تُ triggered الالتهاب بواسطة إهانة أو إصابة لخلايا الجسم وهو خطوة صحية ومنتجة نحو الشفاء. ومع ذلك، فإن الالتهاب المستمر هو استجابة غير منظمة ويمكن أن يؤدي إلى مجموعة من حالات المرض غير المرغوب فيها. من المعروف أن موت الخلايا عن طريق النخر يُ initiates الالتهاب، بينما يحد الموت المبرمج للخلايا عن طريق الاستماتة من الاستجابة الالتهابية. نقترح هنا أن هناك طيفًا من حالات الحياة، والشيخوخة، والموت، والموت للخلايا التي لها تأثيرات مختلفة على الأمراض الالتهابية.
الجزء 1. دورة حياة الالتهاب استجابة شفاء الجروح
يمكن فهم الكثير من إصابات وأمراض الثدييات من خلال عدسة الالتهاب وشفاء الجروح. استجابة شفاء الجروح للإصابة في البشر معروفة جيدًا من حيث الخلايا والجزيئات المعنية، والمدة الزمنية للأحداث المحددة، والعواقب السلبية عندما يتم حظر مثل هذه المكونات. يتم تقسيم شفاء الجروح الصحي إلى أربع مراحل: التجلط، الالتهاب، التكاثر، وإعادة التشكيل. في هذا القسم، سنركز على الالتهاب. يمكن العثور على وصف تفصيلي للمراحل الأخرى في المراجعات المنشورة سابقًا.المنظمون الخلويون الحرجون للالتهاب هم خلايا من جهاز المناعة الفطري، مثل العدلات والبلاعم. بعد الإصابة، في محاولة لإعادة تأسيس التوازن، يتم استقطاب العدلات إلى مواقع الإصابة بعد تحلل الصفائح الدموية لإطلاق السيتوكينات وعوامل النمو. ويساعد في هذا الاستقطاب الخلايا البدينة، التي، بالإضافة إلى إطلاقها للببتيدات المضادة للميكروبات، تطلق جزيئات لتفكيك المصفوفة خارج الخلوية وزيادة نفاذية الأوعية الدموية.تساعد العدلات في تطهير الجرح من خلال البلعمة للبكتيريا والأجسام الغريبة وإفراز مستويات عالية من جزيئات الإشارة المؤيدة للالتهاب لتسريع تجنيد وتنشيط المزيد من خلايا المناعة.تساهم المونوسيتات المجندة والماكروفاجات المقيمة في الأنسجة في البيئة الالتهابية الغنية بالسيتوكينات وتدعم الغالبية العظمى من إزالة الحطام عبر البلعمة.تعتبر جزيئات الإشارة وعرض المستضدات بواسطة هذه الخلايا، بالإضافة إلى الخلايا الشجرية، ضرورية أيضًا لإطلاق استجابة مناعية تكيفية ضد مسببات الأمراض.ثم تخضع العدلات لعملية الموت المبرمج وتتم إزالتها في غضون يوم تقريبًا من وصولها.تعتبر الإيفيروسيتوز، أو إزالة الخلايا المبرمجة للموت، عملية فسيولوجية منظمة بدقة، كما يتضح من أكثر منتتم إزالة الخلايا المبرمجة للموت يوميًا في غياب الإصابة أو المرض بواسطة الجهاز المناعي دون استجابة مناعية التهابية.عندما يتم ابتلاع العدلات بواسطة البلعميات بعد الإصابة، توجه البلعميات الانتقال من الالتهاب نحو الإصلاح والتجديد.هذا الانتقال الظاهري ضروري لحل الالتهاب للحفاظ على وظيفة الأنسجة أو استعادتها..
بينما يعتبر تسلل البلعميات والتفعيل المؤيد للالتهاب أمرًا حيويًا لإصلاح الأنسجة والحفاظ على التوازن الداخليوبالمثل، فإن استقطاب البلعميات نحو نمط ظاهري مضاد للالتهابات ومؤيد للتجديد مطلوب أيضًا لتمكين الشفاء الكامل للجروح.استجابة البلعمة للخلايا الميتة يتم توجيهها بواسطة عدة مستقبلات على البلعميات يمكنها الارتباط بمجموعة متنوعة من العلامات السطحية للخلايا الميتة لتسهيل التعرف، والامتصاص، والإشارات. تعرض الفوسفوليبيد الفوسفاتيديل سيرين (PS) على السطح الخارجي لغشاء الخلية هو الإشارة الأكثر تمييزًا للخلايا الميتة.في معظم الخلايا الحية، يتم حصر الفوسفوتيديل سيرين (PS) داخل الخلية بواسطة بروتين الفليباز، وهو بروتين ناقل للدهون ينقل بعض الدهون من الطبقة الخارجية إلى الطبقة الداخلية، مما ينتج عنه طبقات غير متساوية.عندما تخضع تلك الخلية لعملية الموت المبرمج، تفقد تباين غشائها الخلوي، مما يكشف عن الفوسفاتيديل سيرين على غشائها الخارجي. إن تعرض الفوسفاتيديل سيرين يعمل كإشارة “كلوني” للبلعميات، بما في ذلك البلعميات الكبيرة، لابتلاع الخلية المحتضرة.. هذا الانغماس يبدأ تحولًا في استقطاب البلعميات نحو نمط ظاهري مؤيد للحل، مما يقلل الالتهاب، ويوقف الأضرار الإضافية للأنسجة، ويبدأ تجديد الأنسجة .
الشفاء الطبيعي مقابل الالتهاب المزمن
تظهر البلعميات البلاستيكية الكبيرة مرونة كبيرة لتعزيز كل من الالتهاب وحل الالتهاب بناءً على الإشارات خارج الخلوية.. في استجابة شفاء صحية، يمكن أن تؤدي هذه الإشارات إلى إنتاج السيتوكينات المثبطة للمناعة، بما في ذلك TGF- و IL-10 لاستعادة التوازن بعد المرحلة الالتهابية الأولية؛ ومع ذلك، دون وجود إشارة للاستقطاب، أو مع استمرار التحفيز بواسطة المستضد، تستمر الالتهابات إلى أجل غير مسمى في الأمراض الالتهابية المزمنة.في الحالات الالتهابية المزمنة أو غير المنضبطة، تستمر الأعداد المرتفعة من العدلات، وحيدات النواة، والبلاعم.تستمر العدلات بشكل غير طبيعي، مطلقة كميات مفرطة من البروتيازات، وأنواع الأكسجين التفاعلية (ROS)، والفخاخ خارج الخلوية للعدلات (NETs)، مما يتسبب في تلف الأنسجة واستمرار الالتهاب، بينما تميل البلعميات نحو حالة التهابية (M1)، مما يؤدي إلى فشلها في الانتقال إلى نمط إصلاحي (M2). هذا الخلل يعزز الالتهاب. يؤخر الشفاء في الحالات المزمنة مثل الجروح السكرية غير الشافية. يمكن أن يرتبط استمرار البلعميات في مواقع الالتهاب بشدة المرض وتطوره، كما هو الحال في التهاب المفاصل الروماتويدي.تنتج خلايا المناعة المسببة للالتهابات بشكل مفرط الميتالوبروتينازات (MMPs) ، والجذور الحرة للأكسجين (ROS) ، والسيتوكينات الالتهابية ، مما يؤدي إلى زيادة تلف الأنسجة. يحدث استقطاب إضافي لمكروفاجات محدثة للالتهابات مع زيادة إفراز السيتوكينات الالتهابية ، بما في ذلك TNF-IL-1و IL-6يمكن رؤية نتائج مماثلة من زيادة تسلل وتنشيط البلعميات عبر مجموعة متنوعة من الأمراض الالتهابية وأنظمة الأعضاء.على النقيض من ذلك، يرتبط الشفاء الطبيعي بالموت المبرمج وإزالة العدلات، مما يبدأ سلسلة من التفاعلات المضادة للالتهابات في الأنسجة..
الجزء 2. موت الخلايا
التوقيت الدقيق، ونمط، ومدى موت العدلات وإزالتها من بيئة الجرح يحدد الالتهاب والشفاء اللاحقين.كلا المرحلتين الالتهابية الأولية والمرحلة التالية المضادة للالتهابات والمحفزة للتجديد ضرورية للشفاء الكامل للجروح. هنا نصف أوضاع مختلفة لموت العدلات والتأثيرات الناتجة على الالتهاب، مع التركيز على التأثير على البلعميات. (الشكل 1).
نخر وخلايا ميتة
للحفاظ على التوازن الداخلي الطبيعي، تتطلب فترات نصف الحياة القصيرة للخلايا المتعادلة عادةً برنامجًا منظمًا جيدًا من الموت الخلوي المبرمج الذي يبدأ من خلال مسارات داخلية أو خارجية. تبدأ السلسلة الداخلية من خلال إطلاق السيتوكروم C من الميتوكوندريا إلى الفضاء السيتوبلازمي، مما يؤدي إلى تنشيط الكاسبيز-3. في المقابل، تبدأ السلسلة الخارجية من خلال تفاعل مستقبلات موت الخلايا على الأسطح، مما يؤدي إلى تنشيط الكاسبيز-8 / الكاسبيز-10 والتقارب على الكاسبيز-بينما يعتبر أكسيد النيتريك مهمًا في استجابة شفاء الجروح كوسيط التهابي، فإنه يلعب أيضًا دورًا في موت الخلايا المبرمج للعدلات. تتضمن كل من مسارات الموت الخلوي الداخلية والخارجية زيادة تنظيم إنزيم أكسيد النيتريك الصناعي (iNOS) وإنتاج أكسيد النيتريك الذي ينشط الكاسبيز-3 و-8، مما يؤدي إلى موت الخلايا.آلية الموت الخلوي تؤدي إلى سلسلة من الأحداث النشطة التي تعدل الالتهاب في المراحل اللاحقة. بالإضافة إلى تعرض الفوسفاتيديل سيرين على أسطح الخلايا الميتة، يتم التعبير عن مجموعة من جزيئات الإشارة “كلني” على أسطح الخلايا مع انخفاض في تنظيم إشارات “لا تأكلني”، مثل CD47، مما يؤدي إلى الإيفوريتوسيس وإزالة الخلايا بواسطة البلعميات.
تُعتبر الخلايا المتعادلة أكثر خلايا المناعة عددًا في جسم الإنسان، حيث يتم إنتاج أكثر من مليار خلية يوميًا لكل كيلوغرام. وهذا، بالإضافة إلى عمرها الافتراضي الذي لا يتجاوز بضعة أيام، يعني أن الغالبية العظمى من هذه الخلايا تمر بعملية الاستماتة يوميًا، بشكل أساسي في نخاع العظام والكبد والطحال. ونتيجة لهذه العمليات هي إنتاج الإنترلوكين-23 (IL-23)، وانخفاض مستويات عامل تحفيز مستعمرات الكريات البيضاء (G-CSF)، وانخفاض في إنتاج الخلايا المتعادلة.. تشكل هذه العملية بعد ذلك حلقة تنظيم ذاتي لإنتاج العدلات، وتحريكها، وانخفاضها. لذلك، يُعتبر الموت المبرمج (الموت الخلوي المبرمج) نمطًا غير التهابي من موت الخلايا، على عكس آليات موت العدلات الأخرى.
ومع ذلك، هناك أنماط من الاستماتة (apoptosis) تكون مناعية وتؤدي إلى الإفراج النشط عن أنماط جزيئية مرتبطة بالضرر (DAMPs، الشكل 1). تعمل DAMPs التي يتم إفرازها من الخلايا الميتة والميتة على الإشارة إلى الوسائط الالتهابية اللاحقة، بما في ذلك البلعميات (macrophages) للتجنيد إلى مواقع الإصابة وتعزيز سلسلة الالتهاب.تشمل DAMPs الكلاسيكية ATP، والبروتينات عالية الحركة المجموعة 1 (HMGB1)، والبروتينات المرتبطة بـ RNA القابلة للتبريد خارج الخلية (eCIRP)، والهستونات، وبروتينات الصدمة الحرارية (HSPs)، وRNA خارج الخلية (exRNAs)، وDNA خالي من الخلايا (cfDNA). كما يتم إفراز DAMPs بشكل سلبي عبر نخر الخلايا، والذي يتمثل في فقدان سلامة غشاء الخلية. تختلف التعبئة والهوية لـ DAMPs المفرزة من آليات الموت المبرمج مقارنة بالنخر. يمكن أن تفرز الكريات البيضاء المت apoptotic DAMPs عبر الليزوزومات، وعبر إفراز الحويصلات الإكسوسومية والإكتوسومية، والأجسام الميتة، وعبر الفخاخ خارج الخلوية..
نظرًا لأن النخر هو عملية سلبية بشكل عام، فإن إطلاق العوامل النشطة بيولوجيًا يتم عبر تحلل الغشاء (الشكل 1). وبالتالي، توجد اختلافات في تلك الجزيئات. على سبيل المثال، يتم إطلاق cfDNA من الخلايا النخرية بشكل عام لفترة أطول بكثير من cfDNA المجزأ الذي يتم إطلاقه من الخلايا المبرمجة للموت.. يمكن تعديل DAMPs أخرى، مثل HMGB1، خلال بعض أنماط موت الخلايا، بينما بعد النخر، لا يمكن ذلك.تُطلق بعض DAMPs في مراحل مختلفة؛ على وجه الخصوص، يتم إطلاق ATP في وقت مبكر خلال عملية الموت الخلوي المبرمج، بينما يتم إطلاق HMGB1 في وقت لاحق عندما تتحرك الخلايا نحو النخر الثانوي، أو خلال النخر الأولي.يمكن أن يكون تمزق الغشاء أيضًا عملية منظمة من قبل الخلية، تُسمى
الشكل 1 | مخطط للمنتجات الخلوية المميزة الناتجة عن أوضاع مختلفة من موت الخلايا. (PS الفسفوليبيد سيرين؛ CASP3 كاسبيز-3؛ TLR4 مستقبل شبيه التول 4؛ TNFR1 مستقبل عامل نخر الورم 1؛ GSDMD غازدرمين D؛
HMGB1 مجموعة البروتينات عالية الحركة الصندوق 1؛ eCIRP بروتين ربط RNA القابل للتحفيز بالبرودة خارج الخلية؛ cfDNA الحمض النووي الخالي من الخلايا؛ exRNA RNAs خارج الخلية؛ Hsp بروتين الصدمة الحرارية) النخر الخلوي، الذي يحدث بعد التفاعلات المعتمدة على المستقبلات. وهذا يؤدي إلى الإفراج غير المنضبط عن DAMPs السيتوسولية (الشكل 1).
أشكال أخرى من موت الخلايا المبرمج
تم تحديد عدد من أنماط موت العدلات الالتهابية الأخرى التي تعتبر مهمة بشكل خاص للعدلات. الموت النخرى هو نمط شديد الالتهاب من موت الخلايا يتم تحفيزه داخل الخلايا عبر بروتين 3 المحتوي على مجال ربط النوكليوتيدات، وتكرار غني بالليوسين، ومجال بيرين (NLRP3) أو الليبوساكاريد (LPS) (الشكل 1).تفعيل الكاسبازات ومشاركة غازدرمين D هما علامتان مميزتان لهذه العملية مما يؤدي إلى إطلاق DAMP. يعزز غازدرمين D تشكيل الثقوب في أغشية العدلات مما يسبب IL-سيتم إطلاقها من خلال المسام الناتجة جنبًا إلى جنب مع DAMPs الأخرى. الفيروبتوز هو نمط آخر من موت الخلايا مصحوبًا بفقدان سلامة غشاء الخلية (الشكل 1)تراكم الحديد داخل الخلايا يزيد من الإجهاد التأكسدي في الخلية، مما يؤدي إلى زيادة في تأكسد الدهون واضطراب الغشاء الدهني. تشمل المواد المسببة للالتهاب المهمة التي يتم إطلاقها من خلال هذه العملية HMGB1 و cfDNA. أخيرًا، آلية موت الخلايا الالتهابية المتعمدة هي تلك الخاصة بالشبكات خارج الخلوية للعدلات (NETosis، الشكل 1).يمكن ربط إنتاج NET بالخلايا الحية أو الميتة ويتضمن إطلاق هياكل الكروماتين غير المنسوجة، الشبيهة بالشبكة، التي تحتوي على الحمض النووي، والهستونات، والببتيدات المضادة للميكروبات إلى الفضاء خارج الخلوي. يتم تحفيز هذه العملية بواسطة منتجات ميكروبية والصفائح الدموية المنشطة، والهياكل الناتجة لها تأثير مضاد للميكروبات وتسبب التهابًا شديدًا..
موت الخلايا في الأمراض الالتهابية
هناك أدلة في مختلف أمراض الباثولوجيا أن موت العدلات يتعرض للاضطراب ويمكن أن يؤدي إلى زيادة تلف الأنسجة. عندما يتم تقييد الاستماتة بسبب المرض، يمكن أن يؤدي بقاء العدلات الناتج إلى زيادة تلف الأنسجة. على سبيل المثال، في مرض الانسداد الرئوي المزمن والتليف الكيسي، يرتبط تنشيط العدلات المستمر بمستويات عالية من الالتهاب في الرئتين.يمكن أن تؤدي المبالغة في أنماط موت الخلايا الالتهابية أيضًا إلى تفاقم حالات المرض. إن إطلاق الحمض النووي والهستونات بعد عملية النيتوز يمكن أن يؤدي إلى تحفيز استجابات مناعية ذاتية.تم تحديد الأجسام المضادة الذاتية التي تستهدف NETs في الذئبة الحمامية الجهازية والتهاب المفاصل الروماتويدي. في حالات العدوى أو الإصابة الشديدة، يمكن أن يؤدي إطلاق DAMP بعد نخر العدلات الواسع إلى الالتهاب المفرط الذي يحدث في الإنتان، ومتلازمة الضائقة التنفسية الحادة، وعاصفة السيتوكين المرتبطة بكوفيد.ظهرت استراتيجيات علاجية تستهدف موت العدلات، إما من خلال التركيز على تعزيز المسارات المبرمجة للموت غير الالتهابي أو تقليل مدى التوليد المسبب للالتهابات للشبكات الخارجية (NETs) وإطلاق جزيئات التنبيه (DAMPs).
موت خلايا السدى كنموذج
أعمالنا الخاصة التي تفحص موت الخلايا الداعمة قد أظهرت أن نمط موت الخلايا مهم بالنسبة للتأثيرات اللاحقة على خلايا المناعة.قمنا بتحفيز موت الخلايا الداعمة من خلال بروتوكولين: 1) دورات التجميد والإذابة و 2) التسخين. من كل من هذه المجموعات من الخلايا الميتة، عزلنا قسمين: 1) راسب الخلايا، الذي احتوى على أجسام مبرمجة للموت وفضلات الخلايا، و 2) السائل الخلوي، الذي احتوى على مكونات قابلة للذوبان مثل DAMPs والسيتوكينات. اختبرنا هذه الأقسام لتأثيرها على نمط الماكروفاجات في المختبر وعلى قدرتها على تعزيز استعادة العضلات في نموذج الفأر لقصور الدم في الأطراف الخلفية. كانت فرضيتنا أن إطلاق DAMPs، بما في ذلك محتوى البروتين والأحماض النووية، سيكون أكبر بكثير في السائل الخلوي بينما سيكون قسم الراسب غنيًا بمكونات الغشاء المرتبطة بـ PS. لذلك افترضنا أن السائل الخلوي سيكون أكثر التهابيًا بغض النظر عن طريقة موت الخلايا.
وجدنا أن المكونات الناتجة عن عملية التجميد والإذابة (كلاً من السائل العلوي والرواسب) أنتجت استجابة واضحة مضادة للالتهابات ومشجعة على التجديد في الماكروموجرافات المستمدة من نخاع العظام المزروعة. كما أن السائل القابل للذوبان من عملية التجميد والإذابة حسّن بشكل ملحوظ تدفق الدم النسبي في القدم المتأثرة في نموذج الفأر لدينا إلى مستوىمن القدم السليمة عند مقارنتها بالعلاج غير المعالج، محلول ملحي، والمستخلص الناتج عن التسخين. أنتجت العضلات المعالجة بالمستخلص الناتج عن التجميد والذوبان قوة توتر متوسطة أكبر بكثير من تلك الخاصة بالعضلات غير المعالجة وعضلات المعالجة بالمستخلص الناتج عن التسخين. وهذا يشير إلى أن نمط موت الخلايا كان أكثر أهمية في الالتهاب من الكسر الخلوي. في مختبر منفصل، تم تحميل عوامل قابلة للذوبان معزولة من خلايا ميتة نتيجة للتجميد والذوبان في رغوة هيدروجيل لاستخدام تأثيراتها المناعية والوعائية لتحسين شفاء الجروح المزمنة..
مساهمون في الالتهاب
مع تقدم البشر في العمر، يؤدي تراكم آثار العدوى المتكررة أو منخفضة الدرجة، والأمراض الالتهابية المزمنة، والسمنة، والعوامل البيئية إلى تغييرات في عدد ووظيفة الخلايا النخاعية. هذه المجموعة من التغييرات هي أحد مكونات “الالتهاب المرتبط بالشيخوخة” – تراكم العيوب في جهاز المناعة المرتبطة بالشيخوخة التي تؤدي إلى حالة التهابية مستمرة ومنخفضة الدرجة.تظهر الخلايا المتعادلة، على وجه الخصوص، قدرة مخفضة على ابتلاع البكتيريا المعدية وقدرة ميكروبية مخفضة لدى الأفراد المسنين. تتأثر قدرة الخلايا على إجراء المراقبة المناعية بسبب انخفاض الحساسية للعوامل المضادة للاستماتة. لدى المسنين، هناك احتمال أكبر لحدوث الاستماتة دون تحفيز الالتهاب. وبالتالي، يمكن أن يؤدي التقدم في العمر إلى تعرض الفرد للعدوى الضارة وتقليل الاستجابة للقاحات. وعلى العكس، فإن الالتهاب منخفض الدرجة الذي يحدث خلال الشيخوخة الالتهابية يتميز بمستويات أعلى من السيتوكينات المؤيدة للالتهاب مثل IL-6 وTNF-وبروتين سي التفاعلي، وتراكم DAMPsتحت هذه الظروف، يمكن أن تساهم العدلات في تقصير التيلوميرات وإنتاج أنواع الأكسجين التفاعلية، مما يزيد من الإجهاد التأكسدي وشيخوخة الخلايا. كما أن إزالة العدلات الميتة والميتة تتعطل بشكل مشابه. على سبيل المثال، يمكن أن تقوم أنواع الأكسجين التفاعلية بقطع مستقبلات الفوسفاتيديل سيرين على البلعميات وتقليل قدرتها على البلعمة.قد يؤدي تقليل الكفاءة في إزالة العدلات المبرمجة للموت والحد من التأثيرات المضادة للالتهابات والمُحَصِّنة على البلعميات إلى تفاقم دورة الشيخوخة والالتهاب.
الجزء 3. حياة الخلية
السبات مقابل الشيخوخة
يمكن لخلايا الجسم الدخول في حالة من السكون، وهي حالة قابلة للعكس من توقف دورة الخلية. يسمح ذلك للخلايا بأن تصبح خاملة مع الاحتفاظ بالقدرة على إعادة الدخول في دورة الخلية عند الحاجة للنمو أو إصلاح الأنسجة. يحدث توقف دورة الخلية في مرحلة G0 حيث تحتفظ الخلايا فقط بصيانة الوظائف الأساسية. يتم تقليل علامات التكاثر بينما تزداد مثبطات دورة الخلية، مثل p27.، تكون مرتفعة. تحت المؤثرات المناسبة، عادةً وجود عوامل النمو، يمكن للخلايا استئناف حالتها التكاثرية وبدء انقسام الخلايا. على النقيض من ذلك، فإن شيخوخة الخلايا هي حالة من التوقف الدائم لدورة الخلية.في الشيخوخة، تتوقف الخلايا في مرحلة G1. علامات تثبيط دورة الخلية هي p16. و p21 تحدث الشيخوخة في الثقافة وفي الكائن الحي السليم من خلال تراكم الأضرار بما في ذلك تلف الحمض النووي، والإجهاد التأكسدي، وتنشيط الجينات المسرطنة. على عكس الخلايا الساكنة، يعني التوقف في مرحلة G1 أن الخلايا لا تزال نشطة أيضياً وتقوم بتخليق وإفراز جزيئات نشطة بيولوجياً، تُسمى النمط الإفرازي المرتبط بالشيخوخة (SASP، الجدول 1). مثبطات دورة الخلية، وSASP، والشيخوخة المرتبطة-جالاكتوزيداز (SA--غال) يميز شيخوخة الخلايا.
ميزات ونتائج الشيخوخة
تشمل مكونات SASP السيتوكينات المؤيدة للالتهابات، ولا سيما IL-6 و IL-1، وTNF- ؛ الكيموكينات، مثل CCL2 و CXCL8؛ والبروتيازات، بما في ذلك MMPsتعمل هذه البروتينات المفرزة بشكل كبير على تعزيز الاستجابة الالتهابية من خلال تجنيد وتفعيل خلايا المناعة وإعادة تشكيل الأنسجة المحيطة. في السرطان، يمكن أن تكون الخلايا المسنّة داخل الأورام الصلبة مثبطة للورم من خلال الحد من انقسام الخلايا التالفة التي قد تساهم في نمو الورم.على العكس، يمكن أن يؤدي SASP وظيفة مؤيدة للورم من خلال إزعاج خلايا المناعة وتعزيز غزو الخلايا للMatrix خارج الخلوية المحيطة.تم اقتراح تدخلات علاجية لتقليل عدد الخلايا الشيخوخة من خلال السينوبيوتيك (قتل الخلايا) أو السينوماورفوبكس (تعديل الخلايا). تم اقتراح العديد من هذه
الجدول 1 | النمط الإفرازي المرتبط بالشيخوخة (SASP) مقابل نمط البلعميات
SASP
M1
M2a
M2b
M2c
M2d
TNF-α
TGF-
إنترفيرون-
IL-1α
IL-1
IL-1RA
IL-6
IL-7
IL-8/CXCL8
IL-10
IL-12
IL-13
IL-15
IL-23
IL-27
GM-CSF
VEGF
CCL1/I-309
CCL2/MCP-1
/
CCL3/MIP-1α
CCL4/MIP-1
CCL5/RANTES
CCL8/MCP-2
CCL11/إيوتاكسين-1
CCL13/MCP-4
CCL15/ليوكوتاكتين-1
CCL16/LEC
/
CCL17/TARC
CCL18/PARC
CCL20/MIP-3α
/
CCL22/MDC
CCL24/إيوتاكسين-2
CCL26/إيوتاكسين-3
CXCL1/GRO-
CXCL2/GRO-
CXCL3/GRO-
CXCL5/ENA-78
CXCL9/MIG
CXCL10/IP-10
/
CXCL11/I-TAC
/
CXCL13/BCA-1
CXCL16/SR-PSOX
M1، ماكروفاج مؤيد للالتهابات؛ M2، ماكروفاج مضاد للالتهابات (M2a، شفاء الجروح؛ M2b، تنظيمي؛ M2c، إصلاح الأنسجة؛ M2d، مؤيد لتكوين الأوعية الدموية)؛ TNF-عامل نخر الورم; TGF- عامل النمو المحول; IFN- إنترفيرون-; IL، إنترلوكين؛ IL-1RA، مضاد مستقبلات إنترلوكين-1؛ GM-CSF، عامل تحفيز مستعمرات العدلات والماكروفاجات؛ VEGF، عامل نمو البطانة الوعائية؛ CCL، كيموكين (C-C motif) ligand؛ MCP، بروتين جذب المونوسيت؛ MIP، بروتين الالتهاب الماكروفاجي؛ RANTES، منظم عند التنشيط يعبر عنه ويُفرز من خلايا T الطبيعية؛ LEC، كيموكين معبر عنه في الكبد؛ TARC، كيموكين منظم في الغدة الصعترية والتنشيط؛ PARC، كيموكين منظم في الرئة والتنشيط؛ MDC، كيموكين مشتق من الماكروفاجات؛ CXCL، كيموكين (C-XC motif) ligand؛ GRO، جين الأورام المنظم للنمو؛ ENA-78، ببتيد منشط للعدلات مشتق من الظهارة 78؛ MIG، مونوكين مستحث بواسطة IFN-; IP-10، IFN- -بروتين 10 المستحث؛ I-TAC، كيمياء جذب الخلايا التائية المستحثة بواسطة الإنترفيرون؛ BCA-1، كيمياء جذب الخلايا البائية 1؛ SR-PSOX، مستقبل قاذف يرتبط بالفوسفاتيديل سيرين والدهون المؤكسدة. تشير العلامات إلى إنتاج السيتوكين أو الكيمياء الجذابة في كل نمط خلية.
مضاد أكسدة يثبط الاستقطاب الناتج عن LPS/ يميل التوازن نحو M2
↓ M1
↑ م2
٧٦، ٧٧
داساتينيب
مثبط كيناز التيروزين يستخدم بالاشتراك مع الكيرسيتين/ يستنفد البلعميات المرتبطة بالورم
↓ M1
↑ م2
↓ إجمالي البلعميات في الورم
78,79
فيستين
يقلل من الاستقطاب المؤيد للالتهابات
↓ M1
٨٠
البيولوجيات
مضاد-CD47
يعمل على تمييع الخلايا الشيخوخة لتقليل التهرب المناعي
↑ إزالة العدلات الشيخوخة
81
MFG-E8
جسور PS على الخلايا المبرمجة للموتعلى البلعميات لزيادة الإيفيروسيتوز
↑ تكوين الأوعية الدموية
↓ وقت إغلاق الجرح
82,83
الجسيمات النانوية
ملاحظة
يبدأ عملية الإيفيروسيتوز المعتمدة على الفوسفاتيديل سيرين وسلسلة مضادة للالتهابات في البلعميات
↓ M1
↑ م2
↑ امتصاص البلعميات
٨٤، ٨٩، ٩١
غشاء العدلات
يرتبط بجزيئات التهابية ليعمل كطُعم للعدلات
↓ تنشيط الخلايا الغضروفية والموت الخلوي
↓ قطر الركبة، تلف الغضروف، و TNF- في المصل و IL-1 في نموذج RA
٨٨
siRNA TNF-α
يعيق ترجمة TNF-a
↓ فقدان العظام وسمك الكف في نموذج التهاب المفاصل الروماتويدي
97
M1، البلعميات المسببة للالتهابات؛ M2، البلعميات المضادة للالتهابات؛ LPS، الليبوسكرياتيد؛ CD، مجموعة التمايز؛ MGF-E8، عامل نمو البشرة لكرات الدهون الحليب 8؛ PS، الفوسفاتيديل سيرين؛ TNF، عامل نخر الورم؛ IL، الإنترلوكين؛ RA، التهاب المفاصل الروماتويدي؛ siRNA، RNA صغير متداخل. يمكن أن تؤثر الاستراتيجيات أيضًا على خلايا المناعة والتفاعل بين الخلايا المسنّة وخلايا المناعة.
الشيخوخة والجهاز المناعي الفطري
بينما يُعرف الكثير عن كيفية شيخوخة خلايا الأنسجة المختلفة خلال المرض والشيخوخة، فإن ما يتعلق بخلايا الجهاز المناعي الفطري أقل وضوحًا. يُعتقد أن الطبيعة المؤيدة للالتهابات لمتلازمة الإفرازات المرتبطة بالشيخوخة (SASP) هي واحدة من المساهمين الرئيسيين في الالتهاب المرتبط بالشيخوخة. عادةً، يمكن لخلايا الجهاز المناعي الفطري، بما في ذلك البلعميات، والخلايا المتعادلة، وخلايا القاتل الطبيعي، التعرف على الخلايا الشيخوخة وإزالتها. خلال الشيخوخة، تتراكم الخلايا الشيخوخة، على الأرجح نتيجة لنقص في وظائف إزالة الخلايا المناعية. وقد تم إثبات أن الفئران التي تعاني من انخفاض في وظيفة السمية للخلايا المناعية كانت لديها مستويات أعلى من الخلايا الشيخوخة بالإضافة إلى زيادة في مستويات علامات الالتهاب المزمن.لقد أظهرت الدراسات أيضًا أن الخلايا الشيخوخة تثبط إزالة الجثث بواسطة البلعميات من خلال زيادة تعبير CD47 الذي يعمل كإشارة “لا تأكلني” للبلعميات.لذلك من الممكن أن تؤدي التأثيرات التراكمية للالتهاب المرتبط بالشيخوخة على خلايا المناعة إلى تشكيل حلقة تغذية راجعة إيجابية تحد من إزالة الخلايا المسنّة، وتزيد من متلازمة الإفراز المرتبطة بالشيخوخة، وتساهم في مستويات أعلى من الالتهاب المزمن، والشيخوخة، والالتهاب المرتبط بالشيخوخة..
شيخوخة البلعميات
بينما كانت هناك الآن عدد من التقارير حول شيخوخة البلعمياتمن المهم التمييز بين الشيخوخة الخلوية والانخفاض الوظيفي لخلايا المناعة في الكائن الحي المتقدم في العمربينما من الواضح أن وظيفة المناعة (مثل البلعمة، وإزالة الخلايا، وعرض المستضدات، وما إلى ذلك) تتناقص مع تقدم العمر في الكائن الحي، فإن الطبيعة الدقيقة لشيخوخة الخلايا البالعة غير واضحة. مثلما هو الحال في الكثير من الأدبيات المتعلقة بالخلايا البالعة، هناك عدد من التقارير التي تشير إلى أنه في البشر والفئران المسنين، تظهر الخلايا البالعة نمطًا مضادًا للالتهابات ومؤيدًا للتجديد، بينما في دراسات أخرى يكون العكس صحيحًا.في زراعة الخلايا، يؤدي تحفيز البلعميات بواسطة LPS أو الإشعاع أو الإجهاد التأكسدي إلى العلامات الكلاسيكية للشيخوخة، أي – توقف دورة الخلية، زيادة حجم الليزوزومات، التعبير عن SASP، وزيادة تنظيم علامات الشيخوخة مثل SA--غال و p16لقد ارتبطت شيخوخة الفئران أيضًا بـ SA--غال و p16تراكم البلعميات الإيجابيما يثير القلق بشكل خاص هو أنه لا يوجد خط واضح يميز بين هذه الأمور. خصائص من البلعمة المؤيدة للالتهابات التي تم تحفيزها بواسطة LPS أو IFN- (الجدول 1) .
الشيخوخة كتشابه غير تكيفي للبلاعم
إذا كان هناك القليل أو لا شيء يميز بشكل حصري البلعوم الالتهابي عن البلعوم المتقدم في السن، فمن الجدير بالذكر أن العديد من تلك الميزات موجودة أيضًا في خلايا متقدمة في السن أخرى غير مناعية.لقد تم إظهار أن SASP، وتقليل التكاثر، وزيادة حجم الليزوزومات، وقدرة الخلايا غير الاحترافية على البلعمة قد زادت جميعها عند الشيخوخة. على سبيل المثال، تظهر الخلايا الظهارية الشيخوخة زيادة في امتصاص البكتيريا عند التعرض للإجهاد التأكسدي.إنه نهج مثير للاهتمام لدراسة الشيخوخة أن نفكر فيها كعملية تتكيف فيها الخلايا لأداء بعض وظائف خلايا المناعة.على سبيل المثال، من الممكن أنه عند حدوث ضرر مرتبط بالشيخوخة أو الأمراض المزمنة أو التأثيرات البيئية، تتقلص وظائف إزالة الخلايا والابتلاع الخلوي للماكروفاجات، مما يؤدي إلى التهاب منخفض المستوى مستمر، والتقدم نحو استجابة غير تكيفية في خلايا الأنسجة الأخرى.. في الواقع، يقلد النمط الظاهري للبلاعم الالتهابية التي تحاول تعويض فقدان وظيفتها. ثم يخدم هذه العملية في حلقة تغذية راجعة إيجابية لتسريع شيخوخة الجهاز المناعي.
الجزء 4. الآثار العلاجية (الجدول 2)
سينوليتكس
استنادًا إلى التشابه بين الخلايا الشيخوخة والماكروفاجات الالتهابية، من المنطقي أن تكون للأدوية السنوبيوتية أو السنو مرفولوجية تأثيرات قوية على خلايا الجهاز المناعي، وبالفعل هذا صحيح. لقد أبلغت أول أدوية سنوليتية في التجارب السريرية، الكيرسيتين والداساتينيب، عن تأثيرات على تعزيز استقطاب الماكروفاجات المؤيدة للالتهاب إلى ماكروفاجات مؤيدة للتجديد/ مضادة للالتهاب.تم الإبلاغ أيضًا عن أن الداساتينيب يقلل من عدد البلعميات في الأورام الصلبة.تم إظهار أن مادة فيسيتين، وهي مادة سنوليتك أخرى، تقوم بتوجيه البلعميات في الثقافة بعيدًا عن النمط الظاهري المؤيد للالتهابات الذي تسببه LPS.بينما تعتبر هذه مساحة غنية لاكتشاف الأدوية، من المهم أن نلاحظ أن استنفاد البلعميات أو تأثيراتها الالتهابية يمكن أن يكون له آثار سلبية واسعة النطاق على شفاء الجروح، ومراقبة الأورام، ومقاومة العدوى، وفعالية اللقاحات.
استهداف تفاعل العدلات والبلعميات
تشمل الأساليب البيولوجية تجاه السينوبيوتيك الأجسام المضادة ضد الجزيئات السطحية مثل CD47. هذا يحد من قدرة الخلايا الشيخوخة على التهرب من التعرف عليها من قبل جهاز المناعة لكي تتمكن البلعميات من إزالتها، ولكن قد يكون له آثار جانبية مرتبطة بالتعرف على الذات من غير الذات. على نهج مشابه، قد يكون من المفيد التركيز على التفاعل المحوري بين العدلات المبرمجة للموت والبلعميات التي تقوم بعملية الإخراج. على سبيل المثال، هناك العشرات من التفاعلات المعتمدة على المستقبلات المعروفة التي تسهل التعرف والابتلاع. على وجه الخصوص، يتم التعرف على PS على أسطح الخلايا المبرمجة للموت من خلال عائلة مستقبلات TAM (Tyro3/Axl/MerTK) والاندماجات على البلعميات. تشارك هذه العائلات المستقبلية من خلال بروتينات الربط، حيث يربط عامل الحليب الدهني الكروي-EGF 8 (MFG-E8) PS وتربط الإنترغينات بينما يقوم بروتين 6 المحدد لتوقف النمو (Gas6) وبروتين S بربط PS ومستقبلات TAM. قد تكون الإدارة الخارجية للإصدارات المؤتلفة من هذه البروتينات أو قطعها قادرة على تعزيز الإيفيروسيتوز وتسهيل الانتقال إلى نمط ظاهري مضاد للالتهابات مع تقليل الآثار السلبية. على وجه الخصوص، لقد ثبت أن MFG-E8 يقلل الالتهاب بعد الإصابة.تم إثبات الإمكانات العلاجية من خلال استخدام MFG-E8 المؤتلف الموضعي الذي سرع من حل التهاب الجروح، وزاد من تكوين الأوعية الدموية، وقلل من الوقت اللازم لشفاء الجروح في نموذج جرح السكري..
استراتيجية مستوحاة من الإيفيروسيتوزيس للخلايا الميتة
من الواضح أن التفاعل وإزالة العدلات الميتة والميتة بواسطة البلعميات أمر حاسم للحفاظ على التوازن والسيطرة على تسلسل الأحداث التي تعزز شفاء الجروح بشكل فعال. لاستكشاف هذه العملية لاستخدامها المحتمل في العلاج، قامت مجموعتنا بتطوير طلاء نانوي مشتق من غشاء الخلية لمحاكاة الميزات المميزة لسطح الخلية الميتة، وفي النهاية تسهيل تقليل استجابة البلعميات الالتهابية من خلال المسارات الفسيولوجية الموجودة.بدأنا بحمض البوليلكتيك-الجليكوليك (PLGA) كحامل للجسيمات النانوية لطلائنا المستمد من الغشاء، الذي تم اختياره لخصائصه الخاملة والقابلة للتحلل الحيوي والمتوافقة حيوياً. قمنا بتغطية سطح جسيمات PLGA النانوية بغشاء بلازما الخلايا المعزولة المضاف إليها PS اصطناعي خارجي. لقد تم استخدام أغشية الخلايا سابقاً لتغطية الجسيمات النانوية، على الرغم من أنها قدمت فائدة من خلال البروتينات المحددة الموجودة على سطح الغشاء التي يمكن أن تتفاعل مع بيئتها أو مع خلايا قريبة.. في عملنا، ركزنا على تأثير الفوسفوليبيد النشط بيولوجيًا PS، المقدم في سياق غشاء الخلايا الداعمة العامة، لمحاكاة جسم ميتة الخلايا. أظهرنا أن الجمع بين البروتينات المرتبطة بالغشاء وPS أدى إلى تحول في نمط الماكروفاج من الالتهابي نحو التجديد أكثر من حبيبات PS وحدها. في نظام جزيئات نانوية منفصل، أظهرنا أنه حتى بدون البروتينات المرتبطة بالغشاء، فإن تقديم PS على سطح جزيئات الدهون-بوليمر زاد بشكل كبير من البلعمة بواسطة الماكروفاجات مقارنة بالجزيئات النانوية بدون .
تم اقتراح الحويصلات المحتوية على الفوسفatidylserine كوسائل توصيل الأدوية لاستهداف البلعميات وخلايا تقديم المستضدات الأخرى.تم اقتراح PS أيضًا كمكون من جزيئات النانو ذات الدهون الصلبة لتوصيل لقاحات RNA. إحدى المخاوف بشأن هذه الاستراتيجيات، بالإضافة إلى جزيئات النانو البوليمرية المغلفة بغشاء الخلايا، هي الدور الذي يلعبه PS في تجلط الدم. يمكن أن يرتبط PS ببروتينات التجلط ويشارك في سلسلة التجلط. قامت مختبرنا، لترجمة جزيئات النانو المغلفة بالغشاء والتي تحتوي على PS إلى نموذج حيوي، بتطوير استراتيجية PEGylation لتحسين كل من استقرار جزيئات النانو على المدى الطويل واستهداف مواقع الالتهاب من خلال التعديل السطحي..
يمكن استخدام ثنائي طبقة الدهون في جزيئات PLGA الخاصة بنا لتثبيت الجزيئات على سطح الجسيم من خلال التفاعلات الكارهة للماء. تم ربط الجزيئات المترافقة مع مجموعات محبة للدهون بأسطح الخلايا وأشباح الأغشية ببساطة عن طريق حضن الجزيء والغشاء معًا والسماح للتفاعلات الكارهة للماء بدفع إدخال المجموعة المحبة للدهون في الجزء المحب للدهون من الغشاء.. تتطلب هذه العملية، التي تُسمى أحيانًا طلاء الغشاء، عدم وجود تفاعلات اقتران إضافية. استغللنا انخفاض الرقم الهيدروجيني في المواقع الملتهبة بشكل مزمن مقارنةً بالظروف الفسيولوجية الطبيعية. يمكن لجزيئات النانو المجهزة بسطح حساس للأحماض وقابلة للإزالة من بولي إيثيلين جلايكول (PEG) أن تتجمع بشكل تفضيلي في هذه المواقع.. تظهر نتائج عملنا أنه من خلال نهج طلاء الغشاء، زادت موضع الجسيمات في مواقع الالتهاب في الجسم الحي. كما تم امتصاص هذه الجسيمات بشكل تفضيلي بواسطة البلعميات داخل مناطق الالتهاب. أيضًا، في نموذج الالتهاب المزمن الناتج عن الليبوسكريات (LPS)، قللت هذه الجسيمات النانوية الالتهاب وعززت نمط تجديد أفضل من الجسيمات النانوية الضابطة.
الاستنتاجات
عند فحص دورة حياة وتفاعلات العدلات والبلعميات خلال التوازن الطبيعي، وشفاء الجروح الفعال، وأمراض الالتهاب المزمن، والشيخوخة، من الواضح أن موت العدلات، وإزالتها وبلعها بواسطة البلعميات، تلعب أدوارًا رئيسية في الصحة والمرض. يمكن القول إن إزالة العدلات بواسطة البلعميات هي أكثر تفاعل خلايا المناعة تكرارًا في جسم الإنسان، وهذا التفاعل غالبًا ما يكون غير مرئي من الناحية المناعية. يمكن أن تؤثر التغيرات في نمط ونتيجة موت العدلات بشكل عميق على نمط البلعميات والبيئة النسيجية الناتجة. وبالمثل، يمكن أن يؤدي انخفاض وظيفة نظام المناعة الفطرية مع تقدم العمر إلى إرباك هذا النظام، مما يؤدي إلى التهاب مزمن منخفض المستوى، وشيخوخة الخلايا، ونقص في إزالة الخلايا، وبالتالي يساهم في دورة مفرغة من الالتهاب. من المحتمل أن يكون استغلال الآلية التي تتعرف بها البلعميات على الخلايا وتزيلها وتستقطبها نتيجة لموت الخلايا عملية بحث غنية لتطوير أدوية جديدة واستراتيجيات توصيل الأدوية.
توفر البيانات
لم يتم إنتاج أو تحليل أي مجموعات بيانات خلال الدراسة الحالية.
تاريخ الاستلام: 22 أكتوبر 2024؛ تاريخ القبول: 23 يناير 2025؛
تاريخ النشر على الإنترنت: 04 مارس 2025
References
Rodrigues, M., Kosaric, N., Bonham, C. A. & Gurtner, G. C. Wound Healing: A Cellular Perspective. Physiol. Rev. 99, 665-706 (2019).
Singh, S., Young, A. & McNaught, C.-E. The physiology of wound healing. Surg. (Oxf.) 35, 473-477 (2017).
Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front Physiol. 9, 113 (2018).
Tidball, J. G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 1, 2029-2062 (2011).
Fullerton, J. N. & Gilroy, D. W. Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov. 15, 551-567 (2016).
Iwasaki, A. & Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 327, 291-295 (2010).
Savill, J. S. et al. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest 83, 865-875 (1989).
Sica, A. & Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest 122, 787-795 (2012).
Hammers, D. W. et al. Anti-inflammatory macrophages improve skeletal muscle recovery from ischemia-reperfusion. J. Appl Physiol. 118, 1067-1074 (2015).
Tidball, J. G. & Villalta, S. A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1173-R1187 (2010).
Patel, U. et al. Macrophage polarization in response to epigenetic modifiers during infection and inflammation. Drug Discov. Today 22, 186-193 (2017).
Chazaud, B. et al. Dual and beneficial roles of macrophages during skeletal muscle regeneration. Exerc Sport Sci. Rev. 37, 18-22 (2009).
Tidball, J. G. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R345-R353 (2005).
Novak, M. L. & Koh, T. J. Phenotypic transitions of macrophages orchestrate tissue repair. Am. J. Pathol. 183, 1352-1363 (2013).
Kharraz, Y., Guerra, J., Mann, C. J., Serrano, A. L. & MunozCanoves, P. Macrophage Plasticity and the Role of Inflammation in Skeletal Muscle Repair. Mediat Inflamm. (2013).
Fadok, V. A. et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207-2216 (1992).
Li, M. O., Sarkisian, M. R., Mehal, W. Z., Rakic, P. & Flavell, R. A. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 302, 1560-1563 (2003).
Segawa, K. & Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 25, 639-650 (2015).
Huynh, M.-L. N., Fadok, V. A. & Henson, P. M. Phosphatidylserinedependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Invest. 109, 41-50 (2002).
Mantovani, A. et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677-686 (2004).
Reimold, A. M. TNFa as Therapeutic Target: New Drugs, More Applications. Curr. Drug Targets – Inflamm. Allergy 1, 377-392 (2002).
Herrero-Cervera, A., Soehnlein, O. & Kenne, E. Neutrophils in chronic inflammatory diseases. Cell Mol. Immunol. 19, 177-191 (2022).
Kapellos, T. S. et al. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front Immunol. 10, 2035 (2019).
Parisi, L. et al. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 8917804 (2018).
Guo, Q. et al. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 6, 15 (2018).
Jain, S., Tran, T. H. & Amiji, M. Macrophage repolarization with targeted alginate nanoparticles containing IL-10 plasmid DNA for the treatment of experimental arthritis. Biomaterials 61, 162-177 (2015).
Li, J., Hsu, H. C. & Mountz, J. D. Managing macrophages in rheumatoid arthritis by reform or removal. Curr. Rheumatol. Rep. 14, 445-454 (2012).
Yanni, G., Whelan, A., Feighery, C. & Bresnihan, B. Synovial tissue macrophages and joint erosion in rheumatoid arthritis. Ann. Rheum. Dis. 53, 39-44 (1994).
Kinne, R. W., Brauer, R., Stuhlmuller, B., Palombo-Kinne, E. & Burmester, G. R. Macrophages in rheumatoid arthritis. Arthritis Res. 2, 189-202 (2000).
Benveniste, E. N. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J. Mol. Med. (Berl.) 75, 165-173 (1997).
Clark, R. A. & Kupper, T. S. Misbehaving macrophages in the pathogenesis of psoriasis. J. Clin. Invest 116, 2084-2087 (2006).
Mahida, Y. R. The key role of macrophages in the immunopathogenesis of inflammatory bowel disease. Inflamm. Bowel Dis. 6, 21-33 (2000).
Loh, W. & Vermeren, S. Anti-Inflammatory Neutrophil Functions in the Resolution of Inflammation and Tissue Repair. Cells 11, 4076 (2022).
Tu, H. et al. Dying to Defend: Neutrophil Death Pathways and their Implications in Immunity. Adv. Sci. (Weinh.) 11, e2306457 (2024).
Perez-Figueroa, E., Alvarez-Carrasco, P., Ortega, E. & MaldonadoBernal, C. Neutrophils: Many Ways to Die. Front Immunol. 12, 631821 (2021).
Maianski, N. A., Maianski, A. N., Kuijpers, T. W. & Roos, D. Apoptosis of neutrophils. Acta Haematol. 111, 56-66 (2004).
Dubey, M. et al. Nitric oxide-mediated apoptosis of neutrophils through caspase- 8 and caspase-3-dependent mechanism. Cell Death Dis. 7, e2348 (2016).
Lawrence, D. W., King, S. B., Frazier, W. A. & Koenig, J. M. Decreased CD47 expression during spontaneous apoptosis targets neutrophils for phagocytosis by monocyte-derived macrophages. Early Hum. Dev. 85, 659-663 (2009).
Von Vietinghoff, S. & Ley, K. Homeostatic regulation of blood neutrophil counts. J. Immunol. 181, 5183-5188 (2008).
Denning, N. L., Aziz, M., Gurien, S. D. & Wang, P. DAMPs and NETs in Sepsis. Front Immunol. 10, 2536 (2019).
Murao, A., Aziz, M., Wang, H., Brenner, M. & Wang, P. Release mechanisms of major DAMPs. Apoptosis 26, 152-162 (2021).
Marki, A. & Ley, K. The expanding family of neutrophil-derived extracellular vesicles. Immunol. Rev. 312, 52-60 (2022).
de Miranda, F. S. et al. Properties and Application of Cell-Free DNA as a Clinical Biomarker. Int J. Mol. Sci. 22, 9110 (2021).
Yang, H., Antoine, D. J., Andersson, U. & Tracey, K. J. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 93, 865-873 (2013).
Dosch, M., Gerber, J., Jebbawi, F. & Beldi, G. Mechanisms of ATP Release by Inflammatory Cells. Int J. Mol. Sci. 19, 1222 (2018).
Hou, J., Hsu, J. M. & Hung, M. C. Molecular mechanisms and functions of pyroptosis in inflammation and antitumor immunity. Mol. Cell 81, 4579-4590 (2021).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060-1072 (2012).
Guimaraes-Costa, A. B. et al. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl Acad. Sci. USA 106, 6748-6753 (2009).
Thiam, H. R., Wong, S. L., Wagner, D. D. & Waterman, C. M. Cellular Mechanisms of NETosis. Annu Rev. Cell Dev. Biol. 36, 191-218 (2020).
Vorobjeva, N. V. & Chernyak, B. V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology.Biochemistry (Mosc) 85, 1178-1190 (2020).
Ding, L., Yang, J., Zhang, C., Zhang, X. & Gao, P. Neutrophils Modulate Fibrogenesis in Chronic Pulmonary Diseases. Front Med. (Lausanne) 8, 616200 (2021).
Huang, S. U. & O’Sullivan, K. M. The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs. Int J. Mol. Sci. 23, 3793 (2022).
Land, W. G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome-with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 22, 141-160 (2021).
Huang, W., Kraynak, C. A., Bender, E. C., Farrar, R. P. & Suggs, L. J. Soluble components from mesenchymal stromal cell processing exert anti-inflammatory effects and facilitate ischemic muscle regeneration. Cytotherapy 25, 375-386 (2023).
Lan, Z. et al. Hydrogel foam dressings with angiogenic and immunomodulatory factors from mesenchymal stem cells. J. Biomed. Mater. Res. A 112, 1388-1398 (2024).
Franceschi, C. & Bonafe, M. Centenarians as a model for healthy aging. Biochem Soc. Trans. 31, 457-461 (2003).
Byrne, T., Cooke, J., Bambrick, P., McNeela, E. & Harrison, M. Circulating inflammatory biomarker responses in intervention trials in frail and sarcopenic older adults: A systematic review and metaanalysis. Exp. Gerontol. 177, 112199 (2023).
Naeini, M. B., Bianconi, V., Pirro, M. & Sahebkar, A. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell Mol. Biol. Lett. 25, 23 (2020).
Campisi, J. Aging, cellular senescence, and cancer. Annu Rev. Physiol. 75, 685-705 (2013).
Huang, W., Hickson, L. J., Eirin, A., Kirkland, J. L. & Lerman, L. O. Cellular senescence: the good, the bad and the unknown. Nat. Rev. Nephrol. 18, 611-627 (2022).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853-2868 (2008).
Coppe, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev. Pathol. 5, 99-118 (2010).
Haston, S. et al. Clearance of senescent macrophages ameliorates tumorigenesis in KRAS-driven lung cancer. Cancer Cell 41, 1242-1260.e1246 (2023).
Ovadya, Y. et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 9, 5435 (2018).
Schloesser, D. et al. Senescent cells suppress macrophagemediated corpse removal via upregulation of the CD47-QPCT/L axis. J. Cell Biol. 222, e202207097 (2023).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100-105 (2021).
Sharma, R. Perspectives on the dynamic implications of cellular senescence and immunosenescence on macrophage aging biology. Biogerontology 22, 571-587 (2021).
Wang, L. et al. Macrophage senescence in health and diseases. Acta Pharm. Sin. B 14, 1508-1524 (2024).
McQuattie-Pimentel, A. C. et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J. Clin. Invest 131, e140299 (2021).
Hall, B. M. et al. p16(lnk4a) and senescence-associated betagalactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY) 9, 1867-1884 (2017).
Wang, H. et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosisassociated lipid uptake. Aging (Albany NY) 12, 9240-9259 (2020).
Suzuki, K., Susaki, E. A. & Nagaoka, I. Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis. Int J. Mol. Sci. 23, 11148 (2022).
Robledo, E., Benito Rodriguez, P. G., Vega, I. A., Colombo, M. I. & Aguilera, M. O. Staphylococcus aureus phagocytosis is affected by senescence. Front Aging 4, 1198241 (2023).
Behmoaras, J. & Gil, J. Similarities and interplay between senescent cells and macrophages. J. Cell Biol. 220, e202010162 (2021).
Ogata, Y. et al. SASP-induced macrophage dysfunction may contribute to accelerated senescent fibroblast accumulation in the dermis. Exp. Dermatol 30, 84-91 (2021).
Tsai, C. F. et al. Regulatory Effects of Quercetin on M1/M2 Macrophage Polarization and Oxidative/Antioxidative Balance. Nutrients 14, 67 (2021).
. et al. Quercetin inhibited LPS-induced cytokine storm by interacting with the AKT1-FoxO1 and Keap1-Nrf2 signaling pathway in macrophages. Sci. Rep. 14, 20913 (2024).
Brownlow, N., Mol, C., Hayford, C., Ghaem-Maghami, S. & Dibb, N. J. Dasatinib is a potent inhibitor of tumour-associated macrophages, osteoclasts and the FMS receptor. Leukemia 23, 590-594 (2009).
Hickson, L. J. et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446-456 (2019).
Hada, Y., Uchida, H. A. & Wada, J. Fisetin Attenuates Lipopolysaccharide-Induced Inflammatory Responses in Macrophage. Biomed. Res. Int. 2021, 5570885 (2021).
Yang, H., Xun, Y. & You, H. The landscape overview of CD47-based immunotherapy for hematological malignancies. Biomark. Res. 11, 15 (2023).
Deroide, N. et al. MFGE8 inhibits inflammasome-induced IL-1beta production and limits postischemic cerebral injury. J. Clin. Invest 123, 1176-1181 (2013).
Das, A. et al. Correction of MFG-E8 Resolves Inflammation and Promotes Cutaneous Wound Healing in Diabetes. J. Immunol. 196, 5089-5100 (2016).
Kraynak, C. A., Yan, D. J. & Suggs, L. J. Modulating inflammatory macrophages with an apoptotic body-inspired nanoparticle. Acta Biomater. 108, 250-260 (2020).
Fang, R. H. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 14, 2181-2188 (2014).
Hu, C. M. et al. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl Acad. Sci. USA 108, 10980-10985 (2011).
Thamphiwatana, S. et al. Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc. Natl Acad. Sci. USA 114, 11488-11493 (2017).
Zhang, Q. et al. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 13, 1182-1190 (2018).
Bender, E. C., Sircar, A. J., Taubenfeld, E. K. & Suggs, L. J. Modulating Lipid-Polymer Nanoparticles’ Physicochemical Properties to Alter Macrophage Uptake.ACS Biomater. Sci. Eng. 10, 2911-2924 (2024).
Bender, E. C., Kraynak, C. A., Huang, W. & Suggs, L. J. Cell-Inspired Biomaterials for Modulating Inflammation. Tissue Eng. Part B Rev. 28, 279-294 (2022).
Kraynak, C. A. et al. Apoptotic body-inspired nanoparticles target macrophages at sites of inflammation to support an antiinflammatory phenotype shift. Int J. Pharm. 618, 121634 (2022).
Chen, H. et al. Lipid insertion enables targeted functionalization of paclitaxel-loaded erythrocyte membrane nanosystem by tumorpenetrating bispecific recombinant protein. Int J. Nanomed. 13, 5347-5359 (2018).
Shi, G., Mukthavaram, R., Kesari, S. & Simberg, D. Distearoyl anchor-painted erythrocytes with prolonged ligand retention and circulation properties in vivo. Adv. Health. Mater. 3, 142-148 (2014).
Gerry, A. B. & Leake, D. S. Effect of low extracellular pH on NF-kappaB activation in macrophages. Atherosclerosis 233, 537-544 (2014).
Okajima, F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal 25, 2263-2271 (2013).
Rajamaki, K. et al. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J. Biol. Chem. 288, 13410-13419 (2013).
Aldayel, A. M. et al. Lipid nanoparticles with minimum burst release of TNF-alpha siRNA show strong activity against rheumatoid arthritis unresponsive to methotrexate. J. Control Release 283, 280-289 (2018).
Kalia, J. & Raines, R. T. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int Ed. Engl. 47, 7523-7526 (2008).
Wang, J. L. et al. Aerosolizable siRNA-encapsulated solid lipid nanoparticles prepared by thin-film freeze-drying for potential pulmonary delivery. Int J. Pharm. 596, 120215 (2021).
Ouvrier, B., Ismael, S. & Bix, G. J. Senescence and SASP Are Potential Therapeutic Targets for Ischemic Stroke. Pharmaceuticals 17, 312 (2024).
Arabpour, M., Saghazadeh, A. & Rezaei, N. Anti-inflammatory and M2 macrophage polarization-promoting effect of mesenchymal stem cellderived exosomes. Int. Immunopharmacol. 97, 107823 (2021).
Arango Duque, G. & Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 5, 491 (2014).
Gharavi, A. T., Hanjani, N. A., Movahed, E. & Doroudian, M. The role of macrophage subtypes and exosomes in immunomodulation. Cell. Mol. Biol. Lett. 27, 1-18 (2022).
Abdelaziz, M. H. et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J. Transl. Med. 18, 58 (2020).
Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat Inflamm. 2015, 816460 (2015).
Wang, B., Han, J., Elisseeff, J. H. & Demaria, M. The senescenceassociated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell Biol. 1-21 (2024).
Gessani, S. & Belardelli, F. IFN- Expression in Macrophages and Its Possible Biological Significance. Cytokine Growth Factor Rev. 9, 117-123 (1998).
Sims, J. E. & Smith, D. E. The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10, 89-102 (2010).
Stow, J. L. & Murray, R. Z. Intracellular trafficking and secretion of inflammatory cytokines. Cytokine Growth Factor Rev. 24, 227-239 (2013).
Ma, X. et al. Regulation of IL-10 and IL-12 production and function in macrophages and dendritic cells. F1000 Res. 4, F1000-Faculty Rev1465 (2015).
Orecchioni, M., Ghosheh, Y., Pramod, A. B. & Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 10, 1084 (2019).
Tang, C., Chen, S., Qian, H. & Huang, W. Interleukin-23: as a drug target for autoimmune inflammatory diseases. Immunology 135, 112-124 (2012).
الشكر والتقدير
تم دعم هذا العمل من خلال منحة من المعاهد الوطنية للصحة (R56EB032211-01) إلى لورا سوجز.
مساهمات المؤلفين
E.C.B. حرر المخطوطة وأعد الشكل. H.S.T. جمع الجدول. L.J.S. وضعت وكتبت المسودة الأولية.
المصالح المتنافسة
L.J.S. هي محررة ضيف في مجموعة ‘منظمون تنظيم المناعة، والتوازن، والشيخوخة’، ومع ذلك، لم تكن متورطة في عملية مراجعة الأقران لهذه المقالة.
معلومات إضافية
يجب توجيه المراسلات والطلبات للحصول على المواد إلى لورا ج. سوجز.
معلومات إعادة الطبع والتصاريح متاحة على http://www.nature.com/reprints
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
Inflammation: a matter of immune cell life and death
Elizabeth C. Bender, Halah S. Tareq & Laura J. Suggs
Inflammation is triggered by an insult or injury to the cells of the body and is a healthy and productive step towards healing. Persistent inflammation, however, is a dysregulated response and can lead to a host of unwanted disease states. The death of cells by necrosis is known to initiate inflammation, while the programmed death of cells by apoptosis limits the inflammatory response. We propose here that there exists a spectrum of cell living, aging, dying, and death states that have differential effects on inflammatory disease.
Part 1. The life cycle of inflammation
The wound-healing response
Much of mammalian injury and disease can be understood through the lens of inflammation and wound healing. The wound healing response to injury in humans is well known with respect to the cells and molecules involved, the time course for specific events, and the negative consequences when such components are blocked. Healthy wound healing is broken into four phases: hemostasis, inflammation, proliferation, and remodeling. In this section, we will focus on inflammation. A detailed description of the other phases can be found in previously published reviews . Critical cellular orchestrators of inflammation are cells of the innate immune system, such as neutrophils and macrophages. Following injury, in an effort to reestablish homeostasis, neutrophils are recruited to sites of injury following platelet degranulation of cytokines and growth factors. This recruitment is aided by mast cells, which, in addition to releasing antimicrobial peptides, release molecules to break down the extracellular matrix and increase the permeability of blood vessels . Neutrophils help to decontaminate the wound by phagocytosis of bacteria and foreign bodies and secretion of high levels of pro-inflammatory signaling molecules to accelerate further immune cell recruitment and activation . Recruited monocytes and tissue-resident macrophages contribute to the cytokine-rich inflammatory environment and support the majority of debris clearance via phagocytosis . Signaling molecules and antigen presentation by these cells, as well as dendritic cells, are also critical for mounting an adaptive immune response to pathogens . Neutrophils then undergo apoptosis and are cleared within approximately one day of their arrival . Efferocytosis, or apoptotic cell clearance, is a tightly regulated physiologic process, as demonstrated by more than apoptotic cells cleared daily in the absence of injury or disease by the immune system without an inflammatory immune response . When neutrophils are efferocytosed by macrophages following injury, the macrophages direct a transition from inflammation towards repair and regeneration . This phenotypic transition is necessary for the resolution of inflammation to maintain or restore tissue function .
While macrophage infiltration and pro-inflammatory activation is crucial for tissue repair and homeostasis maintenance , subsequent macrophage polarization towards an anti-inflammatory, pro-regenerative phenotype is similarly required to enable complete wound repair . Macrophage response to apoptotic cell engulfment is mediated by several receptors on the macrophage that can bind to a variety of apoptotic surface markers to facilitate recognition, uptake, and signaling. Exposure of the phospholipid phosphatidylserine (PS) onto the outer surface of the cell membrane is the most well-characterized apoptotic cell signal . In most living cells, PS is confined to the inside of the cell by a flippase, a lipid transporter protein that moves certain lipids from the outer leaflet to the inner leaflet, producing anisotropic layers . When that cell undergoes apoptosis, it loses its cell membrane anisotropy, exposing PS on its outer membrane. The exposure of PS acts as an “eat-me” signal to phagocytes, including macrophages, to engulf the dying cell . This engulfment initiates a shift in macrophage polarization toward a pro-resolving phenotype, which reduces inflammation, stops additional damage to the tissue, and begins tissue regeneration .
Normal healing vs chronic inflammation
Macrophages display significant plasticity to promote both inflammation and resolution based on extracellular signals . In a healthy healing response, these cues can lead to the production of immunosuppressive cytokines, including TGF- and IL-10 to restore homeostasis following an initial inflammatory phase; however, without a signal to polarize, or with continued stimulation by antigen, inflammation continues indefinitely in chronic inflammatory diseases . In chronic or dysregulated inflammatory conditions, elevated numbers of neutrophils, monocytes, and macrophages persist . Neutrophils persist abnormally, releasing excessive proteases, reactive oxygen species (ROS), and neutrophil extracellular traps (NETs), which damage tissue and sustain inflammation, while macrophages are skewed toward a pro-inflammatory (M1) state, failing to transition to a reparative (M2) phenotype. This imbalance perpetuates inflammation,
delays healing in chronic conditions like non-healing diabetic wounds. Macrophage persistence at sites of inflammation can be associated with disease severity and progression, such as in rheumatoid arthritis . Proinflammatory immune cells overproduce matrix metalloproteinases (MMPs), ROS, and inflammatory cytokines, leading to increased tissue damage. Additional pro-inflammatory macrophage polarization occurs with further upregulated secretion of inflammatory cytokines, including TNF- , IL-1 , and IL-6 . Similar outcomes from increased macrophage infiltration and activation can be seen across a variety of inflammatory diseases and organ systems . In contrast, normal healing is associated with the programmed death and clearance of neutrophils, which initiates an anti-inflammatory cascade in the tissue .
Part 2. Cell death
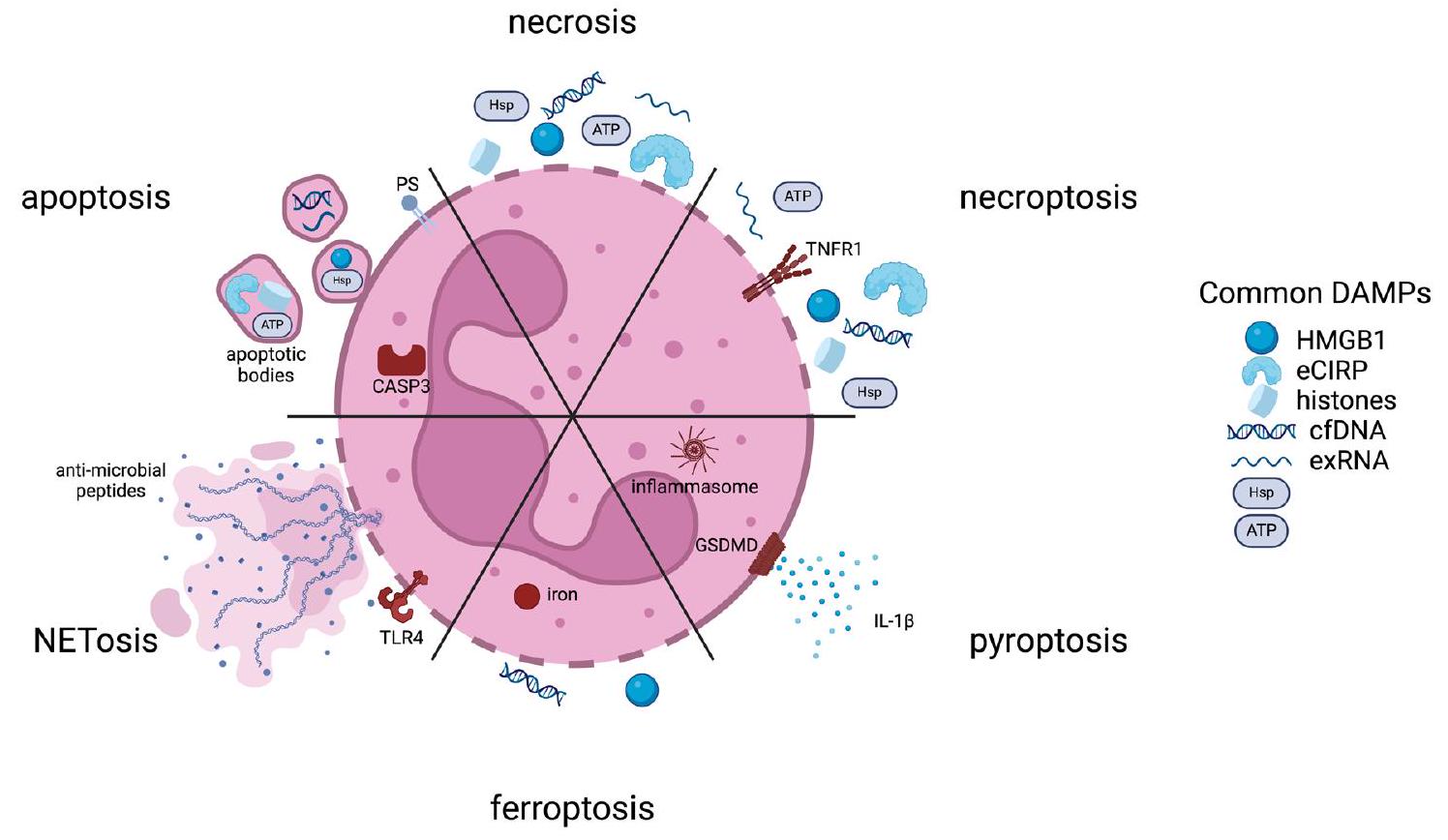
The precise timing, mode, and extent of neutrophil death and removal from the wound environment dictates subsequent inflammation and healing . Both the initial inflammatory phase and the subsequent anti-inflammatory, pro-regenerative phase are necessary for full wound healing. Here we describe various modes of neutrophil death and the resulting effects on inflammation, focusing on the impact on macrophages (Fig. 1).
Necrosis and apoptosis
To maintain normal homeostasis, the short half-lives of neutrophils ordinarily require a well-regulated program of apoptosis initiated through either intrinsic or extrinsic pathways. The intrinsic cascade begins through cytochrome C release from the mitochondrial to the cytoplasmic space, leading to the activation of caspase-3. In contrast, the extrinsic cascade begins with the engagement of cell death receptors on surfaces, subsequent activation of caspase- 8 /caspase-10 and convergence on caspase- . While nitric oxide is important in the wound healing response as an inflammatory mediator, it also plays a role in neutrophil apoptosis. Both the intrinsic and extrinsic apoptosis pathways involve the upregulation of inducible nitric oxide synthase (iNOS) and the production of nitric oxide which activate caspases-3 and -8, resulting in cell death . The mechanism of apoptosis leads to a series of active events that modulation inflammation downstream. In addition to the exposure of PS on apoptotic cell surfaces, a host of “eat me” signaling molecules are expressed on cell surfaces along with a downregulation of
“don’t eat me” signals, e.g. CD47, leading to efferocytosis and clearance by phagocytes .
Neutrophils are the most numerous immune cells in the human body with over 1 billion produced per day per kilogram. This, coupled with a lifespan of a few days at most, means that a majority of these cells are undergoing apoptosis daily, primarily in the bone marrow, liver, and spleen. The result of these processes are a production of interleukin-23 (IL-23), a reduction in granulocyte colony stimulating factor (G-CSF) levels, and a reduction in neutrophil production . This process then forms a self-regulatory loop of neutrophil production, mobilization, and decline. Apoptosis is therefore considered a noninflammatory mode of cell death in contrast with other neutrophil death mechanisms.
There are modes of apoptosis, however, that are immunogenic and result in the active release of damage-associated molecular patterns (DAMPs, Fig. 1). DAMPs released from dead and dying cells serve to signal downstream inflammatory mediators, including macrophages to recruit to sites of injury and enhance the inflammatory cascade . Canonical DAMPs include ATP, high mobility group box 1 (HMGB1), extracellular cold-inducible RNA-binding protein (eCIRP), histones, heat shock proteins (HSPs), extracellular RNAs (exRNAs), and cell-free DNA (cfDNA). DAMPs are also released passively via cell necrosis, that is exemplified by loss of cell membrane integrity. The packaging and identity of DAMPs released from programmed death mechanisms versus necrosis differ. Apoptosing neutrophils can secrete DAMPs via lysosomes, via release exosomal and ectosomal vesicles, apoptotic bodies, and via extracellular traps .
As necrosis is generally a passive process, release of biologically active agents is via membrane lysis (Fig. 1). As such, differences in those molecules exist. For example, cfDNA release from necrotic cells is generally much longer than fragmented cfDNA released from apoptotic cells . Other DAMPs, such as HMGB1, can be modified during certain modes of cell death, while following necrosis, it is not . Certain DAMPs are released at different stages; in particular, ATP is released early during apoptosis, while HMGB1 is released later as cell move towards secondary necrosis, or during primary necrosis . Membrane rupture can also be a cell-regulated process, termed
Fig. 1 | Schematic of characteristic cellular products produced from different modes of cell death. (PS phosphatidylserine; CASP3 caspase-3; TLR4 toll-like receptor 4; TNFR1 tumor necrosis factor receptor 1; GSDMD Gasdermin D;
HMGB1 high mobility group box 1; eCIRP extracellular cold-inducible RNAbinding protein; cfDNA cell-free DNA; exRNA extracellular RNAs; Hsp heat shock protein)
necroptosis, that occurs following receptor-mediated interactions. This results in the uncontrolled release of cytosolic DAMPs (Fig. 1).
Other forms of programmed cell death
A number of other inflammatory modes of neutrophil death have been identified that are particularly important for neutrophils. Pyroptosis is a highly inflammatory mode of cell death that is triggered intracellularly via the nucleotide-binding domain, leucine-rich repeat, and pyrin domaincontaining protein 3 (NLRP3) inflammasome or lipopolysaccharide (LPS) (Fig. 1) . Activation of caspases and involvement of Gasdermin D are hallmarks of this process resulting in DAMP release. Gasdermin D promotes pore formation in neutrophil membranes causing IL- to be released through the resulting pores along with other DAMPs. Ferroptosis is another cell death mode accompanied by loss of cell membrane integrity (Fig. 1) . Intracellular iron accumulation increases oxidative stress in the cell, leading to an increase in lipid peroxidation and disruption of the lipid bilayer. Important DAMPs released through this process include HMGB1 and cfDNA. Finally, a purposefully inflammatory mechanism of cell death is that of neutrophil extracellular traps (NETosis, Fig. 1) . NET production can be associated with both living or dying cells and involves the release of web-like, unraveled, chromatin structures containing DNA, histones, and antimicrobial peptides into the extracellular space. This process is triggered by microbial products and activated platelets, and the resulting structures are microbiocidal and highly inflammatory .
Cell death in inflammatory disease
There is evidence in various disease pathologies that neutrophil death is perturbed and can lead to increased tissue damage. When apoptosis is restricted due to disease, the resulting neutrophil survival can lead to increased tissue damage. For example, in chronic obstructive pulmonary disease and cystic fibrosis, persistent neutrophil activation is associated with high levels of inflammation in the lungs . Excesses of inflammatory modes of cell death can similarly lead to exacerbation of disease conditions. The DNA and histone release following NETosis have the potential to trigger autoimmune responses . Autoantibodies have been identified to target NETs in systemic lupus erythematosus and rheumatoid arthritis. In severe cases of infection or injury, the DAMP release following widespread neutrophil necrosis can lead to the excessive inflammation that is present in sepsis, acute respiratory distress syndrome, and the COVID-associated cytokine storm . Therapeutic strategies targeting neutrophil death have emerged either focusing on promoting the non-inflammatory apoptotic pathways or reducing the extent of pro-inflammatory generation of NETs and the release of DAMPs.
Stromal cell death as a model
Our own work examining stromal cell death has demonstrated that the mode of cell death matters with respect to downstream effects on immune cells . We initiated stromal cell death through two protocols: 1) cycles of freezing and thawing and 2) heating. From each of these populations of dead cells, we isolated two fractions: 1) the cell pellet, which contained apoptotic bodies and cell debris, and 2) the cell supernatant, which contained soluble components such as DAMPs and cytokines. We tested these fractions for their effect on macrophage phenotype in vitro and on their ability to enhance muscle recovery in a mouse model of hindlimb ischemia. Our hypothesis was that the release of DAMPs, including protein and nucleic acid content would be significantly greater in supernatants while the pellet fraction would be rich in PS-bound membrane components. We therefore hypothesized that the supernatant would be more inflammatory regardless of the mode of cell death.
We found that components from the freeze-and-thaw process (both the supernatant and the pellet) produced a clear anti-inflammatory and proregenerative response on cultured bone marrow-derived macrophages. The soluble supernatant from the freeze-and-thaw process also remarkably improved the relative blood flow in the affected foot in our mouse model to the level of of the intact foot when compared to the untreated, saline,
and heat supernatant. Freeze-and-thaw supernatant-treated muscles produced a significantly greater average tetanic force than that of untreated muscles and heat supernatant-treated muscles. This indicates that the mode of cell death was more important in inflammation than the cell fraction. In a separate laboratory, soluble factors isolated from freeze-and-thaw killed cells were loaded into a hydrogel foam to use their immunomodulatory and angiogenic effects to improve chronic wound healing .
Contributors to inflammation
As humans age, the accumulation of effects from repeated or low-grade infection, chronic inflammatory disease, obesity, and environmental factors leads to changes in the number and function of myeloid cells. This set of changes is one component of “inflammaging” – the accumulation of defects in the immune system associated with aging that lead to a persistent, low grade inflammatory state . Neutrophils in particular show a reduced ability to phagocytose infectious bacteria and a reduced microbiocidal ability in aged individuals. The cells’ ability to conduct immune surveillance is impaired due to a decreased sensitivity to antiapoptotic factors. Neutrophils in the elderly have a greater likelihood to undergo apoptosis without triggering inflammation. Advanced age can therefore predispose an individual to harmful infections and reduce the responsiveness to vaccines. Conversely, the low-grade inflammation that results during inflammaging is characterized by higher levels of proinflammatory cytokines such as IL-6, TNF- , and C-reactive protein, and the accumulation of DAMPs . Under these conditions, neutrophils can contribute to telomere shortening and the production of ROS, thus adding to oxidative stress and cellular aging. The clearance of dead and dying neutrophils is similarly perturbed. For example, ROS can cleave PS receptors on macrophages and reduce their efferocytosis ability . The reduction in efficient clearance of apoptotic neutrophils and the limitation on anti-inflammatory and tolerizing effects on macrophages may exacerbate the cycle of aging and inflammation.
Part 3. Cell Life
Quiescence vs senescence
Cells of the body can enter a state of quiescence, which is a reversible state of cell cycle arrest. This allows cells to become dormant while retaining the ability to reenter the cell cycle when required for growth or tissue repair. Cell cycle arrest occurs in the G0 phase with cells retaining only the maintenance of essential functions. Markers of proliferation are reduced while cell cycle inhibitors, like p27 , are elevated. Under the appropriate stimuli, typically the presence of growth factors, cells can resume their proliferative state and initiate cell division. In contrast, cell senescence is a state of permanent cell cycle arrest . In senescence, cells are arrested in the G1 phase. Markers of cell cycle inhibition are p16 and p21 . Senescence in culture and in the intact organism occurs through the accumulation of damage including DNA damage, oxidative stress, and activation of oncogenes. Unlike quiescent cells, arrest in the G1 phase means that cells are still metabolically active and synthesize and secrete biologically active molecules, termed the senescence-associated secretory phenotype (SASP, Table 1). Cell cycle inhibitors, the SASP, and senescence-associated -galactosidase (SA--gal) characterize cell senescence.
Features and consequences of senescence
The components of the SASP include pro-inflammatory cytokines, notably IL-6, IL-1 , and TNF- ; chemokines, like CCL2 and CXCL8; and proteases, including MMPs . These secreted proteins largely serve to augment the inflammatory response by recruiting and activating immune cells and remodeling the surrounding tissue. In cancer, senescent cells within solid tumors can be tumor-suppressive by limiting cell division of damaged cells that would contribute to tumor growth . Conversely, the SASP can serve a pro-tumor function by perturbing immune cells and promoting cellular invasion of the surrounding extracellular matrix . Therapeutic interventions have been proposed to reduce the number of senescent cells through senolytics (cell killing) or senomorphopics (cell modulation). Several such
Table 1 | Senescence-associated secretory phenotype (SASP) vs. Macrophage Phenotype
SASP
M1
M2a
M2b
M2c
M2d
TNF-a
TGF-
IFN-
IL-1a
IL-1
IL-1RA
IL-6
IL-7
IL-8/CXCL8
IL-10
IL-12
IL-13
IL-15
IL-23
IL-27
GM-CSF
VEGF
CCL1/I-309
CCL2/MCP-1
/
CCL3/MIP-1a
CCL4/MIP-1
CCL5/RANTES
CCL8/MCP-2
CCL11/Eotaxin-1
CCL13/MCP-4
CCL15/Leukotactin-1
CCL16/LEC
/
CCL17/TARC
CCL18/PARC
CCL20/MIP-3a
/
CCL22/MDC
CCL24/Eotaxin-2
CCL26/Eotaxin-3
CXCL1/GRO-
CXCL2/GRO-
CXCL3/GRO-
CXCL5/ENA-78
CXCL9/MIG
CXCL10/IP-10
/
CXCL11/I-TAC
/
CXCL13/BCA-1
CXCL16/SR-PSOX
M1, pro-inflammatory macrophage; M2, anti-inflammatory macrophage (M2a, wound healing; M2b, regulatory; M2c, tissue repair; M2d, pro-angiogenic); TNF- , tumor necrosis factor ; TGF- , transforming growth factor ; IFN- , interferon- ; IL, interleukin; IL-1RA, interleukin-1 receptor antagonist; GM-CSF, granulocyte-macrophage colony-stimulating factor; VEGF, vascular endothelial growth factor; CCL, chemokine (C-C motif) ligand; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; RANTES, regulated on activation normal T-cell expressed and secreted; LEC, liver-expressed chemokine; TARC, thymus- and activation-regulated chemokine; PARC, pulmonary- and activation-regulated chemokine; MDC, macrophage-derived chemokine; CXCL, chemokine(C-XC motif) ligand; GRO, growth-regulated oncogene; ENA-78, epithelial-derived neutrophil-activating peptide 78; MIG, monokine induced by IFN- ; IP-10, IFN- -induced protein 10; I-TAC, IFN-inducible Tcell a chemoattractant; BCA-1, B cell-attracting chemokine 1; SR-PSOX, scavenger receptor that binds phosphatidylserine and oxidized lipids. Check marks indicate production of cytokine or chemokine in each cell phenotype.
Table 2 | Selected therapeutic approaches to chronic inflammation
Immune Target
Change in Function
Ref
Senolytics
quercetin
Antioxidant that inhibits LPS-induced polarization/ skews balance towards M2
↓ M1
↑ M2
76,77
dasatinib
Tyrosine kinase inhibitor used in combination with quercetin/ depletes tumor-associated macrophages
↓ M1
↑ M2
↓ total macrophages in tumor
78,79
fisetin
Reduces pro-inflammatory polarization
↓ M1
80
Biologics
Anti-CD47
Opsonizes senescent cells to reduce immune evasion
↑ clearance of senescent neutrophils
81
MFG-E8
Bridges PS on apoptotic cell and on macrophage to increase efferocytosis
↑ angiogenesis
↓ time to wound closure
82,83
Nanoparticles
PS
Initiates PS-dependent efferocytosis and anti-inflammatory cascade in macrophages
↓ M1
↑ M2
↑ macrophage uptake
84,89,91
Neutrophil membrane
Binds inflammatory molecules to act as a neutrophil decoy
↓ chondrocyte activation and apoptosis
↓ knee diameter, cartilage damage, and serum TNF- and IL-1 in RA model
88
TNF-a siRNA
Inhibits translation of TNF-a
↓ bone loss and paw thickness in RA model
97
M1, pro-inflammatory macrophage; M2, anti-inflammatory macrophage; LPS, lipopolysaccharide; CD, cluster of differentiation; MGF-E8, Milk fat globule epidermal growth factor 8; PS, phosphatidylserine; TNF, tumor necrosis factor; IL, interleukin; RA, rheumatoid arthritis; siRNA, small interfering RNA.
strategies can also affect immune cells and the interaction between senescent and immune cells.
Senescence and the innate immune system
While much is known about how cells of various tissues senesce during disease and aging, less is clear about cells of the innate immune system. The pro-inflammatory nature of the SASP is thought to be one of the primary contributors towards inflammaging. Ordinarily, the cells of the innate immune system, including macrophages, neutrophils, and natural killer cells can recognize and remove senescent cells. During aging, senescent cells accumulate, likely a result from deficiencies in immune clearance functions. It has been demonstrated that mice with reduced immune cell cytotoxic function had higher levels of senescent cells along with increased levels of chronic inflammatory markers . It has also been shown that senescent cells suppress macrophagemediated corpse removal via upregulation of CD47 which serves as a “don’t eat me” signal to macrophages . It is therefore possible that the cumulative effects of inflammaging on immune cells form a positive feedback loop that limits senescent cell removal, increases the SASP, and contributes towards higher levels of chronic inflammation, senescence, and inflammaging .
Macrophage senescence
While there have now been a number of reports regarding macrophage senescence , it is important to distinguish between cellular senescence and the functional reduction of immune cells in the aging organism . While it is clear that immune function (e.g. phagocytosis, cell removal, antigen presentation, etc.) is reduced upon aging in the organism, the exact nature of macrophage cellular senescence is unclear. Like much of the macrophage literature, there are a number of reports that in aged humans and mice, the macrophage exhibits an anti-inflammatory, pro-regenerative phenotype, while in other studies the converse is true . In cell culture, stimulation of macrophages with LPS, radiation, or oxidative stress results in the classic hallmarks of senescence, i.e. – cell cycle arrest, increase of lysosomal volume, expression of the SASP, and upregulation of senescence markers such as SA--gal and p16 . The aging of mice has also been associated with SA- -gal and p16 positive macrophage accumulation . What is particularly disconcerting is that there is no bright line differentiating these
characteristics from a pro-inflammatory macrophage that has been stimulated by LPS or IFN- (Table 1) .
Senescence as a mal-adaptive macrophage mimicry
If there is little or nothing that exclusively differentiates the proinflammatory macrophage from a senescent macrophage, it is worth noting that many of those features are also present in other senescent cells of non-immune origin . The SASP, reduction in proliferation, increase in lysosomal volume, and ability of non-professional phagocytotic cells to phagocytose cargo have all been shown to increase upon senescence. For example, senescent epithelial cells demonstrate increased uptake of bacteria when under oxidative stress . It is an interesting approach to studying senescence to think of it as a process where cells adapt to perform some of the functions of immune cells . For example, it is possible that upon damage associated with aging, chronic diseases or environmental effects, the cell clearance and efferocytosis functions of macrophages are reduced, leading to persistent low-level inflammation, and proceeding towards a mal-adaptive response in other tissue cells . In effect, mimicking the phenotype of inflammatory macrophages trying to make up for their lost function. This process then serves in a positive feedback loop to accelerate aging of the immune system.
Part 4. Therapeutic implications (Table 2)
Senolytics
Based on the similarity between senescent cells and inflammatory macrophages, it stands to reason that senolytic or senomorphic drugs would have potent effects on cells of the immune system and indeed this is true. The first senolytic drugs in clinical trials, quercetin and dasatinib, have both reported effects on enhancing the polarization of pro-inflammatory macrophages to pro-regenerative/ anti-inflammatory macrophages . Dasatinib has also been reported to deplete macrophages in solid tumors . Another senolytic, fisetin, has been shown to polarize macrophages in culture away from a pro-inflammatory, LPS-induced phenotype . While this is a rich drug discovery space, it is important to note that depleting macrophages or their inflammatory effects can have potentially widespread negative impacts on wound healing, tumor surveillance, infection resistance, and vaccine potency.
Targeting the neutrophil-macrophage interaction
Biologic approaches towards senolytics include antibodies against surface molecules like CD47 . This limits the senescent cells from evading immune system recognition in order for macrophages to remove them, but may have side effects associated with recognition of self from non-self. Along a similar approach, it may be helpful to focus on the pivotal interaction between apoptotic neutrophils and efferocytosing macrophages. For example, there are dozens of known receptor-mediated interactions facilitating recognition and engulfment. In particular, PS is recognized on apoptotic cell surfaces through the TAM family of receptors (Tyro3/Axl/MerTK) and integrins on macrophages. These receptor families engage via binding proteins, milk fat globule-EGF factor 8 (MFG-E8) bridges PS and integrins while growth arrest-specific protein 6 (Gas6) and protein S bridge PS and the TAM receptors. Exogenous administration of recombinant versions of these proteins or their fragments may be able to enhance efferocytosis and facilitate the transition to an anti-inflammatory phenotype while minimizing negative effects. In particular, MFG-E8 has been shown to attenuate inflammation following injury . Therapeutic potential was demonstrated through the use of a topical recombinant MFG-E8 that accelerated the resolution of wound inflammation, enhanced angiogenesis, and decreased the time to wound closure in a diabetic wound model .
Strategy inspired by efferocytosis of apoptotic cells
Clearly, the interaction and clearance of dead and dying neutrophils by macrophages is critical for maintenance of homeostasis and controlling the sequence of events that promote productive wound healing. To explore this process for potential therapeutic use, our group has developed a cell membrane-derived nanoparticle coating to mimic characteristic features of the apoptotic cell surface and ultimately facilitate a reduction in inflammatory macrophage response through existing physiologic pathways . We began with poly(lactic-co-glycolic) acid (PLGA) as a nanoparticle carrier for our membrane-derived coating, chosen for its inert, biodegradable, and biocompatible properties. We coated the surface of PLGA nanoparticles with isolated cell plasma membrane doped with exogenous, synthetic PS. Cell membranes have previously been used to coat nanoparticles, although they have provided utility through specific proteins present on the membrane surface that can interact with their environment or nearby cells . In our work, we focused on the impact of the bioactive phospholipid PS, presented in the context of a generic stromal cell membrane, to mimic an apoptotic body. We demonstrated that the combination of membraneassociated proteins and PS caused a shift in macrophage phenotype from pro-inflammatory toward pro-regenerative more than PS liposomes alone. In a separate nanoparticle system, we demonstrated that even without membrane-associated proteins, PS presentation on the surface of lipidpolymer nanoparticles significantly increased phagocytosis by macrophages over nanoparticles without .
PS-containing liposomes have been proposed as drug delivery vehicles to target macrophages and other antigen-presenting cells . PS has also been proposed as a component of solid-lipid nanoparticles for RNA vaccine delivery. A concern for these strategies, as well as our cell membrane-coated polymeric nanoparticles, is the role that PS plays in blood coagulation. PS can bind coagulation proteins and participate in the coagulation cascade. Our laboratory, to translate membrane coated, PS-containing PLGA nanoparticles to an in vivo model, developed a PEGylation strategy to improve both long-term nanoparticle stability and targeting to sites of inflammation through surface functionalization .
The lipid bilayer of our PLGA nanoparticles can be used to anchor molecules to the particle surface through hydrophobic interactions. Molecules conjugated to lipophilic moieties have been attached to cell and membrane ghost surfaces by simply incubating the molecule and membrane together and allowing hydrophobic interactions to drive insertion of the lipophilic moiety into the lipophilic portion of the membrane . This process, sometimes called membrane painting, requires no additional conjugation reactions. We took advantage of the low pH of chronically inflamed sites relative to normal physiological . Surface-functionalized nanoparticles with acid-sensitive sheddable polyethylene glycol (PEG) release can then preferentially collect/accumulate in these sites . Results from our work show that through a membrane painting approach, particle localization to sites of inflammation in vivo was increased. These particles were also preferentially taken up by macrophages within areas of inflammation. Also, in a model of lipopolysaccharide (LPS)-induced chronic inflammation, these nanoparticles reduced inflammation and promoted a regenerative phenotype better than control nanoparticles.
Conclusions
In examining the life cycle and interactions of neutrophils and macrophages during normal homeostasis, productive wound healing, chronic inflammatory disease, and aging, it is clear that the death of neutrophils, and their clearance and efferocytosis by macrophages, play major roles in health and disease. Arguably, the clearance of neutrophils by macrophages is the most frequent immune cell interaction in the human body, and this interaction is largely immunologically invisible. Alterations in the mode and result of neutrophil death can have profound effects on macrophage phenotype and the resulting tissue environment. Similarly, a reduction in the function of the innate immune system with aging can perturb this system, resulting in lowlevel chronic inflammation, cell senescence, and lack of cell removal, thus contributing to a vicious cycle of inflammation. Harnessing the mechanism whereby macrophages recognize, eliminate, and polarize as a result of cell apoptosis is likely a rich search space for the development of novel drugs and drug delivery strategies.
Data Availability
No datasets were generated or analysed during the current study.
Received: 22 October 2024; Accepted: 23 January 2025;
Published online: 04 March 2025
References
Rodrigues, M., Kosaric, N., Bonham, C. A. & Gurtner, G. C. Wound Healing: A Cellular Perspective. Physiol. Rev. 99, 665-706 (2019).
Singh, S., Young, A. & McNaught, C.-E. The physiology of wound healing. Surg. (Oxf.) 35, 473-477 (2017).
Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front Physiol. 9, 113 (2018).
Tidball, J. G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 1, 2029-2062 (2011).
Fullerton, J. N. & Gilroy, D. W. Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov. 15, 551-567 (2016).
Iwasaki, A. & Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 327, 291-295 (2010).
Savill, J. S. et al. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest 83, 865-875 (1989).
Sica, A. & Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest 122, 787-795 (2012).
Hammers, D. W. et al. Anti-inflammatory macrophages improve skeletal muscle recovery from ischemia-reperfusion. J. Appl Physiol. 118, 1067-1074 (2015).
Tidball, J. G. & Villalta, S. A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1173-R1187 (2010).
Patel, U. et al. Macrophage polarization in response to epigenetic modifiers during infection and inflammation. Drug Discov. Today 22, 186-193 (2017).
Chazaud, B. et al. Dual and beneficial roles of macrophages during skeletal muscle regeneration. Exerc Sport Sci. Rev. 37, 18-22 (2009).
Tidball, J. G. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R345-R353 (2005).
Novak, M. L. & Koh, T. J. Phenotypic transitions of macrophages orchestrate tissue repair. Am. J. Pathol. 183, 1352-1363 (2013).
Kharraz, Y., Guerra, J., Mann, C. J., Serrano, A. L. & MunozCanoves, P. Macrophage Plasticity and the Role of Inflammation in Skeletal Muscle Repair. Mediat Inflamm. (2013).
Fadok, V. A. et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207-2216 (1992).
Li, M. O., Sarkisian, M. R., Mehal, W. Z., Rakic, P. & Flavell, R. A. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 302, 1560-1563 (2003).
Segawa, K. & Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 25, 639-650 (2015).
Huynh, M.-L. N., Fadok, V. A. & Henson, P. M. Phosphatidylserinedependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Invest. 109, 41-50 (2002).
Mantovani, A. et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677-686 (2004).
Reimold, A. M. TNFa as Therapeutic Target: New Drugs, More Applications. Curr. Drug Targets – Inflamm. Allergy 1, 377-392 (2002).
Herrero-Cervera, A., Soehnlein, O. & Kenne, E. Neutrophils in chronic inflammatory diseases. Cell Mol. Immunol. 19, 177-191 (2022).
Kapellos, T. S. et al. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front Immunol. 10, 2035 (2019).
Parisi, L. et al. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 8917804 (2018).
Guo, Q. et al. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 6, 15 (2018).
Jain, S., Tran, T. H. & Amiji, M. Macrophage repolarization with targeted alginate nanoparticles containing IL-10 plasmid DNA for the treatment of experimental arthritis. Biomaterials 61, 162-177 (2015).
Li, J., Hsu, H. C. & Mountz, J. D. Managing macrophages in rheumatoid arthritis by reform or removal. Curr. Rheumatol. Rep. 14, 445-454 (2012).
Yanni, G., Whelan, A., Feighery, C. & Bresnihan, B. Synovial tissue macrophages and joint erosion in rheumatoid arthritis. Ann. Rheum. Dis. 53, 39-44 (1994).
Kinne, R. W., Brauer, R., Stuhlmuller, B., Palombo-Kinne, E. & Burmester, G. R. Macrophages in rheumatoid arthritis. Arthritis Res. 2, 189-202 (2000).
Benveniste, E. N. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J. Mol. Med. (Berl.) 75, 165-173 (1997).
Clark, R. A. & Kupper, T. S. Misbehaving macrophages in the pathogenesis of psoriasis. J. Clin. Invest 116, 2084-2087 (2006).
Mahida, Y. R. The key role of macrophages in the immunopathogenesis of inflammatory bowel disease. Inflamm. Bowel Dis. 6, 21-33 (2000).
Loh, W. & Vermeren, S. Anti-Inflammatory Neutrophil Functions in the Resolution of Inflammation and Tissue Repair. Cells 11, 4076 (2022).
Tu, H. et al. Dying to Defend: Neutrophil Death Pathways and their Implications in Immunity. Adv. Sci. (Weinh.) 11, e2306457 (2024).
Perez-Figueroa, E., Alvarez-Carrasco, P., Ortega, E. & MaldonadoBernal, C. Neutrophils: Many Ways to Die. Front Immunol. 12, 631821 (2021).
Maianski, N. A., Maianski, A. N., Kuijpers, T. W. & Roos, D. Apoptosis of neutrophils. Acta Haematol. 111, 56-66 (2004).
Dubey, M. et al. Nitric oxide-mediated apoptosis of neutrophils through caspase- 8 and caspase-3-dependent mechanism. Cell Death Dis. 7, e2348 (2016).
Lawrence, D. W., King, S. B., Frazier, W. A. & Koenig, J. M. Decreased CD47 expression during spontaneous apoptosis targets neutrophils for phagocytosis by monocyte-derived macrophages. Early Hum. Dev. 85, 659-663 (2009).
Von Vietinghoff, S. & Ley, K. Homeostatic regulation of blood neutrophil counts. J. Immunol. 181, 5183-5188 (2008).
Denning, N. L., Aziz, M., Gurien, S. D. & Wang, P. DAMPs and NETs in Sepsis. Front Immunol. 10, 2536 (2019).
Murao, A., Aziz, M., Wang, H., Brenner, M. & Wang, P. Release mechanisms of major DAMPs. Apoptosis 26, 152-162 (2021).
Marki, A. & Ley, K. The expanding family of neutrophil-derived extracellular vesicles. Immunol. Rev. 312, 52-60 (2022).
de Miranda, F. S. et al. Properties and Application of Cell-Free DNA as a Clinical Biomarker. Int J. Mol. Sci. 22, 9110 (2021).
Yang, H., Antoine, D. J., Andersson, U. & Tracey, K. J. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 93, 865-873 (2013).
Dosch, M., Gerber, J., Jebbawi, F. & Beldi, G. Mechanisms of ATP Release by Inflammatory Cells. Int J. Mol. Sci. 19, 1222 (2018).
Hou, J., Hsu, J. M. & Hung, M. C. Molecular mechanisms and functions of pyroptosis in inflammation and antitumor immunity. Mol. Cell 81, 4579-4590 (2021).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060-1072 (2012).
Guimaraes-Costa, A. B. et al. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl Acad. Sci. USA 106, 6748-6753 (2009).
Thiam, H. R., Wong, S. L., Wagner, D. D. & Waterman, C. M. Cellular Mechanisms of NETosis. Annu Rev. Cell Dev. Biol. 36, 191-218 (2020).
Vorobjeva, N. V. & Chernyak, B. V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology.Biochemistry (Mosc) 85, 1178-1190 (2020).
Ding, L., Yang, J., Zhang, C., Zhang, X. & Gao, P. Neutrophils Modulate Fibrogenesis in Chronic Pulmonary Diseases. Front Med. (Lausanne) 8, 616200 (2021).
Huang, S. U. & O’Sullivan, K. M. The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs. Int J. Mol. Sci. 23, 3793 (2022).
Land, W. G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome-with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 22, 141-160 (2021).
Huang, W., Kraynak, C. A., Bender, E. C., Farrar, R. P. & Suggs, L. J. Soluble components from mesenchymal stromal cell processing exert anti-inflammatory effects and facilitate ischemic muscle regeneration. Cytotherapy 25, 375-386 (2023).
Lan, Z. et al. Hydrogel foam dressings with angiogenic and immunomodulatory factors from mesenchymal stem cells. J. Biomed. Mater. Res. A 112, 1388-1398 (2024).
Franceschi, C. & Bonafe, M. Centenarians as a model for healthy aging. Biochem Soc. Trans. 31, 457-461 (2003).
Byrne, T., Cooke, J., Bambrick, P., McNeela, E. & Harrison, M. Circulating inflammatory biomarker responses in intervention trials in frail and sarcopenic older adults: A systematic review and metaanalysis. Exp. Gerontol. 177, 112199 (2023).
Naeini, M. B., Bianconi, V., Pirro, M. & Sahebkar, A. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell Mol. Biol. Lett. 25, 23 (2020).
Campisi, J. Aging, cellular senescence, and cancer. Annu Rev. Physiol. 75, 685-705 (2013).
Huang, W., Hickson, L. J., Eirin, A., Kirkland, J. L. & Lerman, L. O. Cellular senescence: the good, the bad and the unknown. Nat. Rev. Nephrol. 18, 611-627 (2022).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853-2868 (2008).
Coppe, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev. Pathol. 5, 99-118 (2010).
Haston, S. et al. Clearance of senescent macrophages ameliorates tumorigenesis in KRAS-driven lung cancer. Cancer Cell 41, 1242-1260.e1246 (2023).
Ovadya, Y. et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 9, 5435 (2018).
Schloesser, D. et al. Senescent cells suppress macrophagemediated corpse removal via upregulation of the CD47-QPCT/L axis. J. Cell Biol. 222, e202207097 (2023).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100-105 (2021).
Sharma, R. Perspectives on the dynamic implications of cellular senescence and immunosenescence on macrophage aging biology. Biogerontology 22, 571-587 (2021).
Wang, L. et al. Macrophage senescence in health and diseases. Acta Pharm. Sin. B 14, 1508-1524 (2024).
McQuattie-Pimentel, A. C. et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J. Clin. Invest 131, e140299 (2021).
Hall, B. M. et al. p16(lnk4a) and senescence-associated betagalactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY) 9, 1867-1884 (2017).
Wang, H. et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosisassociated lipid uptake. Aging (Albany NY) 12, 9240-9259 (2020).
Suzuki, K., Susaki, E. A. & Nagaoka, I. Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis. Int J. Mol. Sci. 23, 11148 (2022).
Robledo, E., Benito Rodriguez, P. G., Vega, I. A., Colombo, M. I. & Aguilera, M. O. Staphylococcus aureus phagocytosis is affected by senescence. Front Aging 4, 1198241 (2023).
Behmoaras, J. & Gil, J. Similarities and interplay between senescent cells and macrophages. J. Cell Biol. 220, e202010162 (2021).
Ogata, Y. et al. SASP-induced macrophage dysfunction may contribute to accelerated senescent fibroblast accumulation in the dermis. Exp. Dermatol 30, 84-91 (2021).
Tsai, C. F. et al. Regulatory Effects of Quercetin on M1/M2 Macrophage Polarization and Oxidative/Antioxidative Balance. Nutrients 14, 67 (2021).
. et al. Quercetin inhibited LPS-induced cytokine storm by interacting with the AKT1-FoxO1 and Keap1-Nrf2 signaling pathway in macrophages. Sci. Rep. 14, 20913 (2024).
Brownlow, N., Mol, C., Hayford, C., Ghaem-Maghami, S. & Dibb, N. J. Dasatinib is a potent inhibitor of tumour-associated macrophages, osteoclasts and the FMS receptor. Leukemia 23, 590-594 (2009).
Hickson, L. J. et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446-456 (2019).
Hada, Y., Uchida, H. A. & Wada, J. Fisetin Attenuates Lipopolysaccharide-Induced Inflammatory Responses in Macrophage. Biomed. Res. Int. 2021, 5570885 (2021).
Yang, H., Xun, Y. & You, H. The landscape overview of CD47-based immunotherapy for hematological malignancies. Biomark. Res. 11, 15 (2023).
Deroide, N. et al. MFGE8 inhibits inflammasome-induced IL-1beta production and limits postischemic cerebral injury. J. Clin. Invest 123, 1176-1181 (2013).
Das, A. et al. Correction of MFG-E8 Resolves Inflammation and Promotes Cutaneous Wound Healing in Diabetes. J. Immunol. 196, 5089-5100 (2016).
Kraynak, C. A., Yan, D. J. & Suggs, L. J. Modulating inflammatory macrophages with an apoptotic body-inspired nanoparticle. Acta Biomater. 108, 250-260 (2020).
Fang, R. H. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 14, 2181-2188 (2014).
Hu, C. M. et al. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl Acad. Sci. USA 108, 10980-10985 (2011).
Thamphiwatana, S. et al. Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc. Natl Acad. Sci. USA 114, 11488-11493 (2017).
Zhang, Q. et al. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 13, 1182-1190 (2018).
Bender, E. C., Sircar, A. J., Taubenfeld, E. K. & Suggs, L. J. Modulating Lipid-Polymer Nanoparticles’ Physicochemical Properties to Alter Macrophage Uptake.ACS Biomater. Sci. Eng. 10, 2911-2924 (2024).
Bender, E. C., Kraynak, C. A., Huang, W. & Suggs, L. J. Cell-Inspired Biomaterials for Modulating Inflammation. Tissue Eng. Part B Rev. 28, 279-294 (2022).
Kraynak, C. A. et al. Apoptotic body-inspired nanoparticles target macrophages at sites of inflammation to support an antiinflammatory phenotype shift. Int J. Pharm. 618, 121634 (2022).
Chen, H. et al. Lipid insertion enables targeted functionalization of paclitaxel-loaded erythrocyte membrane nanosystem by tumorpenetrating bispecific recombinant protein. Int J. Nanomed. 13, 5347-5359 (2018).
Shi, G., Mukthavaram, R., Kesari, S. & Simberg, D. Distearoyl anchor-painted erythrocytes with prolonged ligand retention and circulation properties in vivo. Adv. Health. Mater. 3, 142-148 (2014).
Gerry, A. B. & Leake, D. S. Effect of low extracellular pH on NF-kappaB activation in macrophages. Atherosclerosis 233, 537-544 (2014).
Okajima, F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal 25, 2263-2271 (2013).
Rajamaki, K. et al. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J. Biol. Chem. 288, 13410-13419 (2013).
Aldayel, A. M. et al. Lipid nanoparticles with minimum burst release of TNF-alpha siRNA show strong activity against rheumatoid arthritis unresponsive to methotrexate. J. Control Release 283, 280-289 (2018).
Kalia, J. & Raines, R. T. Hydrolytic stability of hydrazones and oximes. Angew. Chem. Int Ed. Engl. 47, 7523-7526 (2008).
Wang, J. L. et al. Aerosolizable siRNA-encapsulated solid lipid nanoparticles prepared by thin-film freeze-drying for potential pulmonary delivery. Int J. Pharm. 596, 120215 (2021).
Ouvrier, B., Ismael, S. & Bix, G. J. Senescence and SASP Are Potential Therapeutic Targets for Ischemic Stroke. Pharmaceuticals 17, 312 (2024).
Arabpour, M., Saghazadeh, A. & Rezaei, N. Anti-inflammatory and M2 macrophage polarization-promoting effect of mesenchymal stem cellderived exosomes. Int. Immunopharmacol. 97, 107823 (2021).
Arango Duque, G. & Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 5, 491 (2014).
Gharavi, A. T., Hanjani, N. A., Movahed, E. & Doroudian, M. The role of macrophage subtypes and exosomes in immunomodulation. Cell. Mol. Biol. Lett. 27, 1-18 (2022).
Abdelaziz, M. H. et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J. Transl. Med. 18, 58 (2020).
Roszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat Inflamm. 2015, 816460 (2015).
Wang, B., Han, J., Elisseeff, J. H. & Demaria, M. The senescenceassociated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell Biol. 1-21 (2024).
Gessani, S. & Belardelli, F. IFN- Expression in Macrophages and Its Possible Biological Significance. Cytokine Growth Factor Rev. 9, 117-123 (1998).
Sims, J. E. & Smith, D. E. The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10, 89-102 (2010).
Stow, J. L. & Murray, R. Z. Intracellular trafficking and secretion of inflammatory cytokines. Cytokine Growth Factor Rev. 24, 227-239 (2013).
Ma, X. et al. Regulation of IL-10 and IL-12 production and function in macrophages and dendritic cells. F1000 Res. 4, F1000-Faculty Rev1465 (2015).
Orecchioni, M., Ghosheh, Y., Pramod, A. B. & Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 10, 1084 (2019).
Tang, C., Chen, S., Qian, H. & Huang, W. Interleukin-23: as a drug target for autoimmune inflammatory diseases. Immunology 135, 112-124 (2012).
Acknowledgements
This work was supported by a grant from the National Institutes of Health (R56EB032211-01) to L. Suggs.
Author contributions
E.C.B. edited the manuscript and prepared the figure. H.S.T. compiled the table. L.J.S. conceived of and wrote the initial draft.
Competing interests
L.J.S. is a guest editor for the ‘Orchestrators of immune regulation, homeostasis and aging’ collection, however, she was not involved in the peer review process of this article.
Additional information
Correspondence and requests for materials should be addressed to Laura J. Suggs.
Reprints and permissions information is available at http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.