إعادة تكوين الميكروبيئة لذرات النيكل الفردية عالية النشاط على Mo2C المدمج بالأكسجين لتفكيك الماء Microenvironment reconstitution of highly active Ni single atoms on oxygen-incorporated Mo2C for water splitting
إن التصميم العقلاني لمحفزات كهربائية أحادية الذرة ثنائية الوظيفة الفعالة لتفكيك الماء الصناعي والفهم الشامل لآلياتها التحفيزية المعقدة لا يزال يمثل تحديًا. هنا، نبلغ عن ذرات النيكل الأحادية المدعومة على الأكسجين المدمج.عبرروابط الجسر، التي تعطي نشاطًا ثنائي الوظيفة عاليًا في تفاعل تطور الأكسجين (OER) وتفاعل تطور الهيدروجين (HER). من خلال مطيافية الامتصاص بالأشعة السينية باستخدام السنكروترون والمجهر الإلكتروني، وجدنا أنه بعد تفاعل HER، فإن عدد التنسيق وأطوال الروابط لـ و تم تغيير جميعها، ومع ذلك لا تزال أنواع النيكل موزعة على المستوى الذري. على النقيض من ذلك، بعد عملية OER، تم تجمع النيكل الموزع ذريًا في مجموعات صغيرة جدًا مع جديدة ( ظهرت الروابط. من خلال دمج النتائج التجريبية وحسابات DFT، نستنتج درجة الأكسدة لـوتعتبر تكوينات ذرات النيكل الفردية ضرورية لكل من تفاعل تقليل الهيدروجين (HER) أو تفاعل أكسدة الأكسجين (OER). توفر هذه الدراسة استراتيجية ونموذجًا قابلين للتطبيق لتصميم محفزات كهربائية عالية الكفاءة لتحليل الماء.
يُعتبر التحليل الكهربائي للماء على نطاق واسع واحدًا من أهم وأعدّ الطرق الواعدة لتحويل الطاقة المتجددة إلى هيدروجين وأكسجين عاليي النقاء.لعملية فصل الماء المستمرة بشكل عام في التطبيق العملي، فإن دمج تفاعلات الأقطاب الكهربائية معًا في جهاز التحليل الكهربائي المتكامل يعد أمرًا صعبًا بسبب عدم التوافق في نطاقات الرقم الهيدروجيني التي تظل فيها هذه المحفزات مستقرة وتبقى الأكثر نشاطًا.بالإضافة إلى ذلك، يتطلب إنتاج محفزات مختلفة لكل من المحفزات أحادية الوظيفة معدات وعمليات مختلفة، مما قد يزيد من التكلفة مقارنة بالمحفزات ثنائية الوظيفة. لذلك، بالمقارنة مع المحفزات أحادية الوظيفة، تعتبر المحفزات ثنائية الوظيفة خيارات مفضلة نسبيًا لتفكيك الماء بشكل عام.حالياً، تعتبر المواد المعتمدة على البلاتين والمواد المعتمدة على الإيريديوم/الروديوم هي المعايير في محفزات تفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER) على التوالي. ومع ذلك، فإن ندرتها وارتفاع أسعارها تعيق تطويرها وتطبيقاتها الواسعة.. في هذه الأثناء، هناك عدد قليل فقط من المحفزات OER (مثل و مستقرة في المحاليل الحمضية، وبالتالي تصميم حلول فعالة من حيث التكلفة و المحفزات الكهربائية ثنائية الوظيفة المتوفرة بكثرة على الأرض والتي تتمتع بكفاءة عالية في عملية التحليل الكهربائي للماء في نفس الظروف القلوية لها أهمية وضرورة كبيرة..
تعتبر المحفزات ذات الذرة الواحدة (SACs) التي تعظم معدلات استخدام الذرات وتأثيرات التآزر بين مواقع الذرات الواحدة والدعم الخاص بها الاستراتيجية الأكثر وعدًا لتصميم المحفزات الكهربائية ثنائية الوظيفة الفعالة من حيث التكلفة والمتوفرة بكثرة في الطبيعة.. حاليًا، تعتمد معظم المحفزات الكهروكيميائية على الركائز الكربونية، مثل الكربون غير المتبلور، والجرافين، مع ذرات معدنية معزولة منسقة بواسطة عيوب داخلية أو ذرات غير متجانسة. ومع ذلك، تظهر هذه المحفزات نشاطًا غير كافٍ عند تطبيقها في تفاعل تقليل الهيدروجين، وخاصة في تفاعل أكسدة الأكسجين، بينما أظهرت تحسينًا واضحًا في الكهروكيميائية.تفاعلات الاختزال. لذا، فإنها تمثل تحديًا حاسمًا لتطوير المحفزات الأحادية الذرة مع دعم جديد عالي التوصيل. الكربيدات القائمة على المعادن الانتقالية، مثل كربيد الموليبدينوم ( )، كانت تتمتع بتوصيل كهربائي شبيه بالمعادن وحظيت باهتمام خاص كمواد ثنائية الوظيفة المحتملة
المحفزات الكهروكيميائية لتفكيك الماء. ومع ذلك، فإن أداء التحفيز لتفكيك الماء للمواد النقيةبعيد عن الرضا بسبب ضعف قوة الامتزاز لعينات الأكسجين وبطء حركية التفككو intermediates تفاعل أخرىلمواجهة هذا التحدي، واحدة من أكثر الاستراتيجيات فعالية هي التزيين بالمعادن الانتقالية.، و W ) . بالإضافة إلى ذلك، فإن السند الجسريمعدنيمكن أن يؤدي التفاعل بين مادة الركيزة والذرات الفردية إلى إنتاج ارتباط إلكتروني قوي عبر السطح، مما ينتج عنه نقل فعال للشحنات، والذي يمكن أن يحسن سلوك الامتزاز/التحرر لموقع النشاط للوسائط الأكسجينية ووسائط الهيدروجين.. على سبيل المثال، أفادت مجموعة سوريانتو أن السطح فريد من نوعهيمكن أن تؤثر التكوينات على امتصاص الطاقة الحرة لجيبس للوسط التفاعلي و ثم كسر العلاقة النسبية وتعزيز أنشطة OER و HERلذا، من الممكن وتحدي بناء مراكز معدنية موزعة ذريًا على السطح الغني بالأكسجين.لتعديل سلوك الامتزاز/التحرر بشكل فعال للوسائط الرئيسية خلال عملية تحليل الماء.
علاوة على ذلك، مع الرؤى حول العملية الكهروكيميائية، أظهرت الدراسات أن العديد من المحفزات الكهروكيميائية تخضع لإعادة التكوين خلال ما يسمى بعملية التنشيط أو قياس الفولتمترية الدورية الأولية. أما بالنسبة لمثال HER، فقد أظهر لي وآخرون أن الـ “حدث تغيير هيكلي خلال عملية تنشيط HER، مما أدى إلى نشاط تحفيزي عالي بالإضافة إلى الاستقرار.. بالإضافة إلى ذلك،تم توليد المواقع النشطة الفعلية لـ HER بواسطة هيكل النواة@القشرة لـالمحفز الكهربائي المسبق من خلال إعادة التكوين المدفوعة بالجهد. بالإضافة إلى ذلك، أظهرت مجموعة وانغ أن الأنواع النشطة لـ HER من نشأت NBs من تكوينات P-Co-O-Fe-P المعاد بناؤها في الموقع مع مواقع المعادن ذات القيمة المنخفضة.)، مما يمكن أن يقلل من حواجز الطاقة لتفكك الماء وامتصاصه المتوسطاتعلى النقيض الحاد، نشأت نشاطاتهم في مجال الموارد التعليمية المفتوحة من الجسور الأكسجينية، ذات التكافؤ العاليالأجزاء. بسبب عملية الأكسدة والاختزال في تفاعل الأكسدة الكهربائي، من المرجح أن تكون إعادة تكوين سطح المحفز مصاحبة. معظم المحفزات تتأكسد بالكامل إلى أكاسيد أو هيدروكسيدات المعادن الانتقالية، أو يمكن إعادة بناء أسطحها إلى بيئات تنسيق مختلفة. حدد زو وآخرون أنتم تحويل المحفز إلى الشكل منخفض البلوريةخلال عملية OER ووجدت أنتعمل كنوع تفاعلي لتفاعل أكسدة الأكسجين بدلاً من الأصلنوع. في هذه الأثناء، أفادت مجموعة لي بإعادة تشكيل تم تشكيل المحفزات، أي، وحدات FeNi المربوطة بواسطة الأكسجين بعد المعالجة الكهروكيميائية، مما وفر النشاط التحفيزي العالي لتطور الأكسجين.لقد أظهرت جميع الأعمال الرائدة المذكورة أعلاه أن قدرة المحفز على “التطور” خلال التفاعل قد تكون سمة رئيسية من سمات المحفزات النشطة للغاية لتفاعل أكسدة الماء. لذا، فإن فهم التحول الهيكلي للمحفزات أمر حاسم لكشف الآلية الحقيقية للتفاعل التحفيزي وتصميم محفزات كهربائية جديدة، على الرغم من أنها نادراً ما تم دراستها بالنسبة للمحفزات الأحادية الذرة..
هنا، نحن مدفوعون أساسًا لاستخداممع السطح المؤكسد جزئيًا كمواد دعم لتثبيت ذرات النيكل الفردية. تمتلك المواد الناتجة طبقة رقيقة من مواقع النيكل الذرية، التي تفاعلت مع السطح المؤكسد لـعبرسندات الجسر ) وأظهرت نشاطًا تحفيزيًا جيدًا لتفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER) والانقسام الكلي للماء. من خلال استخدام مطيافية الامتصاص بالأشعة السينية في السنكروترون والملاحظات المجهرية الإلكترونية، وجدنا أن تكوين النيكل الموزع ذريًا فيالمحفز الكهربائي خضع لتحول والدعم كان مفرط الأكسدة أو مذاب بدرجات متفاوتة. بعد HER، كان عدد التنسيق لـوطول الرابطة لـ ( تم زيادة جميع الأنواع (Mo) ، لكن أنواع Ni لا تزال في شكل موزع ذري. ومع ذلك، بعد OER، زاد عدد تنسيق Ni O، وزاد عدد التنسيق لـ ( ) انخفضت ولكن جديدة ( ظهرت روابط )، مما يدل على أن النيكل الموزع ذريًا قد تجمّع في مجموعات صغيرة جدًا. تحت الجهود المطبقة. من خلال دمج النتائج التجريبية وحسابات DFT، وجدنا أن المادة مؤكسدة جزئيًاسيكون مفيدًا لأدائها. ومع ذلك، بالمقارنة مع النسخة الأصليةالمجموعات الصغيرة المعاد تشكيلها من النيكل المدفوعة بالقدرة مع الأمثل و الهياكل التنسيقية أكثر ملاءمة لتحفيز إنتاج.
النتائج
تركيب المواد وخصائصها
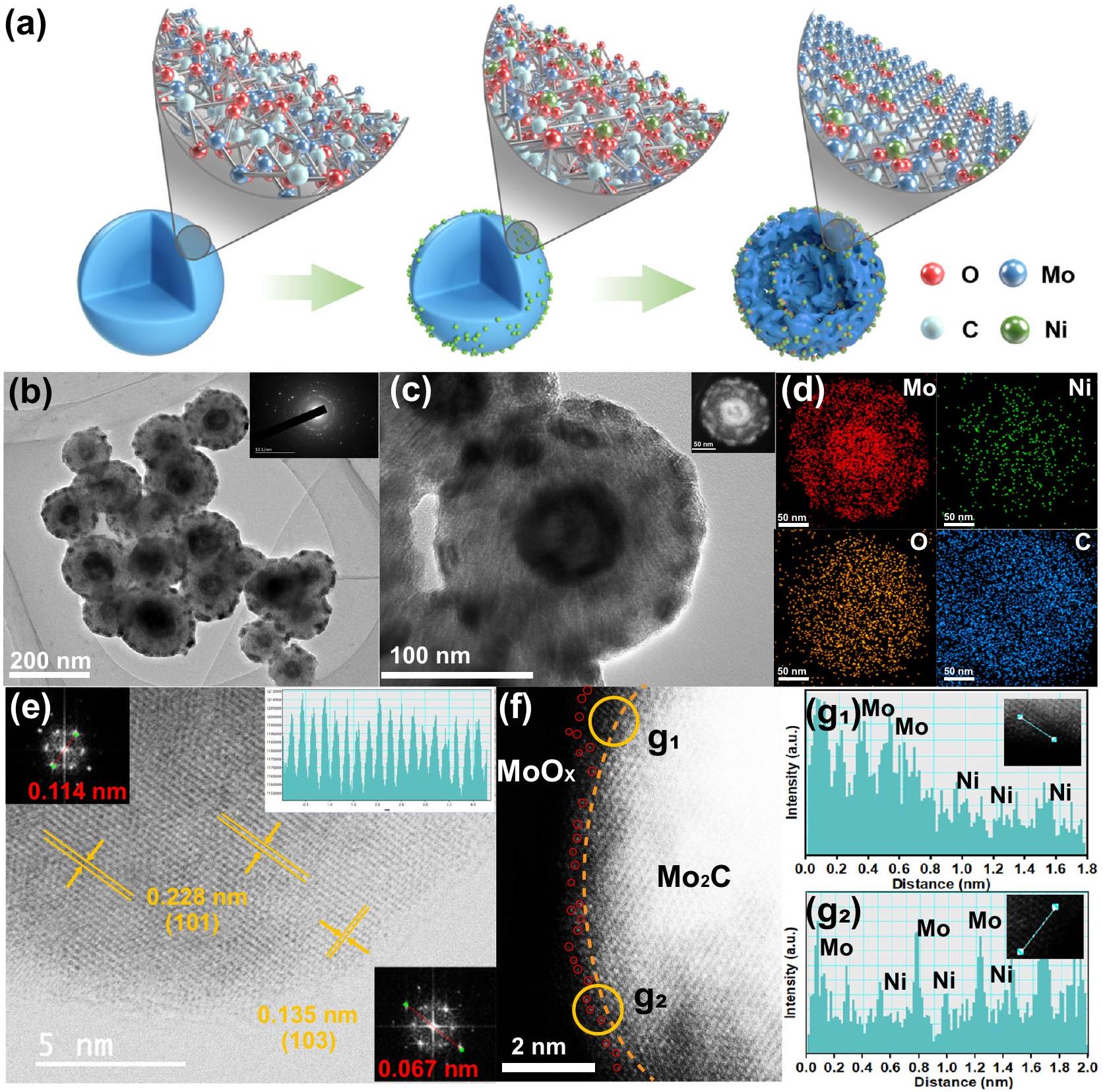
التم تصنيع الكرات النانوية من خلال الخطوات التالية: (1) تحضير سوائل نانوية عضوية قائمة على الموليبدينوم بطريقة هيدروحرارية سهلة؛ (2) امتصاص أيونات معدن النيكل عبر طريقة النقع؛ (3) معالجة حرارية عند درجات حرارة معينة تحت الهواء والجو. خلال عملية التحلل الحراري، تم تحويل المواد الأولية العضوية المعتمدة على الموليبدينوم في الوقت نفسه إلىبلورات نانوية تحتوي على أيونات النيكل مثبتة على أسطحها، والتي تم تضمينها في ركيزة كربونية موحدة. تظهر صور المجهر الإلكتروني الناقل (TEM) في الشكل S1 أن المادة السابقة غير المتبلورة المعتمدة على الموليبدينوم هي هيكل كروي نانوي ذو أسطح خشنة بقطر حوالي 200 نانومتر، ولا يتغير الشكل بشكل ملحوظ بعد امتصاص أيونات النيكل على السطح (الشكل S2). بعد التحلل الحراري، يمكن ملاحظة هيكل مجوف ذو قشرتين بوضوح (الشكل 1a، b). تم دراسة آلية تكوين الهيكل المجوف ذو القشرتين بالتفصيل (الأشكال S3-S6) ورسم توضيحي محتمل له موضح في الشكل S7، والذي يعتمد بشكل أساسي على الأكسدة المبكرة للهواء والانكماش غير المتوازن وغير المتجانس الناتج عن المعالجة الحرارية.أثناء تشكيل الهيكل المجوف ذو القشرة المزدوجة، كانت ظروف الهواء والجوين مهمان للغاية. بالإضافة إلى ذلك، فإن المساحات السطحية المحددة المستمدة منتظهر منحنيات الامتزاز-التحرر زيادة ملحوظة من 13.9 لـإلىلـ (الشكل S8)، والذي يرجع على الأرجح إلى تطاير النترات من مصادر النيكل. تشير أنماط حيود الأشعة السينية (XRD) (الشكل S9) إلى أن حيود يمكن أن يتم فهرسته بشكل جيد إلى النقي المرحلة (رقم بطاقة JCPDS 35-0787) لا يمكن ملاحظة قمم لمعدن النيكل، ولا أكسيد المعدن. نمط حيود الإلكترونات في المنطقة المختارة (المُدرج في الشكل 1ب) يشير إلى تعدد الأشكال لـ.
صور خرائط التحليل الطيفي بالأشعة السينية المشتتة للطاقة (EDS) تكشف أن، وعناصر O تقريبًا لها نفس الشكل المجوف ذو القشرة المزدوجة، وعناصر C موزعة بشكل موحد في فرد واحد الكرات النانوية (أدخل في الشكل 1c، والشكل 1d). بالاقتران مع الصورة المكبرة بتقنية HR-TEM في الشكل S10، نؤكد أكثر أن تم تضمين النانو كريستالات مع أيونات النيكل في ركيزة كربونية موحدة. لتحديد الموقع الدقيق لذرات النيكل الفردية بشكل أكبر فيفي النانوسفيرات، قمنا بإجراء تصوير مجهر إلكتروني نافذ (STEM) بتصحيح تشوهات المجال المظلم الحلقي العالي الزاوية (HAADF) بدقة تحت أنغستروم (الشكل 1e، f). الشكل 1e اكتشف بوضوح خطوط الشبكة مع مسافة بين الطبقات تبلغ 0.228 نانومتر و0.135 نانومتر، والتي تتوافق مع المستوى (101) و(103) من الشكل السداسي.، على التوالي. تكشف الملاحظة التفصيلية لحافة مجموعة ذرات الموليبدينوم اللامعة عن أكسيد الموليبدينوم غير المتبلور أو ذو البلورية المنخفضة (طبقة (حول الخط الأصفر في الشكل 1f) بسبب الأكسدة السطحيةبالإضافة إلى ذلك، بسبب التباين المختلف في Z، يمكن تحديد البقع الداكنة (الدوائر الحمراء في الشكل 1f) على حافة البقع الأكثر سطوعًا (ذرات الموليبدينوم) على أنها ذرات نيكل مفردة معزولة.ملفات شدة الضوء في موقع الموضع g1 و g2 المحددين بدوائر صفراء في الشكل 1f بما في ذلك البقع الداكنة والبقع الأكثر سطوعًا على السطحطبقة تؤكد بشكل أكبر وجود معزولذرات مفردة (الشكل g1 و. للمقارنة، المحفزات الكهربائية الضابطة لـ و تم تحضيرها بكميات متفاوتة من النيكل. تعرض أنماط حيود الأشعة السينية وصور المجهر الإلكتروني نفس التركيب البلوري والأشكال كما هو الحال في (الأشكال S9، S11). تم الكشف عن محتويات تحميل النيكل الخاصة بهم بواسطة مطيافية الانبعاث الذري الناتجة عن البلازما المقترنة بالحث (ICP-AES) هي و على التوالي (الجدول S1).
الشكل 1 | الشكل والمكونات للمواد. أ رسم تخطيطي لعملية تصنيع الـمحفز كهربائيصورة HR-TEM لـ. الإدخالات في (ب): نمط SAED. د رسم الخرائط العنصرية لـ Mo و Ni و O و C لعنصر فرديالكرات النانوية (الموضحة في الشكل 1c)، على التوالي. e HAADF-STEM
صورة وعرض مكبر مطابق لـ. النقاط الساطعة المميزة باللون الأحمر في (f) تشير إلى ذرات النيكل الفردية فيتحليل التباين الخطي للمنطقة المختارة من g1 و g2 عند حافة الشكل 1f.
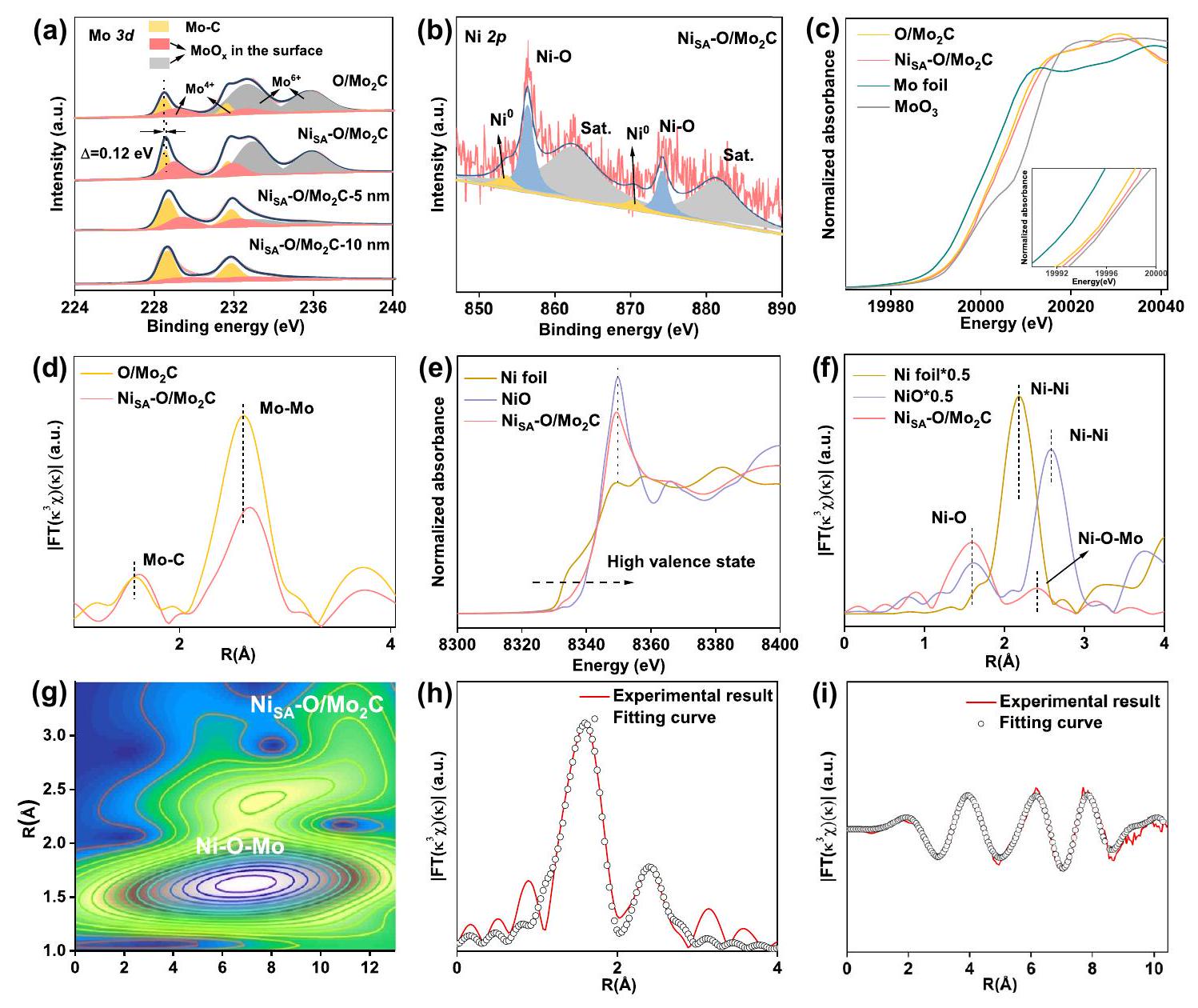
نظرًا لأن بيئة التنسيق تحدد الهيكل الإلكتروني، فقد تم دراسة حالات التكافؤ لمعدني الموليبدينوم (Mo) والنيكل (Ni) بواسطة مطيافية الأشعة السينية للأشعة السينية (XPS) (الأشكال S12-13). كما هو موضح في الشكل 2a، فإن طيف Mo 3d عالي الدقة للنقيتم تفكيكها إلى ستة قمم، تتوافق مع ) و نوع و يمكن أن يُنسب إلى أكاسيد الموليبدينوم بسبب الأكسدة السطحية لأنواع الموليبدينوم، مما يشير إلى وجود طبقة رقيقة على السطح منتكونت خارجالمحفز الكهربائي (المشار إليه بـ. هذا يتماشى جيدًا مع تحليل HAADF-STEM (الشكل 1e-f). ومن الجدير بالذكر أن طبقة الأكسيد السطحية يمكن التحكم فيها من خلال الجو ودرجة الحرارة التي يتم التعرض لها.مكشوف. على سبيل المثال، عندما يتم تقليلتم إخراجها من فرن الأنبوب عند 60في الهواء، ستكون سطحه مؤكسدًا بشكل أكبر بكثير مقارنةً بالعينة الرئيسية لـ. بينما، عندما تم تقليص كانت محمية في جو الأرجون لمدة 12 ساعة قبل إخراجها من الفرن، ستكون سطحها مؤكسد بشكل أقل بكثير مقارنة بالعينة الرئيسية لـ. (الشكل S13a، b). بالإضافة إلى ذلك، بعد نقش طبقة الأكسيد السطحية، فإن المرحلة الرئيسية لـيتسرب تدريجياً، كما يتضح من نتائج XPS مع أعماق نقش مختلفة (الشكل 2a، الشكل S13c). تم تركيب طيف XPS لـ C 1s (الشكل S14a) إلى أربعة إشارات مختلفة عند، و 288.9 إلكترون فولت، والتي تُنسب إلى، و على التواليبعد تحميل ذرات النيكل، فإن طاقة الربط لـطيف لـيعرض تحولًا إيجابيًا حوالي 0.12 إلكترون فولت مقارنةً بذلك الخاص بالنقي، مما يشير إلى هجرة الشحنة الناتجة عن إدخال النيكل الذري. وهذا يتأكد أكثر من خلال التحول الإيجابي للقمة الرئيسية لـ O XPS لـمقارنةً مع ذلك من النقي (الشكل S14b). النيكل يمكن تقسيم طيف XPS إلى ستة قمم، حيث تُنسب القمم عند 856.4 و 874.2 إلكترون فولت إلىناجم عن الـ
الشكل 2 | التوصيف الهيكلي. أ طيف XPS لمستوى Mo 3d. ب طيف XPS لمستوى Ni. طيف XANES عند حافة كاف الموليبدينوم. الصورة المصغرة في (ج) هي تكبير جزئي لقمة ما قبل الحافة. د منحنيات FT-EXAFS للعينات المحضرة. هـ طيف حافة كاف النيكل.ال منحنى FT-EXAFS المقابل.تحويل الموجة (WT) لـ. EXAFS حافة النيكل Kفي فضاءات R و k. الرابطة بين النيكل والأكسجين الممتص، والقمم عند 862.4 و 881.4 إلكترون فولت تتوافق مع قمم الأقمار الصناعية للنيكل (الشكل 2ب). فقط كمية صغيرة منيمكن ملاحظته عند 853.5 و 870.5 إلكترون فولت، مما يشير إلى أن ذرات النيكل مرتبطة بشكل رئيسي مع ذرات الأكسجين المحيطة.يجب أن نلاحظ أنه، بسبب محتوى النيكل فيأقل منذروةليس واضحًا، بينمايلاحظ عند 862.4 و 881.4 إلكترون فولت، مما يشير إلى أن النيكل فيالعينة مرتبطة تقريبًا بالكامل بذرات الأكسجين المحيطة. بينما، نسبةذروة فيأكبر بكثير من ذلك في (الشكل S15)، والذي يرجع على الأرجح إلى تجمعات النيكل المفرطة في كتل أو جزيئات نانوية من النيكل.
لتقييم التركيب الإلكتروني والبيئة المحلية للنيكل والموليبدينوم فيتمت دراسة طيف الامتصاص القريب من حافة الأشعة السينية (XANES) وبنية الامتصاص الدقيق الممتدة للأشعة السينية (EXAFS). تظهر طيف XANES عند حافة Mo K في الشكل 2c. بالمقارنة مع النقيالحافة السابقة لـكان أعلى، مما يشير إلى أن متوسط حالة الأكسدة لمعدن الموليبدينوم في الـكان أكثر إيجابية، وهو ما يتماشى مع نتائج XPS المذكورة أعلاه. تحويل فورييه (FT) لـ EXAFS الممتد (الشكل 2d) للنقي و يمتلك قمتين. القمة الأقوى تقع في، الذي يتوافق مع روابط مو-مو. القمة الأخرى تقع عندالتي ترتبط بروابط Mo-C. بالمقارنة مع O النقي/، فإن إدخال النيكل سيعمل على توسيع و طول الرابطة بسبب التفاعل بين النيكل والحدود السابقة لحد Ni K يقع بين Ni و NiO (الشكل 2e)، مما يشير إلى أن حالة أكسدة Ni في هو موجود بين 0 و +2. طيف XANES عند حافة النيكل K والطيف المقابل في فضاء k لـتشبه بشكل وثيق مرجع NiO (الشكل )، مما يشير إلى تشكيل السطح التحويل فورييه (FT)طيف EXAFS الموزون في فضاء R أكد بشكل إضافي على تشكيلعلى الطبقة الرقيقة غير المتبلورةعلى سطحكما هو موضح في الشكل 2f، هناك قمة ملحوظة عندمساهم من قبليتم ملاحظة الذروة. الـ FTمن رقائق النيكل تظهر قمة عندوNiO يظهر قمة عندكلاهما يتوافق مع تفاعل.لا تحتوي على هذه القمم، مما يدل على أن ذرات النيكل يجب أن توجد في شكل ذري مفرد على الطبقة الرقيقة غير المتبلورة.. بالإضافة إلى ذلك، هناك قمة تشتت عند حوالي ، والذي قد يكون بسبب ارتباطبالإضافة إلى النتائج المذكورة أعلاه لسطح المؤكسديجب أن تسود الروابط على السطح.
لقد قمنا أيضًا بإجراء تحليل تحويل الموجات (WT) لـللتحقق من بيئات التنسيق لـالترابط، الذي يسمح بعرض المعلومات في كل من فضاء R وفضاء K. كما هو موضح في الشكل 2g، إشارة WT قوية مركزة عند، الذي تم اشتقاقه من مساهمة Ni-O-Mo. للحصول على التكوين الدقيق لتنسيق ذرات النيكل في ، يتم إجراء ملاءمة EXAFS الكمية لاستخراج المعلمات الهيكلية وتقديم النتائج في الشكل 2h و i وتلخيصها في الجدول S3. تكشف النتيجة الأفضل ملاءمة أن ذرة النيكل مرتبطة بالأكسجين برقم تنسيق يبلغ 3.9 عندوكذلك تم التنسيق مع Mo برقم تنسيق 5.0 فيفي القشرة الثانية أو الأعلى. بالإضافة إلى ذلك،مع محتوى أعلى من النيكل لديه ضعف
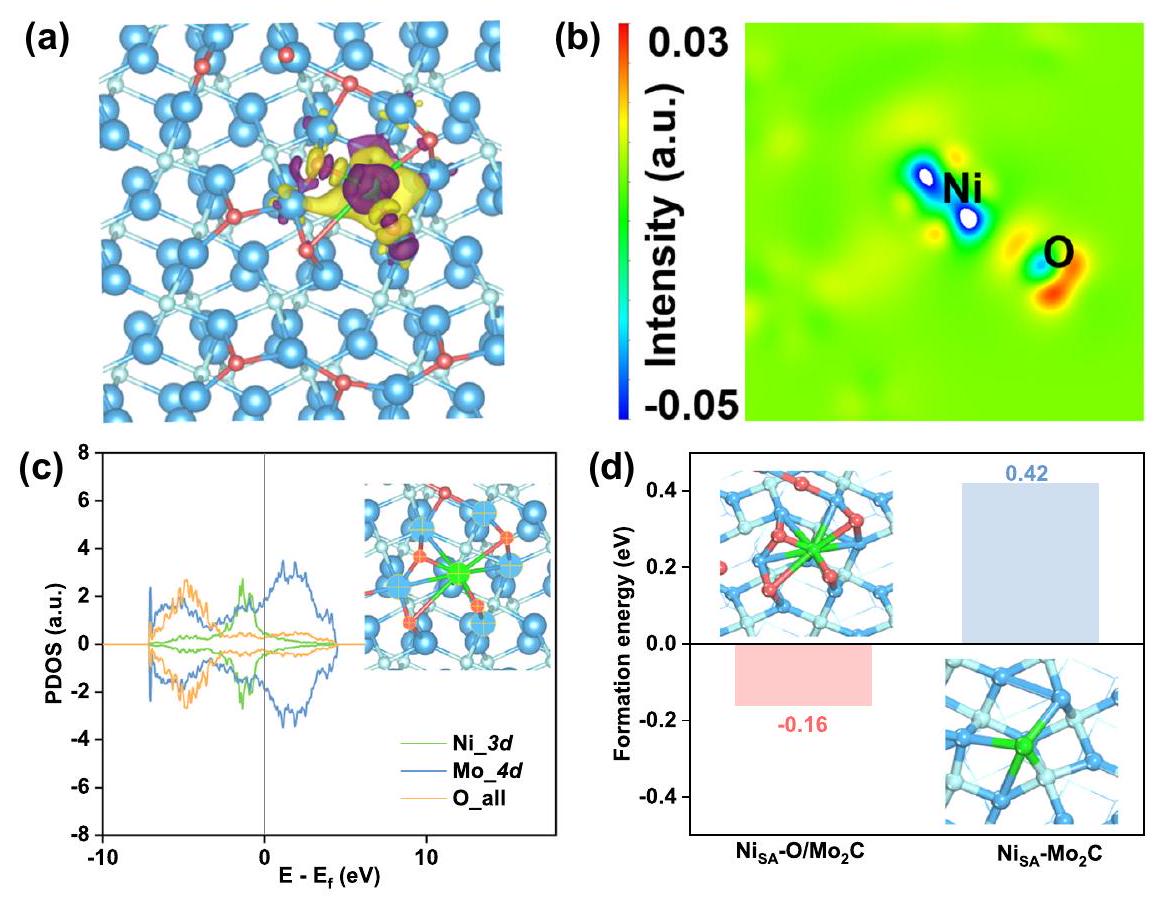
الشكل 3 | حساب DFT. الرسمان البيانيان لاختلاف كثافة الشحنة لـ : عرض علوي للرسم ثلاثي الأبعاد (أ) وعرض ثنائي الأبعاد (ب). مستوى السطح المتساوي مضبوط على 0.003 e، حيث تم تصوير استنفاد الشحنات وتراكمها باللون الأرجواني و
الأصفر، على التوالي. رسم بياني لـ PDOS لـمداراتمدارات الموليبدينوم، وجميع مدارات ذرات الأكسجين المحددة بعلامات الصليب في. د طاقات الكيمياء السطحية لذرة النيكل على أسطح و . ذروة فيالمقابلإشارة (الشكل S17)، والتي تتوافق بشكل جيد مع نتائج XPS.
من أجل دراسة آلية ارتباط ذرات النيكل مع الأكسجين المدمج، قمنا بإجراء سلسلة من حسابات DFT. وفقًا للنتائج السابقة للتحليل الهيكلي، اخترنا المستوى (101) منكسطح تفاعل. ثم تم إجراء حسابات على امتصاص ذرات الأكسجين على السطح، وحددنا الهيكل الأكثر استقرارًا (الشكل S18) من خلال مقارنة قيم طاقتها الكلية. لذلك، قمنا بإرفاق بعض ذرات الأكسجين بالوجه من خلال أقوى امتصاص لمحاكاة سطح مؤكسد جزئيًا منوفقًا للبيانات أعلاه، قمنا بامتصاص ذرة النيكل المرتبطة بأربع ذرات أكسجين وذرة كربون واحدة على السطح، للحصول على نماذج حسابية. و (الشكل S19). أولاً، قمنا بإجراء حسابات فرق كثافة الشحنة بين النيكل والوجه، كما هو موضح في الشكل 3a و 3b. يمكن رؤية ميل واضح لنقل الإلكترونات من موقع النيكل إلى الذرات المحيطة، مما يشير إلى التفاعل بين النيكل والوجه. وهذا ما تم تأكيده أيضًا من خلال الشكل 3c، بسبب التداخلات بين المدارات لذرات النيكل والأكسجين والموليبدينوم (المعلمة بعلامة X) حول مستوى فيرمي. لذا، استنتجنا أن ذرات النيكل يمكن أن تُمتص بشكل مستقر على الوجه المؤكسد جزئيًا.. بالإضافة إلى ذلك، فإن قيمة طاقة الامتزاز عندكان أقل بكثير من (الشكل 3د)، مما يشير بشكل أكبر إلى أن ارتباط Ni-O-Mo كان أكثر استقرارًا.
أداء التحفيز الكهربائي
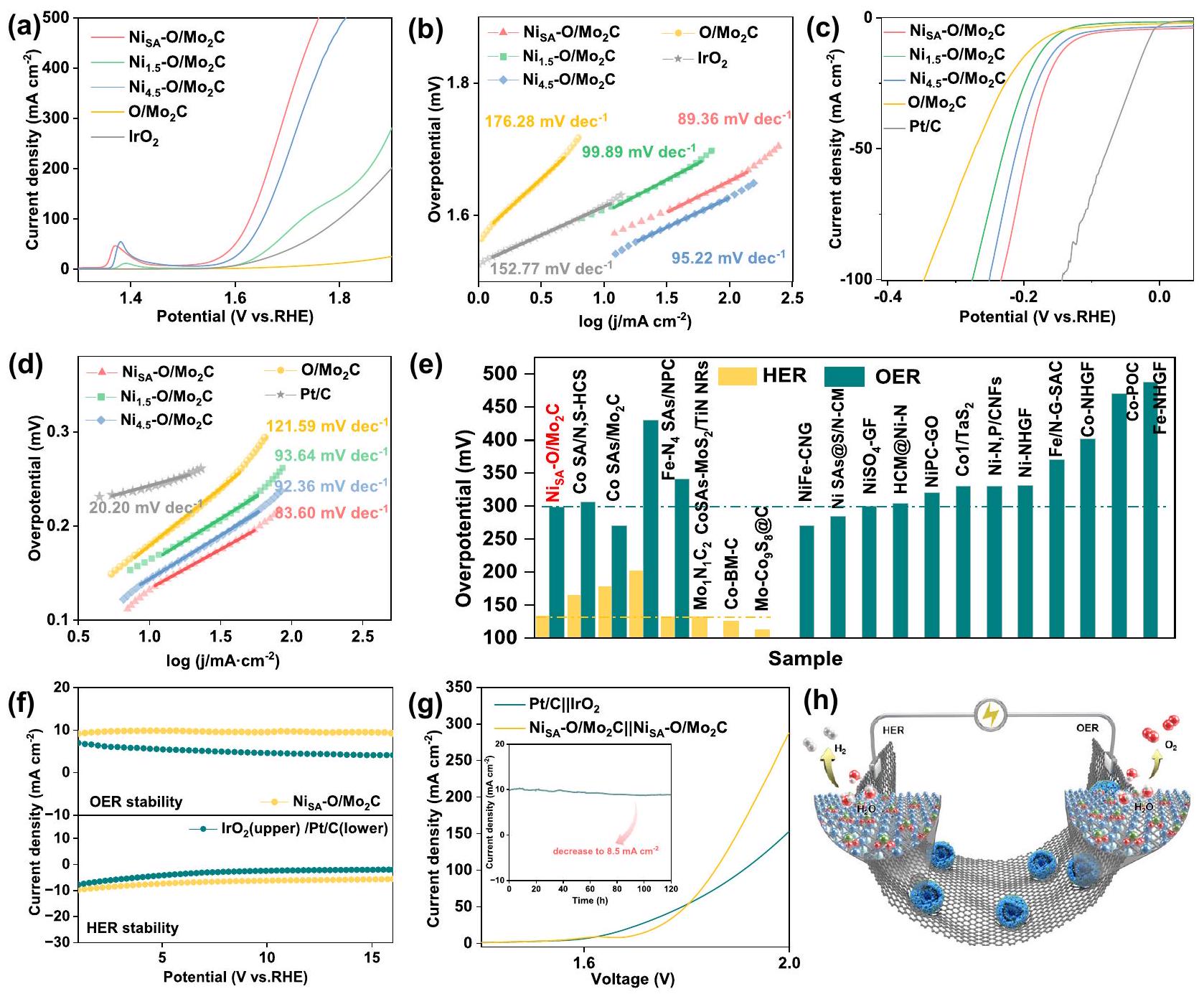
لفحص الأنشطة التحفيزية للتفاعل الكهربائي OER في 1 M KOH باستخدام إعداد ثلاثي الأقطاب. كما هو موضح في الشكل 4a، يظهر جهد زائد ( ) من 299 مللي فولت عند ، والتي تتجاوز بشكل كبير تلك الخاصة بـمللي فولت)، نقيوتجاريالمحفز (386 مللي فولت). من هذهالقيم، يمكننا أن نرى أن التحميلات الأعلى من النيكل تؤدي إلى أداء أفضل بسبب الكثافة الأعلى للمراكز التفاعلية الحفازة. ومع ذلك، لا يمكن أن تؤدي المحفزات بشكل أفضل حتى عندما يكون تحميل النيكل مفرطًا. بالإضافة إلى ذلك، فإن الجهد الزائد لـالمحفز يتطلب فقط 430 مللي فولت عند كثافة التيار، حتى أقل بكثير من 670 مللي فولت المطلوبة من قبل التجاريةمحفز يظهر تحسناً كبيراً في نشاط OER تحت كثافة التيار العالية. بالإضافة إلى ذلك، توضح الشكل 4a ذروة أكسدة النيكل، التي تتوافق مع أكسدة النيكل من حالات التكافؤ المنخفضة.إلى حالات عالية القيمةتم حساب انحدارات الجدول لكشف أنيمنح ميل تافل أصغر )، أقل بكثير من تلك الخاصة بـ نقي وحتى التجارية ( )، وهو علامة على سرعة نقل الإلكترونات وخصائص النقل الكتلي المتفوقة (الشكل 4ب) مدفوعًا بالأداء الواعد للموارد التعليمية المفتوحةتمت دراسة تفاعل الهيدروجين الكهربائي (HER) أيضًا في نفس بيئة 1.0 M KOH. تُظهر الشكل 4 ج أن محفز Pt/C يظهر أفضل نشاط لـ HER، بينمايظهر أداءً تنافسياً مع جهد زائد قدره 133 مللي فولت عند كثافة تيار، وهو أقل من تلك الخاصة بـوطاهر. علاوة على ذلك، فإن ميل تافل لـ هو أقل من تلك الخاصة بـ ) ونقي (الشكل 4 د). من الجدير بالذكر أنه من أجل استبعاد دور ذرات النيكل على الركيزة الكربونية، قمنا بإعداد عينتين ضابطتين: ذرات النيكل على الركيزة الكربونية التي تم الحصول عليها من خلال النقش.جزيئات في وذرات النيكل على كرات الكربون النقية ( تمت تخليقه بنفس الطريقة معكما يتضح من أنماط XRD، TEM، ورسم العناصر (الشكل S20)، فإن كلا العينتين تظهر فقط قمم الكربون وتوزيع موحد لعنصر النيكل. أظهرت الاختبارات الكهروكيميائية أنه بالمقارنة معيمكن تجاهل نشاطها التحفيزي في تفاعلات HER و OER (الشكل S21)، مما يشير إلى النشاط التحفيزي لـيأتي من ذرات النيكل الفردية المثبتة على الأكسجين المدمجبشكل مثير للإعجاب،تفوقت على معظم SACs غير الثمينة في الوسط القلوي (الشكل 4e، الجداول S4-5)، مما يثبت هو محفز ثنائي الوظيفة عالي الكفاءة بشكل محتمل. للتحقيق في حركيات القطب تحت عملية المحفز، تم إجراء قياسات طيف الامتزاز الكهربائي (EIS). الشكل S22 يعرض مقاومة نقل الشحنة المواتية ( ) من مقارنةً مع عينات التحكم الأخرى بالإضافة إلى ذلك، كانت المساحة السطحية النشطة كهربائياً (ECSA) لجميع العينات
الشكل 4 | أداء HER و OER وأداء التحليل الكهربائي العام للماءعامل مساعد فيمع تعويض iR. أ curves الاستقطاب و b مخططات Tafel لـوطاهرتجاريلـ OER (كمية التحميل ). منحنى الاستقطاب و d مخططات تافل لـ وطاهرلها (مبلغ التحميل ). مقارنة الفائض الكهربائي لـ وتم الإبلاغ مؤخرًا عن SACs من المعادن غير الثمينة عندفي 1.0 م
OH. تمثل المدرجات في اللونين الأصفر والأخضر أداء HER و OER على التوالي.كثافة التيار مقابل الزمن ) منحنيات القطبية لـ HER و OER. g منحنيات القطبية لـ و الأزواج لتفكيك الماء بشكل عام في 1.0 م كوه (كمية التحميل ). إدراج هو المتانة على المدى الطويل لعملية تحليل الماء بشكل عام عند كثافة تيار. ه توضيح لـأقطاب كهربائية لتفكيك الماء بشكل عام. المعكوس من سعة الطبقة المزدوجة ( ) (الشكل S23). العامل المساعد لديه الأعلىالقيمة، التي تشير إلى أنلديها مساحة سطح نشطة مكشوفة أكبر من العينات الأخرى. نظرًا لأن النشاط النوعي يعكس النشاط الجوهري للتفاعل الحفزي، تم عرض منحنيات LSV المعدلة حسب ECSA لكل من HER وOER في الشكل S24. يظهر النشاط النوعي (SA) المعدل حسب ECSA أنتظهر أفضل نشاط كهرومحفز تجاه كل من OER و HER مقارنةً بالمواد المحفزة الأخرى وفقًا للترتيب التالي. بشكل خاص بالنسبة لتفاعل أكسدة الماء، عند جهد زائد قدره 370 مللي فولت (الشكل S24d)، يعرض SA بمقدار 8.71، وهو أعلى من تلك الخاصة بـ و تشير هذه النتائج إلى أن دمج النيكل لا يحسن فقط كثافة المواقع النشطة ولكن أيضًا يحسن بشكل جوهري نشاط تفاعل الهيدروجين (HER) وخاصة نشاط تفاعل الأكسدة (OER).المحفز. تعتبر متانة المحفز مؤشرًا مهمًا، خاصة بالنسبة لـ SACs. بالنسبة لتفاعل الأكسدة، تم إجراء التحليل الكهربائي على المدى الطويل عند تيار ثابت تحت تيار ثابت منعن طريق الكرونوأمبرو متري لمدة 16 ساعة (الشكل 4f)، مما أثبت أن التيارات لـزيادة أولاً ثم الحفاظ على الاستقرار لفترة طويلة. بالمقابل، بعد 16 ساعة، تجاريالمحفز يظهر اختلافًا واضحًا في الجهد الزائد المطلوب. بالنسبة لتفاعل تقليل الهيدروجين، على الرغم من أنه كان أكثر استقرارًا بكثير منكثافة الحالية لـانخفض المحفز قليلاً بعد 16 ساعة.
علاوة على ذلك، قمنا بتجميع جهاز تحليل كهربائي باستخدام محلول كOH بتركيز 1.0 م.كأقطاب ثنائية الوظيفة ( ). تم اختبارها أيضًا للمقارنة. الشكل 4 ج يقدم مقارنة بين منحنيات LSV لـ و جهد الخلية المطلوب للوصول إلى 10 و 100 و 150 مللي أمبيرلـهم، و 1.90 فولت، بينمايتطلبو 1.93 فولت، على التوالي. تُظهر النتيجة أن الأداء الكهروكيميائي العام للتفكيك لـيمكن مقارنته بـخصوصًا تحت كثافة التيار العالية وتفوق على معظم المحفزات ذات الذرة الواحدة المبلغ عنها (الجدول S6). في اختبار الاستقرار، انخفضت كثافة التيار فقط منإلىبعد 120 ساعة، مما يدل على استقراره الجيد كعامل حفاز ثنائي الوظيفة (المربع في الشكل 4 ج). علاوة على ذلك، تم ملاحظة الفقاعات الواضحة عند القطب السالب والقطب الموجب خلال اختبار الاستقرار الطويل الأمد، مما يتوافق مع و ، على التوالي (الشكل 4 هـ والشكل S25).
جيل جديد من الواجهات النشطة المدفوعة بالقدرة
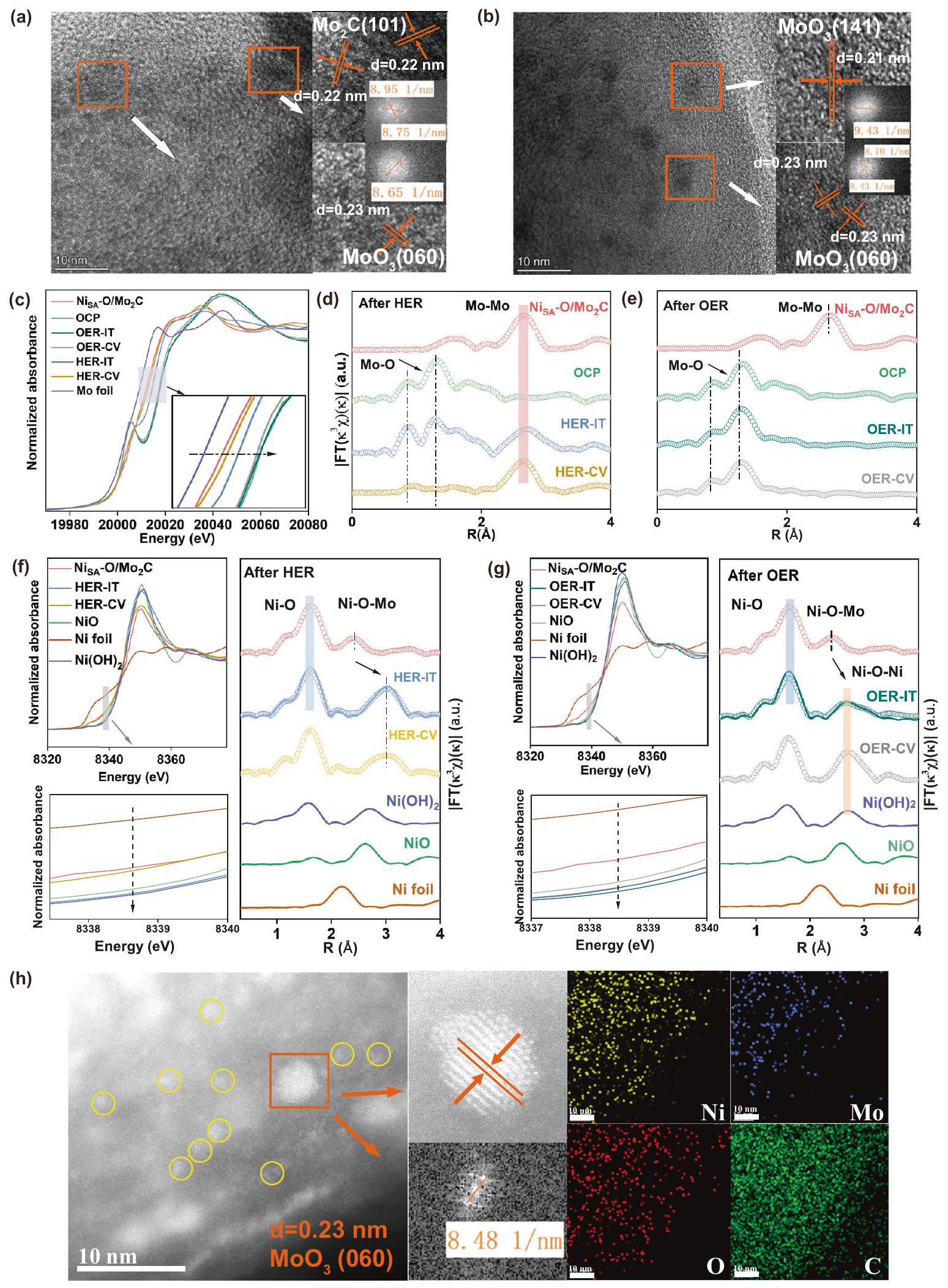
لكي نكشف عن الظواهر المختلفة للاستقرار في تفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER)، تم إجراء سلسلة من التوصيفات لاستكشاف هوية الأنواع النشطة كيميائيًا الحقيقية. يشير نمط حيود الأشعة السينية (الشكل S26a) إلى أن سطحتخضع لإعادة تنظيم هيكلي لتشكيل مراحل مختلفة بعد التفاعلات الكهروكيميائية المختلفة. بعد تفاعل الهيدروجين الكهربائي،المحفز يحافظ على قمم الحيود، ولكن قمم أخرى أضعف من (بطاقة JCPDS رقم 35-0609) و (رقم بطاقة JCPDS 29-1021) تظهر. بالمقابل، بالنسبة لتفاعل تطور الأكسجين (OER)، يمكن تعيين جميع قمم XRD إلى الهياكل البلورية لـ (رقم بطاقة JCPDS 05-0508 و 35-0609) و بدونالقمم، تشير إلىتم تفكيكها بالكامل. الشكل S 27 يظهر الـطيفبعد التفاعلات، يمكن تعيين القمم الرئيسية لمعدن الموليبدينوم إلىدون حالة الفالنس المنخفضة لمعدن الموليبدينوم، مما يشير إلى أن متوسط حالة الأكسدة لمعدن الموليبدينوم أعلى من تلك الخاصة بـأي أن سطح المحفزات كلاهما يظهر سلوك أكسدة واضح. ومع ذلك، بعد رد فعلها، تم نقل القمة إلى موضع أدنى قليلاً، وهو ما يرجع على الأرجح إلى التفاعل بين ذرة النيكل الفردية والركيزة المعاد تشكيلها. لم يتغير شكل الهيكل المجوف ذو القشرة المزدوجة بشكل واضح بعد التفاعلات الحفزية (الشكل S2830). ومع ذلك، في حالةبعدها، تُظهر الصورة المكبرة بتقنية HRTEM الأصلية الكبيرةتم سحق الجسيمات النانوية إلى جزيئات أصغر، وت correspond إلى شبكتين مختلفتين بمسافات d تبلغ 0.22 نانومتر و0.23 نانومتر، والتي تتوافق مع مستويات (101) و(060) من و ، على التوالي (الشكل 5 أ، ب). بالإضافة إلى ذلك، في بعد OER، تظهر الصورة الكبيرةتم تغيير الجسيمات النانوية بوضوح إلى جسيمات أصغر، وتتناسب المسافات بين المستويات 0.21 و0.23 نانومتر مع المستوى (141) و(060) من. تشير هذه النتائج إلى أن ت undergoes إعادة التكوين في عملية HER و OER. لذلك، من أجل تحديد الأنواع النشطة الحقيقية المعنية في هاتين التفاعلتين، تم تسجيل طيف XANES و FT-EXAFS خارج الموقع بعد اختبار CV واختبار التيار الزمني i-t لـ HER و OER. كما هو موضح في الشكل 5c، تظهر أطياف XANES لحافة Mo K للمواد الحفازة بعد تنشيط HER و OER تحولات إيجابية في حافة الامتصاص مقارنةً بتلك الخاصة بـ، مما يشير إلى أن حالات أكسدة الموليبدينوم قد زادت. ومع ذلك، بالنسبة للمحفزات بعد تفاعل الأكسدة الكهربائية للماء، تظهر طيف XANES عند حافة K للموليبدينوم تحولات إيجابية أكثر مقارنة بتلك الخاصة بالمحفزات بعد تفاعل اختزال الهيدروجين، مما يشير إلى أن المحفزات بعد تفاعل الأكسدة الكهربائية للماء لديها حالة تكافؤ أعلى من الموليبدينوم، وهو ما يتوافق مع نتائج XPS. بالإضافة إلى ذلك، تم إظهار قمة بارزة جديدة في منطقة ما قبل الحافة لتفاعل الأكسدة الكهربائية للماء، مما يشير إلى تكوين تشوه قوي.أوكتاهيدرا. الشكلأظهر مو-طيف FT-EXAFS الموزون بـبعد HER و OER، على التوالي. بعد HER، يظهر الذروة الرئيسية المركزية عند، الذي يتوافق مع تشتت في. على وجه الخصوص، هناك قمتان أصغر تقعان حوالي و يمكن العثور عليها، وهي خصائص نموذجية لروابط Mo-O، مما يشير إلى سطحتم أكسدته بشكل أكبر. على النقيض من ذلك، بعد OER، تظهر الأطياف وجود رابطة Mo-O ولكن لا توجد رابطة Mo-Mo، وشكلها مشابه لشكل الموليبدات، مما يدل، باستثناء البلورةتم تشكيل موليبدات غير متبلور أو منخفض التبلور أيضًا تلاه التفاعل المحتمل لـمن أجل توضيح ما إذا كانت التغييراتيكون تلقائيًا في إلكتروليت KOH عند جهد الدائرة المفتوحة (OCP). كما هو موضح في الشكل. قمنا باختبار XRD، ومنحنى Mo k-edge XANES و FTEXAFS لـالمحفزات الكهربائية بعد غمرها في إلكتروليت KOH عند جهد الدائرة المفتوحة (OCP). كما هو موضح في الشكل S26b،المحفزات الكهربية بعد غمرها في محلول KOH عند جهد الدائرة المفتوحةلها قمم مميزة مشابهة لـبعدها باستثناء القليل من الضعف و القمم. استنادًا إلى ذوبانفينحن نفترض السطح الأصلييتفاعل مع KOH لتكوين، ومع استهلاك السطحتتعرض السطح للأكسدة المستمرة لتكوين سطح جديدحتىيتم إذابته بالكامل في KOH. ومع ذلك، بالمقارنة معبعد يمتلك قممًا مميزة لـ. بالإضافة إلى ذلك، مقارنة بـ و بعدها، حافة الامتصاص لـكان أعلى، مما يشير إلى أن حالة الأكسدة المتوسطة لمعدن الموليبدينوم كانت أكثر إيجابية. في الوقت نفسه، شكل حافة كا مو XANES وطيف FT-EXAFS الموزون بـ k3 لـتختلف عنبعدها ولكن مشابه لذلكبعد OER. تشير هذه النتائج إلى الذوبان والأكسدة لـفي KOH يكون تلقائيًا في الظروف القلوية. ستعزز الجهد المؤكسد المطبق لعملية OER هذين العمليتين المستمرتين، بينما ستعيق عملية HER إلى حد ما أكسدة الطبقة الداخلية.ثم تمنع جزئيًا تشكيلمن الطبقة الخارجية لـ. استنادًا إلى حل في KOH، الشكل 5f و g يقدم مقارنات لـالعوامل المساعدة في XANES عند حافة النيكل و FT-EXAFS بعد تفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER). في حالة تفاعل الهيدروجين (الشكل 5f)، يظهر موقع الحافة السابقة لطيف XANES تحولاً طفيفاً نحو قيم طاقة أعلى مقارنةً بالقيم الأصلية.، مما يشير إلى أن حالة أكسدة النيكل تزيد قليلاً لكنها لا تزال بين و . بعد مقارنة طيف FT-EXAFS بعناية قبل وبعدها، وجدنا ذروةلم يتغير O، ومع ذلك تم نقل موضع Ni-O-Mo إلى R أعلى، وذلك بسبب إعادة التكوين لـالركيزة. بعد ملاءمة EXAFS، وجدنا المسافة بينتقريبًا نفس الشيء كما هو الحال مع و تم زيادته من 2.40 إلى (الجدول S7). علاوة على ذلك، زاد عدد تنسيق النيكل-الأكسجين إلى 5.90 بينما انخفض عدد تنسيق النيكل-الموليبدينوم إلى 4.50. ولم تُلاحظ أي قمم نموذجية لروابط النيكل-النيكل أيضًا. تشير هذه النتائج إلى أن النيكل لا يزال موجودًا كذرات مفردة بينما تغيرت هيكله التنسيقي قليلاً. تم الإشارة إلى هذا المحفز بعد تفاعل تقليل الهيدروجين بشكل جماعي باسم . بالمقابل، بعد OER (الشكل 5 ج)، تكون موضع حافة الامتصاص أعلى من تلك الخاصة بـالمرجع، يشير إلى أن النيكل لديه أكسدة أعلى من +2، والتي ربما تعود إلى وجود NiOOH أو التفاعل القوي بين الإلكترونات.والمادة الأساسية المعاد بناؤها (الشكل S31). بالنسبة لطيف FT-EXAFS لحافة Ni K بعد OER، تتطابق هذه الأشكال جيدًا مع تلك الخاصة بـ. بالإضافة إلى ذلك، تختفي قمة التنسيق Ni-O-Mo تقريبًا وتظهر تشتت جديدة مميزة منيظهر عندتظهر نتائج EXAFS المناسبة مسافات الروابطإذا هو و (ني-أو-مو) هو على التوالي. تُظهر صور HAADF-STEM (الشكل 5h) المستوى (060) من البلورةوعناقيد نانوية أصغر من النيكل (دوائر صفراء). بالإضافة إلى ذلك، تؤكد خرائط العناصر المختارة على مستوى ذري توزيع النيكل بشكل موحد وتوزيع الموليبدينوم بشكل غير متساوٍ على السطح. تم الإشارة إلى هذا المحفز بعد عملية OER بشكل جماعي باسم. تشير هذه النتائج، بعد OER، إلى توزيع موحد للنيكل فيتخضع لتحول في التكوين وقد تم تجميعها في مجموعات صغيرة جدًا بواسطة الجهد المطبق.
محاكاة DFT
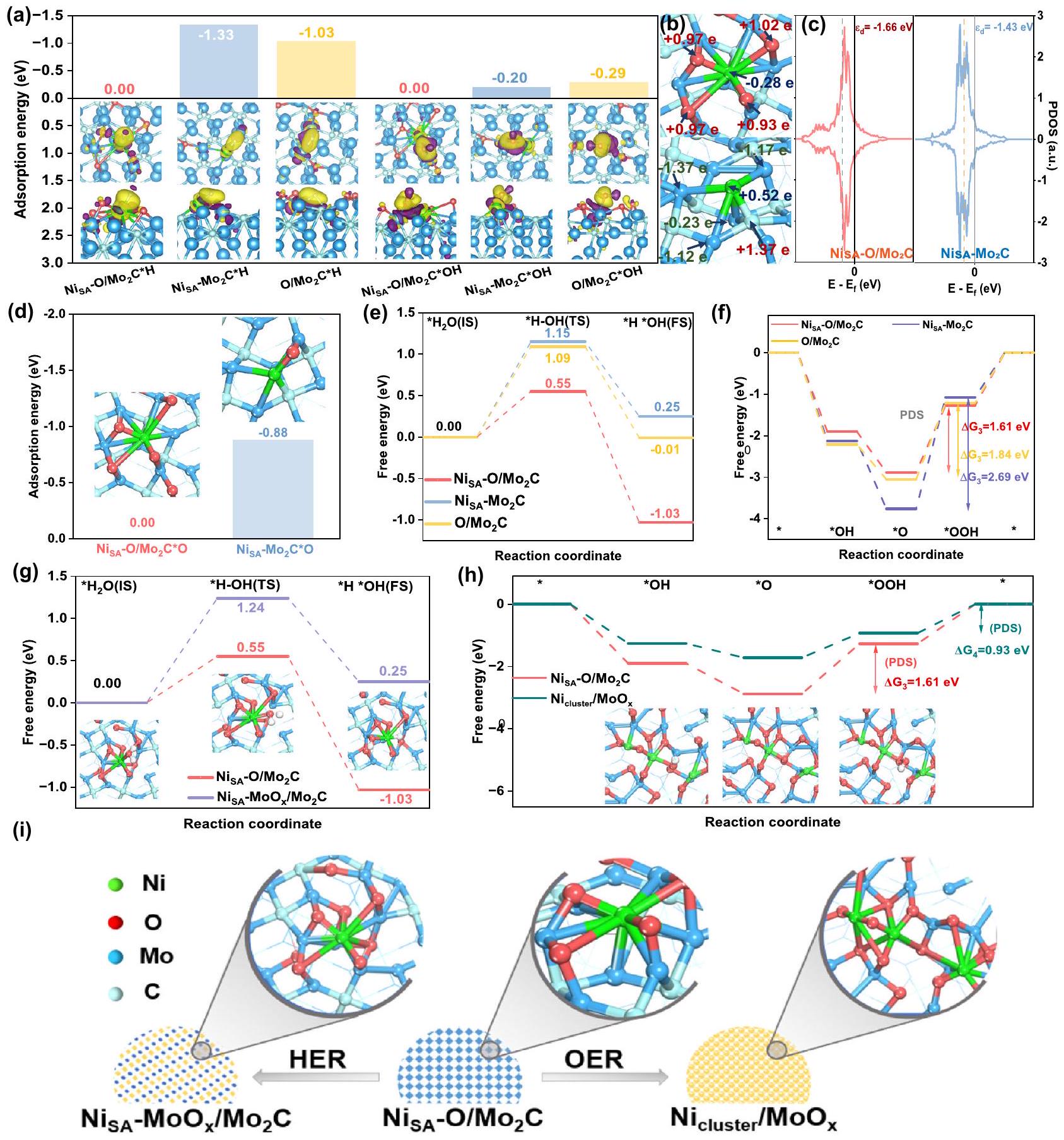
من أجل استكشاف تأثير أشكال الترابط المختلفة على الخصائص الكهروكيميائية لتفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER)، قمنا أولاً بتحليل فرق كثافة الشحنة وشحنة بادير. من المهم أن جميع حسابات طاقة الامتصاص تم الحصول عليها على أساستصحيح إلى الصفر. كما هو موضح في الجزء المرفق من الشكل 6a والشكل S32، هناك ميل لنقل الإلكترونات من موقع النيكل إلى المواد الممتصة، ويمكننا أن نرى بوضوح أن عدد الإلكترونات التكافؤية للنيكل قد زاد عند التكوين.رابطة علىوجه (الشكل 6ب) مقارنةً مع الشكل المتكونرابطة فيوجه. لذلك، تدمج ذرات النيكل مباشرةً المستوى (101) منيمتلك امتصاصًا قويًا للبروتونات والهيدروكسيل، بينما الأكسجين المدمج في السطحسوف يتم تعديل عدد الإلكترونات التكافؤية لـ Ni، مما يؤدي إلى ضعف قدرة الامتزاز. لذا، فإن طاقات الامتزاز لـللبروتون والهيدروكسيل أقل بكثير من تلك الخاصة بـ، مما يقلل بشكل ملحوظ من احتمال انسداد المواقع النشطة، وبالتالي تحسين الخصائص الكهروكيميائية لتفاعل الهيدروجين (الشكل 6أ). بالإضافة إلى ذلك، وجدنا مستوى الطاقة لمركز نطاق d -band من أقل بكثير من
الشكل 5 | التحليل الهيكلي لـبعد HER/OER لدراسة المواقع النشطة. أ، ب صور HRTEM لـبعد اختبارات استقرار HER/OER. c-e منحنى XANES عند حافة Mo خارج الموقع ومنحنى FT-EXAFS المقابل لـ
الشكل 6 | حساب DFT لتفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER). أ طاقات الكيمياء السطحية لـ *H و *OH في مواقع النيكل و الرسوم البيانية ثلاثية الأبعاد لاختلافات كثافة الشحنة: عرض علوي وعرض جانبي.عدد نقل شحنات بادير عند المحددوذرات الموليبدينوم في و . رسومات PDOS لـمدارات ذرات النيكل في و تظهر الخطوط المنقطة السوداء مركز نطاق d، وتم اعتبار مستوى فيرمي كصفر للطاقة. طاقة الامتصاص الكيميائي للأكسجين في مواقع النيكل و . مخطط الطاقة الحرة لجيبس لخطوة فولمر. الإطار في (ج): النماذج المثلى لمراحل مختلفة منتغير الطاقة الحرة لجيبس لمسارات OER على مواقع النيكل. الإدراج في ( ): التكوينات الذرية لمراحل مختلفة من إعادة هيكلة هيكلية لـالعامل المساعد بعد عملية تفاعل مختلفة. الماء الأزرقأخضررماديأحمر. (الشكل 6ج). وفقًا لنظرية مركز نطاق d، فإن انخفاض كثافة الإلكترونات بالقرب من مستوى فيرمي سيسهل إزالة المادة الماصة. لذا، بالمقارنة مع ، الـكان لديه قدرة أقوى بكثير على امتصاص الأكسجين (الشكل 6د)، مما سيؤدي إلى زيادة الجهد الزائد. ثم قمنا بحساب الحاجز المحتمل لإنتاج الهيدروجين وحاجز الطاقة لخطوة فولمر، بالإضافة إلى الجهد الزائد لتفاعل الأكسدة. كما هو موضح في الشكل S33 والشكل 6e، فإن الطاقة للخطوة المحددة المحتملة المطلوبة من لإنتاج الهيدروجين هو 0.5 إلكترون فولت، وهو أقل بكثير من تلك الخاصة بـ و . بالإضافة إلى ذلك، فإن حاجز الطاقة لتفكك الماء البطيء لـ هو 0.55 إلكترون فولت، وهو أقل من 1.15 إلكترون فولت من و 1.09 إلكترون فولت من، مما يشير إلى تحميل النيكل على أكسيد جزئييمكن أن يحسن أدائها. تُظهر العمليات في خطوة فولمر في الشكل S34. في الوقت نفسه، فإن الخطوة المحددة للإمكانات (PDS) لتفاعل الأكسدة الكهربائية (OER) هي الخطوة الكهروكيميائية الثالثة منإلىكما يمكننا أن نرى،يقلل بشكل ملحوظ من حاجز الطاقة في هذه الخطوة إلى 1.61 إلكترون فولت، وهو أقل من ذلك لـ و في، مما يدل على لديه أداء أفضل بكثير، حيث يتماشى مع الملاحظات التجريبية. تم عرض مخطط خطوة تفاعل OER في الشكل S35.
استنادًا إلى النتائج المذكورة أعلاه، سيتبين أن السطح المؤكسد جزئيًا لـسهلت ربط ذرات النيكل عبر و روابط كيميائية (Ni-O-Mo)، مما يعزز الخصائص الكهروكيميائية لتفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER). ومع ذلك، فإنركيزة لـبعد أن خضعت HER و OER للأكسدة وذابت.بعد HER (الشكل 6g، S36)، يمكننا أن نرى أن حاجز الطاقة لتفكك الماء هو 1.24 إلكترون فولت، والذي يصبح أكبر من ذلك لـ، مما يشير إلى أن الأكسدة الزائدة للركيزة يمكن أن تؤدي إلى بعض الآثار السلبية لتطور الهيدروجين. ولكن بالنسبة لـبعد OER، يتغير PDS من خطوة *O إلى *OOH إلى خطوة *OOH إلىإن طاقة حرية الخطوة المحددة بمعدلها هي 0.93 إلكترون فولت (الشكل 6h)، والتي تصبح أقل من تلك الخاصة بـفي. بالإضافة إلى ذلك، مقارنة بـالمعاد بناءهيظهر قدرة امتصاص أضعف لـ (الشكل S37)، مما يشير إلى المعاد بناءه يسرع تفاعل تطور الأكسجين. تشير هذه النتائج إلى أن الأكسدة الزائدة للركيزة تسهم في تجمع النيكل الأحادي الذرة في كتل وتحسن أداء تطور الأكسجين. يوضح الشكل 6i إعادة تشكيل الهيكل لـالمحفز بعد عمليات التفاعل المختلفة. هذه النتائج تتوافق جيدًا مع الملاحظات التجريبية.
باختصار، قمنا بتصميم محفز ثنائي الوظيفة من ذرات النيكل غير الثمينة محملاً على سطح يحتوي على الأكسجين.عبرسندات الجسر. تم الحصول علىأظهر المحفز جهدًا زائدًا قدره 133 مللي فولت و299 مللي فولت عند كثافة التيارلـ HER و OER، على التوالي. أشارت التوصيفات الشاملة وحسابات DFT إلى أن ذرات الأكسجين السطحية لـكانت تسهم في استقرار الذرة الفردية Ni من خلال الربط الجسري لـ. الـتؤدي بيئة الترابط O-Mo إلى تنظيم الهياكل الإلكترونية لذرة النيكل الفردية نحو تحسين البروتون والهيدروكسيل وطاقة الامتزاز المعتدلة للأكسجين، مما يحدد الأداء المتفوق للكاتاليس. علاوة على ذلك، وجدنا أن تكوين النيكل الموزع ذريًا فيالمحفز الكهربائي خضع لتحول وتم أكسدة ودوبان الدعم بدرجات متفاوتة بسبب عدم الاستقرار في KOH وتأثير الجهد المطبق. بعد تفاعل تقليل الهيدروجين، لا تزال أنواع النيكل تحتفظ بشكلها الموزع ذريًا. ومع ذلك، بعد تفاعل أكسدة الأكسجين، تم تجمع النيكل الموزع ذريًا في كتل صغيرة جدًا. بالإضافة إلى ذلك، وجدنا أن هناك أكسدة مفرطة.سوف يؤدي إلى إعادة تشكيل تكوينات ذرة النيكل الفردية، مما سيؤدي إلى تدهور أدائها في تفاعل الهيدروجين. ومع ذلك، مقارنةً بالأصلتعتبر الكتل الصغيرة المعاد تشكيلها من النيكل المدفوعة بالقدرة، والتي تتمتع بهياكل تنسيق مثالية، أكثر ملاءمة لتفاعل أكسدة الماء. باختصار، تُظهر هذه الدراسة استراتيجية سهلة وقابلة للتطبيق لتصنيع محفز كهربائي أحادي الذرة ثنائي الوظيفة منخفض التكلفة وعالي الكفاءة، كما توفر إرشادات لتصميم محفزات فعالة لتفكيك الماء في المستقبل.
طرق
تركيب سلف مو غير المتبلور
تم تخليق المركب السابق القائم على الموليبدينوم لأول مرة بواسطة طريقة هيدروحرارية من خطوة واحدة. تم إذابة 1 جرام منمن، و 0.132 جرام من في 50 مل من الماء المنزوع الأيونات، عندما تغير لون المحلول من الأخضر المصفر إلى الأزرق، تم نقله إلى أوتوكلاف مبطن بتفلون سعة 100 مل بينما كان ساخنًا. ثم تم الاحتفاظ به في فرن كهربائي عند درجة حرارةلمدة 48 ساعة. بعد أن تم تبريد الأوتوكلاف إلى درجة حرارة الغرفة، تم طرده ثلاث مرات باستخدام الماء والإيثانول بالتناوب. تم تجفيف العينة في فرن مفرغ من الهواء عندلمدة 12 ساعة.
تركيبالمحفزات الكهربائية
0.20 جرام من سلف مو القائم علىمنتم إذابتها في 10 مل من الماء المنزوع الأيونات مختلطًا مع 15 مل من الإيثانول. تم إذابتها بالموجات فوق الصوتية لمدة 40 دقيقة، في حمام مائي بدرجة حرارة ثابتة.لمدة ليلة واحدة مع التحريك المستمر. ثم تم غسلها بالإيثانول ثلاث مرات وتجفيفها للحصول على النيكل الممتص على سطح الكرات النانوية المعتمدة على الموليبدينوم. أخيرًا، تم تسخين العينة عندلمدة ساعة واحدة تحت جو هوائي مع معدل تسخين، لضمان أن الكربون في هيكل الكرات النانوية المعتمدة على الموليبدينوم يمكن أن يهرب مع الهواء، تلاه لمدة 3 ساعات تحت جو من الهيدروجين والأرجون بنفس معدل التسخين. تم الحصول عليه بعد التلدين.
بالمثل، و تم تخليقه بنفس الطريقة باستثناء كمية التعديل من نترات النيكل (1.5 م و 4.5 م، على التوالي). تمت تخليقه عن طريق الكلسنة المباشرة للمواد الأولية المعتمدة على الموليبدينوم دون.
التحليل الهيكلي
تم اختبار التركيب البلوري ونقاء الطور بواسطة نمط حيود الأشعة السينية (XRD، ألتيم، اليابان) معالإشعاع في درجة حرارة الغرفة. تم إجراء قياسات المجهر الإلكتروني الناقل (TEM) على جهاز Hitachi H-7650 الذي يعمل عند 2 كيلوفولت. تم قياس ICP-OES بواسطة نظام Thermo Fisher IRIS Intrepid، والذي يمكن استخدامه لتحديد محتويات العناصر في العينات. تم إجراء المجهر الإلكتروني الناقل عالي الدقة (HR-TEM) والمجهر الإلكتروني الناقل الماسح ذو الحقل المظلم الزاوي العالي (HAADF-STEM) بواسطة جهاز Talos F200X G2 وجهاز JEOL JEM2100 F الذي يعمل عند 200 كيلوفولت، على التوالي. تم اختبار تجارب مطيافية الأشعة السينية (XPS) على جهاز PHI 5000 Versaprobe III. تم إجراء قياسات المساحات السطحية المحددة لمنتجات نموذجية باستخدام جهاز Belsorp-max للكشف عن المساحة السطحية عند 77 كيلفن.تم إجراء تجارب الامتزاز-التحرر عند 77 كلفن على جهاز Micromeritics ASAP 2460. قياس وتحليل بيانات طيف الامتصاص بالأشعة السينية (XAS): تم الحصول على طيف الامتصاص بالأشعة السينية بالقرب من حافة Mo K عند محطة 1W1B في BSRF (مرفق الإشعاع السنكروتروني في بكين، جمهورية الصين الشعبية) التي تعمل عند 2.5 جيجا فولت مع تيار أقصى قدره 250 مللي أمبير. تم قياس حافة الكربون وحافة النيتروجين عند خط الشعاع BL12B من مختبر الإشعاع السنكروتروني الوطني (NSRL) في الصين وتم إيداع العينات على شريط كربوني مزدوج الجوانب.
القياسات الكهروكيميائية
تم جمع جميع الأطياف في ظروف محيطة. تم إجراء قياسات نشاط تفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER) للمواد المحفزة الكهربائية باستخدام نظام تقليدي ثلاثي الأقطاب (CHI-760E)، مع استخدام قضيب كربوني جرافيتي كقطب مضاد.كمرجع للقطب، وكربون زجاجي (GC) أو ورق كربوني مع العينات التي تم تحضيرها كقطب عمل. 3.0 ملغ من العينة ومنتم توزيع محلول نافيون فيمنماء/إيثانول، ثم أضيف 1.5 ملغ من الكربون الأسود وتمت الموجات فوق الصوتية لمدة 30 دقيقة لتشكيل حبر متجانس. ثمتم توزيع المحلول على قطب كهربائي GC بقطر 3 مم بكمية تحميل قدرها. بالنسبة للنشاط OER المنفذ، 10.0 ملغ من العينة ومنمحلول نافيون في 1 ملماء/إيزوبروبانول، ثمتم توزيع المحلول علىورق كربون بتحميل 0.74 ملغتم إجراء جميع تجارب OER وHER الكهروكيميائية في محلول 1.0 M KOH، وتم تصحيح منحنيات الاستقطاب لـ iR. تم الإشارة إلى جميع الجهود بالنسبة للقطب الهيدروجيني القابل للعكس (RHE)، وفقًا للمعادلة أدناه:
تم تقدير المساحة السطحية النشطة كهربائياً (ECSA) للعينات التي تم تحضيرها بدون كربون أسود من خلال قياسات الفولتمترية الدورية (CV).تم اختبار سعة الطبقة المزدوجة الكهروكيميائية من 0.86 إلى 0.96 فولت (مقابل RHE) في محلول 1 م KOH لعملية HER أو OER عند سرعات مسح مختلفة.“، 100). تم رسم الفرق بين تيارات الأنود والكاثود بشكل خطي مقابل معدل المسح، مع كون الميل مطابقًا لسعة الطبقة الثنائية الكهروكيميائية ( ).
تم تقدير ECSA بعد ذلك باستخدام المعادلة التالية:
حسابات نظرية الكثافة الوظيفية
تم إجراء سلسلة من حسابات نظرية الكثافة (DFT) باستخدام حزمة المحاكاة الأولية فيينا (VASP). تم اعتبار وتقدير التدوير المغناطيسي باستخدام نموذج هوبارد. تم وصف تفاعل الإلكترون والأيون باستخدام موجة معززة بالمشاريع (PAW)، وتم تعيين حد الطاقة الحركية لتوسعات الموجات المسطحة إلى 450 إلكترون فولت. تم التعامل مع طاقات تبادل الإلكترونات والتداخل ضمن تقريب التدرج العام (GGA) في تبادل-تداخل بيردو-بورك-إرنزرهوف (PBE). تم استخدام مخطط DFT-D3 لتصحيح التشتت لوصف تفاعلات فان der Waals (vdW) في امتصاص الجزيئات. تم أخذ عينات من منطقة بريلوان باستخدام طريقة مونكهورست-باك.أخذ العينات ومعايير التقارب كانتفي تحسين الهيكل، ومعيار تقارب القوةعرض تلطيف الإلكترونتم استخدام تقنية ميثفيسل-باكستون. لتجنب التفاعلات بين صورتين دوريتين متجاورتين، تم ضبط سمك الفراغ ليكونولمحاكاة تأثير داخل مادة صلبة، قمنا بتثبيت ذرات من طبقتين في الأسفل.
تم استخدام طريقة حزام المطاط المرن المدفوع بالصورة المتصاعدة (CI-NEB) لتحديد حالات الانتقال (TS)، مع معايير تقارب الطاقة لـومعايير تقارب القوةتم استخدام الترددات الاهتزازية المحسوبة لوصف حالة الحد الأدنى دون ترددات تخيلية أو حالة انتقالية حقيقية تحتوي على تردد تخيلي واحد فقط. حاجز الطاقة الحركيةيتم استخدام خطوة فولمر في تفاعل الهيدروجين القلوي كموصوف للنشاط للأداء التحفيزي، والتي يمكن حسابها على النحو التالي:
أين و هي الطاقة الحرة لحالة الانتقال والحالة الأولية، على التوالي.
تم الحصول على تصحيح الطاقة الحرة من خلال تضمين طاقة النقطة الصفرية (ZPE) والمساهمات الانتروبية من درجات الحرية الاهتزازية المحسوبة مع تثبيت الركيزة، والقيمة المكتسبة باستخدام Vaspkit.1.2.4.
طاقة الامتزاز (تم حسابها عن طريق طرح طاقات الممتزّ من العوامل المساعدة من الطاقة الكلية للنظام الممتز:
أين هو إجمالي طاقة المادة الممتصة كما تم امتصاصها بثبات في الموقع النشط. و هي الطاقة الكلية للسطح الممتص والممتزات المعزولة، على التوالي.
في الوسط القلوي (قد تسير مسار OER رباعي الإلكترونات من خلال الخطوات الأولية التالية:
حيث يشير * إلى المحفز، و * يشير إلى الأنواع التي تم امتصاصها على مواقع النشاط.
تجاهل مساهمة الضغط الجزئي في الترجمة للجزيئات الممتصة، تم حساب الطاقة الحرة لكل خطوة وفقًا للمعادلة الخاصة بـ حيث E هي طاقة كل نوع تم الحصول عليها من حسابات DFT، وS هي الإنتروبيا، بينما T هو 298.15 كلفن. و هي التصحيحات الحرارية للإنثالبي والتصحيح الحراري للطاقة الحرة لجيبس، على التوالي. هذه منتم أخذ *O و *OH و *OOH من حسابات تحويل فورييه للتردد (DFT) وتم الحصول على القيم باستخدام Vaspkit.1.2.4.
الطاقة الحرة لجزيئات البروتون-الإلكترون – ( )) المتعلقة بتقدم PECT، في حين أن حقيقة أن أزواج البروتون-إلكترون في حالة توازن مع الغاز عند 0 فولت مقابل القطب الهيدروجيني القياسي ، والضغطبار، ودرجة الحرارةوفقًا لـ Vaspkit.1.2.4، فإن الطاقة الداخلية لجزيئات الغاز تُكتسب من المعادلة:enthalpy الغاز الجزيئي المكتسب من المعادلة:وطاقة غيبس الحرة لجزيئات الغاز المستمدة من المعادلة:كور. حيث E_DFT هي طاقة جزيء الغاز الحر المستخرجة من حسابات DFT، و G_cor’ هو التصحيح الحراري لطاقة غيبس الحرة لجزيء الغاز الحر المستخرج من حسابات تردد DFT وتم الحصول على القيمة باستخدام Vaspkit.1.2.4، مع درجة حرارة 298.15 كلفن، والضغط و كانت 0.035 ضغط جوي و 1 ضغط جوي، على التوالي، وكلاهما و أدخل 1 كقيمة لعدد التدويرات. لاحظ أن الطاقة الحرة لـيجب حساب جزيء الغاز بواسطة هذه المعادلة:.
مركز نطاق d لمدارات 3d لمواقع النيكل المستخرجة من كثافة الحالة الجزئية باستخدام المعادلة:
أين، الـهو PDOS لمستوى طاقة. تم تقييم فرق كثافة الشحنة باستخدام المعادلة (المادة الأساسية + المادة الممتصة) – (المادة الممتصة) – (الركيزة)، ثم تم تحليلها باستخدام كود VESTA.
بناء الهيكل
. أسطح سوبرسيل يتكون من 144 ذرة من سطح انقسام المستوى (101) من الشكل المعينيتم استخدام الخلية البدائية أولاً، وتم تعيين السماكة الجزئية إلى 2.0، ثم إضافةسمك الفراغ لبناء بلورة فراغية. يتم تنفيذ تكامل منطقة بريل باستخدام التشتت الموزع بشكل موحد الذي يمر عبر نقطة غاما لاختيار-شبكة في شبكة مونكهورست-باك لإجراء تحسين الهيكل، ونختار-شبكة لإجراء حسابات كثافة الحالة (DOS). سطح مؤكسد جزئيًا يتم بناؤه عن طريق تحميل ستة ذرات أكسجين في أكثر مواقع الامتصاص استقرارًا على سطح . : يتم تحميل ذرة نيكل تتنسيق مع ذرة كربون في سطح. . الـتم تحميل السطح بمزيد من ذرات الأكسجين لمحاكاة سطح مؤكسد أكثر، ثم تم تحميل ذرة نيكل تتنسق مع أربع ذرات أكسجين على السطح. . الـتم تحميل السطح بمزيد من ذرات الأكسجين لمحاكاة سطح مؤكسد أكثر، وتم استبدال بعض ذرات الكربون بذرات الأكسجين لتشكيل حالة غير منظمة جزئيًا.ثم يتم تحميل ذرة نيكل تتنسق مع ست ذرات أكسجين على السطح. . الـتم تحميل السطح بمزيد من ذرات الأكسجين لمحاكاة سطح مؤكسد بشكل أكبر وتم استبدال بعض ذرات الكربون بذرات الأكسجين لتشكيل حالة غير منظمة جزئيًا.السطح، ثم يتم تحميل ثلاثة ذرات نيكل بالتنسيق مع ست ذرات أكسجين، على السطح.
لتمثيل التأثير داخل مادة صلبة، تم تثبيت الذرات الموجودة في النصف السفلي من اللوح لجميع نماذج الحساب.
توفر البيانات
البيانات التي تدعم نتائج هذه الدراسة متاحة من المؤلف المراسل عند الطلب المعقول.
References
Wang, Q. et al. Ultrahigh-loading of ir single atoms on nio matrix to dramatically enhance oxygen evolution reaction. J. Am. Chem. Soc. 142, 7425-7433 (2020).
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294-303 (2012).
Roger, I., Shipman, M. A. & Symes, M. D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 1, 0003 (2017).
Zhang, L. et al. Graphene defects trap atomic ni species for hydrogen and oxygen evolution reactions. Chem 4, 285-297 (2018).
Wang, H. et al. Bifunctional non-noble metal oxide nanoparticle electrocatalysts through lithium-induced conversion for overall water splitting. Nat. Commun., 6, 7261 (2015).
Zhao, X. et al. Bifunctional electrocatalysts for overall water splitting from an iron/nickel-based bimetallic metal-organic framework/ dicyandiamide composite. Angew. Chem., Int. Ed. 57, 8921-8926 (2018).
Shi, H., Liang, H., Ming, F. & Wang, Z. Efficient overall water-splitting electrocatalysis using lepidocrocite vooh hollow nanospheres. Angew. Chem., Int. Ed. 56, 573-577 (2017).
Luo, W. et al. A review: research progress of neural probes for brain research and brain-computer interface. ACS Catal 12, 1167-1179 (2022).
Yang, Y. et al. Hierarchical nanoassembly of as a highly efficient electrocatalyst for overall water splitting in a wide ph range. J. Am. Chem. Soc. 141, 10417-10430 (2019).
Wang, M. et al. Interfacial water activation by single-atom co-n3sites coupled with encapsulated co nanocrystals for accelerating electrocatalytic hydrogen evolution. ACS Catalysis 12, 10771-10780 (2022).
Yin, J. et al. Iridium single atoms coupling with oxygen vacancies boosts oxygen evolution reaction in acid media. J. Am. Chem. Soc. 142, 18378-18386 (2020).
Jiang, K. et al. Dynamic active-site generation of atomic iridium stabilized on nanoporous metal phosphides for water oxidation. Nat. Commun. 11, 2701 (2020).
Wang, W. et al. Confining zero-valent platinum single atoms in -moc1-xfor ph-universal hydrogen evolution reaction. Adv. Funct. Mater. 32, 2108464 (2021).
Kuang, M., Han, P., Wang, Q., Li, J. & Zheng, G. CuCo hybrid oxides as bifunctional electrocatalyst for efficient water splitting. Adv. Funct. Mater. 26, 8555-8561 (2016).
Zhu, Y. et al. A perovskite nanorod as bifunctional electrocatalyst for overall water splitting. Adv. Energy Mater. 7, 1602122 (2017).
Li, M. et al. Proximity electronic effect of ni/co diatomic sites for synergistic promotion of electrocatalytic oxygen reduction and hydrogen evolution. Adv. Funct. Mater. 33, 2210867 (2022).
Yang, H., Wu, Y., Zhuang, Z., Li, Y. & Chen, C. Computational probing of temperature-dependent unfolding of a small globular protein: from cold to heat denaturation. Chin. J. Chem. 40, 515-523 (2021).
Yuan, S. et al. Dual synergistic effects between Co and Mo 2 C in Mo2C heterostructure for electrocatalytic overall water splitting. Chem. Eng. J. 430, 132697 (2022).
Yu, Y., Zhou, J. & Sun, Z. Novel 2D transition-metal carbides: ultrahigh performance electrocatalysts for overall water splitting and oxygen reduction. Adv. Funct. Mater. 30, 2000570 (2020).
Anjum, M. A. R., Lee, M. H. & Lee, J. S. Boron- and nitrogen-codoped molybdenum carbide nanoparticles imbedded in a bcn network as a bifunctional electrocatalyst for hydrogen and oxygen evolution reactions. ACS Catal 8, 8296-8305 (2018).
Das, D., Santra, S. & Nanda, K. K. In situ fabrication of a nickel/ molybdenum carbide-anchored n-doped graphene/cnt hybrid: an efficient (Pre)catalyst for OER and HER. ACS Appl. Mater. Interfaces 10, 35025-35038 (2018).
Li, M. et al. Ni strongly coupled with mo2c encapsulated in nitrogendoped carbon nanofibers as robust bifunctional catalyst for overall water splitting. Adv. Energy Mater. 9, 1803185 (2019).
Xiao, M. et al. Molten-salt-mediated synthesis of an atomic nickel co-catalyst on tio2for improved photocatalytic h2evolution. Angew. Chem., Int. Ed. 59, 7230-7234 (2020).
Suryanto, B. H. R., Wang, Y., Hocking, R. K., Adamson, W. & Zhao, C. Overall electrochemical splitting of water at the heterogeneous interface of nickel and iron oxide. Nat. Commun. 10, 5599 (2019).
Hu, F. et al. Lattice-Matching formed mesoporous transition metal oxide heterostructures advance water splitting by active bridges. Adv. Energy Mater. 12, 2200067 (2022).
Tang, C. et al. Electronic Coupling of Single Atom and Boosts Water Electrolysis. Energy Environ. Sci. 5, 899-905 (2022).
Li, G. et al. The synergistic effect of Hf-O-Ru bonds and oxygen vacancies in for enhanced hydrogen evolution. Nat. Commun. 13, 1270 (2022).
Li, R. et al. Potential-dependent reconstruction of Ni-based cuboid arrays for highly efficient hydrogen evolution coupled with electrooxidation of organic compound. Chem. Eng. J. 453, 139797 (2023).
Zhao, Y. et al. Dynamics and control of active sites in hierarchically nanostructured cobalt phosphide/chalcogenide-based electrocatalysts for water splitting. Energy Environ. Sci. 15, 727-739 (2022).
Zhou, H. et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and fuel. Nat. Commun. 12, 4679 (2021).
Li, S. et al. Oxygen-evolving catalytic atoms on metal carbides. Nat. Mater. 20, 1240-1247 (2021).
Zhang, P. et al. Bifunctional single atom catalysts for rechargeable zinc-air batteries: from dynamic mechanism to rational design. Adv. Mater. 35, 2303243 (2023).
Zhou, L., Zhao, D. & Lou, X. W. Double-shelled CoMn 2 O 4 hollow microcubes as high-capacity anodes for lithium-ion batteries. Adv. Mater. 24, 745-748 (2012).
Guan, J., Mou, F., Sun, Z. & Shi, W. Preparation of hollow spheres with controllable interior structures by heterogeneous contraction. Chem. Commun. 46, 6605-6607 (2010).
Han, L. et al. Atomically dispersed molybdenum catalysts for efficient ambient nitrogen fixation. Angew. Chem., Int. Ed. 58, 2321-2325 (2019).
Wang, Q. et al. Single iridium atom doped ni(2)p catalyst for optimal oxygen evolution. J. Am. Chem. Soc. 143, 13605-13615 (2021).
Wei, H. et al. Molybdenum carbide nanoparticles coated into the graphene wrapping -doped porous carbon microspheres for highly efficient electrocatalytic hydrogen evolution both in acidic and alkaline media. Adv. Sci. 5, 1700733 (2018).
Yu, L., Yang, J. F., Guan, B. Y., Lu, Y. & Lou, X. W. D. Hierarchical hollow nanoprisms based on ultrathin ni-fe layered double hydroxide nanosheets with enhanced electrocatalytic activity towards oxygen evolution. Angew. Chem., Int. Ed. 57, 172-176 (2018).
Han, W. et al. Ultra-small Mo2C nanodots encapsulated in nitrogendoped porous carbon for pH-universal hydrogen evolution: insights into the synergistic enhancement of HER activity by nitrogen doping and structural defects. J. Mater. Chem. A 7, 4734-4743 (2019).
Jin, H. et al. In situ cobalt-cobalt oxide/N-doped carbon hybrids as superior bifunctional electrocatalysts for hydrogen and oxygen evolution. J. Am. Chem. Soc. 137, 2688-2694 (2015).
Zhuang, Z. et al. Atomically dispersed nonmagnetic electron traps improve oxygen reduction activity of perovskite oxides. Energy Environ. Sci. 14, 1016-1028 (2021).
Qin, J. et al. Activating edge-Mo of 2H-MoS2via coordination with pyridinic N-C for pH-universal hydrogen evolution electrocatalysis. ACS Catal 11, 4486-4497 (2021).
Chen, W.-F. et al. Reply to comments on “synthesis, characterization, and structures of persistent aniline radical cation”. Energy Environ. Sci. 6, 943-945 (2013).
Aracena, A., Sanino, A. & Jerez, O. Dissolution kinetics of molybdite in KOH media at different temperatures. T NONFERR METAL SOC 28, 177-185 (2018).
He, Z. et al. Activating lattice oxygen in NiFe-based (oxy)hydroxide for water electrolysis. Nat. Commun. 13, 2191 (2022).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886-17892 (2004).
Norskov, J. K., Abild-Pedersen, F., Studt, F. & Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl Acad Sci USA. 108, 937-943 (2011).
شكر وتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2023YFB4005100)، ومؤسسة العلوم الطبيعية الوطنية في الصين (22309011، 52272186، 22105116، 21872008، 52173232، 21925202، U22B2071)، ومشروع العلوم والتكنولوجيا في مقاطعة يونان في مدرسة الدراسات العليا المتحدة في الجنوب الغربي (202302AO370017)، والمهمة الدولية المشتركة بشأن تغير المناخ والحياد الكربوني، وصندوق أبحاث معهد بكين للتكنولوجيا للباحثين الشباب. نشكر محطة 1W1B على قياسات XAFS في منشأة الإشعاع المتزامن في بكين (BSRF) والدكتور فنج زانغ من مركز التحليل والاختبار، معهد بكين للتكنولوجيا.
مساهمات المؤلفين
قام D.Z. و J.T.Z. و M.H.C. و C.C. بالإشراف على المشروع. صمم M.Y.H. و D.Z. العمل وأجروا معظم التجارب. قام L.R.Z. بتوجيه قياسات XANES و XAS خارج الموقع، وحلل نتائج XAS. ساعد W.Y.F. و X.T. و J.T. و F.T. و X.W. في إعداد العينات وتوصيف العينات باستخدام TEM و XPS. ناقش جميع المؤلفين النتائج وساعدوا خلال إعداد المخطوطة.
يجب توجيه المراسلات وطلبات المواد إلى دي زهاو، مينهوا كاو، جياتا زانغ أو تشين تشين.
معلومات مراجعة الأقران تشكر Nature Communications أوداي مايتي والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة الأقران لهذا العمل. يتوفر ملف مراجعة الأقران.
معلومات إعادة الطبع والتصاريح متاحة على http://www.nature.com/reprints
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
المختبر الرئيسي لعلوم الكتل، وزارة التعليم في الصين، مختبر بكين الرئيسي لمواد تحويل الفوتون/الكهرباء الضوئية، كلية الكيمياء والهندسة الكيميائية، معهد بكين للتكنولوجيا، بكين 100081، الصين.منشأة الإشعاع المتزامن في بكين، معهد الفيزياء عالية الطاقة، الأكاديمية الصينية للعلوم، بكين 100049، الصين.مركز أبحاث الهندسة لمواد الأرض النادرة المتقدمة، قسم الكيمياء، جامعة تسينغhua، بكين 100084، الصين.ساهم هؤلاء المؤلفون بالتساوي: مينغيون هو، ليرونغ تشنغ.البريد الإلكتروني:dizhao@bit.edu.cn; caomh@bit.edu.cn; zhangjt@bit.edu.cn; cchen@mail.tsinghua.edu.cn
The rational design of efficient bifunctional single-atom electrocatalysts for industrial water splitting and the comprehensive understanding of its complex catalytic mechanisms remain challenging. Here, we report a Ni single atoms supported on oxygen-incorporated via bridge bonds, that gives high oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) bifunctional activity. By ex situ synchrotron X-ray absorption spectroscopy and electron microscopy, we found that after HER, the coordination number and bond lengths of and were all altered, yet the Ni species still remain atomically dispersed. In contrast, after OER, the atomically dispersed Ni were agglomerated into very small clusters with new ( bonds appeared. Combining experimental results and DFT calculations, we infer the oxidation degree of and the configuration of single-atom Ni are both vital for HER or OER. This study provides both a feasible strategy and model to rational design highly efficient electrocatalysts for water electrolysis.
Electrochemical water splitting is widely regarded as one of the most important and promising pathways to convert renewable energy into high-purity hydrogen and oxygen . For a continuous overall water splitting process in practical, pairing the two electrode reactions together in an integrated electrolyser is difficult because of the mismatch of pH ranges in which these catalysts are stable and remain the most active . In addition, producing different catalysts for both monofunctional catalysts requires different equipment and processes, which could increase the cost compared to bifunctional catalysts. Therefore, compared to monofunctional catalysts, bifunctional catalysts are relatively favorable options for overall water decomposition . Currently, Pt-based materials and Ir/Ru-based materials are the benchmark in HER and OER catalysts, respectively. Nevertheless, their scarcity and high price hinder their development and wide applications . Meanwhile, only a few OER catalysts (e.g. and ) are stable in acidic solutions, thus designing cost-effective and
earth-abundant bifunctional electrocatalysts with high efficiency of overall water splitting in the same alkaline conditions are of great significance and urgency .
Single-atom catalysts (SACs) featuring maximizing the atomic utilization rates and the synergistic effects between single-atom sites and its support are considered as the most promising strategy for designing cost-effective and earth-abundant bifunctional electrocatalysts . Currently, most SACs are based on carbonaceous substrates, such as amorphous carbon, graphene, with isolated metal atoms coordinated by intrinsic defects or heteroatoms. However, such catalysts show insufficient activity when applied in HER, especially in OER, while they exhibited obvious improvement in electrocatalytic reduction reactions. So, it is a critical challenge to develop SACs with new highly conductive support. Transition metal-based carbides, such as molybdenum carbide ( ), had metal-like electric conductivity and received special attention as potential bifunctional
electrocatalysts for water splitting. However, the water-splitting catalytic performance of pure is far from satisfactory due to the weak adsorption strength of oxygen species and the slow dissociation kinetics of and other reaction intermediates . To address this challenge, one of the most effective strategies is to decorate with transition metals ( , and W ) . In addition, the bridging bond metal between the substrate material and single atoms can produce strong electronic coupling via the surface, resulting in effective charge transfer, which can optimize adsorption/desorption behavior of the active site for oxygen intermediates, hydrogen intermediates . For example, Suryanto group reported that the surface unique configuration could modify Gibbs free energy absorption of reaction intermediates ( and ), then breaking the scale relationship and promoting OER and HER activities . So, it is feasible and challenging to construct atomically distributed metal centers on surface-oxygen-abundant to effectively modulate the adsorption/desorption behavior of the key intermediates during the water splitting process.
Furthermore, with insights into the electrocatalytic process, studies have evidenced that many electrocatalysts undergoes reconstitution during the so-called activation process or the initial cyclic voltammetry measurement. As for the HER example, Li et al. demonstrated that the occurred structural change during the HER activation process, resulting in high catalytic activity as well as stability . In addition, the actual active sites for HER were generated by core@shell structure of pre-electrocatalyst through the potential-driven reconstitution . Besides, Wang group had demonstrate that the active HER species of NBs arose from in situ reconstructed P-Co-O-Fe-P configurations with lowvalence metal sites ( ), which can decrease the energy barriers for water dissociation and adsorption of intermediates . In sharp contrast, their OER activity arose from the oxygen-bridged, highvalence moieties. Due to the redox process in OER, the catalyst surface reconstitution is more likely to accompanied. Most of the catalysts are fully oxidized into transition metal oxide or hydroxides, or their surfaces can be reconstructed into different coordination environments. Zhou et al. identified that the catalyst was transformed into the low-crystalline during the OER process and found that the serve as the reactive species for OER rather than the original species . Meanwhile, Li group reported the reconstitution of catalysts, that is, O-bridged FeNi moieties were formed after electrochemical treatment, which provided the high oxygen-evolving catalytic activity . All the pioneering works mentioned above have demonstrated, that the ability of the catalyst to “evolve” during the reaction may be a key feature of highly active OER catalysts. So, understanding of the structural transformation of the catalysts is crucial to better unveil the real catalytic mechanism and design novel electrocatalysts, although they have rarely been studies for SACs .
Here, we primarily motivated to use with the partially oxidized surface as a support material to stabilize Ni single atoms. The resulting materials possess a thin layer of atomic Ni sites, which interacted with the oxidized surface of via bridge bonds ( ) and exhibited good catalytic activity for HER, OER, and overall water splitting. By ex situ synchrotron X-ray absorption spectroscopy and electron microscopy observations, we found, the configuration of atomically dispersed Ni in electrocatalyst underwent transformation and support were overoxidized or dissolved to varying degrees. After HER, the coordination number of and the bond length of ( Mo) were all increased, but Ni species still remained atomically dispersed form. However, after OER, the coordination number of Ni O increased, the coordination number of ( ) decreased but new ( ) bonds appeared, indicating the atomically dispersed Ni were agglomerated into very small clusters
under applied potentials. Combining the experimental results and DFT calculations, we found partially oxidized will be beneficial to HER performance. However, compared with the original , the potential-driven reconstituted small Ni clusters with optimal and coordination structures are more conducive to catalyzing the production of .
Results
Material synthesis and characterization
The nanospheres were synthesized by the following steps: (1) preparation of Mo-based organic nanosphere precursors by a facile hydrothermal method; (2) adsorption of the Ni metal ions via impregnation method; (3) heat treatment at certain temperatures under air and atmospheres. During the pyrolysis process, the Mo-based organic precursors were simultaneously converted to nanocrystals with Ni ions fixed on their surfaces, which embedded in a uniform carbon substrate. Transmission electron microscopy (TEM) images of Fig. S1 shows that the amorphous Mo-based precursor is a nanosphere structure with rough surfaces with a diameter of about 200 nm , and the morphology does not change significantly after the adsorption of Ni ions on the surface (Fig. S2). After pyrolysis, an obvious double-shelled hollow structure can be observed (Fig. 1a, b). The mechanism of double-shelled hollow structure formation has been studied in detail (Figs. S3-S6) and its possible schematic illustration is shown in Fig. S7, which is mainly based on the early oxidation of air and non-equilibrium non-uniform shrinkage caused by heat treatment . During the formation of double-shelled hollow structure, the conditions of air and atmospheres are both vitally important. In addition, the specific surface areas obtained from adsorption-desorption isotherms show a significant increase from 13.9 for to for (Fig. S8), which is probably due to the volatilization of nitrate from Ni sources. X-ray diffraction (XRD) patterns (Fig. S9) indicate that the diffraction of can be well indexed to pure phase (JCPDS card no.35-0787) . No peaks of Ni metals, metal oxide can be observed. The selected area electron diffraction (SAED) pattern (insert in Fig. 1b) indicates the polymorphism of the .
Energy-dispersive X-ray spectroscopy (EDS) mapping images reveal that , and O elements almost have the same doubleshelled hollow shape and C elements is uniformly distributed in an individual nanospheres (insert in Fig. 1c, and Fig. 1d). Combined with the HR-TEM magnified image in Fig. S10, we further confirm that nanocrystals with Ni ions were embedded in a uniform carbon substrate. To further identify the exact location of single Ni atoms in the nanospheres, we performed sub-angstrom-resolution high-angle annular dark field aberrationcorrected (HAADF)-scanning TEM (STEM) (Fig. 1e, f). Figure 1e clearly detected the lattice fringes with an interplanar distance of 0.228 nm and 0.135 nm , corresponding to the (101) and (103) plane of a hexagonal , respectively. Detailed observation of the edge of the bright Mo atom array reveals an amorphous or low crystallinity molybdenum oxide ( ) layer (around the yellow line in Fig. 1f) due to surface oxidation . In addition, due to the different Z-contrast, the darker spots (red circles in Fig. 1f) on the edge of brighter spots (Mo atoms) can be identified as isolated Ni single atoms . The intensity profiles at the location of position g 1 and g 2 marked by yellow circles in Fig. 1f including darker spots and brighter spots of surface layer further confirm the presence of isolated single atoms (Fig. g1 and . For comparison, the control electrocatalysts of and were prepared with varying amounts of Ni . XRD patterns and TEM images display the same crystal structure and morphologies as those of (Figs. S9, S11). Their Ni-loading contents detected by inductively coupled plasma atomic emission spectroscopy (ICP-AES) are and , respectively (Table S1).
Fig. 1 | Morphology and structure of the materials. a Schematic illustration of the fabrication process of the electrocatalyst. HR-TEM image of . Insets in (b): SAED pattern. d Elemental mapping of Mo, Ni, O, C for an individual nanospheres (inset in Fig. 1c), respectively. e HAADF-STEM
image and corresponding enlarged view of the . The bright dots highlighted with red in (f) marks the Ni single-atoms in . g Lineal contrast analysis for selected area of g1 and g2 at the edge of Fig. 1f.
Since the coordination environment determines the electronic structure, the valence states of Mo, and Ni have been studied by X-ray photoelectron spectroscopy (XPS) (Figs. S12-13). As shown in Fig. 2a, the high-resolution Mo 3d spectra of pure were deconvoluted into six peaks, corresponding to ) and species and can be assigned to molybdenum oxides due to surface oxidation of Mo species, indicating a thin surface layer of were formed outside electrocatalyst (denoted as . This is well consistent with HAADF-STEM analysis (Fig. 1e-f). It is worth mentioning that, the surface oxide layer can be controlled by the atmosphere and temperature to which the is exposed. For example, when the newly reduced were taken out of the tube furnace at 60 in air, its surface would be much more oxidized compared with the main sample of . While, when the newly reduced were protected in Ar atmosphere for 12 h before taking out the furnace, its surface would be much lower oxidized compared with the main sample of . (Fig. S13a, b). In addition, after etching the surface oxide layer, the main phase of gradually leaks out, which is demonstrated by XPS results with different etching depths (Fig. 2a, Fig. S13c). The XPS spectrum of C 1s (Fig. S14a) is fitted into four different signals at , and 288.9 eV , which are attributed to , and , respectively . After loading of Ni atoms, the binding energy of spectra for display a positive shift about 0.12 eV compared with that of pure , indicating the charge migration caused by the atomic Ni incorporation . This is further proved by the positive shift of the main peak of O XPS for compared with that of pure (Fig. S14b). The Ni XPS spectrum can be split into six peaks, of which the peaks at 856.4 and 874.2 eV are assigned to caused by the
Fig. 2 | Structural characterization. a XPS spectra of Mo 3 d. b XPS spectra of Ni . c Mo K-edge XANES spectra. Inset in (c) is the partial enlargement of the pre-edge peak. d The FT-EXAFS curves for the as-prepared samples. e Ni K-edge spectra. The
corresponding FT-EXAFS curve. The wavelet transform (WT) of the . Ni K-edge EXAFS of in R and k spaces.
bonding between Ni and adsorbed oxygen, and the peaks at 862.4 and 881.4 eV correspond to the satellite peaks of Ni (Fig. 2b). Only a small amount of can be observed at 853.5 and 870.5 eV , which indicates that the Ni atoms are mainly coordinated with the surrounding oxygen atoms . It should be noted that, due to the Ni content in the is lower than , the peak of is not obvious, while is observed at 862.4 and 881.4 eV , indicating that Ni in the sample is almost entirely bonded to the surrounding oxygen atoms. While, the proportion of peak in is significantly larger than that in (Fig. S15), which is probably due to the excessive Ni aggregates into Ni clusters or nanoparticles.
To assess the electronic structure and local environment of the Ni and Mo in , X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) were investigated. The Mo K-edge XANES spectra are shown in Fig. 2c. Compared with pure , the pre-edge of was higher, suggesting that the average oxidation state of Mo in the was more positive, which is consistent with the aforementioned XPS results. The Fourier transform (FT) extended EXAFS (Fig. 2d) of the pure and possessed two peaks. The strongest peak is located at , corresponding to the Mo-Mo bonds. The other peak is located at , which is associated with the Mo-C bonds . Compared with pure O/ , the introduction of Ni would extend the and bonding length due to the interaction between Ni and . The Ni K-edge pre-edge of is located between the Ni and NiO (Fig. 2e), which indicates that the valence of Ni in is
situated between 0 and +2 . The Ni K-edge XANES spectra and the corresponding k -space spectra of closely resemble that of NiO reference (Fig. ), suggesting the formation of surface . The Fourier transformed (FT) -weighted EXAFS spectrum in R space further confirmed the formation of on the amorphous thin layer on the surface of . As shown in Fig. 2f, one notable peak at contributed by the peak is observed. The FT of Ni foil shows a peak at and NiO displays a peak at , both corresponding to the interaction. does not have these peaks, which demonstrates that Ni atoms should exist in the single-atomic form on the amorphous thin layer . In addition, there is a scattering peak at around , which could be due to the bonding of . Combined with aforementioned results of the oxidized surface of bonding should dominate on the surface.
We have also performed wavelet transform (WT) analysis of for verifying the coordination environments of bonding, which allows information to be displayed in both R -space and K-space. As shown in Fig. 2 g , a strong WT signal focused at , which was derived from Ni-O-Mo contribution. To acquire the precise coordination configuration of Ni atoms in , the quantitative EXAFS fitting is performed to extract the structural parameters and the results are presented in Fig. 2h, i and summarized in Table S3. The best-fitted result reveals Ni atom is coordinated with O with a coordination number of 3.9 at and as well coordinated with Mo with a coordination number of 5.0 at in the second or higher shell. In addition, the with higher Ni content has a weak
Fig. 3 | DFT calculation. The two plots of charge density differences of : top view of 3D plot (a) and 2D display (b). The isosurface level set to 0.003 e , where charge depletion and accumulation were depicted by purple and
yellow, respectively. c PDOS graph of orbitals of orbitals of Mo, and all orbitals of O atoms specified by cross marks in . d The chemisorption energies of Ni atom on surfaces of and .
peak at corresponding signal (Fig. S17), which is well consistent with the results of XPS.
In order to further study the mechanism of Ni atoms binding on the oxygen-incorporated , we did a series of DFT calculations. According to the previous results of structural analysis, we chose plane (101) of as the reaction surface. Then, calculations were made on the absorption of oxygen atoms on the surface, and we determined the most stable structure (Fig. S18) by comparing the values of their total energy. So, we attached some oxygen atoms to the facet by the most solid adsorption to simulate a partially oxidized surface of . According to the above data, we adsorbed Ni atom bonding with four oxygen atoms and one carbon atom at the facet, respectively, to obtain calculation models and (Fig. S19). Firstly, we performed calculations of the charge density difference between Ni and facet, as shown in Fig. 3a, b. It could be clearly seen that obvious tendency to transfer electrons from Ni site to surrounding atoms, indicating the interaction between Ni with facet. This is also confirmed by Fig. 3c, because of overlaps between the orbitals of Ni, O, and Mo atoms (marked by cross) around the Fermi level. So, we concluded that Ni atoms could be stably adsorbed on the partially oxidized facet of . In addition, the value of adsorption energy at was significantly lower than (Fig. 3d), further indicating that the bonding of Ni-O-Mo was more stable.
Electrocatalytic performance
To examine the electrochemical OER catalytic activities in 1 M KOH using a three-electrode set-up. As shown in Fig. 4a, the exhibits an overpotential ( ) of 299 mV at , which significantly exceeds those of mV ), pure and commercial catalyst ( 386 mV ). From these values, we can see that the higher loadings of Ni results in better performance due to the higher density of catalytic reactive centers. However, the catalysts cannot perform even better when the Ni loading is excessive. In addition, the overpotential of catalyst requires only 430 mV at the current density of , even far less than 670 mV required by commercial catalyst,
demonstrating a huge improvement of the OER activity under the high current density. In addition, Fig. 4a shows the Ni oxidation peak, corresponding to the oxidation Ni from low valence states to high valence states . Tafel slopes were calculated to unveil that endows a smaller Tafel slope ( ), much lower than those of ), pure and even commercial ( ), which is a sign of its fast electron transfer and superior mass transport properties (Fig. 4b) . Motivated by the promising OER performance of , the electrocatalytic HER was also investigated in the same 1.0 M KOH environment. Figure 4 c shows the Pt/C catalyst exhibits the best HER activity, whereas shows competitive performance with an overpotential of 133 mV at a current density of , which is lower than those of and pure . Furthermore, the Tafel slope of the is , less than those of ) and pure (Fig. 4 d ). It is worth noting that in order to exclude the role of Ni atoms on carbon substrate, we set up two control samples: Ni atoms on carbon substrate obtained via etching particles in and Ni atoms on pure carbon spheres ( ) synthesized by the same method with . As can be seen from XRD patterns, TEM, and element mappings (Fig. S20), both samples only show carbon peaks and uniform Ni element distribution. Electrochemical tests showed that compared with , their HER and OER catalytic activity can be ignored (Fig. S21), indicating the catalytic activity of comes from the Ni single atoms anchored on oxygen-incorporated . Impressively, outperformed most of the non-precious metal SACs in alkaline media (Fig. 4e, Tables S4-5), proving is a potentially highly efficient bifunctional catalyst. To investigate the electrode kinetics under the catalyst process, electrochemical impedance spectroscopy (EIS) measurements were carried out. Fig. S22 displays a favorable charge transfer resistance ( ) of as compared with other control samples . Besides, the electrochemical active surface area (ECSA) of all samples was
Fig. 4 | HER, OER and overall water splitting performance of the catalyst in with iR compensation. a Polarization curves and b Tafel plots for and pure , commercial for OER (loading amount ). c Polarization curve and d Tafel plots for and pure for HER (loading amount ). e Comparison of the overpotentials of and recently reported non-precious metal SACs at in 1.0 M
KOH. The histograms in yellow and green denote the HER, OER performance, respectively. Current density versus time ( ) curves for HER and OER. g Polarization curves of and couples for overall water splitting in 1.0 M KOH (loading amount ). Inset is the long-term durability of overall water splitting at a current density of . h Illustration of the electrodes for overall water splitting.
reflected from the double layer capacitance ( ) (Fig. S23). catalyst has the highest value, which indicates that has more exposed active surface area than other samples. Since the specific activity reflects the intrinsic activity of the catalysis, ECSAnormalized LSV curves for both HER and OER were shown in Fig. S24. The specific activity (SA) normalized by ECSA shows that exhibits the best electrocatalytic activity toward both OER and HER compared to other catalysts following the order of . In particular for OER, at the overpotential of 370 mV (Fig. S24d), exhibits the SA of 8.71 , which is higher than those of and ECSA). These results indicate incorporation of Ni not only improves the density of active sites but also intrinsically improves HER and especially OER activity of catalyst. The durability of the catalyst is an important indicator, especially for SACs. For OER, longterm electrolysis at a constant current has been performed under a constant current of by chronoamperometry for 16 h (Fig. 4f), which proved that the currents of first increase and then maintain stability for a long time. In contrast, after 16 h , the
commercial catalyst shows distinctly difference in the required overpotential. For HER, although it was much more stable than , the current density of the catalyst decreased slightly after 16 h .
Furthermore, we assembled an electrolyzer with 1.0 M KOH electrolyte using as bifunctional electrodes ( ). was also tested for comparison. Figure 4 g gives a comparison of LSV curves of and . The cell voltage required to reach 10,100 , and 150 mA for are , and 1.90 V , while requires and 1.93 V , respectively. The result shows that the electrocatalytic overall splitting performance of is comparable to that of especially under the high current density and superior to most reported single-atom catalysts (Table S6). In the stability test, the current density has only decreased from to after 120 h , indicating its good stability as a bifunctional electrocatalyst (inset in Fig. 4 g ). Moreover, the obvious bubbles were observed at the cathode and anode in the longtime stability testing, corresponding to and , respectively (Fig. 4 h and Fig. S25).
Potential-driven new active interfaces generation
In order to reveal the different stability phenomena of HER and OER, a series of characterizations were carried out to probe the identity of the real catalytically active species. The XRD pattern (Fig. S26a) indicates that the surface of undergoes structural reorganization to form different phases after the different electrochemical reactions. After electrochemical HER, the catalyst maintains the diffraction peaks of , but other weaker peaks of (JCPDS card no.35-0609) and (JCPDS card no.29-1021) appear. In contrast, for OER, all the XRD peaks could be assigned to the crystal structures of (JCPDS card no. 05-0508 and 35-0609) and without peaks, indicating the were completely disintegrated. Fig. S 27 shows the spectra of after reactions, the major Mo peaks can be assigned to without low valence state of Mo, suggesting the average oxidation state of Mo are both higher than that of , that is to say the surface of the catalysts both have obvious oxidation behavior . However, after HER reaction, the peak shifted to a little lower position, which is probably due to the interaction between single atom Ni and the reconstructed substrate. The morphology of double-shelled hollow structure was not obviously changed after catalytic reactions (Fig. S2830). However, in the case of after HER, the enlarged HRTEM image shows original large nanoparticles were crushed into smaller ones and two different lattices of 0.22 nm and 0.23 nm correspond to the d -spacing of (101) and (060) planes of the and , respectively (Fig. 5a, b). Besides, in the after OER, the image exhibits the big nanoparticles were changed obviously to smaller ones and the interplanar spacings of 0.21 and 0.23 nm correspond to the (141) and (060) plane of . These results indicate that undergoes reconstitution in the process of HER and OER. So, in order to determine the real active species involved in these two reactions, ex-situ XANES and FT-EXAFS spectra after CV and i-t chronoamperometric test for HER and OER were further recorded. As displayed in Fig. 5c, the Mo K-edge XANES spectra for catalysts after HER and OER activation show both positive shifts in the absorption edge compared with that of , indicating the valences Mo oxidation states were increased. However, for catalysts after OER, the Mo K-edge XANES spectra show more positive shifts compared with that of catalysts after HER, indicating catalysts after OER has higher valence state of Mo, which consistent with XPS results. In addition, a new notable shoulder peak in the pre-edge region of OER were shown, suggesting the formation of strongly distorted octahedra. Figure show the Mo -weighted FT-EXAFS spectra of after HER and OER, respectively. After HER, it shows the main peak centered at , corresponding to the scattering in . In particular, two smaller peaks located at around and can be found, which are typical characteristic of Mo-O bonds, indicating the surface of was further oxidized . In contrast, after OER, the spectra display the existence of the Mo-O bond but no Mo-Mo bond, and its shape is similar with that of molybdate, indicting, except for crystal , amorphous or low crystallinity molybdate was also formed followed by the possible reaction of . In order to clary whether the changes of is automatic in KOH KOH electrolyte at open circuit potential (OCP). As shown in Fig. conditions, we tested the XRD, Mo k-edge XANES and FTEXAFS curve for electrocatalysts after being dipped into KOH electrolyte at open circuit potential (OCP). As shown in Fig. S26b, the electrocatalysts after being dipped into KOH electrolyte at OCP ( ) has similar characteristic peaks with after HER except the a little weaker and peaks. Based on the dissolution of in , we speculate the original surface reacts with KOH to form , and with the consumption of surface surface is continuously oxidized to form new surface until the is completely dissolved in KOH . However, compared with after has characteristic peaks of . In addition, compared with and after HER, the absorption edge of was higher, suggesting its average oxidation state of Mo was more positive. Meanwhile, the Mo k-edge XANES shape and the Mo k3-weighted FT-EXAFS spectra of are different from after HER but similar with that of after OER. These results indicate the dissolution and oxidation of in KOH is automatic in alkaline conditions. The applied oxidation potential of OER process will intensify these two continuous reaction processes, while HER process will inhibit to a certain extent the oxidation of the inner layer of and then partially prevent the formation of from the outer layer of . Based on the dissolution of in KOH , Fig. 5f, g present comparisons of catalysts in Ni K-edge XANES and FT-EXAFS after HER and OER. In the case of HER (Fig. 5f), the pre-edge position of XANES spectra show a slight shift to higher energy values compared with the original , indicating that the Ni oxidation state slightly increases but still between and . After carefully compared the FT-EXAFS spectra of before and after HER, we found the peak of O did not change, however the position of Ni-O-Mo was shift to higher R , which due to the reconstitution of substrate. After EXAFS fitting, we found the distance of is almost the same as that of and is increased from 2.40 to (Table S7). Moreover, the Ni-O coordination number increased to 5.90 while the Ni-Mo decreased to 4.50 . And no typical peaks for NiNi bonds are observed as well. These results indicated the Ni was still exists as single atoms while its coordination structure had changed slightly. This catalyst after HER was collectively denoted as . In contrast, after OER (Fig. 5 g ), the position of the absorption edge is higher than that of the reference, indicating the Ni has the higher oxidation than +2 , which probably due to the existence of NiOOH or the strong electron interaction between and the reconstructed substrate (Fig. S31). For the FT-EXAFS spectra of the Ni K-edge after OER, these shapes match well with that of . In addition, the Ni-O-Mo coordinated peak almost disappears and new distinct scattering of emerges at . The corresponding fitting EXAFS results display the bond distances if is and (Ni-O-Mo) is , respectively. The HAADF-STEM images (Fig. 5h) demonstrate the (060) plane of crystal and a smaller nanocluster of Ni (yellow circles). In addition, the selected-area elemental mappings at an atomic level further confirm the uniformly distributed Ni and uneven distribution Mo on the surface. This catalyst after OER was collectively denoted as . These results indicated, after OER, uniformly distributed Ni in undergoes transformation of the configuration and has been agglomerated into very small clusters by applied potential.
DFT simulations
In order to further explore the influence of various bonding forms on the electrocatalytic properties of HER and OER, we first analyzed the charge density difference and bader charge. Importantly, all adsorption energy calculations are obtained on the basis of correction to zero. As shown in the inset of Fig. 6a and Fig. S32, there is a tendency of transferring electrons from Ni site to adsorbates, and we could see explicitly the number of valence electrons of Ni increased when formed bond on facet (Fig. 6b) compared with formed bond in facet. Therefore, Ni atoms directly embed the plane (101) of has strong adsorption of protons and hydroxyl, while the surface incorporated O of would adjust the number of valence electrons of Ni , then weakening the adsorption capacity. So, the adsorption energies of for proton and hydroxyl are much lower than those of , which markedly reduces the likelihood of blocking active sites, thus improving the electrocatalytic properties of HER (Fig. 6a). In addition, we found the energy level of the d -band center of is much lower than that of
Fig. 5 | Structural analysis of the after HER/OER to study active sites. a, b HRTEM images of after HER/OER stability tests. c-e Ex situ Mo k-edge XANES and the corresponding FT-EXAFS curve of
Fig. 6 | DFT calculation of HER and OER. a The chemisorption energies of *H and * OH in Ni sites of and . Insets are their 3D plots of charge density differences: top view and side view. The number of bader charges transfer at specified , and Mo atoms in and . c PDOS graphs of orbitals of Ni atoms in and . The black dash lines show the d-band center , and the Fermi level was taken as zero of energy.
d The chemisorption energies of O in Ni sites of and . Gibbs free energy diagram of Volmer step. Inset in (g): the optimal models of different stage of Gibbs free energy change of OER pathways on Ni sites. Inset in ( ): the atomic configurations of different stage of . i Structural reconstitution of catalyst after different reaction process. Water blue , green , grey , red . (Fig. 6c). According to the d-band center theory, the lower electron density near of Fermi level will facilitate the desorption of adsorbate. So, compared with , the had much stronger capacity of adsorbed O (Fig. 6d), which will lead to the higher overpotential of . We then calculated the potential barrier for hydrogen production and the energy barrier for Volmer step, as well as the overpotential of OER. As shown in Fig. S33 and Fig. 6e, the energy of the potential-determining step required by for hydrogen generation is 0.5 eV , much lower than those of and . In addition, the energy barrier of sluggish water dissociation for is 0.55 eV , which is lower than 1.15 eV of and 1.09 eV of , indicating Ni loaded on partially oxidized can improve HER performance. The processes of Volmer step are shown in Fig. S34. Meanwhile, the Potential Determining Step (PDS) for OER is the third electrochemical step from to , as we can see, remarkably decreases the energy barrier in this step to 1.61 eV , lower than that of and at , indicating has much better performances, being consistent with the experimental observations. The diagram of OER reaction step was shown in Fig. S35.
Based on the above results, it would be found that the partially oxidized surface of facilitated the binding of Ni atoms via and (Ni-O-Mo) chemical bonds, thus promoting the electrocatalytic properties of HER and OER. However, the substrate of after HER and OER underwent oxidation and dissolved. For after HER (Fig. 6g, S36), we can see that its energy barrier of water dissociation is 1.24 eV , which becomes larger than that of , indicating the overoxidation of the substrate can lead to some adverse hydrogen evolution effects. But for after OER, the PDS changes from the step pf *O to *OOH to the step of *OOH to . Its free energy of the rate-determining step is 0.93 eV (Fig. 6h), which becomes smaller than that of at . In addition, compared with , the reconstructed shows a weaker adsorption capacity of (Fig. S37), indicating the reconstructed accelerates the oxygen evolution reaction. These results indicate the overoxidation of the substrate is conducive to the aggregation of monatomic Ni into clusters and the improvement of oxygen evolution performance. Figure 6i shows the structural reconstitution of catalyst after different reaction processes. These results are consistent well with the experimental observations.
In summary, we engineered a non-precious Ni single-atom bifunctional catalyst loaded on the surface of oxygen-incorporated via bridge bonds . The obtained catalyst exhibited an overpotential of 133 mV and 299 mV at the current density of for HER and OER, respectively. Comprehensive characterizations and DFT calculations indicated that the surface oxygen atoms of were conducive to stabilizing the single atom Ni via the bridge bonding of . The O-Mo bonding environment results in the regulation of electronic structures of single atom Ni towards an optimized proton and hydroxyl and moderate adsorption energy of O , which determines superior performance of the catalyst. Moreover, we found, that the configuration of atomically dispersed Ni in electrocatalyst underwent transformation and support were oxidized and dissolved to varying degrees due to instability in KOH and the influence of applied potential. After HER, Ni species still maintain atomically dispersed form. However, after OER, the atomically dispersed Ni were agglomerated into very small clusters. In addition, we found overoxidized will lead to the configurations of single atom Ni to reconstruct, which will degrade its HER performance. However, compared with the original , the potential-driven reconstituted small Ni clusters with optimal coordination structures are more favorable for OER. In a word, this work demonstrates a facile and feasible strategy to fabricate a low-cost and high-efficient bifunctional single-atom electrocatalyst and also provides guidance for further design of effective water-splitting electrocatalysts.
Methods
Synthesis of amorphous Mo-based precursor
The precursor Mo-based was first synthesized by a one-step hydrothermal method. Heat dissolved 1 g of of , and 0.132 g of in 50 mL of deionized water, when the solution changed from yellow-green to blue, it was transferred to a 100 mL Teflon-lined autoclave while hot. Then kept in an electric oven at the temperature of for 48 h . After the autoclave was cooled down to room temperature, it was centrifuged three times with alternating water and ethanol, respectively. The sample was dried in a vacuum oven at for 12 h .
Synthesis of electrocatalysts
0.20 g of Mo-based precursor and of were dissolved in 10 mL of deionized water mixed with 15 mL of ethanol. Ultrasonically sonication dissolved for 40 min , constant temperature water bath at for one night with continuous stirring. Then washed with ethanol for three times and dried to obtain the surface adsorbed Ni of Mo-based nanospheres. Finally, the sample was annealed at for 1 h under an air atmosphere with a heating rate of , to ensure that the C in the Mo-based nanospheres structure could escape with air, followed by for 3 h under a hydrogenargon atmosphere at the same rate of warming. The was obtained after annealing.
Similarly, and was synthesized with the same method except for the doping amount of nickel nitrate ( 1.5 M and 4.5 M , respectively). was synthesized by direct calcination of Mo-based precursors without .
Structural characterization
The crystalline structure and phase purity were tested by X-ray diffraction pattern (XRD, Ultima, Japan) with radiation at room temperature. The transmission electron microscopy (TEM) measurements were performed on Hitachi H-7650 operated at 2 kV . The Thermo Fisher IRIS Intrepid a system measured ICP-OES, which can be used to determine the element contents of the samples. Highresolution transmission electron microscopy (HR-TEM) and highangle annular dark-field scanning transmission electron microscopy (HAADF-STEM) were carried out by Talos F200X G2 and JEOL JEM2100 F field emission electron microscope working at 200 kV , respectively. The X-ray photoelectron spectroscopy (XPS) experiments were tested on the PHI 5000 Versaprobe III spectrometer. The Brunauer-Emmett-Teller (BET) specific surface areas of typical products were performed at 77 K in a Belsorp-max surface area detecting instrument. adsorption-desorption experiments were carried out at 77 K on a Micromeritics ASAP 2460 Instrument. X-ray absorption spectroscopy (XAS) measurement and data analysis: X-ray absorption near-edge spectra (XANES) at the Mo K-edge obtained at 1W1B station in BSRF (Beijing Synchrotron Radiation Facility, P. R. China) operated at 2.5 GeV with a maximum current of 250 mA . The C-edge and N-edge were measured at beamline BL12B of the National Synchrotron Radiation Laboratory (NSRL) of China and the samples were deposited onto a double-sided carbon tap.
Electrochemical measurements
All spectra were collected in ambient conditions. Measurements of HER and OER activity of the electrocatalysts were performed with a traditional three-electrode system (CHI-760E), using a graphitic carbon rod as the counter electrode, as the reference electrode, and a glassy carbon (GC) or carbon paper with the as-synthesized samples as the working electrode. 3.0 mg of sample and of Nafion solution were dispersed in of water/ethanol, then added 1.5 mg carbon black and sonicated for 30 min to form a homogeneous ink. Then of the solution was dispersed on a GC electrode of 3 mm diameter with a loading amount of . For the OER activity performed, 10.0 mg of sample and of Nafion solution in 1 mL of water/isopropanol, then of solution was dispersed on carbon paper with loading 0.74 mg . All electrochemical OER and HER experiments were performed in a 1.0 M KOH solution, and polarization curves were iR-corrected. All potentials were referenced to the reversible hydrogen electrode (RHE), following the below equation:
The electrochemically active surface area (ECSA) of the assynthesized samples without carbon black was estimated by the cyclic voltammetry (CV) measurements . The electrochemical doublelayer capacitance was tested from 0.86 to 0.96 V (vs RHE) in 1 M KOH solution for the HER or OER process at different scan rates ( , 100). The difference between the anode and cathode currents was plotted linearly against the scan rate, with the slope corresponding to the electrochemical double-layer capacitance ( ).
The ECSA was then estimated using the following equation:
Density Functional Theory Calculations
A series of density functional theory (DFT) calculations were all done with the Vienna Ab initio Simulation Package (VASP). The spin polarized was considered and calculated with Hubbard model. The electronion interaction was described using the projector augmented wave (PAW), and the kinetic energy cutoff for plane wave expansions was set to 450 eV . The electron exchange and correlation energies were treated within a generalized gradient approximation (GGA) in the Perdew-Burke-Ernzerhof (PBE) exchange-correlation. A DFT-D3 scheme of dispersion correction was used to describe the van der Waals (vdW) interactions in molecule adsorption. The Brillouin zone was sampled using the Monkhorst-Pack sampling and the convergence criteria were in structure optimization, and force convergence criterion of . The electron smearing width of was employed according to the Methfessel-Paxton technique. To avoid the interactions between two adjacent periodic images, the vacuum thickness was set to be , and to simulate the effect of inside a solid, we fixed two-layer atoms at the bottom.
The climbing image-nudged elastic band (CI-NEB) method was used to determine transition states (TS), with the energy convergence criteria of and force convergence criteria of . The computed vibrational frequencies were used to characterize a minimum state without imaginary frequencies or an authentic transition state with only one imaginary frequency. The kinetic energy barrier of the Volmer step at alkaline HER is applied as an activity descriptor for catalytic performance, which can be calculated as follows:
where and are the free energy of the transition state and the initial state, respectively.
The free energy correction was obtained by including the zeropoint energy (ZPE) and entropic contributions from vibrational degrees of freedom calculated with the substrate fixed, and the value gained by using Vaspkit.1.2.4
The adsorption energy ( ) was calculated by subtracting the energies of the isolated adsorbate and the catalyst from the total energy of the adsorbed system:
Where is the total energy of adsorbate as adsorbed steadily at the active site. and are the total energy of the adsorbed surface and isolated adsorbate, respectively.
In alkaline media ( ), the four-electron OER pathway may proceed through the following elementary steps:
Where the *refers to the catalytic, and the *one refers to the species that adsorbed on the activity sites.
Neglect PV contribution to translation for adsorbed molecules, the free energy of every step was calculated according to the equation of , where E is the energy of every species obtained from DFT calculations, and S are entropy, while T is 298.15 K . The and are the thermal corrections to enthalpy and the thermal correction to Gibbs free energy, respectively. These of , *O, *OH, and *OOH were taken from the frequency DFT calculation and got value by using Vaspkit.1.2.4.
The Gibbs free energy of the proton-electron pairs ( – ( )) related in the PECT progress, whereas the fact that the proton-electron pairs is in equilibrium with gaseous at 0 V versus standard hydrogen electrode ( , and pressure bar, and temperature . According to Vaspkit.1.2.4, the internal energy of gas molecular gained from the formula: , the enthalpy of gas molecular gained from the formula: , and the Gibbs free energy of gas molecular gained from the formula: cor . Where E_DFT is the energy of the free gas molecule obtained from DFT calculations, G_cor’ is the thermal correction to Gibbs free energy of the free gas molecule obtained from the frequency DFT calculation and got value by using Vaspkit.1.2.4, with the temperature of 298.15 K , the pressure of and were 0.035 atm and 1 atm , respectively, and both and input 1 as the value of spin multiplicity. Note that the free energy of an gas molecule should be calculated by this equation: .
The d -band center of the 3 d orbitals of Ni sites obtained from their PDOS by using equation:
where, the is the PDOS of an energy level of .
The charge density difference was evaluated using the formula (substrate + adsorbate) – (adsorbate) – (substrate), then analyzed by using the VESTA code.
Construction of structure
. A supercell surface consisting of 144 atoms from a cleaving surface of plane (101) of orthorhombic primitive cell was used firstly, and the fractional thickness set to 2.0 , then adding vacuum thickness to build vacuum slab crystal. The Brillouin zone integration is performed using the uniformly distributed scattering of going through the Gamma point to select a -mesh in the Monkhorst-Pack grid to make structure optimization, and we select -mesh to do density of state (DOS) calculations. . A partially oxidized surface of is constructed by loading six oxygen atoms at the most stable adsorption site on the surface of . : A Ni atom coordinating with C atom is loaded in surface. . The surface is loaded with more oxygen atoms to simulate a further oxidized surface, and then a nickel atom coordinating with four oxygen atoms is loaded on the surface. . The surface is loaded with more oxygen atoms to simulate a further oxidized surface and some of the carbon atoms are replaced by oxygen atoms to form a partially disordered surface, then a nickel atom coordinating with six oxygen atoms is loaded on the surface. . The surface is loaded with more oxygen atoms to simulate a further oxidized surface and some of the carbon atoms are replaced by oxygen atoms to form a partially disordered surface, then three nickel atoms coordinating with six oxygen atoms, respectively, are loaded on the surface.
To simulate the effect inside a solid, the atoms located in the lower half of the slab were fixed for all calculation models.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Wang, Q. et al. Ultrahigh-loading of ir single atoms on nio matrix to dramatically enhance oxygen evolution reaction. J. Am. Chem. Soc. 142, 7425-7433 (2020).
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294-303 (2012).
Roger, I., Shipman, M. A. & Symes, M. D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 1, 0003 (2017).
Zhang, L. et al. Graphene defects trap atomic ni species for hydrogen and oxygen evolution reactions. Chem 4, 285-297 (2018).
Wang, H. et al. Bifunctional non-noble metal oxide nanoparticle electrocatalysts through lithium-induced conversion for overall water splitting. Nat. Commun., 6, 7261 (2015).
Zhao, X. et al. Bifunctional electrocatalysts for overall water splitting from an iron/nickel-based bimetallic metal-organic framework/ dicyandiamide composite. Angew. Chem., Int. Ed. 57, 8921-8926 (2018).
Shi, H., Liang, H., Ming, F. & Wang, Z. Efficient overall water-splitting electrocatalysis using lepidocrocite vooh hollow nanospheres. Angew. Chem., Int. Ed. 56, 573-577 (2017).
Luo, W. et al. A review: research progress of neural probes for brain research and brain-computer interface. ACS Catal 12, 1167-1179 (2022).
Yang, Y. et al. Hierarchical nanoassembly of as a highly efficient electrocatalyst for overall water splitting in a wide ph range. J. Am. Chem. Soc. 141, 10417-10430 (2019).
Wang, M. et al. Interfacial water activation by single-atom co-n3sites coupled with encapsulated co nanocrystals for accelerating electrocatalytic hydrogen evolution. ACS Catalysis 12, 10771-10780 (2022).
Yin, J. et al. Iridium single atoms coupling with oxygen vacancies boosts oxygen evolution reaction in acid media. J. Am. Chem. Soc. 142, 18378-18386 (2020).
Jiang, K. et al. Dynamic active-site generation of atomic iridium stabilized on nanoporous metal phosphides for water oxidation. Nat. Commun. 11, 2701 (2020).
Wang, W. et al. Confining zero-valent platinum single atoms in -moc1-xfor ph-universal hydrogen evolution reaction. Adv. Funct. Mater. 32, 2108464 (2021).
Kuang, M., Han, P., Wang, Q., Li, J. & Zheng, G. CuCo hybrid oxides as bifunctional electrocatalyst for efficient water splitting. Adv. Funct. Mater. 26, 8555-8561 (2016).
Zhu, Y. et al. A perovskite nanorod as bifunctional electrocatalyst for overall water splitting. Adv. Energy Mater. 7, 1602122 (2017).
Li, M. et al. Proximity electronic effect of ni/co diatomic sites for synergistic promotion of electrocatalytic oxygen reduction and hydrogen evolution. Adv. Funct. Mater. 33, 2210867 (2022).
Yang, H., Wu, Y., Zhuang, Z., Li, Y. & Chen, C. Computational probing of temperature-dependent unfolding of a small globular protein: from cold to heat denaturation. Chin. J. Chem. 40, 515-523 (2021).
Yuan, S. et al. Dual synergistic effects between Co and Mo 2 C in Mo2C heterostructure for electrocatalytic overall water splitting. Chem. Eng. J. 430, 132697 (2022).
Yu, Y., Zhou, J. & Sun, Z. Novel 2D transition-metal carbides: ultrahigh performance electrocatalysts for overall water splitting and oxygen reduction. Adv. Funct. Mater. 30, 2000570 (2020).
Anjum, M. A. R., Lee, M. H. & Lee, J. S. Boron- and nitrogen-codoped molybdenum carbide nanoparticles imbedded in a bcn network as a bifunctional electrocatalyst for hydrogen and oxygen evolution reactions. ACS Catal 8, 8296-8305 (2018).
Das, D., Santra, S. & Nanda, K. K. In situ fabrication of a nickel/ molybdenum carbide-anchored n-doped graphene/cnt hybrid: an efficient (Pre)catalyst for OER and HER. ACS Appl. Mater. Interfaces 10, 35025-35038 (2018).
Li, M. et al. Ni strongly coupled with mo2c encapsulated in nitrogendoped carbon nanofibers as robust bifunctional catalyst for overall water splitting. Adv. Energy Mater. 9, 1803185 (2019).
Xiao, M. et al. Molten-salt-mediated synthesis of an atomic nickel co-catalyst on tio2for improved photocatalytic h2evolution. Angew. Chem., Int. Ed. 59, 7230-7234 (2020).
Suryanto, B. H. R., Wang, Y., Hocking, R. K., Adamson, W. & Zhao, C. Overall electrochemical splitting of water at the heterogeneous interface of nickel and iron oxide. Nat. Commun. 10, 5599 (2019).
Hu, F. et al. Lattice-Matching formed mesoporous transition metal oxide heterostructures advance water splitting by active bridges. Adv. Energy Mater. 12, 2200067 (2022).
Tang, C. et al. Electronic Coupling of Single Atom and Boosts Water Electrolysis. Energy Environ. Sci. 5, 899-905 (2022).
Li, G. et al. The synergistic effect of Hf-O-Ru bonds and oxygen vacancies in for enhanced hydrogen evolution. Nat. Commun. 13, 1270 (2022).
Li, R. et al. Potential-dependent reconstruction of Ni-based cuboid arrays for highly efficient hydrogen evolution coupled with electrooxidation of organic compound. Chem. Eng. J. 453, 139797 (2023).
Zhao, Y. et al. Dynamics and control of active sites in hierarchically nanostructured cobalt phosphide/chalcogenide-based electrocatalysts for water splitting. Energy Environ. Sci. 15, 727-739 (2022).
Zhou, H. et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and fuel. Nat. Commun. 12, 4679 (2021).
Li, S. et al. Oxygen-evolving catalytic atoms on metal carbides. Nat. Mater. 20, 1240-1247 (2021).
Zhang, P. et al. Bifunctional single atom catalysts for rechargeable zinc-air batteries: from dynamic mechanism to rational design. Adv. Mater. 35, 2303243 (2023).
Zhou, L., Zhao, D. & Lou, X. W. Double-shelled CoMn 2 O 4 hollow microcubes as high-capacity anodes for lithium-ion batteries. Adv. Mater. 24, 745-748 (2012).
Guan, J., Mou, F., Sun, Z. & Shi, W. Preparation of hollow spheres with controllable interior structures by heterogeneous contraction. Chem. Commun. 46, 6605-6607 (2010).
Han, L. et al. Atomically dispersed molybdenum catalysts for efficient ambient nitrogen fixation. Angew. Chem., Int. Ed. 58, 2321-2325 (2019).
Wang, Q. et al. Single iridium atom doped ni(2)p catalyst for optimal oxygen evolution. J. Am. Chem. Soc. 143, 13605-13615 (2021).
Wei, H. et al. Molybdenum carbide nanoparticles coated into the graphene wrapping -doped porous carbon microspheres for highly efficient electrocatalytic hydrogen evolution both in acidic and alkaline media. Adv. Sci. 5, 1700733 (2018).
Yu, L., Yang, J. F., Guan, B. Y., Lu, Y. & Lou, X. W. D. Hierarchical hollow nanoprisms based on ultrathin ni-fe layered double hydroxide nanosheets with enhanced electrocatalytic activity towards oxygen evolution. Angew. Chem., Int. Ed. 57, 172-176 (2018).
Han, W. et al. Ultra-small Mo2C nanodots encapsulated in nitrogendoped porous carbon for pH-universal hydrogen evolution: insights into the synergistic enhancement of HER activity by nitrogen doping and structural defects. J. Mater. Chem. A 7, 4734-4743 (2019).
Jin, H. et al. In situ cobalt-cobalt oxide/N-doped carbon hybrids as superior bifunctional electrocatalysts for hydrogen and oxygen evolution. J. Am. Chem. Soc. 137, 2688-2694 (2015).
Zhuang, Z. et al. Atomically dispersed nonmagnetic electron traps improve oxygen reduction activity of perovskite oxides. Energy Environ. Sci. 14, 1016-1028 (2021).
Qin, J. et al. Activating edge-Mo of 2H-MoS2via coordination with pyridinic N-C for pH-universal hydrogen evolution electrocatalysis. ACS Catal 11, 4486-4497 (2021).
Chen, W.-F. et al. Reply to comments on “synthesis, characterization, and structures of persistent aniline radical cation”. Energy Environ. Sci. 6, 943-945 (2013).
Aracena, A., Sanino, A. & Jerez, O. Dissolution kinetics of molybdite in KOH media at different temperatures. T NONFERR METAL SOC 28, 177-185 (2018).
He, Z. et al. Activating lattice oxygen in NiFe-based (oxy)hydroxide for water electrolysis. Nat. Commun. 13, 2191 (2022).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886-17892 (2004).
Norskov, J. K., Abild-Pedersen, F., Studt, F. & Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl Acad Sci USA. 108, 937-943 (2011).
Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFB4005100), National Natural Science Foundation of China (22309011, 52272186, 22105116, 21872008, 52173232, 21925202, U22B2071), Yunnan Provincial Science and Technology Project at Southwest United Graduate School (202302AO370017), International Joint Mission On Climate Change and Carbon Neutrality, Beijing Institute of Technology Research Fund Program for Young Scholars. We thank the 1W1B station for XAFS measurements in the Beijing Synchrotron Radiation Facility (BSRF) and Dr. Fang Zhang from Analysis & Testing center, Beijing Institute of Technology.
Author contributions
D.Z., J.T.Z., M.H.C., and C.C. supervised the project. M.Y.H. and D.Z. designed the work and carried out most of the experiments. L. R. Z. guided the XANES, ex situ XAS measurements, and analyzed XAS results. W.Y.F., X.T., J.T. F,T. X.W. helped to prepare samples and character the samples with TEM and XPS. All the authors discussed the results and assisted during the manuscript preparation.
Correspondence and requests for materials should be addressed to Di Zhao, Minhua Cao, Jiatao Zhang or Chen Chen.
Peer review information Nature Communications thanks Uday Maiti and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Reprints and permissions information is available at http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key Laboratory of Cluster Science, Ministry of Education of China, Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China. Engineering Research Center of Advanced Rare Earth Materials, Department of Chemistry, Tsinghua University, Beijing 100084, China. These authors contributed equally: Mengyun Hou, Lirong Zheng. e-mail: dizhao@bit.edu.cn; caomh@bit.edu.cn; zhangjt@bit.edu.cn; cchen@mail.tsinghua.edu.cn