DOI: https://doi.org/10.1038/s41467-024-45534-2
PMID: https://pubmed.ncbi.nlm.nih.gov/38341446
تاريخ النشر: 2024-02-10
التحويل المستدام للنترات القلوية إلى الأمونيا عند أنشطة تزيد عن 2 A cm−2
تم القبول: 25 يناير 2024
نُشر على الإنترنت: 10 فبراير 2024
(د) التحقق من التحديثات
الملخص
نترات
لذرات البالاديوم لبناء محلي
النتائج
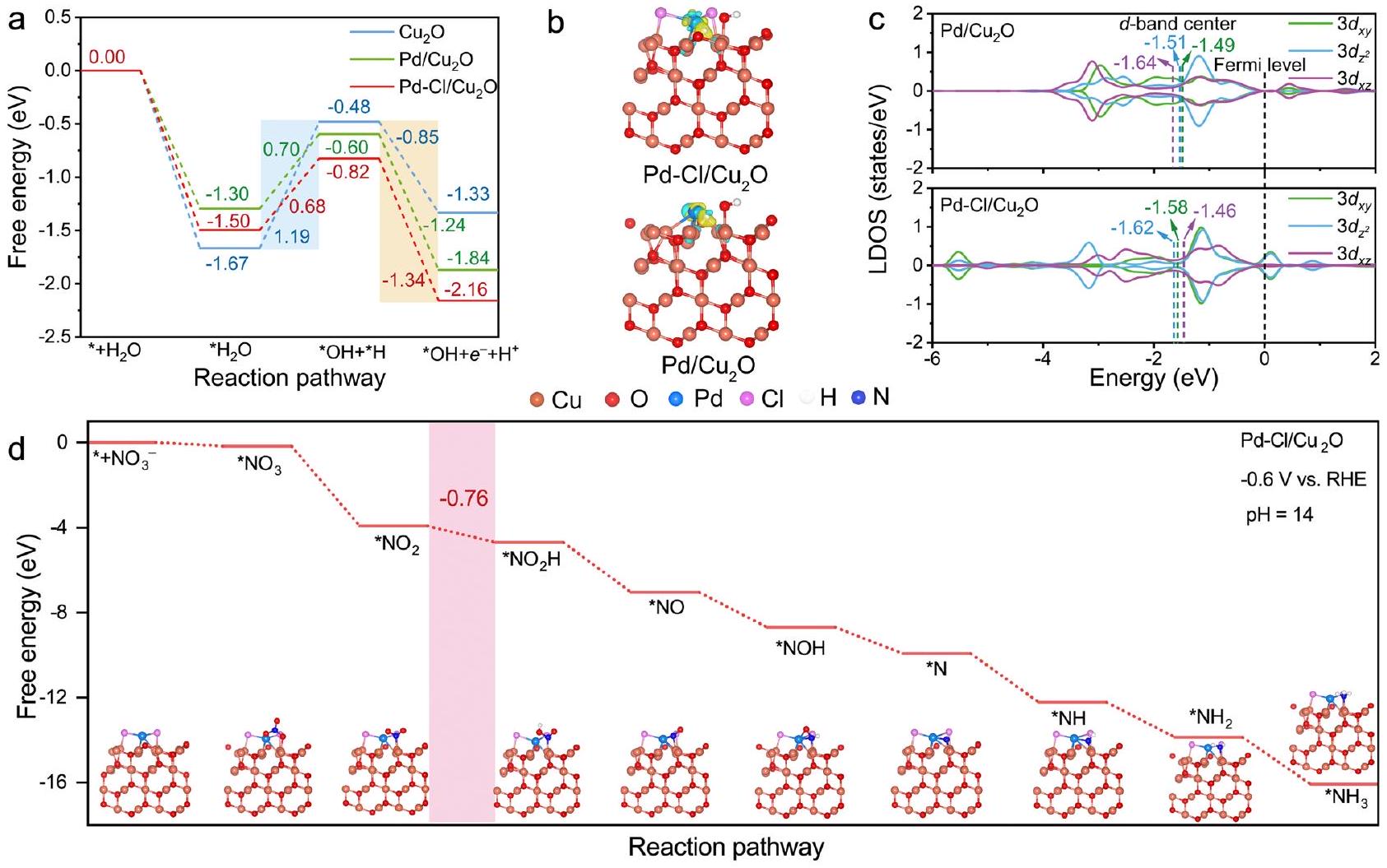
التنبؤ النظري
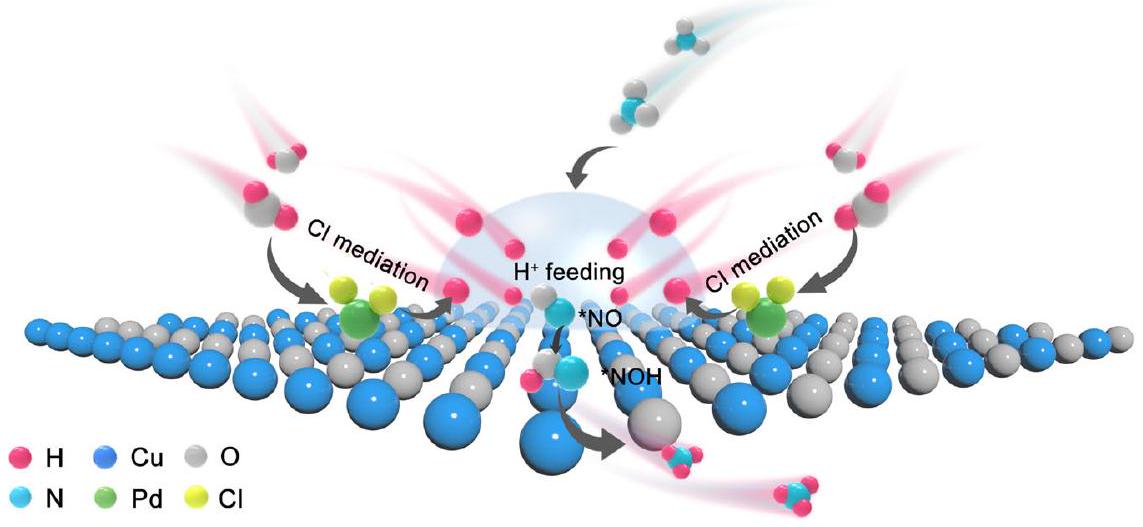
تم النقل من
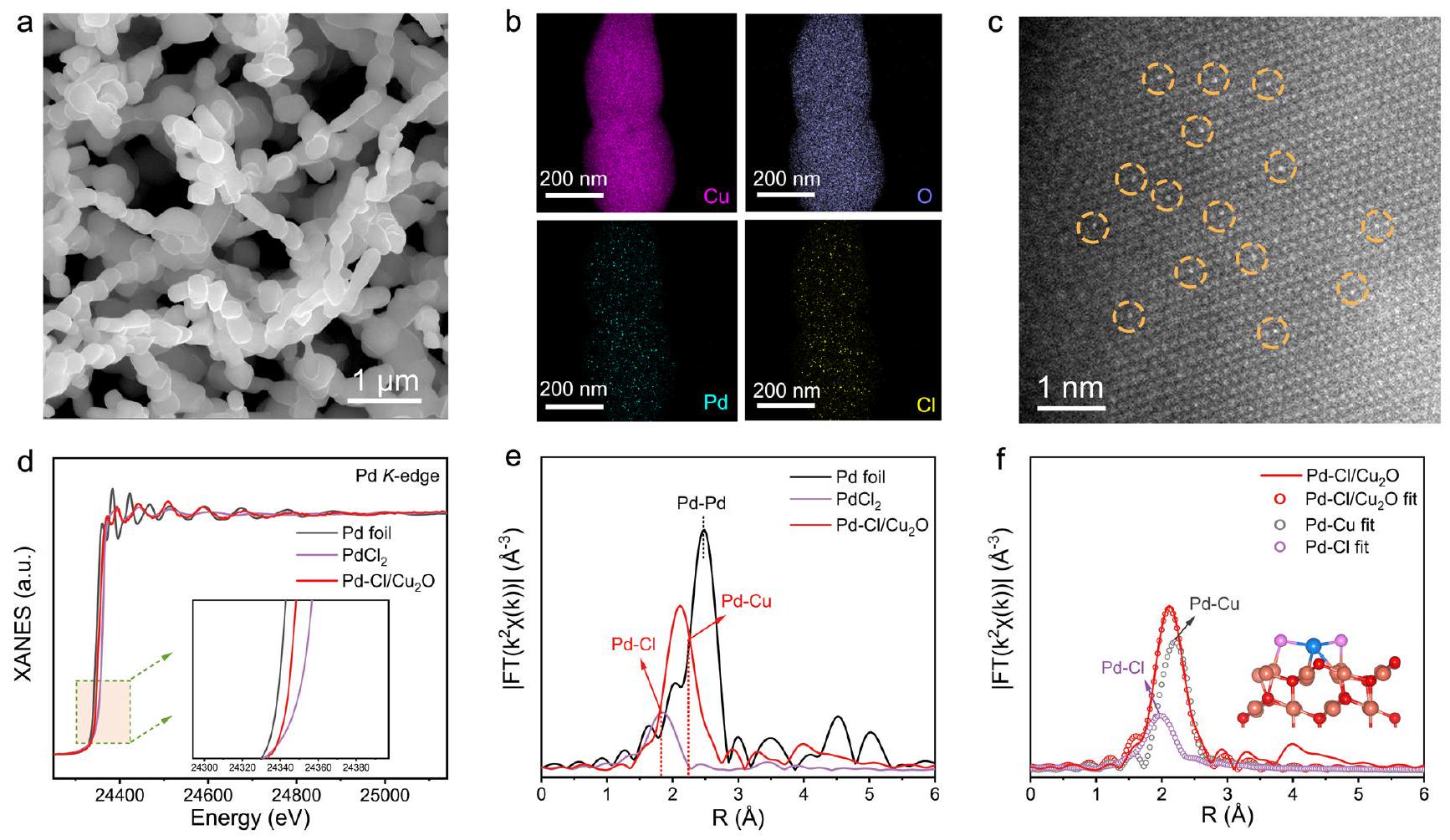
تركيب وتوصيف هيكلي للعوامل المساعدة
من Pd إلى Cl في
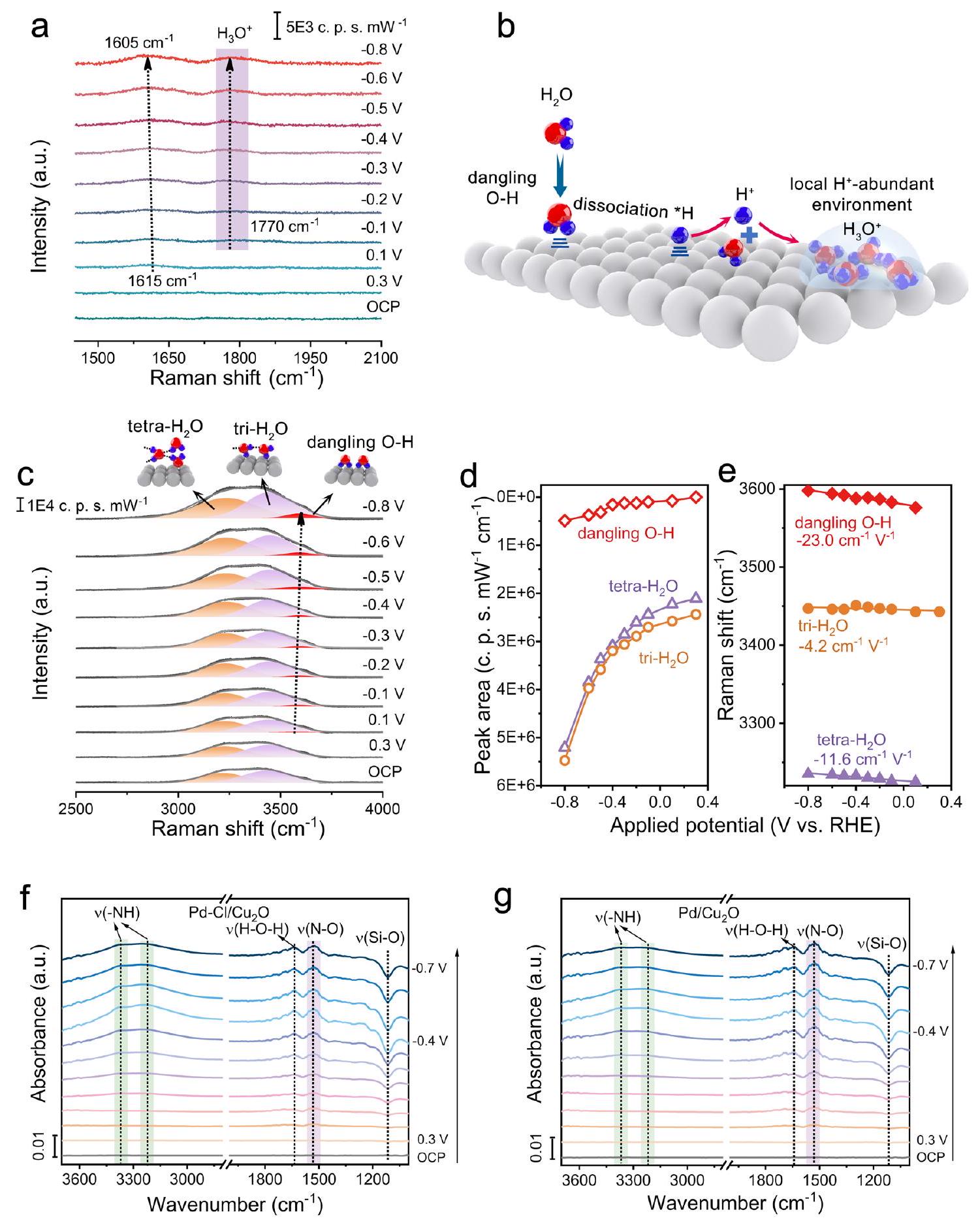
-إشارات NH (الشكل 4f، g). هذه النتائج أثبتت أن الوساطة بواسطة Cl
قلوي
الارتباط القوي لـ *H على Pd، الذي أدى إلى التفاعل الجانبي لتكوين ثنائي الهيدروجين (الأشكال التكميلية 41، 42). تحت تأثير الكلور.
معدل التحويل لـ
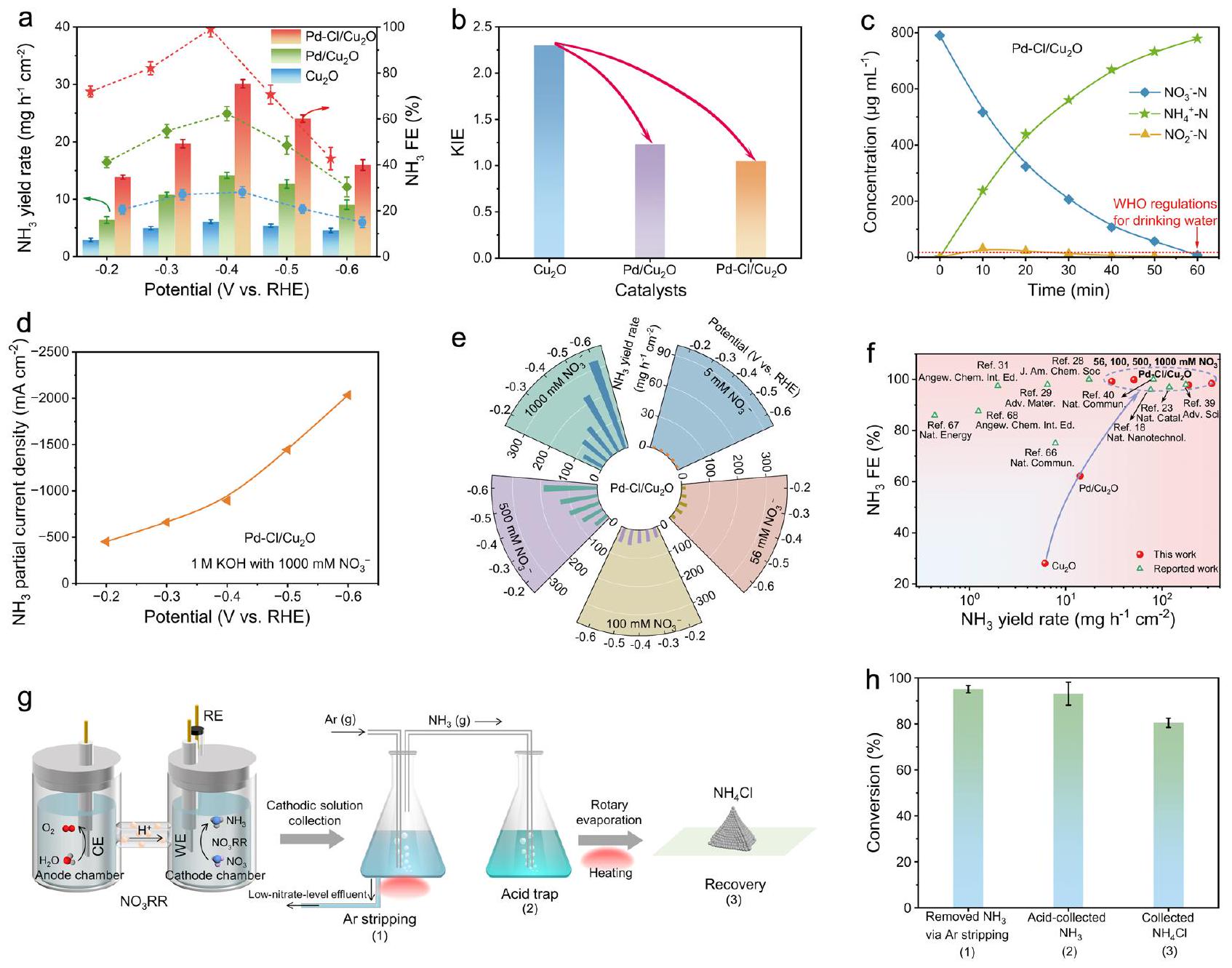
وصلت الانتقائية المقابلة
مياه الصرف الصناعي الثقيل (الشكل التوضيحي التكميلي 55، 56). لم يُظهر المحفز فقط أداءً ممتازًا
تشير إلى نزع النيتروجين من مصدر المياه بكفاءة عالية وإنتاج متزامن لمنتجات ذات قيمة مضافة عالية
طرق
المواد الكيميائية
ثنائي هيدروكلوريد الإيثيلين ديامين (
تحضير
تحضير
تحضير
تحضير
الاختبار الكهروكيميائي
تم الإشارة إلى العمل على مقياس RHE من خلال المعايرة بواسطة المعادلة التالية:
كشف وقياس
كشف وقياس
كشف وقياس
كشف وقياس
طيف رامان الكهروكيميائي في الموقع
طيف الأشعة تحت الحمراء ATR-IR الكهروكيميائي في الموقع
اختبارات DEMS الكهربائية الكيميائية عبر الإنترنت
تركيب منتج الأمونيا المباشر
تفاصيل الحسابات باستخدام تحويل فورييه السريع
حساب التصحيحات الديناميكية الحرارية
يتم حساب طاقة النقطة الصفرية لكل نوع بواسطة
محسوب بواسطة
الطور عند 300 كلفن، كما تم حسابه من ما يلي:
توفر البيانات
References
- Katsounaros, I., Dortsiou, M. & Kyriacou, G. Electrochemical reduction of nitrate and nitrite in simulated liquid nuclear wastes. J. Hazard. Mater. 171, 323-327 (2009).
- Burow, K. R., Nolan, B. T., Rupert, M. G. & Dubrovsky, N. M. Nitrate in groundwater of the united states, 1991-2003. Environ. Sci. Technol. 44, 4988-4997 (2010).
- Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, eaar6611 (2018).
- Galloway, J. N. et al. Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science 320, 889-892 (2008).
- Smolders, A. J. P., Lucassen, E. C. H. E. T., Bobbink, R., Roelofs, J. G. M. & Lamers, L. P. M. How nitrate leaching from agricultural lands provokes phosphate eutrophication in groundwater fed wetlands: The sulphur bridge. Biogeochemistry 98, 1-7 (2009).
- Picetti, R. et al. Nitrate and nitrite contamination in drinking water and cancer risk: A systematic review with meta-analysis. Environ. Res. 210, 112988 (2022).
- Knobeloch, L., Salna, B., Hogan, A., Postle, J. & Anderson, H. Blue babies and nitrate-contaminated well water. Environ. Health Persp. 108, 675-678 (2000).
- Shrimali, M. & Singh, K. P. New methods of nitrate removal from water. Environ. Pollut. 112, 351-359 (2001).
- Rezvani, F., Sarrafzadeh, M. H., Ebrahimi, S. & Oh, H. M. Nitrate removal from drinking water with a focus on biological methods: A review. Environ. Sci. Pollut. Res. 26, 1124-1141 (2019).
- Meng, G. et al. NiFe layered double hydroxide nanosheet array for high-efficiency electrocatalytic reduction of nitric oxide to ammonia. Chem. Commun. 58, 8097-8100 (2022).
- Zhang, S. et al. Electrocatalytic reduction of NO to
in ionic liquids by P-doped nanotubes. Front. Chem. Sci. Eng. 17, 726-734 (2023). - Zhang, W. et al. Single atomic cerium sites anchored on nitrogendoped hollow carbon spheres for highly selective electroreduction of nitric oxide to ammonia. J. Colloid Interface Sci. 638, 650-657 (2023).
- Duca, M. & Koper, M. T. M. Powering denitrification: The perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726 (2012).
- Xu, H., Ma, Y., Chen, J., Zhang, W. X. & Yang, J. Electrocatalytic reduction of nitrate – a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710-2758 (2022).
- Qi, D. et al. High-efficiency electrocatalytic NO reduction to
by nanoporous VN. Nano Res. Energy 1, e9120022 (2022). - Wu, X. et al. Contrasting capability of single atom palladium for thermocatalytic versus electrocatalytic nitrate reduction reaction. ACS Catal. 13, 6804-6812 (2023).
- Van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290-294 (2021).
- Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759-767 (2022).
- Liao, W. et al. Boosting nitrogen activation via Ag nanoneedle arrays for efficient ammonia synthesis. ACS Nano 17, 411-420 (2023).
- Liao, W. et al. Interfacial engineering promoting electrosynthesis of ammonia over Mo/phosphotungstic acid with high performance. Adv. Funct. Mater. 31, 2009151 (2021).
- Ding, J. et al. Iron-doping strategy promotes electroreduction of nitrate to ammonia on
nanosheets. Inorg. Chem. Commun. 151, 110621 (2023). - Dhamole, P. B., Nair, R. R., D’Souza, S. F. & Lele, S. S. Denitrification of highly alkaline nitrate waste using adapted sludge. Appl. Biochem. Biotechnol. 151, 433-440 (2008).
- Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 6, 402-414 (2023).
- Gao, Q. et al. Breaking adsorption-energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine-learned insights. Nat. Commun. 13, 2338 (2022).
- Pérez-Gallent, E., Figueiredo, M. C., Katsounaros, I. & Koper, M. T. M. Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions. Electrochim. Acta 227, 77-84 (2017).
- Song, Z. et al. Efficient electroreduction of nitrate into ammonia at ultralow concentrations via an enrichment effect. Adv. Mater. 34, e2204306 (2022).
- Li, P., Jin, Z., Fang, Z. & Yu, G. A single-site iron catalyst with preoccupied active centers that achieves selective ammonia electrosynthesis from nitrate. Energy Environ. Sci. 14, 3522-3531 (2021).
- Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036-7046 (2020).
- Gao, W. et al. Alloying of Cu with Ru enabling the relay catalysis for reduction of nitrate to ammonia. Adv. Mater. 35, 2202952 (2023).
- Wang, Y. et al. Enhanced nitrate-to-ammonia activity on coppernickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702-5708 (2020).
- Wang, Y. et al. Structurally disordered
nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 61, e202202604 (2022). - Jiang, M. et al. Batch-scale synthesis of nanoparticle-agminated three-dimensional porous
microspheres for highly selective electrocatalysis of nitrate to ammonia. Environ. Sci. Technol. 56, 10299-10307 (2022). - Yin, D. et al. Synergistic active phases of transition metal oxide heterostructures for highly efficient ammonia electrosynthesis. Adv. Funct. Mater. 33, 2303803 (2023).
- Hu, C., Zhang, L. & Gong, J. Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 12, 2620-2645 (2019).
- Valenti, G. et al. Co-axial heterostructures integrating palladium/ titanium dioxide with carbon nanotubes for efficient electrocatalytic hydrogen evolution. Nat. Commun. 7, 13549 (2016).
- Zhu, K. et al. Unraveling the role of interfacial water structure in electrochemical semihydrogenation of alkynes. ACS Catal. 12, 4840-4847 (2022).
- Li, Y. et al. Near-surface dilution of trace Pd atoms to facilitate Pd-H bond cleavage for giant enhancement of electrocatalytic hydrogen evolution. Nano Energy 34, 306-312 (2017).
- Zhang, T. et al. Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction. Nat. Commun. 13, 6875 (2022).
- Deng, X., Yang, Y., Wang, L., Fu, X. Z. & Luo, J. L. Metallic Co nanoarray catalyzes selective
production from electrochemical nitrate reduction at current densities exceeding . Adv. Sci. 8, 2004523 (2021). - Fang, J.-Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
- He, D. et al. Regulation of the electrocatalytic nitrogen cycle based on sequential proton-electron transfer. Nat. Catal. 5, 798-806 (2022).
- Koper, M. T. M. Theory of multiple proton-electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 4, 2710-2723 (2013).
- Zhang, Y. et al. Photoelectrocatalytic reduction of
to syngas via -enhanced nanowires photocathodes. Adv. Funct. Mater. 32, 2109600 (2021). - Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350-5354 (2020).
- Du, P. et al. Single-atom-driven dynamic carburization over
catalyst boosting conversion. Chem 8, 3252-3262 (2022). - Han, Y. et al. Electronic structure engineering to boost oxygen reduction activity by controlling the coordination of the central metal. Energy Environ. Sci. 11, 2348-2352 (2018).
- Zhang, B. et al. Manganese acting as a high-performance heterogeneous electrocatalyst in carbon dioxide reduction. Nat. Commun. 10, 2980 (2019).
- Li, C.-Y. et al. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 18, 697-701 (2019).
- Cai, C. et al. Atomically local electric field induced interface water reorientation for alkaline hydrogen evolution reaction. Angew. Chem. Int. Ed. 62, e202300873 (2023).
- Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81-85 (2021).
- Wang, X., Xu, C., Jaroniec, M., Zheng, Y. & Qiao, S. Z. Anomalous hydrogen evolution behavior in high-pH environment induced by locally generated hydronium ions. Nat. Commun. 10, 4876 (2019).
- Tan, H. et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 13, 2024 (2022).
- Hu, B. & Li, J. One electron makes differences:From heme
to . Angew. Chem. Int. Ed. 54, 10579-10582 (2015). - Yao, Y., Zhu, S., Wang, H., Li, H. & Shao, M. A spectroscopic study of electrochemical nitrogen and nitrate reduction on rhodium surfaces. Angew. Chem. Int. Ed. 59, 10479-10483 (2020).
- Wirz, R., Ferri, D. & Baiker, A. ATR-IR spectroscopy of pendant
groups on silica involved in the knoevenagel condensation. Langmuir 22, 3698-3706 (2006). - Wiles, D. M. & Suprunchuk, T. The infrared absorption spectra of thiosemicarbazide and related compounds:
and NH vibrations. Can. J. Chem. 47, 1087-1089 (1969). - Milligan, D. E. & Jacox, M. E. Matrix-isolation infrared spectrum of the free radical
. J. Chem. Phys. 43, 4487-4493 (1965). - Zheng, W. et al. Self-activated Ni cathode for electrocatalytic nitrate reduction to ammonia: From fundamentals to scale-up for treatment of industrial wastewater. Environ. Sci. Technol. 55, 13231-13243 (2021).
- Albina, P. et al. Influence of hydrogen electron donor, alkaline pH , and high nitrate concentrations on microbial denitrification: A review. Int. J. Mol. Sci. 20, 5163 (2019).
- Ahmadi, M. T., Bodaghzadeh, M., Rahimian Koloor, S. S. & Petru, M. Graphene nanoparticle-based, nitrate ion sensor characteristics. Nanomaterials Basel 11, 150 (2021).
- Liu, H. et al. Electrocatalytic nitrate reduction on oxide-derived silver with tunable selectivity to nitrite and ammonia. ACS Catal. 11, 8431-8442 (2021).
- Ma, W. et al. Promoting electrocatalytic
reduction to formate via sulfur-boosting water activation on indium surfaces. Nat. Commun. 10, 892 (2019). - Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped Fe-N-C single-atom catalyst for enhanced
electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022). - Su, L. et al. Electrochemical nitrate reduction by using a novel
cathode. Water Res. 120, 1-11 (2017). - Chauhan, R. & Srivastava, V. C. Electrochemical denitrification of highly contaminated actual nitrate wastewater by
anode and iron cathode. Chem. Eng. J. 386, 122065 (2020). - Wu, Z. Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
- Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper-molecular solid catalyst. Nat. Energy 5, 605-613 (2020).
- Du, H. et al. Durable electrocatalytic reduction of nitrate to ammonia over defective pseudobrookite
nanofibers with abundant oxygen vacancies. Angew. Chem. Int. Ed. 62, e202215782 (2023). - Liao, P. H., Chen, A. & Lo, K. V. Removal of nitrogen from swine manure wastewaters by ammonia stripping. Bioresour. Technol. 54, 17-20 (1995).
- Yuan, M.-H., Chen, Y.-H., Tsai, J.-Y. & Chang, C.-Y. Ammonia removal from ammonia-rich wastewater by air stripping using a rotating packed bed. Process Saf. Environ. Prot. 102, 777-785 (2016).
- Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209-2244 (2009).
- Searle, P. L. The berthelot or indophenol reaction and its use in the analytical chemistry of nitrogen. Analyst 109, 549-568 (1984).
- Carvalho, A. P., Meireles, L. A. & Malcata, F. X. Rapid spectrophotometric determination of nitrates and nitrites in marine aqueous culture media. Analusis 26, 347-351 (1998).
- Polatides, C. & Kyriacou, G. Electrochemical reduction of nitrate ion on various cathodes – reaction kinetics on bronze cathode. J. Appl. Electrochem. 35, 421-427 (2005).
- Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15-50 (1996).
- Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
- Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
- Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
- Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
- Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouinzone integration in metals. Phys. Rev. B 40, 3616-3621 (1989).
- Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886-17892 (2004).
- Duan, Z. & Xiao, P. Simulation of potential-dependent activation energies in electrocatalysis: Mechanism of O-O bond formation on
. J. Phys. Chem. C 125, 15243-15250 (2021). - Ochterski, J. W. Thermochemistry in Gaussian (Gaussian inc, 2000).
- Gupta, S., Rivera D. J., Shaffer M., Chismar A., Muhich C. Behavior of cupric single atom alloy catalysts for electrochemical nitrate reduction: An ab initio study. ACS EST Engg. https://doi.org/10. 1021/acsestengg.1023c00207 (2023).
- Calle-Vallejo, F., Huang, M., Henry, J. B., Koper, M. T. M. & Bandarenka, A. S. Theoretical design and experimental implementation of
Au electrodes for the electrochemical reduction of nitrate. Phys. Chem. Chem. Phys. 15, 3196 (2013). - Liu, J.-X., Richards, D., Singh, N. & Goldsmith, B. R. Activity and selectivity trends in electrocatalytic nitrate reduction on transition metals. ACS Catal. 9, 7052-7064 (2019).
- Qiao, M., Liu, J., Wang, Y., Li, Y. & Chen, Z. PdSeO
monolayer: Promising inorganic 2D photocatalyst for direct overall water splitting without using sacrificial reagents and cocatalysts. J. Am. Chem. Soc. 140, 12256-12262 (2018).
شكر وتقدير
ممتنون للموارد من مركز الحوسبة عالية الأداء في جامعة جنوب الوسطى. نود أن نعرب عن شكرنا للمساعدة من خطوط الشعاع BL01C1 في مركز أبحاث الإشعاع السنكروتروني الوطني (NSRRC، هسينتشو، تايوان) للقياسات المختلفة المعتمدة على السنكروترون.
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية المتاحة على
https://doi.org/10.1038/s41467-024-45534-2.
http://www.nature.com/reprints
© المؤلفون 2024
مركز الأبحاث الدولية المشترك في هونان لاستغلال موارد ثاني أكسيد الكربون، المختبر الوطني الرئيسي لتكنولوجيا المساحيق، كلية الفيزياء، جامعة جنوب الوسطى، تشانغشا 410083، جمهورية الصين الشعبية. مركز أبحاث وابتكار المواد الميتامادية، قسم الهندسة، جامعة إكستر، إكستر EX4 4QF، المملكة المتحدة. كلية الكيمياء والهندسة الكيميائية، جامعة جنوب الوسطى، تشانغشا 410083، جمهورية الصين الشعبية. مركز أبحاث الإشعاع السنكروتروني الوطني، هسينتشو 300092، تايوان. كلية علوم وهندسة المواد، جامعة هونان، تشانغشا 410082، جمهورية الصين الشعبية. كلية علوم وهندسة الغذاء، جامعة جنوب الوسطى للغابات والتكنولوجيا، تشانغشا 410004، جمهورية الصين الشعبية. كلية البيئة والطاقة، المختبر الرئيسي لمقاطعة قوانغدونغ للتحكم في تلوث النفايات الصلبة وإعادة التدوير، جامعة جنوب الصين للتكنولوجيا، قوانغتشو 510006، جمهورية الصين الشعبية. المختبر الرئيسي للدولة لمعايير البيئة وتقييم المخاطر، الأكاديمية الصينية للعلوم البيئية، 100012 بكين، جمهورية الصين الشعبية. ساهم هؤلاء المؤلفون بالتساوي: وانرو لياو، جون وانغ. البريد الإلكتروني:fujunwei@csu.edu.cn; xibd@craes.org.cn; minliu@csu.edu.cn
DOI: https://doi.org/10.1038/s41467-024-45534-2
PMID: https://pubmed.ncbi.nlm.nih.gov/38341446
Publication Date: 2024-02-10
Sustainable conversion of alkaline nitrate to ammonia at activities greater than
Accepted: 25 January 2024
Published online: 10 February 2024
(D) Check for updates
Abstract
Nitrate
of Pd atoms to construct local
Results
Theoretical prediction
transferred from
Synthesis and structural characterizations of catalysts
from Pd to Cl in
-NH signals (Fig. 4f, g). These results evidenced that Cl -mediated
Alkaline
the strong binding of *H on Pd, which induced the side reaction of hydrogen-hydrogen dimerization (Supplementary Figs. 41, 42). Under the Cl -mediated
conversion rate of
the corresponding selectivity reached
heavy-industry wastewater (Supplementary Fig. 55, 56). The catalyst not only exhibited an excellent
indicating water source denitrification with high efficiency and simultaneous production of high value-added
Methods
Chemicals
ethylenediamine dihydrochloride (
Preparation of
Preparation of
Preparation of
Preparation of
Electrochemical testing
work were referred to RHE scale via calibration by the following equation:
Detection and quantification of
Detection and quantification of
Detection and quantification of
Detection and quantification of
Electrochemical in situ Raman spectroscopy
Electrochemical in situ ATR-IR spectroscopy
Electrochemical online DEMS tests
Direct ammonia product synthesis
DFT computational details
Calculation of thermodynamic corrections
The zero-point energy for each species is calculate by
calculated by
phase at 300 K , as calculated from the following:
Data availability
References
- Katsounaros, I., Dortsiou, M. & Kyriacou, G. Electrochemical reduction of nitrate and nitrite in simulated liquid nuclear wastes. J. Hazard. Mater. 171, 323-327 (2009).
- Burow, K. R., Nolan, B. T., Rupert, M. G. & Dubrovsky, N. M. Nitrate in groundwater of the united states, 1991-2003. Environ. Sci. Technol. 44, 4988-4997 (2010).
- Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, eaar6611 (2018).
- Galloway, J. N. et al. Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science 320, 889-892 (2008).
- Smolders, A. J. P., Lucassen, E. C. H. E. T., Bobbink, R., Roelofs, J. G. M. & Lamers, L. P. M. How nitrate leaching from agricultural lands provokes phosphate eutrophication in groundwater fed wetlands: The sulphur bridge. Biogeochemistry 98, 1-7 (2009).
- Picetti, R. et al. Nitrate and nitrite contamination in drinking water and cancer risk: A systematic review with meta-analysis. Environ. Res. 210, 112988 (2022).
- Knobeloch, L., Salna, B., Hogan, A., Postle, J. & Anderson, H. Blue babies and nitrate-contaminated well water. Environ. Health Persp. 108, 675-678 (2000).
- Shrimali, M. & Singh, K. P. New methods of nitrate removal from water. Environ. Pollut. 112, 351-359 (2001).
- Rezvani, F., Sarrafzadeh, M. H., Ebrahimi, S. & Oh, H. M. Nitrate removal from drinking water with a focus on biological methods: A review. Environ. Sci. Pollut. Res. 26, 1124-1141 (2019).
- Meng, G. et al. NiFe layered double hydroxide nanosheet array for high-efficiency electrocatalytic reduction of nitric oxide to ammonia. Chem. Commun. 58, 8097-8100 (2022).
- Zhang, S. et al. Electrocatalytic reduction of NO to
in ionic liquids by P-doped nanotubes. Front. Chem. Sci. Eng. 17, 726-734 (2023). - Zhang, W. et al. Single atomic cerium sites anchored on nitrogendoped hollow carbon spheres for highly selective electroreduction of nitric oxide to ammonia. J. Colloid Interface Sci. 638, 650-657 (2023).
- Duca, M. & Koper, M. T. M. Powering denitrification: The perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726 (2012).
- Xu, H., Ma, Y., Chen, J., Zhang, W. X. & Yang, J. Electrocatalytic reduction of nitrate – a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710-2758 (2022).
- Qi, D. et al. High-efficiency electrocatalytic NO reduction to
by nanoporous VN. Nano Res. Energy 1, e9120022 (2022). - Wu, X. et al. Contrasting capability of single atom palladium for thermocatalytic versus electrocatalytic nitrate reduction reaction. ACS Catal. 13, 6804-6812 (2023).
- Van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290-294 (2021).
- Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759-767 (2022).
- Liao, W. et al. Boosting nitrogen activation via Ag nanoneedle arrays for efficient ammonia synthesis. ACS Nano 17, 411-420 (2023).
- Liao, W. et al. Interfacial engineering promoting electrosynthesis of ammonia over Mo/phosphotungstic acid with high performance. Adv. Funct. Mater. 31, 2009151 (2021).
- Ding, J. et al. Iron-doping strategy promotes electroreduction of nitrate to ammonia on
nanosheets. Inorg. Chem. Commun. 151, 110621 (2023). - Dhamole, P. B., Nair, R. R., D’Souza, S. F. & Lele, S. S. Denitrification of highly alkaline nitrate waste using adapted sludge. Appl. Biochem. Biotechnol. 151, 433-440 (2008).
- Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 6, 402-414 (2023).
- Gao, Q. et al. Breaking adsorption-energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine-learned insights. Nat. Commun. 13, 2338 (2022).
- Pérez-Gallent, E., Figueiredo, M. C., Katsounaros, I. & Koper, M. T. M. Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions. Electrochim. Acta 227, 77-84 (2017).
- Song, Z. et al. Efficient electroreduction of nitrate into ammonia at ultralow concentrations via an enrichment effect. Adv. Mater. 34, e2204306 (2022).
- Li, P., Jin, Z., Fang, Z. & Yu, G. A single-site iron catalyst with preoccupied active centers that achieves selective ammonia electrosynthesis from nitrate. Energy Environ. Sci. 14, 3522-3531 (2021).
- Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036-7046 (2020).
- Gao, W. et al. Alloying of Cu with Ru enabling the relay catalysis for reduction of nitrate to ammonia. Adv. Mater. 35, 2202952 (2023).
- Wang, Y. et al. Enhanced nitrate-to-ammonia activity on coppernickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702-5708 (2020).
- Wang, Y. et al. Structurally disordered
nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 61, e202202604 (2022). - Jiang, M. et al. Batch-scale synthesis of nanoparticle-agminated three-dimensional porous
microspheres for highly selective electrocatalysis of nitrate to ammonia. Environ. Sci. Technol. 56, 10299-10307 (2022). - Yin, D. et al. Synergistic active phases of transition metal oxide heterostructures for highly efficient ammonia electrosynthesis. Adv. Funct. Mater. 33, 2303803 (2023).
- Hu, C., Zhang, L. & Gong, J. Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 12, 2620-2645 (2019).
- Valenti, G. et al. Co-axial heterostructures integrating palladium/ titanium dioxide with carbon nanotubes for efficient electrocatalytic hydrogen evolution. Nat. Commun. 7, 13549 (2016).
- Zhu, K. et al. Unraveling the role of interfacial water structure in electrochemical semihydrogenation of alkynes. ACS Catal. 12, 4840-4847 (2022).
- Li, Y. et al. Near-surface dilution of trace Pd atoms to facilitate Pd-H bond cleavage for giant enhancement of electrocatalytic hydrogen evolution. Nano Energy 34, 306-312 (2017).
- Zhang, T. et al. Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction. Nat. Commun. 13, 6875 (2022).
- Deng, X., Yang, Y., Wang, L., Fu, X. Z. & Luo, J. L. Metallic Co nanoarray catalyzes selective
production from electrochemical nitrate reduction at current densities exceeding . Adv. Sci. 8, 2004523 (2021). - Fang, J.-Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
- He, D. et al. Regulation of the electrocatalytic nitrogen cycle based on sequential proton-electron transfer. Nat. Catal. 5, 798-806 (2022).
- Koper, M. T. M. Theory of multiple proton-electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 4, 2710-2723 (2013).
- Zhang, Y. et al. Photoelectrocatalytic reduction of
to syngas via -enhanced nanowires photocathodes. Adv. Funct. Mater. 32, 2109600 (2021). - Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350-5354 (2020).
- Du, P. et al. Single-atom-driven dynamic carburization over
catalyst boosting conversion. Chem 8, 3252-3262 (2022). - Han, Y. et al. Electronic structure engineering to boost oxygen reduction activity by controlling the coordination of the central metal. Energy Environ. Sci. 11, 2348-2352 (2018).
- Zhang, B. et al. Manganese acting as a high-performance heterogeneous electrocatalyst in carbon dioxide reduction. Nat. Commun. 10, 2980 (2019).
- Li, C.-Y. et al. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 18, 697-701 (2019).
- Cai, C. et al. Atomically local electric field induced interface water reorientation for alkaline hydrogen evolution reaction. Angew. Chem. Int. Ed. 62, e202300873 (2023).
- Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81-85 (2021).
- Wang, X., Xu, C., Jaroniec, M., Zheng, Y. & Qiao, S. Z. Anomalous hydrogen evolution behavior in high-pH environment induced by locally generated hydronium ions. Nat. Commun. 10, 4876 (2019).
- Tan, H. et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 13, 2024 (2022).
- Hu, B. & Li, J. One electron makes differences:From heme
to . Angew. Chem. Int. Ed. 54, 10579-10582 (2015). - Yao, Y., Zhu, S., Wang, H., Li, H. & Shao, M. A spectroscopic study of electrochemical nitrogen and nitrate reduction on rhodium surfaces. Angew. Chem. Int. Ed. 59, 10479-10483 (2020).
- Wirz, R., Ferri, D. & Baiker, A. ATR-IR spectroscopy of pendant
groups on silica involved in the knoevenagel condensation. Langmuir 22, 3698-3706 (2006). - Wiles, D. M. & Suprunchuk, T. The infrared absorption spectra of thiosemicarbazide and related compounds:
and NH vibrations. Can. J. Chem. 47, 1087-1089 (1969). - Milligan, D. E. & Jacox, M. E. Matrix-isolation infrared spectrum of the free radical
. J. Chem. Phys. 43, 4487-4493 (1965). - Zheng, W. et al. Self-activated Ni cathode for electrocatalytic nitrate reduction to ammonia: From fundamentals to scale-up for treatment of industrial wastewater. Environ. Sci. Technol. 55, 13231-13243 (2021).
- Albina, P. et al. Influence of hydrogen electron donor, alkaline pH , and high nitrate concentrations on microbial denitrification: A review. Int. J. Mol. Sci. 20, 5163 (2019).
- Ahmadi, M. T., Bodaghzadeh, M., Rahimian Koloor, S. S. & Petru, M. Graphene nanoparticle-based, nitrate ion sensor characteristics. Nanomaterials Basel 11, 150 (2021).
- Liu, H. et al. Electrocatalytic nitrate reduction on oxide-derived silver with tunable selectivity to nitrite and ammonia. ACS Catal. 11, 8431-8442 (2021).
- Ma, W. et al. Promoting electrocatalytic
reduction to formate via sulfur-boosting water activation on indium surfaces. Nat. Commun. 10, 892 (2019). - Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped Fe-N-C single-atom catalyst for enhanced
electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022). - Su, L. et al. Electrochemical nitrate reduction by using a novel
cathode. Water Res. 120, 1-11 (2017). - Chauhan, R. & Srivastava, V. C. Electrochemical denitrification of highly contaminated actual nitrate wastewater by
anode and iron cathode. Chem. Eng. J. 386, 122065 (2020). - Wu, Z. Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
- Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper-molecular solid catalyst. Nat. Energy 5, 605-613 (2020).
- Du, H. et al. Durable electrocatalytic reduction of nitrate to ammonia over defective pseudobrookite
nanofibers with abundant oxygen vacancies. Angew. Chem. Int. Ed. 62, e202215782 (2023). - Liao, P. H., Chen, A. & Lo, K. V. Removal of nitrogen from swine manure wastewaters by ammonia stripping. Bioresour. Technol. 54, 17-20 (1995).
- Yuan, M.-H., Chen, Y.-H., Tsai, J.-Y. & Chang, C.-Y. Ammonia removal from ammonia-rich wastewater by air stripping using a rotating packed bed. Process Saf. Environ. Prot. 102, 777-785 (2016).
- Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209-2244 (2009).
- Searle, P. L. The berthelot or indophenol reaction and its use in the analytical chemistry of nitrogen. Analyst 109, 549-568 (1984).
- Carvalho, A. P., Meireles, L. A. & Malcata, F. X. Rapid spectrophotometric determination of nitrates and nitrites in marine aqueous culture media. Analusis 26, 347-351 (1998).
- Polatides, C. & Kyriacou, G. Electrochemical reduction of nitrate ion on various cathodes – reaction kinetics on bronze cathode. J. Appl. Electrochem. 35, 421-427 (2005).
- Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15-50 (1996).
- Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
- Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
- Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
- Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
- Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouinzone integration in metals. Phys. Rev. B 40, 3616-3621 (1989).
- Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886-17892 (2004).
- Duan, Z. & Xiao, P. Simulation of potential-dependent activation energies in electrocatalysis: Mechanism of O-O bond formation on
. J. Phys. Chem. C 125, 15243-15250 (2021). - Ochterski, J. W. Thermochemistry in Gaussian (Gaussian inc, 2000).
- Gupta, S., Rivera D. J., Shaffer M., Chismar A., Muhich C. Behavior of cupric single atom alloy catalysts for electrochemical nitrate reduction: An ab initio study. ACS EST Engg. https://doi.org/10. 1021/acsestengg.1023c00207 (2023).
- Calle-Vallejo, F., Huang, M., Henry, J. B., Koper, M. T. M. & Bandarenka, A. S. Theoretical design and experimental implementation of
Au electrodes for the electrochemical reduction of nitrate. Phys. Chem. Chem. Phys. 15, 3196 (2013). - Liu, J.-X., Richards, D., Singh, N. & Goldsmith, B. R. Activity and selectivity trends in electrocatalytic nitrate reduction on transition metals. ACS Catal. 9, 7052-7064 (2019).
- Qiao, M., Liu, J., Wang, Y., Li, Y. & Chen, Z. PdSeO
monolayer: Promising inorganic 2D photocatalyst for direct overall water splitting without using sacrificial reagents and cocatalysts. J. Am. Chem. Soc. 140, 12256-12262 (2018).
Acknowledgements
grateful for resources from the High Performance Computing Center of Central South University. We would like to acknowledge the help from Beam Lines BL01C1 in the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan) for various synchrotron-based measurements.
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-45534-2.
http://www.nature.com/reprints
© The Author(s) 2024
Hunan Joint International Research Center for Carbon Dioxide Resource Utilization, State Key Laboratory of Powder Metallurgy, School of Physics, Central South University, Changsha 410083, PR China. Centre for Metamaterial Research & Innovation, Department of Engineering, University of Exeter, Exeter EX4 4QF, UK. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, PR China. National Synchrotron Radiation Research Center, Hsinchu 300092, Taiwan. College of Materials Science and Engineering, Hunan University, Changsha 410082 , PR China. College of Food Science and Engineering, Central South University of Forestry and Technology, Changsha 410004, PR China. School of Environment and Energy, Guangdong Provincial Key Laboratory of Solid Wastes Pollution Control and Recycling, South China University of Technology, Guangzhou 510006, PR China. State Key Laboratory of Environmental Criteria and Risk Assessment, Chinese Research Academy of Environmental Sciences, 100012 Beijing, PR China. These authors contributed equally: Wanru Liao, Jun Wang. e-mail: fujunwei@csu.edu.cn; xibd@craes.org.cn; minliu@csu.edu.cn