DOI: https://doi.org/10.1038/s41467-024-46752-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38499522
تاريخ النشر: 2024-03-18
الانتقائية المتوسطة بواسطة جهد النبض للأكسدة الكهروكيميائية للجلسرين إلى حمض الجلسريك
تاريخ القبول: 7 مارس 2024
تاريخ النشر على الإنترنت: 18 مارس 2024
(أ) تحقق من التحديثات
الملخص
منع تعطيل المحفزات القائمة على المعادن النبيلة بسبب الأكسدة الذاتية والامتصاص السام هو تحدٍ كبير في الأكسدة الكهروكيميائية العضوية. في هذه الدراسة، نستخدم استراتيجية التحليل الكهربائي بالجهد النبضي للأكسدة الكهروكيميائية الانتقائية للجلسرين إلى حمض الجلسريك على محفز قائم على البلاتين. تكشف تقنيات الطيف الكهرومغناطيسي بالأشعة تحت الحمراء وتحليل الطيف الضوئي للأشعة السينية شبه في الموقع، والمحاكاة باستخدام العناصر المحدودة أن الجهد النبضي يمكن أن يخصص أكسدة المحفز والبيئة الدقيقة السطحية. هذا يمنع التراكم المفرط للأنواع الوسيطة السامة ويحرر المواقع النشطة لإعادة امتصاص مادة OH و الجلسرين. تؤدي استراتيجية التحليل الكهربائي بالجهد النبضي إلى انتقائية أعلى لحمض الجلسريك (81.8%) مقارنة بالأكسدة الكهروكيميائية ذات الجهد الثابت مع
النتائج
قيود CE على GEOR باستخدام محفز قائم على البلاتين
نظام الإحداثيات السلبية. هذه ظاهرة تسمم المحفز النموذجية للمحفزات القائمة على البلاتين
الأداء الكهروتحفيزي لـ GEOR مع PE
تأثير الوضع الكهربائي على الخصائص الفيزيائية والكيميائية Pt@G خلال GEOR
على التوالي. اختيارية ومعدل تحويل GLY مع 20 مللي مول من GLY تحت وضع التحليل الكهربائي لـ PE3. النسب المئوية باللون الأسود تشير إلى الاختيارية المقابلة لـ GLA.
أثر الوضع الكهربائي على البيئة الدقيقة السطحية المحيطة بـ Pt@G أثناء GEOR
وقت التحليل الكهربائي (الشكل 4 أ والشكل التكميلي 18 ب). وصلت شدة هذه المناطق تحت CE إلى القيمة القصوى بشكل أسرع في
مجموعات الجليسرول، زادت كـ
محاكاة العناصر المحدودة والفهم الميكانيكي للبولي إيثيلين لجهاز GEOR
إلكتروليت قلوي. الصور الثلاثة المدخلة تعرض الهندسة المثلى لامتصاص الأنواع الثلاثة المقابلة على سطح Pt (111). اللون لكل عنصر هو أصفر برتقالي لـ H، أزرق فاتح لـ O، بنفسجي لـ C، ورمادي لـ Pt، على التوالي.
الجهد وتركيز المتفاعلات والمنتجات داخل الطبقة المزدوجة الكهربائية (EDL)
و
إلى
نقاش
طرق
تحضير Pt@G
بـ HF و
التوصيفات
القياسات الكهروكيميائية
تحليل المنتج
دراسة FTIR في الموقع
بروتوكول PE3. أ، ب، ج البيانات التي تم الحصول عليها من خلال حزمة المحاكاة الفيزيائية المتعددة COMSOL. يتم تمثيل المحفز بـ
محملة بالمحفز، قضيب الجرافيت و
حساب طاقة الامتزاز
محاكاة العناصر المحدودة للطبقة المزدوجة المنتشرة
محاكاة العناصر المحدودة لـ GEOR
توفر البيانات
References
- Sharninghausen, L. S., Campos, J., Manas, M. G. & Crabtree, R. H. Efficient selective and atom economic catalytic conversion of glycerol to lactic acid. Nat. Commun. 5, 5084 (2014).
- Alisson Dias da Silva, R., Ana Luíza Freitas, F., Antônio Ésio, B., Rita Maria de Brito, A. & Luiz Antônio Magalhães, P. Market prospecting and assessment of the economic potential of glycerol from biodiesel. In Biotechnological Applications of Biomass (eds Thalita Peixoto B., Thiago Olitta B., Luiz Carlos B.). IntechOpen (2020).
- Sheng, H. et al. Linear paired electrochemical valorization of glycerol enabled by the electro-Fenton process using a stable
cathode. Nat. Catal. 5, 716-725 (2022). - Katryniok, B. et al. Selective catalytic oxidation of glycerol: perspectives for high-value chemicals. Green Chem. 13, 1960-1979 (2011).
- Zhou, C., Beltramini, J. N., Fan, Y. & Lu, G. Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 37, 527-549 (2008).
- González-Cobos, J., Baranton, S. & Coutanceau, C. A systematic in situ infrared study of the electrooxidation of C3 alcohols on carbon-supported Pt and Pt-Bi catalysts. J. Phys. Chem. C 120, 7155-7164 (2016).
- Casebolt, R., Levine, K., Suntivich, J. & Hanrath, T. Pulse check: potential opportunities in pulsed electrochemical
reduction. Joule 5, 1987-2026 (2021). - Zhang, X. et al. Asymmetric low-frequency pulsed strategy enables ultralong
reduction stability and controllable product selectivity. J. Am. Chem. Soc. 145, 2195-2206 (2023). - Timoshenko, J. et al. Steering the structure and selectivity of
electroreduction catalysts by potential pulses. Nat. Catal. 5, 259-267 (2022). - Kim, C. et al. Tailored catalyst microenvironments for
electroreduction to multicarbon products on copper using bilayer ionomer coatings. Nat. Energy 6, 1026-1034 (2021). - Li, P. et al. Pulsed nitrate-to-ammonia electroreduction facilitated by tandem catalysis of nitrite intermediates. J. Am. Chem. Soc. 145, 6471-6479 (2023).
- Guo, M. et al. Pulsed electrocatalysis enabling high overall nitrogen fixation performance for atomically dispersed Fe on
. Angew. Chem. 135, e202217635 (2023). - Bu, Y. et al. Electrical pulse-driven periodic self-repair of Cu-Ni tandem catalyst for efficient ammonia synthesis from nitrate. Angew. Chem. Int. Ed. 62, e202217337 (2023).
- Ding, Y. et al. Pulsed electrocatalysis enables an efficient 2-electron oxygen reduction reaction for
production. J. Mater. Chem. A 9, 15948-15954 (2021). - Vincent, I. et al. Pulsed current water splitting electrochemical cycle for hydrogen production. Int. J. Hydrogen Energy 43, 10240-10248 (2018).
- Zhao, M. et al. Pd-Ru alloy nanocages with a face-centered cubic structure and their enhanced activity toward the oxidation of ethylene glycol and glycerol. Small Methods 4, 1900843 (2020).
- Fan, L. et al. Recent progress in electrocatalytic glycerol oxidation. Energy Technol. 9, 2000804 (2021).
- Zhou, Y., Shen, Y. & Xi, J. Seed-mediated synthesis of PtxAuy@Ag electrocatalysts for the selective oxidation of glycerol. Appl. Catal. B: Environ. 245, 604-612 (2019).
- Li, Z. et al. Alcohols electrooxidation coupled with
production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022). - Zhang, Y. et al. Potential-dynamic surface chemistry controls the electrocatalytic processes of ethanol oxidation on gold surfaces. ACS Energy Lett. 4, 215-221 (2019).
- Gao, F. et al. Shape-control of one-dimensional PtNi nanostructures as efficient electrocatalysts for alcohol electrooxidation. Nanoscale 11, 4831-4836 (2019).
- Zhang, Z. et al. Engineering phase and surface composition of
nanocatalysts: a strategy for enhancing CO tolerance. Nano Energy 34, 224-232 (2017). - Ren, Z. et al. Pt/ZrO2 catalyst with metal-support synergistic effect towards glycerol selective oxidation. Chem. Eng. J. 468, 143623 (2023).
- Li, J. et al. Tuning the product selectivity toward the high yield of glyceric acid in Pt-CeO2/CNT electrocatalyzed oxidation of glycerol. ChemCatChem 14, e202200509 (2022).
- Wu, G., Liu, Y., He, Y., Feng, J. & Li, D. Reaction pathway investigation using in situ Fourier transform infrared technique over Pt/CuO and
for selective glycerol oxidation. Appl. Catal. B: Environ. 291, 120061 (2021). - Dong, J. et al. Direct in situ raman spectroscopic evidence of oxygen reduction reaction intermediates at high-index
surfaces. J. Am. Chem. Soc. 142, 715-719 (2020). - Marković, N. M., Marinković, N. S. & Adžić, R. R. Electrosorption of hydrogen and sulphuric acid anions on single-crystal platinum stepped surfaces: Part I. The [110] zone. J. Electroanal. Chem. Interfacial Electrochem. 241, 309-328 (1988).
- Wang, Y. et al. Efficient electrocatalytic oxidation of glycerol via promoted
generation over single-atom-bismuth-doped spinel . ACS Catal 12, 12432-12443 (2022). - Sandrini, R. M. L. M. et al. Mechanistic aspects of glycerol electrooxidation on
electrode in alkaline media. Electrochem. Commun. 86, 149-152 (2018). - Hong, W., Shang, C., Wang, J. & Wang, E. Bimetallic PdPt nanowire networks with enhanced electrocatalytic activity for ethylene glycol and glycerol oxidation. Energy Environ. Sci. 8, 2910-2915 (2015).
- Zhou, P. et al. Heterogeneous-interface-enhanced adsorption of organic and hydroxyl for biomass electrooxidation. Adv. Mater. 34, 2204089 (2022).
- Yan, Y. et al. Electrocatalytic upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J. Am. Chem. Soc. 145, 6144-6155 (2023).
- Liu, X. & Yang, C. Electrocatalytic selective oxidation of glycerol to glyceric acid over efficient Pt/CNTs-CeO2 catalysts. Mater. Lett. 324, 132658 (2022).
- Zhou, Y., Shen, Y., Xi, J. & Luo, X. Selective electro-oxidation of glycerol to dihydroxyacetone by PtAg skeletons. ACS Appl. Mater. Interfaces 11, 28953-28959 (2019).
- Zhou, Y., Shen, Y. & Piao, J. Sustainable conversion of glycerol into value-added chemicals by selective electro-oxidation on Pt-based catalysts. ChemElectroChem 5, 1636-1643 (2018).
- Kim, Y. et al. The role of ruthenium on carbon-supported PtRu catalysts for electrocatalytic glycerol oxidation under acidic conditions. ChemCatChem 9, 1683-1690 (2017).
- Dai, C. et al. Electrochemical production of lactic acid from glycerol oxidation catalyzed by AuPt nanoparticles. J. Catal. 356, 14-21 (2017).
- Inoue, H. et al. Mechanism of glycerol oxidation reaction on silvermodified palladium electrode in alkaline medium. Int. J. Hydrogen Energy 43, 18664-18671 (2018).
- Copeland, J. R., Santillan, I. A., Schimming, S. M., Ewbank, J. L. & Sievers, C. Surface interactions of glycerol with acidic and basic metal oxides. J. Phys. Chem. C 117, 21413-21425 (2013).
- An, Z. et al. Pt1 enhanced C-H activation synergistic with
catalysis for glycerol cascade oxidation to glyceric acid. Nat. Commun. 13, 5467 (2022). - Mello, G. A. B., Busó-Rogero, C., Herrero, E. & Feliu, J. M. Glycerol electrooxidation on Pd modified Au surfaces in alkaline media: effect of the deposition method. J. Chem. Phys. 150, 041703 (2018).
- Xu, L. et al. Salting-out aldehyde from the electrooxidation of alcohols with 100% selectivity. Angew. Chem. Int. Ed. 61, e202210123 (2022).
- Zhang, L., Li, C. & Huang, J. A beginners’ guide to modelling of electric double layer under equilibrium, nonequilibrium and AC conditions. J. Electrochem. 28, 2108471 (2022).
- Zhang, L. et al. In vivo activation of pH-responsive oxidase-like graphitic nanozymes for selective killing of Helicobacter pylori. Nat. Commun. 12, 2002 (2021).
- Zhang, L. et al. A magnetocatalytic propelled cobalt-platinum@graphene navigator for enhanced tumor penetration and theranostics. CCS Chem. 4, 2382-2395 (2022).
- Obst, S. & Bradaczek, H. Molecular dynamics study of the structure and dynamics of the hydration shell of alkaline and alkaline-earth metal cations. J. Phys. Chem. 100, 15677-15687 (1996).
- Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
- Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية المتاحة على
https://doi.org/10.1038/s41467-024-46752-4.
يجب توجيه المراسلات وطلبات المواد إلى هوان بانغ أو يوقين زو.
http://www.nature.com/reprints
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
© المؤلفون 2024
المختبر الوطني الرئيسي للكيمياء/الاستشعار الحيوي والكيميوميتري، كلية الكيمياء والهندسة الكيميائية، جامعة هونان، تشانغشا 410000، جمهورية الصين الشعبية. مدرسة الكيمياء والهندسة الكيميائية، جامعة يانغتشو، يانغتشو 225009، جمهورية الصين الشعبية. المختبر الرئيسي للجلود في مقاطعة تشجيانغ، معهد المواد الجديدة والتقنيات الصناعية، جامعة ونتشو، ونتشو، تشجيانغ 325035، جمهورية الصين الشعبية. ساهم هؤلاء المؤلفون بالتساوي: وي تشن، ليانغ تشانغ، ليتاو شيو. البريد الإلكتروني: panghuan@yzu.edu.cn; yuqin_zou@hnu.edu.cn
DOI: https://doi.org/10.1038/s41467-024-46752-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38499522
Publication Date: 2024-03-18
Pulse potential mediated selectivity for the electrocatalytic oxidation of glycerol to glyceric acid
Accepted: 7 March 2024
Published online: 18 March 2024
(A) Check for updates
Abstract
Preventing the deactivation of noble metal-based catalysts due to selfoxidation and poisonous adsorption is a significant challenge in organic electro-oxidation. In this study, we employ a pulsed potential electrolysis strategy for the selective electrocatalytic oxidation of glycerol to glyceric acid over a Pt-based catalyst. In situ Fourier-transform infrared spectroscopy, quasi-in situ X-ray photoelectron spectroscopy, and finite element simulations reveal that the pulsed potential could tailor the catalyst’s oxidation and surface micro-environment. This prevents the overaccumulation of poisoning intermediate species and frees up active sites for the re-adsorption of OH adsorbate and glycerol. The pulsed potential electrolysis strategy results in a higher glyceric acid selectivity (81.8%) than constant-potential electrocatalysis with
Results
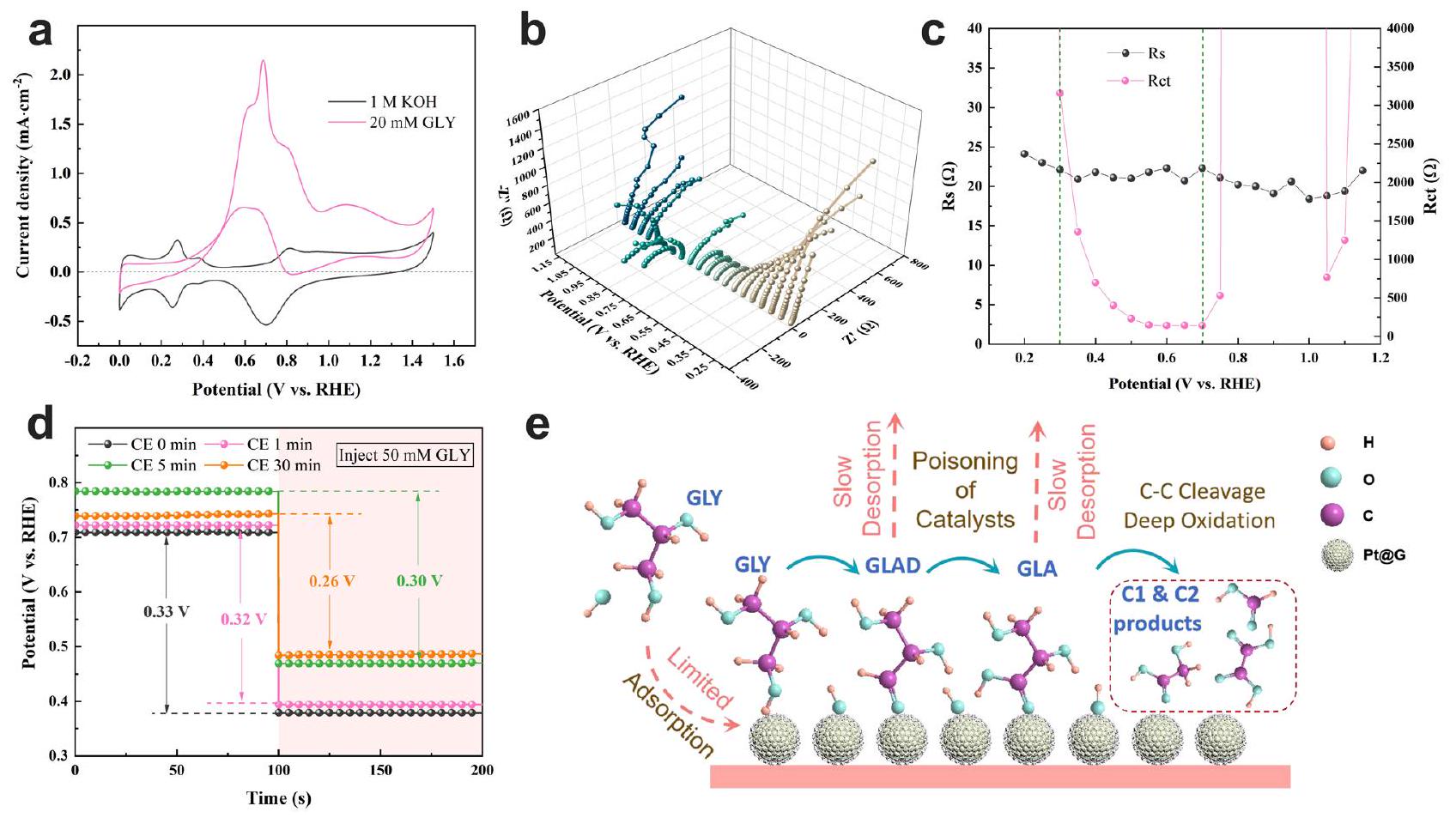
Limitations of CE for GEOR over a Pt-based catalyst
negative coordinate system. This is a typical catalyst poisoning phenomenon of Pt-based catalysts
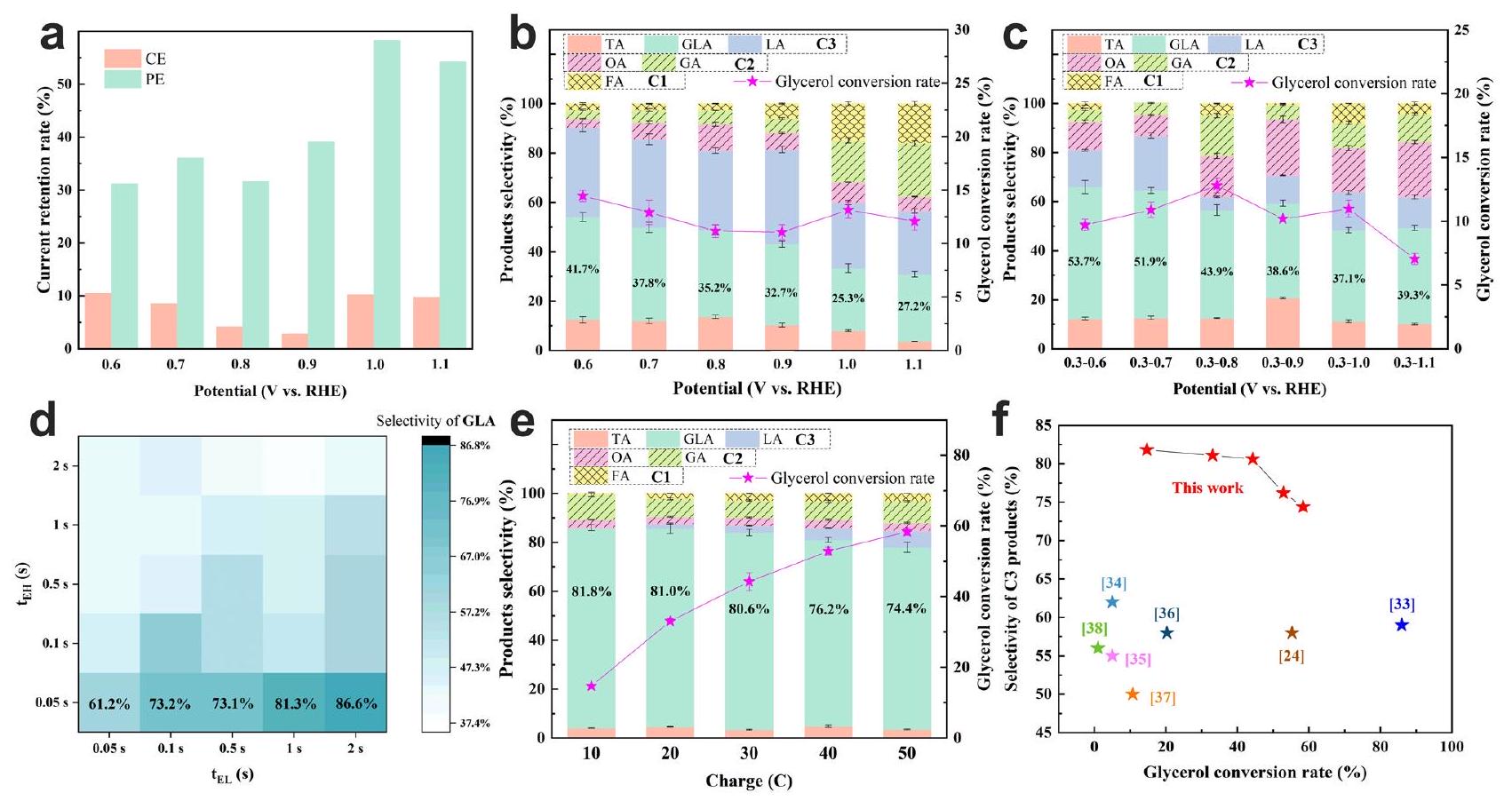
Electrocatalytic performance of GEOR with PE
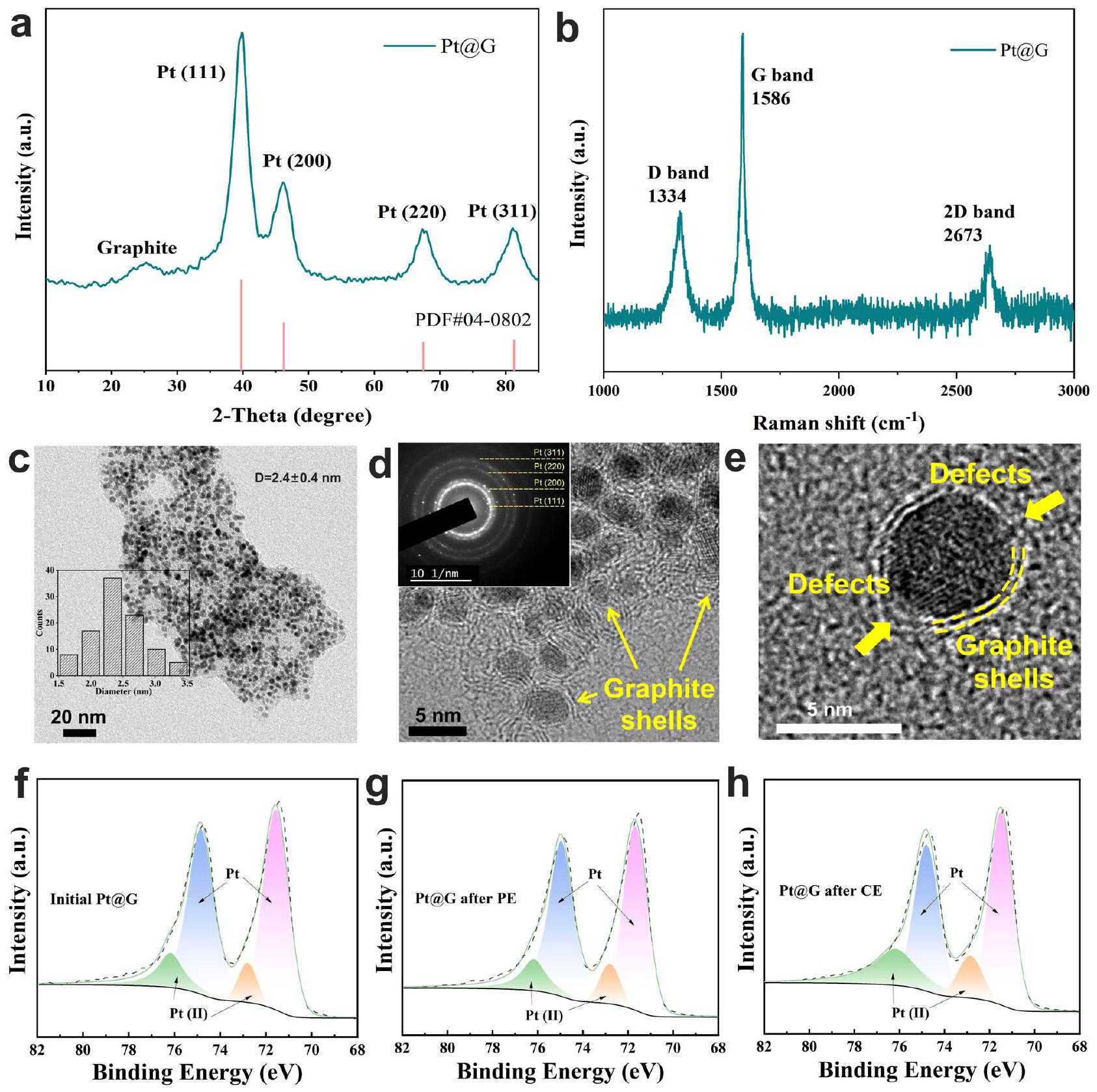
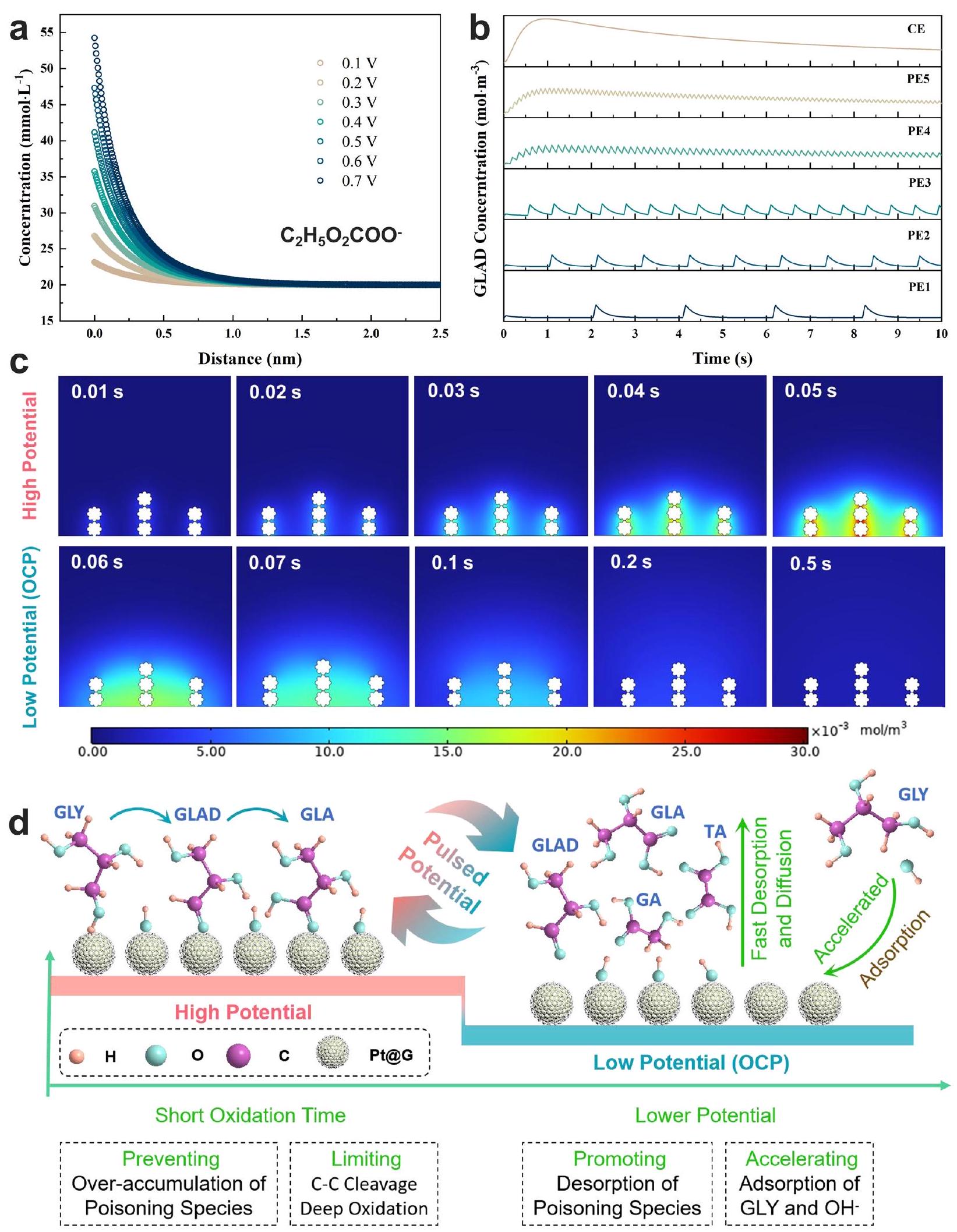
Effect of electrolytic mode on physical and chemical properties Pt@G during GEOR
respectively. e Selectivity and GLY conversion rate with 20 mM GLY under the electrolytic mode of PE3. The percentages in black indicate the corresponding selectivity of GLA.
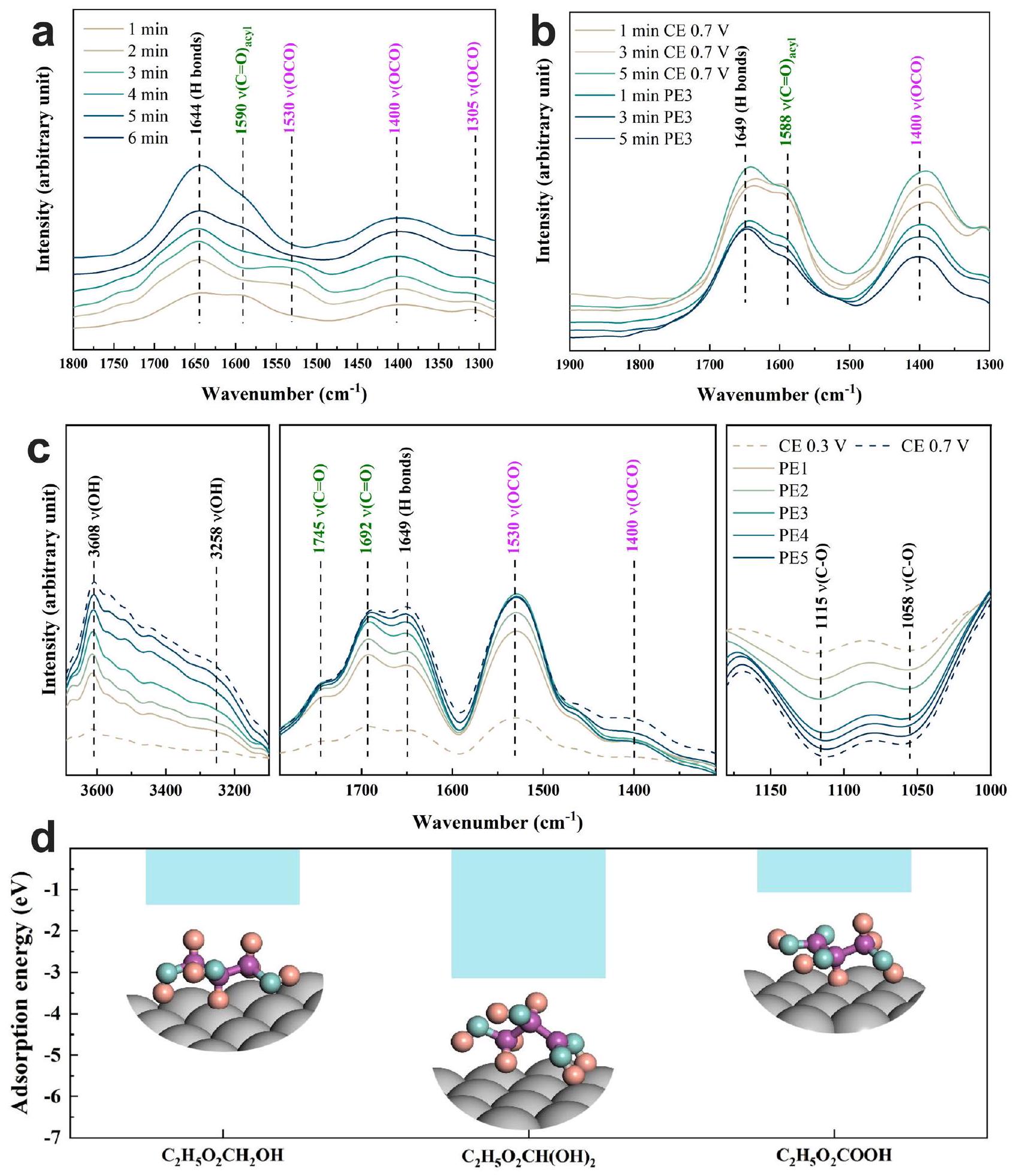
Effect of electrolytic mode on surface micro-environment surrounding Pt@G during GEOR
electrolysis time (Fig. 4a and Supplementary Fig. 18b). The intensity of these regions under CE reached the maximum value faster at
groups of glycerol, increased as
Finite element simulation and mechanistic understanding of PE for GEOR
alkaline electrolyte. The three inset images present the optimized adsorption geometry of the corresponding three species on the Pt (111) surface. The color for each element is tangerine yellow for H , light blue for O , purple for C , and gray for Pt , respectively.
potential and the concentration of reactants and products within the electric double layer (EDL)
and
to
Discussion
Methods
Preparation of Pt@G
with HF and
Characterizations
Electrochemical measurements
Product analysis
In situ FTIR study
the PE3 protocol. a, b, c Data obtained through COMSOL multiple physical quantities. The catalyst of
loaded with catalyst, graphite rod and
Adsorption energy calculation
Finite element simulations of the diffuse double layer
Finite element simulations of the GEOR
Data availability
References
- Sharninghausen, L. S., Campos, J., Manas, M. G. & Crabtree, R. H. Efficient selective and atom economic catalytic conversion of glycerol to lactic acid. Nat. Commun. 5, 5084 (2014).
- Alisson Dias da Silva, R., Ana Luíza Freitas, F., Antônio Ésio, B., Rita Maria de Brito, A. & Luiz Antônio Magalhães, P. Market prospecting and assessment of the economic potential of glycerol from biodiesel. In Biotechnological Applications of Biomass (eds Thalita Peixoto B., Thiago Olitta B., Luiz Carlos B.). IntechOpen (2020).
- Sheng, H. et al. Linear paired electrochemical valorization of glycerol enabled by the electro-Fenton process using a stable
cathode. Nat. Catal. 5, 716-725 (2022). - Katryniok, B. et al. Selective catalytic oxidation of glycerol: perspectives for high-value chemicals. Green Chem. 13, 1960-1979 (2011).
- Zhou, C., Beltramini, J. N., Fan, Y. & Lu, G. Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 37, 527-549 (2008).
- González-Cobos, J., Baranton, S. & Coutanceau, C. A systematic in situ infrared study of the electrooxidation of C3 alcohols on carbon-supported Pt and Pt-Bi catalysts. J. Phys. Chem. C 120, 7155-7164 (2016).
- Casebolt, R., Levine, K., Suntivich, J. & Hanrath, T. Pulse check: potential opportunities in pulsed electrochemical
reduction. Joule 5, 1987-2026 (2021). - Zhang, X. et al. Asymmetric low-frequency pulsed strategy enables ultralong
reduction stability and controllable product selectivity. J. Am. Chem. Soc. 145, 2195-2206 (2023). - Timoshenko, J. et al. Steering the structure and selectivity of
electroreduction catalysts by potential pulses. Nat. Catal. 5, 259-267 (2022). - Kim, C. et al. Tailored catalyst microenvironments for
electroreduction to multicarbon products on copper using bilayer ionomer coatings. Nat. Energy 6, 1026-1034 (2021). - Li, P. et al. Pulsed nitrate-to-ammonia electroreduction facilitated by tandem catalysis of nitrite intermediates. J. Am. Chem. Soc. 145, 6471-6479 (2023).
- Guo, M. et al. Pulsed electrocatalysis enabling high overall nitrogen fixation performance for atomically dispersed Fe on
. Angew. Chem. 135, e202217635 (2023). - Bu, Y. et al. Electrical pulse-driven periodic self-repair of Cu-Ni tandem catalyst for efficient ammonia synthesis from nitrate. Angew. Chem. Int. Ed. 62, e202217337 (2023).
- Ding, Y. et al. Pulsed electrocatalysis enables an efficient 2-electron oxygen reduction reaction for
production. J. Mater. Chem. A 9, 15948-15954 (2021). - Vincent, I. et al. Pulsed current water splitting electrochemical cycle for hydrogen production. Int. J. Hydrogen Energy 43, 10240-10248 (2018).
- Zhao, M. et al. Pd-Ru alloy nanocages with a face-centered cubic structure and their enhanced activity toward the oxidation of ethylene glycol and glycerol. Small Methods 4, 1900843 (2020).
- Fan, L. et al. Recent progress in electrocatalytic glycerol oxidation. Energy Technol. 9, 2000804 (2021).
- Zhou, Y., Shen, Y. & Xi, J. Seed-mediated synthesis of PtxAuy@Ag electrocatalysts for the selective oxidation of glycerol. Appl. Catal. B: Environ. 245, 604-612 (2019).
- Li, Z. et al. Alcohols electrooxidation coupled with
production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022). - Zhang, Y. et al. Potential-dynamic surface chemistry controls the electrocatalytic processes of ethanol oxidation on gold surfaces. ACS Energy Lett. 4, 215-221 (2019).
- Gao, F. et al. Shape-control of one-dimensional PtNi nanostructures as efficient electrocatalysts for alcohol electrooxidation. Nanoscale 11, 4831-4836 (2019).
- Zhang, Z. et al. Engineering phase and surface composition of
nanocatalysts: a strategy for enhancing CO tolerance. Nano Energy 34, 224-232 (2017). - Ren, Z. et al. Pt/ZrO2 catalyst with metal-support synergistic effect towards glycerol selective oxidation. Chem. Eng. J. 468, 143623 (2023).
- Li, J. et al. Tuning the product selectivity toward the high yield of glyceric acid in Pt-CeO2/CNT electrocatalyzed oxidation of glycerol. ChemCatChem 14, e202200509 (2022).
- Wu, G., Liu, Y., He, Y., Feng, J. & Li, D. Reaction pathway investigation using in situ Fourier transform infrared technique over Pt/CuO and
for selective glycerol oxidation. Appl. Catal. B: Environ. 291, 120061 (2021). - Dong, J. et al. Direct in situ raman spectroscopic evidence of oxygen reduction reaction intermediates at high-index
surfaces. J. Am. Chem. Soc. 142, 715-719 (2020). - Marković, N. M., Marinković, N. S. & Adžić, R. R. Electrosorption of hydrogen and sulphuric acid anions on single-crystal platinum stepped surfaces: Part I. The [110] zone. J. Electroanal. Chem. Interfacial Electrochem. 241, 309-328 (1988).
- Wang, Y. et al. Efficient electrocatalytic oxidation of glycerol via promoted
generation over single-atom-bismuth-doped spinel . ACS Catal 12, 12432-12443 (2022). - Sandrini, R. M. L. M. et al. Mechanistic aspects of glycerol electrooxidation on
electrode in alkaline media. Electrochem. Commun. 86, 149-152 (2018). - Hong, W., Shang, C., Wang, J. & Wang, E. Bimetallic PdPt nanowire networks with enhanced electrocatalytic activity for ethylene glycol and glycerol oxidation. Energy Environ. Sci. 8, 2910-2915 (2015).
- Zhou, P. et al. Heterogeneous-interface-enhanced adsorption of organic and hydroxyl for biomass electrooxidation. Adv. Mater. 34, 2204089 (2022).
- Yan, Y. et al. Electrocatalytic upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J. Am. Chem. Soc. 145, 6144-6155 (2023).
- Liu, X. & Yang, C. Electrocatalytic selective oxidation of glycerol to glyceric acid over efficient Pt/CNTs-CeO2 catalysts. Mater. Lett. 324, 132658 (2022).
- Zhou, Y., Shen, Y., Xi, J. & Luo, X. Selective electro-oxidation of glycerol to dihydroxyacetone by PtAg skeletons. ACS Appl. Mater. Interfaces 11, 28953-28959 (2019).
- Zhou, Y., Shen, Y. & Piao, J. Sustainable conversion of glycerol into value-added chemicals by selective electro-oxidation on Pt-based catalysts. ChemElectroChem 5, 1636-1643 (2018).
- Kim, Y. et al. The role of ruthenium on carbon-supported PtRu catalysts for electrocatalytic glycerol oxidation under acidic conditions. ChemCatChem 9, 1683-1690 (2017).
- Dai, C. et al. Electrochemical production of lactic acid from glycerol oxidation catalyzed by AuPt nanoparticles. J. Catal. 356, 14-21 (2017).
- Inoue, H. et al. Mechanism of glycerol oxidation reaction on silvermodified palladium electrode in alkaline medium. Int. J. Hydrogen Energy 43, 18664-18671 (2018).
- Copeland, J. R., Santillan, I. A., Schimming, S. M., Ewbank, J. L. & Sievers, C. Surface interactions of glycerol with acidic and basic metal oxides. J. Phys. Chem. C 117, 21413-21425 (2013).
- An, Z. et al. Pt1 enhanced C-H activation synergistic with
catalysis for glycerol cascade oxidation to glyceric acid. Nat. Commun. 13, 5467 (2022). - Mello, G. A. B., Busó-Rogero, C., Herrero, E. & Feliu, J. M. Glycerol electrooxidation on Pd modified Au surfaces in alkaline media: effect of the deposition method. J. Chem. Phys. 150, 041703 (2018).
- Xu, L. et al. Salting-out aldehyde from the electrooxidation of alcohols with 100% selectivity. Angew. Chem. Int. Ed. 61, e202210123 (2022).
- Zhang, L., Li, C. & Huang, J. A beginners’ guide to modelling of electric double layer under equilibrium, nonequilibrium and AC conditions. J. Electrochem. 28, 2108471 (2022).
- Zhang, L. et al. In vivo activation of pH-responsive oxidase-like graphitic nanozymes for selective killing of Helicobacter pylori. Nat. Commun. 12, 2002 (2021).
- Zhang, L. et al. A magnetocatalytic propelled cobalt-platinum@graphene navigator for enhanced tumor penetration and theranostics. CCS Chem. 4, 2382-2395 (2022).
- Obst, S. & Bradaczek, H. Molecular dynamics study of the structure and dynamics of the hydration shell of alkaline and alkaline-earth metal cations. J. Phys. Chem. 100, 15677-15687 (1996).
- Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
- Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-46752-4.
Correspondence and requests for materials should be addressed to Huan Pang or Yuqin Zou.
http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
© The Author(s) 2024
State Key Laboratory of Chem/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410000, P. R. China. School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225009, P. R. China. Key Laboratory of Leather of Zhejiang Province, Institute of New Materials and Industrial Technologies, Wenzhou University, Wenzhou, Zhejiang 325035, P. R. China. These authors contributed equally: Wei Chen, Liang Zhang, Leitao Xu. e-mail: panghuan@yzu.edu.cn; yuqin_zou@hnu.edu.cn