الاختزال الكهربائي الفعال للنترات إلى الأمونيا من خلال اقتران ذرات النحاس المفردة مع Co3O4 المجاورة Efficient tandem electroreduction of nitrate into ammonia through coupling Cu single atoms with adjacent Co3O4
النتراتالاختزال الكهربائي إلى الأمونيايمثل نهجًا واعدًا للاستدامةالتركيب. ومع ذلك، فإن تنوع تكوينات الامتزاز يسبب صعوبات كبيرة في التحسين المتزامن لطاقة الربط للوسائط. على الرغم من أن المحفزات الكهربية المعتمدة على النحاس التي تم الإبلاغ عنها بشكل واسع تستفيدالامتصاص، واحدة من القضايا الرئيسية تكمن في تراكم النيتريت () بسبب امتصاصه الضعيف، مما يؤدي إلى التفعيل السريع للعوامل الحفازة وبطء الحركيات في خطوات الهدرجة اللاحقة. هنا نبلغ عن محفز كهربائي متسلسل من خلال دمج محفزات ذرات النحاس الفردية مع أوراق نانوية لتعزيز الاختزال الكهربائي لـإلىيظهر المحفز المتسلسل الناتج معدل إنتاج لـمن، التي تتجاوز القيم السابقة للمحفزات القائمة على النحاس المبلغ عنها. تكشف التحقيقات في الآلية أن الجمع بين تنظم تكوين الامتزاز لـويعزز الربط بـوبذلك تسريع الاختزال الكهربائي لـإلى.
كنوع من الأنواع المحتوية على النيتروجين، النترات (يوجد بشكل واسع في مياه الصرف الصناعي والزراعي بتركيز عالٍ، ويعود ذلك بشكل رئيسي إلى انبعاث النفايات النووية منخفضة المستوى والاستخدام المكثف للأسمدة.مفرطهدد بشكل كبير التوازن البيئي، مما أدى إلى الأمطار الحمضية والدخان الضوئي. بالإضافة إلى ذلك،في جسم الإنسان يتم تحويله بسهولة إلى نيتريت ساممما يؤدي إلى مشاكل صحية خطيرة. من بين الطرق لإزالةتعتبر عملية الاختزال الكهربائي باستخدام الكهرباء المتجددة تقنية جذابة في ظل ظروف معتدلة.. المنتجات القابلة للتحكم بما في ذلك النيتروجين غير السام ( ) والأمونيا القيمة ( يمكن الحصول عليه بعدالاختزال الكهربائي.
منذ هو مركب كيميائي أساسي وناقل واعد للهيدروجين الأخضر، الاختزال الكهربائي إلىبدلاً منأكثر رغبة. معًا، من الضروري للغاية تحقيق اختزال كهربائي فعال لـإلىمن منظور حماية البيئة والتنمية المستدامةتركيب.
نظرًا للعديد من الوسائط المحتوية على النيتروجين (مثل *، * ، و *لا) متورط في الاختزال الكهربائي، يجب أن يلبي المحفز الأمثل الامتزاز المحسن في الوقت نفسه للوسائط. إن طاقة الربط المعتدلة للوسائط تعتبر واحدة من العوامل الرئيسية للكفاءة.الاختزال الكهربائي إلىتقليديًا، نظرًا لتنسيق ذرة النيتروجين فيهو
مشبعة بثلاث ذرات أكسجين،يميل إلى الارتباط بالمواقع النشطة من خلال ذرات الأكسجين. بينما، * يتم امتصاصه بشكل تفضيلي على المواقع النشطة من خلال ذرات النيتروجين والأكسجين. أما بالنسبة لـ *NO، فإن ذرة النيتروجين في *NO تميل إلى الاتصال بالمواقع النشطة. إن تغير تكوينات الامتصاص يسبب صعوبات كبيرة في التحسين المتزامن لطاقة الربط للوسائط. مثال نموذجي هو المحفزات الكهربية المعتمدة على النحاس التي تم الإبلاغ عنها بشكل واسع من أجل الاختزال الكهربائي. على الرغم من أن المحفزات الكهربية القائمة على النحاس تستفيدالامتصاص، واحدة من القضايا الرئيسية تكمن في تراكم“، مما يؤدي إلى تعطيل المحفزات بسرعة وبطء في حركيات خطوات الهدرجة اللاحقة لـإنتاج. ومع ذلك، لا يزال من التحديات الكبرى تصميم محفز فعال لتلبية الامتصاص المحسن في الوقت نفسه للوسائط ذات التكوينات المختلفة.
هنا، نبلغ عن محفز كهربائي مزدوج من خلال دمج ذرات النحاس المفردة المثبتة على الكربون المخدر بالنيتروجين مع الجوارأوراق نانوية (يشار إليها بـلزيادة الاختزال الكهربائي لـإلى. تم الحصول علىيظهر المحفز معدل إنتاج ملحوظ لـمن، والتي تتجاوز القيم السابقة لجميع المحفزات القائمة على النحاس المبلغ عنها. تكشف التحقيقات في الآلية أن الجمع بين تنظم تكوين الامتزاز لـويعزز الربط بـ، مما يسرع من الاختزال الكهربائي لـإلى.
النتائج
تحضير المحفزات وخصائصها
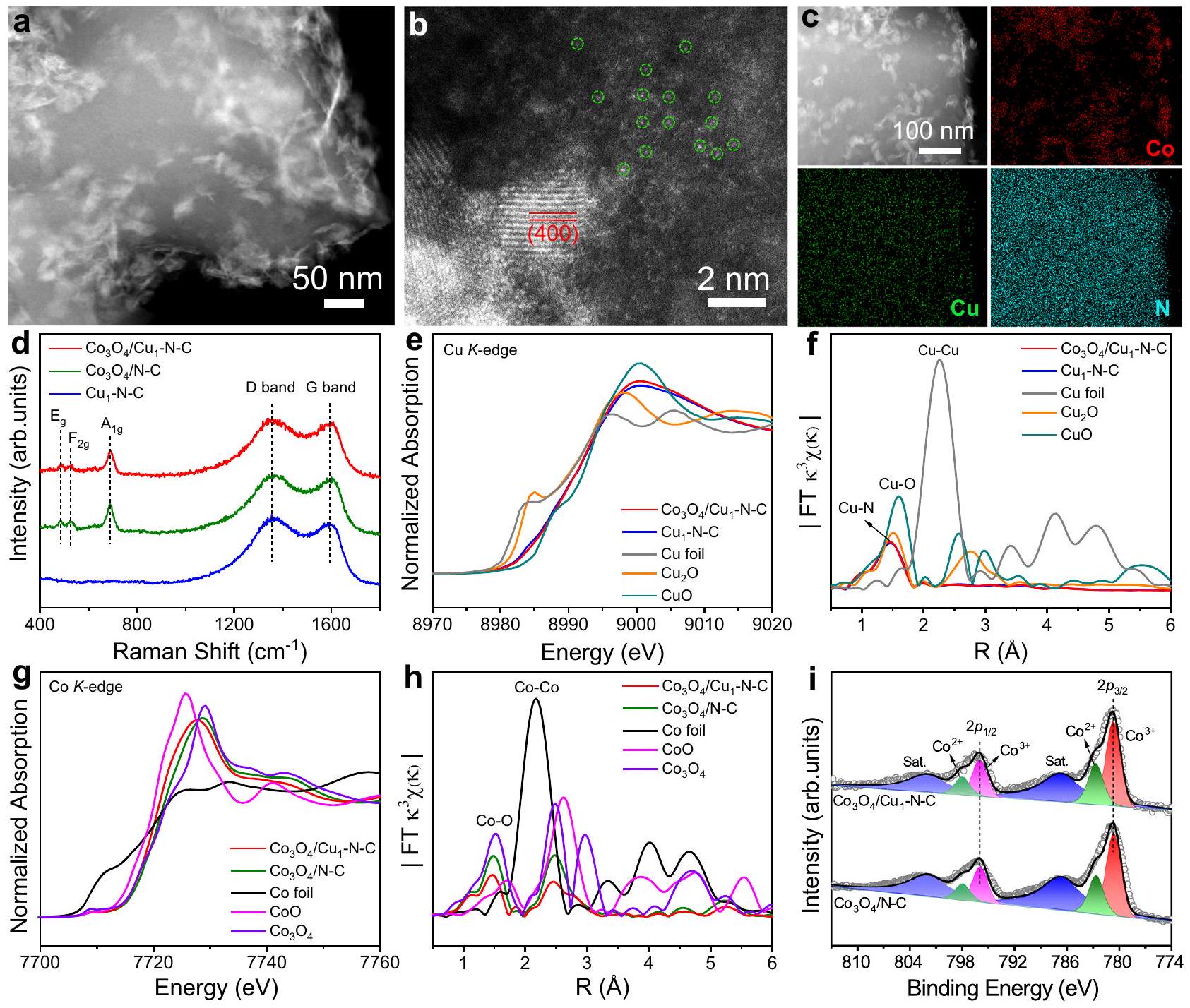
تم تخليق المحفز عن طريق إضافة بوروهيدريد الصوديوم إلى الخليط الذي يحتوي على محفزات ذرات النحاس الفردية ونترات الكوبالت. تم توزيع ذرات النحاس الفردية على الكربون المدعوم بالنيتروجين (المشار إليه بـ –تم تحضيرها عن طريق التحلل الحراري لـ ZIF-8 المدعوم بالنحاس في تحت جو الأرجون (الأشكال التكميلية 1 و 2). الشكل 1أ يظهر صورة المجهر الإلكتروني الناقل بتقنية الحقل الداكن الحلقي بزاوية عالية (HAADF-STEM) لـ-N-C. كما هو موضح في صورة المجهر الإلكتروني عالي الدقة (HRTEM) ونمط حيود الإلكترون في المنطقة المختارة (SAED)،تم ترسيب النانوصفائح بنجاح على سطح (الشكل التكميلي 3). يوضح الشكل 1 ب صورة HAADF-STEM المصححة للانحرافات لـتم نسب الحواف الشبكية بفاصل بين الطبقات قدره 0.201 نانومتر إلى وجه (400) منبالإضافة إلى ذلك، تم ملاحظة وفرة من ذرات النحاس الفردية حولالأغشية النانوية. استنادًا إلى رسم الخرائط العنصرية باستخدام مطياف الأشعة السينية المشتتة للطاقة (EDS)،، و ن
الشكل 1 | التوصيفات الهيكلية. أ صورة HAADF-STEM،صورة HAADF-STEM مصححة للانحراف، وخرائط العناصر باستخدام EDSطيف رامان لـ، و . e -حافة XANES طيفطيف EXAFS لـرقائق، و CuO . ج Co -طيف XANES الحافة وطيف EXAFS لـرقائق، CoO، و. أنا كو طيف XPS لـ و . كانت العناصر موزعة بشكل موحد في جميع أنحاء الهيكل (الشكل 1c). التوزيع الموحد لمواقع النحاس وتشكلت الأنواع من المراكز الحفازة المجاورة. كان محتوى المعادن من النحاس والكوبالت فيتم تحديدها على أنها و على التوالي، بواسطة تحليل طيف الانبعاث الضوئي البلازمي المقترن بالحث (ICP-OES). للمقارنة،أوراق نانوية موزعة على كربون مشوب بالنيتروجين (المشار إليه بـ ) تم تحضيرها بإجراء اصطناعي مشابه لـ -N-C باستثناء إضافة سلفيد النحاس (الشكل التكميلي 4). الشكل 1d يوضح طيف رامان لـ، و . عرضت جميع طيف رامان ذروتين تقعان عند 1356 والمخصصة لشريط D وشريط G من الكربون الجرافيتي، على التوالينسب الشدة المماثلة لفرقة D إلى فرقة Gللعينات الثلاثة أشارت إلى أن الدعم الكربوني يمتلك درجة مشابهة من الاضطراب الهيكلي (الشكل التوضيحي 5). بالمقارنة معثلاثة قمم مميزة تقع عند 482 و 527 وتمت ملاحظتهما لكل من و ، مما يتوافق مع ، و أنماط الاهتزاز البلورات، على التوالي تم تأكيد هيكل دعائم الكربون الجرافيتي بشكل أكبر من خلال أنماط حيود الأشعة السينية (الشكل التوضيحي 6). يوضح الشكل 1eطيف امتصاص الأشعة السينية بالقرب من حافة الهيكل (XANES) لـ و من الواضح أن ملفات حافة امتصاص الطاقة لكل من و كانت تقع بين تلك الخاصة بـ CuO و، موضحًا أن حالة التكافؤ لأنواع النحاس في المحفزين كانت بين +1 إلى +2. كما هو موضح في الشكل 1f، هناك قمة سائدة عندتم ملاحظته في طيف الامتصاص الدقيق للأشعة السينية الممتدة (EXAFS) لـ-حافة لـ و ج ، التي نُسبت إلى رابطة. غيابالرابطة في المحفزين أكدت بشكل أكبر التشتت الذري لأنواع النحاس. بالإضافة إلى ذلك، تشير نتائج تركيب EXAFS إلى أن أعداد التنسيق لـصدفة في كلاهما و كان حوالي 4.0 (الشكل التوضيحي 7 والجدول 1). بعد ترسيبالأغشية النانوية، هيكل التنسيق لذرات النحاس الفرديةفيظل دون تغيير. بالإضافة إلى ذلك، كانت طيفيات EXAFS المحولة بواسطة الموجات (WT-EXAFS) لـ-ن-ك وأيضًا أكدالرابطة في المحفزين (الشكل التوضيحي 8). بالنسبة لـ Co-طيف XANES الحافة، طاقة الحافة لكل من و كانت مشابهة لتلك لـ المرجع (الشكل 1g). الشكل 1h يظهر أن تنسيق Co-O في و كانت تقريبية لذلك في، مصدقًا على هيكل التنسيق المماثل لـ الأنواع في المحفزين (الشكل التكميلي 9 والجدول 2). يوضح الشكل 1i الكوبالت طيف التحليل الطيفي للأشعة السينية (XPS). على وجه التحديد، القمم عند، و 780.8 إلكترون فولت في و توافق مع، و على التواليالتحول غير القابل للاكتشاف لـ Coأظهرت القمم أنحيث لم يؤثر الدعم بشكل كبير على حالة التكافؤ للكوبالت.
الأداء التحفيزي تجاهالاختزال الكهربائي
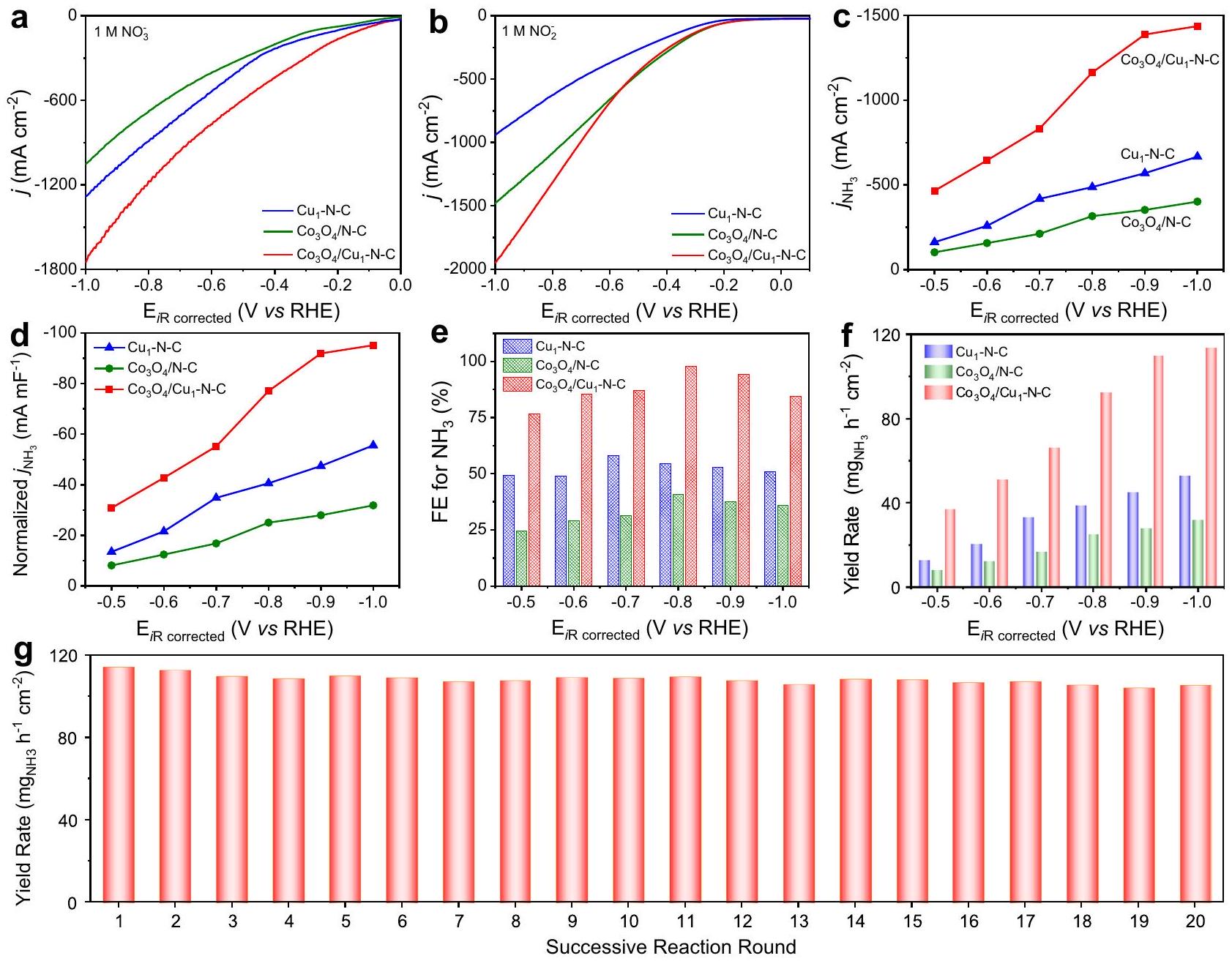
تمت دراسة الأداء التحفيزي للعوامل الحفازة في خلية من نوع H ثلاثية الأقطاب تجاهالاختزال الكهربائي (الشكل التوضيحي 10). تركيزتم قياس المنتج بواسطة طريقة الإندوفينول الأزرق (الشكل التكميلي 11). لاستكشاف عملية التحفيز المتسلسل بشكل أولي، قمنا بإجراء منحنيات الفولتمترية المسحية الخطية (LSV) لـ، و مععلى التوالي. كما هو موضح في الشكل 2أ، كثافة التيار لـفي حضوركان أعلى من، مما يشير إلى أن امتلكت نشاطًا أعلى تجاهالاختزال الكهربائي. بينما،عرضت كثافة تيار أكبر بالنسبة لـفي(الشكل 2ب). النشاط المتفوق لـالأنواع نحوتم إثبات الاختزال الكهربائي بشكل أكبر من خلال كفاءة فاراداي الأعلى (FE) ومعدل العائد لـمنفيالاختزال الكهربائي بالنسبة إلى (الشكل التوضيحي 12). بالنظر إلى أن هو أحد العناصر الحيوية المتوسطات، مزيج من و سيربط الوظائف المنفصلة لمواقع النحاس وأنواع للتقليل المتسلسل منوكما هو متوقع،عرضت أعلى كثافة تيار بين المحفزات الثلاثة في الإلكتروليت لـبالإضافة إلى ذلك، الفجوة الهائلة في منحنيات LSV لـفي 1 م KOH مع/بدونكما أشار إلى النشاط المتفوق لـنحوالاختزال الكهربائي (الشكل التكميلي 13).
الشكل 2ج يوفر كثافة التيار الجزئي لـمن، و عند إمكانيات تطبيقية مختلفة نحو الاختزال الكهربائي. الـمنتجاوزت تلك الخاصة بـ و . خاصة، عند -1.0 فولت مقابل القطب الهيدروجيني القابل للعكس (RHE)، الـمنوصلت، والتي كانت 2.2 مرة و 3.6 مرة أعلى من تلك الخاصة بـ و على التوالي. علاوة على ذلك، فإن المعايير المعياريةاستنادًا إلى سعة الطبقة المزدوجة ) لـ كان الأكبر بين المحفزات الثلاثة، مما يدل على أعلى نشاط داخلي لـنحوالاختزال الكهربائي (الشكل 2d والشكل التكميلي 14). بالإضافة إلى ذلك، فإن الكفاءة من أجلمنكان أعلى بالنسبة إلى النظيرين الآخرين عند جميع الجهود المطبقة (الشكل 2e). خاصةً،حقق الحد الأقصى من FE لـمن عند -0.8 فولت مقابل RHE. علاوة على ذلك، عند -1.0 فولت مقابل RHE، فإن معدل العائد لـ لـوصل إلىالذي تجاوز جميع القيم المبلغ عنها لمحفزات القائمة على النحاس (الشكل 2f والجدول التكميلي 3). معدل العائد من في المحلول الكهربائي بعد عملية الاختزال الكهربائي تم تحديده أيضًا بواسطةتحليل الرنين المغناطيسي النووي (NMR)، الذي تم تقريبه إلى النتائج المكتشفة من خلال طريقة الإندوفينول الأزرق (الشكل التكميلي 15 والجدول 4). منتجات سائلة وغازية أخرى بما في ذلك، و لـتم قياسها أيضًا (الأشكال التكميلية 16-18).كان المنتج الرئيسي الوحيد بعدالاختزال الكهربائي (الجدول التكميلي 5). بالإضافة إلى ذلك، فإن الكفاءة الكاملة لـمنمعتركيزات تتراوح من 10 مللي مول إلى 500 مللي مول جميعها تجاوزت، مما يشير إلى نطاق واسع من التسامح لتركيز (الشكل التوضيحي 19). متانة تم فحصه من خلال 20 جولة من التفاعلات المتعاقبة. أظهر الانخفاض الضئيل في معدل العائد المتانة المرضية لـ (الشكل 2g). قياسات رامان و XAFS لـ بعد أن أظهرت التحليل الكهربائي أنأنواع وتم الحفاظ على الروابط (الأشكال التكميلية 20 و21). استقرار ذرات النحاس الفردية فيتم استكشاف ذلك خلال التحليل الكهربائي من خلال قياسات EXAFS في الموقع، مما يشير إلى أن ذرات النحاس ظلت في حالة متفرقة على المستوى الذري في-N-C خلال الـالاختزال الكهربائي (الأشكال التكميلية 22 و 23).
لتوضيح تأثير التآزر بشكل أكبرحول التحويل من، قمنا بإجراء سلسلة من التجارب الضابطة. أداء التحفيز لمركبات أكسيد المعادن الأخرى (مثل، و موزعة على الكربون المضاف إليه النيتروجين نحوكانت جميع عمليات الاختزال الكهربائي أقل من ذلك على، مما يشير إلى القدرة الضعيفة لهذه الأكاسيد المعدنية على تسهيلالخفض (الأشكال التكميلية 24 و 25). بالإضافة إلى ذلك، كمية التحميل منعلىكان حيويًا للتحويل الفعال للمخزون المتراكم(الشكل التوضيحي 26). بالإضافة إلى ذلك، فإن الخلط الفيزيائي البسيط لـ و لم يكن بالإمكان ضمان الارتباط المكاني للمواقع المجاورة بشكل كافٍ، مما يحد من الهيدروجين الفعال لـإلىخلالالاختزال الكهربائي (الشكل التوضيحي 27). كما نستبعد إمكانية تلوث الأمونيا من التحليل الكهربائي الذاتي لـ، الإلكتروليت، وورق الكربون، على التوالي (الشكل التوضيحي 28). بالإضافة إلى ذلك، كانت النشاط التحفيزي للكربون المضاف له النيتروجين أقل بكثير مقارنة بـج (الشكل التكميلية 29). التدخل المحتمل لذرات الكوبالت الفردية علىيمكن اعتبار الدعم غير مهم لأداء التحفيز لـ (الأشكال التكميلية 30-33). الاختزال الكهربائي لـتأثرت أيضًا بانتشار
الشكل 2 | الأداء التحفيزي. منحنيات LSV لـ، و مع (أ)و (ب)مُعَايَراستنادًا إلىFE لـ، و معدل العائد لـمن، و عند جهد كهربائي مختلف مع معدل العائد لـمنفي -1.0 فولت مقابل RHE تحت 20 جولة من التفاعلات المتعاقبة. تم تحديد مقاومة المحلول على أنهافي الإلكتروليتات بواسطة طيف الامتصاص الكهروكيميائي الثابت الجهد. المتفاعلات (الشكل التوضيحي 34). علاوة على ذلك،قياسات الوسم النظائري لـتم إجراءه معتحليل الرنين المغناطيسي النووي. فقط قمم مزدوجة نموذجية تُنسب إلىتم جمعها معكمصدر للنيتروجين بينما قمم الثلاثيات لـتم الكشف عنها بـكمصدر للنيتروجين (الشكل التوضيحي 35). أشارت هذه النتائج إلى أنتم الكشف عنه في الإلكتروليت الناتج عن الاختزال الكهربائي لـ.
دراسة ميكانيكية حولالاختزال الكهربائي
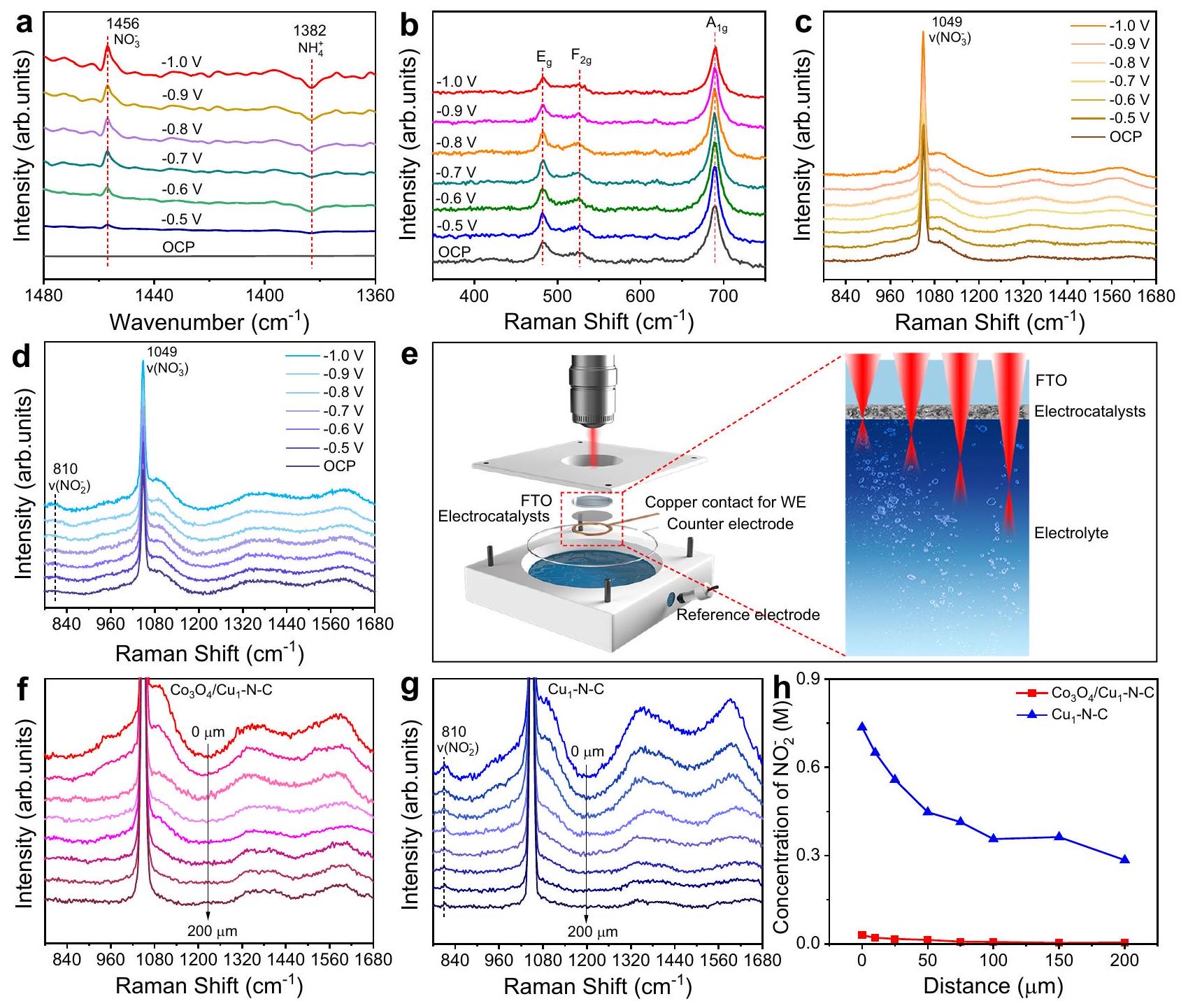
للحصول على مزيد من الفهم حول العملية التحفيزية لـالاختزال الكهربائي على-N-C، قمنا بإجراء مطيافية الأشعة تحت الحمراء التحويلية الكهروكيميائية (FTIR) ومطيافية رامان في الموقع لمراقبة عملية التفاعل (الأشكال التكميلية 36 و37). تعرض الشكل 3a أطياف FTIR في الموقع لـ عند الجهود المطبقة من OCP إلى -1.0 فولت مقابل RHE. القمم السلبية عند نُسِبَت إلى استهلاكنوع. بالإضافة إلى ذلك، ظهور قمم إيجابية تقع عندأكد أنتم إنشاؤه خلالالاختزال الكهربائي. بالإضافة إلى ذلك، هناك قمتان عند 1541 وتم الكشف عنها، والتي تم تخصيصها إلى نطاق الاهتزاز لـ *NO و *NOH، على التوالي (الشكل التوضيحي 38). يوضح الشكل 3b طيف رامان في الموقع لـفي جميع الجهود المطبقة. القمم التي تتوافق مع، و أنماط الاهتزازظل دون تغيير، مما يشير إلى أن كانت الأنواع مستقرة خلالالاختزال الكهربائي. خلال الـالاختزال الكهربائي، فقط القمة عندتمت ملاحظته لـفي جميع الجهود المطبقة، المخصصة للاهتزاز التمددي المتماثل لـ(الشكل 3ج). في حالة بصرف النظر عن إشارةذروة جديدة عند المنسوبة إلى اهتزاز الانحناء لـ ظهرت تدريجياً مع زيادة الجهد المطبق، مما يدل على تراكملـخلالالاختزال الكهربائي (الشكل 3د). لمزيد من التحقيق في تغير التركيز المحلي لـبالقرب من سطح المحفزات، صممنا خلية رامان تسمح لليزر رامان بالكشف من سطح المحفزات إلى كتلة الإلكتروليت. تم تصميم ليزر رامان ليكون متجهًا من خلف المحفزات لتقليل التداخل الناتج عن الامتصاص القوي لـفي الإلكتروليت. كما هو موضح في الشكل 3e، تم ترسيب المحفزات الكهربائية على زجاج مطلي بأكسيد القصدير الفلوري (FTO) كقطب عمل (WE). يتم التحكم في المسافة من شعاع الليزر إلى سطح القطب بواسطة منصة العينة الميكانيكية. تعرض الأشكال 3f و 3g طيف رامان في الموقع لـ و عند -0.8 فولت مقابل RHE عندما تم وضع شعاع الليزر من 0 إلىبعيدًا عن سطح المحفزات، على التوالي. مع زيادة المسافة بين المستوى البؤري لليزر وسطح المحفزات، انخفضت شدة إشارة الكربون الجرافيتي للمحفزات تدريجيًا. قمة ملحوظة عندمُعين إلىظهر بالقرب من سطح. بينما، إشارة لـكان ضئيلاً,
الشكل 3 | التوصيفات في الموقع. أ طيف FTIR في الموقع لـمن OCP إلى -1.0 فولت مقابل RHE في. ب طيف رامان في الموقع لـمن OCP إلى -1.0 فولت مقابل RHE في. طيف رامان في الموقع لـ (ج) و (د) من OCP إلى -1.0 فولت مقابل RHE فيمخطط خلية رامان المصممة للكشف من سطح المحفزات إلى كتلة الإلكتروليت. رامان في الموقع طيف لـ (ف) و (ز) عند -0.8 فولت مقابل RHE فيبمسافات مختلفة تتراوح من 0 إلى التركيز المحسوب لـ لـ و بمسافات مختلفة تتراوح من 0 إلى. التي كانت مستقلة عن المسافة. علاوة على ذلك، حددنا التركيز المحلي لـقرب سطح المحفزات المستندة إلى المساحات المتكاملة لـو، أخذ نسبة المساحات المدمجة لـوالحلول كعامل تصحيح (الشكل التوضيحي 39). مع زيادة بعد شعاع الليزر عن سطح المحفزات إلى الإلكتروليت، زادت تركيزلـانخفضت تدريجياً من 0.74 إلى 0.29 م (الشكل 3h). تشير هذه الاتجاهات إلى أنتم إنشاؤه فيواجهة الإلكتروليت انتشرت في الإلكتروليت بسبب التفاعل البطيء للتقليل من. من الواضح أن تركيز لـكان أقل بكثير بالنسبة لـتجسيد التخفيف الميسر لـبفضل.
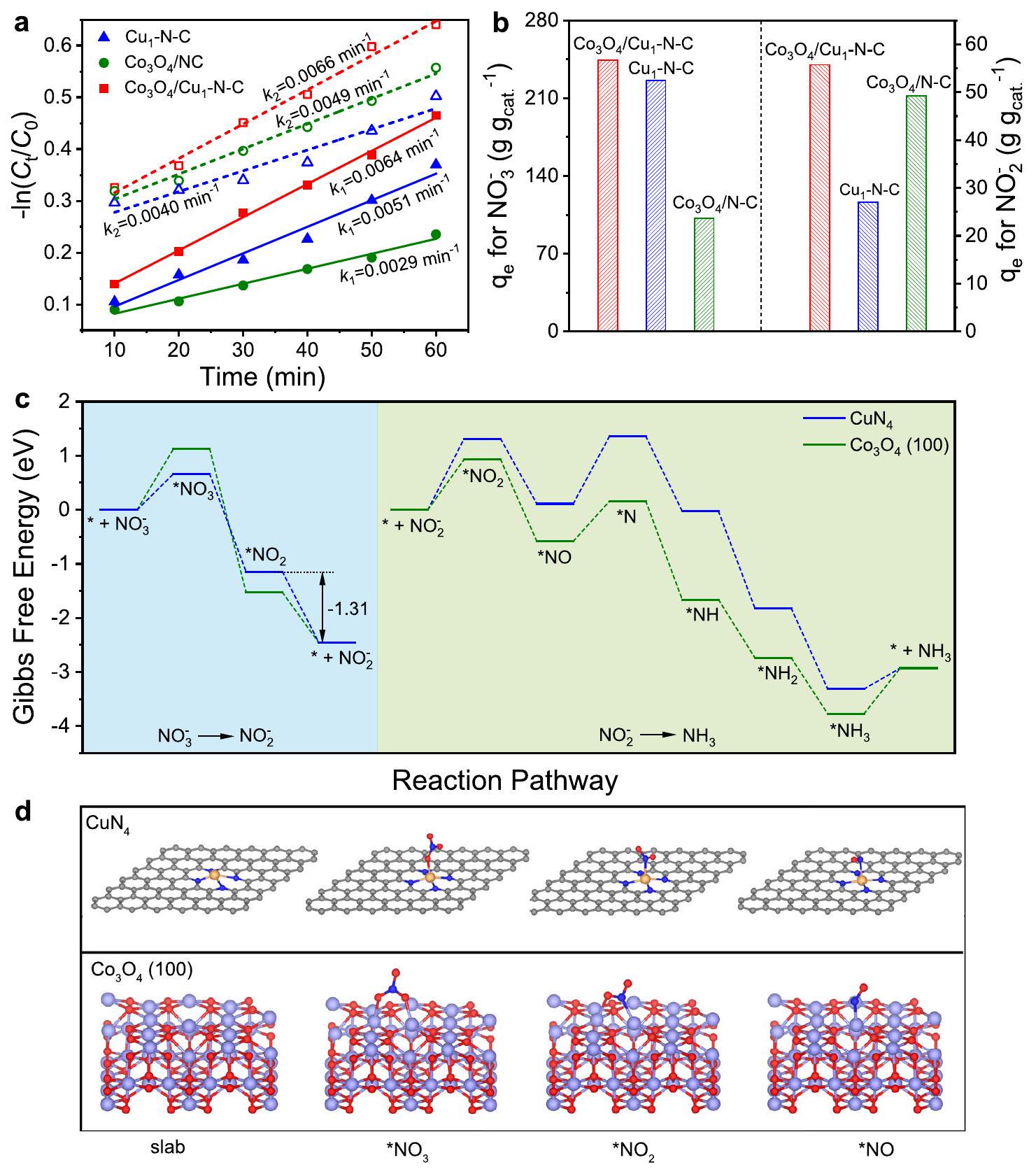
لفهم الدور التكاملي بشكل أفضل و في العملية الحفزية، قمنا بحساب ثوابت المعدل لـالاختزال الكهربائي ) و الاختزال الكهربائي )، على التوالي (الشكل 4أ). تركيز المتبقي بعد أن تم تحديد عملية الاختزال الكهربائي بواسطة مطيافية الأشعة فوق البنفسجية والمرئية (الشكل التكميلي 40). بالمقارنة معالأكبرقيمة لـيشير إلى أنكان أكثر ملاءمة للتحويل منإلى، لكن الـ أصغر القيمة تظهر الحركيات الأبطأ لـ تقليل. وبناءً عليه، مفرطسيتم امتصاصه في الإلكتروليت لـ. من الجدير بالذكر أن الأعلى و منأظهرت التسارع المتزامن للتحويل منإلىوإلى. بالإضافة إلى ذلك، فإن منحدرات الطاولة ، و فيوتشير إلى أن مجموعة منمعتسهيل الحركيات لـوالخفض خلال العملية التحفيزية (الشكل التكميلي 41). يوضح الشكل 4 ب سعات الامتزازمن، و لـوعلى التوالي. من الواضح أنعرضت الأكبرلكليهماوبين الثلاثة محفزات. ونتيجة لذلك، دمجمعكان مهيئًا لتحويل كل منو.
تم إجراء حسابات نظرية الكثافة (DFT) لتفسير آلية التفاعل بشكل أعمقالاختزال الكهربائي علىالعوامل المساعدة. استنادًا إلى نتائج التحليل الهيكلي، اعتمدنا و (100) ألواح كنماذج لحساب طاقات جيبس الحرة ( ) لكل خطوة متضمنة في الاختزال الكهربائي، على التوالي (الشكل التكميلية 42). بعد
الشكل 4 | دراسة آلية علىالاختزال الكهربائي. ملفات حركية خطية مزيفة من الدرجة الأولى لـ، و فيعلى التوالي.سعات الامتزاز لـ، و لـرسم بياني للطاقة الحرةالاختزال الكهربائي علىو (100) ألواح. * تمثل موقع الامتزاز. د نماذج هيكلية للوسطاء الرئيسيين على و الألواح. تمثل الكرات الرمادية والزرقاء والحمراء والصفراء والبنفسجيةوذرات الكوبالت، على التوالي. تحسين الهيكلسيتم امتصاصه على مع ذرة الأكسجين. كما هو موضح في الشكل 4c، فإن تغييرات الطاقة الحرة غيبس ( ) ل الامتزاز على أقل بكثير من ذلك فوق (100)، مما يدل على الارتباط الأقوى لـفوق. ومع ذلك، الـمن *إزالة الامتصاص على هو -1.31 إلكترون فولت، مما قد يكون أكثر ملاءمة من الناحية الديناميكية الحرارية من اختزال إلى. في هذا الصدد، يتم إزالة *سوف يؤدي إلى تراكمفوق، وهو ما يتماشى مع العائد المرتفع لـفوقفيالاختزال الكهربائي (الشكل التوضيحي 17). بالنسبة لتحويلسيتم امتصاصه علىمن خلال ذرة النيتروجين بينما *يمكن أن يكون مرتبطًا بـ (100) من خلال ذرات N و O بعد تحسين الهيكل، مما يؤدي إلى انخفاض لـالامتزاز على (100) (الشكل 4د). بالإضافة إلى ذلك، فإن النسبة العالية نسبياً لـ الامتزاز على و كشفت عن ضعف الامتزاز لـ *H، مما يشير إلى حدوث تنافسقد تكون التطورات مكتئبة (الأشكال التكميلية 43 و 44). نتيجة لذلك،سوف ينظم تكوين الامتزازوتمتلك ارتباطًا أسهل معتسهيل تقليلإلى.
نقاش
باختصار، قمنا بتطوير محفز عالي الكفاءة من خلال دمج الوظائف المنفصلة لـ و للتقليل المتسلسل لـإلىوإلى. تم الحصول علىأظهر المحفز معدل إنتاجية متفوقمن، التي تجاوزت جميع القيم المبلغ عنها لمحفزات القائمة على النحاس. كشفت التحقيقات في الآلية أن الجمع بين نظمت تكوين الامتزاز لـوقوّت الـ ربط مع، مما يسرع من الاختزال الكهربائي لـإلى. يقدم هذا العمل إرشادات جديدة لبناء محفزات مت tandem عالية الكفاءة نحو الاختزال الكهربائي.
طرق
المواد الكيميائية والمواد
نيترات الزنك سداسي الماء2-ميثيل إيميدازول (2-MeIM، 99.0%)، خلات النحاس (II) أحادي الهيدرات، نترات الكوبالت سداسي الهيدراتنترات الحديد غير المائيةنيترات النيكل سداسي الهيدراتنترات النحاس ثلاثي الهيدراتالميثانول )، الإيثانول ( ) ، نترات البوتاسيوم ( ) ، نيتريت البوتاسيوم ( )، هيدروكسيد البوتاسيوم ( ) ، كبريتات الأمونيوم ( )، هيدروكسيد الصوديوم ( )، حمض الساليسيليك ( محلول هيبوكلوريت الصوديوم (NaClO، الكلور المتاحمحلول مائي)، سترات الصوديوم الثلاثي المائي ( ) ، نيتروفيريسيانيد الصوديوم ثنائي الهيدرات ( ) ، حمض الهيدروكلوريك ( )، حمض السلفاميك ( ) ، p -أمينوبنزين سلفوناميد ( ), ن -( ثنائي هيدروكلوريد الإيثيلينديامين (98.0%)، حمض الفوسفوريك ( ) وحمض 1-بروبان سلفونيك 3-(ثلاثي ميثيل سيليل) ملح الصوديوم (DSS) تم شراؤه من شركة سينوفارم للمواد الكيميائية المحدودة. محلول حمض الجلايوكسيل ( )، ثنائي ميثيل سلفوكسيد – (دي ميثيل سلفوكسيد – ذرة د)، ( 99.0 ذرة )، و ( 99.0 ذرة تم شراء ( ) من شركة Aladdin Chemistry Co., Ltd (شنغهاي، الصين). تم شراء الغشاء ثنائي القطب (TRJBM) من شركة Beijing Tingrun Membrane Technology Development Co., Ltd (بكين، الصين). تم إنتاج الماء المنزوع الأيونات (DI) باستخدام جهاز Millipore MilliQ، مع مقاومة قدرهاتم استخدام جميع المواد الكيميائية دون أي تنقية إضافية.
الأدوات
تم التقاط صور TEM باستخدام مجهر إلكتروني نافذ Hitachi HT7700 عند جهد تسريع قدره 100 كيلو فولت. تم إجراء HAADF-STEM ورسم الخرائط العنصرية EDS المقابلة على مجهر إلكتروني نافذ بتقنية الانبعاث الميداني Talos F200X يعمل عند جهد تسريع قدره 200 كيلو فولت باستخدام شبكات TEM قائمة على الموليبدينوم. تم إجراء صور HAADF-STEM مصححة للانحراف على مجهر إلكتروني نافذ بتقنية الانبعاث الميداني Themis Z يعمل عند جهد تسريع قدره 300 كيلو فولت باستخدام شبكات TEM قائمة على الموليبدينوم. تم جمع أنماط XRD باستخدام جهاز قياس حيود الأشعة السينية Rikagu MiniFlex مع النحاس.إشعاعتم استخدام تحليل ICP-OES (Avio 220 MAX، PerkinElmer) لقياس تركيز أنواع المعادن. تم إجراء قياسات XPS باستخدام جهاز قياس الأشعة السينية Kratos Axis supra+ مع Al-K.الإشعاع. تم إجراء طيف رامان عبر نظام رامان LabRAM HR Evolution (هوربا) باستخدام ليزر تحفيز بطول موجي 532 نانومتر. تم قياس بيانات الامتصاص على مطياف UV-vis (أجيلة تكنولوجيز، كاري 60). تم الحصول على أطياف FTIR في الموقع بواسطة مطياف FTIR نيكوليت iS50 مع كاشف MCT مدمج.
تركيب
خليط من و (0.28 مليمول) تم إذابته في 80 مل من الميثانول، والذي تمت إضافته بعد ذلك إلى 80 مل من الميثانول الذي يحتوي على 3.70 جرام من 2-MelM. ثم تم الاحتفاظ بالمحلول المختلط فيلمدة 12 ساعة. تم فصل الراسب الناتج (المشار إليه باسم ZIF-8 المضاف إليه النحاس) بواسطة الطرد المركزي وغسله بعد ذلك بالميثانول خمس مرات، وأخيرًا تم تجفيفه عندتحت فراغ طوال الليل. بعد ذلك، تم تسخين المشتق المحتوي على النحاس من ZIF-8 إلىمعدل تسخين قدرهفي فرن أنبوبي وتم الاحتفاظ به عندتحت تدفق غاز الأرجون لمدة 3 ساعات. بعد أن تم تبريد فرن الأنبوب بشكل طبيعي إلى درجة حرارة الغرفة،تم الحصول عليها واستخدامها مباشرة كعامل حفاز دون معالجة إضافية. للمقارنة، تم تثبيت ذرات الكوبالت الفردية على الكربون المخدر بالنيتروجين (المشار إليه بـ ) وذرات مفردة من Co مُرَكَّز على (المشار إليه بـ تم الحصول عليها عن طريق التحلل الحراري لـ ZIF-8 المضاف إليه الكوبالت و-المخدرة ZIF-8، على التوالي.ZIF-8 المخدر-المخلوط مع ZIF-8 تم تحضيره بنفس الإجراء الذي تم استخدامه فيباستثناءومزيج من و كمقدمة معدنية، على التوالي.
تركيب و
160 ملغ منتم تفريقه في 20 مل من الإيثانول عن طريق الموجات فوق الصوتية لمدة 30 دقيقة. بعد ذلك، 5 مل من يحتوي على 45 ملغ من تم إضافة إلى المحلول أعلاه، الذي تم الاحتفاظ به في حمام من الماء المثلج لمدة ساعة مع التحريك القوي. ثم، 40 مل من المحلول المحضر حديثًامع الثلج الباردتمت الإضافة قطرة قطرة إلى التعليق المذكور أعلاه، تلاها التحريك لمدة ساعة واحدة. تم فصل الراسب الناتج عن طريق الترشيح وغسله بعد ذلك بالماء خمس مرات. أخيرًا،تم الحصول عليه عن طريق التجفيف عندتحت فراغ طوال الليل.تم إعداده كمقارنة مع الإجراء المماثل لذلكباستثناء إضافة الكربون المخدر بالنيتروجين بدلاً منتم تحضير الكربون المضاف بطريقة تركيبية مشابهة لتلك الخاصة بـدون إضافة. للمقارنة، تشمل أكاسيد المعادن الأخرى ، و موزعة على الكربون المخدر بالنيتروجين (المشار إليه بـ، و على التوالي) تم تحضيرها بنفس الإجراء كما فيباستثناء إضافة، و كمقدمة معدنية، على التوالي (المشار إليها بـ، و ، على التوالي).
قياسات هيكل الامتصاص الدقيق للأشعة السينية (XAFS)
طيف XAFS عند-حافة و-تمت التجارب عند خط الشعاع 1W1B في منشأة الإشعاع السنكروتروني في بكين وخط الشعاع BL11B في منشأة الإشعاع السنكروتروني في شنغهاي. تم الحصول على البيانات في ظروف محيطة تحت وضع الفلورية لـ-وضع الحافة والنقل لـ Co-الحافة، على التوالي.
تم استخدام وحدة ATHENA وأكواد ARTEMIS في حزم برامج IFEFFIT لاستخراج البيانات وتناسب الملفات.. الـتم الحصول على طيف EXAFS الموزون من خلال معايرة الطاقة وتطبيع الطيف. بالنسبة لجزء EXAFS، تم تحويل البيانات إلى الفضاء R باستخدام تحويل فورييه.-حافة وتم تحليل الحواف من خلال تطبيق نوافذ هانينغ ) لتمييز تذبذب EXAFS من قذائف التنسيق المختلفة. بعد ذلك، قمنا بإجراء ملاءمة معلمات منحنى المربعات الصغرى للحصول على المعلمات الهيكلية حول الذرات المركزية. كانت النطاقات الملائمة لـ تم تحديد المساحة عندمعنطاق منتشمل المعلمات الأربعة عدد التنسيق (CN) وطول الرابطة (R) وعامل ديباي-والر ( )، و تحويل (تم تركيبها دون أن يتم تثبيت أو تقييد أو ربط أي شخص.
في الموقعتم إجراء قياسات XAFS عند حافة -edge باستخدام خلية XAFS مصنوعة في المنزل. عادةً، تم استخدام 8 ملغ من المحفزات وتم dispersing Nafion في 2 مل من الإيثانول عن طريق الموجات فوق الصوتية لمدة ساعة. ثم تم تحميل الحبر الموحد على ورق الكربون بمساحةتم حساب تحميل الكتلة ليكونالمحفزات المحضرة،تم استخدام إلكترود، وسلك بلاتينيوم كإلكترود عمل، وإلكترود مرجعي، وإلكترود مضاد، على التوالي. تم قياس جميع الاختبارات الكهروكيميائية في محلول إلكتروليتي 1 م KOH معوتتحكم بها محطة عمل كهربائية كيميائية CHI 1140 C.
تحضير الأقطاب الكهربائية العاملة
تم توزيع 8 ملغ من المحفزات في 2 مل من الإيثانول عن طريق الموجات فوق الصوتية لمدة ساعة.تم إضافة محلول نافيون إلى الخليط وتم استخدام الموجات فوق الصوتية لمدة 30 دقيقة للحصول على حبر موحد. أخيرًا، تم تحميل الحبر الموحد على ورق الكربون بمساحةتم حساب تحميل الكتلة ليكونمساحة الأقطاب الكهربائية العاملة المستخدمة في القياسات الكهروكيميائية كانت.
القياسات الكهروكيميائية
تم إجراء القياسات الكهروكيميائية في نظام خلية H تم فصله بواسطة غشاء ثنائي القطب باستخدام محطة عمل كهروكيميائية CHI 1140 C (تشينهوا، شنغهاي).تم استخدام القطب الكهربائي وقضيب الجرافيت كقطب مرجعي وقطب مضاد، على التوالي. من أجلالاختزال الكهربائي، 1 م هيدروكسيد البوتاسيومتم توزيع المحلول (60 مل) بالتساوي على حجيرات الكاثود والأنود. تم تحديد قيمة pH للكهارل لتكون 14 بواسطة جهاز قياس pH FiveEasy Plus (METTLER TOLEDO). تم قياس الجهود المطبقة مقابلإلكترود مرجعي معالتعويض وتحويله إلى مقياس مرجعي RHE بواسطةتم تحديد مقاومة المحلول على أنهاأوم في الإلكتروليتات بواسطة طيف الامتصاص الكهروكيميائي بتقنية الجهد الثابت عند ترددات تتراوح من 10 هرتز إلى 100 كيلوهرتز، والذي تم إجراؤه في نظام ثلاثي الأقطاب قياسي في ظروف محيطية. قبل اختبار الاختزال الكهربائي، تم إجراء منحنيات الجهد حتى تحقق منحنيات الاستقطاب حالة مستقرة بمعدل مسحقبل التحليل الكهربائي، تم توصيل غاز الأرجون إلى حجرة الكاثود بمعدللإزالة المذابتم تسجيل LSVs للعوامل الحفازة بمعدل مسحفي 1 م كوه يحتوي علىتم إجراء التحليل الكهربائي عند جهد مطبق لـتم إجراء الاختزال الكهربائي بنفس الظروف باستثناء أن محلول 1 م كOH يحتوي علىتم استخدامه كإلكتروليت. تم إعداد خلية امتصاص تحتوي على 30 مل من 1 م HCl لتمتص بشكل فعال ما قد يهرب.من خلية الكاثود. بعد التحليل الكهربائي عند كل جهد مطبق، تركيزفي خلية الامتصاص كانت أقل من. في هذه الحالة، يتم تطايرمن الإلكتروليتات قد تكون ضئيلة. تم إجراء قياسات الفولتمترية الدورية في نافذة جهد غير فارادائية مع معدلات مسح مختلفة من 50 إلىتم حسابه عن طريق رسم ( ) في منتصف نافذة الجهد المقابلة مقابل معدلات المسح. الـ و كانت كثافات التيار الأنودي والكاثودي، على التوالي. كان الميل ضعف.
طريقة حساب FE
التقييم المالي للمنتج ( و تم حساب ) عند جهد معين على النحو التالي:
: التركيز المقاس للمنتج ( ) ، : حجم المحلول الكهربائي ( مل ) ، : عدد الإلكترونات المنقولة للمنتج، وهو 8 لـ و 2 لـ ،
F: ثابت فاراداي،، : الشحنة الكهربائية الكلية (كولوم) م: الكتلة الجزيئية النسبية، والتي هيلـ و لـ.
طريقة حساب معدل العائد لـمنتج
معدل العائد لـتم حساب المنتج عند إمكانيات معينة على النحو التالي:
معدل العائد ( ) ، : التركيز المقاس لـ ، : حجم المحلول الكهربائي ( مل ) ، : مساحة المحفز ( ) ، : وقت تفاعل الاختزال (دقيقة).
تحديد تركيز الأيونات
تحديدتركيز بطريقة إندوفينول الأزرق. بعد عملية الاختزال الكهربائي، تم أخذ كمية معينة من المحلول الكهربائي من الخلية الكهربائية وتخفيفها إلى نطاق الكشف. ثم، تم إضافة 2 مل من محلول هيدروكسيد الصوديوم 1 م الذي يحتوي على حمض الساليسيليك ( ) وسيتريت الصوديوم ( ) تم إضافتها إلى المحلول المذكور أعلاه، تلتها إضافة 1 مل من 0.05 م NaClO و 0.2 مل من بعد الوقوف في الظلام لمدة ساعتين، تم قياس طيف الامتصاص باستخدام مطياف الأشعة فوق البنفسجية والمرئية. تم تحديد تركيز الأزرق الإندوفينول باستخدام الامتصاص عند الطول الموجي 650 نانومتر. تم معايرة منحنى التركيز-الامتصاص باستخدام معيار.حل مع سلسلة من التركيزات.
تحديدتركيز معطريقة الرنين المغناطيسي النووي. بعدتم أخذ كمية معينة من الإلكتروليت لإجراء مزيد من التقدير بواسطةتم إجراء جميع التحليلات باستخدام جهاز NMR (Bruker AVANCE AV III 400) مع 128 مسح زمني. تم معايرة منحنى مساحة التكامل التركيز باستخدام معيار.حل. عادةً،تم إذابته في 20 مل من محلول كOH بتركيز 1 مولي كإحدى السلاسل القياسيةمحاليل بتركيزات مختلفة. بعد ذلك، 0.1 مل من ثنائي ميثيل سلفوكسيد- (دي ميثيل سلفوكسيد – تم إضافة 0.1 مل من محلول 6 مليمول من ملح الصوديوم 1-بروبان سلفونيك أسيد 3-(تريميثيل سيلان) (DSS) و0.08 مل من حمض الهيدروكلوريك 6 م إلى 0.32 مل منمحاليل قياسية بتركيزات مختلفة. ظهر الإشارة عند، و 6.97 جزء في المليون كانت تُنسب إلىالمناطق التكاملية لإشارةتم استخدامها لتحديد تركيزمقارنةً مع مرجع DSS المعروف.
تحديدتركيزتم تخفيف كمية معينة من الإلكتروليت إلى نطاق الكشف. ثم، 0.1 مل من 1 م HCl و 0.01 مل من تم خلط محلول حمض السلفاميك مع الإلكتروليت المخفف، تلاه الاهتزاز لمدة 10 دقائق. باستخدام مطياف الأشعة فوق البنفسجية والمرئية، تم جمع طيف الامتصاص، والحصول على شدة الامتصاص عند طول موجي 220 و 275 نانومتر. أخيرًا، تم حساب الامتصاص (تم الحصول عليه. تم معايرة منحنى التركيز-الامتصاص باستخدام معيارمحاليل بسلسلة من التركيزات.
تحديدتركيز. تم خلط 4 جرام من p-أمينوبنزين سلفوناميد، 0.2 جرام من N -(1-نافثيل) إيثيلين دياامين ثنائي الهيدروكلوريد، و10 مل من حمض الفوسفوريك مع 50 مل من الماء ككاشف لوني. تم أخذ كمية معينة من الإلكتروليت من الخلية الكهربية وتخفيفها إلى نطاق الكشف. 1 مل منتمت إضافة إلى 4 مل من المحاليل الكهربية المخففة بعد التحليل الكهربائي لضبط الرقم الهيدروجيني، تلاها إضافة 0.1 مل من كاشف اللون. بعد الوقوف لمدة 20 دقيقة، تم قياس طيف الامتصاص باستخدام مطياف الأشعة فوق البنفسجية والمرئية. تم تسجيل شدة الامتصاص عند طول موجي 540 نانومتر. تم معايرة منحنى التركيز-الامتصاص باستخدام معيار.حل مع سلسلة من التركيزات.
تحديد المنتجات السائلة والغازية الأخرى
مقدارتم تحديده بواسطةبعدتم القبض عليه بسبب كمية زائدة منمن خلال عملية الأكسيم. على وجه التحديد، 0.4 مل من الإلكتروليت بعدتم خلط الاختزال الكهربائي معمنمحلول، يتبعه إضافة 0.1 مل من DMSO-و0.1 مل من محلول DSS بتركيز 6 مللي مولار. تم استخدام المساحة التكاملية للإشارة التي ظهرت عند 7.46 جزء في المليون لتحديد التركيز من مقارنةً بمرجع DSS المعروف. في هذا العمل، كمية كان دون حد الكشف لكلاهماوتقليل على.
أكاسيد النيتروجين بما في ذلك، و تم الكشف عنها بواسطة محلل غاز بالأشعة تحت الحمراء (THA100S). و تم الكشف عنها بواسطة جهاز كروماتوغرافيا الغاز على الإنترنت (GC-2014) المزود بكاشف تأين اللهب وكاشف الموصلية الحرارية.
تم حساب الكفاءة للمنتجات الغازية بواسطة المعادلة التالية:
: الكسر المولي المقاس للمنتج، : الحجم الكلي للغاز (لتر), : عدد الإلكترونات المنقولة للمنتج، وهو 3 لـ لـلـ و 10 لـ ،
F: ثابت فاراداي،، : الشحنة الكهربائية الكلية (C)، : الحجم المولي للغاز، .
تجارب وسم النظائر
تجربة الوسم النظائري استخدمتمعإثراءكمصدر غذائي للنيتروجين لتوضيح مصدر الأمونيا. تم استخدام 1 م كوه كإلكتروليت وتم إضافة 1 م إلى حجرة الكاثود كمادة متفاعلة. بعد عملية التحليل الكهربائي، تم إضافة 0.1 مل من DMSO-وتم إضافة 0.1 مل من محلول DSS بتركيز 6 مللي مولار إلى 0.4 مل من الإلكتروليت، تلاها إضافة 0.05 مل منلتعديل درجة الحموضة في المحاليل. ثم تم الحصول علىتم التعرف عليه باستخدام مطياف الرنين المغناطيسي النووي Varian 400 ميجاهرتز (Bruker AVANCE AV III 400).
تقييم حركي
تم إجراء التحليل الكهربائي عند -1.0 فولت مقابل RHE لمدد زمنية مختلفة للحصول على ثابت المعدل في 1 م كOH يحتوي علىأو“. ثابت التفاعل (لـتقليل ولـتم حساب (الخفض) من خلال رسم تركيزأوضد زمن التفاعل، على فرض أن تركيزاتأوانخفض بشكل أسي وفقًا لمعدل من الدرجة الأولى.
: التركيز الابتدائي لـ أو، : تركيز أوفي الوقت، : زمن التفاعل (دقيقة).
تجارب الامتزاز
لتحديد سعات الامتزاز لـ، و من المحفزات أُضيفت إلى كل 25 مل منأومحاليل بتركيز ابتدائي قدره 1 م تحت التحريك لمدة ساعتين، على التوالي. تم فصل المحاليل عن طريق الترشيح باستخدامفلتر غشاء ميكروبوري. لتركيز عالٍ من أو تم تخفيف المحلول قبل قياسات الامتصاص. تم حساب سعة الامتزاز باستخدام المعادلة التالية:
” : سعة الامتزاز ( ) ، : التركيز الابتدائي لـ أو، : التركيز المقاس لـ أوبعد الامتزاز ) ، : حجم الإلكتروليت (مل) ، : كتلة المحفز ( جرام ).
قياسات FTIR في الموقع
باستخدام مطياف FTIR من نوع Nicolet iS50 (Thermo Scientific) مزود بكاشف MCT مدمج، حصلنا على طيف FTIR الكهروكيميائي في الموقع. عادةً، تم استخدام 2 ملغ من المحفزات ومن نافيوني تم تفريقه في 2 مل من الإيثانول وتم تعريضه للموجات فوق الصوتية لمدة ساعة. ثم تم تحميل الخليط على منشور سيليكون مغطى بالذهب لتغطية فيلم الذهب بالكامل. تم استخدام المنشور المحضر كقطب عمل بعد أن جف طبيعياً. كان القطب المرجعي وقطب العدّاد هو
قطب AgCl وسلك Pt، على التوالي. تم عرض صورة خلية FTIR الكهروكيميائية في الموقع في الشكل التكميلية 36. تم قياس جميع الاختبارات الكهروكيميائية في محلول إلكتروليتي 1 M KOH مع وتم التحكم بها بواسطة محطة عمل كهربائية كيميائية CHI 1140 C. تم إجراء جميع التجارب في درجة حرارة الغرفة. تم الحصول على طيف الخلفية للقطب العامل عند جهد مفتوح قبل الاختبارات الكهروكيميائية. تم جمع جميع الأطياف في الامتصاص عن طريق متوسط 32 مسحًا بدقة .
قياسات رامان في الموقع
تم إجراء تحليل رامان في الموقع باستخدام جهاز Lab RAM HR Evolution (هوربا) المزود بـعدسة المجهر. كانت طول موجة الإثارة 532 نانومتر مع الكثافة. تم عرض صورة الخلية الكهروكيميائية لقياس رامان في الموقع في الشكل التكميلية 37. عادةً، 2 ملغ من المحفزات وتم توزيع Nafion في 2 مل من الإيثانول عن طريق الموجات فوق الصوتية لمدة ساعة واحدة. ثم تم ترسيب المحفزات الكهربائية على زجاج مطلي بأكسيد القصدير الفلوري كقطب عمل.تم استخدام إلكترود مرجعي، وسلك بلاتيني كإلكترود مضاد، على التوالي. تم قياس جميع الاختبارات الكهروكيميائية في محلول إلكتروليتي 1 م KOH معوتم التحكم فيه بواسطة محطة عمل كهربائية كيميائية CHI 1140 C. تم إجراء جميع التجارب في درجة حرارة الغرفة. تم جمع كل طيف من خلال التكامل مرتين، 60 ثانية لكل تكامل. لتحديد التركيز المحلي لـبالقرب من سطح المحفزات، استخدمنا طريقة المعيار الداخلي خلال قياسات رامان في الموقع.
حسابات DFT
تم إجراء حسابات DFT باستخدام حزمة المحاكاة فيينا Ab-Initio (VASP) على مستوى GGA ضمن صيغة PAW-PBE.تم استخدام طريقة DFT-D3 مع تخفيف بيك-جونسن لتصحيح فان دير فالز. الطبقة الثلاثيةتم اعتماد نموذج (100) لوح مع فراغ منتم إجراء حسابات الطاقة الكلية باستخدامشبكة وطاقة قطع للموجة المستوية تبلغ 400 إلكترون فولت. تم تثبيت الذرات في الطبقتين السفلتين. تم تطبيق قيمة U تبلغ 3.5 إلكترون فولت علىحالات الكوبالت لوصف التفاعلات الكولومبية القوية في الموقع بسبب توطين الكوبالتدول. لنموذج (لم يتم تثبيت أي ذرات)، تم إجراء حسابات الطاقة الكلية باستخدام شبكة وطاقة قطع للموجة المستوية تبلغ 400 إلكترون فولت. تم السماح لجميع الذرات، التي لم تكن ثابتة، بما في ذلك المواد الماصة، بالاسترخاء حتى كانت القوة على كل أيون أصغر منÅ.
قمنا بحساب الطاقة الحرة لجيبس ( ) لكل نوع على النحو التالي:
أين، و تمثل الطاقة الكلية المحسوبة بواسطة تحويل فورييه السريع (DFT) والطاقة الصفرية (ZPE) ومساهمة الإنتروبيا، على التوالي (T هي درجة الحرارة، 298.15 كلفن). يُفترض أنلكل الأنواع الممتصة.
توفر البيانات
البيانات التي تدعم نتائج هذه الدراسة متاحة من المؤلف المراسل عند الطلب. البيانات المصدرية التي تستند إليها الأشكال 1-4 والأشكال التكميلية 1-44 مقدمة كملف بيانات مصدر. يتم توفير البيانات المصدرية مع هذه الورقة.
References
Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209-2244, (2009).
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
Wang, Y., Wang, C., Li, M., Yu, Y. & Zhang, B. Nitrate electroreduction: Mechanism insight, in situ characterization, performance evaluation, and challenges. Chem. Soc. Rev. 50, 6720-6733 (2021).
Duca, M. & Koper, M. T. M. Powering denitrification: The perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726-9742 (2012).
Xu, H., Ma, Y., Chen, J., Zhang, W. X. & Yang, J. Electrocatalytic reduction of nitrate – a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710-2758 (2022).
Fan, K. et al. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 13, 7958 (2022).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036-7046 (2020).
Wang, Y. et al. Structurally disordered nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 61, e202202604 (2022).
Deng, X., Yang, Y., Wang, L., Fu, X. Z. & Luo, J. L. Metallic Co nanoarray catalyzes selective production from electrochemical nitrate reduction at current densities exceeding . Adv. Sci. 8, 2004523 (2021).
Zhang, X. et al. Recent advances in non-noble metal electrocatalysts for nitrate reduction. Chem. Eng. J. 403, 126269 (2021).
Wang, X. et al. Free-standing membrane incorporating single-atom catalysts for ultrafast electroreduction of low-concentration nitrate. Proc. Natl Acad. Sci. USA 120, e2217703120 (2023).
Zhang, G. et al. Tandem electrocatalytic nitrate reduction to ammonia on Mbenes. Angew. Chem. Int. Ed. 62, e202300054 (2023).
Fu, Y. F. et al. Enhancing electrochemical nitrate reduction to ammonia over Cu nanosheets via facet tandem catalysis. Angew. Chem. Int. Ed. 62, e202303327 (2023).
Fang, J. Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
Zhu, T. et al. Single-atom Cu catalysts for enhanced electrocatalytic nitrate reduction with significant alleviation of nitrite production. Small 16, e2004526 (2020).
Yang, J. et al. Potential-driven restructuring of cu single atoms to nanoparticles for boosting the electrochemical reduction of nitrate to ammonia. J. Am. Chem. Soc. 144, 12062-12071 (2022).
Li, P. et al. Pulsed nitrate-to-ammonia electroreduction facilitated by tandem catalysis of nitrite intermediates. J. Am. Chem. Soc. 145, 6471-6479 (2023).
Cheng, X. F. et al. Coordination symmetry breaking of single-atom catalysts for robust and efficient nitrate electroreduction to ammonia. Adv. Mater. 34, 2205767 (2022).
He, W. et al. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 13, 1129 (2022).
Wang, W., Chen, J. & Tse, E. C. M. Synergy between Cu and Co in a layered double hydroxide enables close to nitrate-toammonia selectivity. J. Am. Chem. Soc. 145, 26678-26687 (2023).
Liu, Y. et al. A highly efficient metal-free electrocatalyst of f-doped porous carbon toward electroreduction. Adv. Mater. 32, 1907690 (2020).
Li, R. et al. Insights into correlation among surface-structure-activity of cobalt-derived pre-catalyst for oxygen evolution reaction. Adv. Sci. 7, 1902830 (2020).
Wang, J. et al. Electrocatalytic reduction of nitrate to ammonia on low-cost ultrathin nanosheets. ACS Catal. 11, 15135-15140 (2021).
Gao, Q. et al. Synthesis of core/shell nanocrystals with ordered intermetallic single-atom alloy layers for nitrate electroreduction to ammonia. Nat. Synth. 2, 624-634 (2023).
Song, Z. et al. Efficient electroreduction of nitrate into ammonia at ultralow concentrations via an enrichment effect. Adv. Mater. 34, 2204306 (2022).
Gao, W. et al. Alloying of Cu with Ru enabling the relay catalysis for reduction of nitrate to ammonia. Adv. Mater. 35, e2202952 (2023).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on coppernickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702-5708 (2020).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper-supported rhodium cluster and single-atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759-767 (2022).
Wang, Y. T., Zhou, W., Jia, R. R., Yu, Y. F. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350-5354 (2020).
Hao, R. et al. Pollution to solution: A universal electrocatalyst for reduction of all -based species to . Chem. Catal. 2, 622-638 (2022).
Newville, M. IFEFFIT: Interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 8, 322-324 (2001).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537-541, (2005).
Meng, Q. et al. Sustainable production of benzene from lignin. Nat. Commun. 12, 4534 (2021).
Song, Z. et al. Promoting electroreduction into over porous carbon by introducing oxygen-containing groups. Chem. Eng. J. 434, 134636 (2022).
Bai, Y. et al. FCF-LDH/BIVO4 with synergistic effect of physical enrichment and chemical adsorption for efficient reduction of nitrate. Green. Energy Environ. https://doi.org/10.1016/j.gee.2023. 05.011, (2023).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15-50 (1996).
Zasada, F. et al. Periodic DFT and HR-STEM studies of surface structure and morphology of cobalt spinel nanocrystals. Retrieving 3d shapes from 2d images. J. Phy. Chem. C. 115, 6423-6432 (2011).
شكر وتقدير
تم دعم هذا العمل من قبل برنامج البحث الاستراتيجي ذي الأولوية للأكاديمية الصينية للعلوم (XDBO450401)، البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2021YFA1500500، 2019YFA0405600)، NSF (22209161، 22302184، 22322901، 22221003، 22250007، و22361162655)، صندوق العلوم الوطنية للعلماء الشباب المتميزين (21925204)، مشروع الأكاديمية الصينية للعلوم للعلماء الشباب في البحث الأساسي (YSBR-022، YSBR-051)، برنامج ما بعد الدكتوراه في الصين للمواهب المبتكرة (BX20200324)، برنامج الابتكار التعاوني لمركز هيفي للعلوم، الأكاديمية الصينية للعلوم (2022HSC-CIP004)، برنامج الشراكة الدولية للأكاديمية الصينية للعلوم. )، صندوق مشترك من جامعة يولين ومختبر داليان الوطني للطاقة النظيفة (صندوق YLU-DNL 2022012)، وصناديق البحث الأساسية للجامعات المركزية، ومؤسسة أنهوي للعلوم الطبيعية للباحثين الشباب (2208085QB41)، وزمالة مؤسسة العلوم الصينية لما بعد الدكتوراه (2021M693058). يقر J.Z. بالدعم من مؤسسة تينسنت من خلال جائزة XPLORER. تم تنفيذ هذا العمل جزئيًا في مركز أدوات الفيزياء
العلوم، جامعة العلوم والتكنولوجيا في الصين. تم تنفيذ هذا العمل جزئيًا أيضًا في مركز USTC للبحث والتصنيع على المقياس الميكروي والنانو.
مساهمات المؤلفين
Z.G. و J.Zeng. أشرفا على هذا المشروع. Y.L. أجرت معظم التجارب وحللت البيانات التجريبية. J.W. أجرت قياسات رامان في الموقع. Z.Y. و J.Zhao أجريا حسابات DFT وحللا البيانات الحاسوبية. L.Z. أجرت قياسات XAFS وحللت النتائج. Z.S. و Y.Z. و J.C. و J.M. قدموا المساعدة في تخليق المواد وتوصيفها. Y.L. و J.W. و Z.G. و J.Zeng. كتبوا المخطوطة. ناقش جميع المؤلفين النتائج وساعدوا خلال إعداد المخطوطة.
يجب توجيه المراسلات وطلبات المواد إلى Zhigang Geng أو Jie Zeng.
معلومات مراجعة الأقران Nature Communications تشكر Aiqin Wang والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة الأقران لهذا العمل. يتوفر ملف مراجعة الأقران.
معلومات إعادة الطبع والتصاريح متاحة على http://www.nature.com/reprints
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
مركز هيفي الوطني للبحوث في العلوم الفيزيائية على المقياس المجهري، جامعة العلوم والتكنولوجيا في الصين، هيفي 230026 آنهوي، جمهورية الصين الشعبية. معهد الفيزياء عالية الطاقة، الأكاديمية الصينية للعلوم، 100049 بكين، جمهورية الصين الشعبية. قسم الفيزياء، جامعة العلوم والتكنولوجيا في الصين، هيفي 230026 آنهوي، جمهورية الصين الشعبية. مختبر CAS الرئيسي لفيزياء المادة الكمومية المرتبطة بقوة، جامعة العلوم والتكنولوجيا في الصين، هيفي 230026 آنهوي، جمهورية الصين الشعبية. المختبر الرئيسي لكيمياء السطح والواجهة وتحفيز الطاقة في مؤسسات التعليم العالي في آنهوي، قسم الفيزياء الكيميائية، جامعة العلوم والتكنولوجيا في الصين، هيفي 230026 آنهوي، جمهورية الصين الشعبية. كلية الكيمياء والهندسة الكيميائية، جامعة آنهوي للتكنولوجيا، ما’انشان 243002 آنهوي، جمهورية الصين الشعبية. ساهم هؤلاء المؤلفون بالتساوي: يان ليو، جي وي، زينغ وو يانغ.
-البريد الإلكتروني: gengzg@ustc.edu.cn; zengj@ustc.edu.cn
The nitrate electroreduction into ammonia represents a promising approach for sustainable synthesis. However, the variation of adsorption configurations renders great difficulties in the simultaneous optimization of binding energy for the intermediates. Though the extensively reported Cu -based electrocatalysts benefit adsorption, one of the key issues lies in the accumulation of nitrite ( ) due to its weak adsorption, resulting in the rapid deactivation of catalysts and sluggish kinetics of subsequent hydrogenation steps. Here we report a tandem electrocatalyst by combining Cu single atoms catalysts with adjacent nanosheets to boost the electroreduction of to . The obtained tandem catalyst exhibits a yield rate for of , which exceeds the previous values for the reported Cu -based catalysts. Mechanism investigations unveil that the combination of regulates the adsorption configuration of and strengthens the binding with , thus accelerating the electroreduction of to .
As one of the nitrogen-containing species, nitrate ( ) widely exists in industrial and agricultural wastewater with a high concentration, mainly caused by the emission of low-level nuclear waste and intensive usage of fertilizers . Excessive has significantly threatened ecological balance, inducing acid rain and photochemical smog . Additionally, in human body is easily converted into toxic nitrite ( ), leading to serious health issues . Among the methods for removing , electroreduction process using renewable electricity is regarded as an appealing technology under mild conditions . The controllable products including nontoxic nitrogen ( ) and valuable ammonia ( ) could be obtained after electroreduction .
Since is a fundamental chemical compound and a promising green hydrogen carrier, electroreduction into instead of is more desirable. Taken together, it is highly imperative to achieve efficient electroreduction of into from the perspective of environmental protection and sustainable synthesis.
In view of the multiple nitrogen-containing intermediates (e.g. * , * , and *NO) involved in the electroreduction, an optimal catalyst should satisfy the simultaneously optimized adsorption of intermediates. The moderate binding energy of intermediates serves as one of the key factors for efficient electroreduction into . Classically, given that the coordination of N atom in is
saturated by three O atoms, tends to bond with active sites through O atoms. Whereas, * is preferentially adsorbed on active sites through N and O atoms. As for *NO, N atom in *NO is inclined to connect with active sites. The variation of adsorption configurations renders great difficulties in the simultaneous optimization of binding energy for the intermediates. A typical instance is Cu-based electrocatalysts which have been reported extensively for electroreduction . Though Cu -based electrocatalysts benefit adsorption, one of the key issues lies in the accumulation of , resulting in the rapid deactivation of catalysts and sluggish kinetics of the subsequent hydrogenation steps for production . However, it remains a grand challenge to design an efficient catalyst to satisfy the simultaneously optimized adsorption of intermediates with different configurations.
Herein, we report a tandem electrocatalyst by combining Cu single atoms anchored on N -doped carbon with adjacent nanosheets (denoted as ) to boost the electroreduction of to . The obtained catalyst exhibits a remarkable yield rate for of , which exceeds the previous values for all of the reported Cu-based catalysts. Mechanism investigations unveil that the combination of
regulates the adsorption configuration of and strengthens the binding with , thus accelerating the electroreduction of to .
Results
Catalyst synthesis and characterizations
catalyst was synthesized by adding sodium borohydride to the mixture containing Cu single-atom catalysts and cobalt nitrate. Cu single atoms dispersed on N -doped carbon (denoted as – ) were prepared via pyrolyzing Cu-doped ZIF- 8 at under Ar atmosphere (Supplementary Figs. 1 and 2). Figure 1a shows the highangle annular dark field scanning transmission electron microscopy (HAADF-STEM) image of -N-C. As displayed in the high resolution TEM (HRTEM) image and the corresponding selected area electron diffraction pattern (SAED), nanosheets were successfully deposited on the surface of (Supplementary Fig. 3). Figure 1 b shows the aberration-corrected HAADF-STEM image of . The lattice fringes with an interplanar spacing of 0.201 nm were ascribed to the (400) facet of . Besides, abundant Cu single atoms were observed around nanosheets. Based on energydispersive X-ray spectroscopy (EDS) elemental mapping, , and N
Fig. 1 | Structural characterizations. a HAADF-STEM image, aberrationcorrected HAADF-STEM image, and EDS elemental mappings of . d Raman spectra of , and . e -edge XANES
spectra and EXAFS spectra for foil, , and CuO . g Co -edge XANES spectra and EXAFS spectra for foil, CoO , and . i Co XPS spectra for and .
elements were uniformly distributed throughout the whole structure (Fig. 1c). The uniform distribution of Cu sites and species constituted the adjacent catalytic centers. The metal content of Cu and Co in were determined to be and , respectively, by inductively coupled plasma-optical emission spectroscopy analysis (ICP-OES). For comparison, nanosheets dispersed on N -doped carbon (denoted as ) were prepared with a similar synthetic procedure of -N-C except for the addition of Cu precursor (Supplementary Fig. 4). Figure 1d shows the Raman spectra for , and . All of the Raman spectra displayed two peaks located at 1356 and , assigned to the D band and G band of graphite carbon, respectively . The similar intensity ratios of D band to G band for the three samples indicated that the carbon support possessed similar degree of structural disorder (Supplementary Fig. 5). Compared with , three distinguishable peaks located at 482,527 , and were observed for both and , corresponding to , and vibration modes of crystals, respectively . The structure of graphite carbon supports was further confirmed by X-ray diffraction patterns (Supplementary Fig. 6). Figure 1e shows the edge X-ray absorption near edge structure (XANES) spectra of and . Obviously, the energy absorption edge profiles for both and were located between those of CuO and , elucidating that the valence state of Cu species in the two catalysts were between +1 to +2 . As shown in Fig. 1f, a dominant peak at was observed in the extended X -ray absorption fine structure (EXAFS) spectra of -edge for and C , which were attributed to the bond. The absence of bond in the two catalysts further confirmed the atomic dispersion of Cu species. Besides, the EXAFS fitting results indicate that the coordination numbers of shell in both and were approximately 4.0 (Supplementary Fig. 7 and Table 1). After the deposition of nanosheets, the coordination structure of Cu single atoms in was unchanged. Besides, the wavelet transformed EXAFS (WT-EXAFS) spectra of -N-C and also confirmed the bonding in the two catalysts (Supplementary Fig. 8). For the Co -edge XANES spectra, the edge energy for both and were similar to that for reference (Fig. 1g). Figure 1h shows that the Co-O coordination in and were approximate to that in , certifying the similar coordination structure of species in the two catalysts (Supplementary Fig. 9 and Table 2). Figure 1i shows the Co X-ray photoelectron spectroscopy (XPS) spectra. Specifically, the peaks at , and 780.8 eV in and corresponded to , and , respectively . The indiscernible shift of Co peaks demonstrated that as the support did not significantly affect the valence state of Co.
Catalytic performance toward electroreduction
The catalytic performance of the catalysts was investigated in a threeelectrode H-type cell toward electroreduction (Supplementary Fig. 10). The concentration of product was quantified by the indophenol blue method (Supplementary Fig. 11). To preliminarily explore the process of tandem catalysis, we conducted the linear sweep voltammetry (LSV) curves of , and with , respectively. As shown in Fig. 2a, the current density of in the presence of was higher than that of , suggesting that possessed higher activity toward electroreduction. Whereas, exhibited a larger current density relative to in (Fig. 2b). The superior activity of species toward electroreduction was further demonstrated by the higher Faradaic efficiency (FE) and yield rate for of in electroreduction relative to (Supplementary Fig. 12). Considering that is one of the vital
intermediates, the combination of and would couple the separate functions of Cu sites and species for the sequential reduction of and . As expected, displayed the highest current density among the three catalysts in the electrolyte of . Besides, the tremendous discrepancy of the LSV curves of in 1 M KOH with/without also implied the superior activity of toward electroreduction (Supplementary Fig. 13).
Figure 2c provides the partial current density for of , and at various applied potentials toward electroreduction. The of exceeded those of and . Especially, at -1.0 V vs reversible hydrogen electrode (RHE), the of reached , which was 2.2 times and 3.6 times as high as that of and , respectively. Moreover, the normalized based on double-layer capacitance ( ) for was the largest among the three catalysts, indicating the highest intrinsic activity for toward electroreduction (Fig. 2d and Supplementary Fig. 14). In addition, the FE for of was higher with respect to the other two counterparts at all applied potentials (Fig. 2e). Especially, achieved the maximum FE for of at -0.8 V vs RHE. Furthermore, at -1.0 V vs RHE, the yield rate of for reached up to , which exceeded all of the reported value for Cu-based catalysts (Fig. 2f and Supplementary Table 3). The yield rate of in the electrolyte after the electroreduction process was also determined by nuclear magnetic resonance (NMR) analysis, which was approximated to the results detected via the indophenol blue method (Supplementary Fig. 15 and Table 4). Other liquid and gaseous products including , and for were also measured (Supplementary Figs. 16-18). were the only main product after electroreduction (Supplementary Table 5). Besides, the FE for of with concentrations ranging from 10 mM to 500 mM all exceeded , indicating a wide tolerance range for the concentration of (Supplementary Fig. 19). The durability of was examined by 20 rounds of successive reactions. The negligible decay of the yield rate demonstrated the satisfactory durability of (Fig. 2g). The Raman and XAFS measurements for after the electrolysis indicated that the species and bonding were preserved (Supplementary Figs. 20 and 21). The stability of Cu single atoms in during the electrolysis was further explored by in situ EXAFS measurements, indicating that Cu atoms remained the atomically dispersed state in -N-C during the electroreduction (Supplementary Figs. 22 and 23).
To further clarify the synergy effect of on the conversion of , we conducted a series of control experiments. The catalytic performance of other metal oxides (such as , and ) dispersed on N -doped carbon toward electroreduction were all lower than that over , suggesting the inferior ability of these metal oxide to facilitate reduction (Supplementary Figs. 24 and 25). In addition, the loading amount of on was vital to the efficient conversion of the accumulated (Supplementary Fig. 26). Besides, the simply physical mixing of and could not sufficiently assure the spatial couple of the adjacent sites, thereby limiting the effective hydrogenation of into during electroreduction (Supplementary Fig. 27). We also exclude the possible ammonia contamination from the selfelectrolysis of , electrolyte, and carbon paper, respectively (Supplementary Fig. 28). Besides, the catalytic activity of N -doped carbon was much lower compared with that of C (Supplementary Fig. 29). The possible interference of Co single atoms on support could be considered insignificant to the catalytic performance of (Supplementary Figs. 30-33). The electroreduction of was also affected by the diffusion of
Fig. 2 | Catalytic performance. LSV curves of , and with (a) and (b) normalized based on FE for , and yield rate for of , and at different applied potentials with Yield rate for of at
-1.0 V vs RHE under 20 rounds of successive reactions. The solution resistance was determined to be in the electrolytes by potentiostatic electrochemical impedance spectroscopy.
reactants (Supplementary Fig. 34). Moreover, isotopic labeling measurements for was conducted with NMR analysis. Only typical doublet peaks attributed to were collected with as the N source whereas the triplet peaks of were detected with as the N source (Supplementary Fig. 35). These results indicated that the detected in the electrolyte originated from the electroreduction of .
Mechanistic study on electroreduction
To gain more insight into the catalytic process of electroreduction over -N-C, we conducted in situ electrochemical Fourier transform infrared spectroscopy (FTIR) and Raman spectroscopy to monitor the reaction process (Supplementary Figs. 36 and 37). Figure 3a displays the in situ FTIR spectra of at applied potentials from OCP to -1.0 V vs RHE. The negative peaks at were ascribed to the consumption of species . In addition, the emergence of positive peaks located at confirmed that was generated during the electroreduction . Besides, two peaks at 1541 and were detected, which were assigned to the vibration band of *NO and *NOH, respectively (Supplementary Fig. 38). Figure 3b shows the in situ Raman spectra of at all applied potentials. The peaks corresponding to , and vibration modes of remained unchanged, suggesting that the
species was stable during electroreduction. During the electroreduction, only the peak at was observed for at all applied potentials, assigned to the symmetric stretching vibration of (Fig. 3c). In the case of , apart from the signal of , a new peak at ascribed to the bending vibration of gradually appeared as the applied potential increased, indicating the accumulation of for during electroreduction (Fig. 3d). To further probe the variation of local concentration for near the surface of the catalysts, we designed a Raman cell that allows Raman laser to detect from the surface of catalysts to the electrolyte bulk. The Raman laser was designed to be incident from the back of catalysts to diminish the interference from the strong absorbance of in the electrolyte. As illustrated in Fig. 3e, the electrocatalysts were deposited on fluorine tin oxide-coated glass (FTO) as the working electrode (WE). The distance from the laser beam to electrode surface is controlled by the mechanical sample stage. Figure 3f, g display the in situ Raman spectra of and at -0.8 V vs RHE when the laser beam was positioned 0 to away from the surface of catalysts, respectively. With the increment of the distance between the focal plane of laser and the surface of catalysts, the signal intensity of graphite carbon for the catalysts gradually decreased. A noticeable peak at assigned to arose near the surface of . Whereas, the signal of for was negligible,
Fig. 3 | In situ characterizations. a In situ FTIR spectra for from OCP to -1.0 V vs RHE in . b In situ Raman spectra for from OCP to -1.0 V vs RHE in . In situ Raman spectra for (c) and (d) from OCP to -1.0 V vs RHE in . e Scheme of the designed Raman cell for detecting from the surface of catalysts to the electrolyte bulk. In situ Raman
spectra for (f) and (g) at -0.8 V vs RHE in with different distances ranging from 0 to The calculated concentration of for and with different distances ranging from 0 to .
which was independent of the distance. Furthermore, we determined the local concentration of near the surface of catalysts based on the integrated areas of and , taking the ratio of integrated areas for and solutions as a correction factor (Supplementary Fig. 39). As the laser beam was set further far away from the surface of catalysts into the electrolyte, the concentration of for gradually decreased from 0.74 to 0.29 M (Fig. 3h). This trend indicates that the generated at electrolyte interface diffused into the electrolyte due to the sluggish reduction of . Clearly, the concentration of for was much lower relative to , manifesting the facilitated reduction of with the favor of .
To further understand the synergetic role of and in the catalytic process, we calculated the rate constants for electroreduction ( ) and electroreduction ( ), respectively (Fig. 4a). The concentration of residual after the electroreduction process was quantified by UV-Vis spectrophotometry (Supplementary Fig. 40). Compared with , the larger value of suggests that was more favorable for the conversion of to , but the
smaller value shows the slower kinetics for reduction. Accordingly, excessive would be desorbed into the electrolyte for . Notably, the highest and of manifested the simultaneous acceleration of the conversion of to and to . Besides, the tafel slopes of , and in and imply that the combination of with facilitate the kinetics of and reduction during the catalytic process (Supplementary Fig. 41). Figure 4 b shows the adsorption capacities of , and for and , respectively. It is obvious that exhibited the largest for both and among the three catalysts. As a consequence, combining with was conducive to the conversion of both and .
The density functional theory (DFT) calculations were conducted to further interpret the reaction mechanism of electroreduction over catalysts. Based on the results of structural analysis, we adopted and (100) slabs as the models to calculate the Gibbs free energies ( ) for each step involved in electroreduction, respectively (Supplementary Fig. 42). After the
Fig. 4 | Mechanistic study on electroreduction. a Linearized pseudo firstorder kinetic profiles of , and in , respectively. Adsorption capacities of , and for . c Free energy diagram of electroreduction over and (100) slabs. * represents an adsorption site. d Structure models of key intermediates on and slabs. The gray, blue, red, yellow, and purple spheres represent , and Co atoms, respectively.
structure optimization, would be adsorbed on with O atom. As presented in Fig. 4c, the Gibbs free-energy changes ( ) for adsorption over is much lower than that over (100), indicating the stronger binding of over . Nevertheless, the of * desorption over is -1.31 eV , which could be more thermodynamically favorable than the reduction of to . In this regard, the desorption of * would give rise to the accumulation of over , which was consistent with the high yield for over in electroreduction (Supplementary Fig. 17). As for the conversion of would be adsorbed on through N atom whereas * could be connected with (100) through N and O atoms after the structure optimization, leading to the lower for adsorption over (100) (Fig. 4d). In addition, the relative high for adsorption over and
revealed the weak adsorption of *H, indicating that the occurrence of competitive evolution could be depressed (Supplementary Figs. 43 and 44). As a result, would regulate the adsorption configuration of and possesse an easier binding with , facilitating the reduction of to .
Discussion
In summary, we developed a highly efficient catalyst by coupling the separate functions of and for the sequential reduction of to and to . The obtained catalyst exhibited a superior yield rate for of , which exceeded all of the reported values for Cu-based catalysts. The mechanism investigations unveiled that the combination of regulated the adsorption configuration of and strengthened the
binding with , thus accelerating the electroreduction of to . This work offers a novel guideline for the construction of highly efficient tandem catalysts toward electroreduction.
Methods
Chemicals and materials
Zinc nitrate hexahydrate , 2-methyl imidazole (2-MeIM, 99.0%), copper(II) acetate monohydrate , ), cobalt nitrate hexahydrate , iron nitrate nonahydrate , nickel nitrate hexahydrate , copper nitrate trihydrate , methanol ( ), ethanol ( ), potassium nitrate ( ), potassium nitrite ( ), potassium hydroxide ( ), ammonium sulfate ( ), sodium hydroxide ( ), salicylic acid ( ), sodium hypochlorite solution ( NaClO , available chlorine of aqueous solution), trisodium citrate dihydrate ( ), sodium nitroferricyanide dihydrate ( ), hydrochloric acid ( ), sulfamic acid ( ), p -aminobenzenesulfonamide ( ), N -( Naphthyl) ethylenediamine dihydrochloride (98.0%), phosphoric acid ( ), and 1-propanesulfonic acid 3-(trimethylsilyl) sodium salt (DSS) were purchased from Sinopharm Chemical Reagent Co. Ltd. Glyoxylic acid solution ( ), dimethyl sulfoxide- (DMSO- atom D), ( 99.0 atom ), and ( 99.0 atom ) were purchased from Aladdin Chemistry Co., Ltd (Shanghai, China). Bipolar membrane (TRJBM) were purchased from Beijing Tingrun Membrane Technology Development Co., Ltd (Beijing, China). The deionized (DI) water was produced using a Millipore MilliQ grade, with a resistivity of . All of the chemicals were used without any further purification.
Instrumentations
TEM images were taken using a Hitachi HT7700 transmission electron microscope at an acceleration voltage of 100 kV . HAADF-STEM and the corresponding EDS elemental mapping were carried out on a Talos F200X field-emission transmission electron microscope operated at an accelerating voltage of 200 kV using Mo-based TEM grids. Aberrationcorrected HAADF-STEM images were carried out on Themis Z fieldemission transmission electron microscope operating at an accelerating voltage of 300 kV using Mo-based TEM grids. XRD patterns were collected using a Rikagu MiniFlex X-ray diffractometer with Cu radiation ( ). ICP-OES (Avio 220 MAX, PerkinElmer) analysis was employed to measure the concentration of metal species. XPS measurements were performed using a Kratos Axis supra+ diffractometer with Al-K radiation. The Raman spectra were conducted via LabRAM HR Evolution (Horiba) Raman system with a 532 nm excitation laser. The absorbance data was measured on a UV-vis spectrophotometer (Agilent Technologies, Cary 60). The in situ FTIR spectra were acquired by a Nicolet iS50 FTIR spectrometer with a builtin MCT detector.
Synthesis of
A mixture of and ( 0.28 mmol ) was dissolved in 80 mL of methanol, which was subsequently added into 80 mL of methanol containing 3.70 g of 2-MelM. Then the mixed solution was kept at for 12 h . The as-obtained precipitate (denoted as Cu-doped ZIF-8) was separated by centrifugation and washed subsequently with methanol for five times, and finally dried at under vacuum overnight. Next, the obtained Cucontaining derivative of ZIF- 8 was heated to with a heating rate of in a tube furnace and kept at under flowing Ar gas for 3 h . After the tube furnace was naturally cooled to room temperature, was obtained and directly used as the catalyst without further treatment. For comparison, Co single atoms anchored on N -doped carbon (denoted as ) and Co single atoms
anchored on (denoted as ) were obtained via pyrolyzing the Co -doped ZIF-8 and -doped ZIF-8, respectively. doped ZIF- 8 and -doped ZIF- 8 prepared with the similar procedure with that of except the and the mixture of and as the metal precursors, respectively.
Synthesis of and
160 mg of was dispersed in 20 mL of ethanol by sonication for 30 min . Afterwards, 5 mL of containing 45 mg of was added into the above solution, which was maintained in an icewater bath for 1 h with vigorous stirring. Then, 40 mL of freshly prepared with ice-cold was added dropwise into the above suspension, followed by further stirring for 1 h . The as-obtained precipitate was separated by filtration and washed subsequently with water for five times. Finally, was obtained by being dried at under vacuum overnight. was prepared as a comparison with the similar procedure with that of except for the addition of N -doped carbon instead of -doped carbon was prepared with a similar synthetic procedure with that of without the addition of . For comparison, other metal oxides including , and dispersed on N -doped carbon (denoted as , and , respectively) were prepared with the similar procedure with that of except for the addition of , and as the metal precursors, respectively (denoted as , and , respectively).
X-ray absorption fine structure (XAFS) measurements
The XAFS spectra at -edge and -edge were performed at 1W1B beamline of Beijing Synchrotron Radiation Facility and BL11B beamline of Shanghai Synchrotron Radiation Facility. The data were obtained in ambient conditions under fluorescence mode for -edge and transmission mode for Co -edge, respectively.
The ATHENA module and ARTEMIS codes in the IFEFFIT software packages were employed to extract the data and fitted the profiles . The -weighted EXAFS spectra were acquired by energy calibration and spectral normalization. For the EXAFS part, the Fourier transformed data in R space of -edge and -edge were analyzed by applying a hanning windows ( ) to differentiate the EXAFS oscillation from different coordination shells. Subsequently, we performed the least-squares curve parameter fitting to attain the structural parameters around central atoms. The fitted ranges of space were set at with range of . The four parameters including coordination number (CN), bond length (R), Debye-Waller factor ( ), and shift ( ) were fitted without anyone being fixed, constrained, or correlated.
The in situ -edge XAFS measurements were conducted were collected with a home-made XAFS cell. Typically, 8 mg of the catalysts and of Nafion were dispersed in 2 mL of ethanol by sonication for 1 h . Then the uniform ink was loaded onto carbon paper with an area of . The mass loading was calculated to be . The prepared catalysts, a electrode, and a Pt wire were used as the working electrode, reference electrode, and counter electrode, respectively. All electrochemical tests were measured in 1 M KOH electrolyte with and controlled by a CHI 1140 C electrochemical workstation.
Preparation of the working electrodes
8 mg of the catalysts were dispersed in 2 mL of ethanol by sonication for 1 h . Then of Nafion solution was added to the mixture and sonicated for 30 min to obtain a uniform ink. Finally, the uniform ink was loaded onto carbon paper with an area of . The mass loading was calculated to be . The area of working electrodes used in the electrochemical measurements was .
Electrochemical measurements
The electrochemical measurements were carried out in an H-cell system which was separated by a bipolar membrane with a CHI 1140 C electrochemical workstation (Chenhua, Shanghai). electrode and graphite rod were used as the reference electrode and counter electrode, respectively. For electroreduction, 1 M KOH containing solution ( 60 mL ) was evenly distributed to the cathode and anode compartments. The pH value of the electrolyte was determined to be 14 by a FiveEasy Plus pH Meter (METTLER TOLEDO). The applied potentials were measured against the reference electrode with compensation and converted to the RHE reference scale by . The solution resistance was determined to be ohm in the electrolytes by potentiostatic electrochemical impedance spectroscopy at frequencies ranging from 10 Hz to 100 kHz , which was conducted in a standard three-electrode system at ambient conditions. Before the electroreduction test, CV curves were performed until the polarization curves achieved steady-state ones with a scan rate of . Before the electrolysis, Ar gas was delivered into the cathodic compartment at a rate of to remove dissolved . The LSVs of the catalysts were recorded at a scan rate of in 1 M KOH containing . The controlled potential electrolysis was performed at applied potentials for electroreduction was conducted with the same conditions except that the solution of 1 M KOH containing was used as the electrolyte. An absorption cell containing 30 mL of 1 M HCl was set to effectively absorb the possible escaped from the cathode cell. After the electrolysis at each applied potential, the concentration of in the absorption cell was lower than . In this case, the volatilization of from the electrolytes could be negligible. Cyclic voltammetric measurements were conducted in a non-faradaic potential window with various scan rates from 50 to was calculated by plotting the ( ) at the middle of the corresponding potential window against scan rates. The and were the anodic and cathodic current densities, respectively. The slope was twice of .
The calculation method for FE
The FE for the product ( and ) was calculated at a given potential as follows:
: the measured concentration of product ( ), : the volume of the electrolyte ( mL ), : the number of electrons transferred for the product, which is 8 for and 2 for ,
F: Faraday constant, , : total electric charge (C),
M: the relative molecular mass, which is for and for .
The calculation method for the yield rate of product
The yield rate of product was calculated at a given potential as follows:
: the yield rate ( ), : the measured concentration of , : the volume of the electrolyte ( mL ), : the area of the catalyst ( ), : the reduction reaction time ( min ).
Determination of ion concentration
Determination of concentration with indophenol blue method . After the electroreduction process, a certain amount of electrolyte was taken out from the electrolytic cell and diluted to the detection range. Then, 2 mL of 1 M NaOH solution containing salicylic acid ( ) and sodium citrate ( ) were added into the aforementioned solution, followed by the addition of 1 mL of 0.05 M NaClO and 0.2 mL of . After standing in darkness for 2 h , the absorption spectra were measured using a UV-vis spectrophotometer. The concentration of indophenol blue was determined using absorbance at the wavelength of 650 nm . The concentration-absorbance curve was calibrated using standard solution with a series of concentrations.
Determination of concentration with NMR method. After electroreduction, a certain amount of electrolyte was taken out for further quantification by NMR (Bruker AVANCE AV III 400). All analyses were performed with 128 -time scans. The concentrationintegral area curve was calibrated using a standard solution. Typically, was dissolved in 20 mL of 1 M KOH electrolyte as a series of standard solutions with different concentrations. Subsequently, 0.1 mL of dimethyl sulfoxide- (DMSO- ), 0.1 mL of 6 mM 1-propanesulfonic acid 3-(trimethylsilyl) sodium salt (DSS) solution, and 0.08 mL of 6 M HCl to adjust the pH value were added into 0.32 mL of standard solutions with different concentrations. The signal appeared at , and 6.97 ppm were attributed to . The integral areas of the signal of were used to determine the concentration of compared with the asknown DSS reference.
Determination of concentration . A certain amount of electrolyte was diluted to the detection range of . Then, 0.1 mL of 1 M HCl and 0.01 mL of sulfamic acid solution were mixed with the diluted electrolyte, followed by shaking for 10 min . Using a UV-vis spectrophotometer, the absorption spectra were collected, obtaining the absorption intensities at a wavelength of 220 and 275 nm . Finally, the calculated absorbance ( ) was acquired. The concentration-absorbance curve was calibrated using standard solutions with a series of concentrations.
Determination of concentration. 4 g of p-aminobenzenesulfonamide, 0.2 g of N -(1-Naphthyl) ethylenediamine dihydrochloride, and 10 mL of phosphoric acid were mixed with 50 mL of water as the color reagent. A certain amount of electrolyte was taken out from the electrolytic cell and diluted to the detection range. 1 mL of was added to the 4 mL of diluted post-electrolysis electrolytes to adjust the pH , followed by the addition of 0.1 mL of color reagent. After standing for 20 min , the absorption spectra were measured using a UV-vis spectrophotometer. The absorption intensity at a wavelength of 540 nm was recorded. The concentrationabsorbance curve was calibrated using standard solution with a series of concentrations.
Determination of other liquid and gaseous product
The amount of was determined by after was captured by excess amount of through oximation process. Specifically, 0.4 mL of the electrolyte after the electroreduction was mixed with of solution, followed by the addition of 0.1 mL of DMSO- and 0.1 mL of 6 mM DSS solution. The integral area of the signal appeared at 7.46 ppm were used to determine the concentration of compared with the as-known DSS reference. In this work, the amount of was below the detection limit for both and reduction over .
Nitrogen oxides including , and have been detected by an infrared gas analyzer (THA100S). and have been detected by an on-line gas chromatograph (GC-2014) equipped with a flame ionization detector and a thermal conductivity detector.
The FE for gaseous products were calculated by the following equation:
: the measured mole fraction of product, : the total volume of the gas (L), : the number of electrons transferred for the product, which is 3 for for for and 10 for ,
F: Faraday constant, , : total electric charge ( C ), : the molar volume of the gas, .
Isotope labeling experiments
The isotopic labeling experiment used with enrichment of as the feeding N-source to clarify the source of ammonia. 1 M KOH was used as the electrolyte and with a concentration of 1 M was added into the cathode compartment as the reactant. After the electrolysis, 0.1 mL of DMSO- and 0.1 mL of 6 mM DSS solution were added into 0.4 mL of the electrolyte, followed by adding 0.05 mL of to adjust the pH of the solutions. Then the obtained was identified on a Varian 400 MHz NMR spectrometer (Bruker AVANCE AV III 400).
Kinetic evaluation
The electrolysis at -1.0 V vs RHE were conducted for different time to acquire the rate constant in 1 M KOH containing or . The reaction constant ( for reduction and for reduction) was calculated by plotting the concentration of or against the time of reaction, supposing that the concentrations of or declined exponentially as per first-order rate.
: initial concentration of or , : the concentration of or at time , : the time of reaction (min).
Adsorption experiments
To determine the adsorption capacities of , and of catalysts were added to each 25 mL of or solutions with the initial concentration of 1 M under stirring for 2 h , respectively. The solutions were separated by filtration using the microporous membrane filter. For high concentration of or , the solution was diluted before absorbance measurements. The adsorption capacity was calculated using the following equation:
: the adsorption capacity ( ), : the initial concentration of or , : the measured concentration of or after the adsorption ( ), : the volume of the electrolyte ( mL ), : the mass of the catalyst ( g ).
In situ FTIR measurements
Using a Nicolet iS50 FTIR spectrometer (Thermo Scientific) with a built-in MCT detector, we obtained the in situ electrochemical FTIR spectra. Typically, 2 mg of catalysts and of Nafion dispersed in 2 mL of ethanol were sonicated for 1 h . Then the mixture was loaded onto the Au-coated Si prism to completely cover the Au film. The prepared prism was used as the working electrode after being dried naturally. The reference electrode and counter electrode was a
AgCl electrode and a Pt wire, respectively. The photograph of the in situ FTIR electrochemical cell was shown in Supplementary Fig. 36. All electrochemical tests were measured in 1 M KOH electrolyte with and controlled by a CHI 1140 C electrochemical workstation. All experiments were conducted at room temperature. The background spectra of the working electrode were obtained at an open-circuit potential before the electrochemical tests. All of the spectra were collected in absorbance by averaging 32 scans at a resolution of .
In situ Raman measurements
In situ Raman was carried out using Lab RAM HR Evolution (Horiba) equipped with a microscope objective. The excitation wavelength was 532 nm with intensity. The photograph of electrochemical cell for in situ Raman measurement was shown in Supplementary Fig. 37. Typically, 2 mg of catalysts and of Nafion were dispersed in 2 mL of ethanol by sonication for 1 h . Then the electrocatalysts were deposited on fluorine tin oxide-coated glass as the working electrode. electrode, and a Pt wire were used as the reference electrode, and counter electrode, respectively. All electrochemical tests were measured in 1 M KOH electrolyte with and controlled by a CHI 1140 C electrochemical workstation. All experiments were conducted at room temperature. Each spectrum was collected by integration twice, 60 s per integration. To determine the local concentration of near the surface of catalysts, we employed an internal standard method during the in situ Raman measurements.
DFT calculations
DFT calculations were performed using the Vienna Ab-Initio Simulation Package (VASP) code at the GGA level within the PAW-PBE formalism . DFT-D3 method with Becke-Jonson damping is performed for the van der Waals correction. The three-layer (100) slab model was adopted with a vacuum of . The total energy calculations were performed using a grid and a plane wave cut-off energy of 400 eV . Atoms in the bottom two layers were fixed. A U value of 3.5 eV was applied to the states of Co to describe the strong onsite Coulomb interactions due to the localization of the Co states . For the model of (no atoms were fixed), the total energy calculations were performed using a grid and a plane wave cut-off energy of 400 eV . All atoms, which were not fixed, including adsorbates were allowed to relax until the force on each ion was smaller than Å.
We calculated the Gibbs free energy ( ) for each species as follows:
where , and represent the DFT-optimized total energy, zero point energy (ZPE), and entropy contribution, respectively ( T is the temperature, 298.15 K ). It is assumed that for all the adsorbed species.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. The source data underlying Figs. 1-4 and Supplementary Figs. 1-44 are provided as a Source Data file. Source data are provided with this paper.
References
Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209-2244, (2009).
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
Wang, Y., Wang, C., Li, M., Yu, Y. & Zhang, B. Nitrate electroreduction: Mechanism insight, in situ characterization, performance evaluation, and challenges. Chem. Soc. Rev. 50, 6720-6733 (2021).
Duca, M. & Koper, M. T. M. Powering denitrification: The perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726-9742 (2012).
Xu, H., Ma, Y., Chen, J., Zhang, W. X. & Yang, J. Electrocatalytic reduction of nitrate – a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710-2758 (2022).
Fan, K. et al. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 13, 7958 (2022).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036-7046 (2020).
Wang, Y. et al. Structurally disordered nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 61, e202202604 (2022).
Deng, X., Yang, Y., Wang, L., Fu, X. Z. & Luo, J. L. Metallic Co nanoarray catalyzes selective production from electrochemical nitrate reduction at current densities exceeding . Adv. Sci. 8, 2004523 (2021).
Zhang, X. et al. Recent advances in non-noble metal electrocatalysts for nitrate reduction. Chem. Eng. J. 403, 126269 (2021).
Wang, X. et al. Free-standing membrane incorporating single-atom catalysts for ultrafast electroreduction of low-concentration nitrate. Proc. Natl Acad. Sci. USA 120, e2217703120 (2023).
Zhang, G. et al. Tandem electrocatalytic nitrate reduction to ammonia on Mbenes. Angew. Chem. Int. Ed. 62, e202300054 (2023).
Fu, Y. F. et al. Enhancing electrochemical nitrate reduction to ammonia over Cu nanosheets via facet tandem catalysis. Angew. Chem. Int. Ed. 62, e202303327 (2023).
Fang, J. Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
Zhu, T. et al. Single-atom Cu catalysts for enhanced electrocatalytic nitrate reduction with significant alleviation of nitrite production. Small 16, e2004526 (2020).
Yang, J. et al. Potential-driven restructuring of cu single atoms to nanoparticles for boosting the electrochemical reduction of nitrate to ammonia. J. Am. Chem. Soc. 144, 12062-12071 (2022).
Li, P. et al. Pulsed nitrate-to-ammonia electroreduction facilitated by tandem catalysis of nitrite intermediates. J. Am. Chem. Soc. 145, 6471-6479 (2023).
Cheng, X. F. et al. Coordination symmetry breaking of single-atom catalysts for robust and efficient nitrate electroreduction to ammonia. Adv. Mater. 34, 2205767 (2022).
He, W. et al. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 13, 1129 (2022).
Wang, W., Chen, J. & Tse, E. C. M. Synergy between Cu and Co in a layered double hydroxide enables close to nitrate-toammonia selectivity. J. Am. Chem. Soc. 145, 26678-26687 (2023).
Liu, Y. et al. A highly efficient metal-free electrocatalyst of f-doped porous carbon toward electroreduction. Adv. Mater. 32, 1907690 (2020).
Li, R. et al. Insights into correlation among surface-structure-activity of cobalt-derived pre-catalyst for oxygen evolution reaction. Adv. Sci. 7, 1902830 (2020).
Wang, J. et al. Electrocatalytic reduction of nitrate to ammonia on low-cost ultrathin nanosheets. ACS Catal. 11, 15135-15140 (2021).
Gao, Q. et al. Synthesis of core/shell nanocrystals with ordered intermetallic single-atom alloy layers for nitrate electroreduction to ammonia. Nat. Synth. 2, 624-634 (2023).
Song, Z. et al. Efficient electroreduction of nitrate into ammonia at ultralow concentrations via an enrichment effect. Adv. Mater. 34, 2204306 (2022).
Gao, W. et al. Alloying of Cu with Ru enabling the relay catalysis for reduction of nitrate to ammonia. Adv. Mater. 35, e2202952 (2023).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on coppernickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702-5708 (2020).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper-supported rhodium cluster and single-atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759-767 (2022).
Wang, Y. T., Zhou, W., Jia, R. R., Yu, Y. F. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350-5354 (2020).
Hao, R. et al. Pollution to solution: A universal electrocatalyst for reduction of all -based species to . Chem. Catal. 2, 622-638 (2022).
Newville, M. IFEFFIT: Interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 8, 322-324 (2001).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537-541, (2005).
Meng, Q. et al. Sustainable production of benzene from lignin. Nat. Commun. 12, 4534 (2021).
Song, Z. et al. Promoting electroreduction into over porous carbon by introducing oxygen-containing groups. Chem. Eng. J. 434, 134636 (2022).
Bai, Y. et al. FCF-LDH/BIVO4 with synergistic effect of physical enrichment and chemical adsorption for efficient reduction of nitrate. Green. Energy Environ. https://doi.org/10.1016/j.gee.2023. 05.011, (2023).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15-50 (1996).
Zasada, F. et al. Periodic DFT and HR-STEM studies of surface structure and morphology of cobalt spinel nanocrystals. Retrieving 3d shapes from 2d images. J. Phy. Chem. C. 115, 6423-6432 (2011).
Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDBO450401), National Key Research and Development Program of China (2021YFA1500500, 2019YFA0405600), NSFC (22209161, 22302184, 22322901, 22221003, 22250007, and 22361162655), National Science Fund for Distinguished Young Scholars (21925204), CAS project for young scientists in basic research (YSBR-022, YSBR-051), China Postdoctoral Program for Innovative Talents (BX20200324), Collaborative Innovation Program of Hefei Science Center, CAS (2022HSC-CIP004), International Partnership Program of Chinese Academy of Sciences ( ), the Joint Fund of the Yulin University and the Dalian National Laboratory for Clean Energy (YLU-DNL Fund 2022012), Fundamental Research Funds for the Central Universities, the Anhui Natural Science Foundation for Young Scholars (2208085QB41), and the Fellowship of China Postdoctoral Science Foundation (2021M693058). J.Z. acknowledges support from the Tencent Foundation through the XPLORER PRIZE. This work was partially carried out at the Instruments Center for Physical
Science, University of Science and Technology of China. This work was also partially carried out at the USTC Center for Micro and Nanoscale Research and Fabrication.
Author contributions
Z.G. and J.Zeng. supervised this project. Y.L. performed most of the experiments and analyzed the experimental data. J.W. conducted the in situ Raman measurements. Z.Y. and J.Zhao carried out DFT calculations and analyzed the computational data. L.Z. conducted the XAFS measurements and analyzed the results. Z.S., Y.Z., J.C., and J.M. provided help in materials synthesis and characterizations. Y.L., J.W., Z.G., and J.Zeng. wrote the manuscript. All authors discussed the results and assisted during manuscript preparation.
Correspondence and requests for materials should be addressed to Zhigang Geng or Jie Zeng.
Peer review information Nature Communications thanks Aiqin Wang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Reprints and permissions information is available at http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei 230026 Anhui, PR China. Institute of High Energy Physics, Chinese Academy of Sciences, 100049 Beijing, PR China. Department of Physics, University of Science and Technology of China, Hefei 230026 Anhui, PR China. CAS Key Laboratory of Strongly-Coupled Quantum Matter Physics, University of Science and Technology of China, Hefei 230026 Anhui, PR China. Key Laboratory of Surface and Interface Chemistry and Energy Catalysis of Anhui Higher Education Institutes, Department of Chemical Physics, University of Science and Technology of China, Hefei 230026 Anhui, PR China. School of Chemistry & Chemical Engineering, Anhui University of Technology, Ma’anshan 243002 Anhui, PR China. These authors contributed equally: Yan Liu, Jie Wei, Zhengwu Yang.
-e-mail: gengzg@ustc.edu.cn; zengj@ustc.edu.cn