إعادة هيكلة ديناميكية لكبريتيدات النيكل لتفاعل تطور الهيدروجين الكهروكيميائي Dynamic restructuring of nickel sulfides for electrocatalytic hydrogen evolution reaction
تم التعرف على الكالكوجينيدات المعدنية الانتقالية كعوامل تحفيز كهربائية منخفضة التكلفة وفعالة لتعزيز تفاعل تطور الهيدروجين في الوسائط القلوية. ومع ذلك، لا تزال عملية تحديد المواقع النشطة والآلية التحفيزية الأساسية غامضة. في هذا العمل، نستخدم مطيافية امتصاص الأشعة السينية أثناء التشغيل ومطيافية الإلكترون الضوئي تحت ضغط قريب من البيئة لتوضيح أن NiS يخضع لانتقال طور في الموقع إلى طور مختلط بشكل وثيق منو NiO، مما يولد مواقع ثنائية نشطة للغاية ومتآزرة فيالواجهة. النيكل على الواجهة هو الموقع النشط لتفكك الماء والامتزاز بينما تعمل الواجهة S كموقع نشط لـ الامتزاز و التطور. وبناءً عليه، التكوين في الموقع لـتتيح الواجهات لمحفزات NiS الكهربائية تحقيق جهد زائد قدره فقطعند كثافة تيارأبرز عملنا أن كيمياء الكالكوجينيدات المعدنية الانتقالية ديناميكية للغاية، وأن التحكم الدقيق في ظروف العمل قد يؤدي إلى التكوين في الموقع للأنواع الحفازة التي تعزز أدائها الحفاز.
تحليل الماء بالكهرباء المدعوم بالطاقة المتجددة لإنتاج الهيدروجين الأخضر يُعتبر على نطاق واسع مسارًا واعدًا نحو مستقبل طاقة نظيفة ومستدامة عالميًا.المفتاح لتمكين هذه التكنولوجيا هو تطوير محفزات كهربائية منخفضة التكلفة وعالية الكفاءة لتسريع تفاعلات تحليل الماء، أي تفاعل تطور الأكسجين (OER) وتفاعل تطور الهيدروجين (HER).على وجه الخصوص، حظي تصميم المحفزات الكهربائية عالية الأداء لتفاعل تقليل الهيدروجين في الظروف القلوية باهتمام مكثف، لأن التحليل الكهربائي للماء القلوي هو الطريق الأكثر استخدامًا في الصناعة، كما أن تفاعل تقليل الهيدروجين القلوي هو أيضًا خطوة رئيسية في عملية الكلور-قلوي.. ومع ذلك، لا تزال تفاعل الهيدروجين القلوي يعاني من بطء في الديناميكا الحركية مقارنةً بتلك في الوسط الحمضي حل، بسبب خطوة تفكك الماء الإضافية (خطوة فولمر: من أجل تزويد الممتصمتوسط لللاحقجيلإن تفكك الماء له حاجز تنشيط أعلى ويعتبر الخطوة المحددة لمعدل التفاعل التي تحد من التفاعل الكلي.حتى المحفزات القائمة على البلاتين المتطورة تظهر نشاطًا أقل في تفاعل تطور الهيدروجين بحوالي مرتبتين من حيث الحجم في الوسط القلوي مقارنة بالوسط الحمضي.تم إحراز تقدم كبير لتحسين نشاط HER للبلاتين والمعادن النبيلة الأخرى في البيئات القلوية من خلال بناء هياكل غير متجانسة ذات مواقع نشطة مزدوجة، على سبيل المثال،، حيث أن يعمل كمنشط لتسريع تفكك الماءتحسين نشاط المعادن الثمينة القائمة على
لقد كانت المحفزات ذات موقع نشط واحد تمثل تحديًا بشكل رئيسي بسبب علاقات القياس في طاقة تشكيل الوسائط، مما يؤثر ليس فقط على تفاعل تقليل الهيدروجين ولكن أيضًا على تفاعل الأكسدة (OER) وتفاعل اختزال الأكسجين (ORR).تقدم المحفزات ذات المواقع النشطة المزدوجة آليات تفاعل بديلة قد تكسر مثل هذه العلاقات المتدرجة، مما يوفر فرصة أكبر لتحقيق أداء تحفيزي أفضل، مقارنةً بالمحفزات ذات الموقع النشط الواحد..
ومع ذلك، فإن ندرة وارتفاع تكلفة مواد مجموعة البلاتين تحد من تطبيقها على نطاق واسع.في هذا الصدد، ظهرت مؤخرًا المركبات السلفيدية والفوسفيدية والنيتريدية والكربيدية المستندة إلى المعادن الانتقالية الوفيرة في الأرض كعوامل تحفيز كهربائية فعالة من حيث التكلفة لتفاعل الهيدروجين القلوي.تظهر العديد من المحفزات المعتمدة على TM نشاطًا مشابهًا في تفاعل الهيدروجين القلوي كما هو الحال مع مواد مجموعة البلاتين.. بالإضافة إلى ذلك، تم استكشاف التلاعب بالمواد، والعيوب، وهندسة الإجهاد بشكل أكبر لتعديل هيكلها الإلكتروني من أجل تعزيز النشاط والاستقراركبريتيدات النيكل مثل، و لقد جذبت اهتمامًا كبيرًا بسبب نشاطها التحفيزي العالي، بالإضافة إلى الطرق السهلة والقابلة للتوسع للتحضير.على الرغم من هذا التطور السريع في محفزات تفاعل الهيدروجين القلوي، لا تزال آلية التفاعل الأساسية والمواقع النشطة لتفاعل الهيدروجين القلوي، التي تختلف عن تفاعل الهيدروجين الحمضي الموثق جيدًا، قيد النقاش الكبير.علاوة على ذلك، تم إثبات أن المحفزات القائمة على المركبات المعدنية الانتقالية (TM) لتفاعل الأكسدة (OER) تخضع لإعادة بناء هيكلية أو تركيبية، أو حتى تتحول إلى مرحلة جديدة تحت ظروف عمل تفاعل الأكسدة.من ناحية أخرى، تم استكشاف التحويل الهيكلي والكيميائي للعوامل المساعدة تحت ظروف تفاعل الهيدروجين بشكل أقل، وقد تم الافتراض حتى أن العوامل المساعدة لتفاعل الهيدروجين مستقرة خلال عملية تفاعل الهيدروجين.. ومع ذلك، أشارت الأعمال الأخيرة إلى أن قد تخضع بعض كبريتيدات المعادن الانتقالية لانتقال طور تحت ظروف العمل القلوية لتفاعل الهيدروجين.لذلك، فإن المراقبة الفورية للعمليات الحفزية وتحديد المواقع النشطة الحقيقية تحت ظروف تشغيل تفاعل الهيدروجين هي أمر بالغ الأهمية لكشف الآلية الحفزية.
في هذه الدراسة، نقدم توضيحًا متعمقًا لعملية تحويل الطور استنادًا إلى مطيافية امتصاص الأشعة السينية أثناء التشغيل (XAS) ومطيافية رامان أثناء التشغيل تحت ظروف عمل تفاعل الهيدروجين. وجدنا أن أقطاب NiS تشهد تحسينًا كبيرًا في أداء تفاعل الهيدروجين نتيجة لتحول في الموقع إلى طور مختلط من و NiO ، الذي تم التعرف عليه كونه المرحلة الفعالة الحقيقية للحفاز في تفاعل الهيدروجين القلوي. تكشف الدراسات الطيفية الخارجية والتحقيقات النظرية بشكل أكبر أن النيكل على الواجهة (س)يوفر المواقع النشطة لتسريع تفكك الماء بينما السطح الخارجي S (يسهل امتصاص الوسائط الهيدروجينية وترابطها اللاحق لتكوين الهيدروجين الجزيئي. ونتيجة لذلك، يتم تشكيلتظهر نشاطًا متفوقًا في تفاعل تطور الهيدروجين، مما يتطلب جهدًا زائدًا منخفضًا منللوصول إلى كثافة تيارفي اختبارات الأداء لدينا.
النتائج والمناقشة
الانتقال الطوري لـ NiS إلىيحفز نشاط HER المحسن
تم تخليق عينات NiS على ورق الكربون باستخدام طريقة الهيدروحرارية ذات الخطوة الواحدة (الشكل التكميلي 1a). تم تقديم تفاصيل التخليق والتوصيف في قسم الطرق. نمط حيود الأشعة السينية (XRD) الموضح في الشكل 1a وصورة الإلكترون بالأشعة السينية
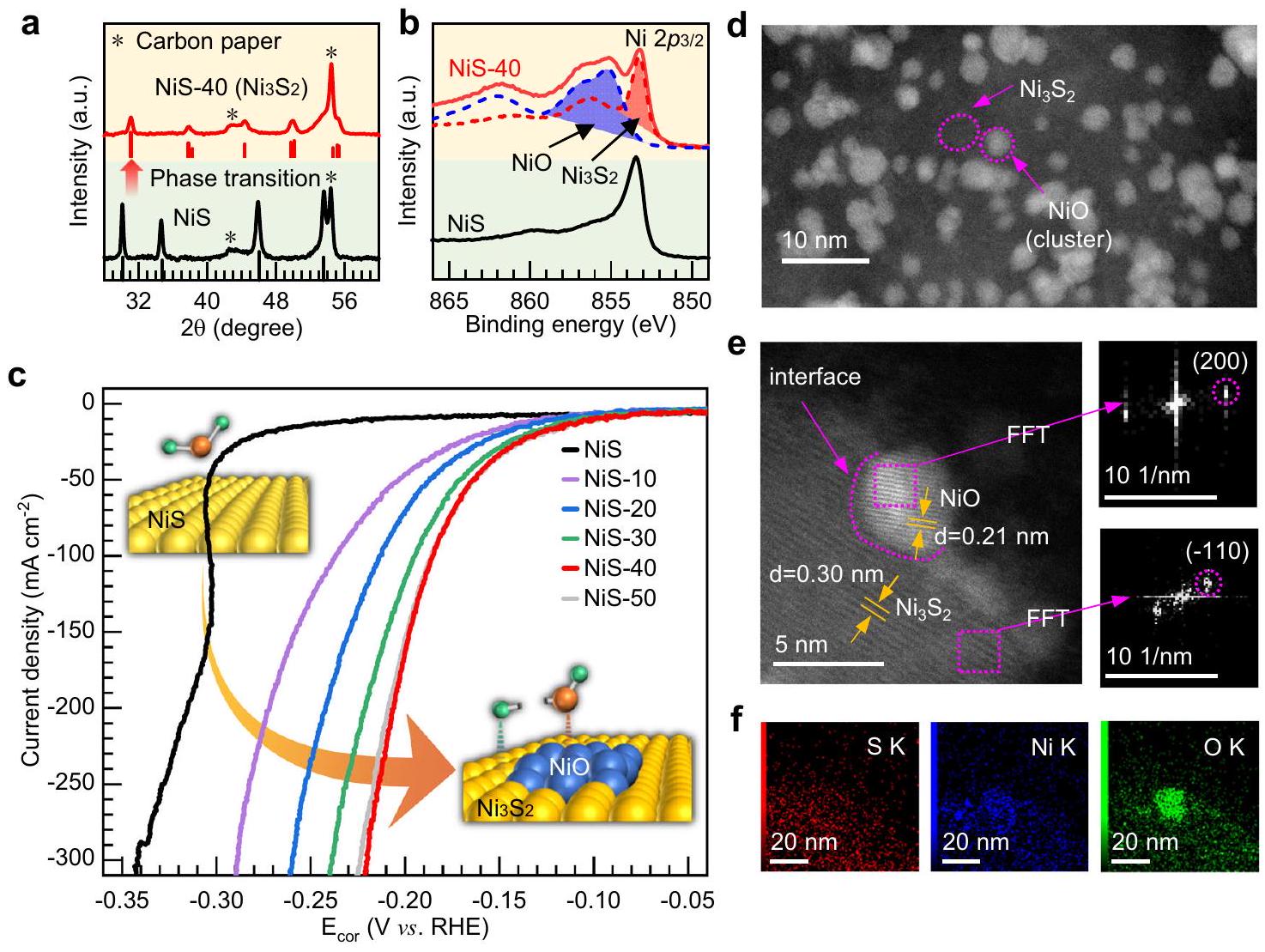
الشكل 1 | أداء HER وخصائص الهيكلية لمحفز NiS قبل وبعد القياس. أ أنماط XRD لـ NiS الذي تم تصنيعه حديثًا وNiS بعد قياس HER (المعلمة كـ NiS-40). ب Niطيف XPS لـ NiS و NiS بعد قياس HER (مُعَلَّم كـ )، جنبًا إلى جنب مع NiO و كمرجع. ج-منحنيات الاستقطاب لقياس الفولتامترية ذات المسح الخطي المصححة (LSV) لعينة NiS، NiS-10، NiS-20، NiS-30، NiS-40، وNiS-50 التي تم قياسها في
محلول 1 م KOH. NiS-10 و NiS-20 و NiS-30 و NiS-40 و NiS-50 هي عينات NiS بعدوتم قياس الكرونوأمبيرومترية لمدة 50 دقيقة عند -1.00 فولت مقابل RHE، على التوالي.صورة TEM ذات المساحة الكبيرة،صورة TEM عالية الدقة (تظهر اللوحة اليمنى صور FFT لكلا المنطقتين) والتخطيط العنصري المقابل لعينة NiS-40 بعد إجراء قياس الجهد الزمني لمدة 25 ساعة عند كثافة تيار. الطيفية (XPS) للنيكلمستوى النواة في الشكل 1ب يشير إلى أن العينات التي تم تحضيرها تتميز بالشكل السداسي-مرحلة منتظهر صور المجهر الإلكتروني الماسح (SEM) والمجهر الإلكتروني الناقل (TEM) في الشكل التكميلية 1 أن العينات تتكون من جزيئات NiS بحجمتغطية موحدة على ركيزة ورق الكربون.
تم تقييم نشاط الهيدروجين التطبيقي (HER) في 1 م KOH باستخدام نظام ثلاثي الأقطاب (التفاصيل في قسم الطرق). تم إجراء قياسات الفولتمترية الدورية (CV) من أجل ضبط الظروف لتقييم النشاط التحفيزي لـ HER (الشكل التكميلي 2a، b). في الدورة الأولى من العينات التي تم تصنيعها، هناك بداية حادة لكثافة التيار لـ HER عند حوالي -0.30 فولت مقابل RHE، والتي تستمر في الزيادة بشكل ثابت مع استمرار ت polarization العينة إلى إمكانيات سالبة. بعد تلك البداية الحادة لكثافة التيار، يكون ملف CV قابلاً للتكرار في الدورة الثانية، مع حد أعلى للإمكان عند 0.05 فولت مقابل RHE. ومع ذلك، مع تمديد الحد الأعلى للإمكان إلى 0.40 فولت مقابل RHE في و دورات، هناك موجة أكسدة بحد أقصى عند +0.27 فولت مقابل RHE وزيادة كثافة التيار عندما يتم استقطاب العينة إلى إمكانيات سالبة. من المRemarkably، تستمر كثافة التيار في الزيادة بينما تتعرض العينة للكرونوأمبيرومترية عند -1.00 فولت مقابل RHE (الشكل التوضيحي 2c). تظهر قياسات الفولتامترية ذات المسح الخطي (LSV) للعينة بعد قياس الكرونوأمبيرومترية أن كثافة التيار تزداد.مقارنةً بالعينة النقية، هناك أيضًا تحول واضح في بداية الجهد إلى جهد أقل (الشكل التكميلي 2d). توضح هذه الدراسات الكهروكيميائية بوضوح أن التعديلات في الموقع على أقطاب NiS تؤدي إلى زيادة كبيرة في نشاط التحفيز لتفاعل تطور الهيدروجين (HER).
استنادًا إلى هذه الأسس، قمنا بإجراء دراسة أكثر تفصيلًا لظروف الكرونوأمبروغرافيا لأقطاب NiS على نشاطها التحفيزي في تفاعل تقليل الهيدروجين. توضح الشكل 1c منحنيات LSV لـ NiS الذي تم تصنيعه، بالإضافة إلى عينات NiS بعد قياسات الكرونوأمبروغرافيا لمدة 10 و20 و30 و40 و50 دقيقة عند -1.00 فولت مقابل RHE، والموسومة بـ NiS-10 وNiS-20 وNiS-30 وNiS-40 وNiS-50، على التوالي. لتجنب الانحرافات الناتجة عن السقوط الأوم، تم تعديل الجهد بواسطة عامل (الشكل التكميلي 3 أ، ب). يظهر NiS الذي تم تصنيعه حديثًا في البداية نشاطًا منخفضًا في تفاعل تقليل الهيدروجين (HER)، ولكن نشاط HER يزيد تدريجيًا مع مرور وقت قياس الكرونوأمبيرومتر حتى يتم إجراء القياس لمدة 40 دقيقة. يتم ملاحظة اتجاه مشابه لنشاط HER أيضًا عندما يتم تطبيع كثافة التيار بواسطة المساحة السطحية النشطة كهربائيًا (ECSA) (الأشكال التكميلية 4 و 5). كما هو موضح في الشكل 1 ج، يظهر عينة NiS-40 نشاطًا عاليًا في التحفيز لتفاعل HER، مما يتطلب جهدًا زائدًا قدرهللوصول إلى كثافة تياريمكن رؤية الحصول على شريط الخطأ في الشكل التكميلي 3c. أظهرت المزيد من التوصيفات الكهروكيميائية أن ميل تافل ومقاومة نقل الشحنة (الشكل التكميلي 6a، b) تتناقص بالترتيب NiS > NiS-10 > NiS-20 > NiS-30 > NiS-50 > NiS-40، مما يشير إلى أن تحسين الأداء بعد المعالجات الكرونوأمبيرومترية ناتج عن حركيات أسرع للتفاعل الهيدروجيني. بالمقارنة مع المحفزات الهيدروجينية التي تم الإبلاغ عنها سابقًا، فإن المحفزات الكهروكيميائية المعالجة NiS تظهر نشاطًا تنافسيًا في تفاعل الهيدروجين من حيث الفائض في الجهد.وميل تافل (الشكل التوضيحي 7 والجدول التكميلي 1). بالإضافة إلى ذلك، فإن النشاط التحفيزي لعينة NiS-40 مستقر جداً، حيث لا يظهر أي زيادة في الجهد الزائد على مدى 25 ساعة من التشغيل المستمر عند كثافة تيارفي 1 م كوه (الشكل التوضيحي التكميلي 6c).
تم إجراء سلسلة من التوصيفات خارج الموقع للعينات النشطة للغاية بعد إجراء اختبارات الأداء. يتم تحويل NiS الذي تم تصنيعه إلى مرحلة مختلطة من و NiO بعد قياس HER. الشكل 1a يظهر نمط XRD لعينة NiS-40، والتي يظهر منها نمط مميز لـ يمكن تحديده، مما يشير إلى تحويل كامل لنيكل كبريتيد إلى شكل بلوري. الـ XPS في النيكلالمنطقة لـ NiS-40 تظهر العينة ميزة طيفية بارزة قد تنشأ من NiO (الشكل 1b). بالإضافة إلى ذلك، يمكن ملاءمة طيف XAS لحافة Ni للعينة NiS-40 (الشكل التكميلية 8) من خلال تركيبة خطية من الأطياف لـو NiO، مما يؤكد بشكل أكبر تشكيلو NiO. لقد قمنا بمزيد من توصيف الـعينة بعدقياس الكرونوبوتنشيومتر عند كثافة تيار، تؤكد تقنيات XRD وXPS وXAS أن المحفزات بعد عملية تقليل الهيدروجين موجودة في شكل و NiO (الشكل التوضيحي 9). تم استخدام مجهر الإلكترون الناقل المصحح للانحراف الكروي (SAC-TEM) لفحص البنية الدقيقة لمحفزات HER-post بشكل أكبر. كما هو موضح في الشكل 1d، تكشف صورة TEM ذات المساحة الكبيرة عن العديد من الكتل الصغيرة من NiO المثبتة على الكبيرة تظهر صورة HR-TEM واجهة مميزة تقسم الصورة إلى منطقتين (الشكل 1e). من خلال تحليل صور FFT المرتبطة بهاتين المنطقتين، تحتوي منطقة الكتلة على تباعد بين الطبقات يبلغ 0.21 نانومتر يتوافق جيدًا مع مستوى الشبكة (200) لـ NiO، بينما تحتوي المنطقة الأخرى على تباعد بين الطبقات يبلغ 0.30 نانومتر يتطابق جيدًا مع مستوى الشبكة (-110).الأهم من ذلك، يُظهر التخطيط العنصري الثراء التفضيلي لعناصر الأكسجين على الكتلة الصغيرة (الشكل 1f)، مما يكشف بقوة أن الكتلة الصغيرة تُعزى إلى NiO. يجب ملاحظة أن NiO المتكون قد يوجد في شكل كتلة صغيرة جدًا، مما يصعب تحديده من خلال أنماط XRD. للتحقيق في تأثير نسبةإلى NiO على النشاط التحفيزي، تم استلام عينات NiS بواسطة، وتم إعداد 50 LSVs في النطاق من 0.00 فولت إلى -1.00 فولت مقابل RHE. يتكون XAS عند حافة النيكل لهذه العينات، الموضحة في الأشكال التكميلية 10a-d، من تركيبة خطية من طيف و NiO، مما يشير إلى أن التحول الكامل لـ NiS يحدث بعد LSV واحدة فقط. ومع ذلك، فإن NiO إلىيزداد النسبة عند زيادة عدد LSVs. قمنا باستخراج NiO إلىنسبة مستندة إلى طيف XAS (الأشكال التكميلية 10a-d والجدول التكميلية 2) ووجدنا أن نشاط HER يزداد مع كمية NiO (الشكل التكميلية 10e)، مما يشير بوضوح إلى التأثير الإيجابي لأنواع أكسيد النيكل على عملية HER.
ديناميات انتقال الطور التي كشفتها تقنية الأشعة السينية الممتصة في الموقع (XAS) وتقنية رامان الطيفية في الموقع
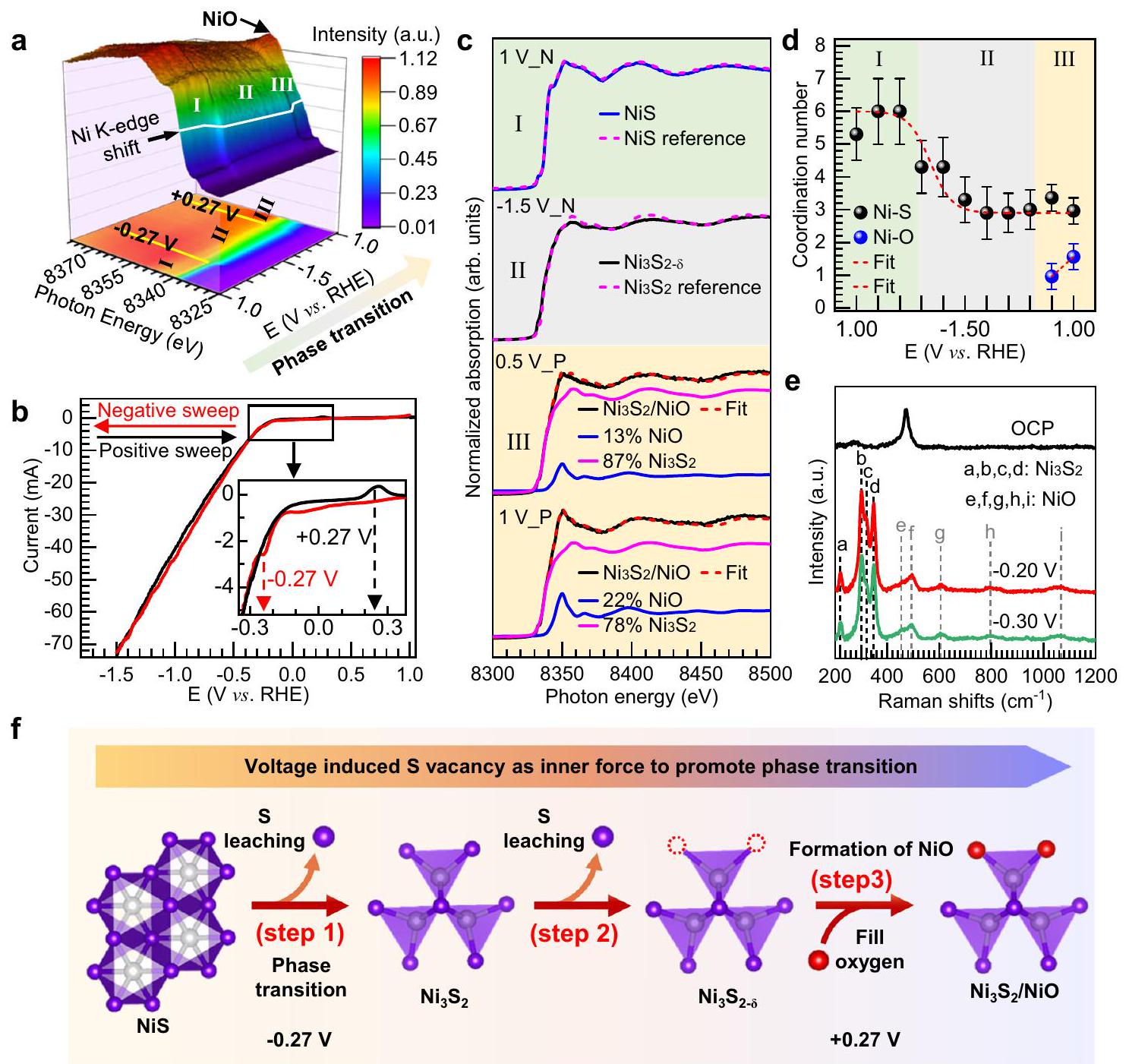
تم استخدام مطيافية XAS أثناء التشغيل للحصول على رؤى مفصلة حولتتم عملية الانتقال، وتفاصيل الطريقة التجريبية موضحة في قسم الطرق. يوضح الشكل 2أ رسمًا ثلاثي الأبعاد (3D) لامتصاص الأشعة السينية عند حافة النيكل (Ni K-edge XAS) لقطب النيكل سلفيد (NiS) كدالة للجهود المطبقة أثناء قياس الفولتية الدورية (الشكل 2ب). لاحظ أن هناك تغييرين مهمين في الملف الطيفي يحدثان عند الجهود المطبقة -0.27 فولت و +0.27 فولت مقابل RHE، مما يقسم الرسم ثلاثي الأبعاد إلى ثلاث مناطق هي I (الجهد المطبق من +1.00 فولت إلى -0.27 فولت)، II (-0.27 فولت إلى -1.50 فولت والعودة إلى +0.27 فولت) و III (من +0.27 فولت إلى 1.00 فولت). يتم عرض طيف Ni K-edge XAS المختار عند جهود محددة ضمن المناطق I و II و III في الشكل 2ج. في المنطقة I، مع الجهد المطبق من +1.00 فولت إلى -0.27 فولت، يبقى طيف العينة كما هو لطيف مرحلة NiS. يمكن تحديد الانتقال من المنطقة I إلى المنطقة II عند -0.27 فولت كمرحلة انتقالية.، استنادًا إلى الملاءمة الطيفية والمقارنات مع عينات مرجعية من و (المنطقة I و II في الشكل 2c). الانخفاض المرتبط في الحالة التأكسدية الرسمية لـفي NiS إلىفييمكن ملاحظته كقمة موجة كاثودية في ملف CV عند بداية تيار HER الموضح في الشكل 2b (انظر الإطار)، مما يظهر بوضوح أن النشاط التحفيزي لـ HER مرتبط بتكوين. بعد الانتقال الطوري، الـالطور مستقر تحت ظروف HER. ومع ذلك، مع تطبيق جهد أكثر سلبية (المنطقة II في الشكل 2a)، يشير الانخفاض المستمر في شدة الطيف لخط الأبيض من طيف XAS لحد Ni K إلى مزيد من تقليل حالة الأكسدة للنيكل. وبشكل متسق، كشفت عملية تركيب معاملات تحويل فورييه للطيف في المنطقة II عن انخفاض في عدد التنسيق المتوسط للنيكل المنسق بواسطة الكبريت (الشكل 2d، الأشكال التكميلية 11 و12، والجداول التكميلية 3 و4، و
الشكل 2 | التوصيفات أثناء التشغيل وآلية الانتقال الطوري. أ. التوصيفات باستخدام الأشعة السينية عند حافة النيكل (Ni K-edge XAS) لمحفز NiS كدالة للجهود المطبقة. اللوحة العلوية تظهر عرضًا ثلاثي الأبعاد لطيف الأشعة السينية عند حافة النيكل؛ بينما اللوحة السفلية تظهر إسقاطًا ثنائي الأبعاد من العرض الثلاثي الأبعاد. الملف الشخصي المقابل المستخدم لقياس XAS في حالة التشغيل. يظهر الشكل المصغر المنطقة المكبرة المميزة في المربع المستطيل. ج طيف XAS عند حافة Ni K عند فولتية معينة مطبقة من يعني +1.00 فولت في المسح السالب لـ CV)، (يعني -1.50 فولت في المسح السالب للجهد الكهربائي)”، و و +1.00 فولت _P تعني +0.50 فولت و +1.00 فولت في المسح الإيجابي لـ CV ) في المنطقة I و II و III على التوالي، المستخرجة من الشكل 2a، وتركيبتها الخطية من و عدد التنسيق للنيكل كدالة للجهود المطبقة.طيف رامان التشغيلي لـ NiS عند فولتages مختلفة.رسم توضيحي تخطيطي لانتقال الطور لعنصر NiS المحفز أثناء قياس تفاعل الهيدروجين. إجراءات المحاكاة في الملاحظة التكميلية 1)، مما يشير إلى تشكيل فراغات S، أي، . مع استقطاب العينة من الجهد السالب إلى الجهد الموجب، يمكن ربط الانتقال من II إلى III عند +0.27 فولت بعملية أكسدة تم ملاحظتها في ملف CV الموضح في الشكل 2b، وهو ما يتماشى مع الزيادة المستمرة في الخط الأبيض للطيف مع استقطاب العينة بشكل أكبر (المنطقة III في الشكل 2a). يمكن ملاءمة الطيف مع تركيبة خطية منوزيادة مساهمة NiO (المنطقة III في الشكل 2c)، وتفاصيل التوفيق تناقش في الملاحظات التكميلية 2-5. استنادًا إلى هذا التحليل، نقدر أن مساهمة NiO عند +0.50 فولت تبلغ حوالي، مما يزيد أيضًا إلى عندما يصل جهد العينة إلى +1.00 فولت (الشكل 2c والجدول التكميلي 5). وبالتالي، يحدث تنسيق النيكل بواسطة الأكسجين تدريجياً في المنطقة III (الشكل 2d)، ربما بسبب ملء فراغات الكبريت فيمع الماء أوالأيونات، مما يؤدي إلى مرحلة مختلطة بشكل وثيق منو NiO، الذي تم التحقق منه بشكل إضافي من خلال تحليل تحويل الموجات (WT) لهيكل الامتصاص بالأشعة السينية الدقيق الممتد (EXAFS) وتحويل فورييه-EXAFS الموزون عند جهد مطبق قدره +1.00 فولت في المنطقة III (الشكل التكميلي 13). تم عرض النتائج المقابلة للتناسب عند +1.00 فولت في الجدول التكميلي 4، ويظهر Ni-O طول رابطة قدرهNiO عالي التبلور له خاصيةالمسافات حول؛ ومع ذلك، فإن الترتيب قصير المدى في NiO يكشف أنقد تقل المسافات للنيكل أكسيد غير المتبلور و/أو النانوي إلى. هذه النتيجة تؤكد أكثر أن النيكل أكسيد المتكون هو في شكل مجموعة صغيرة جداً.
تكوين المرحلة المختلطة من وتم التحقق من NiO بشكل إضافي من خلال دراسة طيف رامان أثناء التشغيل (تفاصيل التجربة في قسم الطرق). يمكن تعيين طيف رامان العلوي في الشكل 2e إلى NiS النقي، مع ظهور نطاقات مميزة عند انزياحات رامان. و . مع ت polarizing العينة إلى إمكانيات سلبية، تختفي القمم المميزة المخصصة لـ NiS، بينما تظهر القمم المميزة المخصصة لـيظهر في، و (المعلمة بـ a و b و c و d، على التوالي)، مما يشير إلى الانتقال الكامل للطور من . بالإضافة إلى ذلك، فإن القمم الجديدة الناشئة e و f و g و h و i تقع عند 453 و 493 و 600 و 800 ويمكن تعيينه إلى، مما يشير إلى
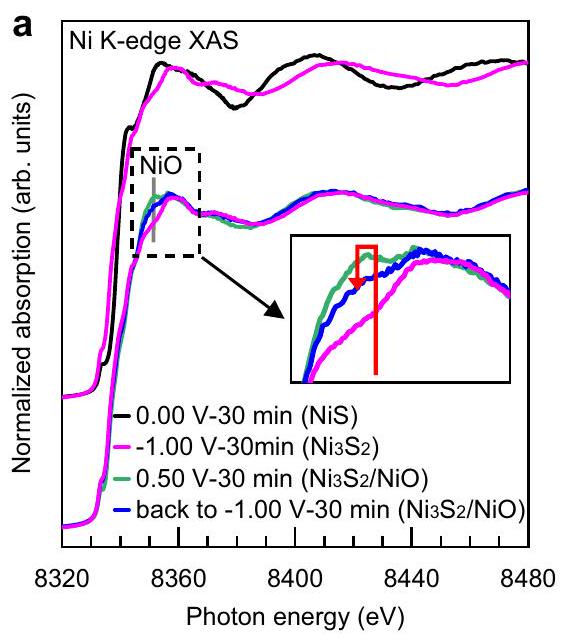
الشكل 3 | الوجود الطويل الأمد لإعادة البناءتم إثباته من خلال التوصيفات أثناء التشغيل. أ. XAS أثناء التشغيل عند حافة Ni K لقطب NiS في الكرونوأمبيرومترية لمدة 30 دقيقة عند 4 جهدات مطبقة بشكل متتالي: 0.00 فولت،
تكوين NiO تحت ظروف HER. ميزات طيف رامان المرتبطة بـو NiO مفصولة بشكل جيد للغاية بحيث يمكن حتى اكتشاف تركيزات صغيرة من أنواع NiO.
استنادًا إلى تحليل بيانات XAS و Raman الطيفية أثناء التشغيل، نقترح آلية من ثلاث خطوات لانتقال الطور لـتحت ظروف HER القلوية، كما هو موضح بشكل تخطيطي في الشكل.الاختزال الكهروكيميائي لـ NiS إلىيمكن أن يتم تحفيز عند -0.27 فولت مقابل RHE بواسطة عدم الاستقرار الديناميكي الحراري لـ NiS تحت ظروف HER (الخطوة 1). مع ضبط إمكانيات أكثر سلبية للوصول إلى كثافات تيار HER أعلى، يؤدي تسرب الكبريت إلى تكوين فراغات الكبريت في (الخطوة 2). مع استقطاب العينة إلى إمكانيات أكثر إيجابية، يحدث أكسدة كبيرة عند +0.27 فولت مقابل RHE، وتُشغل فراغات S بواسطة الأنواع الأكسجينية من الوسط (الماء/ الأيونات)، مما يؤدي إلى تشكيل مرحلة مختلطة بشكل وثيق من (الخطوة 3).
استقرار، وتمت دراسة NiO تحت ظروف مختلفة ذات صلة بأداء التحفيز في تفاعل تقليل الهيدروجين (HER) باستخدام مطيافية الامتصاص بالأشعة السينية في الموقع (operando XAS). يوضح الشكل 3a مطيافية XAS في حافة النيكل K لقطب NiS في قياس التيار الزمني (chronoamperometry) لمدة 30 دقيقة عند جهد متتالي مطبق:“، والعودة إلى -1.00 فولت (جميع الجهود مقابل RHE). كما هو متوقع، يحتفظ العينة بهيكل NiS الأصلي عند 0.00 فولت وتتحول إلىعندما يتم استقطابه إلى -1.00 فولت (الشكل 3أ)، بسبب الاختزال الكهروكيميائي عند -0.27 فولت كما تم مناقشته أعلاه. ثم، عندما يتم استقطاب العينة إلى +0.50 فولت، يظهر الخط الأبيض عند 8350 إلكترون فولت المخصص لـيزداد بشكل ملحوظ (الإطار في الشكل 3أ) ويمكن ملاءمة الطيف مع تركيبة خطية منمنميزات وميزات طيف NiO (الشكل التكميلي 14a والجدول التكميلي 6)، بسبب الأكسدة الكتلية التي تحدث عند +0.27 فولت كما تم مناقشته سابقًا. عندما يتم إعادة العينة إلى ظروف HER عند -1.00 فولت، يبدو أن محتوى NiO يتناقص ولكنه لا يزال موجودًا (الإطار في الشكل 3a) حيث يمكن ملاءمة الطيف مع تركيبة خطية منمنالميزات الطيفية وميزات طيف NiO بعد 30 دقيقة تحت التفاعل (الشكل التكميلي 14b والجدول التكميلي 6). بالإضافة إلى ذلك، يظهر الشكل 3b طيف رامان في الوضع التشغيلي لقطب NiS في قياس التيار الزمني عند -0.30 فولت مقابل RHE لمدة 30 دقيقة، مما يؤكد بشكل أكبر وجود NiO تحت عملية HER. من أجل إثبات الاستقرار على المدى الطويل لـفي المرحلة المختلطة، قمنا بتمديد تجربة رامان العملية عند جهد مطبق قدره -0.30 فولت مقابل RHE لمدة 25 ساعة. تظهر النتائجو NiO تظل مستقرة تحت ظروف HER لفترة طويلة من الزمن (الشكل التكميلي 15). ، والعودة إلى طيف رامان العامل لقطب NiS في الكرونوأمبرو متري لمدة 30 دقيقة عند جهد مطبق ثابت قدره -0.30 فولت. (جميع الجهود مقابل RHE).
رؤية في آلية HER
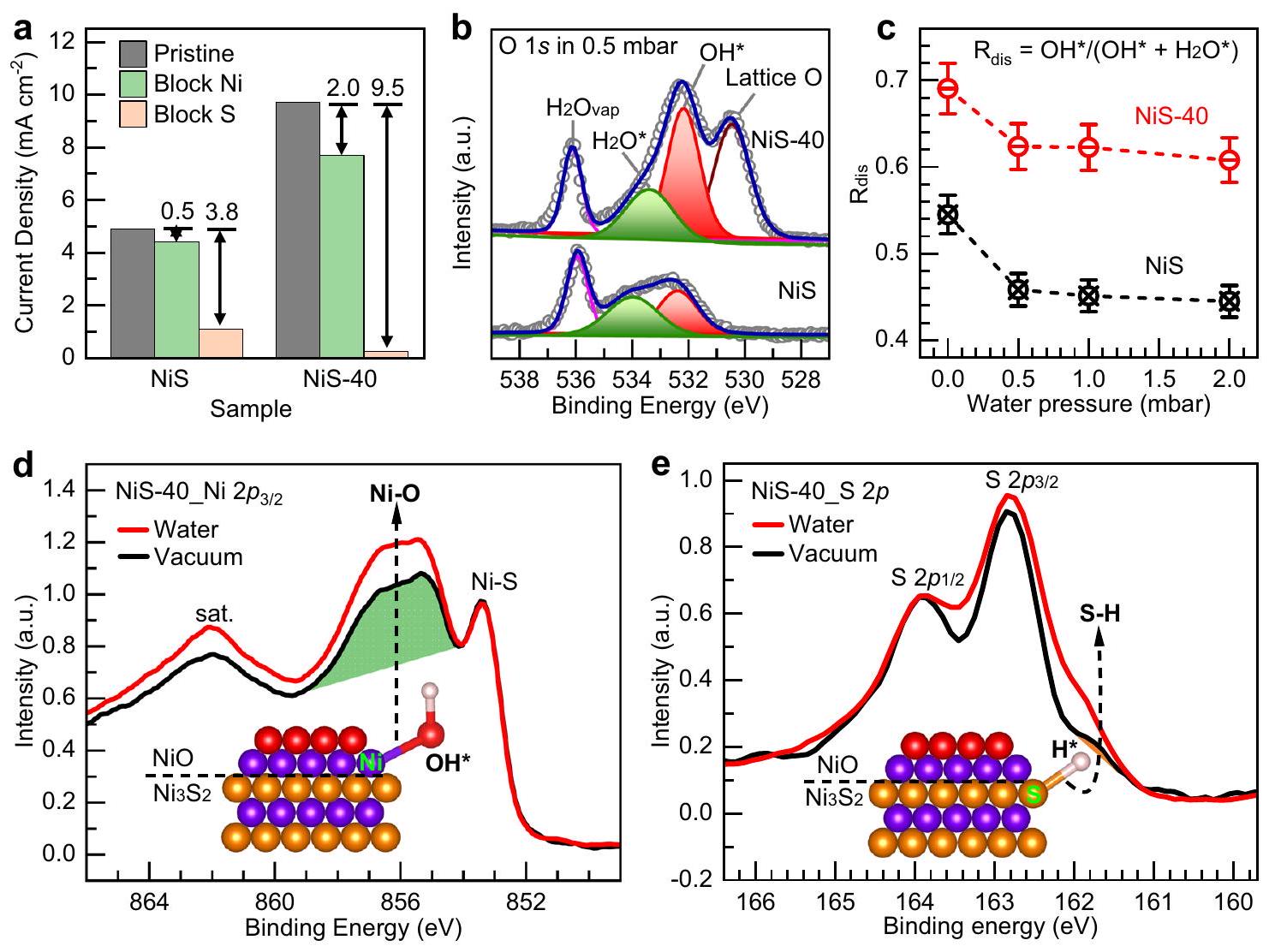
تتضمن عملية تقليل الهيدروجين في الوسط القلوي بشكل رئيسي الخطوة الخاصة بتفكك الماء لتكوين و ، أي، خطوة فولمر، تليها اقتران لتشكيلفي خطوات هيوروفسكي وتافل. لقد أظهرت خطوة فولمر أنها الخطوة المحددة لمعدل التفاعل في القلويات.. لذلك، لتحديد المواقع النشطة التي تشكلت بعد المعالجة الكهروكيميائية للعينات، قمنا بإجراء دراسة لامتصاص الماء التفكيكي على NiS والمحفزات بواسطة تقنية الأشعة السينية تحت ضغط قريب من الضغط الجوي (NAP-XPS) (تفاصيل التجربة في قسم الطرق). تُعتبر NAP-XPS تقنية طيفية مستخدمة بشكل شائع لدراسة الامتصاص التفكيكي للماء على المحفزات في بيئة رطبة.الشكل 4 ب يوضح طيف XPS لـ NiS وفيالمنطقة في وجود 0.5 مللي بار من بخار الماء. الأنواع المرتبطة بالأكسجين بما في ذلك الأكسجين الشبكي في NiO (الأكسجين الشبكي)، الممتصممتصوبخاريمكن تحديدها، ويمكن قياس كمياتها النسبية من خلال ملاءمة مساحة الذروة (الشكل التكميلي 16 والجدول التكميلي 7). على وجه الخصوص، تم امتصاصهايعود إلى الانفصالية الامتزاز، وبالتالي يمكن استخدامه كمقياس لدرجة تفكك الماء على سطح المحفز، أي كلما زادت القيمة، زادت القدرة على تفكك الماء. كما هو موضح في الشكل 4c، النسبة ( ) من إلىفيأعلى بكثير من ذلك في NiS في وجود ضغوط بخار الماء المختلفة، مما يشير إلى قدرة أقوى على الامتصاص التفكيكي للماء على NiS-40. ). بالإضافة إلى ذلك، تم التحقيق في كيمياء سطح المحفز من خلال مراقبة التغيرات في النيكل و س مستويات النواة عند تعرضها للماء. بالنسبة لـعينة، تعرض الماء يؤدي إلى تغيير ملحوظ في و . كما هو موضح في الشكل 4 د، مقارنةً بـالطيف في الفراغ، الكثافة الطيفية المرتبطة بـيزداد بشكل كبير عند التعرض للماء، وهو ما قد ينتج عن الامتزاز التفضيلي لـفي مواقع النيكل. من ناحية أخرى، بالنسبة لـكما هو موضح في الشكل 4e، بعد التعرض للماء، تظهر ميزة الكتف عند جانب طاقة الربط المنخفضة، والتي قد تكون ناتجة عن تقليل أيونات الكبريت عندمايمتص عند مواقع S في. بالمقابل، بالنسبة لـ NiS (الشكل التوضيحي 17)، لا يوجد تغيير واضح في و بعد التعرض للماء، مما يشير إلى أن هناك أقل بكثير و المتوسطات الناتجة على أسطح NiS، وفقًا لقدرة NiS الضعيفة على تفكك الماء التي لوحظت في هذه العينة.
بالنظر إلى دور مواقع النيكل والكبريت في عملية تفكك الماء، تم دراسة تأثير كل موقع على تفاعل الهيدروجين القلوي من خلال تجربة سمّية منهجية (تفاصيل التجربة)
الشكل 4 | تحديد المواقع النشطة من خلال تجارب التسمم وNAP-XPS. أ كثافة التيار لـ NiS وعند جهد مطبق ثابت قدره -0.15 فولت مقابل RHE قبل وبعد حجب مواقع النيكل ومواقع الكبريت.طيف XPS وتناسب قممه لـ NiS و NiS-40 عند ضغط ماء 0.5 مbar. نسبةإلىكنتيجة لضغوط مياه مختلفة (0.0 مbar، 0.5 mbar، 1.0 mbar و 2.0 mbar). د النيكلطيف XPS لـ NiS-40 في الفراغ وبخار الماء عند 0.5 بار. e طيف S2p XPS لـ NiS-40 في الفراغ وبخار الماء عند 0.5 بار. في قسم الطرق). على وجه التحديد، استخدمنا أيونات الثيوسيانات () ودوكادينثيول لحجب مواقع النيكل والكبريت بشكل انتقائي، على التواليتظهر الشكل 4a التغير في كثافة التيار عند -0.15 فولت مقابل RHE قبل وبعد حجب مواقع النيكل والكبريت في NiS الذي تم تصنيعه.التغيير في كثافة التيار هو مقياس لنشاط موقع معين؛ بشكل أساسي، كلما كان التغيير أكبر بعد حجب موقع، زادت النشاط التحفيزي للموقع في الحالة غير المسمومة. بالنسبة لـ NiS الذي تم تصنيعه، هناك فقط انخفاض في كثافة تيار HER بعد حجب مواقع النيكل، بينمابعد حجب مواقع S؛ مما يشير إلى أن مواقع S هي المواقع النشطة. من ناحية أخرى، بالنسبة لـعينة، يمكن ملاحظة أن التغيير هوبعد حظر مواقع Ni، وبعد حجب مواقع S. تشير هذه النتيجة إلى أن كل من مواقع Ni و S هي المواقع النشطة في.
تشير التجارب المذكورة أعلاه إلى التأثير التآزري لـو NiO في المرحلة المتحولة مما يؤدي إلى تعزيز نشاط HER، حيث تلعب مواقع النيكل على الواجهة دورًا حاسمًا في تسريع الخطوة المحددة لمعدل تفكك الماء وإنتاجالوسطاء بينما يسهل السطح الخارجيالامتصاص لتوليد.
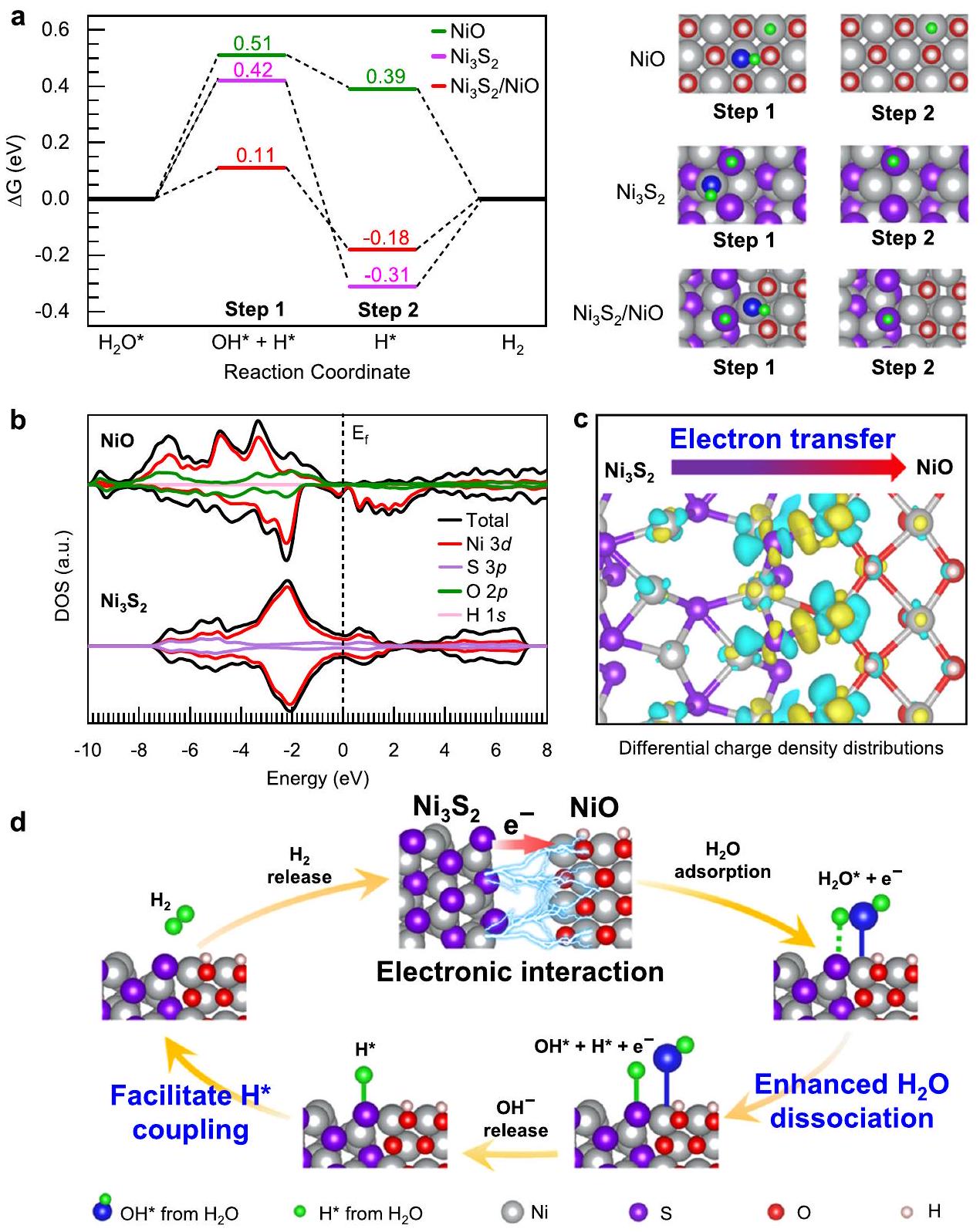
من أجل دعم دراساتنا التجريبية الميكانيكية لنشاط HER المحسن بعد تشكيل المرحلة المختلطة منNiO، قمنا بإجراء حسابات نظرية استنادًا إلى نظرية الوظائف الكثافة (DFT) (طريقة حسابية في قسم الطرق). في نموذجنا، نستخدم هيكلًا غير متجانس منو NiO مع مواقع O المغطاة بـ H (مُعلمة كـ ). نحن أيضًا نبني نماذج للأفراد و NiO للمقارنة. المخططات وتفاصيل النماذج موضحة في الشكل التكميلي 18، والذري تم توفير إحداثيات النماذج المحسّنة في البيانات التكميلية 1. استخدمنا NiO بلوري مع هيكل بلوري FCC في نموذجنا لأن تحليلاتنا الديناميكية الحرارية تشير إلى أن الحالة المستقرة لـ NiO هي بلورية (انظر الشكل التكميلية 19، والإحداثيات الذرية المقابلة موفرة في البيانات التكميلية 1).يمكن تبرير النموذج الذي يحتوي على مواقع O مغطاة بـ H لأن امتصاص H عند مواقع الأكسجين غير المشبعة في NiO يكون تلقائيًا وقويًا جدًا، كما يتضح من القيم الكبيرة لـحتى -0.86 إلكترون فولت كما هو موضح في الشكل التوضيحي 20. قمنا بإجراء دراستنا الميكانيكية باستخدام نظرية الكثافة الوظيفية بناءً على مقارنات بين هذه الأنماط الثلاثة، مستخلصين الاستنتاجات من الاتجاهات التي وجدناها بدلاً من القيم الفردية.
باستخدام هذه النماذج، قمنا بحساب تغير طاقة جيبس نحو تفاعل الهيدروجين.الهياكل غير المتجانسة والمراحل الفردية المقابلة، والنتائج موضحة بشكل بياني في الشكل 5a. بالنسبة لـحاجز الطاقة لخطوة تفكك الماء مرتفع (0.42 إلكترون فولت)، وهو ما يرجع أساسًا إلى الضعف في الامتزازفي مواقع النيكل (الشكل التوضيحي التكميلي 21). وبالمثل، فإن طاقة تفكك الماء لعنصر محفز NiO الفردي تصل إلى 0.51 إلكترون فولت، بسبب ضعف قوة الترابط بين النيكل والامتزاز.. من ناحية أخرى، بالنسبة لـ الهياكل غير المتجانسة، من خلال الاستفادة التكميلية من الامتصاص الأقوى لـفي مواقع النيكل عند جانب NiO وامتصاص أقوى لـفي مواقع S فيجانب، يمكن تقليل حاجز الطاقة لتفكك الماء بشكل كبير إلى 0.11 إلكترون فولت. علاوة على ذلك،الهياكل غير المتجانسة تسهل أيضًا اقترانلإنتاجطاقة الامتزازعلى مواقع S فيالهيكل غير المتجانس هو -0.18 إلكترون فولت، وهو أقرب إلى القيمة المثلى (0 إلكترون فولت) للاقتران بـلتشكيل، مقارنة بالقيمة المقابلة
الشكل 5 | التحقيقات النظرية حول آلية تفاعل الهيدروجين. أ مخططات الطاقة الحرة لجيبس لمسار تفاعل الهيدروجين القلوي عندمواقعمواقعوالواجهةمواقع . ب الكثافة الكلية والجزئية للحالات المحسوبة NiO و. توزيع كثافة الشحنة التفاضلية لـتمثل الخطوط الصفراء تراكم الإلكترونات وتمثل الخطوط السماوية نقص الإلكترونات. د الرسم التخطيطي لآلية تفاعل الهيدروجين.. (-0.31 إلكترون فولت) للفرد (الشكل 5أ). من المثير للاهتمام أن نتائج DFT لدينا تكشف عن وجود تفاعل إلكتروني قوي بين و NiO عند الواجهة توفر طاقة ملائمة لربطلتشكيلتظهر الشكل 5ب كثافة الحالات (DOS) لـ NiO ولديه كثافة حالات منخفضة عند مستوى فيرمي، ويمتلك حالة معدنية، تظهر كثافة عالية من الإلكترونات المشغولة حول مستوى فيرمي. لذلك، يمكن للإلكترونات الانتقال بسهولة منإلى الحالة الفارغة من NiO. كما هو موضح في الشكل 5c، تحليل الفرق في الشحنة لـالواجهة غير المتجانسة تكشف بوضوح أن الإلكترونات تنتقل من مواقع S لـجانب إلى جانب NiO. نقص الإلكترونات في مواقع S منيمكن أن تضبط الجوانب الإلكترونية لذرات الكبريت الطاقة الناتجة عن الامتصاص بشكل أكبرمن -0.31 إلكترون فولت إلى -0.18 إلكترون فولت، مما يوفر طاقة ملائمة للاقتران بـلتشكيل. لذلك، كما هو موضح في النموذج التخطيطي في الشكل 5d، تكشف نتائج دراستنا التجريبية والنظرية بوضوح أن واجهة NiO غير المتجانسة توفر مواقع نشطة مزدوجة لتعزيز تفكك الماء، والتفاعل الإلكتروني القوي بينو NiO تعدل الواجهة أيضًا الحالة الإلكترونية لمواقع S البينية وبالتالي توفر طاقة محسّنة لربطلتشكيل.
باختصار، تم استخدام تقنية XAS أثناء التشغيل ورامان أثناء التشغيل للتحقيق في إعادة هيكلة النيكل سلفيد الديناميكية تحت ظروف تفاعل الهيدروجين القلوي. وُجد أن النيكل سلفيد تم اختزاله كيميائيًا إلى، الذي تأكسد بعد ذلك مع الماء وأيونات من الإلكتروليت، مما يؤدي إلى تكوين مادة نشطة للغايةواجهة غير متجانسة. وقد كشفت مجموعة من الطيفية التشغيلية والتحقيقات النظرية بشكل أكبر أنستعمل واجهة NiO غير المتجانسة كمواقع نشطة فعلية، أي أن مواقع النيكل على الواجهة توفر المواقع النشطة لتسريع تفكك الماء بينما تسهل مواقع الكبريت على الواجهة الامتزاز.المتوسطات وارتباطها اللاحق لتشكيلتظهر أعمالنا أن كيمياء الكالكوجينيدات من المعادن الانتقالية ديناميكية للغاية، وأن التحكم الدقيق في ظروف العمل قد يؤدي إلى التكوين في الموقع للأنواع الحفازة التي تعزز أدائها الحفاز.
طرق
تركيب عينات NiS
تم استخدام ورق الكربون كركيزة لـ NiS. تم غسل ورق الكربون بشكل متتابع بـوماء منزوع الأيونات تحت تأثير الموجات فوق الصوتية لمدة 15 دقيقة في كل محلول لإزالة الشوائب بشكل كامل وتحسين المحبة للماء. 0.2448 جرام من خلات النيكل (، ألفا أيسر) و 0.2300 جرام من ثيوأسيتيمايد (تم إذابة (ألفا أيسار) في 35 مل من الماء المقطر. بعد التحريك، تم صب المزيج في وعاء ضغط سعة 50 مل مع قطعة من ورق الكربون.تمت عملية النمو فيفي فرن كهربائي لمدة 12 ساعة. بعد أن تبرد الأوتوكلاف بشكل طبيعي إلى درجة حرارة الغرفة، تم إزالة العينات، وغسلها بالماء المقطر، ثم تجفيفها في فرن مفرغ عند درجة حرارة الغرفة.
تحضير الإلكتروليت والقطب الكهربائي
يمكن الحصول على 1 م من KOH عن طريق إذابة 32.0058 جرام منسينوفارم كيميائي) في 500 مل من الماء المقطر. قم بتخزينه في زجاجة بلاستيكية. عادةً ما نقوم بتحضيره طازجًا حسب الحاجة. كانت قيمة الرقم الهيدروجينيتم قياسه بواسطة مقياس الرقم الهيدروجيني (التفاصيل في الجدول التكميلي 8 والشكل التكميلي 22). كما هو موضح في الشكل التكميلي 1a، جزء من ورق الكربون معمن الأبعاد الهندسية مغطاة بعينة NiS، بينما جزء آخر معتُترك الأبعاد الهندسية عارية للاتصال بقطب الكربون الزجاجي. يكوّن تثبيت الجزء المكشوف من ورق الكربون مع القطب الكربوني الزجاجي القطب العامل.
خصائص المواد
تم تحليل التركيب البلوري باستخدام حيود الأشعة السينية (XRD) باستخدامالإشعاع. تم تقييم شكل السطح باستخدام مجهر إلكتروني مسح ميداني (SEM) من ZEISS Sigma. لتحليل باستخدام مجهر إلكتروني نافذ (TEM) ومجهر إلكتروني نافذ مصحح للانحراف الكروي (SAC-TEM)، تم تعليق العينات في إيثانول مطلق ثم تم إيداعها على شبكة نحاسية. تم إجراء قياسات طيف الكترون الأشعة السينية عالية الدقة (XPS) باستخدام أشعة كهرومغناطيسية أحادية اللون من الألمنيوم K.مصدر الأشعة السينية مزودًا بمحلل طاقة الإلكترون SPECS PHOIBOS 150، محققًا دقة طاقة إجمالية تبلغ 0.50 إلكترون فولت. تم معايرة طاقة الربط باستخدام ذروة.
القياسات الكهروكيميائية
تم إجراء جميع القياسات الكهروكيميائية باستخدام تكوين ثلاثي الأقطاب يتم التحكم فيه بواسطة محطة عمل كهروكيميائية CHI 750E، والتي تتكون من عينة تم إعدادها مسبقًا على ورق الكربون كقطب عمل،كقطب مرجعي وعمود الجرافيت كقطب مضاد (الشكل التكميلي 23). تم معايرة القطب المرجعي من خلال مقارنته بالمعيارتم الشراء من شركة تيانجين أيدا المحدودة. تم تطبيع الجهود المبلغ عنها في هذا العمل مقابل RHE باستخدامقمنا بإجراء جميع قياسات HER في محلول إلكتروليتي 1 M KOH. قبل القياسات، تم نفخ جميع المحاليل الإلكترونية الجديدة بالنيتروجين النقي لمدة 30 دقيقة. تم استخدام تقنية الفولتميترية ذات المسح الخطي (LSV) للحصول على منحنيات الاستقطاب عن طريق مسح الجهد من 0.00 فولت إلى -1.00 فولت مقابل RHE بمعدل مسحتم إجراء طيف الامتياز الكهربائي (EIS) بنفس التكوين عند جهد -0.20 فولت مقابل RHE على مدى تردد من 100 كيلوهرتز إلى 0.1 هرتز عند سعة جهد جيبي قدرها 5 مللي فولت. لتصحيح السقوط الأومى، تم معايرة الجهود المقاسة باستخدام المعادلة، حيث تم تصحيح الإمكانيات، E كانت الإمكانية المقاسة،كان ساريًا وتم تحديد مقاومة الاتصال إما من خلالالقياس أو من خلال تحليل منحنيات EIS. في هذا العمل،تم إجراء ذلك. تم إعادة رسم منحنيات الاستقطاب كفائض جهد ضد الحاليللحصول على مخططات تافل لتقييم حركيات تفاعل الهيدروجين المدروس للكواشف.
قياسات السعة في منطقة الجهد التي لا توجد فيها عملية فارادائية عند معدلات مسح مختلفةويمكن استخدام 120 مللي فولت في الثانية -1 لتحديد المساحة السطحية النشطة كهربائياً (ECSA).
تجربة امتصاص الأشعة السينية الكهروكيميائية أثناء التشغيل
تم إجراء قياسات XAS أثناء التشغيل على عينات NiS المزروعة على ورق الكربون كقطب عمل في خط شعاع CLAESS في مصادم ALBA في إسبانيا، باستخدام إعداد خلية كيميائية كهربائية مصممة خصيصًا في المنزل (انظر الشكل التكميلية 24). كانت سلك البلاتين بمثابة القطب المضاد، وتم استخدام القطب الكهربائي كقطب مرجعي في محلول 1 م كOH. تم إجراء هذه التجارب الكهروكيميائية في محطة عمل كهروكيميائية تتحكم بها الكمبيوتر. تم جمع الأطياف عند حافة النيكل K بشكل مستمر بينما كانت العينات تخضع لعملية الفولتمترية الدورية (CV) من جهد الدائرة المفتوحة إلى -1.50 فولت مقابل RHE ومعدل المسح لـتم إجراء اكتساب الطيف باستخدام مقياس الطيف أحادي اللون Si (311) الذي قدم دقة طاقة حادثة تبلغ 0.3 إلكترون فولت. تم اختيار الزاوية والتغطية المناسبة للمرآتين الموجهتين والمركّزتين لضمان رفض التوافقيات. تم إجراء القياسات في وضع الفلورية، وتم استخدام كل من أقراص NiO وأوراق Ni لمعايرة الطاقة. تم معالجة بيانات XAS باستخدام حزمة البرمجيات ATHENA..
تجربة رامان التشغيلية
تم الحصول على طيف رامان باستخدام نظام رامان التداخلي Jobin-Yvon Horiba Xplora. تم إجراء جميع التجارب باستخدام طول موجي للتحفيز قدره 638 نانومتر وعدسة المجهر ذات فتحة عددية تبلغ 0.55. تم الحفاظ على قوة الليزر عندتمت التجارب التشغيلية في خلية رامان K006 (انظر الشكل التوضيحي 25)، وتم تسجيل طيف رامان عند إمكانيات تطبيقية مختلفة.
تجربة طيف الانبعاث الضوئي عند ضغط قريب من الضغط الجوي
تم تسجيل طيف الانبعاث الضوئي تحت ضغط قريب من الضغط الجوي (NAP-XPS) على جهاز طيفي قائم في المختبر (SPECS GmbH، برلين) باستخدام أشعة كهرومغناطيسية مونوكروماتية من الألمنيوم K. مصدر ( يعمل عند 50 واط. المحلل هو SPECS PHOIBOS 150 NAP،محلل طاقة نصف كروي بمتوسط نصف قطر 150 مم. مدخل المحلل هو فوهة معقطر الفتحة. يتم تشغيل المحلل في وضع نقل المحلل الثابت (FAT). تم ضبط طاقة المرور على 40 إلكترون فولت لعمليات المسح العامة و20 إلكترون فولت للمناطق عالية الدقة. تم إجراء جرعة بخار الماء عبر صمام تسرب، ني، و تم تسجيل NAP-XPS عند جميع الضغوط بعد استقرار الضغط لمدة لا تقل عن 15 دقيقة.
تجربة التسمم
من أجل استكشاف ما هو الموقع النشط خلال عملية تقليل الهيدروجين، تأثير أيون الثيوسيانات) وتأثير الدودكانثيول على نشاط HER للمواد المحفزة التي تم التحقيق فيها تم تقييمه من خلال إضافة و 10 مللي مول من دوديكانثيول في الإلكتروليت على التوالي، حيث وتمت معرفة أن الدودكانثيول يسبب تسمم مواقع النيكل ومواقع الكبريت، على التوالي..
حسابات نظرية الكثافة الوظيفية
لإجراء حسابات نظرية الكثافة (DFT)، يتم استخدام طريقة بيردو-بورك-إرنزنهوف (PBE) للحصول على دالة التبادل والتصحيح.، وتستخدم إمكانيات الموجات المعززة بواسطة البروجيكتور (PAW)“تم إدخال Hubbard-U و DFT-D3 (B-J) أيضًا في الحسابات لتصحيح تداخل الإلكترونات. تم تنفيذ جميع حسابات DFT في حزمة المحاكاة الأولية فيينا (VASP)”.تم تعيين قيمة مصطلح U-J كـ 4.0 إلكترون فولت لأيونات النيكل.. أشبكة مونكهورست-باك المتمركزة معتم استخدام شبكة ذات نقاط. كانت دوال الموجة للإلكترونات التكافؤية تم توسيعها باستخدام مجموعة أساس الموجة المسطحة مع طاقة قطع تبلغ 500 إلكترون فولت. تم تعيين معيار التقارب علىبين التحسين الإلكتروني. بالنسبة لدورات التحسين الهيكلي، كانت القوة القصوى على كل ذرة أقل من. كانت جميع الحسابات مستقطبة للدوران.
حسابات الطاقة الحرة لجيبس
الخطوات المتضمنة في تفاعل الهيدروجين في الوسط القلوي مدرجة في المعادلات (1)-(3) كما هو موضح أدناه:
هيروفيسكي :طاولة :
هنا، قمنا بتطبيق طريقة تم تطويرها سابقًا لنمذجة الديناميكا الحرارية للتفاعلات الكهروكيميائية استنادًا إلى حسابات الوظائف الكثافة.بشكل محدد، تعتبر الطاقة الحرة لجيبس للتفاعلات، التي تُستخدم كمعيار لتحديد تلقائية التفاعلات، قابلة للاكتساب بناءً على نظرية دالة الكثافة (DFT) مع تصحيحات تشمل المساهمات الانتروبية (TS) وطاقة النقطة الصفرية (ZPE) من خلال المعادلة التالية:
هنا،يمثل طاقة DFT المحسوبة للأنظمة المعنية، ZPE تشير إلى التغير في الطاقة الصفرية المحددة من الترددات الاهتزازية، و هو التغير في الإنتروبيا. لحسابات الطاقة الحرة لجيبس، فإن الطاقة الحرة لبروتون ( ) يتم استبداله بنصف جزيء هيدروجين في الحالة الغازية ( لكل خطوة من التفاعلات الكهروكيميائية. تم الحصول على إنتروبيا الأنواع المقابلة من الترددات الاهتزازية. بالنسبة لجزيئات الغاز مثل يمكن وصف قيمة الإنتروبيا في مجموع الأقسام الانتقالية والاهتزازية والدورانية. علاوة على ذلك، لم يتم اعتبار قيم الإنتروبيا الانتقالية والدورانية بسبب المساهمات الضئيلة. يتم الحصول على الإنتروبيا من خلال المعادلة التالية:
أين يدل على ثابت الغاز، هو ثابت بولتزمان، يمثل ثابت بلانك،يتوافق مع عدد أفوجادرو، تشير إلى التردد و هو عدد الذرات الممتصة. بالإضافة إلى ذلك، بالنسبة لحسابات التردد، تم تثبيت جميع ذرات الركيزة وتم حساب فقط الوسائط التفاعلية المتعلقة بالمضافات، والمساهمات من مرحلة المحفز إلى ZPE ولا تشارك في طاقة جيبس الحرة لتلك التفاعلات. يتم توفير دقة الحسابات في الملاحظة التكميلية 6.
توفر البيانات
البيانات الإضافية التي تدعم نتائج هذه الدراسة متاحة من المؤلف المراسل عند الطلب. يتم تقديم بيانات المصدر مع هذه الورقة.
References
Chu, S., Cui, Y. & Liu, N. The path towards sustainable energy. Nat. Mater. 16, 16-22 (2017).
Li, X. et al. Latest approaches on green hydrogen as a potential source of renewable energy towards sustainable energy: Spotlighting of recent innovations, challenges, and future insights. Fuel 334, 126684 (2023).
Seh, Zhi, W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
Zhang, L.-N. et al. Advanced hydrogen evolution electrocatalysts promising sustainable hydrogen and chlor-alkali co-production. Energy Environ. Sci. 14, 6191-6210 (2021).
Chatenet, M. et al. Water electrolysis: from textbook knowledge to the latest scientific strategies and industrial developments. Chem. Soc. Rev. 51, 4583-4762 (2022).
Hu, C., Zhang, L. & Gong, J. Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 12, 2620-2645 (2019).
Mahmood, N. et al. Electrocatalysts for hydrogen evolution in alkaline electrolytes: mechanisms, challenges, and prospective solutions. Adv. Sci. 5, 1700464 (2018).
Durst, J. et al. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 7, 2255-2260 (2014).
Wei, J. et al. Heterostructured electrocatalysts for hydrogen evolution reaction under alkaline conditions. Nanomicro Lett. 10, 75 (2018).
Subbaraman, R. et al. Enhancing hydrogen evolution activity in water splitting by tailoring Pt interfaces. Science 334, 1256-1260 (2011).
You, B. et al. Enhancing electrocatalytic water splitting by strain engineering. Adv. Mater. 31, 1807001 (2019).
Shen, X . et al. Dual-site cascade oxygen reduction mechanism on for promoting reaction kinetics. J. Am. Chem. Soc. 141, 9463-9467 (2019).
Zhang, W. et al. Emerging dual-atomic-site catalysts for efficient energy catalysis. Adv. Mater. 33, 2102576 (2021).
Wang, Y.-J. et al. Unlocking the door to highly active ORR catalysts for PEMFC applications: polyhedron-engineered Pt-based nanocrystals. Energy Environ. Sci. 11, 258-275 (2018).
Hu, J. et al. Interface modulation of /metal oxide heterostructures for efficient hydrogen evolution electrocatalysis. Small 16, 2002212 (2020).
Zhong, W. et al. Coupled vacancy pairs in Ni-doped CoSe for improved electrocatalytic hydrogen production through topochemical deintercalation. Angew. Chem. Int. Ed. 59, 22743-22748 (2020).
. et al. Controllable surface reorganization engineering on cobalt phosphide nanowire arrays for efficient alkaline hydrogen evolution reaction. Adv. Mater. 30, 1703322 (2018).
Duan, J., Chen, S., Ortíz-Ledón, C. A., Jaroniec, M. & Qiao, S.-Z. Phosphorus vacancies that boost electrocatalytic hydrogen evolution by two orders of magnitude. Angew. Chem. Int. Ed. 59, 8181-8186 (2020).
Lin, F. et al. Electrocatalytic hydrogen evolution of ultrathin Co heterojunction with interfacial electron redistribution. Adv. Energy Mater. 10, 2002176 (2020).
Sun, H. et al. Boosting activity on porous nanosheet by coupling for efficient electrochemical overall water splitting at high current densities. Adv. Funct. Mater. 30, 1910596 (2020).
Cui, Y. et al. Tungsten oxide/carbide surface heterojunction catalyst with high hydrogen evolution activity. ACS Energy Lett. 5, 3560-3568 (2020).
He, W. et al. Fluorine-anion-modulated electron structure of nickel sulfide nanosheet arrays for alkaline hydrogen evolution. ACS Energy Lett. 4, 2905-2912 (2019).
Kim, B.-J. et al. Functional role of Fe-doping in Co-based perovskite oxide catalysts for oxygen evolution reaction. J. Am. Chem. Soc. 141, 5231-5240 (2019).
Zhou, S. et al. Engineering electrocatalytic activity in nanosized perovskite cobaltite through surface spin-state transition. Nat. Commun. 7, 11510 (2016).
Petrie, J. R., Jeen, H., Barron, S. C., Meyer, T. L. & Lee, H. N. Enhancing perovskite electrocatalysis through strain tuning of the oxygen deficiency. J. Am. Chem. Soc. 138, 7252-7255 (2016).
Du, J. et al. Nonstoichiometric perovskite for oxygen electrocatalysis with high activity. Inorg. Chem. 53, 9106-9114 (2014).
Wang, J. et al. Stabilizing the oxygen vacancies and promoting water-oxidation kinetics in cobalt oxides by lower valence-state doping. Nano Energy 53, 144-151 (2018).
Jiang, N. et al. Nickel sulfides for electrocatalytic hydrogen evolution under alkaline conditions: a case study of crystalline , and nanoparticles. Catal. Sci. Technol. 6, 1077-1084 (2016).
Hung, T.-F. et al. Nickel sulfide nanostructures prepared by laser irradiation for efficient electrocatalytic hydrogen evolution reaction and supercapacitors. Chem. Eng. J. 367, 115-122 (2019).
da Silva, M. G. S., Leite, C. M., Cordeiro, M. A. L., Mastelaro, V. R. & Leite, E. R. One-step synthesis of nickel sulfides and their electrocatalytic activities for hydrogen evolution reaction: a case study of crystalline h-NiS and o-Ni, nanoparticles. ACS Appl. Energy Mater. 3, 9498-9503 (2020).
Zheng, Y., Jiao, Y., Jaroniec, M. & Qiao, S. Z. Advancing the electrochemistry of the hydrogen-evolution reaction through combining experiment and theory. Angew. Chem., Int. Ed. 54, 52-65 (2015).
Jin, S. Are metal chalcogenides, nitrides, and phosphides oxygen evolution catalysts or bifunctional catalysts? ACS Energy Lett. 2, 1937-1938 (2017).
Wygant, B. R., Kawashima, K. & Mullins, C. B. Catalyst or precatalyst? the effect of oxidation on transition metal carbide, pnictide, and chalcogenide oxygen evolution catalysts. ACS Energy Lett. 3, 2956-2966 (2018).
Ding, X. et al. An Fe stabilized metallic phase of for the highly efficient oxygen evolution reaction. Nanoscale 11, 23217-23225 (2019).
Wan, G. et al. Amorphization mechanism of electrocatalyst: how oxygen redox initiates ionic diffusion and structural reorganization. Sci. Adv. 7, eabc7323 (2021).
Fabbri, E. et al. Dynamic surface self-reconstruction is the key of highly active perovskite nano-electrocatalysts for water splitting. Nat. Mater. 16, 925-931 (2017).
Zhang, G. et al. Enhanced catalysis of electrochemical overall water splitting in alkaline media by Fe doping in nanosheet arrays. ACS Catal. 8, 5431-5441 (2018).
Ma, Q. et al. Identifying the electrocatalytic sites of nickel disulfide in alkaline hydrogen evolution reaction. Nano Energy 41, 148-153 (2017).
Zhu, Y. et al. Operando unraveling of the structural and chemical stability of P-substituted electrocatalysts toward hydrogen and oxygen evolution reactions in alkaline electrolyte. ACS Energy Lett. 4, 987-994 (2019).
Zhai, L. et al. In situ phase transformation on nickel-based selenides for enhanced hydrogen evolution reaction in alkaline medium. ACS Energy Lett. 5, 2483-2491 (2020).
Fleet, M. E. The crystal structure of heazlewoodite, and metallic bonds in sulfide minerals. Am. Mineral. 62, 341-345 (1977).
Vershinin, A. D., Selivanov, E. N., Gulyaeva, R. I. & Sel’menskikh, N. I. Thermal expansion of in -Ni alloys. Inorg. Mater. 41, 882-887 (2005).
Li, R. et al. Short-range order in amorphous nickel oxide nanosheets enables selective and efficient electrochemical hydrogen peroxide production. Cell Rep. Phys. Sci. 3, 100788 (2022).
Denny, Y. R. et al. Ni K-edge XAFS analysis of NiO thin film with multiple scattering theory. Surf. Interface Anal. 46, 997-999 (2014).
Cheng, Z., Abernathy, H. & Liu, M. Raman spectroscopy of nickel sulfide . J. Phys. Chem. C 111, 17997-18000 (2007).
Fan, L. et al. Molecular functionalization of NiO nanocatalyst for enhanced water oxidation by electronic structure engineering. ChemSusChem 13, 5901-5909 (2020).
Bala, N., Singh, H. K., Verma, S. & Rath, S. Magnetic-order induced effects in nanocrystalline NiO probed by Raman spectroscopy. Phys. Rev. B 102, 024423 (2020).
Sunny, A. & Balasubramanian, K. Raman spectral probe on sizedependent surface optical phonon modes and magnon properties of NiO nanoparticles. J. Phys. Chem. C 124, 12636-12644 (2020).
Radinger, H. et al. Importance of nickel oxide lattice defects for efficient oxygen evolution reaction. Chem. Mater. 33, 8259-8266 (2021).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272-1276 (2011).
Stoerzinger, K. A. et al. Water reactivity on the (001) surface: an ambient pressure X-ray photoelectron spectroscopy study. J. Phys. Chem. C 118, 19733-19741 (2014).
Stoerzinger, K. A., Hong, W. T., Crumlin, E. J., Bluhm, H. & ShaoHorn, Y. Insights into electrochemical reactions from ambient pressure photoelectron spectroscopy. Acc. Chem. Res. 48, 2976-2983 (2015).
Yin, J. et al. Ni-C-N nanosheets as catalyst for hydrogen evolution reaction. J. Am. Chem. Soc. 138, 14546-14549 (2016).
Makarova, M., Okawa, Y. & Aono, M. Selective adsorption of thiol molecules at sulfur vacancies on (0001), followed by vacancy repair via S-C dissociation. J. Phys. Chem. C 116, 22411-22416 (2012).
Zhang, J. et al. Copper dopants improved the hydrogen evolution activity of earth-abundant cobalt pyrite catalysts by activating the electrocatalytically inert sulfur sites. J. Mater. Chem. A 5, 17601-17608 (2017).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537-541 (2005).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558-561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251-14269 (1994).
Peng, L. et al. Atomic cation-vacancy engineering of NiFe-layered double hydroxides for improved activity and stability towards the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 24612-24619 (2021).
Ma, Y. et al. High-entropy energy materials: challenges and new opportunities. Energy Environ. Sci. 14, 2883-2905 (2021).
Yu, B., Jiang, H. & Zhang, Y. Superior specific capacity and energy density simultaneously achieved by Sr/In co-deposition behavior of ternary alloys as anodes for Mg-Air cells. Preprint at https://doi.org/10.1016/j.jma.2024.02.005 (2024).
شكر وتقدير
يقر المؤلفون بدعم التمويل من المؤسسة الوطنية للعلوم الطبيعية في الصين بموجب أرقام المنح 22075232 و 22275154 و 52225104. كما يقر K.H.L.Z. و J.P.H. و X.D. ببرنامج التنقل لمركز الترويج للبحث الصيني الألماني (رقم المنحة M0377). R.W. ممتن لدعم التمويل من “برنامج شوجوانغ” المدعوم من مؤسسة تطوير التعليم في شنغهاي ولجنة التعليم البلدية في شنغهاي (رقم 20SGO3) ولجنة العلوم والتكنولوجيا لبلدية شنغهاي (رقم 22520710600). F.E.O. يشكر MINECO وصندوق NextGenerationEU/PRTR الأوروبي على عقد رامون ي كاجال (RyC2O21-034254-I). V.A.P.O. ممتنة للدعم المالي المقدم من الاتحاد الأوروبي (مشروع HYSOLCHEM مع اتفاقية منحة رقم 101017928) وAEI الإسبانية (SOLFuture PLEC2021-007906). يعترف جيوفيو غورني بمنحة FJC2O20-044866-I الممولة من MCIN/AEI/ 10.13039/501100011033، ومن خطة التعافي والتحول والمرونة، ومن “الاتحاد الأوروبي NextGenerationEU/PRTR”. تعترف مريم براوي بمشروع NovaCO2 (PID2020-118593RB-C22) ومنحة RYC2O22-038157-I الممولة من MCIN/AEI/ 10.13039/501100011033. يعترف م.غ.-ت. بمشروع PEC2Change (TED2021-129999A-C33) الممول من MCIN/AEI/ 10.13039/501100011033 ومن الاتحاد الأوروبي Next Generation EU/ PRTR. بالإضافة إلى ذلك، حصل المشروع الذي أدى إلى هذه النتائج على دعم من منحة من مؤسسة “لا كايسا” (ID 100010434). رمز المنحة هو LCF/BQ/PR23/11980046. تم إجراء جزء من هذه التجارب في خط شعاع CLAESS في مسرع ALBA (رقم الاقتراح AV-2021035069) بمساعدة موظفي ALBA. يود الباحثون أن يشكروا إيفان غارسيا دومينغيز وفرانسيسكو مارتينيز لوبيز، منسقي الطابق في مسرع ALBA، على مساعدتهم في تصميم وطباعة الخلايا الكهروكيميائية المستخدمة في قياسات امتصاص الأشعة السينية في الموقع.
مساهمات المؤلفين
صمم K.H.L.Z. و R.W. و X.D. وأشرفوا على البحث. أعد X.D. و P.Z. العينات. قام X.D. بإجراء القياسات الكهروكيميائية وتوصيف الهيكل، وحلل البيانات. أنجز D.L. حسابات DFT. ساهم X.D. و F.E.O. و G.G. و X.C. و V.A.P.O. في جميع التجارب التشغيلية وتحليل البيانات. شارك D.L. و H.W. و M.B. و M.G.-T. و J.P.H. و J.L. و J.K. و S.C. في مناقشة البيانات. قام K.H.L.Z. و R.W. و F.E.O. بمراجعة المخطوطة. كتب X.D. و D.L. المخطوطة. ساهم جميع المؤلفين في النسخة النهائية.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى فريدي إي. أوروبيزا، رينبينغ وو أو كيلفين إتش. إل. تشانغ.
معلومات مراجعة الأقران تشكر مجلة Nature Communications يوجين تشين، أليكسي كوزمين والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
معلومات إعادة الطباعة والتصاريح متاحة على http://www.nature.com/reprints ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
المختبر الوطني الرئيسي للكيمياء الفيزيائية للأسطح الصلبة، كلية الكيمياء والهندسة الكيميائية، جامعة شيامن، شيامن 361005، الصين. قسم علوم المواد، جامعة فودان، شنغهاي 200433، الصين.وحدة العمليات المعتمدة على الضوء، معهد إيمديا للطاقة، منتزه موستوليس التكنولوجي، Avenida Ramón de la Sagra 3، 28935 موستوليس، مدريد، إسبانيا.مجموعة معالجة الليزر، معهد البصريات (CSIC)، شارع سيرانو 121، 28006 مدريد، إسبانيا.CELLS-ALBASynchrotron، شارع اللوم 2-26، 08290 سيردانيولا ديل فاييس، إسبانيا.مختبر علوم السطح، قسم المواد وعلوم الأرض، الجامعة التقنية في دارمشتات، شارع أوتو-بيرندت 3، 64287 دارمشتات، ألمانيا.قسم علوم المواد والهندسة، المعهد الوطني للعلوم والتكنولوجيا في أولسان (يونست)، أولسان 44919، جمهورية كوريا.ساهم هؤلاء المؤلفون بالتساوي: شينغيو دينغ، دا ليو. – البريد الإلكتروني:freddy.oropeza@imdea.org; rbwu@fudan.edu.cn; kelvinzhang@xmu.edu.cn
Transition metal chalcogenides have been identified as low-cost and efficient electrocatalysts to promote the hydrogen evolution reaction in alkaline media. However, the identification of active sites and the underlying catalytic mechanism remain elusive. In this work, we employ operando X-ray absorption spectroscopy and near-ambient pressure X-ray photoelectron spectroscopy to elucidate that NiS undergoes an in-situ phase transition to an intimately mixed phase of and NiO , generating highly active synergistic dual sites at the interface. The interfacial Ni is the active site for water dissociation and adsorption while the interfacial S acts as the active site for adsorption and evolution. Accordingly, the in-situ formation of interfaces enables NiS electrocatalysts to achieve an overpotential of only at a current density of . Our work highlighted that the chemistry of transition metal chalcogenides is highly dynamic, and a careful control of the working conditions may lead to the in-situ formation of catalytic species that boost their catalytic performance.
Electrolysis of water powered by renewable electricity to produce green hydrogen is widely considered as a promising pathway to a global clean and sustainable energy future . The key to enable this technology is the development of low-cost and highly efficient electrocatalysts to accelerate the water splitting reactions, i.e., oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) . In particular, the design of high-performance electrocatalysts for HER in alkaline conditions has received intensive attention, because the alkaline water electrolysis is the most commonly used route in the industry, and the alkaline HER is also a key step in the chlor-alkali process . However, the alkaline HER still suffers from sluggish reaction kinetics compared to that in acidic
solution, due to the additional water dissociation step (Volmer step: in order to supply the absorbed intermediate for subsequent generation . The water dissociation has a higher activation barrier and is considered to be the rate determining step that limits the overall reaction . Even the state-of-the-art Pt-based catalysts show about two orders of magnitude lower HER activity in alkaline than in acidic media . Significant progress has been made to improve the HER activity of Pt and other noble metals in alkaline environments by constructing dual active site heterostructures, e.g., , in which the functions as a promotor to accelerate the water dissociation . Improving the activity of noble metal-based
catalysts with a single active site has been challenging mainly due to the scaling relationships in the formation energy of intermediates, which affects not only the HER but also the OER and the oxygen reduction reaction (ORR) . Dual active site catalysts provide alternative reaction mechanisms that may break such scaling relationships, offering more chance for achieving better catalytic performance, compared with single active site catalysts .
Nevertheless, the scarcity and high cost of Pt group materials restrict their large-scale application . In this regard, earth-abundant transition metal (TM)-based chalcogenides, phosphides, nitrides, and carbides have been recently emerging as cost-effective alkaline HER electrocatalysts . Many TM-based catalysts exhibit comparable alkaline HER activity as Pt group materials . In addition, doping, defect, and strain engineering have been further explored to modulate their electronic structure to enhance the activity and stability . Nickel sulfides such as , and have drawn significant attention because of their high catalytic activity, as well as easy and scalable methodologies for the preparation . Despite this rapid development of alkaline HER catalysts, the underlying reaction mechanism and the active sites for the alkaline HER, which are different from the well-documented acidic HER, are still under considerable debate . Furthermore, it has been demonstrated that TM-based compounds catalysts for the OER undergo structural or compositional reconstruction, or even transform into a new phase under OER working condition . On the other hand, the structural and chemical conversion of catalysts under HER conditions has been less explored, and it has been even assumed HER catalysts are stable during the HER process . However, recent works indicated that
some transition metal chalcogenides may indeed undergo a phase transition under alkaline HER working condition . Therefore, realtime monitoring of the catalytic processes and identifying the real active sites under HER operation conditions are of vital importance to uncover the catalytic mechanism.
In this study, we present an in-depth elucidation of the phase conversion process based on operando X-ray absorption spectroscopy (XAS) and operando Raman spectroscopy under HER working conditions. We found that NiS electrodes undergo a significant HER performance enhancement due to an in-situ transformation into a mixed phase of and NiO , which is identified to be the actual active catalyst phase for alkaline HER. The ex-situ spectroscopy and theoretical investigations further reveal that the interfacial Ni ( S) of provides the active sites to accelerate water dissociation while the interfacial S ( ) facilitates the adsorption of hydrogen intermediates and its subsequent association to form molecular hydrogen. As a result, the in-situ formed exhibits a superior HER activity, requiring a low overpotential of to reach a current density of in our performance tests.
Results and discussion
Phase transition of NiS to induces enhanced HER activity
The NiS samples were synthesized on carbon paper using one-step hydrothermal method (Supplementary Fig. 1a). Details of the synthesis and characterization are provided in the Methods section. X-ray diffraction (XRD) pattern shown in Fig. 1a and X-ray photoelectron
Fig. 1 | HER performance and structural characterizations of NiS catalyst before and after measurement. a XRD patterns of the as-synthesized NiS and NiS after HER measurement (marked as NiS-40). b Ni XPS spectra for NiS and NiS after HER measurement (marked as ), along with NiO and as reference. c -corrected Linear sweep voltammetry (LSV) polarization curves of assynthesized NiS, NiS-10, NiS-20, NiS-30, NiS-40, and NiS-50 samples measured in
1 M KOH solution. NiS-10, NiS-20, NiS-30, NiS-40, and NiS-50 are NiS samples after , and 50 minute chronoamperometry measurements at -1.00 V vs. RHE, respectively. The large-area TEM image, high-resolution TEM image (the right panel shows the FFT images of both regions) and the corresponding elemental mapping of the NiS-40 sample after conducting 25-h chronopotentiometric measurement at a current density of .
spectroscopy (XPS) of Ni core level in Fig. 1b suggests that the assynthesized samples are characteristic of the hexagonal -phase of . Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images in Supplementary Fig. 1 show that the samples consist of NiS particles with a size of uniformly coating on the carbon paper substrate.
The HER activity was evaluated in 1 M KOH using a threeelectrode system (details in the Methods section). Cyclic voltammetry (CV) measurements were carried out in order to adjust the conditions for the evaluation of the HER catalytic activity (Supplementary Fig. 2a, b). In the first cycle of as-synthesized samples, there is a sharp onset of the current density for the HER at around -0.30 V vs. RHE, which continues to increase steadily as the sample is further polarized to negative potentials. After that sharp onset of the current density, the CV profile is reproducible in the second cycle, with an upper limit of the potential at 0.05 V vs. RHE. However, as the upper limit of the potential is extended to 0.40 V vs. RHE in the and cycles, there is an oxidation wave with a peak at +0.27 V vs. RHE and a increase of the current density when the sample is polarized back to negative potentials. Remarkably, the current density keeps on increasing as the sample is subject to chronoamperometry at -1.00 V vs. RHE (Supplementary Fig. 2c). Linear sweep voltammetry (LSV) of the sample after the chronoamperometry measurement shows that the current density increases compared with the pristine sample, and there is also a clear shift of the onset to lower overpotential (Supplementary Fig. 2d). These electrochemical studies clearly demonstrate that insitu modifications of NiS electrodes lead to a significantly enhanced HER catalytic activity.
On these bases, we carried out a more detailed study of the chronoamperometric conditions of NiS electrodes on their HER catalytic activity. Figure 1c shows the LSV curves of the assynthesized NiS, as well as NiS samples after 10, 20, 30, 40, and 50 minute chronoamperometry measurements at -1.00 V vs. RHE, labeled as NiS-10, NiS-20, NiS-30, NiS-40, and NiS-50, respectively. In order to avoid deviations caused by ohmic drops, the potential was adjusted by an factor (Supplementary Fig. 3a, b). The assynthesized NiS initially exhibits a low HER activity, but the HER activity gradually increases with the chronoamperometry measurement time until it is conducted for 40 minutes. A similar trend of HER activity is also observed when the current density is normalized by electrochemical active surface area (ECSA) (Supplementary Figs. 4 and 5). As shown in Fig. 1c, the NiS-40 sample exhibits a high HER catalytic activity, requiring an overpotential of to reach a current density of (acquisition of error bar can be seen in Supplementary Fig. 3c.). Further electrochemical characterizations showed that the Tafel slope and charge transfer resistance (Supplementary Fig. 6a, b) decrease in the order of NiS > NiS-10 > NiS-20 > NiS-30 > NiS-50 > NiS-40, indicating that the performance enhancement after the chronoamperometric treatments results from an intrinsic faster HER kinetics. Compared with previously reported HER catalysts, electrochemically treated NiS electrocatalysts exhibit competitive HER activity in terms of overpotentials at and Tafel slope (Supplementary Fig. 7 and Supplementary Table 1). Additionally, the catalytic activity of the NiS-40 sample is very stable, showing no overpotential increase over 25 h of continuous operation at a current density of in 1 M KOH (Supplementary Fig. 6c).
A series of ex-situ characterizations of the highly active samples after performance tests were conducted. The assynthesized NiS is converted into a mixed phase of and NiO after HER measurement. Figure 1a shows an XRD pattern of the NiS-40 sample, from which a pattern characteristic of can be identified, suggesting a full-phase conversion of NiS to crystalline . The XPS in the Ni region for the NiS-40
sample exhibits a pronounced spectral feature possibly arising from NiO (Fig. 1b). Additionally, the Ni K-edge XAS of sample NiS-40 (Supplementary Fig. 8) can be fitted by a linear combination of spectra of and NiO , which further confirms the formation of and NiO . We further characterized the sample after chronopotentiometric measurement at a current density of , the XRD, XPS and XAS confirm the HER-post catalysts are present in the form of and NiO (Supplementary Fig. 9). Spherical Aberration Corrected Transmission Electron Microscope (SAC-TEM) was used to further examine the microstructure of the HER-post catalysts. As shown in Fig. 1d, the large-area TEM image reveals numerous small NiO clusters anchored onto the large . The HR-TEM image exhibits a distinct interface that partitions the image into two regions (Fig. 1e). By analyzing the FFT images corresponding to these two regions, the cluster region has an interplanar spacing of 0.21 nm corresponding well to the (200) lattice plane of NiO, while the other region has an interplanar spacing of 0.30 nm which matches well with (-110) lattice plane of . More importantly, the elemental mapping shows the preferential enrichment of oxygen elements on the small cluster (Fig. 1f), which strongly reveals the small cluster is assigned to NiO . It should be noted that the formed NiO may exist in the form of a very small cluster, which is hard to determine by XRD patterns. To investigate the influence of the ratio of to NiO on the catalytic activity, NiS samples received by , and 50 LSVs in the range from 0.00 V to -1.00 V vs. RHE were prepared. The Ni K-edge XAS of these samples, shown in Supplementary Figs. 10a-d, consists of a linear combination of spectra of and NiO , which indicates that a full transformation of NiS occurs after a single LSV . However, the NiO to ratio increases upon increasing the number of LSVs. We extracted the NiO to ratio based on the XAS spectra (Supplementary Figs. 10a-d and Supplementary Table 2) and found that the HER activity increases with the amount of NiO (Supplementary Fig. 10e), which clearly indicates the beneficial effect of the Ni oxide species on the HER process.
Phase transition dynamics revealed by operando XAS and operando Raman spectroscopy
An operando XAS spectroscopy was employed to gain detailed insights into the transition process, and details of the experimental method are provided in the Methods section. Figure 2a shows a three-dimensional (3D) plot of the Ni K-edge XAS of the NiS electrode as a function of the applied potentials during cyclic CV measurement (Fig. 2b). Note that two important changes in the spectral profile occur at applied voltages of -0.27 V and +0.27 V vs. RHE, dividing the 3D plot into three regions as I (applied voltage from +1.00 V to -0.27 V ), II ( -0.27 V to -1.50 V and back to +0.27 V ) and III (from +0.27 V to 1.00 V ). Selected Ni K-edge XAS spectra at specific applied voltages within regions I, II and III are shown in Fig. 2c. In region I, with applied voltage from +1.00 V to -0.27 V , the spectrum of the sample remains as that of the NiS phase. The transition from region I to region II at -0.27 V can be identified as the phase transition , based on spectral fitting and comparisons with reference samples of and (region I and II in Fig. 2c). The associated reduction of the formal oxidation state of in NiS to in can be observed as a cathodic wave peak in the CV profile just at the onset of the HER current shown in Fig. 2b (see the inset), clearly showing that the HER catalytic activity is associated with the formation of . After the phase transition, the phase is stable under HER conditions. However, with a more negative voltage applied (region II in Fig. 2a), a continuous decrease of the spectral intensity of the white line of the Ni K-edge XAS spectra suggests a further reduction of the oxidation state of Ni . Consistently, the fitting of Fourier Transform moduli of spectra in region II revealed a decrease of the average coordination number of Ni coordinated by S (Fig. 2d, Supplementary Figs. 11 and 12, and Supplementary Tables 3 and 4, and
Fig. 2 | Operando characterizations and the mechanism of phase transition. a Operando Ni K-edge XAS for NiS catalyst as a function of applied voltages. Top panel shows a 3-dimensional view of Ni K-edge XAS spectra; bottom shows a 2-dimensional projection from a 3 -dimensional view. The corresponding CV profile used for the operando XAS measurement. The inset shows the magnified region marked in the rectangular box. c The Ni K-edge XAS spectra at specific applied voltages of means +1.00 V in negative sweep of CV),
( means -1.50 V in negative sweep of CV ), and and +1.00 V _P mean +0.50 V and +1.00 V in positive sweep of CV ) in region I, II, and III respectively, extracted from Fig. 2a, and their linear combination of and The coordination number of Ni as a function of applied voltages. Operando Raman spectroscopy of NiS at different applied voltages. Schematic illustration for phase transition of NiS catalyst during HER measurement.
the simulations procedure in Supplementary Note 1), suggesting the formation of S vacancies, i.e., . As the sample is polarized from negative back to positive potentials, the transition from II to III at +0.27 V can be associated with an oxidation process observed in the CV profile shown in Fig. 2b, consistent with a constant increase of the white line of the spectrum as the sample is further polarized (region III in Fig. 2a). The spectrum can be fitted with a linear combination of and an increasing contribution of NiO (region III in Fig. 2c), and details of the fitting are discussed in Supplementary Notes 2-5. Based on this analysis, we estimate that the NiO contribution at +0.50 V is about , which further increases to when the sample potential reaches +1.00 V (Fig. 2c and Supplementary Table 5). Thus, the coordination of Ni by O occurs gradually in region III (Fig. 2d), probably caused by the filling of the sulfur vacancies in with water or ions, leading to an intimately mixed phase of and NiO , which is further verified by the Wavelet transform (WT) analysis of extended X-ray absorption fine structure (EXAFS) and Fourier-transformed -weighted EXAFS at applied voltage of +1.00 V in region III
(Supplementary Fig. 13). The corresponding results of fitting at +1.00 V were shown in Supplementary Table 4, and the Ni-O show a bond length of . Highly crystalline NiO has a characteristic distances around ; however, the short-range order in NiO reveals that the distances for amorphous and/or nanocrystalline NiO may decrease to . This result further confirms the formed NiO is in the form of a very small cluster.
The formation of mixed-phase of and NiO was further verified by an operando Raman spectroscopy study (the details of the experiment in the Methods section). The top Raman spectrum in Fig. 2e can be assigned to pristine NiS, with characteristic bands appearing at Raman shifts and . As the sample was polarized to negative potentials, the characteristic peaks assigned to NiS disappear, while the characteristic peaks assigned to appear at , and (marked as a, b, c, and d, respectively), indicating the complete phase transition from . In addition, the newly emerged peaks e, f, g, h, and i located at 453, 493,600, 800 , and can be assigned to , suggesting the
Fig. 3 | The long-time existence of reconstructed proven by operando characterizations. a Operando XAS at Ni K-edge of a NiS electrode in chronoamperometry for 30 minutes at 4 consecutively applied potentials: 0.00 V ,
formation of NiO under HER conditions. Raman spectra features associated with , and NiO are very well resolved so that even small trace concentrations of NiO species can be detected.
Based on the analysis of the operando XAS and operando Raman spectroscopy data, we propose a three-step mechanism for the phase transition of under alkaline HER conditions, as schematically shown in Fig. . The electrochemical reduction of NiS to at -0.27 V vs. RHE can be driven by the thermodynamic instability of NiS under HER conditions (step 1). As more negative potentials are set to reach higher HER current densities, S leaching leads to the formation of S vacancies in (step 2). As the sample is polarized to more positive potentials, substantial oxidation occurs at +0.27 V vs. RHE, and S vacancies are occupied by the oxygen species from the media (water/ ions), leading to the formation of an intimately mixed phase of (step 3).
The stability of , and NiO under different conditions relevant to the HER catalytic performance was further investigated by operando XAS spectroscopy. Figure 3a shows operando XAS at Ni K-edge of the NiS electrode in chronoamperometry for 30 minutes at consecutively applied potentials: , and back to -1.00 V (all potentials are vs. RHE). As expected, the sample retains its pristine NiS structure at 0.00 V and converts to when polarized to -1.00 V (Fig. 3a), due to the electrochemical reduction at -0.27 V as discussed above. Then, as the sample is polarized to +0.50 V , the white line of 8350 eV assigned to increases noticeably (the inset in Fig. 3a) and the spectrum can be fitted with a linear combination of of features and of NiO spectral features (Supplementary Fig. 14a and Supplementary Table 6), due to the bulk oxidation that occurs at +0.27 V as previously discussed. When the sample is brought back to HER conditions at -1.00 V , the content of NiO seems to decrease but still exist (the inset in Fig. 3a) as the spectrum can be fitted with a linear combination of of spectral features and of NiO spectral features after 30 minutes under the reaction (Supplementary Fig. 14b and Supplementary Table 6). In addition, Fig. 3b shows operando Raman spectra of a NiS electrode in chronoamperometry at -0.30 V vs. RHE for 30 minutes, further confirming the existence of NiO under the HER process. In order to demonstrate the long-term stability of the mixed phase, we extended the operando Raman experiment at an applied potential of -0.30 V vs. RHE to 25 h . The results demonstrate and NiO remain stable under HER conditions over an extended period of time (Supplementary Fig. 15). , and back in Operando Raman spectra of NiS electrode in chronoamperometry for 30 minutes at a constant applied potential of -0.30 V . (All potentials vs. RHE).
Insight into the HER mechanism
The HER in alkaline media mainly involves the step for water dissociation to form and , i.e., Volmer step, followed by the coupling of to form in the Heyrovsky and Tafel steps. The Volmer step has been shown to be the rate-determining step for alkaline . Therefore, to identify the active sites formed after the electrochemical treatment of the samples, we carried out a study for the dissociative adsorption of water on NiS and catalysts by means of near-ambient-pressure (NAP)-XPS (experimental details in the Methods section). NAP-XPS is a commonly used spectroscopic technique to study the dissociative adsorption of water on catalysts in a humid environment . Figure 4 b shows the XPS spectra of NiS and in the region in the presence of 0.5 mbar water vapor. Oxygen related species including lattice oxygen in NiO (lattice O ), adsorbed , adsorbed and vapor can be identified, and their relative amounts can be quantified by fitting the peak area (Supplementary Fig. 16 and Supplementary Table 7). In particular, the adsorbed originates from the dissociative adsorption, and therefore can be used as a measure of the degree of water dissociation on the catalyst surface, i.e., the higher the value, the better the ability to dissociate water. As shown in Fig. 4c, the ratio ( ) of to in is much higher than that in NiS in the presence of different water vapor pressure, suggesting a stronger ability of dissociative adsorption of water on the NiS-40 ( ). In addition, the surface chemistry of the catalyst was further investigated by monitoring changes in the Ni and S core levels upon water exposure. For the sample, water exposure induces a pronounced change in the and . As shown in Fig. 4 d, compared with the spectrum in vacuum, the spectral intensity associated with increases significantly upon water exposure, which may result from the preferential adsorption of at Ni sites. On the other hand, for shown in Fig. 4e, after water exposure, a shoulder feature appears at the lower binding energy side, which may result from the reduction of sulfur ions when adsorbs at S sites in . In contrast, for NiS (Supplementary Fig. 17), there is no obvious change in and after water exposure, which suggests that there is much less and intermediates generated on NiS surfaces, in accordance with the weak water dissociation ability of NiS observed for this sample.
Considering the involvement of Ni and S sites in the water dissociation process, the influence of each site on the alkaline HER was investigated by a systematic poison experiment (experimental details
Fig. 4 | Identification of the active sites by poisoning and NAP-XPS experiments. a The current density of NiS and at a constant applied potential of -0.15 V vs. RHE before and after blocking the Ni sites and S sites. XPS spectra and their peak fitting for NiS and NiS-40 at 0.5 mbar water pressure.
c The ratio of to as a function of different water pressure ( 0.0 mbar, 0.5 mbar, 1.0 mbar and 2.0 mbar). d The Ni XPS spectra of NiS-40 in vacuum and 0.5 mbar water vapor. e The S2p XPS spectra of NiS-40 in vacuum and 0.5 mbar water vapor.
in the Methods section). Specifically, we used thiocyanate ions ( ) and dodecanethiol to selectively block the Ni and S sites, respectively . Figure 4a shows the change in current density at -0.15 V vs. RHE before and after blocking Ni and S sites in assynthesized NiS and . The change in current density is a measure of the activity of a specific site; essentially, the greater the change after blocking a site, the higher the catalytic activity of the site in the non-poisoned state. For as-synthesized NiS, there is only a decrease in the HER current density after blocking Ni sites, while after blocking S sites; suggesting that S sites are the active sites. On the other hand, for the sample, it can be seen that the change is after blocking Ni sites, and after blocking S sites. This result indicates that both the Ni and S sites are the active sites in .
The above experiments indicate the synergistic effect of and NiO in the transformed phase leading to the enhanced HER activity, in which the interfacial Ni sites play a crucial role in accelerating the rate determining step of water dissociation and producing intermediates while the interfacial S facilitates adsorption to generate .
In order to support our experimental mechanistic studies for the enhanced HER activity after the formation of the mixed phase of NiO , we performed theoretical calculations based on density functional theory (DFT) (computational method in the Methods section). In our model, we employ a heterostructure of and NiO with O sites covered by H (marked as ). We also construct models for individual and NiO for comparison. Schematics and details of the models are shown in Supplementary Fig. 18, and the atomic
coordinates of the optimized models are supplied in Supplementary Data 1. We used crystalline NiO with an FCC crystal structure in our model because our thermodynamic analyses suggest that the stable state of NiO is crystalline (see Supplementary Fig. 19, and corresponding atomic coordinates are supplied in Supplementary Data 1). The model with O sites covered by H can be rationalized because the adsorption of H at the unsaturated oxygen sites of NiO is spontaneous and very strong, as indicated by the large values of of up to -0.86 eV shown in Supplementary Fig. 20. We carried out our DFT mechanistic study based on comparisons of these three modes, drawing conclusion from the trends we found rather than from individual values.
Using these models, we calculated the Gibbs energy change toward HER for the heterostructure and the corresponding individual phases, and the results are graphically shown in Fig. 5a. For individual , the energy barrier for the water dissociation step is high ( 0.42 eV ), which is mainly due to the weak adsorption of on Ni sites (Supplementary Fig. 21). Similarly, the water dissociation energy for individual NiO catalyst is as high as 0.51 eV , due to the weak bonding strength between Ni and adsorption . On the other hand, for the heterostructure, by taking complementary advantages of stronger adsorption of at Ni sites at the NiO side and stronger adsorption of at S sites at the side, the energy barrier for water dissociation can be dramatically reduced to 0.11 eV . Furthermore, heterostructures also facilitate the coupling of to produce . The adsorption energy of on S sites at the heterostructure is -0.18 eV , which is closer to the optimal value ( 0 eV ) for coupling of to form , compared with the corresponding value
Fig. 5 | Theoretical investigations on the HER mechanism. a Gibbs free energy diagrams of alkaline HER pathway at sites of sites of and the interfacial sites of . b The calculated total and partial density of states
of NiO and . c Differential charge density distributions of . Yellow contours represent electron accumulation and cyan contours represent electron depletion. d The schematic diagram for HER mechanism of .
( -0.31 eV ) for individual (Fig. 5a). Interestingly, our DFT results reveals that a strong electronic interaction between and NiO at the interface provide favorable energetics for coupling of to form . Figure 5b shows the density of states (DOS) of NiO and has low DOS at the Fermi level, and has a metallic state, showing a high density of occupied electrons around the Fermi level. Therefore, electrons can easily transfer from to the empty state of NiO . As shown in Fig. 5c, the charge differential analysis for the hetero-interface clearly reveals that electrons transfer from S sites of the side to the NiO side. The depletion of electrons at S sites of the side can tune the electronic states of S atoms further weaken the adsorption energy of from -0.31 eV to -0.18 eV , which provides favorable energetics for coupling of to form . Therefore, as shown in the schematic model in Fig. 5d, our experimental and theoretical mechanistic study results clearly reveal that the in-situ formed NiO hetero-interface provides dual active sites to promote water dissociation, and the strong electronic interaction between and NiO
interface also modifies the electronic state of interfacial S sites and thus provide optimized energetics for coupling of to form .
In summary, an operando XAS and operando Raman were employed to investigate the dynamic restructuring of NiS under alkaline HER conditions. It was found that NiS was electrochemically reduced to , which subsequently oxidized with water and ions from the electrolyte, leading to the formation of a highly active hetero-interface. The combination of operando spectroscopy and theoretical investigations further unveiled that the NiO hetero-interface would function as the actual active sites, i.e., the interfacial Ni sites provide the active sites to accelerate water dissociation while the interfacial S sites facilitate the adsorption of intermediates and its subsequent association to form . Our work shows that the chemistry of transition metal chalcogenides is highly dynamic, and careful control of the working conditions may lead to the in-situ formation of catalytic species that boot their catalytic performance.
Methods
Synthesis of NiS samples
Carbon paper was used as the substrate for NiS. Carbon paper was consecutively washed with and deionized water under sonication for 15 minutes in each solution to thoroughly remove impurities and improve hydrophilicity. 0.2448 g nickel acetate ( , Alfa Aesar) and 0.2300 g thioacetamide ( , Alfa Aesar) were dissolved in 35 mL deionized water. After stirring, the mixture was poured into a 50 ml autoclave with a piece of carbon paper . The growth was carried out at in an electric oven for 12 h . After the autoclave cooled down naturally to room temperature, the samples were removed, washed with deionized water, and then dried in a vacuum oven at room temperature.
Preparation of electrolyte and electrode
1 M KOH can be obtained by dissolving 32.0058 g of , Sinopharm Chemical Reagent) in 500 ml of deionized water. Store it in a plastic bottle. We usually prepare it freshly as needed. The pH value was measured by pH meter (details in the Supplementary Table 8 and Supplementary Fig. 22). As shown in Supplementary Fig. 1a, a portion of the carbon paper with of geometric dimensions is covered by the NiS sample, while another portion with of geometric dimensions is left bare for connection to the glassy carbon electrode. Clamping the exposed portion of the carbon paper with the glassy carbon electrode forms the working electrode.
Material characterizations
The crystal structure was analyzed using X-ray diffraction (XRD) employing radiation. Surface morphology was assessed using a ZEISS Sigma field emission scanning electron microscope (SEM). For analysis with transmission electron microscopy (TEM) and spherical aberration-corrected transmission electron microscopy (SAC-TEM), samples were suspended in absolute ethanol and subsequently deposited onto a Cu grid. High-resolution X-ray photoelectron spectroscopy (XPS) measurements were conducted using a monochromatic Al K X-ray source ( ) equipped with a SPECS PHOIBOS 150 electron energy analyzer, achieving a total energy resolution of 0.50 eV . The binding energy was calibrated using the peak .
Electrochemical measurements
All electrochemical measurements were carried out using a threeelectrode configuration controlled by a CHI 750E electrochemical workstation, consisting of an as-prepared sample on carbon paper as working electrode, as the reference electrode and graphite rode as the counter electrode (Supplementary Fig. 23). The reference electrode was calibrated by comparing it with the standard purchased from Tianjin Aida Co., LTD. The potentials reported in this work were normalized versus the RHE using . We conducted all HER measurements in 1 M KOH electrolyte. Prior to measurements, all fresh electrolytes were bubbled with pure nitrogen for 30 minutes. Linear sweep voltammetry (LSV) was employed to obtained polarization curves by sweeping the potential from 0.00 V to -1.00 V vs. RHE at a scan rate of . The electrochemical impedance spectroscopy (EIS) was performed in the same configuration at -0.20 V vs. RHE applied potential over frequency range from 100 kHz to 0.1 Hz at the amplitude of the sinusoidal voltage of 5 mV . To correct the ohmic drop, the measured potentials were calibrated using equation , where was corrected potentials, E was measured potential, was current and was the contact resistance determined either through measurement or by analyzing EIS curves. In this work, was conducted. The polarization curves were replotted as overpotential versus current to get Tafel plots for assessing the HER kinetics of investigated catalysts.
Capacitance measurements in the potential region of no faradaic process at different scan rates of , and 120 mV s -1 can be used to determine electrochemical active surface area (ECSA).
Operando XAS measurements were carried out on NiS samples grown on carbon paper as the working electrode at the CLAESS beamline of the ALBA synchrotron in Spain, using a specially designed in-house electrochemical cell setup (see Supplementary Fig. 24). A platinum wire served as the counter electrode, and an electrode was used as the reference electrode in a 1 M KOH solution. These electrochemical experiments were done in a computer-controlled electrochemical workstation. Spectra at the Ni K-edge were continuously collected as the samples were subject to cyclic voltammetry (CV) from the open circuit potential to -1.50 V versus RHE and scan rate of . Spectra acquisition was performed using a Si (311) monochromator that offered an incident energy resolution of 0.3 eV . Appropriate angle and coating for the collimating and focusing mirrors were selected to ensure harmonic rejection. The measurements were done in fluorescence mode, and both NiO pellets and Ni foils were employed for energy calibration. XAS data processing was conducted using the ATHENA software package .
Operando Raman experiment
Raman spectra were acquired using a Jobin-Yvon Horiba Xplora confocal Raman system. All experiments were done with an excitation wavelength of 638 nm and a microscope objective with a numerical aperture of 0.55 . Laser power was maintained at . Operando experiments took place in a K006 Raman cell (see Supplementary Fig. 25), and Raman spectra were recorded at various applied potentials.
Near-ambient-pressure photoemission spectra (NAP-XPS) were recorded on a lab-based spectrometer (SPECS GmbH, Berlin) using a monochromated Al K source ( ) operating at 50 W . The analyzer is a SPECS PHOIBOS 150 NAP, a hemispherical energy analyzer with a 150 mm mean radius. The entrance to the analyzer is a nozzle with a diameter orifice. The analyzer is run in fixed analyzer transmission (FAT) mode. The pass energy was set to 40 eV for survey scans and 20 eV for high-resolution regions. Water vapor dosing was carried out via a leak valve, Ni , and NAP-XPS at all pressures were recorded after at least 15 minutes of pressure stabilization.
Poisoning experiment
In order to explore what is the active site during the HER process, the influence of thiocyanate ion ( ) and dodecanethiol on the HER activity of investigated catalysts was evaluated by adding and 10 mM dodecanethiol in the electrolyte respectively, where and dodecanethiol are known to poison the Ni sites and S sites, respectively .
Density functional theory calculations
For the density functional theory calculations (DFT), Perdew-BurkeErnzenhof (PBE) method is used to obtain exchange correlation functional , and projector-augmented wave (PAW) pseudopotentials are employed . Hubbard-U and DFT-D3 (B-J) are also introduced in the calculations to correctthe electron correlation. The whole DFT calculations are implemented in the Vienna Ab initio Simulation Package (VASP) code . The value of U-J term was set as 4.0 eV for Ni cations . A -centered Monkhorst-Pack grid with a -point mesh was used. The wave functions of valence electrons were
expanded using a plane wave basis set with a cut-off energy of 500 eV . The convergence criterion was set as between for electronic optimization. For the structural optimization loops, the maximum force on each atom was less than . All calculations were spin-polarized.
Gibbs free energy calculations
The steps involved in HER in alkaline media are listed in Eqs. (1)-(3), as shown below:
Heyrovsky : Tafel :
Here, we applied a method developed previously for modeling the thermochemistry of electrochemical reactions based on density functional calculations . Specifically, the Gibbs free energy of reactions, served as the criterion to determine the spontaneity of reactions, can be acquired based on DFT with corrections including entropic (TS) and zero-point energy (ZPE) contributions by the following equation:
Here, the represents the DFT energy computed for the respective systems, ZPE refers to the change in zero-point energy determined from the vibrational frequencies, and is the change in the entropy. For Gibbs free energy calculations, the free energy of a proton ( ) is replaced by half of a hydrogen molecule in gaseous ( ) for every step of electrochemical reactions . The entropies of corresponding species were obtained from the vibrational frequencies. For gas molecules such as , the value of entropy can be described in the sum of the translational, vibrational, and rotational sections. Furthermore, the translational and rotational entropic values were not considered due to negligible contributions. The entropy is obtained by the following equation:
where denotes the gas constant, is the Boltzmann constant, represents Plank’s constant, corresponds to Avogadro’s number, signifies the frequency and is the number of adsorbed atoms. Additionally, for frequency calculations, all substrate atoms were fixed and only adducts-related reaction intermediates are calculated, and the contributions from the catalyst phase to ZPE and are not involved in the Gibbs free energy of those reactions. The accuracy of calculations is provided in Supplementary Note 6.
Data availability
Additional data supporting this study’s findings are available from the corresponding author upon request. Source data are provided with this paper.
References
Chu, S., Cui, Y. & Liu, N. The path towards sustainable energy. Nat. Mater. 16, 16-22 (2017).
Li, X. et al. Latest approaches on green hydrogen as a potential source of renewable energy towards sustainable energy: Spotlighting of recent innovations, challenges, and future insights. Fuel 334, 126684 (2023).
Seh, Zhi, W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
Zhang, L.-N. et al. Advanced hydrogen evolution electrocatalysts promising sustainable hydrogen and chlor-alkali co-production. Energy Environ. Sci. 14, 6191-6210 (2021).
Chatenet, M. et al. Water electrolysis: from textbook knowledge to the latest scientific strategies and industrial developments. Chem. Soc. Rev. 51, 4583-4762 (2022).
Hu, C., Zhang, L. & Gong, J. Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 12, 2620-2645 (2019).
Mahmood, N. et al. Electrocatalysts for hydrogen evolution in alkaline electrolytes: mechanisms, challenges, and prospective solutions. Adv. Sci. 5, 1700464 (2018).
Durst, J. et al. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 7, 2255-2260 (2014).
Wei, J. et al. Heterostructured electrocatalysts for hydrogen evolution reaction under alkaline conditions. Nanomicro Lett. 10, 75 (2018).
Subbaraman, R. et al. Enhancing hydrogen evolution activity in water splitting by tailoring Pt interfaces. Science 334, 1256-1260 (2011).
You, B. et al. Enhancing electrocatalytic water splitting by strain engineering. Adv. Mater. 31, 1807001 (2019).
Shen, X . et al. Dual-site cascade oxygen reduction mechanism on for promoting reaction kinetics. J. Am. Chem. Soc. 141, 9463-9467 (2019).
Zhang, W. et al. Emerging dual-atomic-site catalysts for efficient energy catalysis. Adv. Mater. 33, 2102576 (2021).
Wang, Y.-J. et al. Unlocking the door to highly active ORR catalysts for PEMFC applications: polyhedron-engineered Pt-based nanocrystals. Energy Environ. Sci. 11, 258-275 (2018).
Hu, J. et al. Interface modulation of /metal oxide heterostructures for efficient hydrogen evolution electrocatalysis. Small 16, 2002212 (2020).
Zhong, W. et al. Coupled vacancy pairs in Ni-doped CoSe for improved electrocatalytic hydrogen production through topochemical deintercalation. Angew. Chem. Int. Ed. 59, 22743-22748 (2020).
. et al. Controllable surface reorganization engineering on cobalt phosphide nanowire arrays for efficient alkaline hydrogen evolution reaction. Adv. Mater. 30, 1703322 (2018).
Duan, J., Chen, S., Ortíz-Ledón, C. A., Jaroniec, M. & Qiao, S.-Z. Phosphorus vacancies that boost electrocatalytic hydrogen evolution by two orders of magnitude. Angew. Chem. Int. Ed. 59, 8181-8186 (2020).
Lin, F. et al. Electrocatalytic hydrogen evolution of ultrathin Co heterojunction with interfacial electron redistribution. Adv. Energy Mater. 10, 2002176 (2020).
Sun, H. et al. Boosting activity on porous nanosheet by coupling for efficient electrochemical overall water splitting at high current densities. Adv. Funct. Mater. 30, 1910596 (2020).
Cui, Y. et al. Tungsten oxide/carbide surface heterojunction catalyst with high hydrogen evolution activity. ACS Energy Lett. 5, 3560-3568 (2020).
He, W. et al. Fluorine-anion-modulated electron structure of nickel sulfide nanosheet arrays for alkaline hydrogen evolution. ACS Energy Lett. 4, 2905-2912 (2019).
Kim, B.-J. et al. Functional role of Fe-doping in Co-based perovskite oxide catalysts for oxygen evolution reaction. J. Am. Chem. Soc. 141, 5231-5240 (2019).
Zhou, S. et al. Engineering electrocatalytic activity in nanosized perovskite cobaltite through surface spin-state transition. Nat. Commun. 7, 11510 (2016).
Petrie, J. R., Jeen, H., Barron, S. C., Meyer, T. L. & Lee, H. N. Enhancing perovskite electrocatalysis through strain tuning of the oxygen deficiency. J. Am. Chem. Soc. 138, 7252-7255 (2016).
Du, J. et al. Nonstoichiometric perovskite for oxygen electrocatalysis with high activity. Inorg. Chem. 53, 9106-9114 (2014).
Wang, J. et al. Stabilizing the oxygen vacancies and promoting water-oxidation kinetics in cobalt oxides by lower valence-state doping. Nano Energy 53, 144-151 (2018).
Jiang, N. et al. Nickel sulfides for electrocatalytic hydrogen evolution under alkaline conditions: a case study of crystalline , and nanoparticles. Catal. Sci. Technol. 6, 1077-1084 (2016).
Hung, T.-F. et al. Nickel sulfide nanostructures prepared by laser irradiation for efficient electrocatalytic hydrogen evolution reaction and supercapacitors. Chem. Eng. J. 367, 115-122 (2019).
da Silva, M. G. S., Leite, C. M., Cordeiro, M. A. L., Mastelaro, V. R. & Leite, E. R. One-step synthesis of nickel sulfides and their electrocatalytic activities for hydrogen evolution reaction: a case study of crystalline h-NiS and o-Ni, nanoparticles. ACS Appl. Energy Mater. 3, 9498-9503 (2020).
Zheng, Y., Jiao, Y., Jaroniec, M. & Qiao, S. Z. Advancing the electrochemistry of the hydrogen-evolution reaction through combining experiment and theory. Angew. Chem., Int. Ed. 54, 52-65 (2015).
Jin, S. Are metal chalcogenides, nitrides, and phosphides oxygen evolution catalysts or bifunctional catalysts? ACS Energy Lett. 2, 1937-1938 (2017).
Wygant, B. R., Kawashima, K. & Mullins, C. B. Catalyst or precatalyst? the effect of oxidation on transition metal carbide, pnictide, and chalcogenide oxygen evolution catalysts. ACS Energy Lett. 3, 2956-2966 (2018).
Ding, X. et al. An Fe stabilized metallic phase of for the highly efficient oxygen evolution reaction. Nanoscale 11, 23217-23225 (2019).
Wan, G. et al. Amorphization mechanism of electrocatalyst: how oxygen redox initiates ionic diffusion and structural reorganization. Sci. Adv. 7, eabc7323 (2021).
Fabbri, E. et al. Dynamic surface self-reconstruction is the key of highly active perovskite nano-electrocatalysts for water splitting. Nat. Mater. 16, 925-931 (2017).
Zhang, G. et al. Enhanced catalysis of electrochemical overall water splitting in alkaline media by Fe doping in nanosheet arrays. ACS Catal. 8, 5431-5441 (2018).
Ma, Q. et al. Identifying the electrocatalytic sites of nickel disulfide in alkaline hydrogen evolution reaction. Nano Energy 41, 148-153 (2017).
Zhu, Y. et al. Operando unraveling of the structural and chemical stability of P-substituted electrocatalysts toward hydrogen and oxygen evolution reactions in alkaline electrolyte. ACS Energy Lett. 4, 987-994 (2019).
Zhai, L. et al. In situ phase transformation on nickel-based selenides for enhanced hydrogen evolution reaction in alkaline medium. ACS Energy Lett. 5, 2483-2491 (2020).
Fleet, M. E. The crystal structure of heazlewoodite, and metallic bonds in sulfide minerals. Am. Mineral. 62, 341-345 (1977).
Vershinin, A. D., Selivanov, E. N., Gulyaeva, R. I. & Sel’menskikh, N. I. Thermal expansion of in -Ni alloys. Inorg. Mater. 41, 882-887 (2005).
Li, R. et al. Short-range order in amorphous nickel oxide nanosheets enables selective and efficient electrochemical hydrogen peroxide production. Cell Rep. Phys. Sci. 3, 100788 (2022).
Denny, Y. R. et al. Ni K-edge XAFS analysis of NiO thin film with multiple scattering theory. Surf. Interface Anal. 46, 997-999 (2014).
Cheng, Z., Abernathy, H. & Liu, M. Raman spectroscopy of nickel sulfide . J. Phys. Chem. C 111, 17997-18000 (2007).
Fan, L. et al. Molecular functionalization of NiO nanocatalyst for enhanced water oxidation by electronic structure engineering. ChemSusChem 13, 5901-5909 (2020).
Bala, N., Singh, H. K., Verma, S. & Rath, S. Magnetic-order induced effects in nanocrystalline NiO probed by Raman spectroscopy. Phys. Rev. B 102, 024423 (2020).
Sunny, A. & Balasubramanian, K. Raman spectral probe on sizedependent surface optical phonon modes and magnon properties of NiO nanoparticles. J. Phys. Chem. C 124, 12636-12644 (2020).
Radinger, H. et al. Importance of nickel oxide lattice defects for efficient oxygen evolution reaction. Chem. Mater. 33, 8259-8266 (2021).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272-1276 (2011).
Stoerzinger, K. A. et al. Water reactivity on the (001) surface: an ambient pressure X-ray photoelectron spectroscopy study. J. Phys. Chem. C 118, 19733-19741 (2014).
Stoerzinger, K. A., Hong, W. T., Crumlin, E. J., Bluhm, H. & ShaoHorn, Y. Insights into electrochemical reactions from ambient pressure photoelectron spectroscopy. Acc. Chem. Res. 48, 2976-2983 (2015).
Yin, J. et al. Ni-C-N nanosheets as catalyst for hydrogen evolution reaction. J. Am. Chem. Soc. 138, 14546-14549 (2016).
Makarova, M., Okawa, Y. & Aono, M. Selective adsorption of thiol molecules at sulfur vacancies on (0001), followed by vacancy repair via S-C dissociation. J. Phys. Chem. C 116, 22411-22416 (2012).
Zhang, J. et al. Copper dopants improved the hydrogen evolution activity of earth-abundant cobalt pyrite catalysts by activating the electrocatalytically inert sulfur sites. J. Mater. Chem. A 5, 17601-17608 (2017).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537-541 (2005).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558-561 (1993).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251-14269 (1994).
Peng, L. et al. Atomic cation-vacancy engineering of NiFe-layered double hydroxides for improved activity and stability towards the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 24612-24619 (2021).
Ma, Y. et al. High-entropy energy materials: challenges and new opportunities. Energy Environ. Sci. 14, 2883-2905 (2021).
Yu, B., Jiang, H. & Zhang, Y. Superior specific capacity and energy density simultaneously achieved by Sr/In co-deposition behavior of ternary alloys as anodes for Mg-Air cells. Preprint at https://doi.org/10.1016/j.jma.2024.02.005 (2024).
Acknowledgements
The authors acknowledge funding supports from National Natural Science Foundation of China under Grant Nos. 22075232, 22275154, and 52225104. K.H.L.Z., J.P.H., and X.D. also acknowledge the Mobility Program of the Sino-German Center for Research Promotion (Grant No. M0377). R.W. is grateful for funding support by “Shuguang Program” supported by the Shanghai Education Development Foundation and Shanghai Municipal Education Commission (No. 20SGO3), and the Science and Technology Commission of Shanghai Municipality (No. 22520710600). F.E.O. thanks to MINECO and European NextGenerationEU/PRTR Fund for the Ramón y Cajal contract (RyC2O21-034254-I).
V.A.P.O. is grateful for the funding supported by the EU (HYSOLCHEM project with grant agreement No. 101017928) and Spanish AEI (SOLFuture PLEC2021-007906). Giulio Gorni acknowledges Grant FJC2O20-044866-I funded by MCIN/AEI/ 10.13039/501100011033, by Plan de recuperación, transformación y resiliencia, and by the “European Union NextGenerationEU/PRTR”. Mariam Barawi acknowledges NovaCO2 project (PID2020-118593RB-C22) and the RYC2O22-038157-I Grant funded by MCIN/AEI/ 10.13039/501100011033. M.G.-T. acknowledges PEC2Change project (TED2021-129999A-C33) funded by MCIN/AEI/ 10.13039/501100011033 and the European Union Next Generation EU/ PRTR. Additionally, the project that gave rise to these results received the support of a fellowship from “la Caixa” Foundation (ID 100010434). The fellowship code is LCF/BQ/PR23/11980046. Part of these experiments was performed at the CLAESS beamline of the ALBA synchrotron (proposal number AV-2021035069) with the help of the ALBA staff. The researchers would like to thank Iván García Dominguez and Francisco Martínez López, floor coordinators at ALBA Synchrotron, for their help with the design and 3D printing of the electrochemical cell used for in situ X-ray absorption measurements.
Author contributions
K.H.L.Z., R.W., and X.D. designed and supervised the research. X.D. and P.Z. prepared the samples. X.D. carried out electrochemical measurements and structure characterization, and analyzed the data. D.L. accomplished the DFT calculations. X.D., F.E.O., G.G., X.C., and V.A.P.O. contributed to all operando experiments and data analysis. D.L., H.W., M.B., M.G.-T., J.P.H., J.L., J.K., and S.C. participated in the discussion of the data. K.H.L.Z., R.W., and F.E.O. revised the manuscript. X.D. and D.L. wrote the manuscript. All authors contributed to the final version.
Correspondence and requests for materials should be addressed to Freddy E. Oropeza, Renbing Wu or Kelvin H. L. Zhang.
Peer review information Nature Communications thanks Yujin Chen, Alexei Kuzmin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Reprints and permissions information is available at http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
State Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. Department of Materials Science, Fudan University, Shanghai 200433, China. Photoactivated Processes Unit, IMDEA Energy Institute, Parque Tecnológico de Móstoles, Avda. Ramón de la Sagra 3, 28935 Móstoles, Madrid, Spain. Laser Processing Group, Institute of Optics (CSIC), C/Serrano 121, 28006 Madrid, Spain. CELLS-ALBASynchrotron, Carrer de la Llum 2-26, 08290 Cerdanyola del Vallès, Spain. Surface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, Otto-Berndt-Strasse 3, 64287 Darmstadt, Germany. Department of Materials Science and Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. These authors contributed equally: Xingyu Ding, Da Liu. – e-mail: freddy.oropeza@imdea.org; rbwu@fudan.edu.cn; kelvinzhang@xmu.edu.cn