في هذا البحث، نطور طريقة بسيطة، خالية من المعادن، وخالية من الأحماض/القلويات للتفاعل الكهروكيميائي الانتقائي C4-ديتيرation لمشتقات البيريدين بطريقة اقتصادية ومريحة.في درجة حرارة الغرفة. تتميز هذه الاستراتيجية بنهج فعال وصديق للبيئة مع انتقائية كيميائية وإقليمية عالية، مما يوفر مجموعة واسعة من المركبات D، مثل البيريدينات، الكوينولونات،الليغاندات والمركبات ذات الصلة البيولوجية. ومن الجدير بالذكر أن التجارب الميكانيكية ودراسات الفولتمترية الدورية (CV) تظهر أنيُعتبر يوديد 2-فينيلبيريدينيوم -بوتيل- مادة وسيطة حاسمة خلال التحول الكهروكيميائي، والذي يوفر طريقة عامة وفعالة لتدريج مشتقات البيريدين.
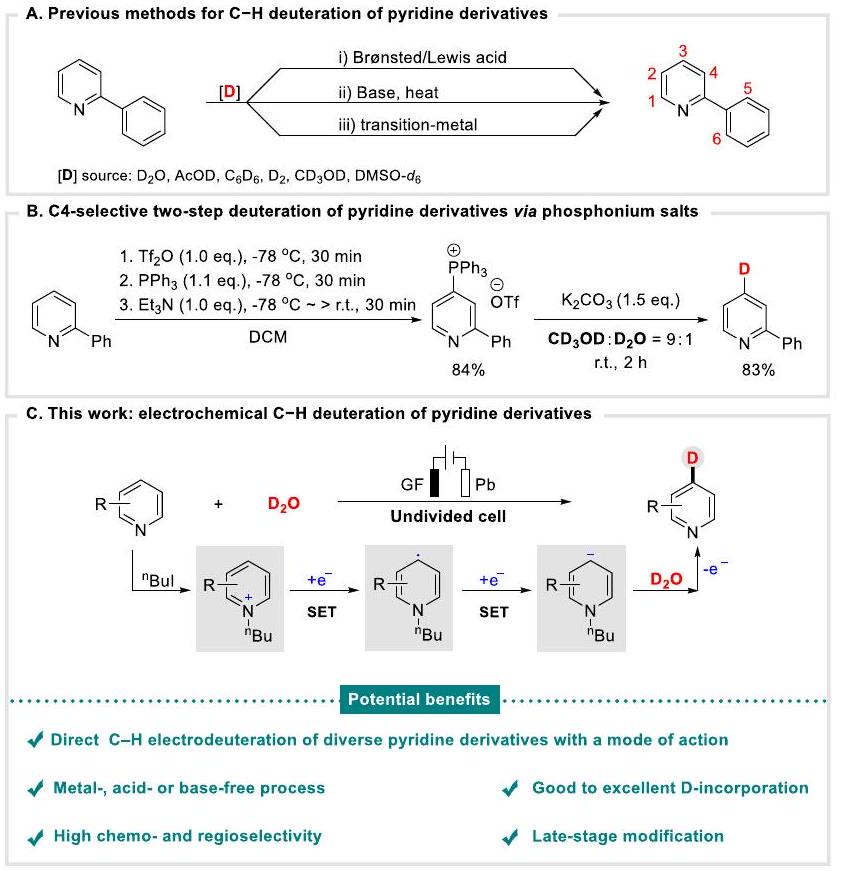
المركبات الموسومة بالديتيريوم هي نوع حاسم من المركبات العضوية، والتي تم تطبيقها على نطاق واسع في مجالات بحثية متنوعة، مثل توضيح آليات التفاعل.تقنيات تتبع النظائروكيمياء الأدوية. على سبيل المثال، -الهيدروأرينات، التي تُستخدم بشكل متكرر كجزيئات حيوية نشطة وأدوية لاستكشاف الخصائص الدوائية الحركية والدوائية الديناميكية (PK/PD)“. لذلك، ليس من المستغرب أن تكون عملية تخليق واستخدام المركبات الموسومة بالديتيريوم (لقد جذبت الهيدروكربونات العطرية (الهيدروكربونات غير المتجانسة) اهتمامًا كبيرًا، خاصة لمشتقات البيريدين المعلّمة بـ Dمن بين البروتوكولات المختلفة التي تم تطويرها، فإن استراتيجية تبادل الهيدروجين/الديوتيريوم المباشرة والبسيطة والفعالةيظهر قبل التثديت الدهالي للهليدات والهليدات الزائفةنظرًا لتوفر المواد الأولية بسهولة، وتكلفتها، بالإضافة إلى الاقتصاد العالي في الذرات، فإن التحدي سيكون في التحكم في الانتقائية للتحولات. على الرغم من أن ذلك قد تم تأسيسهتدريج الديوتيريوم لمشتقات البيريدين في وجود حمض برونستيد/لويسقاعدةأو المعادن الانتقالية (الشكل 1A)، لا يزال السعي نحو منهجيات ذات انتقائية أعلى وإدماج D تحت ظروف أكثر اعتدالًا مع إجراءات تشغيل سهلة مستمرًا. ومن الجدير بالذكر أن مكناي وزملاءه طوروا انتقائية C4-تدريج الديوتيريوم لمشتقات البيريدين من خلال عملية من خطوتين، حيث تمر عبر أملاح الفوسفونيوم الحلقية كوسيط، ثم تتشكل مشتقات البيريدين المديوتيرة النهائية بمساعدة قاعدة، مما يحقق ديوتيريوم انتقائي عالي لمشتقات البيريدين (الشكل 1B).. علاوة على ذلك، تطوير أساليب للاختيار الانتقائي للموقعتثبيت الديوتيريوم لمشتقات البيريدين بطريقة مباشرة ومستدامة وفعالة لا يزال أمرًا مرغوبًا فيه للغاية وتحديًا.
في السنوات الأخيرة، أدى ظهور الكيمياء الكهربائية إلى فرص وتطور في التخليق العضوي.تزداد شعبية التخليق الكهربائي ويعتبر واحدة من المنهجيات المستدامة والمرغوبة التي يمكن أن تحل محل بعض الطرق التقليدية للتخليق. نتخيل أن يكون هناك تخليق مباشر وانتقائييمكن تحقيق الديوتريشن لمشتقات البيريدين من خلال استخدام الكيمياء الكهربائية. ومع ذلك، مقارنةً بالتأكسد الكهربائي المتطور جيدًاتفعيلمدفوع كهربائياً بالتقليللم يتم توضيح وظيفة الأرينات بشكل جيد حتى الآن. خاصة، انتقائيلم يتحقق بعد تديتر مشتقات البيريدين تحت ظروف الاختزال الكهربائي.
في تناقض حاد، مع اهتماماتنا المستمرة في التحفيز الكهربائي المستدامتفعيلوالديتيرation الكهروكيميائيةنود هنا أن نبلغ عن جهودنا في تطوير عملية كيميائية كهربائية عامة ومباشرة وفعالة لاختيار C4.تثبيت الديوتيريوم لمشتقات البيريدين من خلال التنشيط الاختزالي بطريقة اقتصادية“عبر “حاسموسيط يوديد -بيوتيل-2-فينيل بيريدينيوم (الشكل 1C). قدمت هذه البروتوكولات مجموعة واسعة من مشتقات D-بيريدين مع انتقائية كيميائية وإقليمية عالية في عوائد ممتازة. تشمل الميزات البارزة لهذا التحول: (أ) التثبيط الكهربائي المدفوع لتفاعل C-H بالديتيريوم؛ (ب) دمج جيد إلى ممتاز للديتيريوم؛ (ج) عملية خالية من المعادن أو الأحماض أو القواعد؛ (د) انتقائية كيميائية وإقليمية عالية؛ (هـ) تعديل في المراحل المتأخرة من-الليغاندات والمركبات ذات الصلة الحيوية.
الشكل 1 | الخلفية والعمل الحالي للتدريج الديوتيري للمواد المشتقة من البيريدين. أ الطرق السابقة لتدريج C-H للمواد المشتقة من البيريدين. ب التدريج الديوتيري الانتقائي C4 على مرحلتين للمواد المشتقة من البيريدين عبر أملاح الفوسفونيم. ج هذا العمل: كهربائياًتثبيت الديوتيريوم لمشتقات البيريدين.
النتائج
تحسين ظروف التفاعل
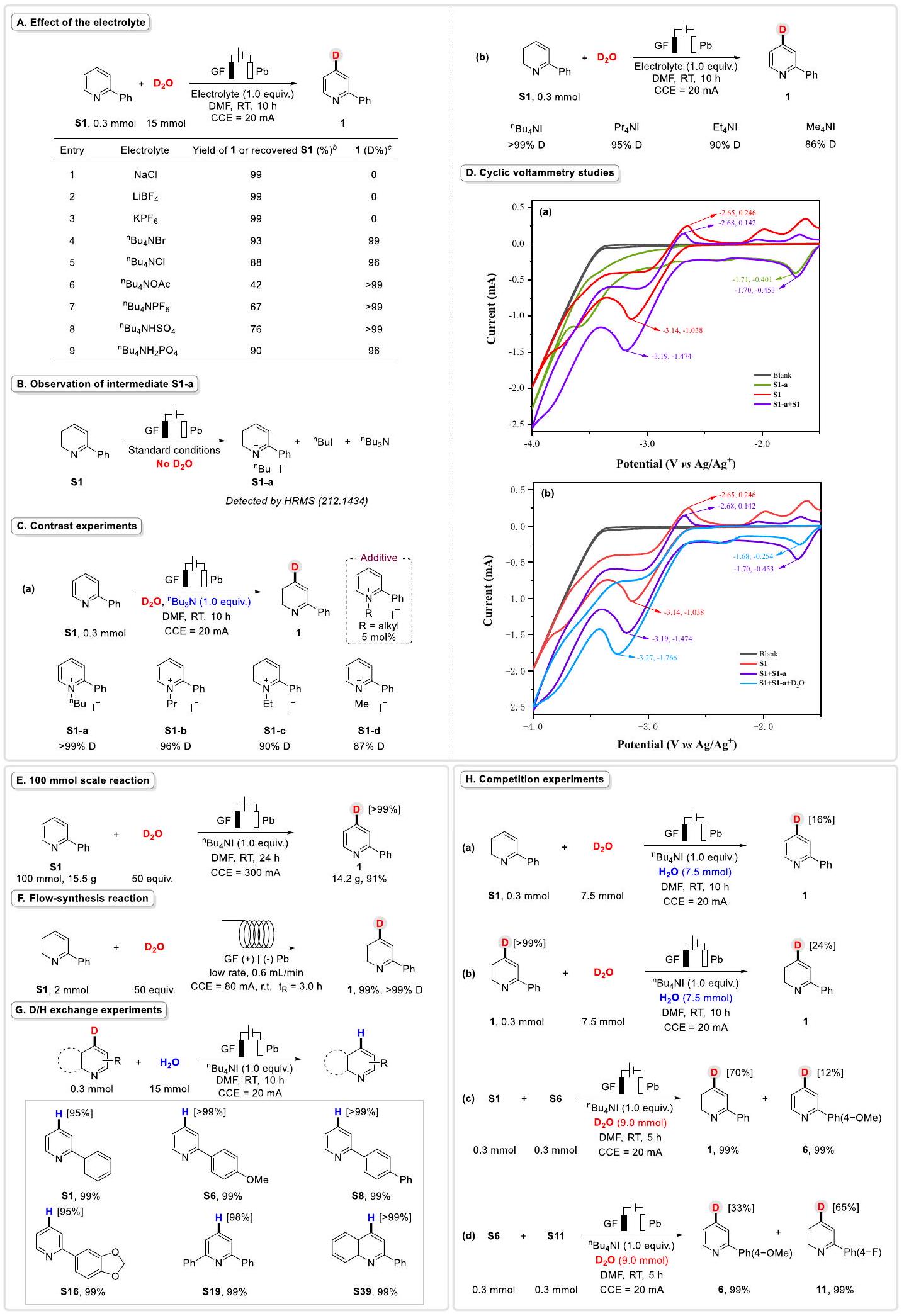
بدأت تحقيقاتنا بتقييم الـتثبيت الديوتيريوم لمواد البيريدين باستخدام أكسيد الديوتيريوم ). بعد بعض التجارب الأولية، تم اختيار 2-فينيل بيريدين (S1) كنموذج للركيزة الأساسية لتحسين ظروف التفاعل. ومن المشجع أنه تم ملاحظة منتج واحد فقط 1، مما أثبت أن التفاعل يتمتع بانتقائية مكانية عالية. بعد اختيار دقيق لمتغيرات النظام، حصلنا على الظروف المثلى للتديوتراة لـمعدمج المتفاعلات مع الإلكتروليتفي خلية غير مقسمة مع DMF تحت تيار ثابت (20 مللي أمبير) في درجة حرارة الغرفة لمدة 10 ساعات، أسفر عنعائد وإدماج الديوتيريوم (المختصر D-inc فيما بعد) (الجدول 1، المدخل 1). في البداية، تم دراسة مجموعة متنوعة من الإلكتروليتات. و عمل بشكل جيد وأسفر عن و انخفاض D للمنتج 1 على التوالي (الجدول 1، الإدخالات 2 و 3)، ولكن لم يتم الكشف عن أي منتج عندما أو تم استخدام NaI (الجدول 1، الإدخالات 4 و 5). ثم، أدى محاولة استخدام NaOAc كقاعدة إلى انخفاض في الديوتريشن (الجدول 1، الإدخال 6). كما قمنا بفحص مذيبات مختلفة، مثل DMA و MeCN، التي وفرت و تغير المنتج 1 على التوالي (الجدول 1، الإدخالات 7 و 8). بعد ذلك، قمنا بدراسة أقطاب كهربائية مختلفة، بما في ذلك، و (الجدول 1، الإدخالات 9-11). ومع ذلك، كانت D-inc . علاوة على ذلك، قمنا بالتحقيق في تأثير التيار على هذه التفاعل. مع تيار أقل، كانت الديوتريشن لـانخفض بشكل ملحوظ (الجدول 1، الإدخالات 12-13). كما عمل النظام بسلاسة عند درجات حرارة أعلى (الجدول 1، الإدخال 14،بالإضافة إلى ذلك، وجدنا أن هذا النظام كان غير حساس للجو وما زال يعطي عائدًا ممتازًا وD-inc تحت جو الأرجون (الجدول 1، المدخل 15). أخيرًا، أثبتت بعض التجارب الضابطة أن وجود الكهرباء والإلكتروليت كان ضروريًا لهذه التحويلة (الجدول 1، المدخلين 16 و17).
نطاق الركيزة
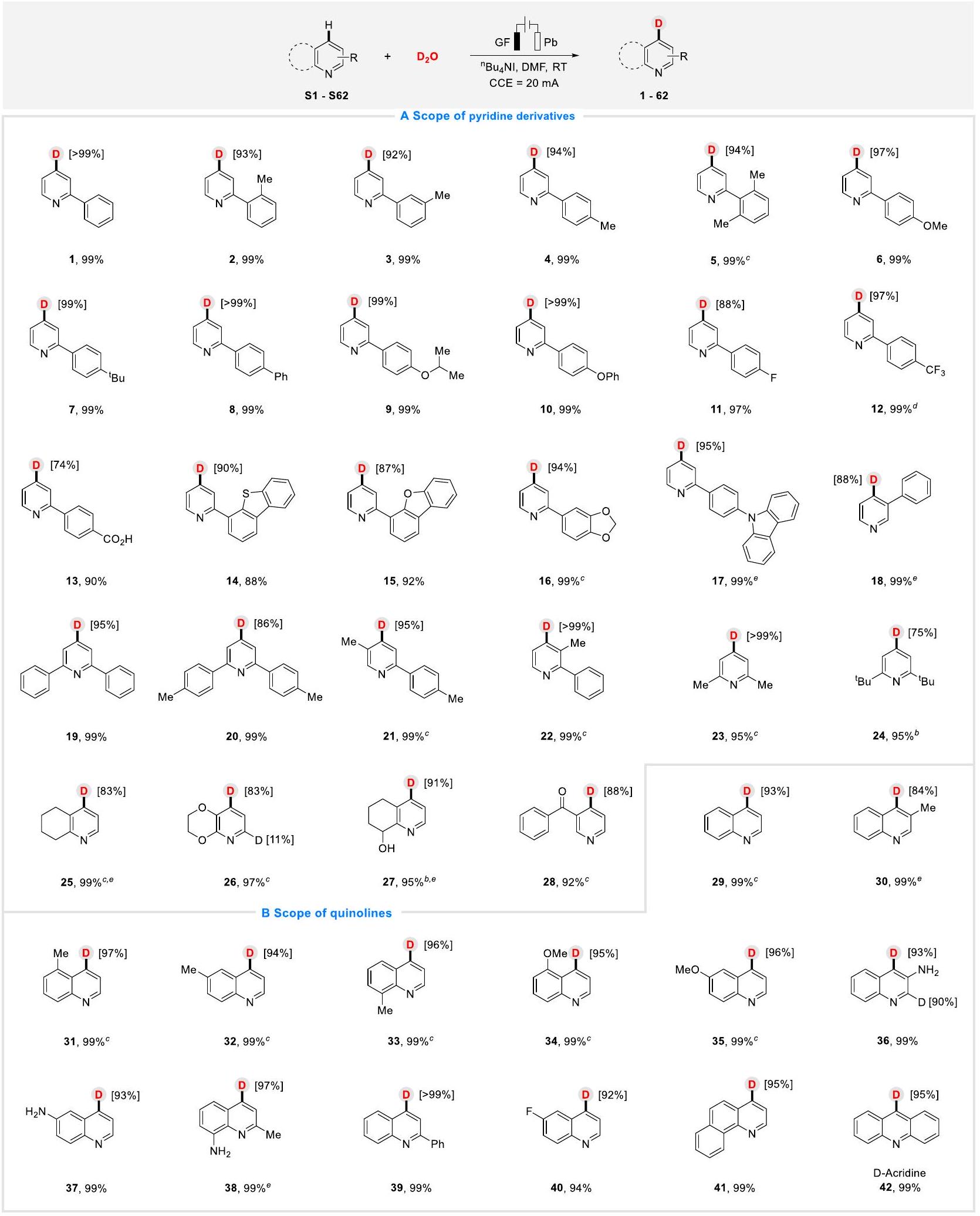
مع توفر الظروف المثلى، قمنا بعد ذلك بالتحقيق في نطاق الركيزة وعمومية هذه العملية الكهروكيميائية الفعالة.تحويل الديوتيريوم. كما هو موضح في الشكل 2A، مجموعة من الأريل/الألكيل- تم تقييم البيريدينات ذات المجموعات الفرعية المتنوعة. كان للتأثير الستيري للمجموعة الفرعية تأثير ضئيل على هذا التحول، على سبيل المثال، 2-فينيلبيريدينات التي تحمل، أو أدى substituent -methyl إلى الحصول على المنتجات المرغوبة بعوائد جيدة إلى ممتازة و D-inc، بالإضافة إلى ذلك الذي يحمل مجموعتين ميثيل (5). مجموعات أخرى، مثل، و تمت toleration بسلاسة. كانت الركائز التي تحمل الفلور (11) والمجموعة ثلاثية الفلورو ميثيل (12) ومجموعة الكربوكسيل (13) متوافقة وعملت بنجاح، مما أدى إلى الحصول على المنتجات المرغوبة بكفاءة عالية. علاوة على ذلك، كانت المنتجات الديوتيرية من الفينيل بيريدين مع أساسات الهترو أريل (تم الحصول أيضًا على مجموعات ( ) بعوائد ممتازة وD-inc (14-17). علاوة على ذلك، تم تحمل الركائز البديلة والمتعددة الاستبدال وتفاعلت بسلاسة في النظام، مما أدى إلى نتائج مرضية (18-22، حتى 99% عوائد، 86%->99% D-inc). بالإضافة إلى البيريدينات الأريلية، قمنا أيضًا بدراسة مشتقات البيريدين الألكيلية المتنوعة. يمكن أن تتفاعل مجموعة واسعة من الركائز مع مجموعات مانحة وإلكترونية مختلفة بشكل جيد معوإنتاج المنتجات الديوتيريوم المقابلة بنسبة 75% إلى أكثر من 99% من د-التركيز (23-28).
الكينولين هو فئة حيوية من الهتروسيكل التي توجد على نطاق واسع في المنتجات الطبيعية والأدوية والأصباغ والمواد.لذلك، حاولنا استكشاف نطاق الكوينولينات ذات المجموعات الوظيفية المتنوعة لإظهار قابلية تطبيق هذه البروتوكول بشكل أكبر (الشكل 2B). وجدنا أن الكوينولينات التي تحمل مجموعات وظيفية محايدة الإلكترون (29) ومانحة (30-39) وسالبة (40) أدت بشكل جيد في هذا التحول، مما أدى إلى الحصول على المنتجات المقابلة بعوائد جيدة (94%-99%) مع تميز ممتاز في D-inc (84%->99%) والانتقائية. بالإضافة إلى ذلك، تم أيضًا استخدام 7,8-بنزوكوينولين (41) والأكرين (42، اللقاحات ضد العدوى والحساسية) بنجاح في هذا البروتوكول، مما أسفر عن نتائج مرضية.
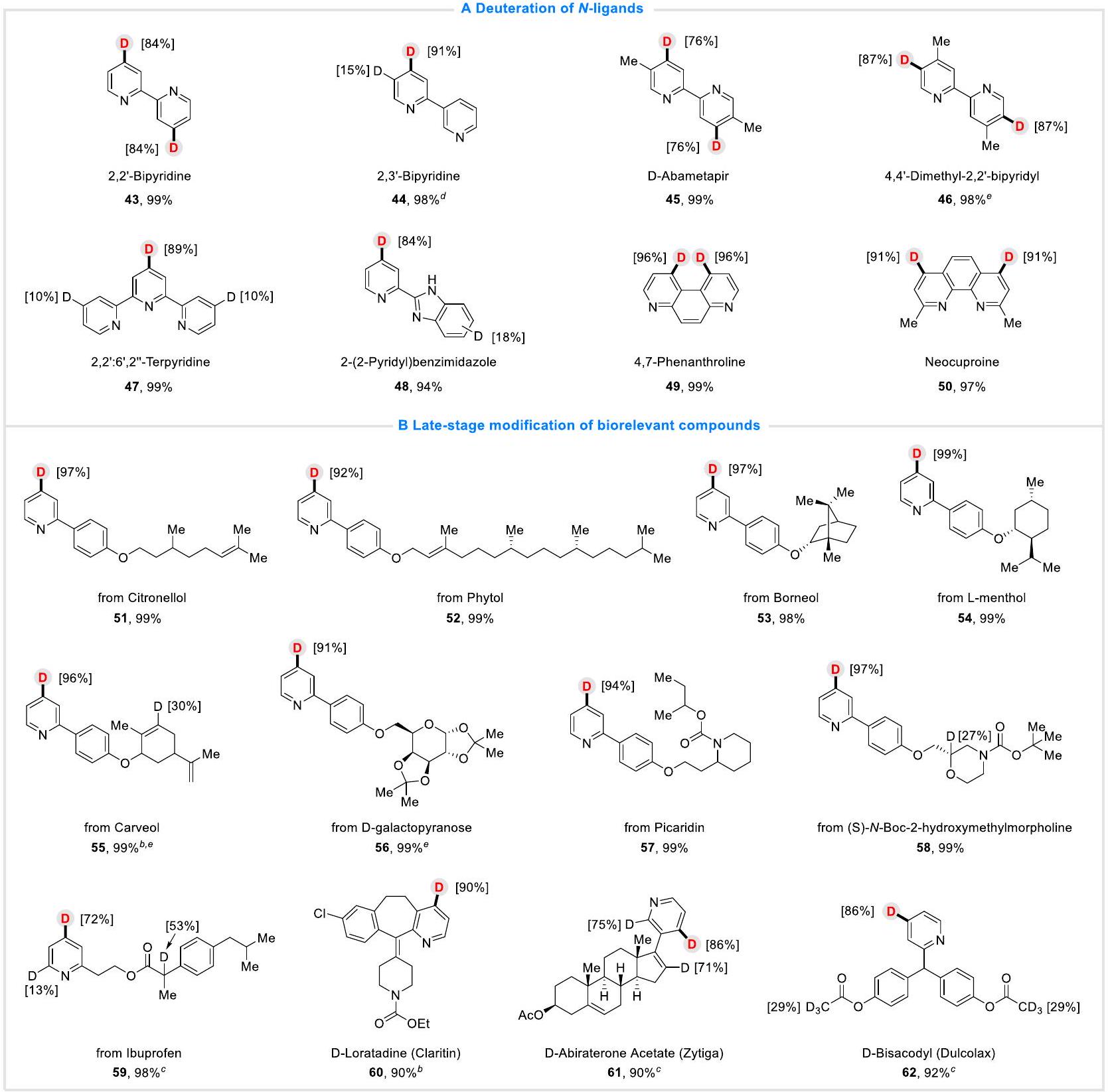
الأهم من ذلك، وجهنا انتباهنا إلى-الليغاندات (الشكل 3A)، تم استيعاب مجموعة متنوعة منها، مما أدى إلى الحصول على الجزيئات المرغوبة بكلا العائدين الجيدين وD-inc (43-50)، بما في ذلك البيبيريدين (43-47، 45، أباميتابير، وهو مبيد حشري لعدوى قمل الرأس)، البنزيميدازول (48) والفينانثرولين (49-50)، مما يدل على توافق كبير وقابلية عملية للبروتوكول. وقدمت طريقة جديدة للتعديل الوظيفي في المراحل المتأخرة من-الليغاندات.
استكشفنا بعد ذلك عملية الديوتريشن في المراحل المتأخرة للمركبات البيولوجية ذات الصلة وبعض الجزيئات الصيدلانية باستخدام هذه الطريقة الكهرواختزالية (الشكل 3B). ومن المفرح أن المركبات البيريدينية المشتقة من السيتروينول (51)، الفيتول (52)، البورنول (53)، ال-مينثول (54)، الكارفول (55)، الد-غالكتوبيرانووز (56)، البيكاريدين (57)، (S)-N-Boc-2-هيدروكسي ميثيل مرفولين (58)، الإيبوبروفين (59) وبعض الجزيئات الصيدلانية، بما في ذلك اللوراتادين (كلارينتين (دواء مضاد للحساسية)، أبراتيرون أسيتات (دواء زيديجا لسرطان البروستاتا) وبيساكوديل (دولكولاكس، ملين)، أظهرت جميعها توافقًا كبيرًا وتفاعلًا مع هذا النظام الكهربائي الاختزالي، مما أدى إلى الحصول على المنتجات الديوتيرية المرغوبة بكلا من العوائد الممتازة ونسبة D-inc%. تشير هذه النتائج إلى أن هذه البروتوكولات الكهروكيميائية لديها إمكانيات وآفاق كبيرة في تطبيق وتعديل المركبات ذات الصلة حيويًا.
الدراسات الميكانيكية
من أجل التحقق بشكل أكبر من منطقية هذا التحول، قمنا بإجراء سلسلة من التجارب للتحقيق في الآلية (الشكل 4). أولاً، استكشفنا تأثير الإلكتروليتات (الشكل 4A). كما هو متوقع، لم يتم إنتاج أي منتجتمت ملاحظته عندما أو تم استخدامه كالإلكتروليت (الشكل 4A، الإدخالات 1-3). ومع ذلك، عندما استخدمنا إلكتروليتات أخرى تحتوي علىأيون، د-إنك الممتازتم الحصول عليه (الشكل 4A، الإدخالات 4-9). في الوقت نفسه، أدت الأنيونات المختلفة من الأملاح إلى عوائد مختلفة (عائد منأو تم التعافي ). هذه النتائج توضح أن كان الأيون حاسمًا لهذه التفاعل الكهروكيميائي. علاوة على ذلك، تحت الظروف المحسّنة، تم الكشف عن يوديد N-butyl-2-phenylpyridinium (S1-a، الذي تم الكشف عنه بواسطة HRMS، 212.1434)، و تمت إتاحتها في غياب، مما أظهر بشكل أكبر الدور المهم لـ(الشكل 4B). علاوة على ذلك، افترضنا أن S1a قد تكون وسيطًا رئيسيًا في هذه العملية. لذلك، (1.0 مكافئ)
الجدول 1 | فحص ظروف التفاعل
S1
1
دخول
الانحراف عن الظروف القياسية
عائد 1 أو استرداد S1 (%)
1 (نسبة مئوية)
1
لا شيء
99
>99
2
بدلاً من
83
99
٣
بدلاً من
99
90
٤
بدلاً من
99
–
٥
NaI بدلاً من
99
–
٦
NaOAc (1.0 مكافئ)
99
16
٧
DMA كمذيب
99
82
٨
MeCN كمذيب
99
<5
9
CF (+)| (-) الرصاص
90
٨٠
10
GF (+)| (-) GF
99
٣٥
11
GF (+) | (-) Pt
99
53
12
10 مللي أمبير
99
60
١٣
15 مللي أمبير
99
81
14
99
99
15
عربى
99
99
16
بدون كهرباء
99
—
17
بدون إلكتروليت
99
—
التنسيق الغامق يُظهر أن الإدخال 1 هو ظروف التفاعل المثلى. فelt الكربون CF، DMF N، N-dيميثيلفورماميد، DMA N، N-dيميثيلأسيتاميد. DMF (4.0 مل)، درجة حرارة الغرفة، هواء، 10 ساعات. العائد المعزول. تم تحديد نسب دمج الديوتيريوم بواسطةطيف الرنين المغناطيسي النووي. بدلاً من، ثم كمية تحفيزية ( ) من تمت إضافتها إلى 2-فينيل بيريدين ) تحت الظروف القياسية، توفير المنتج مع، ) و ( ) على التوالي (الشكل 4C، أ). من ناحية أخرى، تم استخدام الإلكتروليتات المقابلة للتفاعل النموذجي تحت ظروف قياسية وأنتجت المركب مع ) ، ، و على انفراد (الشكل 4C، ب). كانت النتائج متطابقة تقريبًا مع النتائج السابقة. بعد ذلك، أجرينا عدة دراسات للجهد الدوري (CV) (للمزيد من التفاصيل، يرجى مراجعة المعلومات التكميلية في الصفحة 32). تجارب CV علىيوديد 2-فينيل-بروبان-1-يونيوم (S1-a) أعطى قمة اختزالية عند مقابل تحت جو الأرجون (الشكل 4D، أ، الخط الأخضر). قمة أكسدة عكسية واضحة عند مقابل وقمة اختزالية قابلة للعكس منفي مقابل تمت ملاحظتها تحت جو الأرجون (الشكل 4D، أ، الخط الأحمر). بعد خلط المكونين، انتقل الذروة الاختزالية لـ S1-a إلى مقابل (الشكل 4D، أ، الخط الأرجواني). قمم الأكسدة والاختزال لـتغير إلى مقابل و مقابل على التوالي (الشكل 4D، أ، الخط الأرجواني). عندماتمت الإضافة، ذروة الاختزال لـتم تقليله إلى -1.68 فولت مقابل (الشكل 4D، ب، الخط الأزرق). تغير الذروة الاختزالية للمزيج منإلى مقابل تحت جو الأرجون (الشكل 4D، ب، الخط الأزرق). ومع ذلك، اختفى الذروة التأكسدية للمزيج تمامًا (الشكل 4D، ب، الخط الأزرق). أظهرت جميع النتائج أن تيارًا تحفيزيًا تم توليده بسبب S1-a، مما عزز نجاح البروتوكول. بالإضافة إلى ذلك، أثبتت التجارب الضابطة ودراسات CV أيضًا المشاركة ثنائية الوظيفة لـبما في ذلك تحسين الموصلية وتخليق S1-a في هذا البروتوكول.
لإظهار قوة وفائدة هذه التفاعل، حاولنا إجراء تجربة على نطاق 10 مليمول و100 مليمول لتخليق المنتج 1 (الشكل 4E). ومن المدهش أن التفاعل حافظ على انتقائية جيدة مع عائد ممتاز ونسبة Dinc%. )، الذي أظهر التفوق العالي والكفاءة لهذه البروتوكول الكهربائي الاختزالي (للحصول على التفاصيل، يرجى مراجعة المعلومات التكميلية في الصفحات 14-15). في الوقت نفسه، قمنا بتحقيق تفاعل كهربائي مستمر باستخدام معدل تدفق قدره وزمن الإقامة لمدة 3 ساعات. المنتجتم الحصول عليه بعائد ممتاز ونسبة D-inc% (الشكل 4F)، مما أظهر المزيد من الإمكانية لتطبيق هذه التحويلة. ثم استخدمنا عدة منتجات مدهورة لإجراء تجارب تبادل D/H مع (الشكل 4G)، كما هو موضح، قدمت جميع جزيئات D المواد الأولية المقابلة بعوائد عالية (الشكل 4G، S1، S6، S8، S16، S19 و S39، ). وقد أشار إلى أن هذه التحويلة الكهروكيميائية لتثبيت الكربون-هيدروجين كانت قابلة للعكس. بعد ذلك، تم إجراء عدة تجارب تنافسية (الشكل 4 H ). عندما تم تنفيذ هذا النظام في خليط من 7.5 مللي مول)، فقطتم إنتاج المنتج 1 (الشكل 4 H، أ)، عندماتم استبداله بـتحت نفس الظروف،-نسبة %تم توفيره (الشكل.، الذي أظهر أن قدرة أيونات البيريدين على التقاطأفضل منبالإضافة إلى ذلك، تم إجراء المزيد من التجارب لاستكشاف معدل تبادل H/D ومعدل تبادل D/H (للحصول على التفاصيل، يرجى مراجعة المعلومات التكميلية في الصفحات 18-20). بالإضافة إلى ذلك، أظهر مزيج من ركيزتين تحملان مجموعات فرعية متنوعة نتائج مختلفة تحت نفس الظروف (الشكل 4H، ج، د). على سبيل المثال، كان من الواضح أن نسبة D-inc للمنتجات التي تحمل -F كانت أفضل من تلك التي تحمل -OMe.
الشكل 2 | نطاق الركيزة. أ مشتقات البيريدين. ب الكوينولونات. ظروف التفاعل:كهربائي كيميائيتأين البيريدينات والكينولونات في خلية غير مقسمة، GF كأنود وPb ككاثود، تيار ثابتمشتقات البيريدين (0.3 ملليمول)،ما يعادل ذلك)، DMF (4.0 مل)، درجة حرارة الغرفة،
10 ساعات، عائد معزول. تم تحديد نسب دمج الديوتيريوم بواسطةطيف الرنين المغناطيسي النووي.تم إجراء التفاعل تحت تيار ثابت قدره 25 مللي أمبير..
الشكل 3 | نطاق الركيزة. أ-الليغاندات. ب المركبات ذات الصلة الحيوية. ظروف التفاعل: كيمياء كهربائيةتأين البيريدينات والكينولونات في خلية غير مقسمة، GF كأنود وPb ككاثود، تيار ثابت (20 مللي أمبير)، مشتقات البيريدين (0.3 مللي مول)،ما يعادل ذلك)، DMF (4.0 مل)، في درجة حرارة الغرفة درجة الحرارة، 10 ساعة، العائد المعزول. تم تحديد نسب دمج الديوتيريوم بواسطةطيف الرنين المغناطيسي النووي.تم إجراء التفاعل تحت تيار ثابت قدره 25 مللي أمبير..
دراسات الفولتمترية الدائرية
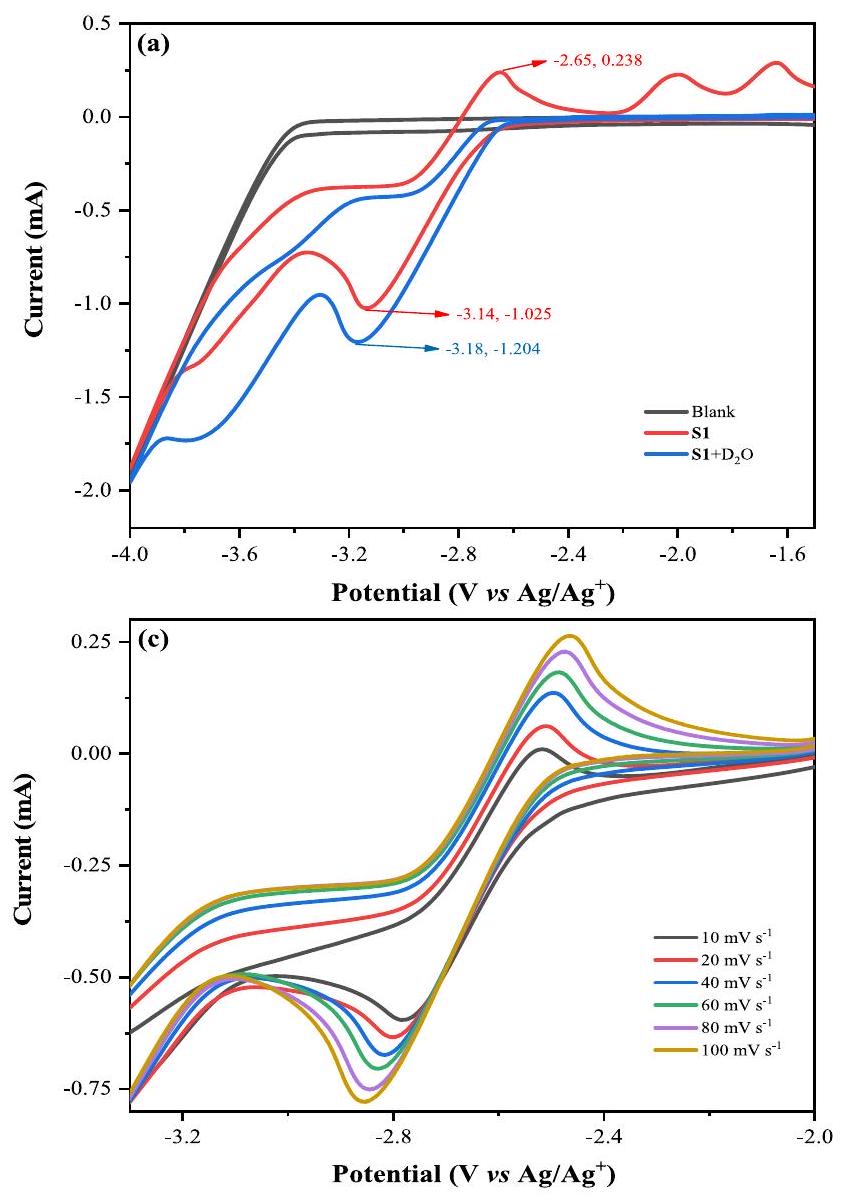
للحصول على استكشاف أعمق لآلية التفاعل، تم إجراء دراسات متعمقة من خلال دراسات الفولتمترية الدورية المفصلة. كان هناك قمة أكسدة عكسية واضحة عند مقابل وقمة انخفاض قابلة للعكس منفي مقابل تمت ملاحظتها تحت جو الأرجون (الشكل 5، الخط الأحمر). في وجودقمة الأكسدة القابلة للعكس فياختفى و shifted peak الانخفاض منإلى مقابل (الشكل 5، الخط الأزرق). في خليط من و التيار الاختزالي لـ زادت بشكل طفيف، مما قد يُعزى إلى التغير الطفيف في الموصلية (الشكل 5، الخط الأخضر). ثم، تمت إضافته إلى المحلول المختلط واختفى ذروة الأكسدة لـ S1 مرة أخرى (الشكل 5، الخط الأزرق الداكن)، وهو ما يتماشى مع النتائج السابقة. ومع ذلك، نظرًا لعدم وجود المنتج المطلوب 1 تم الكشف عنه في ظل ظروف قياسية في غياب، وقد أوضح أن لعبت دورًا حاسمًا في تعزيز التفاعل (الجدول 1، الإدخال 17). ومن الجدير بالذكر أننا أجرينا تجارب CV على S1 بمعدلات مسح مختلفة (الشكل 5c)، كما أن تحليل الانحدار الخطي أوضح أيضًا أن عملية تحكم الانتشار قد تكون متورطة في التحويل (الشكل 5d).
استنادًا إلى التجارب الميكانيكية ودراسات CV، اقترحنا آلية محتملة (الشكل 6). أولاً، (أنا) ينقسم إلى (II) و (III). ثم (II) تم أكسدته إلى كاتيون جذري IV على الأنود لتقديم الإلكترونات. بعد ذلك، بول (III) يشكل مركباً معلتحمل الوسيط (S1-a). بعد ذلك، (S1-a) يخضع لانتقال إلكترون واحد على الكاثود، لتكوين الوسيط الجذري VI. تولد عملية اختزال إلكترون واحد أخرى الوسيط الأنيوني VII. قد يتم تحديد الانتقائية الإقليمية الفريدة والعالية بواسطة هذه.
الشكل 4 | دراسات ميكانيكية. أ تأثير الإلكتروليت. ب ملاحظة الوسيط S1-a. ج تجارب تباين. د دراسات الفولتمترية الدورية، باستخدام الكربون الزجاجي كإلكترود عمل، وصفيحة بلاتينية وكأقطاب مضادة ومرجعية. معدل المسح:المذيب: DMF/nBu4 NBF ( 0.1 م ) أو ( 0.01 م )تم إجراء التجارب تحت غاز الأرجون ما لم يُذكر خلاف ذلك. أ. منحنيات الجهد الكهربائي لـ S1-a و S1 ومزيج منهما. ب. منحنيات الجهد الكهربائي لـ S1 و S1 مع S1-a وكلاهما مع. تفاعل بمقياس 100 مللي مول. F تفاعل تخليق تدفق.تجارب التبادل.تجارب المنافسة.
الشكل 5 | تجارب الفولتمترية الدورية. باستخدام الكربون الزجاجي كإلكترود عمل، وصفيحة بلاتينية وكأقطاب مضادة ومرجعية. معدل المسح:المذيب: DMF/ أو ( 0.01 م ). تم إجراء التجارب تحت غاز الأرجون ما لم يُذكر خلاف ذلك. أ CVs لـ S1
ومع. سيرة ذاتية لـ S1، S1 مع ونظام التفاعل. تم إجراء منحنيات الجهد المتناوب لـ S1 بمعدلات مسح متغيرة تتراوح منإلىتحليل الملاءمة الخطية لـ و .
الشكل 6 | آلية التفاعل المقترحة. الأنود: تفاعل أكسدة. الكاثود: تفاعل اختزال. S1 (2-فينيل بيريدين).
الإجراءات. بعد ذلك، يتفاعل الأنيون الوسيط VII معوينتج الوسيط الديوتيريوم الثامن. أخيرًا، تم أكسدة الوسيط الثامن على الأنود، مما أدى إلى الحصول على المنتج المستهدف..
نقاش
في الختام، أبلغنا عن التثبيت الانتقائي المباشر والفعال للديتيريوم في مشتقات البيريدين من خلال طريقة التحفيز الكهربائي.تفعيل معفي درجة حرارة الغرفة، دون أي معدن أو حمض أو قاعدة. تمت هذه التحويلة بسلاسة، كما تم إثبات ذلك مع مجموعة واسعة من الركائز، مما أدى إلى تشكيل المنتجات المرغوبة مع انتقائية مكانية عالية ودمج جيد إلى ممتاز للـ D. تم أيضًا إظهار فائدة هذه البروتوكول في تركيب الديوتيريد-الليغاندات والتعديل في المراحل المتأخرة من المركبات ذات الصلة البيولوجية. علاوة على ذلك، أظهرت التجارب الآلية ودراسات الفولتامترية الدائرية أن-بيوتيل-2-فينيلبيريدينيوم يوديد يعزز نجاح الاختزال الكهربائيإجراء الديوتريشن. التحويل الكهربائي الإضافي وتطبيقات أملاح البيريدين والمشتقات مستمرة في مختبرنا.
طرق الإجراء العام للاختزال الكهربائيتثبيت الديوتيريوم لمشتقات البيريدين
تم إجراء التحفيز الكهربائي في خلية غير مقسمة باستخدام شعيرات الجرافيت (GF، ) كأنود و ) ككاثود. تم إضافة مادة عضوية إلى خلية كهربائية كيميائية غير مقسمة ( 15 مل ) مجففة في الفرن ومزودة بشريط مغناطيسي.-هتروأرينات (ما يعادل ذلك، ( 0.3 مليمول ، المعادل ) و المكافئ)، ثم أضيف DMF اللامائي (4.0 مل) عبر حقنة. تم تنفيذ نظام التحفيز الكهربائي عندتيار ثابت لمدة 10 ساعات عند درجة حرارة الغرفة. بعد ذلك، تم استخراج خليط التفاعل باستخدام إيثيل أسيتات ( ) وتم تجفيف الطور العضوي المدمج بواسطة اللامائية تم تصفية المنتج وتركيزه في الفراغ. تم تنقية المنتج الخام بواسطة كروماتوغرافيا العمود للحصول على المنتجات الديوتيرية.
توفر البيانات
يعلن المؤلفون أن البيانات التي تدعم نتائج هذه الدراسة متاحة ضمن المقال وملفات المعلومات التكميلية الخاصة به.
البيانات الإضافية متاحة من المؤلف عند الطلب. تم توفير بيانات المصدر مع هذه الورقة.
References
Simmons, E. M. & Hartwig, J. F. On the interpretation of deuterium kinetic isotope effects in C-H bond functionalizations by transitionmetal complexes. Angew. Chem. Int. Ed. 51, 3066-3072 (2012).
Giagou, T. M. & Meyer, P. Kinetic isotope effects in asymmetric reactions. Chem. Eur. J. 16, 10616-10628 (2010).
Kao, C. & Giese, R. W. Measurement of N7-(2′-hydroxyethyl)guanine in human DNA by gas chromatography electron capture mass spectrometry. Chem. Res. Toxicol. 18, 70-75 (2005).
Atzrodt, J. & Derdau, V. Pd- and Pt-catalyzed H/D exchange methods and their application for internal MS standard preparation from a Sanofi-Aventis perspective. J. Label. Compd Radiopharm. 53, 674-685 (2010).
Allais, C., Grassot, J. M., Rodriguez, J. & Constantieux, T. Metal-free multicomponent syntheses of pyridines. Chem. Rev. 114, 10829-10868 (2014).
Katsnelson, A. Heavy drugs draw heavy interest from pharma backers. Nat. Med. 19, 656 (2013).
Mullard, A. Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 15, 219-221 (2016).
Mullard, A. FDA approves first deuterated drug. Nat. Rev. Drug Discov. 16, 305 (2017).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276-5297 (2019).
Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 35, 493-494 (2017).
Sanderson, K. Big interest in heavy drugs. Nature 458, 269 (2009).
Nag, S. et al. Development of a novel fuorine-18 labeled deuterated fluororasagiline ( Fluororasagiline- ) radioligand for PET studies of monoamino oxidase B (MAO-B). Bioorg. Med. Chem. 21, 6634-6641 (2013).
Kopf, S. et al. Recent Developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634-6718 (2022).
Derdau, V., Atzrodt, J., Zimmermann, J., Kroll, C. & Brückner, F. Hydrogen-deuterium exchange reactions of aromatic compounds and heterocycles by -activated rhodium, platinum and palladium catalysts. Chem. Eur. J. 15, 10397-10404 (2009).
Pieters, G. et al. Regioselective and stereospecific deuteration of bioactive aza compounds by the use of ruthenium nanoparticles. Angew. Chem. Int. Ed. 53, 230-234 (2014).
Tlahuext, A. A. & Hartwig, J. F. Site-selective silver-catalyzed C-H bond deuteration of five-membered aromatic heterocycles and pharmaceuticals. ACS Catal. 11, 1119-1127 (2021).
Li, W. et al. Scalable and selective deuteration of (hetero)Arenes. Nat. Chem. 14, 334-341 (2022).
Farizyan, M., Mondal, A., Mal, S., Deufel, F. & van Gemmeren, M. Palladium-catalyzed nondirected late-stage C-H deuteration of arenes. J. Am. Chem. Soc. 143, 16370-16376 (2021).
Zarate, C., Yang, H., Bezdek, M. J., Hesk, D. & Chirik, P. J. Ni(I)-X complexes bearing a bulky -diimine ligand: synthesis, structure, and superior catalytic performance in the hydrogen isotope exchange in pharmaceuticals. J. Am. Chem. Soc. 141, 5034-5044 (2019).
Daniel-Bertrand, M. et al. Multiple site hydrogen isotope labelling of pharmaceuticals. Angew. Chem. Int. Ed. 59, 21114-21120 (2020).
Mai, V. H., Gadzhiev, O. B., Ignatov, S. K. & Nikonov, G. I. H/D exchange in N-heterocycles catalysed by an NHC-supported ruthenium complex. Catal. Sci. Technol. 9, 3398-3407 (2019).
Junk, T. & Catallo, W. J. Hydrogen isotope exchange reactions involving C-H (D, T) bonds. Chem. Soc. Rev. 26, 401-406 (1997).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. Deuterium- and tritiumlabelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758-1784 (2018).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. C-H functionalisation for hydrogen isotope exchange. Angew. Chem. Int. Ed. 57, 3022-3047 (2018).
Liu, C. et al. Controllable deuteration of halogenated compounds by photocatalytic splitting. Nat. Commun. 9, 80 (2018).
Liu, C., Han, S., Li, M., Chong, X. & Zhang, B. Electrocatalytic deuteration of halides with as the deuterium source over a copper nanowire arrays cathode. Angew. Chem. Int. Ed. 59, 18527-18531 (2020).
Li, Y. et al. Organophotocatalytic selective deuterodehalogenation of aryl or alkyl chlorides. Nat. Commun. 12, 2894 (2021).
Garnett, J. L., Long, M. A., Vining, R. & Mole, T. New simple method for rapid, selective aromatic deuteration using organoaluminum dihalide catalysts. J. Am. Chem. Soc. 94, 5913-5914 (1972).
Martins, A., Lautens, M. & Simple, A. A simple, cost-effective method for the regioselective deuteration of anilines. Org. Lett. 10, 4351-4353 (2008).
Zoltewicz, J. A., Grahe, G. & Smith, C. L. Unusual influence of nitrogen on rates of anion formation. Hydrogen-deuterium exchange of pyridine and the diazines. J. Am. Chem. Soc. 91, 5501-5505 (1969).
Kebede, N. & Pavlik, J. W. Hydrogen-deuterium exchange in dimethylpyridines. J. Heterocycl. Chem. 34, 685-686 (1997).
Alexakis, E., Jones, J. R. & Lockley, W. J. S. One-step exchangelabelling of pyridines and other N-heteroaromatics using deuterium gas: catalysis by heterogeneous rhodium and ruthenium catalysts. Tetrahedron Lett. 47, 5025-5028 (2006).
Yang, H. et al. Site-selective Nickel-catalyzed hydrogen isotope exchange in N-Heterocycles and its application to the tritiation of pharmaceuticals. ACS Catal. 8, 10210-10218 (2018).
Koniarczyk, J. L. et al. A general strategy for site-selective incorporation of deuterium and tritium into pyridines, diazines, and pharmaceuticals. J. Am. Chem. Soc. 140, 1990-1993 (2018).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 117, 13230-13319 (2017).
Wiebe, A. et al. Electrifying organic synthesis. Angew. Chem. Int. Ed. 57, 5594-5619 (2018).
Gandeepan, P., Finger, L. H., Meyer, T. H. & Ackermann, L. 3d metallaelectrocatalysis for resource economical syntheses. Chem. Soc. Rev. 49, 4254-4272 (2020).
Novaes, L. et al. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 50, 7941-8002 (2021).
Jutand, A. Contribution of electrochemistry to organometallic catalysis. Chem. Rev. 108, 2300-2347 (2008).
Yoshida, J., Kataoka, K., Horcajada, R. & Nagaki, A. Modern strategies in electroorganic synthesis. Chem. Rev. 108, 2265-2299 (2008).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492-2521 (2014).
Yuan, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 52, 3309-3324 (2019).
Xiong, P. & Xu, H. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 52, 3339-3350 (2019).
Kingston, C. et al. A survival guide for the “Electro-Curious”. Acc. Chem. Res. 53, 72-83 (2020).
Wang, F. & Stahl, S. Electrochemical oxidation of organic molecules at lower overpotential: accessing broader functional group compatibility with electron-proton transfer mediators. Acc. Chem. Res. 53, 561-574 (2020).
Röckl, J. L., Pollok, D., Franke, R. & Waldvogel, S. R. A decade of electrochemical dehydrogenative -coupling of aryls. Acc. Chem. Res. 53, 45-61 (2020).
Siu, J. C., Fu, N. & Lin, S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res. 53, 547-560 (2020).
Shi, S., Liang, Y. & Jiao, N. Electrochemical oxidation induced selective C-C bond cleavage. Chem. Rev. 121, 485-505 (2021).
Liu, C., Wu, Y., Zhao, B. & Zhang, B. Designed nanomaterials for electrocatalytic organic hydrogenation using water as the hydrogen source. Acc. Chem. Res. 56, 1872-1883 (2023).
Wang, Y.-W., Wang, Q. & Qiu, Y.-A. Electroreduction of unactivated alkenes using water as hydrogen source. Nat. Commun. 15, 2780 (2024).
Horn, E. J. et al. Scalable and sustainable electrochemical allylic C -H oxidation. Nature 533, 77-81 (2016).
Sauermann, N., Meyer, T. H., Tian, C. & Ackermann, L. Electrochemical cobalt-catalyzed oxygenation at room temperature. J. Am. Chem. Soc. 139, 18452-18455 (2017).
Xiong, P. et al. Electrochemically enabled carbohydroxylation of alkenes with and organotrifluoroborates. J. Am. Chem. Soc. 140, 16387-16391 (2018).
Liang, Y. et al. Electrochemically induced nickel catalysis for oxygenation reactions with water. Nat. Catal. 4, 116-123 (2021).
Shen, T. & Lambert, T. H. Electrophotocatalytic diamination of vicinal C-H bonds. Science 371, 620-626 (2021).
Chung, D. S., Park, S. H., Lee, S. G. & Kim, H. Electrochemically driven stereoselective approach to syn-1,2-diol derivatives from vinylarenes and DMF. Chem. Sci. 12, 5892-5897 (2021).
Chung, D. S., Park, S. H., Lee, S. G. & Kim, H. Enantioselective electrochemical cobalt-catalyzed aryl C-H activation reactions. Science 379, 1036-1042 (2023).
Shen, T., Li, Y. L., Ye, K. Y. & Lambert, T. H. Electrophotocatalytic oxygenation of multiple adjacent C-H bonds. Nature 614, 275-280 (2023).
Sauermann, N., Meyer, T. H., Qiu, Y. & Ackermann, L. Electrocatalytic C-H activation. ACS Catal. 8, 7086-7103 (2018).
Jiao, K.-J., Xing, Y.-K., Yang, Q.-L., Qiu, H. & Mei, T.-S. Site-selective C -H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 53, 300-310 (2020).
Waldvogel, S. R., Lips, S., Selt, M., Riehl, B. & Kampf, C. J. Electrochemical arylation reaction. Chem. Rev. 118, 6706-6765 (2018).
Wang, H., Gao, X., Lv, Z., Abdelilah, T. & Lei, A. Recent advances in oxidative cross-coupling with hydrogen evolution via photo-/electrochemistry. Chem. Rev. 119, 6769-6787 (2019).
Liu, M., Feng, T. & Qiu, Y. Metal-free electrochemical dihydroxylation of unactivated alkenes. Nat. Commun. 14, 6467 (2023).
Zhao, Z.-W. et al. Site-selective electrochemical C-H carboxylation of arenes with . Angew. Chem. Int. Ed. 62, e202214710 (2023).
Sun, G.-Q. et al. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 615, 67-72 (2023).
Wang, M. et al. Room temperature construction of vicinal amino alcohols via electroreductive cross-coupling of N-heteroarenes and carbonyls. J. Am. Chem. Soc. 145, 10967-10973 (2023).
Wang, P. et al. Electrochemical arylation of electron-deficient arenes through reductive activation. Angew. Chem. Int. Ed. 58, 15747-15751 (2019).
Li, P. et al. Facile and general electrochemical deuteration of unactivated alkyl halides. Nat. Commun. 13, 3774 (2022).
Yang, K., Feng, T. & Qiu, Y. Organo-mediator enabled electrochemical deuteration of styrenes. Angew. Chem. Int. Ed. 62, e202312803 (2023).
Hughes, G. & Bryce, M. R. Electron-transporting materials for organic electroluminescent and electrophosphorescent devices. J. Mater. Chem. 15, 94-107 (2005).
Michael, J. P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 25, 166-187 (2008).
Mu, L., Shi, W., Chang, J. C. & Lee, S. T. Silicon nanowires-based fluorescence sensor for Cu(II). Nano Lett. 8, 104-109 (2008).
Solomon, V. R. & Lee, H. Quinoline as a privileged scaffold in cancer drug discovery. Curr. Med. Chem. 18, 1488-1508 (2011).
شكر وتقدير
يُعرب عن الشكر للدعم المالي من البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2023YFA1507203)، ومؤسسة العلوم الطبيعية الوطنية في الصين (رقم المنحة 22371149، 22188101)، وصناديق البحث الأساسية للجامعات المركزية (رقم 63223015)، ومركز العلوم الحدودية للمواد العضوية الجديدة، جامعة نانكاي (رقم المنحة 63181206)، وجامعة نانكاي.
مساهمات المؤلفين
قام Y.Q. بالإشراف على المشروع، وقدم التوجيه بشأن المشروع. قام Y.Q. و Z.W.Z. بتصميم الدراسة وكتابة المخطوطة. قام Z.W.Z. و R.Z. و Y.L. و Q.W. و Y.Q. بإجراء التجارب والدراسات الآلية، وراجعوا المخطوطة. قام Z.L.Z. بإجراء حسابات DFT. ساهم جميع المؤلفين في تحليل البيانات وتفسيرها.
المختبر الرئيسي للدولة ومعهد الكيمياء العضوية العنصرية، مركز العلوم الحدودية للمواد العضوية الجديدة، كلية الكيمياء، جامعة نانكاي، تيانجين، الصين. البريد الإلكتروني:qywang@nankai.edu.cn; qiuyouai@nankai.edu.cn
Herein, we develop a straightforward, metal-free, and acid-/base-free electrochemical C4-selective C-H deuteration of pyridine derivatives with economic and convenient at room temperature. This strategy features an efficient and environmentally friendly approach with high chemo- and regioselectivity, affording a wide range of D-compounds, such as pyridines, quinolones, ligands and biorelevant compounds. Notably, the mechanistic experiments and cyclic voltammetry (CV) studies demonstrate that -butyl-2-phenylpyridinium iodide is a crucial intermediate during the electrochemical transformation, which provides a general and efficient way for deuteration of pyridine derivatives.
Deuterium-labelled molecules are a critical kind of organic compounds, which have been widely applied into various research areas, such as elucidating the reaction mechanisms , isotopic tracer techniques , and pharmaceutical chemistry . For example, -heteroarenes, which are frequently employed as bioactive molecules and drugs for exploring the pharmacokinetic and pharmacodynamic (PK/ PD) properties . Therefore, it is unsurprising that the synthesis and application of deuterium-labeled ( -hetero)arenes has attracted significant attentions , especially for the D-labeled pyridine derivatives . Among the various protocols that have been developed, the direct, simple and efficient H/D exchange strategy stands prior to dehalogenative deuteration of halides and pseudohalides , owing to the readily available starting materials, their cost, as well as high atom economy, however, the challenge would be the selectivity control of the transformations. Despite the established deuteration of pyridine derivatives in the presence of Brønsted/Lewis acid , base , or transition-metal (Fig. 1A), the pursuance of methodologies with higher selectivity and D-incorporation under milder conditions with easy operation procedures is still on the way. Notably, McNally and co-workers developed a C4-selective deuteration of pyridine derivatives through twostep process, which undergoing the heterocyclic phosphonium salts intermediator, and then forming the final deterateted pyridine derivatives with assistance of base, achieving high selective deuteration of pyridine derivatives (Fig. 1B) . Furthermore, the development of approaches for site-selective deuteration of pyridine derivatives
in a straightforward, sustainable, and efficient way is still highly desirable and challenging.
In recent years, the emergence of electrochemistry has brought opportunities and development in organic synthesis . Electrosynthesis is becoming increasingly popular and considered as one of sustainable and desirable methodology that could substitute some traditional synthetic methods. We envisage that direct and selective deuteration of pyridine derivatives can be achieved through the usage of electrochemistry. However, compared with welldeveloped electrooxidative functionalization , electroreductively driven functionalization of arenes have thus not been well elucidated . Especially, selective deuteration of pyridine derivatives under electroreductive conditions has not achieved yet.
In sharp contrast, with our continuous interests in sustainable electroreductively driven functionalization , and electrochemical deuteration , herein, we would like to report our effort in developing a general, direct and efficient electrochemical C4-selective deuteration of pyridine derivatives through reductive activation with economical , via a crucial -butyl-2-phenylpyridinium iodide intermediate (Fig. 1C). This protocol offered a wide range of D-pyridine derivatives with high chemo- and regioselectivity in excellent yields. The salient features of this transformation including: (a) electroreductively driven C-H deuteration; (b) good to excellent D-incorporation; (c) metal-, acid-, or base-free process; (d) high chemo- and regioselectivity; (e) late-stage modification of -ligands and biorelevant compounds.
Fig. 1 | Background and current work of deuteration for pyridine derivatives. A Previous methods for C-H deuteration of pyridine derivatives. B C4-selective two-step deuteration of pyridine derivatives via phosphonium salts. C This work: electrochemical deuteration of pyridine derivatives.
Results
Optimization of reaction conditions
Our investigations began by evaluating the deuteration of pyridine substrates with deuterium oxide ( ). After some preliminary experiments, 2-phenylpyridine (S1) was selected as the model starting substrate for optimizing the reaction conditions. Encouragingly, only a single product 1 was observed, which proved that the reaction has high regioselectivity. After a careful selection of the system parameters, we obtained the optimal conditions for deuteration of with . Combining the reactants with electrolyte in an undivided cell with DMF under constant current ( 20 mA ) at room temperature for 10 hours, resulted in yield and deuterium incorporation (abbreviated as D-inc hereafter) (Table 1, entry 1). Initially, a variety of electrolytes were investigated. and worked well and resulted in and D-inc of product 1 respectively (Table 1, entries 2 and 3), but no product was detected when or NaI was employed (Table 1, entries 4 and 5). Then, an attempted to use NaOAc as a base, led to a decrease in deuteration (Table 1, entry 6). We also examined various solvents, such as DMA and MeCN, which afforded and D-inc of product 1 respectively (Table 1, entries 7 and 8). Next, we investigated various electrodes, including , and (Table 1, entries 9-11). However, the D-inc were . Furthermore, we probed the effect of the current on this reaction. With a lower current, deuteration of decreased significantly (Table 1, entries 12-13). The system also worked smoothly at higher temperatures (Table 1, entry 14, ). In addition, we found that this system was insensitive to the atmosphere and still gave excellent yield and D-inc under argon atmosphere (Table 1, entry 15). Finally, some control experiments proved that the presence of electricity and electrolyte were essential for this transformation (Table 1, entries 16 and 17).
Substrate scope
With optimal conditions in hand, we subsequently investigated the substrate scope and generality of this efficient electrochemical deuteration transformation. As shown in Fig. 2A, a range of aryl-/alkyl-
pyridines with diverse substituents were evaluated. The steric effect of the substituent exerted little impact on this transformation, for example, 2-phenylpyridines bearing , or -methyl substituent furnished the desired products in good to excellent yields and D-inc , as well as that bearing two methyl (5). Other substituents, such as , and were tolerated smoothly. The substrates that bearing fluorine (11), trifluoromethyl (12) and carboxy (13) groups were compatible and worked successfully, providing the desired products with high efficiency. Moreover, the deuterated products from phenylpyridine with heteroaryl-based ( ) groups were also obtained in excellent yields and D-inc (14-17). Moreover, alternate and multiple substituted substrates were all tolerated and reacted smoothly in the system, providing satisfactory results (18-22, up to 99% yields, 86%->99% D-inc). In addition to aryl pyridines, we also investigated diverse alkyl pyridine derivatives. A wide range of substrates with various electron-donating and electronwithdrawing groups could all react well with and produce the corresponding deuterium products in 75% to >99% D-inc (23-28).
Quinoline is a crucial class of heterocycles that are widely found in natural products, pharmaceuticals, dyes, and materials . Therefore, we tried to probe the scope of quinolines with diverse functional groups to further exhibit the applicability of this protocol (Fig. 2B). We found that quinolines bearing electron-neutral (29), -donating (30-39), and -withdrawing (40) functional groups performed well in this transformation, affording the corresponding products in good yields (94%-99%) with excellent D-inc (84%->99%) and selectivity. In addition, 7,8-Benzoquinoline (41) and acridine (42, vaccines against infection and allergy) were also successfully worked in this protocol, resulting in satisfying results.
More importantly, we turned our attention to -ligands (Fig. 3A), various of them were also accommodated, leading to the desired molecules in both good yields and D-inc (43-50), including bipyridine (43-47, 45, Abametapir, a pediculicide for head lice infestation), benzimidazole (48) and phenanthroline (49-50), which indicated great compatibility and practicability of the protocol. It provided a new method for late-stage functional modification of -ligands.
We next explored the late-stage deuteration of biorelevant compounds and some pharmaceutical molecules with this electroreductive method (Fig. 3B). Gratifyingly, pyridine compounds derived from Citronellol (51), Phytol (52), Borneol (53), l-menthol (54), Carveol (55), d-galactopyranose (56), Picaridin (57), (S)-N-Boc-2-hydroxymethylmorpholine (58), Ibuprofen (59) and some pharmaceutical molecules, including Loratadine ( , Claritin, an anti-allergic drug), Abiraterone acetate (61, Zytiga, a prostate cancer drug) and Bisacodyl (62, Dulcolax, a laxative), all showed great compatibility and reactivity with this electro-reductive system, delivering the desired deuterated products in both excellent yields and D-inc%. These results indicated that this electrochemical protocol has great potential and prospect in application and modification of biorelevant compounds.
Mechanistic studies
In order to further verify the rationality of this transformation, we performed a series of experiments to investigate the mechanism (Fig. 4). Firstly, we explored the effect of electrolytes (Fig. 4A). As expected, no product was observed when or was employed as the electrolyte (Fig. 4A, entries 1-3). However, when we used other electrolytes containing ion, excellent D-inc of was obtained (Fig. 4A, entries 4-9). Meanwhile, various anions from salts led to different yields (yield of or recovered ). These results illustrated that ion was crucial for this electrochemical reaction. Moreover, under the optimized conditions, N-butyl-2-phenylpyridinium iodide (S1-a, detected by HRMS, 212.1434), and were afforded in the absence of , which further demonstrated the important role of (Fig. 4B). Furthermore, we speculated that S1a might be a key intermediate in this process. Hence, (1.0 equiv.)
Table 1 | Screening of reaction conditions
S1
1
Entry
Variation from standard conditions
Yield of 1 or recover S1 (%)
1 (D%)
1
None
99
>99
2
instead of
83
99
3
instead of
99
90
4
instead of
99
–
5
NaI instead of
99
–
6
NaOAc (1.0 equiv.)
99
16
7
DMA as solvent
99
82
8
MeCN as solvent
99
<5
9
CF (+)| (-) Pb
90
80
10
GF (+)| (-) GF
99
35
11
GF (+) | (-) Pt
99
53
12
10 mA
99
60
13
15 mA
99
81
14
99
99
15
Ar
99
99
16
w/o electricity
99
—
17
w/o electrolyte
99
—
Bold formatting shows that entry 1 is the optimal reaction conditions.
CF carbon felt, DMF N, N-dimethylformamide, DMA N, N-dimethylacetamide. DMF ( 4.0 mL ), room temperature, air, 10 h . Isolated yield. Deuterium incorporation percentages were determined by NMR spectroscopy.
instead of , then a catalytic amount ( ) of were added to 2-phenylpyridine ( ) under standard conditions, furnishing the product with , ) and ( ) respectively (Fig. 4C, a). On the other hand, the corresponding electrolytes were employed for the model reaction with standard conditions and gave compound with ), , and severally (Fig. 4C, b). The results were almost identical to the previous ones. Next, we conducted several cyclic voltammetry (CV) studies (for details, please see the Supplementary Information on page 32). CV experiments on -butyl-2-phenylpyridinium iodide (S1-a) gave a reductive peak at vs. under Ar atmosphere (Fig. 4D, a, green line). An obvious reversible oxidative peak at vs. and a reversible reductive peak of at vs. were observed under Ar atmosphere (Fig. 4D, a, red line). After mixing the two ingredients, the reductive peak of S1-a moved to vs. (Fig. 4D, a, purple line). The oxidative and reductive peaks of changed to vs. and vs. respectively (Fig. 4D, a, purple line). When was added, the reductive peak of reduced to -1.68 V vs. (Fig. 4D, b, blue line). The reductive peak of the mixture changed from to vs. under Ar atmosphere (Fig. 4D, b, blue line). However, the oxidative peak of the mixture was fully disappeared (Fig. 4D, b, blue line). All the results showed that a catalytic current was generated because of S1-a, which promoted the success of the protocol. Additionally, control experiments and CV studies also proved the bifunctional participation of including the improvement of conductivity and the synthesis of S1-a in this protocol.
To show the robustness and utility of this reaction, we attempted to carry out a 10 mmol gram-scale experiment and a 100 mmol scale experiment to synthesize the product 1 (Fig. 4E). Surprisingly, the reaction maintained good selectivity with excellent yield and Dinc% ( ), which illustrated the high superiority and efficiency of this electroreductive protocol (For details, please see the Supplementary Information on pages 14-15). Meanwhile, we realized an elctrochemical continuous-flow reaction using a flow rate of and a residence time for 3 h . The product was obtained in excellent yield and D-inc% (Fig. 4F), which further demonstrated the potential application of this transformation. Then, we used several deuterated products to perform D/H exchange experiments with (Fig. 4G), as shown, all D-molecules provided the corresponding initial materials in high yields (Fig. 4G, S1, S6, S8, S16, S19 and S39, ). It indicated that this electrochemical C-H deuteration transformation was reversible. Next, several competition experiments were performed (Fig. 4 H ). When this system was carried out in a mixture of 7.5 mmol ), only of product 1 was produced (Fig. 4 H , a), when the was replaced by under the same conditions, -inc% of was afforded (Fig. , which demonstrated that the ability of pyridine anions to capture is better than . In addition, more experiments were conducted to explore the H/D exchange and D/H exchange rate (For details, please see the Supplementary Information on pages 18-20). In addition, a mixture of two substrates bearing diverse substituents performed different results under the same conditions (Fig. 4H, c, d). For example, it was obvious that the D-inc % of products that bearing -F was superior to that bearing -OMe.
Fig. 2 | Substrate scope. A Pyridine derivatives. B Quinolones. Reaction conditions: Electrochemical deuteration of pyridines and quinolones in an undivided cell, GF as anode and Pb as cathode, constant current , pyridine derivatives ( 0.3 mmol ), equiv.), DMF ( 4.0 mL ), room temperature,
10 h , isolated yield. Deuterium incorporation percentages were determined by NMR spectroscopy. The reaction was conducted under 25 mA constant current. .
Fig. 3 | Substrate scope. A -ligands. B Biorelevant compounds. Reaction conditions: Electrochemical deuteration of pyridines and quinolones in an undivided cell, GF as anode and Pb as cathode, constant current ( 20 mA ), pyridine derivatives ( 0.3 mmol ), equiv.), DMF ( 4.0 mL ), room
temperature, 10 h , isolated yield. Deuterium incorporation percentages were determined by NMR spectroscopy. The reaction was conducted under 25 mA constant current. .
Cyclic voltammetry studies
To gain further exploration into the reaction mechanism, in-depth studies were carried out through detailed cyclic voltammetry studies. An obvious reversible oxidation peak at vs. and a reversible reduction peak of at vs. were observed under Ar atmosphere (Fig. 5, red line). In the presence of , the reversible oxidation peak at disappeared and the reduction peak shifted from to vs. (Fig. 5, blue line). In the mixture of and , the reductive current of increased slightly, which might be attributed to the slight variation in conductivity (Fig. 5, green line). Then, was added to the mixed solution and the oxidation peak of S1 disappeared again (Fig. 5, dark blue line), which was consistent with the previous results. However, since no desired product 1
was detected with standard conditions in the absence of , it illustrated that played a crucial role in promoting the reactivity (Table 1, entry 17). Notably, we conducted CV experiments on S1 with various scan rates (Fig. 5c), and the linear fit analysis also explained that a diffusion-control process might be involved in the conversion (Fig. 5d).
Based on the mechanistic experiments and CV studies, we proposed a possible mechanism (Fig. 6). Firstly, (I) splits into (II) and (III). Then (II) was oxidized to radical cation IV on the anode to offer electrons. Next, Bul (III) forms a complex with to afford intermediate (S1-a). Subsequently, (S1-a) undergoes a single electron transfer on the cathode, to form the radical intermediate VI. Another single electron reduction generates the anion intermediate VII. The unique and high regioselectivity might be determined by these
Fig. 4 | Mechanistic studies. A Effect of the electrolyte. B Observation of intermediate S1-a. C Contrast experiments. D Cyclic voltammetry studies, using glass carbon as work electrode, Pt plate and as counter and reference electrodes. Scan rate: . Solvent: DMF/nBu4 NBF ( 0.1 M ) or
( 0.01 M ), . Experiments were conducted under Ar unless otherwise noted. a CVs of S1-a, S1 and the mixture of them. b CVs of S1, S1 with S1-a and both of them with . E 100 mmol scale reaction. F Flow-synthesis reaction. exchange experiments. Competition experiments.
Fig. 5 | Cyclic voltammetry experiments. Using glass carbon as work electrode, Pt plate and as counter and reference electrodes. Scan rate: . Solvent: DMF/ or ( 0.01 M ). Experiments were conducted under Ar unless otherwise noted. a CVs of S1
and with . b CVs of S1, S1 with and the reaction system. c CVs of S1 performed at variable scan rates ranging from to . d Linear fit analysis of and .
procedures. Subsequently, the anion intermediate VII reacts with and produces the deuteration intermediate VIII. Finally, intermediate VIII was oxidized on the anode, affording the target product .
Discussion
In conclusion, we reported the direct and efficient C4-selective deuteration of pyridine derivatives via the mode of electroreductively driven functionalization with at room temperature, without any metal, acid, and base. This transformation proceeded smoothly, as demonstrated with a wide range of substrates, forming the desired products with high regioselectivity and good to excellent D-incorporation. The utility of this protocol was also shown in the
synthesis of deuterated -ligands and late-stage modification of biorelevant compounds. Moreover, the mechanistic experiments and CV studies showed that -butyl-2-phenylpyridinium iodide promotes the success of the electroreductive deuteration procedure. Further electrochemical transformation and applications of pyridine salts and derivatives are ongoing in our laboratory.
Methods
General procedure of electroreductive deuteration of pyridine derivatives
The electrocatalysis was carried out in an undivided cell with graphite felt (GF, ) as anode and ) as cathode. To an oven-dried undivided electrochemical cell ( 15 mL ) equipped with a magnetic bar was added organic -heteroarenes ( equiv.), ( 0.3 mmol , equiv.) and equiv.), then anhydrous DMF ( 4.0 mL ) was added via a syringe. The electrocatalysis system was performed at of constant current for 10 h at room temperature. After that, the reaction mixture was extracted with EtOAc ( ) and the combined organic phase was dried by anhydrous , filtered, and concentrated in vacuo. The crude product was purified by column chromatography to furnish the deuterated products.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files.
Extra data are available from the author upon request. Source data are provided with this paper.
References
Simmons, E. M. & Hartwig, J. F. On the interpretation of deuterium kinetic isotope effects in C-H bond functionalizations by transitionmetal complexes. Angew. Chem. Int. Ed. 51, 3066-3072 (2012).
Giagou, T. M. & Meyer, P. Kinetic isotope effects in asymmetric reactions. Chem. Eur. J. 16, 10616-10628 (2010).
Kao, C. & Giese, R. W. Measurement of N7-(2′-hydroxyethyl)guanine in human DNA by gas chromatography electron capture mass spectrometry. Chem. Res. Toxicol. 18, 70-75 (2005).
Atzrodt, J. & Derdau, V. Pd- and Pt-catalyzed H/D exchange methods and their application for internal MS standard preparation from a Sanofi-Aventis perspective. J. Label. Compd Radiopharm. 53, 674-685 (2010).
Allais, C., Grassot, J. M., Rodriguez, J. & Constantieux, T. Metal-free multicomponent syntheses of pyridines. Chem. Rev. 114, 10829-10868 (2014).
Katsnelson, A. Heavy drugs draw heavy interest from pharma backers. Nat. Med. 19, 656 (2013).
Mullard, A. Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 15, 219-221 (2016).
Mullard, A. FDA approves first deuterated drug. Nat. Rev. Drug Discov. 16, 305 (2017).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276-5297 (2019).
Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 35, 493-494 (2017).
Sanderson, K. Big interest in heavy drugs. Nature 458, 269 (2009).
Nag, S. et al. Development of a novel fuorine-18 labeled deuterated fluororasagiline ( Fluororasagiline- ) radioligand for PET studies of monoamino oxidase B (MAO-B). Bioorg. Med. Chem. 21, 6634-6641 (2013).
Kopf, S. et al. Recent Developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634-6718 (2022).
Derdau, V., Atzrodt, J., Zimmermann, J., Kroll, C. & Brückner, F. Hydrogen-deuterium exchange reactions of aromatic compounds and heterocycles by -activated rhodium, platinum and palladium catalysts. Chem. Eur. J. 15, 10397-10404 (2009).
Pieters, G. et al. Regioselective and stereospecific deuteration of bioactive aza compounds by the use of ruthenium nanoparticles. Angew. Chem. Int. Ed. 53, 230-234 (2014).
Tlahuext, A. A. & Hartwig, J. F. Site-selective silver-catalyzed C-H bond deuteration of five-membered aromatic heterocycles and pharmaceuticals. ACS Catal. 11, 1119-1127 (2021).
Li, W. et al. Scalable and selective deuteration of (hetero)Arenes. Nat. Chem. 14, 334-341 (2022).
Farizyan, M., Mondal, A., Mal, S., Deufel, F. & van Gemmeren, M. Palladium-catalyzed nondirected late-stage C-H deuteration of arenes. J. Am. Chem. Soc. 143, 16370-16376 (2021).
Zarate, C., Yang, H., Bezdek, M. J., Hesk, D. & Chirik, P. J. Ni(I)-X complexes bearing a bulky -diimine ligand: synthesis, structure, and superior catalytic performance in the hydrogen isotope exchange in pharmaceuticals. J. Am. Chem. Soc. 141, 5034-5044 (2019).
Daniel-Bertrand, M. et al. Multiple site hydrogen isotope labelling of pharmaceuticals. Angew. Chem. Int. Ed. 59, 21114-21120 (2020).
Mai, V. H., Gadzhiev, O. B., Ignatov, S. K. & Nikonov, G. I. H/D exchange in N-heterocycles catalysed by an NHC-supported ruthenium complex. Catal. Sci. Technol. 9, 3398-3407 (2019).
Junk, T. & Catallo, W. J. Hydrogen isotope exchange reactions involving C-H (D, T) bonds. Chem. Soc. Rev. 26, 401-406 (1997).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. Deuterium- and tritiumlabelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758-1784 (2018).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. C-H functionalisation for hydrogen isotope exchange. Angew. Chem. Int. Ed. 57, 3022-3047 (2018).
Liu, C. et al. Controllable deuteration of halogenated compounds by photocatalytic splitting. Nat. Commun. 9, 80 (2018).
Liu, C., Han, S., Li, M., Chong, X. & Zhang, B. Electrocatalytic deuteration of halides with as the deuterium source over a copper nanowire arrays cathode. Angew. Chem. Int. Ed. 59, 18527-18531 (2020).
Li, Y. et al. Organophotocatalytic selective deuterodehalogenation of aryl or alkyl chlorides. Nat. Commun. 12, 2894 (2021).
Garnett, J. L., Long, M. A., Vining, R. & Mole, T. New simple method for rapid, selective aromatic deuteration using organoaluminum dihalide catalysts. J. Am. Chem. Soc. 94, 5913-5914 (1972).
Martins, A., Lautens, M. & Simple, A. A simple, cost-effective method for the regioselective deuteration of anilines. Org. Lett. 10, 4351-4353 (2008).
Zoltewicz, J. A., Grahe, G. & Smith, C. L. Unusual influence of nitrogen on rates of anion formation. Hydrogen-deuterium exchange of pyridine and the diazines. J. Am. Chem. Soc. 91, 5501-5505 (1969).
Kebede, N. & Pavlik, J. W. Hydrogen-deuterium exchange in dimethylpyridines. J. Heterocycl. Chem. 34, 685-686 (1997).
Alexakis, E., Jones, J. R. & Lockley, W. J. S. One-step exchangelabelling of pyridines and other N-heteroaromatics using deuterium gas: catalysis by heterogeneous rhodium and ruthenium catalysts. Tetrahedron Lett. 47, 5025-5028 (2006).
Yang, H. et al. Site-selective Nickel-catalyzed hydrogen isotope exchange in N-Heterocycles and its application to the tritiation of pharmaceuticals. ACS Catal. 8, 10210-10218 (2018).
Koniarczyk, J. L. et al. A general strategy for site-selective incorporation of deuterium and tritium into pyridines, diazines, and pharmaceuticals. J. Am. Chem. Soc. 140, 1990-1993 (2018).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 117, 13230-13319 (2017).
Wiebe, A. et al. Electrifying organic synthesis. Angew. Chem. Int. Ed. 57, 5594-5619 (2018).
Gandeepan, P., Finger, L. H., Meyer, T. H. & Ackermann, L. 3d metallaelectrocatalysis for resource economical syntheses. Chem. Soc. Rev. 49, 4254-4272 (2020).
Novaes, L. et al. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 50, 7941-8002 (2021).
Jutand, A. Contribution of electrochemistry to organometallic catalysis. Chem. Rev. 108, 2300-2347 (2008).
Yoshida, J., Kataoka, K., Horcajada, R. & Nagaki, A. Modern strategies in electroorganic synthesis. Chem. Rev. 108, 2265-2299 (2008).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492-2521 (2014).
Yuan, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 52, 3309-3324 (2019).
Xiong, P. & Xu, H. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 52, 3339-3350 (2019).
Kingston, C. et al. A survival guide for the “Electro-Curious”. Acc. Chem. Res. 53, 72-83 (2020).
Wang, F. & Stahl, S. Electrochemical oxidation of organic molecules at lower overpotential: accessing broader functional group compatibility with electron-proton transfer mediators. Acc. Chem. Res. 53, 561-574 (2020).
Röckl, J. L., Pollok, D., Franke, R. & Waldvogel, S. R. A decade of electrochemical dehydrogenative -coupling of aryls. Acc. Chem. Res. 53, 45-61 (2020).
Siu, J. C., Fu, N. & Lin, S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res. 53, 547-560 (2020).
Shi, S., Liang, Y. & Jiao, N. Electrochemical oxidation induced selective C-C bond cleavage. Chem. Rev. 121, 485-505 (2021).
Liu, C., Wu, Y., Zhao, B. & Zhang, B. Designed nanomaterials for electrocatalytic organic hydrogenation using water as the hydrogen source. Acc. Chem. Res. 56, 1872-1883 (2023).
Wang, Y.-W., Wang, Q. & Qiu, Y.-A. Electroreduction of unactivated alkenes using water as hydrogen source. Nat. Commun. 15, 2780 (2024).
Horn, E. J. et al. Scalable and sustainable electrochemical allylic C -H oxidation. Nature 533, 77-81 (2016).
Sauermann, N., Meyer, T. H., Tian, C. & Ackermann, L. Electrochemical cobalt-catalyzed oxygenation at room temperature. J. Am. Chem. Soc. 139, 18452-18455 (2017).
Xiong, P. et al. Electrochemically enabled carbohydroxylation of alkenes with and organotrifluoroborates. J. Am. Chem. Soc. 140, 16387-16391 (2018).
Liang, Y. et al. Electrochemically induced nickel catalysis for oxygenation reactions with water. Nat. Catal. 4, 116-123 (2021).
Shen, T. & Lambert, T. H. Electrophotocatalytic diamination of vicinal C-H bonds. Science 371, 620-626 (2021).
Chung, D. S., Park, S. H., Lee, S. G. & Kim, H. Electrochemically driven stereoselective approach to syn-1,2-diol derivatives from vinylarenes and DMF. Chem. Sci. 12, 5892-5897 (2021).
Chung, D. S., Park, S. H., Lee, S. G. & Kim, H. Enantioselective electrochemical cobalt-catalyzed aryl C-H activation reactions. Science 379, 1036-1042 (2023).
Shen, T., Li, Y. L., Ye, K. Y. & Lambert, T. H. Electrophotocatalytic oxygenation of multiple adjacent C-H bonds. Nature 614, 275-280 (2023).
Sauermann, N., Meyer, T. H., Qiu, Y. & Ackermann, L. Electrocatalytic C-H activation. ACS Catal. 8, 7086-7103 (2018).
Jiao, K.-J., Xing, Y.-K., Yang, Q.-L., Qiu, H. & Mei, T.-S. Site-selective C -H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 53, 300-310 (2020).
Waldvogel, S. R., Lips, S., Selt, M., Riehl, B. & Kampf, C. J. Electrochemical arylation reaction. Chem. Rev. 118, 6706-6765 (2018).
Wang, H., Gao, X., Lv, Z., Abdelilah, T. & Lei, A. Recent advances in oxidative cross-coupling with hydrogen evolution via photo-/electrochemistry. Chem. Rev. 119, 6769-6787 (2019).
Liu, M., Feng, T. & Qiu, Y. Metal-free electrochemical dihydroxylation of unactivated alkenes. Nat. Commun. 14, 6467 (2023).
Zhao, Z.-W. et al. Site-selective electrochemical C-H carboxylation of arenes with . Angew. Chem. Int. Ed. 62, e202214710 (2023).
Sun, G.-Q. et al. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 615, 67-72 (2023).
Wang, M. et al. Room temperature construction of vicinal amino alcohols via electroreductive cross-coupling of N-heteroarenes and carbonyls. J. Am. Chem. Soc. 145, 10967-10973 (2023).
Wang, P. et al. Electrochemical arylation of electron-deficient arenes through reductive activation. Angew. Chem. Int. Ed. 58, 15747-15751 (2019).
Li, P. et al. Facile and general electrochemical deuteration of unactivated alkyl halides. Nat. Commun. 13, 3774 (2022).
Yang, K., Feng, T. & Qiu, Y. Organo-mediator enabled electrochemical deuteration of styrenes. Angew. Chem. Int. Ed. 62, e202312803 (2023).
Hughes, G. & Bryce, M. R. Electron-transporting materials for organic electroluminescent and electrophosphorescent devices. J. Mater. Chem. 15, 94-107 (2005).
Michael, J. P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 25, 166-187 (2008).
Mu, L., Shi, W., Chang, J. C. & Lee, S. T. Silicon nanowires-based fluorescence sensor for Cu(II). Nano Lett. 8, 104-109 (2008).
Solomon, V. R. & Lee, H. Quinoline as a privileged scaffold in cancer drug discovery. Curr. Med. Chem. 18, 1488-1508 (2011).
Acknowledgements
Financial support from the National Key R&D Program of China (2023YFA1507203), National Natural Science Foundation of China (Grant No. 22371149, 22188101), the Fundamental Research Funds for the Central Universities (No. 63223015), Frontiers Science Center for New Organic Matter, Nankai University (Grant No. 63181206), and Nankai University are gratefully acknowledged.
Author contributions
Y.Q. supervised the project, and provided guidance on the project. Y.Q. and Z.W.Z. conceived and designed the study and wrote the manuscript. Z.W.Z., R.Z., Y.L., Q.W. and Y.Q. performed the experiments, mechanistic studies, and revised the manuscript. Z.L.Z. conducted DFT calculations. All authors contributed to the analysis and interpretation of the data.
State Key Laboratory and Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, Tianjin, China. e-mail: qywang@nankai.edu.cn; qiuyouai@nankai.edu.cn
 DMF (4.0 مل)، درجة حرارة الغرفة، هواء، 10 ساعات.
DMF (4.0 مل)، درجة حرارة الغرفة، هواء، 10 ساعات.