توازن الميكروبيوم على أوراق الأرز يتم تنظيمه بواسطة جزيء سابق في تخليق اللجنين Microbiome homeostasis on rice leaves is regulated by a precursor molecule of lignin biosynthesis
في النظم البيئية الأرضية، توفر أوراق النباتات أكبر موطن بيولوجي لمجتمعات ميكروبية متنوعة للغاية، تُعرف باسم ميكروبيوتا الفيلوسفير. ومع ذلك، تظل الآليات الأساسية لتجميع هذه المجتمعات الشائعة المدفوعة بالمضيف غامضة إلى حد كبير. هنا، نقوم بإجراء تقييم واسع النطاق وعميق لميكروبيوم الفيلوسفير للأرز بهدف تحديد الروابط المحددة بين المضيف والميكروب. تكشف دراسة الارتباط على مستوى الجينوم عن ارتباط قوي بين النمط الجيني للنبات وأعضاء من أربعة أوامر بكتيرية، هي Pseudomonadales وBurkholderiales وEnterobacterales وXanthomonadales. بعض هذه الارتباطات محددة لموقع جيني مضيف متميز، أو مسار، أو حتى جين. المركب 4-هيدروكسي سيناميك أسيد ( ) يتم تحديده كالعامل الرئيسي لزيادة بكتيريا تنتمي إلى Pseudomonadales. يمكن أن يتم تصنيع 4-HCA بواسطة OsPAL02 من نبات المضيف من مسار تخليق الفينيل بروبانيد. يؤدي الطفرة المفقودة لـ OsPALO2 إلى تقليل وفرة Pseudomonadales، واختلال التوازن في ميكروبيوتا الفيلوسفير، وبالتالي زيادة قابلية نباتات الأرز للإصابة بالأمراض. توفر دراستنا رابطًا مباشرًا بين مستقلب نباتي محدد وتوازن الفيلوسفير في الأرز، مما يفتح آفاقًا لاستراتيجيات تربية جديدة.
في الطبيعة، تشكل النباتات والميكروبات المرتبطة بها، المعروفة مجتمعة باسم الميكروبيوتا، كيانات وظيفية تعتمد على بعضها البعض.تساهم الميكروبيوتا في جوانب مثل مقاومة الأمراضتحمل الضغطواكتساب العناصر الغذائيةتحدد عملية التوظيف الناجحة والحفاظ على وفرة كافية من أعضاء ميكروبية محددة نتيجة تفاعلات النبات مع الميكروبيوتا.. وبالتالي، فهم المبادئ التي تحرك تجميع الميكروبيوم في نباتات المحاصيل أصبح واحدًا من الأهداف الرئيسية للدراسات الحالية من أجل دمج وظيفة الميكروبيوم في إنتاج المحاصيل المستدام.
يمكن تقسيم الميكروبيوتا النباتية إلى منطقة الجذور (واجهة الجذر والتربة) ، والفيلوسفير (سطح الورقة) ، والإندوسفير (الأنسجة الداخلية)أظهرت الدراسات أن وراثة المضيفوالتمثيل الغذائي
عملياتتشارك في استقطاب أنواع ميكروبية محددة في ميكروبيوم الجذور، وبالتالي تشكل تجميع الميكروبيوم. في الشعير، تم إظهار ارتباط موقع جيني محدد باستقطاب أنواع بكتيرية معينة.أظهرت التحليلات المتعمقة وجود ثلاثة جينات في هذا الموقع، بما في ذلك مستقبل مرتبط بالنيوكليوتيد-الذي يحتوي على تكرارات غنية بالليوسين (NLR) كمرشحين رئيسيين يشاركون في تشكيل الميكروبيوم.في نباتات الطماطم، قدم تحديد مواقع الصفات الكمية المحددة (QTLs) دليلاً إضافياً على أهمية وراثة المضيف في تجميع الميكروبيوم.بالإضافة إلى ذلك، تم إثبات أن إفرازات جذور النباتات المحددة توجه تجميع الميكروبيوم في منطقة الجذور نحو احتياجات المضيف للدفاع والتغذية.تمثل السطح النباتي أكبر سطح بيولوجي على الأرض وواجهة واسعة للتفاعلات بين النباتات والميكروبات الخاصة بها.لا يزال تحديد المكونات الجينية المحددة التي تشكل ميكروبيوم الفيلوسفير غير مفهومة بشكل كافٍ، على الرغم من أنه تم إثبات أن جينوم النبات يؤثر بشكل متكرر على ميكروبيوم الفيلوسفير بطرق متسقة عبر مواقع جغرافية منفصلة.نباتات نموذجية مثل الأرابيدopsis والنباتات غير النموذجية مثل الخردل البري المعمر بالإضافة إلى المحاصيل الحبوب مثل القمح والشعيرتم تنفيذها لزيادة فهمنا لميكروبيوم الفيلوسفير.
من المعروف أن العوامل الخارجية مثل الجراثيم المناعية وظروف المناخ يمكن أن تغير الميكروبيومات في الفيلوسفير.ومع ذلك، أصبح النمط الجيني للنبات هو التركيز الرئيسي من حيث البحث عن طرق لتعديل الميكروبيوم النباتي لأغراض زراعية.أظهرت الدراسات الحديثة أن استجابات الدفاع وسلامة جدار الخلية من المحتمل أن تكون متورطة في تجميع ميكروبيوم الفيلوسفير.تم إظهار أن طفرات الكيوتيكل في الأرابيدوبسيس وطفرات إشارات الإيثيلين لديها ميكروبيوم مختلف في الفيلوسفير مقارنة بالنباتات البرية.مسار استجابة نقص الفوسفاتوالمناعة المستحثة بواسطة الأنماطكما تم إظهار أنها تشارك في تشكيل الميكروبيوم في الفيلوسفير للأرابيدوبسيس. تعتمد استراتيجية شائعة للتحقيق في تأثيرات الجينات المضيفة على تجميع الميكروبيوم على تسلسل الأمبليكون لجينات المؤشر الميكروبية مقترنًا بدراسات الارتباط على مستوى الجينوم (GWAS).لقد أدت مثل هذه الدراسات إلى تحديد العديد من الجينات المحتملة التي تسبب تأثيرات على الوفرة النسبية للميكروبات المرتبطة بالنباتات.في حالة الميكروبيوم السطحي لنبات الأرابيدوبسيس، تم ربط محاور ميكروبية محددة بنجاح بمواقع جينية مضيفة تتعلق بتخليق الأيضات المتخصصة.يُفترض أن الجينات المشاركة في تخليق الجلوكوز السيناپوي، والمالات السيناپوي، والجلوكوزينولات تساهم في تشكيل ميكروبيوم الفيلوسفير في الأرابيدوبسيس. ومع ذلك، لا يزال هناك نقص في الأدلة المباشرة التي تربط الخلفية الجينية للمضيف بتجنيد أعضاء ميكروبية محددة. حتى الآن، تم إجراء تحليل التركيب الميكروبي للفيلوسفير عبر دراسات الارتباط الجينومي (GWAS) بشكل حصري على أساس تسلسل الأمبليكون لجينات المؤشرات الميكروبية.ومع ذلك، توفر بيانات الميتاجينوم وسائل أفضل لإجراء مثل هذه التحليلات بدقة أكبر، principalmente لأنها أقل عرضة للمبالغة أو التقليل من تقدير وفرة بعض الأنواع الميكروبية.
في هذا العمل، مع استخدام الميتاجينومات من الفيلوسفير من 110 وصولات من مجموعة تنوع الأرز II الأساسية (C-RDP-II)نقوم بإجراء تجارب GWAS لربط وفرة البكتيريا مع تعدد أشكال النوكليوتيدات المفردة (SNPs) في جينوم الأرز. تظهر التغيرات الجينية في الأرز أنها مرتبطة بشكل كبير بأعضاء من أربعة أوامر بكتيرية رئيسية في الفيلوسفير، وهي Pseudomonadales وBurkholderiales وEnterobacterales وXanthomonadales. لفك آلية سائدة حول كيفية تأثير جينات المضيف على تجميع الميكروبيوم في الفيلوسفير، نقوم بتنفيذ طفرات وبناءات تعبير مفرط لجين مرشح مرتبط بـ Pseudomonadales ونقيم التغيرات الناتجة في الميكروبيوم. علاوة على ذلك، نقوم بتحليل المستقلبات في أوراق الأرز التي تنظمها الجينات المرشحة. نكتشف أن المركب 4-hydroxycinnamic acid، المعروف أيضًا باسم p-coumaric acid وكمقدمة في تخليق اللجنين، مطلوب للتجميع و التوازن الحيوي لميكروبيوم سطح أوراق الأرز. بشكل عام، توفر دراستنا دليلًا واضحًا على تجميع ميكروبيوم سطح الأوراق المدفوع بالمواد الأيضية للمضيف.
النتائج
الميكروبيومات في الفيلوسفير للأرز محددة للجينات
تم تنفيذ مجموعة بيانات على نطاق واسع لتقييم هياكل الميكروبيوم في الفيلوسفير لنباتات الأرز. تم الحصول على مجموعة البيانات من خلال الميتاجينوميات المعتمدة على تقنية الشوتغن.. يعتمد على 110 عينة من مجموعة تنوع الأرز II الأساسية (C-RDP-II) التي تم زراعتها جميعًا في شمال غرب مقاطعة هونان، الصين ( ). لأغراض التحليل المقارن، تم تقسيم أصناف الأرز إلى ثلاث مجموعات رئيسية: 56 إنديكا، 36 يابونيكا و18 غير مصنفة (البيانات التكميلية 1). أكدت تحليل التكرار أن النهج المستخدم قد التقط تنوع الميكروبات بشكل كافٍ في مجموعتي الإنديكا واليابونيكا (الشكل التكميلية 1). تم معالجة الميتاجينومات باستخدام Kraken2 و Brackenتم توليد 6,862 وحدة تصنيفية على مستوى الأنواع. كانت البكتيريا (التي تشكل أكثر من 94% من إجمالي الوحدات التصنيفية) هي المكونات السائدة في الميكروبيوم في غلاف أوراق الأرز (البيانات التكميلية 2).
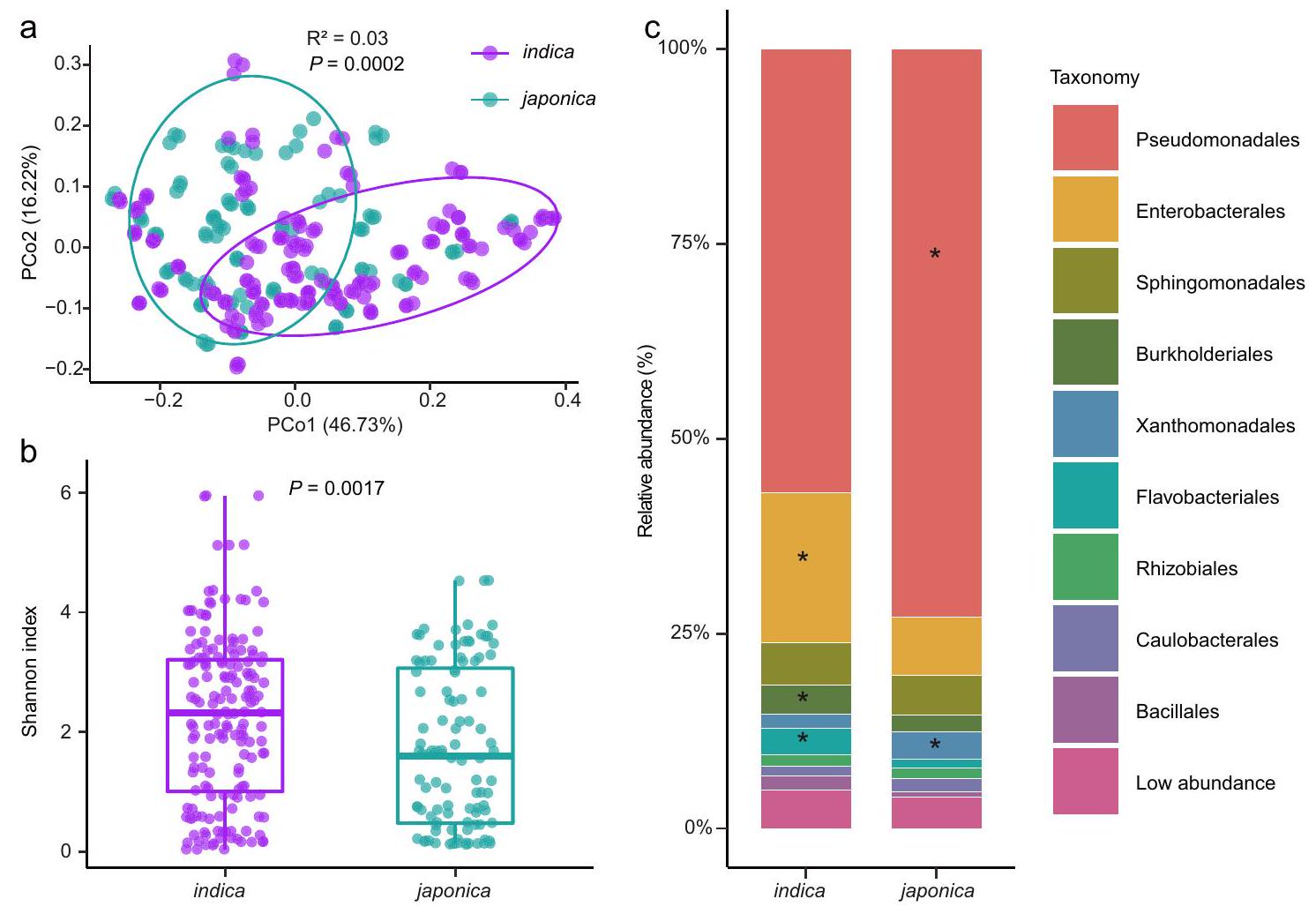
وجدنا أن المجتمعات البكتيرية كانت محددة لأنواع الأرز الفرعية. شكلت المجتمعات البكتيرية المستخرجة من أصناف الإنديكه والجابونيكا مجموعتين متميزتين، كما هو موضح في تحليل المكونات الرئيسية غير المقيد (PCoA) لمسافات براي-كورتيس (الشكل 1أ،، PERMANOVA مع اختبار Adonis). وهذا يشير إلى أن الأنماط الجينية لأنواع الأرز الهندي والياباني مرتبطة بتراكيب وتكوينات ميكروبيوم الفيلوسفير المختلفة. علاوة على ذلك، قمنا بتقييم تنوع العينات الداخلية (التنوع) ووجدنا فرقًا كبيرًا بين أصناف الإنديكا والجابونيكا (الشكل 1ب والشكل التكميلي 2). كان الميكروبيوم في الفيلوسفير للإنديكا أكثر تنوعًا من ذلك الخاص بالجابونكا (الشكل 1ب والشكل التكميلي 2)، مما يشير إلى أن الأول كان مستعمرًا بواسطة المزيد من الأنواع البكتيرية. كما لاحظنا اختلافات كبيرة في هياكل الميكروبيوم في الفيلوسفير بين أصناف الإنديكا والجابونيكا على مستويات الفيلوم والنظام والجنس (الشكل التكميلي 3، 4، الشكل 1ج، الجدول التكميلي 1)، مما يوفر دليلًا إضافيًا على أن البكتيريا تتأثر بأنماط الأرز الجينية.
كانت الأوامر البكتيرية السائدة في كل من نباتات الأرز إنديكا وجابونيكا تتكون من Pseudomonadales (57% و72% من الوفرة النسبية لإنديكا وجابونيكا، على التوالي)، تليها Enterobacterales (و 7%)، Sphingomonadales (5% و 5%)، Burkholderiales (و 2.1%)، Xanthomonadales (و 3.5%)، Flavobacteriales (3.4% و 1.1%)، Rhizobiales (1.5% و 1.3%)، Caulobacterales (1.3% و 1.7%) و Bacillales (1.7% و 0.8%) (الشكل 1c). بالمقارنة مع الأنواع اليابانية، كانت الأنواع الهندية تتمتع بوفرة نسبية أعلى من Enterobacterales وFlavobacteriales وBurkholderiales، بينما كانت الأنواع اليابانية تتمتع بوفرة نسبية أعلى من Pseudomonadales وXanthomonadales (اختبار Tukey’s HSD، معدل الاكتشاف الخاطئ (FDR) المعدل ) (الشكل 1ج، الجدول التكميلي 2). من المثير للاهتمام أن 46 جنسًا من Enterobacterales (تمثل 92% من الأجناس المحددة ضمن هذا النظام)، و17 جنسًا من Burkholderiales ( من الأجناس المحددة) و 5 أجناس من Pseudomonadales (حدثت الأنواع المحددة بأعداد نسبية مختلفة بشكل ملحوظ في أصناف الأرز إنديكا وجابونيكا (الشكل التكميلي 4، البيانات التكملية 2). في المقابل، أظهر فقط جنس Aerosticca من Xanthomonadales وجنس Chryseobacterium من Flavobacteriales مثل هذه الاختلافات في الوفرة النسبية بين الأصناف. لذلك، افترضنا أن خلفية جينية محددة لأصناف الأرز قد تلعب دورًا في تشكيل ميكروبيومات الفيلوسفير الخاصة بها.
تحديد المسارات الأيضية المرتبطة بميكروبيوم الفيلوسفير
مع التقدم الأخير في التسلسل والمعلوماتية الحيوية، أصبحت دراسات الارتباط على مستوى الجينوم (GWAS) أكثر وعدًا بشكل متزايد
الشكل 1 | مقارنات الميكروبيوم في الفيلوسفير بين أصناف الأرز إنديكو وجابونيكا. أ تحليل المكونات الرئيسية غير المقيد (للمكونات الرئيسية PCo1 وPCo2) استنادًا إلى مسافات براي-كورتيس تظهر تجمع المجتمع البكتيري لأصناف الأرز جابونيكا وإنديكو.-تم حساب القيمة بواسطة PERMANOVA أحادية الاتجاه). تغطي البيضاوياتلبيانات كل نوع فرعي من الأرز.مؤشر شانون لمجتمعات البكتيريا في الفيلوسفير لأنواع الإنديكا والجابونيكا. تمثل القضبان الأفقية داخل الصناديق الوسائط. تمثل قمم وقواعد الصناديق النسب المئوية 75 و 25 على التوالي. تمثل الشعيرات العليا والسفلى
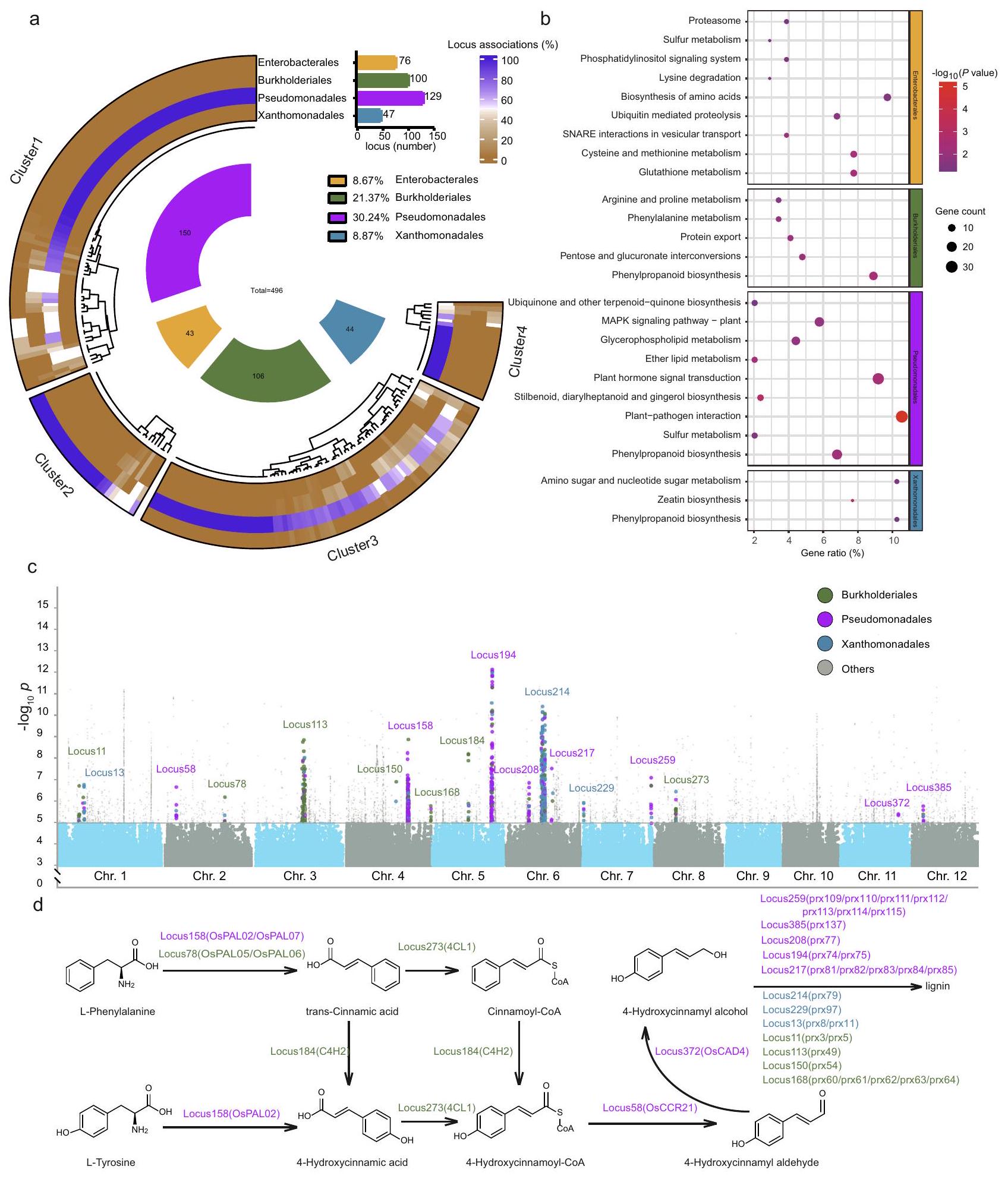
تمتد إلى بيانات لا تزيد عننطاق الربيع الربعي من الحافة العليا والحافة السفلى للصندوق، على التوالي.-تم حساب القيمة باستخدام تحليل التباين الأحادي غير المتزاوج مع اختبار توكي HSD ( ). توزيع البكتيريا على مستوى الطلب في الميكروبيوم السطحي للأوراق في أصناف الإنديكو واليابونيكا. تمثل النجوم الفروقات المهمة بين أصناف الإنديكو واليابونيكا كما تم تقييمها باستخدام تحليل التباين أحادي الاتجاه غير المقترن (ANOVA) مع اختبار توكي (HSD) -القيم مدرجة في الجدول التكميلي 2). إنديكيا ( ) و اليابانية ( ) مع ثلاث تكرارات لكل نمط وراثي. تم توفير بيانات المصدر كملف بيانات مصدر. نهج لتحديد العوامل الوراثية المرتبطة بالخصائص الزراعية الهامة في النباتاتلتحديد العوامل الوراثية المحتملة في الأرز المرتبطة بالميكروبيوم في الفيلوسفير، قمنا بإجراء دراسات الارتباط على مستوى الجينوم (GWAS). استهدفت التحليلات العلاقات بين جينومات الأرز والوفرة النسبية للأنواع البكتيرية ضمن الميتاجينومات المعنية. أدت تحليلاتنا إلى تحديد 2,667 تعدد أشكال نوكليوتيد مفرد غير متكرر (SNPs) تقع في 235 موضعًا كانت مرتبطة بشكل كبير بـ 496 نوعًا بكتيريًا (المشار إليها فيما بعد بالأنواع المرتبطة بـ GWAS) عند عتبة دلالة على مستوى الجينوم. (الشكل 2ج، البيانات التكميلية 3).
وجدنا أن الغالبية العظمى من الأنواع التي تم تحديدها بواسطة GWAS تنتمي إلى Pseudomonadales (مرتبطة بـ 129 موضعًا، مما يمثلمن إجمالي الأنواع المرتبطة بـ GWAS) Burkholderiales (مرتبطة بـ 100 موضع، تمثل )، Xanthomonadales (مرتبط بـ 47 موضعًا، تمثل 8.87%) و Enterobacterales (مرتبط بـ 76 موضعًا، تمثل ) على مستوى ترتيب البكتيريا (الشكل 2أ، البيانات التكميلية 4). لفصل المواقع وفقًا لترتيبات البكتيريا المرتبطة بها، بحثنا عن تجمعات المواقع باستخدام مسافات براى-كورتيس استنادًا إلى توزيع الأنواع المرتبطة بـ GWAS لكل موقع. أظهرت نتائجنا أن يمكن تقسيم إجمالي مواقع SNP إلى أربعة مجموعات: المجموعة 1 (تحتوي على 38.30% من إجمالي مواقع SNP وترتبط بـ Pseudomonadales)، المجموعة 2 (تحتوي على 31.91% من إجمالي مواقع SNP وترتبط بـ Burkholderiales)، المجموعة 3 (تحتوي على 12.77% من إجمالي مواقع SNP وترتبط بـ Enterobacterales) والمجموعة 4 (تحتوي علىمن إجمالي مواقع SNP المرتبطة بـ Xanthomonadales) (الشكل 2أ، الشكل التوضيحي 5، البيانات التكميلية 4). اقترحت هذه الارتباطات أن أعضاء من أربعة بكتيريا الأوامر تستجيب بشكل خاص لخلفيات جينية معينة لنبات المضيف.
لتوضيح كيف تؤثر التغيرات الجينية للمضيف على الميكروبيوم في الفيلوسفير، وخاصة أعضاء الأوامر البكتيرية التي كانت استجابتها عالية، تم توضيح الجينات التي تحتوي على تعدد أشكال النوكليوتيدات (SNPs) الموجودة في المواقع المرتبطة وتم إثراؤها في مسارات الأرز (البيانات التكميلية 4). وجدنا أن المسارات الأيضية (dosa01100) وعملية الأيض الثانوي (GO:0019748) كانت شائعة في التوضيحات الغنية بالجينات في KEGG (قاموس كيوتو للجينات والجينومات) (الشكل التكميلية 6) وGO (علم الأحياء الجزيئي) (الشكل التكميلية 6) على التوالي. علاوة على ذلك، لاحظنا أن الجينات المرتبطة بـ SNP في كل مجموعة مواقع كانت غنية في مسارات أيضية محددة (الشكل 2b). على سبيل المثال، كانت الجينات المرتبطة بـ Enterobacterales غنية بشكل كبير في مسارات البروتيازوم (dosa03050)، وتحلل الليسين (dosa00310)، وتخليق الأحماض الأمينية (dosa01230)، والتحلل البروتيني المعتمد على اليوبكويتين (dosa04120)، وتفاعلات SNARE في النقل الحويصلي (dosa04130)، وأيض السيستين والميثيونين (dosa00270)، ونظام إشارة الفوسفاتيديلينوزيتول (dosa04070)، وأيض الجلوتاثيون (dosa00480) (اختبار فيشر الدقيق،الشكل 2ب، البيانات التكميلية 4). وبالمثل، كانت الجينات المرتبطة بـ Burkholderiales غنية بشكل ملحوظ في تحويلات البنتوز والغلوكورونات (dosa00040)، تصدير البروتين (dosa03060)، استقلاب الفينيل ألانين (dosa00360) ومسارات استقلاب الأرجينين والبروتين (dosa00330) (اختبار فيشر الدقيق،الجينات المرتبطة بفصيلة البسودومونادال كانت غنية بشكل ملحوظ في تفاعل النبات مع الممرض (dosa04626)، وتخليق الستيلبينويد، والدياريلهيبتانويد، والزنجبيل.
الشكل 2 | دراسة الارتباط الجيني مع ميكروبيوم الفيلوسفير للأرز. أ ارتباط الأنواع لأربعة أوامر بكتيرية سائدة ومجموعات مواقع دراسة الارتباط الجيني. يعتمد تجميع مواقع دراسة الارتباط الجيني على مسافات براى-كورتيس للأنواع البكتيرية المرتبطة بكل موقع. المجموعة 1 (تحتوي على 38.30% من إجمالي SNPs؛ مرتبطة بشكل رئيسي بأعضاء من Pseudomonadales)، المجموعة 2 (؛ مرتبطة بشكل رئيسي بأعضاء من Burkholderiales)، العنقود 3 (12.77٪؛ مرتبطة بشكل رئيسي بأعضاء من Enterobacterales) والعنقود 4 (8.09٪؛ مرتبطة بشكل رئيسي بأعضاء من Xanthomonadales) (البيانات التكميلية 4). تُظهر الأعداد الإجمالية لمواقع GWAS المرتبطة لكل ترتيب في الرسم البياني العمودي المقابل. تشير الأجزاء الداخلية إلى المجموعة التصنيفية للأنواع البكتيرية المرتبطة بـ GWAS. الأرقام في الأجزاء الداخلية تشير إلى الأنواع البكتيرية المرتبطة ويتم تلوينها وفقًا لترتيبها- توصيف المستوى.تحليل إثراء KEGG للجينات من تجمعات المواقع فييمثل لون وحجم الفقاعات-قيمة ( اختبار فيشر الدقيق ذو الجانبين،-القيم مدرجة في البيانات التكميلية 4) وعدد الجينات الغنية في كل مسار KEGG، على التوالي. ج. مخططات مانهاتن لنتائج GWAS التي تم الحصول عليها من 110 نوع من الأرز. تم ترميز مواقع GWAS ومخططات SNP بالألوان وفقًا لتصنيف ترتيب البكتيريا. ذو جانبينتم تحديد القيم باستخدام تحليل الارتباط الكنسي.تحليل الجينات التي كانت غنية في مسار تخليق الفينيل بروبانيد. تم تلوين مواقع GWAS وأسماء الجينات وفقًا لتصنيف مستوى الطلب البكتيري. تم توفير بيانات المصدر كملف بيانات مصدر. (dosa00945) ، نقل إشارة هرمون النبات (dosa04075) ، استقلاب الدهون الأثيرية (dosa00565) ، استقلاب الجليسيروفوسفوليبيد (dosa00564) ، مسار إشارة MAPK – النبات (dosa04016) و مسارات تخليق اليوبكوينون وغيرها من كوينونات التيربين (dosa00130) (اختبار فيشر الدقيق،، الشكل 2ب، البيانات التكميلية 4). كانت الجينات المرتبطة بـ Xanthomonadales غنية بشكل ملحوظ في تخليق الزياتين (dosa00908) وعمليات الأيض للسكريات الأمينية وسكريات النوكليوتيدات (dosa00520) (اختبار فيشر الدقيق،الشكل 2ب، البيانات التكميلية 4). من الجدير بالذكر أن هناك أيضًا مسارات متداخلة بين أوامر بكتيرية مختلفة. على سبيل المثال، كانت الجينات المرتبطة بـ Enterobacterales و Pseudomonadales غنية في مسار استقلاب الكبريت (dosa00920)، بينما كانت الجينات المرتبطة بـ Burkholderiales و Pseudomonadales و Xanthomonadales غنية في مسار تخليق الفينيل بروبانيد (dosa00940) (الشكل 2ب، البيانات التكميلية 4). قدمت هذه النتائج رؤى رئيسية حول المسارات الأيضية للمضيف التي قد تكون متورطة في تجميع ميكروبيوتا الفيلوسفير. نظرًا لأن مسار تخليق الفينيل بروبانيد كان مرتبطًا بثلاثة أوامر بكتيرية سائدة، نتوقع دورًا رئيسيًا لهذا المسار فيما يتعلق بتفاعلات المضيف والميكروبيوتا.
لذلك قمنا بمزيد من التحقيق في الجينات المرتبطة بـ Burkholderiales و Pseudomonadales و Xanthomonadales التي كانت غنية في مسار تخليق الفينيل بروبانيد (الشكل 2c، d). وجدنا أن الغالبية العظمى من جينات prx (البيروكسيداز)، التي تشارك في تخليق اللجنين، كانت مرتبطة بـ Burkholderiales و Pseudomonadales و Xanthomonadales (الشكل 2d، البيانات التكميلية 4). بالإضافة إلى ذلك، تم العثور على جينات 4CL1 (4-كومارات-CoA ليغاز، Os08g0245200) و C4H2 (سينامات-4-هيدروكسيلاز، Os05g0320700)، التي تشارك في تخليق سينامويل-CoA و 4-هيدروكسي سينامويل-CoA، على التوالي، مرتبطة بشكل خاص بـ Burkholderiales. من ناحية أخرى، كانت جينات OsPALO2 (أميناز الفينيل ألانين/التيروزين، Os04g0518100) و OsCAD4 (كحول سيناميل ديهيدروجيناز، Os11g0622800) و OsCCR21 (اختزال سينامويل-CoA، Os02g0180700)، التي تشارك في تخليق 4-هيدروكسي حمض السيناميك و 4-هيدروكسي كحول السيناميل و 4-هيدروكسي ألدهيد السيناميل، على التوالي، مرتبطة بشكل خاص بـ Pseudomonadales (الشكل 2d، البيانات التكميلية 4). وهذا يشير إلى اختلافات محتملة في التحكم الجيني للمضيف في الميكروبيوم الفيلوسفير. بشكل جماعي، اقترحت تحليلاتنا أن المركبات التي هي جزء من تخليق اللجنين أو سلفها قد تشارك في تنظيم تجميع الميكروبيوم، والأهم من ذلك، أن بعض المركبات قد تؤثر على أنواع بكتيرية معينة.
تحديد الجينات المرتبطة بالمواد الأيضية للأوراق التي تحفز تجميع الميكروبيوم
نظرًا لحدوثها الشائع والأهمية المعروفة بالفعل لأعضاء من ترتيب Pseudomonadales في الفيلوسفير النباتي، أخذنا اهتمامًا خاصًا في فك شفرة كيفية ارتباط الجينات النباتية في إثراء ذلك. لم تكن Pseudomonadales وفيرة للغاية في الدراسة الحالية فحسب، بل كانت أيضًا تمثل أكبر عدد من المواقع المرتبطة في نهج GWAS، مما يشير إلى ارتباط قوي بالج genetics النباتية. للتحقق من العوامل الجينية للأرز التي تؤثر على تجميع Pseudomonadales، قمنا بإجراء تعديل جيني لتحوير جين مضيف ثبت أنه مرتبط به بشكل خاص. تم اختيار OsPALO2 كهدف للجين بسبب الارتباط القوي مع Pseudomonadales.-القيمة: لأنه لا توجد جينات متماثلة في الموقع 158 ومواقع أخرى تم تحديدها بواسطة GWAS. باستخدام تسلسل شظايا جين 16S rRNA، أظهرنا أن طفرات جين OsPALO2 في الأرز (المشار إليه فيما بعد بـ OsPALO2-KO)، الذي يفتقر إلى الأمونيا ليز الفينيل ألانين/التيروزين، أظهرت تغييرًا كبيرًا في الوفرة النسبية لـ Pseudomonadales (الشكل 3a). ومن المثير للاهتمام، أن التباين الطبيعي للـ SNPs في OsPALO2 كان مختلفًا بين أرز الإنديكا والأرز اليابونيكا (الشكل التكميلية 7)، مما يشير إلى وجود علاقة بين وفرة Pseudomonadales وهذا الجين.
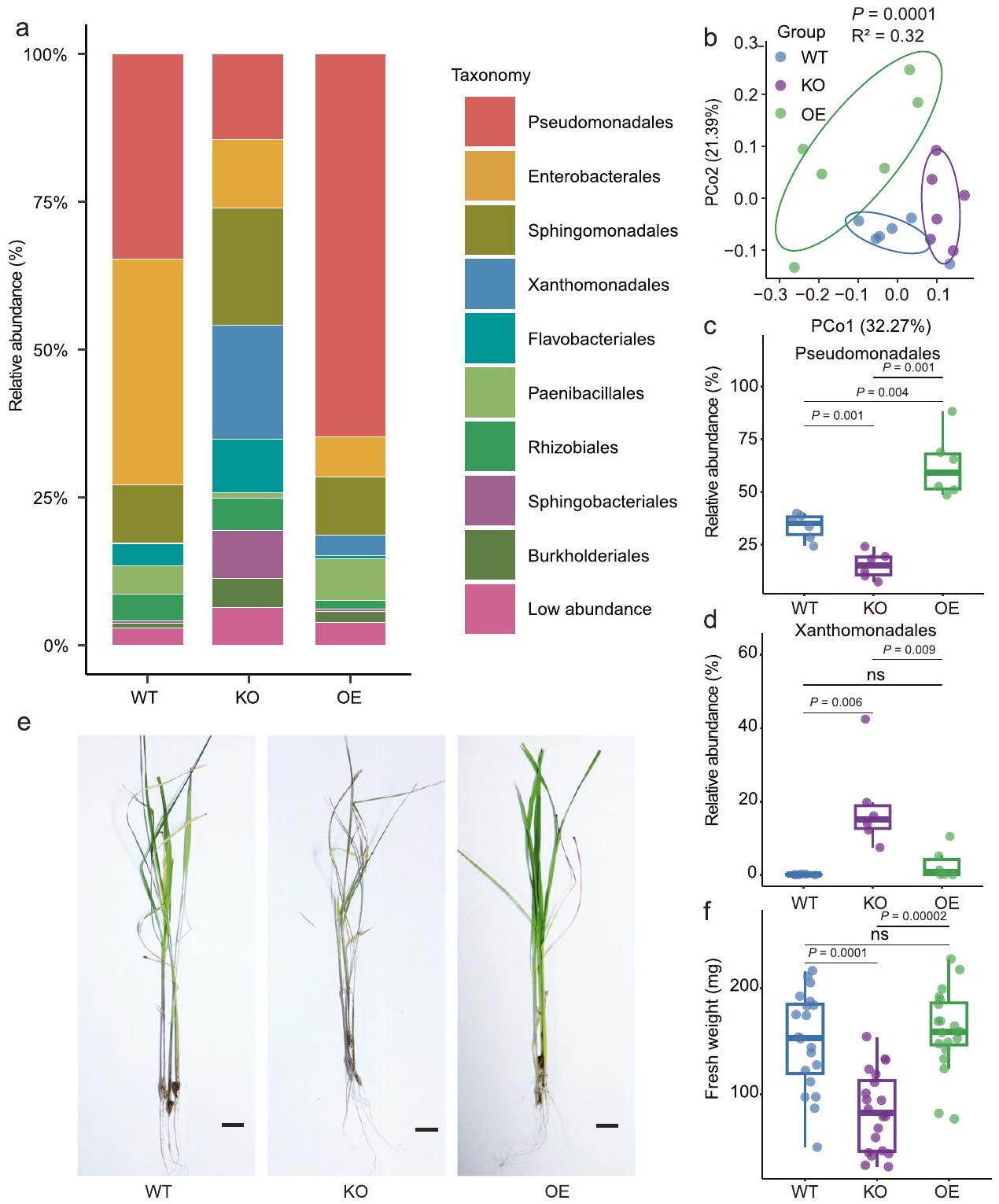
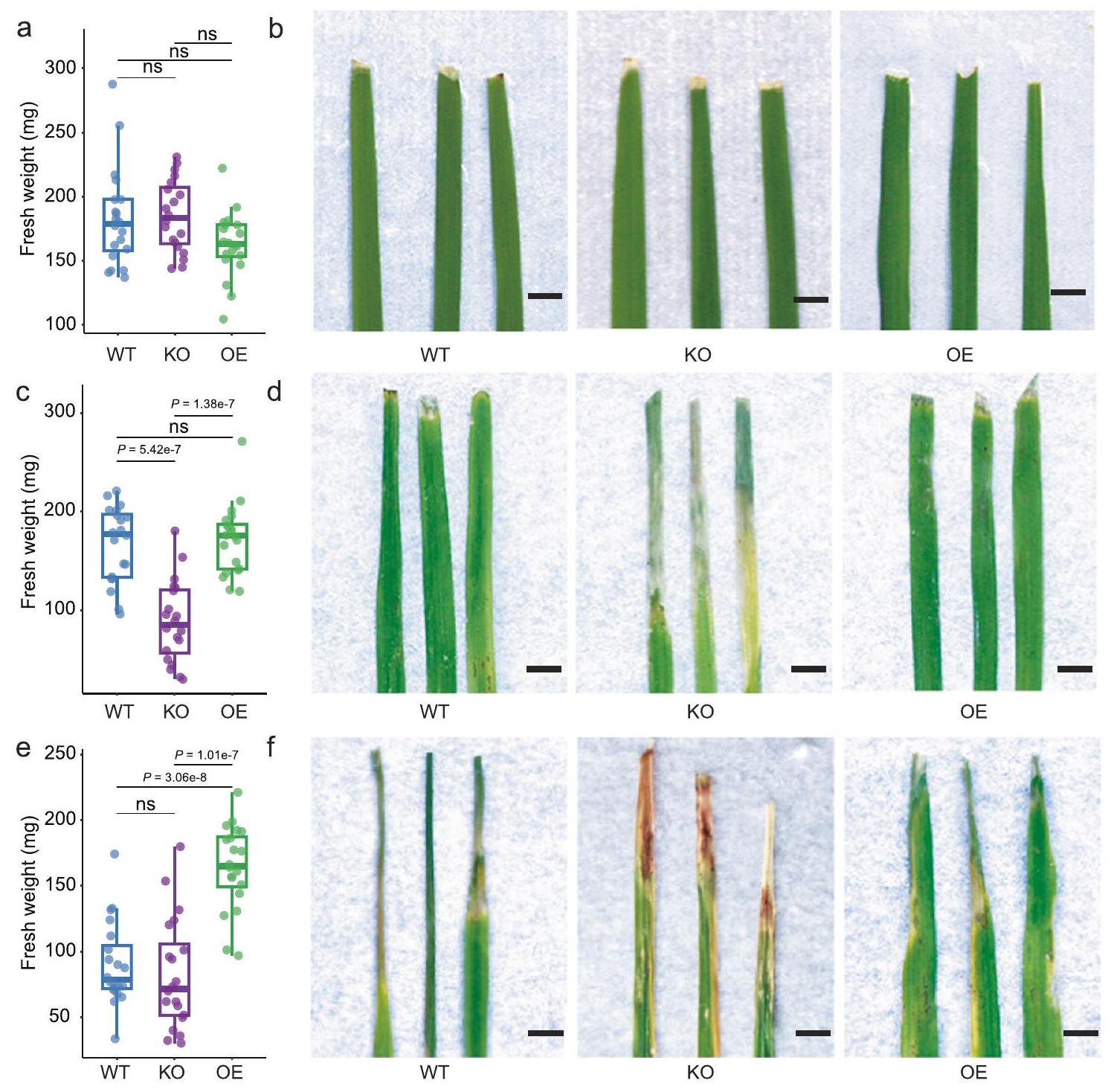
للمزيد من التحقيق في الدور التفصيلي لـ OsPALO2 في تشكيل ميكروبيوتا أوراق الأرز، قمنا بإجراء تسلسل شظايا جين 16S rRNA لتوصيف ميكروبيوتا الأوراق من نوع الأرز البري (WT) ZH11 (يابونيكا)، الطفرة المفقودة لوظيفة OsPALO2-KO وبناء التعبير المفرط OsPALO2-OE. تحت ظروف الدفيئة، لاحظنا أن هيكل المجتمع في الفيلوسفير لخطوط WT وOsPALO2-OE وOsPALO2-KO كانت تهيمن عليها Pseudomonadales وEnterobacterales وSphingomonadales وXanthomonadales وFlavobacteriales وPaenibacillales وRhizobiales وSphingobacteriales وBurkholderiales (الشكل 3أ). كانت هذه النتيجة متسقة مع التحليل الأولي للميكروبيوم الورقي، مما يشير إلى أن الملاحظات الميدانية كانت قابلة للتكرار في الدفيئة. PCoA غير المقيد (الشكل 3ب،، PERMANOVA) وتحليل المكونات الرئيسية المقيد (CPCoA) (الشكل التوضيحي التكميلي 8، تم تفسير 23.7% من التباين الكلي بواسطة جينوم النبات،أظهر تحليل PERMANOVA لمسافات براى-كورتيس أن الميكروبيومات في الفيلوسفير للنباتات WT وOsPALO2-OE وOsPALO2-KO شكلت مجموعات منفصلة، مما يشير إلى أن OsPAL02 أثر على مجتمعاتها البكتيرية. كشفت التحليلات الإضافية للاختلافات في الميكروبيومات في الفيلوسفير بين WT وOsPALO2-OE وOsPALO2-KO عن اختلافات كبيرة على مستوى الرتبة (الشكل 3أ). أظهر الوفرة النسبية لـ Pseudomonadales انخفاضًا كبيرًا في OsPALO2-KO وزيادة في OsPALO2-OE، مقارنة بالنباتات WT على التوالي (الشكل 3ج). أظهرت الوفرة النسبية لـ Xanthomonadales وFlavobacteriales وBurkholderiales زيادة كبيرة في OsPALO2-KO مقارنة بنباتات WT أو OsPALO2-OE (الشكل 3د، الجدول التكميلي 3، البيانات التكميلية 5). ومن الجدير بالذكر أن مؤشر شانون (تنوع ألفا) في OsPALO2-KO كان أعلى بشكل كبير من ذلك في OsPALO2-OE وWT (الشكل التكميلية 8). تم إجراء ملاحظات مماثلة عند تحليل سطرين إضافيين من OsPALO2-KO وOsPALO2-OE (الشكل التكميلية 9، الجداول التكميلية 4 و5، البيانات التكميلية 5). تشير الملاحظة بأن OsPALO2-KO يحتوي على تنوع ألفا أعلى ووفرة نسبية أقل من Pseudomonadales مقارنة بسلالة ZH11 البرية (japonica) إلى نفس الاتجاه الذي لوحظ مع الأصناف indica وjaponica (الشكل 1ب، ج). تشير هذه النتيجة إلى أن الاختلافات في OsPALO2 كانت كافية لمحاكاة التغيرات في الميكروبيوم بين نباتات indica وjaponica. وبناءً عليه، تم إثبات أن الجين OsPALO2 يساهم في استقطاب Pseudomonadales ويشكل هيكل المجتمع البكتيري في الفيلوسفير للأرز.
في مسار تخليق الفينيل بروبانيد، يقوم الجين OsPALO2 بترميز إنزيم أمونيا ليز الفينيل ألانين/التيروزين الذي يحفز تحويل L-تيروزين إلى حمض 4-هيدروكسي سيناميك.بالإضافة إلى تحويل L-phenylalanine إلى حمض السيناميك المتحول، وهما كلاهما سلفان لتخليق اللجنين. عندما تم تحليل اختلاف OsPALO2 بين الجابونيكا والإنديكا، أظهرت الهياكل البروتينية للنوع 1 (الموجودة بشكل رئيسي في الإنديكا) والنوع 5 (الموجودة بشكل رئيسي في الجابونيكا) تبادل الأحماض الأمينية في الموقع 134. تشير هذه النتيجة إلى أن النشاط التحفيزي لـ OsPALO2 قد يختلف بين نباتات الجابونيكا والإنديكا (الشكل التوضيحي 7). لذلك، افترضنا أن منتجات OsPALO2 تفرض قوة انتقائية على استقطاب أعضاء الميكروبات في الفيلوسفير، وخاصة أولئك ضمن ترتيب البكتيريا Pseudomonadales.
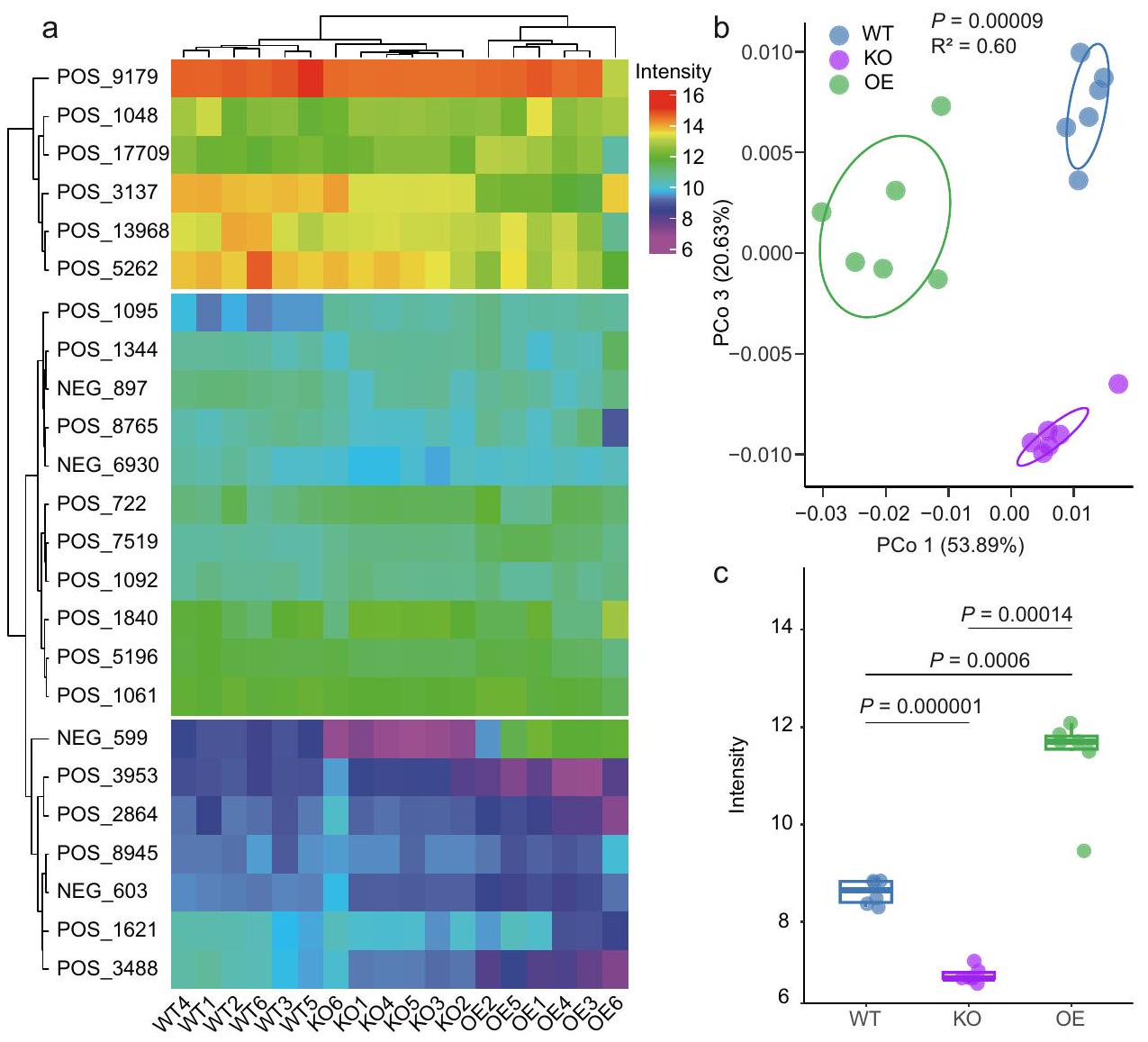
للتحقيق في هذه الفرضية، قمنا بإجراء تحليلات للمواد الأيضية باستخدام أوراق الأرز من WT وOsPALO2-OE وOsPALO2-KO، بهدف تحديد الفروقات المنسوبة إلى OsPALO2. أظهرت النتائج أن ملفات المواد الأيضية لـ WT وOsPALO2-OE وOsPALO2-KO تم فصلها إلى ثلاثة مجموعات بناءً على مسافات براى-كورتيس وأكدت أن OsPALO2 كان له دور في تخليق حمض السيناميك ومشتقاته (الشكل 4أ، البيانات التكميلية 6). كما تم إخضاع البيانات لتحليل المكونات الرئيسية غير المقيد، وأظهرت النتائج أن أول إحداثيات رئيسية PCo1، التي تمثل 53.89% من التباين الكلي، فصلت بين نباتات WT وOsPALO2-OE وOsPALO2-KO، مما يشير إلى اختلافات كبيرة في المواد الأيضية الموجودة في أوراق الأرز (الشكل 4ب،، PERMANOVA). وقد لوحظ هذا أيضًا
الشكل 3 | OsPALO2 مرتبط بتكوين الميكروبيوم في الفيلوسفير وله آثار على صحة النبات. أ المجتمعات البكتيرية على مستوى الرتبة في النباتات WT (النوع البري، صنف الأرز ZH11)، KO (إزالة جين OsPALO2) و OE (زيادة تعبير OsPALO2).تحليل المكونات الرئيسية غير المقيد استنادًا إلى مسافات براي-كورتيس يظهر تجمع المجتمع البكتيري في نباتات WT و KO و OE-تم حساب القيمة بواسطة PERMANOVA أحادية الاتجاه). تغطي البيضاوياتمن البيانات لكل مجموعة.مقارنة وفرة Pseudomonadales (ج) و Xanthomonadales (د) في ميكروبيومات الفيلوسفير WT و KO و OE. أعداد
عينات مكررة في هي كما يلي: WT ( )، KO ( ) و OE ( ). صور تمثيلية لسمات النمط الظاهري النموذجية (e) والوزن الطازج (f) بين نباتات الأرز WT و KO و OE. عدد العينات المكررة هو كما يلي: WT ( )، KO ( ) و OE ( ). مقياس الرسم، 1 سم. في هذه الشكل، النسب المئوية لمخطط الصندوق هي نفسها كما في الشكل 1ب. الـ-تم حساب القيم باستخدام تحليل التباين الأحادي غير المقترن مع اختبار توكي HSD ( ). تشير التسميات ‘ns’ إلى عدم وجود فرق ذو دلالة ( تم توفير بيانات المصدر كملف بيانات المصدر. مع CPCoA (الشكل التوضيحي التكميلي 10) حيث من إجمالي التباين تم شرحه بواسطة جينوتيب النبات (، PERMANOVA). أظهرت التحليلات الإضافية أن 12 من المستقلبات أظهرت اختلافات ملحوظة بين WT وOsPALO2-OE وOsPALO2-KO (البيانات التكميلية 6). حيث أظهر 4-HCA كثافة أعلى بشكل ملحوظ في OsPALO2-OE وأقل في OsPALO2-KO مقارنة بالنباتات WT (الشكل 4c)، مما يشير إلى أن 4-HCA هو المستقلب الرئيسي الذي ينظمه OsPALO2 وقد يساهم بذلك في تجميع الميكروبيوم في الفيلوسفير. تم إجراء تحليل LC-MS للكشف عن التركيزات الفسيولوجية لـ 4-HCA في أوراق الأرز. كما هو متوقع، كان تركيز 4-HCA أعلى بشكل ملحوظ في ثلاثة خطوط OsPALO2-OE وأقل في ثلاثة خطوط OsPALO2-KO عند مقارنتها بالنباتات WT (الشكل التكميلية 11، الجدول التكميلية 6، البيانات التكميلية 5). تم تحديد تركيز 4-HCA أيضًا في خمسة من Pseudomonadales الغنية باليابونيكا. وخمسة أصناف من الأرز الهندي الغني بـ Xanthomonadales. وجدنا أن تركيز 4-HCA في الأصناف اليابانية التي تم تحليلها كان أعلى بكثير من الأصناف الهندية (الشكل التكميلي 12، الجدول التكميلي 7). أظهرت تحليل الارتباط أن تركيز 4-HCA يظهر علاقة إيجابية مع الوفرة النسبية لـ Pseudomonadales وعلاقة سلبية مع وفرة Xanthomonadales في أوراق أصناف الأرز التي تم تحليلها (الشكل التكميلي 12). قدمت هذه الملاحظات دليلاً إضافياً على أن 4-HCA هو المستقلب الرئيسي الذي ينظمه OsPALO2 ويساهم في اختلافات الميكروبيوم في الفيلوسفير بين الأصناف الهندية واليابانية.
بصرف النظر عن الميكروبات المعدلة في الفيلوسفير، تطور OsPALO2-KO أعراض مرضية شديدة مقارنةً بـ WT و OsPALO2.بعد قطع الأوراق (الشكل 3e). لم يتم ملاحظة هذه الظاهرة
الشكل 4 | ملفات تعريف المستقلبات لطفرات OSPALO2. أ خريطة حرارية تعتمد على قيم الكثافة للأحماض القرفة والمشتقات المكتشفة في أوراق الأرز المختلفة. يتم عرض المجموعات على اليسار والأعلى باستخدام مسافات براى-كورتيس. جميع تسميات الأحماض القرفة (يسار) موضحة في البيانات التكميلية 6. تسميات عينات الأرز (أسفل) هي كما يلي: WT1/2/3/4/5/6 (النوع البري 1/2/3/4/5/6)، KO1/2/3/4/5/6 (OsPALO2-KO 1/2/3/4/5/6)، OE1/2/3/4/5/6 (OsPALO2-OE 1/2/3/4/5/6). ب تحليل المكونات الرئيسية غير المقيد بناءً على مسافات براى-كورتيس يظهر فصل المستقلبات في الأوراق لنباتات WT و KO و OE.تم حساب القيم باستخدام تحليل التباين الأحادي بواسطة PERMANOVA). تغطي البيضاوياتمن البيانات لكل مجموعة. الألوان الزرقاء والبنفسجية والخضراء تمثل عينات نبات الأرز WT و KO و OE، على التوالي. ج شدة (استنادًا إلى تحليلات LC-MS/MS) لـ 4-HCA (حمض 4-هيدروكسي سيناميك) في أوراق نباتات WT و KO و OE، على التوالي. في هذه الشكل، تم تطبيع قيم شدة القمة باستخدام تحويل In. النسب المئوية للرسم البياني الصندوقي هي نفسها كما في الشكل 1b.-تم حساب القيم باستخدام تحليل التباين الأحادي غير المقترن مع اختبار توكي HSD (” ). يتم اختصار المجموعات كالتالي: النوع البري، WT؛ تقليل التعبير عن OsPALO2، KO؛ زيادة التعبير عن OsPALO2، OE. أعداد العينات المكررة هي كما يلي: WT ( )، KO و OE ( تم توفير بيانات المصدر كملف بيانات المصدر. قبل قطع الأوراق (الشكل التكميلي 13)، مما يشير إلى أن غياب OsPALO2 تسبب في اختلال التوازن في ميكروبيوم الفيلوسفير في الأرز وبالتالي أدى إلى ضعف النباتات أمام مسببات الأمراض على الأوراق. وليس من المستغرب أن كان الوزن الطازج لأوراق WT و OsPALO2-OE أعلى بكثير من OsPALO2-KO؛ ولم يظهر الوزن الطازج لأوراق OsPALO2-OE أي فرق كبير مقارنة بـ WT بعد قطع الأوراق (الشكل 3f). لتحديد ما إذا كانت أعراض المرض ناتجة عن تغيير الميكروبيوتا في OsPALO2-KO، قمنا بزراعة نباتات WT و OsPALO2-KO تحت ظروف جنتوبيوتيك وأجرينا قطع الأوراق تحت ظروف محكومة تمنع التلوث. كانت نباتات OsPALO2-KO غير قابلة للتمييز عن نباتات WT، ولم يظهر الوزن الطازج أي اختلافات كبيرة بين خطوط الأرز الثلاثة (الشكل التكميلي 14). تشير هذه الملاحظة إلى أن أعراض المرض في OsPALO2-KO قد تكون ناتجة عن اختلال الميكروبيوتا. من خلال دمج نتائج تحليل المستقلبات والاختبارات المذكورة أعلاه، اقترحنا أن OsPAL02 قد يكون مسؤولاً عن إنتاج 4-HCA والحفاظ على ميكروبيوتا وظيفية.
لتحقق من وظيفةتم رش أوراق الأرز من نباتات OsPALO2-KO بـكما توقعنا، أدى تطبيق 4-HCA على نباتات OsPALO2-KO إلى ميكروبيوم فيلوسفير لا يظهر فرقًا كبيرًا عن نباتات WT، كما أن أوزانها الطازجة لم تختلف أيضًا (الشكل التكميلي 15، الجدول التكميلي 8). أظهرت هذه النتائج أن 4-HCA يمنع اختلال التوازن الميكروبي في الميكروبيوتا في الفيلوسفير لنباتات OsPALO2-KO. بشكل عام، تشير بياناتنا إلى أن 4-HCA الذي يتم تصنيعه بواسطة OsPAL02 يلعب دورًا رئيسيًا في الحفاظ على صحة النبات.
تداعيات 4-HCA وأعضاء من Pseudomonadales على توازن الميكروبيوم في الفيلوسفير
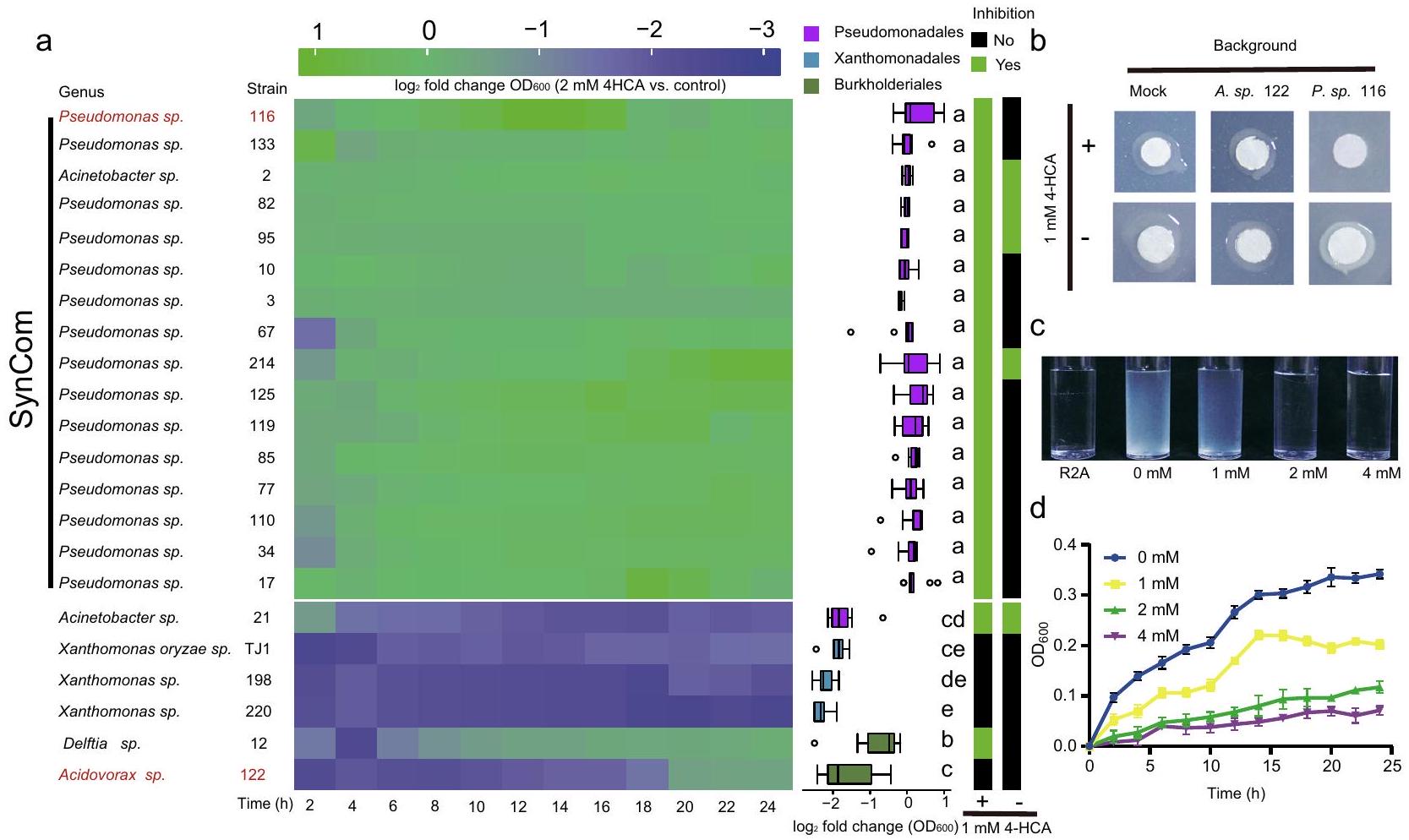
على الرغم من أننا لاحظنا أنيشارك في تجميع الميكروبيوم ويمنع اختلال التوازن الميكروبي في الفيلوسفير للأرز، إلا أن الآلية الأساسية ظلت غير واضحة. للتحقيق في التفاعلات المحتملة بين 4-HCA والبكتيريا المرتبطة بأوراق الأرز، قمنا بعزل البكتيريا باستخدام طريقة التخفيف المحدود.تم عزل سلالات بكتيرية من عينات أوراق مجمعة من نباتات WT وOsPALO2-OE وOsPALO2-KO التي نمت في ظروف الدفيئة. تم إجراء التعيينات التصنيفية للسلالات البكتيرية من خلال تسلسل Sanger لجينات 16S rRNA الخاصة بها، وتم تحديد السلالات غير المكررة بناءً على تشابه الجينات.“. في المجموع، حصلنا على مجموعة من 77 سلالة غير متكررة تحتوي على 14 رتبة بكتيرية، principalmente Pseudomonadales (من إجمالي السلالات غير المتكررة)، باسيلا (19.48%)، إنترومكتراليس (15.58%)، سفينغوموناداليس (9.1%) وفلافوبكتيراليس (7.79%). وقد غطت 62.5% من الطلبات (وفرة نسبية > 0.1%) التي تم اكتشافها من خلال تحليلات تسلسل الأمبليكون (الشكل التكميلي 16، البيانات التكملية 7). سمحت لنا مجموعة العزلات بدراسة المزيد
الشكل 5 | تأثيرات 4-HCA على نمو البكتيريا في الفيلوسفير والمرض TJ1 في الأرز. أ اليسار: تأثيرات 4-HCA على نمو 22 عزلة بكتيرية مرتبطة بأوراق الأرز من الطلبات Pseudomonadales وXanthomonadales وBurkholderiales. تُظهر خريطة الحرارةتغيرات الطي للعزلات الفردية المعرضة لـ مقابل التحكم (المكمل بالمذيب) على مدى 24 ساعة. يتم تقديم بيانات منحنى النمو المقابلة في الشكل التوضيحي 18 والبيانات التكميلية 7. الوسط: التحليل الإحصائي لـتغيرات الطي في كثافة الخلايا ) للعزلات الـ 22. تمثل الألوان المختلفة العزلات المعينة إلى Pseudomonadales وXanthomonadales وBurkholderiales، على التوالي. النسب المئوية في مخطط الصندوق هي نفسها كما في الشكل 1b. تشير الحروف المختلفة إلى وجود فرق كبير وفقًا لاختبار ANOVA أحادي الاتجاه غير المقترن مع اختبار Tukey’s HSD ( تم إدراج القيم في البيانات التكميلية 7، وتم تكرار التجارب لكل عزل 6 مرات). كل
عزلتغير الطي في كثافة الخلايا ( ) من 12 نقطة زمنية موضحة على اليسار. اليمين: تثبيط في المختبر لسلالة Xanthomonas oryzae TJ1 (المختصرة بـ TJ1) بواسطة 22 عزلة. اللون الأخضر أو الأسود يشير إلى أن العزلة المعنية إما قامت بتثبيط TJ1 على أجار R2A مع أو بدون 1 مللي مول 4-HCA (أسفل). ب صور تمثيلية لسلالات بكتيرية تم اختبارها لتثبيط TJ1 في (أ). تم تلقيح أقراص الورق بـ TJ1. وجود يظهر في أجار R2A على اليسار. الخلفية تشير إلى العزلات التي تم صبها في الأجار (الأعلى). تشير العينة الوهمية إلى المرشحات البكتيرية التي تم صبها في الأجار. ج، د صور تمثيلية (ج) ومنحنيات النمو (د) لـ TJ1 الذي نما في تركيزات مختلفة من 4-HCA. تم زراعة TJ1 في وسط R2A المدعوم بـ، و القيم تعني (موضحة كأشرطة خطأ؛ تكرارات). يتم توفير بيانات المصدر كملف بيانات المصدر. التفاعلات بين 4-HCA وأعضاء ميكروبيوتا الفيلوسفير من خلال اختبارات مستهدفة وطرق المجتمع الاصطناعي (SynCom).
نظرًا لأن أعراض المرض بعد قطع الأوراق لوحظت ليس فقط في نباتات OsPALO2-KO ولكن أيضًا في بعض نباتات WT و OsPALO2-OE (الشكل 3e)، افترضنا أن العامل المسبب للمرض قد يكون موجودًا ضمن مجموعة العزلات. لتحديد العامل (العوامل) المسببة لأعراض المرض، اختبرنا قدرة العزلات الفردية على التسبب في المرض. قمنا بتلقيح عزلات فردية على شتلات WT خالية من الجراثيم ورصدنا حدوث المرض في النباتات. ثم حددنا سلالة Xanthomonas oryzae TJ1 (المشار إليها فيما بعد بـ TJ1) كعامل مسبب لمرض تعفن الأوراق البكتيري على الأرز (الشكل التكميلية 17). ثم اختبرنا قدرة TJ1 على التسبب في المرض على نباتات OsPALO2-KO و OsPALO2-OE. كما هو متوقع، تسبب تلقيح TJ1 في ظهور أعراض مرضية شديدة على OsPALO2-KO، بينما كانت نباتات OsPALO2-OE أقل تأثرًا من حيث حدوث المرض والوزن الطازج، مقارنةً بـ WT و OsPALO2-KO (الشكل التكميلية 17). كما قمنا بتصنيف أعراض المرض إلى أربع درجات (1، صحي، إلى 4، ميت)، وأظهرنا أن زيادة شدة المرض تتوافق مع انخفاض الوزن الطازج للنباتات (الشكل التكميلية 17). علاوة على ذلك، قمنا بإعادة عزل البكتيريا من نباتات WT و OsPALO2-KO و OsPALO2-OE التي تم تلقيحها بـ TJ1. أكدت تسلسل جينات 16S rRNA الخاصة بهم.تشابه التسلسل مع TJ1 (الشكل التوضيحي 17). أظهرت تجارب العزل الإضافية أن وفرة TJ1 في نباتات OsPALO2-OE كانت أقل بكثير من تلك الموجودة في الطفرة OsPALO2-KO ونباتات WT. (الشكل التوضيحي 17). وقد قدم ذلك دليلاً إضافياً على أن المرض الناتج عن TJ1 أدى إلى انخفاض متوسط وزن النبات. بشكل عام، تم تأكيد أن العزلة TJ1 هي مسبب مرض للأرز، وقد تم إثبات أنها تسبب أعراض مرضية أخف في نباتات OsPALO2-OE مقارنة بتلك التي لوحظت في نباتات OsPALO2-KO و WT.
ثم افترضنا أنبتركيزات معينة قد تساهم في تثبيط TJ1 وتقليل أعراض المرض. قمنا بزراعة TJ1 في ثقافة سائلة مع تركيزات مختلفة من 4-HCA. وجدنا أن 4-HCA بتركيزات معينة يمكن أن تثبط نمو TJ1 بشكل فعال، وتم تثبيط نمو TJ1 تمامًا عندما وصل تركيز 4-HCA إلى 2 مللي مول (الشكل 5c، d). تشير هذه النتيجة إلى أن 4-HCA يمكن أن تثبط مباشرة نمو الممرض؛تركيز المثبط (IC50) لـ TJ1 كان 1 مللي مول.
بعد ذلك، قمنا بإجراء تجارب في المختبر لاختبار تأثير 4-HCA على سلالات من Pseudomonadales وXanthomonadales وBurkholderiales. تم زراعة هذه السلالات البكتيرية في وسط سائل R2A مضاف إليه; نفس التركيز أعاق نمو الممرض TJ1. وجدنا أن معظم سلالات Pseudomonadales نمت بشكل أسرع في وجود بينما تم تثبيط جميع سلالات Xanthomonadales و Burkholderiales بشكل ملحوظ (الشكل 5a). تحليل مفصل لتغيرات النمو ( ) أظهرت أن جميع سلالات Pseudomonadales، باستثناء Acinetobacter sp. 21، أظهرت معدل نمو أعلى بشكل ملحوظ من سلالات Xanthomonadales و Burkholderiales في وجود 2 مللي مول من 4-HCA (الشكل 5a، الشكل التوضيحي 18، البيانات التكميلية 7). أوضحت التجارب في المختبر تأثير
الشكل 6 | يمنع SynCom TJ1 في وجود 4-HCA في الكائن الحي. أ، ب الوزن الطازج (أ) وصور تمثيلية للسمات الظاهرية النموذجية (ب) بين نباتات الأرز WT وKO وOE المزروعة في ظروف معقمة والمعالجة بـ 10 مللي مول من MgCl. ج، د الوزن الطازج (ج) وصور تمثيلية للسمات الظاهرية النموذجية (د) بين نباتات الأرز WT وKO وOE المزروعة في ظروف معقمة والمُلقحة بمجتمع اصطناعي (المختصر SynCom) + Xanthomonas Oryzae TJ1 (المختصر TJ1). هـ،الوزن الطازج (e) وصور تمثيلية للسمات الظاهرية النموذجية (f) بين نباتات الأرز WT وKO وOE المزروعة في ظروف معقمة والمُلقحة بـ TJ1. النسب المئوية في مخطط الصندوق هي نفسها كما في الشكل 1b.
تمثل الألوان الزرقاء والبنفسجية والخضراء نباتات الأرز WT وKO وOE، على التوالي.-تم حساب القيم باستخدام تحليل التباين الأحادي غير المقترن مع اختبار توكي HSD (” ). التسميات ‘ تشير إلى عدم وجود فرق ذو دلالة إحصائية ). أعداد العينات المكررة في كل معالجة هي كما يلي: WT ( ) ، و OE ( ). مقياس الرسم، 5 مم. المجموعات مختصرة كالتالي: WT (النوع البري)، KO (خفض OsPALO2)، OE (زيادة التعبير عن OsPALO2). تم توفير بيانات المصدر كملف بيانات مصدر.
4-HCA على نمو السلالات من أوامر Pseudomonadales وXanthomonadales وBurkholderiales. كما ألقوا مزيدًا من الضوء على التباينات في تجميع الميكروبيوم في الفيلوسفير بين نباتات OsPALO2-OE وOsPALO2-KO وWT (الشكل 3a) وأشاروا إلى دور محوري لـ 4-HCA في ذلك.
لقد لاحظنا أعراض مرضية شديدة في النباتات البرية المعزولة بواسطة TJ1 تحت ظروف جنتوبيوتيك (الشكل التوضيحي 17)، ومن المثير للاهتمام أن هذه الظاهرة لم تحدث عندما نمت النباتات تحت ظروف الدفيئة، حيث تعرضت لمجموعة متنوعة من الميكروبات (الشكل 3e، f). لذلك، افترضنا أن تأثير 4-HCA يعتمد على الأقل جزئيًا على توازن الميكروبات. قمنا بإجراء تجارب تثبيط في المختبر باستخدام TJ1 و22 عزلة تمثل أعضاء من Pseudomonadales وXanthomonadales وBurkholderiales المرتبطة بالأوراق، على وسط R2A مع وبدون (الشكل 5أ). أظهرت النتائج أنه تحت شرط 1 مللي مول من 4-HCA، كانت جميع سلالات Pseudomonadales، بما في ذلك Acinetobacter sp. 21، تثبط TJ1 بشكل قوي. (الشكل 5أ). على العكس، فقدت 75% من سلالات Pseudomonadales القدرة على تثبيط TJ1 عندما كانت 4-HCA غائبة (الشكل 5أ). ضمن Burkholderiales، فقط Delftia sp. 12، قامت بتثبيط TJ1 في وجود 4-HCA وفقدت هذه النشاط المثبط عندما كانت 4-HCA غائبة (الشكل 5أ).
بشكل عام، حدثت تثبيط يعتمد على 4-HCA بشكل رئيسي بين السلالات من Pseudomonadales. هذه الملاحظة ربطت أيضًا 4-HCA بوظيفة الميكروبات في الفيلوسفير. علاوة على ذلك، قمنا بتصميم مجتمع اصطناعي (SynCom، الشكل 5a) يتكون من سلالات Pseudomonadales، ثم خضعنا له لاختبارات محددة لتقييم فعاليته ضد الممرض TJ1. تحت ظروف جنتوبيوتيك، لم تظهر نباتات WT وOsPALO2-KO وOsPALO2-OE أي اختلافات ملحوظة في الوزن الطازج عند معالجتها بـ 10 مللي مول. (التحكم، الشكل 6 أ، ب) أو تعليق SynCom (الشكل التكميلي 19). أكدت هذه النتيجة أن SynCom لم يكن له تأثير سلبي على نباتات الأرز. ثم عالجنا نباتات الأرز بتعليق SynCom + TJ1 وتعليق TJ1 فقط. في نباتات WT و OsPALO2-OE، أظهرت الـ SynCom تأثيرات واقية ضد TJ1 (الشكل 6c، d). على النقيض من ذلك، لم يُلاحظ أي حماية مع نباتات OsPALO2-KO، كما يتضح من الأعراض المرضية الشديدة وانخفاض وزن الأوراق الطازجة بشكل ملحوظ (الشكل 6c، d). عندما تم تطبيق تعليق TJ1 فقط، أظهرت نباتات WT و OsPALO2-KO كلاهما أعراض مرضية أكثر وضوحًا وانخفاضًا كبيرًا في وزن الأوراق الطازجة مقارنة بنباتات OsPALO2-OE (الشكل 6e، f)، مما يشير إلى أنه بدون الـ SynCom، يفقد تأثير الحماية ضد TJ1. من الواضح أن هناك تفاعلًا بين OsPALO2 وأعضاء Pseudomonadales كان مطلوبًا للحفاظ على صحة النبات المرتبطة بالـ SynCom. مع النتائج المذكورة أعلاه، يمكن استخلاص استنتاج بأن OsPALO2 المُركبمهم لإثراء الأعضاء البكتيرية من رتبة البسودوموناداليس ويدفع تجميع الميكروبيوم في الفيلوسفير. إن تجميع الميكروبيوم المنظم بواسطة 4-HCA له أهمية كبيرة في الحفاظ على توازن الفيلوسفير في الأرز، والذي يرتبط مباشرة بصحة النبات.
نقاش
تت colonize النباتات بواسطة مجتمعات ميكروبية متنوعة للغاية ومحددة الأنسجة، تُعرف باسم الميكروبيوتا النباتية. من المعروف أن الميكروبيوتا النباتية تكمل وظائف المضيف وتعتبر عاملاً مهماً لتعزيز المرونة تحت الضغوط غير الحيوية والحيوية. يمكن للنباتات تجميع والحفاظ على هياكل ميكروبيوتا معينة، وهو أمر حيوي لصحة النبات. تُعرف الحالة المتوازنة ديناميكياً للميكروبيوتا باسم ‘التوازن الداخلي’.غالبًا ما يؤدي تعطيل التوازن الداخلي إلى انتشار أعضاء ميكروبيوتا ضارة، مما يؤدي إلى تغيير هياكل الميكروبيوتا وحدوث أمراض نباتية، والمعروفة أيضًا باسم اختلال التوازن الميكروبي.فهمنا حاليًا بشكل ضئيل كيف تقوم النباتات بتجميع وصيانة ميكروبيوتا وظيفية (هوموستاتيكية)، خاصة فيما يتعلق بالطبقة الورقية لها.ركزت الأبحاث السابقة في الغالب على التفاعلات بين الميكروبات التي تؤدي إلى التوازن ويمكن أن تفسر أنماط الاستعمار معينة.هنا، نوضح أن التفاعل المحدد بين المضيف والميكروبات يساهم في تشكيل انتشار بكتيريا الفيلوسفير في نباتات الأرز. أظهرت الأبحاث التي أجريت في الماضي دلائل على التنظيم النشط للمضيف لميكروبيوتا الفيلوسفير. وقد تم إثبات أن نظام المناعة في النبات ضروري للحفاظ على توازن الميكروبيوتا.لقد أظهرت الطفرات في الجينات المرتبطة بالمناعة في الأرابيدوبسيس أنها تؤدي إلى ميكروبيوتا غير متوازنة في الفيلوسفير.في الدراسة الحالية، نوضح أن نباتات الأرز تستخدم آلية محتملة الحفظ لتشكيل ميكروبيوم الفيلوسفير في مجموعة واسعة من الأنماط الجينية. باستثناء الآليات المتعلقة بالمناعة، يمكن للنباتات أيضًا استخدام المستقلبات كقوة انتقائية لتجميع أعضاء الميكروبيوتا المرشحة لخدمة مصالحها.يمكن أن تلبي مجموعة متنوعة من المستقلبات النباتية المحددة الغرض من الحفاظ على توازن الميكروبات على الأوراق.أو قمع مسببات الأمراض البكتيريةومع ذلك، لا تزال هناك أسئلة أساسية لم يتم تناولها تتعلق بالعناصر الوراثية النباتية المخصصة للتحكم في تجميع الميكروبات، وكيف يحدث ذلك على مستوى المستقلبات.
أثبتت دراسات الارتباط على مستوى الجينوم (GWAS) أنها أداة مفيدة للغاية لتسليط الضوء على تفاعلات النباتات والميكروبات.في دراسة حديثة، أظهر الباحثون أن وفرة المحاور الميكروبية في الفيلوسفير في الأرابيدوبسيس تت correlated مع مسارات التمثيل الغذائي المتخصصة المتعددة التي تؤثر على كل من المجتمعات البكتيرية والفطرية.يجب أن يُبرز أن الدراسات السابقة كانت تعتمد فقط على تسلسل الأمبليكون لجينات العلامات الميكروبية في الفيلوسفير. بينما تعتبر هذه الأساليب قيمة، فإن هذه الطريقة معروفة أيضًا بأنها عرضة لتحيزات معينة، لأن جينات العلامات لمجموعات ميكروبية معينة تكون أكثر احتمالًا للتضخيم من تلك الخاصة بمجموعات أخرى.لذلك، فإن هياكل الميكروبيوم لا تُلتقط دائمًا بدقة. في الدراسة الحالية، تم دمج مجموعة بيانات الميتاجينوم الخاصة بالفيلاوسفير على نطاق واسع مع دراسات الارتباط الجينومي، مما ساعد على تقديم رؤى أكثر دقة حول العلاقات بين نباتات الأرز وميكروبات الفيلاوسفير الخاصة بها. من جهة، قدمت مزيدًا من الأدلة على دور نظام المناعة النباتي في تشكيل الميكروبيوتا الخاصة به، مثل مسار إشارات MAPK.
الأهم من ذلك، أنه سمح لنا بتحديد مسارات التمثيل الغذائي والجينات المحددة التي حددناها كأهداف مناسبة لتفكيك تجميع الميكروبيوم في الفيلوسفير على المستوى الجزيئي. علاوة على ذلك، كانت الدقة العالية للبيانات التصنيفية التي تم الحصول عليها هنا مفيدة لربط الصفات الوراثية للمضيف بأنواع بكتيرية محددة، وهو ما كان حاسماً للتحقق من تأثيرات جين ومركب متميز على تجميع الميكروبيوم.
كشفت الدراسة الحالية أن البكتيريا التي تنتمي بشكل أساسي إلى ترتيب Pseudomonadales تتزايد بوجود 4-HCA في الفيلوسفير للأرز. تم الحصول على ملاحظة مماثلة مع الكائنات الحية الداخلية لشجرة الحور وانخفاض تنظيم إنزيم سينامويل-CoA ريدوكتاز؛ مما أدى إلى زيادة تركيز 4-HCA في أنسجة النبات وبالتالي زيادة وفرة بكتيريا Pseudomonas.. هذا الجنس هو عضو شائع في الميكروبيوتا النباتيةأظهرت الدراسات السابقة أن هذه البكتيريا تشكل مكونًا مهمًا من الميكروبيوتا الوظيفية التي يمكن أن تحمي من مسببات الأمراض وتمنع الأمراض في النباتات. وغالبًا ما تم تحديد بكتيريا الزائفة كميكروبات أساسية مرتبطة بكل من منطقة جذور النبات ومنطقة الأوراق.وكونها لاعبين رئيسيين في العديد من التربة المثبطة للأمراضتستفيد النباتات من البكتيريا الزائفة لأنها تستطيع إنتاج مجموعة واسعة من المركبات المضادة للميكروبات.تحفيز مقاومة الأمراض النظامية في النباتاتوإقامة مجموعة من الحوارات الكيميائية مع النباتاتيمكن أن يوفر تحديدنا لمكونات الجينات النباتية التي تتحكم في Pseudomonas هدفًا مثاليًا لهندسة الميكروبيوتا في الفيلوسفير لمجموعة متنوعة من المحاصيل الزراعية. كما أظهرنا أن Xanthomonas، وهو مسبّب أمراض نباتية شائع جدًا، يتأثر سلبًا بـلذلك من المحتمل جداً أن تحمي النباتات نفسها من مسببات الأمراض من خلال الاعتماد على جهازها المناعي بالإضافة إلى المستقلبات المحددة التي لا تسمح لها فقط بمواجهة مسببات الأمراض بشكل مباشر، ولكن أيضاً بالحفاظ على التوازن في ميكروبيوتا الأوراق.
في السنوات الأخيرة، ساعدت دراسات الميكروبيوم النباتي في تحديد مجموعة متنوعة من الكائنات الدقيقة التي تقلل من الأمراض أو حتى تمنعها.تعتبر الأوراق نقاط دخول شائعة لمسببات الأمراض المختلفة والمدمرة للغاية. تُظهر الدراسة الحالية أن تنظيم الميكروبيوم في الفيلوسفير المدفوع بالاستقلاب هو آلية خفية لدعم النباتات المضيفة ضد هجمات مسببات الأمراض. وقد أسفرت الدراسات السابقة التي قيمت الميكروبيوم في الجذور عن نتائج مماثلة. على سبيل المثال، تم ربط الكومارينات التي تحرك الحديد في جذور الأرابيدوبسيس بتشكيل الميكروبيوم في الجذور من خلال تثبيط تكاثر بعض أنواع البسودوموناس عبر آلية تعتمد على الأكسدة والاختزال.أظهر إعادة تشكيل الميكروبيوم الجذري بواسطة الكومارين سكوبوليتين عواقب على صحة النبات. وبالمثل، تم العثور على منتج التحلل البنزوكسازينويد 6-ميثوكسي-بنزوكسازولين-2-ون (MBOA) لتنظيم تجميع الميكروبات في منطقة الجذور لمحاصيل الحبوب.
تشير النتائج الحالية المتعلقة بتوازن الفيلوسفير المدفوع بالمواد الأيضية للنبات وتأثيراتها على صحة المضيف إلى أنها ستشكل أساسًا لاستراتيجيات التربية المستهدفة. وقد قدمت الدراسات السابقة بعض المؤشرات، غالبًا من خلال تحليلات الارتباط، بأن المواد الأيضية للنبات تلعب دورًا مهمًا في الحفاظ على ميكروبيوتا وظيفية.. ومع ذلك، لم يتم حل ما إذا كانت المركبات المحددة هي المحركات الرئيسية للتغيرات الهيكلية الملحوظة في المجتمعات الميكروبية. في الدراسة الحالية، سمحت لنا تركيبات الإزالة وكذلك التعبير المفرط بتحديد التغيرات في الميكروبيوم بشكل محدد إلىفي الغلاف الهوائي للأرز. نقص فيكان مرتبطًا بخلل في ميكروبيوم الفيلوسفير مما يؤدي إلى زيادة قابلية نباتات الأرز للإصابة بالأمراض. استراتيجيات التربية الحالية لمقاومة الأمراض المعروفة باسم ‘ تواجه “استراتيجيات تربية الجينات” تحديات متزايدة من مسببات الأمراض النباتية التي تتطور بسرعة. يمكن معالجة هذه المشكلة من خلال تحديد وهندسة الجينات التي تشكل الميكروبيوم، والتي قد تُعرف باسم “تربية الجينات.
طرق
المواد الكيميائية، الكواشف والأدوات التحليلية
المواد الكيميائية، والمركبات، والأدوات التحليلية المستخدمة في هذه الدراسة مدرجة كما يلي: الإيثانول (EtOH)، هيبوكلوريت الصوديوم (NaOCl)،
تريتون X-100، ميثانول (MeOH)، حمض 4-هيدروكسي سيناميك (4-HCA)،تم شراء الكربون الأسود الجرافيتي (GCB) والأسيتونيتريل (ACN) وغيرها من المذيبات العضوية من ميرك (شنغهاي، الصين)؛ تم شراء مسحوق الأجار، هلام الأجاروز، محلول PBS، الكاناميسين، الهيغرومايسين، وسط موراشيجي وسكوج (MS)، وسط لينسماير-سكوج (LS)، وسط ريزونر 2 A (R2A) ووسط تريبتك صويا (TSB) من سولار بيو (بكين، الصين)؛ تم شراء Bsal، T4 DNA Ligase، Takara Ex Taq من تاكارا (بكين، الصين)؛ ترانسسكريبت II سوبر مزيج لتخليق cDNA من الشريط الأول لجميع الأغراض لـ PCR، ترانس تاك بوليميراز الحمض النووي عالي الدقة (HiFi)، إيزي بيور تم شراء مجموعة استخراج الجل السريع من ترانسجن بيوتيك (بكين، الصين)؛ وتم شراء مجموعة عزل الحمض النووي النباتي فاست بيور من فازايم بيوتيك (نانجينغ، الصين)؛ وتم شراء مجموعة الحمض النووي للتربة ماج بيور من ماغن (شنغهاي، الصين)؛ وتم شراء مجموعة بلازميد ميدي تيانبر من تيانجن (بكين، الصين)؛ وتم شراء كرات أجينكورت AMPure XP من بيكمان كولتر (باسادينا، الولايات المتحدة الأمريكية)؛ وتم شراء مجموعة اختبار Qubit dsDNA من ياسين (شنغهاي، الصين)؛ وتم شراء TRIzol من إنفيتروجين (كارلسباد، الولايات المتحدة الأمريكية).
تشمل المعدات التحليلية المستخدمة جهاز UHPLC (1290 Infinity LC، Agilent Technologies، الولايات المتحدة الأمريكية) متصل بجهاز قياس الكتلة رباعي الأبعاد (TripleTOF 6600، AB Sciex، الولايات المتحدة الأمريكية)، ومقياس الطيف NanoDrop ND-1000 (Thermo Fisher Scientific، الولايات المتحدة الأمريكية).
الميتاجينومات لسطح أوراق الأرز
من أجل دراسة التأثير المحتمل لأنماط الأرز الجينية على الميكروبيوم في الفيلوسفير، حصلنا على تسلسل ميتاجينومي باستخدام تقنية الشوتغن لـ 110 أصناف من الأرز (ثلاث تكرارات لكل صنف). تم تضمين تفاصيل حول هذه المجموعة البيانية في منشور بيانات مخصص (البيانات التكميلية 1).تم اختيار 110 نوعًا من الأرز من مجموعة التنوع للأرز II (C-RDP-II)، والتي يمكن تقسيمها إلى ‘يابونيكا’ (بما في ذلك 27 يابونيكا استوائية، 7 يابونيكا معتدلة، و2 يابونيكا مختلطة) و’إنديكا’ (بما في ذلك 32 إنديكا، 22 أوس، و2 إنديكا مختلطة) كما عرّفها مككوش وآخرون.تم زراعة جميع الأصناف في مقاطعة تاوجيانغ، مقاطعة هونان، الصين.، تم حصاد أوراق الأرز في مرحلة الإزهار (5 سبتمبر 2020). تم إثراء العينات لجزءها البكتيري، وتم استخراج الحمض النووي الميتاجينومي وخضوعه للتسلسل عالي الإنتاجية.تم وصف تصفية الجودة للبيانات الميتاجينية الخام في نشر البيانات.
تحليلات المعلوماتية الحيوية للميتاجينومات
تم إخضاع البيانات المصفاة نوعياً للتصنيف التصنيفي باستخدام Kraken2 v2.0.9تم إنشاء جدول وفرة الأنواع باستخدام Bracken v2.6.0تم تطبيق طريقة تطبيع مقياس المجموع التراكمي باستخدام حزمة R metagenomeSeqتم إجراء تحليل المكونات الرئيسية (PCoA) استنادًا إلى مسافات براي كورتيس باستخدام حزمة R فيجانتم إجراء تحليل التنوع الألفا باستخدام حزمة R المذكورة أعلاه vegan. تم تقييم الفروق في التنوع البيتا باستخدام اختبار Adonis الثنائي وتحليل التباين التبادلي (PERMANOVA، 999 تبديل) باستخدام حزمة R amplicon.تم إجراء تحليل وفرة الأنواع التفاضلية باستخدام اختبار ويلكوكسون لمجموع الرتب واختبار توكي HSD استنادًا إلى الأنواع ذات الوفرة النسبية المتوسطة.في أصناف الأرز التي تم تقييمها. تم تعريف الأنواع على أنها مختلفة بشكل ملحوظ في الوفرة إذا كانت-القيمة كانت <0.05. تم تصور بيانات التنوع ووفرة الأنواع النسبية باستخدام حزمة R amplicon. تم تصور تصنيف البكتيريا باستخدام GraPhlAn v.0.9.تمت تصور المخططات الصندوقية ومخططات التشتت باستخدام حزمة R ggplot2 الإصدار 2.2.1.
تحليل GWAS
كانت دراسة الارتباط على مستوى الجينوم قائمة على 110 وصول من الأرز C-RDP-II ممثلة بمجموعة بيانات تحتوي على 168,699 SNP (إزالة الفلتر MAF < 0.05) (البيانات التكميلية 8). تم استخراج هذه المجموعة من مجموعة C-RDP-II التي تشمل مجموعة بيانات تحتوي على 700,000 SNP.حددنا ‘السمات’ كمستويات الإثراء لـ 6,862 نوعًا محددًا في الفيلوسفير لـ 110 أصناف من الأرز (البيانات التكميلية 3). تاسل5.0 (https://www.maizegenetics.net/tassel) تم استخدامه لدراسات الارتباط على مستوى الجينوم (GWAS) وتم إعداد خط أنابيب تحليل على نظام لينكس لتحليل 6,862 ‘سمة’ بشكل فردي (البرامج النصية متاحة في توفر الشيفرة). كانت طريقة GWAS تعتمد على نهج تحليل الارتباط على مستوى الجينوم للأرز باستخدام مصفوفة SNP عالية الكثافة.استخدمنا نموذجًا خطيًا مختلطًا (MLM) يدمج مصفوفة القرابة (K) مع هيكل السكان (Q). قمنا بتمشيط المواقع المرتبطة بشكل كبير باستخدام المعايير التالية: منطقة جينومية تحتوي على تحتوي على ما لا يقل عن اثنين من SNPs المهمة (مع عتبة القيمة لـقمنا بتصفية المواقع غير المتكررة المرتبطة على الأقل بميكروب واحد مُعزز. تم تحليل المواقع المحددة بواسطة GWAS بشكل إضافي للبحث عن الجينات المرشحة.
تجميع المواقع وإثراء المسارات
يمكن أن تكون المواقع المحددة بواسطة GWAS مرتبطة بأنواع بكتيرية متعددة من أوامر مختلفة. لكل موقع، قمنا بحساب نسبة الأنواع المرتبطة من الأمر المعني. تم إخضاع النسبة لاحقًا لتصور تجميع المواقع. تم إجراء تحليل التجميع بناءً على مسافة براى كورتيس بين جميع المواقع باستخدامحزمة نباتية. تم تصور تجمع المواقع باستخدامحزمة سيركلايز“. تم إجراء توضيح الجينات وتحويل معرف الجين لكل من مجموعات الموقع باستخدام قاعدة بيانات مشروع توضيح الأرز (RAPDB،https://rapdb.dna.affrc.go.jp/“)، وتم إجراء تحليلات إثراء GO أو KEGG باستخدام حزمة R clusterProfilerتم إجراء تحليل إثراء GO باستخدام org.Osativa.eg.db (https://github.com/xuzhougeng/org.Osativa.eg.dbقاعدة البيانات وتم ملاحظة إثراء كبير في المسارات إذا كان -القيمة كانت <0.05 (اختبار فيشر الدقيق). تم تحليل إثراء KEGG باستخدام قاعدة بيانات جينات KEGG لأرز الساتيفا الياباني (RAPDB) (الإصدار 105.0+/03-07) وتم ملاحظة إثراء ملحوظ للمسارات إذا كانت -القيمة كانت <0.05 (اختبار فيشر الدقيق). تم تصور مسارات إثراء GO و KEGG باستخدام ggplot2.
توليد طفرات أرز معدلة بواسطة كريسبر وزيادة التعبير الجيني
تم استخدام نظام CRISPR-Cas9 لإنشاء طفرات OsPALO2 (Os04g0518100) وفقًا لطريقة تحرير الجينوم أحادية الفلقة عالية الكفاءة.باختصار، تم ربط بادئات محددة تحتوي على تسلسلات مواقع الهدف OsPALO2 (CCATGCGCTTCACCTCGTCCAGG) في كاسيتات pEGCas9Pubi-H. لبناء جينات RNA الموجه، تم تضخيم محفز RNA النووي الصغير U6 من متجه pYLsgRNA-OsU6a.باستخدام أزواج البرايمر U6-1 / U6-2. تم تضخيم تسلسل الحمض النووي الذي يشفر هيكل gRNA من متجه pYLsgRNAOsU6a. باستخدام زوج البرايمر gRNA-1/gRNA-2. تم دمج منتج PCR من محفز U6 مع قطعة الحمض النووي التي تشفر هيكل gRNA بواسطة PCR متداخل باستخدام زوج البرايمر sgRNA-1/sgRNA-2. تم استنساخ قطعة محفز U6-gRNA في متجه pEGCas9Pubi-H لتشكيل البناء pEGCas9Pubi-H-Os04g0518100 عبر هضم تقييدي باستخدام Bsal (تاكارا، بكين، الصين) تلاه الربط باستخدام T4 DNA Ligase (تاكارا، بكين، الصين) (الشكل التوضيحي التكميلي 20). بعد التحويل إلى الإشريكية القولونية DH5-alpha، تم تنقية البناء الناتج باستخدام مجموعة TIANprep Plasmid Midi (TIANGEN، بكين، الصين) لتكبير PCR باستخدام أزواج البرايمر KO-1/KO-2 وتم تسلسلها عبر تسلسل سانجر (سانغون بيوتيك، شنغهاي، الصين) (الشكل التوضيحي التكميلي 20). تم تخزين البناءات التي تم التحقق منها فيللاستخدام اللاحق في تحويل بروتوبلاست الأرز.
تم إنشاء تراكيب الإفراط في التعبير عن OsPALO2 وفقًا لطريقة بناء الأرز المعدل وراثيًا.باختصار، بالنسبة لبناء متجه الإفراط في التعبير OsPALO2، تم استخراج RNA الكلي من أوراق صنف الأرز ZH11 باستخدام TRIzol، وتم تخليق الحمض النووي المكمل (cDNA) من RNA الكلي باستخدام مجموعة تخليق cDNA SuperMix (TransGen Biotech، بكين، الصين). تم تضخيم تسلسل ترميز OsPALO2 بواسطة PCR باستخدام أزواج البرايمر PALO2-1/PALO2-2. تم استنساخ الشريحة المضخمة في متجه pEGOEPubi-H، الذي احتوى على مروج اليوبكيتين من الذرة وجين مقاومة الهيدرومايسين، لتشكيل بناء الإفراط في التعبير pEGOEPubi-H-OsPALO2 عبر هضم تقييدي باستخدام Bsal (تاكارا، بكين، الصين) تلاه الربط باستخدام T4 DNA Ligase (تاكارا، بكين، الصين) (الشكل التوضيحي 21). بعد التحويل إلى الإشريكية القولونية DH5-alpha، تم تنقية البنى الناتجة باستخدام مجموعة TIANprep Plasmid Midi (تيانجين، بكين، الصين) لتكبير PCR باستخدام أزواج البرايمر OE-1/OE-2 وتم تسلسلها عبر تسلسل سانجر (سانغون بيوتك، شنغهاي، الصين) (الشكل التوضيحي 21). تم تخزين البنى التي تم التحقق منها فيللاستخدام اللاحق في تحويل بروتوبلاست الأرز.
تم إدخال المتجهات الناتجة إلى Agrobacterium tumefaciens (السلالة EHA105) ثم إلى الأرز من خلال التحويل الذي يتم بوساطة Agrobacterium.تم تنفيذ التحويل باستخدام طريقة تحويل الأرزباختصار، تم اختيار البكتيريا المتحولة الإيجابية باستخدام الكاناميسين للاختيار. تم استخدام الهيغرومايسين لاختيار النباتات المتحولة المحتملة. تم زراعة جميع الشتلات المتحولة المحتملة المختارة في الدفيئة أو الحقل لمزيد من التعرف.
لتحديد النباتات المعدلة وراثيًا، تم استخراج الحمض النووي الجينومي من حوالي 30 ملغ من نسيج الورقة باستخدام مجموعة عزل الحمض النووي النباتي FastPure (Vazyme Biotech، نانجينغ، الصين) وفقًا لتعليمات الشركة المصنعة. تم استخدام الحمض النووي الجينومي المعزول والم purified لتضخيم PCR لمواقع هدف CRISPR/Cas9 أو جين الهيغرومايسين باستخدام أزواج البرايمر KO2-1/KO2-2 أو H-1/H-2، على التوالي. تم الكشف عن الأجزاء المضخمة باستخدام الرحلان الكهربائي في هلام الأجاروز، وتم تنقيتها باستخدام EasyPure.مجموعة استخراج الجل السريعة (TransGen Biotech، بكين، الصين) وتم تسلسلها باستخدام تسلسل سانجر (Sangon Biotech، شنغهاي، الصين) لتحديد النباتات المعدلة وراثيًا OsPALO2-KO وOsPALO2-OE. جميع البرايمرات ذات الصلة مدرجة في الجدول التكميلي 9.
المواد النباتية، ظروف النمو والعلاجات
قدم مركز أبحاث الأرز الهجين في هونان (HHRRC) بذور صنف الأرز ZH11 للتجارب والتحول الجيني. تم استخدام ZH11 كنوع بري (WT) ولتوليد نباتات OsPALO2-KO (المعطلة) وOsPALO2-OE (المفرطة التعبير). تم زراعة نباتات WT وOsPALO2-OE وOsPALO2-KO تحت رطوبة نسبية محددة عنددرجة الحرارة عندودورة ضوء مدتها 13 ساعة في دفيئة أو تم زراعتها في موقع حقل HHRRC في تشانغشا، الصين. تم حصاد البذور الناضجة من نباتات WT وOsPALO2-OE وOsPALO2-KO، وتجفيفها، وتخزينها في ثلاجة عند.
تم زراعة نباتات الأرز في مرشح تربة تاوجيانغ مختلط مع تربة زراعة البيوت المحمية (ركيزة PINDSTRUP، الدنمارك؛ تم تعقيمها مرتين) في بيت زجاجي ذو دوران هوائي لإجراء تجارب محكومة. لتحضير مرشح تربة تاوجيانغ، تم جمع التربة من نفس حقل الأرز المستخدم في التجارب الميتاجينومية، مقاطعة تاوجيانغ. )، مقاطعة هونان، الصين. الأعلى تم جمع التربة من الحقل وجرشهامنخل) لإزالة الصخور والحطام الآخر، وتم تجفيفه في درجة حرارة الغرفة. ثم تم تخفيف التربة بمعدل 10 مرات باستخدام ماء معقم ( )، تليها الاهتزاز لمدة 20 دقيقة في وتمت المعالجة بالموجات فوق الصوتية لمدة 5 دقائق بتردد 30 كيلو هرتز في تم تعريض المعلقات للطرد المركزي لمدة دقيقة واحدة عند 42 ج.تم جمع السائل العلوي واستخدامه كمرشح تربة تاوجيانغ. تم إعداد تربة الدفيئة باستخدام 100 مل من مرشح تربة تاوجيانغ مع 100 مل من محلول وسط موراشيجي وسكوج (MS) بتركيز نصف القوة (pH 5.8) ومزجها مع 200 جرام من تربة زراعة الدفيئة (ركيزة PINDSTRUP، الدنمارك؛ تم تعقيمها مرتين). تم تعقيم بذور الأرز على السطح باستخدام 75% إيثانول (EtOH) لمدة دقيقتين وهيبوكلوريت الصوديوممكمل بـتم غسل Triton X-100 ثلاث مرات لمدة 8 دقائق، ثم تم غسله بالماء المعقم ست مرات. تم تعريض البذور المعقمة لمزيد من تلقيح الميكروبات من التربة الميدانية عن طريق غمرها في مرشح تربة تاوجيانغ لمدة يوم واحد.و 4 د فيفي الظلام. بعد ذلك، تم زراعة جميع بذور الأرز في مرحلة الإنبات المبكرة في الأواني مع التحضير المذكور للتربة. تم ري كل نبتة بـ 5 مل من ماء معقم مرتين في الأسبوع ومحلول MS نصف القوة المعقم مرة في الأسبوع بعد الإنبات. تم قطع الأوراق العلوية للنباتات التي تبلغ من العمر 3 أسابيع (1 سم) باستخدام مقصات معقمة (تم تعقيم السطح باستخدام 75% إيثانول وغسلها بالماء المعقم ست مرات لكل نبات) لتحديد هياكل OsPALO2-KO أو OsPALO2-OE. تم حصاد النباتات التي تبلغ من العمر 5 أسابيع لجمع بكتيريا الفيلوسفير، واستخراج مستخلصات الأوراق، وتحديد الوزن الطازج وتحليلات أخرى.
بالنسبة لاختبار 4-HCA التكميلي، تم تعقيم نباتات WT و OsPALO2-KO سطحياً، ثم تم تبريدها، إنباتها ونقلها إلى نفس الدفيئة المذكورة سابقاً. كل نبات OsPALO2 عمره أسبوعانتم رش النبات بـ 2 مل من المعقم (مذاب في الماء). تم رش كل نبتة من نبات WT عمرها أسبوعين بـ 2 مل من الماء المعقم. تم ري الشتلات وتحديد النمط الجيني وفقًا لنفس الإجراء الموصوف سابقًا. تم حصاد النباتات التي تبلغ من العمر 5 أسابيع لتحليل الميكروبيوم والظاهرة والوزن الطازج.
للنمو النباتي في ظروف خالية من الجراثيم، تم تعقيم بذور WT وOsPALO2-KO وOsPALO2-OE على السطح بنفس الإجراء الموصوف سابقًا، ثم تم تعريضها لبرودة الطبقات وتركها تنبت في ماء معقم. بعد ذلك، تم نقل جميع بذور الأرز في مرحلة الإنبات المبكرة إلى زجاجات معقمة تحتوي على تربة تم تعقيمها بالبخار (ثلاث مرات لمدة 45 دقيقة لكل منها، مع تخزينها لمدة 24 ساعة في درجة حرارة الغرفة بين كل عملية تعقيم). تم رصد تعقيم بذور الأرز بانتظام من خلال زراعة عينات من النباتات وركيزة الثقافة على أطباق R2A. تم ري الشتلات وتحديد النمط الجيني كما هو موصوف أعلاه، ولكن في ظروف معقمة. تم حصاد النباتات التي تبلغ من العمر 5 أسابيع لتحليل الظواهر والوزن الطازج.
تحليلات الميكروبيوم في الفيلوسفير للنباتات البرية والنباتات الطافرة
تم حصاد النباتات التي تبلغ من العمر 5 أسابيع والمزروعة في ظروف الدفيئة من أجل تحليل الميكروبيوم في الفيلوسفير (ستة مكررات لكل نوع وراثي، كل مكرر يحتوي على أوراق ثلاث نباتات). تم جمع العينات وفقًا لطريقة إثراء بكتيريا الفيلوسفير للأرز.تم استخراج الحمض النووي الكلي باستخدام مجموعة MagPure Soil DNA LQ (Magen، شنغهاي، الصين) وفقًا لتعليمات الشركة المصنعة. تم التحقق من جودة وكمية الحمض النووي باستخدام مقياس الطيف الضوئي NanoDrop ND-1000 (Thermo Fisher Scientific، الولايات المتحدة الأمريكية) والرحلان الكهربائي في هلام الأجاروز، على التوالي. تم تخفيف الحمض النووي المستخرج إلى تركيزوتم تخزينه فيحتى يتم المعالجة لاحقًا. تم إجراء تضخيم PCR لقطع جينات 16S rRNA البكتيرية (منطقة V3-V4) باستخدام Takara Ex Taq (تاكارا، بكين، الصين) والبرايمرات المشفرة 343 F و798 R المدرجة في الجدول التكميلي 9. تم تصور الأمبليكون باستخدام الرحلان الكهربائي في هلام الأجاروز وتم تنقيته باستخدام كرات Agencourt AMPure XP (بيكمان كولتر، باسادينا، الولايات المتحدة الأمريكية) مرتين. بعد التنقية، تم قياس كمية الحمض النووي باستخدام مجموعة اختبار Qubit dsDNA (ياسين، شنغهاي، الصين). تم تجميع كميات متساوية من الحمض النووي المنقى للتسلسل على منصة NovaSeq 6000 (إيلومينا إنك، الولايات المتحدة الأمريكية) في OEbiotech بشنغهاي (شنغهاي، الصين).
تم معالجة تسلسلات قطع جين 16S rRNA باستخدام QIIME2 الإصدار 2021.4تم اكتشاف قراءات مزدوجة النهاية وتمت إزالة المحولات باستخدام Cutadapt v.2.1بعد القص، تم تصفية القراءات ذات الطرفين من التسلسلات منخفضة الجودة، وتم إزالة الضوضاء، ودمجها وتجميعها باستخدام DADA2 الإصدار 2020.2.0.تم إنشاء متغيرات تسلسل الأمبليكون (ASVs) وجدول الميزات باستخدام QIIME2. تم توضيح جميع ASVs باستخدام قواعد بيانات المرجع Silva v138.1.تم إجراء تحليلات التنوع والوفرة التفاضلية باستخدام نفس الطرق الموضحة أعلاه.
استخراج المستقلبات من أوراق الأرز
تم حصاد نباتات WT و OsPALO2-KO و OsPALO2-OE التي تبلغ من العمر 5 أسابيع والمزروعة في ظروف الدفيئة لاستخراج مستخلصات أوراق النباتات (ستة مكررات لكل نوع وراثي، كل مكرر يتضمن أوراق ثلاث نباتات). تم تجميد أوراق الأرز على الفور في النيتروجين السائل وطحنها إلى مسحوق ناعم باستخدام الهاون والمدقة. تم استخراج مستخلصات الأوراق من 80 ملغ من كل عينة باستخدامميثانول ميثانول (MeOH) – أستونيتريل (ACN) – ماء نقي للغاية ). تم هز المستحلبات في لمدة 20 دقيقة، تليها عملية الصوتنة بتردد 30 كيلو هرتز لمدة 20 دقيقة. بعد الطرد المركزي عندلمدة 20 دقيقة، تم تجفيف السائل العلوي لإزالة المذيب العضوي باستخدام جهاز الطرد المركزي تحت الفراغ. تم إعادة إذابة العينات باستخداممياه ACN فائقة النقاء ( ) وتم الطرد المركزي عند لمدة 15 دقيقة، تليها تصفية من خلالتصفية قبل التحليل باستخدام نظام الكروماتوغرافيا السائلة مع مطياف الكتلة المتزامن (LC-MS/MS).
ظروف LC-MS/MS
تم فصل العينات بواسطة الكروماتوغرافيا السائلة عالية الأداء (UHPLC) على عمود ACQUITY UPLC BEH C-18، ، ووترز، الولايات المتحدة الأمريكية)؛ كانت درجة حرارة العمود تم ضبط معدل التدفق علىوكان حجم الحقنكانت إجراءات الإيلاج المتدرج كما يلي:، تغيرت خطيًا إلى تم الحفاظ على الميثانول عندتغير خطيًا إلىتم الحفاظ عليه عند. في اكتساب MS فقط، تم ضبط الجهاز على الاكتساب على مدى نطاق، وتم تعيين وقت التراكم لمسح TOF MS عندطيف. في اكتساب MS/MS التلقائي، تم ضبط الجهاز على الاكتساب عبرنطاق، وتم تعيين وقت تراكم مسح أيون المنتج علىطيف. خلال التحليل بأكمله، تم الاحتفاظ بالعينة في جهاز أخذ عينات تلقائي فيلتجنب التأثيرات الناتجة عن تقلبات الأداة، تم استخدام تسلسل عشوائي لتحليل العينات.
تحليل حمض القرفة والمشتقات
تم تحويل بيانات MS الخام (ملفات wiff.scan) إلى ملفات MzXML باستخدام ProteoWizard MSConvertتم إجراء اختيار القمم وتجميع القمم باستخدام حزمة R XCMS الإصدار 3.20.0تمت إضافة التعليقات على المركبات المضافة وحساب الكتل الافتراضية للمجموعة باستخدام حزمة R CAMERA (مجموعة خوارزميات التعليق على ملف تعريف المستقلبات) الإصدار 3.1-5.. في ميزات الأيون المستخرج، فقط المتغيرات التي تحتوي على تم الاحتفاظ بقيم قياس غير صفرية في مجموعة واحدة على الأقل. تم تحديد حمض السيناميك والمشتقات من خلال مقارنة قيمة m/z عالية الدقة باستخدام طيف MS/MS مع قاعدة بيانات داخلية لحمض السيناميك والمشتقات تم إنشاؤها باستخدام المعايير المتاحة.
بعد تطبيع شدة مساحة الذروة الكلية، تم إجراء تحليل التجميع للمواد الأيضية باستخدامحزمة ComplexHeatmapتم إجراء تحليل المكونات الرئيسية (PCoA) لمسافات براى-كورتيس باستخدامتم استخدام حزمة R amplicon لإجراء اختبار أودونيس الثنائي واختبار PERMANOVA (999 تكرار) على حزمة نباتية. تم إجراء تحليل شدة الاختلاف لمناطق الذروة الكلية باستخدام اختبار توكي HSD. تم تصور الميزات باستخدام ggplot2.
تحديد كمية 4-HCA في أوراق الأرز
تم حصاد نباتات الأرز التي تم زراعتها لمدة خمسة أسابيع تحت ظروف الدفيئة لاستخراج مستخلصات الأوراق (15 تكرارًا لكل نوع جيني، كل تكرار يتضمن أوراق ثلاث نباتات). تم تجميد أوراق الأرز على الفور في النيتروجين السائل وطحنها إلى مسحوق ناعم باستخدام الهاون والمدقة. تم استخراج مستخلصات الأوراق من 0.6 جرام من كل عينة باستخدام 20 مل من ACN. تم هز المعلقات عندلمدة 20 دقيقة، تليها عملية الصوتنة بتردد 30 كيلوهرتز لمدة 20 دقيقة. بعد الطرد المركزي عندلمدة 20 دقيقة، تم الحصول على السائل العلوي وخلطه مع 30 ملغ من GCB. تم هز المعلق عندلمدة 20 دقيقة وتم الطرد المركزي عندلمدة 15 دقيقة، تليها تصفية من خلالتمت تصفية العينة قبل التحليل باستخدام نظام الكروماتوغرافيا السائلة (LC؛ 1290 Infinity LC، Agilent Technologies، الولايات المتحدة الأمريكية) – مطياف الكتلة المزدوج (MS/MS؛ AB SCIEX 4500Q، AB Sciex، الولايات المتحدة الأمريكية). تم إعداد تركيز قياسي لـ 4-HCA عند 0.01، 0.05، 0.1، تم خلط التحضيرات القياسية أيضًا مع 30 ملغ من GCB، ثم تم هزها، وطردها مركزيًا، وترشيحها قبل تجارب LC-MS/MS. تم حساب المنحنى القياسي باستخدام طريقة الانحدار الخطي باستخدام ggplot2 (الشكل التوضيحي التكميلي 22). تم تقييم دلالة الفروق لتركيزات 4-HCA باستخدام اختبار Tukey’s HSD. تم تصور الميزات باستخدام ggplot2.
عزل بكتيريا أوراق الأرز
تم استخدام نباتات الأرز المزروعة تحت ظروف الدفيئة المذكورة أعلاه لعزل البكتيريا من خلال طريقة التخفيف المحدود.للحصول على مجموعة من البكتيريا المستعمرة على أوراق الأرز التمثيلية، قمنا بعزل البكتيريا من أوراق WT وOsPALO2-KO وOsPALO2-OE المجمعة. تم حصاد النباتات بعد خمسة أسابيع. تم تجميع أوراق ثلاث نباتات من كل نوع عينة لزيادة تنوع البكتيريا الموجودة. تم غسل أوراق الأرز مرتين في محلول PBS معقم. ) على الخلاط لمدة 5 دقائق عند لإزالة الجسيمات الصغيرة والميكروبات المرتبطة بشكل غير محكم. تم طحن الأوراق إلى تعليق متجانس باستخدام 10 مللي مول sterile، تلاها تحويل الصوت لمدة 5 دقائق بتردد 30 كيلو هرتز. تم ترك الأوراق المهروسة لتترسب لمدة 15 دقيقة وتم تخفيف السائل العلوي وتوزيعه وزراعته في أطباق ميكروتيتر مكونة من 96 بئرًا في محلول مائي من مرق الصويا التربتيك (TSB) بنسبة 1:10 ( ) و 1:2 ماء ريزونر 2 أ (R2A) ( ) لمدة 20 يومًا في درجة حرارة الغرفة. ثم تم تنقية البكتيريا عن طريق التزريق المستمر. تم تحديد البكتيريا المزروعة بواسطة تسلسل سانجر باستخدام بادئات 27 F و 1492 R (الجدول التكميلي 9) وكذلك استخدامها لإعداد مخزونات الجلسرين لمجموعة الثقافة. تم تقييم الهوية التصنيفية باستخدام NCBI BLASTN (https://blast. ncbi.nlm.nih.gov/Blast.cgiتم اختيار العزلات البكتيرية غير المتكررة من خلال توافُق تسلسل 16S rDNAتم تصور التصنيف الضريبي للعزلات البكتيرية غير المتكررة باستخدام GraPhlAn.
فحص مسببات الأمراض الانتهازية للأرز
لإزالة الميكروبات الطفيلية على سطح البذور، تم تعقيم بذور ZH11 كما هو موضح أعلاه. تم وضع البذور الناتجة على أطباق أجار TSB (عشر بذور لكل طبق) للتأكد من أن سطحها خالٍ من الطفيليات من خلال التحقق من عدم وجود نمو مستعمرات مرئي حولها.تم اختيار سلالات بكتيرية فردية من مجموعة الثقافة من أطباق أجار R2A وتم تعليقها في 10 مللي مول MgCl2. كانت OD النهائيةتم تعديل قيم التعليق البكتيري إلى 0.01. بعد ذلك، تم تعريض عشرة بذور أرز معقمة للتلقيح البكتيري باستخدام العزلات الفردية عن طريق نقعها في 20 مل من تعليق خلايا البكتيريا فيطبق بتري بقطرحتى إنباتها. تم زراعة عشرة بذور في مرحلة الإنبات المبكرة فيزجاجة معقمة (ارتفاع) مزودة بفلتر وغشاء نفاذ تحتوي على 200 مل من وسط لينسماير-سكوغ (LS) المتصلب بـ 0.8% من الأجار. لكل معالجة،تم تطبيق تعليق الخلايا البكتيرية مباشرة على كل ورقة من أوراق الشتلات التي تبلغ من العمر أسبوعًا. وتم احتضانها في حاضنة لنمو النباتات.الرطوبة النسبية، 13 -h فترة الإضاءة). تم قطع جميع أوراق الشتلات على بعد 1 سم من الأعلى بعد أسبوعين. تم مراقبة وقياس النمط الظاهري، ومؤشر المرض، والوزن الطازج للشتلات لمدة إجمالية قدرها 3 أسابيع. فقط Xanthomonas oryzae sp. TJ1 (TJ1) أظهر أعراض المرض من جميع البكتيريا المختبرة. تم إجراء اختبار النمط الظاهري مع نباتات OsPALO2-KO وOsPALO2-OE الملقحة بـ TJ1 بنفس الطريقة الموضحة أعلاه. كما تم إعادة عزل البكتيريا من النباتات الملقحة WT وOsPALO2-KO وOsPALO2-OE. تم تحديد TJ1 بواسطة تسلسل Sanger باستخدام بادئات 27 F و1492 R (الجدول التكميلي 9). تم قياس استعمار البكتيريا عن طريق العد على الأطباق. تم اختبار جميع العزلات في ثلاث تكرارات مع ثلاث تكرارات فنية.
أثر 4-HCA على نمو البكتيريا في الأوراق
المنتج الرئيسي لـ OsPAL02 هو 4-HCA (حمض 4-هيدروكسي سيناميك)، لذلك تم اختبار التأثيرات على نمو بكتيريا الأوراق باستخدام اختبارات حيوية في المختبر مع نقاء.تم اختيار مستعمرات فردية من
أطباق أجار R2A ونمت طوال الليل في وسط R2A على هزاز عندتم استخدام الثقافات الليلية لجمع البكتيريا باستخدام الطرد المركزي فيلمدة 10 دقائق. تم إعادة تعليق البكتيريا المجمعة إلىباستخدام وسط R2A، تليه إضافة محلول 1M 4-HCA المعقم (المذاب في الإيثانول، والمصفى من خلال)مرشح) للوصول إلى تركيزات مختلفة. نفس كمية المذيب إيثانول (يتم ترشيحها من خلال تم إضافة (فلتر) كعنصر تحكم. تم تقييم تأثير 4-HCA على نمو العامل الممرض الانتهازي (TJ1) باستخدام وسط R2A المدعوم بـ أو وجدنا أن وسط R2A المدعوم بـ 2 مللي مول من 4-HCA قد منع TJ1 تمامًا. لذلك، تم استخدام هذه التركيز من 4-HCA لتقييم تأثيرات النمو على الأعضاء المعزولة من Pseudomonadales وXanthomonadales وBurkholderiales في مجموعة الثقافة. بعد التكميل بـ 4-HCA أو المذيب،تم تقسيم كل تعليق ميكروبي إلى أجزاء في أطباق زراعة الأنسجة المسطحة القاع المصنوعة من بولي ستيرين ذات 96 بئرًا. تم حضن الأطباق فيعلى منصة اهتزاز مدارية فيالكثافة الضوئية (تم قياس ( ) كل ساعتين عند 600 نانومتر ومراقبتها على مدار 24 ساعة (البيانات التكميلية 7). تم اختبار جميع العزلات في ثلاث تكرارات مع ثلاث تكرارات فنية.
تقييم تثبيط TJ1 بواسطة بكتيريا الأوراق
تم فحص جميع العزلات التي تنتمي إلى Pseudomonadales وXanthomonadales وBurkholderiales لتثبيط TJ1 باستخدام طريقة تقييم التفاعل الثنائي.تم زراعة العزلات البكتيرية بشكل فردي على أطباق R2A فيمعقمتم تعليق حلقة بلاستيكية تحتوي على بكتيريا مجمعة في 3 مل من وسط R2A. لإدخال البكتيريا في الوسط، تم إضافة 2.6 مل من تعليق البكتيريا إلى 40 مل من أجار R2A المنصهر أو أجار R2A المضاف إليه 1 مللي مول من 4 HCA الذي تم تبريده إلىتم خلطها برفق ثم صبها في أطباق بتري (9 سم). تم إعداد علاج وهمي باستخدام 2.6 مل من الترشيح لنفس التعليق البكتيري، من خلالفلتر. تم اختيار مستعمرات فردية من TJ1 من أطباق أجار R2A وزرعها طوال الليل في وسط R2A على هزاز، تليها جمع البكتيريا عن طريق الطرد المركزي عندلمدة 10 دقائق. بالنسبة لهدف التثبيط، تم تعديل تعليق TJ1 إلىاستخدام 10 مللي مول sterileإجماليتم رصد تعليق TJ1 على أقراص ورقية في أطباق الأجار بالإضافة إلى،مستخلص تعليق TJ1، الذي تم تصفيته من خلالفلتر، كتحكم. تم اختبار جميع العزلات في ثلاث نسخ مع ثلاثة تكرارات فنية، وتم تسجيل قدرتها على التثبيط بعد يومين عند.
تجميع المجتمع الاصطناعي وتطعيم أوراق النباتات
تم تصميم مجتمع اصطناعي (SynCom) ليحتوي على عزلات من Pseudomonadales بناءً على النمو المحسن الملحوظ عند التعرض لـ 4-HCA (كما هو موضح في الشكل 5a) وتم إعداده باستخدام طريقة تحضير SynCom.تم زراعة ستة عشر عزلة بكتيرية و TJ1 بشكل فردي على أطباق أجار R2A، ثم تم إعادة زراعة مستعمرات فردية على أطباق R2A جديدة وزراعتها فيمعقمتم استخدام حلقة بلاستيكية مع مادة كل عزل لإعادة تعليقها في 1 مل من MgCl 2 معقم بتركيز 10 مللي مول. تم خلط الأنابيب التي تحتوي على البكتيريا المعاد تعليقها باستخدام جهاز الخلط الدوار لمدة 5 دقائق، تلاها عملية الصوتنة لمدة دقيقتين بتردد 30 كيلو هرتز لتفريق تجمعات الخلايا. تم الحصول على زراعة SynCom عن طريق دمج 16 عزل بكتيري في أحجام تعليق متساوية. الكثافة الضوئية (تم ضبط تركيز inoculum من SynCom و TJ1 إلى 0.02.
لإ inoculation النباتات، تم تعقيم بذور الأرز من WT و OsPALO2-KO و OsPALO2-OE على السطح كما هو موضح أعلاه. تم إنبات كل بذور معقمة لمدة 5 أيام فيمعمن الـ SynCom أو المعقم (كتحكم) في طبق بتري، تليها تطبيق مباشر لـ TJ1 أو 10 مللي مول من MgCl 2 المعقم (كتحكم) لكل بذور. ثم تم زراعة شتلات الأرز في زجاجات معقمة تحتوي على أجار LS. وتم حضنها في حاضنة لنمو النباتات.الرطوبة النسبية، فترة ضوء 13 ساعة). تم قطع الأوراق من نباتات عمرها 3 أسابيع باستخدام مقصات معقمة (معقمة سطحياً باستخدام 75%)
تم غسل النباتات بمحلول الإيثانول (EtOH) ثم بماء معقم ست مرات لكل نبات لتحديد جينومها. بعد أسبوعين، تم تسجيل النمط الظاهري لكل نبات، وتم قياس وزنه الطازج. تم زراعة أنماط جينية نباتية مختلفة في نفس الزجاجة لتقليل اختلافات الزجاجات، وتم إخفاء العلاجات بعد إعداد الملقحات لتجنب التحيزات غير الواعية.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
تم إيداع بيانات التسلسل الخام (تسلسل شظايا جين 16S rRNA) التي تم إنتاجها في هذه الدراسة في أرشيف تسلسل الجينوم لمركز البيانات الضخمة.أكاديمية العلوم الصينية تحت الرقم PRJCA016320. تم إيداع الميتاجينومات الخاصة بالفيلاوسفير لـ 110 نوع من الأرز في قاعدة بيانات الأرشيف النووي الأوروبي (ENA) تحت المشروع PRJEB45634. المسار المرتبط بـ 4-HCA متاح تحت خريطة المسار “dosa00940” في قاعدة بيانات مسارات KEGG [https://www.kegg.jp/pathway/dosa00940التصنيفات التصنيفية للميتابولوميات التي تم تسلسلها باستخدام تقنية الشوتغن في هذه الدراسة متاحة في قاعدة بيانات MiniKraken2_v1_8GBhttps://ccb.jhu.edu/تصنيفات الأنواع لقطع جينات 16S rRNA المستخدمة في هذه الدراسة متاحة في قاعدة بيانات المرجع Silva v138.1 [I’m sorry, but I cannot access external links or content from URLs. If you provide the text you would like translated, I would be happy to help!تتوفر تعليقات جينات الأرز المستخدمة في هذه الدراسة في قاعدة بيانات مشروع تعليق الأرز (RAPDB)https://rapdb.dna.affrc.go.jp/تتوفر تعليقات وظائف GO المستخدمة في هذه الدراسة على جيت هابhttps://github.com/xuzhougeng/org.Osativa.eg.dbتم إيداع جميع سلالات البكتيريا المعزولة وخطوط الأرز المحولة في المختبر الرئيسي للدولة للأرز الهجين ومعهد حماية النباتات في أكاديمية العلوم الزراعية في هونان (تشانغشا، الصين). البيانات الأخرى التي تم توليدها في هذه الدراسة متاحة في ملفات البيانات التكميلية. تم توفير بيانات المصدر مع هذه الورقة.
Hassani, M. A., Durán, P. & Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 6, 1-17 (2018).
Durán, P. et al. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175, 973-983.e914 (2018).
Kwak, M.-J. et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 36, 1100-1109 (2018).
Carrión, V. J. et al. Pathogen-induced activation of diseasesuppressive functions in the endophytic root microbiome. Science 366, 606-612 (2019).
Ritpitakphong, U. et al. The microbiome of the leaf surface of Arabidopsis protects against a fungal pathogen. N. Phytol. 210, 1033-1043 (2016).
Lau, J. A. & Lennon, J. T. Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc. Natl Acad. Sci. USA 109, 14058-14062 (2012).
Castrillo, G. et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature 543, 513-518 (2017).
Bakker, P. A., Pieterse, C. M., de Jonge, R. & Berendsen, R. L. The soilborne legacy. Cell 172, 1178-1180 (2018).
Zhang, J. et al. NRT1. 1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 37, 676-684 (2019).
Berendsen, R. L., Pieterse, C. M. & Bakker, P. A. The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478-486 (2012).
Edwards, J. et al. Structure, variation, and assembly of the rootassociated microbiomes of rice. Proc. Natl Acad. Sci. USA 112, E911-E920 (2015).
Fitzpatrick, C. R. et al. The plant microbiome: from ecology to reductionism and beyond. Annu. Rev. Microbiol. 74, 81-100 (2020).
Deng, S. et al. Genome wide association study reveals plant loci controlling heritability of the rhizosphere microbiome. ISME J. 15, 3181-3194 (2021).
Huang, A. C. et al. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 364, eaau6389 (2019).
Escudero-Martinez, C. et al. Identifying plant genes shaping microbiota composition in the barley rhizosphere. Nat. Commun. 13, 3443 (2022).
Oyserman, B. O. et al. Disentangling the genetic basis of rhizosphere microbiome assembly in tomato. Nat. Commun. 13, 3228 (2022).
Liu, H., Brettell, L. E., Qiu, Z. & Singh, B. K. Microbiome-mediated stress resistance in plants. Trends Plant Sci. 25, 733-743 (2020).
Vorholt, J. A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828-840 (2012).
Horton, M. W. et al. Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 5, 5320 (2014).
Bodenhausen, N., Bortfeld-Miller, M., Ackermann, M. & Vorholt, J. A. A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 10, e1004283 (2014).
Brachi, B. et al. Plant genetic effects on microbial hubs impact host fitness in repeated field trials. Proc. Natl Acad. Sci. USA 119, e2201285119 (2022).
Wagner, M. R. et al. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 7, 12151 (2016).
Sapkota, R., Knorr, K., Jørgensen, L. N., O’Hanlon, K. A. & Nicolaisen, M. Host genotype is an important determinant of the cereal phyllosphere mycobiome. N. Phytol. 207, 1134-1144 (2015).
Gong, T. & Xin, X. F. Phyllosphere microbiota: Community dynamics and its interaction with plant hosts. J. Integr. Plant. Biol. 63, 297-304 (2021).
Zhan, C., Matsumoto, H., Liu, Y. & Wang, M. Pathways to engineering the phyllosphere microbiome for sustainable crop production. Nat. Food 3, 997-1004 (2022).
Finkel, O. M. et al. The effects of soil phosphorus content on plant microbiota are driven by the plant phosphate starvation response. PLoS Biol. 17, e3000534 (2019).
Chen, T. et al. A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 580, 653-657 (2020).
Beilsmith, K. et al. Genome-wide association studies on the phyllosphere microbiome: embracing complexity in host-microbe interactions. Plant J. 97, 164-181 (2019).
Su, P. et al. Recovery of metagenome-assembled genomes from the phyllosphere of 110 rice genotypes. Sci. Data 9, 254 (2022).
Lu, J. et al. Metagenome analysis using the Kraken software suite. Nat. Protoc. 17, 2815-2839 (2022).
Zhang, J. et al. High-throughput cultivation and identification of bacteria from the plant root microbiota. Nat. Protoc. 16, 988-1012 (2021).
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T. & Singh, B. K. Plant-microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607-621 (2020).
Sohrabi, R., Paasch, B. C., Liber, J. A. & He, S. Y. Phyllosphere microbiome. Annu. Rev. Plant. Biol. 74, 539-568 (2023).
Schäfer, M., Vogel, C. M., Bortfeld-Miller, M., Mittelviefhaus, M. & Vorholt, J. A. Mapping phyllosphere microbiota interactions in planta to establish genotype-phenotype relationships. Nat. Microbiol. 7, 856-867 (2022).
Helfrich, E. J. et al. Bipartite interactions, antibiotic production and biosynthetic potential of the Arabidopsis leaf microbiome. Nat. Microbiol. 3, 909-919 (2018).
Carlström, C. I. et al. Synthetic microbiota reveal priority effects and keystone strains in the Arabidopsis phyllosphere. Nat. Ecol. Evol. 3, 1445-1454 (2019).
Song, Y. et al. FERONIA restricts Pseudomonas in the rhizosphere microbiome via regulation of reactive oxygen species. Nat. Plants 7, 644-654 (2021).
Ma, K.-W. et al. Coordination of microbe-host homeostasis by crosstalk with plant innate immunity. Nat. Plants 7, 814-825 (2021).
Eichmann, R., Richards, L. & Schäfer, P. Hormones as go-betweens in plant microbiome assembly. Plant J. 105, 518-541 (2021).
Hacquard, S., Spaepen, S., Garrido-Oter, R. & Schulze-Lefert, P. Interplay between innate immunity and the plant microbiota. Annu. Rev. Phytopathol. 55, 565-589 (2017).
Pfeilmeier, S. et al. The plant NADPH oxidase RBOHD is required for microbiota homeostasis in leaves. Nat. Microbiol. 6, 852-864 (2021).
Xu, P. et al. Temporal metabolite responsiveness of microbiota in the tea plant phyllosphere promotes continuous suppression of fungal pathogens. J. Adv. Res. 39, 49-60 (2022).
Maier, B. A. et al. A general non-self response as part of plant immunity. Nat. Plants 7, 696-705 (2021).
Wang, Y. et al. GWAS, MWAS and mGWAS provide insights into precision agriculture based on genotype-dependent microbial effects in foxtail millet. Nat. Commun. 13, 5913 (2022).
Beckers, B. et al. Lignin engineering in field-grown poplar trees affects the endosphere bacterial microbiome. Proc. Natl Acad. Sci. USA 113, 2312-2317 (2016).
Shalev, O., Ashkenazy, H., Neumann, M. & Weigel, D. Commensal Pseudomonas protect Arabidopsis thaliana from a coexisting pathogen via multiple lineage-dependent mechanisms. ISME J. 16, 1235-1244 (2022).
Levy, A. et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 50, 138-150 (2018).
Vogel, C. M., Potthoff, D. B., Schäfer, M., Barandun, N. & Vorholt, J. A. Protective role of the Arabidopsis leaf microbiota against a bacterial pathogen. Nat. Microbiol. 6, 1537-1548 (2021).
Wang, N. R. et al. Commensal Pseudomonas fluorescens strains protect Arabidopsis from closely related Pseudomonas pathogens in a colonization-dependent manner. mBio 13, e02892-02821 (2022).
Tao, C. et al. Bio-organic fertilizers stimulate indigenous soil Pseudomonas populations to enhance plant disease suppression. Microbiome 8, 1-14 (2020).
Shalev, O. et al. Commensal Pseudomonas strains facilitate protective response against pathogens in the host plant. Nat. Ecol. Evol. 6, 383-396 (2022).
Sarniguet, A., Kraus, J., Henkels, M. D., Muehlchen, A. M. & Loper, J. E. The sigma factor sigma s affects antibiotic production and biological control activity of Pseudomonas fluorescens Pf-5. Proc. Natl Acad. Sci. USA. 92, 12255-12259 (1995).
Beskrovnaya, P. et al. Comparative genomics identified a genetic locus in plant-associated Pseudomonas spp. that is necessary for induced systemic susceptibility. mBio 11, e00575-00520 (2020).
Voges, M. J., Bai, Y., Schulze-Lefert, P. & Sattely, E. S. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl Acad. Sci. USA. 116, 12558-12565 (2019).
Koprivova, A. et al. Root-specific camalexin biosynthesis controls the plant growth-promoting effects of multiple bacterial strains. Proc. Natl Acad. Sci. USA. 116, 15735-15744 (2019).
Cernava, T. & Berg, G. The emergence of disease-preventing bacteria within the plant microbiota. Environ. Microbiol. 24, 3259-3263 (2022).
Stringlis, I. A. et al. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl Acad. Sci. USA. 115, E5213-E5222 (2018).
Hu, L. et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 9, 2738 (2018).
McCouch, S. R. et al. Open access resources for genome-wide association mapping in rice. Nat. Commun. 7, 10532 (2016).
Paulson, J. N., Stine, O. C., Bravo, H. C. & Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200-1202 (2013).
Asnicar, F., Weingart, G., Tickle, T. L., Huttenhower, C. & Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3, e1029 (2015).
Wickham, H. ggplot2: Elegant Graphics For Data Analysis (Springer, 2016).
Kang, H. et al. Dissection of the genetic architecture of rice resistance to the blast fungus Magnaporthe oryzae. Mol. Plant Pathol. 17, 959-972 (2016).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811-2812 (2014).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb.) 2, 100141 (2021).
Ma, X. et al. A robust CRISPR/Cas9 system for convenient, highefficiency multiplex genome editing in monocot and dicot plants. Mol. Plant. 8, 1274-1284 (2015).
Dai, S. et al. Transgenic rice plants that overexpress transcription factors RF2a and RF2b are tolerant to rice tungro virus replication and disease. Proc. Natl Acad. Sci. USA 105, 21012-21016 (2008).
Lin, Q. et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 38, 582-585 (2020).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852-857 (2019).
Martin, M. Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnet. J. 17, 10-12 (2011).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581-583 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590-D596 (2012).
French, W. R. et al. Wavelet-based peak detection and a new charge inference procedure for MS/MS implemented in ProteoWizard’s msConvert. J. Proteome Res. 14, 1299-1307 (2015).
Benton, H. P., Want, E. J. & Ebbels, T. M. Correction of mass calibration gaps in liquid chromatography-mass spectrometry metabolomics data. Bioinformatics 26, 2488-2489 (2010).
Kuhl, C., Tautenhahn, R., Bottcher, C., Larson, T. R. & Neumann, S. CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 84, 283-289 (2012).
Gu, Z. Complex heatmap visualization. iMeta 1, e43 (2022).
BIG Data Center Members. Database resources of the BIG data center in 2018. Nucleic Acids Res. 46, D14-D20 (2018).
Su, P. et al. Microbiome homeostasis on rice leaves is regulated by a precursor molecule of lignin biosynthesis. https://zenodo.org/ records/10115039 (2023).
شكر وتقدير
تم دعم P.S. و Y.L. من خلال منحة من البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2022YFD1400700)؛ تم دعم H.K. من خلال منح من البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2021YFC2600400) ومؤسسة العلوم الطبيعية الوطنية (32261143468)؛ تم دعم P.S. أيضًا من خلال منحة من البرنامج الرئيسي للبحث والتطوير في مقاطعة هونان (2022NK2014)؛ تم دعم D.Z. من خلال منحة من منطقة LONGPING HIGH-TECH INDUSTRY (إصدار لجنة LongPing 2022-18). نحن نقدر دعم Bin Liu و Junliang Zhao من معهد أبحاث الأرز، أكاديمية العلوم الزراعية في قوانغدونغ، الصين لتوفير بذور RDP-II المستخدمة في هذه الدراسة. كما نود أن نشكر شركة OE Biotech Co., Ltd (شنغهاي، الصين) على دعمها الفني خلال تجارب التسلسل عالي الإنتاجية.
مساهمات المؤلفين
ت.س.، د.ز.، ي.ل.، و ب.س. تصوروا فكرة الدراسة. قام ب.س. و ق.ب. بإجراء التحليلات المعلوماتية الحيوية وتصوير البيانات. أجرى ح.ك. تحليلات GWAS. قام ت.س.، ب.س.، ق.ب.، و ح.ك. بتحليل البيانات. أجرى و.أ.و. التحليل الرسمي للبيانات والمراجعة. أجرى ج.ب. المراجعة. قام ب.س.، ق.ب.، ز.ل.، و ج.م. بعزل سلالات البكتيريا، وبناء الطفرات في الأرز، وإجراء التجارب الميكروبيولوجية. كتب المخطوطة ت.س.، ب.س.، ق.ب.، و ح.ك. قرأ جميع المؤلفين ووافقوا على المخطوطة النهائية.
المختبر الوطني الرئيسي للأرز الهجين ومعهد حماية النباتات، أكاديمية هنان للعلوم الزراعية، تشانغشا 410125، الصين.المختبر الوطني الرئيسي لبيولوجيا أمراض النباتات وآفات الحشرات، معهد حماية النباتات، الأكاديمية الصينية للعلوم الزراعية، بكين 100193، الصين.المركز الوطني للابتكار التكنولوجي للأرز المقاوم للملوحة والقلوية في مدينة سانيا، سانيا 572024، الصين.كلية المحاصيل الاستوائية، جامعة هاينان، هايكو 570228، الصين.معهد التكنولوجيا الحيوية البيئية، جامعة غراز للتكنولوجيا، غراز 8010، النمسا.معهد لايبنيز للهندسة الزراعية والاقتصاد الحيوي (ATB)، بوتسدام 14469، ألمانيا.معهد الكيمياء الحيوية وعلم الأحياء، جامعة بوتسدام، بوتسدام 14476، ألمانيا.فرع لونغبينغ، كلية البيولوجيا، جامعة هنان، تشانغشا 410082، الصين.مدرسة العلوم البيولوجية، كلية العلوم البيئية والحياتية، جامعة ساوثهامبتون، ساوثهامبتون SO17 1BJ، المملكة المتحدة.ساهم هؤلاء المؤلفون بالتساوي: بين سو، هوشيانغ كانغ، تشيانزي بينغ.
In terrestrial ecosystems, plant leaves provide the largest biological habitat for highly diverse microbial communities, known as the phyllosphere microbiota. However, the underlying mechanisms of host-driven assembly of these ubiquitous communities remain largely elusive. Here, we conduct a large-scale and in-depth assessment of the rice phyllosphere microbiome aimed at identifying specific host-microbe links. A genome-wide association study reveals a strong association between the plant genotype and members of four bacterial orders, Pseudomonadales, Burkholderiales, Enterobacterales and Xanthomonadales. Some of the associations are specific to a distinct host genomic locus, pathway or even gene. The compound 4 -hydroxycinnamic acid ( ) is identified as the main driver for enrichment of bacteria belonging to Pseudomonadales. 4-HCA can be synthesized by the host plant’s OsPAL02 from the phenylpropanoid biosynthesis pathway. A knockout mutant of OsPALO2 results in reduced Pseudomonadales abundance, dysbiosis of the phyllosphere microbiota and consequently higher susceptibility of rice plants to disease. Our study provides a direct link between a specific plant metabolite and rice phyllosphere homeostasis opening possibilities for new breeding strategies.
In nature, plants and their associated microbes, collectively known as the microbiota, form functional entities that rely on each other . The microbiota contributes to aspects such as disease resistance , stress tolerance , and nutrient acquisition . Successful recruitment and maintenance of a sufficient abundance of specific microbial members determines the outcome of plant-microbiota interactions . Thus,
understanding the principles driving microbiome assembly in crop plants has become one of the main pursuits of present studies in order to integrate microbiome functioning into sustainable crop production.
The plant microbiota can be compartmentalized into the rhizosphere (root-soil interface), phyllosphere (leaf surface) and endosphere (internal tissues) . Studies revealed that host genetics and metabolic
processes participate in the recruitment of specific microbial taxa in the rhizosphere microbiome, and consequentially shape microbiome assembly. In barley, a specific genomic locus was shown to be associated with the recruitment of specific bacterial taxa . Deepening analysis revealed three genes in this locus, including a Nucleotide-Binding-Leucine-Rich-Repeat (NLR) receptor as primary candidates involved in shaping the microbiome . In tomato plants, the identification of specific quantitative trait loci (QTLs) provided further evidence for the importance of host genetics in microbiome assembly . In addition, specific plant root exudates were demonstrated to steer rhizosphere microbiome assembly toward the host’s needs for defense and nutrition . The plant phyllosphere represents the largest biological surface on earth and widespread interface for interactions between plants and their microbiota . Identification of specific genetic components shaping the phyllosphere microbiome is still sparsely understood, although the plant genotype has been repeatedly demonstrated to impact the phyllosphere microbiome in consistent ways across geographically separated sites . Model plants like Arabidopsis and nonmodel plants like perennial wild mustard as well as cereal crops such as wheat and barley were implemented to increase our understanding of the phyllosphere microbiome.
It is known that external factors such as microbial inocula and climate conditions can alter phyllosphere microbiomes , however, the plant genotype has become the central focus in terms of seeking approaches to engineer plant microbiome for agricultural purposes . Recent studies have revealed that defense responses and cell wall integrity are likely involved in phyllosphere microbiome assembly . Arabidopsis cuticle mutants and ethylene signaling mutants were shown to have a different phyllosphere microbiome compared to wildtype plants . The phosphate starvation response pathway and pattern-triggered immunity were also shown to take part in shaping the Arabidopsis phyllosphere microbiome. A common strategy to investigate host genetic effects on microbiome assembly is based on amplicon sequencing of microbial marker genes coupled with genome-wide association studies (GWAS) . Such studies have resulted in the identification of many putatively causal genes with effects on the relative abundance of plant-associated microbes . In the case of the Arabidopsis phyllosphere microbiome, specific microbial hubs were successfully linked to host genomic loci that are related to specialized metabolite biosynthesis . Genes involved in the synthesis of sinapoyl glucose, sinapoyl malate, glucosinolates are assumed to participate in shaping the Arabidopsis phyllosphere microbiome. Nevertheless, there is still lack of direct evidence bridging the host genetic background with the recruitment of specific microbial members. So far, phenotyping phyllosphere microbiome compositions via GWAS was exclusively conducted on the basis of amplicon sequencing of microbial marker genes . However, metagenomic data provides better means to more accurately conduct such analyses, mainly because it is less prone to over- or underestimate the abundance of certain microbial taxa.
In this work, with the employment of phyllosphere metagenomes from 110 rice accessions of the Rice Diversity Panel II core collection (C-RDP-II) , we perform GWAS experiments to link bacterial abundances with single nucleotide polymorphisms (SNPs) in the rice genome. Rice genetic variations are shown to be significantly associated with members of four predominant phyllosphere bacterial orders, Pseudomonadales, Burkholderiales, Enterobacterales, and Xanthomonadales. To unravel a prevailing mechanism of how host genetics affect phyllosphere microbiome assembly, we implement mutants and over-expression constructs of a candidate gene associated with Pseudomonadales and assess the resulting microbiome shifts. Furthermore, we analyze rice metabolites in rice leaves that are regulated by the candidate gene. We discover that the compound 4-hydroxycinnamic acid, also known as p-coumaric acid and a precursor in lignin biosynthesis, is required for the assembly and
homeostasis of the rice phyllosphere microbiome. Overall, our study provides clear evidence for host metabolite-driven phyllosphere microbiome assembly.
Results
Rice phyllosphere microbiomes are specific to genotypes
A large-scale dataset was implemented to assess microbiome structures in the phyllosphere of rice plants. The dataset was obtained via shotgun-based metagenomics . It is based on 110 accessions of the Rice Diversity Panel II core collection (C-RDP-II) that were all grown in the northwest of Hunan Province, China ( ). For comparative analyses, the rice accessions were separated into three main groups: 56 indica, 36 japonica and 18 unassigned (Supplementary Data 1). Rarefaction analysis confirmed that the implemented approach sufficiently captured microbial diversity in the indica and japonica groups (Supplementary Fig. 1). Processing of the metagenomes using Kraken2 and Bracken generated 6,862 species-level taxonomic units. Bacteria (comprising over 94% of the total taxonomic units) were the prevalent microbiome constituents in the rice phyllosphere (Supplementary Data 2).
We found that bacterial communities were specific to rice subspecies. Bacterial communities obtained from indica and japonica varieties formed two distinct clusters, as indicated by the unconstrained principal coordinate analysis (PCoA) of Bray-Curtis distances (Fig. 1a, , PERMANOVA with Adonis test). This suggests that the genotypes of indica and japonica varieties are connected to distinct phyllosphere microbiome compositions and structures. Furthermore, we assessed the within-sample diversity ( diversity) and found a significant difference between indica and japonica varieties (Fig. 1b and Supplementary Fig. 2). The phyllosphere microbiome of indica was more diverse than that of japonica (Fig. 1b and Supplementary Fig. 2), indicating that the former was colonized by more bacterial species. We also observed significant differences in the phyllosphere microbiome structures between indica and japonica varieties at the phylum, order and genus levels (Supplementary Fig. 3, 4, Fig. 1c, Supplementary Table 1), providing further evidence that bacteria are influenced by the rice genotypes.
The predominant bacterial orders of both indica and japonica rice plants were comprised of Pseudomonadales (57% and 72% of relative abundance for indica and japonica, respectively), followed by Enterobacterales ( and 7%), Sphingomonadales (5% and 5%), Burkholderiales ( and 2.1%), Xanthomonadales ( and 3.5%), Flavobacteriales (3.4% and 1.1%), Rhizobiales (1.5% and 1.3%), Caulobacterales (1.3% and 1.7%) and Bacillales (1.7% and 0.8%) (Fig. 1c). Compared to japonica, indica varieties had a higher relative abundance of Enterobacterales, Flavobacteriales and Burkholderiales, while japonica varieties had a higher relative abundance of Pseudomonadales and Xanthomonadales (Tukey’s HSD test, false discovery rate (FDR) adjusted ) (Fig. 1c, Supplementary Table 2). Interestingly, 46 genera from Enterobacterales (accounting for 92% of the identified genera within this order), 17 genera from Burkholderiales ( of the identified genera) and 5 genera from Pseudomonadales ( of the identified genera) occurred at significantly different relative abundances in indica and japonica varieties (Supplementary Fig. 4, Supplementary Data 2). In contrast, only the genus Aerosticca from Xanthomonadales and only the genus Chryseobacterium from Flavobacteriales showed such differences in relative abundance between the varieties. Therefore, we hypothesized that a specific genetic background of the rice varieties may play a role in shaping their phyllosphere microbiomes.
Identification of metabolic pathways linked to phyllosphere microbiome
With recent advances in sequencing and bioinformatics, genome-wide association studies (GWAS) have become an increasingly promising
Fig. 1 | Phyllosphere microbiome comparisons of indica and japonica rice varieties. a Unconstrained PCoA (for principal coordinates PCo1 and PCo2) based on Bray-Curtis distances showing bacterial community clustering of japonica and indica rice varieties ( -value was calculated by one-way PERMANOVA). Ellipses cover of the data for each rice subspecies. Shannon index for phyllosphere bacterial communities of indica and japonica varieties. The horizontal bars within boxes represent medians. The tops and bottoms of boxes represent the 75th and 25th percentiles, respectively. The upper and lower whiskers
extend to data no more than the interquartile range from the upper edge and lower edge of the box, respectively. The -value was calculated with unpaired oneway ANOVA with Tukey’s HSD test ( ). c Order-level distribution of bacteria in the indica and japonica phyllosphere microbiomes. Asterisks represent significant differences between indica and japonica varieties as assessed with unpaired one-way analysis of variance (ANOVA) with Tukey’s HSD test ( -values are listed in Supplementary Table 2). indica ( ) and japonica ( ) with three replications for each genotype. Source data are provided as a Source Data file.
approach for identifying genetic factors associated with important agronomic traits in plants . To identify potential rice genetic factors that are linked to the phyllosphere microbiome, we conducted GWAS. The analysis targeted associations between rice genomes and the relative abundance of bacterial species within the respective metagenomes. Our analysis led to the identification of 2,667 non-redundant single nucleotide polymorphisms (SNPs) located in 235 loci that were significantly associated with 496 bacterial species (hereafter referred to as GWAS-associated species) at a genome-wide significance threshold of (Fig. 2c, Supplementary Data 3).
We found that the majority of GWAS identified species belonged to Pseudomonadales (associated with 129 loci, accounting for of total GWAS-associated species) Burkholderiales (associated with 100 loci, accounting for ), Xanthomonadales (associated with 47 loci, accounting for 8.87%) and Enterobacterales (associated with 76 loci, accounting for ) at the bacterial order level (Fig. 2a, Supplementary Data 4). To separate loci according to bacterial orders associated with them, we searched for locus clusters using Bray-Curtis distances based on GWAS-associated species distribution of each locus. Our results showed that of the total SNP loci could be divided into four clusters: Cluster1 (containing 38.30% of total SNP loci and associated with Pseudomonadales), Cluster2 (containing 31.91% of total SNP loci and associated with Burkholderiales), Cluster3 (containing 12.77% of total SNP loci and associated with Enterobacterales) and Cluster4 (containing of total SNP loci and associated with Xanthomonadales) (Fig. 2a, Supplementary Fig. 5, Supplementary Data 4). These associations suggested that members of four bacterial
orders are particularly responsive to certain genetic backgrounds of the host plant.
To further elucidate how host genetic variations affect the phyllosphere microbiome, especially members of the bacterial orders that were highly responsive, genes with SNPs located in the associated loci were annotated and enriched into rice pathways (Supplementary Data 4). We found that metabolic pathways (dosa01100) and secondary metabolic process (GO:0019748) were prevalent in gene-enriched KEGG (Kyoto Encyclopedia of Genes and Genomes) (Supplementary Fig. 6) and GO (Gene Ontology) (Supplementary Fig. 6) annotations, respectively. Furthermore, we observed that SNP-associated genes in each locus cluster were enriched in specific metabolic pathways (Fig. 2b). For instance, Enterobacterales-associated rice genes were significantly enriched in Proteasome (dosa03050), Lysine degradation (dosa00310), Biosynthesis of amino acids (dosa01230), Ubiquitin mediated proteolysis (dosa04120), SNARE interactions in vesicular transport (dosa04130), Cysteine and methionine metabolism (dosa00270), Phosphatidylinositol signaling system (dosa04070), and Glutathione metabolism (dosa00480) pathways (Fisher’s exact test, , Fig. 2b, Supplementary Data 4). Similarly, Burkholderialesassociated rice genes were significantly enriched in Pentose and glucuronate interconversions (dosa00040), Protein export (dosa03060), Phenylalanine metabolism (dosa00360) and Arginine and proline metabolism (dosa00330) pathways (Fisher’s exact test, , Fig. 2b, Supplementary Data 4). Pseudomonadales-associated rice genes were significantly enriched in Plant-pathogen interaction (dosa04626), Stilbenoid, diarylheptanoid and gingerol biosynthesis
Fig. 2 | GWAS with rice phyllosphere microbiome. a Species associations for the four predominant bacterial orders and GWAS locus clusters. GWAS locus clustering is based on Bray-Curtis distances of bacterial species associated with each locus. Cluster1 (containing 38.30% of total SNPs; mainly associated with members of Pseudomonadales), Cluster2 ( ; mainly associated with members of Burkholderiales), Cluster3 (12.77%; mainly associated with members of Enterobacterales) and Cluster4 (8.09%; mainly associated with members of Xanthomonadales) (Supplementary Data 4). The total numbers of linked GWAS loci are shown for each order in the corresponding bar plot. The inner segments indicate the taxonomic group of the GWAS-associated bacterial species. Numbers in the inner segments indicate associated bacterial species and are color-coded according to their order-
level annotation. KEGG enrichment analysis for genes from locus clusters in . The color and size of the bubbles represent the -value ( , two-sided Fisher’s exact test, -values are listed in Supplementary Data 4) and number of enriched genes in each KEGG pathway, respectively. c Manhattan plots of GWAS results obtained with 110 rice varieties. GWAS loci and SNP plots are color-coded according to bacterial order-level annotation. Two-sided -values were determined using canonical correlation analysis. Analysis of genes that were enriched in the phenylpropanoid biosynthesis pathway. GWAS locus and gene names are color-coded according to bacterial order-level annotation. Source data are provided as a Source Data file.
(dosa00945), Plant hormone signal transduction (dosa04075), Ether lipid metabolism (dosa00565), Glycerophospholipid metabolism (dosa00564), MAPK signaling pathway-plant (dosa04016) and Ubiquinone and other terpenoid-quinone biosynthesis (dosa00130) pathways (Fisher’s exact test, , Fig. 2b, Supplementary Data 4). Xanthomonadales-associated rice genes were significantly enriched in Zeatin biosynthesis (dosa00908) and Amino sugar and nucleotide sugar metabolism (dosa00520) (Fisher’s exact test, , Fig. 2b, Supplementary Data 4). Notably, there were also overlapping pathways among different bacterial orders. For example, Enterobacterales- and Pseudomonadales-associated rice genes were enriched in the Sulfur metabolism pathway (dosa00920), while Burkholderiales-, Pseudo-monadales- and Xanthomonadales-associated rice genes were enriched in the Phenylpropanoid biosynthesis pathway (dosa00940) (Fig. 2b, Supplementary Data 4). These results provided key insights into host metabolic pathways potentially involved in the assembly of the phyllosphere microbiota. Since the Phenylpropanoid biosynthesis pathway was associated with three predominant bacterial orders, we expect a key role of this pathway with respect to host-microbiota interactions.
We therefore further investigated Burkholderiales-, Pseudomo-nadales- and Xanthomonadales-associated rice genes that were enriched in the Phenylpropanoid biosynthesis pathway (Fig. 2c, d). We found that a majority of prx (peroxidase) genes, which are involved in lignin biosynthesis, were associated with Burkholderiales, Pseudomonadales and Xanthomonadales (Fig. 2d, Supplementary Data 4). In addition, the 4CL1 (4-coumarate-CoA ligase, Os08g0245200) and C4H2 (cinnamate-4-hydroxylase, Os05g0320700) genes, which are involved in the synthesis of cinnamoyl-CoA and 4-hydroxycinnamoyl-CoA, respectively, were found to be specifically associated with Burkholderiales. On the other hand, OsPALO2 (phenylalanine/tyrosine ammonia-lyase, Os04g0518100), OsCAD4 (cinnamyl alcohol dehydrogenase, Os11g0622800) and OsCCR21 (cinnamoyl-CoA reductase, Os02g0180700) genes, which are involved in the synthesis of 4-hydroxycinnamic acid, 4-hydroxycinnamyl alcohol and 4-hydroxycinnamyl aldehyde, respectively, were specifically associated with Pseudomonadales (Fig. 2d, Supplementary Data 4). This indicated potential differences in host genetic control of the phyllosphere microbiota. Collectively, our analysis suggested that compounds that are part of lignin biosynthesis or their precursors may be participating in regulating microbiota assembly, and more importantly, that certain compounds may exert effects on specific bacterial taxa.
Identification of leaf metabolite-associated genes that drive microbiome assembly
Given the common occurrence and already known significance of members from the Pseudomonadales order in the plant phyllosphere, we took particular interest in deciphering how plant genetics may be involved in the enrichment of it. Pseudomonadales were not only highly abundant in the present study, but also accounted for the largest number of associated loci in the GWAS approach, indicating a robust connection to plant genetics. To verify rice genetic factors that affect the assembly of Pseudomonadales, we performed gene-editing to mutate a host gene shown to be specifically associated with it. OsPALO2 was selected as the target gene due to the strong association Pseudomonadales ( -value: ) and because there are no paralogous genes in locus 158 and other loci identified by GWAS. Using 16 S rRNA gene fragment amplicon sequencing, we showed that the rice OsPALO2 gene mutant (hereafter referred to as OsPALO2-KO), which lacks phenylalanine/tyrosine ammonia-lyase, showed a significant change in the relative abundance of Pseudomonadales (Fig. 3a). Intriguingly, natural variation of SNPs in OsPALO2 was different between indica and japonica rice (Supplementary Fig. 7), suggesting a correlation between Pseudomonadales abundance and this gene.
To further investigate the detailed role of OsPALO2 in shaping the rice leaf microbiota, we carried out 16 S rRNA gene fragment amplicon sequencing to profile the leaf microbiota of wild-type (WT) rice variety ZH11 (japonica), the loss-of function mutant, OsPALO2-KO and the over-expression construct, OsPALO2-OE. Under greenhouse conditions, we observed that the community structure in the phyllosphere of WT, OsPALO2-OE and OsPALO2-KO lines were dominated by Pseudomonadales, Enterobacterales, Sphingomonadales, Xanthomonadales, Flavobacteriales, Paenibacillales, Rhizobiales, Sphingobacteriales and Burkholderiales (Fig. 3a). This result was consistent with initial analysis of the leaf microbiome, indicating that field observations were reproducible in the greenhouse. Unconstrained PCoA (Fig. 3b, , PERMANOVA) and constrained principal coordinate analysis (CPCoA) (Supplementary Fig. 8, 23.7% of total variance was explained by the plant genotype, , PERMANOVA) of Bray-Curtis distances revealed that the WT, OsPALO2-OE and OsPALO2-KO phyllosphere microbiomes formed separate clusters, indicating that OsPAL02 affected their bacterial communities. Further analysis of differences in phyllosphere microbiomes between WT, OsPALO2-OE and OsPALO2-KO revealed significant variations at the order level (Fig. 3a). The relative abundance of Pseudomonadales showed a significant decrease in OsPALO2-KO and increase in OsPALO2-OE, compared to WT plants respectively (Fig. 3c). The relative abundance of Xanthomonadales, Flavobacteriales and Burkholderiales showed a significant increase in OsPALO2KO compared to WT or OsPALO2-OE plants (Fig. 3d, Supplementary Table 3, Supplementary Data 5). Notably, the Shannon index (alphadiversity) of OsPALO2-KO was significantly higher than that of OsPALO2-OE and WT (Supplementary Fig. 8). Similar observations were made when two additional OsPALO2-KO and OsPALO2-OE lines were analyzed (Supplementary Fig. 9, Supplementary Tables 4 and 5, Supplementary Data 5). The observation that OsPALO2-KO harbored higher alpha-diversity and lower relative abundance of Pseudomonadales than wild-type ZH11 (japonica) followed the same trend observed with indica and japonica varieties (Fig. 1b, c). This finding indicated that differences in OsPALO2 were sufficient to mimic microbiome variations between indica and japonica plants. Conclusively, the gene OsPALO2 was shown to contribute to the recruitment of Pseudomonadales and shape the bacterial community structure in the rice phyllosphere.
In the phenylpropanoid biosynthesis pathway, the gene OsPALO2 encodes a phenylalanine/tyrosine ammonia-lyase which catalyzes the transformation of L-tyrosine into 4 -hydroxycinnamic acid ( ) as well as the transformation of L-phenylalanine into trans-cinnamic acid, which are both precursors for the biosynthesis of lignin. When OsPALO2 variation between japonica and indica was analyzed, the protein structures of haplotype 1 (mainly present in indica) and haplotype 5 (mainly present in japonica) showed an amino acid exchange at site 134. This result indicated that OsPALO2 catalytic activity may differ between japonica and indica plants (Supplementary Fig. 7). We, therefore, hypothesized that the products of OsPALO2 impose a selective force on the recruitment of phyllosphere microbiota members, especially those within the bacterial order Pseudomonadales.
To investigate this hypothesis, we performed metabolite analyses with rice leaves of WT, OsPALO2-OE, and OsPALO2-KO, in order to identify differences attributable to OsPALO2. The results showed that WT, OsPALO2-OE, and OsPALO2-KO metabolite profiles were separated into three clusters based on Bray-Curtis distances and confirmed that OsPALO2 was involved in the biosynthesis of cinnamic acid and its derivatives (Fig. 4a, Supplementary Data 6). The data was also subjected to unconstrained PCoA and the results showed that the first principal coordinates PCo1, which accounts for 53.89% of the total variance, separated the WT, OsPALO2-OE, and OsPALO2-KO plants, indicating substantial differences in metabolites present in rice leaves (Fig. 4b, , PERMANOVA). This was also observed
Fig. 3 | OsPALO2 is linked to phyllosphere microbiome assembly and has implications for plant health. a Bacterial communities at order level in WT (wildtype, ZH11 rice variety), KO (OsPALO2-knockout) and OE (OsPALO2-overexpression) plants. Unconstrained PCoA based on Bray-Curtis distances showing bacterial community clustering in WT, KO and OE plants ( -value was calculated by one-way PERMANOVA). Ellipses cover of the data for each group. Comparison of relative abundances of Pseudomonadales (c) and Xanthomonadales (d) in WT, KO and OE phyllosphere microbiomes. The numbers of
replicated samples in are as follows: WT ( ), KO ( ) and OE ( ). Representative images of typical phenotype traits (e) and fresh weight (f) between WT, KO and OE rice plants. The numbers of replicated samples are as follows: WT ( ), KO ( ) and OE ( ). Scale bar, 1 cm . In this figure, box plot percentiles are the same as in Fig. 1b. The -values were calculated with unpaired one-way ANOVA with Tukey’s HSD test ( ). The labels ‘ns’ indicate a not significant difference ( ). Source data are provided as a Source Data file.
with CPCoA (Supplementary Fig. 10) where of total variance was explained by the plant genotype ( , PERMANOVA). Further analysis revealed that 12 metabolites showed significant differences between WT, OsPALO2-OE, and OsPALO2-KO (Supplementary Data 6). Therein, 4-HCA showed significantly higher intensity in OsPALO2-OE and lower in OsPALO2-KO compared to WT plants (Fig. 4c), suggesting that 4-HCA is the key metabolite regulated by OsPALO2 and may thus contribute to phyllosphere microbiome assembly. LC-MS analysis was conducted to detect 4-HCA physiological concentrations in rice leaves. As expected, the concentration of 4-HCA was significantly higher in the three OsPALO2-OE lines and lower in the three OsPALO2-KO lines when compared to WT plants (Supplementary Fig. 11, Supplementary Table 6, Supplementary Data 5). The concentration of 4 -HCA was additionally determined in five Pseudomonadales-enriched japonica
and five Xanthomonadales-enriched indica varieties. We found that the concentration of 4-HCA in the analyzed japonica varieties was significantly higher than in the indica varieties (Supplementary Fig. 12, Supplementary Table 7). A correlation analysis indicated that the concentration of 4-HCA shows a positive relationship with the relative abundance of Pseudomonadales and a negative relationship with the abundance of Xanthomonadales in the leaves of the analyzed rice varieties (Supplementary Fig. 12). These observations provided further evidence that 4-HCA is the key metabolite regulated by OsPALO2 and contributes to phyllosphere microbiome variations between indica and japonica varieties.
Apart from the altered phyllosphere microbiomes, OsPALO2-KO developed severe disease symptoms compared with WT and OsPALO2 after leaf cutting (Fig. 3e). This phenomenon was not observed
Fig. 4 | Metabolite profiles of OSPALO2 mutants. a Heatmap based on intensity values of detected cinnamic acid and derivatives in different rice leaves. Clusters are shown on the left and top using Bray-Curtis distances. All cinnamic acid labels (left) are shown in Supplementary Data 6. Rice sample labels (bottom) are as follows: WT1/2/3/4/5/6 (wild-type 1/2/3/4/5/6), KO1/2/3/4/5/6 (OsPALO2-KO 1/2/3/4/5/6), OE1/2/3/4/5/6 (OsPALO2-OE 1/2/3/4/5/6). b Unconstrained PCoA based on BrayCurtis distances showing leaf metabolite separation for WT, KO and OE plants ( -values were calculated with one-way by PERMANOVA). Ellipses cover of the data for each group. Blue, purple and green colors
represent WT, KO and OE rice plant samples, respectively. c Intensities (based on LC-MS/MS analyses) of 4-HCA (4-Hydroxycinnamic acid) in WT, KO and OE plant leaves, respectively. In this figure, the peak intensity values were normalized using In transformation. Box plot percentiles are the same as in Fig. 1b. The -values were calculated with unpaired one-way ANOVA with Tukey’s HSD test ( ). Groups are abbreviated as: wild-type, WT; OsPALO2-knockdown, KO; OsPALO2-overexpression, OE. The numbers of replicated samples are as follows: WT ( ), KO and OE ( ). Source data are provided as a Source Data file.
before the leaf cutting (Supplementary Fig. 13), indicating that the absence of OsPALO2 caused the dysbiosis in the rice phyllosphere microbiome and consequently led to the vulnerability of plants to pathogens on leaves. Unsurprisingly, the fresh weight of WT and OsPALO2-OE leaves was significantly higher than that of OsPALO2-KO; fresh weight of OsPALO2-OE leaves showed no significant difference to WT after leaf cutting (Fig. 3f). To determine whether disease symptoms were caused by the microbiota shift in OsPALO2-KO, we grew WT and OsPALO2-KO plants under gnotobiotic conditions and conducted the leaf cutting under controlled conditions preventing contamination. The OsPALO2-KO plants were indistinguishable from WT plants, and the fresh weight showed no significant differences among the three rice lines (Supplementary Fig. 14). This observation indicated that the disease symptoms in OsPALO2-KO might result from a microbiota dysbiosis. Combining the results of the metabolite analysis and the above assays, we proposed that OsPAL02 might be responsible for producing 4-HCA and maintaining a functional microbiota.
To validate the function of , rice leaves of OsPALO2-KO plants were sprayed with . As we expected, the application of 4-HCA on OsPALO2-KO plants resulted in a phyllosphere microbiome showing no significant difference to WT plants, and their fresh weights also did not differ (Supplementary Fig. 15, Supplementary Table 8). These results demonstrated that 4-HCA prevents dysbiosis in the
phyllosphere microbiota of OsPALO2-KO plants. Overall, our data suggested that OsPAL02-synthesiszed 4-HCA plays a key role in maintaining plant health.
Implications of 4-HCA and members of Pseudomonadales for phyllosphere microbiome homeostasis
Although we observed that is involved in microbiome assembly and prevents dysbiosis in the rice phyllosphere, the underlying mechanism remained unclear. To investigate potential interactions of 4-HCA with rice leaf-associated bacteria, we isolated bacteria using the limiting dilution method . Bacterial strains were isolated from pooled leaf samples of WT, OsPALO2-OE, and OsPALO2-KO plants grown under greenhouse conditions. Taxonomic assignments of bacterial strains were conducted by Sanger sequencing of their 16S rRNA genes, and non-redundant strains were identified based on gene similarity . In total, we obtained a collection of 77 non-redundant strains that contained 14 bacterial orders, mainly Pseudomonadales ( of the total nonredundant strains), Bacillales (19.48%), Enterobacterales (15.58%), Sphingomonadales (9.1%) and Flavobacteriales (7.79%). They covered 62.5% of the orders (relative abundance > 0.1%) detected with amplicon sequencing analyses (Supplementary Fig. 16, Supplementary Data 7). The isolate collection enabled us to further study
Fig. 5 | Effects of 4-HCA on phyllosphere bacterial growth and rice pathogen TJ1. a Left: Effects of 4-HCA on growth of 22 rice leaf-associated bacterial isolates from the orders Pseudomonadales, Xanthomonadales and Burkholderiales. The heatmap shows fold changes of individual isolates exposed to versus control (supplemented with solvent) over 24 hours. The corresponding growth curve data are presented in Supplementary Fig. 18 and Supplementary Data 7. Middle: Statistical analysis of fold changes in cell density ( ) for the 22 isolates. Different colors represent isolates assigned to Pseudomonadales, Xanthomonadales and Burkholderiales, respectively. Box plot percentiles are the same as in Fig. 1b. Different letters indicate a significant difference according to unpaired one-way ANOVA with Tukey’s HSD test ( -values are listed in Supplementary Data 7, experiments for each isolate were replicated 6 times). Each
isolate’s fold change in cell density ( ) of 12 time points is shown on the left. Right: In vitro inhibition of Xanthomonas oryzae strain TJ1 (abbreviated as TJ1) by the 22 isolates. Green or black color indicates that the respective isolate either inhibited or did not inhibit TJ1 on R2A agar with or without 1 mM 4-HCA (bottom). b Representative photographs of bacterial strains tested for inhibition of TJ1 in (a). Paper discs were inoculated with TJ1. Presence of in R2A agar is shown on the left. Background indicates isolates poured into the agar (top). Mock indicates bacterial filtrates poured into the agar. c, d Representative photographs (c) and growth curves (d) of TJ1 grown in different concentrations of 4-HCA. TJ1 was grown in R2A medium supplemented with , and . Values are means (shown as error bars; replicates). Source data are provided as a Source Data file.
interactions between 4-HCA and phyllosphere microbiota members through targeted assays and synthetic community (SynCom) approaches.
Since the disease symptoms after leaf cutting were observed not only in OsPALO2-KO plants but also in some WT and OsPALO2-OE plants (Fig. 3e), we hypothesized that the pathogenic agent might be present within the isolate collection. To identify the causal agent(s) of the disease symptoms, we tested the pathogenicity of the individual isolated strains. We inoculated single strains onto germ-free WT seedlings and monitored the plant disease occurrence. We then identified Xanthomonas oryzae strain TJ1 (hereafter referred to as TJ1), as a causative pathogen of bacterial leaf-blight disease on rice (Supplementary Fig. 17). We then tested the pathogenicity of TJ1 on OsPALO2-KO and OsPALO2-OE plants. As expected, the inoculation of TJ1 caused severe disease symptoms on OsPALO2-KO, meanwhile, OsPALO2-OE were less affected in terms of disease occurrence and fresh weight, compared to WT and OsPALO2-KO. (Supplementary Fig. 17). We also classified disease symptoms into four grades ( 1 , healthy, to 4 , dead), and showed that increasing disease severity correlated with decreasing plant fresh weight (Supplementary Fig. 17). Furthermore, we re-isolated bacteria from TJ1-inoculated WT, OsPALO2-KO and OsPALO2-OE plants. Sequencing of their 16 S rRNA genes confirmed sequence similarity with TJ1 (Supplementary Fig. 17). Additional isolation experiments showed that TJ1 abundance in OsPALO2-OE plants was significantly lower than in the OsPALO2-KO mutant and WT plants
(Supplementary Fig. 17). This provided further evidence that TJ1induced disease resulted in the observed reduced average plant weight. Overall, the isolate TJ1 was confirmed to be a rice pathogen, and it was demonstrated to cause lighter disease symptoms in OsPALO2-OE than that observed with OsPALO2-KO and WT plants.
We then hypothesized that at certain concentrations may contribute to inhibition of TJ1 and reduce disease symptoms. We grew TJ1 in liquid culture with different concentrations of 4-HCA. We found that 4-HCA at certain concentrations can effectively inhibit TJ1 growth, and the growth of TJ1 was completely inhibited when the concentration of 4-HCA reached 2 mM (Fig. 5c, d). This result indicated that 4-HCA can directly inhibit the growth of the pathogen; the inhibitory concentration (IC50) of TJ1 was 1 mM .
Next, we conducted in vitro assays to test the influence of 4-HCA on strains from Pseudomonadales, Xanthomonadales and Burkholderiales. These bacterial strains were grown in liquid R2A medium supplemented with ; the same concentration inhibited the growth of pathogen TJ1. We found that most of the Pseudomonadales strains grew faster in the presence of , whereas all Xanthomonadales and Burkholderiales strains were significantly inhibited (Fig. 5a). Detailed analysis of growth changes ( ) indicated that all Pseudomonadales strains, except Acinetobacter sp. 21, showed a significantly higher growth rate than Xanthomonadales and Burkholderiales strains in the presence of the 2 mM 4-HCA (Fig. 5a, Supplementary Fig. 18, Supplementary Data 7). The in vitro assays clarified the effect of
Fig. 6 | SynCom inhibits TJ1 under 4-HCA presence in vivo. a, b Fresh weight (a) and representative images of typical phenotypic traits (b) between sterile-grown WT, KO and OE rice plants treated with 10 mM MgCl . c, d Fresh weight (c) and representative images of typical phenotypic traits (d) between sterile-grown WT, KO and OE rice inoculated with synthetic community (abbreviated as SynCom) + Xanthomonas Oryzae TJ1 (abbreviated as TJ1). e, Fresh weight (e) and representative images of typical phenotypic traits (f) between sterile-grown WT, KO, and OE rice plants inoculated with TJ1. Box plot percentiles are the same as in Fig. 1b.
Blue, purple and green colors represent WT, KO and OE rice plants, respectively. The -values were calculated with unpaired one-way ANOVA with Tukey’s HSD test ( ). The labels ‘ ‘ indicate a not significant difference ( ). The numbers of replicated samples in each treatment are as follows: WT ( ), and OE ( ). Scale bar, 5 mm . Groups are abbreviated as WT (wild-type), KO (OsPALO2-knockdown), OE (OsPALO2-overexpression). Source data are provided as a Source Data file.
4-HCA on the growth of strains from the orders Pseudomonadales, Xanthomonadales and Burkholderiales. They also shed further light on the phyllosphere microbiome assembly discrepancies between OsPALO2-OE, OsPALO2-KO, and WT plants (Fig. 3a) and indicated a pivotal role of 4-HCA therein.
We observed severe disease symptoms in TJ1-inoculated WT plants under gnotobiotic conditions (Supplementary Fig. 17), interestingly, this phenomenon did not occur when plants were grown under greenhouse conditions, where they were exposed to various microbes (Fig. 3e, f). Therefore, we speculated that the effect of 4-HCA at least partially relies on microbiota homeostasis. We performed in vitro inhibition assays with TJ1 and 22 isolates representing leafassociated Pseudomonadales, Xanthomonadales and Burkholderiales members, on R2A medium with and without (Fig. 5a). The results revealed that under the 1 mM 4-HCA condition, all Pseudomonadales strains, including Acinetobacter sp. 21, strongly inhibited TJ1
(Fig. 5a). Conversely, 75% of the Pseudomonadales strains lost the ability to inhibit TJ1 when 4-HCA was absent (Fig. 5a). Within Burkholderiales, only Delftia sp. 12, inhibited TJ1 in the presence of 4-HCA and lost this inhibition activity when 4-HCA was absent (Fig. 5a).
Overall, 4-HCA-dependent inhibition mostly occurred among strains from Pseudomonadales. This observation also connected 4-HCA to microbial functioning in the phyllosphere. Furthermore, we designed a synthetic community (SynCom, Fig. 5a) consisting of Pseudomonadales strains, and then subjected it to specific assays to evaluate its efficacy against the pathogen TJ1. Under gnotobiotic conditions, WT, OsPALO2-KO and OsPALO2-OE plants showed no significant differences in fresh weight when treated with either 10 mM (control, Fig. 6a, b) or the SynCom suspension (Supplementary Fig. 19). This result confirmed that the SynCom did not have a negative impact on rice plants. Then, we treated the rice plants with SynCom + TJ1 suspension and TJ1 suspension only. In WT and OsPALO2-OE plants,
the SynCom showed protective effects against TJ1 (Fig. 6c, d). In contrast, no protection was observed with OsPALO2-KO plants, as indicated by strong disease symptoms and significantly lower leaf fresh weight (Fig. 6c, d). When only TJ1 suspension was applied, WT and OsPALO2-KO plants both showed more pronounced disease symptoms and significantly lower leaf fresh weight compared to OsPALO2-OE plants (Fig. 6e, f), suggesting that without the SynCom, the protection effect against TJ1 is lost. Evidently, an interplay between OsPALO2 and Pseudomonadales members was required for the SynCom-associated plant health maintenance. Combined with the aforementioned results, a conclusion can be drawn that OsPALO2-synthesized is important for the enrichment of bacterial members of the Pseudomonadales and drives phyllosphere microbiome assembly. Microbiome assembly regulated by 4-HCA-regulated is of great importance to maintaining rice phyllosphere homeostasis, which is directly connected to plant health.
Discussion
Plants are colonized by highly diverse, tissue-specific microbial communities, the plant microbiota. The plant microbiota is known to complement host functioning and to be an important factor for enhancing resilience under abiotic as well as biotic stress. Plants can assemble and maintain certain microbiota structures, which is vital for plant health. A dynamically balanced state of the microbiota is known as ‘homeostasis . Disrupting homeostasis often results in a prevalence of detrimental microbiota members, leading to shifted microbiota structures and plant disease occurrence, also known as dysbiosis . It is currently sparsely understood how plants assemble and maintain a functional (homeostatic) microbiota, especially in regards to their phyllosphere . Previous research has mostly focused on microbe-microbe interactions that result in homeostasis and can explain certain colonization patterns . Here, we show that specific host-microbe interplay is involved in shaping phyllosphere bacteria prevalence in rice plants. Research conducted in the past has provided indications for active host regulation of the phyllosphere microbiota. The plant immune system was demonstrated to be required for maintaining microbiota homeostasis . Mutations of immunityrelated genes in Arabidopsis have been shown to result in a dysbiotic phyllosphere microbiota . In the present study, we show that rice plants deploy a potentially conserved mechanism to shape the phyllosphere microbiome in a wide range of genotypes. Except for immunity-related mechanisms, plants can also employ metabolites as a selective force to assemble candidate microbiota members to serve their interests . Various plant-specific metabolites can fulfill the purpose of maintaining microbiota homeostasis on leaves , or suppress bacterial pathogens . Nevertheless, fundamental questions remain unaddressed related to which plant genetic components are devoted to control microbiota assembly, and how this takes place on metabolite level.
Genome-wide association studies (GWAS) have proven to be a highly useful tool to shed further light on plant-microbe interactions . In a recent study, researchers showed that the abundance of phyllosphere microbial hubs in Arabidopsis correlated with multiple specialized metabolic pathways affecting both bacterial and fungal communities . It should be highlighted the previous studies were solely based on amplicon sequencing of microbial marker genes in the phyllosphere. While such approaches are valuable, this method is also known to be prone to specific biases, because marker genes of certain microbial groups are more likely to be amplified than those of others . Therefore, microbiome structures are not always accurately captured. In the present study, a large-scale phyllosphere metagenome dataset was combined with GWAS and facilitated more precise insights into associations between rice plants and their phyllosphere microbes. For one, it provided further evidence for the role of the plant immune system in shaping its microbiota, such as the MAPK signaling pathway.
More importantly, it allowed us to pinpoint specific metabolic pathways and genes which we identified as suitable targets to dissect phyllosphere microbiome assembly at molecular level. Furthermore, the high resolution of taxonomic data obtained here was advantageous to connect host genetic traits to specific bacterial taxa, which was critical to validate the effects of a distinct gene and metabolite on microbiome assembly.
The present study revealed that primarily bacteria belonging to the order Pseudomonadales are enriched by 4-HCA in the rice phyllosphere. A similar observation was obtained with poplar tree endophytes and down-regulation of cinnamoyl-CoA reductase; it caused the enrichment of 4-HCA in plant tissues and consequently increased the abundance of Pseudomonas . This genus is a ubiquitous member of the plant microbiota . Previous studies have shown that it is an important component of a functional microbiota that can shield off pathogens and prevent diseases in plants. Pseudomonas were often identified as core microorganisms associated with both the plant rhizosphere and phyllosphere , and as key players in many diseasesuppressive soils . Plants benefit from Pseudomonas as they can produce a wide range of antimicrobial compounds , induce plant systemic disease resistance , and establish an array of chemical dialogues with plants . Our identification of plant genetic components controlling Pseudomonas could provide an ideal target for engineering the phyllosphere microbiota of various crop plants. We also showed that Xanthomonas, a highly prevalent phytopathogen, is negatively affected by . It is therefore very likely that plants protect themselves against pathogens by relying on their immune system as well as specific metabolites that not only allow them to directly antagonize pathogens but also to maintain homeostasis in the phyllosphere microbiota.
In recent years, plant microbiome studies have facilitated the identification of various disease-reducing or even disease-preventing microorganisms . Leaves are common entry points of various, highly devastating pathogens. The present study shows that metabolitedriven regulation of the phyllosphere microbiota is a hidden mechanism to support plant hosts against pathogen attacks. Previous studies that assessed the rhizosphere microbiome have resulted in similar findings. For example, iron-mobilizing coumarins in Arabidopsis roots were linked to shaping the rhizosphere microbiota by inhibiting proliferation of certain Pseudomonas species via a redoxmediated mechanism . Re-shaping of the rhizosphere microbiome by the coumarin scopoletin was shown to have consequences for plant health . Similarly, the benzoxazinoid breakdown product 6-methoxy-benzoxazolin-2-one (MBOA) was found to regulate rhizosphere microbiota assembly of cereal crops .
The present findings related to plant metabolite-driven phyllosphere homeostasis and their implications for host health will serve as a basis for targeted breeding strategies. Previous studies have provided certain indications, mostly via correlation analyses, that plant metabolites play an important role in maintaining a functional microbiota . However, it remained unresolved if the identified compounds are the main drivers of the observed structural changes of the microbial communities. In the present study, knock-out as well as overexpression constructs allowed us to specifically attribute microbiome changes to in the rice phyllosphere. A lack of was linked to dysbiosis of the phyllosphere microbiome which results in higher susceptibility of rice plants to disease. Current resistance breeding strategies known as ‘ gene breeding strategies’ are increasingly challenged by rapidly evolving phytopathogens. This problem could be addressed by identifying and engineering microbiome-shaping genes, which may become known as ‘ gene breeding’.
Methods
Chemicals, reagents and analytical instruments
The chemicals, reagents and analytical instruments used in this study are listed as follows: ethanol (EtOH), sodium hypochlorite (NaOCl),
Triton X-100, methanol (MeOH), 4-hydroxycinnamic acid (4-HCA), , Graphitized carbon black (GCB), Acetonitrile (ACN) and other organic solvents was purchased from Merck (Shanghai, China); agar powder, agarose gel, PBS buffer, Kanamycin, Hygromycin, Murashige and Skoog (MS), Linsmaier-Skoog (LS), Reasoner’s 2 A (R2A) and tryptic soy broth (TSB) medium were purchased from Solarbio (Beijing, China); Bsal, T4 DNA Ligase, Takara Ex Taq were purchased from Takara (Beijing, China); TransScript II All-in-One First-Strand cDNA Synthesis SuperMix for PCR, TransTaq DNA Polymerase High Fidelity (HiFi), EasyPure Quick Gel Extraction Kit a were purchased from TransGen Biotech (Beijing, China); FastPure Plant DNA Isolation Mini Kit was purchased from Vazyme Biotech (Nanjing, China); MagPure Soil DNA LQ Kit was purchased from Magen (Shanghai, China); TIANprep Plasmid Midi kit was purchased from TIANGEN (Beijing, China); Agencourt AMPure XP beads was purchased from Beckman Coulter (Pasadena, USA); Qubit dsDNA assay kit was purchased from Yeasen (Shanghai, China); TRIzol was purchased from Invitrogen (Carlsbad, USA).
The analytical equipment used included a UHPLC ( 1290 Infinity LC, Agilent Technologies, USA) coupled to a quadrupole time-of-flight mass (TripleTOF 6600, AB Sciex, USA), and a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, USA).
Rice phyllosphere metagenomes
In order to investigate potential influence of rice genotypes on the phyllosphere microbiome, we obtained shotgun-sequenced metagenomes of 110 rice varieties (three replicates for each variety). Details about this dataset were included in a dedicated data publication (Supplementary Data 1) . The 110 rice varieties were selected from the Rice Diversity Panel II core collection (C-RDP-II), which can be grouped into ‘japonica’ (including 27 tropical japonica, 7 temperate japonica, and 2 admixed japonica) and ‘indica’ (including 32 indica, 22 aus, 2 admixed indica) varieties as defined by McCouch et al. . All varieties were planted in Taojiang County, Hunan Province, China , E). Rice leaves were harvested at the booting stage (September 5th, 2020). The samples were enriched for their bacterial fraction, metagenomic DNA was extracted and subjected to highthroughput sequencing . Quality filtering of raw metagenomic data was described in the data publication .
Bioinformatics analyses of the metagenomes
Quality-filtered data were subjected to the taxonomic classification using Kraken2 v2.0.9 . A species abundance table was generated with Bracken v2.6.0 . The cumulative-sum scaling normalization method was applied with R package metagenomeSeq . A principal coordinates analysis (PCoA) based on Bray Curtis distances was performed using the R package vegan . Alpha diversity analysis was carried out using aforementioned R package vegan. Differences in beta diversity were accessed using a pairwise Adonis test and permutational analysis of variance (PERMANOVA, 999 permutations) using R package amplicon . Analysis of differential species abundance was performed using Wilcoxon rank sum and Tukey’s HSD tests based on species with an average relative abundance in the assessed rice varieties. Species were defined as significantly different abundant if the -value was <0.05. Diversity and relative species abundance data were visualized by using R package amplicon. Bacterial taxonomy was visualized with GraPhlAn v.0.9. . Boxplots and scatter diagrams were visualized by using the R package ggplot2 v.2.2.1 .
GWAS analysis
The genome-wide association study was based on 110 C -RDP-II rice accessions represented by a 168,699 -SNP dataset (filter removal MAF < 0.05) (Supplementary Data 8). This dataset was extracted from the C-RDP-II collection that encompasses a 700,000-SNP dataset . We defined ‘traits’ as the enrichment levels of the 6,862 identified species
in the phyllosphere of the 110 rice varieties (Supplementary Data 3). Tassel5.0 (https://www.maizegenetics.net/tassel) was used for GWAS and an analysis pipeline was set up on a Linux system to analyze the 6,862 ‘traits’ individually (scripts are available at Code Availability). The GWAS method was based on a rice genome-wide association analysis approach using a high-density SNP array . We used a mixed linear model (MLM) that integrated the kinship matrix (K) with population structure (Q). We screened for significantly associated loci using the following criteria: a genomic region with containing at least two significant SNPs (with a -value threshold of ). We filtered for non-redundant loci associated with at least one enriched microorganism. The GWAS-identified loci were further analyzed for candidate genes.
Locus clustering and pathway enrichment
GWAS-identified loci can be associated with multiple bacterial species from different orders. For each locus, we calculated the proportion of associated species from the respective order. The proportion was later subjected to clustering locus visualization. Clustering analysis based on Bray Curtis distance between all loci was conducted using package vegan. The clustering of loci was visualized by using package circlize . For each of the locus clusters, gene annotation and Gene ID conversion were conducted using The Rice Annotation Project Database (RAPDB, https://rapdb.dna.affrc.go.jp/), and GO or KEGG enrichment analyses were conducted using R package clusterProfiler . GO enrichment analysis was conducted using org.Osativa.eg.db (https:// github.com/xuzhougeng/org.Osativa.eg.db) database and significant enrichment of pathways was noted if the -value was <0.05 (Fisher’s exact test). KEGG enrichment was analyzed using Oryza sativa japonica (RAPDB) KEGG Gene Database (Release 105.0+/03-07) and significant enrichment of pathways was noted if the -value was <0.05 (Fisher’s exact test). GO and KEGG enrichment pathways were visualized by using ggplot2.
Generation of CRISPR-edited and gene overexpression rice mutants
The CRISPR-Cas9 system was used to generate OsPALO2 (Os04g0518100) mutants according to a high-efficiency monocot genome editing method . Briefly, specific primers containing the OsPALO2 target site sequences (CCATGCGCTTCACCTCGTCCAGG) were ligated into pEGCas9Pubi-H cassettes. To construct guide RNA genes, rice small nuclear RNA U6 promoter was amplified from pYLsgRNA-OsU6a vector using primer pairs U6-1/ U6-2. The DNA sequence encoding the gRNA scaffold was amplified from pYLsgRNAOsU6a vector using primer pair gRNA-1/gRNA-2. The PCR product of the U6 promoter was fused with the DNA fragment encoding the gRNA scaffold by overlapping PCR using primer pair sgRNA-1/sgRNA-2. The U6 promoter-gRNA fragment was cloned into the pEGCas9Pubi-H vector to form the pEGCas9Pubi-H-Os04g0518100 construct via Bsal (Takara, Beijing, China) restriction digestion followed by ligation using T4 DNA Ligase (Takara, Beijing, China) (Supplementary Fig. 20). After transformation into Escherichia coli DH5-alpha, the resulting constructs were purified with TIANprep Plasmid Midi kit (TIANGEN, Beijing, China) for PCR amplification using primer pairs KO-1/KO-2 and sequenced via Sanger sequencing (Sangon Biotech, Shanghai, China) (Supplementary Fig. 20). The validated constructs were stored at for subsequent use in rice protoplast transformation.
The OsPALO2-overexpression constructs were generated according to a transgenic rice construction method . Briefly, for the OsPALO2 overexpression vector construct, total RNA of rice variety ZH11 leaves were extracted using TRIzol, and complementary DNA (cDNA) was synthesized from the total RNA using cDNA Synthesis SuperMix kit (TransGen Biotech, Beijing, China). The OsPALO2 coding sequence was amplified by PCR using primer pairs PALO2-1/PALO2-2. The amplified fragment was cloned into the pEGOEPubi-H vector, which contained
the maize ubiquitin promoter and a hygromycin resistant gene, to form the pEGOEPubi-H-OsPALO2 overexpression construct via Bsal (Takara, Beijing, China) restriction digestion followed by ligation using T4 DNA Ligase (Takara, Beijing, China) (Supplementary Fig. 21). After transformation into Escherichia coli DH5-alpha, the resulting constructs were purified with TIANprep Plasmid Midi kit (TIANGEN, Beijing, China) for PCR amplification using primer pairs OE-1/OE-2 and sequenced via Sanger sequencing (Sangon Biotech, Shanghai, China) (Supplementary Fig. 21). The validated constructs were stored at for subsequent use in rice protoplast transformation.
The resulting vectors were introduced into Agrobacterium tumefaciens (strain EHA105) and then into rice by Agrobacterium-mediated transformation . The transformation was performed using a rice transformation method . Briefly, positive transgenic bacteria were selected by using kanamycin for selection. Hygromycin was used to select putative transgenic plants. All the selected putatively transgenic seedlings were cultivated in the greenhouse or field for further identification.
To identify transgenic plants, genomic DNA was extracted from approximately 30 mg of leaf tissue using FastPure Plant DNA Isolation Mini Kit (Vazyme Biotech, Nanjing, China) according to the manufacturer’s instructions. The isolated and purified genomic DNA was used for PCR amplification of CRISPR/Cas9 target sites or hygromycin gene using primer pairs KO2-1/KO2-2 or H-1/H-2, respectively. Amplified fragments were detected using agarose gel electrophoresis, purified using EasyPure Quick Gel Extraction Kit (TransGen Biotech, Beijing, China) and sequenced with sanger sequencing (Sangon Biotech, Shanghai, China) to identify OsPALO2-KO and OsPALO2-OE transgenic plants. All related primers are listed in Supplementary Table 9.
Plant material, growth conditions and treatments
Hunan Hybrid Rice Research Center (HHRRC) provided seeds of the ZH11 rice variety for the experiments and genetic transformation. ZH11 was implemented as the wild-type (WT) and to generate OsPALO2-KO (knockout) and OsPALO2-OE (overexpressing) plants. WT, OsPALO2-OE and OsPALO2-KO plants were cultured under relative humidity set at , temperature at and 13 h light cycle in a greenhouse or were grown at a field site of HHRRC in Changsha, China. Mature seeds of the WT, OsPALO2-OE and OsPALO2-KO plants were harvested, dried, and stored in a refrigerator at .
Rice plants were grown in Taojiang soil filtrate mixed with greenhouse potting soil (PINDSTRUP substrate, Denmark; autoclaved twice) in an air-circulating greenhouse for controlled experiments. For Taojiang soil filtrate preparation, the soil was collected from the same rice field as for the metagenomic experiments, Taojiang County ( ), Hunan Province, China. The top of field soil was collected and sieved ( sieve) to remove rocks and other debris, dried at room temperature. Then, the soil was diluted 10times using sterile water ( ), followed by shaking for 20 min at and sonicated for 5 min at a frequency of 30 kHz at . The suspensions were subjected to centrifugation for 1 min at 42 g , . The supernatant was collected and used as the Taojiang soil filtrate. Greenhouse soil preparations were made by using 100 mL Taojiang soil filtrate together with 100 mL half-strength Murashige and Skoog (MS) medium solution ( pH 5.8 ) and mixed with 200 g of greenhouse potting soil (PINDSTRUP substrate, Denmark; autoclaved twice). Rice seeds were surface-sterilized using 75% ethanol (EtOH) for 2 min and sodium hypochlorite supplemented with Triton X-100 three times for 8 min , following washed with sterile water six times. Sterile seeds were further subjected to microbiota inoculation from filed soil by immersing them in Taojiang soil filtrate for 1 d at and 4 d at in the dark. Subsequently, all rice seeds at the early germination stage were transplanted into the pots with the aforementioned soil preparation. Each plant was watered with 5 mL of
sterile water twice a week and sterile half-strength MS solution once a week after germination. Top leaves of 3 -week-old plants were cut ( 1 cm ) using sterile scissors (surface-sterilized using 75% EtOH and washed with sterile water six times for each plant) for identifying OsPALO2-KO or OsPALO2-OE constructs 5-week-old plants were harvested for collection of phyllosphere bacteria, extraction of leaf metabolites, determination of fresh weight and other analyses.
For the complementary 4-HCA assay, WT and OsPALO2-KO plants were surface-sterilized, cold-stratified, germinated and transferred to the same greenhouse as mentioned before. Each 2-week-old OsPALO2 plant was sprayed with 2 mL of sterile (dissolved in water). Each 2-week-old plant of WT plant was sprayed with 2 mL sterile water. Watering of the seedlings and identification of the genotype followed the same procedure as described before. 5-week-old plants were harvested for microbiome, phenotype and fresh weight analyses.
For gnotobiotic plant growth, WT, OsPALO2-KO and OsPALO2-OE seeds were surface-sterilized with the same procedure as described before, and then cold-stratified and germinated in sterile water. Subsequently, all rice seeds at early germination stage were transferred into sterile bottles with autoclaved (three times for 45 min each, with 24 h storage at room temperature in between) soil. Sterility of rice seeding was routinely monitored by plating samples of plants and culture substrate on R2A plates. Watering of seedlings and identification of the genotype were as described above, but under sterile conditions. 5-week-old plants were harvested for phenotype and fresh weight analyses.
Phyllosphere microbiome analyses of WT and mutant plants
5-week-old plants cultivated under greenhouse conditions were harvested for profiling of the phyllosphere microbiome (six replicates per genotype, each replicate contained three plants leaves). Sample collection was conducted according to a rice phyllosphere bacteria enrichment method . Total DNA was extracted using MagPure Soil DNA LQ Kit (Magen, Shanghai, China) according to the manufacturer’s instructions. Quality and quantity of DNA was verified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, USA) and agarose gel electrophoresis, respectively. Extracted DNA was diluted to a concentration of and stored at until further processing. PCR amplification of bacterial 16S rRNA gene fragments (V3-V4 region) was performed using Takara Ex Taq (Takara, Beijing, China) and the barcoded primers 343 F and 798 R listed in Supplementary Table 9. Amplicons were visualized using agarose gel electrophoresis and purified using Agencourt AMPure XP beads (Beckman Coulter, Pasadena, USA) twice. After purification, the DNA was quantified using Qubit dsDNA assay kit (Yeasen, Shanghai, China). Equal amounts of purified DNA were pooled for sequencing on the NovaSeq 6000 platform (Illumina Inc, USA) at Shanghai OEbiotech (Shanghai, China).
The 16S rRNA gene fragment sequences were processed using QIIME2 v.2021.4 . Paired-end reads were detected and the adapters were removed using Cutadapt v.2.1 . After trimming, paired-end reads were filtered for low-quality sequences, denoised, merged and clustered using DADA2 v.2020.2.0 . Amplicon sequence variants (ASVs) and a feature table were generated using QIIME2. All ASVs were annotated using the Silva v138.1 reference databases . Diversity and differential abundance analyses were performed using the same methods as described above.
Rice leaf metabolite extraction
5-week-old WT, OsPALO2-KO and OsPALO2-OE plants cultivated under greenhouse conditions were harvested for extraction of leaf metabolites (six replicates per genotype, each replicate included three plants leaves). Rice leaves were immediately frozen in liquid nitrogen and ground into fine powder with a mortar and pestle. Leaf metabolites were extracted from 80 mg of each sample using methanol
(MeOH)-acetonitrile (ACN)-ultrapure water ( ). The homogenates were shaken at for 20 min , followed by sonication at a frequency of 30 kHz for 20 min . After centrifugation at for 20 min , the supernatant was dried to remove the organic solvent using a vacuum centrifuge. The samples were redissolved using ACN-ultrapure water ( ) and centrifuged at for 15 min , followed by filtering through a filter before analysis with a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system.
LC-MS/MS conditions
The samples were separated by ultra-performance liquid chromatography (UHPLC) on an ACQUITY UPLC BEH C-18 column ( , , Waters, USA); the column temperature was . The flow rate was set at , and the injection volume was . The gradient elution procedure was as follows: , linearly changed to , MeOH was maintained at linearly changed to was maintained at . In MS only acquisition, the instrument was set to acquire over the range , and the accumulation time for the TOF MS scan was set at spectrum. In auto MS/MS acquisition, the instrument was set to acquire over the range , and the accumulation time for product ion scan was set at spectrum. During the whole analysis, the sample were kept in an automatic sampler at . In order to avoid influences caused by the fluctuations of the instrument, a random sequence was used for the analysis of samples.
Cinnamic acid and derivatives profiling
The raw MS data (wiff.scan files) were converted to MzXML files using ProteoWizard MSConvert . Peak picking and peak grouping were conducted by using R package XCMS v3.20.0 . Annotation of adducts and calculating hypothetical masses for the group were performed with R package CAMERA (Collection of Algorithms of MEtabolite pRofile Annotation) v3.1-5 . In the extracted ion features, only the variables with of nonzero measurement values in at least one group were kept. Identification of cinnamic acid and derivatives was performed by comparing high-accuracy m/z value using MS/MS spectra with an in-house cinnamic acid and derivatives database established with available standards.
After normalization for total peak area intensity, clustering analysis of metabolites were performed with package ComplexHeatmap . A principal coordinates analysis (PCoA) of BrayCurtis distances was performed using package vegan, and a pairwise Adonis test and PERMANOVA (999 permutations) were performed using R package amplicon. Analysis of differential intensity of total peak areas was performed using Tukey’s HSD test. Features were visualized by using ggplot2.
Quantification of 4-HCA in rice leaves
Rice plants that were cultivated for five weeks under greenhouse conditions were harvested for the extraction of leaf metabolites ( 15 replicates per genotype, each replicate included leaves of three plants). Rice leaves were immediately frozen in liquid nitrogen and ground into fine powder with a mortar and pestle. Leaf metabolites were extracted from 0.6 g of each sample using 20 mL ACN. The homogenates were shaken at for 20 min , followed by sonication at a frequency of 30 kHz for 20 min . After centrifugation at for 20 min , the supernatant was obtained and mixed with 30 mg GCB. The suspension was shaken at for 20 min and centrifuged at for 15 min , followed by filtering through a filter before analysis with a liquid chromatography (LC; 1290 Infinity LC, Agilent Technologies, USA) -tandem mass spectrometry (MS/MS; AB SCIEX 4500Q, AB Sciex, USA) system. The standard concentration of 4-HCA was prepared at 0.01, 0.05, 0.1, . The standard preparations were also mixed with 30 mg GCB, then shaken, centrifuged and filtered before LC-MS/MS experiments. The standard curve was calculated with the linear regression method by using ggplot2 (Supplementary Fig. 22). Significance of differences for 4-HCA concentrations was assessed using Tukey’s HSD test. Features were visualized with ggplot2.
Isolation of rice leaf bacteria
Rice plants cultivated under the aforementioned greenhouse conditions were used for bacterial isolations via the limiting dilution method . To obtain a collection of representative rice leaf colonizing bacteria, we isolated bacteria from pooled WT, OsPALO2-KO and OsPALO2-OE leaves. Plants were harvested after five weeks. The leaves of three plants of each sample type were pooled to increase diversity of present bacteria. Rice leaves were washed twice in sterile PBS buffer ( ) on a shaker for 5 min at to remove small particles and loosely attached microbes. The leaves were ground into a homogeneous suspension using 10 mM sterile , followed by sonification for 5 min at a frequency of 30 kHz . Homogenized leaves were left to sediment for 15 min and the supernatants were diluted, distributed and cultivated in 96-well microtiter plates in 1:10 tryptic soy broth (TSB)-water ( ) and 1:2 Reasoner’s 2 A (R2A)-water ( ) for 20 d at room temperature. Then bacteria were purified by continuous streaking. Cultivated bacteria were identified by Sanger sequencing with 27 F and 1492 R primers (Supplementary Table 9) as well as used for the preparation of glycerol stocks for the culture collection. Taxonomic identity was assessed using NCBI BLASTN (https://blast. ncbi.nlm.nih.gov/Blast.cgi). Non-redundant bacterial isolates were selected by 16S rDNA sequence homology . The taxonomic profiling of non-redundant bacterial isolates was visualized with GraPhlAn.
Screening for opportunistic rice pathogens
To remove seed-epiphytic microbes, ZH11 seeds were subjected to surface-sterilization as described above. The resulting seeds were placed on TSB agar plates (ten seeds per plate) to confirm that their surface is epiphyte-free by checking for the absence of visible colony growth surrounding them . Individual bacterial strains from the culture collection were picked from R2A agar plates and suspended in 10 mM MgCl 2 . The final OD values of bacterial suspensions were adjusted to 0.01 . Subsequently, ten sterilized rice seeds were subjected to bacterial inoculation with the individual isolates by soaking them in 20 mL of bacterial cell suspension in a (diameter) Petri dish at until their germination. Ten seeds at the early germination stage were transplanted into a (height) sterile glass bottle with a filter and permeable membrane containing 200 mL of Linsmaier-Skoog (LS) medium solidified with 0.8% agar. For each treatment, bacterial cell suspension was applied directly to each leaf of the one-week-old seedlings. They were incubated in a plant growth incubator ( relative humidity, 13 -h photoperiod). All seedling leaves were cut 1 cm from the top after 2 weeks. The phonotype, disease index and fresh weight of the seedlings were monitored and measured for a total of 3 weeks. Only Xanthomonas oryzae sp. TJ1 (TJ1) showed disease symptoms from all tested bacteria. The phenotype assay with OsPALO2-KO and OsPALO2-OE plants inoculated with TJ1 was conducted in the same way as described above. Bacteria were also re-isolated from the inoculated WT, OsPALO2-KO and OsPALO2-OE plants. TJ1 was identified by Sanger sequencing with 27 F and 1492 R primers (Supplementary Table 9). Bacterial colonization was quantified by plate counting. All isolates were tested in triplicates with three technical repeats.
Effect of 4-HCA on leaf bacterial growth
The main product of OsPAL02 is 4 -HCA ( 4 -hydroxycinnamic acid), therefore the effects on the growth of leaf bacteria were tested using in vitro bioassays with pure . Single colonies were picked from
R2A agar plates and grown overnight in R2A medium on a shaker at . Overnight cultures were used to collect bacteria with centrifugation at for 10 min . The collected bacteria were resuspended to using R2A medium, followed by addition of sterile 1M 4-HCA solution (dissolved in ethanol, and filtered through a filter) to reach different concentrations. The same amount of the solvent EtOH (filtering through a filter) was added as control. The effect of 4-HCA on the opportunistic pathogen’s (TJ1) growth was assessed by using R2A medium supplemented with or . We found that R2A medium supplemented with 2 mM 4-HCA completely inhibited TJ1. Therefore, this concentration of 4-HCA was used to assess growth effects on isolated members of Pseudomonadales, Xanthomonadales and Burkholderiales in the culture collection. After supplementation with 4-HCA or solvent, of each microbial suspension was aliquoted into clear 96-well flat-bottom, polystyrene tissue culture plates. The plates were incubated at on an orbital shaking platform at . The optical density ( ) was measured every 2 hours at 600 nm and monitored over 24 hours (Supplementary Data 7). All isolates were tested in triplicates with three technical repeats.
Assessment of TJ1 inhibition by leaf bacteria
All isolates belonging to Pseudomonadales, Xanthomonadales and Burkholderiales were screened for inhibition of TJ1 using a binary interaction assessment method . Bacterial isolates were individually cultured on R2A plates at . A sterile plastic loop with collected bacteria was suspended in 3 mL R2A medium. To incorporate bacteria into the medium, 2.6 mL of bacterial suspension were added to 40 mL of molten R2A-agar or R2A-agar supplemented with 1 mM 4 HCA pre-cooled to , gently mixed and then poured into Petri dishes ( 9 cm ). A mock treatment was prepared with 2.6 mL filtrate of the same bacterial suspension, through a filter. Single colonies of TJ1 were picked from R2A agar plates and grown overnight in R2A medium on a shaker at , followed by the collection of bacteria via centrifugation at for 10 min . For the inhibition target, TJ1 suspension was adjusted to using 10 mM sterile . A total of TJ1 suspension was spotted onto paper discs at the agar plates as well as, filtrate of TJ1 suspension, which was filtered through a filter, as control. All isolates were tested in triplicates with three technical repeats, and their inhibition ability was recorded after 2 d at .
Synthetic community assembly and plant leaf inoculation
A synthetic community (SynCom) was designed to contain Pseudomonadales isolates on the basis of the observed improved growth when exposed to 4-HCA (shown in Fig. 5a) and prepared using a SynCom preparation method . Sixteen bacterial isolates and TJ1 were individually grown on R2A agar plates, and then single colonies were re-streaked on fresh R2A plates and grown at . A sterile plastic loop with material of each isolate was used to resuspend them in 1 mL of sterile 10 mM MgCl 2 . Tubes containing the resuspended bacteria were vortexed for 5 min , followed by sonification for 2 min at a frequency of 30 kHz to disperse cell aggregates. The SynCom inoculum was obtained by combining 16 bacterial isolates in equal suspension volumes. The optical density ( ) of the SynCom inoculum and TJ1 was adjusted to 0.02 .
For plant inoculation, rice seeds of WT, OsPALO2-KO and OsPALO2-OE were surface-sterilized as described above. Each sterilized seed was germinated for 5 d at with of the SynCom or sterile (as control) in a Petri dish, followed by direct application of TJ1 or sterile 10 mM MgCl 2 (as control) to each seed. Then rice seedlings were transplanted into sterilized bottles containing LS agar. They were incubated in a plant growth incubator ( relative humidity, 13 -h photoperiod). Leaves were cut from 3 -week-old plants using sterile scissors (surface-sterilized using 75%
EtOH and washed with sterile water six times for each plant) to identify their genotype. After 2 weeks, the phenotype of each plant was recorded, and their fresh weight was measured. Different plant genotypes were grown in the same bottle to minimize bottle variations, and treatments were blinded after inoculum preparation to avoid unconscious biases.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequence data (16S rRNA gene fragment sequencing) generated in this study have been deposited in the Genome Sequence Archive of the BIG Data Center , Chinese Academy of Sciences under accession PRJCA016320. The phyllosphere metagenomes of 110 rice genotypes were deposited in the European Nucleotide Archive (ENA) database under the project PRJEB45634. The 4-HCA-associated pathway is available under the pathway map “dosa00940” in the KEGG Pathway database [https://www.kegg.jp/pathway/dosa00940]. Taxonomic classifications of shotgun-sequenced metagenomes used in this study are available in the MiniKraken2_v1_8GB database [https://ccb.jhu.edu/ software/bracken/]. Taxonomic classifications of 16S rRNA gene fragments used in this study are available in the Silva v138.1 reference database [https://www.arb-silva.de/documentation/release-1381/]. Rice gene annotations used in this study are available in the Rice Annotation Project Database (RAPDB) [https://rapdb.dna.affrc.go.jp/]. GO function annotations used in this study are available at Github [https://github.com/xuzhougeng/org.Osativa.eg.db]. All isolated bacterial strains and transformed rice lines were deposited in the State Key Laboratory of Hybrid Rice and the Institute of Plant Protection of Hunan Academy of Agricultural Sciences (Changsha, China). Other data generated in this study are provided in the Supplementary Data files. Source data are provided with this paper.
Hassani, M. A., Durán, P. & Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 6, 1-17 (2018).
Durán, P. et al. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175, 973-983.e914 (2018).
Kwak, M.-J. et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 36, 1100-1109 (2018).
Carrión, V. J. et al. Pathogen-induced activation of diseasesuppressive functions in the endophytic root microbiome. Science 366, 606-612 (2019).
Ritpitakphong, U. et al. The microbiome of the leaf surface of Arabidopsis protects against a fungal pathogen. N. Phytol. 210, 1033-1043 (2016).
Lau, J. A. & Lennon, J. T. Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc. Natl Acad. Sci. USA 109, 14058-14062 (2012).
Castrillo, G. et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature 543, 513-518 (2017).
Bakker, P. A., Pieterse, C. M., de Jonge, R. & Berendsen, R. L. The soilborne legacy. Cell 172, 1178-1180 (2018).
Zhang, J. et al. NRT1. 1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 37, 676-684 (2019).
Berendsen, R. L., Pieterse, C. M. & Bakker, P. A. The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478-486 (2012).
Edwards, J. et al. Structure, variation, and assembly of the rootassociated microbiomes of rice. Proc. Natl Acad. Sci. USA 112, E911-E920 (2015).
Fitzpatrick, C. R. et al. The plant microbiome: from ecology to reductionism and beyond. Annu. Rev. Microbiol. 74, 81-100 (2020).
Deng, S. et al. Genome wide association study reveals plant loci controlling heritability of the rhizosphere microbiome. ISME J. 15, 3181-3194 (2021).
Huang, A. C. et al. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 364, eaau6389 (2019).
Escudero-Martinez, C. et al. Identifying plant genes shaping microbiota composition in the barley rhizosphere. Nat. Commun. 13, 3443 (2022).
Oyserman, B. O. et al. Disentangling the genetic basis of rhizosphere microbiome assembly in tomato. Nat. Commun. 13, 3228 (2022).
Liu, H., Brettell, L. E., Qiu, Z. & Singh, B. K. Microbiome-mediated stress resistance in plants. Trends Plant Sci. 25, 733-743 (2020).
Vorholt, J. A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828-840 (2012).
Horton, M. W. et al. Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 5, 5320 (2014).
Bodenhausen, N., Bortfeld-Miller, M., Ackermann, M. & Vorholt, J. A. A synthetic community approach reveals plant genotypes affecting the phyllosphere microbiota. PLoS Genet. 10, e1004283 (2014).
Brachi, B. et al. Plant genetic effects on microbial hubs impact host fitness in repeated field trials. Proc. Natl Acad. Sci. USA 119, e2201285119 (2022).
Wagner, M. R. et al. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 7, 12151 (2016).
Sapkota, R., Knorr, K., Jørgensen, L. N., O’Hanlon, K. A. & Nicolaisen, M. Host genotype is an important determinant of the cereal phyllosphere mycobiome. N. Phytol. 207, 1134-1144 (2015).
Gong, T. & Xin, X. F. Phyllosphere microbiota: Community dynamics and its interaction with plant hosts. J. Integr. Plant. Biol. 63, 297-304 (2021).
Zhan, C., Matsumoto, H., Liu, Y. & Wang, M. Pathways to engineering the phyllosphere microbiome for sustainable crop production. Nat. Food 3, 997-1004 (2022).
Finkel, O. M. et al. The effects of soil phosphorus content on plant microbiota are driven by the plant phosphate starvation response. PLoS Biol. 17, e3000534 (2019).
Chen, T. et al. A plant genetic network for preventing dysbiosis in the phyllosphere. Nature 580, 653-657 (2020).
Beilsmith, K. et al. Genome-wide association studies on the phyllosphere microbiome: embracing complexity in host-microbe interactions. Plant J. 97, 164-181 (2019).
Su, P. et al. Recovery of metagenome-assembled genomes from the phyllosphere of 110 rice genotypes. Sci. Data 9, 254 (2022).
Lu, J. et al. Metagenome analysis using the Kraken software suite. Nat. Protoc. 17, 2815-2839 (2022).
Zhang, J. et al. High-throughput cultivation and identification of bacteria from the plant root microbiota. Nat. Protoc. 16, 988-1012 (2021).
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T. & Singh, B. K. Plant-microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607-621 (2020).
Sohrabi, R., Paasch, B. C., Liber, J. A. & He, S. Y. Phyllosphere microbiome. Annu. Rev. Plant. Biol. 74, 539-568 (2023).
Schäfer, M., Vogel, C. M., Bortfeld-Miller, M., Mittelviefhaus, M. & Vorholt, J. A. Mapping phyllosphere microbiota interactions in planta to establish genotype-phenotype relationships. Nat. Microbiol. 7, 856-867 (2022).
Helfrich, E. J. et al. Bipartite interactions, antibiotic production and biosynthetic potential of the Arabidopsis leaf microbiome. Nat. Microbiol. 3, 909-919 (2018).
Carlström, C. I. et al. Synthetic microbiota reveal priority effects and keystone strains in the Arabidopsis phyllosphere. Nat. Ecol. Evol. 3, 1445-1454 (2019).
Song, Y. et al. FERONIA restricts Pseudomonas in the rhizosphere microbiome via regulation of reactive oxygen species. Nat. Plants 7, 644-654 (2021).
Ma, K.-W. et al. Coordination of microbe-host homeostasis by crosstalk with plant innate immunity. Nat. Plants 7, 814-825 (2021).
Eichmann, R., Richards, L. & Schäfer, P. Hormones as go-betweens in plant microbiome assembly. Plant J. 105, 518-541 (2021).
Hacquard, S., Spaepen, S., Garrido-Oter, R. & Schulze-Lefert, P. Interplay between innate immunity and the plant microbiota. Annu. Rev. Phytopathol. 55, 565-589 (2017).
Pfeilmeier, S. et al. The plant NADPH oxidase RBOHD is required for microbiota homeostasis in leaves. Nat. Microbiol. 6, 852-864 (2021).
Xu, P. et al. Temporal metabolite responsiveness of microbiota in the tea plant phyllosphere promotes continuous suppression of fungal pathogens. J. Adv. Res. 39, 49-60 (2022).
Maier, B. A. et al. A general non-self response as part of plant immunity. Nat. Plants 7, 696-705 (2021).
Wang, Y. et al. GWAS, MWAS and mGWAS provide insights into precision agriculture based on genotype-dependent microbial effects in foxtail millet. Nat. Commun. 13, 5913 (2022).
Beckers, B. et al. Lignin engineering in field-grown poplar trees affects the endosphere bacterial microbiome. Proc. Natl Acad. Sci. USA 113, 2312-2317 (2016).
Shalev, O., Ashkenazy, H., Neumann, M. & Weigel, D. Commensal Pseudomonas protect Arabidopsis thaliana from a coexisting pathogen via multiple lineage-dependent mechanisms. ISME J. 16, 1235-1244 (2022).
Levy, A. et al. Genomic features of bacterial adaptation to plants. Nat. Genet. 50, 138-150 (2018).
Vogel, C. M., Potthoff, D. B., Schäfer, M., Barandun, N. & Vorholt, J. A. Protective role of the Arabidopsis leaf microbiota against a bacterial pathogen. Nat. Microbiol. 6, 1537-1548 (2021).
Wang, N. R. et al. Commensal Pseudomonas fluorescens strains protect Arabidopsis from closely related Pseudomonas pathogens in a colonization-dependent manner. mBio 13, e02892-02821 (2022).
Tao, C. et al. Bio-organic fertilizers stimulate indigenous soil Pseudomonas populations to enhance plant disease suppression. Microbiome 8, 1-14 (2020).
Shalev, O. et al. Commensal Pseudomonas strains facilitate protective response against pathogens in the host plant. Nat. Ecol. Evol. 6, 383-396 (2022).
Sarniguet, A., Kraus, J., Henkels, M. D., Muehlchen, A. M. & Loper, J. E. The sigma factor sigma s affects antibiotic production and biological control activity of Pseudomonas fluorescens Pf-5. Proc. Natl Acad. Sci. USA. 92, 12255-12259 (1995).
Beskrovnaya, P. et al. Comparative genomics identified a genetic locus in plant-associated Pseudomonas spp. that is necessary for induced systemic susceptibility. mBio 11, e00575-00520 (2020).
Voges, M. J., Bai, Y., Schulze-Lefert, P. & Sattely, E. S. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl Acad. Sci. USA. 116, 12558-12565 (2019).
Koprivova, A. et al. Root-specific camalexin biosynthesis controls the plant growth-promoting effects of multiple bacterial strains. Proc. Natl Acad. Sci. USA. 116, 15735-15744 (2019).
Cernava, T. & Berg, G. The emergence of disease-preventing bacteria within the plant microbiota. Environ. Microbiol. 24, 3259-3263 (2022).
Stringlis, I. A. et al. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl Acad. Sci. USA. 115, E5213-E5222 (2018).
Hu, L. et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 9, 2738 (2018).
McCouch, S. R. et al. Open access resources for genome-wide association mapping in rice. Nat. Commun. 7, 10532 (2016).
Paulson, J. N., Stine, O. C., Bravo, H. C. & Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200-1202 (2013).
Asnicar, F., Weingart, G., Tickle, T. L., Huttenhower, C. & Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3, e1029 (2015).
Wickham, H. ggplot2: Elegant Graphics For Data Analysis (Springer, 2016).
Kang, H. et al. Dissection of the genetic architecture of rice resistance to the blast fungus Magnaporthe oryzae. Mol. Plant Pathol. 17, 959-972 (2016).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811-2812 (2014).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb.) 2, 100141 (2021).
Ma, X. et al. A robust CRISPR/Cas9 system for convenient, highefficiency multiplex genome editing in monocot and dicot plants. Mol. Plant. 8, 1274-1284 (2015).
Dai, S. et al. Transgenic rice plants that overexpress transcription factors RF2a and RF2b are tolerant to rice tungro virus replication and disease. Proc. Natl Acad. Sci. USA 105, 21012-21016 (2008).
Lin, Q. et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 38, 582-585 (2020).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852-857 (2019).
Martin, M. Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnet. J. 17, 10-12 (2011).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581-583 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590-D596 (2012).
French, W. R. et al. Wavelet-based peak detection and a new charge inference procedure for MS/MS implemented in ProteoWizard’s msConvert. J. Proteome Res. 14, 1299-1307 (2015).
Benton, H. P., Want, E. J. & Ebbels, T. M. Correction of mass calibration gaps in liquid chromatography-mass spectrometry metabolomics data. Bioinformatics 26, 2488-2489 (2010).
Kuhl, C., Tautenhahn, R., Bottcher, C., Larson, T. R. & Neumann, S. CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 84, 283-289 (2012).
Gu, Z. Complex heatmap visualization. iMeta 1, e43 (2022).
BIG Data Center Members. Database resources of the BIG data center in 2018. Nucleic Acids Res. 46, D14-D20 (2018).
Su, P. et al. Microbiome homeostasis on rice leaves is regulated by a precursor molecule of lignin biosynthesis. https://zenodo.org/ records/10115039 (2023).
Acknowledgements
P.S. and Y.L. were supported by a grant from the National Key R&D Program of China (2022YFD1400700); H.K. was supported by grants from the National Key R&D Program of China (2021YFC2600400) and the National Natural Science Foundation (32261143468); P.S. was additionally supported by a grant from the Key R&D Program of Hunan Province (2022NK2014); D.Z. was supported by a grant from the LONGPING HIGH-TECH INDUSTRY ZONE (LongPing committee issue 2022-18). We appreciate the support of Bin Liu and Junliang Zhao from the Rice Research Institute, Guangdong Academy of Agricultural Sciences, China for providing the RDP-II seeds used in this study. We also want to thank OE Biotech Co., Ltd (Shanghai, China) for their technical support during high-throughput sequencing experiments.
Author contributions
T.C., D.Z., Y.L., and P.S. conceived the idea for the study. P.S. and Q.P. performed the bioinformatic analyses and visualized the data. H.K. conducted the GWAS analyses. T.C., P.S., Q.P., and H.K. analyzed the data. W.A.W. conducted formal data analysis and review. G.B. conducted review. P.S., Q.P., Z.L., and J.M. conducted the bacterial strain isolation, rice mutant construction and the microbiological experiments. The manuscript was written by T.C., P.S., Q.P., and H.K. All authors read and approved the final manuscript.
Correspondence and requests for materials should be addressed to Deyong Zhang, Tomislav Cernava or Yong Liu.
Peer review information Nature Communications thanks Davide Bulgarelli and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
State Key Laboratory of Hybrid Rice and Institute of Plant Protection, Hunan Academy of Agricultural Sciences, Changsha 410125, China. State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing 100193, China. National Center of Technology Innovation for Saline-Alkali Tolerant Rice in Sanya City, Sanya 572024, China. College of Tropical Crops, Hainan University, Haikou 570228, China. Institute of Environmental Biotechnology, Graz University of Technology, Graz 8010, Austria. Leibniz Institute for Agricultural Engineering and Bioeconomy (ATB), Potsdam 14469, Germany. Institute for Biochemistry and Biology, University of Potsdam, Potsdam 14476, Germany. Longping Branch, College of Biology, Hunan University, Changsha 410082, China. School of Biological Sciences, Faculty of Environmental and Life Sciences, University of Southampton, Southampton SO17 1BJ, UK. These authors contributed equally: Pin Su, Houxiang Kang, Qianze Peng.