locus for PET. Yet, its mutation to threonine, armed with a less pronounced steric feature, may release the space for PET binding with a more-flexible state. More importantly, without the steric profile of the aromatic ring, T243 may beckon PET deeper into the cleft, drawing the substrate’s labile carbonyl closer to the catalytic serine, with the interstitial distances contracting from to (Supplementary Fig. 14). Concurrently, the enhanced flexibility might compromise the protein’s stability, which is consistent with the observed decrease in the melting temperatures of the single point mutations. Synthesizing our structural analysis with the kinetic data, we postulated that the greatly increased flexibility along the PETbinding groove may provide more space to accommodate a variety of attack conformations through dynamic binding, which may be crucial for the formation of catalytically competent complexes on different surface structures (Fig. 3D). Based on the above results, we reasoned that TurboPETase is probably more promiscuous with respect to the conformation of the PET strand it attacks. Nevertheless, detailed
analysis of the mechanism requires further efforts through more indepth research.
Performance of TurboPETase on alternative substrates and in dual-enzyme systems
Previous study has demonstrated limited biodegradation efficiency of ISPETase and its variant towards other semiaromatic polyesters, specifically PBT, at . Compared to PET, PBT has a lower glass transition temperature ( ), which ranges between 37 and . In this study, even though surpasses the of PBT, thus significantly enhancing the mobility of PBT polymer chains, all of the examined enzymes exhibited substantially reduced degradation efficiency towards PBT films at with respect to the degradation of PET (Supplementary Fig. 15). Specifically, TurboPETase yielded higher amounts of hydrolytic products ( ) than both BhrPETase and LCC . These results suggested that the active sites of current PET-degrading enzymes were less efficient in binding with the extended aliphatic chains in PBT compared to PET. Consequently, dedicated efforts in enzyme discovery or tailored engineering are still needed for further improving the depolymerization of new classes of semiaromatic polyesters.
Various efforts have been made to develop dual enzyme systems to remove the intermediate products BHET and MHET , which are known inhibitors of PET hydrolases. Haugwitz et al. reported a dual enzyme system combining an engineered TfCa mutant and PETase PM enzyme that yielded a 4 -fold increase in overall products towards GfPET films compared to PETase PM alone at over . We further explored the application potential of TurboPETase by coupling it with the recently reported thermostable BHET hydrolyzing enzyme, BHETase , in a dual-enzyme system. At a low substrate loading ( ), the dual-enzyme system effectively doubled the overall yield of the products, relative to the singular use of TurboPETase (Supplementary Fig. 16). However, an intriguing observation emerged at an elevated PET loading of . TurboPETase alone surpassed the yields from most enzyme ratios in the dual-enzyme system at , the only exception being the ratio. Nevertheless, in the current investigation, only degradation of amorphous PET materials at were evaluated. Future research could expand to examine the efficacy of the dual-enzyme system on different MHET hydrolases and PET substrates with varying physical properties under diverse reaction conditions, including varied temperatures.
Nearly complete degradation of postconsumer PET products at industrially relevant high-solids loading
To better evaluate the depolymerization performance of TurboPETase at industrially relevant levels of solids loading, we have compared the depolymerization of pretreated postconsumer coloured-flake PET (PcPET) wastes with TurboPETase and LCC at a substrate loading of . TurboPETase achieved nearly complete depolymerization ( , calculated from the HPLC data) of PcPET wastes in 8 h (Fig. 4A), with a maximum production rate of 61.3 ghydrolyzed PET (Supplementary Fig. 17 and Supplementary Tables 9 and 10). A slight change in the crystallinity of PcPET was observed (reaching after 8 h ), which allows an immediate reuse of the remaining PcPET waste for depolymerization. In contrast, LCC required 16 h to reach depolymerization (calculated from the HPLC data) at , and the remaining PcPET exhibited a similar crystallinity of , demonstrating that the “physical aging” process was suppressed at lower temperatures. At the previously reported optimal reaction temperature of , LCC reached its maximal conversion of (calculated from the HPLC data) over 12 h , and no further increase was obtained after prolonged reaction time due to the higher deformability of PET chains. The remaining PcPET showed a high level of
Fig. 4 | Depolymerization of PcPET with TurboPETase and LCC in bioreactors. A Comparison of PcPET depolymerization kinetics for TurboPETae at and at and . Reactions were performed with substrate loading and enzyme loading at pH 8.0. PETdepolymerization percentages were calculated based on the released products measured by HPLC. Data are presented as mean s.d. ( biologically independent experiments). B Time courses of crystallinity, and changes during the
enzymatic hydrolysis. Data are presented as mean s.d. ( biologically independent experiments). C Nearly complete degradation of pretreated PcPET bottles by TurboPETase at with substrate loading and enzyme loading in water solution in a 7.5 L bioreactor. PET-depolymerization percentages were calculated based on NaOH consumption ( pH of the reaction mixture was regulated at 8.0). Data are from one independent experiment. Source data are provided as a Source Data file.
crystallinity, estimated at , which is in accordance with a previous report .
Many factors can affect enzymatic attack against the plastics: e.g., crystallinity, chain mobility, molecular size, surface topography and hydrophobicity . Among these, the role of low crystallinity in PET degradation has been extensively studied. A crystallinity exceeding has been previously proposed to significantly impede the enzymatic degradation process. In the depolymerization of non-meltquenched PET powders ( crystallinity), we found a significant reduction in degradation performance (Supplementary Fig. 18 and Supplementary Table 11). However, during the depolymerization of pretreated PcPET, even when the crystallinity reached after 4 h of degradation by at , we did not observe a rapid decline in degradation rate. Pfaff et al. showed that LCC is more efficient in hydrolyzing shorter polymers . Recent studies also demonstrated that heavily pretreated PET, with a degree of polymerization (DP) less than 20 and high crystallinity, can remain highly degradable . These findings underscored the importance of factors other than crystallinity in enzymatic degradation, and promoted further investigation through gel permeation chromatography (GPC) analysis, which revealed notably low weight-average ( ) and number-average ( ) molecular masses in PcPET powders compared to non-melt-quenched PET powders (Supplementary Table 12). From the GPC results, we speculate that the low molecular weight of pretreated PcPET may contribute to the maintained degradation rate by at during the first several reaction hours, but a further increase in crystallinity to led to a substantial reduction in the amorphous
regions, impeding further enzymatic catalysis. Moreover, during the depolymerization process, we also observed a slight decrease in and moderate changes in , despite achieving over depolymerization (Fig. 4B). This is consistent with previous studies on PET degradation hydrolyzed by Cut190 . It has been suggested that the enzymatic hydrolysis of PET typically initiates with an endo-type chain scission after the enzyme binds to the PET surface. Further hydrolysis of the neighboring ester bonds within the amorphous regions occurs through an exo-type chain scission, leading to the release of soluble products . For polymers undergoing random scission, the probability of exo-scission inversely correlates with chain length . For PET with a DP around 200, the probability for exo-scission is substantially less than for endo-scission. In this study, the DP of the pretreated PcPET, approximately 50, is much lower than that of typical raw PET materials. This resulted in a significantly increased number of chain ends available for exo-scissions. Consequently, endo-type scission, which would reduce the DP of PET, no longer held a dominant proportion, which may potentially explain the minimal changes in molecular size observed during the PET depolymerization process.
To explore the industrial viability, we scaled up the reaction and performed it in a 7.5 L bioreactor with a PET loading of in water solution at pH 8.0 . Enzyme loading is of particular concern to the industry since enzyme cost is among the essential factors in the biomass depolymerization process. In contrast to using high enzyme concentrations up to in the biomass conversion process enzyme loading is saturated in PET degradation. Based on the TEA model , a change from to enzyme loading levels only results in a increase in MSP (US ), whereas the capital and operating costs would increase by at least to maintain the prolonged reaction duration when low enzyme loading levels are used. More importantly, in large-scale reactions with substrate loading, the reduction in enzyme activity and depolymerization efficiency was more significant at lower enzyme loading levels . In PET degradation process reported by Tournier et al., a final conversion of was achieved by LCC using enzyme loading compared to the conversion with enzyme loading . Hence, a enzyme loading was chosen to attain a balance between productivity and enzyme cost. As shown in Fig. 4C, the scaledup reaction progression is almost linear for the first 2 h with approximately 57% depolymerization achieved, and followed by a slower phase from 2 to 8 h . According to the kinetic parameters obtained from Fig. 3, we suggested that the reduced reaction rate may be attributed to the substantial decrease in PET loading at high conversion levels. Despite the decreased hydrolytic efficiency, the approximately depolymerization ( calculated from the HPLC data, calculated from the consumed NaOH and calculated from the weight loss, as listed in Supplementary Table 10) achieved within 8 h during the scaled-up reaction makes pilot-scale production feasible. Further process refinement could potentially optimize the depolymerization efficiency for lower concentrations of PET waste.
Discussion
Biocatalytic PET depolymerization offers a sustainable and energyefficient approach to PET recycling, presenting a more environmentally friendly alternative to current disposal methods such as landfills and incineration. While significant progress has been made to this end, the ultimate goal is to develop enzymes and processes suitable for industrial-scale applications. Both high substrate solids content and conversion benefit the economics of PET biodegradation, as it reduces both capital and operational expenditures. However, the material slurry exhibits a high apparent viscosity due to the high solids loading, leading to limited mass and heat transfer, which reduces the efficiency of enzymes in the early stages of hydrolysis. More importantly, increasing the solids loading to industrially relevant levels would lower the depolymerization yield due to the inhibition by high product concentrations . Hence, mere thermostability of the enzyme may not suffice for industrial PET degradation. Multiple factors interplay, influencing enzyme efficacy in real-world scenarios. An aspect often overlooked by the scientific community is the variance in heterogeneous enzymatic hydrolysis between laboratory experiments and industrial production. For instance, the depolymerization activity of PET can be enhanced in the laboratory by fusion with noncatalytic binding modules to increase the enzyme concentration on the PET surface. However, this advantage is completely lost at an industrial solids loading level . Despite recent advancements in engineering PET hydrolases to improve their performance in PET depolymerization, efforts are still needed in the quest of new PET hydrolases to address conversion loss under industrially relevant conditions.
Here, we employed a hybrid computational strategy to redesign a PET hydrolase that significantly outperforms other well-known PET hydrolases. Nearly full degradation of postconsumer PET bottles was achieved at an industrially relevant level of solids loading, rendering this highly efficient, optimized enzyme a good candidate for future applications in industrial plastic recycling processes. The mechanism responsible for enhancing enzyme performance has been demonstrated via kinetic analyses derived from an inverse Michaelis-Menten reaction regime as well as structural analysis, highlighting the importance of improving the specific polymer interactions on specific attack sites rather than general nonspecific surface adsorption. The results may help further knowledge on heterogeneous reaction catalysis and shall be beneficial for designing industrial viable plastic-degrading
enzymes to address the challenges associated with other more abundant plastics, such as polyurethanes with hydrolysable backbones. While the potential of enzyme design drives further developments in the improvement of enzyme performance, it is crucial to assess their depolymerization efficiency under industrially relevant conditions to demonstrate practical feasibility.
Methods
Training the Transformer model
Unsupervised training datasets. Homologous sequences of BhrPETase and LCC were searched from Uniclust30 (version 2018_08) and the BFD database with HHblits (the number of iterations was set as 4, and other parameters were left as default values) using 15 seed sequences in Pfam family PF01083 as queries. All searched sequences were clustered at identity using CD-HIT to obtain 10847 sequences. Since BFD and Uniclust30 were clustered at very low similarity, they may have been undersampled for fitness modelling in very close regions. Previous work by Frazer et al. sampled MSA built with more similar proteins to predict disease variants. We also retrieved 15051 sequences belonging to PF01083 from the UniProt database.
Training details and the prediction of “less-fitted” candidates. To model the fitness distance of sequences, we trained a neural network with an encoder-decoder from starch. We used a Transformer encoder to process input amino acid sequences with absolute position embedding. Unlike other models such as recurrent and convolutional networks, the Transformer made no assumption on sequence ordering and was more powerful at capturing long-distance relationships in sequence because of the attention mechanism (Eq. 1):
We applied multi-head self-attention as described by ref. 60. The encoder consists of 3 Transformer layers with 8 heads using an embedding size of 512 . Based on the encoder embeddings, the decoder generates probabilities of each token. The model was trained with a masked language modelling objective to predict the real amino acid at the masked position. In this study, of tokens were replaced with mask tokens during training. We used the Adam optimizer with a learning rate set to . Models were trained for 20 epochs using a batch size of 32 . Residues were filtered by sorting by the logits assigned to the WT amino acid. The top ten residue positions with the highest average scores from the prediction of each model were selected. The average score was calculated using the following equation:
where is the average score of the predicted residue position, and are the predicted logits of the single point mutation in the residue position and the wild-type amino acid, respectively. Excluding the duplicated positions, a total of 18 residue positions were generated, among which W104, H164, M166, W190, H191, H218, and F/I243 were suggested to be located on the PET-binding groove.
Design of stabilizing mutations by the GRAPE strategy
The GRAPE strategy reported in our previous study was used to improve the protein stability. The sequence of BhrPETase was submitted to the GRAPE-WEB online server (https://nmdc.cn/grape-web/). Based on the sequence information of BhrPETase , AlphaFold2 was used to predict the structure model. Subsequently, energy calculations with FoldX , Rosetta_cartesian_ddg , and ABACUS were used to predict stabilizing mutations. The DDD algorithm was used to predict the
suitable disulfide bonds. The thresholds for ABACUS, FoldX, and Rosetta were set to and -1.5 REU , respectively.
Molecular docking and MD simulations
TurboPETase was modelled by AlphaFold2. Then, the protein was simulated for 20 ns . The representative structure obtained from the MD simulations and the crystal structure of BhrPETase (PDB ID: 7EOA) was used for further docking. A model substrate comprising three consecutive PET units, mimicking the N-terminal and C-terminal groups to cap the polymer chain at both ends, was generated. YASARA was employed to make molecular docking of PET to TurboPETase and BhrPETase. The highest binding energy model was selected. The selected model was subjected to local docking for 999 runs. The optimized model of TurboPETase-PET and BhrPETase-PET complexes were simulated in AMBER using the ff16SB force field. The His242 residue was protonated in the HID state. ions were added to maintain system neutrality. The systems were then solvated in a truncated octahedron box using the TIP3P water model with an distance around the solute. Following solvation, the systems underwent a 12,000-step minimization and were gradually heated from 0 K to 338 K with a collision frequency of . After equilibration, 100 ns MD simulations were conducted for each complex, employing a time step of 2 fs .
Cloning
Genes encoding BhrPETase (GenBank accession number: GBD22443), LCC (GenBank accession number: AEV21261), LCC (LCC S283C/1127G), ICCG (ICCG ), FastPETase (ISPETase , ISPETase GenBank accession number: BBYR01000074), HotPETase (IsPETase S214Y/O119K/S213E/R90T/O182M/N212K/R224L/S58A/S61V/K95N/M154G/N241/K252M/T270Q), DepoPET ase (ISPETase ), PES-H1 and CaPETase (CaPETase , PETase GenBank accession number: SHM40309) were synthesized and optimizated for expression in Escherichia coli (General Biosystems, Anhui, China). The signal peptide of BhrPETase, LCC, LCC , ICCG and PETase were removed from the synthetic DNA. The synthesized genes for BhrPETase, LCC, LCC , PES-H1 and FastPETase were cloned into the Nhel and Xhol sites of the pBAD vector (containing an N-terminal His-tag), whereas the gene for ICCG , DepoPETase, CaPETase and HotPETase were cloned into the Ndel and Xhol sites of the pET-21a(+) vector (containing a C-terminal His-tag). A list of nucleotide sequences is provided in Supplementary Table 13.
Site-directed mutagenesis
BhrPETase mutants were generated using the QuickChange sitedirected mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). The PCR products were subsequently treated with DpnI (New England Biolabs, Ipswich, MA, USA) to digest the original DNA template and then introduced into E. coli TOP10 cells. Verification of the introduced mutations was carried out through DNA sequencing (Tianyi Huiyuan, Beijing, China).
Protein purification
Plasmids containing the genes of BhrPETase, , FastPETase and their variants were transformed into E. coli BW25113. Plasmids of ICCG , DepoPETase and CaPETase were transformed into E. coli C41(DE3), while the plasmid of HotPETase was transformed into . coli Rosetta gami-B cells (competent cells of the expression strains were purchased from Zoman Biotech, Beijing, China). The cells were cultured in medium at to an of , and then protein expression was induced by adding L-arabinose (BhrPETase, LCC, LCC , PES-H1 , FastPETase and their variants) or 1 mM isopropyl -D-thiogalactopyranoside (ICCG , DepoPETase, CaPETase and HotPETase). The cells were cultured for 20 h at , harvested by
centrifugation ( ) and suspended in lysis buffer ( and 20 mM imidazole, pH 7.5 ). Cell disruption was carried out through ultrasonication on ice. The resulting cell extracts were obtained after centrifugation at for 60 min at , followed by filtration through a Millex filter to remove precipitates. The unbound proteins were washed away in a 5 mL HisTrap HP column with washing buffer ( , and 60 mM imidazole, pH 7.5 ), the target protein was eluted with an elution buffer ( and 300 mM imidazole, pH 7.5 ). Afterwards, the buffer was exchanged for a storage buffer and 100 mM NaCl , pH 7.5) by using a HiPrep Desalting column. The purified enzyme was stored at . Protein concentrations were determined by the BCA method with bovine serum albumin as the reference.
PET depolymerization assay using Gf-PET films
Evaluating the activity of the mutants during BhrPETase engineering. The amorphous Gf-PET film (Goodfellow, thickness, product number ES301445, , approximately 15 mg ) was soaked in of 1 M potassium phosphate buffer ( solids loading) with of purified enzyme ( enzyme loading) at for 3 h . In HPLC analysis, the mobile phase consisted of buffer and buffer (Buffer A: formic acid in distilled water; Buffer B: acetonitrile). The flow rate was and the separation was carried out at with detection performed at 260 nm .
Comparing TurboPETase with other PET hydrolases under identical reaction conditions. The initial activities of the PET hydrolases at different temperatures were estimated. The Gf-PET film ( , approximately 15 mg ) was soaked in of 100 mM potassium phosphate buffer ( solids loading) with ( 0.3 enzyme loading) or ( enzyme loading) of purified enzyme at 50,60 or for 3 h , and then the supernatant was analysed by HPLC to quantify the concentration of released PET monomers.
Due to the high extent of conversion after prolonged reactions, the time-course analysis was performed in 1 M potassium phosphate buffer ( pH 8.0 ) to provide sufficient buffering capacity. The Gf-PET film ( , approximately 15 mg ) was soaked in of 1 M potassium phosphate buffer ( solids loading) with ( enzyme loading) of purified enzyme at 50 , 60 or for 12 h .
Comparing TurboPETase with other PET hydrolases under their reported reaction conditions. The Gf-PET film ( , approximately 15 mg ) was soaked in buffer ( solids loading) for 12 h . For comparison with FastPETase, the Gf-PET film was soaked in 100 mM potassium phosphate buffer ( pH 8.0 ) at with of purified enzyme ( g enzyme loading). For comparison with HotPETase, the Gf-PET film was soaked in 50 mM Glycine-NaOH buffer ( pH 9.2 ) at with of purified enzyme ( enzyme loading). For comparison with LCC , the Gf-PET film was soaked in 100 mM potassium phosphate buffer ( pH 8.0 ) at with of purified enzyme ( enzyme loading) for 3 h .
Depolymerization of the pretreated PET bottles
PET bottles pretreatment. Postconsumer PET bottles were collected from the garbage collection station in Beijing, China. The bottles were washed with deionized water and dried and then micronized into small flakes using a crusher JZ-T-005 (Shiyan Precision Instruments, Dongguan, China). The flakes were subsequently amorphized using a twinscrew extruder SY-6219-20/32 (Shiyan Precision Instruments, Dongguan, China). The set temperatures were in the extruder zones, in the melt pump, and in the screen changer zones. The obtained amorphous fibres of PET were further micronized by a DFY-1000D grinder (DingLi, Wenzhou, China) at room temperature. After sieving, PET powders with particle sizes less than were obtained (11.1% crystallinity on average).
Comparing the depolymerization performance of TurboPETase and LCC . Twenty grams of pretreated PET powder and 80 mL of 100 mM potassium phosphate buffer ( pH 8.0 ) containing 40 mg of purified enzyme were mixed in a 200 mL bioreactor (Kusnc, Shanghai, China). A magnetic stirrer was utilized to maintain constant agitation at 300 rpm . The temperature was regulated at (or for ) by water bath immersion, and the pH was regulated at 8.0 by adding 4 M NaOH solution. Assays were performed in triplicate and evaluated accordingly.
During the reaction, the percentage of PET depolymerization was evaluated by analysing the supernatant through HPLC. The final depolymerization yield was additionally verified by investigating the weight of residual PET. The reaction mixture was filtered through Whatman grade 1 qualitative filter papers (GE Healthcare, USA). The residue was washed twice with deionized water and then dried at overnight. To evaluate the proportion of PET in the residue, 200 mg of the dried residue was incubated in 20 mL of 4 M NaOH solution at for 3 h . Afterwards, the amount of released TPA was measured by HPLC analysis. The overall weight of the residue solids and the proportion of PET are shown in Supplementary Table 9.
Estimating the depolymerization performance of TurboPETase at a larger scale. A total of 0.5 kg pretreated PET powder, 1.9 L of deionized water and 0.1 L of TurboPETase solution (containing 1 g of purified enzyme) were combined in a 7.5 L bioreactor (New Brunswick Scientific, Edison, NJ, USA). The stirring rate was maintained at 300 rpm , the temperature was regulated at , and the pH was regulated at 8.0 by adding 4 M NaOH solution. The percentage of PET depolymerization during the reaction was evaluated according to base consumption. Additionally, the final depolymerization yield was verified by analysing the residue (alkaline hydrolysis and HPLC analysis methods were identical to the protocol above), which was collected by centrifugation ( ), washed twice with deionized water and dried at overnight.
Determination of apparent melting temperatures, PET crystallinity and molecular weights
To determine apparent melting temperatures, a fluorescence-based thermal stability assay was employed. A -fold diluted SYPRO Orange dye (Molecular Probes, Life Technologies, USA) was added in a protein solution. The mixture was placed in a thin-walled 96-well PCR plate, sealed with optical-quality sealing tape. The mixture was heated in a CFX 96 real-time polymerase chain reaction (PCR) system (BioRad, Hercules, CA, USA) from 25 to at a heating rate of . The crystallinity of PET and PBT was analysed by differential scanning calorimetry (DSC). The parameters were set following a previously reported procedure . For molecular weight analysis of PET, gel permeation chromatography (GPC) analysis was applied. GPC measurements were carried out using an Agilent 1260 system (Agilent 1260, Agilent Technologies Inc., U.S.A.) with a Refractive Index (RI) detector. The mobile phase consisted of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), and the analysis was performed at with a flow rate of .
Kinetics analysis
For PET depolymerisation, initial rate measurements were collected for the and datasets. For the dataset, Gf-PET films over a range of were treated in of 100 mM potassium phosphate buffer ( pH 8.0 ) with enzyme loadings of , and . For the dataset, , , and Gf-PET films were treated in of 100 mM potassium phosphate buffer ( pH 8.0 ) with enzyme loading levels ranging from 0 to . The reaction mixture was incubated at for 1 h . All reactions were performed in triplicate and terminated by heating to for 10 min . The supernatant obtained by centrifugation ( ) was then analysed by HPLC. The data were fitted using Matplotlib in python.
For 4-Nitrophenol butyrate (pNPB), all reactions were performed in 96 -well plates in a total reaction volume of . The final concentrations of range from 0.2 to 1.4 mM , and the final concentration of enzymes was of the solution of NPB (in anhydrous ethanol), of 10 mM potassium phosphate buffer ( pH 8.0 ), and of enzymes were incubated at for 3 min . Then, of anhydrous ethanol was added to quench the enzymatic reactions. Each reaction was conducted in triplicate. The nitrophenol products were measured at 405 nm using an Infinite PRO microplate reader (Tecan, Switzerland). One unit of enzyme activity was defined as the amount of enzyme required to convert of NPB per min. The data were fitted using Matplotlib in python.
For MHET, the reactions were performed in a total volume of . The final concentrations of MHET range from 6 to 60 mM (in 100 mM potassium phosphate buffer, pH 8.0 ), and the final concentration of enzymes was . The reactions were conducted at in triplicate for 15 min , then were quenched by adding of dimethylsulfoxide. The hydrolysis extent was measured by HPLC. The data were fitted using Matplotlib in python.
Binding isotherms
Adsorption measurements for TurboPETase, BhrPETase, LCC and bovine serum albumin (BSA) were conducted in a 100 mM potassium phosphate buffer ( pH 8.0 ) using low-binding Eppendorf tubes . A GfPET film ( , approximately 15 mg ) was soaked in of buffers containing protein concentrations ranging from 0 to , maintained at for 1 h equilibration period. Afterward, of the supernatant was mixed with of freshly prepared BCA working solution. This mixture underwent incubation at for 30 min , followed by absorbance measurements at 562 nm using a plate reader to determine the concentration of free proteins within the solution. The quantities of bound proteins were then deduced by calculating the difference between the total protein concentrations and the concentrations of free proteins.
Determination of free cysteine residues
To identify the free cysteine residues in the solution of TurboPETase, 5,5′-Dithiobis-(2-nitrobenzoic acid) (DTNB) was used of enzyme solution ( of BhrPETase or TurboPETase) or L-cysteine solution ( and ), of freshly prepared DTNB solution ( DTNB in 100 mM potassium phosphate buffer, pH 8.0) and of EDTA solution ( 100 mM ethylene diamine tetraacetic acid in 100 mM potassium phosphate buffer, pH 8.0 ) were mixed thoroughly and incubated for 15 min . Afterward, the absorption of samples at 412 nm was measured using the spectrophotometer (MAPADA V-1100D, Shanghai, China).
Depolymerization of PBT
The PBT films were soaked in of 100 mM potassium phosphate buffer ( solids loading) with of purified enzyme ( enzyme loading) at for 3 h .
Depolymerization with a dual-enzyme system
The Gf-PET film ( , approximately 2 mg ) was soaked in 1 mL of 100 mM potassium phosphate buffer ( solids loading) containing a two-enzyme system ( TurboPETase
and BHETase loading ranging between 0 and ) and incubated at for 9 h . For a substrate loading of , the GfPET film ( , approximately 15 mg ) was soaked in of 100 mM potassium phosphate buffer ( solids loading) containing the two-enzyme system. The mixture was incubated at for 1 h with TurboPETase and BHETase concentrations ranged from 0 to . The supernatant was analysed by HPLC.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data that support the findings of this study are presented within the article and its supplementary files. For further inquiries or requests for additional information, please contact the corresponding authors. Source data are provided with this paper.
Jambeck, J. R. et al. Plastic waste inputs from land into the ocean. Science 347, 768-771 (2015).
MacLeod, M. et al. The global threat from plastic pollution. Science 373, 61-65 (2021).
Wei, R. & Zimmermann, W. Microbial enzymes for the recycling of recalcitrant petroleumbased plastics: how far are we? Microb. Biotechnol. 10, 1308-1322 (2017).
Bornscheuer, U. T. Feeding on plastic. Science 351, 1154-1155 (2016).
Ellis, L. D. et al. Chemical and biological catalysis for plastics recycling and upcycling. Nat. Catal. 4, 539-556 (2021).
Kakadellis, S. & Rosetto, G. Achieving a circular bioeconomy for plastics. Science 373, 49-50 (2021).
Wei, R. et al. Possibilities and limitations of biotechnological plastic degradation and recycling. Nat. Catal. 3, 867-871 (2020).
Wei, R. et al. Mechanism-based design of efficient PET hydrolases. ACS Catal. 12, 3382-3396 (2022).
Taniguchi, I. et al. Biodegradation of PET: current status and application aspects. ACS Catal. 9, 4089-4105 (2019).
Kawai, F., Kawabata, T. & Oda, M. Current knowledge on enzymatic PET degradation and its possible application to waste stream management and other fields. Appl. Microbiol. Biotechnol. 103, 4253-4268 (2019).
Modenbach, A. A. & Nokes, S. E. Enzymatic hydrolysis of biomass at high-solids loadings-a review. Biomass-. Bioenergy 56, 526-544 (2013).
Singh, A. et al. Techno-economic, life-cycle, and socioeconomic impact analysis of enzymatic recycling of poly(ethylene terephthalate). Joule 5, 2479-2503 (2021).
Graham, R. et al. The role of binding modules in enzymatic poly (ethylene terephthalate) hydrolysis at high-solids loadings. Chem. Catal. 2, 2644-2657 (2022).
Ayla, S. D. S. et al. Constraints and advances in high-solids enzymatic hydrolysis of lignocellulosic biomass: a critical review. Biotechnol. Biofuels 13, 58 (2020).
Tournier, V. et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 580, 216-219 (2020).
Bell, E. L. et al. Directed evolution of an efficient and thermostable PET depolymerase. Nat. Catal. 5, 673-681 (2022).
Wei, R. et al. Biocatalytic degradation efficiency of postconsumer polyethylene terephthalate packaging determined by their polymer microstructures. Adv. Sci. 6, 1900491 (2019).
Cui, Y., Sun, J. & Wu, B. Computational enzyme redesign: large jumps in function. Trends Chem. 4, 409-419 (2022).
Sulaiman, S. et al. Isolation of a novel cutinase homolog with polyethylene terephthalate-degrading activity from leaf-branch compost by using a metagenomic approach. Appl. Environ. Microbiol. 78, 1556-1562 (2012).
Ding, Z. et al. Rational redesign of thermophilic PET hydrolase LCCICCG to enhance hydrolysis of high crystallinity polyethylene terephthalates. J. Hazard. Mater. 453, 131386 (2023).
Xi, X. et al. Secretory expression in Bacillus subtilis and biochemical characterization of a highly thermostable polyethylene terephthalate hydrolase from bacterium HR29. Enzym. Microb. Technol. 143, 109715 (2021).
Lu, H. et al. Machine learning-aided engineering of hydrolases for PET depolymerization. Nature 604, 662-667 (2022).
Shi, L. et al. Complete depolymerization of PET wastes by an evolved PET hydrolase from directed evolution. Angew. Chem. Int. Ed. Engl. 62, e202218390 (2023).
Hong, H. et al. Discovery and rational engineering of PET hydrolase with both mesophilic and thermophilic PET hydrolase properties. Nat. Commun. 14, 4556 (2023).
Pfaff, L. et al. Multiple substrate binding mode-guided engineering of a thermophilic PET hydrolase. ACS Catal. 12, 9790-9800 (2022).
Joho, Y. et al. Ancestral sequence reconstruction identifies structural changes underlying the evolution of Ideonella Sakaiensis PETase and variants with improved stability and activity. Biochemistry 62, 437-450 (2023).
Erickson, E. et al. Sourcing thermotolerant poly (ethylene terephthalate) hydrolase scaffolds from natural diversity. Nat. Commun. 13, 7850 (2022).
Wei, R. et al. Conformational fitting of a flexible oligomeric substrate does not explain the enzymatic PET degradation. Nat. Commun. 10, 5581 (2019).
Ding, X. & Zou, Z. & Brooks lii, C. L. Deciphering protein evolution and fitness landscapes with latent space models. Nat. Commun. 10, 5644 (2019).
Alley, E. C. et al. Unified rational protein engineering with sequencebased deep representation learning. Nat. Methods 16, 1315-1322 (2019).
Biswas, S. et al. Low-N protein engineering with data-efficient deep learning. Nat. Methods 18, 389-396 (2021).
Zeng, W. et al. Substrate-binding mode of a thermophilic PET hydrolase and engineering the enzyme to enhance the hydrolytic efficacy. ACS Catal. 12, 3033-3040 (2022).
Chen, C.-C. et al. General features to enhance enzymatic activity of poly(ethylene terephthalate) hydrolysis. Nat. Catal. 4, 425-430 (2021).
Cui, Y. et al. Computational redesign of a PETase for plastic biodegradation under ambient condition by the GRAPE strategy. ACS Catal. 11, 1340-1350 (2021).
Delgado, J., Radusky, L. G., Cianferoni, D. & Serrano, L. FoldX 5.0: working with RNA, small molecules and a new graphical interface. Bioinformatics 35, 4168-4169 (2019).
Park, H. et al. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J. Chem. Theory Comput. 12, 6201-6212 (2016).
Xiong, P. et al. Protein design with a comprehensive statistical energy function and boosted by experimental selection for foldability. Nat. Commun. 5, 5330 (2014).
Arabnejad, H. et al. A robust cosolvent-compatible halohydrin dehalogenase by computational library design. Protein Eng. Des. Sel. 30, 173-187 (2017).
Arnal, G. et al. Assessment of four engineered PET degrading enzymes considering large-scale industrial applications. ACS Catal. 13, 13156-13166 (2023).
Kari, J., Andersen, M., Borch, K. & Westh, P. An inverse Michaelis-Menten approach for interfacial enzyme kinetics. ACS Catal. 7, 4904-4914 (2017).
Schiano-di-Cola, C. et al. Systematic deletions in the cellobiohydrolase (CBH) Cel7A from the fungus Trichoderma reesei reveal flexible loops critical for CBH activity. J. Biol. Chem. 294, 1807-1815 (2019).
Baath, J. A. et al. Comparative biochemistry of four polyester (PET) hydrolases. Chem. Bio. Chem. 22, 1627-1637 (2021).
Erickson, E. et al. Comparative performance of PETase as a function of reaction conditions, substrate properties, and product accumulation. Chem. Sus. Chem. 15, e202101932 (2022).
Badino, S. et al. Adsorption of enzymes with hydrolytic activity on polyethylene terephthalate. Enzym. Microb. Technol. 152, 109937 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583-589 (2021).
Heidrich, D. & Gehde, M. The 3-phase structure of polyesters (PBT, PET) after isothermal and non-isothermal crystallization. Polymers 14, 793 (2022).
Barth, M. et al. A dual enzyme system composed of a polyester hydrolase and a carboxylesterase enhances the biocatalytic degradation of polyethylene terephthalate films. Biotechnol. J. 11, 1082-1087 (2016).
Knott, B. C. et al. Characterization and engineering of a two-enzyme system for plastics depolymerization. Proc. Natl Acad. Sci. USA. 117, 25476-25485 (2020).
Mrigwani, A. et al. Conversion of polyethylene terephthalate into pure terephthalic acid through synergy between a solid-degrading cutinase and a reaction intermediate-hydrolysing carboxylesterase. Green. Chem. 24, 6707-6719 (2022).
Haugwitz, G. et al. Structural insights into (tere) phthalate-ester hydrolysis by a carboxylesterase and its role in promoting PET depolymerization. ACS Catal. 12, 15259-15270 (2022).
Tarazona, N. A. et al. Rapid depolymerization of poly (ethylene terephthalate) thin films by a dual-enzyme system and its impact on material properties. Chem. Catal. 2, 3573-3589 (2022).
Li, A. et al. Discovery and mechanism-guided engineering of BHET hydrolases for improved PET recycling and upcycling. Nat. Commun. 14, 4169 (2023).
Guo, B. et al. Conformational selection in biocatalytic plastic degradation by PETase. ACS Catal. 12, 3397-3409 (2022).
Guo, B. et al. Fast depolymerization of PET bottle mediated by microwave pre-treatment and an engineered PETase. Chem. Sus. Chem. 16, e202300742 (2023).
Kawai, F. et al. Efficient depolymerization of polyethylene terephthalate (PET) and polyethylene furanoate by engineered PET hydrolase Cut190. AMB Express 12, 134 (2022).
Resch, M. G. et al. Clean fractionation pretreatment reduces enzyme loadings for biomass saccharification and reveals the mechanism of free and cellulosomal enzyme synergy. ACS Sustain. Chem. Eng. 2, 1377-1387 (2014).
Frazer, J. et al. Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91-95 (2021).
Vaswani, A. et al. Attention is all you need. In 31st Conference on Neural Information Processing Systems. 5998-6008 (Association of Computational Machinery, 2017).
Case, D. et al. AMBER 14, University of California, San Francisco. (2014).
Acknowledgements
This work was supported by the National Key R&D Program of China (grant no. 2021YFC2103600 to C.L.L.), the National Natural Science Foundation of China (32225002 to B.W., 31822002 to B.W. and 32170033 to Y.L.C.), the Key Research Program of Frontier Sciences (ZDBS-LY-SM014 to B.W.), the Biological Resources Program (KFJ-BRP009 to B.W. and KFJ-BRP-017-58 to Y.L.C.) from the Chinese Academy of Sciences, the Informatization Plan of Chinese Academy of Sciences (CAS-WX2021SF-0111 to B.W.), the Youth Innovation Promotion Association CAS (2022086 to Y.L.C.), and the Postdoctoral Fellowship Program of CPSF (GZC2O232929 to T.Z.).
Author contributions
B.W. initiated the project. Y.L.C. and J.Y.S. performed the computational work, Y.C.C., H.P., and C.L.L. performed biochemical and biocatalytic experiments, T.Z. and W.C.G. performed polymer degradation experiments, Y.L.C. and B.W. drafted the manuscript, which was revised and approved by all authors.
Correspondence and requests for materials should be addressed to Yinglu Cui or Bian Wu.
Peer review information Nature Communications thanks Pedro Alexandrino Fernandes, and the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
AIM Center, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China. University of Chinese Academy of Sciences, Beijing, China. College of Chemistry, Nankai University, Tianjin, China. These authors contributed equally: Yinglu Cui, Yanchun Chen, Jinyuan Sun, Tong Zhu.
-e-mail: cuiyinglu@im.ac.cn; wub@im.ac.cn