DOI: https://doi.org/10.1038/s41467-024-44896-x
PMID: https://pubmed.ncbi.nlm.nih.gov/38242907
تاريخ النشر: 2024-01-19
الفهم الجزيئي للدور الحاسم لأيونات المعادن القلوية في بدء اختزال ثاني أكسيد الكربون على سطح Cu(100)
تم القبول: 9 يناير 2024
نُشر على الإنترنت: 19 يناير 2024
(أ) التحقق من التحديثات
الملخص
فهم الجزيئات لواجهة الصلب-السائل هو أمر صعب ولكنه ضروري لتوضيح دور البيئة على حركية التفاعلات الكهروكيميائية. أيونات المعادن القلوية (
تم بذل العديد من الجهود لتحسين أدائها الكهروكيميائي بشكل أكبر، خاصة من أجل المزيد من القيمة.
النتائج والمناقشة
استجابة الطبقة المزدوجة الكهربائية للجهد المطبق
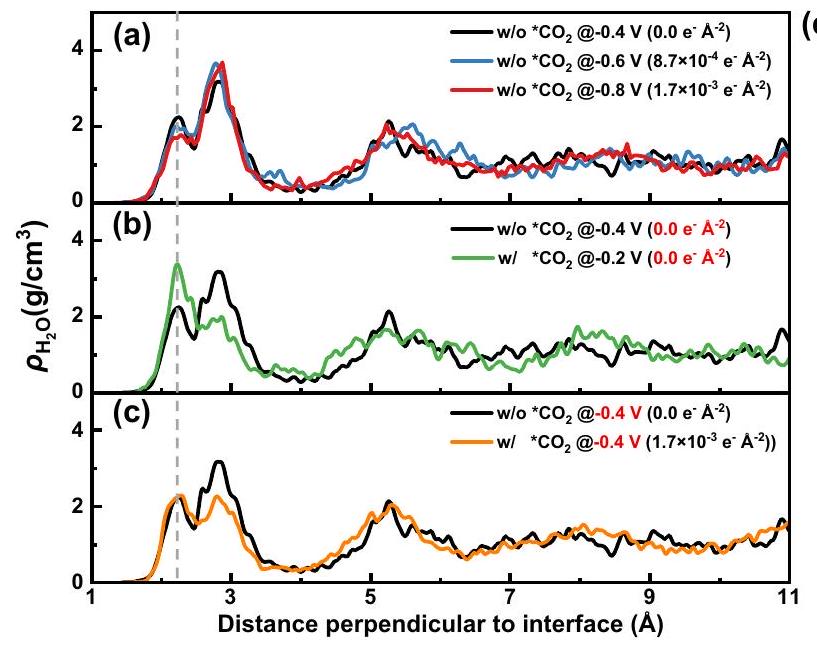
توزيع كثافة الماء على طول الاتجاه العمودي على
سطح، مما يسبب
تعزيز تأثيرات كاتيونات المعادن القلوية على
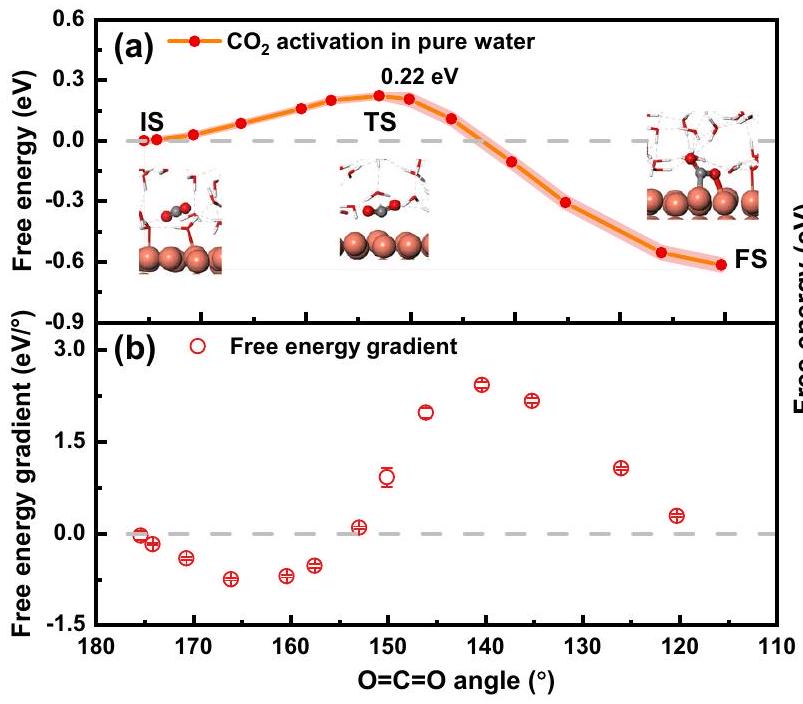
مع كاتيونات المعادن القلوية بسبب تحولها من تكوين غير قطبي خطي إلى تكوين قطبي منحنٍ. درسنا الـ
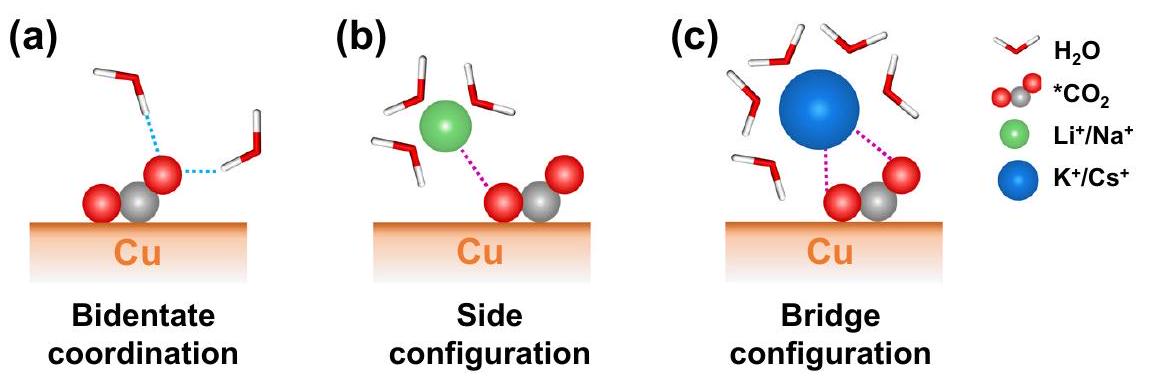
التي بالكاد يمكن أن تقبل المزيد من جزيئات الإذابة مع البيئة، كما هو موضح في المنصة في دالة التوزيع الشعاعي المتكاملة (RDF) عند نصف قطر الغلاف الأول للإذابة في الشكل التكميلية 5. ومع ذلك، بالنسبة لـ
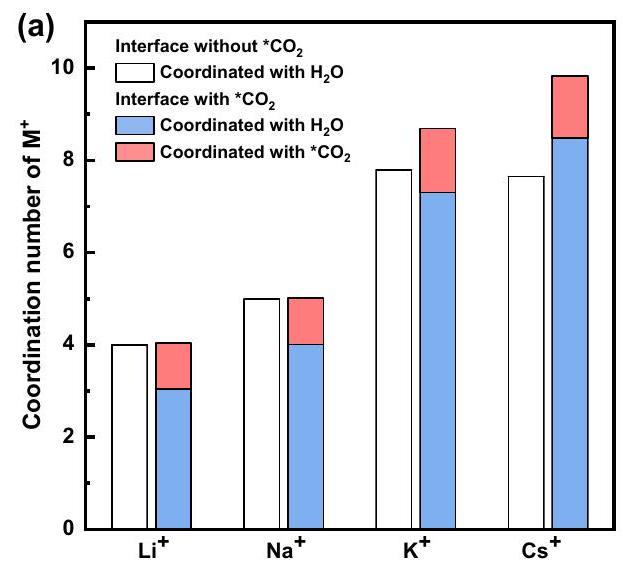
توزيع المسافة بين كاتيونات المعادن القلوية والأكسجينين
هذا يشير إلى تنسيق أكثر ديناميكية حول
التأثير الضروري لـ
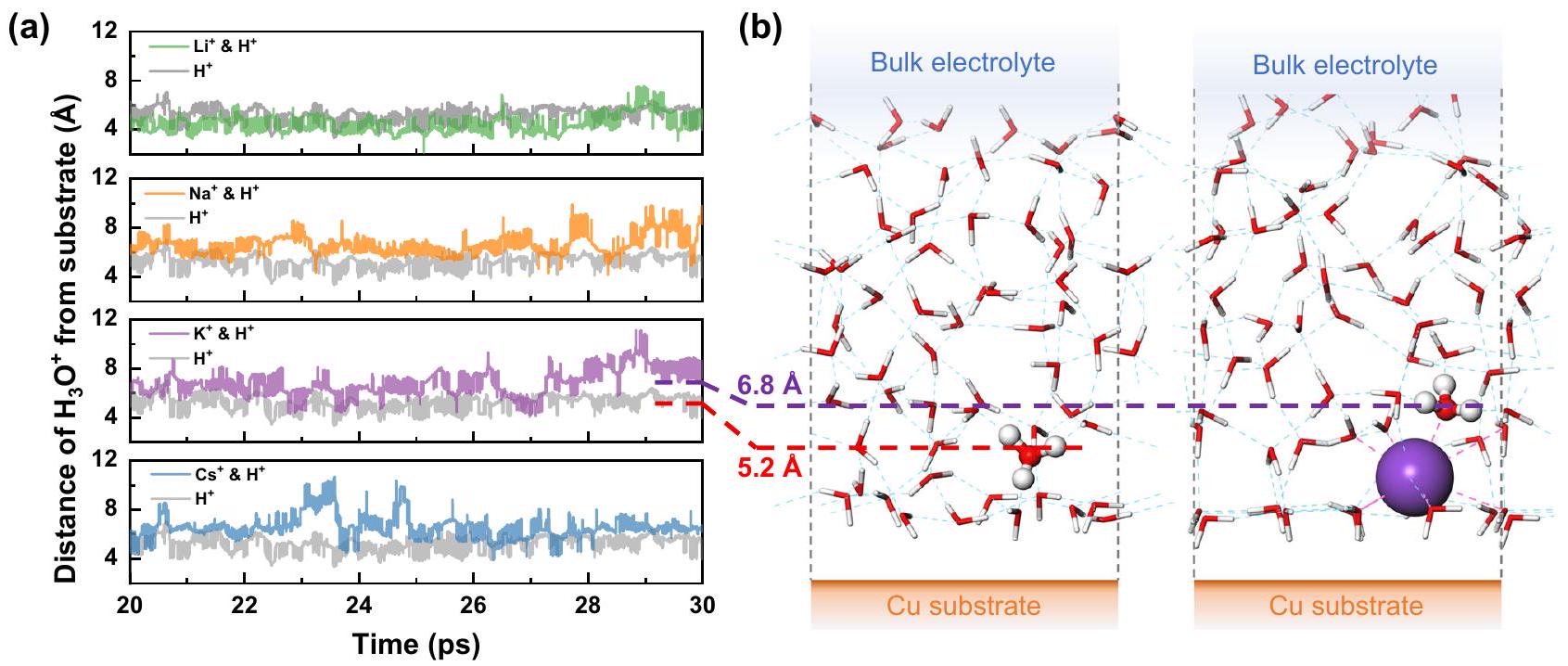
(ب).
بين الطبقتين الأولى والثانية من الماء على الرغم من النبذ الكهروستاتيكي بينهما. علاوة على ذلك، بعد إدخال أيونات المعادن القلوية، تزداد كثافة الشحنة السلبية عند سطح
تساعد نتائجنا في فهم الأصل الجزيئي للدور المحتمل للبيئة الكهروكيميائية، وتؤكد على أهمية الترطيب الصريح، والأيونات، والاستكشاف المنهجي للتفاعلات المتنافسة في المحاكاة الكهروكيميائية، وتظهر فعالية الأدوات النظرية في استكشاف واجهة الصلب-الإلكتروليت.
طرق
نموذج القطبين مزدوجي الإلكتروليت الصلب
محاكاة AIMD
توفر البيانات
References
- Nitopi, S. et al. Progress and perspectives of electrochemical
reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610-7672 (2019). - Huang, J. E. et al.
electrolysis to multicarbon products in strong acid. Science 372, 1074-1078 (2021). - He, M. et al. Selective enhancement of methane formation in electrochemical
reduction enabled by a Raman-inactive oxygen-containing species on Cu. ACS Catal. 12, 6036-6046 (2022). - Li, J. et al. Constraining CO coverage on copper promotes highefficiency ethylene electroproduction. Nat. Catal. 2, 1124-1131 (2019).
- Dinh, C. T. et al.
electroreduction to ethylene via hydroxidemediated copper catalysis at an abrupt interface. Science 360, 783-787 (2018). - Timoshenko, J. et al. Steering the structure and selectivity of
electroreduction catalysts by potential pulses. Nat. Catal. 5, 259-267 (2022). - Wang, L. et al. Electrochemical carbon monoxide reduction on polycrystalline copper: effects of potential, pressure, and pH on selectivity toward multicarbon and oxygenated products. ACS Catal. 8, 7445-7454 (2018).
- Chen, X. et al. Electrochemical
-to-ethylene conversion on polyamine-incorporated Cu electrodes. Nat. Catal. 4, 20-27 (2021). - Wang, R. et al. Partial coordination-perturbed bi-copper sites for selective electroreduction of
to hydrocarbons. Angew. Chem. Int. Ed. 60, 19829-19835 (2021). - Bagger, A. et al. Electrochemical
reduction: classifying Cu facets. ACS Catal. 9, 7894-7899 (2019). - Scholten, F. et al. Identifying structure-selectivity correlations in the electrochemical reduction of
: a comparison of wellordered atomically clean and chemically etched copper singlecrystal surfaces. Angew. Chem. Int. Ed. 60, 19169-19175 (2021). - Wu, Z.-Z. et al. Identification of
interfaces as superior active sites for CO dimerization during electroreduction. J. Am. Chem. Soc. 144, 259-269 (2021). - Akira, M. & Yoshio, H. Product selectivity affected by cationic species in electrochemical reduction of
and CO at a Cu electrode. Bull. Chem. Soc. Jpn 64, 123-127 (1991). - Hori, Y. et al. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc. Faraday Trans. 85, 2309-2326 (1989).
- Resasco, J. et al. Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J. Am. Chem. Soc. 139, 11277-11287 (2017).
- Ren, W. et al. A cation concentration gradient approach to tune the selectivity and activity of
electroreduction. Angew. Chem. Int. Ed. 61, e202214173 (2022). - Ayemoba, O. & Cuesta, A. Spectroscopic evidence of size-dependent buffering of interfacial pH by cation hydrolysis during
electroreduction. ACS Appl. Mater. Interfaces 9, 27377-27382 (2017). - Singh, M. R. et al. Hydrolysis of electrolyte cations enhances the electrochemical reduction of
over Ag and Cu . J. Am. Chem. Soc. 138, 13006-13012 (2016). - Malkani, A. S. et al. Cation effect on interfacial
concentration in the electrochemical reduction reaction. ACS Catal. 10, 14871-14876 (2020). - Gu, J. et al. Modulating electric field distribution by alkali cations for
electroreduction in strongly acidic medium. Nat. Catal. 5, 268-276 (2022). - Ringe, S. et al. Understanding cation effects in electrochemical
reduction. Energy Environ. Sci. 12, 3001-3014 (2019). - Chen, L. D. et al. Electric field effects in electrochemical
reduction. ACS Catal. 6, 7133-7139 (2016). - Monteiro, M. C. O. et al. Absence of
electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 4, 654-662 (2021). - Qin, H.-G. et al. Quantitative understanding of cation effects on the electrochemical reduction of
and in acidic solution. ACS Catal. 13, 916-926 (2022). - Zhu, Q. et al. The solvation-induced Onsager reaction field rather than the double-layer field controls
reduction on gold. JACS Au 2, 472-482 (2022). - Groß, A. & Sakong, S. Ab initio simulations of water/metal interfaces. Chem. Rev. 122, 10746-10776 (2022).
- Qin, X. et al. Cation-coordinated inner-sphere
electroreduction at Au-water interfaces. J. Am. Chem. Soc. 145, 1897-1905 (2023). - Liu, H. et al. Promotional role of a cation intermediate complex in
formation from electrochemical reduction of over Cu . ACS Catal. 11, 12336-12343 (2021). - Yu, M. & Trinkle, D. R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 134, 064111 (2011).
- Chan, K. & Nørskov, J. K. Electrochemical barriers made simple. J. Phys. Chem. Lett. 6, 2663-2668 (2015).
- Kronberg, R. & Laasonen, K. Reconciling the experimental and computational hydrogen evolution activities of Pt(111) through DFTbased constrained MD simulations. ACS Catal. 11, 8062-8078 (2021).
- Zhao, X. & Liu, Y. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc. 143, 9423-9428 (2021).
- Le, J.-B. et al. Molecular origin of negative component of Helmholtz capacitance at electrified Pt(111)/water interface. Sci. Adv. 6, eabb1219 (2020).
- Vijay, S. et al. Unified mechanistic understanding of
reduction to CO on transition metal and single atom catalysts. Nat. Catal. 4, 1024-1031 (2021). - Cao, H. et al. Potential-dependent free energy relationship in interpreting the electrochemical performance of
reduction on single atom catalysts. ACS Catal. 12, 6606-6617 (2022). - Shin, S.-J. et al. A unifying mechanism for cation effect modulating
and productions from electroreduction. Nat. Commun. 13, 5482 (2022). - Le, D. & Rahman, T. S. On the role of metal cations in
electrocatalytic reduction. Nat. Catal. 5, 977-978 (2022). - Li, P. et al. Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt. Nat. Catal. 5, 900-911 (2022).
- Li, X.-Y. et al. Mechanism of cations suppressing proton diffusion kinetics for electrocatalysis. Angew. Chem. Int. Ed. 62, e202218669 (2023).
- Le, J.-B. et al. Molecular understanding of cation effects on double layer and its significance to CO-CO dimerization. Natl. Sci. Rev. 10, nwad105 (2023).
- Hussain, G. et al. How cations determine the interfacial potential profile: relevance for the
reduction reaction. Electrochim. Acta 327, 135055 (2019). - Garlyyev, B. et al. Influence of the nature of the alkali metal cations on the electrical double-layer capacitance of model Pt(111) and Au(111) electrodes. J. Phys. Chem. Lett. 9, 1927-1930 (2018).
- Xue, S. et al. How the nature of the alkali metal cations influences the double-layer capacitance of
, and Pt single-crystal electrodes. J. Phys. Chem. C 124, 12442-12447 (2020). - Qin, X. et al. Cation-induced changes in the inner- and outer-sphere mechanisms of electrocatalytic
reduction. Nat. Commun. 14, 7607 (2023). - Kwon, S. et al. Dramatic change in the step edges of the
electrocatalyst upon exposure to CO: operando observations by electrochemical STM and explanation using quantum mechanical calculations. ACS Catal. 11, 12068-12074 (2021). - Surendralal, S. et al. First-principles approach to model electrochemical reactions: understanding the fundamental mechanisms behind Mg corrosion. Phys. Rev. Lett. 120, 246801 (2018).
- Surendralal, S. et al. Impact of water coadsorption on the electrode potential of H-Pt(111)-liquid water interfaces. Phys. Rev. Lett. 126, 166802 (2021).
- Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
- Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
- Hammer, B. et al. Improved adsorption energetics within densityfunctional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413-7421 (1999).
- Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
- Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188-5192 (1976).
- Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695-1697 (1985).
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511-519 (1984).
- Woo, T. K. et al. A combined Car-Parrinello QM/MM implementation for ab initio molecular dynamics simulations of extended systems: application to transition metal catalysis. J. Phys. Chem. B 101, 7877-7880 (1997).
- Carter, E. A. et al. Constrained reaction coordinate dynamics for the simulation of rare events. Chem. Phys. Lett. 156, 472-477 (1989).
- Ryckaert, J.-P. et al. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327-341 (1977).
- Neugebauer, J. & Scheffler, M. Adsorbate-substrate and adsorbateadsorbate interactions of Na and K adlayers on Al (111). Phys. Rev. B 46, 16067-16080 (1992).
- Chan, K. & Nørskov, J. K. Potential dependence of electrochemical barriers from ab initio calculations. J. Phys. Chem. Lett. 7, 1686-1690 (2016).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-44896-x.
http://www.nature.com/reprints
© المؤلف(ون) 2024
مركز جرافين شنتشن ومعهد أبحاث المواد، المدرسة الدولية للدراسات العليا بجامعة تسينغhua، شنتشن 518055، جمهورية الصين الشعبية. المختبر الوطني الرئيسي للفيزياء الكمومية ذات الأبعاد المنخفضة وقسم الفيزياء، جامعة تسينغhua، بكين 100084، جمهورية الصين الشعبية. معهد الدراسات المتقدمة، جامعة تسينغhua، بكين 100084، جمهورية الصين الشعبية. مركز العلوم الحدودية لمعلومات الكم، بكين 100084، جمهورية الصين الشعبية. قسم تحويل وتخزين الطاقة، الجامعة التقنية في الدنمارك، كنجس، لينغبي 2800، الدنمارك. البريد الإلكتروني:fykang@sz.tsinghua.edu.cn; li.jia@sz.tsinghua.edu.cn
DOI: https://doi.org/10.1038/s41467-024-44896-x
PMID: https://pubmed.ncbi.nlm.nih.gov/38242907
Publication Date: 2024-01-19
Molecular understanding of the critical role of alkali metal cations in initiating
Accepted: 9 January 2024
Published online: 19 January 2024
(A) Check for updates
Abstract
Molecular understanding of the solid-liquid interface is challenging but essential to elucidate the role of the environment on the kinetics of electrochemical reactions. Alkali metal cations (
many efforts have been made to improve their electrochemical performance further, especially for more valuable
Results and discussion
Electrical double-layer response of applied potential
a-c Distribution of water density along the direction perpendicular to the
surface, causing the
Promoting effects of alkali metal cations on
with alkali metal cations due to its transformation from a linear nonpolar configuration to a bent polar one. We studied the
which can hardly accept more solvation molecules with the environment, as shown by the platform in the integrated radial distribution function (RDF) at the radius of the first solvation shell in Supplementtary Fig. 5. However, for
distance distribution between alkali metal cations and two oxygens of
d). This suggests a more dynamic coordination around
Necessary effect of
(b).
between the first and second water layers despite of the electrostatic repulsion between them. Furthermore, after the introduction of alkali metal cations, the negative charge density at the
and Au. Our results help to understand the molecular origin of the possible role of the electrochemical environment emphasize the importance of explicit solvation, ions, and systematic exploration of competing reactions in electrochemical simulations, and show the effectiveness of theoretical tools in exploring the solid-electrolyte interface.
Methods
Solid-electrolyte-Ne double electrode model
AIMD simulations
Data availability
References
- Nitopi, S. et al. Progress and perspectives of electrochemical
reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610-7672 (2019). - Huang, J. E. et al.
electrolysis to multicarbon products in strong acid. Science 372, 1074-1078 (2021). - He, M. et al. Selective enhancement of methane formation in electrochemical
reduction enabled by a Raman-inactive oxygen-containing species on Cu. ACS Catal. 12, 6036-6046 (2022). - Li, J. et al. Constraining CO coverage on copper promotes highefficiency ethylene electroproduction. Nat. Catal. 2, 1124-1131 (2019).
- Dinh, C. T. et al.
electroreduction to ethylene via hydroxidemediated copper catalysis at an abrupt interface. Science 360, 783-787 (2018). - Timoshenko, J. et al. Steering the structure and selectivity of
electroreduction catalysts by potential pulses. Nat. Catal. 5, 259-267 (2022). - Wang, L. et al. Electrochemical carbon monoxide reduction on polycrystalline copper: effects of potential, pressure, and pH on selectivity toward multicarbon and oxygenated products. ACS Catal. 8, 7445-7454 (2018).
- Chen, X. et al. Electrochemical
-to-ethylene conversion on polyamine-incorporated Cu electrodes. Nat. Catal. 4, 20-27 (2021). - Wang, R. et al. Partial coordination-perturbed bi-copper sites for selective electroreduction of
to hydrocarbons. Angew. Chem. Int. Ed. 60, 19829-19835 (2021). - Bagger, A. et al. Electrochemical
reduction: classifying Cu facets. ACS Catal. 9, 7894-7899 (2019). - Scholten, F. et al. Identifying structure-selectivity correlations in the electrochemical reduction of
: a comparison of wellordered atomically clean and chemically etched copper singlecrystal surfaces. Angew. Chem. Int. Ed. 60, 19169-19175 (2021). - Wu, Z.-Z. et al. Identification of
interfaces as superior active sites for CO dimerization during electroreduction. J. Am. Chem. Soc. 144, 259-269 (2021). - Akira, M. & Yoshio, H. Product selectivity affected by cationic species in electrochemical reduction of
and CO at a Cu electrode. Bull. Chem. Soc. Jpn 64, 123-127 (1991). - Hori, Y. et al. Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution. J. Chem. Soc. Faraday Trans. 85, 2309-2326 (1989).
- Resasco, J. et al. Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J. Am. Chem. Soc. 139, 11277-11287 (2017).
- Ren, W. et al. A cation concentration gradient approach to tune the selectivity and activity of
electroreduction. Angew. Chem. Int. Ed. 61, e202214173 (2022). - Ayemoba, O. & Cuesta, A. Spectroscopic evidence of size-dependent buffering of interfacial pH by cation hydrolysis during
electroreduction. ACS Appl. Mater. Interfaces 9, 27377-27382 (2017). - Singh, M. R. et al. Hydrolysis of electrolyte cations enhances the electrochemical reduction of
over Ag and Cu . J. Am. Chem. Soc. 138, 13006-13012 (2016). - Malkani, A. S. et al. Cation effect on interfacial
concentration in the electrochemical reduction reaction. ACS Catal. 10, 14871-14876 (2020). - Gu, J. et al. Modulating electric field distribution by alkali cations for
electroreduction in strongly acidic medium. Nat. Catal. 5, 268-276 (2022). - Ringe, S. et al. Understanding cation effects in electrochemical
reduction. Energy Environ. Sci. 12, 3001-3014 (2019). - Chen, L. D. et al. Electric field effects in electrochemical
reduction. ACS Catal. 6, 7133-7139 (2016). - Monteiro, M. C. O. et al. Absence of
electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 4, 654-662 (2021). - Qin, H.-G. et al. Quantitative understanding of cation effects on the electrochemical reduction of
and in acidic solution. ACS Catal. 13, 916-926 (2022). - Zhu, Q. et al. The solvation-induced Onsager reaction field rather than the double-layer field controls
reduction on gold. JACS Au 2, 472-482 (2022). - Groß, A. & Sakong, S. Ab initio simulations of water/metal interfaces. Chem. Rev. 122, 10746-10776 (2022).
- Qin, X. et al. Cation-coordinated inner-sphere
electroreduction at Au-water interfaces. J. Am. Chem. Soc. 145, 1897-1905 (2023). - Liu, H. et al. Promotional role of a cation intermediate complex in
formation from electrochemical reduction of over Cu . ACS Catal. 11, 12336-12343 (2021). - Yu, M. & Trinkle, D. R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 134, 064111 (2011).
- Chan, K. & Nørskov, J. K. Electrochemical barriers made simple. J. Phys. Chem. Lett. 6, 2663-2668 (2015).
- Kronberg, R. & Laasonen, K. Reconciling the experimental and computational hydrogen evolution activities of Pt(111) through DFTbased constrained MD simulations. ACS Catal. 11, 8062-8078 (2021).
- Zhao, X. & Liu, Y. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc. 143, 9423-9428 (2021).
- Le, J.-B. et al. Molecular origin of negative component of Helmholtz capacitance at electrified Pt(111)/water interface. Sci. Adv. 6, eabb1219 (2020).
- Vijay, S. et al. Unified mechanistic understanding of
reduction to CO on transition metal and single atom catalysts. Nat. Catal. 4, 1024-1031 (2021). - Cao, H. et al. Potential-dependent free energy relationship in interpreting the electrochemical performance of
reduction on single atom catalysts. ACS Catal. 12, 6606-6617 (2022). - Shin, S.-J. et al. A unifying mechanism for cation effect modulating
and productions from electroreduction. Nat. Commun. 13, 5482 (2022). - Le, D. & Rahman, T. S. On the role of metal cations in
electrocatalytic reduction. Nat. Catal. 5, 977-978 (2022). - Li, P. et al. Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt. Nat. Catal. 5, 900-911 (2022).
- Li, X.-Y. et al. Mechanism of cations suppressing proton diffusion kinetics for electrocatalysis. Angew. Chem. Int. Ed. 62, e202218669 (2023).
- Le, J.-B. et al. Molecular understanding of cation effects on double layer and its significance to CO-CO dimerization. Natl. Sci. Rev. 10, nwad105 (2023).
- Hussain, G. et al. How cations determine the interfacial potential profile: relevance for the
reduction reaction. Electrochim. Acta 327, 135055 (2019). - Garlyyev, B. et al. Influence of the nature of the alkali metal cations on the electrical double-layer capacitance of model Pt(111) and Au(111) electrodes. J. Phys. Chem. Lett. 9, 1927-1930 (2018).
- Xue, S. et al. How the nature of the alkali metal cations influences the double-layer capacitance of
, and Pt single-crystal electrodes. J. Phys. Chem. C 124, 12442-12447 (2020). - Qin, X. et al. Cation-induced changes in the inner- and outer-sphere mechanisms of electrocatalytic
reduction. Nat. Commun. 14, 7607 (2023). - Kwon, S. et al. Dramatic change in the step edges of the
electrocatalyst upon exposure to CO: operando observations by electrochemical STM and explanation using quantum mechanical calculations. ACS Catal. 11, 12068-12074 (2021). - Surendralal, S. et al. First-principles approach to model electrochemical reactions: understanding the fundamental mechanisms behind Mg corrosion. Phys. Rev. Lett. 120, 246801 (2018).
- Surendralal, S. et al. Impact of water coadsorption on the electrode potential of H-Pt(111)-liquid water interfaces. Phys. Rev. Lett. 126, 166802 (2021).
- Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
- Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
- Hammer, B. et al. Improved adsorption energetics within densityfunctional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413-7421 (1999).
- Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
- Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188-5192 (1976).
- Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695-1697 (1985).
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511-519 (1984).
- Woo, T. K. et al. A combined Car-Parrinello QM/MM implementation for ab initio molecular dynamics simulations of extended systems: application to transition metal catalysis. J. Phys. Chem. B 101, 7877-7880 (1997).
- Carter, E. A. et al. Constrained reaction coordinate dynamics for the simulation of rare events. Chem. Phys. Lett. 156, 472-477 (1989).
- Ryckaert, J.-P. et al. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327-341 (1977).
- Neugebauer, J. & Scheffler, M. Adsorbate-substrate and adsorbateadsorbate interactions of Na and K adlayers on Al (111). Phys. Rev. B 46, 16067-16080 (1992).
- Chan, K. & Nørskov, J. K. Potential dependence of electrochemical barriers from ab initio calculations. J. Phys. Chem. Lett. 7, 1686-1690 (2016).
Acknowledgements
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-44896-x.
http://www.nature.com/reprints
© The Author(s) 2024
Shenzhen Geim Graphene Center and Institute of Materials Research, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, People’s Republic of China. State Key Laboratory of Low Dimensional Quantum Physics and Department of Physics, Tsinghua University, Beijing 100084, People’s Republic of China. Institute for Advanced Study, Tsinghua University, Beijing 100084, People’s Republic of China. Frontier Science Center for Quantum Information, Beijing 100084, People’s Republic of China. Department of Energy Conversion and Storage, Technical University of Denmark, Kgs, Lyngby 2800, Denmark. e-mail: fykang@sz.tsinghua.edu.cn; li.jia@sz.tsinghua.edu.cn