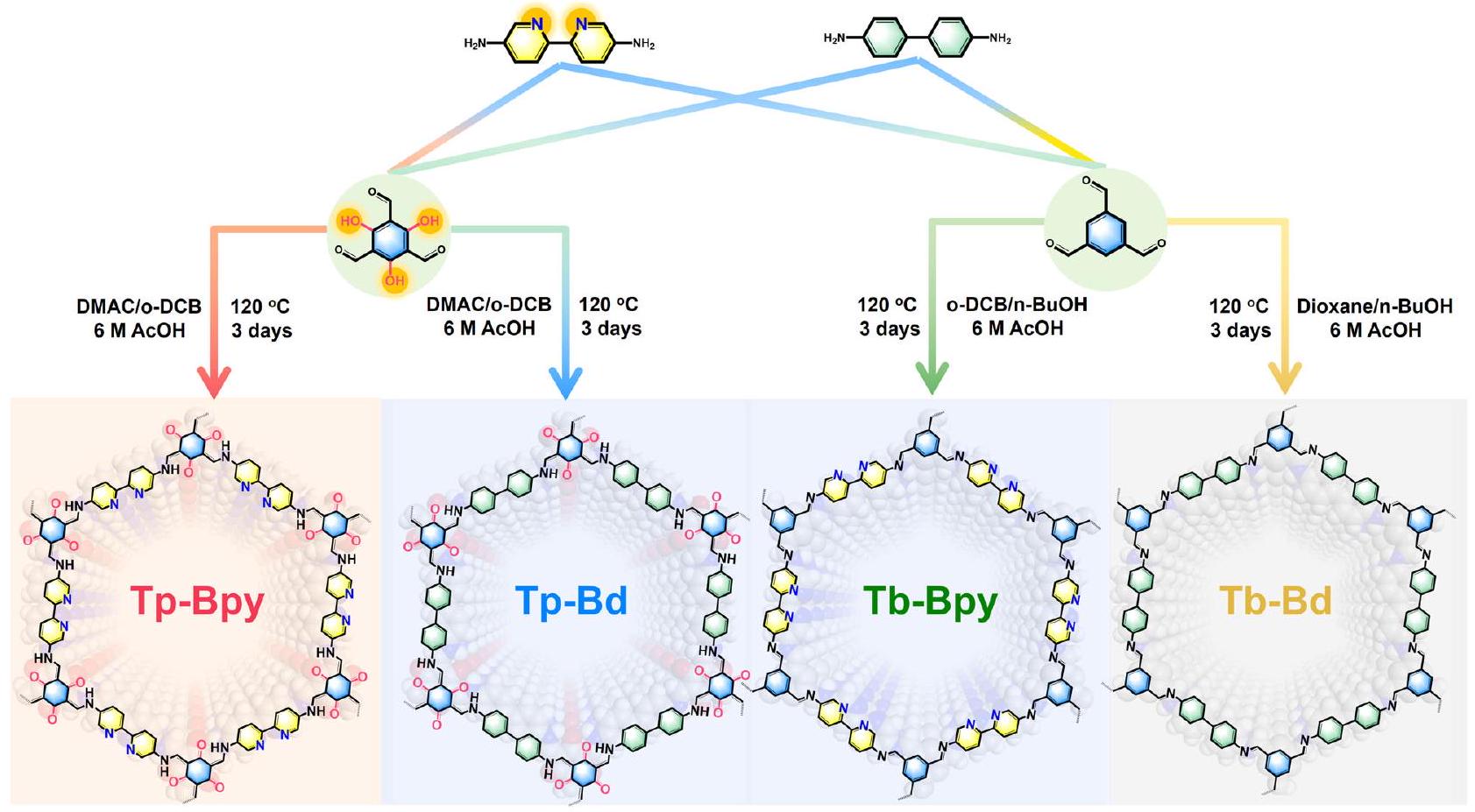
التحسين الفائق الكفاءة لاستخلاص اليورانيوم من مياه البحر عبر دورة نمو وإزالة نقاط النانو ستودتايت Ultra-highly efficient enrichment of uranium from seawater via studtite nanodots growth-elution cycle
الاستخراج المتتالي لليورانيوم من مياه البحر هو نهج واعد لضمان الإمداد طويل الأمد باليورانيوم واستدامة الطاقة النووية. هنا، نبلغ عن استراتيجية فائقة الكفاءة من خلال نمو نقاط النانو من الستودتايت بقدرة استيعاب مثيرة للإعجاب لليورانيوم.من مياه البحر الطبيعية في 12 يومًا متتاليًا (أي، متوسط لـيمكن استخراج اليورانيوم على شكل ستودتايت تحت الضوء المرئي من خلال التفاعل بين اليورانيوم الممتص والضوء المتولد.مع المحفزات الضوئية القائمة على الإيمين من إطار العمل العضوي التساهمي. بالتفصيل، يتم استخراج اليورانيوم من Tp-Bpy وTp-Bpy-2 وTp-Py مع مواقع متعددة لت chelation اليورانيوم على شكل نقاط نانوية من الستودتايت يمكن إزالتها بسهولة، بينما يتم تحويل اليورانيوم في Tp-Bd وTb-Bpy إلى قضبان نانوية من الستودتايت التي تكون أكثر ثباتًا للإزالة. تم إثبات أن وفرة مواقع chelation لليورانيوم من خلال التنظيم الهيكلي لمحفزات COF تسهل تشكيل وإزالة فعالة لنقاط نانوية من الستودتايت.
مع تزايد الطلب العالمي على الطاقة وزيادة الحاجة إلى انتقال طاقة نظيفةمن المتوقع أن تكون الطاقة النووية مصدر طاقة واعد في المستقبل القريب، لذا تم تطويرها بشكل مكثف.اليورانيوم هو العنصر الأكثر أهمية لتحفيز تفاعل الانشطار.. بالنظر إلى المصدر المحدود من اليورانيوم في اليابسة والكمية الكبيرة من موارد اليورانيوم في البحر (أكثر من 4 مليارات طن) إن تخصيب اليورانيوم من مياه البحر له أهمية كبيرة في التنمية المستدامة للصناعة النووية.. ومع ذلك، بسبب التركيز المنخفض لليورانيوم في مياه البحر (جزء في المليار) والبيئة البحرية المعقدةيبقى تطوير نهج فعال لإعادة تدوير اليورانيوم تحديًا.
المواد المسامية المتقدمة مثل الإطارات العضوية المعدنية (MOFs)البوليمرات العضوية المسامية (POPs)وأطر الكربون العضوي التساهمي (COFs)تم تطويرها بشكل مكثف لاستخراج اليورانيوم من مياه البحر الطبيعية، بسبب كتلها البنائية الوظيفية وخصائص المسام القابلة للتعديل.تضمن تنوع المجموعات الوظيفية والمواد الماصة/المحفزات الضوئية المصممة بشكل جيد قدرة عالية على الاستخراج بفضل الامتزازو/أو الاختزال الضوئي لليورانيوم المنفصل في مواقع الربطفي مواقع الربط الغنية. على سبيل المثال، صمم يانغ وزملاؤه غشاءً مساميًا هرميًا باستخدام PIM-1 المفعّل بالأميدوكسي، مما يسمح بالانتشار السريع لأنواع اليورانيوم وحقق زيادة ملحوظة بمقدار 20 ضعفًا في قدرة امتصاص اليورانيوم مقارنةً بالغشاء الذي يحتوي فقط على المسامية الدقيقة الجوهرية.نجح ليو وآخرون في تثبيت النحاس الموزع ذريًا على UiO-66-الدعم من خلال مسار اختزال ضوئي بمساعدة ligand بسيط، مما يحقق قدرة عالية على استخراج اليورانيوم.-إعلانات) من مياه البحر الطبيعية. في عام 2023، أبلغ يانغ وآخرون عن سلسلة من أنظمة المانح-المستقبل (D-A) الداخلية الجديدة من خلال إدخال مجموعات مانحة وسالبة مختلفة للإلكترونات في مسام هيكل COF، والتي تظهر قدرة على امتصاص اليورانيوم.يوم في مياه البحر الحقيقية تحت الضوء المرئي. ومع ذلك، بسبب المواقع المحدودة المتاحة للربط داخل مثل هذه المواد الإطارية، من الصعب تجاوز سعة الامتصاص بناءً على الاستخراج عبر الربط المنفصل لليورانيوم.
في دراستنا السابقةالمتحمسيمكن استخدامه للإنتاج الضوئي لبيروكسيد الهيدروجين )، التي يمكن أن تتفاعل مع لتشكيل ستودتايت صلب. تم تطبيق هذه العملية الضوئية لاستخراج اليورانيوم الانتقائي من مياه البحر دون استخدام أي مواد صلبة.. ومع ذلك، بسبب غياب مركز النواة، يجب أن تستغرق عملية الاستخراج وقتًا طويلاً (على الأقل 24 ساعة). وقد أبلغ كيم وآخرون عن ذلك أيضًا.. أن المعادلة (1) بين و اليورانيوم عند الظروف المحيطة قابل للعكس.
عندما تكون قيمة pH منخفضة بما فيه الكفاية (على سبيل المثال،يمكن إذابة مادة الستوديت الصلبة بكفاءة كحر.استلهمًا من هذه الاستنتاجات، اقترحنا بناء سلسلة من المحفزات الضوئية الفعالةنشاط الإنتاج ومراكز الربط لتكوين الستودتايت. والأهم من ذلك، يجب أن تكون هذه المواد العضوية الإطارية مقاومة للأحماض حتى يمكن إذابة الستودتايت الذي قد يتكون في ظل ظروف حمضية ويمكن تجديد المواد العضوية الإطارية في نفس الوقت للاستخدام على المدى الطويل. هو مؤكسد أخضر أساسي مشهور جدًا ولكنه يواجه صعوبة في التخزين على المدى الطويل بسبب التحلل الذاتي.تُستخدم طريقة الأنثراكوينون على نطاق واسع في الإنتاج الصناعي لـ، مما يساهم في أكثر منمنالإخراجتشمل طريقة الأنثراكوينون استخدام مذيبات عضوية كبيرة، وتولد نواتج ثانوية سامة، وتكون عرضة لتعطيل المحفز، مما يؤدي إلى استهلاك طاقة مرتفع وتلوث كبير.. على الرغم من لا يعتبر قابلًا للاشتعال بنفسه، لكن خصائصه المؤكسدة العالية تجعله مادة خام صناعية خطرة. بشكل خاص، خلال النقل المغلق، تتركز كميات عالية من (فوقيمكن أن يؤدي إلى زيادة سريعة في درجة الحرارة، مما قد يتسبب في انفجار الطور الغازي. وبالتالي، فإن استخدام استراتيجية التخليق الأخضر لإنتاجلقد أصبح الاستخدام المحلي موضوع بحث مؤخرًا. نظرًا لأن COFs المعتمدة على الإيمين معروفة جيدًا كعوامل تحفيز ضوئي لعملية التخليق قمنا بتصميم وتصنيع أربعة محفزات ضوئية قائمة على الإيمين (Tp-Bpy وTp-Bd وTb-Bpy وTb-Bd) مع مجموعات خالطة لليورانيوم مثل مجموعات الكيتو والبيبيريدين. (الشكل 1). وُجد أنه بالمقارنة مع COFs التي تحتوي على مجموعة كيتو أو ثنائي بيريدين N مخلبية فردية، فإن مواقع الربط الوفيرة على Tp-Bpy التي تحتوي على كل من الكيتو المجموعات وبيبيريدين N مفيدة لتكوين نقاط نانوية من ستودتايت القابلة للذوبان، والتي يمكن أن تحقق دورة نمو-إزالة فعالة لنقاط ستودتايت النانوية. لتوسيع نطاق المحفزات الضوئية وتوضيح الافتراض بأن الجمع بين مواقع الامتزاز الوفيرة وقد تؤدي القدرة الإنتاجية للمواد الحفازة الضوئية إلى إثراء اليورانيوم بكفاءة عالية من خلال نمو نقاط النانو من مادة الستودتايت ودورة الإزالة، قمنا بتحضير نوعين آخرين من المواد الحفازة الضوئية القائمة على الإيمين مع مجموعات كيتو ومواقع بيريدين-ن (Tp-Bpy-2 و Tp-Py). (الشكل التكميلي 1، 2)، كلاهما أظهر إثراءً فعالاً لليورانيوم في دورات نمو وإزالة النانو دوتس للدراستيت.
النتائج
تركيب وتوصيف المواد الإطارية العضوية
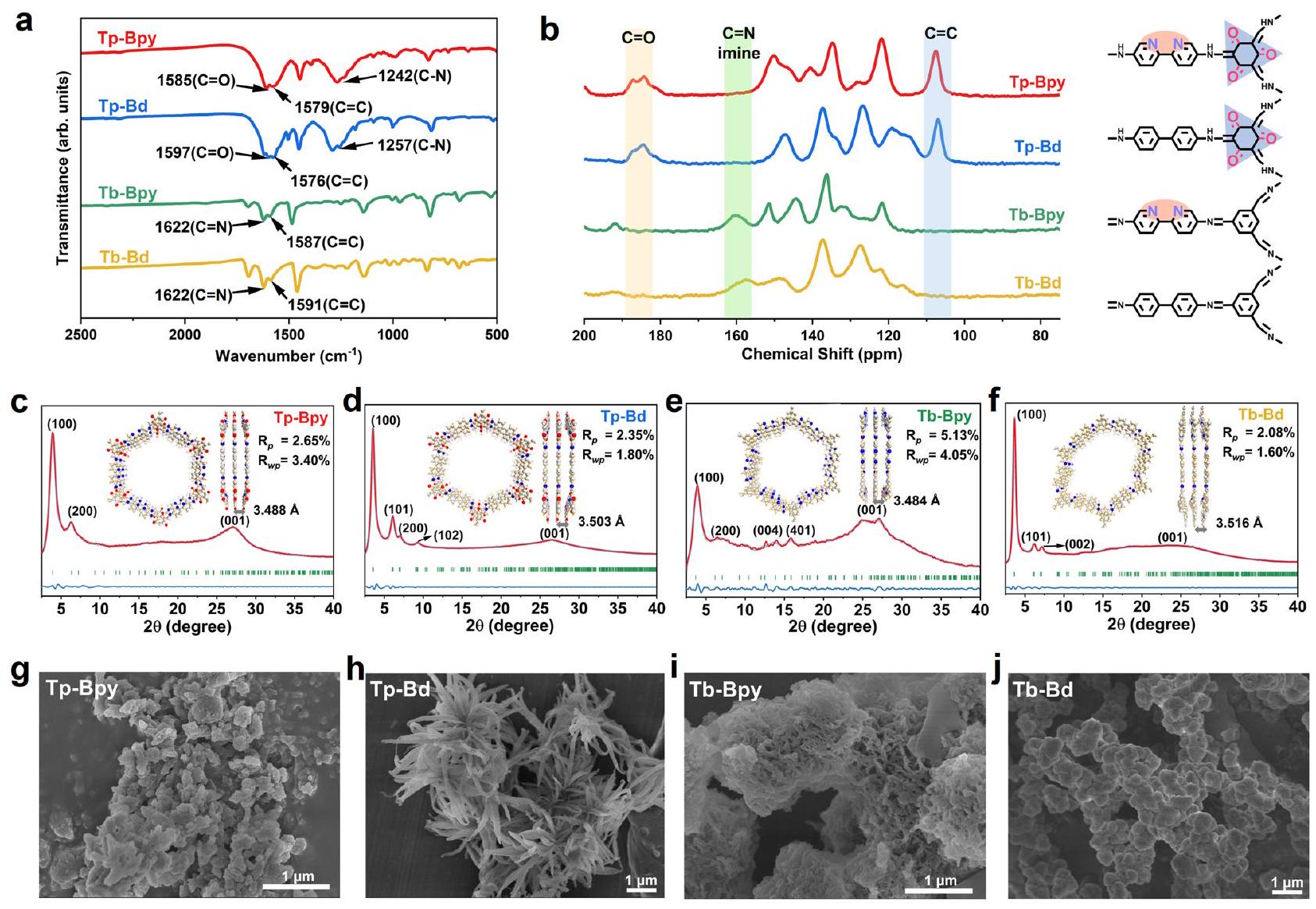
في هذا العمل، تم استخدام مجموعة متنوعة من الروابط لتخليق COFs مع مجموعات وظيفية مختلفة، مما يمنحها سعة مختلفة.الإنتاج. في البداية،تم تخليق COFs -ketoenamine عن طريق تغيير المونومر ثنائي البيريدين، والحصول على Tp-Bpy و Tp-Bd من خلال تفاعل التكثيف في خليط من-ثنائي ميثيل أسيتاميد (DMAC) و o-dichlorobenzene (o-DCB) مع حمض الأسيتيك كعامل حفاز في. بعد ذلك، تم تخليق COFs بدون أي مجموعات كربونيل من خلال تفاعل حلقي مشابه، والتي تم تعيينها إلى Tb-Bpy و Tb-Bd. تم تحديد هيكل الأربعة COFs بواسطة حيود الأشعة السينية البودرة (PXRD)، الصلبةطيف الرنين المغناطيسي النووي وطيف تحويل فورييه للأشعة تحت الحمراء (FT-IR).
كما هو موضح بوضوح في الشكل 2 أ والشكل التكميلي 3، اختفاء الـشريط تمدد ) في و يمتد عندأكدت الاستهلاك الكامل لمجموعات الألدهيد والديامين في المتفاعلات. أما بالنسبة لـ COFs المعتمدة على Tp، فإن القمم الجديدة المميزة لتمدد C-N التي ظهرت و و رابطة عند و ، على التوالي، تم التحقق من وجود الشكل الكيتوني بدلاً من الشكل الإينولي. أما بالنسبة لـ COFs المعتمدة على Tb، فإنتمدد عندأظهر بوضوح تكوين روابط الإيمين. في الوقت نفسه، طيف الإلكترون الضوئي بالأشعة السينية (XPS) Nحدد بوضوح ثلاثة أشكال من Nورابط الإيمين) للأربعة (الشكل التوضيحي 2)، والتي كانت متوافقة مع نتائج FT-IR المذكورة أعلاه. الحالة الصلبةأكدت طيفيات NMR لـ CP/MAS بالتساوي
الشكل 1 | التخليق والهياكل. الرسم التخطيطي لتخليق وهياكل أربعة من COFs.
الشكل 2 | التركيب الكيميائي والتوصيف لـ Tp-Bpy و Tp-Bd و Tb-Bpy و Tb-Bd. أ طيف FT-IR. ب الحالة الصلبةطيف NMR لـ CP/MAS. c-f توصيف PXRD (الملف التجريبي لـ PXRD: أحمر، الملف المصقول: أزرق رفيع، براج)
المواقع: الأخضر والفرق: الأزرق. تُظهر الإضافات النماذج الهيكلية والمسافات بين الطبقات لكل COF بافتراض وضع تكديس AA.صور SEM. التحضير الناجح لـأظهرت Tp-Bpy و Tp-Bd إشارات مميزة لذرات الكربون الكربونيل. ) حوالي والكربونات الخارجية ( ) عند حوالي 107 جزء في المليون (الشكل 2ب). من ناحية أخرى، روابط الإيمين ( ) في ظهر 157-162 جزء في المليون في Tb-Bpy و Tb-Bd. تجمع FTIR وأظهر تحليل الرنين المغناطيسي النووي (NMR) نجاح تخليق أربعة هياكل تنظيمية كيميائية (COFs) على مستوى التركيب الكيميائي.
علاوة على ذلك، تم فحص بلورية كل COF بواسطة تحليل PXRD وتحليل باولي. كما هو موضح في الأشكال 2c-f، هناك أقوى قمم حيود في كل نمط PXRD عند، و ، مما يشير إلى أن القناة المفتوحة للأربعة COFs قد تشكلت بنجاح. في الوقت نفسه، كان لديهم جميعًا قمة واسعة حول بسبب – التكديس بين طبقات COF التي تتوافق مع المستوى (001). علاوة على ذلك، تم إجراء تحسين باولي لبيانات PXRD التجريبية لمعرفة تفاصيل معلمات وحدة الخلية (الجدول التكميلي 1-4)، والعوامل التفصيلية والبقايا الضئيلة. و ) والتي كانت متوافقة مع التوقعات. تم إجراء دراسة الخصائص الشكلية السطحية لأربعة من COFs باستخدام المجهر الإلكتروني الماسح (SEM). الأشكال أظهر أنبيأظهر Bd هيكلًا كتليًا مكونًا من كريات مجمعة، بينما أظهر Tp-Bd أشكالًا شبيهة بالعشب تتكون من ألياف نانوية مرتبة بشكل جيد، وأظهر Tb-Bpy تجميعًا وثيقًا لهياكل على شكل قضبان.
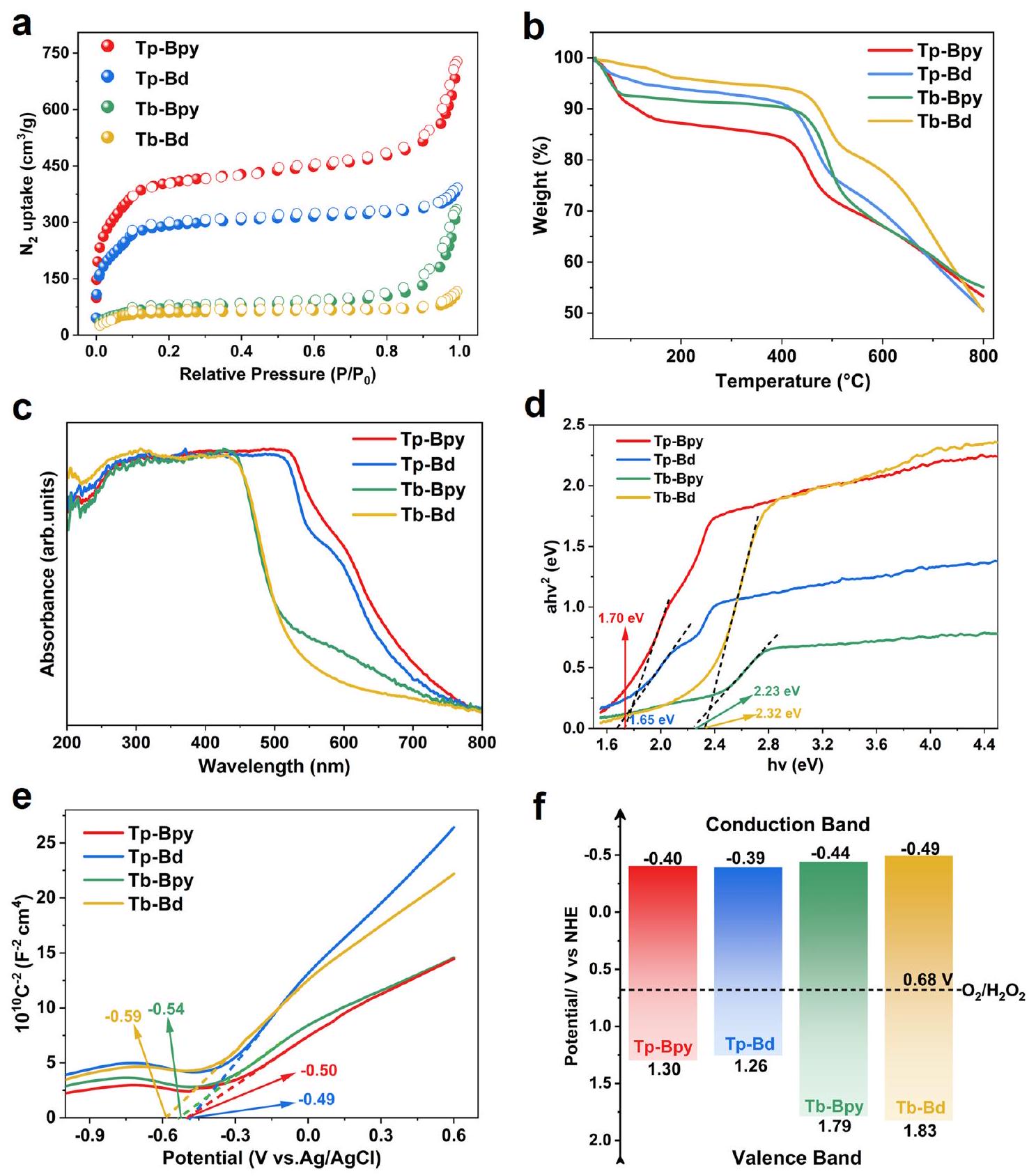
تجعل المساحة السطحية المحددة العالية والتمعدن القابل للتعديل لمركبات الإطار العضوي (COFs) منها محفزات ضوئية مثالية مع المزيد من المواقع النشطة المكشوفة. للحصول على المسامية،تم إجراء تحليلات الامتزاز-الامتزاز على العينات المفعلة بالكامل عند 77 كلفن. تم تحديد منحنيات الامتزاز-الامتزاز على أنها منحنيات من النوع الثاني ذات خصائص مسامية متوسطة. (الشكل 3أ). تم تقييم إجمالي أحجام المسام لـ كن، و لـ Tp-Bpy و Tp-Bd و Tb-Bpy و Tb-Bd، على التوالي. تم حساب توزيعات حجم المسام بواسطة BJH لتكون مركزة عند 2.06 نانومتر لـ Tp-Bpy، 2.09 نانومتر لـ Tp-Bd، 2.24 نانومتر لـ Tb-Bpy و Tb-Bd، على التوالي، والتي كانت جميعها متوافقة بشكل جيد مع القيمة المحسوبة (الشكل التكميلي 5). علاوة على ذلك، كانت المساحات السطحية لبراوناور-إيميت-تيلر (BET) لـ Tp-Bpy و Tp-Bd و Tb-Bpy و هي 1346.5، و على التوالي. تم تقدير الاستقرارية الحرارية لأربعة من COFs من خلال التحليل الحراري الوزني (TGA) تحت جو من النيتروجين فيكشفت منحنيات TGA أن هذه COFs تظهر متانة حرارية استثنائية، حيث تتحمل درجات حرارة تصل إلىتقريبًا بدون تحلل (الشكل 3ب).
توصيف الأربعة COFs في الخصائص الضوئية الكهربائية
تمت دراسة قدرة جمع الضوء المرئي وهياكل نطاق الطاقة لـ TpBpy و Tp-Bd و Tb-Bpy و Tb-Bd بواسطة مطيافية الانعكاس المنتشر للأشعة فوق البنفسجية-visible (UV-vis DRS) ومنحنيات موت-شوتكي (M-S). كما هو موضح في الشكل 3c، تظهر جميع COFs امتصاصًا قويًا في المنطقة المرئية. أظهر التحليل من خلال معادلة تحويل كوبيلكا-مونك بالتزامن مع مخططات تاوك (الشكل 3d) أن فجوات الطاقة ( ) من Tp-Bpy و Tp-Bd و Tb-Bpy و Tb-Bd كانت 1.70 و 1.65 و 2.23 و 2.32 إلكترون فولت، على التوالي. وفقًا لمنحنيات M-S (الشكل 3e)، كان من الواضح أن جميع COFs كانت أشباه موصلات من النوع n النموذجي بسبب الميل الإيجابي. علاوة على ذلك، كانت قيم الشريط المسطح ( تم تقديرها بـو -0.59 إلكترون فولت لـ Tp-Bpy، Tp-Bd، و ، على التوالي. جهد نطاق التوصيل (CB) عادة ما يكون أقل بحوالي 0.1 فولت من جهد الشريط المسطح لأشباه الموصلات من النوع nدمج مع قيمة و جهد نطاق التكافؤ (VB)يمكن حسابه بوضوح (الشكل 3f).
الشكل 3 | هيكل المسام والاختبارات الضوئية الكهروكيميائية لـ Tp-Bpy و Tp-Bd و Tb-Bpy و Tb-Bd. أمنحنيات الامتصاصطيف التحليل الجاذبي الحراري. ج طيف الانعكاس المنتشر للأشعة فوق البنفسجية-المرئية. د طيف الانعكاس المحول بواسطة كوبيلكا-مونك. هـ منحنيات موت-شوتكي. و توضيح مخطط لهياكل النطاقات الإلكترونية.
علاوة على ذلك، كان مستوى الطاقة متوافقًا جيدًا مع المتطلبات اللازمة للاختزالإلى، مما يكشف أنهم جميعًا محفزات ضوئية واعدة لـالإنتاج.
تحقيقات استخراج اليورانيوم بالتصوير
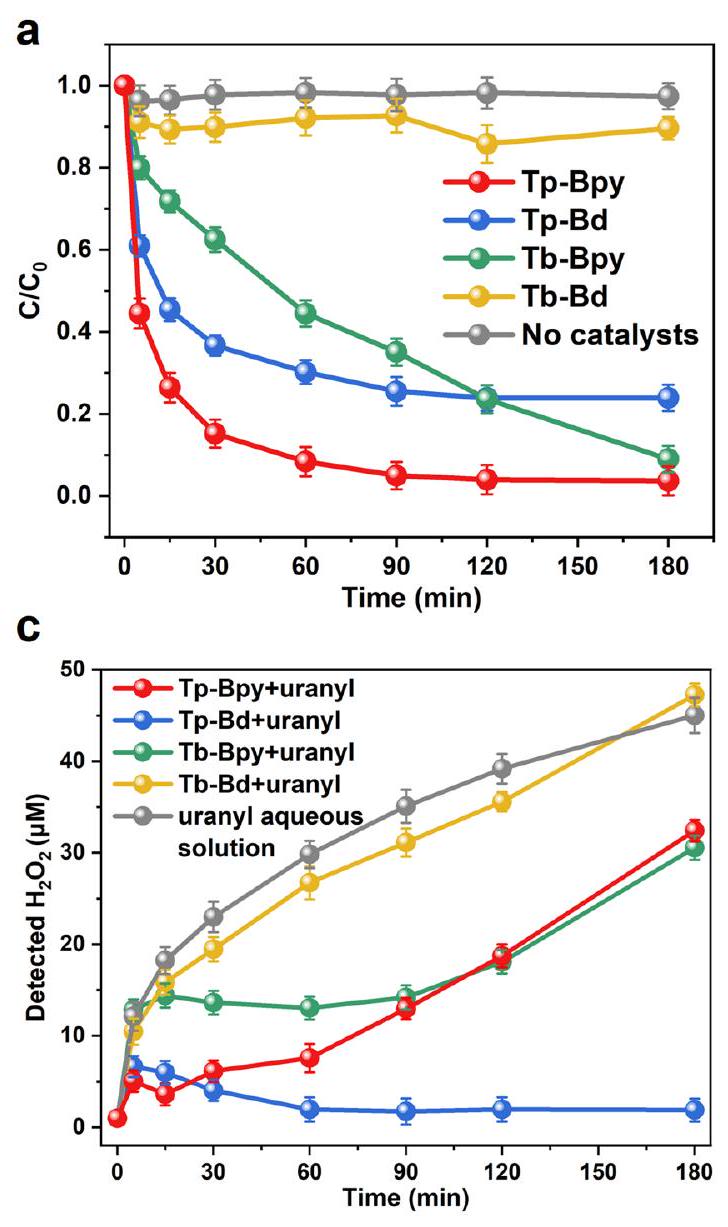
لتقييم الأداء الضوئي التحفيزي، تم اختبار الاستخراج الضوئي لليورانيوم. و 5. عند كما هو موضح في الشكل التوضيحي 6، حوالييمكن استخراج اليورانيوم مع TpBpy، وهو بالتأكيد أعلى من ذلك مع الثلاثة COFs الأخرى. أما بالنسبة لـ Tp-Bd، فقد كان هناك جمود في الاستخراج إلىعندما كانت مدة التفاعل تصل إلى ثلاث ساعات. بشكل مختلف، وجدنا أن Tb-Bpy أظهر معدل استخراج بطيء نسبيًا ولكنه ثابت.، تم زيادة سرعة التفاعل العامة بشكل ملحوظ، مع زيادة كفاءة الاستخراج النهائية لـ Tp-Bpy و Tp-Bd و Tb-Bpy إلى و على التوالي (الشكل 4أ). وبالمثل، يظهر Tb-Bpy استخراجًا ثابتًا لليورانيوم.، بينما كانت نسبة الاستخراج على Tp-Bpy و
تم إبطاء Tp-Bd بشكل واضح أو حتى تجميده. معأو في غياب المحفزات، كانت إزالة اليورانيوم ضئيلة. بالمقارنة مع استخراج اليورانيوم في ظروف الظلام، يُستنتج أن استخراج اليورانيوم بكفاءة عبر Tp-Bpy و Tp-Bd و Tb-Bpy مرتبط إلى حد كبير بالتفاعل الضوئي على الرغم من أن امتصاص اليورانيوم عبر Tp-Bpy يساهم بحواليإلى الكفاءة الكلية للاستخراج (الشكل 4ب والشكل التوضيحي 7). إن امتصاص اليورانيوم على Tb-Bd هو الأدنى، مما قد يكون مرتبطًا بضعف قدرة التعقيد لأيونات اليورانيوم في غياب مجموعات الخلب.
الكميات منتم قياس الإنتاج بطريقة الكروماتومتر (الشكل 4c، d والشكل التكميلية 8، 9).معدل الإنتاج على Tp-Bpy و Tp-Bd و Tb-Bpy و Tb-Bd هو حوالي 123.89 و 8.79 و 23.48 وعلى التوالي. بدون محفز، لا يوجدتم إنتاجه فيمحلول مائي من الميثانول. الشكل 3 ج أظهر الاتجاهات المختلفة لـديناميكية الإنتاج مع، الذي كان مرتبطًا بالتفاعل المعقد لـالإنتاج و
الشكل 4 | الأداء الضوئي التحفيزي لـ Tp-Bpy و Tp-Bd و Tb-Bpy و Tb-Bd لاستخراج اليورانيومالإنتاج. ظروف التفاعل: الضوء المرئي،و 3 ساعات. أشرطة الخطأ هي الانحرافات المعيارية لثلاثة تكرارات
القياسات. أ ديناميكية استخراج الصورة. ب كفاءة استخراج اليورانيوم تحت ظروف الظلام والضوء.ديناميكا الإنتاج في وجود اليورانيوم.ديناميكية الإنتاج في غياب اليورانيوم.
عملية الاستهلاك مع محفزات ضوئية مختلفة. أولاً، بدون محفز، يمكن أن يتم تحفيز اليورانيوم تحت الضوء المرئي لإنتاج، بمعدل إنتاج مشابه لذلك الخاص بـ Tb-Bpy (الشكل 4c). ثانيًا، يمكن أن يؤدي امتصاص اليورانيوم على COFs إلى حجب الـ نشاط الإنتاج لكل من اليورانيوم وCOFs حيث لا يمكن تحويل الإلكترونات الناتجة عن الضوء مباشرة إلىعندما كانت المواقع النشطة مثل مجموعة الكيتو أو مجموعة البيبيريدين N المخلبية مشغولة باليورانيومثالثًا،يمكن أن يتم استهلاكه من خلال التفاعل بينوأيون اليورانيوم. استنادًا إلى العمليات المذكورة أعلاه، فإن الاتجاهات المختلفة لـيمكن تفسير الإنتاج. في حالة محلول اليورانيوم المائي بدون محفز، تم الكشف عن هو من إنتاج اليورانيوم نفسه دون استهلاك متزامن لـلأن اليورانيوم لا يتحول إلى ستودتايت. في حالةيورانيلي، حيث كان اليورانيلي فقط حوالي.تمت إزالته (الشكل 4أ)،كان تقريبًا غير مستهلك لتشكيل ستودتايت. إنتاجمن اليورانيوم نفسه و، المصحوب بالتدخل المتبادل يؤدي إلى الاتجاه الملحوظ. في حالة Tb-Bpy + يورانيوم، Tp-Bd + يورانيوم و Tp-Bpy + يورانيوم، أدى ارتباط اليورانيوم إلى حجب واضحالإنتاج، حيث تم امتصاص Tb -Bpy لأدنى يورانيوم، وبالتالي تم الكشف عن ذلك في البدايةكان أعلى قليلاً من ذلك مع Tp-Bd و Tp-Bpy بسبب الحد الأدنى من الحجب الناتج عن ارتباط اليورانيوم. مع استخراج اليورانيوم على COFs وانخفاض تركيز اليورانيوم في المحلول، كلاهماكانت الإنتاج والاستهلاك أبطأ، وبالتالي كان هناك نوع من المنصة للكشف عنها.فيعندما كانت تركيزات اليورانيوم منخفضة بما فيه الكفاية، كانت إنتاجيةتجاوز استهلاكالمكتشف المبلغ يرتفع مرة أخرى. في حالة Tp-Bd، في الشكل 4d، أظهر أنكانت قدرة الإنتاج لـ Tp-Bd ضعيفة للغاية وكان امتصاص اليورانيوم عند موقع الربط يمنع بشكل خطير الإنتاج.، وبالتالي ظهرت المنصة فقط في، ولم يكن هناك تقريبًا أي إنتاج لـ بعد 60 دقيقة، لم يكن بالإمكان استخراج أي يورانيوم على شكل ستودتايت كما هو موضح في الشكل 4أ. علاوة على ذلك، في الشكل 4أ، أظهر Tb-Bpy معدل استخراج أبطأ نسبيًا منلكن إنتاجبواسطةكان Bpy أعلى من ذلك بـ، والذي يمكن شرحه بناءً على الاستنتاج التالي. خلال التسعين دقيقة الأولى، على الرغم من أن Tb -Bpy أنتجت كمية أكبر منمنأظهرت Tp-Bd ديناميكية امتصاص أكبر بشكل ملحوظ مقارنةً بـ Bpy كما هو موضح في الشكل التوضيحي 7b. بعد 90 دقيقة، معدل إنتاج Tp-Bd ينخفض بسبب تأثير اليورانيوم الممتص، مما يؤدي إلى ركود في استخراج اليورانيوم. على العكس، استمر Tb-Bpy في الإنتاج.وبالتالي، يستمر تركيز اليورانيوم في الانخفاض بعد 90 دقيقة. في المرحلة الأولى من الاستخراج، تهيمن الديناميات السريعة لامتصاص اليورانيوم على العملية، بينما في المرحلة الجانبية عندما تم شغل مواقع الربط بواسطة اليورانيوم، توقف امتصاص اليورانيوم ببساطة وتوقفت التفاعل بين اليورانيوم ولعبت دورًا مهمًا في الاستخراج. باختصار، تم الكشف عن المختلفالاتجاهات الموضحة في الشكل 4c كانت نتيجة تفاعل معقد بين الإنتاج والاستهلاك لـخلال فترة استخراج اليورانيوم.
التحليل المشترك لديناميكية استخراج اليورانيوم الموضحة في الشكل 4أ والإنتاج الموضح في الشكل 3c و d والشكل التكميلي 8، يشير إلى أن i) المنخفضمعدل الإنتاج والامتصاص الضعيف
الشكل 5 | دراسة منتجات الاستخراج الضوئي لـ Tp-Bpy و Tb-Bpy. أ) طيف XPS بعد الاستخراج الضوئي. ب) مقارنة طيف FT-IR قبل وبعد الاستخراج الضوئي. ج) PXRD بعد الاستخراج الضوئي. د) مخططات WT للكونتور لـ
وصورة HRTEM لـ Tb-Bpy بعد الاستخراج الضوئي.صورة HRTEM لـ Tp-Bpy بعد الاستخراج الضوئي.
لليورانيوم فوقيؤدي إلى استخراج ضئيل لليورانيوم؛ ii) المحدودمعدل الإنتاج بعد 90 دقيقة أدى إلى توقف استخراج اليورانيوم؛ iii) حركية الاستخراج على Tp-Bpy و Tb-Bpy تختلف إلى حد ما، أي أن معدل تدهور الاستخراج على TbBpy أبطأ من ذلك على Tp-Bpy، مما يعني أن مسارات الاستخراج قد تكون مختلفة؛ iv) Tp-Bpy و Tb-Bpy هما المحفزات الضوئية الواعدة للاستخراج الفعال لليورانيوم المائي، على الرغم من أنه يجب توضيح المزيد من الاستخدام المتكرر.
توصيفات منتجات الاستخراج الضوئي
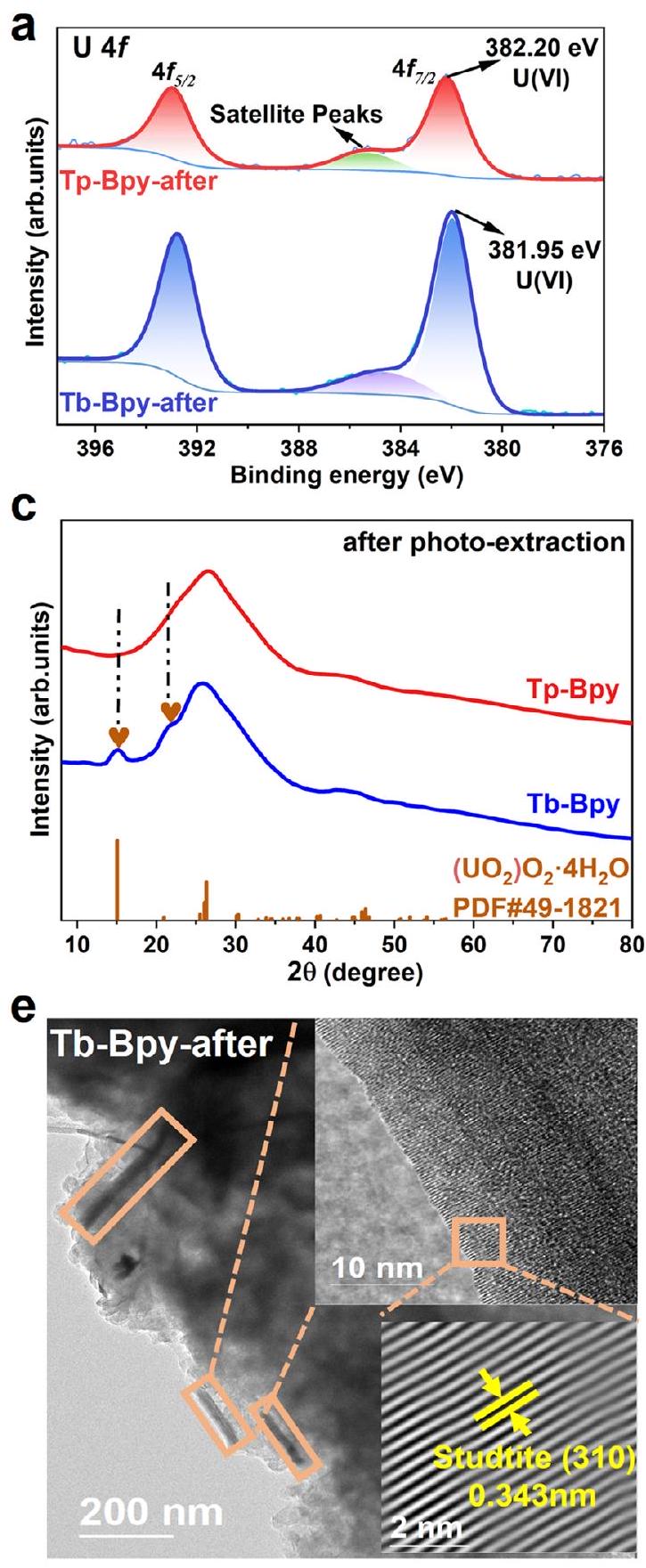
تم تحليل منتجات استخراج الصورة التي تم الحصول عليها باستخدام Tp-Bpy و Tb-Bpy بشكل إضافي من خلال XPS و FT-IR و PXRD و مجهر الإلكترون الناقل عالي الدقة (HRTEM). بالنسبة لكل من Tp-Bpy و
يمكن تعيين القمم عند 382.20 إلكترون فولت و 381.95 إلكترون فولت إلى من (الشكل 5أ). كما هو موضح في الشكل 5ب، هناك قمة جديدة عند تنتمي إلى السندنشأت لكل من Tp -Bpy و Tb -Bpy بعد الاستخراج الضوئي. في هذه الأثناء، الكربونات الكربونية الفريدة (ورابط الإيمين ( ) بالنسبة لـ Tp -Bpy و Tb -Bpy تم الحفاظ عليها جيدًا بعد الاستخراج الضوئي. بالنسبة لـ Tb-Bpy، بعد الاستخراج الضوئي، ظهرت قمم حيود جديدة عند و تقترح تكوين جزيئات ستوديت، بينما لا يمكن ملاحظة أي قمم إضافية لـ Tp-Bpy (الشكل 5c). تتطابق مخططات الكنتور القصوى لـ WT لـ Tp-Bpy بعد استخراج الضوء بشكل وثيق مع الـالرسوم البيانية، التي دعمت بقوة تشكيل نقاط نانوية من الستودتايت (الشكل 5d). تظهر صورة TEM لـ Tb-Bpy بعد الاستخراج الضوئي (الشكل 5e) جزيئات نانوية من الستودتايت على السطح بشكل واضح. بالمقابل، لا يوجد وضوح
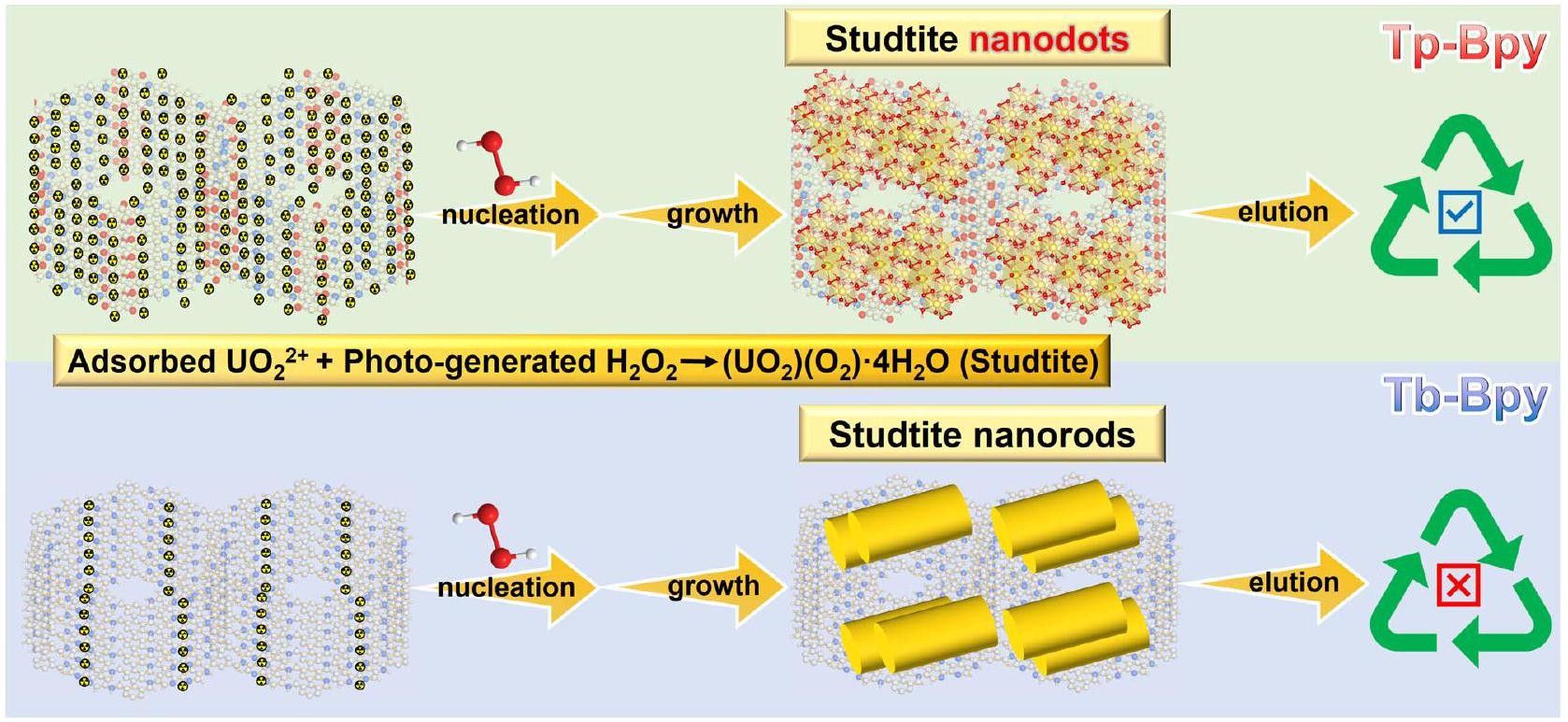
الشكل 6 | المخطط التخطيطي لعملية التفاعل بين Tp-Bpy و Tb-Bpy. إن المواقع الغزيرة للتعقيد لليورانيوم على Tp-Bpy مفيدة لتكوين نقاط نانوية من الستودتايت القابلة للذوبان. إن القدرة المحدودة على الامتصاص لليورانيوم على Tb-Bpy مفيدة لتكوين قضبان نانوية كبيرة وصعبة الذوبان من الستودتايت.
يمكن العثور على الجسيمات النانوية على سطح Tp-Bpy (الشكل 5f). أظهرت صورة HRTEM (الموجودة في الشكل 5f) مسافتين لشبكة البلورات تبلغ 0.265 و 0.325 نانومتر، والتي يمكن أن تُنسب إلى المستويات (021) و (111) في الستودتايت (PDF#49-1821). وهذا يشير إلى أن منتج الاستخراج الضوئي على Tp-Bpy هو نقاط نانوية من الستودتايت، بينما على Tb-Bpy توجد جزيئات ستودتايت على شكل قضبان أكبر بكثير. وبناءً عليه، أكدت خريطة العناصر (الشكل التكميلي 10، 11) توزيعاً متجانساً لعنصر اليورانيوم لكل منهما. بالنسبة لـ Tb-Bpy، كان اليورانيوم موزعاً حول البلورات القضيبية، بينما بالنسبة لـ Tp-Bpy، كان اليورانيوم منتشراً على السطح بالكامل دون نمو كقضبان نانوية كبيرة.
دورة النمو-الإزالة لاستخراج اليورانيوم
لاستعادة سطح Tp-Bpy و Tb-Bpy، تم غسل المحفزات الضوئية المستخدمة بـ لمدة ساعة واحدة. نسبة الإزالة لليورانيوم من Tp-Bpy حوالي ، أعلى من ذلك الناتج من Tb-Bpy (الشكل التكميلي 12). تم استخدام Tp-Bpy الذي تم الحصول عليه بعد الإزالة مرة أخرى لاستخراج اليورانيوم. بعد 7 دورات متتالية من الاستخراج والإزالة، أكثر منيمكن تحقيق كفاءة الاستخراج والإزالة لليورانيوم (الشكل التكميلي 13). تشير خصائص FT-IR وSEM وXRD لـ Tp-Bpy بعد اختبار دورة الاستخراج والإزالة إلى الاستقرار العالي في ظروف الإزالة (الشكل التكميلي 14-16). على النقيض من ذلك، فإن دورات الاستخراج والإزالة المتتالية مع Tb-Bpy ليست مرضية، مما قد يُعزى إلى عدم اكتمال إزالة جزيئات الستودتايت الأكبر. من الجدير بالذكر أن النقاط النانوية للستودتايت المتكونة هي التي تجعل الإزالة القصيرة الزمن ممكنة، مما يمنع التحلل المائي المحتمل لـ imine-COFs في الإزالة الطويلة. بينما لا يمكن إزالة القضبان النانوية للستودتايت المتكونة تمامًا في وقت قصير قبل أن تواجه imine-COFs التحلل.
لتوضيح التأثير الناتج عن وجود أيونات المعادن المتزامنة، تم إجراء استخراج اليورانيوم باستخدام Tp-Bpy في وجود، وبتركيز أعلى بمقدار 50 مرة من تركيز اليورانيوم. أظهرت النتائج أن أكثر منيمكن استخراجها بعد 3 ساعات من الإشعاع، مما يشير إلى أن وجود أيونات المعادن المتزامنة لن يتداخل مع استخراج اليورانيوم (الشكل التوضيحي 17).
استنادًا إلى التحليل المذكور أعلاه، أظهرت الأربعة COFs المعدة ذاتيًا أداءً مختلفًا: i) Tb-Bd لديه نشاط استخراج منخفض جدًا بسبب القدرة المنخفضة على الامتصاص على الرغم منيمكن إنتاجه؛ ii) يظهر Tp-Bd نشاط استخراج غير مُرضٍ بسبب المحدودالإنتاج على الرغم من الامتصاص الكبير لليورانيوم. أكد هذا، لقد أضفنا الإضافة الإضافيةحتىتصل التركيز إلىوأجريت أيضًا اختبار استخراج اليورانيوم مع Tp-Bd (الشكل التكميلي 18). واتضح أن 34.8% المتبقية من اليورانيوم يمكن استخراجها كأنابيب نانوية من الستودتايت، وأقل من ذلك فقطتركت في المحلول بعد تفاعل آخر لمدة ساعتين تحت ظروف مظلمة؛ iii) يظهر Tb-Bpy حركية استخراج تتدهور ببطء مع تكوين جزيئات ستودتايت على شكل قضبان، مما يجعل الإزالة صعبة، ويُفترض أن ذلك بسبب القدرة المحدودة على الامتصاص لليورانيوم؛ iv) تم تحقيق دورة استخراج-إزالة مثالية لتركيز اليورانيوم على Tp-Bpy. الأربعة COFs مختلفة مع مجموعات تعقيد، ثنائي البيريدين ومجموعات كيتو، مما يمنحها أيضًا أداءً ضوئيًا مختلفًا.الإنتاج. لقد تم تحديد أن التفاعل بينويتميز اليورانيوم بسرعة حركية منخفضة نسبيًا، مما يعني إنتاجعلى Tp-Bpy و Tb-Bpy ليست الخطوة المحددة للسرعةلذلك، يُفترض أن الأداء المختلف بين Tp-Bpy و TbBpy مرتبط بتكوين الستودتايت على سطح COFs، والذي يعتمد بشكل كبير على موقع التChelating لليورانيوم (الشكل 6).
لتوسيع نطاق المحفزات الضوئية وتوضيح الافتراض بأن الجمع بين مواقع التChelating الوفيرة وقد تؤدي القدرة الإنتاجية للمواد المحفزة الضوئية إلى إثراء اليورانيوم بكفاءة عالية من خلال نمو نقاط النانو من مادة ستودتايت ودورة الإزالة، قمنا بتحضير نوعين آخرين من المواد المحفزة الضوئية القائمة على الإيمين مع مجموعات كيتو ومواقع ربط بيريدين-ني (Tp-Bpy-2 و Tp-Py). تم إثبات التحضير الناجح لهياكلها الكيميائية بواسطة PXRD (الشكل التكميلي 19)، FTIR (الشكل التكميلي 20)،الرنين المغناطيسي النووي (الشكل التوضيحي 21) والتحليل الطيفي للأشعة السينية (الشكل التوضيحي 22). إمكانيةتم التحقق من الإنتاج من خلال سلسلة من التوصيفات الفيزيائية والضوئية (الشكل التكميلي 23-25). اتجاه فعال مشابه لامتصاص اليورانيوم (الشكل التكميلي 26)،تم العثور على دورات نمو وإزالة studtite nanodots (الشكل التكميلي 27، 28) مع Tp-Py وTp-Bpy-2 كعامل حفاز ضوئي. أكدت دورات النمو والإزالة الممتازة لـ Tp-Bpy-2 وTp-Py أن مواقع التChelation المتعددة لليورانيوم عززت نمو studtite nanodots وسهلت الاستخراج الفعال لليورانيوم (الشكل التكميلي 34-37).
باختصار، المتطلبات لتكوين نقاط نانوية من ستودايت القابلة للذوبان تشمل وجود مواقع خلب لليورانيوم وقدرة التمثيل الضوئي لـ، فضلاً عن الاستقرار في
الشكل 7 | استخراج اليورانيوم من مياه البحر الطبيعية. أ معدل الإزالة عند 10 و 1 جزء في المليون من مياه البحر الملوثة. ب الانتقائية في مياه البحر الملوثة بتركيز 1 جزء في المليون من اليورانيوم. ج مخطط التركيز باستخدام Tp-Bpy. د النشاط المضاد للبكتيريا لـ Tp-Py ضد البكتيريا البحرية. الحجم الدقيق للعينة (ن): الأعلى-، . أسفل قيمة n هي الانحرافات المعيارية لثلاث قياسات مكررة. طيف EPR لـ-DMPO،-DMPO،-معقدات TEMP التي تم إنشاؤها بواسطة Tp-Py تحت إشعاع الضوء المرئي. f المعدات المستخدمة لاستخراج اليورانيوم من مياه البحر الطبيعية بواسطة. شرط الإذابة. هنا، بالإضافة إلى تقديم سلسلة من COFs ذات قدرة استخراج عالية الكفاءة، قمنا بتطوير استراتيجية للتوليد القابل للدورة وإذابة نقاط النانو من ستودايت لاستخراج اليورانيوم على المدى الطويل.
إثراء اليورانيوم في مياه البحر الطبيعية
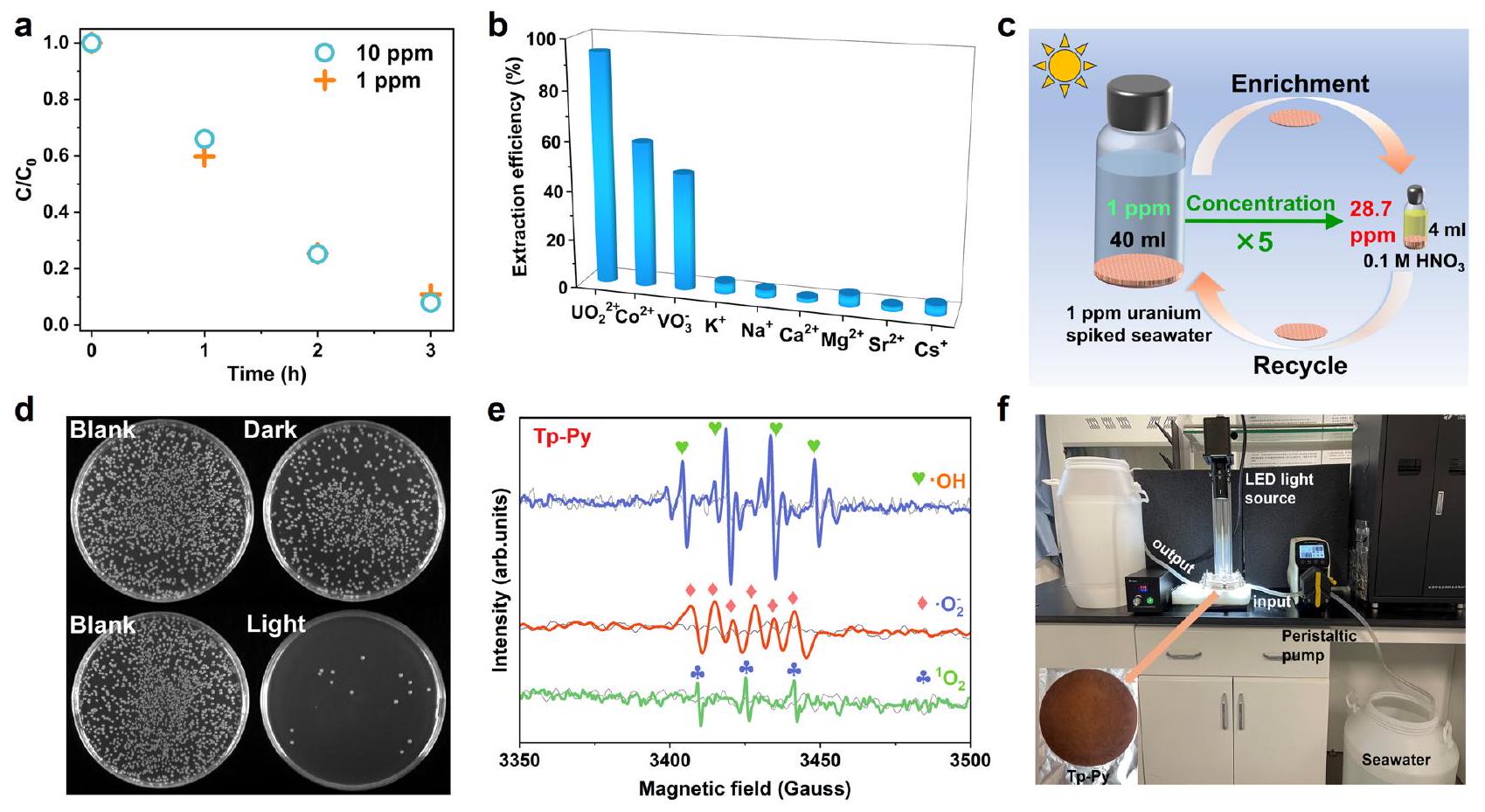
من أجل التحقق مما إذا كانت المواد العضوية الإطارية (COFs) ذات المواقع المتعددة للتعقيد لديها القدرة على استخدامها لاستخراج اليورانيوم من مياه البحر الحقيقية، تم استخدام اليورانيوم بتركيزات منخفضة (10 جزء في المليون و1 جزء في المليون) في مياه البحر الملوثة لاستخراج الضوء باستخدام Tp-Bpy كعامل حفاز ضوئي، حيث…يمكن استخراج اليورانيوم (الشكل 7أ). بالإضافة إلى ذلك،يمكن استخراج 1 جزء في المليون من اليورانيوم في مياه البحر حتى في وجود أيونات المعادن ذات التكافؤ المتغير (الشكل 7ب). بعد خمس دورات متتالية من الاستخراج والإزالة باستخدام 200 مل من مياه البحر الملوثة (1 جزء في المليون من اليورانيوم)، تم الحصول على 4 مل من المحلول المركز الذي يحتوي على 28.7 جزء في المليون من اليورانيوم (الشكل 7ج). للتحقيق بشكل أكبر في القابلية العملية للعملية المذكورة للاستخراج الضوئي، قمنا بفحص كفاءةالاستخراج من مياه البحر الطبيعية تحت الضوء المرئي. بعد حساب التكلفة الأساسية لإعداد COFs، اخترنا المحفز الضوئي Tp-Py الفعال من حيث التكلفة لاستخراج وتركيز اليورانيوم من مياه البحر في اختبار التوسع (الجدول التكميلي 5 و 6). نظرًا للبيئة البحرية المعقدة للميكروبات، يجب أخذ القدرة المضادة للميكروبات الأعلى ضد مجتمع الميكروبات البحرية في الاعتبار لاستخراج اليورانيوم بكفاءة من مياه البحر الطبيعية.تم اختبار نشاط مكافحة التلوث لـ Tp-Py باستخدام البكتيريا البحرية كأهداف (الشكل 7d). أظهرت النتيجة تحت ظروف الظلام (الجدول التكميلي 7)،يمكن تثبيط المجتمع الميكروبي البحري من مياه البحر الطبيعية. بعد التعرض للضوء المرئي، زاد التثبيط بشكل ملحوظتم إدراك ذلك، والذي يُفترض أنه بسبب إنتاج أنواع الأكسجين التفاعلية (ROS) في وجود إشعاع الضوء المرئي، كما تم تأكيده من خلال طيف الرنين المغناطيسي الإلكتروني (EPR) (الشكل 7e والشكل التكميلي 38). أظهرت التجارب مع مياه البحر الطبيعية (3.3 ppb) أنحقق إنجازًا مثيرًا للإعجاب قدرة امتصاص اليورانيومأكثر من 12 يومًا متتاليًا تحت الضوء المرئي، بمتوسطيوم (الشكل 7f، الشكل التكميلية 39 والجدول التكميلية 8، 9).
نقاش
باختصار، أبلغنا عن استراتيجية فعالة ومستدامة لإعادة تدوير مورد اليورانيوم من مياه البحر الطبيعية باستخدام محفزات ضوئية من COF. دراسة مقارنة شاملة لمختلف محفزات COFs Tp-Bpy وTp-Py وTp-Bpy-2 وثلاثة COFs أخرى (Tp-Bd وTb-Bpy وTb-Bd) على سعة الامتصاص،قدرة الإنتاج، أظهرت أداء دورة استخراج وإزالة اليورانيوم أن i) الضوء المتولديمكن أن يتفاعل مع اليورانيوم لتشكيل الستودتايت؛ ii) موقع التChelating لليورانيوم على المحفز الضوئي ضروري لنواة الستودتايت؛ iii) يمكن تحقيق التركيز الفائق الكفاءة لليورانيوم من خلال تشكيل نقاط نانوية قابلة للذوبان من الستودتايت بناءً على مواقع التChelating الوفيرة لليورانيوم، مما تجنب تشكيل قضبان نانوية كبيرة وصعبة الذوبان من الستودتايت. بعد خمس دورات متتالية من الاستخراج والغسل، تم تركيز 200 مل من مياه البحر الملوثة باليورانيوم بتركيز 1 جزء في المليون إلى 4 مل من محلول اليورانيوم بتركيز 28.7 جزء في المليون باستخدام محفز ضوئي قابل للتجديد TpBpy. علاوة على ذلك، فإن سعة امتصاص اليورانيوم المثيرة للإعجاب لـيمكن تحقيق اليوم بتكلفة فعالةفي مياه البحر الحقيقية تحت إشعاع الضوء المرئي. يُعتقد أنه استنادًا إلى نتائجنا، يمكن أن تكون هناك مواد أكثر اقتصادية ومقاومة للأحماض مثل COFs أو حتى مواد أخرى تحتوي على المتطلبات اللازمة، أي مواقع ربط اليورانيوم الوفيرة وقدرة الإنتاج، يمكن إعدادها في مجال هندسة المواد لاستخراج اليورانيوم العملي من مياه البحر.
طرق
المواد
تم شراء جميع المواد الكيميائية من الموردين التجاريين واستخدامها دون مزيد من التنقية ما لم يُذكر خلاف ذلك. 1,3,5-ثلاثي الفورمال فيلوروجلوكزينول (Tp،1,3,5-ثلاثي الفورمال بنزين (Tb، )، 2،2′-بيبيريدين-5،5′-ديامين (Bpy، 97%)، وبنزيدين (Bd، 97%) تم شراؤها من أكاديمية العلوم الصينية في جيلين – يانشين
شركة التكنولوجيا المحدودة. N، N-dيميثيل أسيتاميد (DMAC، 99.8%)، 1،3،5-تريميثيل بنزين كان (ثنائي كلوريد البنزين ( -دي سي بي، )، 1-بيوتانول (99 %، AR)، 1,4-ديكسان (99.5 %)، حمض الأسيتيك (AcOH، )، إيثانول نقي ( الماء ) وثنائي ميثيل الفوران (THF، تم توفير جميعها من قبل علاء الدين. تم تحضير الماء النقي للغاية من نظام ميليبور. ).
تركيب Tp-Bpy و Tp-Bd
-تري فوميل فلوروجلوكزينول ( ) و -بيبيريدين-5,5′-ديامين (Bpy، ) أو بنزيدين (Bd، ) تم وضعها في أنبوب شلنق سعة 15 مل. ثم تم استخدام مزيج من المذيب N، N-dimethylacetamide (DMAC) وثنائي كلور البنزين (3/1، ) تم إضافته أيضًا، وتم تعريضه لعلاج بالموجات فوق الصوتية لتحقيق تشتت متجانس. ثم تم إضافة 0.6 مل من محلول حمض الأسيتيك (AcOH) بتركيز 6 م بسرعة وتمت المعالجة بالموجات فوق الصوتية مرة أخرى. بعد إزالة الغازات، تم إجراء التفاعل عند لمدة 3 أيام. ثم تم غسل المادة الصلبة بكمية كبيرة من DMAC والماء وTHF وتجفيفها تحت الفراغ عندلمدة 12 ساعة.
تركيب Tb-Bpy
ثلاثي الفورمال بنزين ) و 2، 2′-بيبيريدين-5، 5′-ديامين (Bpy، ) تم وضعها في أنبوب شلنق سعة 15 مل. ثم تم استخدام مزيج من المذيبين o-dichlorobenzene و Butanol ( ) ( ) تم إضافتها أيضًا، وتم تعريضها لعلاج بالموجات فوق الصوتية لتحقيق تشتت متجانس. ثم تم إضافة 0.3 مل من محلول حمض الأسيتيك (AcOH) المائي بتركيز 6 م بسرعة وتمت المعالجة بالموجات فوق الصوتية مرة أخرى. بعد المعالجة بالموجات فوق الصوتية لمدة 10 دقائق، تم تجميد الأنبوب بسرعة عبر السائلتم غسلها وإزالة الغازات منها من خلال ثلاث دورات تجميد-ضخ-ذوبان. ثم تم إغلاق الأنبوب وتسخينه عندلمدة 3 أيام. تم غسل المادة الصلبة بكمية كبيرة من الإيثانول وتجفيفها تحت الفراغ عندلمدة 12 ساعة.
تركيب Tb-Bd
ثلاثي الفورمال بنزين ) و بنزيدين (Bd، ) تم وضعها في أنبوب شلنكي سعة 15 مل. ثم تم استخدام مزيج من المذيبات 1,4-دي أوكسان وبيوتانول ( ) تم إضافته، وخضع لعلاج بالموجات فوق الصوتية لتحقيق تشتت متجانس. ثم تم إضافة 0.3 مل من محلول حمض الأسيتيك (AcOH) المائي بتركيز 6 م بسرعة. بعد المعالجة بالموجات فوق الصوتية لمدة 10 دقائق، تم تجميد الأنبوب بسرعة عبر السائلتم غسلها وإزالة الغازات منها من خلال ثلاث دورات تجميد-ضخ-ذوبان. ثم تم إغلاق الأنبوب وتسخينه عندلمدة 3 أيام. تم غسل المادة الصلبة بكمية كبيرة من الإيثانول وتجفيفها تحت الفراغ عندلمدة 12 ساعة.
تركيب Tp-Py و Tp-Bpy-2
-تري فوميل فلوروجلوكزينول ( ) و 2,5 -دايمينوبيريدين (باي، ) أو [ -بيبيريدين]-6،6′-ديامين (Bpy- ) تم وضعها في أنبوب شلنكي سعة 15 مل. ثم تم إضافة مزيج من المذيبات من -ثنائي ميثيل أسيتاميد (DMAC) وثنائي كلور البنزين ( )، وتم إخضاعها لعلاج بالموجات فوق الصوتية لتحقيق تشتت متجانس. ثم تم إضافة 0.6 مل من حمض الأسيتيك 6 M محلول مائي بسرعة وتمت المعالجة بالموجات فوق الصوتية مرة أخرى. بعد إزالة الغازات، تم إجراء التفاعل عند لمدة 3 أيام. ثم تم غسل الصلبة بكميات كبيرة من DMAC والماء وTHF وتجفيفها تحت الفراغ عند لمدة 12 ساعة.
الخصائص
تم إجراء تحليلات حيود الأشعة السينية (PXRD) باستخدام جهاز حيود الأشعة السينية Rigaku SmartLab SE مزود بـ مصدر (تم استخدام بيانات تشتت الأشعة السينية بزاوية صغيرة التي تم جمعها على جهاز حيود Bruker D8 Advance لتصحيح الانحراف) بحجم خطوة قدره . تم تسجيل أطياف تحويل فورييه للأشعة تحت الحمراء (FT-IR) على جهاز SHIMADZU IRTracer-100. تم تحديد مساحات السطح BET من إيزوثرمات الامتصاص/الامتزاز التي تم جمعها عند 77 ك باستخدام Micromeritics TriStar II. تم تسجيل صور المجهر الإلكتروني الماسح (SEM)
على جهاز المجهر الإلكتروني الماسح Hitachi SU 8100. تم تسجيل صور المجهر الإلكتروني الناقل (TEM) وصور المجهر الإلكتروني الناقل عالي الدقة (HRTEM) على جهاز المجهر الإلكتروني الناقل JEOL (JEM-F200) الذي يعمل عند جهد تسريع قدره 200 كيلوفولت. تم جمع أطياف CP/MAS NMR على جهاز BRUKER AVANCE NEO 400 WB. تم إجراء تحليلات طيف الأشعة السينية (XPS) باستخدام جهاز Thermo Scientific ESCALAB 250Xi، مزود بمصدر أشعة سينية أحادي اللون Al K (1486.8 eV). تم إجراء قياسات التجارب الكهروضوئية على جهاز عمل كهربائي (CHI760E، CHI Instruments، شنغهاي، الصين). تم تسجيل أطياف الرنين المغناطيسي الإلكتروني (EPR) عند 293 ك باستخدام جهاز Bruker EMXnano259، الذي يعمل عند 9.62 غيغاهرتز مع طاقة 12.59 مللي واط وتعديل عند . تم تسجيل نتائج طيف الأشعة فوق البنفسجية-المرئية في وضع الانعكاس المنتشر (DR) في درجة حرارة الغرفة على جهاز SHIMADZU UV-2700 مزود بملحق كرة متكاملة. تم إجراء تحليلات الوزن الحراري (TGA) على جهاز NETZSCH STA 2500. تم إجراء تحليلات قياس الطيف الكتلي بالبلاتين المتصل (ICP-MS) على جهاز Agilent 7800.
قياسات الكهروضوئية
تم قياس مخططات Mott-Schottky، استجابة التيار الضوئي والمقاومة الكهروكيميائية للمواد المحفزة الضوئية على جهاز عمل كهربائي (CHI760E، CHI Instruments، شنغهاي، الصين). تم استخدام مصباح Xe بقدرة 300 واط كمصدر للضوء و ، ) محلول مائي تم استخدامه كإلكتروليت داعم طوال قياسات التيار الضوئي. تم استخدام سلك من البلاتين و كإلكترود مضاد وإلكترود مرجعي، على التوالي. تم إذابة نافثول في حوالي 3 مل من الإيثانول مع 1 ملغ من المحفز. ثم، تم إضافته إلى ITO مع طبقة من الشريط اللاصق على الحافة ست مرات، تلاها التجفيف في الهواء. تم تحديد الجهد مقابل NHE باستخدام المعادلة التالية (2):
تجارب استخراج اليورانيوم
تم إجراء جميع التجارب على استخراج في الهواء تحت الضوء المرئي ( ) باستخدام مصباح LED بقدرة 10 واط (Perfect PCX50C Discover) كمصدر للضوء وكان الحد الأقصى التيار الضوئي يتوافق مع 10 واط.
الاستخراج الضوئي للماء U(VI)
تم تفريق 10 ملغ من المحفز الضوئي في 40 مل من مياه البحر الملوثة باليورانيوم ( ) والماء النقي ( )، الذي يحتوي على ميثانول. تم الحفاظ على درجة الحرارة عند عن طريق تدوير الماء المبرد طوال عملية الاستخراج الضوئي. بعد الاستخراج الضوئي، تم تصفية التعليق الناتج بواسطة غشاء 220 نانومتر. تم قياس تركيز باستخدام طيف الأشعة فوق البنفسجية-المرئية عند طول موجي 650 نانومتر باستخدام طريقة Arsenazo III.
الاستخراج الضوئي لليورانيوم من مياه البحر الطبيعية
تم تفريق 10 ملغ من Tp-Py في مفاعل مصمم للتجميع الذاتي. تدفقت مياه البحر عبر المحفز الضوئي بسرعة باستخدام مضخة دائرية. تم تحليل تركيز اليورانيوم في مياه البحر الطبيعية بواسطة قياس الطيف الكتلي بالبلاتين المتصل (ICP-MS).
تصميم الجهاز لاستخراج اليورانيوم من مياه البحر الطبيعية
تم خلط 10 ملغ من COFs مع 7 مل من الإيثانول، 1 مل من الماء النقي و من محلول نافيون. تم إسقاط الخليط على لوحة من الكوارتز. باستخدام المضخة الدائرية، تدفقت مياه البحر الحقيقية عبر لوحة الكوارتز المطلية.
اختبار التركيز
تم وضع المحفز الضوئي المستخدم مع اليورانيوم في 1 مل من محلول الإذابة في درجة حرارة الغرفة. بعد ساعة، تم إخراجه لإجراء استخراج ضوئي آخر في محلول يحتوي على اليورانيوم لمدة 3 ساعات. تم تكرار عملية الإذابة في نفس محلول الإذابة لتركيز اليورانيوم. بعد 5 مرات من الإذابة، تم الكشف عن تركيز اليورانيوم في محلول الإذابة النهائي بواسطة ICP-MS.
أثر الأيونات المتنافسة على الاستخراج الضوئي للماء VI
تم تفريق 10 ملغ من المحفز الضوئي في 40 مل من الماء النقي ( ) مع ، ) أو أملاح الصوديوم ( و ) لمدة 3 ساعات من الإشعاع.
اختبار القابلية لإعادة الاستخدام
بعد إجراء عملية استخراج ضوئي واحدة، تم تجديد المحفز الضوئي بواسطة الإذابة باستخدام محلول ( أو 2.5). ثم تم إعادة استخدام المحفز الضوئي الناتج في تجربة استخراج ضوئي أخرى. بالنسبة لـ 10 ملغ من المحفزات، تم استخدام 40 مل من محلول الإذابة لإذابة اليورانيوم المرتبط لمدة 60 دقيقة في درجة حرارة الغرفة. تم تحديد كفاءة الإذابة (E، %) باستخدام المعادلة التالية (3):
حيث هو تركيز اليورانيوم في محلول الإذابة، هو حجم محلول الإذابة، هو تركيز اليورانيوم في مياه البحر الملوثة باليورانيوم بعد التفاعل الضوئي، ) هو التركيز الأولي لليورانيوم في مياه البحر الملوثة باليورانيوم، (لتر) هو حجم مياه البحر الملوثة باليورانيوم المستخدمة للامتصاص. تم تصفية التعليق الناتج وغسله بالماء النقي حتى أصبح السائل العلوي محايدًا. بعد التجفيف تحت الفراغ، تم استخدام المادة الناتجة في تجربة تفاعل ضوئي أخرى. وُجد أنه بعد ست دورات متتالية، لا يزال Tp-Bpy يظهر امتصاصًا ممتازًا لليورانيوم.
طرق الكشف
تم تأهيل الجيل باستخدام طريقة لونية. عادةً، يتم طرد الخليط بعد التفاعل الضوئي ثم يتم إضافة من التعليق إلى خليط من 2 مل من محلول KI (0.1 M) و محلول (0.01 M). تم قياس الامتصاص المقابل عند 352 نانومتر على طيف الأشعة فوق البنفسجية-المرئية.
حساب ثابت التوازن للمعادلة (1)
تم خلط 42.5 مل من محلول مع 7.5 مل من الميثانول في مفاعل سعة 100 مل. تم ضبط درجة حرارة المفاعل عند باستخدام مضخة دورانية. تم ضبط الرقم الهيدروجيني للخليط عند 4 عن طريق إضافة كميات ضئيلة من محلول NaOH و ( ). تم إضاءة المحلول المختلط تحت الضوء المرئي ( ) باستخدام مصباح Xe بقدرة 300 واط (Microsolar 300، PerfectLight) مزود بفلتر قطع الأشعة فوق البنفسجية، خلال ذلك، يمكن تحفيز أنواع اليورانيوم لإنتاج و يتفاعل الناتج مع اليورانيوم لتشكيل صلبة ستودايت. تم مراقبة تركيز اليورانيوم في المحلول كل ساعة من أجل التأكد من التوازن. وُجد أنه بعد 8 ساعات، توقف تركيز اليورانيوم المتبقي عن الانخفاض. بعد ذلك، يتم تحريك التعليق النهائي في الظلام لمدة ساعة. ثم تم قياس تركيز وقيمة الرقم الهيدروجيني. يمكن حساب ثابت التوازن للتفاعل باستخدام التركيزات المكتشفة لليورانيوم و ، بالإضافة إلى قيمة الرقم الهيدروجيني.
قياسات مقاومة التلوث البيولوجي
تم استخدام سلالة بكتيرية بحرية لاختبار النشاط المضاد للميكروبات للمحفز. تم تفريق Tp-Py في بكتيريا بحرية/مرق لوريا-بيرتاني الطازج بتركيز تفريق قدره . بعد الزراعة عند لمدة 3 ساعات باستخدام مذبذب حراري، تم تحديد قابلية
البكتيريا. تم استخدام طريقة عد الصفائح المخففة وفقًا للمعيار الصيني GB/T20944 لتحديد قابلية الثقافات البكتيرية المعالجة. لمحاكاة ظروف التجربة، تم مقارنة التأثير المضاد للبكتيريا على التوالي تحت حالة الظلام وإشعاع الضوء المرئي. تم حساب معدلات التثبيط (IR) وفقًا للمعادلة التالية (4):
حيث و هما تركيزات الميكروبات في الثقافات قبل وبعد المعالجة مع Tp-Py.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
البيانات التي تدعم نتائج هذه الدراسة متاحة في المقالة وملفات المعلومات التكميلية أو متاحة من المؤلفين المقابلين عند الطلب. القيم العددية للبيانات المعروضة في الأشكال في النص الرئيسي متوفرة في ملف بيانات المصدر المرفق. يتم توفير بيانات المصدر مع هذه الورقة.https://doi. org/10.6084/m9.figshare.26149174). تم توفير بيانات المصدر مع هذه الورقة.
References
Kittner, N., Lill, F. & Kammen, D. M. Energy storage deployment and innovation for the clean energy transition. Nat. Energy 2, 17125 (2017).
Holechek, J. L., Geli, H. M. E., Sawalhah, M. N. & Valdez, R. A global assessment: Can renewable energy replace fossil Fuels by 2050? Sustainability 14, 4792 (2022).
Wang, C., Raza, S. A., Adebayo, T. S., Yi, S. & Shah, M. I. The roles of hydro, nuclear and biomass energy towards carbon neutrality target in China: A policy-based analysis. Energy 262, 125303 (2023).
Meitner, L. & Frisch, O. R. Disintegration of uranium by neutrons: a new type of nuclear reaction. Nature 143, 239-240 (1939).
Liu, Z. et al. Multi-scale computer-aided design and photocontrolled macromolecular synthesis boosting uranium harvesting from seawater. Nat. Commun. 13, 3918 (2022).
Abney, C. W., Mayes, R. T., Saito, T. & Dai, S. Materials for the recovery of uranium from seawater. Chem. Rev. 117, 13935-14013 (2017).
Sun, Q. et al. Bio-inspired nano-traps for uranium extraction from seawater and recovery from nuclear waste. Nat. Commun. 9, 1644 (2018).
Liu, C. et al. A half-wave rectified alternating current electrochemical method for uranium extraction from seawater. Nat. Energy 2, 17007 (2017).
Ku, T.-l, Knauss, K. G. & Mathieu, G. G. Uranium in open ocean: concentration and isotopic composition. Deep Sea Res. 24, 1005-1017 (1977).
Ye, Y. et al. Spontaneous electrochemical uranium extraction from wastewater with net electrical energy production. Nat. Water 1, 887-898 (2023).
Chen, L. et al. Ultrafast and efficient extraction of uranium from seawater using an amidoxime appended metal-organic framework. ACS Appl. Mater. Interf. 9, 32446-32451 (2017).
Yuan, Y. et al. A bio-inspired nano-pocket spatial structure for ttargeting uranyl capture. Angew. Chem. Int. Ed. 59, 4262-4268 (2020).
Li, Z. et al. Constructing amidoxime-modified porous adsorbents with open architecture for cost-effective and efficient uranium extraction. Chem. Sci. 11, 4747-4752 (2020).
Chen, Z. et al. Tuning excited state electronic structure and charge transport in covalent organic frameworks for enhanced photocatalytic performance. Nat. Commun. 14, 1106 (2023).
Cui, W.-R. et al. Regenerable Covalent organic frameworks for photo-enhanced uranium adsorption from seawater. Angew. Chem. Int. Ed. 59, 17684-17690 (2020).
Wang, G.-B. et al. Covalent organic frameworks and their composites as multifunctional photocatalysts for efficient visible-light induced organic transformations. Coord. Chem. Rev. 472, 214774 (2022).
Zhang, Q. Y. et al. Ultra-selective uranium separation by in-situ formation of conjugated 2D uranium-organic framework. Nat. Commun. 15, 453 (2024).
Xie, Y. et al. Uranium extraction from seawater: material design, emerging technologies and marine engineering. Chem. Soc. Rev. 52, 97-162 (2023).
Yuan, Y. et al. DNA nano-pocket for ultra-selective uranyl extraction from seawater. Nat. Commun. 11, 5708 (2020).
Yuan, Y. et al. Selective extraction of uranium from seawater with biofouling-resistant polymeric peptide. Nat. Sustainability 4, 708-714 (2021).
Pan, Z. et al. Nanoscale mechanism of formation through uranium reduction by magnetite. Nat. Commun. 11, 4001 (2020).
Feng, L. et al. Halogen hydrogen-bonded organic framework (XHOF) constructed by singlet open-shell diradical for efficient photoreduction of U(VI). Nat. Commun. 13, 1389 (2022).
Yang, L. et al. Bioinspired hierarchical porous membrane for efficient uranium extraction from seawater. Nat. Sustainability 5, 71-80 (2022).
Liu, T. et al. Ligand-assistant iced photocatalytic reduction to synthesize atomically dispersed Cu implanted metal-organic frameworks for photo-enhanced uranium extraction from seawater. Small 19, 2208002 (2023).
Yang, H. et al. Tuning local charge distribution in multicomponent covalent organic frameworks for dramatically enhanced photocatalytic uranium extraction. Angew. Chem. Int. Ed. 62, e202303129 (2023).
Hu, Y. et al. Photochemically triggered self-extraction of uranium from aqueous solution under ambient conditions. Appl. Catal. B-Environ. 322, 122092 (2023).
Wang, Z. et al. Photo-induced removal of uranium under air without external photocatalysts. Green. Chem. 24, 7092-7099 (2022).
Kim, J., Kim, H., Kim, W.-S. & Um, W. Dissolution of studtite [ ] in various geochemical conditions. J. Environ. Radioact. 189, 57-66 (2018).
Yu, F.-Y., Zhou, Y.-J., Tan, H.-Q., Li, Y.-G. & Kang, Z.-H. Versatile photoelectrocatalysis strategy raising up the green production of hydrogen peroxide. Adv. Energy Mater. 13, 2300119 (2023).
Edwards, J. K. et al. The Direct Synthesis of Hydrogen Peroxide Using Platinum-Promoted Gold-Palladium Catalysts. Angew. Chem. Int. Ed. 53, 2381-2384 (2014).
Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962-6984 (2006).
Liao, Y. et al. Regulating benzene ring number as connector in covalent organic framework for boosting photosynthesis of from seawater. Nano Lett. 24, 3819-3825 (2024).
Li, P. et al. 1D covalent organic frameworks triggering highly efficient photosynthesis of via controllable modular design. Angew. Chem. Int. Ed. 63, e202319885 (2024).
Zhang, T. et al. Atomic-level understanding for the enhanced generation of hydrogen peroxide by the introduction of an aryl amino group in polymeric carbon nitrides. ACS Catal. 11, 14087-14101 (2021).
Liao, Q. et al. Regulating relative nitrogen locations of diazine functionalized covalent organic frameworks for overall photosynthesis. Angew. Chem. Int. Ed. 62, e202310556 (2023).
Daugherty, M. C. et al. Improved synthesis of -ketoenamine-linked covalent organic frameworks via monomer exchange reactions. Chem. Commun. 55, 2680-2683 (2019).
Zhong, W. et al. A covalent organic framework bearing single Ni sites as a synergistic photocatalyst for selective photoreduction of to CO. J. Am. Chem. Soc. 141, 7615-7621 (2019).
Dey, K. et al. Selective molecular separation by interfacially crystallized covalent organic framework thin films. J. Am. Chem. Soc. 139, 13083-13091 (2017).
Xu, L. et al. Surface-confined crystalline two-dimensional covalent organic frameworks via on-surface schiff-base coupling. ACS Nano 7, 8066-8073 (2013).
Wang, Q. et al. N-doping of -ketoenamine based covalent organic frameworks for catalytic conversion of to cyclic carbonate and Knoevenagel condensation. Microporous Mesoporous Mater. 364, 112872 (2024).
Yang, Y. et al. Engineering -ketoamine covalent organic frameworks for photocatalytic overall water splitting. Nat. Commun. 14, 593 (2023).
Chu, F. et al. Regulating keto-enol tautomerism of -ketoenamine covalent-organic frameworks for photocatalytic oxidative coupling of amines. ACS Catal. 13, 13167-13180 (2023).
Wang, H. et al. Integrating suitable linkage of covalent organic frameworks into covalently bridged inorganic/organic hybrids toward efficient photocatalysis. J. Am. Chem. Soc. 142, 4862-4871 (2020).
Kandambeth, S. et al. Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc. 134, 19524-19527 (2012).
Yang, H. et al. MOFs-derived fusiform mesoporous nanorods anchored with ultrafine CdZnS nanoparticles for boosting visible-light photocatalytic hydrogen evolution. Small 17, 2102307 (2021).
Hao, X. et al. Architecture of high efficient zinc vacancy mediated Z-scheme photocatalyst from metal-organic frameworks. Nano Energy 52, 105-116 (2018).
Kou, M. et al. Molecularly engineered covalent organic frameworks for hydrogen peroxide photosynthesis. Angew. Chem. Int. Ed. 61, e202200413 (2022).
. et al. Conjugated organic polymers with anthraquinone redox centers for efficient photocatalytic hydrogen peroxide production from water and oxygen under visible light irradiation without any additives. ACS Catal. 12, 12954-12963 (2022).
Hu, H. et al. Rational modification of hydroxy-functionalized covalent organic frameworks for enhanced photocatalytic hydrogen peroxide evolution. J. Colloid Interf. Sci. 629, 750-762 (2023).
Guo, R. et al. Chelating effect between uranyl and pyridine N containing covalent organic frameworks: A combined experimental and DFT approach. J. Colloid Interf. Sci. 606, 1617-1626 (2022).
Colella, M., Lumpkin, G. R., Zhang, Z., Buck, E. C. & Smith, K. L. Determination of the uranium valence state in the brannerite structure using EELS, XPS, and EDX. Phys. Chem. Miner. 32, 52-64 (2005).
Liu, W. et al. Highly sensitive and selective uranium detection in natural water systems using a luminescent mesoporous metal-organic framework equipped with abundant lewis basic sites: A combined batch, X-ray absorption spectroscopy, and first principles simulation investigation. Environ. Sci. Technol. 51, 3911-3921 (2017).
Li, H. et al. Powerful uranium extraction strategy with combined ligand complexation and photocatalytic reduction by
postsynthetically modified photoactive metal-organic frameworks. Appl. Catal. B-Environ. 254, 47-54 (2019).
Song, Y. et al. Unassisted uranyl photoreduction and separation in a Donor-Acceptor covalent organic framework. Chem. Mater. 34, 2771-2778 (2022).
Li, Z. et al. Exciton dissociation and transfer behavior and surface reaction mechanism in Donor-Acceptor organic semiconductor photocatalytic separation of uranium. Appl. Catal. B-Environ. 332, 122751 (2023).
Yuan, Y. et al. Photoinduced multiple effects to enhance uranium extraction from natural seawater by black phosphorus nanosheets. Angew. Chem. Int. Ed. 59, 1220-1227 (2020).
Zhang, C.-R. et al. Simultaneous sensitive detection and rapid adsorption of based on a post-modified carbonconjugated covalent organic framework. Environ. Sci. Nano. 7, 842-850 (2020).
شكر وتقدير
نحن نعرب عن شكرنا العميق للدعم المالي من المؤسسة الوطنية للعلوم الطبيعية في الصين (رقم 22176054 (جي.-إكس.زد.)، 22306060 (ي.زد.هـ.))، مؤسسة العلوم ما بعد الدكتوراه في الصين (رقم 2022M721134 (ي.زد.هـ.))، البرنامج الوطني الرئيسي للبحث والتطوير في الصين (رقم 2018YFC1900105 (إكس.-كي.دبليو.)) وبرنامج العلماء الشباب المتميزين في بكين (إكس.-كي.دبليو.).
مساهمات المؤلفين
قام P.G. وG.-X.Z. وX.-K.W. وX.-B.H. بتصور البحث وتصميم التجارب. قام P.G. وY.-Z.H. وZ.-W.S. بتنفيذ التجربة. قام جميع المؤلفين بتحليل البيانات. ساهم Z.-Y.J. وR.-Q.C. وF.C. في مناقشات المشروع. شارك P.G. وG.-X.Z. وX.-K.W. وX.-B.H. في صياغة الورقة وأعطوا الموافقة على النسخة النهائية من المخطوطة.
كلية علوم البيئة والهندسة، جامعة شمال الصين للطاقة الكهربائية، بكين 102206، جمهورية الصين الشعبية.مركز بكين المتقدم للابتكار في هندسة جينوم المواد، مختبر بكين الرئيسي للمواد الوظيفية لبناء الجزيئات والهياكل، كلية علوم المواد والهندسة، جامعة العلوم والتكنولوجيا في بكين، بكين 100083، جمهورية الصين الشعبية. البريد الإلكتروني:guixiazhao@ncepu.edu.cn; jizhuoyu@ncepu.edu.cn; xkwang@ncepu.edu.cn; xiubinghuang@ustb.edu.cn
Ultra-highly efficient enrichment of uranium from seawater via studtite nanodots growthelution cycle
Received: 3 February 2024
Accepted: 24 July 2024
Published online: 07 August 2024
(A) Check for updates
Abstract
Peng Gao , Yezi Hu , Zewen Shen , Guixia Zhao , Ruiqing Cai , Feng Chu , Zhuoyu Ji (B) , Xiangke Wang ) & Xiubing Huang ²
Consecutive uranium extraction from seawater is a promising approach to secure the long-term supply of uranium and the sustainability of nuclear energy. Here, we report an ultra-highly efficient strategy via studtite nanodots growth with impressive uranyl uptake capacity of from natural seawater in 12 consecutive days (i.e., average for day ). Uranyl can be extracted as studtite under visible light via the reaction between the adsorbed uranyl and the photogenerated with imine-based CovalentOrganic Framework photocatalysts. In detail, over Tp-Bpy, Tp-Bpy-2 and Tp-Py with multiple uranyl chelating sites, uranyl is found extracted as studtite nanodots which can be eluted readily, while over Tp-Bd and Tb-Bpy, uranyl is transformed into studtite nanorods that is more inert for elution. Abundant chelating sites of uranyl via structural regulation of COF photocatalysts are proved to facilitate the formation and efficient elution of studtite nanodots.
As global energy demand grows and the need for a clean energy transition increases , nuclear power is expected to be a promising new energy in the foreseeable future thus has been developed vigorously . Uranium is the most important component to trigger the fission reaction . Considering the limited uranium source in the land and the considerable amount of uranium resource in the sea (over 4 billion tons) , enrichment of uranium from seawater is of great significance for the sustainable development of nuclear industrial . However, due to the low concentration of uranyl in seawater ( parts per billion) and the complicated marine environment , developing an efficient enrichment approach for uranyl up-recycle remains a challenge.
Advanced porous materials such as Metal-Organic Frameworks (MOFs) , Porous Organic Polymers (POPs) and Covalent-Organic Frameworks (COFs) , have been extensively developed for uranium extraction from natural seawater, due to their functionalized building blocks and tunable pore characteristics . The diverse functional groups and the well-designed adsorbents/photocatalysts ensured the
high extraction capacity by virtue of the adsorption and/or photoreduction of discrete uranyl at binding sites at enriched binding sites. For instance, Yang et al. designed a hierarchical porous membrane utilizing amidoxime-functionalized PIM-1, which allows rapid diffusion of uranium species and realized a remarkable 20 -fold enhancement in uranyl adsorption capacity compared to the membrane with only intrinsic microporosity . Liu et al. successfully anchored atomically dispersed Cu to a UiO-66- support via a simple ligand-assisted ice photocatalytic reduction pathway, achieving a high uranyl extraction capacity ( -Ads) from natural seawater . In 2023, Yang et al. reported a series of novel intramolecular donor-acceptor (D-A) systems by introducing different electron-donating and withdrawing groups in the pores of the COF structure, which exhibit uranyl uptake capacity of day in real seawater under visible light . However, due to the limited available binding sites within such framework material, it is hard to overtake the uptake capacity based on the extraction via the discrete uranyl-binding.
In our previous study , the excited can be utilized for photochemical production of hydrogen peroxide ( ), which can react with to form solid studtite. Such photochemical process was applied for selective uranyl extraction from seawater without using any solid materials . Nevertheless, due to the absence of nucleation center, the extraction must take a long time (at least 24 h ). It was also reported by Kim et al. . that the Eq. (1) between and uranyl at ambient condition are reversible.
When pH value is low enough (e.g., ), solid studtite can be efficiently dissolved as free . Inspired by these conclusions, we proposed to construct a series of photocatalysts with efficient production activity and binding centers for studtite formation. More importantly, such COFs should be acid-resistant so that the possibly formed studtite can be dissolved under acidic condition and COFs can be regenerated simultaneously for long-term utilization. is a very famous quintessential green oxidant but has difficulty for long-term storage because of the self-decomposition . Anthraquinone method is widely employed in industrial production of , contributing to over of output . The anthraquinone method involves the use of substantial organic solvents, generates toxic by-products, and is prone to catalyst deactivation, leading to high energy consumption and substantial pollution . Although is not combustible itself, its high oxidizing properties render it a hazardous industrial raw material. In particular, during closed transportation, high concentrations of (above ) can lead to a rapid temperature increase, potentially causing the gas phase to explode. Consequently, utilizing a green synthesis strategy to produce for local utilization has become a research topic recently . Since iminebased COFs are well-known photocatalysts for the synthesis of , we designed and synthesized four imine-based COFs photocatalysts (Tp-Bpy, Tp-Bd, Tb-Bpy and Tb-Bd) with chelating groups for uranyl such as keto groups and bipyridine (Fig. 1). It is found that compared with COFs with individual keto or bipyridine N chelating group, the abundant binding sites on Tp-Bpy containing both keto
groups and bipyridine N are beneficial for the formation of dissoluble studtite nanodots, which could realize the efficient studtite nanodots growth-elution cycle. To broaden the photocatalysts and clarify the suppose that combination of abundant adsorption sites and the production ability of photocatalysts could lead to the highly efficient uranyl enrichment via studtite nanodots growth and elution cycle, we prepared another two imine-based COF photocatalysts with keto groups and pyridine-N sites (Tp-Bpy-2 and Tp-Py) (Supplementary Fig. 1,2), both of which showed efficient enrichment of uranyl in the studtite nanodots growth-elution cycles.
Results
Synthesis and characterization of COFs
In this work, combination of diverse linkers was used to synthesize COFs with different functional groups, which endow the different capacity of production. Initially, -ketoenamine COFs were synthesized by altering the bipyridine monomer, obtaining Tp-Bpy and Tp-Bd via the condensation reaction in a mixture of -dimethylacetamide (DMAC) and o-dichlorobenzene (o-DCB) with acetic acid as catalyst at . Next, the COFs without any carbonyl groups were synthesized via the similar solvothermal reaction, which were assigned to Tb-Bpy and Tb-Bd. The structure of the four COFs was determined by powder X-ray diffraction (PXRD), solid NMR Spectroscopy and Fourier infrared transform spectroscopy (FT-IR).
As clearly shown in Fig. 2a and Supplementary Fig. 3, the vanish of the stretching band ( ) at and stretches at confirmed complete consumption of the aldehyde and diamine groups in the reactants . As for Tp-based COFs, the newly emerged C-N characteristic stretching peaks and the formed and bond at and , respectively, verified the presence of keto form rather than enol form. As for Tb -based COFs, the stretching at perspicuously manifested the formation of imine bonds. Simultaneously, X-ray photoelectron spectrum (XPS) N distinctly designated three forms of N ( and imine bonds) for the four (Supplementary Fig. 2), which were in line with above-mentioned FT-IR results. Solid-state CP/MAS NMR spectra equally confirmed the
Fig. 1 | Synthesis and structures. The schematic diagram for synthesis and structures of four COFs.
Fig. 2 | Chemical structure and characterization of Tp-Bpy, Tp-Bd, Tb-Bpy and Tb-Bd. a FT-IR spectra. b Solid-state CP/MAS NMR spectra. c-f PXRD characterization (experimental PXRD profile: red, refined profile: thin blue, Bragg
positions: green and difference: blue. The insets show the structural models and interlayer distances of each COF assuming an AA stacking mode. SEM images.
successful preparation of the . Tp-Bpy and Tp-Bd displayed distinctive signals for the carbonyl carbons ( ) at approximately and exocyclic carbons ( ) at approximately 107 ppm (Fig. 2b). On the other side, imine bonds ( ) at 157-162 ppm emerged in Tb-Bpy and Tb-Bd. The combination of FTIR and NMR analysis demonstrated the successful synthesis of four COFs at the chemical structure level.
Furthermore, the crystallinity of each COF was examined by PXRD and Pawley refinement analysis. As shown in Figs. 2c-f, there are the strongest diffraction peaks in each PXRD pattern at , and , indicating that the open channel of the four COFs was formed successfully. Meanwhile, they all had a broad peak at around due to the – stacking between the COF layers corresponding to the (001) plane. Furthermore, the Pawley refinement of the experimental PXRD data was done to find out the details of the unit cell parameters (Supplementary Table 1-4), the detailed factors and negligible residuals ( and ) of which were in accordance with the predictions. The characteristic of the surface morphologies of the four COFs was conducted utilizing Scanning Electron Microscopy (SEM). Figures showed that Bpy and Bd exhibited blocky structure composed of aggregated microspheres, while Tp-Bd exhibited grasslike morphologies that consist of well-bedded nanofibers and Tb-Bpy showed a close aggregation of rod-like structures.
High specific surface area and adjustable porosity of COFs make them optimal photocatalysts with more exposed active sites. To acquire the porosity, adsorption-desorption analyses were conducted on the fully activated samples at 77 K . The adsorptiondesorption isotherms were identified as type-II curves with a mesoporous character (Fig. 3a). The total pore volumes were evaluated to
be , and for Tp-Bpy, Tp-Bd, Tb-Bpy and Tb-Bd, respectively. The pore size distributions were calculated by BJH to be centered at 2.06 nm for Tp-Bpy, 2.09 nm for Tp-Bd, 2.24 nm for Tb-Bpy and Tb-Bd, respectively, which were all in good concordance with the calculation value (Supplementary Fig. 5). Furthermore, the Brunauer-Emmett-Teller (BET) surface areas of Tp-Bpy, Tp-Bd, Tb-Bpy and are 1346.5, and , respectively. The thermal stabilities of the four COFs were estimated by thermogravimetric analysis (TGA) under nitrogen atmosphere at . The TGA curves unveiled that these COFs demonstrate exceptional thermal robustness, enduring temperatures as high as almost without decomposition (Fig. 3b).
Characterization of the four COFs in photoelectric properties
The visible-light-harvesting capacity and energy band structures of TpBpy, Tp-Bd, Tb-Bpy and Tb-Bd were investigated by UV-Visible diffuse reflectance spectroscopy (UV-vis DRS) and Mott-Schottky (M-S) curves. As presented in Fig. 3c, all COFs exhibit intense absorption in the visible region. Analysis via Kubelka-Munk transformation equation in conjunction with Tauc plots (Fig. 3d) displayed that the energy band-gaps ( ) of Tp-Bpy, Tp-Bd, Tb-Bpy and Tb-Bd were 1.70, 1.65, 2.23 and 2.32 eV , respectively. According to the M-S curves (Fig. 3e), it was obvious that all COFs were typical n-type semiconductors for a positive slope. Moreover, the flat band values ( ) were estimated to be and -0.59 eV for Tp-Bpy, Tp-Bd, and , respectively. The conduction band (CB) potential is typically about 0.1 V lower than the flat band potential of n-type semiconductors . Combining with the value of and , the valence band (VB) potential ( ) could be plainly calculated (Fig. 3f).
Fig. 3 | Pore structure and photoelectrochemical tests for Tp-Bpy, Tp-Bd, Tb-Bpy and Tb-Bd. a sorption isotherms. Thermal gravimetric analysis spectra. c UV-vis diffuse reflection spectra. d Kubelka-Munk-transformed reflectance spectra. e Mott-Schottky curves. f Scheme illustration of the electronic band structures.
Furthermore, the energy level matched well with the requirements for the reduction of to , revealing that they are all promising photocatalysts for production.
Photo-extraction uranyl investigations
To evaluate the photocatalytic performance, the photo-extraction of uranyl was tested at and 5. At , as shown in Supplementary Fig. 6, approximately of uranyl can be extracted with TpBpy, obviously higher than that with other three COFs. As for Tp-Bd, there was a stagnation of extraction to when the reaction time was up to three hours. Differently, we found that Tb-Bpy showed a relatively slow but steady extraction rate. At , the overall reaction velocity had been significantly increased, with the final extraction efficiency of Tp-Bpy, Tp-Bd and Tb-Bpy increased to and , respectively (Fig. 4a). Similarly, Tb-Bpy shows a steady uranyl extraction from , while the extraction rate on Tp-Bpy and
Tp-Bd are slowed down obviously or even stagnated. With or in the absence of catalysts, the removal of uranyl was negligible. Compared with the uranyl extraction under dark condition, it’s concluded that the efficient uranyl extraction over Tp-Bpy, Tp-Bd and Tb-Bpy is largely related with photo-reaction although the adsorption of uranyl over Tp-Bpy contributes about to the whole extraction efficiency (Fig. 4b and Supplementary Fig. 7). The adsorption of uranyl on Tb-Bd is the lowest, which may be related with the poor complexation ability for uranyl ions in the absence of chelating groups.
The quantities of production were measured by colorimetric method (Fig. 4c, d and Supplementary Fig. 8, 9). The production rate over Tp-Bpy, Tp-Bd, Tb-Bpy and Tb-Bd is about 123.89, 8.79, 23.48 and , respectively. Without catalyst, there is no produced in the methanol aqueous solution. Figure 3 c showed the different trends of production kinetics with , which was related with the complicated interplay of production and
Fig. 4 | Photocatalytic performance of Tp-Bpy, Tp-Bd, Tb-Bpy and Tb-Bd for uranyl extraction and production. Reaction condition: , visible light, and 3 h . Error bars are the standard deviations of three replicate
measurements. a Photo-extraction kinetics. b Uranyl extraction efficiency under dark and light conditions. production kinetics in the presence of uranyl. d production kinetics in the absence of uranyl.
consumption process with different photocatalysts. Firstly, without catalyst, uranyl itself could be excited under visible light to produce , with similar production rate as that for the Tb-Bpy (Fig. 4c). Secondly, the adsorption of uranyl on COFs could block the production activity of both uranyl and COFs since the photogenerated electrons could not be directly transformed to when the active sites such as keto or bipyridine N chelating group was occupied with uranyl ; Thirdly, could be consumed via the reaction between and uranyl. Based on the above processes, the different trends of production can be explained. For the case of uranyl aqueous solution without catalyst, the detected is from the production of uranyl itself with no simultaneous consumption of since uranyl is not transformed into studtite. For the case of uranyl, since uranyl was only ca. removed (Fig. 4a), was almost not consumed to form studtite. The production of from uranyl itself and , accompanied with mutual intervention results in the observed trend. For the case of Tb-Bpy + uranyl, Tp-Bd + uranyl and Tp-Bpy + uranyl, the uranyl binding leaded to the obviously blocked production, in which Tb -Bpy adsorbed the lowest uranyl, thus the initially detected was a little higher than that with Tp-Bd and Tp-Bpy due to the minimal blocking from uranyl binding. With the uranyl extracted on COFs and the decreased concentration of uranyl in the solution, both production and consumption were slower, thus there were somehow platform for the detected in . When uranyl concentration was low enough, the production of overpass the consumption of , the detected
amount goes up again. For the case of Tp-Bd, in Fig. 4d, it showed that the production ability of Tp-Bd was quite poor and the adsorption of uranyl at the binding site seriously blocked the production of , thus the platform only emerged in , and there was almost no production of after 60 min thus no uranyl could be extracted as studtite in Fig. 4a. Moreover, in Fig. 4a, Tb-Bpy exhibited a relatively slower extraction rate than , but the production of by Bpy was higher than that by , which can be explained based on the following deduction. Within the first 90 minutes, although Tb -Bpy produced a higher quantity of than , Tp-Bd exhibited a significantly greater adsorption kinetics compared to Bpy as shown in Supplementary Fig. 7b. After 90 min , the rate of production of Tp-Bd decreases due to the affection of adsorbed uranyl, leading to a stagnation in uranyl extraction. In contrast, Tb-Bpy continued to produce , thus uranyl concentration continues to decline after 90 min . In the earlier stage of extraction, the quick adsorption kinetics dominates the process, while in the lateral stage when the binding sites were occupied by uranyl, the uranyl adsorption was merely stopped and the reaction between uranyl and played important role to the extraction. In brief, the different detected trends shown in Fig. 4c was the results of complicated interplay of the production and consumption of during the time course of uranyl extraction.
Combined analysis of uranyl extraction kinetics shown in Fig. 4a and production shown in Fig. 3c, d and Supplementary Fig. 8, it suggests that i) the low production rate and the poor adsorption
Fig. 5 | Investigation of photo-extraction products for Tp-Bpy and Tb-Bpy.
a XPS spectra after photo-extraction. b FT-IR spectrum comparison before and after photo-extraction. c PXRD after photo-extraction. d WT contour plots for
and Tp-Bpy after photo-extraction. e HRTEM image of Tb-Bpy after photo-extraction. HRTEM image of Tp-Bpy after photo-extraction.
for uranyl over result in the negligible extraction of uranyl; ii) the limited production rate after 90 min resulted in the stagnation of uranyl extraction; iii) the extraction kinetics over Tp-Bpy and Tb-Bpy is somehow different, i.e., the extraction rate decay over TbBpy is slower than that over Tp-Bpy, which implies that the extraction paths may be different; iv) Tp-Bpy and Tb-Bpy are the promising photocatalysts for the efficient extraction of aqueous uranyl, although further recycling utilization has to be clarified.
Characterizations of photo-extraction products
The photo-extraction products obtained with Tp-Bpy and Tb-Bpy were further analyzed through XPS, FT-IR, PXRD, and High-resolution transmission electron microscopy (HRTEM). For both Tp-Bpy and
Tb -Bpy, peaks at 382.20 eV and 381.95 eV can be assigned to the of (Fig. 5a). As shown in Fig. 5b, a new peak at belonging to the bond arisen for both Tp -Bpy and Tb -Bpy after photo-extraction . Meanwhile, the unique carbonyl carbons ( ) and imine bonds ( ) for respective Tp -Bpy and Tb -Bpy maintained well after photo-extraction. For Tb-Bpy, after photo-extraction, newly emerged diffraction peaks at and suggest the formation studtite particles, while for Tp-Bpy, no additional peaks can be observed (Fig. 5c). The WT maximum contour plots of Tp-Bpy after photoextraction closely matched to the plots, which strongly supported the formation of studtite nanodots (Fig. 5d). TEM image of Tb-Bpy after photo-extraction (Fig. 5e) shows obvious rodlike studtite nanoparticles on the surface. In contrast, no obvious
Fig. 6 | The schematic diagram for the reaction process of Tp-Bpy and Tb-Bpy. The abundant chelating sites for uranyl on Tp-Bpy are beneficial for the formation of dissolvable studtite nanodots. The limited adsorption ability for uranyl on Tb-Bpy is beneficial for the formation of large, hard-to-dissolve studtite nanorods.
nanoparticles can be found on the surface of Tp-Bpy (Fig. 5f). The HRTEM image (inset in Fig. 5f) showed two lattice fringe spacings of 0.265 and 0.325 nm , which can be ascribed to the (021) and (111) planes in studtite (PDF#49-1821). It suggests that the photo-extraction product on Tp-Bpy is studtite nanodots, while that on Tb-Bpy are much bigger studtite rodlike particles. Accordingly, the elemental mapping (Supplementary Fig. 10, 11) confirmed a homogenous distribution of U for each of them. For Tb-Bpy, U was distributed around the rodlike crystals, while for Tp-Bpy, U was dispersed on the whole surface without growth as big nanorods.
Growth-elution cycle for uranyl extraction
To recover the surface of Tp-Bpy and Tb-Bpy, the used photocatalysts were eluted with for 1 h . The elution ratio of U from Tp-Bpy is about , higher than that from Tb-Bpy (Supplementary Fig. 12). The obtained Tp-Bpy after elution was then used again for the photoextraction of uranyl. After 7 consecutive extraction-elution cycles, more than of extraction and elution efficiency for uranyl can be achieved (Supplementary Fig. 13). The FT-IR, SEM and XRD characterizations of the Tp-Bpy after extraction-elution cycling test suggest the high stability in the elution condition (Supplementary Fig. 14-16). In contrast, the consecutive extraction-elution cycles with Tb-Bpy are not satisfied, which may be attributed to the incomplete elution of bigger studtite particles. Notably, it is the formed studtite nanodots that makes the short-time elution feasible, which prevents the possible hydrolysis of imine-COFs in long-time elution. While the formed studtite nanorods cannot be completely eluted in short time before imine-COFs encounter hydrolysis.
To clarify the effect from co-existing metal ions, the photoextraction of uranyl with Tp-Bpy was conducted in the presence of , and with concentration 50 times as high as that of uranyl. Results showed that more than can be extracted after 3 h irradiation, indicating that co-existing metal ions would not interfere the uranyl extraction (Supplementary Fig. 17).
Based on the analysis mentioned above, the four self-prepared COFs showed different performance: i) Tb-Bd has quite low extraction activity due to the low adsorption ability although can be produced; ii) Tp-Bd shows unsatisfied extraction activity due to the limited production despite of the considerable adsorption of uranyl. To
confirm this, we supplemented the extra addition of until the concentration reaches up to and further conducted the uranyl extraction test with Tp-Bd (Supplementary Fig. 18). It turned out that the remained 34.8 % uranyl could further be extracted as studtite nanorods and only less than left in the solution after another 2 h reaction under dark condition; iii) Tb-Bpy shows a slowly decayed extraction kinetics with the formation of rodlike studtite particles that is difficult for elution, which is supposed due to the limited adsorption ability for uranyl; iv) the perfect extraction-elution cycling for uranyl enrichment was realized on Tp-Bpy. The four COFs are different with complexation groups, bipyridine and keto groups, which would also endow them with different photocatalytic performance for production. It has been identified that the reaction between and uranyl has a relatively low kinetics, which means the production of on Tp-Bpy and Tb-Bpy is not the rate-determinedstep . Therefore, the different performance between Tp-Bpy and TbBpy is supposed related with the nucleation of studtite on the surface of COFs, which is largely dependent on the chelating site for uranyl (Fig. 6).
To broaden the photocatalysts and clarify the suppose that combination of abundant chelating sites and the production ability of photocatalysts could lead to the highly efficient uranyl enrichment via studtite nanodots growth and elution cycle, we prepared another two imine-based COF photocatalysts with keto groups and pyridine-N chelating sites (Tp-Bpy-2 and Tp-Py). The successful preparation of their chemical structures was demonstrated by PXRD (Supplementary Fig. 19), FTIR (Supplementary Fig. 20), NMR (Supplementary Fig. 21) and XPS (Supplementary Fig. 22). The possibility of production was verified by a series of physical and photoelectric characterizations (Supplementary Fig. 23-25). Similar efficient tendency of uranyl adsorption (Supplementary Fig. 26), production (Supplementary Fig. 27, 28), studtite nanodots growthelution cycle (Supplementary Fig. 29-33) were found with Tp-Py and Tp-Bpy-2 as photocatalyst. The excellent growth-elution cycles of Tp-Bpy-2 and Tp-Py revalidated that multiple uranyl chelating sites promoted the growth of studtite nanodots and facilitated the efficient extraction of uranyl (Supplementary Fig. 34-37).
In summary, the requisites for the formation of dissolvable studtite nanodots include the presence of chelating sites for uranyl and the photosynthetic ability for , as well as the stability in
Fig. 7 | Uranium extraction from natural seawater. a Removal rate at 10 and 1 ppm spiked seawater over Tp-Bpy. b Selectivity in 1 ppm uranyl spiked seawater over Tp-Bpy. c Diagram of enrichment with Tp-Bpy. d The antibacterial activity of Tp-Py against marine bacteria. The exact sample size ( n ): Top- , . Down . The value of n is the standard deviations of three replicate measurements. e EPR spectra for -DMPO, -DMPO, -TEMP complexes generated by Tp-Py under visible light irradiation. f Equipment used for uranyl extraction from natural seawater by .
elution condition. Herein, more than introducing a series of COFs with highly efficient extraction capacity, we developed a strategy for cyclable generation and elution of studtite nanodots for long-term uranyl extraction.
Enrichment of uranyl in natural seawater
In order to verify whether the COFs with multiple chelating sites has the potential to be applied for uranyl extraction from real seawater, uranyl with low concentrations ( 10 ppm and 1 ppm ) in spiked seawater was used for photo-extraction with Tp-Bpy as photocatalyst, in which above of uranyl can be extracted (Fig. 7a). In addition, of 1 ppm uranyl in seawater can be extracted even in the presence of variable valency metal ions (Fig. 7b). After five consecutive extraction-elution cycles with 200 mL spiked seawater ( 1 ppm uranyl), 4 mL of concentrated solution with 28.7 ppm uranyl was obtained (Fig. 7c). To further investigate the practical applicability of the mentioned photo-extraction process, we examined the efficiency of extraction from natural seawater under visible light. After calculating the prime cost for the preparation of the COFs, we choose the cost-efficient Tp-Py photocatalyst for the extraction and enrichment of uranyl from seawater in scale-up test (Supplementary Table 5 and 6). Owing to the complex marine environment of microorganisms, the higher antimicrobial ability against the marine microorganism community must be considered for efficient uranyl extraction in natural seawater . The antifouling activity of Tp-Py is tested by using marine bacteria as targets (Fig. 7d). The result showed under dark condition (Supplementary Table 7), of the marine microbial community from natural seawater can be inhibited. After exposure to visible light, a markedly increased inhibition of was realized, which is supposed due to the production of reactive oxygen species (ROS) in the presence of visible light irradiation, as confirmed by the electron paramagnetic resonance (EPR) spectra (Fig. 7e and Supplementary Fig. 38). The experiments with natural seawater ( 3.3 ppb ) demonstrated that achieved an impressive
uranyl uptake capacity of over 12 consecutive days under visible light, averaging day (Fig. 7f, Supplementary Fig. 39 and Supplementary Table 8, 9).
Discussion
In summary, we reported an efficient and sustainable strategy to upcycle the uranyl resource from natural seawater with COF photocatalysts. Comprehensive comparison study of different COFs Tp-Bpy, Tp-Py, Tp-Bpy-2 and other three COFs (Tp-Bd, Tb-Bpy, and Tb-Bd) photocatalysts on the adsorption capacity, the production ability, the uranyl extraction-elution cycling performance showed that i) the photogenerated can react with uranyl to form studtite; ii) the chelating site for uranyl on the photocatalyst is necessary for nucleation of studtite; iii) the ultra-highly efficient enrichment of uranyl can be realized via the formation of dissolvable studtite nanodots based on the abundant chelating sites for uranyl, which avoided the formation of large, hard-to dissolve studtite nanorod. After five consecutive extraction-elution cycles, 200 mL of 1 ppm uranyl-spiked seawater was concentrated to 4 mL of 28.7 ppm uranyl solution with regenerable TpBpy photocatalyst. Furthermore, an impressive uranyl uptake capacity of day can be achieved with cost-efficient in real seawater under visible light irradiation. It’s believed that based on our results, more economical and acid-resistant COFs or even other materials with the necessary requisites, i.e., abundant uranyl-chelating sites and production ability, could be prepared in the field of material engineering for the practical uranyl extraction from seawater.
Methods
Materials
All reagents were purchased from commercial suppliers and used without further purification unless stated otherwise. 1,3,5-Triformylphloroglucinol (Tp, ), 1,3,5-Triformylbenzene (Tb, ), 2,2′-bipyridine-5,5′-diamine (Bpy, 97 %), and Benzidine (Bd, 97 %) were purchased from Jilin Chinese Academy of Sciences-Yanshen
Technology Co., Ltd. N, N-dimethylacetamide (DMAC, 99.8 %), 1,3,5Trimethylbenzenewas ( , AR ), o-dichlorobenzene ( -DCB, ), 1-Butanol (99 %, AR), 1,4-Dioxane (99.5 %), acetic acid (AcOH, ), ethanol absolute ( , Water ) and Tetrahydrofuran (THF, , AR) were all provided by Aladdin. Ultrapure water was prepared from the Millipore system ( ).
Synthesis of Tp-Bpy and Tp-Bd
-Triformylphloroglucinol ( ) and -bipyr-idine-5,5′-diamine (Bpy, ) or Benzidine (Bd, ) were put into a 15 mL Schlenk tube. Then a mixed solvent of N, N-dimethylacetamide (DMAC) and dichlorobenzene (3/1, ) was also added, and subjected to ultrasonic treatment to achieve uniform dispersion. Then 0.6 mL of 6 M acetic acid (AcOH) aqueous solution was quickly added and sonicated again. After degassing, the reaction was conducted at for 3 days. The solid was then washed away with a large amount of DMAC, water and THF and dried under vacuum at for 12 h .
Synthesis of Tb-Bpy
-Triformylbenzene ( ) and 2, 2′-bipyridine-5, 5’diamine (Bpy, ) were put into a 15 mL Schlenk tube. Then a mixed solvent of o-dichlorobenzene and Butanol ( ) ( ) were also added, and subjected to ultrasonic treatment to achieve uniform dispersion. Then 0.3 mL of 6 M acetic acid (AcOH) aqueous solution was quickly added and sonicated again. After sonicating for 10 min , the tube was then flash frozen via liquid bath and degassed by three freeze-pump-thaw cycles. Then the tube was sealed off and heated at for 3 days. The solid was washed away with a large amount of ethanol and dried under vacuum at for 12 h .
Synthesis of Tb-Bd
-Triformylbenzene ( ) and Benzidine (Bd, ) were put into a 15 mL Schlenk tube. Then a mixed solvent of 1,4-Dioxane and Butanol ( ) was added, and subjected to ultrasonic treatment to achieve uniform dispersion. Then 0.3 mL of 6 M acetic acid (AcOH) aqueous solution was quickly added. After sonicating for 10 min , the tube was flash frozen via liquid bath and degassed by three freeze-pump-thaw cycles. Then the tube was sealed off and heated at for 3 days. The solid was washed away with a large amount of ethanol and dried under vacuum at for 12 h .
Synthesis of Tp-Py and Tp-Bpy-2
-Triformylphloroglucinol ( ) and 2,5 -Diaminopyridine (Py, ) or [ -Bipyridine]-6,6′-diamine (Bpy- ) were put into a 15 mL Schlenk tube. Then a mixed solvent of -dimethylacetamide (DMAC) and dichlorobenzene ( ) was also added, and subjected to ultrasonic treatment to achieve uniform dispersion. Then 0.6 mL of 6 M acetic acid aqueous solution was quickly added and sonicated again. After degassing, the reaction was conducted at for 3 days. The solid was then washed away with a large amount of DMAC, water and THF and dried under vacuum at for 12 h .
Characterizations
Powder X-ray diffraction (PXRD) analyses were performed using a Rigaku SmartLab SE X-ray diffractometer equipped with a source (small angle X-ray scattering data collected on a Bruker D8 Advance diffractometer was used to correct the deviation) with a step size of . Fourier transform infrared spectra (FT-IR) were recorded on a SHIMADZU IRTracer-100. BET surface areas were determined from adsorption/desorption isotherms collected at 77 K using Micromeritics TriStar II. Scanning electron microscopy (SEM) images
were recorded on a Hitachi SU 8100 Scanning Electron Microscope. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were recorded on a JEOL transmission electron microscope (JEM-F200) operating at an accelerating voltage of 200 kV . Solid-state CP/MAS NMR spectra were collected on a BRUKER AVANCE NEO 400 WB spectrometer. X-ray photoelectron spectroscopy (XPS) analyses were performed using a Thermo Scientific ESCALAB 250Xi spectrometer, equipped with a monochromatic Al K X-ray source ( 1486.8 eV ). Photoelectrochemical experiments measurements were performed on an electrochemical workstation (CHI760E, CHI Instruments, Shanghai, China). Electron paramagnetic resonance (EPR) spectra were recorded at 293 K with a Bruker EMXnano259 spectrometer, operated at 9.62 GHz with 12.59 mW power and modulation at . UV-vis spectroscopy results were recorded in diffuse reflectance (DR) mode at room temperature on a SHIMADZU UV-2700 spectrophotometer equipped with an integrating sphere attachment. Thermogravimetric analyses (TGA) were carried out on a NETZSCH STA 2500 instrument. Inductively coupled plasma mass spectrometry (ICP-MS) analyses were performed on an Agilent 7800 spectrometer.
Photoelectrochemical measurements
The Mott-Schottky plots, photocurrent response and electrochemical impedance of the photo-catalysts were measured on an electrochemical workstation (CHI760E, CHI Instruments, Shanghai, China). A 300 W Xe lamp was utilized as the light source and , ) aqueous solution was used as the supporting electrolyte throughout the photocurrent measurements. A platinum wire and electrode were used as counter electrode and reference electrode, respectively. naphthol was dissolved in about 3 mL ethanol with 1 mg catalyst. Then, was added to ITO with a layer of adhesive tape on the edge for six times, followed by drying in air. The potential vs NHE was determined by using the following Eq. (2):
Uranyl extraction experiments
All experiments on the extraction of were carried out in air under visible light ( ) using 10 W LED lamp (Perfect PCX50C Discover) as light source and the maximum photocurrent was corresponding to 10 W .
Photo-extraction of aqueous U(VI)
10 mg photocatalyst was dispersed in 40 mL of a uranyl spiked seawater ( ) and ultra-pure water ( ), which contains methanol. The temperature was kept at by circulating cooling water throughout this photo-extraction process. After photo-extraction, the resulted suspension was filtrated by 220 nm membrane. The concentration of was measured by UV-vis spectrophotometry at a wavelength of 650 nm using the Arsenazo III method.
Photo-extraction uranyl from natural seawater
10 mg Tp-Py was dispersed in self-assembly designed reactor. The seawater flowed through the photocatalyst at the speed of by using peristaltic pump. The concentration of uranyl in the natural seawater was analyzed by inductively coupled plasma-Mass Spectrometry (ICP-MS).
Design of the device for uranyl extraction from natural seawater
10 mg COFs was mixed with 7 mL ethanol, 1 mL ultra-pure water and of Nafion solution. The mixture was drop-casted onto quartz panel. With the peristaltic pump, the real seawater flowed through the coated quartz panel.
Enrichment Test
The used photocatalyst with uranyl uploaded was put into 1 mL of elution solution at room temperature. One hour later, it was taken out for another photo-extraction in uranyl-containing object solution for 3 h . The elution operation was repeated in the same elution solution to enrich uranyl. After 5 times of elution, the uranyl concentration of the final elution solution was detected by ICP-MS.
Effect of competing ions on the photo-extraction of aqueous VI
10 mg photocatalyst was dispersed in 40 mL of ultra-pure water ( ) with , ) or sodium salts ( and ) for 3 h irradiation.
Recyclability test
After one run of photo-extraction, the photocatalyst was regenerated by elution with solution ( or 2.5). The resulted photocatalyst was then reused for another photo-extraction experiment. For 10 mg catalysts, 40 mL elution solution was used to elute the binding uranium for 60 min at room temperature. The elution efficiency (E, %) was determined by using the following Eq. (3):
where is the uranium concentration in elution solution, is the volume elution solution, is the uranium concentration in uranium-spiked seawater after photo-reaction, ) is the initial uranium concentration of uranium-spiked seawater, (L) is the volume of uranium-spiked seawater used for adsorption. The resulting suspension was filtered and washed with ultra-pure water till the supernatant became neutral. After being dried under vacuum, the resultant material was used for another photo-reaction experiment. It was found that after six consecutive cycles Tp-Bpy still showed excellent uranium uptake.
detection methods
The generation was qualified by using a colorimetric method. Typically, the mixture after photo-reaction was centrifuged and then of the suspension was added to a mixture of 2 mL KI solution ( 0.1 M ) and solution ( 0.01 M ). Corresponding absorption at 352 nm was measured on a UV-Vis spectrometry.
Calculation of the equilibrium constant for the Eq. (1)
42.5 mL of solution was mixed with 7.5 mL MeOH in a 100 mL reactor. The reactor was thermostated at with a cycling pump. The pH of the mixture was adjusted to 4 by adding negligible volumes of NaOH and solutions ( ). The mixed solution was illuminated under visible light ( ) using a 300 W Xe lamp (Microsolar 300, PerfectLight) equipped with a UV cutoff filter, during which, uranyl species could be excited to produce and the produced then reacts with uranyl to form studtite solid. The concentration of uranyl in the solution was monitored every one hour in order to ascertain the equilibrium. It was found after 8 h , the concentration of remaining uranyl stopped decreasing. Subsequently, the final suspension stirs in the dark for an hour. Then the concentration of and pH value were measured. The equilibrium constant for the reaction can be calculated with the detected concentrations of uranyl and , as well as pH value.
Anti-biofouling measurements
The marine bacterial strain was used to test the antimicrobial activity of the catalyst. The Tp-Py was dispersed into marine bacteria/fresh Luria-Bertani broth at a dispersion concentration of . After cultivated at for 3 h with thermostatic oscillator, the viability of
the bacterium was determined. The dilution plate counting method was used according to the Chinese standard GB/T20944 to determine the viability of the adsorbents treated bacterial cultures. In order to simulate the experimental conditions, the antibacterial effect was compared respectively under dark state and visible light irradiation. Inhibition rates (IR) were calculated according to the following Eq. (4):
Where and are the microbial concentrations in cultures before and after treatment with Tp-Py.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available in the article and Supplementary Information files or available from the corresponding authors upon request. The numerical values for the data shown in the Figures in the main text are provided in the attached Source Data file. Source data are provided with this paper (https://doi. org/10.6084/m9.figshare.26149174). Source data are provided with this paper.
References
Kittner, N., Lill, F. & Kammen, D. M. Energy storage deployment and innovation for the clean energy transition. Nat. Energy 2, 17125 (2017).
Holechek, J. L., Geli, H. M. E., Sawalhah, M. N. & Valdez, R. A global assessment: Can renewable energy replace fossil Fuels by 2050? Sustainability 14, 4792 (2022).
Wang, C., Raza, S. A., Adebayo, T. S., Yi, S. & Shah, M. I. The roles of hydro, nuclear and biomass energy towards carbon neutrality target in China: A policy-based analysis. Energy 262, 125303 (2023).
Meitner, L. & Frisch, O. R. Disintegration of uranium by neutrons: a new type of nuclear reaction. Nature 143, 239-240 (1939).
Liu, Z. et al. Multi-scale computer-aided design and photocontrolled macromolecular synthesis boosting uranium harvesting from seawater. Nat. Commun. 13, 3918 (2022).
Abney, C. W., Mayes, R. T., Saito, T. & Dai, S. Materials for the recovery of uranium from seawater. Chem. Rev. 117, 13935-14013 (2017).
Sun, Q. et al. Bio-inspired nano-traps for uranium extraction from seawater and recovery from nuclear waste. Nat. Commun. 9, 1644 (2018).
Liu, C. et al. A half-wave rectified alternating current electrochemical method for uranium extraction from seawater. Nat. Energy 2, 17007 (2017).
Ku, T.-l, Knauss, K. G. & Mathieu, G. G. Uranium in open ocean: concentration and isotopic composition. Deep Sea Res. 24, 1005-1017 (1977).
Ye, Y. et al. Spontaneous electrochemical uranium extraction from wastewater with net electrical energy production. Nat. Water 1, 887-898 (2023).
Chen, L. et al. Ultrafast and efficient extraction of uranium from seawater using an amidoxime appended metal-organic framework. ACS Appl. Mater. Interf. 9, 32446-32451 (2017).
Yuan, Y. et al. A bio-inspired nano-pocket spatial structure for ttargeting uranyl capture. Angew. Chem. Int. Ed. 59, 4262-4268 (2020).
Li, Z. et al. Constructing amidoxime-modified porous adsorbents with open architecture for cost-effective and efficient uranium extraction. Chem. Sci. 11, 4747-4752 (2020).
Chen, Z. et al. Tuning excited state electronic structure and charge transport in covalent organic frameworks for enhanced photocatalytic performance. Nat. Commun. 14, 1106 (2023).
Cui, W.-R. et al. Regenerable Covalent organic frameworks for photo-enhanced uranium adsorption from seawater. Angew. Chem. Int. Ed. 59, 17684-17690 (2020).
Wang, G.-B. et al. Covalent organic frameworks and their composites as multifunctional photocatalysts for efficient visible-light induced organic transformations. Coord. Chem. Rev. 472, 214774 (2022).
Zhang, Q. Y. et al. Ultra-selective uranium separation by in-situ formation of conjugated 2D uranium-organic framework. Nat. Commun. 15, 453 (2024).
Xie, Y. et al. Uranium extraction from seawater: material design, emerging technologies and marine engineering. Chem. Soc. Rev. 52, 97-162 (2023).
Yuan, Y. et al. DNA nano-pocket for ultra-selective uranyl extraction from seawater. Nat. Commun. 11, 5708 (2020).
Yuan, Y. et al. Selective extraction of uranium from seawater with biofouling-resistant polymeric peptide. Nat. Sustainability 4, 708-714 (2021).
Pan, Z. et al. Nanoscale mechanism of formation through uranium reduction by magnetite. Nat. Commun. 11, 4001 (2020).
Feng, L. et al. Halogen hydrogen-bonded organic framework (XHOF) constructed by singlet open-shell diradical for efficient photoreduction of U(VI). Nat. Commun. 13, 1389 (2022).
Yang, L. et al. Bioinspired hierarchical porous membrane for efficient uranium extraction from seawater. Nat. Sustainability 5, 71-80 (2022).
Liu, T. et al. Ligand-assistant iced photocatalytic reduction to synthesize atomically dispersed Cu implanted metal-organic frameworks for photo-enhanced uranium extraction from seawater. Small 19, 2208002 (2023).
Yang, H. et al. Tuning local charge distribution in multicomponent covalent organic frameworks for dramatically enhanced photocatalytic uranium extraction. Angew. Chem. Int. Ed. 62, e202303129 (2023).
Hu, Y. et al. Photochemically triggered self-extraction of uranium from aqueous solution under ambient conditions. Appl. Catal. B-Environ. 322, 122092 (2023).
Wang, Z. et al. Photo-induced removal of uranium under air without external photocatalysts. Green. Chem. 24, 7092-7099 (2022).
Kim, J., Kim, H., Kim, W.-S. & Um, W. Dissolution of studtite [ ] in various geochemical conditions. J. Environ. Radioact. 189, 57-66 (2018).
Yu, F.-Y., Zhou, Y.-J., Tan, H.-Q., Li, Y.-G. & Kang, Z.-H. Versatile photoelectrocatalysis strategy raising up the green production of hydrogen peroxide. Adv. Energy Mater. 13, 2300119 (2023).
Edwards, J. K. et al. The Direct Synthesis of Hydrogen Peroxide Using Platinum-Promoted Gold-Palladium Catalysts. Angew. Chem. Int. Ed. 53, 2381-2384 (2014).
Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962-6984 (2006).
Liao, Y. et al. Regulating benzene ring number as connector in covalent organic framework for boosting photosynthesis of from seawater. Nano Lett. 24, 3819-3825 (2024).
Li, P. et al. 1D covalent organic frameworks triggering highly efficient photosynthesis of via controllable modular design. Angew. Chem. Int. Ed. 63, e202319885 (2024).
Zhang, T. et al. Atomic-level understanding for the enhanced generation of hydrogen peroxide by the introduction of an aryl amino group in polymeric carbon nitrides. ACS Catal. 11, 14087-14101 (2021).
Liao, Q. et al. Regulating relative nitrogen locations of diazine functionalized covalent organic frameworks for overall photosynthesis. Angew. Chem. Int. Ed. 62, e202310556 (2023).
Daugherty, M. C. et al. Improved synthesis of -ketoenamine-linked covalent organic frameworks via monomer exchange reactions. Chem. Commun. 55, 2680-2683 (2019).
Zhong, W. et al. A covalent organic framework bearing single Ni sites as a synergistic photocatalyst for selective photoreduction of to CO. J. Am. Chem. Soc. 141, 7615-7621 (2019).
Dey, K. et al. Selective molecular separation by interfacially crystallized covalent organic framework thin films. J. Am. Chem. Soc. 139, 13083-13091 (2017).
Xu, L. et al. Surface-confined crystalline two-dimensional covalent organic frameworks via on-surface schiff-base coupling. ACS Nano 7, 8066-8073 (2013).
Wang, Q. et al. N-doping of -ketoenamine based covalent organic frameworks for catalytic conversion of to cyclic carbonate and Knoevenagel condensation. Microporous Mesoporous Mater. 364, 112872 (2024).
Yang, Y. et al. Engineering -ketoamine covalent organic frameworks for photocatalytic overall water splitting. Nat. Commun. 14, 593 (2023).
Chu, F. et al. Regulating keto-enol tautomerism of -ketoenamine covalent-organic frameworks for photocatalytic oxidative coupling of amines. ACS Catal. 13, 13167-13180 (2023).
Wang, H. et al. Integrating suitable linkage of covalent organic frameworks into covalently bridged inorganic/organic hybrids toward efficient photocatalysis. J. Am. Chem. Soc. 142, 4862-4871 (2020).
Kandambeth, S. et al. Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc. 134, 19524-19527 (2012).
Yang, H. et al. MOFs-derived fusiform mesoporous nanorods anchored with ultrafine CdZnS nanoparticles for boosting visible-light photocatalytic hydrogen evolution. Small 17, 2102307 (2021).
Hao, X. et al. Architecture of high efficient zinc vacancy mediated Z-scheme photocatalyst from metal-organic frameworks. Nano Energy 52, 105-116 (2018).
Kou, M. et al. Molecularly engineered covalent organic frameworks for hydrogen peroxide photosynthesis. Angew. Chem. Int. Ed. 61, e202200413 (2022).
. et al. Conjugated organic polymers with anthraquinone redox centers for efficient photocatalytic hydrogen peroxide production from water and oxygen under visible light irradiation without any additives. ACS Catal. 12, 12954-12963 (2022).
Hu, H. et al. Rational modification of hydroxy-functionalized covalent organic frameworks for enhanced photocatalytic hydrogen peroxide evolution. J. Colloid Interf. Sci. 629, 750-762 (2023).
Guo, R. et al. Chelating effect between uranyl and pyridine N containing covalent organic frameworks: A combined experimental and DFT approach. J. Colloid Interf. Sci. 606, 1617-1626 (2022).
Colella, M., Lumpkin, G. R., Zhang, Z., Buck, E. C. & Smith, K. L. Determination of the uranium valence state in the brannerite structure using EELS, XPS, and EDX. Phys. Chem. Miner. 32, 52-64 (2005).
Liu, W. et al. Highly sensitive and selective uranium detection in natural water systems using a luminescent mesoporous metal-organic framework equipped with abundant lewis basic sites: A combined batch, X-ray absorption spectroscopy, and first principles simulation investigation. Environ. Sci. Technol. 51, 3911-3921 (2017).
Li, H. et al. Powerful uranium extraction strategy with combined ligand complexation and photocatalytic reduction by
postsynthetically modified photoactive metal-organic frameworks. Appl. Catal. B-Environ. 254, 47-54 (2019).
Song, Y. et al. Unassisted uranyl photoreduction and separation in a Donor-Acceptor covalent organic framework. Chem. Mater. 34, 2771-2778 (2022).
Li, Z. et al. Exciton dissociation and transfer behavior and surface reaction mechanism in Donor-Acceptor organic semiconductor photocatalytic separation of uranium. Appl. Catal. B-Environ. 332, 122751 (2023).
Yuan, Y. et al. Photoinduced multiple effects to enhance uranium extraction from natural seawater by black phosphorus nanosheets. Angew. Chem. Int. Ed. 59, 1220-1227 (2020).
Zhang, C.-R. et al. Simultaneous sensitive detection and rapid adsorption of based on a post-modified carbonconjugated covalent organic framework. Environ. Sci. Nano. 7, 842-850 (2020).
Acknowledgements
We sincerely acknowledge the financial supports by National Natural Science Foundation of China (No. 22176054 (G.-X.Z.), 22306060 (Y.Z.H.)), China Postdoctoral Science Foundation (No. 2022M721134 (Y.Z.H.)), National Key Research and Development Program of China (No. 2018YFC1900105 (X.-K.W.)) and Beijing Outstanding Young Scientist Program (X.-K.W.).
Author contributions
P.G., G.-X.Z., X.-K.W. and X.-B.H. conceived the research and designed the experiments. P.G., Y.-Z.H. and Z.-W.S. carried out the experiment. All authors analyzed the data. Z.-Y.J., R.-Q.C., and F.C. contributed to the project discussions. P.G., G.-X.Z., X.-K.W. and X.-B.H. participated in drafting the paper and gave approval to the final version of the manuscript.
Correspondence and requests for materials should be addressed to Guixia Zhao, Zhuoyu Ji, Xiangke Wang or Xiubing Huang.
Peer review information Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
College of Environmental Science and Engineering, North China Electric Power University, Beijing 102206, P. R. China. Beijing Advanced Innovation Center for Materials Genome Engineering, Beijing Key Laboratory of Function Materials for Molecule & Structure Construction, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, P. R. China. e-mail: guixiazhao@ncepu.edu.cn; jizhuoyu@ncepu.edu.cn; xkwang@ncepu.edu.cn; xiubinghuang@ustb.edu.cn