DOI: https://doi.org/10.1021/acscatal.3c05388
PMID: https://pubmed.ncbi.nlm.nih.gov/38449527
تاريخ النشر: 2024-02-15
يرجى الإشارة إلى النسخة المنشورة
الناشر: الجمعية الكيميائية الأمريكية (ACS)
الإصدار: النسخة المنشورة
تم التنزيل من:https://e-space.mmu.ac.uk/634603/
حقوق الاستخدام:
الاستفسارات:
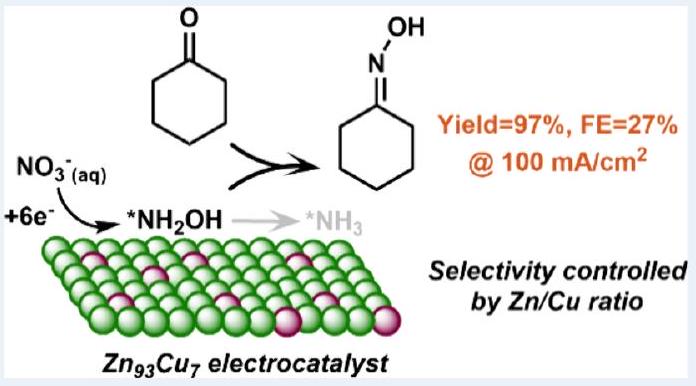
التحليل الكهربائي المستدام لأكسيم سيكلوهكسان من خلال اختزال النترات على محفز سبيكة الزنك والنحاس
اقرأ على الإنترنت
تم التنزيل عبر جامعة مانشستر متروبوليتان في 13 مايو 2024 الساعة 15:26:33 (UTC).
انظرhttps://pubs.acs.org/sharingguidelinesلخيارات حول كيفية مشاركة المقالات المنشورة بشكل قانوني.
الملخص
أوكسي ميثيل سيكلوهكسان هو مقدمة مهمة للنايلون-6 وعادة ما يتم تصنيعه من خلال إضافة النيوكليوفيل-الإزالة للهيدروكسيلامين مع سيكلوهكسان. ومع ذلك، فإن التقنيات الحالية لإنتاج الهيدروكسيلامين ليست صديقة للبيئة بسبب الحاجة إلى ظروف تفاعل قاسية. هنا، نبلغ عن طريقة كيميائية كهربائية لتصنيع أوكسي ميثيل سيكلوهكسان في وعاء واحد تحت ظروف محيطية باستخدام نترات مائية كمصدر للنيتروجين. سلسلة من
 لإنتاج الأوكسيما المقابل. يتم تحقيق أفضل أداء على
لإنتاج الأوكسيما المقابل. يتم تحقيق أفضل أداء على
– المقدمة
درجة الحموضة، ووجود فائض من
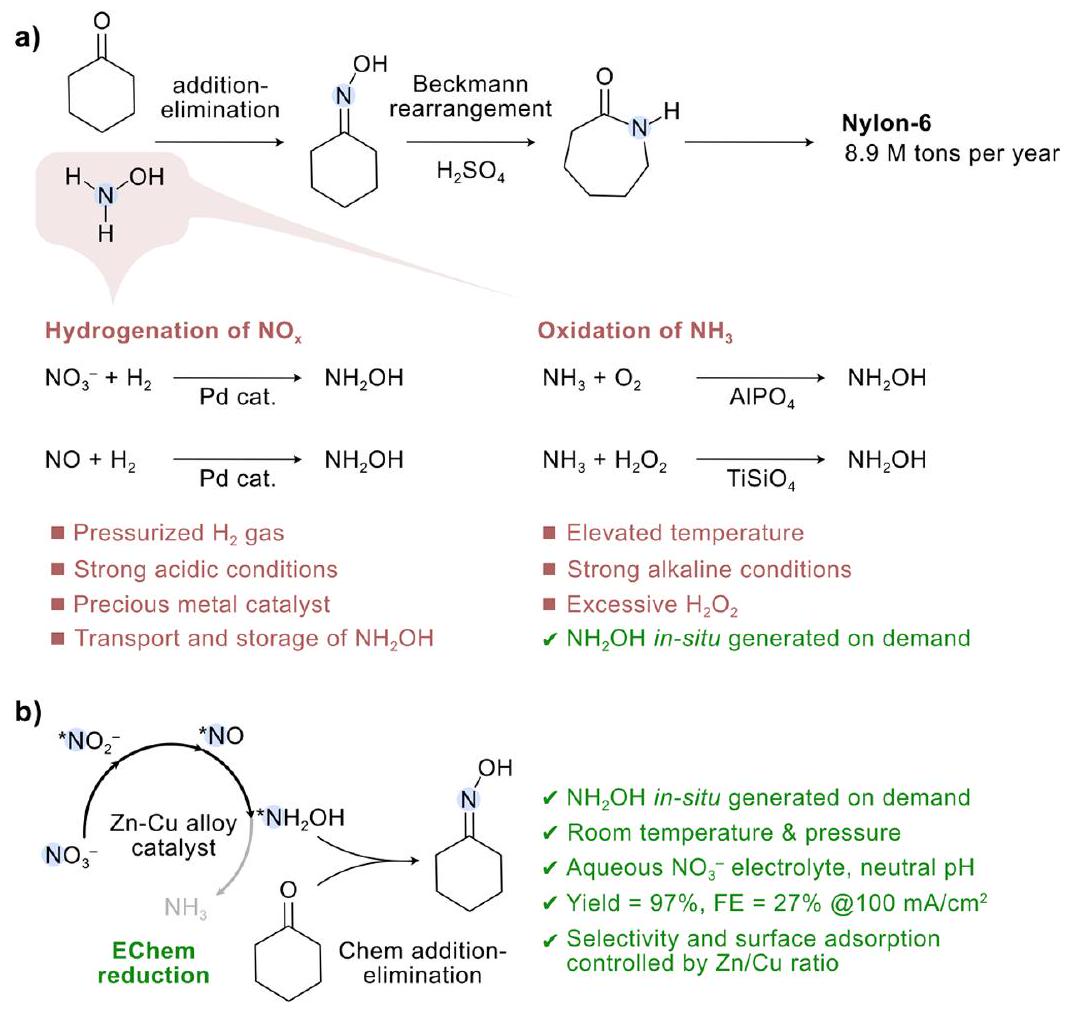
عملية كيميائية-كيميائية (EChem-Chem) للتخليق في وعاء واحد لأوكسيما السيكلوهكسان باستخدام نترات مائية كمصدر للنيتروجين (الشكل 1 ب) وتقديم تحقيق متعمق حول كيفية التحكم في انتقائية المنتج من خلال امتصاص السطح لعينات النيتروجين. سلسلة من
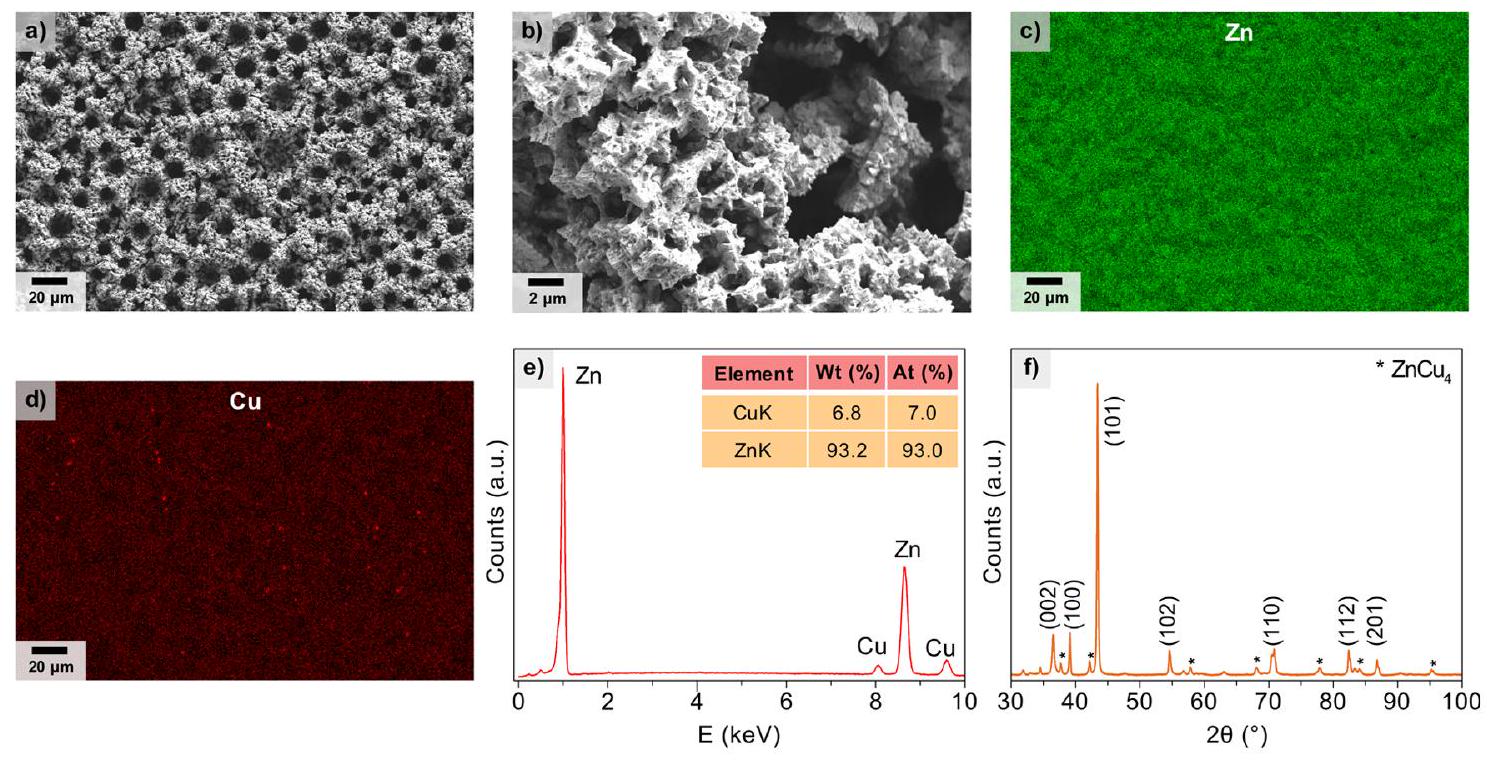
– النتائج والمناقشة
استخدم كقطب كهربائي للعمل لدفع التفاعل الكهروكيميائي
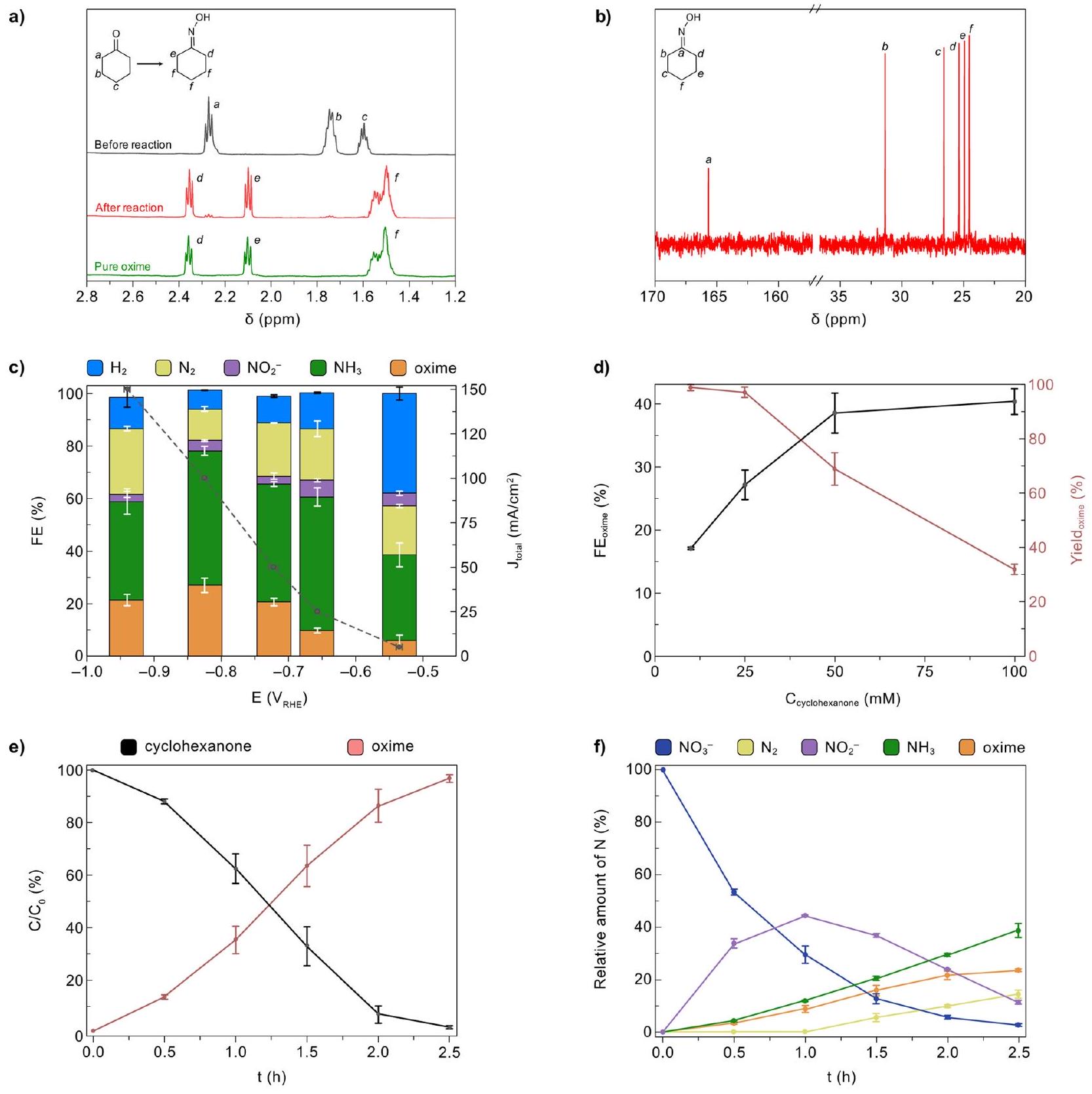
تمت دراسة إضافة (ثابت عند 100 مليمول) إلى الإلكتروليت من خلال تجارب التحليل الكهربائي لمدة 2.5 ساعة في
| مدخل | الكاثود | مصدر C | مصدر ن | التحليل الكهربائي | أوكسي ميثيل سيكلوهكسان | ||||||||
| 1 |
|
سيكلوهكسانون |
|
نعم | نعم | ||||||||
| 2 |
|
سيكلوهكسانون |
|
لا | لا | ||||||||
| ٣ |
|
|
نعم | لا | |||||||||
| ٤ |
|
سيكلوهكسانون | نعم | لا | |||||||||
| ٥ | رقائق النحاس | سيكلوهكسانون |
|
نعم | لا | ||||||||
| ٦ |
|
سيكلوهكسانون |
|
نعم | نعم،
|
||||||||
| ٧ |
|
سيكلوهكسانون |
|
نعم | نعم | ||||||||
| ٨ |
|
سيكلوهكسانون | لا | نعم | نعم | ||||||||
| 9 |
|
سيكلوهكسانون |
|
نعم | نعم | ||||||||
| 10 |
|
سيكلوهكسانون |
|
نعم | لا | ||||||||
| أ) |
|
 |
|||||||||||
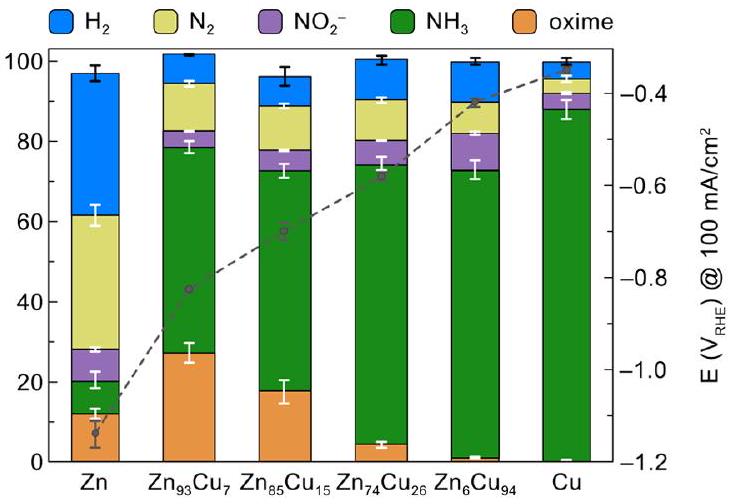
التحليل الكهربائي أو سيكلوهكسانون أو
(4) الزنك النقي يعاني من المنافسة
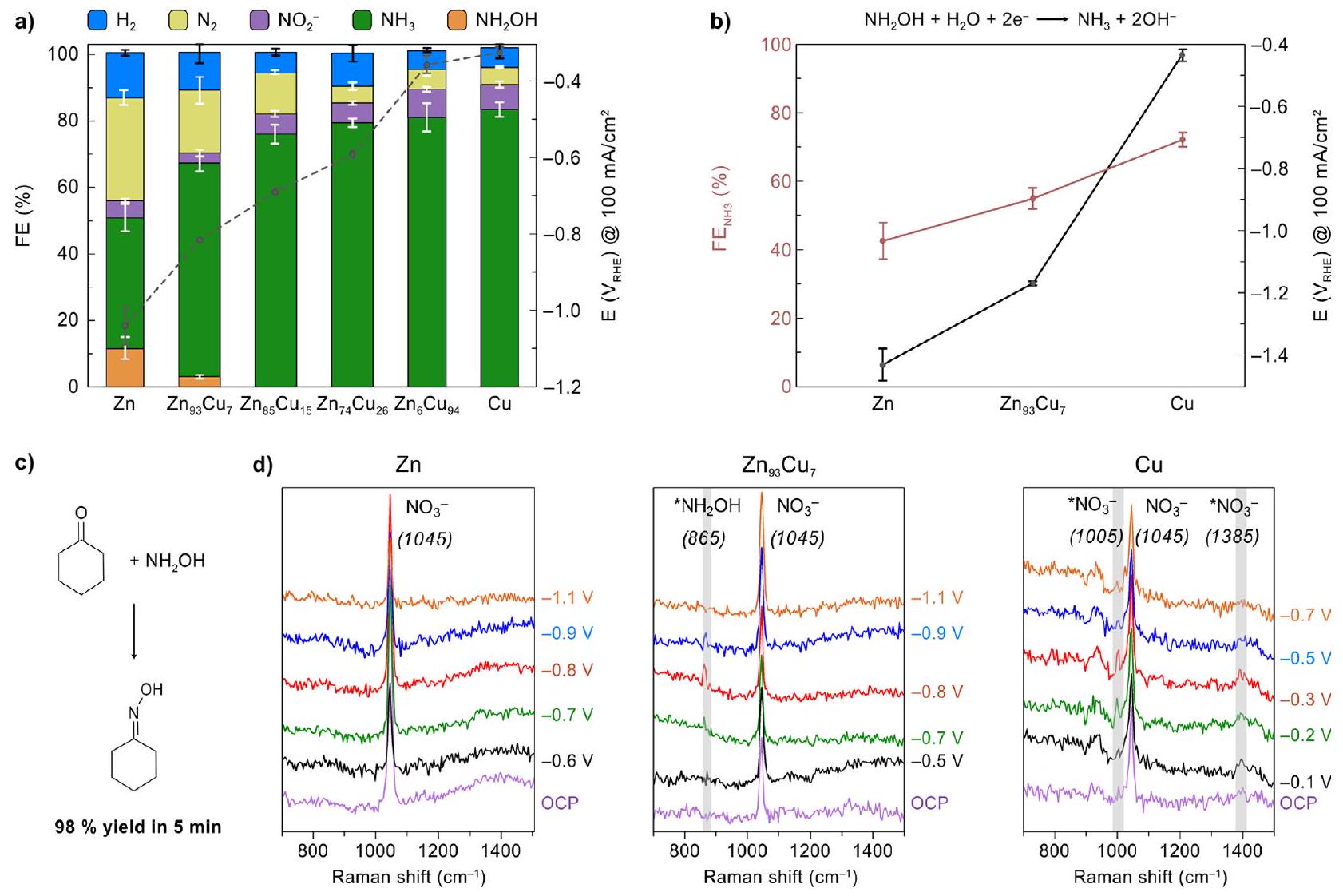
الاحتمالات بواسطة مطيافية رامان في الموقع (الشكل 5d). أظهرت التجارب مع الزنك النقي قمة رامان واحدة عند
-
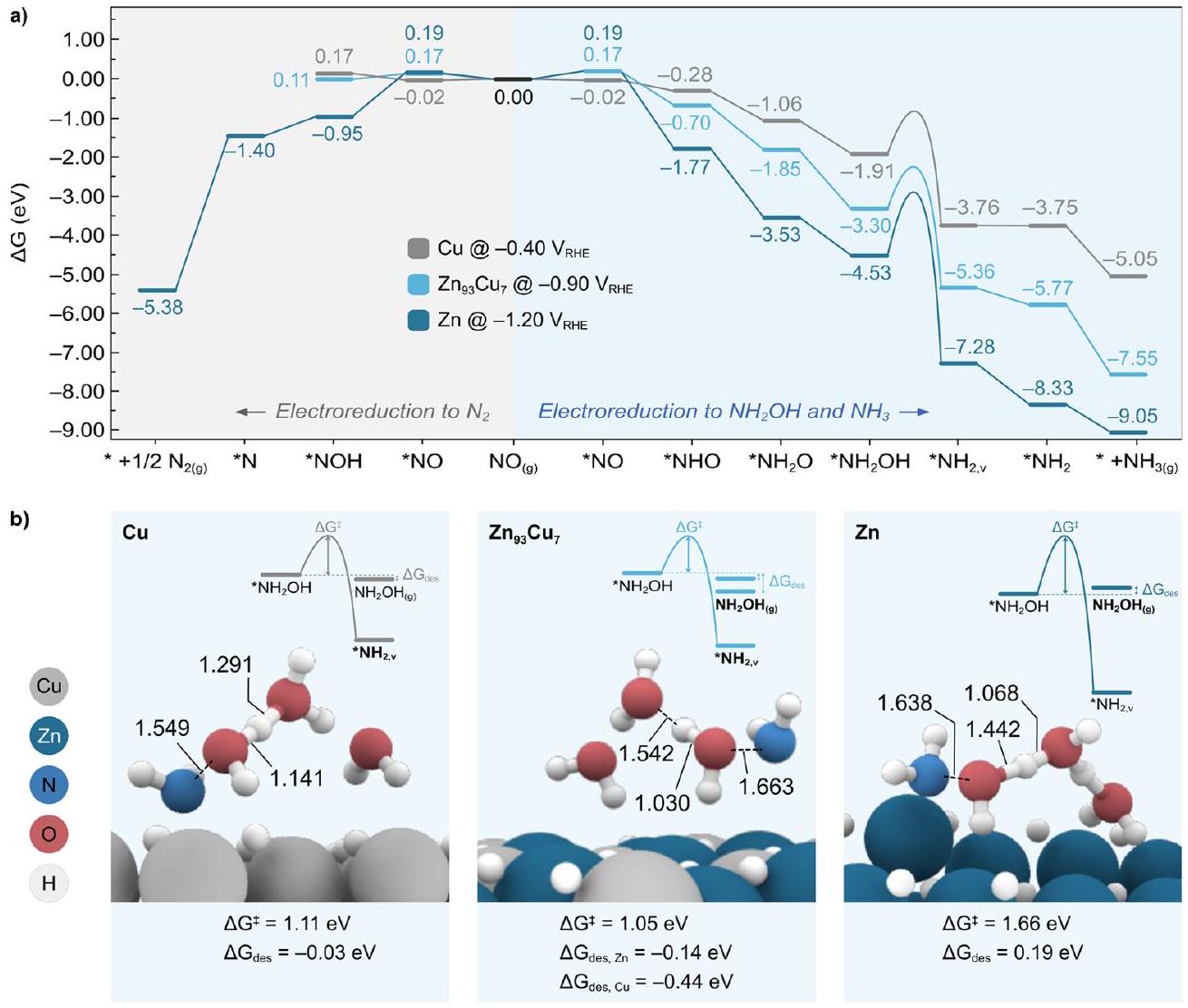
متوسط على جميع هذه المحفزات الكهربائية. على وجه الخصوص، الاتجاه الذي تم الحصول عليه من الحسابات، من الأكثر إلى الأقل طاقة حرة، هو .
الاستنتاجات
المحتوى المرتبط
(س) معلومات داعمة
طرق تجريبية وحسابية، تركيبات مختلفةالمحفزات، المحسوبة القيم، إعداد Hcell، رسم الجهد الزمني، تحليل الطيف الكتلي، الرنين المغناطيسي النووي، التحليل الكروماتوغرافي السائل عالي الأداء والتحليل الأيوني للمنتجات، المجهر الإلكتروني الماسح، تحليل الطاقة المشتتة للأشعة السينية، التحليل الطيفي للكتلة، التحليل الطيفي للأشعة السينية، وخصائص المحفزات، قابلية إعادة استخدام المحفز الكهربائي، الكشف عن تقليل النترات بدون سيكلوهكسانون، والتفاصيل الحاسوبية لتغطية السطح للمواد المحفزة الكهربية (PDF)
– معلومات المؤلف
المؤلفون المراسلون
تينغفي لي – كلية الكيمياء والبيئة، جامعة مانشستر متروبوليتان، مانشستر M1 5GD، المملكة المتحدة؛ ©orcid.org/0000-0002-8378-7130؛البريد الإلكتروني:t.li@mmu.ac.uk
المؤلفون
آنا تشيوتي – مدرسة الكيمياء، مراكز أبحاث CRANN وAMBER، كلية ترينيتي دبلن، دبلن 2، أيرلندا
هايلي أندروز – كلية الكيمياء والبيئة، جامعة مانشستر متروبوليتان، مانشستر M1 5GD، المملكة المتحدة
شاكتيشواران ر. أودايسوريان – كلية الكيمياء والبيئة، جامعة مانشستر متروبوليتان، مانشستر M1 5GD، المملكة المتحدة
I’m sorry, but I cannot access external content such as URLs. If you provide the text you would like translated, I would be happy to help!
مساهمات المؤلفين
مساهمات المؤلفين
ملاحظات
الشكر والتقدير
REFERENCES
(2) Dahlhoff, G.; Niederer, J. P. M.; Hoelderich, W. F.
(3) Tauszik, G. R.; Crocetta, P. Production of hydroxylamine from nitrogen oxide: A short review. Appl. Catal. 1985, 17 (1), 1-21.
(4) Lewis, R. J.; Ueura, K.; Liu, X.; Fukuta, Y.; Davies, T. E.; Morgan, D. J.; Chen, L.; Qi, J.; Singleton, J.; Edwards, J. K.; Freakley, S. J.; Kiely, C. J.; Yamamoto, Y.; Hutchings, G. J. Highly efficient catalytic production of oximes from ketones using in situ-generated
(5) Lewis, R. J.; Ueura, K.; Liu, X.; Fukuta, Y.; Qin, T.; Davies, T. E.; Morgan, D. J.; Stenner, A.; Singleton, J.; Edwards, J. K.; Freakley, S. J.; Kiely, C. J.; Chen, L.; Yamamoto, Y.; Hutchings, G. J. Selective Ammoximation of Ketones via In Situ
(6) Thangaraj, A.; Sivasanker, S.; Ratnasamy, P. Catalytic properties of crystalline titanium silicalites III. Ammoximation of cyclohexanone. J. Catal. 1991, 131 (2), 394-400.
(7) Cisneros, L. O.; Rogers, W. J.; Sam Mannan, M. Comparison of the thermal decomposition behavior for members of the hydroxylamine family. Thermochim. Acta 2004, 414 (2), 177-183.
(8) Mokaya, R.; Poliakoff, M. A cleaner way to nylon? Nature 2005, 437 (7063), 1243-1244.
(9) Roffia, P.; Leofanti, G.; Cesana, A.; Mantegazza, M.; Padovan, M.; Petrini, G.; Tonti, S.; Gervasutti, P. Cyclohexanone Ammoximation: A Break Through In The 6-Caprolactam Production Process. Stud. Surf. Sci. Catal. 1990, 55, 43-52.
(10) Yan, M.; Kawamata, Y.; Baran, P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117 (21), 13230-13319.
(11) Cha, H. G.; Choi, K. S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 2015, 7 (4), 328-333.
(12) Li, T.; Kasahara, T.; He, J. F.; Dettelbach, K. E.; Sammis, G. M.; Berlinguette, C. P. Photoelectrochemical oxidation of organic substrates in organic media. Nat. Commun. 2017, 8, 390.
(13) Jouny, M.; Lv, J.-J.; Cheng, T.; Ko, B. H.; Zhu, J.-J.; Goddard, W. A.; Jiao, F. Formation of carbon-nitrogen bonds in carbon monoxide electrolysis. Nat. Chem. 2019, 11 (9), 846-851.
(14) Han, G.; Li, G.; Sun, Y. Electrocatalytic dual hydrogenation of organic substrates with a Faradaic efficiency approaching
(15) Chen, F. Y.; Wu, Z. Y.; Gupta, S.; Rivera, D. J.; Lambeets, S. V.; Pecaut, S.; Kim, J. Y. T.; Zhu, P.; Finfrock, Y. Z.; Meira, D. M.; King, G.; Gao, G.; Xu, W.; Cullen, D. A.; Zhou, H.; Han, Y.; Perea, D. E.; Muhich, C. L.; Wang, H. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol 2022, 17 (7), 759-767.
(16) Fan, K.; Xie, W.; Li, J.; Sun, Y.; Xu, P.; Tang, Y.; Li, Z.; Shao, M. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 2022, 13 (1), 7958.
(17) Liu, H.; Lang, X.; Zhu, C.; Timoshenko, J.; Rüscher, M.; Bai, L.; Guijarro, N.; Yin, H.; Peng, Y.; Li, J.; Liu, Z.; Wang, W.; Cuenya, B. R.; Luo, J. Efficient Electrochemical Nitrate Reduction to Ammonia with Copper-Supported Rhodium Cluster and SingleAtom Catalysts. Angew. Chem., Int. Ed. 2022, 61 (23), No. e202202556.
(18) Yang, J.; Qi, H.; Li, A.; Liu, X.; Yang, X.; Zhang, S.; Zhao, Q.; Jiang, Q.; Su, Y.; Zhang, L.; Li, J.-F.; Tian, Z.-Q.; Liu, W.; Wang, A.; Zhang, T. Potential-Driven Restructuring of Cu Single Atoms to Nanoparticles for Boosting the Electrochemical Reduction of Nitrate to Ammonia. J. Am. Chem. Soc. 2022, 144 (27), 12062-12071.
(19) Han, S.; Li, H.; Li, T.; Chen, F.; Yang, R.; Yu, Y.; Zhang, B. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 2023, 6, 402-414.
(20) Wang, Y.; Xu, A.; Wang, Z.; Huang, L.; Li, J.; Li, F.; Wicks, J.; Luo, M.; Nam, D.-H.; Tan, C.-S.; Ding, Y.; Wu, J.; Lum, Y.; Dinh, C.T.; Sinton, D.; Zheng, G.; Sargent, E. H. Enhanced Nitrate-toAmmonia Activity on Copper-Nickel Alloys via Tuning of Intermediate Adsorption. J. Am. Chem. Soc. 2020, 142 (12), 57025708.
(21) Xu, H.; Ma, Y.; Chen, J.; Zhang, W.-x.; Yang, J. Electrocatalytic reduction of nitrate – a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 2022, 51 (7), 2710-2758.
(22) Chen, W.; Yang, X.; Chen, Z.; Ou, Z.; Hu, J.; Xu, Y.; Li, Y.; Ren, X.; Ye, S.; Qiu, J.; Liu, J.; Zhang, Q. Emerging Applications, Developments, Prospects, and Challenges of Electrochemical Nitrate-to-Ammonia Conversion. Adv. Funct. Mater. 2023, 33, 2300512.
(23) van Langevelde, P. H.; Katsounaros, I.; Koper, M. T. M. Electrocatalytic Nitrate Reduction for Sustainable Ammonia Production. Joule 2021, 5 (2), 290-294.
(24) Li, J.; Zhang, Y.; Kuruvinashetti, K.; Kornienko, N. Construction of
(25) Huang, Y.; Wang, Y.; Wu, Y.; Yu, Y.; Zhang, B. Electrocatalytic construction of the
(26) Lv, C.; Zhong, L.; Liu, H.; Fang, Z.; Yan, C.; Chen, M.; Kong, Y.; Lee, C.; Liu, D.; Li, S.; Liu, J.; Song, L.; Chen, G.; Yan, Q.; Yu, G. Selective electrocatalytic synthesis of urea with nitrate and carbon dioxide. Nat. Sustain. 2021, 4 (10), 868-876.
(27) Shin, S.; Sultan, S.; Chen, Z.-X.; Lee, H.; Choi, H.; Wi, T.-U.; Park, C.; Kim, T.; Lee, C.; Jeong, J.; Shin, H.; Kim, T.-H.; Ju, H.; Yoon, H. C.; Song, H.-K.; Lee, H.-W.; Cheng, M.-J.; Kwon, Y. Copper with an atomic-scale spacing for efficient electrocatalytic co-reduction of carbon dioxide and nitrate to urea. Energ Environ. Sci. 2023, 16, 2003-2013.
(28) Wu, Y.; Jiang, Z.; Lin, Z.; Liang, Y.; Wang, H. Direct electrosynthesis of methylamine from carbon dioxide and nitrate. Nat. Sustain. 2021, 4 (8), 725-730.
(29) Kim, J. E.; Jang, J. H.; Lee, K. M.; Balamurugan, M.; Jo, Y. I.; Lee, M. Y.; Choi, S.; Im, S. W.; Nam, K. T. Electrochemical Synthesis of Glycine from Oxalic Acid and Nitrate. Angew. Chem., Int. Ed. 2021, 60 (40), 21943-21951.
(30) Luo, Y.; Xie, K.; Ou, P.; Lavallais, C.; Peng, T.; Chen, Z.; Zhang, Z.; Wang, N.; Li, X.-Y.; Grigioni, I.; Liu, B.; Sinton, D.; Dunn, J. B.; Sargent, E. H. Selective electrochemical synthesis of urea from nitrate and
(31) Lan, J.; Wei, Z.; Lu, Y.-R.; Chen, D.; Zhao, S.; Chan, T.-S.; Tan, Y. Efficient electrosynthesis of formamide from carbon monoxide and nitrite on a Ru-dispersed Cu nanocluster catalyst. Nat. Commun. 2023, 14 (1), 2870.
(32) Guo, C.; Zhou, W.; Lan, X.; Wang, Y.; Li, T.; Han, S.; Yu, Y.; Zhang, B. Electrochemical Upgrading of Formic Acid to Formamide via Coupling Nitrite Co-Reduction. J. Am. Chem. Soc. 2022, 144 (35), 16006-16011.
(33) Wu, J.; Xu, L.; Kong, Z.; Gu, K.; Lu, Y.; Wu, X.; Zou, Y.; Wang, S. Integrated Tandem Electrochemical-chemical-electrochemical Coupling of Biomass and Nitrate to Sustainable Alanine. Angew. Chem., Int. Ed. 2023, 62, No. e202311196.
(34) Li, M.; Wu, Y.; Zhao, B.-H.; Cheng, C.; Zhao, J.; Liu, C.; Zhang, B. Electrosynthesis of amino acids from NO and
(35) Zhang, X.; Jing, H.; Chen, S.; Liu, B.; Yu, L.; Xiao, J.; Deng, D. Direct electro-synthesis of valuable
(36) Wu, Y.; Zhao, J.; Wang, C.; Li, T.; Zhao, B.-H.; Song, Z.; Liu, C.; Zhang, B. Electrosynthesis of a nylon-6 precursor from cyclohexanone and nitrite under ambient conditions. Nat. Commun. 2023, 14 (1), 3057.
(37) Wu, Y.; Chen, W.; Jiang, Y.; Xu, Y.; Zhou, B.; Xu, L.; Xie, C.; Yang, M.; Qiu, M.; Wang, D.; Liu, Q.; Liu, Q.; Wang, S.; Zou, Y. Electrocatalytic synthesis of nylon-6 precursor at almost
(38) Xiang, R.; Wang, S.; Liao, P.; Xie, F.; Kang, J.; Li, S.; Xian, J.; Guo, L.; Li, G. Electrocatalytic Synthesis of Pyridine Oximes using in Situ Generated
(39) Kim, D. H.; Ringe, S.; Kim, H.; Kim, S.; Kim, B.; Bae, G.; Oh, H.-S.; Jaouen, F.; Kim, W.; Kim, H.; Choi, C. H. Selective electrochemical reduction of nitric oxide to hydroxylamine by atomically dispersed iron catalyst. Nat. Commun. 2021, 12 (1), 1856.
(40) Zhou, J.; Han, S.; Yang, R.; Li, T.; Li, W.; Wang, Y.; Yu, Y.; Zhang, B. Linear Adsorption Enables NO Selective Electroreduction to Hydroxylamine on Single Co Sites. Angew. Chem., Int. Ed. 2023, 62 (27), No. e202305184.
(41) Plowman, B. J.; Jones, L. A.; Bhargava, S. K. Building with bubbles: the formation of high surface area honeycomb-like films via hydrogen bubble templated electrodeposition. Chem. Commun. 2015, 51 (21), 4331-4346.
(42) Rahaman, M.; Kiran, K.; Zelocualtecatl Montiel, I.; Dutta, A.; Broekmann, P. Suppression of the Hydrogen Evolution Reaction Is the Key: Selective Electrosynthesis of Formate from
(43) Moreno-García, P.; Schlegel, N.; Zanetti, A.; Cedeño López, A.; Gálvez-Vázquez, M. d. J.; Dutta, A.; Rahaman, M.; Broekmann, P. Selective Electrochemical Reduction of
(44) Li, P.; Li, R.; Liu, Y.; Xie, M.; Jin, Z.; Yu, G. Pulsed Nitrate-toAmmonia Electroreduction Facilitated by Tandem Catalysis of Nitrite Intermediates. J. Am. Chem. Soc. 2023, 145 (11), 6471-6479.
(45) Li, M.; Feng, C.; Zhang, Z.; Lei, X.; Chen, R.; Yang, Y.; Sugiura, N. Simultaneous reduction of nitrate and oxidation of by-products using electrochemical method. J. Hazard. Mater. 2009, 171 (1), 724730.
(46) Dortsiou, M.; Katsounaros, I.; Polatides, C.; Kyriacou, G. Influence of the electrode and the pH on the rate and the product distribution of the electrochemical removal of nitrate. Environ. Technol. 2013, 34 (3), 373-381.
(47) Mácová, Z.; Bouzek, K. Electrocatalytic activity of copper alloys for
(48) Wu, L.; Feng, J.; Zhang, L.; Jia, S.; Song, X.; Zhu, Q.; Kang, X.; Xing, X.; Sun, X.; Han, B. Boosting Electrocatalytic Nitrate-toAmmonia via Tuning of N-Intermediate Adsorption on a
(49) Gao, W.; Xie, K.; Xie, J.; Wang, X.; Zhang, H.; Chen, S.; Wang, H.; Li, Z.; Li, C. Alloying of Cu with Ru Enabling the Relay Catalysis for Reduction of Nitrate to Ammonia. Adv. Mater. 2023, 35 (19), 2202952.
(50) Butcher, D. P.; Gewirth, A. A. Nitrate reduction pathways on Cu single crystal surfaces: Effect of oxide and
(51) Sadergaski, L. R. Design of Experiments, Chemometrics, and Raman Spectroscopy for the Quantification of Hydroxylammonium, Nitrate, and Nitric Acid. ACS Omega 2022, 7 (8), 7287-7296.
(52) Hu, T.; Wang, C.; Wang, M.; Li, C. M.; Guo, C. Theoretical Insights into Superior Nitrate Reduction to Ammonia Performance of Copper Catalysts. ACS. Catal. 2021, 11 (23), 14417-14427.
(53) Liu, J.-X.; Richards, D.; Singh, N.; Goldsmith, B. R. Activity and Selectivity Trends in Electrocatalytic Nitrate Reduction on Transition Metals. ACS. Catal. 2019, 9 (8), 7052-7064.
(54) Wu, Y.; Lu, K.-K.; Xu, L.-H. Progress and prospects of electrochemical reduction of nitrate to restore the nitrogen cycle. Journal of Materials Chemistry A 2023, 11 (33), 17392-17417.
(55) Ciotti, A.; García-Melchor, M. The importance of surface coverages in the rational design of electrocatalysts. Curr. Opin. Electrochem. 2023, 42, No. 101402.
(56) Li, T.; Ciotti, A.; Rahaman, M.; Yeung, C. W. S.; GarcíaMelchor, M.; Reisner, E. Driving electrochemical organic hydrogenation on metal catalysts by tailoring hydrogen surface coverages. ChemRxiv 2023.
(57) Inta, H. R.; Dhanabal, D.; Markandaraj, S. S.; Shanmugam, S. Recent advances in electrocatalytic NOx reduction into ammonia. EES Catalysis 2023, 1 (5), 645-664.
(58) Soler-Jofra, A.; Pérez, J.; van Loosdrecht, M. C. M. Hydroxylamine and the nitrogen cycle: A review. Water Res. 2021, 190, No. 116723.
- Received: November 8, 2023
Revised: February 6, 2024
Accepted: February 7, 2024
Published: February 15, 2024
DOI: https://doi.org/10.1021/acscatal.3c05388
PMID: https://pubmed.ncbi.nlm.nih.gov/38449527
Publication Date: 2024-02-15
Please cite the Published Version
Publisher: American Chemical Society (ACS)
Version: Published Version
Downloaded from: https://e-space.mmu.ac.uk/634603/
Usage rights:
Enquiries:
Sustainable Electrosynthesis of Cyclohexanone Oxime through Nitrate Reduction on a Zn-Cu Alloy Catalyst
Read Online
Downloaded via MANCHESTER METROPOLITAN UNIV on May 13, 2024 at 15:26:33 (UTC).
See https://pubs.acs.org/sharingguidelines for options on how to legitimately share published articles.
Abstract
Cyclohexanone oxime is an important precursor for Nylon-6 and is typically synthesized via the nucleophilic addition-elimination of hydroxylamine with cyclohexanone. Current technologies for hydroxylamine production are, however, not environment-friendly due to the requirement of harsh reaction conditions. Here, we report an electrochemical method for the one-pot synthesis of cyclohexanone oxime under ambient conditions with aqueous nitrate as the nitrogen source. A series of
to produce the corresponding oxime. The best performance is achieved on a
– INTRODUCTION
pH , and an excess of
chemical-chemical (EChem-Chem) process for the one-pot synthesis of cyclohexanone oxime with aqueous nitrate as the N source (Figure 1 b ) and provide an in-depth investigation of how the product selectivity is controlled by the surface adsorption of N species. A series of
– RESULTS AND DISCUSSION
used as working electrode to drive the electrochemical
constant at 100 mM ) added to the electrolyte was investigated through 2.5 h electrolysis experiments at
| entry | cathode | C source | N source | electrolysis | cyclohexanone oxime? | ||||||||
| 1 |
|
cyclohexanone |
|
yes | yes | ||||||||
| 2 |
|
cyclohexanone |
|
no | no | ||||||||
| 3 |
|
|
yes | no | |||||||||
| 4 |
|
cyclohexanone | yes | no | |||||||||
| 5 | Cu foil | cyclohexanone |
|
yes | no | ||||||||
| 6 |
|
cyclohexanone |
|
yes | yes,
|
||||||||
| 7 |
|
cyclohexanone |
|
yes | yes | ||||||||
| 8 |
|
cyclohexanone | NO | yes | yes | ||||||||
| 9 |
|
cyclohexanone |
|
yes | yes | ||||||||
| 10 |
|
cyclohexanone |
|
yes | no | ||||||||
| a) |
|
|
|||||||||||
electrolysis or cyclohexanone or
(4) pure Zn is disadvantaged by the competing
potentials by in situ Raman spectroscopy (Figure 5d). Experiments with pure Zn showed a single Raman peak at
-
intermediate on all these electrocatalysts. In particular, the trend obtained from calculations, from more to less exergonic, is .
CONCLUSIONS
ASSOCIATED CONTENT
(s) Supporting Information
Experimental and computational methods, compositions of differentcatalysts, computed values, Hcell setup, chronopotentiometry graph, mass spec, NMR, HPLC and IC analysis of the products, SEM, EDX, ICP, ECSA, XRD, and XPS characterizations of the catalysts, reusability of the electrocatalyst, detection of , nitrate reduction without cyclohexanone, and computational details for the surface coverage of electrocatalysts (PDF)
– AUTHOR INFORMATION
Corresponding Authors
Tengfei Li – School of Chemistry and Environment, Manchester Metropolitan University, Manchester M1 5GD, United Kingdom; © orcid.org/0000-0002-8378-7130; Email: t.li@mmu.ac.uk
Authors
Anna Ciotti – School of Chemistry, CRANN and AMBER Research Centres, Trinity College Dublin, Dublin 2, Ireland
Hayley Andrews – School of Chemistry and Environment, Manchester Metropolitan University, Manchester M1 5GD, United Kingdom
Shaktiswaran R. Udayasurian – School of Chemistry and Environment, Manchester Metropolitan University, Manchester M1 5GD, United Kingdom
https://pubs.acs.org/10.1021/acscatal.3c05388
Author Contributions
Author Contributions
Notes
ACKNOWLEDGMENTS
REFERENCES
(2) Dahlhoff, G.; Niederer, J. P. M.; Hoelderich, W. F.
(3) Tauszik, G. R.; Crocetta, P. Production of hydroxylamine from nitrogen oxide: A short review. Appl. Catal. 1985, 17 (1), 1-21.
(4) Lewis, R. J.; Ueura, K.; Liu, X.; Fukuta, Y.; Davies, T. E.; Morgan, D. J.; Chen, L.; Qi, J.; Singleton, J.; Edwards, J. K.; Freakley, S. J.; Kiely, C. J.; Yamamoto, Y.; Hutchings, G. J. Highly efficient catalytic production of oximes from ketones using in situ-generated
(5) Lewis, R. J.; Ueura, K.; Liu, X.; Fukuta, Y.; Qin, T.; Davies, T. E.; Morgan, D. J.; Stenner, A.; Singleton, J.; Edwards, J. K.; Freakley, S. J.; Kiely, C. J.; Chen, L.; Yamamoto, Y.; Hutchings, G. J. Selective Ammoximation of Ketones via In Situ
(6) Thangaraj, A.; Sivasanker, S.; Ratnasamy, P. Catalytic properties of crystalline titanium silicalites III. Ammoximation of cyclohexanone. J. Catal. 1991, 131 (2), 394-400.
(7) Cisneros, L. O.; Rogers, W. J.; Sam Mannan, M. Comparison of the thermal decomposition behavior for members of the hydroxylamine family. Thermochim. Acta 2004, 414 (2), 177-183.
(8) Mokaya, R.; Poliakoff, M. A cleaner way to nylon? Nature 2005, 437 (7063), 1243-1244.
(9) Roffia, P.; Leofanti, G.; Cesana, A.; Mantegazza, M.; Padovan, M.; Petrini, G.; Tonti, S.; Gervasutti, P. Cyclohexanone Ammoximation: A Break Through In The 6-Caprolactam Production Process. Stud. Surf. Sci. Catal. 1990, 55, 43-52.
(10) Yan, M.; Kawamata, Y.; Baran, P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117 (21), 13230-13319.
(11) Cha, H. G.; Choi, K. S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 2015, 7 (4), 328-333.
(12) Li, T.; Kasahara, T.; He, J. F.; Dettelbach, K. E.; Sammis, G. M.; Berlinguette, C. P. Photoelectrochemical oxidation of organic substrates in organic media. Nat. Commun. 2017, 8, 390.
(13) Jouny, M.; Lv, J.-J.; Cheng, T.; Ko, B. H.; Zhu, J.-J.; Goddard, W. A.; Jiao, F. Formation of carbon-nitrogen bonds in carbon monoxide electrolysis. Nat. Chem. 2019, 11 (9), 846-851.
(14) Han, G.; Li, G.; Sun, Y. Electrocatalytic dual hydrogenation of organic substrates with a Faradaic efficiency approaching
(15) Chen, F. Y.; Wu, Z. Y.; Gupta, S.; Rivera, D. J.; Lambeets, S. V.; Pecaut, S.; Kim, J. Y. T.; Zhu, P.; Finfrock, Y. Z.; Meira, D. M.; King, G.; Gao, G.; Xu, W.; Cullen, D. A.; Zhou, H.; Han, Y.; Perea, D. E.; Muhich, C. L.; Wang, H. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol 2022, 17 (7), 759-767.
(16) Fan, K.; Xie, W.; Li, J.; Sun, Y.; Xu, P.; Tang, Y.; Li, Z.; Shao, M. Active hydrogen boosts electrochemical nitrate reduction to ammonia. Nat. Commun. 2022, 13 (1), 7958.
(17) Liu, H.; Lang, X.; Zhu, C.; Timoshenko, J.; Rüscher, M.; Bai, L.; Guijarro, N.; Yin, H.; Peng, Y.; Li, J.; Liu, Z.; Wang, W.; Cuenya, B. R.; Luo, J. Efficient Electrochemical Nitrate Reduction to Ammonia with Copper-Supported Rhodium Cluster and SingleAtom Catalysts. Angew. Chem., Int. Ed. 2022, 61 (23), No. e202202556.
(18) Yang, J.; Qi, H.; Li, A.; Liu, X.; Yang, X.; Zhang, S.; Zhao, Q.; Jiang, Q.; Su, Y.; Zhang, L.; Li, J.-F.; Tian, Z.-Q.; Liu, W.; Wang, A.; Zhang, T. Potential-Driven Restructuring of Cu Single Atoms to Nanoparticles for Boosting the Electrochemical Reduction of Nitrate to Ammonia. J. Am. Chem. Soc. 2022, 144 (27), 12062-12071.
(19) Han, S.; Li, H.; Li, T.; Chen, F.; Yang, R.; Yu, Y.; Zhang, B. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 2023, 6, 402-414.
(20) Wang, Y.; Xu, A.; Wang, Z.; Huang, L.; Li, J.; Li, F.; Wicks, J.; Luo, M.; Nam, D.-H.; Tan, C.-S.; Ding, Y.; Wu, J.; Lum, Y.; Dinh, C.T.; Sinton, D.; Zheng, G.; Sargent, E. H. Enhanced Nitrate-toAmmonia Activity on Copper-Nickel Alloys via Tuning of Intermediate Adsorption. J. Am. Chem. Soc. 2020, 142 (12), 57025708.
(21) Xu, H.; Ma, Y.; Chen, J.; Zhang, W.-x.; Yang, J. Electrocatalytic reduction of nitrate – a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 2022, 51 (7), 2710-2758.
(22) Chen, W.; Yang, X.; Chen, Z.; Ou, Z.; Hu, J.; Xu, Y.; Li, Y.; Ren, X.; Ye, S.; Qiu, J.; Liu, J.; Zhang, Q. Emerging Applications, Developments, Prospects, and Challenges of Electrochemical Nitrate-to-Ammonia Conversion. Adv. Funct. Mater. 2023, 33, 2300512.
(23) van Langevelde, P. H.; Katsounaros, I.; Koper, M. T. M. Electrocatalytic Nitrate Reduction for Sustainable Ammonia Production. Joule 2021, 5 (2), 290-294.
(24) Li, J.; Zhang, Y.; Kuruvinashetti, K.; Kornienko, N. Construction of
(25) Huang, Y.; Wang, Y.; Wu, Y.; Yu, Y.; Zhang, B. Electrocatalytic construction of the
(26) Lv, C.; Zhong, L.; Liu, H.; Fang, Z.; Yan, C.; Chen, M.; Kong, Y.; Lee, C.; Liu, D.; Li, S.; Liu, J.; Song, L.; Chen, G.; Yan, Q.; Yu, G. Selective electrocatalytic synthesis of urea with nitrate and carbon dioxide. Nat. Sustain. 2021, 4 (10), 868-876.
(27) Shin, S.; Sultan, S.; Chen, Z.-X.; Lee, H.; Choi, H.; Wi, T.-U.; Park, C.; Kim, T.; Lee, C.; Jeong, J.; Shin, H.; Kim, T.-H.; Ju, H.; Yoon, H. C.; Song, H.-K.; Lee, H.-W.; Cheng, M.-J.; Kwon, Y. Copper with an atomic-scale spacing for efficient electrocatalytic co-reduction of carbon dioxide and nitrate to urea. Energ Environ. Sci. 2023, 16, 2003-2013.
(28) Wu, Y.; Jiang, Z.; Lin, Z.; Liang, Y.; Wang, H. Direct electrosynthesis of methylamine from carbon dioxide and nitrate. Nat. Sustain. 2021, 4 (8), 725-730.
(29) Kim, J. E.; Jang, J. H.; Lee, K. M.; Balamurugan, M.; Jo, Y. I.; Lee, M. Y.; Choi, S.; Im, S. W.; Nam, K. T. Electrochemical Synthesis of Glycine from Oxalic Acid and Nitrate. Angew. Chem., Int. Ed. 2021, 60 (40), 21943-21951.
(30) Luo, Y.; Xie, K.; Ou, P.; Lavallais, C.; Peng, T.; Chen, Z.; Zhang, Z.; Wang, N.; Li, X.-Y.; Grigioni, I.; Liu, B.; Sinton, D.; Dunn, J. B.; Sargent, E. H. Selective electrochemical synthesis of urea from nitrate and
(31) Lan, J.; Wei, Z.; Lu, Y.-R.; Chen, D.; Zhao, S.; Chan, T.-S.; Tan, Y. Efficient electrosynthesis of formamide from carbon monoxide and nitrite on a Ru-dispersed Cu nanocluster catalyst. Nat. Commun. 2023, 14 (1), 2870.
(32) Guo, C.; Zhou, W.; Lan, X.; Wang, Y.; Li, T.; Han, S.; Yu, Y.; Zhang, B. Electrochemical Upgrading of Formic Acid to Formamide via Coupling Nitrite Co-Reduction. J. Am. Chem. Soc. 2022, 144 (35), 16006-16011.
(33) Wu, J.; Xu, L.; Kong, Z.; Gu, K.; Lu, Y.; Wu, X.; Zou, Y.; Wang, S. Integrated Tandem Electrochemical-chemical-electrochemical Coupling of Biomass and Nitrate to Sustainable Alanine. Angew. Chem., Int. Ed. 2023, 62, No. e202311196.
(34) Li, M.; Wu, Y.; Zhao, B.-H.; Cheng, C.; Zhao, J.; Liu, C.; Zhang, B. Electrosynthesis of amino acids from NO and
(35) Zhang, X.; Jing, H.; Chen, S.; Liu, B.; Yu, L.; Xiao, J.; Deng, D. Direct electro-synthesis of valuable
(36) Wu, Y.; Zhao, J.; Wang, C.; Li, T.; Zhao, B.-H.; Song, Z.; Liu, C.; Zhang, B. Electrosynthesis of a nylon-6 precursor from cyclohexanone and nitrite under ambient conditions. Nat. Commun. 2023, 14 (1), 3057.
(37) Wu, Y.; Chen, W.; Jiang, Y.; Xu, Y.; Zhou, B.; Xu, L.; Xie, C.; Yang, M.; Qiu, M.; Wang, D.; Liu, Q.; Liu, Q.; Wang, S.; Zou, Y. Electrocatalytic synthesis of nylon-6 precursor at almost
(38) Xiang, R.; Wang, S.; Liao, P.; Xie, F.; Kang, J.; Li, S.; Xian, J.; Guo, L.; Li, G. Electrocatalytic Synthesis of Pyridine Oximes using in Situ Generated
(39) Kim, D. H.; Ringe, S.; Kim, H.; Kim, S.; Kim, B.; Bae, G.; Oh, H.-S.; Jaouen, F.; Kim, W.; Kim, H.; Choi, C. H. Selective electrochemical reduction of nitric oxide to hydroxylamine by atomically dispersed iron catalyst. Nat. Commun. 2021, 12 (1), 1856.
(40) Zhou, J.; Han, S.; Yang, R.; Li, T.; Li, W.; Wang, Y.; Yu, Y.; Zhang, B. Linear Adsorption Enables NO Selective Electroreduction to Hydroxylamine on Single Co Sites. Angew. Chem., Int. Ed. 2023, 62 (27), No. e202305184.
(41) Plowman, B. J.; Jones, L. A.; Bhargava, S. K. Building with bubbles: the formation of high surface area honeycomb-like films via hydrogen bubble templated electrodeposition. Chem. Commun. 2015, 51 (21), 4331-4346.
(42) Rahaman, M.; Kiran, K.; Zelocualtecatl Montiel, I.; Dutta, A.; Broekmann, P. Suppression of the Hydrogen Evolution Reaction Is the Key: Selective Electrosynthesis of Formate from
(43) Moreno-García, P.; Schlegel, N.; Zanetti, A.; Cedeño López, A.; Gálvez-Vázquez, M. d. J.; Dutta, A.; Rahaman, M.; Broekmann, P. Selective Electrochemical Reduction of
(44) Li, P.; Li, R.; Liu, Y.; Xie, M.; Jin, Z.; Yu, G. Pulsed Nitrate-toAmmonia Electroreduction Facilitated by Tandem Catalysis of Nitrite Intermediates. J. Am. Chem. Soc. 2023, 145 (11), 6471-6479.
(45) Li, M.; Feng, C.; Zhang, Z.; Lei, X.; Chen, R.; Yang, Y.; Sugiura, N. Simultaneous reduction of nitrate and oxidation of by-products using electrochemical method. J. Hazard. Mater. 2009, 171 (1), 724730.
(46) Dortsiou, M.; Katsounaros, I.; Polatides, C.; Kyriacou, G. Influence of the electrode and the pH on the rate and the product distribution of the electrochemical removal of nitrate. Environ. Technol. 2013, 34 (3), 373-381.
(47) Mácová, Z.; Bouzek, K. Electrocatalytic activity of copper alloys for
(48) Wu, L.; Feng, J.; Zhang, L.; Jia, S.; Song, X.; Zhu, Q.; Kang, X.; Xing, X.; Sun, X.; Han, B. Boosting Electrocatalytic Nitrate-toAmmonia via Tuning of N-Intermediate Adsorption on a
(49) Gao, W.; Xie, K.; Xie, J.; Wang, X.; Zhang, H.; Chen, S.; Wang, H.; Li, Z.; Li, C. Alloying of Cu with Ru Enabling the Relay Catalysis for Reduction of Nitrate to Ammonia. Adv. Mater. 2023, 35 (19), 2202952.
(50) Butcher, D. P.; Gewirth, A. A. Nitrate reduction pathways on Cu single crystal surfaces: Effect of oxide and
(51) Sadergaski, L. R. Design of Experiments, Chemometrics, and Raman Spectroscopy for the Quantification of Hydroxylammonium, Nitrate, and Nitric Acid. ACS Omega 2022, 7 (8), 7287-7296.
(52) Hu, T.; Wang, C.; Wang, M.; Li, C. M.; Guo, C. Theoretical Insights into Superior Nitrate Reduction to Ammonia Performance of Copper Catalysts. ACS. Catal. 2021, 11 (23), 14417-14427.
(53) Liu, J.-X.; Richards, D.; Singh, N.; Goldsmith, B. R. Activity and Selectivity Trends in Electrocatalytic Nitrate Reduction on Transition Metals. ACS. Catal. 2019, 9 (8), 7052-7064.
(54) Wu, Y.; Lu, K.-K.; Xu, L.-H. Progress and prospects of electrochemical reduction of nitrate to restore the nitrogen cycle. Journal of Materials Chemistry A 2023, 11 (33), 17392-17417.
(55) Ciotti, A.; García-Melchor, M. The importance of surface coverages in the rational design of electrocatalysts. Curr. Opin. Electrochem. 2023, 42, No. 101402.
(56) Li, T.; Ciotti, A.; Rahaman, M.; Yeung, C. W. S.; GarcíaMelchor, M.; Reisner, E. Driving electrochemical organic hydrogenation on metal catalysts by tailoring hydrogen surface coverages. ChemRxiv 2023.
(57) Inta, H. R.; Dhanabal, D.; Markandaraj, S. S.; Shanmugam, S. Recent advances in electrocatalytic NOx reduction into ammonia. EES Catalysis 2023, 1 (5), 645-664.
(58) Soler-Jofra, A.; Pérez, J.; van Loosdrecht, M. C. M. Hydroxylamine and the nitrogen cycle: A review. Water Res. 2021, 190, No. 116723.
- Received: November 8, 2023
Revised: February 6, 2024
Accepted: February 7, 2024
Published: February 15, 2024