DOI: https://doi.org/10.1038/s41467-024-44815-0
PMID: https://pubmed.ncbi.nlm.nih.gov/38228626
تاريخ النشر: 2024-01-16
التفاعل بين المعدن والدعم المحدد بالموقع لتغيير نشاط ذرات الإيريديوم الفردية في تفاعل تطور الأكسجين
تم القبول: 5 يناير 2024
نُشر على الإنترنت: 16 يناير 2024
(أ) التحقق من التحديثات
الملخص
يمكن أن تحدد التفاعلات بين المعدن والداعم (MSI) بشكل كبير الخصائص الإلكترونية لمحفزات الذرات الفردية، مما يؤثر على الأداء التحفيزي. ومع ذلك، فإن الطريقة النموذجية لتنظيم MSI عادة ما تعاني من تداخل تغير الداعمين أو التضحية باستقرار المحفزات. هنا، نقوم بتنظيم MSI المحدد للموقع لذرات Ir الفردية المثبتة على هيدروكسيد النيكل المزدوج الطبقات بشكل فعال من خلال استراتيجية ترسيب كيميائي كهربائي. يدفع الترسيب الكاثودي ذرات Ir لتتواجد في مواقع فراغية ثلاثية الأبعاد ذات مركز مكعب مع MSI قوي، بينما يدفع الترسيب الأنودي ذرات Ir لترسيبها على مواقع فراغ الأكسجين ذات MSI ضعيف. النشاط الكتلي والنشاط الجوهري لمحفزات الذرات الفردية من Ir ذات MSI القوي تجاه تفاعل تطور الأكسجين هما 19.5 و5.2 مرة مقارنة بتلك ذات MSI الضعيف، على التوالي. تكشف دراسة الآلية أن MSI القوي بين ذرات Ir والداعم يحفز نشاط مواقع Ir من خلال تحفيز التحول من مواقع Ni إلى مواقع Ir ويعمل على تحسين قوة الامتصاص للوسائط، مما يعزز النشاط.
دعم وإجراء علاج التخفيض باستخدام الهيدروجين
النتائج
التركيب والتوصيفات
مع رقائق الإيريديوم و
الأداء الكهروتحفيزي تجاه تفاعل تطور الأكسجين
تم استخدامها كعوامل كهربائية لتحفيز تفاعل الأكسدة (OER)، وهو أحد التفاعلات الأساسية لتحويل الطاقة النظيفة وتطبيقات التخزين.
تم استخدامها كمراجع.
فوق الجهد لمختلف المحفزات عند كثافة التيار
بالإضافة إلى ذلك، ظلت نشاط Ni LDH بعد المعالجة الكاثودية والأنيودية دون وجود سلفيد الإريديوم تقريبًا دون تغيير، مما يشير إلى أن الأداء المحسن لـ
(ICP-MS) (الشكل التكميلي 20). بعد اختبار المتانة، تم الحفاظ على التشتت الذري لذرات الإريديوم دون أي تكتل ملحوظ كما هو موضح في صورة HAADF-STEM ونتيجة رسم الخرائط العنصرية EDS (الشكل التكميلي 21a، b). علاوة على ذلك، نمط XRD وطيف XPS لـ
التوصيفات الطيفية في الموقع
تم تصنيفها في البداية بـ
حسابات DFT
حسبت الكثافة المتوقعة للحالات (PDOS) لتوضيح الفروق في التفاعلات الإلكترونية بين
نقاش
حسنت قوة الامتزاز للوسائط المحتوية على الأكسجين ولكنها أيضًا أدت إلى تحويل المواقع النشطة التحفيزية من مواقع النيكل إلى مواقع الإيريديوم، مما يعزز الأداء التحفيزي. يوفر هذا العمل استراتيجية جديدة لتصميم محفزات عالية النشاط مع MSI محدد المواقع من خلال تثبيت انتقائي كهربائيًا للذرات الفردية ويقدم فهمًا عميقًا للعلاقة بين MSI وأداء OER.
طرق
المواد الكيميائية
تركيب هيدروكسيد النيكل المائي
تركيب
توصيف المحفز
تم قياسه على نظام Veeco DI Nanoscope MultiMode V. تم جمع أنماط حيود الأشعة السينية باستخدام جهاز حيود الأشعة السينية (Philips X’Pert Pro Super) مع
القياسات الكهروكيميائية
قضيب كربوني و
طيف رامان في الموقع
طريقة حساب DFT
التقييم النظري لنشاط OER
تم حساب الجزيء على أنه
توفر البيانات
References
- Qiao, B. et al. Single-atom catalysis of CO oxidation using
. Nat. Chem. 3, 634-641 (2011). - Yang, H. et al. Intramolecular hydroxyl nucleophilic attack pathway by a polymeric water oxidation catalyst with single cobalt sites. Nat. Catal. 5, 414-429 (2022).
- Gu, J., Hsu, C. S., Bai, L., Chen, H. M. & Hu, X. Atomically dispersed
sites catalyze efficient electroreduction to CO . Science 364, 1091-1094 (2019). - Yang, J., Li, W., Wang, D. & Li, Y. Electronic metal-support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 32, 2003300 (2020).
- Zhang, J., Liu, C. & Zhang, B. Insights into single-atom metal-support interactions in electrocatalytic water splitting. Small Methods 3, 1800481 (2019).
- Jin, Z. Y. et al. Understanding the inter-site distance effect in singleatom catalysts for oxygen electroreduction. Nat. Catal. 4, 615-622 (2021).
- Sun, H. et al. Atomic metal-support interaction enables reconstruction-free dual-site electrocatalyst. J. Am. Chem. Soc. 144, 1174-1186 (2022).
- Jiang, Z. et al. Modulating the electronic metal-support interactions in single-atom Pt-CuO catalyst for boosting acetone oxidation. Angew. Chem. Int. Ed. 61, e202200763 (2022).
- Li, W. H. et al. Creating high regioselectivity by electronic metal-support interaction of a single-atomic-site catalyst. J. Am. Chem. Soc. 143, 15453-15461 (2021).
- Han, B. et al. Strong metal-support interactions between Pt single atoms and
. Angew. Chem. Int. Ed. 59, 11824-11829 (2020). - Li, J. et al. Highly active and stable metal single-atom catalysts achieved by strong electronic metal-support interactions. J. Am. Chem. Soc. 141, 14515-14519 (2019).
- Yang, J. et al. Modulating the strong metal-support interaction of single-atom catalysts via vicinal structure decoration. Nat. Commun. 13, 4244 (2022).
- Ren, Y. et al. Unraveling the coordination structure-performance relationship in
single-atom catalyst. Nat. Commun. 10, 4500 (2019). - Jeong, H. et al. Controlling the oxidation state of Pt single atoms for maximizing catalytic activity. Angew. Chem. Int. Ed. 59, 20691-20696 (2020).
- Lee, S., Banjac, K., Lingenfelder, M. & Hu, X. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 58, 10295-10299 (2019).
- Zheng, X. et al. Origin of enhanced water oxidation activity in an iridium single atom anchored on NiFe oxyhydroxide catalyst. Proc. Natl Acad. Sci. USA 118, e2101817118 (2021).
- Van Ooij, W. J. & Houtman, J. P. W. Ligand exchange in iridium hexahalocomplexes: Part 2. Influence of the pH on the reactions of the hexachloroiridite and hexachloroiridate ions in aqueous solutions. Radiochim. Acta 20, 47-50 (1973).
- Cui, C. et al. Atomically dispersed hybrid nickel-iridium sites for photoelectrocatalysis. Nat. Commun. 8, 1341 (2017).
- Zhang, Z. et al. Electrochemical deposition as a universal route for fabricating single-atom catalysts. Nat. Commun. 11, 1215 (2020).
- Yao, Y. C. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304-313 (2019).
- Lin, C. et al. In-situ reconstructed Ru atom array on
with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021). - Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: toward boosted and sustained water oxidation catalysis. Joule 5, 2164-2176 (2021).
- Zhang, J. et al. Single-atom Au/NiFe layered double hydroxide electrocatalyst: probing the origin of activity for oxygen evolution reaction. J. Am. Chem. Soc. 140, 3876-3879 (2018).
- Li, P. et al. Boosting oxygen evolution of single-atomic ruthenium through electronic coupling with cobalt-iron layered double hydroxides. Nat. Commun. 10, 1711 (2019).
- Wang, Q. et al. Ultrahigh-loading of Ir single atoms on NiO matrix to dramatically enhance oxygen evolution reaction. J. Am. Chem. Soc. 142, 7425-7433 (2020).
- Cao, L. et al. Dynamic oxygen adsorption on single-atomic ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
- Yin, J. et al. Iridium single atoms coupling with oxygen vacancies boosts oxygen evolution reaction in acid media. J. Am. Chem. Soc. 142, 18378-18386 (2020).
- Wang, Q. et al. Coordination engineering of iridium nanocluster bifunctional electrocatalyst for highly efficient and pH-universal overall water splitting. Nat. Commun. 11, 4246 (2020).
- Louie, M. W. & Bell, A. T. An investigation of thin-film Ni-Fe oxide catalysts for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 135, 12329-12337 (2013).
- Bai, L., Lee, S. & Hu, X. Spectroscopic and electrokinetic evidence for a bifunctional mechanism of the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 3095-3103 (2021).
- Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159-1165 (2011).
- Zheng, Y.-R. et al. Monitoring oxygen production on mass-selected iridium-tantalum oxide electrocatalysts. Nat. Energy 7, 55-64 (2021).
- Kang, J. X. et al. Valence oscillation and dynamic active sites in monolayer NiCo hydroxides for water oxidation. Nat. Catal. 4, 1050-1058 (2021).
- Wu, Z. Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
- Lee, S., Bai, L. & Hu, X. Deciphering iron-dependent activity in oxygen evolution catalyzed by nickel-iron layered double hydroxide. Angew. Chem. Int. Ed. 59, 8072-8077 (2020).
- Stevens, M. B. et al. Measurement techniques for the study of thin film heterogeneous water oxidation electrocatalysts. Chem. Mater. 29, 120-140 (2016).
- Sun, J., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
- Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+ U study. Phys. Rev. B 57, 1505 (1998).
- Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456-1465 (2011).
- Bajdich, M., García-Mota, M., Vojvodic, A., Nørskov, J. K. & Bell, A. T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521-13530 (2013).
- Friebel, D. et al. Identification of highly active Fe sites in (Ni, Fe) OOH for electrocatalytic water splitting. J. Am. Chem. Soc. 137, 1305-1313 (2015).
- Hu, M. et al. Tuning single-atom catalysts of nitrogen-coordinated transition metals for optimizing oxygen evolution and reduction reactions. J. Phys. Chem. C 124, 13168-13176 (2020).
- Bhat, A. et al. Prospects challenges and stability of 2D MXenes for clean energy conversion and storage applications. npj 2D Mater. Appl. 5, 61 (2021).
- Lv, Q. et al. Selectively nitrogen-doped carbon materials as superior metal-free catalysts for oxygen reduction. Nat. Commun. 9, 3376 (2018).
شكر وتقدير
(U19A2015، 22221003، 22250007، و22302184)، صناديق البحث الأساسية للجامعات المركزية، تعليم ك. تشي. وونغ (GJTD-2020-15)، برنامج الابتكار التعاوني لمركز هيفي للعلوم، الأكاديمية الصينية للعلوم (2022HSC-CIP004)، الصندوق المشترك لجامعة يولين والمختبر الوطني داليان للطاقة النظيفة (YLU-DNL Fund 2022012)، صندوق التعاون DNL، الأكاديمية الصينية للعلوم (DNL202003)، برنامج الشراكة الدولية للأكاديمية الصينية للعلوم (
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-44815-0.
© المؤلف(ون) 2024
المركز الوطني للبحوث في العلوم الفيزيائية على المقياس المجهري، جامعة العلوم والتكنولوجيا في الصين، 230026 هيفي، آنهوي، جمهورية الصين الشعبية.
مختبر الإشعاع السنكروتروني الوطني، جامعة العلوم والتكنولوجيا في الصين، 230026 هيفي، آنهوي، جمهورية الصين الشعبية. مختبر المفاتيح التابع للأكاديمية الصينية للعلوم في فيزياء المادة الكمومية المرتبطة بقوة، جامعة العلوم والتكنولوجيا في الصين، 230026 هيفي، آنهوي، جمهورية الصين الشعبية. المختبر الرئيسي لكيمياء السطح والواجهة وتحفيز الطاقة في مؤسسات التعليم العالي في آنهوي، قسم الفيزياء الكيميائية، جامعة العلوم والتكنولوجيا في الصين، 230026 هيفي، آنهوي، جمهورية الصين الشعبية. مدرسة الكيمياء والهندسة الكيميائية، جامعة أنهوي للتكنولوجيا، 243002 ما’انشان، أنهوي، جمهورية الصين الشعبية. معهد التكنولوجيا المتقدمة، جامعة العلوم والتكنولوجيا في الصين، 230031 هيفي، آنهوي، جمهورية الصين الشعبية. ساهم هؤلاء المؤلفون بالتساوي: جي وي، هوا تانغ، لي شينغ. البريد الإلكتروني:zzhirong@ustc.edu.cn; zengj@ustc.edu.cn
DOI: https://doi.org/10.1038/s41467-024-44815-0
PMID: https://pubmed.ncbi.nlm.nih.gov/38228626
Publication Date: 2024-01-16
Site-specific metal-support interaction to switch the activity of Ir single atoms for oxygen evolution reaction
Accepted: 5 January 2024
Published online: 16 January 2024
(A) Check for updates
Abstract
The metal-support interactions (MSI) could greatly determine the electronic properties of single-atom catalysts, thus affecting the catalytic performance. However, the typical approach to regulating MSI usually suffers from interference of the variation of supports or sacrificing the stability of catalysts. Here, we effectively regulate the site-specific MSI of Ir single atoms anchored on Ni layered double hydroxide through an electrochemical deposition strategy. Cathodic deposition drives Ir atoms to locate at three-fold facial center cubic hollow sites with strong MSI, while anodic deposition drives Ir atoms to deposit onto oxygen vacancy sites with weak MSI. The mass activity and intrinsic activity of Ir single-atom catalysts with strong MSI towards oxygen evolution reaction are 19.5 and 5.2 times that with weak MSI, respectively. Mechanism study reveals that the strong MSI between Ir atoms and the support stimulates the activity of Ir sites by inducing the switch of active sites from Ni sites to Ir sites and optimizes the adsorption strength of intermediates, thereby enhancing the activity.
of supports and performing the reduction treatment with hydrogen
Results
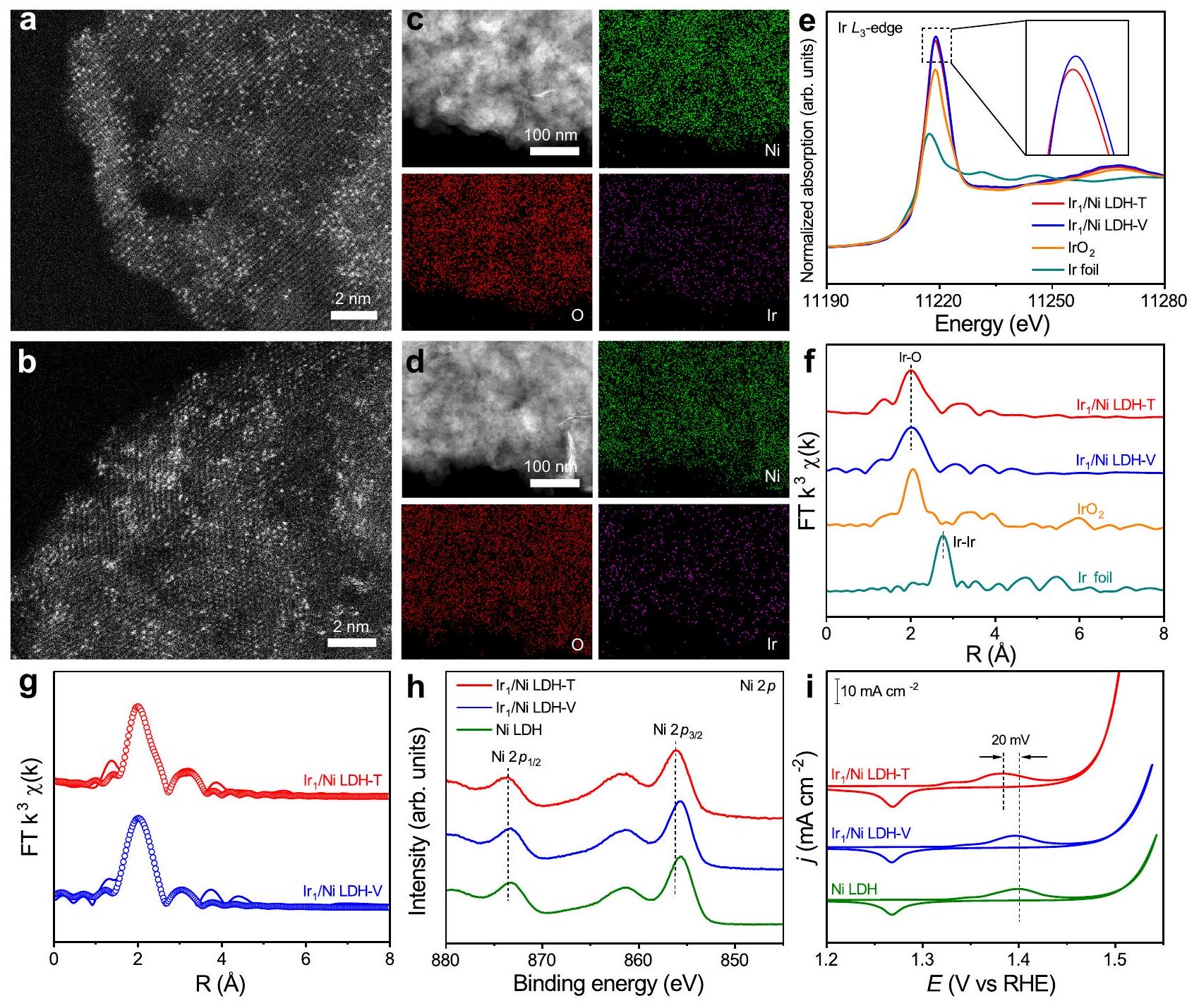
Synthesis and characterizations
with Ir foil and
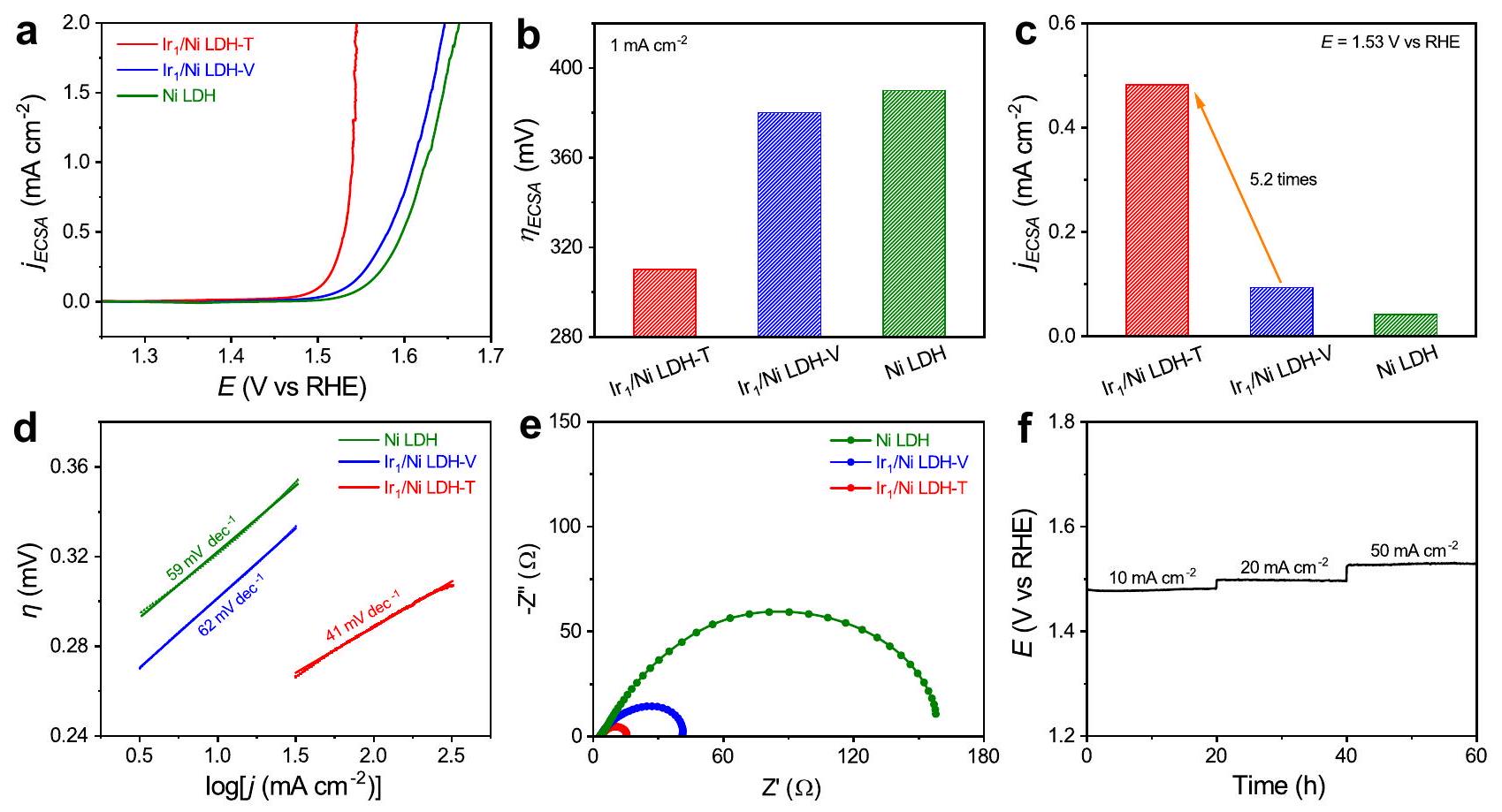
Electrocatalytic performance towards oxygen evolution reaction
were applied as electrocatalysts towards OER, one of the most essential reactions for clean energy conversion and storage application
were used as references.
b Overpotentials of different catalysts at the current density of
addition, the activity of Ni LDH after the cathodic and anodic treatment without Ir precursor remained almost unchanged, suggesting that the enhanced performance of
(ICP-MS) (Supplementary Fig. 20). After the durability test, the atomic dispersion of Ir atoms was maintained without any noticeable aggregation as shown in the HAADF-STEM image and EDS elemental mapping result (Supplementary Fig. 21a, b). Moreover, the XRD pattern and XPS spectra of
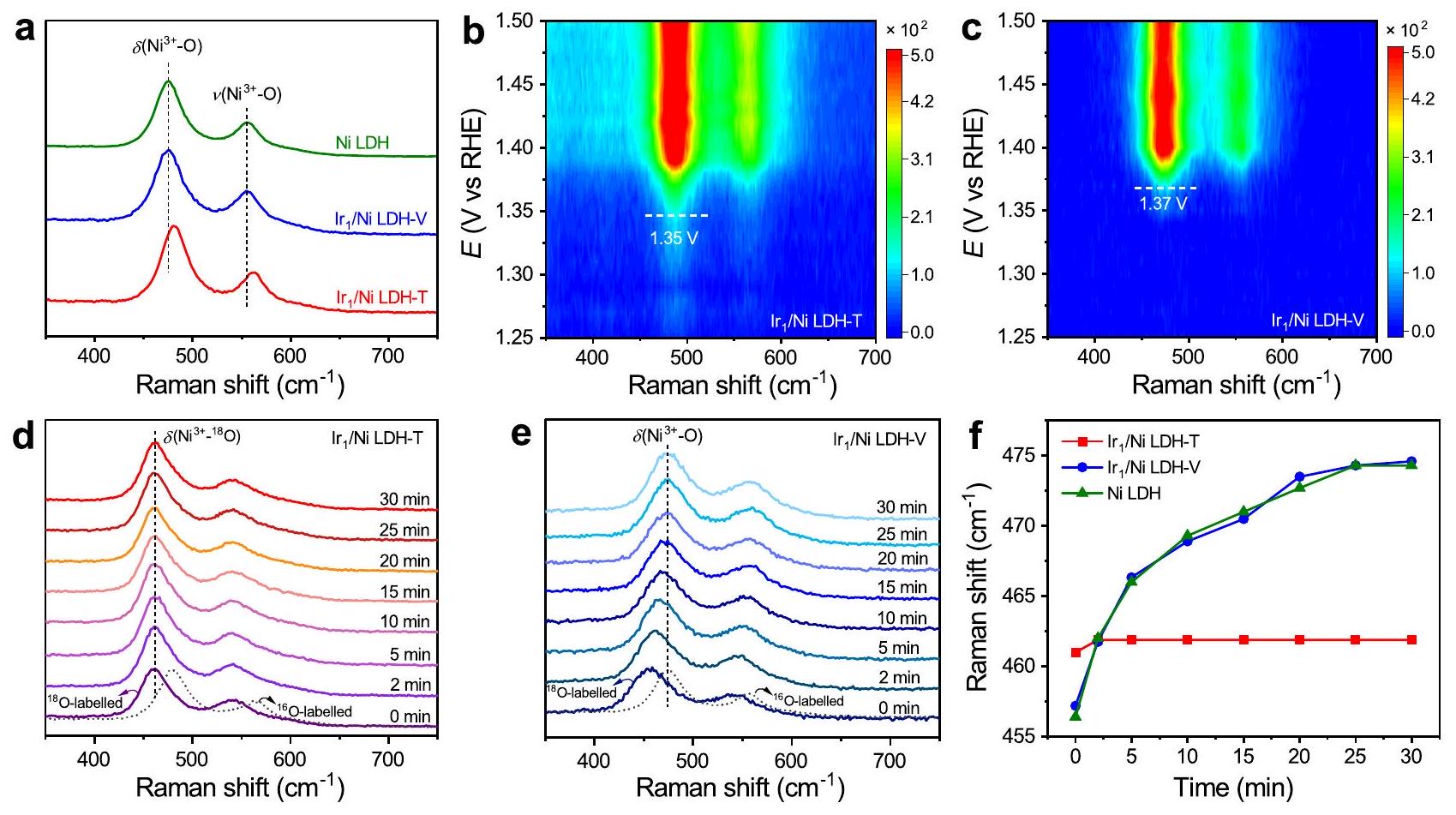
In situ spectroscopic characterizations
were initially labeled with
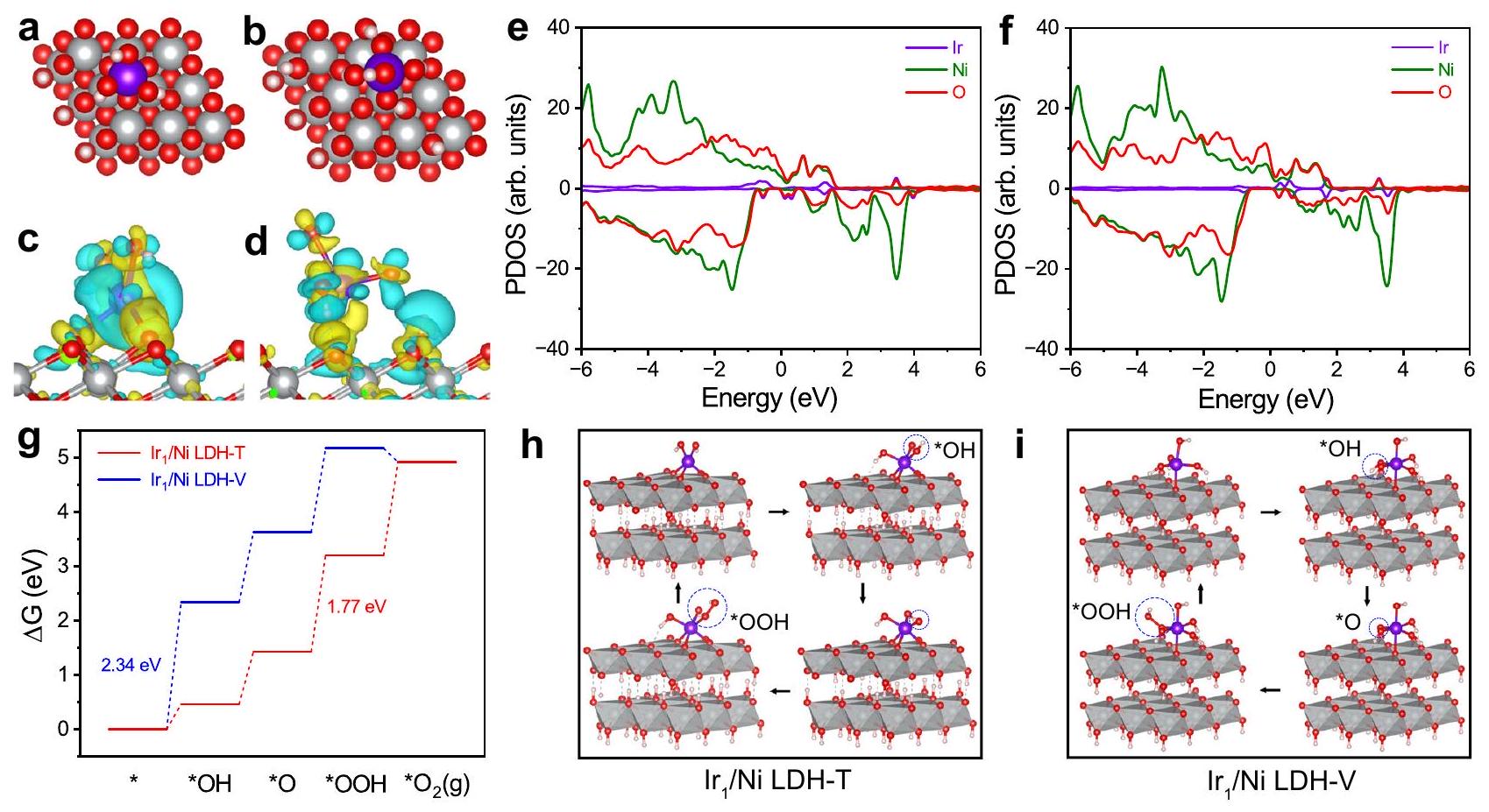
DFT calculations
calculated the projected density of states (PDOS) to elucidate the differences in electronic interactions between
Discussion
optimized the adsorption strength of oxygen-containing intermediates but also induced the switch of the catalytic active sites from Ni sites to Ir sites, thereby boosting the catalytic performance. This work provides a novel strategy to design highly active catalysts with site-specific MSI by electrochemically selective anchoring of single atoms and offers an in-depth understanding of the correlation between MSI and OER performance.
Methods
Chemicals
Synthesis of Ni LDH
Synthesis of
Catalyst characterization
was measured on the Veeco DI Nanoscope MultiMode V system. XRD patterns were collected using an X-ray diffractometer (Philips X’Pert Pro Super) with
Electrochemical measurements
carbon rod and an
In situ Raman spectroscopy
DFT calculation method
Theoretical evaluation of OER activity
molecule was calculated as
Data availability
References
- Qiao, B. et al. Single-atom catalysis of CO oxidation using
. Nat. Chem. 3, 634-641 (2011). - Yang, H. et al. Intramolecular hydroxyl nucleophilic attack pathway by a polymeric water oxidation catalyst with single cobalt sites. Nat. Catal. 5, 414-429 (2022).
- Gu, J., Hsu, C. S., Bai, L., Chen, H. M. & Hu, X. Atomically dispersed
sites catalyze efficient electroreduction to CO . Science 364, 1091-1094 (2019). - Yang, J., Li, W., Wang, D. & Li, Y. Electronic metal-support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 32, 2003300 (2020).
- Zhang, J., Liu, C. & Zhang, B. Insights into single-atom metal-support interactions in electrocatalytic water splitting. Small Methods 3, 1800481 (2019).
- Jin, Z. Y. et al. Understanding the inter-site distance effect in singleatom catalysts for oxygen electroreduction. Nat. Catal. 4, 615-622 (2021).
- Sun, H. et al. Atomic metal-support interaction enables reconstruction-free dual-site electrocatalyst. J. Am. Chem. Soc. 144, 1174-1186 (2022).
- Jiang, Z. et al. Modulating the electronic metal-support interactions in single-atom Pt-CuO catalyst for boosting acetone oxidation. Angew. Chem. Int. Ed. 61, e202200763 (2022).
- Li, W. H. et al. Creating high regioselectivity by electronic metal-support interaction of a single-atomic-site catalyst. J. Am. Chem. Soc. 143, 15453-15461 (2021).
- Han, B. et al. Strong metal-support interactions between Pt single atoms and
. Angew. Chem. Int. Ed. 59, 11824-11829 (2020). - Li, J. et al. Highly active and stable metal single-atom catalysts achieved by strong electronic metal-support interactions. J. Am. Chem. Soc. 141, 14515-14519 (2019).
- Yang, J. et al. Modulating the strong metal-support interaction of single-atom catalysts via vicinal structure decoration. Nat. Commun. 13, 4244 (2022).
- Ren, Y. et al. Unraveling the coordination structure-performance relationship in
single-atom catalyst. Nat. Commun. 10, 4500 (2019). - Jeong, H. et al. Controlling the oxidation state of Pt single atoms for maximizing catalytic activity. Angew. Chem. Int. Ed. 59, 20691-20696 (2020).
- Lee, S., Banjac, K., Lingenfelder, M. & Hu, X. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. Int. Ed. 58, 10295-10299 (2019).
- Zheng, X. et al. Origin of enhanced water oxidation activity in an iridium single atom anchored on NiFe oxyhydroxide catalyst. Proc. Natl Acad. Sci. USA 118, e2101817118 (2021).
- Van Ooij, W. J. & Houtman, J. P. W. Ligand exchange in iridium hexahalocomplexes: Part 2. Influence of the pH on the reactions of the hexachloroiridite and hexachloroiridate ions in aqueous solutions. Radiochim. Acta 20, 47-50 (1973).
- Cui, C. et al. Atomically dispersed hybrid nickel-iridium sites for photoelectrocatalysis. Nat. Commun. 8, 1341 (2017).
- Zhang, Z. et al. Electrochemical deposition as a universal route for fabricating single-atom catalysts. Nat. Commun. 11, 1215 (2020).
- Yao, Y. C. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304-313 (2019).
- Lin, C. et al. In-situ reconstructed Ru atom array on
with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021). - Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: toward boosted and sustained water oxidation catalysis. Joule 5, 2164-2176 (2021).
- Zhang, J. et al. Single-atom Au/NiFe layered double hydroxide electrocatalyst: probing the origin of activity for oxygen evolution reaction. J. Am. Chem. Soc. 140, 3876-3879 (2018).
- Li, P. et al. Boosting oxygen evolution of single-atomic ruthenium through electronic coupling with cobalt-iron layered double hydroxides. Nat. Commun. 10, 1711 (2019).
- Wang, Q. et al. Ultrahigh-loading of Ir single atoms on NiO matrix to dramatically enhance oxygen evolution reaction. J. Am. Chem. Soc. 142, 7425-7433 (2020).
- Cao, L. et al. Dynamic oxygen adsorption on single-atomic ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
- Yin, J. et al. Iridium single atoms coupling with oxygen vacancies boosts oxygen evolution reaction in acid media. J. Am. Chem. Soc. 142, 18378-18386 (2020).
- Wang, Q. et al. Coordination engineering of iridium nanocluster bifunctional electrocatalyst for highly efficient and pH-universal overall water splitting. Nat. Commun. 11, 4246 (2020).
- Louie, M. W. & Bell, A. T. An investigation of thin-film Ni-Fe oxide catalysts for the electrochemical evolution of oxygen. J. Am. Chem. Soc. 135, 12329-12337 (2013).
- Bai, L., Lee, S. & Hu, X. Spectroscopic and electrokinetic evidence for a bifunctional mechanism of the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 3095-3103 (2021).
- Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159-1165 (2011).
- Zheng, Y.-R. et al. Monitoring oxygen production on mass-selected iridium-tantalum oxide electrocatalysts. Nat. Energy 7, 55-64 (2021).
- Kang, J. X. et al. Valence oscillation and dynamic active sites in monolayer NiCo hydroxides for water oxidation. Nat. Catal. 4, 1050-1058 (2021).
- Wu, Z. Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
- Lee, S., Bai, L. & Hu, X. Deciphering iron-dependent activity in oxygen evolution catalyzed by nickel-iron layered double hydroxide. Angew. Chem. Int. Ed. 59, 8072-8077 (2020).
- Stevens, M. B. et al. Measurement techniques for the study of thin film heterogeneous water oxidation electrocatalysts. Chem. Mater. 29, 120-140 (2016).
- Sun, J., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
- Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+ U study. Phys. Rev. B 57, 1505 (1998).
- Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456-1465 (2011).
- Bajdich, M., García-Mota, M., Vojvodic, A., Nørskov, J. K. & Bell, A. T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521-13530 (2013).
- Friebel, D. et al. Identification of highly active Fe sites in (Ni, Fe) OOH for electrocatalytic water splitting. J. Am. Chem. Soc. 137, 1305-1313 (2015).
- Hu, M. et al. Tuning single-atom catalysts of nitrogen-coordinated transition metals for optimizing oxygen evolution and reduction reactions. J. Phys. Chem. C 124, 13168-13176 (2020).
- Bhat, A. et al. Prospects challenges and stability of 2D MXenes for clean energy conversion and storage applications. npj 2D Mater. Appl. 5, 61 (2021).
- Lv, Q. et al. Selectively nitrogen-doped carbon materials as superior metal-free catalysts for oxygen reduction. Nat. Commun. 9, 3376 (2018).
Acknowledgements
(U19A2015, 22221003, 22250007, and 22302184), Fundamental Research Funds for the Central Universities, K.C. Wong Education (GJTD-2020-15), Collaborative Innovation Program of Hefei Science Center, CAS (2022HSC-CIP004), the Joint Fund of the Yulin University and the Dalian National Laboratory for Clean Energy (YLU-DNL Fund 2022012), the DNL Cooperation Fund, CAS (DNL202003), International Partnership Program of Chinese Academy of Sciences (
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-44815-0.
© The Author(s) 2024
Hefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, 230026 Hefei, Anhui, P. R. China.
National Synchrotron Radiation Laboratory, University of Science and Technology of China, 230026 Hefei, Anhui, P. R. China. CAS Key Laboratory of Strongly-Coupled Quantum Matter Physics, University of Science and Technology of China, 230026 Hefei, Anhui, P. R. China. Key Laboratory of Surface and Interface Chemistry and Energy Catalysis of Anhui Higher Education Institutes, Department of Chemical Physics, University of Science and Technology of China, 230026 Hefei, Anhui, P. R. China. School of Chemistry & Chemical Engineering, Anhui University of Technology, 243002 Ma’anshan, Anhui, P. R. China. Institute of Advanced Technology, University of Science and Technology of China, 230031 Hefei, Anhui, P. R. China. These authors contributed equally: Jie Wei, Hua Tang, Li Sheng. e-mail: zzhirong@ustc.edu.cn; zengj@ustc.edu.cn