DOI: https://doi.org/10.1186/s13045-024-01528-7
PMID: https://pubmed.ncbi.nlm.nih.gov/38520006
تاريخ النشر: 2024-03-22
استهداف الالتهاب كعلاج للسرطان
الملخص
لقد رافقت الالتهابات البشر منذ ظهور الجروح والعدوى. في العقود الماضية، تم بذل جهود عديدة لاستكشاف الدور المحتمل للالتهابات في السرطان، بدءًا من تطور الورم، والغزو، والنقائل، إلى مقاومة الأورام للعلاج. لا تُظهر العوامل المستهدفة للالتهابات فقط القدرة على كبح تطور السرطان، ولكن أيضًا على تحسين فعالية أساليب العلاج الأخرى. في هذه المراجعة، نصف البيئة الدقيقة المعقدة والديناميكية للغاية للورم الالتهابي، مع مناقشة الوسائط الرئيسية للالتهابات في السرطان بما في ذلك الخلايا الالتهابية، والسيتوكينات الالتهابية، ومساراتها الداخلية اللاحقة. بالإضافة إلى ذلك، نركز بشكل خاص على دور الالتهابات في تطور السرطان ونبرز آليات عمل العلاجات المستهدفة للالتهابات في الاستجابة المضادة للورم. أخيرًا، نلخص النتائج من الدراسات ما قبل السريرية والسريرية حتى الآن لتوضيح الإمكانية التحويلية للعلاجات المستهدفة للالتهابات.
الخلفية
التي توفر بشكل جماعي بيئة ميكروية معقدة للورم (TME) [8]. غالبًا ما تتميز الأورام بتسلل خلايا المناعة وزيادة تنظيم الوسائط الالتهابية المحيطة بالأورام. قد تؤثر هذه البيئة الالتهابية على تطور الورم في مراحل مختلفة، من بدء الورم إلى تقدمه. في هذه المراجعة، نناقش دور الالتهاب في تطور السرطان، مع التركيز الخاص على الأنشطة المعززة للورم الناتجة عن الالتهاب. نبرز بشكل خاص الآليات الأساسية لفعالية العلاجات المستهدفة للالتهاب في مكافحة الأورام، مع الأدلة السريرية حتى الآن المتعلقة باستراتيجيات استهداف الالتهاب.
وسائط الالتهاب في السرطان
تشير الدراسات الوبائية إلى أن التهاب القولون التقرحي ومرض كرون قد يزيدان من خطر الإصابة بسرطان القولون، وهو أحد أشهر الأمثلة على الالتهاب المرتبط بالأورام. علاوة على ذلك، تم وصف العدوى المسرطنة بواسطة عوامل ميكروبية مثل هيليكوباكتر بيلوري والتهاب الكبد B أيضًا كعوامل خطر لسرطان المعدة والكبد. خلال الالتهاب المزمن الذي تسببه العوامل الميكروبية، تنتج خلايا المناعة مثل البلعميات في مواقع الالتهاب أنواعًا تفاعلية من الأكسجين (ROS)، مما يؤدي إلى تلف مستمر في الحمض النووي وطفرات جينية لاحقة. علاوة على ذلك، يتم إفراز السيتوكينات بواسطة خلايا المناعة مثل عامل نخر الورم-
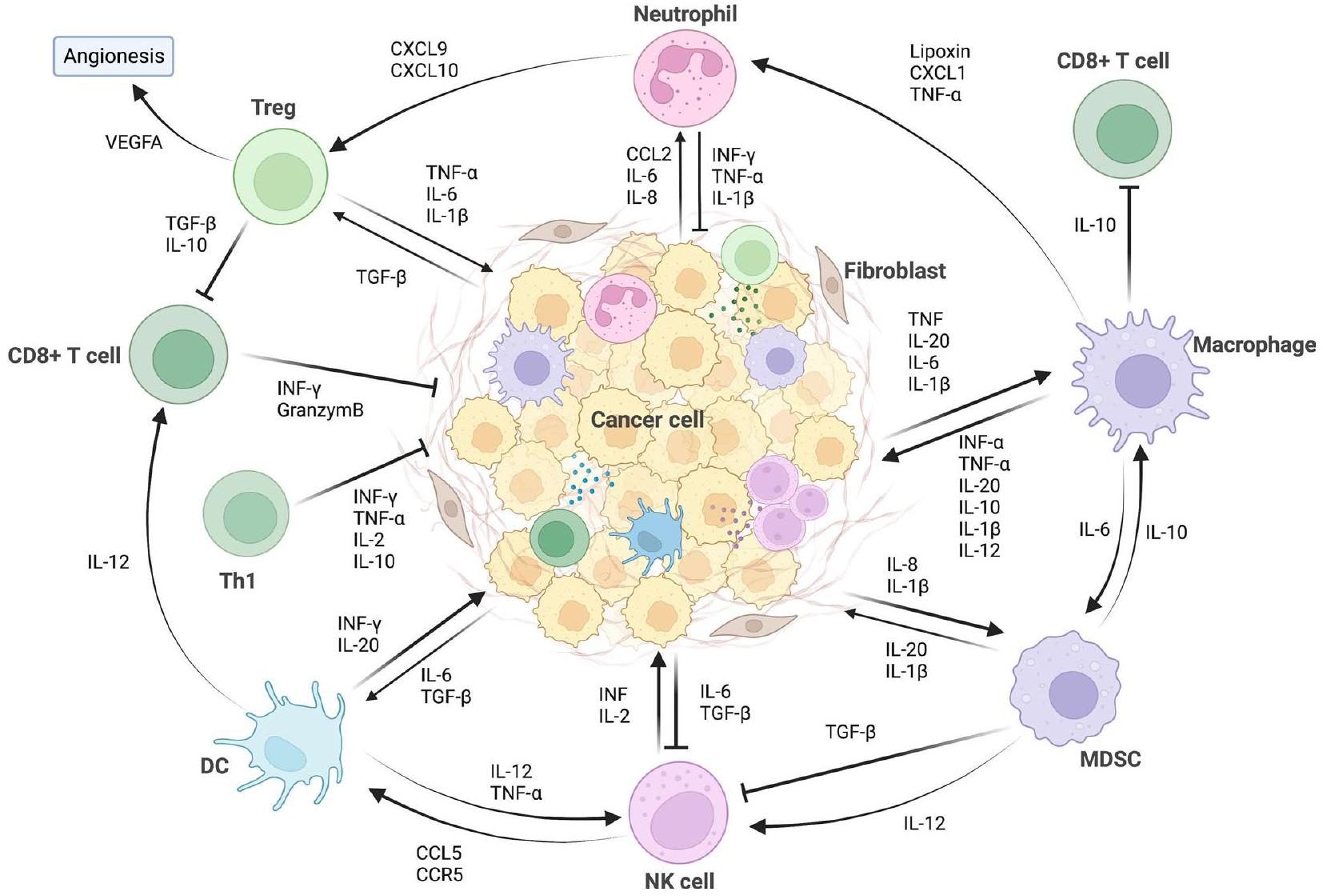
الخلايا الالتهابية الرئيسية في السرطان
مع الأدوار المضادة للأورام أو المساعدة على الأورام المعروضة في الجدول 1.
الخلايا المتعادلة المرتبطة بالورم (TANs)
| نوع الخلية | أنشطة مسببة للورم | أنشطة مضادة للورم | ||||
| العدلات المرتبطة بالورم (TANs) | ||||||
|
-N1 TANs تمارس نشاطًا مضادًا للورم، من خلال السمية الخلوية المباشرة أو غير المباشرة | |||||
| البلاعم المرتبطة بالأورام (TAMs) | ||||||
| الخلايا الشجرية (DCs) | ||||||
|
||||||
|
||||||
ومع ذلك، بناءً على الحالة المختلفة لـ TME، فإن دور NETs متغير. يمكن أن تمارس NETs أيضًا تأثيرًا مضادًا للورم من خلال قتل خلايا الورم مباشرةً ومنع نمو الورم وانتقاله. في سرطان القولون (CRC) وسرطان الخلايا الحرشفية في الرأس والعنق، يمكن أن تعيق NETs المولدة في المختبر نمو الورم من خلال تحفيز موت الخلايا المبرمج ومنع التكاثر [45، 46]. علاوة على ذلك، أدى زراعة خلايا الميلانوما مع NETs إلى نخر خلايا الميلانوما [47]. يرتبط NETosis بإفراز البروتين S100A8/A9، حيث وُجد أن النسبة المتزايدة
بينه وبين CRP ترتبط بالبقاء الجيد لمرضى سرطان المبيض عالي الدرجة (HGSOC) [48].
البلاعم المرتبطة بالورم (TAMs)
الخلايا الشجرية (DCs)
في العقود الأخيرة، حظيت وفاة الخلايا المناعية (ICD) باهتمام بحثي كبير. تُرافق ICD بإطلاق وتعرض مزمن لأنماط الجزيئات المرتبطة بالضرر (DAMPs)، مما يمنح خلايا السرطان الميتة قدرة قوية على تعزيز الاستجابة المناعية. يتطلب إنتاج الجذور الحرة للأكسجين (ROS) وإجهاد الشبكة الإندوبلازمية (ER) انبعاث DAMPs التي ترتبط بمستقبلات التعرف على الأنماط (PRRs) المعبر عنها على خلايا المناعة، وخاصة خلايا التغصن (DCs). غالبًا ما يرتبط هذا التعرف وعملية الارتباط بتوليد استجابة مناعية.
الذاكرة [76، 77]. وصفت دراسات متعددة الدور الحاسم للخلايا التغصنية في الاستجابة المناعية التي ت triggered بواسطة خلايا الورم التي تخضع لعملية الموت الخلوي المبرمج (ICD) [78]، والتي أظهرت أن الاستجابة المناعية القوية للخلايا التائية المضادة للورم التي تحفزها ICD تعتمد إلى حد كبير على الخلايا التغصنية في البيئة المجهرية للورم (TME). وبالتالي، من الممكن أن يكون التلاعب بالخلايا التغصنية في TME يحمل إمكانات كبيرة كاستراتيجيات لمكافحة السرطان. بينما تساهم ICD في نجاح العديد من العلاجات المضادة للسرطان بما في ذلك العلاج الكيميائي، والعلاج الإشعاعي، والعلاجات المستهدفة، فإن المناعية تختلف بين الخلايا ذات أنماط الموت المختلفة. اقترحت دراسة حديثة أن خلايا السرطان التي تخضع لعملية الفيروبتوز قد تعيق نضوج الخلايا التغصنية، مع قدرة ضعيفة على الابتلاع وعرض المستضدات، مما يضيف مخاوف بشأن تطبيقات العلاجات المحفزة للفيروبتوز [79].
خلايا مثبطة مشتقة من النخاع العظمي (MDSCs)
من محور VEGF/VEGFR الذي يحفز MDSCs، تعمل MMPs المؤيدة لتكوين الأوعية الدموية التي تنتجها MDSCs كإشارات ثانوية لتكوين الأوعية الدموية. MMPs هي عائلة من إنزيمات ECM التي تسهل غزو خلايا الورم، ومن بينها يُعتبر MMP9 منظمًا رئيسيًا لتكوين الأوعية الدموية للورم الذي تحفزه PMN-MDSCs.
نظرًا لأن نسبة M-MDSC العالية مرتبطة بتقليل توسع وتنشيط خلايا T المحددة للأورام، أصبحت MDSCs الآن علامة جديدة للتنبؤ باستجابة المرضى لعلاج حجب نقاط التفتيش المناعية (ICB). على سبيل المثال، المرضى الذين لديهم نسب أقل من MDSCs الدائرة يكونون أكثر حساسية لعلاج الإيبيلوماب، خاصة مرضى الميلانوما. عند حجب CTLA-4، تظهر MDSCs المتسللة إلى الورم زيادة في تعبير مستقبل عامل تحفيز المستعمرات-1 (CSF-1R)، والذي يرتبط بدوره بزيادة تسلل MDSC في الأورام. يمكن أن يُستخدم حجب إشارات CSF-1/CSF-1R ليس فقط لتقليل أعداد MDSCs، ولكن أيضًا لتحويل MDSCs المثبطة للمناعة نحو نمط ظاهري مضاد للورم. بالمثل، يمكن أن يزيد IL-10 المفرز من DCs في البيئة المجهرية للورم من عدد MDSCs المتسللة إلى الورم، مما يمنح مقاومة تكيفية لعلاج الأجسام المضادة PD-1. وبالتالي، يصبح استهداف MDSCs عبر مثبطات CSF-1/CSF-1R استراتيجية محتملة للتغلب على مقاومة الورم لـ ICBs. على الرغم من أن عددًا كبيرًا من العوامل التي تستهدف العوامل أو المستقبلات العليا لتراكم MDSC يتم اختبارها لتعزيز فعالية ICB، يجب الإشارة إلى أن الغالبية العظمى من الكيموكينات التي تجذب MDSC يمكن أن تؤثر أيضًا على خلايا مناعية أخرى ذات أنشطة مضادة للورم مثل الخلايا اللمفاوية T وخلايا NK. وبالتالي، فإن حجب هذه الكيموكينات قد يؤدي إلى تأثيرات إيجابية وسلبية على الأورام.
خلايا البطانة الوعائية
تسهل الوصلات على الكريات البيضاء الهجرة عبر جدران الأوعية الدموية.
السيتوكينات الالتهابية الرئيسية في السرطان
عامل نخر الورم ألفا (TNF-a)
| السيتوكينات الالتهابية | المصادر الرئيسية | مستقبلات | الإجراءات الرئيسية في السرطان | ||||
| TNF-α | البلاعم، الخلايا اللمفاوية التائية، خلايا NK، العدلات، الخلايا البدينة، الحمضات والعصبونات | TNF-aR-1، TNF-aR-2 |
|
||||
| TGF-
|
خلايا الورم، مصفوفة العظام | TGF-
|
|
||||
| إنترفيرون نوع 1 | الخلايا الجذعية، خلايا ب، الألياف | IFNAR1، IFNAR2 |
|
||||
| IL-1 | خلايا الورم، خلايا المناعة المثبطة المتعددة (MDSCs)، خلايا المناعة المرتبطة بالورم (TAMs)، خلايا المناعة المرتبطة بالورم (TANs)، خلايا B التنظيمية (Breg) وخلايا Th17 | IL-1R |
|
||||
| IL-6 | خلايا الورم، خلايا T، خلايا B، وحيدات النواة، الخلايا الليفية، الخلايا الكيراتينية، الخلايا البطانية، الخلايا المسنجة، الخلايا الدهنية | IL-6R | -يعزز تقدم الورم من خلال تحفيز تكاثر خلايا الورم، والبقاء، والتحول الظهاري، وتكوين الأوعية الدموية، ومقاومة العلاج الكيميائي | ||||
| IL-10 | خلايا الورم، الكريات البيضاء | IL-10R |
|
في الميلانوما، TNF-
مع التهاب المفاصل الروماتويدي [122، 123]. ومع ذلك، أشارت بعض التقارير إلى أن TNF قادر على زيادة التوسع، والاستقرار، وربما وظيفة Tregs عبر TNFR2 [124]. يتم التعبير عن TNFR2 بشكل كبير على Tregs مما يدعم تكاثرها وأنشطتها المثبطة [125]. تم تحديد TNFR2 كعلامة تعبير حيوية لمجموعة Tregs المثبطة بشكل كبير [125]. وبالتالي، فإن الأجسام المضادة المعادية لـ TNFR2 تعتبر علاجًا محتملاً للأورام. كانت مثبطات TNFR2 قادرة على استهداف TNFR2 السطحي على خلايا سرطان المبيض، مما يمنع تنشيط مسار NF-кB وتكاثر خلايا الورم [126].
عامل النمو المحول بيتا (TGF-
ونوع II (TGF-
من المثير للاهتمام، في سياق الأورام، دور TGF-
الإنترفيرونات (IFNs)
خلال الالتهاب المزمن، توفر العمليات الوقائية المرتدة التي تحفزها IFN-Is بيئة ميكروية داعمة لنمو الورم وتطوره [141، 142]. إلى جانب الإشارات المؤيدة للالتهاب لتقدم الورم، قد تسهل IFN-Is أيضًا هروب الخلايا الورمية من المناعة من خلال زيادة مسارات تثبيط المناعة التي تتراوح من استشعار الخطر إلى
إنتاج السيتوكينات [143، 144]. على سبيل المثال، في سرطان الخلايا الحرشفية في الرأس والعنق (HNSCC)، يؤدي تنشيط IFN-I الخاص بالسرطان إلى تقليل توسع ووظائف خلايا CD8 +T الفعالة ويرتبط بنتائج سريرية سيئة [145].
إنترلوكين-1
من IL-1
إنترلوكين-6
إنترلوكين-10
من قبل الكريات البيضاء، فضلاً عن خلايا الورم البشرية. تتكون هذه العائلة من السيتوكينات من IL-10 وسيتوكينات تحت عائلة IL-20 بما في ذلك IL-19 وIL-20 وIL-22 وIL-24 وIL-26 [181]. يقوم IL-10 بتثبيط الاستجابات الالتهابية غير المنضبطة، مما يحافظ على التوازن [182]. في أورام مثل سرطان المعدة، يساهم IL-10 المنتج من TAM في بيئة ميكروية مثبطة للمناعة تفضل نمو الورم [183]. أظهرت دراسة أكثر حداثة أن التعبير عن IL-10 في خلايا T التنظيمية المتسللة إلى الورم قد يؤدي إلى استنفاد خلايا CD8 + T داخل الورم [184]. من ناحية أخرى، اقترحت بعض الدراسات أن IL-10 يمكن استخدامه كعلاج مناعي في نماذج الأورام [185]. يمكن أن يحفز IL-10 التعبير عن جزيئات CD3 وCD8 على الخلايا التوتية وبالتالي يعزز النشاط السام لخلايا CD8 + T [186]. آلية أخرى للعمل المضاد للورم لـ IL-10 هي زيادة تسلل خلايا CD8 + T ومستوى IFN-
ROS
المؤثرات الخارجية مثل العلاج الكيميائي والعلاج الإشعاعي والأشعة فوق البنفسجية أيضًا إلى تحفيز إنتاج ROS [194].
تحمل خلايا السرطان كميات أكبر من أنواع الأكسجين التفاعلية (ROS) مقارنةً بنظيراتها الطبيعية، وذلك بسبب تنشيط الجينات المسرطنة بشكل غير طبيعي ونشاط الميتوكوندريا. دور ROS في تطور السرطان معقد، مما يجعله سلاحًا ذو حدين. من ناحية، قد يتسبب الضغط المستمر لـ ROS في إلحاق الضرر بالهياكل الخلوية، ويعيق وظائفها البيولوجية، ويسبب الطفرات، مما يزيد بشكل جماعي من مخاطر حدوث السرطان. من ناحية أخرى، قد تتراكم ROS عند التعرض لمحفزات خارجية مثل العلاج الكيميائي والعلاج الإشعاعي، مما يؤدي إلى موت خلايا الورم وبالتالي زيادة حساسية خلايا الورم للعلاجات. سيساعد توضيح الأدوار المعقدة لـ ROS في السرطان في تصميم علاجات تستهدف ROS للسرطان. تشير الدراسات الحديثة إلى أن البيئة منخفضة الأكسجين في الأورام قد تنشط إنتاج ROS. استجابةً لنقص الأكسجين، يعتبر عامل الاستجابة لنقص الأكسجين-1 (HIF-1) من المحفزات النسخية المعروفة التي تنظم توازن الأكسجين. من خلال التفاعل مع عناصر استجابة نقص الأكسجين للجينات المستهدفة، يعزز ROS تنشيط HIF-1.
المسارات الالتهابية الرئيسية في السرطان
إشارات الإيكوسانويد
انتشار خلايا سرطان الرئة، ولكن في نفس الوقت يعزز غزوها وهجرتها [230]، مما يؤدي إلى فرضية أن الدور الدقيق لـ PGD2 في السرطان قد يختلف وفقًا لمرحلة الورم.
قد تكون شريكًا جيدًا محتملًا للعلاج المناعي، مثل مثبطات نقاط التفتيش (الشكل 2).
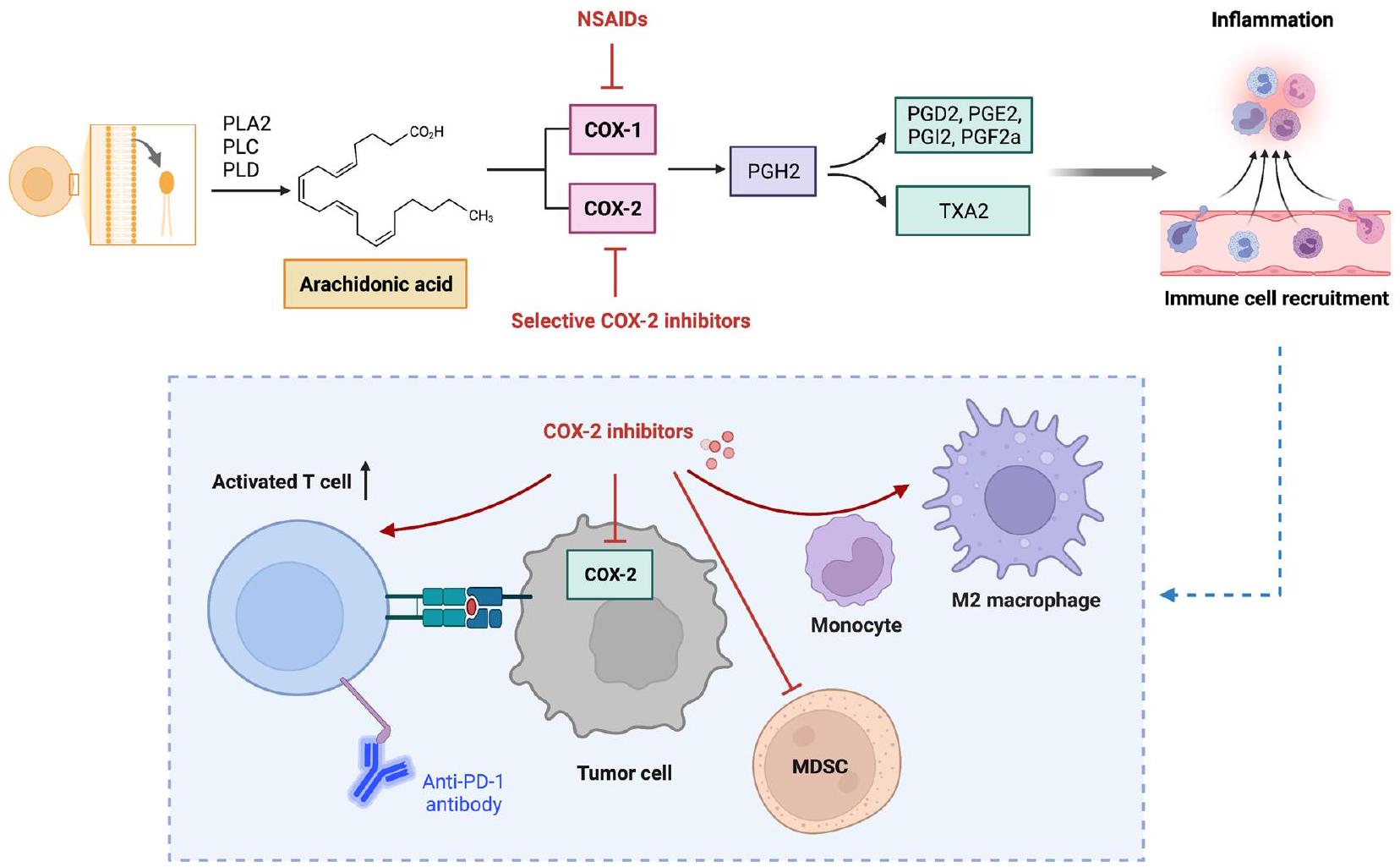
على العكس، يتميز نوع آخر من الإيكوسانويدات المشتقة من LOX، وهو الليبوكسينات (LXs)، بأنها مضادة للورم [250]. تحفز الليبوكسينات وحيدات النوى دون التسبب في إطلاق ROS الالتهابي [251]. قد تعزز الليبوكسينات أيضًا البلعمة للخلايا المتعادلة الميتة بواسطة البلعميات، مما يقلل الالتهاب [252]. تشير الأدلة المتزايدة إلى التأثير المضاد للالتهابات لليبوكسين.
إشارات JAK-STAT
فوسفوريلاسيون JAKs التي تشكل بعد ذلك موقع ربط لـ STATs مما يؤدي إلى فوسفوريلاسيون STAT. كعضو أساسي في عائلة بروتينات STAT، يلعب STAT3 أدوارًا متعددة في الاستجابة الالتهابية وتقدم الورم. تتورط عوامل النمو المتعددة والسيتوكينات في مسارات STAT3 التقليدية، حيث تنظم نسخ جينات STAT3 المستهدفة والعمليات الخلوية اللاحقة مثل تمايز الخلايا، وتكوين الأوعية، وتكون الأورام. لقد تم ربط تنظيم إشارة STAT3 بشكل غير طبيعي بسلسلة من الأمراض الالتهابية مثل التهاب المفاصل الروماتويدي، والتصلب المتعدد، ومرض الأمعاء الالتهابي. علاوة على ذلك، قد تؤدي التنشيط المستمر لإشارة STAT3 إلى تكون الأورام في كل من الأورام الصلبة والأورام الدموية.
أيض المعادن
الذي ينتج الحديد الثلاثي مباشرةً (
الزنك هو العنصر الغذائي الأساسي الثاني الأكثر وفرة في جسم الإنسان، والذي تم توثيقه لأول مرة في الستينيات فيما يتعلق بدوره في صحة الإنسان. يرتبط الزنك بإنتاج وإشارات العديد من السيتوكينات الالتهابية، وعند الاستجابة الحادة لمحفزات الإجهاد، تنخفض تركيزات الزنك في البلازما بسرعة. يرتبط أيض الزنك في البشر ارتباطًا وثيقًا بأنشطة ناقلات الزنك مثل ZIP8. خلال الالتهاب، يزيد NF-кB المنشط من تعبير ZIP8 الذي يتوضع على غشاء الخلية وينظم امتصاص الزنك. بعد دخول الزنك إلى السيتوسول، يقوم الزنك بقمع IKK.
العلاجات المستهدفة للالتهابات في السرطان
مضادات الالتهاب غير الستيرويدية (NSAIDs)
| عميل/هدف | نوع الورم | نظام التركيب | التجربة السريرية الرئيسية | الإجراء المبلغ عنه |
| سيلوكوكسيب | ||||
| كوكس-2 | سرطان الثدي | سليكوكسيد نيوأدجوانت + العلاج الكيميائي / كوليكالسيفيرول / إكسمستين | NCT02429427، NCT01041781 | أدى السيلوكوكسيب إلى تغييرات إيجابية في مؤشرات الدم وعلم الخلايا لدى النساء المعرضات لزيادة خطر الإصابة بسرطان الثدي، لكنه لم يظهر أي فوائد كبيرة للمرضى الذين يعانون من سرطان الثدي السلبي لـ ERBB2. |
| سرطان الرئة | سيلوكوكسيب + العلاج الكيميائي/العلاج الإشعاعي/مثبطات EGFR TKIs | NCT00300729، NCT01503385 | سيليكوكسيب بجرعة قصوى متسامحة من
|
|
| CRC | سيلوكوكسيب + سيتوكسيماب/العلاج الكيميائي (نظام فولي فولي/RT/ | NCT03645187، NCT00005094، NCT00141193، NCT03926338، NCT01150045 | يعتبر الجمع بين سيليكوكسيب والعلاج الكيميائي (نظام FOLFIRI الذي يتكون من 5-فلورويوراسيل، ليوكوفورين، إيرينوتيكان) أو حجب PD-1 توريباليماب بروتوكولًا تآزريًا فعالًا وآمنًا للمرضى الذين يعانون من سرطان القولون المستعصي. | |
| العلاجات المضادة للفيروسات | ||||
| إنتيكافير | ||||
| فيروس التهاب الكبد B | HCC | NCT00388674 | أدى إنتيكافير إلى تقليل خطر الأحداث المرتبطة بفيروس التهاب الكبد B بما في ذلك سرطان الكبد. | |
| تينوفوفير | ||||
| فيروس التهاب الكبد B | HCC | NCT019553458 | أدى التينوفوفير إلى خطر طويل الأمد قابل للمقارنة لسرطان الكبد الأولي وسرطان القنوات الصفراوية في مرضى التهاب الكبد B المزمن الذين يتلقون الإنتيكافير. | |
| لقاح HPV-16 ISA 101 | ||||
| فيروس الورم الحليمي البشري | سرطان عنق الرحم | ISA 101 + جسم مضاد مضاد PD-1 نيفولوماب | NCT02426892 | العلاج المتزامن لـ ISA 101 والأجسام المضادة المضادة لـ PD-1 نيفولوماب زاد من معدلات الاستجابة العامة والبقاء على قيد الحياة في سرطان مرتبط بـ HPV-16 |
| علاجات موجهة بواسطة السيتوكينات | ||||
| إنترفيرون ألفا | RCC | IFN-ألفا + أوبليمرسين/(إيزو)تريتينوين/إيزوتريتينوين/IL-2/العلاج الكيميائي (فلورويوراسيل، كابيسيتابين)/سورافينيب/مثبط VEGF (بيفاسيزوماب، SU5416)/مثبط mTOR (CCI-779)/نابتوموماب إستييفاناتوكس/بازوبانيب/سيلوكوكسيب/ثاليدوميد/العلاج الكيميائي (5-فلورويوراسيل)/بمبروليزوماب | UMIN000002466، CALGB 90206 | علاج IFN-a المطول أدى إلى استجابات كاملة طويلة الأمد ونتائج طويلة الأمد مع سمية مقبولة لدى المرضى الذين يعانون من سرطان الكلى النقيلي. كما أن IFN-a هو علاج تركيبي واعد للعلاجات المستهدفة ومثبطات نقاط التفتيش المناعية مثل علاجات anti-PD-1. |
| الميلانوما | IFN-أ + العلاج الكيميائي المركب (داكارباين، تيموزولوميد، أزاكيتيدين، سيسبلاتين) / IL-12 / ثاليدوميد / بيفاسيزوماب / إيماتينيب / مثبط BRAF (فيمورافينيب) / مثبط CTLA-4 إيبيلوماب / مثبط البروتيازوم (PS-341) / ستبوجلوكونات الصوديوم | NCT00204529، NCT01959633، EORTC 18991، S0008 | يمكن أن يؤدي العلاج المساعد باستخدام IFN-a-2a أو PEG-IFN-a-2b إلى تحسين مستدام في فترة البقاء خالية من المرض (RFS) لدى مرضى الميلانوما من المرحلة الثالثة وقد تم الموافقة عليه من قبل إدارة الغذاء والدواء الأمريكية كعلاج مساعد للميلانوما. | |
| عميل/هدف | نوع الورم | نظام التركيب | التجربة السريرية الرئيسية | الإجراء المبلغ عنه |
| لوكيميا | IFNa-2a + العلاج الكيميائي المركب (ميلفالن، أدرياميسين، بليوميسين، فيلبان، وداكاربازين)/نيلوتينيب/إيماتينيب/ريتوكسيماب/داساتينيب | NCT02328755، NCT02185261 | علاج IFN-α هو استراتيجية فعالة لمرضى اللوكيميا الإيجابيين لمرض الخلايا المتبقية الدنيا (MRD) الذين يتلقون زراعة خلايا جذعية دموية من متبرع (allo-HSCT) | |
| لمفوما | IFN-ألفا + العلاج الكيميائي المركب (ميلفالن، أدرياميسين، بليوميسين، فيلبان، وداكارباين)/بيكساروتين/ريتوكسيماب | NCT01609010 | العلاج المناعي باستخدام IFN-α و rIL-2 يتم تحمله بشكل جيد وقد يعزز الشفاء في مرضى NHL | |
| HCC | إنترفيرون ألفا + العلاج الكيميائي (كابيسيتابين) / سيليكوكسيب + رينتاتوليمود / ثاليدوميد | قد تقلل علاج IFN-a من تكرار سرطان الكبد بعد العلاج بالتبخير الطبي للأورام الأولية. يعتبر IFN-a مع سيسبلاتين فعالاً في المرضى الذين يعانون من سرطان الكبد غير القابل للجراحة. | ||
| غالونيسيرتيب (LY2157299) | ||||
| TGF-
|
سرطان البنكرياس | غالونيسيرتيب + دورفالماب/جمسيتابين | NCT02734160 | تحسنت فترة البقاء على قيد الحياة لدى المرضى الذين يعانون من سرطان البنكرياس غير القابل للجراحة عند استخدام تركيبة الغالونيسيرتيب والجمسيتابين مع سمية إضافية طفيفة. |
| HCC | غالونيسيرتيب + سورافينيب/العلاج الإشعاعي المجسم (SBRT) | NCT01246986 | أظهر الجمع بين جالونيسيرتيب وسورافينيب ملف أمان يمكن التحكم فيه وتحسين توقعات سرطان الكبد. | |
| فريزوليموماب (GC1008) | ||||
| TGF-
|
الميلانوما، سرطان الخلايا الكلوية | NCT00356460 | أظهر فريزوليموماب فعالية مضادة للأورام أولية وملف أمان مقبول عند جرعات متعددة في مرضى الميلانوما المتقدمة وسرطان الكلى RCC. | |
| PF-03446962 | ||||
| TGF-
|
سرطان الكبد، سرطان القولون | ريغورافينيب + PF-03446962 | NCT00557856 | كان لدى PF-03446962 ملفات تعريف أمان ودوائية قابلة للإدارة في سرطان الكبد الخلوي، ولكن الجمع بين ريجورافينيب وPF-03446962 تسبب في سمية غير مقبولة مع نشاط سريري محدود لدى المرضى الذين يعانون من سرطان القولون المستعصي النقيلي. |
| بينترافوسب ألفا (M7824) | ||||
| TGF-
|
سرطان الرئة غير صغير الخلايا | بينترافوسب ألفا + العلاج الكيميائي (دوستكسل، قائم على البلاتين) | NCT02517398 | أظهر بينترافوسب ألفا فعالية واعدة وقابلية تحمل قابلة للإدارة لدى المرضى المصابين بسرطان الرئة غير صغير الخلايا الذين تم علاجهم سابقًا بالبلاتين. |
| سرطان مرتبط بفيروس الورم الحليمي البشري | NCT02517398، NCT02517398، NCT04247282 | أظهر بينترافوسب ألفا نشاطًا سريريًا وسلامة قابلة للإدارة في السرطانات المرتبطة بفيروس الورم الحليمي البشري. | ||
| عميل/هدف | نوع الورم | نظام التركيب | التجربة السريرية الرئيسية | الإجراء المبلغ عنه |
| سرطان المريء | NCT02517398، NCT02699515 | أظهر بينترافوسب ألفا نشاطًا سريريًا مع ملف أمان يمكن التحكم فيه لدى المرضى الذين يعانون من سرطان المريء الغدي المتقدم. | ||
| أنكينرا | ||||
| IL-1 | الورم النقوي المتعدد | أنكينرا + تركيبة الأدوية المناعية ليناليدوميد وديكساميثازون | NCT00635154 | أنكينرا قلل من معدلات تكاثر الورم، مما أدى إلى حالة مرضية مزمنة مع تحسين فترة البقاء بدون تقدم في المرض لدى المرضى الذين يعانون من المايلوما المتعددة والذين هم في خطر مرتفع للتقدم إلى المايلوما النشطة. |
| CRC | أنكينرا + 5-FU + بيفاسيزوماب | كان لدواء 5-FU بالإضافة إلى بيفاسيزوماب وأنكينرا نشاط واعد وملف أمان قابل للإدارة في سرطان القولون المستعصي النقيلي. | ||
| بمبيغالديزليوكين (NKTR-214) | ||||
| IL-2 | الميلانوما | بمبيغالديزليوكين + نيفولوماب/بمبروليزوماب | NCT03635983، PIVOT-02 | يمكن استخدام بيمبيغالديزليوكين بالاشتراك مع نيفولوماب أو بيمبروليزوماب في المرضى الذين يعانون من الميلانوما النقيلي. |
| سرطان الظهارة البولية | بمبيغالدي سليوكين + نيفولوماب | NCT02983045، PIVOT-02 | يُقترح استخدام بيمبيغالديزليوكين مع نيفولوماب كعلاج خط أول للمرضى الذين يعانون من سرطان الظهارة البولية النقيلي مع آثار جانبية قابلة للإدارة. | |
| نيمفاليكين ألفا (LKS 4230) | ||||
| IL-2 | سرطان المبيض | نيمفاليوكين ألفا + بيمبروليزوماب | NCT05092360 | تحت التقييم من حيث الفعالية والسلامة كعلاج أحادي وعلاج مركب مع بيمبروليزوماب في المرضى الذين يعانون من سرطان المبيض المقاوم للبلاتين |
| CNTO 328 | ||||
| IL-6 | الورم النقوي المتعدد | سيلتوكسيماب + بورتزوميب-ميلفالن-بريدنيزون (VMP) | NCT00911859 | لم يُحسن إضافة السيلتوكسيماب إلى نظام البورتزوميب-ميلفالن-بريدنيزون (VMP) معدل الاستجابة الكاملة أو النتائج طويلة الأمد لمرضى الورم النقوي المتعدد. |
| سرطان البروستاتا | سيلتوكسيماب + ميتوكسنترون/بريدنيزون | SWOG S0354 | كان سيلتوكسيماب مقبولاً بشكل جيد وحسن النتائج السريرية، مما أدى إلى معدل استجابة لمستضد البروستاتا النوعي بنسبة 3.8% ومعدل مرض مستقر بنسبة 23% لدى المرضى الذين يعانون من سرطان البروستاتا المقاوم للإخصاء. | |
| توسيليزوماب | ||||
| IL-6R | سرطان المبيض | توسيليزوماب + كاربوبلاتين/دوكسوروبيسين | NCT01637532 | توسيليزوماب في
|
| بيجيلوديكين (LY3500518) | ||||
| عميل/هدف | نوع الورم | نظام التركيب | التجربة السريرية الرئيسية | الإجراء المبلغ عنه |
| IL-10 | الأورام الصلبة | بيغيلوديكين + العلاجات الكيميائية أو حجب PD-1 | NCT02009449 | تم استخدام بيغيلوديكين كعلاج أحادي وفي تركيبة مع العلاجات الكيميائية أو حجب PD-1 لعلاج الأورام مثل الميلانوما، وسرطان الرئة غير صغير الخلايا، وسرطان القولون والمستقيم، وسرطان البنكرياس. |
| علاجات موجهة بواسطة الكيموكينات كارلوماب | ||||
| CCL2 | سرطان البروستاتا | يمكن إعطاء كارلوماب بأمان للمرضى الذين يعانون من سرطان البروستاتا المقاوم للعلاج المتقدم، لكنه فشل في إظهار أنشطة مضادة للورم ذات دلالة إحصائية كعامل وحيد. | ||
| PF-04136309 CCR2 | ||||
| سرطان البنكرياس | PF-04136309 + العلاج الكيميائي (جمسيتابين بالإضافة إلى ناب-باكليتاكسيل) | NCT02732938 | قد يؤدي PF-04136309 بالاشتراك مع ناب-باكليتاكسيل وجيمسيتابين إلى سمية رئوية، دون وجود إشارة فعالية متفوقة ملحوظة مقارنةً بناب-باكليتاكسيل وجيمسيتابين. | |
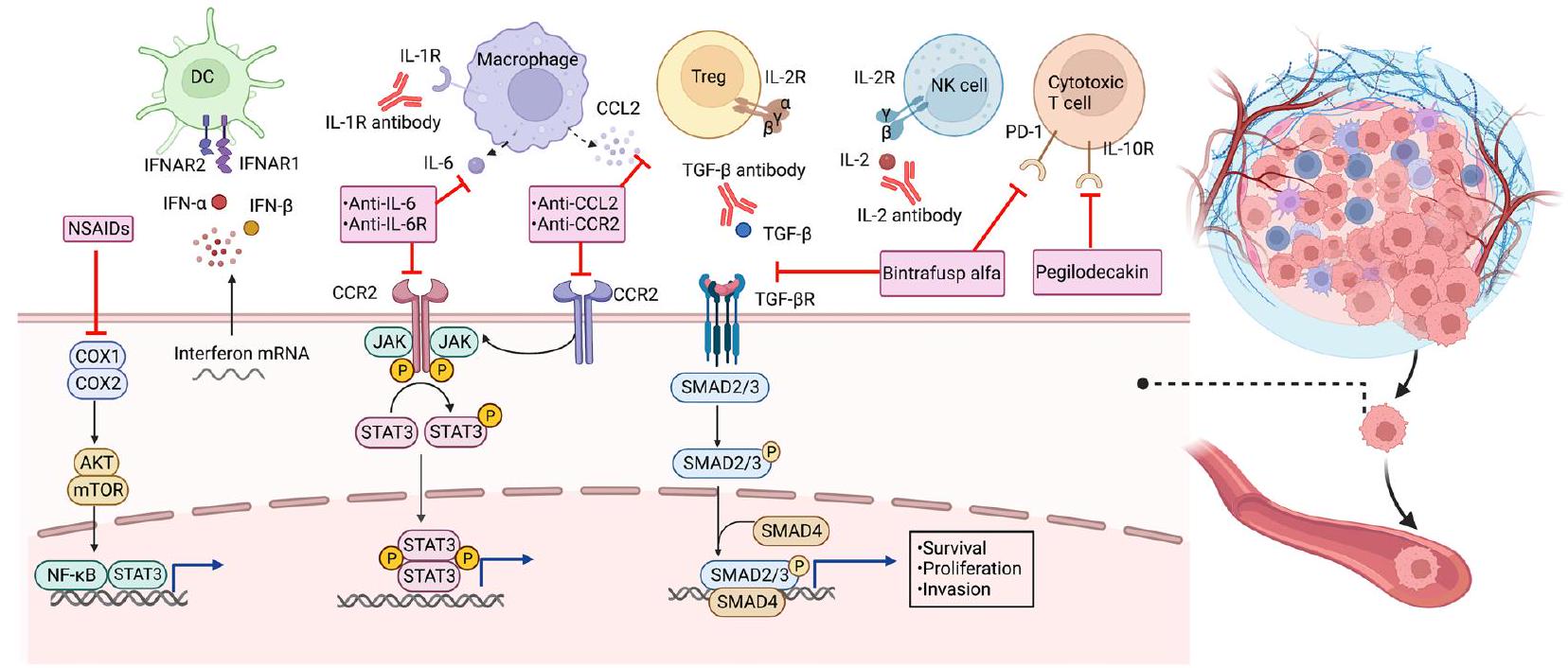
ومع ذلك، أظهرت تجربة سريرية حديثة أن إضافة السيلوكوكسيب إلى نظام العلاج الكيميائي المساعد القياسي لم تحقق فوائد إضافية للمرضى الذين يعانون من سرطان القولون في المرحلة الثالثة (NCT01150045) [299]. وقد قيمت دراسة أخرى فعالية السيلوكوكسيب كعلاج مشترك.
شريك للعلاج التقليدي في سرطان الثدي السلبي لـ ERBB2، والذي أظهر عدم وجود فوائد كبيرة من السيلوكوكسيب من حيث فترة البقاء خالية من المرض بعد علاج لمدة عامين (NCT02429427) [300]. علاوة على ذلك، اقترحت بعض الدراسات أن إضافة السيلوكوكسيب إلى العلاج الكيميائي قد تؤثر سلبًا على تشخيص مرضى سرطان الثدي، خاصة أولئك الذين لديهم أورام منخفضة التعبير عن إنزيم سينثاز البروستاجلاندين-إنهيدروكسي (PTGS2) (NCT01041781) [301]. من المحتمل أن تعكس هذه النتائج المتضاربة تأثير أنظمة العلاج المختلفة أو جرعات السيلوكوكسيب، وملف التعبير عن العلامات الحيوية في الأورام. لذلك، يجب أخذ جميع العوامل المذكورة أعلاه في الاعتبار للتحقيق في الإمكانات العلاجية للسيلوكوكسيب. بالإضافة إلى ذلك، قد يؤدي الاستخدام طويل الأمد لمضادات الالتهاب غير الستيرويدية بما في ذلك COXIBs بجرعات عالية إلى آثار جانبية قلبية وعائية شديدة لدى المرضى، خاصة أولئك الذين لديهم تاريخ من مرض القلب التصلبي [302]. إحدى الطرق لمنع أو تقليل هذه الآثار الجانبية ستكون من خلال استهداف مسار PGE2 السفلي بشكل بديل. قدم بعض الباحثين مركبات طبيعية معروفة بأنشطة مثبطة على COX-2، مثل الفينولات الطبيعية، الفلافونويدات، الستيلبينات، التربينويدات، الكينونات، والقلويدات [303].
العلاجات المضادة للفيروسات
علاجات فيروس التهاب الكبد B (HBV)
(التهاب الكبد المزمن) العدوى، قد تؤدي الاستجابة المناعية للعدوى المستمرة إلى التهاب مزمن وتكوين ألياف كبدية، مما يؤدي إلى تلف لا يمكن عكسه في بنية الكبد. قد تتسبب النسخ المستمر لحمض نووي الفيروس واندماجه في الجينوم المضيف في تغييرات جينية، مما يدفع في النهاية إلى تكوين السرطان في خلايا الكبد [120]. من ناحية أخرى، قد تزيد بروتينات الفيروس مثل بروتين X لفيروس التهاب الكبد B من حساسية المضيف للمواد المسرطنة الكيميائية [304]. لقد حفزت هذه الدراسات ما قبل السريرية تصميم العلاجات المضادة للفيروسات في علاج سرطان الكبد الخلوي المرتبط بفيروس التهاب الكبد B.
تهدف العلاجات المضادة للفيروسات إلى قمع نسخ حمض نووي فيروس التهاب الكبد B، وتعزيز تحويل مصل مستضد التهاب الكبد B (HBeAg)، وتخفيف تطور تليف الكبد. تشمل الأدوية المضادة للفيروسات الشائعة نظائر النوكليوزيد والنوكليوتيد (NAs) والإنترفيرونات (IFNs). من بينها، تم التوصية بالإدارة طويلة الأمد لنظائر النوكليوزيد القوية ذات الحواجز العالية ضد المقاومة مثل إنتيكافير وتينوفوفير ديسوبروكسيل كأدوية خط أول مضادة لفيروس التهاب الكبد B في توافق إدارة الحالات السريرية لالتهاب الكبد المزمن [305]. في تجربة عشوائية محكومة شملت 299 مركزًا في آسيا وأوروبا وأمريكا الشمالية والجنوبية مع متابعة لمدة 10 سنوات، كان لدى المرضى الذين عولجوا بإنتيكافير خطر منخفض من الأحداث المرتبطة بفيروس التهاب الكبد B بما في ذلك سرطان الكبد (NCT00388674) [306]. اقترحت دراسة قائمة على السكان على مستوى البلاد حول مرضى التهاب الكبد المزمن أن علاج تينوفوفير كان له معدل حدوث أقل لسرطان الكبد مقارنة بعلاج إنتيكافير [307]. تم تأكيد تفوق تينوفوفير على إنتيكافير في تقليل معدل حدوث سرطان الكبد في مرضى التهاب الكبد المزمن في عدة دراسات أخرى [303، 308]. ومع ذلك، فشلت بعض الدراسات في تحديد فرق ذي دلالة سريرية في خطر الأحداث المرتبطة بالكبد أو الوفيات بما في ذلك سرطان الكبد بين المجموعات المعالجة بإنتيكافير وتينوفوفير، مما يشير إلى أن الاختيار بين تينوفوفير أو إنتيكافير يجب أن يستند إلى تحمل المرضى (NCT019553458) [309، 310]. اقترحت دراسة حديثة مقارنة الخطر طويل الأمد لتينوفوفير مقابل إنتيكافير على سرطان الكبد وسرطان القناة الصفراوية داخل الكبد (ICC) في مرضى التهاب الكبد المزمن ووجدت خطرًا طويل الأمد قابلًا للمقارنة بين هذين العاملين [311]. مؤخرًا، تم إدخال بعض الأعشاب الصينية المضادة للتليف في صيغ العلاج المضاد للفيروسات لعلاج تليف الكبد المرتبط بالتهاب الكبد المزمن. على سبيل المثال، يتم حاليًا التحقيق في الإمكانات العلاجية لإنتيكافير المدمج مع حبيبات روانغان لعكس تليف الكبد المتقدم في عدد من الدراسات السريرية [312، 313].
علاجات مضادة لفيروس الورم الحليمي البشري (HPV)
تبدأ العدوى المستمرة بفيروس الورم الحليمي البشري سلسلة من التفاعلات التي تنظم إفراز السيتوكينات الالتهابية وتسلل خلايا المناعة [317]. على سبيل المثال، لوحظ ارتفاع مستمر في مستويات السيتوكينات الالتهابية الجهازية في الفئات العمرية الأكبر سناً مع عدوى فيروس الورم الحليمي البشري المزمنة [318]، مما زاد من خطر الإصابة بسرطان عنق الرحم في هذه الفئة العمرية [319، 320].
علاجات موجهة للسيتوكينات والكيموكينات
علاجات موجهة لـ IFN-α
تجارب سريرية [327-330]. تم استخدام IFN-
[344]. وبالمثل، أدى استخدام بيفاسيزوماب مع IFN إلى فوائد متفوقة من حيث فترة البقاء بدون تقدم المرض (PFS) ومعدل الاستجابة الكلي (ORR) لدى المرضى الذين يعانون من سرطان الكلى النقيلي (RCC) مقارنةً بالعلاج الأحادي بـ IFN (CALGB 90206) [345]. وقد ركزت الأبحاث الحديثة على إمكانيات IFN-
تي جي إف-
الفعالية وملف الأمان المقبول عند جرعات متعددة [360]. بالنسبة للمرضى الذين يعانون من الميلانوما الخبيثة المتقدمة وRCC، كان الفريزوليموماب آمناً وأظهر فعالية أولية مضادة للورم (NCT00356460) [360]. دراسة حديثة فحصت فعالية وتأثيرات المناعة للفريزوليموماب في مرضى سرطان الثدي النقيلي خلال علاج الإشعاع، حيث لوحظ استجابة مناعية نظامية إيجابية. ومن الجدير بالذكر أن الفريزوليموماب حسّن من البقاء على قيد الحياة (OS) للمرضى بطريقة تعتمد على الجرعة، مع ملاحظة بقاء متوسط أطول لدى أولئك الذين تم علاجهم بجرعات أعلى [361].
غالونيسيرتيب هو TGF-
PF-03446962 هو جسم مضاد وحيد النسيلة (mAb) يستهدف كيناز مستقبلات الأكتيفين الشبيه-1 (ALK1)، وهو عامل نمو مرتبط بـ TGF-
مستقبل ومضاد جسم مناعي IgG1 يحجب PD-L1 [373].
العلاجات الموجهة نحو IL-1
علاج عدة أنواع من السرطان [387-390]. أظهرت الدراسات ما قبل السريرية أن الجيمسيتابين و5-فلورويوراسيل (5-FU) يمكن أن يعززا IL-1
العلاجات الموجهة نحو IL-2
لقد حفز التعبير التفاضلي لمستقبلات IL-2 تصميم منبهات IL-2R التي تنشط بشكل انتقائي IL-
(TNBC) [403]. عدد من التجارب السريرية جارية لتقييم سلامة وفوائد bempegaldesleukin السريرية عند دمجه مع pembrolizumab في المرضى الذين يعانون من الميلانوما النقيلي (NCT03635983) [404]. يُقترح أيضًا استخدام bempegaldesleukin بالاشتراك مع nivolumab كعلاج خط أول للمرضى الذين يعانون من سرطان الخلايا الانتقالية النقيلي (NCT02983045) أو الميلانوما النقيلي (PIVOT-02)، مع آثار جانبية قابلة للإدارة [405، 406]. Nemvaleukin alfa (nemvaleukin، ALKS 4230) هو شكل مهندَس جديد من IL-2 يرتبط بشكل انتقائي بمستقبل IL-2R على خلايا CD8 + T المضادة للورم وخلايا NK مع تأثير ضئيل على Tregs المثبطة للمناعة [325]. في نموذج فئري جديد لسرطان الرئة صغير الخلايا (SCLC)، أظهر النسخة الفئران من nemvaleukin (mNemvaleukin) تثبيطًا كبيرًا لنمو ورم SCLC الفئري وحسن من بقاء الفئران، مما يدعم تقييم nemvaleukin بمفرده أو بالاشتراك مع العلاج الكيميائي في التجارب السريرية [407]. التجارب السريرية الجارية مثل تجربة ARTISTRY-7 قارنت فعالية وسلامة nemvaleukin كعلاج أحادي وعلاج مشترك مع pembrolizumab في المرضى الذين يعانون من سرطان المبيض المقاوم للبلاatinum (NCT05092360) [408-410].
العلاجات الموجهة نحو IL-6
نظرًا لارتفاع مستويات IL-6 النظامية الناتجة عن الأجسام المضادة الأحادية النسيلة المضادة لـ IL-6 [425]، تم تطوير بعض العلاجات البديلة الموجهة لـ IL-6 مثل الحجب الوظيفي لمستقبلات IL-6 (IL-6R). يتم إعطاء مثبط IL-6R توcilizumab عند
العلاجات الموجهة نحو IL-10
العلاجات الموجهة نحو محور CCL2/CCR2
نظرًا للفعالية السريرية غير المثلى لمثبطات CCR2 كعلاج أحادي، تم تقييم الإمكانات العلاجية لمثبطات CCR2 للعمل بالتآزر مع العلاجات الكيميائية ومثبطات نقاط التفتيش المناعية. PF-04136309 هو مثبط CCR2 جزيئي صغير تم دراسته بشكل رئيسي في سياق سرطان البنكرياس. في تجربة المرحلة الأولى، كان استهداف TAMs باستخدام مجموعة PF-04136309-FOLFIRINOX آمنًا وقابلًا للتحمل لدى المرضى الذين يعانون من سرطان البنكرياس القابل لإعادة الاستئصال الحدودي والمتقدم محليًا. للأسف، أدى دمج PF-04136309 مع ناب-باكليتاكسيل وجيمسيتابين إلى سمية رئوية تآزرية، دون تفوق من حيث الفعالية لدى مرضى PDAC. CCR2i هو مثبط تنافسي يرتبط بشكل انتقائي وبقوة عالية بحجرة ارتباط CCR2، وعند دمجه مع مثبط نقطة تفتيش مناعية، يمكن أن يثبط نمو الأورام اللمفاوية T-cell الجلدية. تم استخدام BMS-687681، وهو مثبط مزدوج يستهدف CCR2 وCCR5، كعلاج مطول بعد
علاجات طبيعية مضادة للالتهابات
زيادة مستوى البلعميات المضادة للالتهابات (M2). يقوم الريسفيراترول بإعاقة تنشيط البلعميات الناتج عن LPS من خلال تثبيط إشارات NF-kB و COX-2 وتنشيط الانفلامازوم [459]. في دراسة سريرية، أدى الاستهلاك اليومي للريسفيراترول إلى تأثير مضاد للورم كبير في 20 مريضًا بسرطان القولون، مما يشير إلى إمكانية استخدام الريسفيراترول كدواء للوقاية الكيميائية من السرطان.
الاستنتاجات وآفاق المستقبل
حتى الآن، تم تطوير مجموعة واسعة من العلاجات الموجهة للالتهابات وهي قيد التقييم سواء في المرحلة ما قبل السريرية أو السريرية في نماذج السرطان. مع التقدم الموضح هنا، أثبتت بعض الأساليب المضادة للالتهابات فعاليتها في الوقاية من السرطان وعلاجه، مما يوفر مبررات علمية قوية لمزيد من تطوير مثل هذه الاستراتيجيات. علاوة على ذلك، فإن بعض الاستجابات الالتهابية بعد علاجات السرطان قد تمنح خلايا السرطان المتبقية مقاومة للعلاجات اللاحقة. تؤدي العلاجات المناعية إلى استجابات دائمة فقط في مجموعة صغيرة من المرضى، حيث يعاني معظم المرضى في النهاية من مقاومة للعلاج الأولي أو المكتسبة. غالبًا ما يُعزى مقاومة العلاج للعلاجات المناعية إلى وجود بيئة الورم المؤيدة للالتهابات والمثبطة للمناعة. أحد الأمثلة على ذلك هو استخدام علاجات مضادة لـ CTLA-4 المرتبطة بحدوث التهاب القولون والتهاب الغدة النخامية، وعلاجات مضادة لـ PD-1 المرتبطة بالتهاب الغدة الدرقية. وبالتالي، فإن إضافة العلاجات المضادة للالتهابات إلى أنظمة علاج السرطان قد تؤدي إلى استجابات سريرية أفضل في بعض الحالات السريرية.
الهدف الأولي من العلاجات المضادة للالتهابات هو قمع الالتهاب المؤيد للورم وفي نفس الوقت تنشيط الاستجابة المناعية المضادة للورم. على عكس العلاجات التي تستهدف علامات ورمية محددة، تفتقر العلاجات المضادة للالتهابات إلى مؤشرات حيوية لاختيارها. ستؤثر الفروق الداخلية بين المرضى مثل العمر، والملف الجزيئي للورم على الاستجابة العلاجية للعلاجات الموجهة نحو الالتهاب. وبالتالي، يُوصى باستخدام طرق عالية الدقة مثل التحليلات متعددة الأوميات، وتحليل الخلايا المفردة، والتحليلات المكانية لتسهيل اتخاذ القرارات الطبية ولتوقع الاستجابة العلاجية للعلاجات الموجهة نحو الالتهاب. بالإضافة إلى ذلك، لا يزال الأمر
من الصعب الحفاظ على توازن الالتهاب في جهاز المناعة. كما أن التباين والمرونة في بيئة الورم الدقيقة تطرح تحديات أمام العلاجات الموجهة نحو الالتهاب من خلال استهداف جزيء واحد أو نوع واحد من خلايا المناعة. على سبيل المثال، قد تؤدي الحلقات الراجعة المعطلة من خلال استهداف سيتوكين التهابي واحد إلى تنشيط تعويضي لمساراته المعنية. هناك حاجة إلى دراسات مستقبلية للتحقيق في دمج العلاجات الموجهة نحو الالتهاب مع خيارات علاجية أخرى للسرطان، مما يسهل تصميم علاج آمن وشخصي.
الاختصارات
ROS أنواع الأكسجين التفاعلية
عامل نخر الورم ألفا
عامل تثبيط الهجرة MIF
ماكروفاجات مرتبطة بالورم (TAMs)
الخلايا المتعادلة المرتبطة بالورم (TANs)
الخلايا الشجرية DCs
خلايا مثبطة مشتقة من النخاع العظمي (MDSCs)
ماتريكس ميتالوبيبتيداز (MMP)
إنترفيرون IFN
تGF-
CXCL عامل كيميائي موجه من نوع C-X-C
أنجيوبويتين-1
فخاخ خارج الخلوية للعدلات (NETs)
الانتقال من الخلايا البطانية إلى الخلايا المتوسطة (EMT)
عامل تحفيز المستعمرات للعدلات والبلعميات GM-CSF
NK القاتل الطبيعي
المصفوفة خارج الخلوية (ECM)
الخلايا التائية التنظيمية (Tregs)
الخلايا التائية المساعدة 17
FLT3 مستقبل كيناز التيروزين المرتبط بـ Fms 3
مستضدات مرتبطة بالأورام (TAAs)
ICD موت الخلايا المناعية
أنماط الجزيئات المرتبطة بالضرر (DAMPs)
الشبكة الإندوبلازمية
مستقبلات التعرف على الأنماط (PRRs)
M-MDSCs خلايا مثبطة مشتقة من المونوسيتات-المايلويد
PBMC خلايا الدم المحيطية الوحيدة النواة
لا أكسيد النيتريك
مستقبلات الخلايا التائية TCR
حجب نقاط التفتيش المناعية ICB
مستقبل عامل تحفيز المستعمرات 1 (CSF-1R)
بروتين البرين
عامل تنشيط الصفائح الدموية (PAF)
مثبطات الأنسجة لمثبطات الماتريكس
سرطان الخلايا الحرشفية في الرأس والعنق (HNSCC)
إنترلوكين IL
LDL-C كوليسترول البروتين الدهني منخفض الكثافة
سرطان الرئة الغدي المتحور K-ras KM-LUAD
بريج التنظيمي ب
سرطان البنكرياس القنوي الغدي
NF-kB عامل النسخ النووي كابا ب
عامل تثبيط اللوكيميا (LIF)
أو إس إم أونكوسيتين م
عامل التغذية العصبية الهدبية CNTF
CT-1 كارديوتروفين-1
السيتوكين الشبيه بكارديوتروفين CLC
كيناز المعتمد على السيكلين (CDK)
XIAP بروتين مثبط الموت المبرمج المرتبط بالكروموسوم X
كيناز جانوس JAK
إشارة STAT الناقل ومفعل النسخ
الأحماض الدهنية المتعددة غير المشبعة (PUFAs)
كوكس سيكلوأوكسيجيناز
لوكس ليبكسجيناز
| PGs | بروستاجلاندينات |
| LXs | ليبوسين |
| mPGES-1 | السينثاز 1 للبروستاجلاندين E2 الميكروسومي |
| LT | ليوكوترين |
| CRC | سرطان القولون والمستقيم |
| IBD | مرض الأمعاء الالتهابي |
| سي أي سي | سرطان القولون المستقيمي المرتبط بالتهاب القولون |
| مضادات الالتهاب غير الستيرويدية | الأدوية غير الستيرويدية المضادة للالتهابات |
| DFS | البقاء على قيد الحياة خالٍ من المرض |
| PFS | البقاء بدون تقدم |
| أيه | الأحداث السلبية |
| MMR | إصلاح عدم التطابق |
| MSI | عدم استقرار الميكروساتلايت |
| HCC | سرطان الخلايا الكبدية |
| فيروس التهاب الكبد B | فيروس التهاب الكبد B |
| نأس | نظائر النوكليوتيدات |
| RFS | البقاء بدون انتكاسة |
| الرابطة الوطنية الهوكي | سرطانات الغدد اللمفاوية غير هودجكين |
| 5-FU | 5-فلورويوراسيل |
| MM | الورم النقوي المتعدد |
شكر وتقدير
مساهمات المؤلفين
تمويل
توفر البيانات والمواد
الإعلانات
موافقة الأخلاقيات والموافقة على المشاركة
موافقة على النشر
المصالح المتنافسة
تم النشر على الإنترنت: 22 مارس 2024
References
- Plytycz B, Seljelid R. From inflammation to sickness: historical perspective. Arch Immunol Ther Exp (Warsz). 2003;51(2):105-9.
- Granger DN, Senchenkova E. In: Inflammation and the Microcirculation. San Rafael (CA); 2010.
- Virchow R. An address on the value of pathological experiments. Br Med J. 1881;2(1075):198-203.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74.
- Haddow A. Addendum to “molecular repair, wound healing, and carcinogenesis: tumor production a possible overhealing”? Adv Cancer Res. 1974;20:343-66.
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650-9.
- Abramovitch R, Marikovsky M, Meir G, Neeman M. Stimulation of tumour angiogenesis by proximal wounds: spatial and temporal analysis by MRI. Br J Cancer. 1998;77(3):440-7.
- Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591-6.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70.
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391-403.
- Krugliak Cleveland N, Torres J, Rubin DT. What does disease progression look like in ulcerative colitis, and how might it be prevented? Gastroenterology. 2022;162(5):1396-408.
- Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. 2022;162(3):715-30.
- Lee YC, Chiang TH, Chou CK, Tu YK, Liao WC, Wu MS, Graham DY. Association between helicobacter pylori eradication and gastric cancer incidence: a systematic review and meta-analysis. Gastroenterology. 2016;150(5):1113-24.
- Tian T, Song C, Jiang L, Dai J, Lin Y, Xu X, Yu C, Ge Z, Ding Y, Wen Y, et al. Hepatitis
virus infection and the risk of cancer among the Chinese population. Int J Cancer. 2020;147(11):3075-84. - Kawanishi S, Ohnishi S, Ma N, Hiraku Y, Murata M. Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. Int J Mol Sci. 2017;18(8).
- Suresh V, Dash P, Suklabaidya S, Murmu KC, Sasmal PK, Jogdand GM, Parida D, Sethi M, Das B, Mohapatra D, et al. MIF confers survival advantage to pancreatic CAFs by suppressing interferon pathwayinduced p53-dependent apoptosis. FASEB J. 2022;36(8): e22449.
- Chen L, Zhou X, Fan LX, Yao Y, Swenson-Fields KI, Gadjeva M, Wallace DP, Peters DJ, Yu A, Grantham JJ, et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest. 2015;125(6):2399-412.
- Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19(4):237-53.
- Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, Wang B, Wang Q, Zhao Y, Cai Y. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol. 2020;88: 106939.
- Li MO, Wolf N, Raulet DH, Akkari L, Pittet MJ, Rodriguez PC, Kaplan RN, Munitz A, Zhang Z, Cheng S, et al. Innate immune cells in the tumor microenvironment. Cancer Cell. 2021;39(6):725-9.
- Aga E, Mukherjee A, Rane D, More V, Patil T, van Zandbergen G, Solbach W, Dandapat J, Tackenberg H, Ohms M, et al. Type-1 interferons prolong the lifespan of neutrophils by interfering with members of the apoptotic cascade. Cytokine. 2018;112:21-6.
- Wu M, Ma M, Tan Z, Zheng H, Liu X. Neutrophil: a new player in metastatic cancers. Front Immunol. 2020;11: 565165.
- Li S, Cong X, Gao H, Lan X, Li Z, Wang W, Song S, Wang Y, Li C, Zhang H , et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J Exp Clin Cancer Res. 2019;38(1):6.
- Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: implications for immunotherapy. Front Immunol. 2018;9:527.
- Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, Quatromoni JG, Stephen TL, Litzky L, Deshpande C, et al. Origin and role of a subset of tumor-associated neutrophils with antigenpresenting cell features in early-stage human lung cancer. Cancer Cell. 2016;30(1):120-35.
- Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, Fan J, Cao Y, Dai Z, Zhou J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150(7):1646-58.
- Mishalian I, Bayuh R, Eruslanov E, Michaeli J, Levy L, Zolotarov L, Singhal S, Albelda SM, Granot Z, Fridlender ZG. Neutrophils recruit regulatory
28. Sasaki S, Baba T, Muranaka H, Tanabe Y, Takahashi C, Matsugo S, Mukaida N. Involvement of prokineticin 2-expressing neutrophil infiltration in 5-fluorouracil-induced aggravation of breast cancer metastasis to lung. Mol Cancer Ther. 2018;17(7):1515-25.
29. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. 2021;61(2):194-211.
30. Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, Quail D, Walsh L, Sangwan V, Bertos N et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight. 2019;5(16).
31. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133-8.
32. Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2019;216(1):176-94.
33. Bates AM, Gomez Hernandez MP, Lanzel EA, Qian F, Brogden KA. Matrix metalloproteinase (MMP) and immunosuppressive biomarker profiles of seven head and neck squamous cell carcinoma (HNSCC) cell lines. Transl Cancer Res. 2018;7(3):533-42.
34. Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, Liu P, Li Z, Xia Y, Jiang W. Neutrophil extracellular traps induced by IL8 promote diffuse large B-cell lymphoma progression via the TLR9 signaling. Clin Cancer Res. 2019;25(6):1867-79.
35. Weiss E, Kretschmer D. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol. 2018;39(10):815-29.
36. Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, de Andrea C, Ochoa MC, Otano I, Etxeberria I, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52(5):856-71.
37. Azevedo PO, Paiva AE, Santos GSP, Lousado L, Andreotti JP, Sena IFG, Tagliati CA, Mintz A, Birbrair A. Cross-talk between lung cancer and bones results in neutrophils that promote tumor progression. Cancer Metastasis Rev. 2018;37(4):779-90.
38. Berger-Achituv S, Brinkmann V, Abed UA, Kuhn LI, Ben-Ezra J, Elhasid R, Zychlinsky A. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. 2013;4:48.
39. Demkow U. Neutrophil extracellular traps (NETs) in cancer invasion, evasion and metastasis. Cancers (Basel). 2021;13(17).
40. Wang Y, Liu F, Chen L, Fang C, Li S, Yuan S, Qian X, Yin Y, Yu B, Fu B, et al. Neutrophil extracellular traps (NETs) promote non-small cell lung cancer metastasis by suppressing IncRNA MIR503HG to activate the NF-kappaB/NLRP3 inflammasome pathway. Front Immunol. 2022;13: 867516.
41. Deng J, Kang Y, Cheng CC, Li X, Dai B, Katz MH, Men T, Kim MP, Koay EA, Huang H et al. DDR1-induced neutrophil extracellular traps drive pancreatic cancer metastasis. JCI Insight. 2021;6(17).
42. Khan U, Chowdhury S, Billah MM, Islam KMD, Thorlacius H, Rahman M. Neutrophil extracellular traps in colorectal cancer progression and metastasis. Int J Mol Sci. 2021;22(14).
43. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, Wang Y, Yang S, Liang C, Liang Y, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39(3):423-37.
44. Yang C, Wang Z, Li L, Zhang Z, Jin X, Wu P, Sun S, Pan J, Su K, Jia F et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J Immunother Cancer. 2021;9(10).
45. Arelaki S, Arampatzioglou A, Kambas K, Papagoras C, Miltiades P, Angelidou I, Mitsios A, Kotsianidis I, Skendros P, Sivridis E, et al. Gradient infiltration of neutrophil extracellular traps in colon cancer and evidence for their involvement in tumour growth. PLoS ONE. 2016;11(5): e0154484.
46. Millrud CR, Kagedal A, Kumlien Georen S, Winqvist O, Uddman R, Razavi R, Munck-Wikland E, Cardell LO. NET-producing CD16(high) CD62L(dim) neutrophils migrate to tumor sites and predict improved survival in patients with HNSCC. Int J Cancer. 2017;140(11):2557-67.
47. Schedel F, Mayer-Hain S, Pappelbaum KI, Metze D, Stock M, Goerge T, Loser K, Sunderkotter C, Luger TA, Weishaupt C. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment Cell Melanoma Res. 2020;33(1):63-73.
48. Muqaku B, Pils D, Mader JC, Aust S, Mangold A, Muqaku L, Slany A, Del Favero G, Gerner C. Neutrophil extracellular trap formation correlates with favorable overall survival in high grade ovarian cancer. Cancers (Basel). 2020;12(2).
49. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445-55.
50. Mackaness GB. Cellular resistance to infection. J Exp Med. 1962;116(3):381-406.
51. Duan
52. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14-20.
53. Eum HH, Kwon M, Ryu D, Jo A, Chung W, Kim N, Hong Y, Son DS, Kim ST, Lee J, et al. Tumor-promoting macrophages prevail in malignant ascites of advanced gastric cancer. Exp Mol Med. 2020;52(12):1976-88.
54. Bernsmeier C, van der Merwe S, Perianin A. Innate immune cells in cirrhosis. J Hepatol. 2020;73(1):186-201.
55. Bruns H, Buttner M, Fabri M, Mougiakakos D, Bittenbring JT, Hoffmann MH, Beier F, Pasemann S, Jitschin R, Hofmann AD, et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci Transl Med. 2015;7(282):282-247.
56. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549-55.
57. Ding P, Wang W, Wang J, Yang Z, Xue L. Expression of tumor-associated macrophage in progression of human glioma. Cell Biochem Biophys. 2014;70(3):1625-31.
58. Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, Ma X. Prognostic significance of tumor-associated macrophages in ovarian cancer: a meta-analysis. Gynecol Oncol. 2017;147(1):181-7.
59. Larionova I, Kazakova E, Gerashchenko T, Kzhyshkowska J. New angiogenic regulators produced by TAMs: perspective for targeting tumor angiogenesis. Cancers (Basel). 2021;13(13).
60. Gurevich DB, Severn CE, Twomey C, Greenhough A, Cash J, Toye AM, Mellor H, Martin P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J 2018;37(13).
61. Ramirez-Pedraza M, Fernandez M. Interplay between macrophages and angiogenesis: a double-edged sword in liver disease. Front Immunol. 2019;10:2882.
62. Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, Wang H, Wang K, Lin Y, Wang
63. Lan J, Sun L, Xu F, Liu L, Hu F, Song D, Hou Z, Wu W, Luo X, Wang J, et al. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. 2019;79(1):146-58.
64. Yin Z, Ma T, Huang B, Lin L, Zhou Y, Yan J, Zou Y, Chen S. Macrophagederived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-beta signaling pathway. J Exp Clin Cancer Res. 2019;38(1):310.
65. Shima T, Shimoda M, Shigenobu T, Ohtsuka T, Nishimura T, Emoto K, Hayashi Y, Iwasaki T, Abe T, Asamura H, et al. Infiltration of tumorassociated macrophages is involved in tumor programmed deathligand 1 expression in early lung adenocarcinoma. Cancer Sci. 2020;111(2):727-38.
66. Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang CL. PD-L1 expression on tumor-infiltrating immune cells is highly associated with M2 TAM and aggressive malignant potential in patients with resected non-small cell lung cancer. Lung Cancer. 2019;136:136-44.
67. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245-52.
68. Ness S, Lin S, Gordon JR. Regulatory dendritic cells, t cell tolerance, and dendritic cell therapy for immunologic disease. Front Immunol. 2021;12: 633436.
69. Tsapogas P, Mooney CJ, Brown G, Rolink A. The cytokine Flt3-ligand in normal and malignant hematopoiesis. Int J Mol Sci. 2017;18(6).
70. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265-77.
71. Fu C, Jiang A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front Immunol. 2018;9:3059.
72. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7-24.
73. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117(11):1583-91.
74. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945-54.
75. Rufo N, Garg AD, Agostinis P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer. 2017;3(9):643-58.
76. Wang Y, Xiang Y, Xin VW, Wang XW, Peng XC, Liu XQ, Wang D, Li N, Cheng JT, Lyv YN, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13(1):107.
77. Hole CR, Wager CML, Castro-Lopez N, Campuzano A, Cai H, Wozniak KL, Wang Y, Wormley FL Jr. Induction of memory-like dendritic cell responses in vivo. Nat Commun. 2019;10(1):2955.
78. Alzeibak R, Mishchenko TA, Shilyagina NY, Balalaeva IV, Vedunova MV, Krysko DV. Targeting immunogenic cancer cell death by photodynamic therapy: past, present and future. J Immunother Cancer. 2021;9(1).
79. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, Vandenabeele P. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13(1):3676.
80. Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C, Maurer D, Ottensmeier C, van der Burg SH, Welters MJ, et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother. 2016;65(2):161-9.
81. Li BH, Garstka MA, Li ZF. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol Immunol. 2020;117:201-15.
82. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloidderived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208-20.
83. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J Biol Chem. 2002;277(24):21123-9.
84. Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor premetastatic niche formation. Front Immunol. 2019;10:172.
85. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: roles in tumor progression. Front Immunol. 2019;10:771.
86. Johnson BW, Achyut BR, Fulzele S, Mondal AK, Kolhe R, Arbab AS. Delineating pro-angiogenic myeloid cells in cancer therapy. Int J Mol Sci 2018;19(9).
87. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19-28.
88. Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM, Maio M, Sucker A, Schilling B, Schadendorf D, et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin Cancer Res. 2014;20(6):1601-9.
89. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E, Speiser DE. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247-57.
90. Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, Shtainberg H, Lotem M, Baniyash M. Clinical significance
of circulating CD33+CD11b+HLA-DR-myeloid cells in patients with stage IV melanoma treated with ipilimumab. Clin Cancer Res. 2016;22(23):5661-72.
91. Martens A, Wistuba-Hamprecht K, Geukes Foppen M, Yuan J, Postow MA, Wong P, Romano E, Khammari A, Dreno B, Capone M, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22(12):2908-18.
92. Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine. 2016;6:50-8.
93. Holmgaard RB, Brachfeld A, Gasmi B, Jones DR, Mattar M, Doman T, Murphy M, Schaer D, Wolchok JD, Merghoub T. Timing of CSF-1/CSF-1R signaling blockade is critical to improving responses to CTLA-4 based immunotherapy. Oncoimmunology. 2016;5(7): e1151595.
94. Lamichhane P, Karyampudi L, Shreeder B, Krempski J, Bahr D, Daum J, Kalli KR, Goode EL, Block MS, Cannon MJ, et al. IL10 release upon PD-1 blockade sustains immunosuppression in ovarian cancer. Cancer Res. 2017;77(23):6667-78.
95. Gomes-Santos IL, Amoozgar Z, Kumar AS, Ho WW, Roh K, Talele NP, Curtis H, Kawaguchi K, Jain RK, Fukumura D. Exercise training improves tumor control by increasing CD8(+) T-cell infiltration via CXCR3 signaling and sensitizes breast cancer to immune checkpoint blockade. Cancer Immunol Res. 2021;9(7):765-78.
96. Holder KA, Grant MD. Human cytomegalovirus IL-10 augments NK cell cytotoxicity. J Leukoc Biol. 2019;106(2):447-54.
97. O’Carroll SJ, Kho DT, Wiltshire R, Nelson V, Rotimi O, Johnson R, Angel CE, Graham ES. Pro-inflammatory TNFalpha and IL-1 beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J Neuroinflammation. 2015;12:131.
98. Hillyer P, Mordelet E, Flynn G, Male D. Chemokines, chemokine receptors and adhesion molecules on different human endothelia: discriminating the tissue-specific functions that affect leucocyte migration. Clin Exp Immunol. 2003;134(3):431-41.
99. Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004;167(2):223-9.
100. Tichet M, Prod’Homme V, Fenouille N, Ambrosetti D, Mallavialle A, Cerezo M, Ohanna M, Audebert S, Rocchi S, Giacchero D, et al. Tumourderived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun. 2015;6:6993.
101. Hiratsuka S, Goel S, Kamoun WS, Maru Y, Fukumura D, Duda DG, Jain RK. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc Natl Acad Sci U S A. 2011;108(9):3725-30.
102. Burdick MM, Henson KA, Delgadillo LF, Choi YE, Goetz DJ, Tees DF, Benencia F. Expression of E-selectin ligands on circulating tumor cells: cross-regulation with cancer stem cell regulatory pathways? Front Oncol. 2012;2:103.
103. Hauselmann I, Roblek M, Protsyuk D, Huck V, Knopfova L, Grassle S, Bauer AT, Schneider SW, Borsig L. Monocyte induction of E-selectinmediated endothelial activation releases VE-cadherin junctions to promote tumor cell extravasation in the metastasis cascade. Cancer Res. 2016;76(18):5302-12.
104. Shea DJ, Li YW, Stebe KJ, Konstantopoulos K. E-selectin-mediated rolling facilitates pancreatic cancer cell adhesion to hyaluronic acid. FASEB J. 2017;31(11):5078-86.
105. Kang SA, Blache CA, Bajana S, Hasan N, Kamal M, Morita Y, Gupta V, Tsolmon B, Suh KS, Gorenstein DG, et al. The effect of soluble E-selectin on tumor progression and metastasis. BMC Cancer. 2016;16:331.
106. Zamarron
107. Amin MN, Siddiqui SA, Ibrahim M, Hakim ML, Ahammed MS, Kabir A, Sultana F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020;8:2050312120965752.
108. Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, Lee SR, Yang SH. The role of tumor necrosis factor alpha (TNF-alpha) in autoimmune
disease and current TNF-alpha inhibitors in therapeutics. Int J Mol Sci. 2021;22(5).
109. Zhang GP, Yue X, Li SQ. Cathepsin C interacts with TNF-alpha/p38 MAPK signaling pathway to promote proliferation and metastasis in hepatocellular carcinoma. Cancer Res Treat. 2020;52(1):10-23.
110. Schroder SK, Asimakopoulou A, Tillmann S, Koschmieder S, Weiskirchen R. TNF-alpha controls lipocalin-2 expression in PC-3 prostate cancer cells. Cytokine. 2020;135: 155214.
111. Jo E, Jang HJ, Yang KE, Jang MS, Huh YH, Yoo HS, Park JS, Jang IS, Park SJ. Cordyceps militaris induces apoptosis in ovarian cancer cells through TNF-alpha/TNFR1-mediated inhibition of NF-kappaB phosphorylation. BMC Complement Med Ther. 2020;20(1):1.
112. Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (TNF-alpha) in breast cancer: molecular insights and therapeutic approaches. Cell Oncol (Dordr). 2020;43(1):1-18.
113. Garcia-Tunon I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. Role of tumor necrosis factor-alpha and its receptors in human benign breast lesions and tumors (in situ and infiltrative). Cancer Sci. 2006;97(10):1044-9.
114. Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, Frahm I, Allemand DH, Deza EG, Ares S, et al. TNFalpha-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clin Cancer Res. 2017;23(3):636-48.
115. Wu C, Fernandez SA, Criswell T, Chidiac TA, Guttridge D, Villalona-Calero M, Bekaii-Saab TS. Disrupting cytokine signaling in pancreatic cancer: a phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas. 2013;42(5):813-8.
116. Yoshimatsu Y, Wakabayashi I, Kimuro S, Takahashi N, Takahashi K, Kobayashi M, Maishi N, Podyma-Inoue KA, Hida K, Miyazono K, et al. TNFalpha enhances TGF-beta-induced endothelial-to-mesenchymal transition via TGF-beta signal augmentation. Cancer Sci. 2020;111(7):2385-99.
117. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014: 149185.
118. Rossi S, Cordella M, Tabolacci C, Nassa G, D’Arcangelo D, Senatore C, Pagnotto P, Magliozzi R, Salvati A, Weisz A, et al. TNF-alpha and metalloproteases as key players in melanoma cells aggressiveness. J Exp Clin Cancer Res. 2018;37(1):326.
119. Bertrand F, Rochotte J, Colacios C, Montfort A, Tilkin-Mariame AF, Touriol C, Rochaix P, Lajoie-Mazenc I, Andrieu-Abadie N, Levade T, et al. Blocking tumor necrosis factor alpha enhances CD8 T-cell-dependent immunity in experimental melanoma. Cancer Res. 2015;75(13):2619-28.
120. Li H, Wang R, Yu Z, Shi R, Zhang J, Gao S, Shao M, Cui S, Gao Z, Xu J, et al. Tumor necrosis factor alpha reduces SNAP29 dependent autolysosome formation to increase prion protein level and promote tumor cell migration. Virol Sin. 2021;36(3):458-75.
121. Nagar M, Jacob-Hirsch J, Vernitsky H, Berkun Y, Ben-Horin S, Amariglio N, Bank I, Kloog Y, Rechavi G, Goldstein I. TNF activates a NF-kappaBregulated cellular program in human CD45RA- regulatory T cells that modulates their suppressive function. J Immunol. 2010;184(7):3570-81.
122. Medler J, Wajant H. Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Expert Opin Ther Targets. 2019;23(4):295-307.
123. Farrugia M , Baron B . The role of TNF-alpha in rheumatoid arthritis: a focus on regulatory T cells. J Clin Transl Res. 2016;2(3):84-90.
124. Salomon BL, Leclerc M, Tosello J, Ronin E, Piaggio E, Cohen JL. Tumor necrosis factor alpha and regulatory T cells in oncoimmunology. Front Immunol. 2018;9:444.
125. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol. 2008;180(10):6467-71.
126. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, Defusco A, Plager S, Warden S, Huang D et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumorassociated Tregs. Sci Signal. 2017;10(462).
127. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50(4):924-40.
128. Baba AB, Rah B, Bhat GR, Mushtaq I, Parveen S, Hassan R, Hameed Zargar M, Afroze D. Transforming growth factor-beta (TGF-beta) signaling in cancer-a betrayal within. Front Pharmacol. 2022;13: 791272.
129. Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF-beta signaling in bone remodeling. J Clin Invest. 2014;124(2):466-72.
130. Hu Q, Hisamatsu T, Haemmerle M, Cho MS, Pradeep S, Rupaimoole R, Rodriguez-Aguayo C, Lopez-Berestein G, Wong STC, Sood AK, et al. Role of platelet-derived Tgfbeta1 in the progression of ovarian cancer. Clin Cancer Res. 2017;23(18):5611-21.
131. Melzer C, Hass R, von der Ohe J, Lehnert H, Ungefroren H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun Signal. 2017;15(1):19.
132. Melzer C, von der Ohe J, Otterbein H, Ungefroren H, Hass R. Changes in uPA, PAI-1, and TGF-beta Production during Breast Cancer Cell Interaction with Human Mesenchymal Stroma/Stem-Like Cells (MSC). Int J Mol Sci. 2019. 20(11).
133. Villalba M, Evans SR, Vidal-Vanaclocha F, Calvo A. Role of TGF-beta in metastatic colon cancer: it is finally time for targeted therapy. Cell Tissue Res. 2017;370(1):29-39.
134. Hao Y, Baker D, Ten Dijke P. TGF-beta-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. 2019;20(11).
135. Tauriello DVF, Sancho E, Batlle E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat Rev Cancer. 2022;22(1):25-44.
136. Tan
137. Esquivel-Velazquez M, Ostoa-Saloma P, Palacios-Arreola MI, NavaCastro KE, Castro JI, Morales-Montor J. The role of cytokines in breast cancer development and progression. J Interferon Cytokine Res. 2015;35(1):1-16.
138. Laine A, Labiad O, Hernandez-Vargas H, This S, Sanlaville A, Leon S, Dalle S, Sheppard D, Travis MA, Paidassi H, et al. Regulatory T cells promote cancer immune-escape through integrin alphavbeta8-mediated TGFbeta activation. Nat Commun. 2021;12(1):6228.
139. Akkaya M, Akkaya B, Miozzo P, Rawat M, Pena M, Sheehan PW, Kim AS, Kamenyeva O, Kabat J, Bolland S, et al. B cells produce type 1 IFNs in response to the TLR9 agonist CpG-A conjugated to cationic lipids. J Immunol. 2017;199(3):931-40.
140. Ali S, Mann-Nuttel R, Schulze A, Richter L, Alferink J, Scheu S. Sources of type I interferons in infectious immunity: plasmacytoid dendritic cells not always in the driver’s seat. Front Immunol. 2019;10:778.
141. Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernan-dez-Hinojal G, Vera R, Smerdou C, Martisova E, Arozarena I, et al. PDL1 signals through conserved sequence motifs to overcome interferonmediated cytotoxicity. Cell Rep. 2017;20(8):1818-29.
142. Chen J, Cao Y, Markelc B, Kaeppler J, Vermeer JA, Muschel RJ. Type I IFN protects cancer cells from CD8+T cell-mediated cytotoxicity after radiation. J Clin Invest. 2019;129(10):4224-38.
143. Lee MS, Kim B, Oh GT, Kim YJ. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat Immunol. 2013;14(4):346-55.
144. Cunningham CR, Champhekar A, Tullius MV, Dillon BJ, Zhen A, de la Fuente JR, Herskovitz J, Elsaesser H, Snell LM, Wilson EB, et al. Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog. 2016;12(1): e1005356.
145. Gong W, Donnelly CR, Heath BR, Bellile E, Donnelly LA, Taner HF, Broses L, Brenner JC, Chinn SB, Ji RR, et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology. 2021;10(1):1997385.
146. Musella M, Guarracino A, Manduca N, Galassi C, Ruggiero E, Potenza A, Maccafeo E, Manic G, Mattiello L, Soliman Abdel Rehim S, et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nat Immunol. 2022;23(9):1379-92.
147. Pidugu VK, Wu MM, Yen AH, Pidugu HB, Chang KW, Liu CJ, Lee TC. IFIT1 and IFIT3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-EGFR recycling. Oncogene. 2019;38(17):3232-47.
148. Boukhaled GM, Harding S, Brooks DG. Opposing roles of type I interferons in cancer immunity. Annu Rev Pathol. 2021;16:167-98.
149. Spaapen RM, Leung MY, Fuertes MB, Kline JP, Zhang L, Zheng Y, Fu YX, Luo X, Cohen KS, Gajewski TF. Therapeutic activity of high-dose intratumoral IFN-beta requires direct effect on the tumor vasculature. J Immunol. 2014;193(8):4254-60.
150. Golomb HM, Ratain MJ, Mick R, Daly K. Interferon treatment for hairy cell leukemia: an update on a cohort of 69 patients treated from 1983-1986. Leukemia. 1992;6(11):1177-80.
151. Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int J Mol Sci. 2018;19(8).
152. Malik A, Kanneganti TD. Function and regulation of IL-1alpha in inflammatory diseases and cancer. Immunol Rev. 2018;281(1):124-37.
153. Fahey E, Doyle SL. IL-1 family cytokine regulation of vascular permeability and angiogenesis. Front Immunol. 2019;10:1426.
154. Xiao
155. Haabeth OA, Lorvik KB, Yagita H, Bogen B, Corthay A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology. 2016;5(1): e1039763.
156. Baker KJ, Houston A, Brint E. IL-1 family members in cancer; two sides to every story. Front Immunol. 2019;10:1197.
157. Zhang W, Borcherding N, Kolb R. IL-1 signaling in tumor microenvironment. Adv Exp Med Biol. 2020;1240:1-23.
158. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, Koski G, Disis ML, Czerniecki BJ, Kodumudi K. Differentiation and regulation of T(H) cells: a balancing act for cancer immunotherapy. Front Immunol. 2021;12: 669474.
159. Lin D, Mei Y, Lei L, Binte Hanafi Z, Jin Z, Liu Y, Song Y, Zhang Y, Hu B, Liu
160. Jiang H, Gebhardt C, Umansky L, Beckhove P, Schulze TJ, Utikal J, Umansky V. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int J Cancer. 2015;136(10):2352-60.
161. Voronov E, Carmi Y, Apte RN. The role IL-1 in tumor-mediated angiogenesis. Front Physiol. 2014;5:114.
162. Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, Abutbul S, Huszar M, Dinarello CA, Apte RN, et al. The role of IL-1 beta in the early tumor cell-induced angiogenic response. J Immunol. 2013;190(7):3500-9.
163. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, Dinarello CA, Voronov E, Apte RN. Blocking IL-1 beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A. 2019;116(4):1361-9.
164. Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. Tumor cell-derived IL1beta promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 2020;80(5):1088-101.
165. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10): a016295.
166. Qin B, Zhou Z, He J, Yan C, Ding S. IL-6 inhibits starvation-induced autophagy via the STAT3/Bcl-2 signaling pathway. Sci Rep. 2015;5:15701.
167. Shi R, Chen M, Litifu B. Serum interleukin-6 and survivin levels predict clinical response to etanercept treatment in patients with established rheumatoid arthritis. Mod Rheumatol. 2018;28(1):126-32.
168. Yao X, Huang J, Zhong H, Shen N, Faggioni R, Fung M, Yao Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141(2):125-39.
169. Ortiz-Montero P, Londono-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/ inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. 2017;15(1):17.
170. Sapochnik M, Haedo MR, Fuertes M, Ajler P, Carrizo G, Cervio A, Sevlever G, Stalla GK, Arzt E. Autocrine IL-6 mediates pituitary tumor senescence. Oncotarget. 2017;8(3):4690-702.
171. Ray K, Ujvari B, Ramana V, Donald J. Cross-talk between EGFR and IL-6 drives oncogenic signaling and offers therapeutic opportunities in cancer. Cytokine Growth Factor Rev. 2018;41:18-27.
172. Gao S, Hu J, Wu X, Liang Z. PMA treated THP-1-derived-IL-6 promotes EMT of SW48 through STAT3/ERK-dependent activation of Wnt/betacatenin signaling pathway. Biomed Pharmacother. 2018;108:618-24.
173. Liu W, Wang H, Bai F, Ding L, Huang Y, Lu C, Chen S, Li C, Yue X, Liang X, et al. IL-6 promotes metastasis of non-small-cell lung cancer by upregulating TIM-4 via NF-kappaB. Cell Prolif. 2020;53(3): e12776.
174. Bharti R, Dey G, Das AK, Mandal M. Differential expression of IL-6/IL-6R and MAO-A regulates invasion/angiogenesis in breast cancer. Br J Cancer. 2018;118(11):1442-52.
175. Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 is a promising therapeutic target for hepatocellular carcinoma. Front Oncol. 2021;11: 760971.
176. Zhang B, Li Y, Wu Q, Xie L, Barwick B, Fu C, Li X, Wu D, Xia S, Chen J, et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat Commun. 2021;12(1):1714.
177. Manore SG, Doheny DL, Wong GL, Lo HW. IL-6/JAK/STAT3 signaling in breast cancer metastasis: biology and treatment. Front Oncol. 2022;12: 866014.
178. Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse Thelper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170(6):2081-95.
179. Vieira P, de Waal-Malefyt R, Dang MN, Johnson KE, Kastelein R, Fiorentino DF, deVries JE, Roncarolo MG, Mosmann TR, Moore KW. Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: homology to Epstein-Barr virus open reading frame BCRFI. Proc Natl Acad Sci U S A. 1991;88(4):1172-6.
180. Macatonia SE, Doherty TM, Knight SC, O’Garra A. Differential effect of IL-10 on dendritic cell-induced T cell proliferation and IFN-gamma production. J Immunol. 1993;150(9):3755-65.
181. Ouyang W, O’Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. 2019;50(4):871-91.
182. Wang
183. Zhang H, Li R, Cao Y, Gu Y, Lin C, Liu X, Lv K, He X, Fang H, Jin K, et al. Poor clinical outcomes and immunoevasive contexture in intratumoral IL-10-producing macrophages enriched gastric cancer patients. Ann Surg. 2022;275(4):e626-35.
184. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, Callahan DJ, Sun Z, Sun T, Tabib T, et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. 2019;20(6):724-35.
185. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, Blaisdell S, Basham B, Dai J, Grein J, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell. 2011;20(6):781-96.
186. Chen WF, Zlotnik A. IL-10: a novel cytotoxic T cell differentiation factor. J Immunol. 1991;147(2):528-34.
187. Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, Rhee SG, Thornalley PJ, Partridge L, Gems D, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011;13(4):361-6.
188. Ochoa CD, Wu RF, Terada LS. ROS signaling and ER stress in cardiovascular disease. Mol Aspects Med. 2018;63:18-29.
189. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020;152:116-41.
190. Violi F, Carnevale R, Loffredo L, Pignatelli P, Gallin JI. NADPH oxidase-2 and atherothrombosis: insight from chronic granulomatous disease. Arterioscler Thromb Vasc Biol. 2017;37(2):218-25.
191. Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8(9):722-8.
192. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829-37.
193. Pei J, Pan X, Wei G, Hua Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front Pharmacol. 2023;14:1147414.
194. Zhang J, Li F, Yin Y, Liu N, Zhu M, Zhang H, Liu W, Yang M, Qin S, Fan X , et al. Alpha radionuclide-chelated radioimmunotherapy promoters enable local radiotherapy/chemodynamic therapy to discourage cancer progression. Biomater Res. 2022;26(1):44.
195. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, Castoria G, Migliaccio A. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52(2):192-203.
196. Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61(4):290-302.
197. Lee HC, Wei YH. Mitochondrial DNA instability and metabolic shift in human cancers. Int J Mol Sci. 2009;10(2):674-701.
198. Yu LM, Zhang WH, Han XX, Li YY, Lu Y, Pan J, Mao JQ, Zhu LY, Deng JJ, Huang W, et al. Hypoxia-induced ROS contribute to myoblast pyroptosis during obstructive sleep apnea via the NF-kappaB/HIF-1alpha signaling pathway. Oxid Med Cell Longev. 2019;2019:4596368.
199. Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47-71.
200. Willson JA, Arienti S, Sadiku P, Reyes L, Coelho P, Morrison T, Rinaldi G, Dockrell DH, Whyte MKB, Walmsley SR. Neutrophil HIF-1alpha stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood. 2022;139(2):281-6.
201. Sun B, Yu L, Xu C, Li YM, Zhao YR, Cao MM, Yang LY. NAD(P)HX epimerase downregulation promotes tumor progression through ROS/HIF-1 alpha signaling in hepatocellular carcinoma. Cancer Sci. 2021;112(7):2753-69.
202. Zhang L, Cao Y, Guo X, Wang X, Han X, Kanwore K, Hong X, Zhou H, Gao D. Hypoxia-induced ROS aggravate tumor progression through HIF-1 alpha-SERPINE1 signaling in glioblastoma. J Zhejiang Univ Sci B. 2023;24(1):32-49.
203. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10(3):181-93.
204. Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002;99(21):13926-31.
205. Esh CJ, Chrismas BCR, Mauger AR, Taylor L. Pharmacological hypotheses: is acetaminophen selective in its cyclooxygenase inhibition? Pharmacol Res Perspect. 2021;9(4): e00835.
206. Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183-8.
207. Hijos-Mallada G, Sostres C, Gomollon F. NSAIDs, gastrointestinal toxicity and inflammatory bowel disease. Gastroenterol Hepatol. 2022;45(3):215-22.
208. Tudor DV, Baldea I, Lupu M, Kacso T, Kutasi E, Hopartean A, Stretea R, Gabriela Filip A. COX-2 as a potential biomarker and therapeutic target in melanoma. Cancer Biol Med. 2020;17(1):20-31.
209. Solanki R, Agrawal N, Ansari M, Jain S, Jindal A. COX-2 expression in breast carcinoma with correlation to clinicopathological parameters. Asian Pac J Cancer Prev. 2018;19(7):1971-5.
210. Khor LY, Bae K, Pollack A, Hammond ME, Grignon DJ, Venkatesan VM, Rosenthal SA, Ritter MA, Sandler HM, Hanks GE, et al. COX-2 expression predicts prostate-cancer outcome: analysis of data from the RTOG 92-02 trial. Lancet Oncol. 2007;8(10):912-20.
211. Guo W, Zhang Z, Li G, Lai X, Gu R, Xu W, Chen H, Xing Z, Chen L, Qian J, et al. Pyruvate kinase M2 promotes prostate cancer metastasis through regulating ERK1/2-COX-2 signaling. Front Oncol. 2020;10: 544288.
212. Du J, Feng J, Luo D, Peng L. Prognostic and clinical significance of COX-2 overexpression in laryngeal cancer: a meta-analysis. Front Oncol. 2022;12: 854946.
213. Hu Z, Yang Y, Zhao Y, Huang Y. The prognostic value of cyclooxyge-nase-2 expression in patients with esophageal cancer: evidence from a meta-analysis. Onco Targets Ther. 2017;10:2893-901.
214. Ren J, Liu J, Sui X. Correlation of COX-2 and MMP-13 expressions with gastric cancer and their effects on prognosis. J BUON. 2019;24(1):187-93.
215. Pomianowska E, Schjolberg AR, Clausen OP, Gladhaug IP. COX-2 overexpression in resected pancreatic head adenocarcinomas correlates with favourable prognosis. BMC Cancer. 2014;14:458.
216. Sun H, Zhang X, Sun D, Jia X, Xu L, Qiao Y, Jin Y. COX-2 expression in ovarian cancer: an updated meta-analysis. Oncotarget. 2017;8(50):88152-62.
217. Wang D, Cabalag CS, Clemons NJ, DuBois RN. Cyclooxygenases and prostaglandins in tumor immunology and microenvironment of gastrointestinal cancer. Gastroenterology. 2021;161(6):1813-29.
218. Li YF, Han CC, Wang Y, Cui DQ, Luo TT, Zhang YW, Ma Y, Wei W. Combined PGE2 with TNF-alpha promotes laryngeal carcinoma progression by enhancing GRK2 and TRAF2 interaction. Neoplasma. 2020;67(2):354-63.
219. Walker OL, Dahn ML, Power Coombs MR, Marcato P. The prostaglandin e2 pathway and breast cancer stem cells: evidence of increased signaling and potential targeting. Front Oncol. 2021;11: 791696.
220. Frejborg E, Salo T, Salem A. Role of cyclooxygenase-2 in head and neck tumorigenesis. Int J Mol Sci. 2020;21(23).
221. Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Xu D, Rosenberg DW. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68(9):3251-9.
222. Dean PT, Hooks SB. Pleiotropic effects of the COX-2/PGE2 axis in the glioblastoma tumor microenvironment. Front Oncol. 2022;12:1116014.
223. Knudsen NH, Manguso RT. Tumor-derived PGE2 gives NK cells a headache. Immunity. 2020;53(6):1131-2.
224. Walker W, Rotondo D. Prostaglandin E2 is a potent regulator of interleu-kin-12- and interleukin-18-induced natural killer cell interferon-gamma synthesis. Immunology. 2004;111(3):298-305.
225. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67(9):4507-13.
226. Tomic S, Joksimovic B, Bekic M, Vasiljevic M, Milanovic M, Colic M, Vucevic D. Prostaglanin-E2 potentiates the suppressive functions of human mononuclear myeloid-derived suppressor cells and increases their capacity to expand IL-10-producing regulatory T cell subsets. Front Immunol. 2019;10:475.
227. Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, Larghi P, Rimoldi M, Tripodo C, Strauss L, et al. Tumor-derived prostaglandin E2 promotes p50 NF-kappaB-dependent differentiation of monocytic MDSCs. Cancer Res. 2020;80(13):2874-88.
228. Zhang B, Bie Q, Wu P, Zhang J, You B, Shi H, Qian H, Xu W. PGD2/PTGDR2 signaling restricts the self-renewal and tumorigenesis of gastric cancer. Stem Cells. 2018;36(7):990-1003.
229. Iwanaga K, Nakamura T, Maeda S, Aritake K, Hori M, Urade Y, Ozaki H, Murata T. Mast cell-derived prostaglandin D2 inhibits colitis and colitisassociated colon cancer in mice. Cancer Res. 2014;74(11):3011-9.
230. Vafaeinik F, Kum HJ, Jin SY, Min DS, Song SH, Ha HK, Kim CD, Bae SS. Regulation of epithelial-mesenchymal transition of A549 cells by prostaglandin D(2). Cell Physiol Biochem. 2022;56(2):89-104.
231. Conejo-Garcia JR. Breaking barriers for T cells by targeting the EPHA2/TGF-beta/COX-2 axis in pancreatic cancer. J Clin Invest. 2019;129(9):3521-3.
232. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, Wang X, Zhang J, Zhang X, Zheng L, et al. A RIPK3-PGE(2) circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res. 2018;78(19):5586-99.
233. Take Y, Koizumi S, Nagahisa A. Prostaglandin E receptor 4 antagonist in cancer immunotherapy: mechanisms of action. Front Immunol. 2020;11:324.
234. Clemente SM, Martinez-Costa OH, Monsalve M, Samhan-Arias AK. Targeting lipid peroxidation for cancer treatment. Molecules. 2020;25(21).
235. Lin H, Weng J, Mei H, Zhuang M, Xiao X, Du F, Lin L, Wu J, Chen Z, Huang Y, et al. 5-Lipoxygenase promotes epithelial-mesenchymal transition through the ERK signaling pathway in gastric cancer. J Gastroenterol Hepatol. 2021;36(2):455-66.
236. Xia C, Sadeghi L, Straat K, Merrien M, Wright AP, Sander B, Xu D, Osterborg A, Bjorkholm M, Claesson HE. Intrinsic 5-lipoxygenase activity regulates migration and adherence of mantle cell lymphoma cells. Prostaglandins Other Lipid Mediat. 2021;156: 106575.
237. Muthuraman S, Sinha S, Vasavi CS, Waidha KM, Basu B, Munussami P, Balamurali MM, Doble M, Saravana Kumar R. Design, synthesis and identification of novel coumaperine derivatives for inhibition of human 5-LOX: Antioxidant, pseudoperoxidase and docking studies. Bioorg Med Chem. 2019;27(4):604-19.
238. Doiphode S, Lokhande KB, Ghosh P, Swamy KV, Nagar S. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) by resveratrol derivatives in cancer therapy: in silico approach. J Biomol Struct Dyn. 2022:1-16.
239. Shi HY, Lv FJ, Zhu ST, Wang QG, Zhang ST. Dual inhibition of 5-LOX and COX-2 suppresses esophageal squamous cell carcinoma. Cancer Lett. 2011;309(1):19-26.
240. Chen S, Zou H. Key role of 12 -lipoxygenase and its metabolite 12-hydroxyeicosatetraenoic acid (12-HETE) in diabetic retinopathy. Curr Eye Res. 2022;47(3):329-35.
241. Liu Q, Tan W, Che J, Yuan D, Zhang L, Sun Y, Yue X, Xiao L, Jin Y. 12-HETE facilitates cell survival by activating the integrin-linked kinase/NF-kappaB pathway in ovarian cancer. Cancer Manag Res. 2018;10:5825-38.
242. Mao F, Wang M, Wang J, Xu WR. The role of 15-LOX-1 in colitis and colitis-associated colorectal cancer. Inflamm Res. 2015;64(9):661-9.
243. Kazan HH, Urfali-Mamatoglu C, Yalcin GD, Bulut O, Sezer A, Banerjee S, Gunduz U. 15-LOX-1 has diverse roles in the resensitization of resistant cancer cell lines to doxorubicin. J Cell Physiol. 2020;235(5):4965-78.
244. Na YJ, Kim BR, Kim JL, Kang S, Jeong YA, Park SH, Jo MJ, Kim JY, Kim HJ, Oh SC et al. Deficiency of 15-LOX-1 induces radioresistance through downregulation of MacroH2A2 in colorectal cancer. Cancers (Basel). 2019;11(11).
245. Berger A. What are leukotrienes and how do they work in asthma? BMJ. 1999;319(7202):90.
246. Bachi AL, Kim FJ, Nonogaki S, Carneiro CR, Lopes JD, Jasiulionis MG, Correa M. Leukotriene B4 creates a favorable microenvironment for murine melanoma growth. Mol Cancer Res. 2009;7(9):1417-24.
247. Wendel A, Tiegs G. Leukotriene D4 mediates galactosamine/endotoxininduced hepatitis in mice. Biochem Pharmacol. 1987;36(12):1867.
248. Zhou Y, Guo D, Li H, Jie S. Circulating LTD4 in patients with hepatocellular carcinoma. Tumour Biol. 2011;32(1):139-44.
249. Arai J, Goto K, Otoyama Y, Nakajima Y, Sugiura I, Kajiwara A, Tojo M, Ichikawa Y, Uozumi S, Shimozuma Y, et al. Leukotriene receptor antagonists enhance HCC treatment efficacy by inhibiting ADAMs and suppressing MICA shedding. Cancer Immunol Immunother. 2021;70(1):203-13.
250. Wang Z, Cheng Q, Tang K, Sun Y, Zhang K, Zhang Y, Luo S, Zhang H, Ye D, Huang B. Lipid mediator lipoxin A4 inhibits tumor growth by targeting IL-10-producing regulatory B (Breg) cells. Cancer Lett. 2015;364(2):118-24.
251. Serhan CN. The resolution of inflammation: the devil in the flask and in the details. FASEB J. 2011;25(5):1441-8.
252. Gleeson MJ, Felix H, Johnsson LG. Ultrastructural aspects of the human peripheral vestibular system. Acta Otolaryngol Suppl. 1990;470:80-7.
253. Liu H, Zeng J, Huang W, Xu Q, Ye D, Sun R, Zhang D. Colorectal cancer is associated with a deficiency of lipoxin A(4), an endogenous anti-inflammatory mediator. J Cancer. 2019;10(19):4719-30.
254. Jia G, Wang X, Wu W, Zhang Y, Chen S, Zhao J, Zhao W, Li W, Sun X, Han B. LXA4 enhances prostate cancer progression by facilitating M2 macrophage polarization via inhibition of METTL3. Int Immunopharmacol. 2022;107: 108586.
255. Yuan J, Lin F, Chen L, Chen W, Pan X, Bai Y, Cai Y, Lu H. Lipoxin A4 regulates M1/M2 macrophage polarization via FPR2-IRF pathway. Inflammopharmacology. 2022;30(2):487-98.
256. Korbecki J, Rebacz-Maron E, Kupnicka P, Chlubek D, BaranowskaBosiacka I. Synthesis and significance of arachidonic acid, a substrate for cyclooxygenases, lipoxygenases, and cytochrome p450 pathways in the tumorigenesis of glioblastoma multiforme, including a pan-cancer comparative analysis. Cancers (Basel). 2023;15(3).
257. Lakshmanan K, Byran G, Bandlamudi S, Krishnamurthy PT. The role of STAT3 signaling in different types of cancers: a comprehensive review. Curr Enzym Inhib. 2020;16(3):189-98.
258. Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol Rev. 2020;72(2):486-526.
259. Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, Ma H, Wei D, Sun S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. 2020;80: 106210.
260. Wong ALA, Hirpara JL, Pervaiz S, Eu JQ, Sethi G, Goh BC. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin Investig Drugs. 2017;26(8):883-7.
261. Hirano T, Hirayama D, Wagatsuma K, Yamakawa T, Yokoyama Y, Nakase H: Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. Int J Mol Sci. 2020;21(9).
262. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119-24.
263. Kasembeli MM, Bharadwaj U, Robinson P, Tweardy DJ. Contribution of STAT3 to inflammatory and fibrotic diseases and prospects for its targeting for treatment. Int J Mol Sci. 2018; 19(8).
264. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103-13.
265. Gargalionis AN, Papavassiliou KA, Papavassiliou AG. Targeting STAT3 signaling pathway in colorectal cancer. Biomedicines. 2021; 9(8).
266. Zhong B, Cheng B, Huang X, Xiao Q, Niu Z, Chen YF, Yu Q, Wang W, Wu XJ . Colorectal cancer-associated fibroblasts promote metastasis by upregulating LRG1 through stromal IL-6/STAT3 signaling. Cell Death Dis. 2021;13(1):16.
267. Wang S, Dong W, Liu L, Xu M, Wang Y, Liu T, Zhang Y, Wang B, Cao H. Interplay between bile acids and the gut microbiota promotes intestinal carcinogenesis. Mol Carcinog. 2019;58(7):1155-67.
268. Roche B, Vanden-Bossche A, Normand M, Malaval L, Vico L, LafageProust MH. Validated laser doppler protocol for measurement of mouse bone blood perfusion-response to age or ovariectomy differs with genetic background. Bone. 2013;55(2):418-26.
269. Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci. 2014;19(2):164-74.
270. D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813-24.
271. Florean
272. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245-313.
273. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. 2019;19(18): e1800311.
274. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, Li B. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12(1):34.
275. Prasad AS, Miale A Jr, Farid Z, Sandstead HH, Schulert AR. Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypognadism. J Lab Clin Med. 1963;61:537-49.
276. Liu MJ, Bao S, Galvez-Peralta M, Pyle CJ, Rudawsky AC, Pavlovicz RE, Killilea DW, Li C, Nebert DW, Wewers MD, et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-kappaB. Cell Rep. 2013;3(2):386-400.
277. Burn J, Sheth H, Elliott F, Reed L, Macrae F, Mecklin JP, Moslein G, McRonald FE, Bertario L, Evans DG, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet. 2020;395(10240):1855-63.
278. Bens A, Cronin-Fenton D, Dehlendorff C, Jensen MB, Ejlertsen B, Kroman N, Friis S, Mellemkjaer L. Nonaspirin NSAIDs and contralateral breast cancer risk. Int J Cancer. 2019;144(6):1243-50.
279. Hao W, Shen Y, Feng M, Wang H, Lin M, Fang Y, Tan L. Aspirin acts in esophageal cancer: a brief review. J Thorac Dis. 2018;10(4):2490-7.
280. Burn J, Bishop DT, Chapman PD, Elliott F, Bertario L, Dunlop MG, Eccles D, Ellis A, Evans DG, Fodde R, et al. A randomized placebocontrolled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev Res (Phila). 2011;4(5):655-65.
281. Ishikawa H, Wakabayashi K, Suzuki S, Mutoh M, Hirata K, Nakamura T, Takeyama I, Kawano A, Gondo N, Abe T, et al. Preventive effects of lowdose aspirin on colorectal adenoma growth in patients with familial adenomatous polyposis: double-blind, randomized clinical trial. Cancer Med. 2013;2(1):50-6.
282. Jankowski JAZ, de Caestecker J, Love SB, Reilly G, Watson P, Sanders S, Ang Y, Morris D, Bhandari P, Brooks C, et al. Esomeprazole and aspirin
in Barrett’s oesophagus (AspECT): a randomised factorial trial. Lancet. 2018;392(10145):400-8.
283. Wu QB, Sun GP. Expression of COX-2 and HER-2 in colorectal cancer and their correlation. World J Gastroenterol. 2015;21(20):6206-14.
284. Sheng J, Sun H, Yu FB, Li B, Zhang Y, Zhu YT. The role of cyclooxyge-nase-2 in colorectal cancer. Int J Med Sci. 2020;17(8):1095-101.
285. Todoric J, Antonucci L, Karin M. Targeting inflammation in cancer prevention and therapy. Cancer Prev Res (Phila). 2016;9(12):895-905.
286. North GL. Celecoxib as adjunctive therapy for treatment of colorectal cancer. Ann Pharmacother. 2001;35(12):1638-43.
287. Mostafa TM, Alm El-Din MA, Rashdan AR. Celecoxib as an adjuvant to chemotherapy for patients with metastatic colorectal cancer: a randomized controlled clinical study. Saudi Med J. 2022;43(1):37-44.
288. Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355(9):873-84.
289. Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J, Zavoral M, Lechuga MJ, Gerletti P, Tang J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355(9):885-95.
290. Hu H, Kang L, Zhang J, Wu Z, Wang H, Huang M, Lan P, Wu X, Wang C, Cao W, et al. Neoadjuvant PD-1 blockade with toripalimab, with or without celecoxib, in mismatch repair-deficient or microsatellite instability-high, locally advanced, colorectal cancer (PICC): a single-centre, parallel-group, non-comparative, randomised, phase 2 trial. Lancet Gastroenterol Hepatol. 2022;7(1):38-48.
291. Ye SY, Li JY, Li TH, Song YX, Sun JX, Chen XW, Zhao JH, Li Y, Wu ZH, Gao P , et al. The efficacy and safety of celecoxib in addition to standard cancer therapy: a systematic review and meta-analysis of randomized controlled trials. Curr Oncol. 2022;29(9):6137-53.
292. Guo Q, Liu X, Lu L, Yuan H, Wang Y, Chen Z, Ji R, Zhou Y. Comprehensive evaluation of clinical efficacy and safety of celecoxib combined with chemotherapy in management of gastric cancer. Medicine (Baltimore). 2017;96(51): e8857.
293. Liao Z, Komaki R, Milas L, Yuan C, Kies M, Chang JY, Jeter M, Guerrero T, Blumenschien G, Smith CM, et al. A phase I clinical trial of thoracic radiotherapy and concurrent celecoxib for patients with unfavorable performance status inoperable/unresectable non-small cell lung cancer. Clin Cancer Res. 2005;11(9):3342-8.
294. Koch A, Bergman B, Holmberg E, Sederholm C, Ek L, Kosieradzki J, Lamberg K, Thaning L, Ydreborg SO, Sorenson S, et al. Effect of celecoxib on survival in patients with advanced non-small cell lung cancer: a double blind randomised clinical phase III trial (CYCLUS study) by the Swedish Lung Cancer Study Group. Eur J Cancer. 2011;47(10):1546-55.
295. Bi N, Liang J, Zhou Z, Chen D, Fu Z, Yang X, Feng Q, Hui Z, Xiao Z, Lv J, et al. Effect of concurrent chemoradiation with celecoxib vs concurrent chemoradiation alone on survival among patients with non-small cell lung cancer with and without cyclooxygenase 2 genetic variants: a phase 2 randomized clinical trial. JAMA Netw Open. 2019;2(12): e1918070.
296. Bayraktar S, Baghaki S, Wu J, Liu DD, Gutierrez-Barrera AM, Bevers TB, Valero V, Sneige N, Arun BK. Biomarker modulation study of celecoxib for chemoprevention in women at increased risk for breast cancer: a phase II pilot study. Cancer Prev Res (Phila). 2020;13(9):795-802.
297. Guo Q, Li Q, Wang J, Liu M, Wang Y, Chen Z, Ye Y, Guan Q, Zhou Y. A comprehensive evaluation of clinical efficacy and safety of celecoxib in combination with chemotherapy in metastatic or postoperative recurrent gastric cancer patients: a preliminary, three-center, clinical trial study. Medicine (Baltimore). 2019;98(27): e16234.
298. De Cremoux P, Hamy AS, Lehmann-Che J, Scott V, Sigal B, Mathieu MC, Bertheau P, Guinebretiere JM, Pierga JY, Giacchetti S, et al. COX2/PTGS2 expression is predictive of response to neoadjuvant celecoxib in HER2negative breast cancer patients. Anticancer Res. 2018;38(3):1485-90.
299. Meyerhardt JA, Shi Q, Fuchs CS, Meyer J, Niedzwiecki D, Zemla T, Kumthekar P, Guthrie KA, Couture F, Kuebler P, et al. Effect of celecoxib vs placebo added to standard adjuvant therapy on disease-free survival among patients with stage III colon cancer: the CALGB/SWOG 80702 (alliance) randomized clinical trial. JAMA. 2021;325(13):1277-86.
300. Coombes RC, Tovey H, Kilburn L, Mansi J, Palmieri C, Bartlett J, Hicks J, Makris A, Evans A, Loibl S, et al. Effect of celecoxib vs placebo as adjuvant therapy on disease-free survival among patients with
breast cancer: the REACT randomized clinical trial. JAMA Oncol. 2021;7(9):1291-301.
301. Hamy AS, Tury S, Wang X, Gao J, Pierga JY, Giacchetti S, Brain E, Pistilli B, Marty M, Espie M, et al. Celecoxib with neoadjuvant chemotherapy for breast cancer might worsen outcomes differentially by COX-2 expression and ER status: exploratory analysis of the REMAGUS02 trial. J Clin Oncol. 2019;37(8):624-35.
302. Schjerning AM, McGettigan P, Gislason G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat Rev Cardiol. 2020;17(9):574-84.
303. Cui J, Jia J. Natural COX-2 inhibitors as promising anti-inflammatory agents: an update. Curr Med Chem. 2021;28(18):3622-46.
304. Yang L, Zou T, Chen Y, Zhao Y, Wu X, Li M, Du F, Chen Y, Xiao Z, Shen J. Hepatitis
305. European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L: EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370-98.
306. Hou JL, Zhao W, Lee C, Hann HW, Peng CY, Tanwandee T, Morozov V, Klinker H, Sollano JD, Streinu-Cercel A, et al. Outcomes of long-term treatment of chronic HBV infection with entecavir or other agents from a randomized trial in 24 countries. Clin Gastroenterol Hepatol. 2020;18(2):457-67.
307. Choi J, Kim HJ, Lee J, Cho S, Ko MJ, Lim YS. Risk of hepatocellular carcinoma in patients treated with entecavir vs tenofovir for chronic hepatitis B: a Korean nationwide cohort study. JAMA Oncol. 2019;5(1):30-6.
308. Zhang Z, Zhou Y, Yang J, Hu K, Huang Y. The effectiveness of TDF versus ETV on incidence of HCC in CHB patients: a meta analysis. BMC Cancer. 2019;19(1):511.
309. Tan DJH, Ng CH, Tay PWL, Syn N, Muthiah MD, Lim WH, Tang ASP, Lim KE, Lim GEH, Tamaki N, et al. Risk of hepatocellular carcinoma with tenofovir vs entecavir treatment for chronic hepatitis B virus: a reconstructed individual patient data meta-analysis. JAMA Netw Open. 2022;5(6): e2219407.
310. Pol S. group AAs: Similar 5-year HCC occurrence in Tenofovir- and Entecavir-treated HBV chronic infection in the French AFEF/ANRS CO22 Hepather cohort. Aliment Pharmacol Ther. 2021;53(5):616-29.
311. Chang TS, Yang YH, Chen WM, Shen CH, Tung SY, Yen CW, Hsieh YY, Lee CP, Tsai ML, Hung CH, et al. Long-term risk of primary liver cancers in entecavir versus tenofovir treatment for chronic hepatitis B. Sci Rep. 2021;11(1):1365.
312. Xing Y, Zhong W, Peng D, Han Z, Zeng H, Wang Y, Feng L, Huang J, Xu L, Chen
313. Ji D, Chen Y, Bi J, Shang Q, Liu H, Wang JB, Tan L, Wang J, Chen Y, Li Q, et al. Entecavir plus Biejia-Ruangan compound reduces the risk of hepatocellular carcinoma in Chinese patients with chronic hepatitis B. J Hepatol. 2022;77(6):1515-24.
314. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394-424.
315. Nahand JS, Taghizadeh-Boroujeni S, Karimzadeh M, Borran S, Pourhanifeh MH, Moghoofei M, Bokharaei-Salim F, Karampoor S, Jafari A, Asemi Z, et al. microRNAs: new prognostic, diagnostic, and therapeutic biomarkers in cervical cancer. J Cell Physiol. 2019;234(10):17064-99.
316. Sadri Nahand J, Moghoofei M, Salmaninejad A, Bahmanpour Z, Karimzadeh M, Nasiri M, Mirzaei HR, Pourhanifeh MH, Bokharaei-Salim F, Mirzaei H, et al. Pathogenic role of exosomes and microRNAs in HPV-mediated inflammation and cervical cancer: a review. Int J Cancer. 2020;146(2):305-20.
317. Laniewski P, Barnes D, Goulder A, Cui H, Roe DJ, Chase DM, HerbstKralovetz MM. Linking cervicovaginal immune signatures, HPV and microbiota composition in cervical carcinogenesis in non-Hispanic and Hispanic women. Sci Rep. 2018;8(1):7593.
318. KempTJ, Hildesheim A, Garcia-Pineres A, Williams MC, Shearer GM, Rodriguez AC, Schiffman M, Burk R, Freer E, Bonilla J, et al. Elevated systemic levels of inflammatory cytokines in older women with persistent
cervical human papillomavirus infection. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1954-9.
319. Yost S, Hoekstra A. Cervical cancer in women over 65: an analysis of screening. Gynecol Oncol Rep. 2018;25:48-51.
320. Sales KJ, Katz AA. Inflammatory pathways in cervical cancer-the UCT contribution. S Afr Med J. 2012;102(6):493-6.
321. Drolet M, Benard E, Perez N, Brisson M. Group HPVVIS: Population-level impact and herd effects following the introduction of human papillomavirus vaccination programmes: updated systematic review and meta-analysis. Lancet. 2019;394(10197):497-509.
322. Lei J, Ploner A, Elfstrom KM, Wang J, Roth A, Fang F, Sundstrom K, Dillner J, Sparen P. HPV vaccination and the risk of invasive cervical cancer. N Engl J Med. 2020;383(14):1340-8.
323. Brisson M, Kim JJ, Canfell K, Drolet M, Gingras G, Burger EA, Martin D, Simms KT, Benard E, Boily MC, et al. Impact of HPV vaccination and cervical screening on cervical cancer elimination: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet. 2020;395(10224):575-90.
324. Komdeur FL, Singh A, van de Wall S, Meulenberg JJM, Boerma A, Hoogeboom BN, Paijens ST, Oyarce C, de Bruyn M, Schuuring E, et al. First-in-human phase I clinical trial of an SFV-based RNA replicon cancer vaccine against HPV-induced cancers. Mol Ther. 2021;29(2):611-25.
325. Porras C, Tsang SH, Herrero R, Guillen D, Darragh TM, Stoler MH, Hildesheim A, Wagner S, Boland J, Lowy DR, et al. Efficacy of the bivalent HPV vaccine against HPV 16/18-associated precancer: longterm follow-up results from the Costa Rica Vaccine Trial. Lancet Oncol. 2020;21(12):1643-52.
326. Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, Feng L, Lee JJ, Tran H, Kim YU, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5(1):67-73.
327. Bernhard H, Jager-Arand E, Bernhard G, Heike M, Klein O, Riemann JF, Meyer zum Buschenfelde KH, Dippold W, Knuth A. Treatment of advanced pancreatic cancer with 5-fluorouracil, folinic acid and interferon alpha-2A: results of a phase II trial. Br J Cancer. 1995;71(1):102-5.
328. David AK, Vaughn DJ, Holroyde CP, Armstead B, Haller DG. A phase II trial of 5-fluorouracil, leucovorin, and interferon alpha 2A (IFN-alpha 2a) in metastatic pancreatic carcinoma: a Penn Cancer Clinical Trials Group (PCCTG) trial. Am J Clin Oncol. 2000;23(1):37-9.
329. Ohman KA, Liu J, Linehan DC, Tan MC, Tan BR, Fields RC, Strasberg SM, Hawkins WG. Interferon-based chemoradiation followed by gemcitabine for resected pancreatic adenocarcinoma: long-term follow-up. HPB (Oxford). 2017;19(5):449-57.
330. Rocha FG, Hashimoto Y, Traverso LW, Dorer R, Kozarek R, Helton WS, Picozzi VJ. Interferon-based adjuvant chemoradiation for resected pancreatic head cancer: long-term follow-up of the Virginia mason protocol. Ann Surg. 2016;263(2):376-84.
331. Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14(1):7-17.
332. Eigentler TK, Gutzmer R, Hauschild A, Heinzerling L, Schadendorf D, Nashan D, Holzle E, Kiecker F, Becker J, Sunderkotter C, et al. Adjuvant treatment with pegylated interferon alpha-2a versus low-dose interferon alpha-2a in patients with high-risk melanoma: a randomized phase III DeCOG trial. Ann Oncol. 2016;27(8):1625-32.
333. Ives NJ, Suciu S, Eggermont AMM, Kirkwood J, Lorigan P, Markovic SN, Garbe C, Wheatley K. International Melanoma Meta-Analysis Collaborative G: Adjuvant interferon-alpha for the treatment of high-risk melanoma: An individual patient data meta-analysis. Eur J Cancer. 2017;82:171-83.
334. Yamazaki N, Uhara H, Wada H, Matsuda K, Yamamoto K, Shimamoto T, Kiyohara Y. Phase I study of pegylated interferon-alpha-2b as an adjuvant therapy in Japanese patients with malignant melanoma. J Dermatol. 2016;43(10):1146-53.
335. Dummer R, Mangana J. Long-term pegylated interferon-alpha and its potential in the treatment of melanoma. Biologics. 2009;3:169-82.
336. Najjar YG, Puligandla M, Lee SJ, Kirkwood JM. An updated analysis of 4 randomized ECOG trials of high-dose interferon in the adjuvant treatment of melanoma. Cancer. 2019;125(17):3013-24.
337. Eggermont AM, Suciu S, Testori A, Santinami M, Kruit WH, Marsden J, Punt CJ, Sales F, Dummer R, Robert C, et al. Long-term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma. J Clin Oncol. 2012;30(31):3810-8.
338. Bottomley A, Coens C, Suciu S, Santinami M, Kruit W, Testori A, Marsden J, Punt C, Sales F, Gore M, et al. Adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma: a phase III randomized controlled trial of health-related quality of life and symptoms by the European Organisation for Research and Treatment of Cancer Melanoma Group. J Clin Oncol. 2009;27(18):2916-23.
339. Flaherty LE, Othus M, Atkins MB, Tuthill RJ, Thompson JA, Vetto JT, Haluska FG, Pappo AS, Sosman JA, Redman BG, et al. Southwest Oncology Group S0008: a phase III trial of high-dose interferon Alfa-2b versus cisplatin, vinblastine, and dacarbazine, plus interleukin-2 and interferon in patients with high-risk melanoma-an intergroup study of cancer and leukemia Group B, Children’s Oncology Group, Eastern Cooperative Oncology Group, and Southwest Oncology Group. J Clin Oncol. 2014;32(33):3771-8.
340. Simeone E, Scognamiglio G, Capone M, Giannarelli D, Grimaldi AM, Mallardo D, Madonna G, Curvietto M, Esposito A, Sandomenico F, et al. A monocentric phase I study of vemurafenib plus cobimetinib plus PEGinterferon (VEMUPLINT) in advanced melanoma patients harboring the V600BRAF mutation. J Transl Med. 2021;19(1):17.
341. Magenau JM, Peltier D, Riwes M, Pawarode A, Parkin B, Braun T, Anand S, Ghosh M, Maciejewski J, Yanik G, et al. Type 1 interferon to prevent leukemia relapse after allogeneic transplantation. Blood Adv. 2021;5(23):5047-56.
342. Mo XD, Zhang XH, Xu LP, Wang Y, Yan CH, Chen H, Chen YH, Han W, Wang FR, Wang JZ, et al. IFN-alpha is effective for treatment of minimal residual disease in patients with acute leukemia after allogeneic hematopoietic stem cell transplantation: results of a registry study. Biol Blood Marrow Transplant. 2017;23(8):1303-10.
343. Kankuri-Tammilehto M, Perasto L, Pyrhonen S, Salminen E. Longterm outcome with prolonged use of interferon-alpha administered intermittently for metastatic renal cell carcinoma: a phase II study. Anticancer Res. 2023;43(6):2645-57.
344. Eto M, Kawano Y, Hirao Y, Mita K, Arai Y, Tsukamoto T, Hashine K, Matsubara A, Fujioka T, Kimura G, et al. Phase II clinical trial of sorafenib plus interferon-alpha treatment for patients with metastatic renal cell carcinoma in Japan. BMC Cancer. 2015;15:667.
345. Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Ou SS, Archer L, Atkins JN, Picus J, Czaykowski P, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol. 2008;26(33):5422-8.
346. Rafique I, Kirkwood JM, Tarhini AA. Immune checkpoint blockade and interferon-alpha in melanoma. Semin Oncol. 2015;42(3):436-47.
347. Wang H, Xia L, Yao CC, Dong H, Yang Y, Li C, Ji WX, Sun RM, Duan HQ, Mengzhou W, et al. NLRP4 negatively regulates type I interferon response and influences the outcome in anti-programmed cell death protein (PD)-1/PD-ligand 1 therapy. Cancer Sci. 2022;113(3):838-51.
348. Brohl AS, Khushalani NI, Eroglu Z, Markowitz J, Thapa R, Chen YA, Kudchadkar R, Weber JS. A phase IB study of ipilimumab with peginterferon alfa-2b in patients with unresectable melanoma. J Immunother Cancer. 2016;4:85.
349. Tarhini A, Lin Y, Lin H, Rahman Z, Vallabhaneni P, Mendiratta P, Pingpank JF, Holtzman MP, Yusko EC, Rytlewski JA, et al. Neoadjuvant ipilimumab (
350. Trinh KR, Vasuthasawat A, Steward KK, Yamada RE, Timmerman JM, Morrison SL. Anti-CD20-interferon-beta fusion protein therapy of murine B-cell lymphomas. J Immunother. 2013;36(5):305-18.
351. Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood. 2010;115(14):2864-71.
352. Li Z, Zhu Y, Li C, Trinh R, Ren X, Sun F, Wang Y, Shang P, Wang T, Wang M, et al. Anti-VEGFR2-interferon-alpha2 regulates the tumor microenvironment and exhibits potent antitumor efficacy against colorectal cancer. Oncoimmunology. 2017;6(3): e1290038.
353. Green DS, Husain SR, Johnson CL, Sato Y, Han J, Joshi B, Hewitt SM, Puri RK, Zoon KC. Combination immunotherapy with IL-4 Pseudomonas exotoxin and IFN-alpha and IFN-gamma mediate antitumor effects in vitro and in a mouse model of human ovarian cancer. Immunotherapy. 2019;11(6):483-96.
354. Duggan MC, Jochems C, Donahue RN, Richards J, Karpa V, Foust E, Paul B, Brooks T, Tridandapani S, Olencki T, et al. A phase I study of recombinant (
355. Sheng
356. Gigante M, Mandic M, Wesa AK, Cavalcanti E, Dambrosio M, Mancini V, Battaglia M, Gesualdo L, Storkus WJ, Ranieri E. Interferon-alpha (IFN-alpha)-conditioned DC preferentially stimulate type-1 and limit Treg-type in vitro T-cell responses from RCC patients. J Immunother. 2008;31(3):254-62.
357. Rozera C, Cappellini GA, D’Agostino G, Santodonato L, Castiello L, Urbani F, Macchia I, Arico E, Casorelli I, Sestili P, et al. Intratumoral injection of IFN-alpha dendritic cells after dacarbazine activates anti-tumor immunity: results from a phase I trial in advanced melanoma. J Transl Med. 2015;13:139.
358. Cox MC, Lapenta C, Santini SM. Advances and perspectives of dendritic cell-based active immunotherapies in follicular lymphoma. Cancer Immunol Immunother. 2020;69(6):913-25.
359. Derynck R, Turley SJ, Akhurst RJ. TGFbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18(1):9-34.
360. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, Hsu FJ, Berzofsky JA, Lawrence DP. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE. 2014;9(3): e90353.
361. Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, Ratikan JA, Felix C, Hwang L, Faull KF, et al. Focal irradiation and systemic TGFbeta blockade in metastatic breast cancer. Clin Cancer Res. 2018;24(11):2493-504
362. Faivre S, Santoro A, Kelley RK, Gane E, Costentin CE, Gueorguieva I, Smith C, Cleverly A, Lahn MM, Raymond E, et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019;39(8):1468-77.
363. Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, Hourmand IO, Cleverly A, Zhao Y, Gueorguieva I, et al. A phase 2 study of galunisertib (TGF-beta1 receptor type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin Transl Gastroenterol. 2019;10(7): e00056.
364. Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, Trojan J, Oettle H, Kozloff M, Cleverly A, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 2018;119(10):1208-14.
365. Melisi D, Oh DY, Hollebecque A, Calvo E, Varghese A, Borazanci E, Macarulla T, Merz V, Zecchetto C, Zhao Y et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer. 2021;9(3).
366. Nadal E, Saleh M, Aix SP, Ochoa-de-Olza M, Patel SP, Antonia S, Zhao Y, Gueorguieva I, Man M, Estrem ST, et al. A phase Ib/II study of galunisertib in combination with nivolumab in solid tumors and non-small cell lung cancer. BMC Cancer. 2023;23(1):708.
367. Yamazaki T, Gunderson AJ, Gilchrist M, Whiteford M, Kiely MX, Hayman A, O’Brien D, Ahmad R, Manchio JV, Fox N, et al. Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: a single-arm, phase 2 trial. Lancet Oncol. 2022;23(9):1189-200.
368. Necchi A, Giannatempo P, Mariani L, Fare E, Raggi D, Pennati M, Zaffaroni N, Crippa F, Marchiano A, Nicolai N, et al. PF-03446962, a fully-human monoclonal antibody against transforming growth-factor beta (TGFbeta) receptor ALK1, in pre-treated patients with urothelial cancer: an open label, single-group, phase 2 trial. Invest New Drugs. 2014;32(3):555-60.
369. Simonelli M, Zucali P, Santoro A, Thomas MB, de Braud FG, Borghaei H, Berlin J, Denlinger CS, Noberasco C, Rimassa L, et al. Phase I study
of PF-03446962, a fully human monoclonal antibody against activin receptor-like kinase-1, in patients with hepatocellular carcinoma. Ann Oncol. 2016;27(9):1782-7.
370. Wheatley-Price P, Chu Q, Bonomi M, Seely J, Gupta A, Goss G, Hilton J, Feld R, Lee CW, Goffin JR, et al. A phase II study of PF-03446962 in patients with advanced malignant pleural mesothelioma. CCTG trial IND207. J Thorac Oncol. 2016;11(11):218-21.
371. Goff LW, Cohen RB, Berlin JD, de Braud FG, Lyshchik A, Noberasco C, Bertolini F, Carpentieri M, Stampino CG, Abbattista A, et al. A phase I study of the anti-activin receptor-like kinase 1 (ALK-1) monoclonal antibody PF-03446962 in patients with advanced solid tumors. Clin Cancer Res. 2016;22(9):2146-54.
372. Clarke JM, Blobe GC, Strickler JH, Uronis HE, Zafar SY, Morse M, Dropkin E, Howard L, O’Neill M, Rushing CN, et al. A phase Ib study of the combination regorafenib with PF-03446962 in patients with refractory metastatic colorectal cancer (REGAL-1 trial). Cancer Chemother Pharmacol. 2019;84(4):909-17.
373. Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, Lan Y, Dussault I, Moustakas A. Dual inhibition of TGF-beta and PD-L1: a novel approach to cancer treatment. Mol Oncol. 2022;16(11):2117-34.
374. Paz-Ares L, Kim TM, Vicente D, Felip E, Lee DH, Lee KH, Lin CC, Flor MJ, Di Nicola M, Alvarez RM, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in second-line treatment of patients with NSCLC: results from an expansion cohort of a phase 1 trial. J Thorac Oncol. 2020;15(7):1210-22.
375. Lin CC, Doi T, Muro K, Hou MM, Esaki T, Hara H, Chung HC, Helwig C, Dussault I, Osada M, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGFbeta and PD-L1, in patients with esophageal squamous cell carcinoma: results from a phase 1 cohort in Asia. Target Oncol. 2021;16(4):447-59.
376. Tan B, Khattak A, Felip E, Kelly K, Rich P, Wang D, Helwig C, Dussault I, Ojalvo LS, Isambert N. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with esophageal adenocarcinoma: results from a phase 1 cohort. Target Oncol. 2021;16(4):435-46.
377. Cho BC, Daste A, Ravaud A, Salas S, Isambert N, McClay E, Awada A, Borel C, Ojalvo LS, Helwig C et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in advanced squamous cell carcinoma of the head and neck: results from a phase I cohort. J Immunother Cancer. 2020;8(2).
378. Strauss J, Gatti-Mays ME, Cho BC, Hill A, Salas S, McClay E, Redman JM, Sater HA, Donahue RN, Jochems C et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with human papillomavirus-associated malignancies. J Immunother Cancer. 2020;8(2).
379. Redman JM, Friedman J, Robbins Y, Sievers C, Yang X, Lassoued W, Sinkoe A, Papanicolau-Sengos A, Lee CC, Marte JL et al. Enhanced neoepitope-specific immunity following neoadjuvant PD-L1 and TGFbeta blockade in HPV-unrelated head and neck cancer. J Clin Invest. 2022;132(18).
380. Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, Lazorchak A, Yeung TL, Hasheminasab SM, Jenkins MH, Meister S, et al. Simultaneous targeting of TGF-beta/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell. 2021;39(10):1388-403.
381. Singh S, Xiao Z, Bavisi K, Roszik J, Melendez BD, Wang Z, Cantwell MJ, Davis RE, Lizee G, Hwu P, et al. IL-1alpha mediates innate and acquired resistance to immunotherapy in melanoma. J Immunol. 2021;206(8):1966-75.
382. Aggen DH, Ager CR, Obradovic AZ, Chowdhury N, Ghasemzadeh A, Mao W, Chaimowitz MG, Lopez-Bujanda ZA, Spina CS, Hawley JE, et al. Blocking IL1 beta promotes tumor regression and remodeling of the myeloid compartment in a renal cell carcinoma model: multidimensional analyses. Clin Cancer Res. 2021;27(2):608-21.
383. Wong CC, Baum J, Silvestro A, Beste MT, Bharani-Dharan B, Xu S, Wang YA, Wang X, Prescott MF, Krajkovich L, et al. Inhibition of IL1 beta by canakinumab may be effective against diverse molecular subtypes of lung cancer: an exploratory analysis of the CANTOS trial. Cancer Res. 2020;80(24):5597-605.
384. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, Group CT. Effect of interleukin-1 beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results
from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833-42.
385. Garrido P, Pujol JL, Kim ES, Lee JM, Tsuboi M, Gomez-Rueda A, Benito A, Moreno N, Gorospe L, Dong T, et al. Canakinumab with and without pembrolizumab in patients with resectable non-small-cell lung cancer: CANOPY-N study design. Future Oncol. 2021;17(12):1459-72.
386. Yuan B, Clowers MJ, Velasco WV, Peng S, Peng Q, Shi Y, Ramos-Castaneda M, Zarghooni M, Yang S, Babcock RL et al. Targeting IL-1 beta as an immunopreventive and therapeutic modality for K-ras-mutant lung cancer. JCl Insight. 2022;7(11).
387. Wu TC, Xu K, Martinek J, Young RR, Banchereau R, George J, Turner J, Kim KI, Zurawski S, Wang X, et al. IL1 Receptor antagonist controls transcriptional signature of inflammation in patients with metastatic breast cancer. Cancer Res. 2018;78(18):5243-58.
388. Lust JA, Lacy MQ, Zeldenrust SR, Witzig TE, Moon-Tasson LL, Dinarello CA, Donovan KA. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am J Hematol. 2016;91(6):571-4.
389. Becerra C, Paulson AS, Cavaness KM, Celinski SA. Gemcitabine, nabpaclitaxel, cisplatin, and anakinra (AGAP) treatment in patients with localized pancreatic ductal adenocarcinoma (PDAC). J Clin Oncol. 2018;36(4_suppl):449-449.
390. Voigt C, May P, Gottschlich A, Markota A, Wenk D, Gerlach I, Voigt S, Stathopoulos GT, Arendt KAM, Heise C, et al. Cancer cells induce inter-leukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc Natl Acad Sci USA. 2017;114(49):12994-9.
391. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, et al. Chemotherapy-triggered cathepsin
392. Isambert N, Hervieu A, Rebe C, Hennequin A, Borg C, Zanetta S, Chevriaux A, Richard C, Derangere V, Limagne E, et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): a single-arm phase 2 study. Oncoimmunology. 2018;7(9): e1474319.
393. Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, Kumar S, Hayman SR, Russell SJ, Buadi FK, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1beta-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009;84(2):114-22.
394. Briukhovetska D, Dorr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in cancer: from biology to therapy. Nat Rev Cancer. 2021;21(8):481-99.
395. Waldmann TA. Cytokines in cancer immunotherapy. Cold Spring Harb Perspect Biol. 2018;10(12).
396. Yang Y, Lundqvist A. Immunomodulatory Effects of IL-2 and IL-15; Implications for Cancer Immunotherapy. Cancers (Basel). 2020;12(12)
397. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451-8.
398. Payne R, Glenn L, Hoen H, Richards B, Smith JW 2nd, Lufkin R, Crocenzi TS, Urba WJ, Curti BD. Durable responses and reversible toxicity of highdose interleukin-2 treatment of melanoma and renal cancer in a Community Hospital Biotherapy Program. J Immunother Cancer. 2014;2:13.
399. Baik AH, Oluwole OO, Johnson DB, Shah N, Salem JE, Tsai KK, Moslehi JJ. Mechanisms of cardiovascular toxicities associated with immunotherapies. Circ Res. 2021;128(11):1780-801.
400. Damoiseaux J. The IL-2—IL-2 receptor pathway in health and disease: the role of the soluble IL-2 receptor. Clin Immunol. 2020;218: 108515.
401. Doberstein SK. Bempegaldesleukin (NKTR-214): a CD-122-biased IL-2 receptor agonist for cancer immunotherapy. Expert Opin Biol Ther. 2019;19(12):1223-8.
402. Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, Tetzlaff MT, Tagliaferri MA, Zalevsky J, Hoch U, et al. A first-inhuman study and biomarker analysis of NKTR-214, a novel IL2Rbet-agamma-biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov. 2019;9(6):711-21.
403. Diab A, Tannir NM, Bentebibel SE, Hwu P, Papadimitrakopoulou V, Haymaker C, Kluger HM, Gettinger SN, Sznol M, Tykodi SS, et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced
solid tumors: phase i dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discov. 2020;10(8):1158-73.
404. Khushalani NI, Diab A, Ascierto PA, Larkin J, Sandhu S, Sznol M, Koon HB, Jarkowski A, Zhou M, Statkevich P, et al. Bempegaldesleukin plus nivolumab in untreated, unresectable or metastatic melanoma: phase III PIVOT IO 001 study design. Future Oncol. 2020;16(28):2165-75.
405. Siefker-Radtke AO, Cho DC, Diab A, Sznol M, Bilen MA, Balar AV, Grignani G, Puente E, Tang L, Chien D, et al. Bempegaldesleukin plus nivolumab in first-line metastatic urothelial carcinoma: results from PIVOT-02. Eur Urol. 2022;82(4):365-73.
406. Diab A, Tykodi SS, Daniels GA, Maio M, Curti BD, Lewis KD, Jang S, Kalinka E, Puzanov I, Spira AI, et al. Bempegaldesleukin plus nivolumab in first-line metastatic melanoma. J Clin Oncol. 2021;39(26):2914-25.
407. Pan Y, Hao Y, Han H, Chen T, Ding H, Labbe KE, Shum E, Guidry K, Hu H , Sherman F et al. Nemvaleukin alfa, a novel engineered IL-2 fusion protein, drives antitumor immunity and inhibits tumor growth in small cell lung cancer. J Immunother Cancer. 2022; 10(9).
408. Boni V, Winer IS, Gilbert L, Vaishampayan UN, Rosen SD, Muzaffar J, Spreafico A, McDermott DF, Chu QS, Dumas O, et al. ARTISTRY-1: Nemvaleukin alfa monotherapy and in combination with pembrolizumab in patients (pts) with advanced solid tumors. J Clin Oncol. 2021;39(15_suppl):2513-2513.
409. Hamid O, Liu SV, Boccia RV, Call JA, Wise-Draper TM, Alistar AT, Powderly JD, Carthon BC, Vaishampayan UN, Olszanski AJ, et al. Selection of the recommended phase 2 dose (RP2D) for subcutaneous nemvaleukin alfa: ARTISTRY-2. J Clin Oncol. 2021;39(15_suppl):2552-2552.
410. Vaishampayan UN, Tomczak P, Muzaffar J, Winer IS, Rosen SD, Hoimes CJ, Chauhan A, Spreafico A, Lewis KD, Bruno DS, et al. Nemvaleukin alfa monotherapy and in combination with pembrolizumab in patients (pts) with advanced solid tumors: ARTISTRY-1. J Clin Oncol. 2022;40(16_suppl):2500-2500.
411. Rech AJ, Vonderheide RH. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann N Y Acad Sci. 2009;1174:99-106.
412. Sampson JH, Schmittling RJ, Archer GE, Congdon KL, Nair SK, Reap EA, Desjardins A, Friedman AH, Friedman HS, Herndon JE 2nd, et al. A pilot study of IL-2Ralpha blockade during lymphopenia depletes regulatory T-cells and correlates with enhanced immunity in patients with glioblastoma. PLoS ONE. 2012;7(2): e31046.
413. Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, Klasen IS, Hilbrands LB, Figdor CG, de Vries IJ, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res. 2010;16(20):5067-78.
414. Solomon I, Amann M, Goubier A, Arce Vargas F, Zervas D, Qing C, Henry JY, Ghorani E, Akarca AU, Marafioti T, et al. CD25-T(reg)-depleting antibodies preserving IL-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat Cancer. 2020;1(12):1153-66.
415. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda Rota E, Dahan R, Georgiou A, Sledzinska A, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory
416. Ji D, Song C, Li Y, Xia J, Wu Y, Jia J, Cui X, Yu S, Gu J. Combination of radiotherapy and suppression of Tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J Immunother Cancer. 2020;8(2).
417. Anderson KC , Jones RM, Morimoto C , Leavitt P , Barut BA. Response patterns of purified myeloma cells to hematopoietic growth factors. Blood. 1989;73(7):1915-24.
418. Klein B, Wijdenes J, Zhang XG, Jourdan M, Boiron JM, Brochier J, Liautard J, Merlin M, Clement C, Morel-Fournier B, et al. Murine anti-interleukin-6 monoclonal antibody therapy for a patient with plasma cell leukemia. Blood. 1991;78(5):1198-204.
419. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448-57.
420. Voorhees PM, Manges RF, Sonneveld P, Jagannath S, Somlo G, Krishnan A, Lentzsch S, Frank RC, Zweegman S, Wijermans PW, et al. A phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2013;161(3):357-66.
421. San-Miguel J, Blade J, Shpilberg O, Grosicki S, Maloisel F, Min CK, Polo Zarzuela M, Robak T, Prasad SV, Tee Goh Y, et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood. 2014;123(26):4136-42.
422. Rossi JF, Negrier S, James ND, Kocak I, Hawkins R, Davis H, Prabhakar U, Qin X, Mulders P, Berns B. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br J Cancer. 2010;103(8):1154-62.
423. Dorff TB, Goldman B, Pinski JK, Mack PC, Lara PN Jr, Van Veldhuizen PJ Jr, Quinn DI, Vogelzang NJ, Thompson IM Jr, Hussain MH. Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin Cancer Res. 2010;16(11):3028-34.
424. Fizazi K, De Bono JS, Flechon A, Heidenreich A, Voog E, Davis NB, Qi M, Bandekar R, Vermeulen JT, Cornfeld M, et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur J Cancer. 2012;48(1):85-93.
425. Lu ZY, Brochier J, Wijdenes J, Brailly H, Bataille R, Klein B. High amounts of circulating interleukin (IL)-6 in the form of monomeric immune complexes during anti-IL-6 therapy. Towards a new methodology for measuring overall cytokine production in human in vivo. Eur J Immunol. 1992;22(11):2819-24.
426. Dijkgraaf EM, Santegoets SJ, Reyners AK, Goedemans R, Wouters MC, Kenter GG, van Erkel AR, van Poelgeest MI, Nijman HW, van der Hoeven JJ, et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferonalpha2b in patients with recurrent epithelial ovarian cancer. Ann Oncol. 2015;26(10):2141-9.
427. Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med. 2020;217(1).
428. Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, Gorman DM, Oft M. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012;72(14):3570-81.
429. Oft M. Immune regulation and cytotoxic T cell activation of IL-10 agonists—preclinical and clinical experience. Semin Immunol. 2019;44: 101325.
430. Autio K, Oft M. Pegylated interleukin-10: clinical development of an immunoregulatory cytokine for use in cancer therapeutics. Curr Oncol Rep. 2019;21(2):19.
431. Naing A, Papadopoulos KP, Autio KA, Ott PA, Patel MR, Wong DJ, Falchook GS, Pant S, Whiteside M, Rasco DR, et al. Safety, antitumor activity, and immune activation of pegylated recombinant human interleu-kin-10 (AM0010) in patients with advanced solid tumors. J Clin Oncol. 2016;34(29):3562-9.
432. Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthelemy P, Porta C, George S, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277-90.
433. Tilg H, Ulmer H, Kaser A, Weiss G. Role of IL-10 for induction of anemia during inflammation. J Immunol. 2002;169(4):2204-9.
434. Naing A, Infante JR, Papadopoulos KP, Chan IH, Shen C, Ratti NP, Rojo B, Autio KA, Wong DJ, Patel MR, et al. PEGylated IL-10 (pegilodecakin) induces systemic immune activation, CD8(+) T cell invigoration and polyclonal T cell expansion in cancer patients. Cancer Cell. 2018;34(5):775-91.
435. Naing A, Wong DJ, Infante JR, Korn WM, Aljumaily R, Papadopoulos KP, Autio KA, Pant S, Bauer TM, Drakaki A, et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): a multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019;20(11):1544-55.
436. Pal S, Hu-Lieskovan S, Agarwal N. Can pegylated IL-10 add to a backbone of PD-1 inhibition for solid tumours? Lancet Oncol. 2019;20(11):1473-4.
437. Spigel D, Jotte R, Nemunaitis J, Shum M, Schneider J, Goldschmidt J, Eisenstein J, Berz D, Seneviratne L, Socoteanu M, et al. Randomized phase 2 studies of checkpoint inhibitors alone or in combination with
pegilodecakin in patients with metastatic NSCLC (CYPRESS 1 and CYPRESS 2). J Thorac Oncol. 2021;16(2):327-33.
438. Tannir NM, Papadopoulos KP, Wong DJ, Aljumaily R, Hung A, Afable M, Kim JS, Ferry D, Drakaki A, Bendell J, et al. Pegilodecakin as monotherapy or in combination with anti-PD-1 or tyrosine kinase inhibitor in heavily pretreated patients with advanced renal cell carcinoma: Final results of cohorts A, G, H and I of IVY Phase I study. Int J Cancer. 2021;149(2):403-8.
439. Hecht JR, Papadopoulos KP, Falchook GS, Patel MR, Infante JR, Aljumaily R, Wong DJ, Autio KA, Wainberg ZA, Bauer TM, et al. Immunologic and tumor responses of pegilodecakin with 5-FU/LV and oxaliplatin (FOLFOX) in pancreatic ductal adenocarcinoma (PDAC). Invest New Drugs. 2021;39(1):182-92.
440. Hecht JR, Lonardi S, Bendell J, Sim HW, Macarulla T, Lopez CD, Van Cutsem E, Munoz Martin AJ, Park JO, Greil R, et al. Randomized phase III study of FOLFOX alone or with pegilodecakin as second-line therapy in patients with metastatic pancreatic cancer that progressed after gemcitabine (SEQUOIA). J Clin Oncol. 2021;39(10):1108-18.
441. Grossman JG, Nywening TM, Belt BA, Panni RZ, Krasnick BA, DeNardo DG, Hawkins WG, Goedegebuure SP, Linehan DC, Fields RC. Recruitment of CCR2(+) tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology. 2018;7(9): e1470729.
442. Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, Wang G, Tromp BJ, Puchalski TA, Balkwill F, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71(4):1041-50.
443. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31(3):760-8.
444. Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, Zhong B, de Boer CJ, Tabernero J, Calvo E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an openlabel, multicenter phase 1b study. Target Oncol. 2015;10(1):111-23.
445. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651-62.
446. Noel M, O’Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, Linehan DC, Belt BA, Gamelin EC, Ganguly B, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2020;38(3):800-11.
447. Wu X, Singh R, Hsu DK, Zhou Y, Yu S, Han D, Shi Z, Huynh M, Campbell JJ, Hwang ST. A small molecule CCR2 antagonist depletes tumor macrophages and synergizes with anti-PD-1 in a murine model of cutaneous T-cell lymphoma (CTCL). J Invest Dermatol. 2020;140(7):1390-400.
448. Wang J, Saung MT, Li K, Fu J, Fujiwara K, Niu N, Muth S, Wang J, Xu Y, Rozich N et al. CCR2/CCR5 inhibitor permits the radiation-induced effector T cell infiltration in pancreatic adenocarcinoma. J Exp Med. 2022;219(5).
449. Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE, Chauca-Diaz A, Costello JC, Dancik GM, Tamburini BAJ, et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun Biol. 2020;3(1):720.
450. Bartkowiak T, Jaiswal AR, Ager CR, Chin R, Chen CH, Budhani P, Ai M, Reilley MJ, Sebastian MM, Hong DS, et al. Activation of
451. Abd Wahab NA, Lajis NH, Abas F, Othman I, Naidu R. Mechanism of anticancer activity of curcumin on androgen-dependent and androgenindependent prostate cancer. Nutrients. 2020;12(3).
452. Ferguson JJA, Abbott KA, Garg ML. Anti-inflammatory effects of oral supplementation with curcumin: a systematic review and meta-analysis of randomized controlled trials. Nutr Rev. 2021;79(9):1043-66.
453. Gupta SC, Patchva S, Aggarwal BB. Therapeutic roles of curcumin: lessons learned from clinical trials. AAPS J. 2013;15(1):195-218.
454. Peng Y, Ao M, Dong B, Jiang Y, Yu L, Chen Z, Hu C, Xu R. Anti-inflammatory effects of curcumin in the inflammatory diseases: status, limitations and countermeasures. Drug Des Devel Ther. 2021;15:4503-25.
455. Sharma RA, Euden SA, Platton SL, Cooke DN, Shafayat A, Hewitt HR, Marczylo TH, Morgan B, Hemingway D, Plummer SM, et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004;10(20):6847-54.
456. Howells LM, Iwuji COO, Irving GRB, Barber S, Walter H, Sidat Z, GriffinTeall N, Singh R, Foreman N, Patel SR, et al. Curcumin combined with FOLFOX chemotherapy is safe and tolerable in patients with metastatic colorectal cancer in a randomized phase lla trial. J Nutr. 2019;149(7):1133-9.
457. Zhan Y, Chen Y, Liu R, Zhang H, Zhang Y. Potentiation of paclitaxel activity by curcumin in human breast cancer cell by modulating apoptosis and inhibiting EGFR signaling. Arch Pharm Res. 2014;37(8):1086-95.
458. Calaf GM, Ponce-Cusi R, Carrion F. Curcumin and paclitaxel induce cell death in breast cancer cell lines. Oncol Rep. 2018;40(4):2381-8.
459. Malaguarnera L. Influence of resveratrol on the immune response. Nutrients. 2019;11(5).
460. Delmas D, Limagne E, Ghiringhelli F, Aires V. Immune Th17 lymphocytes play a critical role in the multiple beneficial properties of resveratrol. Food Chem Toxicol. 2020;137: 111091.
461. Hou J, Karin M, Sun B. Targeting cancer-promoting inflammationhave anti-inflammatory therapies come of age? Nat Rev Clin Oncol. 2021;18(5):261-79.
462. Barnabei A, Carpano S, Chiefari A, Bianchini M, Lauretta R, Mormando M, Puliani G, Paoletti G, Appetecchia M, Torino F. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10: 582394.
463. Kotwal A, Kottschade L, Ryder M. PD-L1 inhibitor-induced thyroiditis is associated with better overall survival in cancer patients. Thyroid. 2020;30(2):177-84.
ملاحظة الناشر
- *المراسلة:
يونغ يوان
yongyuan@scu.edu.cn
شياوي وي
xiaweiwei@scu.edu.cn
مختبر أبحاث الشيخوخة وأهداف أدوية السرطان، المختبر الوطني الرئيسي للعلاج البيولوجي ومركز السرطان، المركز الوطني للبحوث السريرية لكبار السن، مستشفى غرب الصين، جامعة سيتشوان، رقم 17، المبنى 3، طريق رينمين الجنوبي، تشنغدو 610041، سيتشوان، جمهورية الصين الشعبية
قسم جراحة الصدر، مستشفى غرب الصين، جامعة سيتشوان، تشنغدو، جمهورية الصين الشعبية
DOI: https://doi.org/10.1186/s13045-024-01528-7
PMID: https://pubmed.ncbi.nlm.nih.gov/38520006
Publication Date: 2024-03-22
Targeting inflammation as cancer therapy
Abstract
Inflammation has accompanied human beings since the emergence of wounds and infections. In the past decades, numerous efforts have been undertaken to explore the potential role of inflammation in cancer, from tumor development, invasion, and metastasis to the resistance of tumors to treatment. Inflammation-targeted agents not only demonstrate the potential to suppress cancer development, but also to improve the efficacy of other therapeutic modalities. In this review, we describe the highly dynamic and complex inflammatory tumor microenvironment, with discussion on key inflammation mediators in cancer including inflammatory cells, inflammatory cytokines, and their downstream intracellular pathways. In addition, we especially address the role of inflammation in cancer development and highlight the action mechanisms of inflammation-targeted therapies in antitumor response. Finally, we summarize the results from both preclinical and clinical studies up to date to illustrate the translation potential of inflammation-targeted therapies.
Background
which collectively provide a complex tumor microenvironment (TME) [8]. Tumors are often characterized with the infiltration of immune cells and the upregulation of inflammatory mediators surrounding tumors. This inflammatory microenvironment may impact tumor development varying stages, from tumor initiation to progression. In this review, we discuss the role of inflammation in cancer development, with special focus on the tumor-promoting activities of inflammation. We especially highlight the underlying mechanisms of the antitumor efficacy of inflammation-targeted therapies in cancer, with clinical evidence up to date in relation to inflammation-targeting strategies.
Inflammation mediators in cancer
epidemiological studies, the ulcerative colitis and Crohn’s disease could increase the risk of colon cancer, which is one of the best known examples of tumor-associated inflammation [11, 12]. Moreover, oncogenic infection by microbial agents such as Helicobacter pylori [13] and hepatitis B [14] has also been described as risk factors for gastric and hepatic cancer. During the chronic inflammation induced by microbial agents, immune cells such as macrophages at the inflammatory sites produce reactive oxygen species (ROS), leading to persistent DNA damage and subsequent gene mutations [15]. Furthermore, cytokines secreted by immune cells such as tumor necrosis factor-
Key inflammatory cells in cancer
with antitumor or protumoral roles are presented in Table 1.
Tumor-associated neutrophils (TANs)
| Cell type | Protumor activities | Antitumor activities | ||||
| Tumor-associated neutrophils (TANs) | ||||||
|
-N1 TANs exert an antitumor activity, by direct or indirect cytotoxicity | |||||
| Tumor-associated macrophages (TAMs) | ||||||
| Dendritic cells (DCs) | ||||||
|
||||||
|
||||||
However, based on different status of TME, the role of NETs is variable. NETs can also exert an antitumor effect by directly killing tumor cells and inhibiting tumor growth and metastasis. In colorectal cancer (CRC) and head and neck squamous cell carcinoma, in vitro generated NETs could imped tumor growth by inducing apoptosis and inhibiting proliferation [45, 46]. Furthermore, co-culture of melanoma cells with NETs led to necrosis of melanoma cells [47]. NETosis is associated with the release of protein S100A8/A9, the increased ratio
of which to CRP was found to correlate with favorable survival of high-grade serous ovarian cancer (HGSOC) patients [48].
Tumor-associated macrophages (TAMs)
Dendritic cells (DCs)
In recent decades, immunogenic cell death (ICD) has received considerable research attention. ICD is accompanied by the release and chronic exposure of damageassociated molecular patterns (DAMPs), conferring a potent adjuvanticity to dying cancer cells. ROS production and endoplasmic reticulum (ER) stress are required for the emission of DAMPs which bind to the pattern recognition receptors (PRRs) expressed on immune cells, especially DCs [75]. This recognition and binding process is often associated with the generation of immunological
memory [76, 77]. Multiple studies have described the critical role of DCs in the immune response triggered by tumor cells undergoing ICD [78], which demonstrated that the robust antitumor T cell response induced by ICD largely relied on DCs in the TME. It is thus conceivable that manipulating DCs in the TME holds great potential as anticancer strategies. Whereas ICD contributes to the success of many anticancer treatments including chemotherapy, radiotherapy, and target therapies, the immunogenicity varies among cells with different death modalities. A recent study suggested that cancer cells undergoing ferroptosis would impede the maturation of DCs, with poor engulfment and antigen presentation capacity, adding concerns to the applications of ferropto-sis-inducing therapeutics [79].
Myeloid-derived suppressor cells (MDSCs)
from the VEGF/VEGFR axis that stimulates MDSCs, the proangiogenic MMPs produced by MDSCs serve as a secondary angiogenetic signals [86]. MMPs are a family of ECM enzymes that facilitate the invasion of tumor cells, and among them MMP9 is perceived as a key regulator for tumor angiogenesis induced by PMN-MDSCs [87].
Given that high M-MDSC fraction is correlated with decreased expansion and activation of tumor-specific T cells [88], MDSCs have now become a novel marker for predicting patients’ response to immune checkpoint blockade (ICB) therapy. For instance, patients with lower fractions of circulating MDSCs are more sensitive to ipilimumab treatment [89], especially melanoma patients [90, 91]. Upon CTLA-4 blockade, tumor-infiltrating MDSCs exhibit increased expression of colony-stimulating fac-tor-1 receptor (CSF-1R), which in turn is correlated with increased MDSC infiltration in tumors. CSF-1/CSF-1R signaling blockade could not only be used to decrease the numbers of MDSCs, but also convert the immunesuppressive MDSCs toward an antitumor phenotype [92, 93]. Likewise, IL-10 secreted by DCs in the TME could increase the number of tumor-infiltrating MDSCs, conferring adaptive resistance to PD-1 antibody treatment [94]. Targeting MDSCs via CSF-1/CSF-1R inhibitors thus becomes a potential strategy to overcome tumor resistance to ICBs. Though a large number of agents targeting the upstream factors or receptors of MDSC accumulation are being tested to potentiate ICB efficacy, it has to be addressed that the majority of MDSC-recruiting chemokines can also act on other immune cells with antitumor activities such as T lymphocytes [95] and NK cells [96]. Thus, such chemokine blockades would possibly yield both positive and negative effect on tumors.
Vascular endothelial cells
junctions make it easier for leukocytes to migrate through vascular walls.
Key inflammatory cytokines in cancer
Tumor necrosis factor alpha (TNF-a)
| Inflammatory cytokines | Major sources | Receptors | Key actions in cancer | ||||
| TNF-a | Macrophages, T lymphocytes, NK cells, neutrophils, mast cells, eosinophils and neurons | TNF-aR-1, TNF-aR-2 |
|
||||
| TGF-
|
Tumor cells, bone matrix | TGF-
|
|
||||
| IFN-I | DCs, B cells, fibroblasts | IFNAR1, IFNAR2 |
|
||||
| IL-1 | Tumor cells, MDSCs, TAMs, TANs, regulatory B (Breg) cells and Th17 | IL-1R |
|
||||
| IL-6 | Tumor cells, T cells, B cells, monocytes, fibroblasts, keratinocytes, endothelial cells, mesangial cells, adipocytes | IL-6R | -Promotes tumor progression by inducing tumor cell proliferation, survival, EMT, angiogenesis, and chemoresistance | ||||
| IL-10 | Tumor cells, leukocytes | IL-10R |
|
In melanoma, TNF-
with rheumatoid arthritis [122, 123]. However, some reports suggested that TNF is able to increase expansion, stability, and possibly function of Tregs via TNFR2 [124]. TNFR2 is highly expressed on Tregs supporting the proliferation and suppressive activities of Tregs [125]. TNFR2 was identified as a expression biomarker for the highly suppressive subset of Tregs [125]. The antagonistic TNFR2 antibodies are thus potential treatment for tumors. TNFR2 antagonists were capable of targeting surface TNFR2 on ovarian cancer cells, inhibiting NF-кB pathway activation and proliferation of tumor cells [126].
Transforming growth factor-beta (TGF-
and type II (TGF-
Interestingly, in the context of tumors, the role of TGF-
Interferons (IFNs)
During chronic inflammation, the feedback protective processes induced by IFN-Is provide tumor cells with supportive microenvironment for tumor growth and progression [141, 142]. Alongside the proinflammatory signals for tumor progression, IFN-Is may also facilitate the immune evasion of tumor cells by upregulating immunesuppressive pathways ranging from danger sensing to
cytokine production [143, 144]. For instance in head and neck squamous cell carcinoma (HNSCC), cancer-specific IFN-I activation attenuates the expansion and functions of CD8 +T effector cells and is associated with poor clinical outcomes [145].
Interleukin-1
level of IL-1
Interleukin-6
Interleukin-10
produced by leukocytes, as well as human tumor cells. This cytokine family consists of IL-10 and IL-20 subfamily cytokines including IL-19, IL-20, IL-22, IL-24, and IL-26 [181]. IL-10 suppresses uncontrolled inflammatory responses, thereby maintaining homeostasis [182]. In tumors such as gastric cancer, TAM-produced IL-10 contributes to an immunosuppressive microenvironment that favors tumor growth [183]. A more recent study showed that the expression of IL-10 in tumor-infiltrating regulatory T cells may result in the exhaustion of intratumoral CD8 + T cells [184]. Some studies on the other hand suggested that IL-10 can be used as an immunotherapy in tumor models [185]. IL-10 could induce the expression of CD3 and CD8 molecules on thymocytes and thereby promotes the cytotoxic activity of CD8 + T cells [186]. Another mechanism for the antitumor action of IL-10 is the increased CD8 + T cell infiltration and IFN-
ROS
external stimuli such as chemotherapy, radiotherapy, and ultraviolet may also trigger ROS production [194].
Cancer cells carry higher amount of ROS than their normal counterparts, due to aberrant oncogene activation and mitochondrial activity. The role of ROS in cancer development is intricate, making it a double-edged sword [195]. On one hand, the sustained ROS stress may damage cell structures, impede their biological functions, and cause mutagenesis, which collectively increase the risks for oncogenesis [196, 197]. On the contrary, ROS may accumulate upon exogenous stimuli such as chemotherapy and radiotherapy, leading to tumor cell death and thereby sensitizing tumor cells to treatments. Elucidating the complex roles of ROS in cancer will aid the design of ROS-targeting therapies for cancer. Recent studies suggest that hypoxic environment in tumors could activate ROS generation [198]. In response to hypoxia, the hypoxia-inducible factor-1 (HIF-1) is a well-characterized transcriptional activator that modulates oxygen homeostasis [199]. By interacting with hypoxia response elements of target genes, ROS promotes the activation of HIF-1
Key inflammatory pathways in cancer
Eicosanoid signaling
the proliferation of lung cancer cells, but at the same time enhance their invasion and migration [230], leading to the hypothesis that the exact role of PGD2 in cancer may vary according to the tumor stage.
potentially a good combination partner for immunotherapies, such as checkpoint inhibitors (Fig. 2).
On the contrary, another LOX-derived eicosanoids, lipoxins (LXs), are characterized as antitumorigenic [250]. Lipoxins stimulate monocytes without causing the inflammatory release of ROS [251]. Lipoxins may also promote the phagocytosis of apoptotic neutrophils by macrophages, thereby reducing inflammation[252]. Accumulating evidence suggests the anti-inflammatory effect of lipoxin
JAK-STAT signaling
phosphorylation of JAKs which then form a docking site for STATs leading to STAT phosphorylation. As the core member of the STAT protein family, STAT3 plays a with versatile roles in the inflammatory response and tumor progression. Multiple growth factors and cytokines are implicated in the canonical STAT3 pathways, regulating the transcription of STAT3 target genes and downstream cellular processes such as cell differentiation, angiogenesis, and tumorigenesis [258]. The dysregulated STAT3 signaling has been implicated in a series of inflammatory diseases such as rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease [259]. Moreover, the persistent activation of the STAT3 signaling may result in the tumorigenesis of both solid and hematological malignancies [260].
Metal metabolism
which directly generates ferric iron (
Zinc is the second most abundant fundamental nutritional element in human body, which was first documented in the 1960s regarding its role in human health [275]. Zinc is implicated in the production and signaling of numerous inflammatory cytokines, and upon acute response to stress stimuli, plasma concentrations of zinc rapidly drop. Zinc metabolism in humans is tightly associated with the activities of zinc transporters such as ZIP8. During inflammation, activated NF-кB increases the expression of ZIP8 which localizes to cell membrane and regulates zinc uptake. Following the entry of zinc into cytosol, zinc suppresses IKK
Inflammation-targeted therapies in cancer
Non-steroidal anti-inflammatory drugs (NSAIDs)
| Agent/target | Tumor type | Combination regime | Key clinical trial | Reported action |
| Celecoxib | ||||
| COX-2 | Breast cancer | Neoadjuvant celecoxib + chemotherapy/ cholecalciferol/exemestane | NCT02429427, NCT01041781 | Celecoxib induced favorable changes in serum biomarkers and cytology in women with increased risk for breast cancer, but demonstrated no significant benefits for patients with ERBB2-negative breast cancer |
| Lung cancer | Celecoxib + chemotherapy/RT/anti-EGFR TKIs | NCT00300729, NCT01503385 | Celecoxib at a maximal tolerated dose of
|
|
| CRC | Celecoxib + cetuximab/chemotherapy (FOLFIRI regimen)/RT/ | NCT03645187, NCT00005094, NCT00141193, NCT03926338, NCT01150045 | Celecoxib combined with chemotherapy (FOLFIRI regimen consisting of 5-flourouracil, leucovorin, irinotecan) or PD-1 blockade toripalimab represents an effective and safe synergetic protocol for patients with metastatic CRC | |
| Antiviral therapies | ||||
| Entecavir | ||||
| HBV | HCC | NCT00388674 | Entecavir led to a reduced risk of HBV-related events including HCC | |
| Tenofovir | ||||
| HBV | HCC | NCT019553458 | Tenofovir led to a comparable long-term risk of HCC and ICC in CHB patients with entecavir | |
| ISA 101 HPV-16 vaccine | ||||
| HPV | Cervical cancer | ISA 101 + anti-PD-1 antibody nivolumab | NCT02426892 | Concurrent treatment of ISA 101 and anti-PD-1 antibody nivolumab increased both overall response rates and survival of HPV-16-related cancer |
| Cytokine-directed therapies | ||||
| IFN-a | RCC | IFN-a + oblimersen/(iso)tretinoin/isotreti-noin/IL-2/chemotherapy (fluorouracil, capecitabine)/sorafenib/VEGF inhibitor (bevacizumab, SU5416)/mTOR inhibitor (CCI-779)/naptumomab estafenatox/pazo-panib/celecoxib/thalidomide/chemotherapy (5-Fluorouracil) /pembrolizumab | UMIN000002466, CALGB 90206 | The prolonged IFN-a treatment induced long-lasting complete responses and longterm outcome with acceptable toxicity in patients with metastatic RCC. IFN-a is also a promising combination therapy for target therapies and immune checkpoint inhibitors such as anti-PD-1 therapies |
| Melanoma | IFN-a + combination chemotherapy (dacarbazine, temozolomide, azacitidine, cisplatin)/IL-12/thalidomide/bevacizumab/ imatinib/BRAF inhibitor (vemurafenib)/ CTLA-4 inhibitor ipilimumab/proteasome inhibitor (PS-341)/sodium stibogluconateRT | NCT00204529, NCT01959633, EORTC 18991, S0008 | Adjuvant treatment with IFN-a-2a or PEG-IFN-a-2b could induce sustained improvement of RFS in stage III melanoma patients and has been approved by the FDA as adjuvant therapy for melanoma | |
| Agent/target | Tumor type | Combination regime | Key clinical trial | Reported action |
| Leukemia | IFNa-2a + combination chemotherapy (melphalan, adriamycin, bleomycin, velban, and dacarbazine)/nilotinib/imatinib/rituximab/dasatinib | NCT02328755, NCT02185261 | IFN-a treatment is an effective strategy for minimal residual disease (MRD)-positive leukemia patients receiving allogeneic hematopoietic stem cell transplantation (allo-HSCT) | |
| Lymphoma | IFN-a+combination chemotherapy (melphalan, adriamycin, bleomycin, velban, and dacarbazine)/bexarotene/rituximab | NCT01609010 | Immunotherapy with IFN-a and rIL-2 is well tolerated and may intensify remission in NHL patients | |
| HCC | IFN-a + chemotherapy (capecitabine)/ celecoxib + rintatolimod/thalidomide | IFN-a therapy may reduce HCC recurrence after medical ablation therapy for primary tumors. IFN-a plus cis-platinum is effective in patients with inoperable HCC | ||
| Galunisertib (LY2157299) | ||||
| TGF-
|
Pancreatic cancer | Galunisertib + durvalumab/gemcitabine | NCT02734160 | The galunisertib-gemcitabine combination improved OS in patients with unresectable pancreatic cancer with minimal added toxicity |
| HCC | Galunisertib + sorafenib/stereotactic body radiotherapy (SBRT) | NCT01246986 | The combination of galunisertib and sorafenib demonstrated a manageable safety profile and improved prognosis of HCC | |
| Fresolimumab (GC1008) | ||||
| TGF-
|
Melanoma, RCC | NCT00356460 | Fresolimumab displayed preliminary antitumor efficacy and acceptable safety profile at multiple doses in patients with advanced melanoma and RCC | |
| PF-03446962 | ||||
| TGF-
|
HCC, CRC | Regorafenib + PF-03446962 | NCT00557856 | PF-03446962 had manageable safety and pharmacokinetic profiles in HCC, but the combination of regorafenib and PF-03446962 caused unacceptable toxicity with limited clinical activity in patients with refractory metastatic CRC |
| Bintrafusp alfa (M7824) | ||||
| TGF-
|
NSCLC | Bintrafusp alfa + chemotherapy (docetaxel, platinum-based) | NCT02517398 | Bintrafusp alfa induced promising efficacy and manageable tolerability in patients with NSCLC previously treated with platinum |
| HPV-associated cancer | NCT02517398, NCT02517398, NCT04247282 | Bintrafusp alfa showed clinical activity and manageable safety in HPV-associated cancers | ||
| Agent/target | Tumor type | Combination regime | Key clinical trial | Reported action |
| Esophageal cancer | NCT02517398, NCT02699515 | Bintrafusp alfa showed clinical activity with manageable safety profile in patients with advanced esophageal adenocarcinoma | ||
| Anakinra | ||||
| IL-1 | Multiple myeloma | Anakinra + immunomodulatory drug combination lenalidomide and dexamethasone | NCT00635154 | Anakinra decreased the proliferative rates of tumor, leading to a chronic disease state with improved PFS in patients with multiple myeloma at high risk of progression to active myeloma |
| CRC | Anakinra + 5-FU + bevacizumab | 5-FU plus bevacizumab and anakinra had promising activity and a manageable safety profile in refractory metastatic CRC | ||
| Bempegaldesleukin (NKTR-214) | ||||
| IL-2 | Melanoma | Bempegaldesleukin + nivolumab/pembrolizumab | NCT03635983, PIVOT-02 | Bempegaldesleukin can be used in combination with nivolumab or pembrolizumab in patients with metastatic melanomas |
| Urothelial carcinoma | Bempegaldesleukin + nivolumab | NCT02983045, PIVOT-02 | Bempegaldesleukin combined with nivolumab is suggested as the first-line therapy for patients with metastatic urothelial carcinoma with manageable side effects | |
| Nemvaleukin alfa (LKS 4230) | ||||
| IL-2 | Ovarian cancer | Nemvaleukin alfa + pembrolizumab | NCT05092360 | Under evaluation for the efficacy and safety as monotherapy and combination therapy with pembrolizumab in patients with plati-num-resistant ovarian cancer |
| CNTO 328 | ||||
| IL-6 | Multiple myeloma | Siltuximab + bortezomib-melphalan-prednisone (VMP) | NCT00911859 | The addition of siltuximab to the bortezomib-melphalan-prednisone (VMP) regimen did not improve the complete response rate or long-term outcomes of MM patients |
| Prostate cancer | Siltuximab + mitoxantrone/prednisone | SWOG S0354 | Siltuximab was well tolerated and improved clinical outcomes, leading to a PSA response rate of 3.8% and a stable disease rate of 23% in patients with castration-resistant prostate cancer | |
| Tocilizumab | ||||
| IL-6R | Ovarian cancer | Tocilizumab + carboplatin/doxorubicin | NCT01637532 | Tocilizumab at
|
| Pegilodecakin (LY3500518) | ||||
| Agent/target | Tumor type | Combination regime | Key clinical trial | Reported action |
| IL-10 | Solid tumors | Pegilodecakin + chemotherapies or anti-PD-1 blockade | NCT02009449 | Pegilodecakin was used as monotherapy and in combination with chemotherapies or anti-PD-1 blockade to treat tumors such as melanoma, NSCLC, CRC, and pancreatic cancer |
| Chemokine-directed therapies Carlumab | ||||
| CCL2 | Prostate cancer | Carlumab could be safely administered in patients with metastatic CRPC, but failed to demonstrate significant antitumor activities as a single agent | ||
| PF-04136309 CCR2 | ||||
| Pancreatic cancer | PF-04136309+chemotherapy (gemcitabine plus nab-paclitaxel) | NCT02732938 | PF-04136309 in combination with nabpaclitaxel plus gemcitabine may induce pulmonary toxicity, with no significant superior efficacy signal over nab-paclitaxel and gemcitabine | |
However, a recent clinical trial suggested that the addition of celecoxib to the standard adjuvant chemotherapy regime failed to bring more benefits to patients with stage III colon cancer (NCT01150045) [299]. Another study evaluated the efficacy of celecoxib as a combination
partner for conventional therapy in ERBB2-negative breast cancer, which demonstrated no significant benefits from celecoxib in terms of DFS following 2-year treatments (NCT02429427) [300]. Moreover, some studies suggested that the addition of celecoxib to chemotherapy might adversely impact the prognosis of breast cancer patients, especially those with prostaglandin-endoperoxide synthase 2 (PTGS2) low tumors (NCT01041781) [301]. Such conflicting results likely reflect the impact of different treatment regimens or administration doses of celecoxib, and the expression profile of biomarkers in tumors. Thus, all the above factors should be taken into account to investigate the therapeutic potential of celecoxib. In addition, long-term use of NSAIDs including COXIBs at high doses may lead to severe cardiovascular side effects in patients, especially in those with a history of atherosclerotic heart disease [302]. One way to prevent or reduce these side effects would be the alternative targeting of the downstream PGE2 pathway. Some researchers have introduced natural compounds with known inhibitory activities on COX-2, such as natural phenols, flavonoids, stilbenes, terpenoids, quinones, and alkaloids [303].
Antiviral therapies
Antihepatitis B virus (HBV) therapies
(CHB) infection, the immune response to persistent infection may cause chronic inflammation and hepatic fibrogenesis, leading to irreversible damage in the liver structure. The continuous replication of virus DNA and its integration into host genomes may cause genetic alterations, ultimately driving the carcinogenesis of hepatocytes [120]. On the other hand, viral proteins such as hepatitis B virus X protein may increase the sensitivity of the host to chemical carcinogens [304]. These preclinical studies have motivated the design of antiviral therapies in the treatment of HBV-related hepatocellular carcinoma.
The antiviral therapies aim to suppress HBV DNA replication, promote the serum conversion of hepatitis B e antigen ( HBeAg ), and attenuate the development of cirrhosis. Common antiviral drugs include the nucleoside and nucleotide analogs (NAs) and IFNs. Among them, the long-term administration of potent NAs with high barrier to resistance such as entecavir and tenofovir disoproxil, was recommended as first-line anti-HBV drugs in the clinical management consensus of CHB [305]. In a randomized controlled trial involving 299 centers in Asia, Europe, and North and South America with a 10 year of follow-up, patients treated with entecavir had a reduced risk of HBV-related events including HCC (NCT00388674) [306]. A nationwide population-based cohort study on CHB patients suggested that tenofovir treatment had lower incidence of HCC compared with entecavir treatment [307]. The superiority of tenofovir over entecavir in reducing HCC incidence in CHB patients was further confirmed in several other studies [303, 308]. However, some studies failed to identify clinically meaningful difference in the risk of liver-related events or deaths including HCC between entecavir- and tenofovir-treated cohorts, suggesting that the choice between tenofovir or entecavir should be based on patients’ tolerability (NCT019553458) [309, 310]. A recent study compared the long-term risk of tenofovir versus entecavir on HCC and intrahepatic cholangiocarcinoma (ICC) in CHB patients and suggested a comparable long-term risk between these two agents [311]. Recently, some antifibrotic Chinese herbs have been introduced to the antiviral therapy formulas for the treatment of CHB-related liver fibrosis. For instance the therapeutic potential of entecavir combined with Ruangan granule to reverse advanced liver fibrosis is currently being investigated in a number of clinical studies [312, 313].
Antihuman papillomavirus (HPV) therapies
infection initiates a chain of reactions that regulate the secretion of inflammatory cytokines and immune cell infiltration [317]. For instance, the sustained elevation of systemic inflammatory cytokine levels was observed in older populations with chronic HPV infection [318], which potentially increased the risk for cervical cancer in this age group [319, 320].
Cytokine- and chemokine-directed therapies
IFN-a-directed therapies
clinical trials [327-330]. IFN-
[344]. Similarly, bevacizumab plus IFN led to superior benefits in terms of PFS and ORR in patients with metastatic RCC as compared with IFN monotherapy (CALGB 90206) [345]. Recent research has focused on the potential of IFN-
TGF-
efficacy and acceptable safety profile at multiple doses [360]. For patients with advanced malignant melanoma and RCC, Fresolimumab was safe and displayed preliminary antitumor efficacy (NCT00356460) [360]. A recent study examined the efficacy and immune effects of fresolimumab in metastatic breast cancer patients during radiotherapy treatment, where a favorable systemic immune response was observed. Notably, fresolimumab improved the OS of patients in a dose-dependent manner, with longer median OS observed in those treated at higher dose [361].
Galunisertib is a TGF-
PF-03446962 is a monoclonal antibody (mAb) targeting activin receptor like kinase-1 (ALK1), a TGF-
receptor and a PD-L1-blocking immunoglobulin G1 (IgG1) mAb [373].
IL-1-directed therapies
treatment of several cancers [387-390]. Preclinical studies reported that gemcitabine and 5-fluorouracil (5-FU) could promote IL-1
IL-2-directed therapies
The differential expression of IL-2 receptors has motivated the design of IL-2R agonists that selectively activate the IL-
(TNBC) [403]. A number of clinical trials are ongoing to assess the safety and clinical benefits of bempegaldesleukin when combined with pembrolizumab in patients with metastatic melanoma (NCT03635983) [404]. Bempegaldesleukin is also suggested to be used in combination with nivolumab as the first-line therapy for patients with metastatic urothelial carcinoma (NCT02983045) or metastatic melanoma (PIVOT-02), with manageable side effects [405, 406]. Nemvaleukin alfa (nemvaleukin, ALKS 4230) is a novel engineered forms of IL-2 that selectively binds to the IL-2R on antitumor CD8 + T cells and NK cells with minimal effect on immunosuppressive Tregs [325]. In a novel SCLC murine model, the mouse version of nemvaleukin (mNemvaleukin) significantly inhibited murine SCLC tumor growth and improved mouse survival, supporting the evaluation of nemvaleukin alone or in combination with chemotherapy in clinical trials [407]. Ongoing clinical trials such as ARTISTRY-7 trial compared efficacy and safety of nemvaleukin as monotherapy and combination therapy with pembrolizumab in patients with platinum-resistant ovarian cancer (NCT05092360) [408-410].
IL-6-directed therapies
Due to the elevation in systemic IL-6 levels caused by anti-IL-6 mAbs [425], some alternative IL-6-directed therapies have been developed such as functional blocking of IL-6 receptors (IL-6R). Administration of IL-6R inhibitor tocilizumab at
IL-10-directed therapies
CCL2/CCR2 axis-directed therapies
Given the suboptimal clinical efficacy of CCR2 inhibitors as monotherapy, the therapeutic potential of CCR2 inhibitors to work in synergy with chemotherapies and immune checkpoint inhibitors was then evaluated. PF-04136309 is a small-molecule CCR2 inhibitor which was mainly studied in the context of pancreatic cancer. In a phase I trial, the targeting of TAMs with PF-04136309-FOLFIRINOX combination was safe and tolerable in patients with borderline resectable and locally advanced pancreatic cancer [445]. Unfortunately, PF-04136309 combined with nab-paclitaxel plus gemcitabine resulted in synergistic pulmonary toxicity, with no superiority over in terms efficacy in PDAC patients (NCT02732938) [446]. CCR2i is a competitive binding inhibitor with a selective and high affinity for the binding pocket of CCR2 and, when combined with an immune checkpoint inhibitor, could suppress tumor growth of cutaneous T-cell lymphomas [447]. BMS-687681, a dual inhibitor targeting CCR2 and CCR5, was used as a prolonged treatment following
Natural anti-inflammatory therapies
increased level of anti-inflammatory macrophages (M2). Resveratrol impedes LPS-induced macrophage activation by inhibiting NF-kB and COX-2 signaling and inflammasome activation [459]. In a clinical study, daily consumption of resveratrol induced substantial antitumor effect in 20 patients with colorectal cancer, suggesting the potential of resveratrol as a chemopreventive drug in cancer.
Conclusions and future perspectives
To date, a wide array of inflammation-directed therapies has been developed and is under evaluation both preclinically and clinically in cancer models. With the advances outlined herein, some anti-inflammatory approaches have proven rather effective in cancer prevention and treatment, providing solid scientific rationale for further development of such strategies. Moreover, some inflammatory responses following cancer therapies would confer residual cancer cells with resistance to subsequent treatments. Immunotherapies induce durable responses in only a small subset of patients, with the majority of patients eventually experiencing primary or acquired therapy resistance. Treatment resistance to immunotherapies is often attributed to the presence of proinflammatory and immunosuppressive TME [461]. One such example is the use of anti-CTLA-4 therapies that are related to incidence of colitis and hypophysitis [462], and anti-PD-1 therapies are associated with thyroiditis [463]. Thus, the addition of anti-inflammatory therapies into cancer treatment regimes would yield better clinical responses in some clinical cases.
The initial aim of anti-inflammatory therapies is to suppress the protumoral inflammation and at the same time activate antitumor immune response. Unlike therapies that target specific tumor markers, biomarkers for the selection of anti-inflammatory therapies are lacking. Intrinsic differences of patients such as age, and tumor molecular profile would affect the therapeutic response to inflammation-directed treatments. Thus, high-resolution methods such as multiomics, single-cell, and spatial analyses are recommended to facilitate medical decision and to predict the therapeutic response to inflam-mation-directed therapies. In addition, it still remains
challenging to maintain the balance of inflammation in immune system. The heterogeneity and plasticity of the TME also pose challenges to inflammation-directed therapies by targeting a single molecule or immune cell type. For example, the disrupted feedback loops by targeting one inflammatory cytokine may lead to the compensatory activation of its involved pathways. Future studies are warranted to investigate the combination of inflam-mation-directed therapies and other treatment options for cancer, facilitating the design of safe and personalized treatment.
Abbreviations
ROS Reactive oxygen species
TNF-a Tumor necrosis factor-a
MIF Migration inhibitory factor
TAMs Tumor-associated macrophages
TANs Tumor-associated neutrophils
DCs Dendritic cells
MDSCs Myeloid-derived suppressor cells
MMP Matrix metallopeptidase
IFN Interferon
TGF-
CXCL C-X-C motif chemokine ligand
ANG1 Angiopoietin-1
NETs Neutrophil extracellular traps
EMT Endothelial-to-mesenchymal transition
GM-CSF Granulocyte-macrophage colony-stimulating factor
NK Natural killer
ECM Extracellular matrix
Tregs Regulatory T cells
Th17 Thelper 17
FLT3 Fms-related tyrosine kinase receptor 3
TAAs Tumor-associated antigens
ICD Immunogenic cell death
DAMPs Damage-associated molecular patterns
ER Endoplasmic reticulum
PRRs Pattern recognition receptors
M-MDSCs Monocytic-myeloid-derived suppressor cells
PBMC Peripheral blood mononuclear cells
NO Nitric oxide
TCR T cell receptor
ICB Immune checkpoint blockade
CSF-1R Colony-stimulating factor-1 receptor
PrP Prion protein
PAF Platelet-activating factor
TIMPs Tissue inhibitors of MMPs
HNSCC Head and neck squamous cell carcinoma
IL Interleukin
LDL-C Low-density lipoprotein cholesterol
KM-LUAD K-ras-mutant lung adenocarcinoma
Breg Regulatory B
PDAC Pancreatic ductal adenocarcinoma
NF-kB Nuclear factor kappa B
LIF Leukemia inhibitory factor
OSM Oncostatin M
CNTF Ciliary neurotrophic factor
CT-1 Cardiotrophin-1
CLC Cardiotrophin-like cytokine
CDK Cyclin-dependent kinase
XIAP X-linked inhibitor of apoptosis protein
JAK Janus kinase
STAT Signal transducer and activator of transcription
PUFAs Polyunsaturated fatty acids
COX Cyclooxygenase
LOX Lipoxygenase
| PGs | Prostaglandins |
| LXs | Lipoxins |
| mPGES-1 | Microsomal PGE2 synthase 1 |
| LT | Leukotriene |
| CRC | Colorectal cancer |
| IBD | Inflammatory bowel disease |
| CAC | Colitis-associated CRC |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| DFS | Disease-free survival |
| PFS | Progression-free survival |
| AEs | Adverse events |
| MMR | Mismatch repair |
| MSI | Microsatellite instability |
| HCC | Hepatocellular carcinoma |
| HBV | Hepatitis B virus |
| NAs | Nucleotide analogs |
| RFS | Relapse-free survival |
| NHL | Non-Hodgkin lymphomas |
| 5-FU | 5-Fluorouracil |
| MM | Multiple myeloma |
Acknowledgements
Author contributions
Funding
Availability of data and materials
Declarations
Ethics approval and consent to participate
Consent for publication
Competing interests
Published online: 22 March 2024
References
- Plytycz B, Seljelid R. From inflammation to sickness: historical perspective. Arch Immunol Ther Exp (Warsz). 2003;51(2):105-9.
- Granger DN, Senchenkova E. In: Inflammation and the Microcirculation. San Rafael (CA); 2010.
- Virchow R. An address on the value of pathological experiments. Br Med J. 1881;2(1075):198-203.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74.
- Haddow A. Addendum to “molecular repair, wound healing, and carcinogenesis: tumor production a possible overhealing”? Adv Cancer Res. 1974;20:343-66.
- Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650-9.
- Abramovitch R, Marikovsky M, Meir G, Neeman M. Stimulation of tumour angiogenesis by proximal wounds: spatial and temporal analysis by MRI. Br J Cancer. 1998;77(3):440-7.
- Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591-6.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70.
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391-403.
- Krugliak Cleveland N, Torres J, Rubin DT. What does disease progression look like in ulcerative colitis, and how might it be prevented? Gastroenterology. 2022;162(5):1396-408.
- Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. 2022;162(3):715-30.
- Lee YC, Chiang TH, Chou CK, Tu YK, Liao WC, Wu MS, Graham DY. Association between helicobacter pylori eradication and gastric cancer incidence: a systematic review and meta-analysis. Gastroenterology. 2016;150(5):1113-24.
- Tian T, Song C, Jiang L, Dai J, Lin Y, Xu X, Yu C, Ge Z, Ding Y, Wen Y, et al. Hepatitis
virus infection and the risk of cancer among the Chinese population. Int J Cancer. 2020;147(11):3075-84. - Kawanishi S, Ohnishi S, Ma N, Hiraku Y, Murata M. Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. Int J Mol Sci. 2017;18(8).
- Suresh V, Dash P, Suklabaidya S, Murmu KC, Sasmal PK, Jogdand GM, Parida D, Sethi M, Das B, Mohapatra D, et al. MIF confers survival advantage to pancreatic CAFs by suppressing interferon pathwayinduced p53-dependent apoptosis. FASEB J. 2022;36(8): e22449.
- Chen L, Zhou X, Fan LX, Yao Y, Swenson-Fields KI, Gadjeva M, Wallace DP, Peters DJ, Yu A, Grantham JJ, et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest. 2015;125(6):2399-412.
- Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19(4):237-53.
- Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, Wang B, Wang Q, Zhao Y, Cai Y. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol. 2020;88: 106939.
- Li MO, Wolf N, Raulet DH, Akkari L, Pittet MJ, Rodriguez PC, Kaplan RN, Munitz A, Zhang Z, Cheng S, et al. Innate immune cells in the tumor microenvironment. Cancer Cell. 2021;39(6):725-9.
- Aga E, Mukherjee A, Rane D, More V, Patil T, van Zandbergen G, Solbach W, Dandapat J, Tackenberg H, Ohms M, et al. Type-1 interferons prolong the lifespan of neutrophils by interfering with members of the apoptotic cascade. Cytokine. 2018;112:21-6.
- Wu M, Ma M, Tan Z, Zheng H, Liu X. Neutrophil: a new player in metastatic cancers. Front Immunol. 2020;11: 565165.
- Li S, Cong X, Gao H, Lan X, Li Z, Wang W, Song S, Wang Y, Li C, Zhang H , et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J Exp Clin Cancer Res. 2019;38(1):6.
- Albini A, Bruno A, Noonan DM, Mortara L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: implications for immunotherapy. Front Immunol. 2018;9:527.
- Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, Quatromoni JG, Stephen TL, Litzky L, Deshpande C, et al. Origin and role of a subset of tumor-associated neutrophils with antigenpresenting cell features in early-stage human lung cancer. Cancer Cell. 2016;30(1):120-35.
- Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, Fan J, Cao Y, Dai Z, Zhou J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150(7):1646-58.
- Mishalian I, Bayuh R, Eruslanov E, Michaeli J, Levy L, Zolotarov L, Singhal S, Albelda SM, Granot Z, Fridlender ZG. Neutrophils recruit regulatory
28. Sasaki S, Baba T, Muranaka H, Tanabe Y, Takahashi C, Matsugo S, Mukaida N. Involvement of prokineticin 2-expressing neutrophil infiltration in 5-fluorouracil-induced aggravation of breast cancer metastasis to lung. Mol Cancer Ther. 2018;17(7):1515-25.
29. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. 2021;61(2):194-211.
30. Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, Quail D, Walsh L, Sangwan V, Bertos N et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight. 2019;5(16).
31. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133-8.
32. Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2019;216(1):176-94.
33. Bates AM, Gomez Hernandez MP, Lanzel EA, Qian F, Brogden KA. Matrix metalloproteinase (MMP) and immunosuppressive biomarker profiles of seven head and neck squamous cell carcinoma (HNSCC) cell lines. Transl Cancer Res. 2018;7(3):533-42.
34. Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, Liu P, Li Z, Xia Y, Jiang W. Neutrophil extracellular traps induced by IL8 promote diffuse large B-cell lymphoma progression via the TLR9 signaling. Clin Cancer Res. 2019;25(6):1867-79.
35. Weiss E, Kretschmer D. Formyl-peptide receptors in infection, inflammation, and cancer. Trends Immunol. 2018;39(10):815-29.
36. Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, de Andrea C, Ochoa MC, Otano I, Etxeberria I, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52(5):856-71.
37. Azevedo PO, Paiva AE, Santos GSP, Lousado L, Andreotti JP, Sena IFG, Tagliati CA, Mintz A, Birbrair A. Cross-talk between lung cancer and bones results in neutrophils that promote tumor progression. Cancer Metastasis Rev. 2018;37(4):779-90.
38. Berger-Achituv S, Brinkmann V, Abed UA, Kuhn LI, Ben-Ezra J, Elhasid R, Zychlinsky A. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. 2013;4:48.
39. Demkow U. Neutrophil extracellular traps (NETs) in cancer invasion, evasion and metastasis. Cancers (Basel). 2021;13(17).
40. Wang Y, Liu F, Chen L, Fang C, Li S, Yuan S, Qian X, Yin Y, Yu B, Fu B, et al. Neutrophil extracellular traps (NETs) promote non-small cell lung cancer metastasis by suppressing IncRNA MIR503HG to activate the NF-kappaB/NLRP3 inflammasome pathway. Front Immunol. 2022;13: 867516.
41. Deng J, Kang Y, Cheng CC, Li X, Dai B, Katz MH, Men T, Kim MP, Koay EA, Huang H et al. DDR1-induced neutrophil extracellular traps drive pancreatic cancer metastasis. JCI Insight. 2021;6(17).
42. Khan U, Chowdhury S, Billah MM, Islam KMD, Thorlacius H, Rahman M. Neutrophil extracellular traps in colorectal cancer progression and metastasis. Int J Mol Sci. 2021;22(14).
43. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, Wang Y, Yang S, Liang C, Liang Y, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39(3):423-37.
44. Yang C, Wang Z, Li L, Zhang Z, Jin X, Wu P, Sun S, Pan J, Su K, Jia F et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J Immunother Cancer. 2021;9(10).
45. Arelaki S, Arampatzioglou A, Kambas K, Papagoras C, Miltiades P, Angelidou I, Mitsios A, Kotsianidis I, Skendros P, Sivridis E, et al. Gradient infiltration of neutrophil extracellular traps in colon cancer and evidence for their involvement in tumour growth. PLoS ONE. 2016;11(5): e0154484.
46. Millrud CR, Kagedal A, Kumlien Georen S, Winqvist O, Uddman R, Razavi R, Munck-Wikland E, Cardell LO. NET-producing CD16(high) CD62L(dim) neutrophils migrate to tumor sites and predict improved survival in patients with HNSCC. Int J Cancer. 2017;140(11):2557-67.
47. Schedel F, Mayer-Hain S, Pappelbaum KI, Metze D, Stock M, Goerge T, Loser K, Sunderkotter C, Luger TA, Weishaupt C. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment Cell Melanoma Res. 2020;33(1):63-73.
48. Muqaku B, Pils D, Mader JC, Aust S, Mangold A, Muqaku L, Slany A, Del Favero G, Gerner C. Neutrophil extracellular trap formation correlates with favorable overall survival in high grade ovarian cancer. Cancers (Basel). 2020;12(2).
49. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445-55.
50. Mackaness GB. Cellular resistance to infection. J Exp Med. 1962;116(3):381-406.
51. Duan
52. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14-20.
53. Eum HH, Kwon M, Ryu D, Jo A, Chung W, Kim N, Hong Y, Son DS, Kim ST, Lee J, et al. Tumor-promoting macrophages prevail in malignant ascites of advanced gastric cancer. Exp Mol Med. 2020;52(12):1976-88.
54. Bernsmeier C, van der Merwe S, Perianin A. Innate immune cells in cirrhosis. J Hepatol. 2020;73(1):186-201.
55. Bruns H, Buttner M, Fabri M, Mougiakakos D, Bittenbring JT, Hoffmann MH, Beier F, Pasemann S, Jitschin R, Hofmann AD, et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci Transl Med. 2015;7(282):282-247.
56. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549-55.
57. Ding P, Wang W, Wang J, Yang Z, Xue L. Expression of tumor-associated macrophage in progression of human glioma. Cell Biochem Biophys. 2014;70(3):1625-31.
58. Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, Ma X. Prognostic significance of tumor-associated macrophages in ovarian cancer: a meta-analysis. Gynecol Oncol. 2017;147(1):181-7.
59. Larionova I, Kazakova E, Gerashchenko T, Kzhyshkowska J. New angiogenic regulators produced by TAMs: perspective for targeting tumor angiogenesis. Cancers (Basel). 2021;13(13).
60. Gurevich DB, Severn CE, Twomey C, Greenhough A, Cash J, Toye AM, Mellor H, Martin P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J 2018;37(13).
61. Ramirez-Pedraza M, Fernandez M. Interplay between macrophages and angiogenesis: a double-edged sword in liver disease. Front Immunol. 2019;10:2882.
62. Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, Wang H, Wang K, Lin Y, Wang
63. Lan J, Sun L, Xu F, Liu L, Hu F, Song D, Hou Z, Wu W, Luo X, Wang J, et al. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. 2019;79(1):146-58.
64. Yin Z, Ma T, Huang B, Lin L, Zhou Y, Yan J, Zou Y, Chen S. Macrophagederived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-beta signaling pathway. J Exp Clin Cancer Res. 2019;38(1):310.
65. Shima T, Shimoda M, Shigenobu T, Ohtsuka T, Nishimura T, Emoto K, Hayashi Y, Iwasaki T, Abe T, Asamura H, et al. Infiltration of tumorassociated macrophages is involved in tumor programmed deathligand 1 expression in early lung adenocarcinoma. Cancer Sci. 2020;111(2):727-38.
66. Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang CL. PD-L1 expression on tumor-infiltrating immune cells is highly associated with M2 TAM and aggressive malignant potential in patients with resected non-small cell lung cancer. Lung Cancer. 2019;136:136-44.
67. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245-52.
68. Ness S, Lin S, Gordon JR. Regulatory dendritic cells, t cell tolerance, and dendritic cell therapy for immunologic disease. Front Immunol. 2021;12: 633436.
69. Tsapogas P, Mooney CJ, Brown G, Rolink A. The cytokine Flt3-ligand in normal and malignant hematopoiesis. Int J Mol Sci. 2017;18(6).
70. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265-77.
71. Fu C, Jiang A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front Immunol. 2018;9:3059.
72. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7-24.
73. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117(11):1583-91.
74. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945-54.
75. Rufo N, Garg AD, Agostinis P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer. 2017;3(9):643-58.
76. Wang Y, Xiang Y, Xin VW, Wang XW, Peng XC, Liu XQ, Wang D, Li N, Cheng JT, Lyv YN, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13(1):107.
77. Hole CR, Wager CML, Castro-Lopez N, Campuzano A, Cai H, Wozniak KL, Wang Y, Wormley FL Jr. Induction of memory-like dendritic cell responses in vivo. Nat Commun. 2019;10(1):2955.
78. Alzeibak R, Mishchenko TA, Shilyagina NY, Balalaeva IV, Vedunova MV, Krysko DV. Targeting immunogenic cancer cell death by photodynamic therapy: past, present and future. J Immunother Cancer. 2021;9(1).
79. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS, Vandenabeele P. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13(1):3676.
80. Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C, Maurer D, Ottensmeier C, van der Burg SH, Welters MJ, et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother. 2016;65(2):161-9.
81. Li BH, Garstka MA, Li ZF. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol Immunol. 2020;117:201-15.
82. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloidderived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208-20.
83. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J Biol Chem. 2002;277(24):21123-9.
84. Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor premetastatic niche formation. Front Immunol. 2019;10:172.
85. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: roles in tumor progression. Front Immunol. 2019;10:771.
86. Johnson BW, Achyut BR, Fulzele S, Mondal AK, Kolhe R, Arbab AS. Delineating pro-angiogenic myeloid cells in cancer therapy. Int J Mol Sci 2018;19(9).
87. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19-28.
88. Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM, Maio M, Sucker A, Schilling B, Schadendorf D, et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin Cancer Res. 2014;20(6):1601-9.
89. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E, Speiser DE. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247-57.
90. Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, Shtainberg H, Lotem M, Baniyash M. Clinical significance
of circulating CD33+CD11b+HLA-DR-myeloid cells in patients with stage IV melanoma treated with ipilimumab. Clin Cancer Res. 2016;22(23):5661-72.
91. Martens A, Wistuba-Hamprecht K, Geukes Foppen M, Yuan J, Postow MA, Wong P, Romano E, Khammari A, Dreno B, Capone M, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22(12):2908-18.
92. Holmgaard RB, Zamarin D, Lesokhin A, Merghoub T, Wolchok JD. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine. 2016;6:50-8.
93. Holmgaard RB, Brachfeld A, Gasmi B, Jones DR, Mattar M, Doman T, Murphy M, Schaer D, Wolchok JD, Merghoub T. Timing of CSF-1/CSF-1R signaling blockade is critical to improving responses to CTLA-4 based immunotherapy. Oncoimmunology. 2016;5(7): e1151595.
94. Lamichhane P, Karyampudi L, Shreeder B, Krempski J, Bahr D, Daum J, Kalli KR, Goode EL, Block MS, Cannon MJ, et al. IL10 release upon PD-1 blockade sustains immunosuppression in ovarian cancer. Cancer Res. 2017;77(23):6667-78.
95. Gomes-Santos IL, Amoozgar Z, Kumar AS, Ho WW, Roh K, Talele NP, Curtis H, Kawaguchi K, Jain RK, Fukumura D. Exercise training improves tumor control by increasing CD8(+) T-cell infiltration via CXCR3 signaling and sensitizes breast cancer to immune checkpoint blockade. Cancer Immunol Res. 2021;9(7):765-78.
96. Holder KA, Grant MD. Human cytomegalovirus IL-10 augments NK cell cytotoxicity. J Leukoc Biol. 2019;106(2):447-54.
97. O’Carroll SJ, Kho DT, Wiltshire R, Nelson V, Rotimi O, Johnson R, Angel CE, Graham ES. Pro-inflammatory TNFalpha and IL-1 beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J Neuroinflammation. 2015;12:131.
98. Hillyer P, Mordelet E, Flynn G, Male D. Chemokines, chemokine receptors and adhesion molecules on different human endothelia: discriminating the tissue-specific functions that affect leucocyte migration. Clin Exp Immunol. 2003;134(3):431-41.
99. Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004;167(2):223-9.
100. Tichet M, Prod’Homme V, Fenouille N, Ambrosetti D, Mallavialle A, Cerezo M, Ohanna M, Audebert S, Rocchi S, Giacchero D, et al. Tumourderived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun. 2015;6:6993.
101. Hiratsuka S, Goel S, Kamoun WS, Maru Y, Fukumura D, Duda DG, Jain RK. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc Natl Acad Sci U S A. 2011;108(9):3725-30.
102. Burdick MM, Henson KA, Delgadillo LF, Choi YE, Goetz DJ, Tees DF, Benencia F. Expression of E-selectin ligands on circulating tumor cells: cross-regulation with cancer stem cell regulatory pathways? Front Oncol. 2012;2:103.
103. Hauselmann I, Roblek M, Protsyuk D, Huck V, Knopfova L, Grassle S, Bauer AT, Schneider SW, Borsig L. Monocyte induction of E-selectinmediated endothelial activation releases VE-cadherin junctions to promote tumor cell extravasation in the metastasis cascade. Cancer Res. 2016;76(18):5302-12.
104. Shea DJ, Li YW, Stebe KJ, Konstantopoulos K. E-selectin-mediated rolling facilitates pancreatic cancer cell adhesion to hyaluronic acid. FASEB J. 2017;31(11):5078-86.
105. Kang SA, Blache CA, Bajana S, Hasan N, Kamal M, Morita Y, Gupta V, Tsolmon B, Suh KS, Gorenstein DG, et al. The effect of soluble E-selectin on tumor progression and metastasis. BMC Cancer. 2016;16:331.
106. Zamarron
107. Amin MN, Siddiqui SA, Ibrahim M, Hakim ML, Ahammed MS, Kabir A, Sultana F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020;8:2050312120965752.
108. Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, Lee SR, Yang SH. The role of tumor necrosis factor alpha (TNF-alpha) in autoimmune
disease and current TNF-alpha inhibitors in therapeutics. Int J Mol Sci. 2021;22(5).
109. Zhang GP, Yue X, Li SQ. Cathepsin C interacts with TNF-alpha/p38 MAPK signaling pathway to promote proliferation and metastasis in hepatocellular carcinoma. Cancer Res Treat. 2020;52(1):10-23.
110. Schroder SK, Asimakopoulou A, Tillmann S, Koschmieder S, Weiskirchen R. TNF-alpha controls lipocalin-2 expression in PC-3 prostate cancer cells. Cytokine. 2020;135: 155214.
111. Jo E, Jang HJ, Yang KE, Jang MS, Huh YH, Yoo HS, Park JS, Jang IS, Park SJ. Cordyceps militaris induces apoptosis in ovarian cancer cells through TNF-alpha/TNFR1-mediated inhibition of NF-kappaB phosphorylation. BMC Complement Med Ther. 2020;20(1):1.
112. Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (TNF-alpha) in breast cancer: molecular insights and therapeutic approaches. Cell Oncol (Dordr). 2020;43(1):1-18.
113. Garcia-Tunon I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. Role of tumor necrosis factor-alpha and its receptors in human benign breast lesions and tumors (in situ and infiltrative). Cancer Sci. 2006;97(10):1044-9.
114. Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, Frahm I, Allemand DH, Deza EG, Ares S, et al. TNFalpha-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clin Cancer Res. 2017;23(3):636-48.
115. Wu C, Fernandez SA, Criswell T, Chidiac TA, Guttridge D, Villalona-Calero M, Bekaii-Saab TS. Disrupting cytokine signaling in pancreatic cancer: a phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas. 2013;42(5):813-8.
116. Yoshimatsu Y, Wakabayashi I, Kimuro S, Takahashi N, Takahashi K, Kobayashi M, Maishi N, Podyma-Inoue KA, Hida K, Miyazono K, et al. TNFalpha enhances TGF-beta-induced endothelial-to-mesenchymal transition via TGF-beta signal augmentation. Cancer Sci. 2020;111(7):2385-99.
117. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014: 149185.
118. Rossi S, Cordella M, Tabolacci C, Nassa G, D’Arcangelo D, Senatore C, Pagnotto P, Magliozzi R, Salvati A, Weisz A, et al. TNF-alpha and metalloproteases as key players in melanoma cells aggressiveness. J Exp Clin Cancer Res. 2018;37(1):326.
119. Bertrand F, Rochotte J, Colacios C, Montfort A, Tilkin-Mariame AF, Touriol C, Rochaix P, Lajoie-Mazenc I, Andrieu-Abadie N, Levade T, et al. Blocking tumor necrosis factor alpha enhances CD8 T-cell-dependent immunity in experimental melanoma. Cancer Res. 2015;75(13):2619-28.
120. Li H, Wang R, Yu Z, Shi R, Zhang J, Gao S, Shao M, Cui S, Gao Z, Xu J, et al. Tumor necrosis factor alpha reduces SNAP29 dependent autolysosome formation to increase prion protein level and promote tumor cell migration. Virol Sin. 2021;36(3):458-75.
121. Nagar M, Jacob-Hirsch J, Vernitsky H, Berkun Y, Ben-Horin S, Amariglio N, Bank I, Kloog Y, Rechavi G, Goldstein I. TNF activates a NF-kappaBregulated cellular program in human CD45RA- regulatory T cells that modulates their suppressive function. J Immunol. 2010;184(7):3570-81.
122. Medler J, Wajant H. Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Expert Opin Ther Targets. 2019;23(4):295-307.
123. Farrugia M , Baron B . The role of TNF-alpha in rheumatoid arthritis: a focus on regulatory T cells. J Clin Transl Res. 2016;2(3):84-90.
124. Salomon BL, Leclerc M, Tosello J, Ronin E, Piaggio E, Cohen JL. Tumor necrosis factor alpha and regulatory T cells in oncoimmunology. Front Immunol. 2018;9:444.
125. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol. 2008;180(10):6467-71.
126. Torrey H, Butterworth J, Mera T, Okubo Y, Wang L, Baum D, Defusco A, Plager S, Warden S, Huang D et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumorassociated Tregs. Sci Signal. 2017;10(462).
127. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50(4):924-40.
128. Baba AB, Rah B, Bhat GR, Mushtaq I, Parveen S, Hassan R, Hameed Zargar M, Afroze D. Transforming growth factor-beta (TGF-beta) signaling in cancer-a betrayal within. Front Pharmacol. 2022;13: 791272.
129. Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF-beta signaling in bone remodeling. J Clin Invest. 2014;124(2):466-72.
130. Hu Q, Hisamatsu T, Haemmerle M, Cho MS, Pradeep S, Rupaimoole R, Rodriguez-Aguayo C, Lopez-Berestein G, Wong STC, Sood AK, et al. Role of platelet-derived Tgfbeta1 in the progression of ovarian cancer. Clin Cancer Res. 2017;23(18):5611-21.
131. Melzer C, Hass R, von der Ohe J, Lehnert H, Ungefroren H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun Signal. 2017;15(1):19.
132. Melzer C, von der Ohe J, Otterbein H, Ungefroren H, Hass R. Changes in uPA, PAI-1, and TGF-beta Production during Breast Cancer Cell Interaction with Human Mesenchymal Stroma/Stem-Like Cells (MSC). Int J Mol Sci. 2019. 20(11).
133. Villalba M, Evans SR, Vidal-Vanaclocha F, Calvo A. Role of TGF-beta in metastatic colon cancer: it is finally time for targeted therapy. Cell Tissue Res. 2017;370(1):29-39.
134. Hao Y, Baker D, Ten Dijke P. TGF-beta-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. 2019;20(11).
135. Tauriello DVF, Sancho E, Batlle E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat Rev Cancer. 2022;22(1):25-44.
136. Tan
137. Esquivel-Velazquez M, Ostoa-Saloma P, Palacios-Arreola MI, NavaCastro KE, Castro JI, Morales-Montor J. The role of cytokines in breast cancer development and progression. J Interferon Cytokine Res. 2015;35(1):1-16.
138. Laine A, Labiad O, Hernandez-Vargas H, This S, Sanlaville A, Leon S, Dalle S, Sheppard D, Travis MA, Paidassi H, et al. Regulatory T cells promote cancer immune-escape through integrin alphavbeta8-mediated TGFbeta activation. Nat Commun. 2021;12(1):6228.
139. Akkaya M, Akkaya B, Miozzo P, Rawat M, Pena M, Sheehan PW, Kim AS, Kamenyeva O, Kabat J, Bolland S, et al. B cells produce type 1 IFNs in response to the TLR9 agonist CpG-A conjugated to cationic lipids. J Immunol. 2017;199(3):931-40.
140. Ali S, Mann-Nuttel R, Schulze A, Richter L, Alferink J, Scheu S. Sources of type I interferons in infectious immunity: plasmacytoid dendritic cells not always in the driver’s seat. Front Immunol. 2019;10:778.
141. Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernan-dez-Hinojal G, Vera R, Smerdou C, Martisova E, Arozarena I, et al. PDL1 signals through conserved sequence motifs to overcome interferonmediated cytotoxicity. Cell Rep. 2017;20(8):1818-29.
142. Chen J, Cao Y, Markelc B, Kaeppler J, Vermeer JA, Muschel RJ. Type I IFN protects cancer cells from CD8+T cell-mediated cytotoxicity after radiation. J Clin Invest. 2019;129(10):4224-38.
143. Lee MS, Kim B, Oh GT, Kim YJ. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat Immunol. 2013;14(4):346-55.
144. Cunningham CR, Champhekar A, Tullius MV, Dillon BJ, Zhen A, de la Fuente JR, Herskovitz J, Elsaesser H, Snell LM, Wilson EB, et al. Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog. 2016;12(1): e1005356.
145. Gong W, Donnelly CR, Heath BR, Bellile E, Donnelly LA, Taner HF, Broses L, Brenner JC, Chinn SB, Ji RR, et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology. 2021;10(1):1997385.
146. Musella M, Guarracino A, Manduca N, Galassi C, Ruggiero E, Potenza A, Maccafeo E, Manic G, Mattiello L, Soliman Abdel Rehim S, et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nat Immunol. 2022;23(9):1379-92.
147. Pidugu VK, Wu MM, Yen AH, Pidugu HB, Chang KW, Liu CJ, Lee TC. IFIT1 and IFIT3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-EGFR recycling. Oncogene. 2019;38(17):3232-47.
148. Boukhaled GM, Harding S, Brooks DG. Opposing roles of type I interferons in cancer immunity. Annu Rev Pathol. 2021;16:167-98.
149. Spaapen RM, Leung MY, Fuertes MB, Kline JP, Zhang L, Zheng Y, Fu YX, Luo X, Cohen KS, Gajewski TF. Therapeutic activity of high-dose intratumoral IFN-beta requires direct effect on the tumor vasculature. J Immunol. 2014;193(8):4254-60.
150. Golomb HM, Ratain MJ, Mick R, Daly K. Interferon treatment for hairy cell leukemia: an update on a cohort of 69 patients treated from 1983-1986. Leukemia. 1992;6(11):1177-80.
151. Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int J Mol Sci. 2018;19(8).
152. Malik A, Kanneganti TD. Function and regulation of IL-1alpha in inflammatory diseases and cancer. Immunol Rev. 2018;281(1):124-37.
153. Fahey E, Doyle SL. IL-1 family cytokine regulation of vascular permeability and angiogenesis. Front Immunol. 2019;10:1426.
154. Xiao
155. Haabeth OA, Lorvik KB, Yagita H, Bogen B, Corthay A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology. 2016;5(1): e1039763.
156. Baker KJ, Houston A, Brint E. IL-1 family members in cancer; two sides to every story. Front Immunol. 2019;10:1197.
157. Zhang W, Borcherding N, Kolb R. IL-1 signaling in tumor microenvironment. Adv Exp Med Biol. 2020;1240:1-23.
158. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, Koski G, Disis ML, Czerniecki BJ, Kodumudi K. Differentiation and regulation of T(H) cells: a balancing act for cancer immunotherapy. Front Immunol. 2021;12: 669474.
159. Lin D, Mei Y, Lei L, Binte Hanafi Z, Jin Z, Liu Y, Song Y, Zhang Y, Hu B, Liu
160. Jiang H, Gebhardt C, Umansky L, Beckhove P, Schulze TJ, Utikal J, Umansky V. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int J Cancer. 2015;136(10):2352-60.
161. Voronov E, Carmi Y, Apte RN. The role IL-1 in tumor-mediated angiogenesis. Front Physiol. 2014;5:114.
162. Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, Abutbul S, Huszar M, Dinarello CA, Apte RN, et al. The role of IL-1 beta in the early tumor cell-induced angiogenic response. J Immunol. 2013;190(7):3500-9.
163. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, Dinarello CA, Voronov E, Apte RN. Blocking IL-1 beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A. 2019;116(4):1361-9.
164. Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. Tumor cell-derived IL1beta promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 2020;80(5):1088-101.
165. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6(10): a016295.
166. Qin B, Zhou Z, He J, Yan C, Ding S. IL-6 inhibits starvation-induced autophagy via the STAT3/Bcl-2 signaling pathway. Sci Rep. 2015;5:15701.
167. Shi R, Chen M, Litifu B. Serum interleukin-6 and survivin levels predict clinical response to etanercept treatment in patients with established rheumatoid arthritis. Mod Rheumatol. 2018;28(1):126-32.
168. Yao X, Huang J, Zhong H, Shen N, Faggioni R, Fung M, Yao Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141(2):125-39.
169. Ortiz-Montero P, Londono-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/ inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. 2017;15(1):17.
170. Sapochnik M, Haedo MR, Fuertes M, Ajler P, Carrizo G, Cervio A, Sevlever G, Stalla GK, Arzt E. Autocrine IL-6 mediates pituitary tumor senescence. Oncotarget. 2017;8(3):4690-702.
171. Ray K, Ujvari B, Ramana V, Donald J. Cross-talk between EGFR and IL-6 drives oncogenic signaling and offers therapeutic opportunities in cancer. Cytokine Growth Factor Rev. 2018;41:18-27.
172. Gao S, Hu J, Wu X, Liang Z. PMA treated THP-1-derived-IL-6 promotes EMT of SW48 through STAT3/ERK-dependent activation of Wnt/betacatenin signaling pathway. Biomed Pharmacother. 2018;108:618-24.
173. Liu W, Wang H, Bai F, Ding L, Huang Y, Lu C, Chen S, Li C, Yue X, Liang X, et al. IL-6 promotes metastasis of non-small-cell lung cancer by upregulating TIM-4 via NF-kappaB. Cell Prolif. 2020;53(3): e12776.
174. Bharti R, Dey G, Das AK, Mandal M. Differential expression of IL-6/IL-6R and MAO-A regulates invasion/angiogenesis in breast cancer. Br J Cancer. 2018;118(11):1442-52.
175. Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 is a promising therapeutic target for hepatocellular carcinoma. Front Oncol. 2021;11: 760971.
176. Zhang B, Li Y, Wu Q, Xie L, Barwick B, Fu C, Li X, Wu D, Xia S, Chen J, et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat Commun. 2021;12(1):1714.
177. Manore SG, Doheny DL, Wong GL, Lo HW. IL-6/JAK/STAT3 signaling in breast cancer metastasis: biology and treatment. Front Oncol. 2022;12: 866014.
178. Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse Thelper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170(6):2081-95.
179. Vieira P, de Waal-Malefyt R, Dang MN, Johnson KE, Kastelein R, Fiorentino DF, deVries JE, Roncarolo MG, Mosmann TR, Moore KW. Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: homology to Epstein-Barr virus open reading frame BCRFI. Proc Natl Acad Sci U S A. 1991;88(4):1172-6.
180. Macatonia SE, Doherty TM, Knight SC, O’Garra A. Differential effect of IL-10 on dendritic cell-induced T cell proliferation and IFN-gamma production. J Immunol. 1993;150(9):3755-65.
181. Ouyang W, O’Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. 2019;50(4):871-91.
182. Wang
183. Zhang H, Li R, Cao Y, Gu Y, Lin C, Liu X, Lv K, He X, Fang H, Jin K, et al. Poor clinical outcomes and immunoevasive contexture in intratumoral IL-10-producing macrophages enriched gastric cancer patients. Ann Surg. 2022;275(4):e626-35.
184. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, Callahan DJ, Sun Z, Sun T, Tabib T, et al. Adaptive plasticity of IL-10(+) and IL-35(+) T(reg) cells cooperatively promotes tumor T cell exhaustion. Nat Immunol. 2019;20(6):724-35.
185. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, Blaisdell S, Basham B, Dai J, Grein J, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell. 2011;20(6):781-96.
186. Chen WF, Zlotnik A. IL-10: a novel cytotoxic T cell differentiation factor. J Immunol. 1991;147(2):528-34.
187. Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, Rhee SG, Thornalley PJ, Partridge L, Gems D, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011;13(4):361-6.
188. Ochoa CD, Wu RF, Terada LS. ROS signaling and ER stress in cardiovascular disease. Mol Aspects Med. 2018;63:18-29.
189. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020;152:116-41.
190. Violi F, Carnevale R, Loffredo L, Pignatelli P, Gallin JI. NADPH oxidase-2 and atherothrombosis: insight from chronic granulomatous disease. Arterioscler Thromb Vasc Biol. 2017;37(2):218-25.
191. Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8(9):722-8.
192. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829-37.
193. Pei J, Pan X, Wei G, Hua Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front Pharmacol. 2023;14:1147414.
194. Zhang J, Li F, Yin Y, Liu N, Zhu M, Zhang H, Liu W, Yang M, Qin S, Fan X , et al. Alpha radionuclide-chelated radioimmunotherapy promoters enable local radiotherapy/chemodynamic therapy to discourage cancer progression. Biomater Res. 2022;26(1):44.
195. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, Castoria G, Migliaccio A. ROS in cancer therapy: the bright side of the moon. Exp Mol Med. 2020;52(2):192-203.
196. Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61(4):290-302.
197. Lee HC, Wei YH. Mitochondrial DNA instability and metabolic shift in human cancers. Int J Mol Sci. 2009;10(2):674-701.
198. Yu LM, Zhang WH, Han XX, Li YY, Lu Y, Pan J, Mao JQ, Zhu LY, Deng JJ, Huang W, et al. Hypoxia-induced ROS contribute to myoblast pyroptosis during obstructive sleep apnea via the NF-kappaB/HIF-1alpha signaling pathway. Oxid Med Cell Longev. 2019;2019:4596368.
199. Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47-71.
200. Willson JA, Arienti S, Sadiku P, Reyes L, Coelho P, Morrison T, Rinaldi G, Dockrell DH, Whyte MKB, Walmsley SR. Neutrophil HIF-1alpha stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood. 2022;139(2):281-6.
201. Sun B, Yu L, Xu C, Li YM, Zhao YR, Cao MM, Yang LY. NAD(P)HX epimerase downregulation promotes tumor progression through ROS/HIF-1 alpha signaling in hepatocellular carcinoma. Cancer Sci. 2021;112(7):2753-69.
202. Zhang L, Cao Y, Guo X, Wang X, Han X, Kanwore K, Hong X, Zhou H, Gao D. Hypoxia-induced ROS aggravate tumor progression through HIF-1 alpha-SERPINE1 signaling in glioblastoma. J Zhejiang Univ Sci B. 2023;24(1):32-49.
203. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10(3):181-93.
204. Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002;99(21):13926-31.
205. Esh CJ, Chrismas BCR, Mauger AR, Taylor L. Pharmacological hypotheses: is acetaminophen selective in its cyclooxygenase inhibition? Pharmacol Res Perspect. 2021;9(4): e00835.
206. Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183-8.
207. Hijos-Mallada G, Sostres C, Gomollon F. NSAIDs, gastrointestinal toxicity and inflammatory bowel disease. Gastroenterol Hepatol. 2022;45(3):215-22.
208. Tudor DV, Baldea I, Lupu M, Kacso T, Kutasi E, Hopartean A, Stretea R, Gabriela Filip A. COX-2 as a potential biomarker and therapeutic target in melanoma. Cancer Biol Med. 2020;17(1):20-31.
209. Solanki R, Agrawal N, Ansari M, Jain S, Jindal A. COX-2 expression in breast carcinoma with correlation to clinicopathological parameters. Asian Pac J Cancer Prev. 2018;19(7):1971-5.
210. Khor LY, Bae K, Pollack A, Hammond ME, Grignon DJ, Venkatesan VM, Rosenthal SA, Ritter MA, Sandler HM, Hanks GE, et al. COX-2 expression predicts prostate-cancer outcome: analysis of data from the RTOG 92-02 trial. Lancet Oncol. 2007;8(10):912-20.
211. Guo W, Zhang Z, Li G, Lai X, Gu R, Xu W, Chen H, Xing Z, Chen L, Qian J, et al. Pyruvate kinase M2 promotes prostate cancer metastasis through regulating ERK1/2-COX-2 signaling. Front Oncol. 2020;10: 544288.
212. Du J, Feng J, Luo D, Peng L. Prognostic and clinical significance of COX-2 overexpression in laryngeal cancer: a meta-analysis. Front Oncol. 2022;12: 854946.
213. Hu Z, Yang Y, Zhao Y, Huang Y. The prognostic value of cyclooxyge-nase-2 expression in patients with esophageal cancer: evidence from a meta-analysis. Onco Targets Ther. 2017;10:2893-901.
214. Ren J, Liu J, Sui X. Correlation of COX-2 and MMP-13 expressions with gastric cancer and their effects on prognosis. J BUON. 2019;24(1):187-93.
215. Pomianowska E, Schjolberg AR, Clausen OP, Gladhaug IP. COX-2 overexpression in resected pancreatic head adenocarcinomas correlates with favourable prognosis. BMC Cancer. 2014;14:458.
216. Sun H, Zhang X, Sun D, Jia X, Xu L, Qiao Y, Jin Y. COX-2 expression in ovarian cancer: an updated meta-analysis. Oncotarget. 2017;8(50):88152-62.
217. Wang D, Cabalag CS, Clemons NJ, DuBois RN. Cyclooxygenases and prostaglandins in tumor immunology and microenvironment of gastrointestinal cancer. Gastroenterology. 2021;161(6):1813-29.
218. Li YF, Han CC, Wang Y, Cui DQ, Luo TT, Zhang YW, Ma Y, Wei W. Combined PGE2 with TNF-alpha promotes laryngeal carcinoma progression by enhancing GRK2 and TRAF2 interaction. Neoplasma. 2020;67(2):354-63.
219. Walker OL, Dahn ML, Power Coombs MR, Marcato P. The prostaglandin e2 pathway and breast cancer stem cells: evidence of increased signaling and potential targeting. Front Oncol. 2021;11: 791696.
220. Frejborg E, Salo T, Salem A. Role of cyclooxygenase-2 in head and neck tumorigenesis. Int J Mol Sci. 2020;21(23).
221. Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Xu D, Rosenberg DW. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68(9):3251-9.
222. Dean PT, Hooks SB. Pleiotropic effects of the COX-2/PGE2 axis in the glioblastoma tumor microenvironment. Front Oncol. 2022;12:1116014.
223. Knudsen NH, Manguso RT. Tumor-derived PGE2 gives NK cells a headache. Immunity. 2020;53(6):1131-2.
224. Walker W, Rotondo D. Prostaglandin E2 is a potent regulator of interleu-kin-12- and interleukin-18-induced natural killer cell interferon-gamma synthesis. Immunology. 2004;111(3):298-305.
225. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67(9):4507-13.
226. Tomic S, Joksimovic B, Bekic M, Vasiljevic M, Milanovic M, Colic M, Vucevic D. Prostaglanin-E2 potentiates the suppressive functions of human mononuclear myeloid-derived suppressor cells and increases their capacity to expand IL-10-producing regulatory T cell subsets. Front Immunol. 2019;10:475.
227. Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, Larghi P, Rimoldi M, Tripodo C, Strauss L, et al. Tumor-derived prostaglandin E2 promotes p50 NF-kappaB-dependent differentiation of monocytic MDSCs. Cancer Res. 2020;80(13):2874-88.
228. Zhang B, Bie Q, Wu P, Zhang J, You B, Shi H, Qian H, Xu W. PGD2/PTGDR2 signaling restricts the self-renewal and tumorigenesis of gastric cancer. Stem Cells. 2018;36(7):990-1003.
229. Iwanaga K, Nakamura T, Maeda S, Aritake K, Hori M, Urade Y, Ozaki H, Murata T. Mast cell-derived prostaglandin D2 inhibits colitis and colitisassociated colon cancer in mice. Cancer Res. 2014;74(11):3011-9.
230. Vafaeinik F, Kum HJ, Jin SY, Min DS, Song SH, Ha HK, Kim CD, Bae SS. Regulation of epithelial-mesenchymal transition of A549 cells by prostaglandin D(2). Cell Physiol Biochem. 2022;56(2):89-104.
231. Conejo-Garcia JR. Breaking barriers for T cells by targeting the EPHA2/TGF-beta/COX-2 axis in pancreatic cancer. J Clin Invest. 2019;129(9):3521-3.
232. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, Wang X, Zhang J, Zhang X, Zheng L, et al. A RIPK3-PGE(2) circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res. 2018;78(19):5586-99.
233. Take Y, Koizumi S, Nagahisa A. Prostaglandin E receptor 4 antagonist in cancer immunotherapy: mechanisms of action. Front Immunol. 2020;11:324.
234. Clemente SM, Martinez-Costa OH, Monsalve M, Samhan-Arias AK. Targeting lipid peroxidation for cancer treatment. Molecules. 2020;25(21).
235. Lin H, Weng J, Mei H, Zhuang M, Xiao X, Du F, Lin L, Wu J, Chen Z, Huang Y, et al. 5-Lipoxygenase promotes epithelial-mesenchymal transition through the ERK signaling pathway in gastric cancer. J Gastroenterol Hepatol. 2021;36(2):455-66.
236. Xia C, Sadeghi L, Straat K, Merrien M, Wright AP, Sander B, Xu D, Osterborg A, Bjorkholm M, Claesson HE. Intrinsic 5-lipoxygenase activity regulates migration and adherence of mantle cell lymphoma cells. Prostaglandins Other Lipid Mediat. 2021;156: 106575.
237. Muthuraman S, Sinha S, Vasavi CS, Waidha KM, Basu B, Munussami P, Balamurali MM, Doble M, Saravana Kumar R. Design, synthesis and identification of novel coumaperine derivatives for inhibition of human 5-LOX: Antioxidant, pseudoperoxidase and docking studies. Bioorg Med Chem. 2019;27(4):604-19.
238. Doiphode S, Lokhande KB, Ghosh P, Swamy KV, Nagar S. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) by resveratrol derivatives in cancer therapy: in silico approach. J Biomol Struct Dyn. 2022:1-16.
239. Shi HY, Lv FJ, Zhu ST, Wang QG, Zhang ST. Dual inhibition of 5-LOX and COX-2 suppresses esophageal squamous cell carcinoma. Cancer Lett. 2011;309(1):19-26.
240. Chen S, Zou H. Key role of 12 -lipoxygenase and its metabolite 12-hydroxyeicosatetraenoic acid (12-HETE) in diabetic retinopathy. Curr Eye Res. 2022;47(3):329-35.
241. Liu Q, Tan W, Che J, Yuan D, Zhang L, Sun Y, Yue X, Xiao L, Jin Y. 12-HETE facilitates cell survival by activating the integrin-linked kinase/NF-kappaB pathway in ovarian cancer. Cancer Manag Res. 2018;10:5825-38.
242. Mao F, Wang M, Wang J, Xu WR. The role of 15-LOX-1 in colitis and colitis-associated colorectal cancer. Inflamm Res. 2015;64(9):661-9.
243. Kazan HH, Urfali-Mamatoglu C, Yalcin GD, Bulut O, Sezer A, Banerjee S, Gunduz U. 15-LOX-1 has diverse roles in the resensitization of resistant cancer cell lines to doxorubicin. J Cell Physiol. 2020;235(5):4965-78.
244. Na YJ, Kim BR, Kim JL, Kang S, Jeong YA, Park SH, Jo MJ, Kim JY, Kim HJ, Oh SC et al. Deficiency of 15-LOX-1 induces radioresistance through downregulation of MacroH2A2 in colorectal cancer. Cancers (Basel). 2019;11(11).
245. Berger A. What are leukotrienes and how do they work in asthma? BMJ. 1999;319(7202):90.
246. Bachi AL, Kim FJ, Nonogaki S, Carneiro CR, Lopes JD, Jasiulionis MG, Correa M. Leukotriene B4 creates a favorable microenvironment for murine melanoma growth. Mol Cancer Res. 2009;7(9):1417-24.
247. Wendel A, Tiegs G. Leukotriene D4 mediates galactosamine/endotoxininduced hepatitis in mice. Biochem Pharmacol. 1987;36(12):1867.
248. Zhou Y, Guo D, Li H, Jie S. Circulating LTD4 in patients with hepatocellular carcinoma. Tumour Biol. 2011;32(1):139-44.
249. Arai J, Goto K, Otoyama Y, Nakajima Y, Sugiura I, Kajiwara A, Tojo M, Ichikawa Y, Uozumi S, Shimozuma Y, et al. Leukotriene receptor antagonists enhance HCC treatment efficacy by inhibiting ADAMs and suppressing MICA shedding. Cancer Immunol Immunother. 2021;70(1):203-13.
250. Wang Z, Cheng Q, Tang K, Sun Y, Zhang K, Zhang Y, Luo S, Zhang H, Ye D, Huang B. Lipid mediator lipoxin A4 inhibits tumor growth by targeting IL-10-producing regulatory B (Breg) cells. Cancer Lett. 2015;364(2):118-24.
251. Serhan CN. The resolution of inflammation: the devil in the flask and in the details. FASEB J. 2011;25(5):1441-8.
252. Gleeson MJ, Felix H, Johnsson LG. Ultrastructural aspects of the human peripheral vestibular system. Acta Otolaryngol Suppl. 1990;470:80-7.
253. Liu H, Zeng J, Huang W, Xu Q, Ye D, Sun R, Zhang D. Colorectal cancer is associated with a deficiency of lipoxin A(4), an endogenous anti-inflammatory mediator. J Cancer. 2019;10(19):4719-30.
254. Jia G, Wang X, Wu W, Zhang Y, Chen S, Zhao J, Zhao W, Li W, Sun X, Han B. LXA4 enhances prostate cancer progression by facilitating M2 macrophage polarization via inhibition of METTL3. Int Immunopharmacol. 2022;107: 108586.
255. Yuan J, Lin F, Chen L, Chen W, Pan X, Bai Y, Cai Y, Lu H. Lipoxin A4 regulates M1/M2 macrophage polarization via FPR2-IRF pathway. Inflammopharmacology. 2022;30(2):487-98.
256. Korbecki J, Rebacz-Maron E, Kupnicka P, Chlubek D, BaranowskaBosiacka I. Synthesis and significance of arachidonic acid, a substrate for cyclooxygenases, lipoxygenases, and cytochrome p450 pathways in the tumorigenesis of glioblastoma multiforme, including a pan-cancer comparative analysis. Cancers (Basel). 2023;15(3).
257. Lakshmanan K, Byran G, Bandlamudi S, Krishnamurthy PT. The role of STAT3 signaling in different types of cancers: a comprehensive review. Curr Enzym Inhib. 2020;16(3):189-98.
258. Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol Rev. 2020;72(2):486-526.
259. Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, Ma H, Wei D, Sun S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. 2020;80: 106210.
260. Wong ALA, Hirpara JL, Pervaiz S, Eu JQ, Sethi G, Goh BC. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin Investig Drugs. 2017;26(8):883-7.
261. Hirano T, Hirayama D, Wagatsuma K, Yamakawa T, Yokoyama Y, Nakase H: Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. Int J Mol Sci. 2020;21(9).
262. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119-24.
263. Kasembeli MM, Bharadwaj U, Robinson P, Tweardy DJ. Contribution of STAT3 to inflammatory and fibrotic diseases and prospects for its targeting for treatment. Int J Mol Sci. 2018; 19(8).
264. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103-13.
265. Gargalionis AN, Papavassiliou KA, Papavassiliou AG. Targeting STAT3 signaling pathway in colorectal cancer. Biomedicines. 2021; 9(8).
266. Zhong B, Cheng B, Huang X, Xiao Q, Niu Z, Chen YF, Yu Q, Wang W, Wu XJ . Colorectal cancer-associated fibroblasts promote metastasis by upregulating LRG1 through stromal IL-6/STAT3 signaling. Cell Death Dis. 2021;13(1):16.
267. Wang S, Dong W, Liu L, Xu M, Wang Y, Liu T, Zhang Y, Wang B, Cao H. Interplay between bile acids and the gut microbiota promotes intestinal carcinogenesis. Mol Carcinog. 2019;58(7):1155-67.
268. Roche B, Vanden-Bossche A, Normand M, Malaval L, Vico L, LafageProust MH. Validated laser doppler protocol for measurement of mouse bone blood perfusion-response to age or ovariectomy differs with genetic background. Bone. 2013;55(2):418-26.
269. Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci. 2014;19(2):164-74.
270. D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813-24.
271. Florean
272. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245-313.
273. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. 2019;19(18): e1800311.
274. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, Li B. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12(1):34.
275. Prasad AS, Miale A Jr, Farid Z, Sandstead HH, Schulert AR. Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypognadism. J Lab Clin Med. 1963;61:537-49.
276. Liu MJ, Bao S, Galvez-Peralta M, Pyle CJ, Rudawsky AC, Pavlovicz RE, Killilea DW, Li C, Nebert DW, Wewers MD, et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-kappaB. Cell Rep. 2013;3(2):386-400.
277. Burn J, Sheth H, Elliott F, Reed L, Macrae F, Mecklin JP, Moslein G, McRonald FE, Bertario L, Evans DG, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet. 2020;395(10240):1855-63.
278. Bens A, Cronin-Fenton D, Dehlendorff C, Jensen MB, Ejlertsen B, Kroman N, Friis S, Mellemkjaer L. Nonaspirin NSAIDs and contralateral breast cancer risk. Int J Cancer. 2019;144(6):1243-50.
279. Hao W, Shen Y, Feng M, Wang H, Lin M, Fang Y, Tan L. Aspirin acts in esophageal cancer: a brief review. J Thorac Dis. 2018;10(4):2490-7.
280. Burn J, Bishop DT, Chapman PD, Elliott F, Bertario L, Dunlop MG, Eccles D, Ellis A, Evans DG, Fodde R, et al. A randomized placebocontrolled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev Res (Phila). 2011;4(5):655-65.
281. Ishikawa H, Wakabayashi K, Suzuki S, Mutoh M, Hirata K, Nakamura T, Takeyama I, Kawano A, Gondo N, Abe T, et al. Preventive effects of lowdose aspirin on colorectal adenoma growth in patients with familial adenomatous polyposis: double-blind, randomized clinical trial. Cancer Med. 2013;2(1):50-6.
282. Jankowski JAZ, de Caestecker J, Love SB, Reilly G, Watson P, Sanders S, Ang Y, Morris D, Bhandari P, Brooks C, et al. Esomeprazole and aspirin
in Barrett’s oesophagus (AspECT): a randomised factorial trial. Lancet. 2018;392(10145):400-8.
283. Wu QB, Sun GP. Expression of COX-2 and HER-2 in colorectal cancer and their correlation. World J Gastroenterol. 2015;21(20):6206-14.
284. Sheng J, Sun H, Yu FB, Li B, Zhang Y, Zhu YT. The role of cyclooxyge-nase-2 in colorectal cancer. Int J Med Sci. 2020;17(8):1095-101.
285. Todoric J, Antonucci L, Karin M. Targeting inflammation in cancer prevention and therapy. Cancer Prev Res (Phila). 2016;9(12):895-905.
286. North GL. Celecoxib as adjunctive therapy for treatment of colorectal cancer. Ann Pharmacother. 2001;35(12):1638-43.
287. Mostafa TM, Alm El-Din MA, Rashdan AR. Celecoxib as an adjuvant to chemotherapy for patients with metastatic colorectal cancer: a randomized controlled clinical study. Saudi Med J. 2022;43(1):37-44.
288. Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355(9):873-84.
289. Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J, Zavoral M, Lechuga MJ, Gerletti P, Tang J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355(9):885-95.
290. Hu H, Kang L, Zhang J, Wu Z, Wang H, Huang M, Lan P, Wu X, Wang C, Cao W, et al. Neoadjuvant PD-1 blockade with toripalimab, with or without celecoxib, in mismatch repair-deficient or microsatellite instability-high, locally advanced, colorectal cancer (PICC): a single-centre, parallel-group, non-comparative, randomised, phase 2 trial. Lancet Gastroenterol Hepatol. 2022;7(1):38-48.
291. Ye SY, Li JY, Li TH, Song YX, Sun JX, Chen XW, Zhao JH, Li Y, Wu ZH, Gao P , et al. The efficacy and safety of celecoxib in addition to standard cancer therapy: a systematic review and meta-analysis of randomized controlled trials. Curr Oncol. 2022;29(9):6137-53.
292. Guo Q, Liu X, Lu L, Yuan H, Wang Y, Chen Z, Ji R, Zhou Y. Comprehensive evaluation of clinical efficacy and safety of celecoxib combined with chemotherapy in management of gastric cancer. Medicine (Baltimore). 2017;96(51): e8857.
293. Liao Z, Komaki R, Milas L, Yuan C, Kies M, Chang JY, Jeter M, Guerrero T, Blumenschien G, Smith CM, et al. A phase I clinical trial of thoracic radiotherapy and concurrent celecoxib for patients with unfavorable performance status inoperable/unresectable non-small cell lung cancer. Clin Cancer Res. 2005;11(9):3342-8.
294. Koch A, Bergman B, Holmberg E, Sederholm C, Ek L, Kosieradzki J, Lamberg K, Thaning L, Ydreborg SO, Sorenson S, et al. Effect of celecoxib on survival in patients with advanced non-small cell lung cancer: a double blind randomised clinical phase III trial (CYCLUS study) by the Swedish Lung Cancer Study Group. Eur J Cancer. 2011;47(10):1546-55.
295. Bi N, Liang J, Zhou Z, Chen D, Fu Z, Yang X, Feng Q, Hui Z, Xiao Z, Lv J, et al. Effect of concurrent chemoradiation with celecoxib vs concurrent chemoradiation alone on survival among patients with non-small cell lung cancer with and without cyclooxygenase 2 genetic variants: a phase 2 randomized clinical trial. JAMA Netw Open. 2019;2(12): e1918070.
296. Bayraktar S, Baghaki S, Wu J, Liu DD, Gutierrez-Barrera AM, Bevers TB, Valero V, Sneige N, Arun BK. Biomarker modulation study of celecoxib for chemoprevention in women at increased risk for breast cancer: a phase II pilot study. Cancer Prev Res (Phila). 2020;13(9):795-802.
297. Guo Q, Li Q, Wang J, Liu M, Wang Y, Chen Z, Ye Y, Guan Q, Zhou Y. A comprehensive evaluation of clinical efficacy and safety of celecoxib in combination with chemotherapy in metastatic or postoperative recurrent gastric cancer patients: a preliminary, three-center, clinical trial study. Medicine (Baltimore). 2019;98(27): e16234.
298. De Cremoux P, Hamy AS, Lehmann-Che J, Scott V, Sigal B, Mathieu MC, Bertheau P, Guinebretiere JM, Pierga JY, Giacchetti S, et al. COX2/PTGS2 expression is predictive of response to neoadjuvant celecoxib in HER2negative breast cancer patients. Anticancer Res. 2018;38(3):1485-90.
299. Meyerhardt JA, Shi Q, Fuchs CS, Meyer J, Niedzwiecki D, Zemla T, Kumthekar P, Guthrie KA, Couture F, Kuebler P, et al. Effect of celecoxib vs placebo added to standard adjuvant therapy on disease-free survival among patients with stage III colon cancer: the CALGB/SWOG 80702 (alliance) randomized clinical trial. JAMA. 2021;325(13):1277-86.
300. Coombes RC, Tovey H, Kilburn L, Mansi J, Palmieri C, Bartlett J, Hicks J, Makris A, Evans A, Loibl S, et al. Effect of celecoxib vs placebo as adjuvant therapy on disease-free survival among patients with
breast cancer: the REACT randomized clinical trial. JAMA Oncol. 2021;7(9):1291-301.
301. Hamy AS, Tury S, Wang X, Gao J, Pierga JY, Giacchetti S, Brain E, Pistilli B, Marty M, Espie M, et al. Celecoxib with neoadjuvant chemotherapy for breast cancer might worsen outcomes differentially by COX-2 expression and ER status: exploratory analysis of the REMAGUS02 trial. J Clin Oncol. 2019;37(8):624-35.
302. Schjerning AM, McGettigan P, Gislason G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat Rev Cardiol. 2020;17(9):574-84.
303. Cui J, Jia J. Natural COX-2 inhibitors as promising anti-inflammatory agents: an update. Curr Med Chem. 2021;28(18):3622-46.
304. Yang L, Zou T, Chen Y, Zhao Y, Wu X, Li M, Du F, Chen Y, Xiao Z, Shen J. Hepatitis
305. European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L: EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370-98.
306. Hou JL, Zhao W, Lee C, Hann HW, Peng CY, Tanwandee T, Morozov V, Klinker H, Sollano JD, Streinu-Cercel A, et al. Outcomes of long-term treatment of chronic HBV infection with entecavir or other agents from a randomized trial in 24 countries. Clin Gastroenterol Hepatol. 2020;18(2):457-67.
307. Choi J, Kim HJ, Lee J, Cho S, Ko MJ, Lim YS. Risk of hepatocellular carcinoma in patients treated with entecavir vs tenofovir for chronic hepatitis B: a Korean nationwide cohort study. JAMA Oncol. 2019;5(1):30-6.
308. Zhang Z, Zhou Y, Yang J, Hu K, Huang Y. The effectiveness of TDF versus ETV on incidence of HCC in CHB patients: a meta analysis. BMC Cancer. 2019;19(1):511.
309. Tan DJH, Ng CH, Tay PWL, Syn N, Muthiah MD, Lim WH, Tang ASP, Lim KE, Lim GEH, Tamaki N, et al. Risk of hepatocellular carcinoma with tenofovir vs entecavir treatment for chronic hepatitis B virus: a reconstructed individual patient data meta-analysis. JAMA Netw Open. 2022;5(6): e2219407.
310. Pol S. group AAs: Similar 5-year HCC occurrence in Tenofovir- and Entecavir-treated HBV chronic infection in the French AFEF/ANRS CO22 Hepather cohort. Aliment Pharmacol Ther. 2021;53(5):616-29.
311. Chang TS, Yang YH, Chen WM, Shen CH, Tung SY, Yen CW, Hsieh YY, Lee CP, Tsai ML, Hung CH, et al. Long-term risk of primary liver cancers in entecavir versus tenofovir treatment for chronic hepatitis B. Sci Rep. 2021;11(1):1365.
312. Xing Y, Zhong W, Peng D, Han Z, Zeng H, Wang Y, Feng L, Huang J, Xu L, Chen
313. Ji D, Chen Y, Bi J, Shang Q, Liu H, Wang JB, Tan L, Wang J, Chen Y, Li Q, et al. Entecavir plus Biejia-Ruangan compound reduces the risk of hepatocellular carcinoma in Chinese patients with chronic hepatitis B. J Hepatol. 2022;77(6):1515-24.
314. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394-424.
315. Nahand JS, Taghizadeh-Boroujeni S, Karimzadeh M, Borran S, Pourhanifeh MH, Moghoofei M, Bokharaei-Salim F, Karampoor S, Jafari A, Asemi Z, et al. microRNAs: new prognostic, diagnostic, and therapeutic biomarkers in cervical cancer. J Cell Physiol. 2019;234(10):17064-99.
316. Sadri Nahand J, Moghoofei M, Salmaninejad A, Bahmanpour Z, Karimzadeh M, Nasiri M, Mirzaei HR, Pourhanifeh MH, Bokharaei-Salim F, Mirzaei H, et al. Pathogenic role of exosomes and microRNAs in HPV-mediated inflammation and cervical cancer: a review. Int J Cancer. 2020;146(2):305-20.
317. Laniewski P, Barnes D, Goulder A, Cui H, Roe DJ, Chase DM, HerbstKralovetz MM. Linking cervicovaginal immune signatures, HPV and microbiota composition in cervical carcinogenesis in non-Hispanic and Hispanic women. Sci Rep. 2018;8(1):7593.
318. KempTJ, Hildesheim A, Garcia-Pineres A, Williams MC, Shearer GM, Rodriguez AC, Schiffman M, Burk R, Freer E, Bonilla J, et al. Elevated systemic levels of inflammatory cytokines in older women with persistent
cervical human papillomavirus infection. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1954-9.
319. Yost S, Hoekstra A. Cervical cancer in women over 65: an analysis of screening. Gynecol Oncol Rep. 2018;25:48-51.
320. Sales KJ, Katz AA. Inflammatory pathways in cervical cancer-the UCT contribution. S Afr Med J. 2012;102(6):493-6.
321. Drolet M, Benard E, Perez N, Brisson M. Group HPVVIS: Population-level impact and herd effects following the introduction of human papillomavirus vaccination programmes: updated systematic review and meta-analysis. Lancet. 2019;394(10197):497-509.
322. Lei J, Ploner A, Elfstrom KM, Wang J, Roth A, Fang F, Sundstrom K, Dillner J, Sparen P. HPV vaccination and the risk of invasive cervical cancer. N Engl J Med. 2020;383(14):1340-8.
323. Brisson M, Kim JJ, Canfell K, Drolet M, Gingras G, Burger EA, Martin D, Simms KT, Benard E, Boily MC, et al. Impact of HPV vaccination and cervical screening on cervical cancer elimination: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet. 2020;395(10224):575-90.
324. Komdeur FL, Singh A, van de Wall S, Meulenberg JJM, Boerma A, Hoogeboom BN, Paijens ST, Oyarce C, de Bruyn M, Schuuring E, et al. First-in-human phase I clinical trial of an SFV-based RNA replicon cancer vaccine against HPV-induced cancers. Mol Ther. 2021;29(2):611-25.
325. Porras C, Tsang SH, Herrero R, Guillen D, Darragh TM, Stoler MH, Hildesheim A, Wagner S, Boland J, Lowy DR, et al. Efficacy of the bivalent HPV vaccine against HPV 16/18-associated precancer: longterm follow-up results from the Costa Rica Vaccine Trial. Lancet Oncol. 2020;21(12):1643-52.
326. Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, Feng L, Lee JJ, Tran H, Kim YU, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5(1):67-73.
327. Bernhard H, Jager-Arand E, Bernhard G, Heike M, Klein O, Riemann JF, Meyer zum Buschenfelde KH, Dippold W, Knuth A. Treatment of advanced pancreatic cancer with 5-fluorouracil, folinic acid and interferon alpha-2A: results of a phase II trial. Br J Cancer. 1995;71(1):102-5.
328. David AK, Vaughn DJ, Holroyde CP, Armstead B, Haller DG. A phase II trial of 5-fluorouracil, leucovorin, and interferon alpha 2A (IFN-alpha 2a) in metastatic pancreatic carcinoma: a Penn Cancer Clinical Trials Group (PCCTG) trial. Am J Clin Oncol. 2000;23(1):37-9.
329. Ohman KA, Liu J, Linehan DC, Tan MC, Tan BR, Fields RC, Strasberg SM, Hawkins WG. Interferon-based chemoradiation followed by gemcitabine for resected pancreatic adenocarcinoma: long-term follow-up. HPB (Oxford). 2017;19(5):449-57.
330. Rocha FG, Hashimoto Y, Traverso LW, Dorer R, Kozarek R, Helton WS, Picozzi VJ. Interferon-based adjuvant chemoradiation for resected pancreatic head cancer: long-term follow-up of the Virginia mason protocol. Ann Surg. 2016;263(2):376-84.
331. Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14(1):7-17.
332. Eigentler TK, Gutzmer R, Hauschild A, Heinzerling L, Schadendorf D, Nashan D, Holzle E, Kiecker F, Becker J, Sunderkotter C, et al. Adjuvant treatment with pegylated interferon alpha-2a versus low-dose interferon alpha-2a in patients with high-risk melanoma: a randomized phase III DeCOG trial. Ann Oncol. 2016;27(8):1625-32.
333. Ives NJ, Suciu S, Eggermont AMM, Kirkwood J, Lorigan P, Markovic SN, Garbe C, Wheatley K. International Melanoma Meta-Analysis Collaborative G: Adjuvant interferon-alpha for the treatment of high-risk melanoma: An individual patient data meta-analysis. Eur J Cancer. 2017;82:171-83.
334. Yamazaki N, Uhara H, Wada H, Matsuda K, Yamamoto K, Shimamoto T, Kiyohara Y. Phase I study of pegylated interferon-alpha-2b as an adjuvant therapy in Japanese patients with malignant melanoma. J Dermatol. 2016;43(10):1146-53.
335. Dummer R, Mangana J. Long-term pegylated interferon-alpha and its potential in the treatment of melanoma. Biologics. 2009;3:169-82.
336. Najjar YG, Puligandla M, Lee SJ, Kirkwood JM. An updated analysis of 4 randomized ECOG trials of high-dose interferon in the adjuvant treatment of melanoma. Cancer. 2019;125(17):3013-24.
337. Eggermont AM, Suciu S, Testori A, Santinami M, Kruit WH, Marsden J, Punt CJ, Sales F, Dummer R, Robert C, et al. Long-term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma. J Clin Oncol. 2012;30(31):3810-8.
338. Bottomley A, Coens C, Suciu S, Santinami M, Kruit W, Testori A, Marsden J, Punt C, Sales F, Gore M, et al. Adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma: a phase III randomized controlled trial of health-related quality of life and symptoms by the European Organisation for Research and Treatment of Cancer Melanoma Group. J Clin Oncol. 2009;27(18):2916-23.
339. Flaherty LE, Othus M, Atkins MB, Tuthill RJ, Thompson JA, Vetto JT, Haluska FG, Pappo AS, Sosman JA, Redman BG, et al. Southwest Oncology Group S0008: a phase III trial of high-dose interferon Alfa-2b versus cisplatin, vinblastine, and dacarbazine, plus interleukin-2 and interferon in patients with high-risk melanoma-an intergroup study of cancer and leukemia Group B, Children’s Oncology Group, Eastern Cooperative Oncology Group, and Southwest Oncology Group. J Clin Oncol. 2014;32(33):3771-8.
340. Simeone E, Scognamiglio G, Capone M, Giannarelli D, Grimaldi AM, Mallardo D, Madonna G, Curvietto M, Esposito A, Sandomenico F, et al. A monocentric phase I study of vemurafenib plus cobimetinib plus PEGinterferon (VEMUPLINT) in advanced melanoma patients harboring the V600BRAF mutation. J Transl Med. 2021;19(1):17.
341. Magenau JM, Peltier D, Riwes M, Pawarode A, Parkin B, Braun T, Anand S, Ghosh M, Maciejewski J, Yanik G, et al. Type 1 interferon to prevent leukemia relapse after allogeneic transplantation. Blood Adv. 2021;5(23):5047-56.
342. Mo XD, Zhang XH, Xu LP, Wang Y, Yan CH, Chen H, Chen YH, Han W, Wang FR, Wang JZ, et al. IFN-alpha is effective for treatment of minimal residual disease in patients with acute leukemia after allogeneic hematopoietic stem cell transplantation: results of a registry study. Biol Blood Marrow Transplant. 2017;23(8):1303-10.
343. Kankuri-Tammilehto M, Perasto L, Pyrhonen S, Salminen E. Longterm outcome with prolonged use of interferon-alpha administered intermittently for metastatic renal cell carcinoma: a phase II study. Anticancer Res. 2023;43(6):2645-57.
344. Eto M, Kawano Y, Hirao Y, Mita K, Arai Y, Tsukamoto T, Hashine K, Matsubara A, Fujioka T, Kimura G, et al. Phase II clinical trial of sorafenib plus interferon-alpha treatment for patients with metastatic renal cell carcinoma in Japan. BMC Cancer. 2015;15:667.
345. Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Ou SS, Archer L, Atkins JN, Picus J, Czaykowski P, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol. 2008;26(33):5422-8.
346. Rafique I, Kirkwood JM, Tarhini AA. Immune checkpoint blockade and interferon-alpha in melanoma. Semin Oncol. 2015;42(3):436-47.
347. Wang H, Xia L, Yao CC, Dong H, Yang Y, Li C, Ji WX, Sun RM, Duan HQ, Mengzhou W, et al. NLRP4 negatively regulates type I interferon response and influences the outcome in anti-programmed cell death protein (PD)-1/PD-ligand 1 therapy. Cancer Sci. 2022;113(3):838-51.
348. Brohl AS, Khushalani NI, Eroglu Z, Markowitz J, Thapa R, Chen YA, Kudchadkar R, Weber JS. A phase IB study of ipilimumab with peginterferon alfa-2b in patients with unresectable melanoma. J Immunother Cancer. 2016;4:85.
349. Tarhini A, Lin Y, Lin H, Rahman Z, Vallabhaneni P, Mendiratta P, Pingpank JF, Holtzman MP, Yusko EC, Rytlewski JA, et al. Neoadjuvant ipilimumab (
350. Trinh KR, Vasuthasawat A, Steward KK, Yamada RE, Timmerman JM, Morrison SL. Anti-CD20-interferon-beta fusion protein therapy of murine B-cell lymphomas. J Immunother. 2013;36(5):305-18.
351. Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood. 2010;115(14):2864-71.
352. Li Z, Zhu Y, Li C, Trinh R, Ren X, Sun F, Wang Y, Shang P, Wang T, Wang M, et al. Anti-VEGFR2-interferon-alpha2 regulates the tumor microenvironment and exhibits potent antitumor efficacy against colorectal cancer. Oncoimmunology. 2017;6(3): e1290038.
353. Green DS, Husain SR, Johnson CL, Sato Y, Han J, Joshi B, Hewitt SM, Puri RK, Zoon KC. Combination immunotherapy with IL-4 Pseudomonas exotoxin and IFN-alpha and IFN-gamma mediate antitumor effects in vitro and in a mouse model of human ovarian cancer. Immunotherapy. 2019;11(6):483-96.
354. Duggan MC, Jochems C, Donahue RN, Richards J, Karpa V, Foust E, Paul B, Brooks T, Tridandapani S, Olencki T, et al. A phase I study of recombinant (
355. Sheng
356. Gigante M, Mandic M, Wesa AK, Cavalcanti E, Dambrosio M, Mancini V, Battaglia M, Gesualdo L, Storkus WJ, Ranieri E. Interferon-alpha (IFN-alpha)-conditioned DC preferentially stimulate type-1 and limit Treg-type in vitro T-cell responses from RCC patients. J Immunother. 2008;31(3):254-62.
357. Rozera C, Cappellini GA, D’Agostino G, Santodonato L, Castiello L, Urbani F, Macchia I, Arico E, Casorelli I, Sestili P, et al. Intratumoral injection of IFN-alpha dendritic cells after dacarbazine activates anti-tumor immunity: results from a phase I trial in advanced melanoma. J Transl Med. 2015;13:139.
358. Cox MC, Lapenta C, Santini SM. Advances and perspectives of dendritic cell-based active immunotherapies in follicular lymphoma. Cancer Immunol Immunother. 2020;69(6):913-25.
359. Derynck R, Turley SJ, Akhurst RJ. TGFbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18(1):9-34.
360. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, Hsu FJ, Berzofsky JA, Lawrence DP. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE. 2014;9(3): e90353.
361. Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, Ratikan JA, Felix C, Hwang L, Faull KF, et al. Focal irradiation and systemic TGFbeta blockade in metastatic breast cancer. Clin Cancer Res. 2018;24(11):2493-504
362. Faivre S, Santoro A, Kelley RK, Gane E, Costentin CE, Gueorguieva I, Smith C, Cleverly A, Lahn MM, Raymond E, et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019;39(8):1468-77.
363. Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, Hourmand IO, Cleverly A, Zhao Y, Gueorguieva I, et al. A phase 2 study of galunisertib (TGF-beta1 receptor type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin Transl Gastroenterol. 2019;10(7): e00056.
364. Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, Trojan J, Oettle H, Kozloff M, Cleverly A, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 2018;119(10):1208-14.
365. Melisi D, Oh DY, Hollebecque A, Calvo E, Varghese A, Borazanci E, Macarulla T, Merz V, Zecchetto C, Zhao Y et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer. 2021;9(3).
366. Nadal E, Saleh M, Aix SP, Ochoa-de-Olza M, Patel SP, Antonia S, Zhao Y, Gueorguieva I, Man M, Estrem ST, et al. A phase Ib/II study of galunisertib in combination with nivolumab in solid tumors and non-small cell lung cancer. BMC Cancer. 2023;23(1):708.
367. Yamazaki T, Gunderson AJ, Gilchrist M, Whiteford M, Kiely MX, Hayman A, O’Brien D, Ahmad R, Manchio JV, Fox N, et al. Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: a single-arm, phase 2 trial. Lancet Oncol. 2022;23(9):1189-200.
368. Necchi A, Giannatempo P, Mariani L, Fare E, Raggi D, Pennati M, Zaffaroni N, Crippa F, Marchiano A, Nicolai N, et al. PF-03446962, a fully-human monoclonal antibody against transforming growth-factor beta (TGFbeta) receptor ALK1, in pre-treated patients with urothelial cancer: an open label, single-group, phase 2 trial. Invest New Drugs. 2014;32(3):555-60.
369. Simonelli M, Zucali P, Santoro A, Thomas MB, de Braud FG, Borghaei H, Berlin J, Denlinger CS, Noberasco C, Rimassa L, et al. Phase I study
of PF-03446962, a fully human monoclonal antibody against activin receptor-like kinase-1, in patients with hepatocellular carcinoma. Ann Oncol. 2016;27(9):1782-7.
370. Wheatley-Price P, Chu Q, Bonomi M, Seely J, Gupta A, Goss G, Hilton J, Feld R, Lee CW, Goffin JR, et al. A phase II study of PF-03446962 in patients with advanced malignant pleural mesothelioma. CCTG trial IND207. J Thorac Oncol. 2016;11(11):218-21.
371. Goff LW, Cohen RB, Berlin JD, de Braud FG, Lyshchik A, Noberasco C, Bertolini F, Carpentieri M, Stampino CG, Abbattista A, et al. A phase I study of the anti-activin receptor-like kinase 1 (ALK-1) monoclonal antibody PF-03446962 in patients with advanced solid tumors. Clin Cancer Res. 2016;22(9):2146-54.
372. Clarke JM, Blobe GC, Strickler JH, Uronis HE, Zafar SY, Morse M, Dropkin E, Howard L, O’Neill M, Rushing CN, et al. A phase Ib study of the combination regorafenib with PF-03446962 in patients with refractory metastatic colorectal cancer (REGAL-1 trial). Cancer Chemother Pharmacol. 2019;84(4):909-17.
373. Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, Lan Y, Dussault I, Moustakas A. Dual inhibition of TGF-beta and PD-L1: a novel approach to cancer treatment. Mol Oncol. 2022;16(11):2117-34.
374. Paz-Ares L, Kim TM, Vicente D, Felip E, Lee DH, Lee KH, Lin CC, Flor MJ, Di Nicola M, Alvarez RM, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in second-line treatment of patients with NSCLC: results from an expansion cohort of a phase 1 trial. J Thorac Oncol. 2020;15(7):1210-22.
375. Lin CC, Doi T, Muro K, Hou MM, Esaki T, Hara H, Chung HC, Helwig C, Dussault I, Osada M, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGFbeta and PD-L1, in patients with esophageal squamous cell carcinoma: results from a phase 1 cohort in Asia. Target Oncol. 2021;16(4):447-59.
376. Tan B, Khattak A, Felip E, Kelly K, Rich P, Wang D, Helwig C, Dussault I, Ojalvo LS, Isambert N. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with esophageal adenocarcinoma: results from a phase 1 cohort. Target Oncol. 2021;16(4):435-46.
377. Cho BC, Daste A, Ravaud A, Salas S, Isambert N, McClay E, Awada A, Borel C, Ojalvo LS, Helwig C et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in advanced squamous cell carcinoma of the head and neck: results from a phase I cohort. J Immunother Cancer. 2020;8(2).
378. Strauss J, Gatti-Mays ME, Cho BC, Hill A, Salas S, McClay E, Redman JM, Sater HA, Donahue RN, Jochems C et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with human papillomavirus-associated malignancies. J Immunother Cancer. 2020;8(2).
379. Redman JM, Friedman J, Robbins Y, Sievers C, Yang X, Lassoued W, Sinkoe A, Papanicolau-Sengos A, Lee CC, Marte JL et al. Enhanced neoepitope-specific immunity following neoadjuvant PD-L1 and TGFbeta blockade in HPV-unrelated head and neck cancer. J Clin Invest. 2022;132(18).
380. Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, Lazorchak A, Yeung TL, Hasheminasab SM, Jenkins MH, Meister S, et al. Simultaneous targeting of TGF-beta/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell. 2021;39(10):1388-403.
381. Singh S, Xiao Z, Bavisi K, Roszik J, Melendez BD, Wang Z, Cantwell MJ, Davis RE, Lizee G, Hwu P, et al. IL-1alpha mediates innate and acquired resistance to immunotherapy in melanoma. J Immunol. 2021;206(8):1966-75.
382. Aggen DH, Ager CR, Obradovic AZ, Chowdhury N, Ghasemzadeh A, Mao W, Chaimowitz MG, Lopez-Bujanda ZA, Spina CS, Hawley JE, et al. Blocking IL1 beta promotes tumor regression and remodeling of the myeloid compartment in a renal cell carcinoma model: multidimensional analyses. Clin Cancer Res. 2021;27(2):608-21.
383. Wong CC, Baum J, Silvestro A, Beste MT, Bharani-Dharan B, Xu S, Wang YA, Wang X, Prescott MF, Krajkovich L, et al. Inhibition of IL1 beta by canakinumab may be effective against diverse molecular subtypes of lung cancer: an exploratory analysis of the CANTOS trial. Cancer Res. 2020;80(24):5597-605.
384. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, Group CT. Effect of interleukin-1 beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results
from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833-42.
385. Garrido P, Pujol JL, Kim ES, Lee JM, Tsuboi M, Gomez-Rueda A, Benito A, Moreno N, Gorospe L, Dong T, et al. Canakinumab with and without pembrolizumab in patients with resectable non-small-cell lung cancer: CANOPY-N study design. Future Oncol. 2021;17(12):1459-72.
386. Yuan B, Clowers MJ, Velasco WV, Peng S, Peng Q, Shi Y, Ramos-Castaneda M, Zarghooni M, Yang S, Babcock RL et al. Targeting IL-1 beta as an immunopreventive and therapeutic modality for K-ras-mutant lung cancer. JCl Insight. 2022;7(11).
387. Wu TC, Xu K, Martinek J, Young RR, Banchereau R, George J, Turner J, Kim KI, Zurawski S, Wang X, et al. IL1 Receptor antagonist controls transcriptional signature of inflammation in patients with metastatic breast cancer. Cancer Res. 2018;78(18):5243-58.
388. Lust JA, Lacy MQ, Zeldenrust SR, Witzig TE, Moon-Tasson LL, Dinarello CA, Donovan KA. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am J Hematol. 2016;91(6):571-4.
389. Becerra C, Paulson AS, Cavaness KM, Celinski SA. Gemcitabine, nabpaclitaxel, cisplatin, and anakinra (AGAP) treatment in patients with localized pancreatic ductal adenocarcinoma (PDAC). J Clin Oncol. 2018;36(4_suppl):449-449.
390. Voigt C, May P, Gottschlich A, Markota A, Wenk D, Gerlach I, Voigt S, Stathopoulos GT, Arendt KAM, Heise C, et al. Cancer cells induce inter-leukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc Natl Acad Sci USA. 2017;114(49):12994-9.
391. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, et al. Chemotherapy-triggered cathepsin
392. Isambert N, Hervieu A, Rebe C, Hennequin A, Borg C, Zanetta S, Chevriaux A, Richard C, Derangere V, Limagne E, et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): a single-arm phase 2 study. Oncoimmunology. 2018;7(9): e1474319.
393. Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, Kumar S, Hayman SR, Russell SJ, Buadi FK, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1beta-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009;84(2):114-22.
394. Briukhovetska D, Dorr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in cancer: from biology to therapy. Nat Rev Cancer. 2021;21(8):481-99.
395. Waldmann TA. Cytokines in cancer immunotherapy. Cold Spring Harb Perspect Biol. 2018;10(12).
396. Yang Y, Lundqvist A. Immunomodulatory Effects of IL-2 and IL-15; Implications for Cancer Immunotherapy. Cancers (Basel). 2020;12(12)
397. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451-8.
398. Payne R, Glenn L, Hoen H, Richards B, Smith JW 2nd, Lufkin R, Crocenzi TS, Urba WJ, Curti BD. Durable responses and reversible toxicity of highdose interleukin-2 treatment of melanoma and renal cancer in a Community Hospital Biotherapy Program. J Immunother Cancer. 2014;2:13.
399. Baik AH, Oluwole OO, Johnson DB, Shah N, Salem JE, Tsai KK, Moslehi JJ. Mechanisms of cardiovascular toxicities associated with immunotherapies. Circ Res. 2021;128(11):1780-801.
400. Damoiseaux J. The IL-2—IL-2 receptor pathway in health and disease: the role of the soluble IL-2 receptor. Clin Immunol. 2020;218: 108515.
401. Doberstein SK. Bempegaldesleukin (NKTR-214): a CD-122-biased IL-2 receptor agonist for cancer immunotherapy. Expert Opin Biol Ther. 2019;19(12):1223-8.
402. Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, Tetzlaff MT, Tagliaferri MA, Zalevsky J, Hoch U, et al. A first-inhuman study and biomarker analysis of NKTR-214, a novel IL2Rbet-agamma-biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov. 2019;9(6):711-21.
403. Diab A, Tannir NM, Bentebibel SE, Hwu P, Papadimitrakopoulou V, Haymaker C, Kluger HM, Gettinger SN, Sznol M, Tykodi SS, et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced
solid tumors: phase i dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discov. 2020;10(8):1158-73.
404. Khushalani NI, Diab A, Ascierto PA, Larkin J, Sandhu S, Sznol M, Koon HB, Jarkowski A, Zhou M, Statkevich P, et al. Bempegaldesleukin plus nivolumab in untreated, unresectable or metastatic melanoma: phase III PIVOT IO 001 study design. Future Oncol. 2020;16(28):2165-75.
405. Siefker-Radtke AO, Cho DC, Diab A, Sznol M, Bilen MA, Balar AV, Grignani G, Puente E, Tang L, Chien D, et al. Bempegaldesleukin plus nivolumab in first-line metastatic urothelial carcinoma: results from PIVOT-02. Eur Urol. 2022;82(4):365-73.
406. Diab A, Tykodi SS, Daniels GA, Maio M, Curti BD, Lewis KD, Jang S, Kalinka E, Puzanov I, Spira AI, et al. Bempegaldesleukin plus nivolumab in first-line metastatic melanoma. J Clin Oncol. 2021;39(26):2914-25.
407. Pan Y, Hao Y, Han H, Chen T, Ding H, Labbe KE, Shum E, Guidry K, Hu H , Sherman F et al. Nemvaleukin alfa, a novel engineered IL-2 fusion protein, drives antitumor immunity and inhibits tumor growth in small cell lung cancer. J Immunother Cancer. 2022; 10(9).
408. Boni V, Winer IS, Gilbert L, Vaishampayan UN, Rosen SD, Muzaffar J, Spreafico A, McDermott DF, Chu QS, Dumas O, et al. ARTISTRY-1: Nemvaleukin alfa monotherapy and in combination with pembrolizumab in patients (pts) with advanced solid tumors. J Clin Oncol. 2021;39(15_suppl):2513-2513.
409. Hamid O, Liu SV, Boccia RV, Call JA, Wise-Draper TM, Alistar AT, Powderly JD, Carthon BC, Vaishampayan UN, Olszanski AJ, et al. Selection of the recommended phase 2 dose (RP2D) for subcutaneous nemvaleukin alfa: ARTISTRY-2. J Clin Oncol. 2021;39(15_suppl):2552-2552.
410. Vaishampayan UN, Tomczak P, Muzaffar J, Winer IS, Rosen SD, Hoimes CJ, Chauhan A, Spreafico A, Lewis KD, Bruno DS, et al. Nemvaleukin alfa monotherapy and in combination with pembrolizumab in patients (pts) with advanced solid tumors: ARTISTRY-1. J Clin Oncol. 2022;40(16_suppl):2500-2500.
411. Rech AJ, Vonderheide RH. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann N Y Acad Sci. 2009;1174:99-106.
412. Sampson JH, Schmittling RJ, Archer GE, Congdon KL, Nair SK, Reap EA, Desjardins A, Friedman AH, Friedman HS, Herndon JE 2nd, et al. A pilot study of IL-2Ralpha blockade during lymphopenia depletes regulatory T-cells and correlates with enhanced immunity in patients with glioblastoma. PLoS ONE. 2012;7(2): e31046.
413. Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, Klasen IS, Hilbrands LB, Figdor CG, de Vries IJ, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res. 2010;16(20):5067-78.
414. Solomon I, Amann M, Goubier A, Arce Vargas F, Zervas D, Qing C, Henry JY, Ghorani E, Akarca AU, Marafioti T, et al. CD25-T(reg)-depleting antibodies preserving IL-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat Cancer. 2020;1(12):1153-66.
415. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda Rota E, Dahan R, Georgiou A, Sledzinska A, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory
416. Ji D, Song C, Li Y, Xia J, Wu Y, Jia J, Cui X, Yu S, Gu J. Combination of radiotherapy and suppression of Tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J Immunother Cancer. 2020;8(2).
417. Anderson KC , Jones RM, Morimoto C , Leavitt P , Barut BA. Response patterns of purified myeloma cells to hematopoietic growth factors. Blood. 1989;73(7):1915-24.
418. Klein B, Wijdenes J, Zhang XG, Jourdan M, Boiron JM, Brochier J, Liautard J, Merlin M, Clement C, Morel-Fournier B, et al. Murine anti-interleukin-6 monoclonal antibody therapy for a patient with plasma cell leukemia. Blood. 1991;78(5):1198-204.
419. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448-57.
420. Voorhees PM, Manges RF, Sonneveld P, Jagannath S, Somlo G, Krishnan A, Lentzsch S, Frank RC, Zweegman S, Wijermans PW, et al. A phase 2 multicentre study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2013;161(3):357-66.
421. San-Miguel J, Blade J, Shpilberg O, Grosicki S, Maloisel F, Min CK, Polo Zarzuela M, Robak T, Prasad SV, Tee Goh Y, et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood. 2014;123(26):4136-42.
422. Rossi JF, Negrier S, James ND, Kocak I, Hawkins R, Davis H, Prabhakar U, Qin X, Mulders P, Berns B. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br J Cancer. 2010;103(8):1154-62.
423. Dorff TB, Goldman B, Pinski JK, Mack PC, Lara PN Jr, Van Veldhuizen PJ Jr, Quinn DI, Vogelzang NJ, Thompson IM Jr, Hussain MH. Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin Cancer Res. 2010;16(11):3028-34.
424. Fizazi K, De Bono JS, Flechon A, Heidenreich A, Voog E, Davis NB, Qi M, Bandekar R, Vermeulen JT, Cornfeld M, et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur J Cancer. 2012;48(1):85-93.
425. Lu ZY, Brochier J, Wijdenes J, Brailly H, Bataille R, Klein B. High amounts of circulating interleukin (IL)-6 in the form of monomeric immune complexes during anti-IL-6 therapy. Towards a new methodology for measuring overall cytokine production in human in vivo. Eur J Immunol. 1992;22(11):2819-24.
426. Dijkgraaf EM, Santegoets SJ, Reyners AK, Goedemans R, Wouters MC, Kenter GG, van Erkel AR, van Poelgeest MI, Nijman HW, van der Hoeven JJ, et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferonalpha2b in patients with recurrent epithelial ovarian cancer. Ann Oncol. 2015;26(10):2141-9.
427. Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med. 2020;217(1).
428. Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, Gorman DM, Oft M. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012;72(14):3570-81.
429. Oft M. Immune regulation and cytotoxic T cell activation of IL-10 agonists—preclinical and clinical experience. Semin Immunol. 2019;44: 101325.
430. Autio K, Oft M. Pegylated interleukin-10: clinical development of an immunoregulatory cytokine for use in cancer therapeutics. Curr Oncol Rep. 2019;21(2):19.
431. Naing A, Papadopoulos KP, Autio KA, Ott PA, Patel MR, Wong DJ, Falchook GS, Pant S, Whiteside M, Rasco DR, et al. Safety, antitumor activity, and immune activation of pegylated recombinant human interleu-kin-10 (AM0010) in patients with advanced solid tumors. J Clin Oncol. 2016;34(29):3562-9.
432. Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthelemy P, Porta C, George S, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277-90.
433. Tilg H, Ulmer H, Kaser A, Weiss G. Role of IL-10 for induction of anemia during inflammation. J Immunol. 2002;169(4):2204-9.
434. Naing A, Infante JR, Papadopoulos KP, Chan IH, Shen C, Ratti NP, Rojo B, Autio KA, Wong DJ, Patel MR, et al. PEGylated IL-10 (pegilodecakin) induces systemic immune activation, CD8(+) T cell invigoration and polyclonal T cell expansion in cancer patients. Cancer Cell. 2018;34(5):775-91.
435. Naing A, Wong DJ, Infante JR, Korn WM, Aljumaily R, Papadopoulos KP, Autio KA, Pant S, Bauer TM, Drakaki A, et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): a multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019;20(11):1544-55.
436. Pal S, Hu-Lieskovan S, Agarwal N. Can pegylated IL-10 add to a backbone of PD-1 inhibition for solid tumours? Lancet Oncol. 2019;20(11):1473-4.
437. Spigel D, Jotte R, Nemunaitis J, Shum M, Schneider J, Goldschmidt J, Eisenstein J, Berz D, Seneviratne L, Socoteanu M, et al. Randomized phase 2 studies of checkpoint inhibitors alone or in combination with
pegilodecakin in patients with metastatic NSCLC (CYPRESS 1 and CYPRESS 2). J Thorac Oncol. 2021;16(2):327-33.
438. Tannir NM, Papadopoulos KP, Wong DJ, Aljumaily R, Hung A, Afable M, Kim JS, Ferry D, Drakaki A, Bendell J, et al. Pegilodecakin as monotherapy or in combination with anti-PD-1 or tyrosine kinase inhibitor in heavily pretreated patients with advanced renal cell carcinoma: Final results of cohorts A, G, H and I of IVY Phase I study. Int J Cancer. 2021;149(2):403-8.
439. Hecht JR, Papadopoulos KP, Falchook GS, Patel MR, Infante JR, Aljumaily R, Wong DJ, Autio KA, Wainberg ZA, Bauer TM, et al. Immunologic and tumor responses of pegilodecakin with 5-FU/LV and oxaliplatin (FOLFOX) in pancreatic ductal adenocarcinoma (PDAC). Invest New Drugs. 2021;39(1):182-92.
440. Hecht JR, Lonardi S, Bendell J, Sim HW, Macarulla T, Lopez CD, Van Cutsem E, Munoz Martin AJ, Park JO, Greil R, et al. Randomized phase III study of FOLFOX alone or with pegilodecakin as second-line therapy in patients with metastatic pancreatic cancer that progressed after gemcitabine (SEQUOIA). J Clin Oncol. 2021;39(10):1108-18.
441. Grossman JG, Nywening TM, Belt BA, Panni RZ, Krasnick BA, DeNardo DG, Hawkins WG, Goedegebuure SP, Linehan DC, Fields RC. Recruitment of CCR2(+) tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology. 2018;7(9): e1470729.
442. Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, Wang G, Tromp BJ, Puchalski TA, Balkwill F, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71(4):1041-50.
443. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31(3):760-8.
444. Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, Zhong B, de Boer CJ, Tabernero J, Calvo E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an openlabel, multicenter phase 1b study. Target Oncol. 2015;10(1):111-23.
445. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651-62.
446. Noel M, O’Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, Linehan DC, Belt BA, Gamelin EC, Ganguly B, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2020;38(3):800-11.
447. Wu X, Singh R, Hsu DK, Zhou Y, Yu S, Han D, Shi Z, Huynh M, Campbell JJ, Hwang ST. A small molecule CCR2 antagonist depletes tumor macrophages and synergizes with anti-PD-1 in a murine model of cutaneous T-cell lymphoma (CTCL). J Invest Dermatol. 2020;140(7):1390-400.
448. Wang J, Saung MT, Li K, Fu J, Fujiwara K, Niu N, Muth S, Wang J, Xu Y, Rozich N et al. CCR2/CCR5 inhibitor permits the radiation-induced effector T cell infiltration in pancreatic adenocarcinoma. J Exp Med. 2022;219(5).
449. Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE, Chauca-Diaz A, Costello JC, Dancik GM, Tamburini BAJ, et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun Biol. 2020;3(1):720.
450. Bartkowiak T, Jaiswal AR, Ager CR, Chin R, Chen CH, Budhani P, Ai M, Reilley MJ, Sebastian MM, Hong DS, et al. Activation of
451. Abd Wahab NA, Lajis NH, Abas F, Othman I, Naidu R. Mechanism of anticancer activity of curcumin on androgen-dependent and androgenindependent prostate cancer. Nutrients. 2020;12(3).
452. Ferguson JJA, Abbott KA, Garg ML. Anti-inflammatory effects of oral supplementation with curcumin: a systematic review and meta-analysis of randomized controlled trials. Nutr Rev. 2021;79(9):1043-66.
453. Gupta SC, Patchva S, Aggarwal BB. Therapeutic roles of curcumin: lessons learned from clinical trials. AAPS J. 2013;15(1):195-218.
454. Peng Y, Ao M, Dong B, Jiang Y, Yu L, Chen Z, Hu C, Xu R. Anti-inflammatory effects of curcumin in the inflammatory diseases: status, limitations and countermeasures. Drug Des Devel Ther. 2021;15:4503-25.
455. Sharma RA, Euden SA, Platton SL, Cooke DN, Shafayat A, Hewitt HR, Marczylo TH, Morgan B, Hemingway D, Plummer SM, et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004;10(20):6847-54.
456. Howells LM, Iwuji COO, Irving GRB, Barber S, Walter H, Sidat Z, GriffinTeall N, Singh R, Foreman N, Patel SR, et al. Curcumin combined with FOLFOX chemotherapy is safe and tolerable in patients with metastatic colorectal cancer in a randomized phase lla trial. J Nutr. 2019;149(7):1133-9.
457. Zhan Y, Chen Y, Liu R, Zhang H, Zhang Y. Potentiation of paclitaxel activity by curcumin in human breast cancer cell by modulating apoptosis and inhibiting EGFR signaling. Arch Pharm Res. 2014;37(8):1086-95.
458. Calaf GM, Ponce-Cusi R, Carrion F. Curcumin and paclitaxel induce cell death in breast cancer cell lines. Oncol Rep. 2018;40(4):2381-8.
459. Malaguarnera L. Influence of resveratrol on the immune response. Nutrients. 2019;11(5).
460. Delmas D, Limagne E, Ghiringhelli F, Aires V. Immune Th17 lymphocytes play a critical role in the multiple beneficial properties of resveratrol. Food Chem Toxicol. 2020;137: 111091.
461. Hou J, Karin M, Sun B. Targeting cancer-promoting inflammationhave anti-inflammatory therapies come of age? Nat Rev Clin Oncol. 2021;18(5):261-79.
462. Barnabei A, Carpano S, Chiefari A, Bianchini M, Lauretta R, Mormando M, Puliani G, Paoletti G, Appetecchia M, Torino F. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10: 582394.
463. Kotwal A, Kottschade L, Ryder M. PD-L1 inhibitor-induced thyroiditis is associated with better overall survival in cancer patients. Thyroid. 2020;30(2):177-84.
Publisher’s Note
- *Correspondence:
Yong Yuan
yongyuan@scu.edu.cn
Xiawei Wei
xiaweiwei@scu.edu.cn
Laboratory of Aging Research and Cancer Drug Target, State Key Laboratory of Biotherapy and Cancer Center, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, No.17, Block3, Southern Renmin Road, Chengdu 610041, Sichuan, People’s Republic of China
Department of Thoracic Surgery, West China Hospital, Sichuan University, Chengdu, People’s Republic of China