DOI: https://doi.org/10.1186/s40168-024-01766-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38454512
تاريخ النشر: 2024-03-07
التحليل الشامل والمدهش وسرعة جهاز تحليل الكتلة الأستري للعينات المعقدة للغاية هو قفزة نوعية في التحليل الوظيفي للميكروبيومات
الملخص
الخلفية من خلال تحليل البروتينات التي تعتبر العمود الفقري للأنظمة البيولوجية، تتيح الميتابروتيوميات لنا قائمة الأنواع الموجودة في أي ميكروبيوم، ومراقبة الكتلة الحيوية النسبية، وتوصيف وظيفة الأنظمة البيولوجية المعقدة. النتائج هنا، نقدم استراتيجية جديدة لتحديد هيكل المجتمع الميكروبي لعينة معينة وتصميم قاعدة بيانات تسلسل بروتين مخصصة لاستغلال بيانات مطيافية الكتلة الشاملة بشكل مثالي. تستفيد هذه الطريقة من قدرات الجيل الأول من مطياف الكتلة رباعي القطب الذي يتضمن محلل أسترا (Astral) غير المفقود للمسار غير المتماثل، مما يوفر سرعة مسح MS/MS وسرعة حساسية عالية. استغللنا استراتيجيات الاكتساب المعتمد على البيانات والاكتساب المستقل عن البيانات باستخدام مستخلص ببتيد من عينة براز إنسان تم إضافة كميات دقيقة من الببتيدات من نوعين مرجعيين من البكتيريا. الاستنتاجات تثبت طريقتنا، التي تجمع بين كلا طريقتي الاكتساب، أنها فعالة من حيث الوقت أثناء معالجة قواعد البيانات العامة الواسعة والمجموعات الضخمة من البيانات، حيث تحقق تغطية لأكثر من 122,000 ببتيد فريد و38,000 مجموعة بروتينية خلال فترة 30 دقيقة من DIA. يمثل هذا تحولًا كبيرًا عن منهجيات الميتابروتيوميات الحالية، مما يؤدي إلى تغطية أوسع للمسارات الأيضية التي تحكم النظام البيولوجي. معًا، تمثل استراتيجيتنا ومحلل الكتلة أسترا قفزة نوعية في التحليل الوظيفي للميكروبيومات.
*المراسلة:
جان أرمونغو
jean.armengaud@cea.fr
الخلفية
يوفر معلومات حاسمة حول المكونات الهيكلية، والإنزيمات، والرسائل المعلوماتية للميكروبات، بالإضافة إلى استجابة المضيف، إن وجدت [2]. بالإضافة إلى تحديد المسارات الأيضية النشطة وتقييم مستوى نشاطها من خلال كمياتها، تتيح هذه المنهجية تتبعها إلى الكائنات المحددة التي أنتجت البروتينات المقابلة بفضل تسلسلات الببتيد التي تم إنشاؤها بواسطة مطيافية الكتلة المت tandem عالية الدقة. لذلك، تلعب الميتابروتيوميات دورًا رئيسيًا في تعميق معرفتنا بالميكروبيومات، مقارنة بالمنهجيات المحدودة بتصنيف الميكروبات والإمكانات الجينومية. علاوة على ذلك، بفضل سرعتها القصوى، يمكن أن تصبح هذه المنهجية أداة تشخيصية جديدة جذابة للطب البشري والبيئة [3].
تتأثر أبحاث الميكروبيوم بشدة بالتقدم المنهجي. التطورات الأخيرة في مطيافية الكتلة المتزامنة، واستراتيجيات الاكتساب، وأدوات التفسير لديها إمكانيات كبيرة لتحويل الميتابروتيومكس إلى منهجية عالية الأداء لتعميق المعرفة بوظائف الميكروبات. يواجه الميتابروتيومكس كمية هائلة من البيانات المعقدة، بما في ذلك قواعد بيانات ضخمة من تسلسلات البروتينات المبنية على بيانات الميتاجينوم أو أعداد كبيرة من الكائنات الحية المتسلسلة. كما يواجه الميتابروتيومكس عددًا استثنائيًا من البروتينات والمتغيرات من العينة، مما يجعل تحديد الببتيدات الشائعة أسهل من تحديد الببتيدات المحددة. أخيرًا، يميل نقص التغطية الشاملة لقاعدة بيانات تسلسل البروتينات إلى تقليل نتائج التفسير. مؤخرًا، وصف ستيوارت وآخرون تطوير مطياف كتلة جديد يجمع بين رباعي الكتلة القوي Orbitrap، ومعالج الأيونات الجديد فخ الأيونات المستقيمة، ومحلل مفاهيمي ثوري يسمى محلل الخسارة غير المتماثلة (Astral)، مما يتيح اكتسابًا أسرع لطيف MS/MS عالي الدقة وحساسية عالية مقارنة بمطيافات الكتلة الحديثة. أظهرت النتائج التي قدمها هذا الجهاز الجديد في علم البروتينات وعودًا واعدة من حيث عمق التحليل مع تحديد 10,000 مجموعة من مستخلص ببتيدات HeLa خلال تشغيل واحد مدته 48 دقيقة. تم توثيق مثل هذا الأداء بشكل أكبر للتحليل الشامل لتعديلات البروتينات بعد الترجمة، وبروتينات البلازما، والخلايا الدنيا، وعلم البروتينات على مستوى الخلية الواحدة. نظرًا لهذه التحسينات الكبيرة، نستكشف في هذه الدراسة أدائها لتوصيف عينات معقدة للغاية باستخدام معيار محدد من المواد البرازية البشرية المضافة إليها كميات دقيقة من بروتينات بكتيرية. لاستغلال هذه التكنولوجيا الجديدة بالكامل، نقترح سير عمل جديد للميتابروتيومكس، يعتمد على تحديد موثوق للبروتينات الدقيقة من اكتساب يعتمد على البيانات القصيرة.
قاعدة بيانات تختار الجينومات الأكثر قيمة، تسجيل مجموعات بيانات عالية الكثافة في الاكتساب غير المعتمد على البيانات، وتفسير لزيادة تغطية اللاعبين الرئيسيين في الميكروبيوتا.
المواد والطرق
عينة مرجعية من MetaP
مطياف الكتلة أوربيتراب أسترا
تفسير البيانات لتحديد بروتينات الكائنات الحية
إنشاء قاعدة بيانات DB48
تفسير بيانات الميتابروتيوميات
تم إجراء توليد المكتبة في السليكو استنادًا إلى قاعدة بيانات DB48. تم السماح بحد أقصى من 2 انقسام مفقود، و2 تعديل متغير (أكسدة الميثيونين وأستلة الطرف N)، وطول الببتيد يتراوح من 7 إلى 30 بقايا، وشحنة السلف 2 و
التحليل الوظيفي للمضيف والميكروبات والبكتيريا المضافة
تحليل الطيف الكتلي Orbitrap Exploris 480 ومعالجة البيانات
بيانات بروتيوميات مطيافية الكتلة
النتائج
تحديد الأنواع الأكثر وفرة في العينة باستخدام تقنية البروتوتيبينغ المعتمدة على DDA
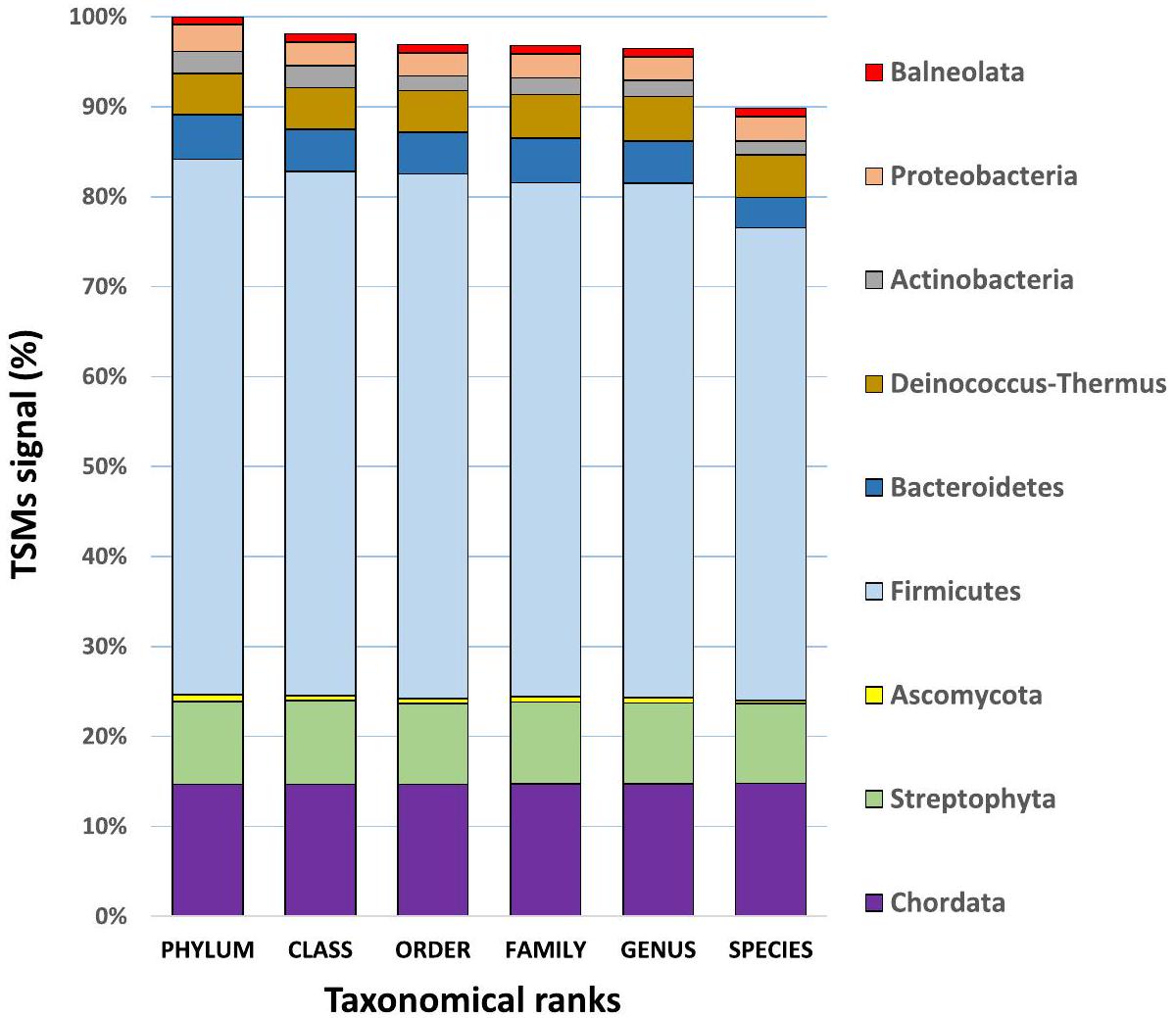
تم استخدامه لإجراء بحث ثانٍ لتحديد الكائنات الحية على مستوى النوع التصنيفي. الأنواع المحددة في هذه التحليلات الست المستقلة مدرجة في الجدول S1، مع العدد المقابل من الببتيدات المحددة لكل نوع ومطابقات الطيف لكل نوع (TSMs) في مختلف الرتب التصنيفية. يوفر الجدول S2 قائمة موثوقة من 9 شعب، و44 جنسًا، و56 نوعًا تم تحديد بروتيناتها من خلال دمج هذه النتائج، مع مساهمتها الخاصة في الكتلة الحيوية للبروتين (الشكل 1). إشارة TSMs العامة تنخفض قليلاً عند الانتقال لأسفل التسلسل الهرمي التصنيفي، من
تم ملاحظة النسبة بالنسبة لـ Ascomycota و Actinobacteria، مما يشير إلى أن الأنواع الممثلة بروتيوتيب داخل هذين الفيلين تمثل بشكل ضعيف فقط. نظرًا لأن مساهماتها الإجمالية صغيرة، فإن هذا له تأثير ضئيل على النتائج. في الواقع، فإن إشارة TSMs العالمية، التي تمثل
من بين الـ 13 حقيقيات النوى، يعتبر المضيف Homo sapiens منطقياً أكثر الأنواع وفرة، حيث يمثل أكثر من
من الإجمالي (الجدول S3).
نظرًا للتنوع العالي للببتيدات الموجودة في معيار MetaP، من المتوقع أن تكون نسبة كبيرة من طيف MS/MS
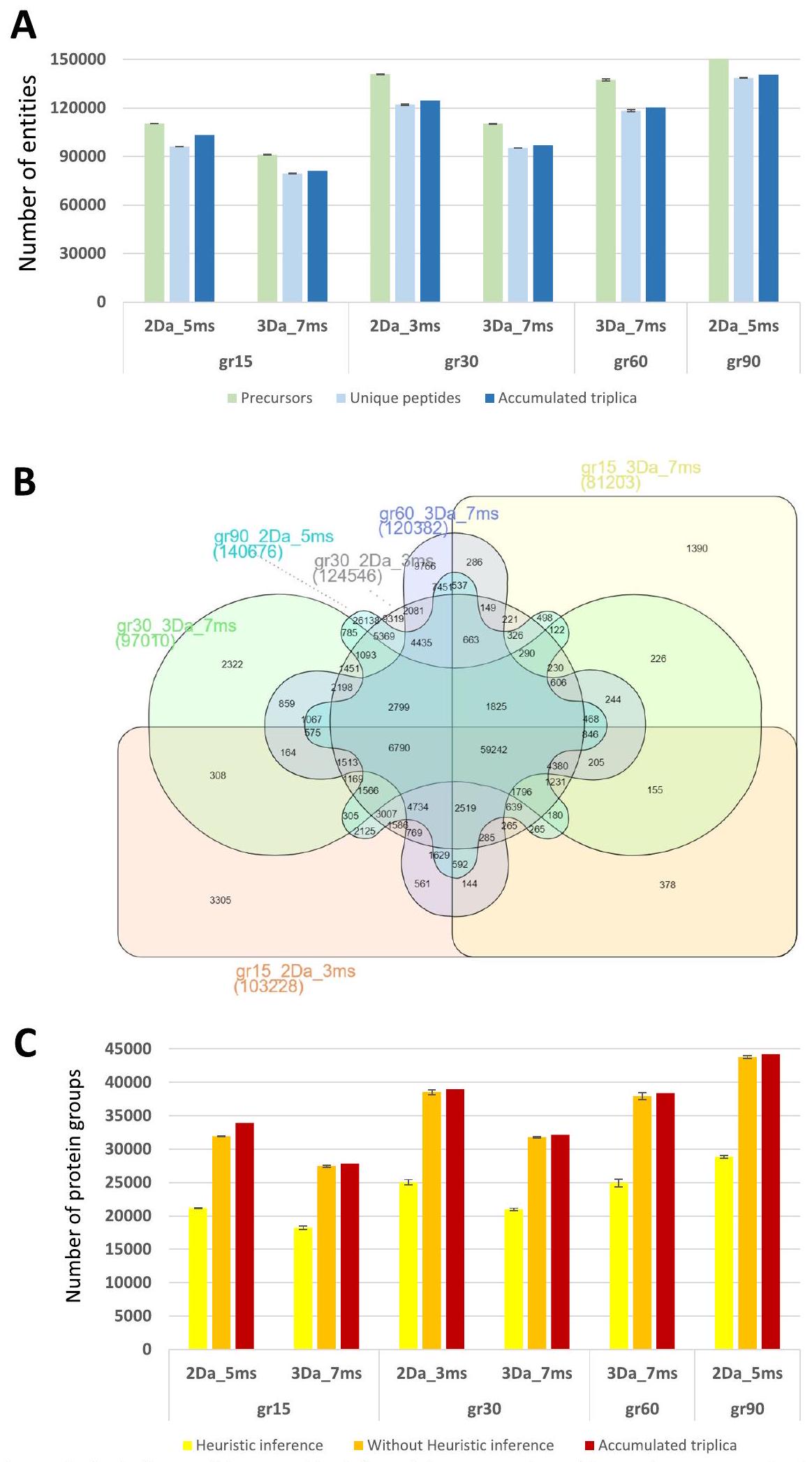
تزيد الميتابروتيوميات DIA بشكل كبير من تغطية الببتيد واكتشاف البروتين
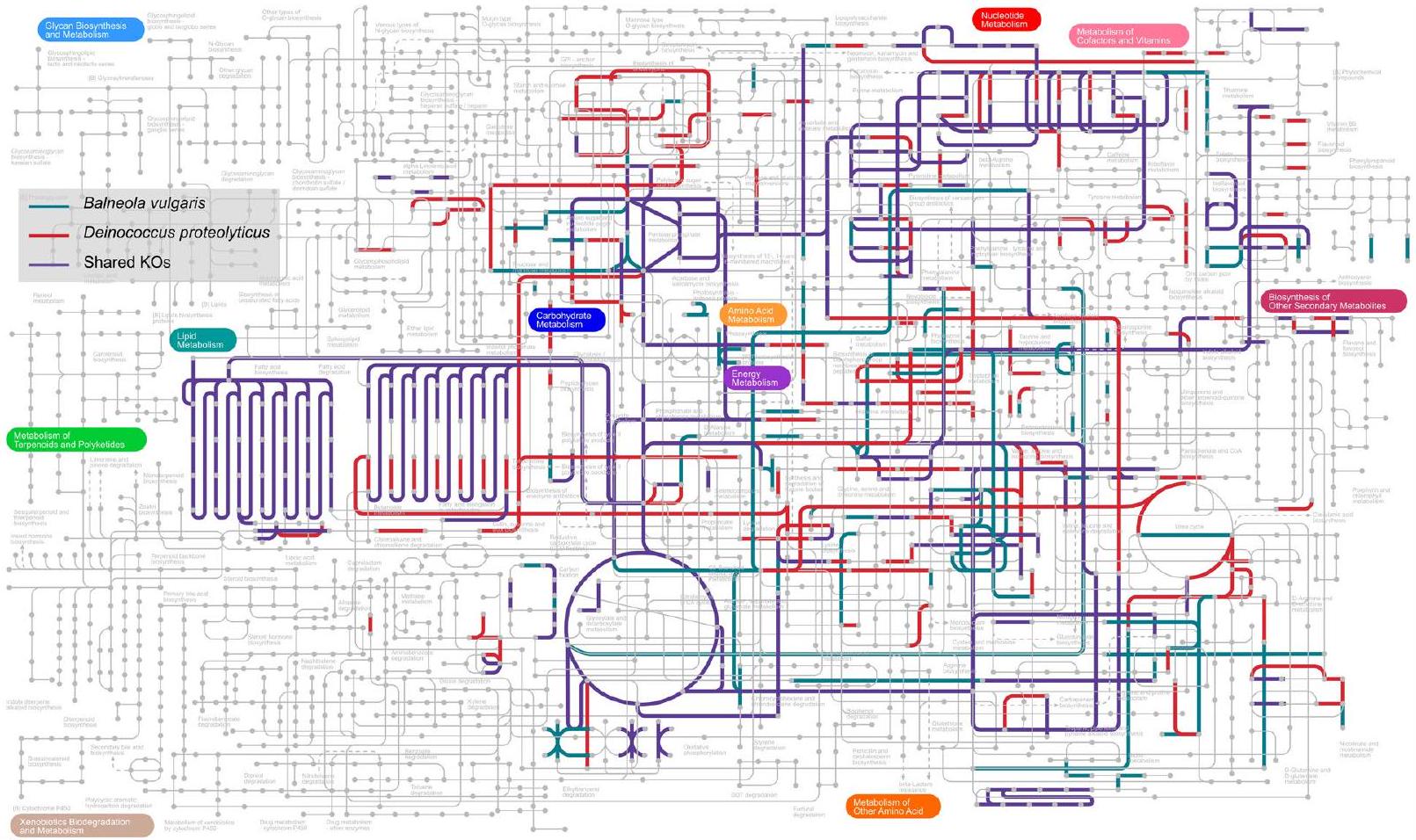
تعتمد المناظر المحسنة بشكل كبير لمسارات الأيض على مشهد DIA للببتيدات كبير جدًا
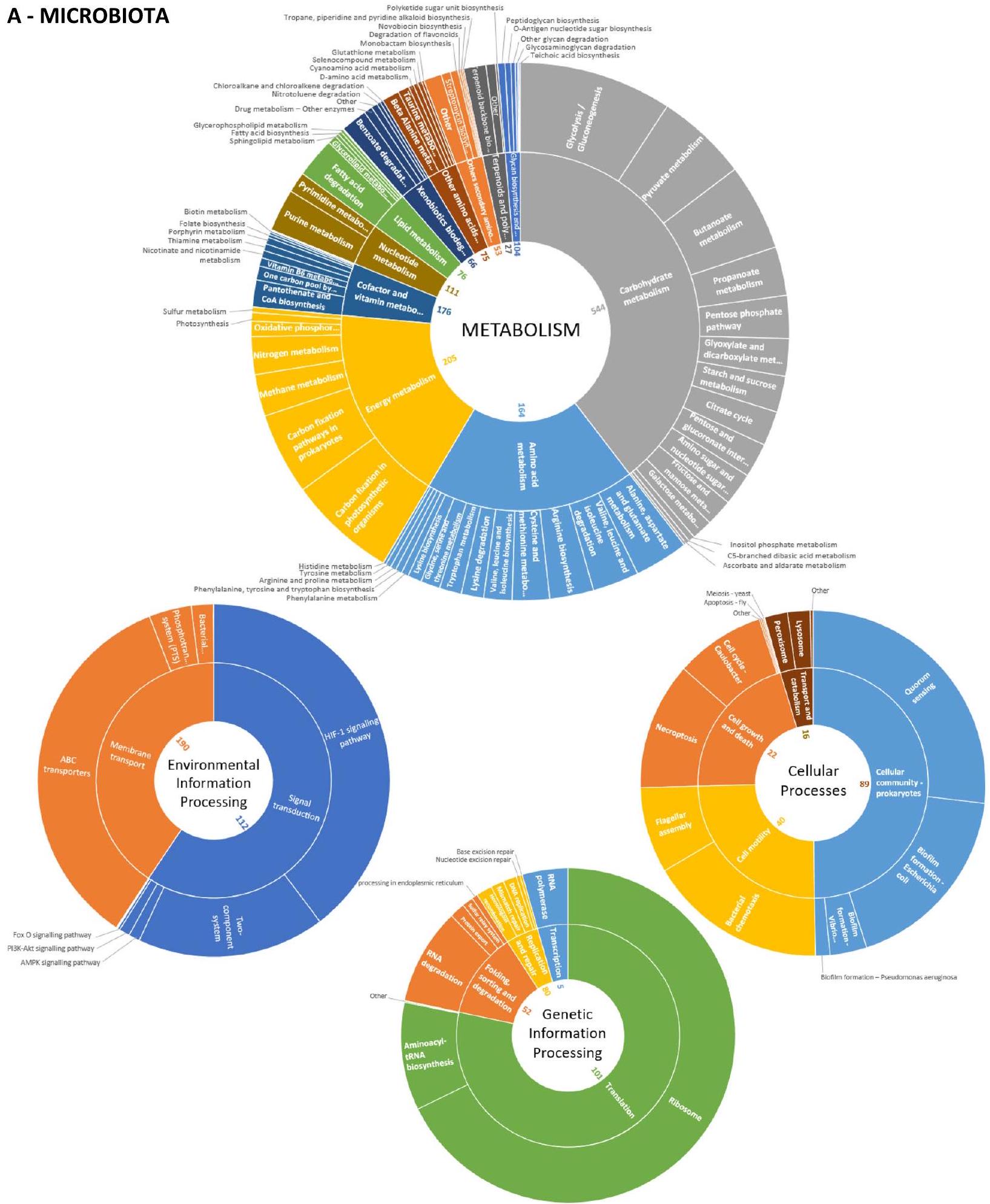
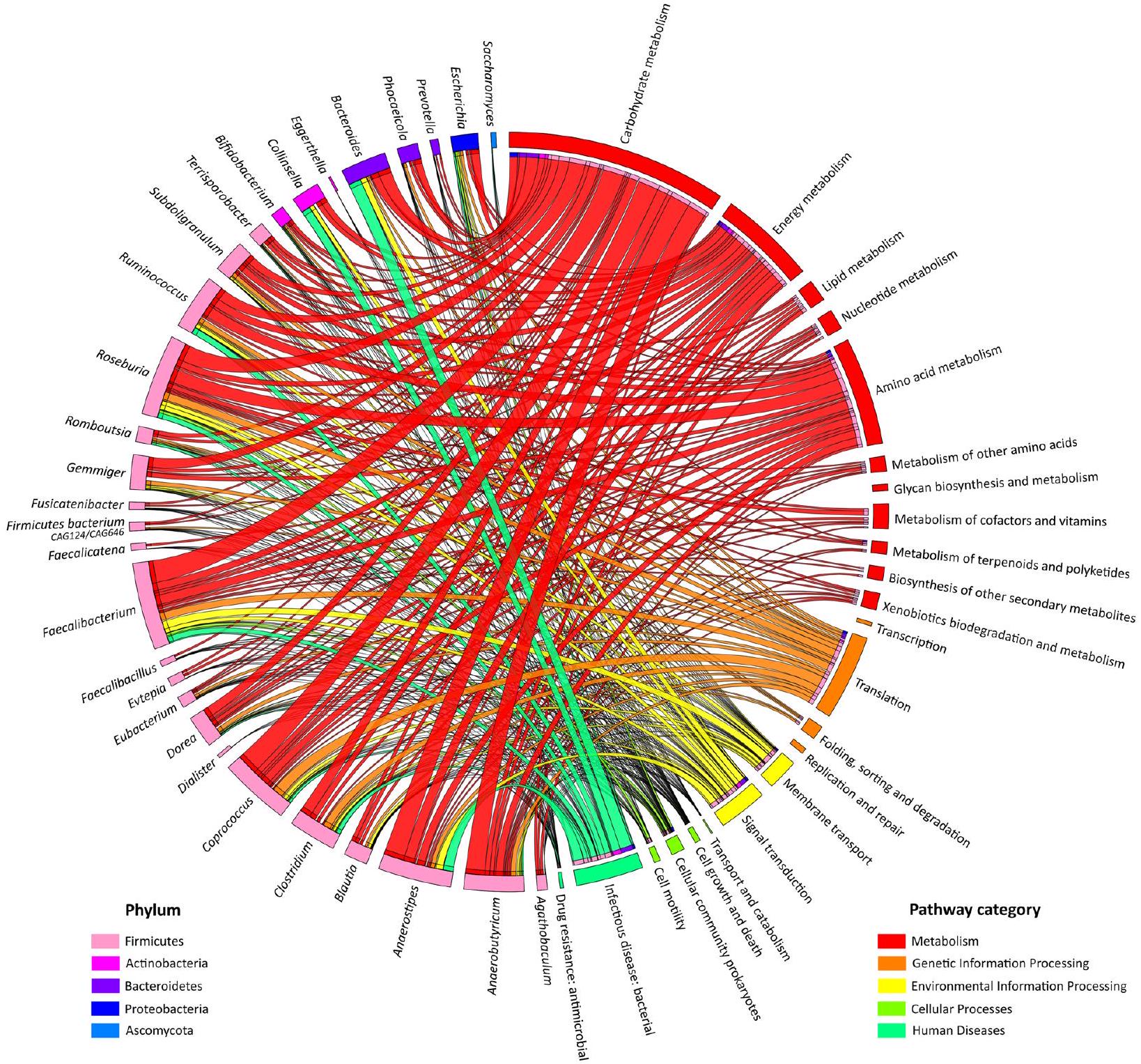
داخل الأنواع الأقل تمثيلًا نسبيًا، مما يدعم إمكانية أداة Orbitrap Astral في تسليط الضوء على الدور الفريد للميكروبيوتا بأكملها (الشكل 5).
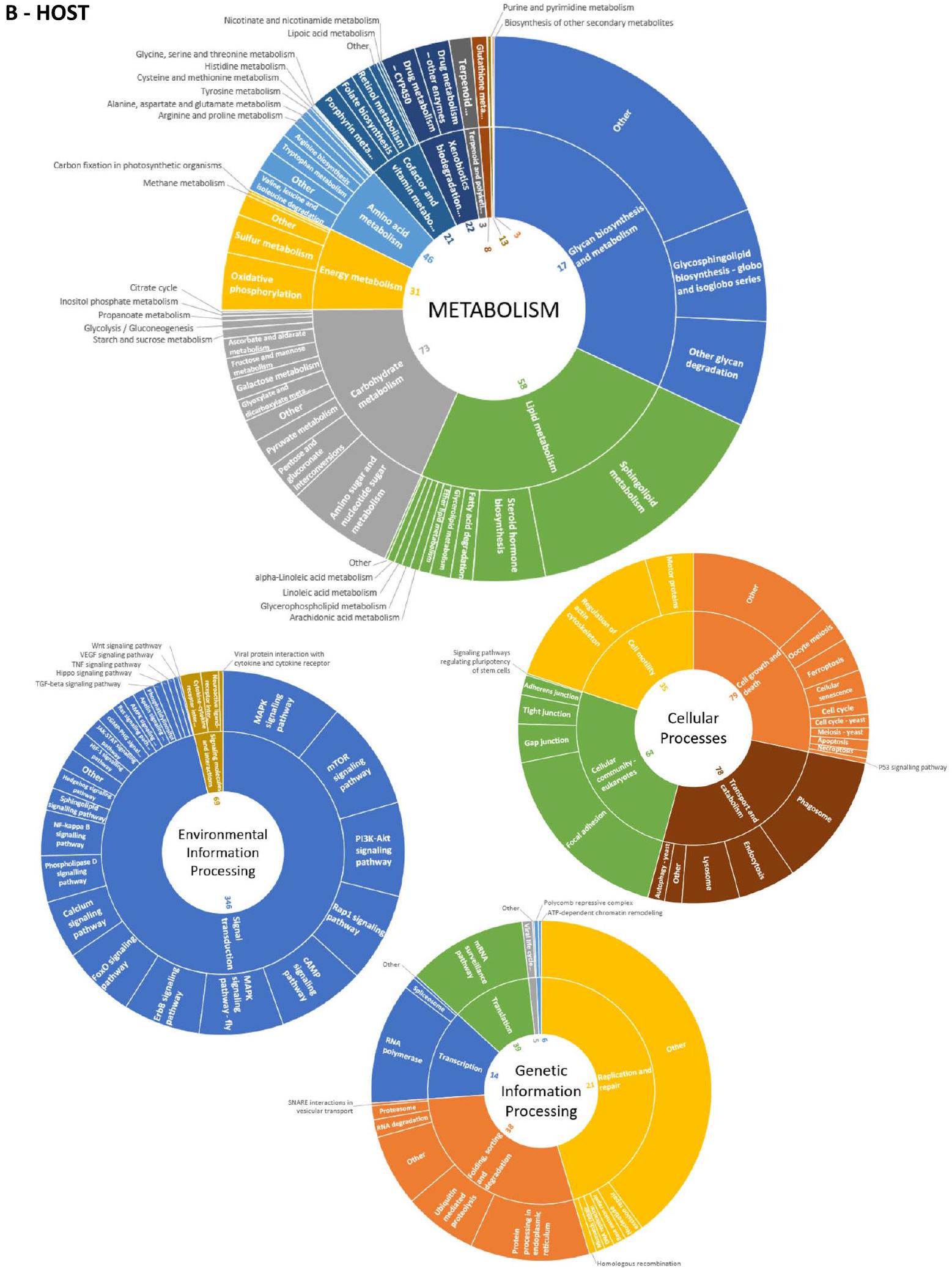
أخيرًا، تم التعليق على البروتينات الخاصة بالمضيف ضمن 367 مسارًا على مستوى KEGG 2، مع تميز نقل الإشارة كواحد من أكثر المسارات كثافة من حيث تعليقات KO. بلغ عمق التغطية
نقاش
كولاي، سيتم اختيار قاعدة البيانات المخصصة للتفسير وفقًا للمعلومات السابقة المتاحة. بدون معرفة مسبقة، سيحدد بروتيوتيب بسهولة ما إذا كانت العينة تحتوي على بروتينات بشرية أو بروتينات معوية، وستكون قاعدة البيانات النهائية لتفسير البروتيوم منطقياً مخصصة. على الرغم من أن قاعدة البيانات لتفسير الميتابروتيوم لعينات ميكروبيوم الأمعاء قد لا تكون شاملة بما يكفي لتشمل جميع تسلسلات المتغيرات البروتينية الموجودة في العينة، فإن الاستراتيجية التي نقترحها فعالة للغاية، كما هو موضح في هذه الدراسة. ستتعزز نتائج مثل هذه الاستراتيجية في المستقبل القريب مع زيادة عدد الجينومات المتاحة في قواعد البيانات العامة مع مرور الوقت. بالتأكيد، فإن إضافة بعد جديد للتفسير، مثل تفسير التسلسل الجديد [29]، أو البحث المتسامح مع الأخطاء، أو البحث عن تعديلات ما بعد الترجمة المتعددة [30]، لاستغلال إشارات MS/MS التي لم يتم تعيينها بعد مع الاستراتيجية الحالية، ستفيد بالتأكيد النتائج. ومع ذلك، يجب رفع القيود الحوسبية للبرامج المقابلة لتكون قابلة للتطبيق على مجموعات بيانات DIA الكبيرة جدًا التي تم الحصول عليها بواسطة جهاز Orbitrap Astral.
استنادًا إلى النتائج المبلغ عنها هنا، يبدو أن وضع DIA متفوق على وضع DDA لتحليل الميكروبيوم، حيث يمكن تحديد وقياس المزيد من الببتيدات والبروتينات، كما تم إثباته بالفعل [37-39]، مما يوفر مزيدًا من المعلومات حول المسارات البيولوجية للنظام. هنا، لاحظنا أن حتى بكتيريا مضافة عند
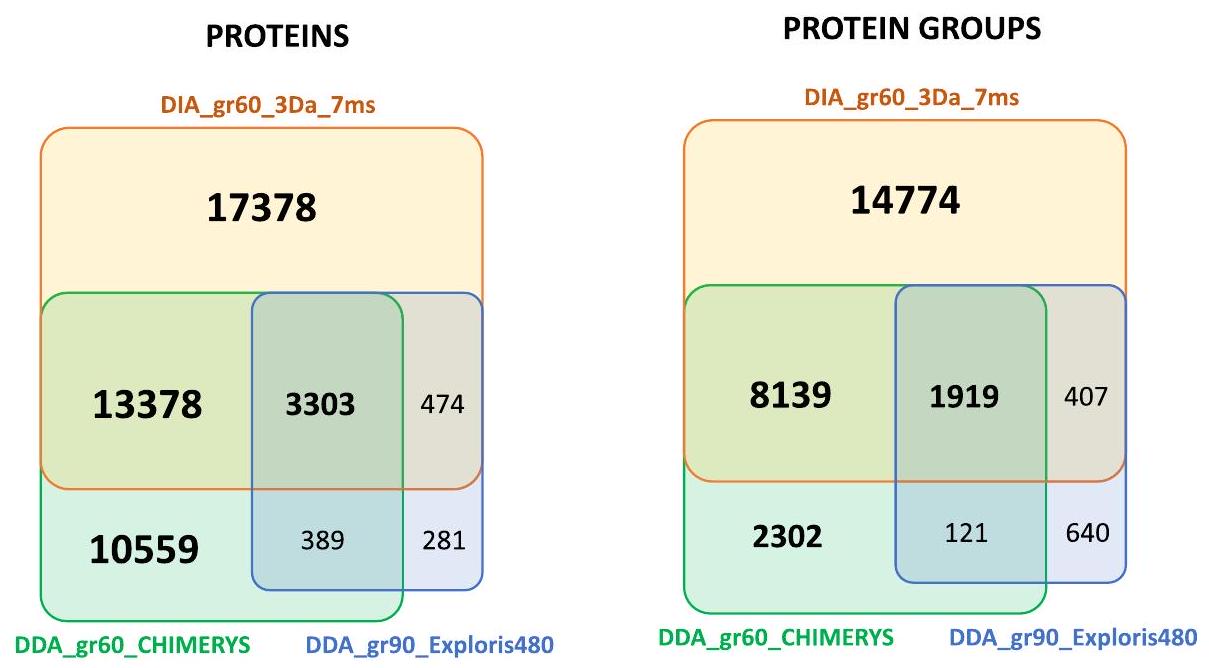
في الختام، نبلغ، من عينة واحدة، عن تحديد وقياس 44,204 مجموعة بروتين في تحليل DIA
جميع التقارير المنشورة حتى الآن حول عينات الميتابروتيوم الحقيقية التي نعلم بها. من المتوقع أن تكون هذه القيمة أعلى بكثير في المستقبل، بمجرد إجراء تحسينات محددة في جميع مراحل الإجراء التحليلي. إن القدرة على تضمين أكثر من 122,000 ببتيد فريد و38,000 مجموعة بروتين ضمن تشغيل DIA لمدة 30 دقيقة، مع الحفاظ على تكرارية جيدة جدًا عبر العمليات التحليلية، يعد أيضًا واعدًا جدًا. هذا الرقم القياسي المحدد من حيث عدد الببتيدات والبروتينات المكتشفة ليس له قيمة في حد ذاته ولكنه يسمح لنا بالتطلع إلى إمكانيات الميتابروتيوميات في المستقبل لمواجهة تحديات أكثر تعقيدًا، مثل مجموعات العينات المتتالية وعمق وظيفي محسّن. في النهاية، فإن محلل الكتلة Astral للعينات المعقدة للغاية يقرب الميتابروتيوميات من الاستخدام الروتيني في التشخيصات السريرية.
معلومات إضافية
الملف الإضافي 5: الجدول S5. قائمة البروتينات المحددة في مجموعات بيانات DIA لمدة 30 دقيقة (2Da-3 ms) (3 تكرارات).
شكر وتقدير
مساهمات المؤلفين
التمويل
توفر البيانات والمواد
الإعلانات
موافقة الأخلاقيات والموافقة على المشاركة
الموافقة على النشر
المصالح المتنافسة
تم النشر على الإنترنت: 07 مارس 2024
References
- Berg G, Rybakova D, Fischer D, Cernava T, Verges MC, Charles T, Chen X, Cocolin L, Eversole K, Corral GH, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. 2020;8:103. https://doi. org/10.1186/s40168-020-00875-0.
- Van Den Bossche T, Arntzen MO, Becher D, Benndorf D, Eijsink VGH, Henry C, Jagtap PD, Jehmlich N, Juste C, Kunath BJ, et al. The Metaproteomics Initiative: a coordinated approach for propelling the functional characterization of microbiomes. Microbiome. 2021;9:243. https://doi.org/10. 1186/s40168-021-01176-w.
- Armengaud J. Metaproteomics to understand how microbiota function: The crystal ball predicts a promising future. Environ Microbiol. 2023;25:115-25. https://doi.org/10.1111/1462-2920.16238.
- Stewart HI, Grinfeld D, Giannakopulos A, Petzoldt J, Shanley T, Garland M, Denisov E, Peterson AC, Damoc E, Zeller M, et al. Parallelized acquisition of orbitrap and astral analyzers enables high-throughput quantitative analysis. Anal Chem. 2023;95:15656-64. https://doi.org/10.1021/acs.analc hem.3c02856.
- Stewart H, Grinfeld D, Wagner A, Kholomeev A, Biel M, Giannakopulos A, Makarov A, Hock C. A conjoined rectilinear collision cell and pulsed extraction ion trap with auxiliary DC electrodes. J Am Soc Mass Spectrom. 2023. https://doi.org/10.1021/jasms.3c00311.
- Guzman UH, Martinez Del Val A, Ye Z, Damoc E, Arrey TN, Pashkova A, Denisov E, Petzoldt J, Peterson AC, Harking F, et al. Narrow-window DIA: Ultra-fast quantitative analysis of comprehensive proteomes with high sequencing depth. In: bioRxiv. 2023.
- Heil LR, Damoc E, Arrey TN, Pashkova A, Denisov E, Petzoldt J, Peterson AC, Hsu C, Searle BC, Shulman N, et al. Evaluating the performance of the Astral mass analyzer for quantitative proteomics using data-independent acquisition. J Proteome Res. 2023. https://doi.org/10.1021/acs.jproteome. 3c00357.
- Ye Z, Sabatier P, Martin-Gonzalez J, Eguchi A, Bekker-Jensen DB, Bache N, Olsen JV. One-Tip enables comprehensive proteome coverage in minimal cells and single zygotes. In: bioRxiv. 2023.
- Petrosius V, Aragon-Fernandez P, Arrey TN, Üresin N, Furtwängler B, Stewart H, Denisov E, Petzoldt J, Peterson AC, Hock C, et al. Evaluating the capabilities of the Astral mass analyzer for single-cell proteomics. In: bioRxiv. 2023.
- Lozano C, Kielbasa M, Gaillard JC, Miotello G, Pible O, Armengaud J. Identification and characterization of marine microorganisms by tandem mass spectrometry proteotyping. Microorganisms. 2022;10. https://doi. org/10.3390/microorganisms10040719.
- Hayoun K, Gouveia D, Grenga L, Pible O, Armengaud J, Alpha-Bazin B. Evaluation of sample preparation methods for fast proteotyping of microorganisms by tandem mass spectrometry. Front Microbiol. 1985;2019:10. https://doi.org/10.3389/fmicb.2019.01985.
- Grenga L, Pible O, Miotello G, Culotta K, Ruat S, Roncato MA, Gas F, Bellanger L, Claret PG, Dunyach-Remy C, et al. Taxonomical and functional changes in COVID-19 faecal microbiome could be related to SARS-CoV-2 faecal load. Environ Microbiol. 2022;24:4299-316. https://doi.org/10.1111/ 1462-2920.16028.
- Ma ZQ, Chambers MC, Ham AJ, Cheek KL, Whitwell CW, Aerni HR, Schilling B, Miller AW, Caprioli RM, Tabb DL. ScanRanker: Quality assessment
of tandem mass spectra via sequence tagging. J Proteome Res. 2011;10:2896-904. https://doi.org/10.1021/pr200118r. - Pible O, Allain F, Jouffret V, Culotta K, Miotello G, Armengaud J. Estimating relative biomasses of organisms in microbiota using “phylopeptidomics.” Microbiome. 2020;8:30. https://doi.org/10.1186/s40168-020-00797-x.
- Demichev V, Messner CB, Vernardis SI, Lilley KS, Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods. 2020;17:41-4. https://doi.org/10.1038/ s41592-019-0638-x.
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27-30. https://doi.org/10.1093/nar/28.1.27.
- Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726-31. https://doi.org/10.1016/j.jmb. 2015.11.006.
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639-45. https://doi.org/10.1101/gr.092759.109.
- Darzi Y, Letunic I, Bork P, Yamada T. iPath3.0: interactive pathways explorer v3. Nucleic Acids Res. 2018;46:W510-3. https://doi.org/10.1093/nar/ gky299.
- Ramos-Nascimento A, Grenga L, Haange SB, Himmelmann A, Arndt FS, Ly YT, Miotello G, Pible O, Jehmlich N, Engelmann B, et al. Human gut microbiome and metabolite dynamics under simulated microgravity. Gut Microbes. 2023;15:2259033. https://doi.org/10.1080/19490976.2023. 2259033.
- Blakeley-Ruiz JA, Kleiner M. Considerations for constructing a protein sequence database for metaproteomics. Comput Struct Biotechnol J. 2022;20:937-52. https://doi.org/10.1016/j.csbj.2022.01.018.
- Bassignani A, Plancade S, Berland M, Blein-Nicolas M, Guillot A, Chevret D, Moritz C, Huet S, Rizkalla S, Clement K, et al. Benefits of iterative searches of large databases to interpret large human gut metaproteomic data sets. J Proteome Res. 2021;20:1522-34. https://doi.org/10.1021/acs.jproteome. 0c00669.
- Sun Z, Ning Z, Cheng K, Duan H, Wu Q, Mayne J, Figeys D. MetaPep: A core peptide database for faster human gut metaproteomics database searches. Comput Struct Biotechnol J. 2023;21:4228-37. https://doi.org/ 10.1016/j.csbj.2023.08.025.
- Grenga L, Pible O, Armengaud J. Pathogen proteotyping: a rapidly developing application of mass spectrometry to address clinical concerns. Clin Mass Spectrom. 2019;14 Pt A:9-17. https://doi.org/10.1016/j.clinms.2019. 04.004.
- Gouveia D, Pible O, Culotta K, Jouffret V, Geffard O, Chaumot A, DegliEsposti D, Armengaud J. Combining proteogenomics and metaproteomics for deep taxonomic and functional characterization of microbiomes from a non-sequenced host. NPJ Biofilms Microbiomes. 2020;6:23. https:// doi.org/10.1038/s41522-020-0133-2.
- Charlier P, Armengaud J. Did Saint Leonard suffer from Madura foot at the time of death? Infectious disease diagnosis by paleo-proteotyping. J Infect. 2023. https://doi.org/10.1016/j.jinf.2023.10.021.
- Oumarou Hama H, Chenal T, Pible O, Miotello G, Armengaud J, Drancourt M. An ancient coronavirus from individuals in France, circa 16th century. Int J Infect Dis. 2023;131:7-12. https://doi.org/10.1016/j.jid.2023.03.019.
- Brooks B, Mueller RS, Young JC, Morowitz MJ, Hettich RL, Banfield JF. Strain-resolved microbial community proteomics reveals simultaneous aerobic and anaerobic function during gastrointestinal tract colonization of a preterm infant. Front Microbiol. 2015;6:654. https://doi.org/10.3389/ fmicb.2015.00654.
- Kleikamp HBC, Pronk M, Tugui C, Guedes da Silva L, Abbas B, Lin YM, van Loosdrecht MCM, Pabst M. Database-independent de novo metaproteomics of complex microbial communities. Cell Syst. 2021;12:375-383 e375. https://doi.org/10.1016/j.cels.2021.04.003.
- Duan H, Zhang X, Figeys D. An emerging field: post-translational modification in microbiome. Proteomics. 2023;23:e2100389. https://doi.org/10. 1002/pmic. 202100389.
- Jiang N, Gao Y, Xu J, Luo F, Zhang X, Chen R. A data-independent acquisition (DIA)-based quantification workflow for proteome analysis of 5000 cells. J Pharm Biomed Anal. 2022;216:114795. https://doi.org/10.1016/j. jpba.2022.114795.
- Steger M, Demichev V, Backman M, Ohmayer U, Ihmor P, Muller S, Ralser M, Daub H. Time-resolved in vivo ubiquitinome profiling by
33. Zhang F, Ge W, Huang L, Li D, Liu L, Dong Z, Xu L, Ding X, Zhang C, Sun Y, et al. A Comparative analysis of data analysis tools for data-independent acquisition mass spectrometry. Mol Cell Proteomics. 2023;22:100623. https://doi.org/10.1016/j.mcpro.2023.100623.
34. Gomez-Varela D, Xian F, Grundtner S, Sondermann JR, Carta G, Schmidt M. Increasing taxonomic and functional characterization of hostmicrobiome interactions by DIA-PASEF metaproteomics. Front Microbiol. 2023;14:1258703. https://doi.org/10.3389/fmicb.2023.1258703.
35. Pietila S, Suomi T, Elo LL. Introducing untargeted data-independent acquisition for metaproteomics of complex microbial samples. ISME Commun. 2022;2:51. https://doi.org/10.1038/s43705-022-00137-0.
36. Van Den Bossche T, Kunath BJ, Schallert K, Schape SS, Abraham PE, Armengaud J, Arntzen MO, Bassignani A, Benndorf D, Fuchs S, et al. Critical Assessment of MetaProteome Investigation (CAMPI): a multi-laboratory comparison of established workflows. Nat Commun. 2021;12:7305. https://doi.org/10.1038/s41467-021-27542-8.
37. Zhao J, Yang Y, Xu H, Zheng J, Shen C, Chen T, Wang T, Wang B, Yi J, Zhao D, et al. Data-independent acquisition boosts quantitative metaproteomics for deep characterization of gut microbiota. NPJ Biofilms Microbiomes. 2023;9:4. https://doi.org/10.1038/s41522-023-00373-9.
38. Aakko J, Pietila S, Suomi T, Mahmoudian M, Toivonen R, Kouvonen P, Rokka A, Hanninen A, Elo LL. Data-independent acquisition mass spectrometry in metaproteomics of gut microbiota-implementation and computational analysis. J Proteome Res. 2020;19:432-6. https://doi.org/ 10.1021/acs.jproteome.9b00606.
39. Long S, Yang Y, Shen C, Wang Y, Deng A, Qin Q, Qiao L. Metaproteomics characterizes human gut microbiome function in colorectal cancer. NPJ Biofilms Microbiomes. 2020;6:14. https://doi.org/10.1038/ s41522-020-0123-4.
ملاحظة الناشر
DOI: https://doi.org/10.1186/s40168-024-01766-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38454512
Publication Date: 2024-03-07
The astounding exhaustiveness and speed of the Astral mass analyzer for highly complex samples is a quantum leap in the functional analysis of microbiomes
Abstract
Background By analyzing the proteins which are the workhorses of biological systems, metaproteomics allows us to list the taxa present in any microbiota, monitor their relative biomass, and characterize the functioning of complex biological systems. Results Here, we present a new strategy for rapidly determining the microbial community structure of a given sample and designing a customized protein sequence database to optimally exploit extensive tandem mass spectrometry data. This approach leverages the capabilities of the first generation of Quadrupole Orbitrap mass spectrometer incorporating an asymmetric track lossless (Astral) analyzer, offering rapid MS/MS scan speed and sensitivity. We took advantage of data-dependent acquisition and data-independent acquisition strategies using a peptide extract from a human fecal sample spiked with precise amounts of peptides from two reference bacteria. Conclusions Our approach, which combines both acquisition methods, proves to be time-efficient while processing extensive generic databases and massive datasets, achieving a coverage of more than 122,000 unique peptides and 38,000 protein groups within a 30 -min DIA run. This marks a significant departure from current state-of-the-art metaproteomics methodologies, resulting in broader coverage of the metabolic pathways governing the biological system. In combination, our strategy and the Astral mass analyzer represent a quantum leap in the functional analysis of microbiomes.
*Correspondence:
Jean Armengaud
jean.armengaud@cea.fr
Background
that provides crucial information on the structural components, enzymes, and informational messengers of microorganisms, as well as on the host response, if any [2]. In addition to identifying the metabolic pathways in action and assessing their level of activity by means of their quantities, the methodology makes it possible to trace them back to the specific organisms that produced the corresponding proteins thanks to peptide sequences established by high-resolution tandem mass spectrometry. Metaproteomics therefore has a key role in deepening our knowledge of microbiomes, compared with methodologies limited to cataloguing microorganisms and genomic potential. Moreover, thanks to its extreme speed, this methodology could become an attractive new diagnostic tool for human medicine and the environment [3].
Microbiome research is strongly influenced by methodological advances. Recent developments in tandem mass spectrometry, acquisition strategies, and interpretation tools have great potential to transform metaproteomics into a high-performance methodology for deepening knowledge of microbial functioning. Metaproteomics grapples with an enormous amount of complex data, including giant databases of protein sequences built from metagenomic data or large numbers of sequenced organisms. Metaproteomics is also confronted with an exceptionally high number of proteins and variants from the sample, making the identification of common peptides easier than specific ones. Last, the lack of comprehensive coverage of the protein sequence database tends to decrease the outcome of the interpretation. Very recently, Stewart et al. [4] described the development of a new mass spectrometer that combines a powerful Orbitrap mass-resolving quadrupole, a novel ion processor rectilinear ion trap [5], and a revolutionary conceptual analyzer called Asymmetric Track Lossless (Astral) analyzer, enabling faster acquisition of high-resolution MS/MS spectra and high sensitivity compared with state-of-theart mass spectrometers. The results demonstrated by this novel instrument for proteomics are promising in terms of depth of analysis with 10,000 groups identified from a HeLa peptide extract over a single 48 -min run [4]. Such performance was further documented for comprehensive analysis of proteome post-translational modifications [6], plasma proteome [7], minimal cells [8], and single-cell proteomics [9]. Given these substantial improvements, in the present study, we explore its performance for profiling highly complex samples using a specific standard of human fecal material spiked with precise amounts of two bacterial proteomes. To fully exploit this new technology, we propose a novel workflow for metaproteomics, based on reliable proteotyping of microorganisms from short data-dependent acquisition (DDA), designing a specific
database selecting the most valuable genomes, recording of high-density datasets in data-independent acquisition, and interpretation for increased coverage of the key players in the microbiota.
Materials and methods
MetaP reference sample
Orbitrap Astral mass spectrometry
Data interpretation for proteotyping organisms
DB48 database creation
Data metaproteomics interpretation
library generation was conducted in silico based on the DB48 database. A maximum of 2 missed cleavages were allowed, 2 variable modifications (oxidation of methionines and acetylation of the N-terminus), peptide length ranging from 7 to 30 residues, precursor charge of 2 and
Functional profiling of the host, microbiota, and spiked bacteria
Orbitrap Exploris 480 mass spectrometry and data processing
Mass spectrometry proteomics data
Results
DDA-based proteotyping to identify the most abundant organisms in the sample
used to perform a second search to identify organisms at the species taxonomical rank. The species identified in these six independent analyses are listed in Table S1, along with the corresponding number of taxon-specific peptides and Taxon-to-Spectrum Matches (TSMs) at the various taxonomical ranks. Table S2 provides a reliable list of the 9 phyla, 44 genera, and 56 species that were proteotyped through the merging of these results, along with their respective contribution to protein biomass (Fig. 1). The overall TSMs signal decreases slightly when moving down the taxonomical hierarchy, from
the ratio is observed for Ascomycota and Actinobacteria, suggesting that the proteotyped species within these two phyla are only weakly representative. Given that their overall contributions are small, this has minimal impact on the results. In fact, the global TSMs signal, which represents
Amongst the 13 Eukaryota, the host Homo sapiens is logically the most abundant species, accounting for over
Astral DDA current state-of-the-art metaproteomics is improved
are expected to be chimeric, leading to a decrease in the assignment ratio. Preliminary testing was performed on a single
DIA metaproteomics increases significantly peptide coverage and protein detection
Significantly improved panorama of metabolic pathways relies on extra-large DIA peptidome landscape
within relatively less represented taxa, supporting the potential of the Orbitrap Astral instrument in shedding light on the peculiar role of the entire microbiota (Fig. 5).
Finally, host-specific proteins were annotated within 367 KEGG-level 2 pathways, with signal transduction standing out as one of the most densely populated pathways in terms of KO annotations. The depth of coverage reached
Discussion
coli sample is processed, the database chosen for interpretation will be selected accordingly on the basis of the prior information available. Without prior knowledge, proteotyping will easily determine whether the sample contains human or enterobacterial proteins and the final database for proteomic interpretation will logically be customized. Although the database for metaproteomic interpretation of gut microbiome samples may not be comprehensive enough to encompass all protein variant sequences present in the sample, the strategy we propose is highly effective, as demonstrated in the present study. The results of such a strategy will be further strengthened in the near future as the number of genomes available in generalist databases increases over time. Certainly, the addition of a new interpretation dimension, such as de novo sequencing interpretation [29], error-tolerant searches, or searches for multiple post-translational modifications [30], to exploit the MS/MS signals that have not yet been assigned with the current strategy, will certainly benefit the results. However, the computational limitations of the corresponding software would need to be lifted in order to be applied to the very large DIA datasets acquired by the Orbitrap Astral instrument.
Based on the results reported here, DIA mode appears superior to DDA mode for microbiome analysis, as more peptides and proteins can be identified and quantified, as has already been established [37-39], providing more information on the biological pathways of the system. Here, we observed that even a bacterium added at
In conclusion, we report, from a single sample, the identification and quantification of 44,204 protein groups in a
all reports published to date on real-life metaproteomic samples of which we are aware. This value is set to be much higher in the future, once specific optimizations have been made at all stages of the analytical procedure. The ability to encompass more than 122,000 unique peptides and 38,000 protein groups within a 30 -min DIA run, while maintaining a very good repeatability across analytical runs, is also very promising. This specific record in terms of the number of peptides and proteins detected is futile in itself but allows us to glimpse the possibilities of metaproteomics for the future to tackle more complex challenges, such as consequent cohorts of samples and improved functional depth. Ultimately, the Astral mass analyzer for highly complex samples brings metaproteomics closer to routine use in clinical diagnostics.
Supplementary Information
Additional file 5: Table S5. List of identified proteins in the 30 min (2Da-3 ms) DIA datasets (3 replicates).
Acknowledgements
Authors’ contributions
Funding
Availability of data and materials
Declarations
Ethics approval and consent to participate
Consent for publication
Competing interests
Published online: 07 March 2024
References
- Berg G, Rybakova D, Fischer D, Cernava T, Verges MC, Charles T, Chen X, Cocolin L, Eversole K, Corral GH, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. 2020;8:103. https://doi. org/10.1186/s40168-020-00875-0.
- Van Den Bossche T, Arntzen MO, Becher D, Benndorf D, Eijsink VGH, Henry C, Jagtap PD, Jehmlich N, Juste C, Kunath BJ, et al. The Metaproteomics Initiative: a coordinated approach for propelling the functional characterization of microbiomes. Microbiome. 2021;9:243. https://doi.org/10. 1186/s40168-021-01176-w.
- Armengaud J. Metaproteomics to understand how microbiota function: The crystal ball predicts a promising future. Environ Microbiol. 2023;25:115-25. https://doi.org/10.1111/1462-2920.16238.
- Stewart HI, Grinfeld D, Giannakopulos A, Petzoldt J, Shanley T, Garland M, Denisov E, Peterson AC, Damoc E, Zeller M, et al. Parallelized acquisition of orbitrap and astral analyzers enables high-throughput quantitative analysis. Anal Chem. 2023;95:15656-64. https://doi.org/10.1021/acs.analc hem.3c02856.
- Stewart H, Grinfeld D, Wagner A, Kholomeev A, Biel M, Giannakopulos A, Makarov A, Hock C. A conjoined rectilinear collision cell and pulsed extraction ion trap with auxiliary DC electrodes. J Am Soc Mass Spectrom. 2023. https://doi.org/10.1021/jasms.3c00311.
- Guzman UH, Martinez Del Val A, Ye Z, Damoc E, Arrey TN, Pashkova A, Denisov E, Petzoldt J, Peterson AC, Harking F, et al. Narrow-window DIA: Ultra-fast quantitative analysis of comprehensive proteomes with high sequencing depth. In: bioRxiv. 2023.
- Heil LR, Damoc E, Arrey TN, Pashkova A, Denisov E, Petzoldt J, Peterson AC, Hsu C, Searle BC, Shulman N, et al. Evaluating the performance of the Astral mass analyzer for quantitative proteomics using data-independent acquisition. J Proteome Res. 2023. https://doi.org/10.1021/acs.jproteome. 3c00357.
- Ye Z, Sabatier P, Martin-Gonzalez J, Eguchi A, Bekker-Jensen DB, Bache N, Olsen JV. One-Tip enables comprehensive proteome coverage in minimal cells and single zygotes. In: bioRxiv. 2023.
- Petrosius V, Aragon-Fernandez P, Arrey TN, Üresin N, Furtwängler B, Stewart H, Denisov E, Petzoldt J, Peterson AC, Hock C, et al. Evaluating the capabilities of the Astral mass analyzer for single-cell proteomics. In: bioRxiv. 2023.
- Lozano C, Kielbasa M, Gaillard JC, Miotello G, Pible O, Armengaud J. Identification and characterization of marine microorganisms by tandem mass spectrometry proteotyping. Microorganisms. 2022;10. https://doi. org/10.3390/microorganisms10040719.
- Hayoun K, Gouveia D, Grenga L, Pible O, Armengaud J, Alpha-Bazin B. Evaluation of sample preparation methods for fast proteotyping of microorganisms by tandem mass spectrometry. Front Microbiol. 1985;2019:10. https://doi.org/10.3389/fmicb.2019.01985.
- Grenga L, Pible O, Miotello G, Culotta K, Ruat S, Roncato MA, Gas F, Bellanger L, Claret PG, Dunyach-Remy C, et al. Taxonomical and functional changes in COVID-19 faecal microbiome could be related to SARS-CoV-2 faecal load. Environ Microbiol. 2022;24:4299-316. https://doi.org/10.1111/ 1462-2920.16028.
- Ma ZQ, Chambers MC, Ham AJ, Cheek KL, Whitwell CW, Aerni HR, Schilling B, Miller AW, Caprioli RM, Tabb DL. ScanRanker: Quality assessment
of tandem mass spectra via sequence tagging. J Proteome Res. 2011;10:2896-904. https://doi.org/10.1021/pr200118r. - Pible O, Allain F, Jouffret V, Culotta K, Miotello G, Armengaud J. Estimating relative biomasses of organisms in microbiota using “phylopeptidomics.” Microbiome. 2020;8:30. https://doi.org/10.1186/s40168-020-00797-x.
- Demichev V, Messner CB, Vernardis SI, Lilley KS, Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods. 2020;17:41-4. https://doi.org/10.1038/ s41592-019-0638-x.
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27-30. https://doi.org/10.1093/nar/28.1.27.
- Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726-31. https://doi.org/10.1016/j.jmb. 2015.11.006.
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639-45. https://doi.org/10.1101/gr.092759.109.
- Darzi Y, Letunic I, Bork P, Yamada T. iPath3.0: interactive pathways explorer v3. Nucleic Acids Res. 2018;46:W510-3. https://doi.org/10.1093/nar/ gky299.
- Ramos-Nascimento A, Grenga L, Haange SB, Himmelmann A, Arndt FS, Ly YT, Miotello G, Pible O, Jehmlich N, Engelmann B, et al. Human gut microbiome and metabolite dynamics under simulated microgravity. Gut Microbes. 2023;15:2259033. https://doi.org/10.1080/19490976.2023. 2259033.
- Blakeley-Ruiz JA, Kleiner M. Considerations for constructing a protein sequence database for metaproteomics. Comput Struct Biotechnol J. 2022;20:937-52. https://doi.org/10.1016/j.csbj.2022.01.018.
- Bassignani A, Plancade S, Berland M, Blein-Nicolas M, Guillot A, Chevret D, Moritz C, Huet S, Rizkalla S, Clement K, et al. Benefits of iterative searches of large databases to interpret large human gut metaproteomic data sets. J Proteome Res. 2021;20:1522-34. https://doi.org/10.1021/acs.jproteome. 0c00669.
- Sun Z, Ning Z, Cheng K, Duan H, Wu Q, Mayne J, Figeys D. MetaPep: A core peptide database for faster human gut metaproteomics database searches. Comput Struct Biotechnol J. 2023;21:4228-37. https://doi.org/ 10.1016/j.csbj.2023.08.025.
- Grenga L, Pible O, Armengaud J. Pathogen proteotyping: a rapidly developing application of mass spectrometry to address clinical concerns. Clin Mass Spectrom. 2019;14 Pt A:9-17. https://doi.org/10.1016/j.clinms.2019. 04.004.
- Gouveia D, Pible O, Culotta K, Jouffret V, Geffard O, Chaumot A, DegliEsposti D, Armengaud J. Combining proteogenomics and metaproteomics for deep taxonomic and functional characterization of microbiomes from a non-sequenced host. NPJ Biofilms Microbiomes. 2020;6:23. https:// doi.org/10.1038/s41522-020-0133-2.
- Charlier P, Armengaud J. Did Saint Leonard suffer from Madura foot at the time of death? Infectious disease diagnosis by paleo-proteotyping. J Infect. 2023. https://doi.org/10.1016/j.jinf.2023.10.021.
- Oumarou Hama H, Chenal T, Pible O, Miotello G, Armengaud J, Drancourt M. An ancient coronavirus from individuals in France, circa 16th century. Int J Infect Dis. 2023;131:7-12. https://doi.org/10.1016/j.jid.2023.03.019.
- Brooks B, Mueller RS, Young JC, Morowitz MJ, Hettich RL, Banfield JF. Strain-resolved microbial community proteomics reveals simultaneous aerobic and anaerobic function during gastrointestinal tract colonization of a preterm infant. Front Microbiol. 2015;6:654. https://doi.org/10.3389/ fmicb.2015.00654.
- Kleikamp HBC, Pronk M, Tugui C, Guedes da Silva L, Abbas B, Lin YM, van Loosdrecht MCM, Pabst M. Database-independent de novo metaproteomics of complex microbial communities. Cell Syst. 2021;12:375-383 e375. https://doi.org/10.1016/j.cels.2021.04.003.
- Duan H, Zhang X, Figeys D. An emerging field: post-translational modification in microbiome. Proteomics. 2023;23:e2100389. https://doi.org/10. 1002/pmic. 202100389.
- Jiang N, Gao Y, Xu J, Luo F, Zhang X, Chen R. A data-independent acquisition (DIA)-based quantification workflow for proteome analysis of 5000 cells. J Pharm Biomed Anal. 2022;216:114795. https://doi.org/10.1016/j. jpba.2022.114795.
- Steger M, Demichev V, Backman M, Ohmayer U, Ihmor P, Muller S, Ralser M, Daub H. Time-resolved in vivo ubiquitinome profiling by
33. Zhang F, Ge W, Huang L, Li D, Liu L, Dong Z, Xu L, Ding X, Zhang C, Sun Y, et al. A Comparative analysis of data analysis tools for data-independent acquisition mass spectrometry. Mol Cell Proteomics. 2023;22:100623. https://doi.org/10.1016/j.mcpro.2023.100623.
34. Gomez-Varela D, Xian F, Grundtner S, Sondermann JR, Carta G, Schmidt M. Increasing taxonomic and functional characterization of hostmicrobiome interactions by DIA-PASEF metaproteomics. Front Microbiol. 2023;14:1258703. https://doi.org/10.3389/fmicb.2023.1258703.
35. Pietila S, Suomi T, Elo LL. Introducing untargeted data-independent acquisition for metaproteomics of complex microbial samples. ISME Commun. 2022;2:51. https://doi.org/10.1038/s43705-022-00137-0.
36. Van Den Bossche T, Kunath BJ, Schallert K, Schape SS, Abraham PE, Armengaud J, Arntzen MO, Bassignani A, Benndorf D, Fuchs S, et al. Critical Assessment of MetaProteome Investigation (CAMPI): a multi-laboratory comparison of established workflows. Nat Commun. 2021;12:7305. https://doi.org/10.1038/s41467-021-27542-8.
37. Zhao J, Yang Y, Xu H, Zheng J, Shen C, Chen T, Wang T, Wang B, Yi J, Zhao D, et al. Data-independent acquisition boosts quantitative metaproteomics for deep characterization of gut microbiota. NPJ Biofilms Microbiomes. 2023;9:4. https://doi.org/10.1038/s41522-023-00373-9.
38. Aakko J, Pietila S, Suomi T, Mahmoudian M, Toivonen R, Kouvonen P, Rokka A, Hanninen A, Elo LL. Data-independent acquisition mass spectrometry in metaproteomics of gut microbiota-implementation and computational analysis. J Proteome Res. 2020;19:432-6. https://doi.org/ 10.1021/acs.jproteome.9b00606.
39. Long S, Yang Y, Shen C, Wang Y, Deng A, Qin Q, Qiao L. Metaproteomics characterizes human gut microbiome function in colorectal cancer. NPJ Biofilms Microbiomes. 2020;6:14. https://doi.org/10.1038/ s41522-020-0123-4.