DOI: https://doi.org/10.1186/s40164-024-00505-7
PMID: https://pubmed.ncbi.nlm.nih.gov/38609997
تاريخ النشر: 2024-04-12
الخلايا المثبطة المشتقة من النخاع في السرطان: أهداف علاجية للتغلب على هروب المناعة من الورم
الملخص
بشكل متناقض، يمكن أن يتم تثبيط وترويج تطور الورم وتقدمه بواسطة الجهاز المناعي. بعد ثلاث مراحل من تحرير المناعة، وهي الإزالة، التوازن، والهروب، لم تعد خلايا الورم مقيدة بمراقبة المناعة وبالتالي تتطور إلى أورام سريرية. تشمل آليات الهروب المناعي الشذوذات في خلايا المناعة المرتبطة بمكافحة الورم، والاختيار لمقاومة المناعة ضد خلايا الورم، وضعف نقل خلايا T، وتكوين بيئة ميكروية مثبطة للمناعة. مجموعة من خلايا المايلويد غير الناضجة المتميزة، وهي خلايا مثبطة مشتقة من المايلويد (MDSCs)، تتوسط الهروب المناعي بشكل أساسي من خلال exerting آثار مثبطة للمناعة والمشاركة في تكوين بيئة ميكروية مثبطة للمناعة. وقد وجدت التجارب السريرية أن مستويات MDSCs في الدم المحيطي لمرضى السرطان مرتبطة ارتباطًا وثيقًا بمرحلة الورم، والانتشار، والتنبؤ. علاوة على ذلك، أكدت التجارب الحيوانية أن إزالة MDSCs تثبط نمو الورم والانتشار إلى حد ما. لذلك، قد تصبح MDSCs هدفًا للعلاج المناعي للعديد من أنواع السرطان، ويمكن أن تساعد إزالة MDSCs في تحسين معدل الاستجابة لعلاج السرطان وبقاء المرضى. ومع ذلك، تفتقر التعريفات الواضحة لـ MDSCs والآلية المحددة المعنية في الهروب المناعي. في هذه الورقة، نستعرض دور مجموعة MDSCs في تطور الورم والآليات المعنية في الهروب المناعي في سياقات ورمية مختلفة. بالإضافة إلى ذلك، نناقش استخدام هذه الخلايا كأهداف للعلاج المناعي للورم. تسهم هذه المراجعة ليس فقط في فهم منهجي وشامل للدور الأساسي لـ MDSCs في ردود فعل الجهاز المناعي ضد الأورام، ولكن أيضًا توفر معلومات لتوجيه تطوير العلاجات السرطانية التي تستهدف MDSCs.
مقدمة
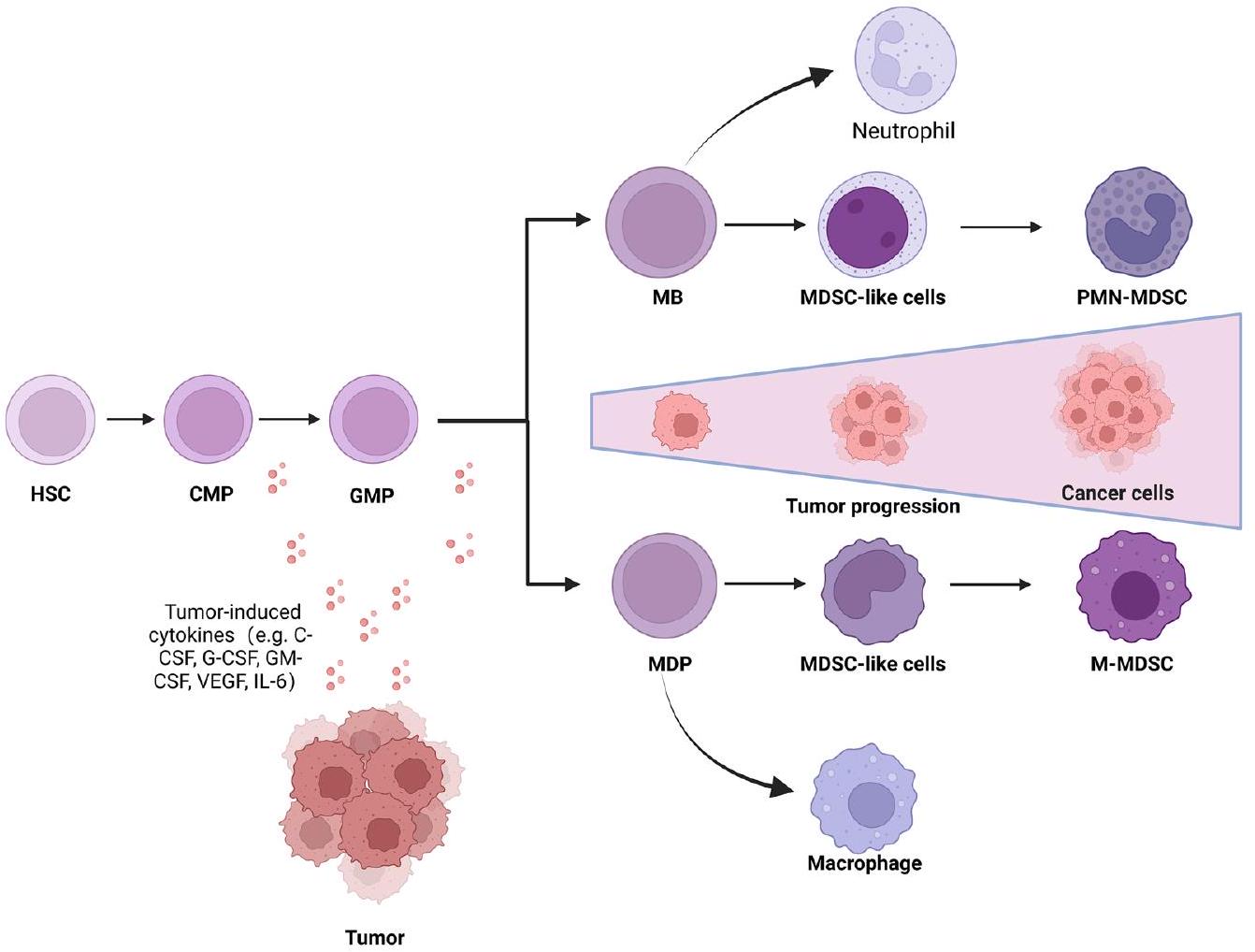
هناك نوعان رئيسيان من خلايا MDSCs يُطلق عليهما MDSCs متعددة الأشكال النوى (PMN-MDSCs) وMDSCs أحادية النواة (M-MDSCs). تشبه هذه الخلايا العدلات والوحيدات من حيث الشكل والخصائص، وبالتالي، فإن الشكل والخصائص وحدهما ليسا كافيين لتحديد MDSCs. بالإضافة إلى النوعين الرئيسيين من الخلايا، تشمل MDSCs مجموعة صغيرة من الخلايا (أقل من
تقوم خلايا MDSCs بتسهيل الهروب المناعي بشكل رئيسي من خلال ممارسة وظائف مثبطة للمناعة. على الرغم من أن خلايا MDSCs هي
المشاركة في قمع خلايا المناعة المختلفة، الهدف الرئيسي لها هو خلايا T. على النقيض من ذلك، فإن استنفاد MDSCs باستخدام أجسام مضادة محددة يعزز
في السنوات الأخيرة، كشفت الأبحاث عن الأهمية السريرية لخلايا النخاع العظمي المثبطة (MDSCs). وقد وثقت دراسات مختلفة انتشار خلايا MDSCs في عدة أنواع من الأورام البشرية، مثل الميلانوما الجلدية [17]، وسرطان الكبد [18]، وسرطان الثدي [19]، وسرطان البروستاتا [20] وسرطان الرئة [21]. بالإضافة إلى ذلك، أظهرت عدد من الدراسات أن خلايا MDSCs تعتبر مؤشرات حيوية تنبؤية هامة لتطور السرطان وأهداف محتملة للعلاج المضاد للسرطان [22]. يمكن لخلايا MDSCs أن تثبط الاستجابة المناعية وتحمي خلايا الورم من هجوم نظام المناعة لدى المضيف، مما يؤدي إلى هروب المناعة من الورم. قد يكون استهداف خلايا MDSCs لتنشيط مناعة الورم وعكس الهروب المناعي خيارًا قابلاً للتطبيق لدى مرضى الأورام.
في هذه المراجعة، نناقش الدور البيولوجي لخلايا MDSCs في هروب الورم من المناعة. بالإضافة إلى ذلك، نستعرض أيضًا الآليات المحددة التي تشارك بها خلايا MDSCs.
في هروب المناعة من الورم في أنواع مختلفة من الأورام ومناقشة بالتفصيل الأساليب المستخدمة لاستهداف خلايا المناعة المثبطة للورم (MDSCs) لعلاج السرطان.
تمييز وتراكم خلايا المناعة المثبطة المتعددة
وظائف خلايا MDSC
انسداد توجيه خلايا T
تنظيم تنازلي لـ
تثبيط وظيفة خلايا T من خلال الإجهاد التأكسدي
استهلاك الأحماض الأمينية اللازمة لوظيفة خلايا T
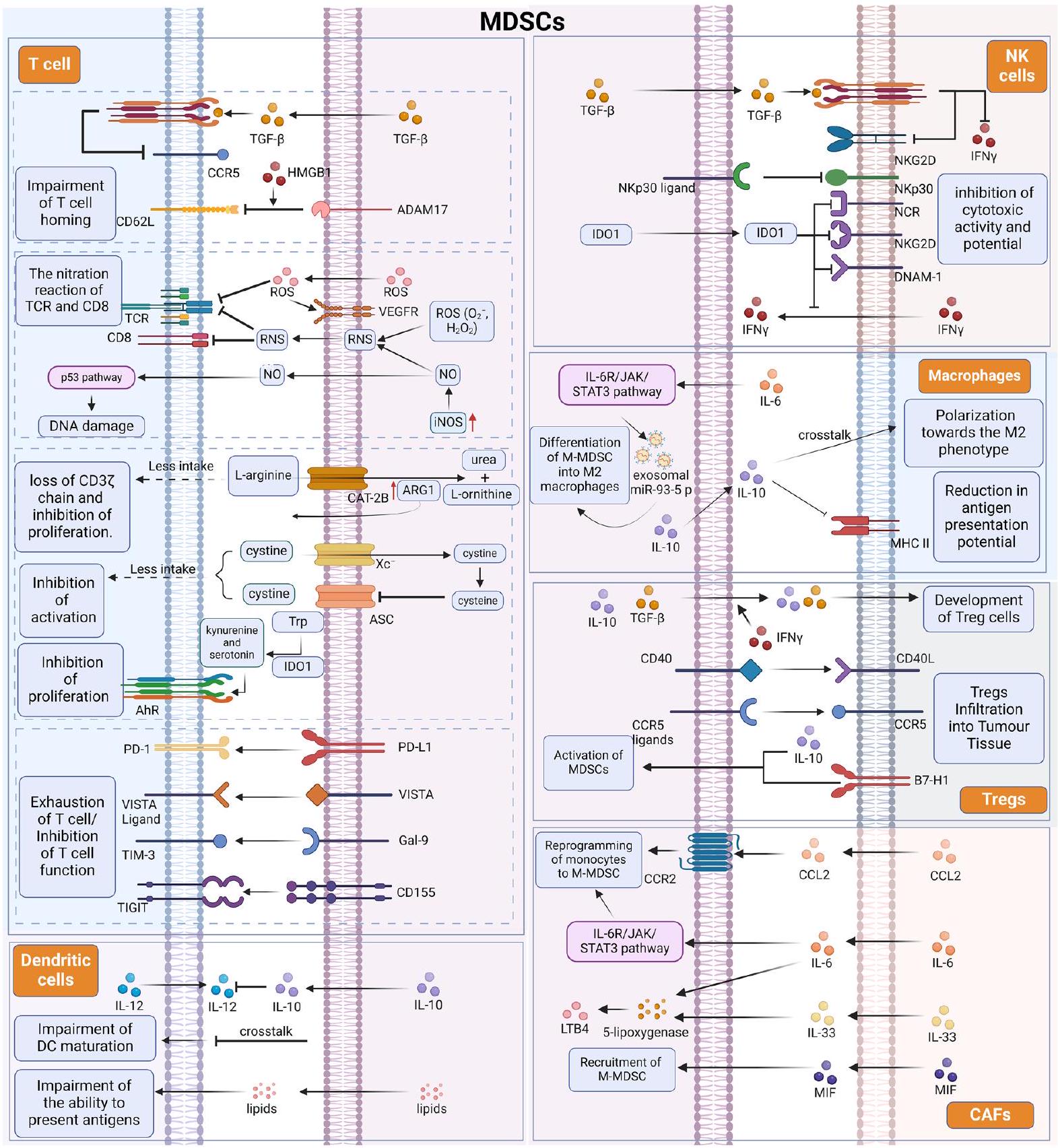
يؤدي نقص التربتوفان إلى تنشيط مستقبل الهيدروكربونات العطرية (AhR) بواسطة السيروتونين لتحفيز إنتاج IDO1 ورد فعل مضاد للالتهابات [45].
تعبير جزيئات نقاط التفتيش المناعية السلبية على خلايا MDSCs
التواصل المتبادل بين خلايا MDSC وغيرها من الخلايا المناعية المشاركة في بيئة الورم
الخلايا. تعتبر خلايا DC من الخلايا المايلويد الرئيسية الأخرى التي تتسلل إلى بيئة الورم. على الرغم من أن الإشارات من بيئة الورم تعزز تدفق خلايا DC غير الناضجة، إلا أن عوامل متعددة، بما في ذلك تراكم الأدينوزين، وتراكم اللاكتات، والظروف الهايبكسية، تؤدي إلى خلل في وظيفة خلايا DC. بالإضافة إلى ذلك، تم اقتراح أن التفاعل بين خلايا DC وMDSCs قد يكون أيضًا مسؤولًا جزئيًا عن انخفاض وظيفة خلايا DC. عندما يتم زراعة MDSCs المشتقة من نخاع العظام مع خلايا DC في المختبر، ينخفض عدد خلايا DC مع زيادة عدد MDSCs. أظهرت الدراسات على MDSCs في مرضى الميلانوما أن الترددات العالية من M-MDSCs تعيق نضوج خلايا DC عن طريق تقليل امتصاص المستضدات، ومنع هجرة خلايا DC غير الناضجة والناضجة، وتحويل إنتاج السيتوكينات في خلايا DC نحو نمط مضاد للالتهابات، وحجب قدرة خلايا DC على تحفيز IFN.
بالإضافة إلى قدرتها على قمع خلايا T المناعية لتدمير الأورام، قد تكون الخلايا المناعية المثبطة المتعددة (MDSCs) أيضًا متورطة في هروب الورم من المناعة من خلال تحفيز خلايا مثبطة مناعية أخرى، مثل البلعميات وخلايا T التنظيمية (Treg). لا تعتبر MDSCs فقط مصدرًا للبلعميات المرتبطة بالورم، ولكنها قد تؤثر أيضًا على حالة تنشيط البلعميات ووظيفتها واستقطابها من خلال الارتباط. مدفوعة بـ IL-6، يتم تنشيط مسار IL-6R/JAK/STAT3 في PMN-MDSCs، مما يؤدي بدوره إلى تخليق وإفراز miR-93-5p الإكسوزومي، مما يدفع تمايز M-MDSCs إلى بلعميات M2. في بيئة الورم الدقيقة،
دور MDSCs في الأورام الشائعة
الميلانوما الجلدية
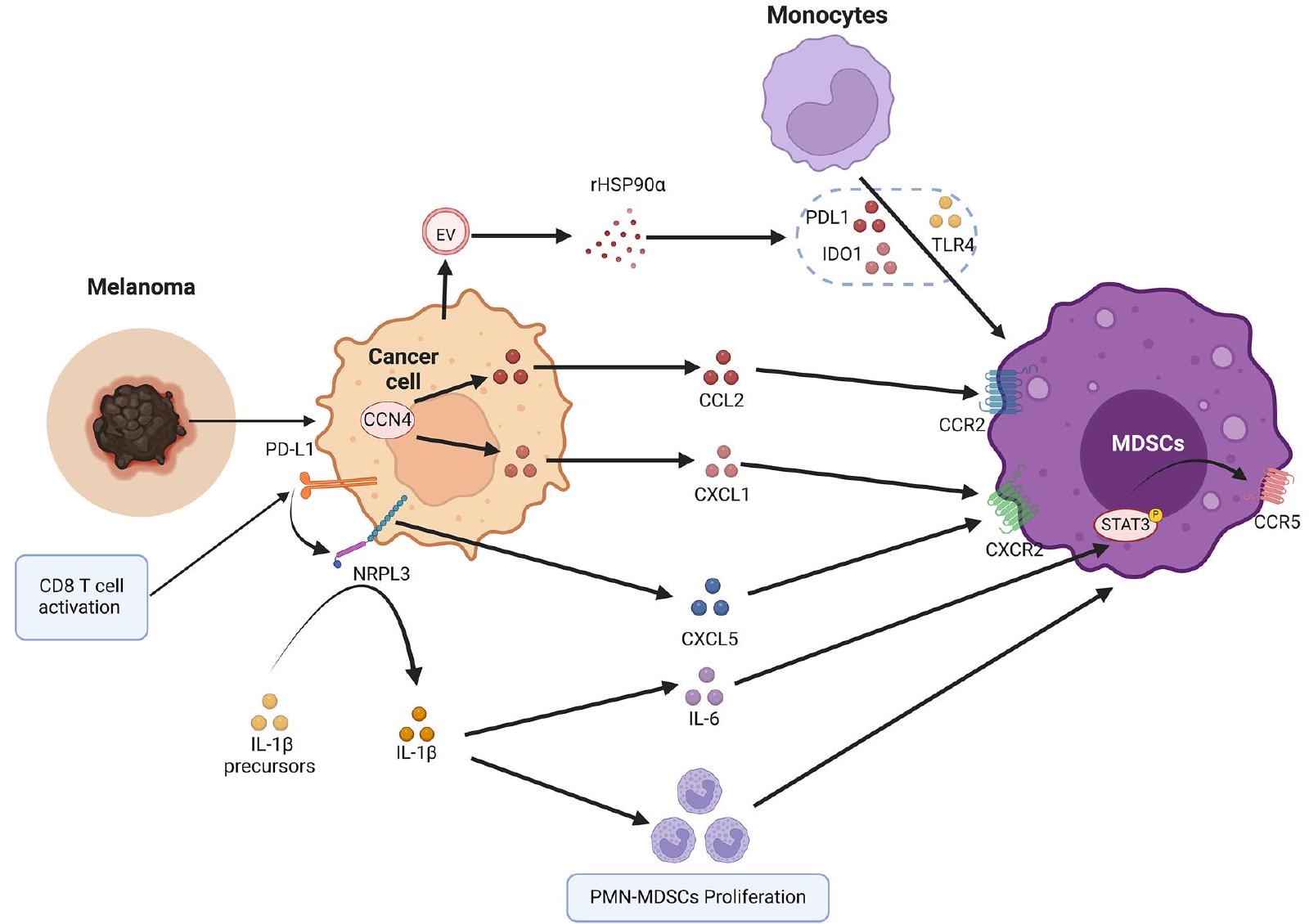
بعد تراكمها وتنشيطها في نخاع العظام، يتم جذب MDSCs إلى موقع الورم بواسطة مجموعة من الكيماوكينات. على الرغم من أن CCL2 وCCL3 وCCL4 مهمة لتجنيد M-MDSCs عبر CCR2 [94]، فإن الروابط CXCR2 وCXCL1 وCXCL2 وCXCL3 وCXCL5 وCXCL6 وCXCL7 تتوسط بشكل رئيسي هجرة PMN-MDSCs [95]. يعتبر تسلل PMN-MDSCs داخل أنسجة الميلانوما مساهمًا كبيرًا في نمو الميلانوما الأولية والانتشار. أظهرت الدراسات أن PMN-MDSCs تتسلل إلى الميلانوما الأولية والانتقالات عبر تفاعلات CXCL1/CXCR2 [96]. في الفئران المصابة بالميلانوما، تنتج PMN-MDSCs عامل نمو الكبد وTGF-
الميلانوما من خلال المشاركة في الانتقال من الظهارة إلى الميزانشيم [98]. بالإضافة إلى ذلك، عندما تم زرع خلايا الميلانوما التي تم حذف CCN4 في فئران مناعية كاملة، تم تقليل تسلل PMN-MDSCs. وذلك لأن التعبير المحلي عن CNN4 منع إفراز IFN من خلايا T CD8 + وزاد من إفراز الورم للكيماوكينات الجاذبة لـ MDSCs مثل CCL2 وCXCL1 [99]. بالإضافة إلى ذلك، في نماذج الأورام قبل السريرية المتعددة وكذلك العينات السريرية، يؤدي تنشيط خلايا T CD8 استجابةً لحجب PD-1 إلى تحفيز سلسلة إشارات التهابية PD-L1/NLRP3 التي تؤدي في النهاية إلى تجنيد PMN-MDSCs إلى أنسجة الميلانوما، مما يؤدي إلى تثبيط المناعة وبالتالي الهروب المناعي [100]. يمكن تثبيط تسلل PMN-MDSCs في الأورام بواسطة حجب NLRP3، مما يحسن بشكل كبير من فعالية العلاج المناعي بالأجسام المضادة المضادة لـ PD-1 [100].
سرطان الكبد الخلوي
تشمع الكبد [101]. أظهرت الدراسات السريرية على مدار العقد الماضي الأهمية السريرية لخلايا المناعة المثبطة المشتقة من النخاع (MDSCs) في المرضى الذين يعانون من سرطان الكبد الخلوي [102-104]. في المرضى الذين يعانون من سرطان الكبد الخلوي، كانت خلايا MDSCs CD14(+)HLA-DR(low/-) مرتفعة بشكل ملحوظ في الدم المحيطي أو نسيج الورم. لم تتمكن خلايا MDSCs من سرطان الكبد الخلوي من تحفيز استجابات خلايا T المتبرعة وكان لديها نشاط عالٍ من الأرجيناز [18]. تشير هذه النتيجة إلى أن سرطان الكبد الخلوي يدفع خلايا MDSCs للتسلل والتجنيد وكبح وظيفة خلايا T الفعالة داخل بيئة الورم (TME) من خلال آليات مختلفة (الشكل 4). أشارت تحليل تسلسل الخلايا المفردة في نماذج سرطان الكبد الخلوي في الفئران وأعضاء سرطان الكبد الخلوي البشرية إلى أن هذا الاختلاف قد يكون بسبب تراكم PMN-MDSCs الذي يتوسطه METTL1 بعد علاج غير كافٍ بترددات الراديو، مما يكبح المناعة المضادة للورم ويعزز تقدم سرطان الكبد الخلوي [105].
في البيئة المجهرية للورم، يؤدي LDL المؤكسد الناتج عن الأيض غير الطبيعي للدهون إلى تنشيط محور أكسدة الدهون/فوسفوريلation p38/CEBP داخل خلايا CAFs CD36، مما يعزز في النهاية إفراز MIF من البلعميات بطريقة تعتمد على CD36.
تحفزها MIF يتم تجنيدها إلى أنسجة HCC وتعزز الإضعاف المناعي في البيئة المجهرية للورم. الفيروبتوز هو نوع من موت الخلايا المعتمد على الحديد الذي يؤدي إلى تدمير غشاء الخلية من خلال تراكم بيروكسيدات الدهون. في HCC، لا يوفر الفيروبتوز قمعًا ذاتيًا للورم ولكنه يحفز تسلل الورم من قبل MDSCs عبر HMGB1، مما يؤدي إلى استجابة مناعية تكيفية. بالإضافة إلى ذلك، تلعب العوامل الالتهابية في البيئة المجهرية للورم أدوارًا أساسية في هروب المناعة الذي تسببه MDSCs في HCC. لقد أظهرت الدراسات أن IL-
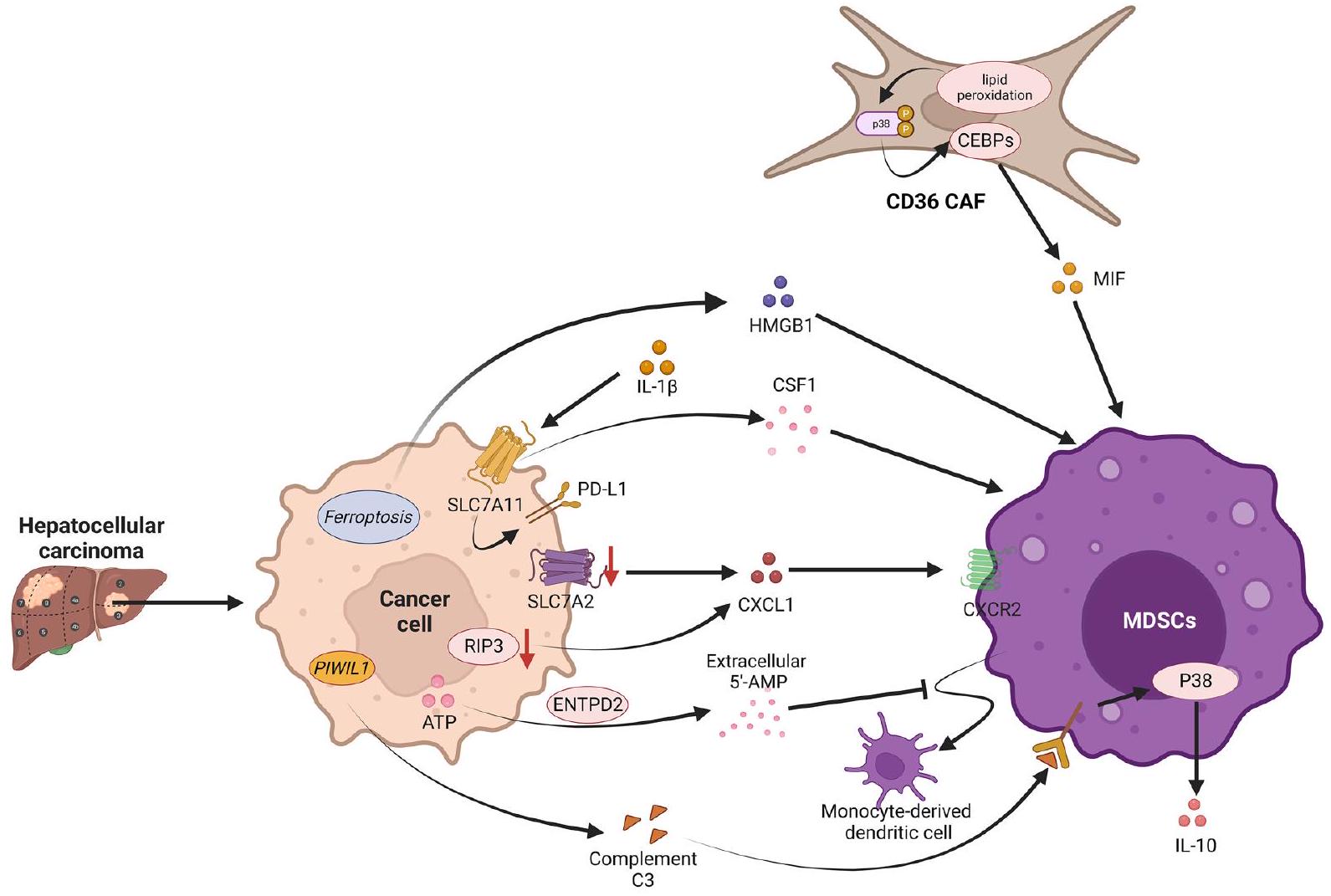
نقص الأكسجين هو عامل بيئي مهم في سرطان الكبد الخلوي. في سرطان الكبد الخلوي، يؤدي نقص الأكسجين إلى تحفيز الإكتودومين، إنزيم ثنائي فوسفات ثلاثي النوكليوزيد 2 (ENTPD2)، بشكل رئيسي من خلال عامل نقص الأكسجين 1 (HIF-1)، مما يؤدي إلى زيادة تعبيره. يقوم ENTPD2 بتحويل الأدينوزين ثلاثي الفوسفات (ATP) الخارجي إلى 5′-أدينوزين أحادي الفوسفات.
لقد أظهرت الدراسات أن المستقلبات التي تنتجها خلايا السرطان يمكن أن تعزز تطور الورم من خلال تعديل النمط الوظيفي لمختلف خلايا المناعة. في سرطان الكبد الخلوي، يزيد RNA المشابه لـ Piwi (PIWIL1) من استقلاب الأحماض الدهنية عبر الأحماض الدهنية الميتوكوندرية.
سرطان الثدي
لقد أظهرت التصنيفات تباينات ملحوظة من حيث البقاء على قيد الحياة خالية من النقائل البعيدة [122]. خلال تطور سرطان الثدي، تفرز خلايا السرطان عددًا كبيرًا من السيتوكينات التي تؤثر على تمايز خلايا نخاع العظام وتعزز تطوير MDSCs [19، 54]. في نموذج الفأر لسرطان الثدي، تضمنت ميزات MDSCs عدة جينات مرتبطة بتنظيم المناعة، مثل الأرجيناز 2 وCd84، وم receptors الكيميائية (مثل Ccr2 وCxcr2)، مما يشير إلى أن MDSCs يمكن توجيهها إلى نسيج الورم بواسطة السيتوكينات [123]. يكشف الجمع بين مستقبلات سطح الخلية CD84 وJAML على MDSCs مع صبغة CD11b/Gr1 عن وجود MDSCs في أنسجة الأورام لدى الفئران أو في البشر مع CD11b/CD14 أو CD15 [123].
يعزز القدرة المثبطة للمناعة لخلايا MDSCs [129]. كما أن GM-CSF المستمد من خلايا السرطان يحفز أيضًا نسخ الجينات التي تشفر لإنزيم كيناز البروتين المنشط بواسطة AMP ألفا (AMPK
علاوة على ذلك، تم تحفيز تطوير خلايا مثبطة مشتقة من النخاع العظمي المبكرة (eMDSCs) في سرطان الثدي بواسطة الميكرو RNA المشتق من الإكسوزومات الورمية miR-9 وmiR-181a، والتي نشطت مسار إشارة Janus kinase (JAK)/STAT من خلال استهداف مثبط إشارة السيتوكين-3 (SOCS3) ومثبط البروتين لـ STAT-3 المنشط (PIAS3)، على التوالي. بالإضافة إلى ذلك، زادت أسيتيلation لعضو عائلة Smad 3 (SMAD3) عند K20 وK117 بواسطة أسيتيل ترانسفيراز الليسين 6A (KAT6A) من ارتباط SMAD3 بمعدل التعديل الكروماتيني المسرطن TRIM24 وأعاقت ارتباط SMAD3 بمثبط الورم TRIM33. بدوره، يؤدي ذلك إلى استقطاب مركب TRIM24-SMAD3 إلى الكروماتين من خلال أسيتيلation KAT6A لهيستون H3 عند الليسين 23، مما يزيد من تعبير السيتوكينات المرتبطة بالمناعة ويؤدي إلى استقطاب MDSCs وهروب المناعة في سرطان الثدي الثلاثي السلبي من خلال تثبيط المناعة. تعزز المستقلبات الأدينوسينرجيك الناتجة عن التعبير العالي عن إكوتونيوكليوتيد بيفوسفاتاز/فوسفوديستراز 1 (Enpp1) في سرطان الثدي تعبير الهبتوجلوبين، الذي يستقطب PMN-MDSCs. تسبب تسلل PMN-MDSCs في تثبيط المناعة، مما يسمح لخلايا الورم الدائرية ذات التعبير العالي عن Enpp1 بالترويج للانتكاس من خلال آلية إعادة البذر الذاتي التي تسبب الفشل المحلي الإقليمي.
سرطان البروستاتا
إنترلوكين-23 (IL-23)، الذي يتم إنتاجه بواسطة خلايا المناعة المثبطة للأورام، يعمل كمنظم للمناعة المؤيدة للورم وينظم البروستاتا
مقاومة سرطان البروستاتا للإخصاء من خلال الحفاظ على إشارات AR. IL-23 الذي تفرزه PMN-MDSCs هو لاعب رئيسي في مقاومة الأدوية الهرمونية في سرطان البروستاتا. لذلك، يمكن أن يؤدي التثبيط المباشر لـ PMN-MDSCs إلى عكس مقاومة ADT لدى المرضى الذين يعانون من سرطان البروستاتا المتقدم. بالإضافة إلى ذلك، تعزز الإكسوزومات المستمدة من MDSCs في بيئة الورم تقدم الورم من خلال استقطاب البلعميات. في سرطان البروستاتا، تحفز الإكسوزومات التي تنقل S100A9 من MDSCs إلى خلايا سرطان البروستاتا تكاثر خلايا سرطان البروستاتا، والغزو، والهجرة من خلال زيادة التعبير عن circMID1 (hsa_circ_0007718)، مما يعزز في النهاية تقدم CRPC.
سرطان الرئة
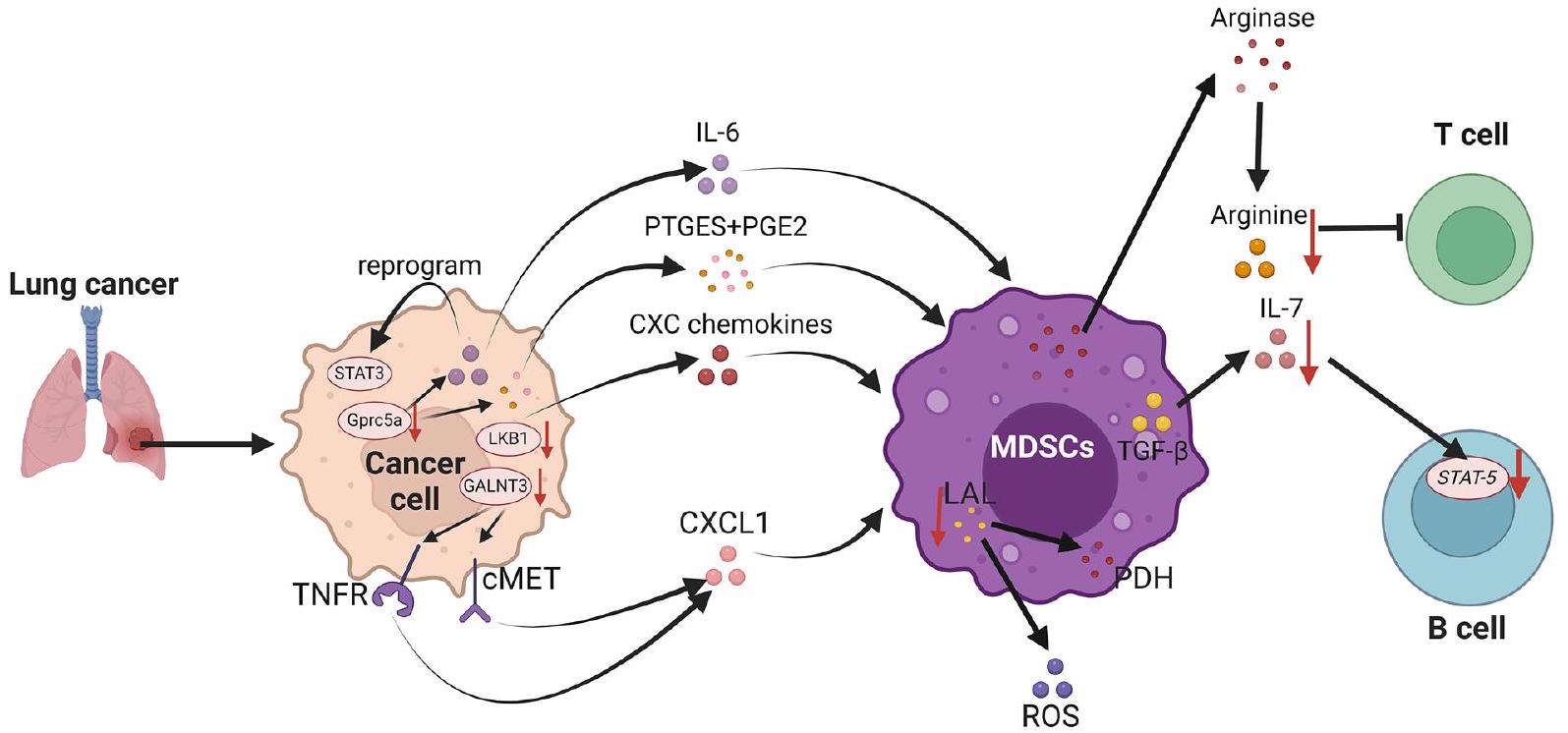
أظهرت الدراسات الحديثة أنه داخل الأورام الغدية الرئوية، يلعب الفطر Aspergillus sydowii، على الرغم من كتلته الحيوية المنخفضة، دورًا مهمًا في التحفيز
البيئة المجهرية المثبطة للمناعة، مما يعزز تقدم أورام الرئة ويرتبط بتوقعات سيئة للمرضى. ويرجع ذلك أساسًا إلى أن Aspergillus sydowii، الذي يتواجد بكثرة داخل الأورام الغدية الرئوية، يمكنه أيضًا إفراز IL-1.
يمكن أن تعيقها GALNT3، التي تمنع تجديد خلايا LC الذاتية وتؤدي إلى تقليل مستوى CXCL1 عن طريق خفض المستوى من
سرطانات أخرى
تساهم هذه البيئة الدقيقة في تطوير نمط أكثر كبتًا للمناعة، مما يسهم في تطور مقاومة الإشعاع في سرطان البنكرياس.
العلاج الرئيسي لـ MDSCs
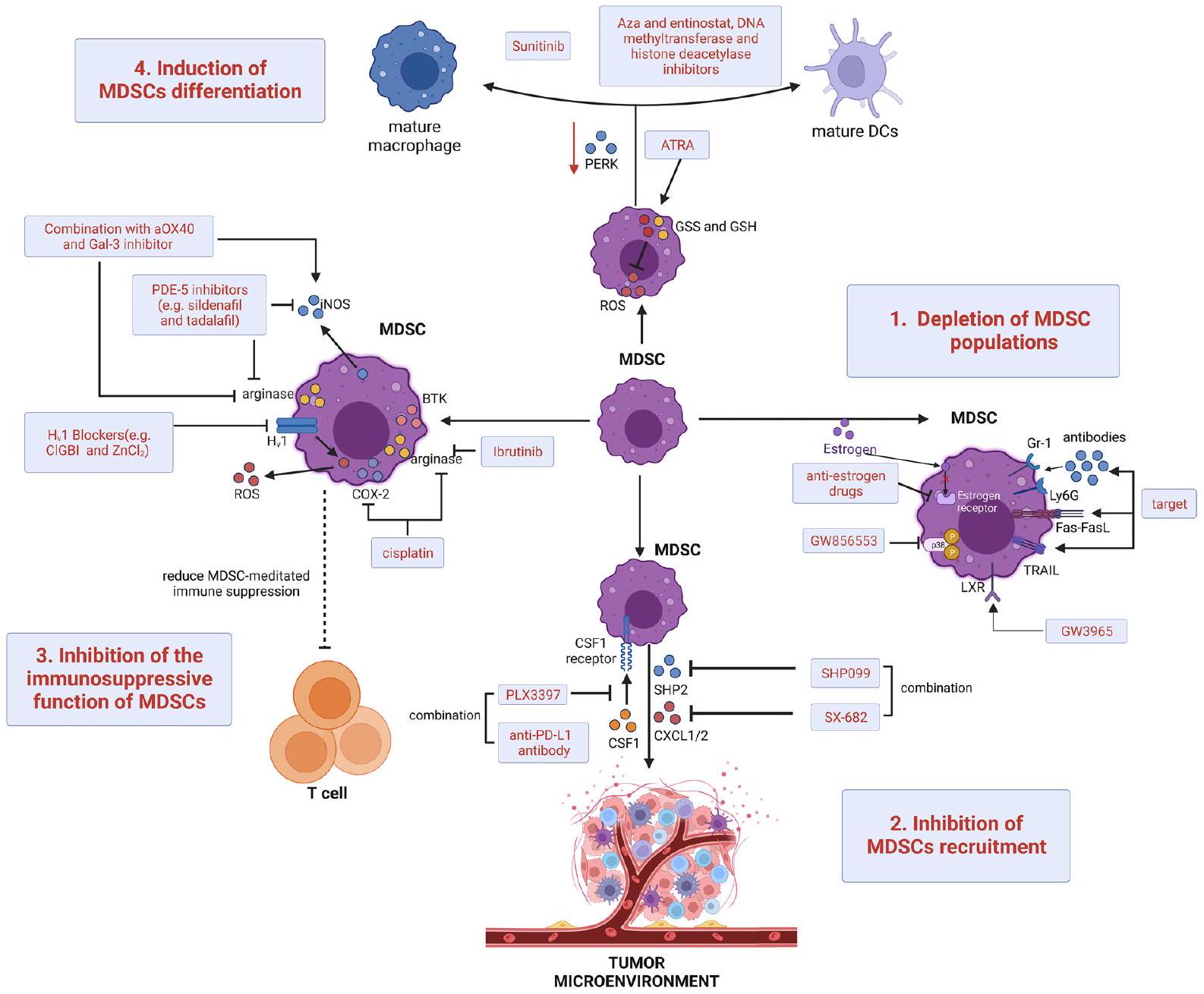
لقد استكشفت الدراسات المكثفة طرقًا جديدة لاستهداف وتقليل خلايا المناعة المثبطة المشتقة من النخاع (MDSCs). في نماذج الفئران، يمكن أن يتم تحفيز موت الخلايا المبرمج في MDSCs من خلال استهداف الأجسام المضادة لعلامات السطح Gr-1 أو Ly6G، مما يؤدي إلى تحفيز Fas-FasL أو استهداف مستقبل TNF المرتبط بموت الخلايا المبرمج (TRAIL) [186-188]. وبالمثل، في نماذج الأورام الخبيثة لدى الإناث، يتسبب الإستروجين وإشارات مستقبل ألفا في تضخيم MDSCs وزيادة النشاط المناعي المثبط من خلال تغيير إشارات pSTAT3، مما يدعم فكرة أن الأدوية المضادة للإستروجين الأكثر تحديدًا يمكن أن تكمل العلاجات المناعية الناشئة [189]. بالإضافة إلى ذلك، فإن استخدام مثبط p38 MAPK GW856553 في نماذج HCC الفئران مع تليف الكبد قد منع بشكل فعال إعادة برمجة المحسن لتطوير M-MDSCs والإ immunosuppression الناتج عن خلايا الكبد النجمية المنشطة [190]. علاوة على ذلك، فإن تنشيط مستقبل الكبد-X العلاجي وهدفه النسخي بروتين الأبوليبوبروتين E من خلال تطبيق المحفز الانتقائي GW3965 قد منع مباشرة بقاء MDSCs في نماذج الفئران وفي المرضى الذين تم علاجهم في تجربة تصعيد الجرعة الأولى في البشر [191]. بعض العلاجات الكيميائية السامة، مثل الكاربوبلاتين والباكليتاكسيل، يمكن أن تقلل أيضًا من عدد MDSCs الدائرة في مرضى الأورام [192]، ويعتقد الآن أن بعضها يدعم التأثيرات المضادة للأورام لبعض الأنظمة. بالإضافة إلى ذلك، أظهرت مجموعة من العلاج الكيميائي مع علاجات أخرى لاستهداف MDSCs تأثيرات مضادة للأورام في الدراسات ما قبل السريرية والسريرية (الجدول 1).
تقليل تجنيد خلايا المناعة المثبطة للأورام (MDSCs) إلى مواقع الأورام هو أحد الأساليب العلاجية الرئيسية لإعادة تأسيس البيئة الدقيقة المناعية وتحسين نجاح العلاج المناعي. يعتبر حجب الكيموكينات وتفاعلاتها مع الروابط هدفًا فعالًا لتقليل نقل خلايا MDSCs. مثبطات بروتين الفوسفاتاز 2 المحتوية على تماثل Src (SHP2) (مثل SHP099) لها تأثيرات مضادة للأورام على نماذج مع طفرات KRAS وEGFR في سرطان الرئة غير صغير الخلايا. ومع ذلك، فإن كل من مثبطات SHP2 ومثبطات مسار RAS/ERK الأخرى تسبب تجنيد خلايا MDSCs عن طريق تحفيز إنتاج الروابط CXCR2 المعتمدة على NF-kB. لذلك، يجب دمج مثبطات SHP2 مع مثبطات CXCR1/2 (مثل SX682) لتحسين البقاء في نماذج متعددة من سرطان الرئة غير صغير الخلايا. العلاج المساعد بالإبيجينيتك باستخدام جرعات منخفضة من ميثيل ترانسفيراز الحمض النووي ومثبطات هيستون ديأسيتيلز مثل إنتينوسات و5-أزاسيتيدين (Aza) بعد استئصال الورم الأساسي في نماذج الفئران، تم تثبيط انتشار خلايا الورم عن طريق تقليل نقل خلايا MDSCs من خلال تقليل CCR2 وCXCR2 وتشجيع تمايز خلايا MDSCs. تم تثبيط نقل PMN-MDSCs بشكل كبير في نماذج الأورام الفئران بعد تطبيق
| هدف | تدخل | الشروط | مرحلة | حالة | عدد المسجلين | NCT | ||
| مضاد مستقبلات الأدينوزين A2B؛ العلاج الكيميائي | PBF-1129 ونيفولوماب | سرطان الرئة غير صغير الخلايا | 。 | التوظيف | 30 | NCT05234307 | ||
| العلاج الكيميائي؛ العلاج الكهربائي الحراري |
|
سرطان الثدي | أنا | التوظيف | ٤٨ | NCT04796220 | ||
| العلاج الكيميائي | تادالافيل | ورم دبقي | أنا | مكتمل | 18 | NCT04757662 | ||
| العلاج الهرموني؛ العلاج الكيميائي | أبيماسيكلب؛ فلفيسترون؛ مثبطات الأروماتاز | سرطان الثدي | || | نشط، لا يجند | ١٨ | NCT04352777 | ||
| مضاد PD-1؛ العلاج الكيميائي | نيفولوماب؛ نيفولوماب وجيمسيتابين | سرطان الرئة غير صغير الخلايا | الثانية | مُنهى | ٣ | NCT03302247 | ||
| العلاج الكيميائي | فلودارابين؛ بوسولفان؛ ميثوتريكسات | لوكيميا | أنا | نشط، لا يتم التوظيف | 20 | NCT02916979 | ||
| مضاد CXCR1/2؛ مضاد PD-1 | إس إكس-682 وبمبروليزوماب | الميلانوما | أنا | التوظيف | 77 | NCT03161431 | ||
| مثبط PDE-5 | تادالافيل | سرطان الخلايا الحرشفية في الرأس والعنق | الثاني | مكتمل | 40 | NCT01697800 | ||
| مضاد PD-1؛ ATRA | بمبروليزوماب مع أترالين | الميلانوما | |/|| | نشط، لا يتم التوظيف | 26 | NCT03200847 | ||
| مضاد PD-1؛ ATRA | أتراترا وأتيزوليزوماب | سرطان الرئة غير صغير الخلايا | أنا | التوظيف | 18 | NCT04919369 | ||
| أATRA؛ مضاد-CTLA-4 | أترا؛ إيبيلوماب | الميلانوما | الثانية | نشط، لا يجند | 10 | NCT02403778 | ||
| مضاد مستقبلات H2 | رانيتيدين | سرطان | الرابع | مكتمل | 30 | NCT03145012 | ||
| منشط TLR9؛ مضاد PD-1 | سي إم بي-001 ونيفولوماب | الميلانوما؛ سرطان الغدد اللمفاوية | الثاني | نشط، لا يتم التوظيف | ٣٤ | NCT03618641 | ||
| العلاج الكيميائي؛ مضاد VEGF | كابيسيتابين؛ بيفاسيزوماب | الورم الدبقي المتكرر | أنا | نشط، لا يجند | 12 | NCT02669173 |
ويقلل من قمع المناعة الذي تسببه M-MDSCs، مما يزيد من تجنيد خلايا CD8 + T. في نماذج الفئران، تم تثبيط قنوات البروتون المعتمدة على الجهد (Hv1) بواسطة 5-كلورو-2-غوانيدينوبنزيميدازول أو
استراتيجية علاجية نهائية لإعادة نشاط خلايا T ونجاح العلاج المناعي هي تحفيز خلايا MDSCs للتمايز إلى خلايا نقي غير مثبطة ناضجة. على الرغم من أن التنشيط المعتدل لإشارات استجابة البروتين غير المطوي (UPR) يساعد خلايا المناعة على التمايز والوظيفة بشكل فسيولوجي، فإن بدء UPR بشكل مستمر وغير تكيفي يسهل نقص المناعة. إن زيادة إشارات كيناز الشبكة الإندوبلازمية الشبيهة بـ PKR هي سمة مميزة لـ UPR في MDSCs لنموذج الفئران الحاملة للأورام، حيث يؤدي حذفها إلى تحويل MDSCs إلى خلايا تنشط مناعة خلايا T من النوع CD8 + في أسرة الأورام. وقد وُجد أن حمض الريتينويك الكلي (ATRA)، وهو مشتق من فيتامين A، نشط للغاية ضد MDSCs. إن إعطاء ATRA في الجسم الحي يحفز MDSCs من الفئران الحاملة للأورام للتمايز إلى خلايا نقي ناضجة. يتضمن تمايز MDSCs المستحث بواسطة ATRA زيادة تعبير إنزيم غلوتاثيون سينثاز والغلوتاثيون في MDSCs، مما يؤدي إلى تحييد ROS ودفع التمايز النقي. في نماذج سرطان الثدي قبل السريرية، وُجد أن ATRA يعزز فعالية العلاج المضاد لتكوين الأوعية الدموية لسرطان الثدي من خلال استنفاد MDSCs. بالإضافة إلى ذلك، من خلال تعزيز تعبير جينات التمايز في PMN-MDSCs البريتونية، يعزز مثبط كيناز التيروزين السونيتينيب تمايز MDSCs إلى MDSCs متعددة النوى الناضجة في نموذج الفئران لبطانة الرحم. أخيرًا، تم استخدام العلاج الجيني المساعد بجرعة منخفضة من Aza، وإنفينوستات، ومثبطات ميثيل ترانسفيراز الحمض النووي ومزيلات الأسيتيل من الهيستون في نماذج الفئران لمنع انتشار خلايا الورم من خلال تعطيل البيئة الدقيقة السابقة للنقائل من خلال تعزيز تمايز MDSCs إلى
ظاهرة شبيهة بالبلاعم الميزنشيمية [197]. لتحفيز تمايز خلايا MDSCs، تركيزات عالية من البلازما (
الخاتمة وآفاق المستقبل
الأورام. يجب أن تساعد التقدمات في هذا المجال في تبرير تصميم استراتيجيات جديدة ضد MDSCs لتعزيز الاستجابة السريرية للعلاجات المناعية الحالية وتحسين توقعات المرضى. يجب أن توضح الدراسات المستقبلية مدى فعالية وفائدة البقاء التي يمكن أن توفرها العلاجات المركبة لمرضى السرطان. هناك حاجة إلى تجربة سريرية واسعة النطاق ودراسة قبل سريرية متعمقة لتأكيد هذه الأسئلة.
الشكر والتقدير
مساهمات المؤلفين
التمويل
توفر البيانات والمواد
الإعلانات
موافقة الأخلاقيات والموافقة على المشاركة
الموافقة على النشر
المصالح المتنافسة
تم النشر عبر الإنترنت: 12 أبريل 2024
References
- Gabrilovich DI, Bronte V, Chen SH, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67(1):425. https:// doi.org/10.1158/0008-5472.
- Bennett JA, Rao VS, Mitchell MS. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells. Proc Natl Acad Sci U S A. 1978;75(10):5142-4. https://doi.org/10.1073/pnas.75.10.5142.
- Bronte V , Brandau
, et al. Recommendations for myeloidderived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. https://doi.org/10.1038/ncomms12150. - Cassetta L, Baekkevold ES, Brandau S, et al. Deciphering myeloidderived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother. 2019;68(4):68797. https://doi.org/10.1007/s00262-019-02302-2.
- Park JA, Wang L, Cheung NV. Modulating tumor infiltrating myeloid cells to enhance bispecific antibody-driven T cell infiltration and antitumor response. J Hematol Oncol. 2021;14(1):142. https://doi.org/10. 1186/s13045-021-01156-5.
- Bronte V, Serafini P, De Santo C, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170(1):2708. https://doi.org/10.4049/jimmunol.170.1.270.
- Kusmartsev S, Nefedova Y, Yoder D, et al. Antigen-specific inhibition of CD8+T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989-99. https://doi. org/10.4049/jimmunol.172.2.989.
- Mazzoni A, Bronte V, Visintin A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168(2):689-95. https://doi.org/10.4049/jimmunol.168.2.689.
- Groth C, Hu X, Weber R, et al. Immunosuppression mediated by mye-loid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120(1):16-25. https://doi.org/10.1038/s41416-018-0333-1.
- Kujawski M, Kortylewski M, Lee H, et al. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118(10):3367-77. https://doi.org/10.1172/JCl35213.
- Murdoch
, Muthana , Coffelt , et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618-31. https://doi.org/10.1038/nrc2444. - Wang D, Sun H, Wei J, et al. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 2017;77(13):3655-65. https://doi.org/10.1158/0008-5472.CAN-16-3199.
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565-70. https://doi.org/10.1126/science. 1203486.
- Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934-49. https://doi.org/10.1016/j.cell.2017.09.028.
- Tang S, Ning Q, Yang L, et al. Mechanisms of immune escape in the cancer immune cycle. Int Immunopharmacol. 2020;86: 106700. https:// doi.org/10.1016/j.intimp.2020.106700.
- FuT, Dai
, et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol. 2021;14(1):98. https://doi.org/10.1186/ s13045-021-01103-4. - de Coana YP, Wolodarski M, Poschke I, et al. Ipilimumab treatment decreases monocytic MDSCs and increases CD8 effector memory T cells in long-term survivors with advanced melanoma. Oncotarget. 2017;8(13):21539-53. https://doi.org/10.18632/oncotarget. 15368.
- Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234-43. https://doi.org/10.1053/j.gastro.2008.03.020.
- Casbon AJ, Reynaud D, Park C, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A. 2015;112(6):E566-75. https://doi.org/10.1073/pnas.1424927112.
- Idorn M, Kollgaard T, Kongsted P, et al. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castrationresistant metastatic prostate cancer. Cancer Immunol Immunother. 2014;63(11):1177-87. https://doi.org/10.1007/s00262-014-1591-2.
- Henick BS, Villarroel-Espindola F, Datar I, et al. Quantitative tissue analysis and role of myeloid cells in non-small cell lung cancer. J Immunother Cancer. 2022;10:7. https://doi.org/10.1136/jitc-2022-005025.
- Okla K, Wertel I, Wawruszak A, et al. Blood-based analyses of cancer: Circulating myeloid-derived suppressor cells – is a new era coming? Crit Rev Clin Lab Sci. 2018;55(6):376-407. https://doi.org/10.1080/10408363. 2018.1477729.
- Yanez A, Ng MY, Hassanzadeh-Kiabi N, et al. IRF8 acts in lineagecommitted rather than oligopotent progenitors to control neutrophil vs monocyte production. Blood. 2015;125(9):1452-9. https://doi.org/10. 1182/blood-2014-09-600833.
- Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. 2015;98(6):913-22. https://doi.org/10.1189/jlb.4RI0515-204R.
- Karin N. The development and homing of myeloid-derived suppressor cells: from a two-stage model to a multistep narrative. Front Immunol. 2020;11: 557586. https://doi.org/10.3389/fimmu.2020.557586.
- Wu Y, Yi M, Niu M, et al. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. 2022;21(1):184. https://doi.org/10.1186/s12943-022-01657-y.
- Pal S, Dey D, Chakraborty BC, et al. Diverse facets of MDSC in different phases of chronic HBV infection: Impact on HBV-specific T-cell response
and homing. Hepatology. 2022;76(3):759-74. https://doi.org/10.1002/ hep.32331. - Hanson EM, Clements VK, Sinha P, et al. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol. 2009;183(2):937-44. https://doi.org/10.4049/jimmunol. 08042 53.
- Parker KH, Sinha P, Horn LA, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014;74(20):5723-33. https://doi.org/10.1158/0008-5472.CAN-13-2347.
- Ku AW, Muhitch JB, Powers CA, et al. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. Elife. 2016;510:17875.
- Liu Y, Wei J, Guo G, et al. Norepinephrine-induced myeloid-derived suppressor cells block T-cell responses via generation of reactive oxygen species. Immunopharmacol Immunotoxicol. 2015;37(4):359-65. https:// doi.org/10.3109/08923973.2015.1059442.
- Kusmartsev S, Eruslanov E, Kubler H, et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol. 2008;181(1):346-53. https://doi.org/10.4049/jimmunol.181.1.346.
- Huang Y, Chen X, Dikov MM, et al. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood. 2007;110(2):624-31. https://doi.org/10.1182/ blood-2007-01-065714.
- Niu M, Yi M, Wu Y, et al. Synergistic efficacy of simultaneous anti-TGF-beta/VEGF bispecific antibody and PD-1 blockade in cancer therapy. J Hematol Oncol. 2023;16(1):94. https://doi.org/10.1186/ s13045-023-01487-5.
- Corzo CA, Cotter MJ, Cheng P, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693-701. https://doi.org/10.4049/jimmunol. 0900092.
- Raber PL, Thevenot P, Sierra R, et al. Subpopulations of myeloid-derived suppressor cells impair
cell responses through independent nitric oxide-related pathways. Int J Cancer. 2014;134(12):2853-64. https://doi. org/10.1002/ijc.28622. - LuT, Ramakrishnan R, Altiok S, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121(10):4015-29. https://doi.org/10.1172/JCl45862.
- Cartwright ANR, Suo S, Badrinath S, et al. Immunosuppressive myeloid cells induce nitric oxide-dependent DNA damage and p53 pathway activation in CD8(+) T Cells. Cancer Immunol Res. 2021;9(4):470-85. https://doi.org/10.1158/2326-6066.CIR-20-0085.
- Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180-91. https://doi.org/10.1111/j. 1600-065X.2008.00608.x.
- Cimen Bozkus C, Elzey BD, Crist SA, et al. Expression of cationic amino acid transporter 2 is required for myeloid-derived suppressor cellmediated control of T cell immunity. J Immunol. 2015;195(11):5237-50. https://doi.org/10.4049/jimmunol. 1500959.
- Rodriguez PC, Ernstoff MS, Hernandez C, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69(4):1553-60. https://doi.org/10.1158/0008-5472.CAN-08-1921.
- Zea AH, Rodriguez PC, Culotta KS, et al. L-Arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol. 2004;232(1-2):21-31. https://doi.org/10.1016/j.cellimm.2005. 01.004.
- Srivastava MK, Sinha P, Clements VK, et al. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68-77. https://doi.org/10.1158/0008-5472.CAN-09-2587.
- Yu J, Du W, Yan F, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783-97. https://doi.org/10.4049/jimmunol.1201449.
- Schafer CC, Wang Y, Hough KP, et al. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget. 2016;7(46):75407-24. https://doi.org/10.18632/oncotarget.12249.
- Liu
, Yu X, Xu L, et al. Current insight into the regulation of PD-L1 in cancer. Exp Hematol Oncol. 2022;11(1):44. https://doi.org/10.1186/ s40164-022-00297-8. - Kim SH, Li M, Trousil S, et al. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J Invest Dermatol. 2017;137(8):1740-8. https://doi.org/10. 1016/j.jid.2017.03.033.
- Wang L, Jia B, Claxton DF, et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology. 2018;7(9): e1469594. https://doi.org/10.1080/2162402X. 2018.1469594.
- Deng J, Li J, Sarde A, et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol Res. 2019;7(7):1079-90. https:// doi.org/10.1158/2326-6066.CIR-18-0507.
- Sakuishi K, Jayaraman P, Behar SM, et al. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011;32(8):345-9. https://doi.org/10.1016/j.it.2011.05.003.
- Zhang CX, Huang DJ, Baloche V, et al. Correction to: Galectin-9 promotes a suppressive microenvironment in human cancer by enhancing STING degradation. Oncogenesis. 2022;11(1):5. https://doi.org/10.1038/ s41389-021-00375-2.
- Kong Y, Zhu L, Schell TD, et al. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin Cancer Res. 2016;22(12):3057-66. https:// doi.org/10.1158/1078-0432.CCR-15-2626.
- Wu L, Mao L, Liu JF, et al. Blockade of TIGIT/CD155 signaling reverses t-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res. 2019;7(10):1700-13. https://doi.org/10.1158/2326-6066.CIR-18-0725.
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253-68. https://doi.org/10.1038/nri3175.
- Sinha P, Clements VK, Fulton AM, et al. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67(9):4507-13. https://doi.org/10.1158/0008-5472. CAN-06-4174.
- Greifenberg V, Ribechini E, Rossner S, et al. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol. 2009;39(10):2865-76. https://doi.org/10. 1002/eji. 200939486.
- Poschke I, Mao Y, Adamson L, et al. Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol Immunother. 2012;61(6):827-38. https://doi.org/10.1007/s00262-011-1143-y.
- Hu CE, Gan J, Zhang RD, et al. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. 2011;46(2):156-64. https://doi.org/10.3109/00365521.2010.516450.
- Ugolini A, Tyurin VA, Tyurina YY, et al. Polymorphonuclear myeloidderived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight. 2020. https://doi.org/10.1172/jci.insight. 138581.
- Li H, Han Y, Guo Q, et al. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182(1):240-9. https://doi.org/10.4049/jimmunol.182.1. 240.
- Di Pace AL, Tumino N, Besi F, et al. Characterization of Human NK CellDerived Exosomes: Role of DNAM1 Receptor In Exosome-Mediated Cytotoxicity Against Tumor. Cancers (Basel). 2020;12:3. https://doi.org/ 10.3390/cancers12030661.
- Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799-807. https://doi. org/10.1002/hep.23054.
- Vaknin I, Blinder L, Wang L, et al. A common pathway mediated through Toll-like receptors leads to T- and natural killer-cell immunosuppression. Blood. 2008;111(3):1437-47. https://doi.org/10.1182/ blood-2007-07-100404.
- Zhang J, Han X, Hu X, et al. IDO1 impairs NK cell cytotoxicity by decreasing NKG2D/NKG2DLs via promoting miR-18a. Mol Immunol. 2018;103:144-55. https://doi.org/10.1016/j.molimm.2018.09.011.
- Della Chiesa M, Carlomagno S, Frumento G, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. 2006;108(13):4118-25. https://doi.org/10.1182/blood-2006-03-006700.
- Sui Q, Zhang J, Sun X, et al. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J Immunol. 2014;193(4):2016-23. https://doi.org/10. 4049/jimmunol. 1302389.
- Tie Y, Tang F, Wei YQ, et al. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15(1):61. https://doi.org/10.1186/s13045-022-01282-8.
- Franklin RA, Liao W, Sarkar A, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921-5. https://doi.org/10.1126/science.1252510.
- Wang Y, Liu H, Zhang Z, et al. G-MDSC-derived exosomes mediate the differentiation of M-MDSC into M2 macrophages promoting colitis-tocancer transition. J Immunother Cancer. 2023;11(6):45. https://doi.org/ 10.1136/jitc-2022-006166.
- DeNardo DG, Barreto JB, Andreu P, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91-102. https://doi. org/10.1016/j.ccr.2009.06.018.
- Thibodeau J, Bourgeois-Daigneault MC, Huppe G, et al. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol. 2008;38(5):1225-30. https://doi.org/10. 1002/eji.200737902.
- Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):112331. https://doi.org/10.1158/0008-5472.CAN-05-1299.
- Serafini P, Mgebroff S, Noonan K, et al. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68(13):5439-49. https://doi.org/10.1158/0008-5472.CAN-07-6621.
- Pan PY, Ma G, Weber KJ, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70(1):99-108. https://doi.org/10.1158/0008-5472.CAN-09-1882.
- Schlecker E, Stojanovic A, Eisen C, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol. 2012;189(12):5602-11. https://doi.org/10.4049/jimmunol. 1201018.
- Fujimura T, Ring S, Umansky V, et al. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol. 2012;132(4):1239-46. https://doi.org/10.1038/jid.2011. 416.
- Yang
, Lin , Shi , et al. FAP promotes immunosuppression by cancerassociated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76(14):4124-35. https://doi.org/10.1158/ 0008-5472.CAN-15-2973. - Xiang H, Ramil CP, Hai J, et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol Res. 2020;8(4):436-50. https://doi.org/10.1158/2326-6066.CIR-19-0507.
- Liang T, Tao T, Wu K, et al. Cancer-associated fibroblast-induced remodeling of tumor microenvironment in recurrent bladder cancer. Adv Sci (Weinh). 2023;10(31): e2303230. https://doi.org/10.1002/advs. 20230 3230.
- Deng Y, Cheng J, Fu B, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene. 2017;36(8):1090-101. https://doi.org/10.1038/onc.2016.273.
- Zhu GQ, Tang Z, Huang R, et al. CD36(+) cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023;9(1):25. https://doi.org/10.1038/s41421-023-00529-z.
- Zhao Q, Huang L, Qin G, et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021;518:35-48. https://doi.org/10.1016/j.canlet.2021.06.009.
- Lin
, Cai , Chen , et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology. 2022;75(1):28-42. https://doi.org/10.1002/ hep.32099. - Lideikaite A, Mozuraitiene J, Letautiene S. Analysis of prognostic factors for melanoma patients. Acta Med Litu. 2017;24(1):25-34. https://doi. org/10.6001/actamedica.v24i1.3460.
- Ross MI, Gershenwald JE. Evidence-based treatment of early-stage melanoma. J Surg Oncol. 2011;104(4):341-53. https://doi.org/10.1002/ jso. 21962.
- Jordan KR, Amaria RN, Ramirez O, et al. Myeloid-derived suppressor cells are associated with disease progression and decreased overall survival in advanced-stage melanoma patients. Cancer Immunol Immunother. 2013;62(11):1711-22. https://doi.org/10.1007/s00262-013-1475-x.
- Sade-Feldman M, Kanterman J, Klieger Y, et al. Clinical Significance of Circulating CD33+CD11b+HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin Cancer Res. 2016;22(23):5661-72. https://doi.org/10.1158/1078-0432.CCR-15-3104.
- Arkhypov I, Lasser S, Petrova V, et al. Myeloid cell modulation by tumorderived extracellular vesicles. Int J Mol Sci. 2020;21(17):34. https://doi. org/10.3390/ijms21176319.
- Arkhypov I, Ozbay Kurt FG, Bitsch R, et al. HSP90alpha induces immunosuppressive myeloid cells in melanoma via TLR4 signaling. J Immunother Cancer. 2022;10:9. https://doi.org/10.1136/jitc-2022-005551.
- Tengesdal IW, Menon DR, Osborne DG, et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc Natl Acad Sci U S A. 2021;118:10. https://doi.org/10.1073/pnas. 2000915118.
- Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397-411. https://doi.org/10.1038/ nri3452.
- Tengesdal IW, Dinarello A, Powers NE, et al. Tumor NLRP3-Derived IL1 beta Drives the IL-6/STAT3 Axis Resulting in Sustained MDSC-Mediated Immunosuppression. Front Immunol. 2021;12: 661323. https://doi.org/ 10.3389/fimmu.2021.661323.
- Weber R, Riester Z, Huser L, et al. IL-6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J Immunother Cancer. 2020;8:2. https://doi.org/10.1136/jitc-2020-000949.
- Chang AL, Miska J, Wainwright DA, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory t cells and myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5671-82. https://doi.org/10.1158/0008-5472.CAN-16-0144.
- Cheng Y, Ma XL, Wei YQ, et al. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim Biophys Acta Rev Cancer. 2019;1871(2):289-312. https://doi.org/10. 1016/j.bbcan.2019.01.005.
- Groth C, Arpinati L, Shaul ME, et al. Blocking migration of polymorphonuclear myeloid-derived suppressor cells inhibits mouse melanoma progression. Cancers (Basel). 2021;13:4. https://doi.org/10.3390/cance rs13040726.
- Toh B, Wang X, Keeble J, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9(9): e1001162. https:// doi.org/10.1371/journal.pbio. 1001162.
- Deng W, Fernandez A, McLaughlin SL, et al. Cell Communication Network Factor 4 (CCN4/WISP1) Shifts Melanoma Cells from a Fragile Proliferative State to a Resilient Metastatic State. Cell Mol Bioeng. 2020;13(1):45-60. https://doi.org/10.1007/s12195-019-00602-2.
- Fernandez A, Deng W, McLaughlin SL, et al. Cell Communication Network factor 4 promotes tumor-induced immunosuppression in melanoma. EMBO Rep. 2022;23(4): e54127. https://doi.org/10.15252/ embr. 202154127.
- Theivanthiran B, Evans KS, DeVito NC, et al. A tumor-intrinsic PD-L1/ NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J Clin Invest. 2020;130(5):2570-86. https://doi.org/10. 1172/JCl133055.
- Rawla P, Sunkara T, Muralidharan P, et al. Update in global trends and aetiology of hepatocellular carcinoma. Contemp Oncol (Pozn). 2018;22(3):141-50. https://doi.org/10.5114/wo.2018.78941.
- Li X, Xing YF, Lei AH, et al. Neutrophil count is associated with myeloid derived suppressor cell level and presents prognostic value of for
hepatocellular carcinoma patients. Oncotarget. 2017;8(15):24380-8. https://doi.org/10.18632/oncotarget. 15456. - Zhou J, Liu M, Sun H, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67(5):93144. https://doi.org/10.1136/gutjnl-2017-314032.
- Elwan N, Salem ML, Kobtan A, et al. High numbers of myeloid derived suppressor cells in peripheral blood and ascitic fluid of cirrhotic and HCC patients. Immunol Invest. 2018;47(2):169-80. https://doi.org/10. 1080/08820139.2017.1407787.
- Zeng X, Liao G, Li S, et al. Eliminating METTL1-mediated accumulation of PMN-MDSCs prevents hepatocellular carcinoma recurrence after radiofrequency ablation. Hepatology. 2023;77(4):1122-38. https://doi. org/10.1002/hep.32585.
- Simpson KD, Cross JV. MIF: metastasis/MDSC-inducing factor? Oncoimmunology. 2013;2(3): e23337. https://doi.org/10.4161/onci.23337.
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an irondependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-72. https://doi.org/10.1016/j.cell.2012.03.042.
- Conche C, Finkelmeier F, Pesic M, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023. https://doi.org/ 10.1136/gutjnl-2022-327909.
- He Q, Liu M, Huang W, et al. IL-1 beta-induced elevation of solute carrier family 7 member 11 promotes hepatocellular carcinoma metastasis through up-regulating programmed death ligand 1 and colony-stimulating factor 1. Hepatology. 2021;74(6):3174-93. https://doi.org/10. 1002/hep.32062.
- Wang W, Wu J, Ji M, et al. Exogenous interleukin-33 promotes hepatocellular carcinoma growth by remodelling the tumour microenvironment. J Transl Med. 2020;18(1):477. https://doi.org/10.1186/ s12967-020-02661-w.
- Xia S, Wu J, Zhou W, et al. SLC7A2 deficiency promotes hepatocellular carcinoma progression by enhancing recruitment of myeloid-derived suppressors cells. Cell Death Dis. 2021;12(6):570. https://doi.org/10. 1038/s41419-021-03853-y.
- Li YM, Liu ZY, Wang JC, et al. Receptor-Interacting Protein Kinase 3 Deficiency Recruits Myeloid-Derived Suppressor Cells to Hepatocellular Carcinoma Through the Chemokine (C-X-C Motif) Ligand 1-Chemokine (C-X-C Motif) Receptor 2 Axis. Hepatology. 2019;70(5):1564-81. https:// doi.org/10.1002/hep.30676.
- Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9(8):122135. https://doi.org/10.1089/ars.2007.1628.
- Chiu DK, Tse AP, Xu IM, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/ CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8(1):517. https://doi.org/10.1038/s41467-017-00530-7.
- Raychaudhuri D, Bhattacharya R, Sinha BP, et al. Lactate induces protumor reprogramming in intratumoral plasmacytoid dendritic cells. Front Immunol. 2019;10:1878. https://doi.org/10.3389/fimmu.2019. 01878.
- Dias AS, Almeida CR, Helguero LA, et al. Metabolic crosstalk in the breast cancer microenvironment. Eur J Cancer. 2019;121:154-71. https://doi.org/10.1016/j.ejca.2019.09.002.
- Wang N, Tan HY, Lu Y, et al. PIWIL1 governs the crosstalk of cancer cell metabolism and immunosuppressive microenvironment in hepatocellular carcinoma. Signal Transduct Target Ther. 2021;6(1):86. https://doi. org/10.1038/s41392-021-00485-8.
- Awuah PK, Monga SP. Cell cycle-related kinase links androgen receptor and beta-catenin signaling in hepatocellular carcinoma: why are men at a loss? Hepatology. 2012;55(3):970-3. https://doi.org/10.1002/hep. 24774.
- Wang SH, Yeh SH, Chen PJ. The driving circuit of HBx and androgen receptor in HBV-related hepatocarcinogenesis. Gut. 2014;63(11):1688-9. https://doi.org/10.1136/gutjnl-2013-306678.
- Feng H, Yu Z, Tian Y, et al. A CCRK-EZH2 epigenetic circuitry drives hepatocarcinogenesis and associates with tumor recurrence and poor survival of patients. J Hepatol. 2015;62(5):1100-11. https://doi.org/10. 1016/j.jhep.2014.11.040.
- Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209-49. https://doi.org/10. 3322/caac.21660.
- Miller LD, Chou JA, Black MA, et al. Immunogenic subtypes of breast cancer delineated by gene classifiers of immune responsiveness. Cancer Immunol Res. 2016;4(7):600-10. https://doi.org/10.1158/2326-6066. CIR-15-0149.
- Alshetaiwi H, Pervolarakis N, McIntyre LL, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol. 2020;5:44. https://doi.org/10.1126/sciim munol.aay6017.
- Zhang R, Dong M, Tu J, et al. PMN-MDSCs modulated by CCL20 from cancer cells promoted breast cancer cell stemness through CXCL2CXCR2 pathway. Signal Transduct Target Ther. 2023;8(1):97. https://doi. org/10.1038/s41392-023-01337-3.
- Peng D, Tanikawa T, Li W, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/ NOTCH Cross-talk signaling. Cancer Res. 2016;76(11):3156-65. https:// doi.org/10.1158/0008-5472.CAN-15-2528.
- Liu M, Wei F, Wang J, et al. Myeloid-derived suppressor cells regulate the immunosuppressive functions of PD-1(-)PD-L1(+) Bregs through PD-L1/PI3K/AKT/NF-kappaB axis in breast cancer. Cell Death Dis. 2021;12(5):465. https://doi.org/10.1038/s41419-021-03745-1.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74. https://doi.org/10.1016/j.cell.2011.02.013.
- Li W, Tanikawa T, Kryczek I, et al. Aerobic Glycolysis Controls MyeloidDerived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018;28(1):87-103. https://doi.org/10.1016/j.cmet.2018.04.022.
- Xiao P, Wan X, Cui B, et al. Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. Oncoimmunology. 2016;5(1): e1063772. https://doi.org/10. 1080/2162402X.2015.1063772.
- Trillo-Tinoco J, Sierra RA, Mohamed E, et al. AMPK Alpha-1 intrinsically regulates the function and differentiation of tumor myeloid-derived suppressor cells. Cancer Res. 2019;79(19):5034-47. https://doi.org/10. 1158/0008-5472.CAN-19-0880.
- Jiang M, Zhang W, Zhang R, et al. Cancer exosome-derived miR-9 and miR-181a promote the development of early-stage MDSCs via interfering with SOCS3 and PIAS3 respectively in breast cancer. Oncogene. 2020;39(24):4681-94. https://doi.org/10.1038/s41388-020-1322-4.
- Yu B, Luo F, Sun B, et al. KAT6A Acetylation of SMAD3 Regulates Myeloid-Derived Suppressor Cell Recruitment, Metastasis, and Immunotherapy in Triple-Negative Breast Cancer. Adv Sci (Weinh). 2021;8(20): e2100014. https://doi.org/10.1002/advs.202100014.
- Ruiz-Fernandez de Cordoba B, Moreno H, Valencia K, et al. Tumor ENPP1 (CD203a)/Haptoglobin Axis Exploits Myeloid-Derived Suppressor Cells to Promote Post-Radiotherapy Local Recurrence in Breast Cancer. Cancer Discov. 2022;12(5):1356-1377.
- Beer TM, Kwon ED, Drake CG, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castrationresistant prostate cancer. J Clin Oncol. 2017;35(1):40-7. https://doi.org/ 10.1200/JCO.2016.69.1584.
- Wang J, McGuire TR, Britton HC, et al. Lenalidomide and cyclophosphamide immunoregulation in patients with metastatic, castration-resistant prostate cancer. Clin Exp Metastasis. 2015;32(2):111-24. https://doi. org/10.1007/s10585-015-9696-3.
- Lu X, Horner JW, Paul E, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543(7647):728-32. https://doi.org/10.1038/nature21676.
- Calcinotto A, Spataro C, Zagato E, et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature. 2018;559(7714):3639. https://doi.org/10.1038/s41586-018-0266-0.
- Burke M, Choksawangkarn W, Edwards N, et al. Exosomes from myeloidderived suppressor cells carry biologically active proteins. J Proteome Res. 2014;13(2):836-43. https://doi.org/10.1021/pr400879c.
- Gao F, Xu Q, Tang Z, et al. Exosomes derived from myeloid-derived suppressor cells facilitate castration-resistant prostate cancer progression
via S100A9/circMID1/miR-506-3p/MID1. J Transl Med. 2022;20(1):346. https://doi.org/10.1186/s12967-022-03494-5. - Hossain DM, Pal SK, Moreira D, et al. TLR9-Targeted STAT3 silencing abrogates immunosuppressive activity of myeloid-derived suppressor cells from prostate cancer patients. Clin Cancer Res. 2015;21(16):377182. https://doi.org/10.1158/1078-0432.CCR-14-3145.
- Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11-22. https://doi. org/10.1016/j.ccr.2010.05.026.
- Zhao D, Cai L, Lu X, et al. Chromatin regulator CHD1 remodels the immunosuppressive tumor microenvironment in PTEN-deficient prostate cancer. Cancer Discov. 2020;10(9):1374-87. https://doi.org/10. 1158/2159-8290.CD-19-1352.
- Jachetti E, Rigoni A, Bongiovanni L, et al. Imatinib Spares cKit-expressing prostate neuroendocrine tumors, whereas kills seminal vesicle epithelial-stromal tumors by targeting PDGFR-beta. Mol Cancer Ther. 2017;16(2):365-75. https://doi.org/10.1158/1535-7163.MCT-16-0466.
- Pittoni P, Tripodo C, Piconese S, et al. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res. 2011;71(18):5987-97. https://doi.org/10.1158/0008-5472.CAN-11-1637.
- Jachetti E, Cancila V, Rigoni A, et al. Cross-talk between myeloid-derived suppressor cells and mast cells mediates tumor-specific immunosuppression in prostate cancer. Cancer Immunol Res. 2018;6(5):552-65. https://doi.org/10.1158/2326-6066.CIR-17-0385.
- Miret JJ, Kirschmeier P, Koyama S, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. J Immunother Cancer. 2019;7(1):32. https://doi.org/10.1186/s40425-019-0504-5.
- Wang Y, Schafer CC, Hough KP, et al. Myeloid-Derived Suppressor Cells Impair B Cell Responses in Lung Cancer through IL-7 and STAT5. J Immunol. 2018;201(1):278-95. https://doi.org/10.4049/jimmunol. 17010 69.
- Lee JK, Won C, Yi EH, et al. Signal transducer and activator of transcription 3 (Stat3) contributes to T-cell homeostasis by regulating prosurvival Bcl-2 family genes. Immunology. 2013;140(3):288-300. https:// doi.org/10.1111/imm. 12133.
- Li Y, Du H, Qin Y, et al. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007;67(18):8494-503. https://doi.org/10.1158/0008-5472. CAN-07-0647.
- Tao Q, Fujimoto J, Men T, et al. Identification of the retinoic acid-inducible Gprc5a as a new lung tumor suppressor gene. J Natl Cancer Inst. 2007;99(22):1668-82. https://doi.org/10.1093/jnci/djm208.
- Zhong S, Yin H, Liao Y, et al. Lung Tumor Suppressor GPRC5A Binds EGFR and Restrains Its Effector Signaling. Cancer Res. 2015;75(9):180114. https://doi.org/10.1158/0008-5472.CAN-14-2005.
- Jing B, Wang T, Sun B, et al. IL6/STAT3 signaling orchestrates premetastatic niche formation and immunosuppressive traits in lung. Cancer Res. 2020;80(4):784-97. https://doi.org/10.1158/0008-5472. CAN-19-2013.
- Wang
, Jing , et al. PTGES/PGE(2) signaling links immunosuppression and lung metastasis in Gprc5a-knockout mouse model. Oncogene. 2020;39(15):3179-94. https://doi.org/10.1038/s41388-020-1207-6. - Liu NN, Yi CX, Wei LQ, et al. The intratumor mycobiome promotes lung cancer progression via myeloid-derived suppressor cells. Cancer Cell. 2023;41(11):1927-44. https://doi.org/10.1016/j.ccell.2023.08.012.
- Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19(9):495-509. https://doi.org/10.1038/s41568-019-0179-8.
- Koyama S, Akbay EA, Li YY, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999-1008. https://doi.org/10.1158/0008-5472.CAN-15-1439.
- Li R, Salehi-Rad R, Crosson W, et al. Inhibition of granulocytic myeloid-derived suppressor cells overcomes resistance to immune checkpoint inhibition in LKB1-deficient non-small cell lung cancer. Cancer Res. 2021;81(12):3295-308. https://doi.org/10.1158/0008-5472. CAN-20-3564.
- Li Y, Qu P, Wu L, et al. Api6/AIM/Spalpha/CD5L overexpression in alveolar type II epithelial cells induces spontaneous lung adenocarcinoma. Cancer Res. 2011;71(16):5488-99. https://doi.org/10.1158/0008-5472. CAN-10-4225.
- Zhao S, Guo T, Li J, et al. Expression and prognostic value of GalNAc-T3 in patients with completely resected small (
) peripheral lung adenocarcinoma after IASLC/ATS/ERS classification. Onco Targets Ther. 2015;8:3143-52. https://doi.org/10.2147/OTT.S93486. - Park MS, Yang AY, Lee JE, et al. GALNT3 suppresses lung cancer by inhibiting myeloid-derived suppressor cell infiltration and angiogenesis in a TNFR and c-MET pathway-dependent manner. Cancer Lett. 2021;521:294-307. https://doi.org/10.1016/j.canlet.2021.08.015.
- Zhao T, Liu S, Hanna NH, et al. LAL deficiency induced myeloidderived suppressor cells as targets and biomarkers for lung cancer. J Immunother Cancer. 2023;11(3): e006272. https://doi.org/10.1136/ jitc-2022-006272.
- Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582-98. https://doi.org/10.1038/nrc.2016.73.
- Porembka MR, Mitchem JB, Belt BA, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61(9):1373-85. https://doi.org/10.1007/s00262-011-1178-0.
- Zhang J, Xu X, Shi M, et al. CD13(hi) Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology. 2017;6(2): e1258504. https://doi.org/10.1080/2162402X.2016.12585 04.
- Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518-27. https://doi.org/10.1158/0008-5472. CAN-07-0175.
- Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769-81. https://doi.org/10.1136/ gutjnl-2013-306271.
- Choueiry F, Torok M, Shakya R, et al. CD200 promotes immunosuppression in the pancreatic tumor microenvironment. J Immunother Cancer. 2020;8(1):10. https://doi.org/10.1136/jitc-2019-000189.
- Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318-48. https://doi.org/10. 3322/caac.21190.
- Yang
, Lu , Hang J, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res. 2020;8(11):1440-51. https:// doi.org/10.1158/2326-6066.CIR-20-0111. - Chun E, Lavoie S, Michaud M, et al. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. 2015;12(2):244-57. https://doi. org/10.1016/j.celrep.2015.06.024.
- Maisonneuve C, Tsang DKL, Foerster EG, et al. Nod1 promotes colorectal carcinogenesis by regulating the immunosuppressive functions of tumor-infiltrating myeloid cells. Cell Rep. 2021;34(4): 108677. https:// doi.org/10.1016/j.celrep.2020.108677.
- Wang Y, Yin K, Tian J, et al. Granulocytic myeloid-derived suppressor cells promote the stemness of colorectal cancer cells through exosomal S100A9. Adv Sci (Weinh). 2019;6(18):1901278. https://doi.org/10. 1002/advs. 201901278.
- Chen H, Pan Y, Zhou Q, et al. METTL3 Inhibits Antitumor Immunity by Targeting m(6)A-BHLHE41-CXCL1/CXCR2 Axis to Promote Colorectal Cancer. Gastroenterology. 2022;163(4):891-907. https://doi.org/10. 1053/j.gastro.2022.06.024.
- Du Z, Su J, Lin S, et al. Hydroxyphenylpyruvate Dioxygenase Is a Metabolic Immune Checkpoint for UTX-deficient Colorectal Cancer. Gastroenterology. 2023;164(7):1165-79. https://doi.org/10.1053/j.gastro.2023. 02.010.
- Zhang
, Zheng , Chen , et al. Gut fungi enhances immunosuppressive function of myeloid-derived suppressor cells by activating PKM2dependent glycolysis to promote colorectal tumorigenesis. Exp Hematol Oncol. 2022;11(1):88. https://doi.org/10.1186/s40164-022-00334-6. - Yu S, Ren X, Li L. Myeloid-derived suppressor cells in hematologic malignancies: two sides of the same coin. Exp Hematol Oncol. 2022;11(1):43. https://doi.org/10.1186/s40164-022-00296-9.
- Jitschin R, Saul D, Braun M, et al. CD33/CD3-bispecific T-cell engaging (BiTE(R)) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer. 2018;6(1):116. https://doi.org/ 10.1186/s40425-018-0432-9.
- Tohumeken S, Baur R, Bottcher M, et al. Palmitoylated proteins on AML-derived extracellular vesicles promote myeloid-derived suppressor cell differentiation via TLR2/Akt/mTOR Signaling. Cancer Res. 2020;80(17):3663-76. https://doi.org/10.1158/0008-5472.CAN-20-0024.
- Cai B, Liu Y, Chong Y, et al. IRAK1-regulated IFN-gamma signaling induces MDSC to facilitate immune evasion in FGFR1-driven hematological malignancies. Mol Cancer. 2021;20(1):165. https://doi.org/10. 1186/s12943-021-01460-1.
- Highfill SL, Cui Y, Giles AJ, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6(237):237-67. https://doi.org/10.1126/scitranslmed.3007974.
- Raychaudhuri B, Rayman P, Ireland J, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13(6):591-9. https://doi.org/10.1093/ neuonc/nor042.
- Gieryng A, Pszczolkowska D, Walentynowicz KA, et al. Immune microenvironment of gliomas. Lab Invest. 2017;97(5):498-518. https://doi.org/ 10.1038/labinvest.2017.19.
- Su MT, Kumata S, Endo S, et al. LILRB4 promotes tumor metastasis by regulating MDSCs and inhibiting miR-1 family miRNAs. Oncoimmunology. 2022;11(1):2060907. https://doi.org/10.1080/2162402X.2022.20609 07.
- Cheng
, Wang , Wang , et al. Tumor-associated myeloid cells in cancer immunotherapy. J Hematol Oncol. 2023;16(1):71. https://doi.org/10. 1186/s13045-023-01473-x. - Wang Y, Johnson KCC, Gatti-Mays ME, et al. Emerging strategies in targeting tumor-resident myeloid cells for cancer immunotherapy. J Hematol Oncol. 2022;15(1):118. https://doi.org/10.1186/ s13045-022-01335-y.
- Bronte V, Serafini P, Apolloni E, et al. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother. 2001;24(6):431-46. https://doi.org/10.1097/00002371-20011 1000-00001.
- Zheng
, et al. Cimetidine suppresses lung tumor growth in mice through proapoptosis of myeloid-derived suppressor cells. Mol Immunol. 2013;54(1):74-83. https://doi.org/10.1016/j.molimm.2012.10. 035. - Condamine T, Kumar V, Ramachandran IR, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124(6):2626-39. https://doi.org/10.1172/JCl74 056.
- Svoronos N, Perales-Puchalt A, Allegrezza MJ, et al. Tumor cellindependent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov. 2017;7(1):72-85. https://doi.org/10.1158/2159-8290.CD-16-0502.
- Liu M, Zhou J, Liu X, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut. 2020;69(2):365-79. https://doi.org/10.1136/ gutjnl-2018-317257.
- Tavazoie MF, Pollack I, Tanqueco R, et al. LXR/ApoE activation restricts innate immune suppression in cancer. Cell. 2018;172(4):825-40. https:// doi.org/10.1016/j.cell.2017.12.026.
- Welters MJ, van der Sluis TC, van Meir H, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med. 2016;8(334):334-52. https://doi.org/10.1126/ scitranslmed.aad8307.
- Homey B, Muller A, Zlotnik A. Chemokines: agents for the immunotherapy of cancer? Nat Rev Immunol. 2002;2(3):175-84. https://doi.org/ 10.1038/nri748.
- Fedele C, Li S, Teng KW, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. 2021. https://doi.org/10.1084/jem. 20201414.
- Quintana E, Schulze CJ, Myers DR, et al. Allosteric Inhibition of SHP2 stimulates antitumor immunity by transforming the
immunosuppressive environment. Cancer Res. 2020;80(13):2889-902. https://doi.org/10.1158/0008-5472.CAN-19-3038. - Tang KH, Li S, Khodadadi-Jamayran A, et al. Combined Inhibition of SHP2 and CXCR1/2 Promotes Antitumor T-cell Response in NSCLC. Cancer Discov. 2022;12(1):47-61. https://doi.org/10.1158/2159-8290. CD-21-0369.
- Lu Z, Zou J, Li S, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature. 2020;579(7798):284-90. https://doi. org/10.1038/s41586-020-2054-X.
- Sun L, Clavijo PE, Robbins Y, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight. 2019;4:7. https://doi.org/10.1172/jci.insight.126853.
- Tap WD, Wainberg ZA, Anthony SP, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373(5):428-37. https://doi.org/10.1056/NEJMoa1411366.
- Cannarile MA, Weisser M, Jacob W, et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5(1):53. https://doi.org/10.1186/s40425-017-0257-y.
- Mok S, Koya RC, Tsui C, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74(1):153-61. https://doi.org/10.1158/0008-5472.CAN-13-1816.
- Zhu Y, Yang J, Xu D, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68(9):1653-66. https://doi.org/10.1136/gutjnl-2019-318419.
- Kumar V, Donthireddy L, Marvel D, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell. 2017;32(5):654-68. https://doi.org/10.1016/j.ccell.2017.10.005.
- Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumourassociated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67(6):1112-23. https:// doi.org/10.1136/gutjnl-2017-313738.
- Hasnis E, Alishekevitz D, Gingis-Veltski S, et al. Anti-Bv8 antibody and metronomic gemcitabine improve pancreatic adenocarcinoma treatment outcome following weekly gemcitabine therapy. Neoplasia. 2014;16(6):501-10. https://doi.org/10.1016/j.neo.2014.05.011.
- Serafini P, Meckel K, Kelso M, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691-702. https://doi. org/10.1084/jem. 20061104.
- Lin S, Wang J, Wang L, et al. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res. 2017;7(1):41-52.
- Yu SJ, Ma C, Heinrich B, et al. Targeting the crosstalk between cytokineinduced killer cells and myeloid-derived suppressor cells in hepatocelIular carcinoma. J Hepatol. 2019;70(3):449-57. https://doi.org/10.1016/j. jhep.2018.10.040.
- Benguigui M, Vorontsova A, Timaner M, et al. Bv8 blockade sensitizes Anti-PD1 therapy resistant tumors. Front Immunol. 2022;13: 903591. https://doi.org/10.3389/fimmu.2022.903591.
- Sturgill ER, Rolig AS, Linch SN, et al. Galectin-3 inhibition with belapectin combined with anti-OX40 therapy reprograms the tumor microenvironment to favor anti-tumor immunity. Oncoimmunology. 2021;10(1):1892265. https://doi.org/10.1080/2162402X.2021.1892265.
- Alvear-Arias JJ, Carrillo C, Villar JP, et al. Expression of H(v)1 proton channels in myeloid-derived suppressor cells (MDSC) and its potential role in T cell regulation. Proc Natl Acad Sci U S A. 2022;119(15): e2104453119. https://doi.org/10.1073/pnas.2104453119.
- Ishfaq M , Pham T, Beaman C, et al. BTK inhibition reverses MDSCmediated immunosuppression and enhances response to Anti-PDL1 therapy in neuroblastoma. Cancers (Basel). 2021;13(4):817. https://doi. org/10.3390/cancers13040817.
- Weed DT, Vella JL, Reis IM, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21(1):39-48. https://doi.org/10.1158/1078-0432.CCR-14-1711.
- Noonan KA, Ghosh N, Rudraraju L, et al. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol Res. 2014;2(8):725-31. https://doi.org/10.1158/2326-6066.CIR-13-0213.
- Van Wigcheren GF, De Haas N, Mulder TA, et al. Cisplatin inhibits frequency and suppressive activity of monocytic myeloid-derived suppressor cells in cancer patients. Oncoimmunology. 2021;10(1):1935557. https://doi.org/10.1080/2162402X.2021.1935557.
- Yan D, Wang HW, Bowman RL, et al. STAT3 and STAT6 signaling pathways synergize to promote cathepsin secretion from macrophages via IRE1alpha activation. Cell Rep. 2016;16(11):2914-27. https://doi.org/10. 1016/j.celrep.2016.08.035.
- Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, et al. ER Stress Sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527-38. https://doi.org/10.1016/j.cell.2015.05. 025.
- Mohamed E, Sierra RA, Trillo-Tinoco J, et al. The Unfolded Protein Response Mediator PERK Governs Myeloid Cell-Driven Immunosuppression in Tumors through Inhibition of STING Signaling. Immunity. 2020;52(4):668-82. https://doi.org/10.1016/j.immuni.2020.03.004.
- Gabrilovich DI, Velders MP, Sotomayor EM, et al. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166(9):5398-406. https://doi.org/10.4049/jimmunol. 166.9.5398.
- Kusmartsev S, Cheng F, Yu B, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63(15):4441-9.
- Nefedova Y, Fishman M, Sherman S, et al. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67(22):11021-8. https://doi.org/10.1158/0008-5472. CAN-07-2593.
- Bauer R, Udonta F, Wroblewski M, et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. 2018;78(12):3220-32. https://doi.org/10.1158/0008-5472.CAN-17-3415.
- He Y, Hung SW, Liang B, et al. Receptor tyrosine kinase inhibitor sunitinib as novel immunotherapy to inhibit myeloid-derived suppressor cells for treatment of endometriosis. Front Immunol. 2021;12: 641206. https://doi.org/10.3389/fimmu.2021.641206.
- Mirza N, Fishman M, Fricke I, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66(18):9299-307. https://doi.org/10.1158/0008-5472. CAN-06-1690.
- Li MO, Wolf N, Raulet DH, et al. Innate immune cells in the tumor microenvironment. Cancer Cell. 2021;39(6):725-9. https://doi.org/10.1016/j. ccell.2021.05.016.
ملاحظة الناشر
- *المراسلة:
ليمين شيا
xialimin@tjh.tjmu.edu.cn
وينجيه هوانغ
huangwenjie@tjh.tjmu.edu.cn
مركز جراحة الكبد، مستشفى تونغجي، كلية تونغجي الطبية، جامعة هوازهونغ للعلوم والتكنولوجيا، مختبر هوبى الرئيسي لأمراض الكبد والبنكرياس والصفراوية، ووهان 430030، هوبى، الصين مركز أبحاث الطب السريري لجراحة الكبد في مقاطعة هوبى، المختبر الرئيسي لزراعة الأعضاء، وزارة التعليم ووزارة الصحة العامة، ووهان 430030، هوبى، الصين
DOI: https://doi.org/10.1186/s40164-024-00505-7
PMID: https://pubmed.ncbi.nlm.nih.gov/38609997
Publication Date: 2024-04-12
Myeloid-derived suppressor cells in cancer:
Check for updates therapeutic targets to overcome tumor immune evasion
Abstract
Paradoxically, tumor development and progression can be inhibited and promoted by the immune system. After three stages of immune editing, namely, elimination, homeostasis and escape, tumor cells are no longer restricted by immune surveillance and thus develop into clinical tumors. The mechanisms of immune escape include abnormalities in antitumor-associated immune cells, selection for immune resistance to tumor cells, impaired transport of T cells, and the formation of an immunosuppressive tumor microenvironment. A population of distinct immature myeloid cells, myeloid-derived suppressor cells (MDSCs), mediate immune escape primarily by exerting immunosuppressive effects and participating in the constitution of an immunosuppressive microtumor environment. Clinical trials have found that the levels of MDSCs in the peripheral blood of cancer patients are strongly correlated with tumor stage, metastasis and prognosis. Moreover, animal experiments have confirmed that elimination of MDSCs inhibits tumor growth and metastasis to some extent. Therefore, MDSCs may become the target of immunotherapy for many cancers, and eliminating MDSCs can help improve the response rate to cancer treatment and patient survival. However, a clear definition of MDSCs and the specific mechanism involved in immune escape are lacking. In this paper, we review the role of the MDSCs population in tumor development and the mechanisms involved in immune escape in different tumor contexts. In addition, we discuss the use of these cells as targets for tumor immunotherapy. This review not only contributes to a systematic and comprehensive understanding of the essential role of MDSCs in immune system reactions against tumors but also provides information to guide the development of cancer therapies targeting MDSCs.
Introduction
There are two primary types of MDSCs called polymorphonuclear-MDSCs (PMN-MDSCs) and monocytic-MDSCs (M-MDSCs). These cells resemble neutrophils and monocytes phenotypically and morphologically and thus, phenotype and morphology alone are not enough to identify MDSCs. Besides the two main types of cells, MDSCs include a small population of cells (less than
MDSCs mediate immune escape mainly by exerting immunosuppressive functions. Although MDSCs are
involved in suppressing various immune cells, their primary target is T cells. In contrast depletion of MDSCs using specific antibodies enhances
In recent years, research has revealed the clinical significance of MDSCs. Various studies have documented the proliferation of MDSCs in several types of human tumors, such as cutaneous melanoma [17], hepatocellular carcinoma [18], breast cancer [19], prostate cancer [20] and lung cancer [21]. In addition, a number of studies have shown that MDSCs are important prognostic biomarkers for cancer development and potential targets for anticancer therapy [22]. MDSCs can suppress the immune response and protect tumor cells from attack by the host immune system, resulting in tumor immune evasion. Targeting MDSCs to activate tumor immunity and reverse immune escape may be a viable option in tumor patients.
In this review, we discuss the biological role of MDSCs in tumor immune escape. In addition, we also review the specific mechanisms by which MDSCs are involved
in tumor immune escape in various types of tumors and discuss in detail the approaches used to target MDSCs for cancer treatment.
Differentiation and accumulation of MDSCs
The functions of MDSCs
Blockage of T-cell homing
downregulation of
Inhibition of T-cell function through oxidative stress
Consumption of amino acids needed for T-cell function
serotonin due to Trp depletion activates the aryl hydrocarbon receptor (AhR) to trigger IDO1 production and an anti-inflammatory reaction [45].
Expression of negative immune checkpoint molecules on MDSCs
Crosstalk between MDSCs and other immune cells involved in the TME
cells. DCs are the other major myeloid cells infiltrating into the TME. Although signals from the TME promote the influx of immature DCs, multiple factors, including adenosine accumulation, lactate accumulation, and hypoxic conditions, induce DC dysfunction [54]. In addition, it has been suggested that crosstalk between DCs and MDSCs may also be partly responsible for the decreased DC function. When bone marrow-derived MDSCs are co-cultured with DCs in vitro, the DC population decreases as the number of MDSCs increases [55, 56]. Studies of MDSCs in melanoma patients have shown that high frequencies of M-MDSCs impair DC maturation by reducing antigen uptake, preventing migration of immature and mature DCs, skewing DC cytokine production toward an anti-inflammatory phenotype, and blocking the ability of DCs to induce IFN
In addition to their ability to suppress immune T cells to destroy tumors, MDSCs may also be involved in tumor immune escape by stimulating other immune suppressor cells, such as macrophages and regulatory T (Treg) cells [67]. MDSCs not only are a source of tumor-associated macrophages but also may influence macrophage activation status, function, and polarization through association [68]. Driven by IL-6, the IL-6R/JAK/ STAT3 pathway is activated in PMN-MDSCs, which in turn causes the synthesis and secretion of exosomal miR-93-5p, driving differentiation of M-MDSCs into M2 macrophages [69]. In the tumor microenvironment,
The role of MDSCs in common tumors
Cutaneous melanoma
After their accumulation and activation in the bone marrow, MDSCs are attracted to the tumor site by a group of chemokines. Although CCL2, CCL3, and CCL4 are important for the recruitment of M-MDSCs through CCR2 [94], the ligands CXCR2, CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, and CXCL7 mainly mediate PMNMDSCs migration [95]. PMN-MDSCs infiltration within melanoma tissues is a significant contributor to primary melanoma growth and metastasis. PMN-MDSCs were shown to infiltrate primary melanoma and metastases via CXCL1/CXCR2 interactions [96]. In mice with melanoma, PMN-MDSCs produce hepatocyte growth factor and TGF-
melanoma by participating in the epithelial-mesenchymal transition [98]. In addition, when CCN4-knockout melanoma cells were implanted into immunocompetent mice, the infiltration of PMN-MDSCs was reduced. This was because local CNN4 expression inhibited the release of IFN from CD8 + T cells and increased tumor secretion of MDSCs-attracting chemokines such as CCL2 and CXCL1 [99]. In addition, in multiple preclinical tumor models as well as clinical specimens, activation of CD8 T cells in answer to PD-1 blockade triggers a PD-L1/ NLRP3 inflammatory signaling cascade that eventually causes PMN-MDSCs recruitment into melanoma tissue, resulting in immune suppression and thus immune escape [100]. PMN-MDSCs infiltration in tumors can be inhibited by NLRP3 blockade, significantly improving the efficacy of anti-PD-1 antibody immunotherapy [100].
Hepatocellular carcinoma
cirrhosis [101]. Clinical studies over the past decade have demonstrated the clinical significance of MDSCs in patients with hepatocellular carcinoma [102-104]. In patients with hepatocellular carcinoma, CD14(+)HLA-DR(low/-)MDSCs were markedly upregulated in the peripheral blood or tumor tissue. MDSCs from HCC were unable to stimulate allogeneic T-cell responses and had high arginase activity [18]. This finding suggested that hepatocellular carcinoma drives MDSCs to infiltrate, recruit and suppress effector T-cell function within the TME through various mechanisms (Fig. 4). Analysis of single-cell sequencing in mouse HCC models and human HCC organoids suggested that this difference may be due to METTL1-mediated accumulation of PMN-MDSCs following insufficient radio frequency treatment, which suppresses antitumor immunity and promotes HCC progression [105].
In the TME, oxidized LDL produced by dyslipidemic metabolism induces activation of the lipid peroxidation/p38 phosphorylation/CEBP axis within CD36 CAFs and ultimately promotes macrophage MIF secretion in a CD36-dependent manner [81]. M-MDSCs
stimulated by MIF are recruited to HCC tissues and enhance immunosuppression in the TME [81, 106]. Ferroptosis is an iron-dependent type of cell death that leads to cell membrane destruction through the accumulation of lipid peroxides [107]. In HCC, ferroptosis does not provide cell-autonomous tumor suppression but triggers tumor infiltration of MDSCs via HMGB1, thereby eliciting an adaptive immune response [108]. In addition, inflammatory factors in the TME play essential roles in MDSCs-mediated immune escape in HCC. It has been shown that IL-
Hypoxia is an important environmental factor in hepatocellular carcinoma [113]. In HCC, hypoxia induces the ectodomain, ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2), mainly through hypoxia-inducible factor-1 (HIF-1), leading to its overexpression [114]. ENTPD2 converts extracellular adenosine triphosphate (ATP) to 5′-Adenosine monophosphate (
It has been shown that metabolites produced by cancer cells can promote tumor development by modulating the functional phenotype of different immune cells [115, 116]. In hepatocellular carcinoma, Piwi Like RNA-Mediated Gene Silencing 1 (PIWIL1) increases fatty acid metabolism via mitochondrial fatty acid
Breast cancer
classifications have exhibited noteworthy variations in terms of distant m0etastasis-free survival [122]. During the development of breast cancer, cancer cells secrete a large number of cytokines that affect the differentiation of bone marrow cells and promote MDSCs development [19, 54]. In a mouse model of breast cancer, MDSCs features included several genes related to immunomodulation, such as arginase 2 and Cd84, and chemokine receptors (e.g., Ccr2 and Cxcr2), suggesting that MDSCs can be directed into tumor tissue by chemokines [123]. The combination of CD84 and JAML cell surface receptors on MDSCs with CD11b/Gr1 staining detects the presence of MDSCs in mouse tumor tissues or in humans with CD11b/CD14 or CD15 [123].
enhances the immunosuppressive ability of MDSCs [129]. Cancer cell-derived GM-CSF also induces transcription of the genes encoding AMP-activated protein kinase alpha (AMPK
Furthermore, the development of early myeloidderived suppressor cells (eMDSCs) in BC was induced by tumor exosome-derived miR-9 and miR-181a, which activated the Janus kinase (JAK)/STAT signaling pathway by targeting suppressor of cytokine signaling-3 (SOCS3) and protein inhibitor of activated STAT-3 (PIAS3), respectively [131]. Additionally, acetylation of Smad family member 3 (SMAD3) at K20 and K117 by lysine acetyltransferase 6A (KAT6A) enhanced SMAD3 binding to the oncogenic chromatin modifier TRIM24 and disrupted the binding of SMAD3 to the tumor suppressor TRIM33 [132]. In turn, this leads to the recruitment of the TRIM24-SMAD3 complex to chromatin through KAT6A acetylation of histone H3 lysine 23, which increases immune-associated cytokine expression and leads to MDSCs recruitment and immune escape in triple-negative breast cancer through immunosuppression [132]. Adenosinergic metabolites produced by high ectonucleotide pyrophosphatase/ phosphodiesterase 1 (Enpp1) expression in breast cancer enhance the expression of haptoglobin, which recruits PMN-MDSCs [133]. PMN-MDSCs infiltration causes immunosuppression, allowing Enpp1high circulating tumor cells to promote relapse through a self-seeding mechanism that causes locoregional failure [133].
Prostate cancer
Interleukin-23 (IL-23), generated by MDSCs, acts as a modulator of pro-tumor immunity and regulates prostate
cancer castration resistance by maintaining AR signaling [137]. IL-23 secreted by PMN-MDSCs is a major player in endocrine drug resistance in prostate cancer [137]. Therefore, direct inhibition of PMN-MDSCs can reverse ADT resistance in patients with advanced prostate cancer [137]. Additionally, MDSCs-derived exosomes in the tumor environment promote tumor progression by polarizing macrophages [138]. In PCa, exosomemediated S100A9 metastasis from MDSCs to PCa cells stimulates PCa cell proliferation, invasion and migration through upregulation of circMID1 (hsa_circ_0007718), which ultimately promotes CRPC progression [139].
Lung cancer
Recent studies have shown that within lung adenocarcinomas, the fungus Aspergillus sydowii, despite its low biomass, plays an important role in stimulating
the immunosuppressive TME, thereby promoting lung tumor progression and is associated with poor patient prognosis. This is mainly due to the fact that Aspergillus sydowii, which is enriched within lung adenocarcinomas, can also secrete IL-1
can be impeded by GALNT3, which blocks the selfrenewal of LC cells and leads to the downregulation of CXCL1 by decreasing the level of
Other cancers
microenvironment to a more immunosuppressive phenotype [169]. This contributes to the development of radioresistance in PDAC.
The main treatment for MDSCs
Intensive studies have explored new ways to target and deplete MDSCs. In mouse models, apoptosis in MDSCs can be mediated by targeting antibodies to the surface markers Gr-1 or Ly6G, inducing Fas-FasL or targeting the TNF-related apoptosis-induced ligand (TRAIL) receptor [186-188]. Similarly, in models of female malignancy estrogen and its receptor alpha signaling cause MDSCs amplification and enhanced immunosuppressive activity through altered pSTAT3 signaling, which supports the idea that more specific anti-estrogen drugs could complement emerging immunotherapies [189]. In addition, the use of the p38 MAPK inhibitor GW856553 in murine HCC models with cirrhosis effectively inhibited the enhancer reprogramming of M-MDSCs development and immunosuppression induced by activated hepatic stellate cells [190]. Furthermore, activation of the therapeutic liver-X nuclear receptor and its transcriptional target apolipoprotein E signaling by the application of the selective agonist GW3965 directly inhibited the survival of MDSCs in murine models and in patients treated in a first-in-human dose escalation phase 1 trial [191]. Some cytotoxic chemotherapies, such as carboplatin and paclitaxel, can also reduce the number of circulating MDSCs in tumor patients [192], some of which are now thought to support the antitumor effects of certain regimens. In addition, the combination of chemotherapy with other therapies to target MDSCs has demonstrated preclinical and clinical antitumor effects (Table 1).
Reducing the recruitment of MDSCs to tumor sites is one of the main therapeutic approaches to re-establish the immune microenvironment and improve the success of immunotherapy. Blocking chemokines and their interactions with ligands is an effective target for reducing the transport of MDSCs [193]. Src homology-2-containing protein tyrosine phosphatase 2 (SHP2) inhibitors (e.g., SHP099) have antitumor effects on Models with KRAS-mutant and EGFR-mutant NSCLC [194, 195]. However, both SHP2 inhibitors and other RAS/ERK pathway inhibitors cause the recruitment of MDSCs by inducing NF-kB-dependent CXCR2 ligand production [196]. Therefore, SHP2 inhibitors need to be combined with CXCR1/2 (e.g., SX682) inhibitors and improve survival in multiple NSCLC models. Adjuvant epigenetic treatment with low-dose DNA methyltransferase and the histone deacetylase inhibitors entinostat and 5-azacytidine (Aza) after primary tumor resection within mouse models inhibited tumor cell dissemination by reducing the transport of MDSCs by downregulating CCR2 and CXCR2 and by encouraging MDSCs differentiation [197]. PMN-MDSCs transport was significantly inhibited in mouse tumor models following the application of
| Target | Intervention | Conditions | Phase | Status | Number Enrolled | NCT | ||
| Adenosine A2B Receptor antagonist; Chemotherapy | PBF-1129 and Nivolumab | NSCLC | । | Recruiting | 30 | NCT05234307 | ||
| Chemotherapy; Electrothermal therapy |
|
Breast Cancer | I | Recruiting | 48 | NCT04796220 | ||
| Chemotherapy | Tadalafil | Astrocytoma | I | Completed | 18 | NCT04757662 | ||
| Endocrine Therapy; Chemotherapy | Abemaciclib; Fulvestrant; Aromatase Inhibitors | Breast Cancer | || | Active, not recruiting | 18 | NCT04352777 | ||
| Anti-PD-1; Chemotherapy | Nivolumab; Nivolumab and Gemcitabine | NSCLC | II | Terminated | 3 | NCT03302247 | ||
| Chemotherapy | Fludarabine; Busulfan; Methotrexate | Leukemia | I | Active, not recruiting | 20 | NCT02916979 | ||
| CXCR1/2 antagonist; Anti-PD-1 | SX-682 and Pembrolizumab | Melanoma | I | Recruiting | 77 | NCT03161431 | ||
| PDE-5 inhibitor | Tadalafil | Head and Neck Squamous Cell Carcinoma | II | Completed | 40 | NCT01697800 | ||
| Anti-PD-1; ATRA | Pembrolizumab with ATRA | Melanoma | |/|| | Active, not recruiting | 26 | NCT03200847 | ||
| Anti-PD-1; ATRA | ATRA and Atezolizumab | NSCLC | I | Recruiting | 18 | NCT04919369 | ||
| ATRA; Anti-CTLA-4 | ATRA; Ipilimumab | Melanoma | II | Active, not recruiting | 10 | NCT02403778 | ||
| H2 receptor antagonist | Ranitidine | Cancer | IV | Completed | 30 | NCT03145012 | ||
| TLR9 agonist; Anti-PD-1 | CMP-001 and Nivolumab | Melanoma; Lymph Node Cancer | II | Active, not recruiting | 34 | NCT03618641 | ||
| Chemotherapy; Anti-VEGF | Capecitabine; Bevacizumab | Recurrent Glioblastoma | I | Active, not recruiting | 12 | NCT02669173 |
and reduces M-MDSCs-mediated immune suppression, thereby increasing CD8 + T-cell recruitment [210]. In mouse models, inhibition of voltage-gated proton channels (Hv1) by 5-chloro-2-guanidinobenzimidazole or
A final therapeutic strategy to re-establish T-cell activity and immunotherapeutic success is the induction of MDSCs to differentiate into mature, non-suppressive myeloid cells. Although moderate activation of unfolded protein response (UPR)-related signaling helps immune cells differentiate and function physiologically, persistent and maladaptive initiation of UPR drivers facilitates immunodeficiency [216, 217]. Increased PKR-like endoplasmic reticulum kinase signaling is characteristic of the UPR in MDSCs of a tumor-bearing mouse model, whose deletion converts MDSCs into cells that activate CD8 + T-cell immunity in tumor beds [218]. All-trans retinoic acid (ATRA), a derivative of vitamin A, has been found to be highly active against MDSCs [219]. In vivo administration of ATRA stimulates MDSCs from tumorbearing mice to differentiate into mature myeloid cells [220]. ATRA-induced MDSCs differentiation involves ATRA specifically upregulating the expression of glutathione synthase and glutathione in MDSCs, thereby neutralizing ROS and driving myeloid differentiation [221]. In preclinical breast cancer models, ATRA was found to enhance the efficacy of antiangiogenic therapy for breast cancer by depleting MDSCs [222]. In addition, by promoting the expression of differentiation genes in peritoneal PMN-MDSCs, the receptor tyrosine kinase inhibitor sunitinib promotes the differentiation of MDSCs into mature polynuclear MDSCs in a mouse model of endometriosis [223]. Finally, adjuvant epigenetic therapy with low-dose Aza, entinostat, DNA methyltransferase and histone deacetylase inhibitors within mouse models was used to inhibit tumor cell dissemination by disrupting the premetastatic microenvironment through the promotion of MDSCs differentiation to a
more mesenchymal macrophage-like phenotype [197]. To induce differentiation of MDSCs, high plasma concentrations (
Conclusion and prospects
tumors. Advances in this area should help rationalize the design of new strategies against MDSCs to enhance the clinical response to current immunotherapies and improve patient prognosis. Future studies should clarify how much efficacy and survival benefit combination therapies can provide to cancer patients. A large-scale clinical trial and in-depth preclinical study are needed to confirm these questions.
Acknowledgements
Author contributions
Funding
Availability of data and materials
Declarations
Ethics approval and consent to participate
Consent for publication
Competing interests
Published online: 12 April 2024
References
- Gabrilovich DI, Bronte V, Chen SH, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67(1):425. https:// doi.org/10.1158/0008-5472.
- Bennett JA, Rao VS, Mitchell MS. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells. Proc Natl Acad Sci U S A. 1978;75(10):5142-4. https://doi.org/10.1073/pnas.75.10.5142.
- Bronte V , Brandau
, et al. Recommendations for myeloidderived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. https://doi.org/10.1038/ncomms12150. - Cassetta L, Baekkevold ES, Brandau S, et al. Deciphering myeloidderived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother. 2019;68(4):68797. https://doi.org/10.1007/s00262-019-02302-2.
- Park JA, Wang L, Cheung NV. Modulating tumor infiltrating myeloid cells to enhance bispecific antibody-driven T cell infiltration and antitumor response. J Hematol Oncol. 2021;14(1):142. https://doi.org/10. 1186/s13045-021-01156-5.
- Bronte V, Serafini P, De Santo C, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol. 2003;170(1):2708. https://doi.org/10.4049/jimmunol.170.1.270.
- Kusmartsev S, Nefedova Y, Yoder D, et al. Antigen-specific inhibition of CD8+T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989-99. https://doi. org/10.4049/jimmunol.172.2.989.
- Mazzoni A, Bronte V, Visintin A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168(2):689-95. https://doi.org/10.4049/jimmunol.168.2.689.
- Groth C, Hu X, Weber R, et al. Immunosuppression mediated by mye-loid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120(1):16-25. https://doi.org/10.1038/s41416-018-0333-1.
- Kujawski M, Kortylewski M, Lee H, et al. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118(10):3367-77. https://doi.org/10.1172/JCl35213.
- Murdoch
, Muthana , Coffelt , et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618-31. https://doi.org/10.1038/nrc2444. - Wang D, Sun H, Wei J, et al. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 2017;77(13):3655-65. https://doi.org/10.1158/0008-5472.CAN-16-3199.
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565-70. https://doi.org/10.1126/science. 1203486.
- Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934-49. https://doi.org/10.1016/j.cell.2017.09.028.
- Tang S, Ning Q, Yang L, et al. Mechanisms of immune escape in the cancer immune cycle. Int Immunopharmacol. 2020;86: 106700. https:// doi.org/10.1016/j.intimp.2020.106700.
- FuT, Dai
, et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol. 2021;14(1):98. https://doi.org/10.1186/ s13045-021-01103-4. - de Coana YP, Wolodarski M, Poschke I, et al. Ipilimumab treatment decreases monocytic MDSCs and increases CD8 effector memory T cells in long-term survivors with advanced melanoma. Oncotarget. 2017;8(13):21539-53. https://doi.org/10.18632/oncotarget. 15368.
- Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234-43. https://doi.org/10.1053/j.gastro.2008.03.020.
- Casbon AJ, Reynaud D, Park C, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci U S A. 2015;112(6):E566-75. https://doi.org/10.1073/pnas.1424927112.
- Idorn M, Kollgaard T, Kongsted P, et al. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castrationresistant metastatic prostate cancer. Cancer Immunol Immunother. 2014;63(11):1177-87. https://doi.org/10.1007/s00262-014-1591-2.
- Henick BS, Villarroel-Espindola F, Datar I, et al. Quantitative tissue analysis and role of myeloid cells in non-small cell lung cancer. J Immunother Cancer. 2022;10:7. https://doi.org/10.1136/jitc-2022-005025.
- Okla K, Wertel I, Wawruszak A, et al. Blood-based analyses of cancer: Circulating myeloid-derived suppressor cells – is a new era coming? Crit Rev Clin Lab Sci. 2018;55(6):376-407. https://doi.org/10.1080/10408363. 2018.1477729.
- Yanez A, Ng MY, Hassanzadeh-Kiabi N, et al. IRF8 acts in lineagecommitted rather than oligopotent progenitors to control neutrophil vs monocyte production. Blood. 2015;125(9):1452-9. https://doi.org/10. 1182/blood-2014-09-600833.
- Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. 2015;98(6):913-22. https://doi.org/10.1189/jlb.4RI0515-204R.
- Karin N. The development and homing of myeloid-derived suppressor cells: from a two-stage model to a multistep narrative. Front Immunol. 2020;11: 557586. https://doi.org/10.3389/fimmu.2020.557586.
- Wu Y, Yi M, Niu M, et al. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. 2022;21(1):184. https://doi.org/10.1186/s12943-022-01657-y.
- Pal S, Dey D, Chakraborty BC, et al. Diverse facets of MDSC in different phases of chronic HBV infection: Impact on HBV-specific T-cell response
and homing. Hepatology. 2022;76(3):759-74. https://doi.org/10.1002/ hep.32331. - Hanson EM, Clements VK, Sinha P, et al. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol. 2009;183(2):937-44. https://doi.org/10.4049/jimmunol. 08042 53.
- Parker KH, Sinha P, Horn LA, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014;74(20):5723-33. https://doi.org/10.1158/0008-5472.CAN-13-2347.
- Ku AW, Muhitch JB, Powers CA, et al. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. Elife. 2016;510:17875.
- Liu Y, Wei J, Guo G, et al. Norepinephrine-induced myeloid-derived suppressor cells block T-cell responses via generation of reactive oxygen species. Immunopharmacol Immunotoxicol. 2015;37(4):359-65. https:// doi.org/10.3109/08923973.2015.1059442.
- Kusmartsev S, Eruslanov E, Kubler H, et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol. 2008;181(1):346-53. https://doi.org/10.4049/jimmunol.181.1.346.
- Huang Y, Chen X, Dikov MM, et al. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood. 2007;110(2):624-31. https://doi.org/10.1182/ blood-2007-01-065714.
- Niu M, Yi M, Wu Y, et al. Synergistic efficacy of simultaneous anti-TGF-beta/VEGF bispecific antibody and PD-1 blockade in cancer therapy. J Hematol Oncol. 2023;16(1):94. https://doi.org/10.1186/ s13045-023-01487-5.
- Corzo CA, Cotter MJ, Cheng P, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693-701. https://doi.org/10.4049/jimmunol. 0900092.
- Raber PL, Thevenot P, Sierra R, et al. Subpopulations of myeloid-derived suppressor cells impair
cell responses through independent nitric oxide-related pathways. Int J Cancer. 2014;134(12):2853-64. https://doi. org/10.1002/ijc.28622. - LuT, Ramakrishnan R, Altiok S, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. 2011;121(10):4015-29. https://doi.org/10.1172/JCl45862.
- Cartwright ANR, Suo S, Badrinath S, et al. Immunosuppressive myeloid cells induce nitric oxide-dependent DNA damage and p53 pathway activation in CD8(+) T Cells. Cancer Immunol Res. 2021;9(4):470-85. https://doi.org/10.1158/2326-6066.CIR-20-0085.
- Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180-91. https://doi.org/10.1111/j. 1600-065X.2008.00608.x.
- Cimen Bozkus C, Elzey BD, Crist SA, et al. Expression of cationic amino acid transporter 2 is required for myeloid-derived suppressor cellmediated control of T cell immunity. J Immunol. 2015;195(11):5237-50. https://doi.org/10.4049/jimmunol. 1500959.
- Rodriguez PC, Ernstoff MS, Hernandez C, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69(4):1553-60. https://doi.org/10.1158/0008-5472.CAN-08-1921.
- Zea AH, Rodriguez PC, Culotta KS, et al. L-Arginine modulates CD3zeta expression and T cell function in activated human T lymphocytes. Cell Immunol. 2004;232(1-2):21-31. https://doi.org/10.1016/j.cellimm.2005. 01.004.
- Srivastava MK, Sinha P, Clements VK, et al. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70(1):68-77. https://doi.org/10.1158/0008-5472.CAN-09-2587.
- Yu J, Du W, Yan F, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783-97. https://doi.org/10.4049/jimmunol.1201449.
- Schafer CC, Wang Y, Hough KP, et al. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget. 2016;7(46):75407-24. https://doi.org/10.18632/oncotarget.12249.
- Liu
, Yu X, Xu L, et al. Current insight into the regulation of PD-L1 in cancer. Exp Hematol Oncol. 2022;11(1):44. https://doi.org/10.1186/ s40164-022-00297-8. - Kim SH, Li M, Trousil S, et al. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J Invest Dermatol. 2017;137(8):1740-8. https://doi.org/10. 1016/j.jid.2017.03.033.
- Wang L, Jia B, Claxton DF, et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology. 2018;7(9): e1469594. https://doi.org/10.1080/2162402X. 2018.1469594.
- Deng J, Li J, Sarde A, et al. Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol Res. 2019;7(7):1079-90. https:// doi.org/10.1158/2326-6066.CIR-18-0507.
- Sakuishi K, Jayaraman P, Behar SM, et al. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011;32(8):345-9. https://doi.org/10.1016/j.it.2011.05.003.
- Zhang CX, Huang DJ, Baloche V, et al. Correction to: Galectin-9 promotes a suppressive microenvironment in human cancer by enhancing STING degradation. Oncogenesis. 2022;11(1):5. https://doi.org/10.1038/ s41389-021-00375-2.
- Kong Y, Zhu L, Schell TD, et al. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin Cancer Res. 2016;22(12):3057-66. https:// doi.org/10.1158/1078-0432.CCR-15-2626.
- Wu L, Mao L, Liu JF, et al. Blockade of TIGIT/CD155 signaling reverses t-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res. 2019;7(10):1700-13. https://doi.org/10.1158/2326-6066.CIR-18-0725.
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253-68. https://doi.org/10.1038/nri3175.
- Sinha P, Clements VK, Fulton AM, et al. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67(9):4507-13. https://doi.org/10.1158/0008-5472. CAN-06-4174.
- Greifenberg V, Ribechini E, Rossner S, et al. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol. 2009;39(10):2865-76. https://doi.org/10. 1002/eji. 200939486.
- Poschke I, Mao Y, Adamson L, et al. Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol Immunother. 2012;61(6):827-38. https://doi.org/10.1007/s00262-011-1143-y.
- Hu CE, Gan J, Zhang RD, et al. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. 2011;46(2):156-64. https://doi.org/10.3109/00365521.2010.516450.
- Ugolini A, Tyurin VA, Tyurina YY, et al. Polymorphonuclear myeloidderived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight. 2020. https://doi.org/10.1172/jci.insight. 138581.
- Li H, Han Y, Guo Q, et al. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182(1):240-9. https://doi.org/10.4049/jimmunol.182.1. 240.
- Di Pace AL, Tumino N, Besi F, et al. Characterization of Human NK CellDerived Exosomes: Role of DNAM1 Receptor In Exosome-Mediated Cytotoxicity Against Tumor. Cancers (Basel). 2020;12:3. https://doi.org/ 10.3390/cancers12030661.
- Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50(3):799-807. https://doi. org/10.1002/hep.23054.
- Vaknin I, Blinder L, Wang L, et al. A common pathway mediated through Toll-like receptors leads to T- and natural killer-cell immunosuppression. Blood. 2008;111(3):1437-47. https://doi.org/10.1182/ blood-2007-07-100404.
- Zhang J, Han X, Hu X, et al. IDO1 impairs NK cell cytotoxicity by decreasing NKG2D/NKG2DLs via promoting miR-18a. Mol Immunol. 2018;103:144-55. https://doi.org/10.1016/j.molimm.2018.09.011.
- Della Chiesa M, Carlomagno S, Frumento G, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. 2006;108(13):4118-25. https://doi.org/10.1182/blood-2006-03-006700.
- Sui Q, Zhang J, Sun X, et al. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J Immunol. 2014;193(4):2016-23. https://doi.org/10. 4049/jimmunol. 1302389.
- Tie Y, Tang F, Wei YQ, et al. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15(1):61. https://doi.org/10.1186/s13045-022-01282-8.
- Franklin RA, Liao W, Sarkar A, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921-5. https://doi.org/10.1126/science.1252510.
- Wang Y, Liu H, Zhang Z, et al. G-MDSC-derived exosomes mediate the differentiation of M-MDSC into M2 macrophages promoting colitis-tocancer transition. J Immunother Cancer. 2023;11(6):45. https://doi.org/ 10.1136/jitc-2022-006166.
- DeNardo DG, Barreto JB, Andreu P, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91-102. https://doi. org/10.1016/j.ccr.2009.06.018.
- Thibodeau J, Bourgeois-Daigneault MC, Huppe G, et al. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol. 2008;38(5):1225-30. https://doi.org/10. 1002/eji.200737902.
- Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):112331. https://doi.org/10.1158/0008-5472.CAN-05-1299.
- Serafini P, Mgebroff S, Noonan K, et al. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68(13):5439-49. https://doi.org/10.1158/0008-5472.CAN-07-6621.
- Pan PY, Ma G, Weber KJ, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70(1):99-108. https://doi.org/10.1158/0008-5472.CAN-09-1882.
- Schlecker E, Stojanovic A, Eisen C, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol. 2012;189(12):5602-11. https://doi.org/10.4049/jimmunol. 1201018.
- Fujimura T, Ring S, Umansky V, et al. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol. 2012;132(4):1239-46. https://doi.org/10.1038/jid.2011. 416.
- Yang
, Lin , Shi , et al. FAP promotes immunosuppression by cancerassociated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76(14):4124-35. https://doi.org/10.1158/ 0008-5472.CAN-15-2973. - Xiang H, Ramil CP, Hai J, et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol Res. 2020;8(4):436-50. https://doi.org/10.1158/2326-6066.CIR-19-0507.
- Liang T, Tao T, Wu K, et al. Cancer-associated fibroblast-induced remodeling of tumor microenvironment in recurrent bladder cancer. Adv Sci (Weinh). 2023;10(31): e2303230. https://doi.org/10.1002/advs. 20230 3230.
- Deng Y, Cheng J, Fu B, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene. 2017;36(8):1090-101. https://doi.org/10.1038/onc.2016.273.
- Zhu GQ, Tang Z, Huang R, et al. CD36(+) cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023;9(1):25. https://doi.org/10.1038/s41421-023-00529-z.
- Zhao Q, Huang L, Qin G, et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021;518:35-48. https://doi.org/10.1016/j.canlet.2021.06.009.
- Lin
, Cai , Chen , et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology. 2022;75(1):28-42. https://doi.org/10.1002/ hep.32099. - Lideikaite A, Mozuraitiene J, Letautiene S. Analysis of prognostic factors for melanoma patients. Acta Med Litu. 2017;24(1):25-34. https://doi. org/10.6001/actamedica.v24i1.3460.
- Ross MI, Gershenwald JE. Evidence-based treatment of early-stage melanoma. J Surg Oncol. 2011;104(4):341-53. https://doi.org/10.1002/ jso. 21962.
- Jordan KR, Amaria RN, Ramirez O, et al. Myeloid-derived suppressor cells are associated with disease progression and decreased overall survival in advanced-stage melanoma patients. Cancer Immunol Immunother. 2013;62(11):1711-22. https://doi.org/10.1007/s00262-013-1475-x.
- Sade-Feldman M, Kanterman J, Klieger Y, et al. Clinical Significance of Circulating CD33+CD11b+HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin Cancer Res. 2016;22(23):5661-72. https://doi.org/10.1158/1078-0432.CCR-15-3104.
- Arkhypov I, Lasser S, Petrova V, et al. Myeloid cell modulation by tumorderived extracellular vesicles. Int J Mol Sci. 2020;21(17):34. https://doi. org/10.3390/ijms21176319.
- Arkhypov I, Ozbay Kurt FG, Bitsch R, et al. HSP90alpha induces immunosuppressive myeloid cells in melanoma via TLR4 signaling. J Immunother Cancer. 2022;10:9. https://doi.org/10.1136/jitc-2022-005551.
- Tengesdal IW, Menon DR, Osborne DG, et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc Natl Acad Sci U S A. 2021;118:10. https://doi.org/10.1073/pnas. 2000915118.
- Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397-411. https://doi.org/10.1038/ nri3452.
- Tengesdal IW, Dinarello A, Powers NE, et al. Tumor NLRP3-Derived IL1 beta Drives the IL-6/STAT3 Axis Resulting in Sustained MDSC-Mediated Immunosuppression. Front Immunol. 2021;12: 661323. https://doi.org/ 10.3389/fimmu.2021.661323.
- Weber R, Riester Z, Huser L, et al. IL-6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J Immunother Cancer. 2020;8:2. https://doi.org/10.1136/jitc-2020-000949.
- Chang AL, Miska J, Wainwright DA, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory t cells and myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5671-82. https://doi.org/10.1158/0008-5472.CAN-16-0144.
- Cheng Y, Ma XL, Wei YQ, et al. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim Biophys Acta Rev Cancer. 2019;1871(2):289-312. https://doi.org/10. 1016/j.bbcan.2019.01.005.
- Groth C, Arpinati L, Shaul ME, et al. Blocking migration of polymorphonuclear myeloid-derived suppressor cells inhibits mouse melanoma progression. Cancers (Basel). 2021;13:4. https://doi.org/10.3390/cance rs13040726.
- Toh B, Wang X, Keeble J, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9(9): e1001162. https:// doi.org/10.1371/journal.pbio. 1001162.
- Deng W, Fernandez A, McLaughlin SL, et al. Cell Communication Network Factor 4 (CCN4/WISP1) Shifts Melanoma Cells from a Fragile Proliferative State to a Resilient Metastatic State. Cell Mol Bioeng. 2020;13(1):45-60. https://doi.org/10.1007/s12195-019-00602-2.
- Fernandez A, Deng W, McLaughlin SL, et al. Cell Communication Network factor 4 promotes tumor-induced immunosuppression in melanoma. EMBO Rep. 2022;23(4): e54127. https://doi.org/10.15252/ embr. 202154127.
- Theivanthiran B, Evans KS, DeVito NC, et al. A tumor-intrinsic PD-L1/ NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J Clin Invest. 2020;130(5):2570-86. https://doi.org/10. 1172/JCl133055.
- Rawla P, Sunkara T, Muralidharan P, et al. Update in global trends and aetiology of hepatocellular carcinoma. Contemp Oncol (Pozn). 2018;22(3):141-50. https://doi.org/10.5114/wo.2018.78941.
- Li X, Xing YF, Lei AH, et al. Neutrophil count is associated with myeloid derived suppressor cell level and presents prognostic value of for
hepatocellular carcinoma patients. Oncotarget. 2017;8(15):24380-8. https://doi.org/10.18632/oncotarget. 15456. - Zhou J, Liu M, Sun H, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67(5):93144. https://doi.org/10.1136/gutjnl-2017-314032.
- Elwan N, Salem ML, Kobtan A, et al. High numbers of myeloid derived suppressor cells in peripheral blood and ascitic fluid of cirrhotic and HCC patients. Immunol Invest. 2018;47(2):169-80. https://doi.org/10. 1080/08820139.2017.1407787.
- Zeng X, Liao G, Li S, et al. Eliminating METTL1-mediated accumulation of PMN-MDSCs prevents hepatocellular carcinoma recurrence after radiofrequency ablation. Hepatology. 2023;77(4):1122-38. https://doi. org/10.1002/hep.32585.
- Simpson KD, Cross JV. MIF: metastasis/MDSC-inducing factor? Oncoimmunology. 2013;2(3): e23337. https://doi.org/10.4161/onci.23337.
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an irondependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-72. https://doi.org/10.1016/j.cell.2012.03.042.
- Conche C, Finkelmeier F, Pesic M, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023. https://doi.org/ 10.1136/gutjnl-2022-327909.
- He Q, Liu M, Huang W, et al. IL-1 beta-induced elevation of solute carrier family 7 member 11 promotes hepatocellular carcinoma metastasis through up-regulating programmed death ligand 1 and colony-stimulating factor 1. Hepatology. 2021;74(6):3174-93. https://doi.org/10. 1002/hep.32062.
- Wang W, Wu J, Ji M, et al. Exogenous interleukin-33 promotes hepatocellular carcinoma growth by remodelling the tumour microenvironment. J Transl Med. 2020;18(1):477. https://doi.org/10.1186/ s12967-020-02661-w.
- Xia S, Wu J, Zhou W, et al. SLC7A2 deficiency promotes hepatocellular carcinoma progression by enhancing recruitment of myeloid-derived suppressors cells. Cell Death Dis. 2021;12(6):570. https://doi.org/10. 1038/s41419-021-03853-y.
- Li YM, Liu ZY, Wang JC, et al. Receptor-Interacting Protein Kinase 3 Deficiency Recruits Myeloid-Derived Suppressor Cells to Hepatocellular Carcinoma Through the Chemokine (C-X-C Motif) Ligand 1-Chemokine (C-X-C Motif) Receptor 2 Axis. Hepatology. 2019;70(5):1564-81. https:// doi.org/10.1002/hep.30676.
- Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9(8):122135. https://doi.org/10.1089/ars.2007.1628.
- Chiu DK, Tse AP, Xu IM, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/ CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8(1):517. https://doi.org/10.1038/s41467-017-00530-7.
- Raychaudhuri D, Bhattacharya R, Sinha BP, et al. Lactate induces protumor reprogramming in intratumoral plasmacytoid dendritic cells. Front Immunol. 2019;10:1878. https://doi.org/10.3389/fimmu.2019. 01878.
- Dias AS, Almeida CR, Helguero LA, et al. Metabolic crosstalk in the breast cancer microenvironment. Eur J Cancer. 2019;121:154-71. https://doi.org/10.1016/j.ejca.2019.09.002.
- Wang N, Tan HY, Lu Y, et al. PIWIL1 governs the crosstalk of cancer cell metabolism and immunosuppressive microenvironment in hepatocellular carcinoma. Signal Transduct Target Ther. 2021;6(1):86. https://doi. org/10.1038/s41392-021-00485-8.
- Awuah PK, Monga SP. Cell cycle-related kinase links androgen receptor and beta-catenin signaling in hepatocellular carcinoma: why are men at a loss? Hepatology. 2012;55(3):970-3. https://doi.org/10.1002/hep. 24774.
- Wang SH, Yeh SH, Chen PJ. The driving circuit of HBx and androgen receptor in HBV-related hepatocarcinogenesis. Gut. 2014;63(11):1688-9. https://doi.org/10.1136/gutjnl-2013-306678.
- Feng H, Yu Z, Tian Y, et al. A CCRK-EZH2 epigenetic circuitry drives hepatocarcinogenesis and associates with tumor recurrence and poor survival of patients. J Hepatol. 2015;62(5):1100-11. https://doi.org/10. 1016/j.jhep.2014.11.040.
- Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209-49. https://doi.org/10. 3322/caac.21660.
- Miller LD, Chou JA, Black MA, et al. Immunogenic subtypes of breast cancer delineated by gene classifiers of immune responsiveness. Cancer Immunol Res. 2016;4(7):600-10. https://doi.org/10.1158/2326-6066. CIR-15-0149.
- Alshetaiwi H, Pervolarakis N, McIntyre LL, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol. 2020;5:44. https://doi.org/10.1126/sciim munol.aay6017.
- Zhang R, Dong M, Tu J, et al. PMN-MDSCs modulated by CCL20 from cancer cells promoted breast cancer cell stemness through CXCL2CXCR2 pathway. Signal Transduct Target Ther. 2023;8(1):97. https://doi. org/10.1038/s41392-023-01337-3.
- Peng D, Tanikawa T, Li W, et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/ NOTCH Cross-talk signaling. Cancer Res. 2016;76(11):3156-65. https:// doi.org/10.1158/0008-5472.CAN-15-2528.
- Liu M, Wei F, Wang J, et al. Myeloid-derived suppressor cells regulate the immunosuppressive functions of PD-1(-)PD-L1(+) Bregs through PD-L1/PI3K/AKT/NF-kappaB axis in breast cancer. Cell Death Dis. 2021;12(5):465. https://doi.org/10.1038/s41419-021-03745-1.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74. https://doi.org/10.1016/j.cell.2011.02.013.
- Li W, Tanikawa T, Kryczek I, et al. Aerobic Glycolysis Controls MyeloidDerived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018;28(1):87-103. https://doi.org/10.1016/j.cmet.2018.04.022.
- Xiao P, Wan X, Cui B, et al. Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. Oncoimmunology. 2016;5(1): e1063772. https://doi.org/10. 1080/2162402X.2015.1063772.
- Trillo-Tinoco J, Sierra RA, Mohamed E, et al. AMPK Alpha-1 intrinsically regulates the function and differentiation of tumor myeloid-derived suppressor cells. Cancer Res. 2019;79(19):5034-47. https://doi.org/10. 1158/0008-5472.CAN-19-0880.
- Jiang M, Zhang W, Zhang R, et al. Cancer exosome-derived miR-9 and miR-181a promote the development of early-stage MDSCs via interfering with SOCS3 and PIAS3 respectively in breast cancer. Oncogene. 2020;39(24):4681-94. https://doi.org/10.1038/s41388-020-1322-4.
- Yu B, Luo F, Sun B, et al. KAT6A Acetylation of SMAD3 Regulates Myeloid-Derived Suppressor Cell Recruitment, Metastasis, and Immunotherapy in Triple-Negative Breast Cancer. Adv Sci (Weinh). 2021;8(20): e2100014. https://doi.org/10.1002/advs.202100014.
- Ruiz-Fernandez de Cordoba B, Moreno H, Valencia K, et al. Tumor ENPP1 (CD203a)/Haptoglobin Axis Exploits Myeloid-Derived Suppressor Cells to Promote Post-Radiotherapy Local Recurrence in Breast Cancer. Cancer Discov. 2022;12(5):1356-1377.
- Beer TM, Kwon ED, Drake CG, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castrationresistant prostate cancer. J Clin Oncol. 2017;35(1):40-7. https://doi.org/ 10.1200/JCO.2016.69.1584.
- Wang J, McGuire TR, Britton HC, et al. Lenalidomide and cyclophosphamide immunoregulation in patients with metastatic, castration-resistant prostate cancer. Clin Exp Metastasis. 2015;32(2):111-24. https://doi. org/10.1007/s10585-015-9696-3.
- Lu X, Horner JW, Paul E, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543(7647):728-32. https://doi.org/10.1038/nature21676.
- Calcinotto A, Spataro C, Zagato E, et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature. 2018;559(7714):3639. https://doi.org/10.1038/s41586-018-0266-0.
- Burke M, Choksawangkarn W, Edwards N, et al. Exosomes from myeloidderived suppressor cells carry biologically active proteins. J Proteome Res. 2014;13(2):836-43. https://doi.org/10.1021/pr400879c.
- Gao F, Xu Q, Tang Z, et al. Exosomes derived from myeloid-derived suppressor cells facilitate castration-resistant prostate cancer progression
via S100A9/circMID1/miR-506-3p/MID1. J Transl Med. 2022;20(1):346. https://doi.org/10.1186/s12967-022-03494-5. - Hossain DM, Pal SK, Moreira D, et al. TLR9-Targeted STAT3 silencing abrogates immunosuppressive activity of myeloid-derived suppressor cells from prostate cancer patients. Clin Cancer Res. 2015;21(16):377182. https://doi.org/10.1158/1078-0432.CCR-14-3145.
- Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11-22. https://doi. org/10.1016/j.ccr.2010.05.026.
- Zhao D, Cai L, Lu X, et al. Chromatin regulator CHD1 remodels the immunosuppressive tumor microenvironment in PTEN-deficient prostate cancer. Cancer Discov. 2020;10(9):1374-87. https://doi.org/10. 1158/2159-8290.CD-19-1352.
- Jachetti E, Rigoni A, Bongiovanni L, et al. Imatinib Spares cKit-expressing prostate neuroendocrine tumors, whereas kills seminal vesicle epithelial-stromal tumors by targeting PDGFR-beta. Mol Cancer Ther. 2017;16(2):365-75. https://doi.org/10.1158/1535-7163.MCT-16-0466.
- Pittoni P, Tripodo C, Piconese S, et al. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res. 2011;71(18):5987-97. https://doi.org/10.1158/0008-5472.CAN-11-1637.
- Jachetti E, Cancila V, Rigoni A, et al. Cross-talk between myeloid-derived suppressor cells and mast cells mediates tumor-specific immunosuppression in prostate cancer. Cancer Immunol Res. 2018;6(5):552-65. https://doi.org/10.1158/2326-6066.CIR-17-0385.
- Miret JJ, Kirschmeier P, Koyama S, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. J Immunother Cancer. 2019;7(1):32. https://doi.org/10.1186/s40425-019-0504-5.
- Wang Y, Schafer CC, Hough KP, et al. Myeloid-Derived Suppressor Cells Impair B Cell Responses in Lung Cancer through IL-7 and STAT5. J Immunol. 2018;201(1):278-95. https://doi.org/10.4049/jimmunol. 17010 69.
- Lee JK, Won C, Yi EH, et al. Signal transducer and activator of transcription 3 (Stat3) contributes to T-cell homeostasis by regulating prosurvival Bcl-2 family genes. Immunology. 2013;140(3):288-300. https:// doi.org/10.1111/imm. 12133.
- Li Y, Du H, Qin Y, et al. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007;67(18):8494-503. https://doi.org/10.1158/0008-5472. CAN-07-0647.
- Tao Q, Fujimoto J, Men T, et al. Identification of the retinoic acid-inducible Gprc5a as a new lung tumor suppressor gene. J Natl Cancer Inst. 2007;99(22):1668-82. https://doi.org/10.1093/jnci/djm208.
- Zhong S, Yin H, Liao Y, et al. Lung Tumor Suppressor GPRC5A Binds EGFR and Restrains Its Effector Signaling. Cancer Res. 2015;75(9):180114. https://doi.org/10.1158/0008-5472.CAN-14-2005.
- Jing B, Wang T, Sun B, et al. IL6/STAT3 signaling orchestrates premetastatic niche formation and immunosuppressive traits in lung. Cancer Res. 2020;80(4):784-97. https://doi.org/10.1158/0008-5472. CAN-19-2013.
- Wang
, Jing , et al. PTGES/PGE(2) signaling links immunosuppression and lung metastasis in Gprc5a-knockout mouse model. Oncogene. 2020;39(15):3179-94. https://doi.org/10.1038/s41388-020-1207-6. - Liu NN, Yi CX, Wei LQ, et al. The intratumor mycobiome promotes lung cancer progression via myeloid-derived suppressor cells. Cancer Cell. 2023;41(11):1927-44. https://doi.org/10.1016/j.ccell.2023.08.012.
- Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19(9):495-509. https://doi.org/10.1038/s41568-019-0179-8.
- Koyama S, Akbay EA, Li YY, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999-1008. https://doi.org/10.1158/0008-5472.CAN-15-1439.
- Li R, Salehi-Rad R, Crosson W, et al. Inhibition of granulocytic myeloid-derived suppressor cells overcomes resistance to immune checkpoint inhibition in LKB1-deficient non-small cell lung cancer. Cancer Res. 2021;81(12):3295-308. https://doi.org/10.1158/0008-5472. CAN-20-3564.
- Li Y, Qu P, Wu L, et al. Api6/AIM/Spalpha/CD5L overexpression in alveolar type II epithelial cells induces spontaneous lung adenocarcinoma. Cancer Res. 2011;71(16):5488-99. https://doi.org/10.1158/0008-5472. CAN-10-4225.
- Zhao S, Guo T, Li J, et al. Expression and prognostic value of GalNAc-T3 in patients with completely resected small (
) peripheral lung adenocarcinoma after IASLC/ATS/ERS classification. Onco Targets Ther. 2015;8:3143-52. https://doi.org/10.2147/OTT.S93486. - Park MS, Yang AY, Lee JE, et al. GALNT3 suppresses lung cancer by inhibiting myeloid-derived suppressor cell infiltration and angiogenesis in a TNFR and c-MET pathway-dependent manner. Cancer Lett. 2021;521:294-307. https://doi.org/10.1016/j.canlet.2021.08.015.
- Zhao T, Liu S, Hanna NH, et al. LAL deficiency induced myeloidderived suppressor cells as targets and biomarkers for lung cancer. J Immunother Cancer. 2023;11(3): e006272. https://doi.org/10.1136/ jitc-2022-006272.
- Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582-98. https://doi.org/10.1038/nrc.2016.73.
- Porembka MR, Mitchem JB, Belt BA, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61(9):1373-85. https://doi.org/10.1007/s00262-011-1178-0.
- Zhang J, Xu X, Shi M, et al. CD13(hi) Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology. 2017;6(2): e1258504. https://doi.org/10.1080/2162402X.2016.12585 04.
- Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518-27. https://doi.org/10.1158/0008-5472. CAN-07-0175.
- Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769-81. https://doi.org/10.1136/ gutjnl-2013-306271.
- Choueiry F, Torok M, Shakya R, et al. CD200 promotes immunosuppression in the pancreatic tumor microenvironment. J Immunother Cancer. 2020;8(1):10. https://doi.org/10.1136/jitc-2019-000189.
- Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318-48. https://doi.org/10. 3322/caac.21190.
- Yang
, Lu , Hang J, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res. 2020;8(11):1440-51. https:// doi.org/10.1158/2326-6066.CIR-20-0111. - Chun E, Lavoie S, Michaud M, et al. CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. 2015;12(2):244-57. https://doi. org/10.1016/j.celrep.2015.06.024.
- Maisonneuve C, Tsang DKL, Foerster EG, et al. Nod1 promotes colorectal carcinogenesis by regulating the immunosuppressive functions of tumor-infiltrating myeloid cells. Cell Rep. 2021;34(4): 108677. https:// doi.org/10.1016/j.celrep.2020.108677.
- Wang Y, Yin K, Tian J, et al. Granulocytic myeloid-derived suppressor cells promote the stemness of colorectal cancer cells through exosomal S100A9. Adv Sci (Weinh). 2019;6(18):1901278. https://doi.org/10. 1002/advs. 201901278.
- Chen H, Pan Y, Zhou Q, et al. METTL3 Inhibits Antitumor Immunity by Targeting m(6)A-BHLHE41-CXCL1/CXCR2 Axis to Promote Colorectal Cancer. Gastroenterology. 2022;163(4):891-907. https://doi.org/10. 1053/j.gastro.2022.06.024.
- Du Z, Su J, Lin S, et al. Hydroxyphenylpyruvate Dioxygenase Is a Metabolic Immune Checkpoint for UTX-deficient Colorectal Cancer. Gastroenterology. 2023;164(7):1165-79. https://doi.org/10.1053/j.gastro.2023. 02.010.
- Zhang
, Zheng , Chen , et al. Gut fungi enhances immunosuppressive function of myeloid-derived suppressor cells by activating PKM2dependent glycolysis to promote colorectal tumorigenesis. Exp Hematol Oncol. 2022;11(1):88. https://doi.org/10.1186/s40164-022-00334-6. - Yu S, Ren X, Li L. Myeloid-derived suppressor cells in hematologic malignancies: two sides of the same coin. Exp Hematol Oncol. 2022;11(1):43. https://doi.org/10.1186/s40164-022-00296-9.
- Jitschin R, Saul D, Braun M, et al. CD33/CD3-bispecific T-cell engaging (BiTE(R)) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer. 2018;6(1):116. https://doi.org/ 10.1186/s40425-018-0432-9.
- Tohumeken S, Baur R, Bottcher M, et al. Palmitoylated proteins on AML-derived extracellular vesicles promote myeloid-derived suppressor cell differentiation via TLR2/Akt/mTOR Signaling. Cancer Res. 2020;80(17):3663-76. https://doi.org/10.1158/0008-5472.CAN-20-0024.
- Cai B, Liu Y, Chong Y, et al. IRAK1-regulated IFN-gamma signaling induces MDSC to facilitate immune evasion in FGFR1-driven hematological malignancies. Mol Cancer. 2021;20(1):165. https://doi.org/10. 1186/s12943-021-01460-1.
- Highfill SL, Cui Y, Giles AJ, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6(237):237-67. https://doi.org/10.1126/scitranslmed.3007974.
- Raychaudhuri B, Rayman P, Ireland J, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13(6):591-9. https://doi.org/10.1093/ neuonc/nor042.
- Gieryng A, Pszczolkowska D, Walentynowicz KA, et al. Immune microenvironment of gliomas. Lab Invest. 2017;97(5):498-518. https://doi.org/ 10.1038/labinvest.2017.19.
- Su MT, Kumata S, Endo S, et al. LILRB4 promotes tumor metastasis by regulating MDSCs and inhibiting miR-1 family miRNAs. Oncoimmunology. 2022;11(1):2060907. https://doi.org/10.1080/2162402X.2022.20609 07.
- Cheng
, Wang , Wang , et al. Tumor-associated myeloid cells in cancer immunotherapy. J Hematol Oncol. 2023;16(1):71. https://doi.org/10. 1186/s13045-023-01473-x. - Wang Y, Johnson KCC, Gatti-Mays ME, et al. Emerging strategies in targeting tumor-resident myeloid cells for cancer immunotherapy. J Hematol Oncol. 2022;15(1):118. https://doi.org/10.1186/ s13045-022-01335-y.
- Bronte V, Serafini P, Apolloni E, et al. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother. 2001;24(6):431-46. https://doi.org/10.1097/00002371-20011 1000-00001.
- Zheng
, et al. Cimetidine suppresses lung tumor growth in mice through proapoptosis of myeloid-derived suppressor cells. Mol Immunol. 2013;54(1):74-83. https://doi.org/10.1016/j.molimm.2012.10. 035. - Condamine T, Kumar V, Ramachandran IR, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124(6):2626-39. https://doi.org/10.1172/JCl74 056.
- Svoronos N, Perales-Puchalt A, Allegrezza MJ, et al. Tumor cellindependent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov. 2017;7(1):72-85. https://doi.org/10.1158/2159-8290.CD-16-0502.
- Liu M, Zhou J, Liu X, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut. 2020;69(2):365-79. https://doi.org/10.1136/ gutjnl-2018-317257.
- Tavazoie MF, Pollack I, Tanqueco R, et al. LXR/ApoE activation restricts innate immune suppression in cancer. Cell. 2018;172(4):825-40. https:// doi.org/10.1016/j.cell.2017.12.026.
- Welters MJ, van der Sluis TC, van Meir H, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med. 2016;8(334):334-52. https://doi.org/10.1126/ scitranslmed.aad8307.
- Homey B, Muller A, Zlotnik A. Chemokines: agents for the immunotherapy of cancer? Nat Rev Immunol. 2002;2(3):175-84. https://doi.org/ 10.1038/nri748.
- Fedele C, Li S, Teng KW, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. 2021. https://doi.org/10.1084/jem. 20201414.
- Quintana E, Schulze CJ, Myers DR, et al. Allosteric Inhibition of SHP2 stimulates antitumor immunity by transforming the
immunosuppressive environment. Cancer Res. 2020;80(13):2889-902. https://doi.org/10.1158/0008-5472.CAN-19-3038. - Tang KH, Li S, Khodadadi-Jamayran A, et al. Combined Inhibition of SHP2 and CXCR1/2 Promotes Antitumor T-cell Response in NSCLC. Cancer Discov. 2022;12(1):47-61. https://doi.org/10.1158/2159-8290. CD-21-0369.
- Lu Z, Zou J, Li S, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature. 2020;579(7798):284-90. https://doi. org/10.1038/s41586-020-2054-X.
- Sun L, Clavijo PE, Robbins Y, et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight. 2019;4:7. https://doi.org/10.1172/jci.insight.126853.
- Tap WD, Wainberg ZA, Anthony SP, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373(5):428-37. https://doi.org/10.1056/NEJMoa1411366.
- Cannarile MA, Weisser M, Jacob W, et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5(1):53. https://doi.org/10.1186/s40425-017-0257-y.
- Mok S, Koya RC, Tsui C, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74(1):153-61. https://doi.org/10.1158/0008-5472.CAN-13-1816.
- Zhu Y, Yang J, Xu D, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68(9):1653-66. https://doi.org/10.1136/gutjnl-2019-318419.
- Kumar V, Donthireddy L, Marvel D, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell. 2017;32(5):654-68. https://doi.org/10.1016/j.ccell.2017.10.005.
- Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumourassociated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67(6):1112-23. https:// doi.org/10.1136/gutjnl-2017-313738.
- Hasnis E, Alishekevitz D, Gingis-Veltski S, et al. Anti-Bv8 antibody and metronomic gemcitabine improve pancreatic adenocarcinoma treatment outcome following weekly gemcitabine therapy. Neoplasia. 2014;16(6):501-10. https://doi.org/10.1016/j.neo.2014.05.011.
- Serafini P, Meckel K, Kelso M, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691-702. https://doi. org/10.1084/jem. 20061104.
- Lin S, Wang J, Wang L, et al. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res. 2017;7(1):41-52.
- Yu SJ, Ma C, Heinrich B, et al. Targeting the crosstalk between cytokineinduced killer cells and myeloid-derived suppressor cells in hepatocelIular carcinoma. J Hepatol. 2019;70(3):449-57. https://doi.org/10.1016/j. jhep.2018.10.040.
- Benguigui M, Vorontsova A, Timaner M, et al. Bv8 blockade sensitizes Anti-PD1 therapy resistant tumors. Front Immunol. 2022;13: 903591. https://doi.org/10.3389/fimmu.2022.903591.
- Sturgill ER, Rolig AS, Linch SN, et al. Galectin-3 inhibition with belapectin combined with anti-OX40 therapy reprograms the tumor microenvironment to favor anti-tumor immunity. Oncoimmunology. 2021;10(1):1892265. https://doi.org/10.1080/2162402X.2021.1892265.
- Alvear-Arias JJ, Carrillo C, Villar JP, et al. Expression of H(v)1 proton channels in myeloid-derived suppressor cells (MDSC) and its potential role in T cell regulation. Proc Natl Acad Sci U S A. 2022;119(15): e2104453119. https://doi.org/10.1073/pnas.2104453119.
- Ishfaq M , Pham T, Beaman C, et al. BTK inhibition reverses MDSCmediated immunosuppression and enhances response to Anti-PDL1 therapy in neuroblastoma. Cancers (Basel). 2021;13(4):817. https://doi. org/10.3390/cancers13040817.
- Weed DT, Vella JL, Reis IM, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21(1):39-48. https://doi.org/10.1158/1078-0432.CCR-14-1711.
- Noonan KA, Ghosh N, Rudraraju L, et al. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol Res. 2014;2(8):725-31. https://doi.org/10.1158/2326-6066.CIR-13-0213.
- Van Wigcheren GF, De Haas N, Mulder TA, et al. Cisplatin inhibits frequency and suppressive activity of monocytic myeloid-derived suppressor cells in cancer patients. Oncoimmunology. 2021;10(1):1935557. https://doi.org/10.1080/2162402X.2021.1935557.
- Yan D, Wang HW, Bowman RL, et al. STAT3 and STAT6 signaling pathways synergize to promote cathepsin secretion from macrophages via IRE1alpha activation. Cell Rep. 2016;16(11):2914-27. https://doi.org/10. 1016/j.celrep.2016.08.035.
- Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, et al. ER Stress Sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527-38. https://doi.org/10.1016/j.cell.2015.05. 025.
- Mohamed E, Sierra RA, Trillo-Tinoco J, et al. The Unfolded Protein Response Mediator PERK Governs Myeloid Cell-Driven Immunosuppression in Tumors through Inhibition of STING Signaling. Immunity. 2020;52(4):668-82. https://doi.org/10.1016/j.immuni.2020.03.004.
- Gabrilovich DI, Velders MP, Sotomayor EM, et al. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166(9):5398-406. https://doi.org/10.4049/jimmunol. 166.9.5398.
- Kusmartsev S, Cheng F, Yu B, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63(15):4441-9.
- Nefedova Y, Fishman M, Sherman S, et al. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67(22):11021-8. https://doi.org/10.1158/0008-5472. CAN-07-2593.
- Bauer R, Udonta F, Wroblewski M, et al. Blockade of myeloid-derived suppressor cell expansion with all-trans retinoic acid increases the efficacy of antiangiogenic therapy. Cancer Res. 2018;78(12):3220-32. https://doi.org/10.1158/0008-5472.CAN-17-3415.
- He Y, Hung SW, Liang B, et al. Receptor tyrosine kinase inhibitor sunitinib as novel immunotherapy to inhibit myeloid-derived suppressor cells for treatment of endometriosis. Front Immunol. 2021;12: 641206. https://doi.org/10.3389/fimmu.2021.641206.
- Mirza N, Fishman M, Fricke I, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66(18):9299-307. https://doi.org/10.1158/0008-5472. CAN-06-1690.
- Li MO, Wolf N, Raulet DH, et al. Innate immune cells in the tumor microenvironment. Cancer Cell. 2021;39(6):725-9. https://doi.org/10.1016/j. ccell.2021.05.016.
Publisher’s Note
- *Correspondence:
Limin Xia
xialimin@tjh.tjmu.edu.cn
Wenjie Huang
huangwenjie@tjh.tjmu.edu.cn
Hepatic Surgery Centre, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Hubei Key Laboratory of Hepato-Pancreato-Biliary Diseases, Wuhan 430030, Hubei, China Clinical Medicine Research Center for Hepatic Surgery of Hubei Province, Key Laboratory of Organ Transplantation, Ministry of Education and Ministry of Public Health, Wuhan 430030, Hubei, China