DOI: https://doi.org/10.1038/s41557-024-01482-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38499849
تاريخ النشر: 2024-03-18
الهروب من القفص يحكم معدلات تفاعلات الفوتوريدوكس وعوائد الكم
تم القبول: 20 فبراير 2024
نُشر على الإنترنت: 18 مارس 2024
(د) التحقق من التحديثات
الملخص
تعتمد التحفيز الضوئي على نقل الإلكترونات المستحثة بالضوء مما يؤدي إلى تكوين زوج جذري يتكون من مانح مؤكسد وقابل مختزل في قفص مذيب. لكي تحدث تفاعلات منتجة، يجب أن يهرب المانح المؤكسد والقابل المختزل من ذلك القفص المذيب قبل أن يخضعا لنقل الإلكترونات العكسي التلقائي. هنا نوضح الدور الحاسم الذي يلعبه هروب القفص في ثلاث تفاعلات ضوئية مرجعية، وهي: الهيدروكسيل الهوائي، وإزالة البروم الاختزالية، وتفاعل أزا-هنري. باستخدام محفزات ضوئية قائمة على الروثينيوم(II) والكروم(III)، والتي توفر عوائد هروب قفص مختلفة جوهريًا، قمنا بتحديد علاقات كمية بين معدلات تكوين منتجات التحفيز الضوئي وعوائد هروب القفص. يمكن تفسير هذه النتائج إلى حد كبير ضمن إطار نظرية ماركوس لنقل الإلكترونات.
التفاعلات
النتائج والمناقشة
نقل الإلكترون المحفز بالضوء لمركبات الروثينيوم والكروم
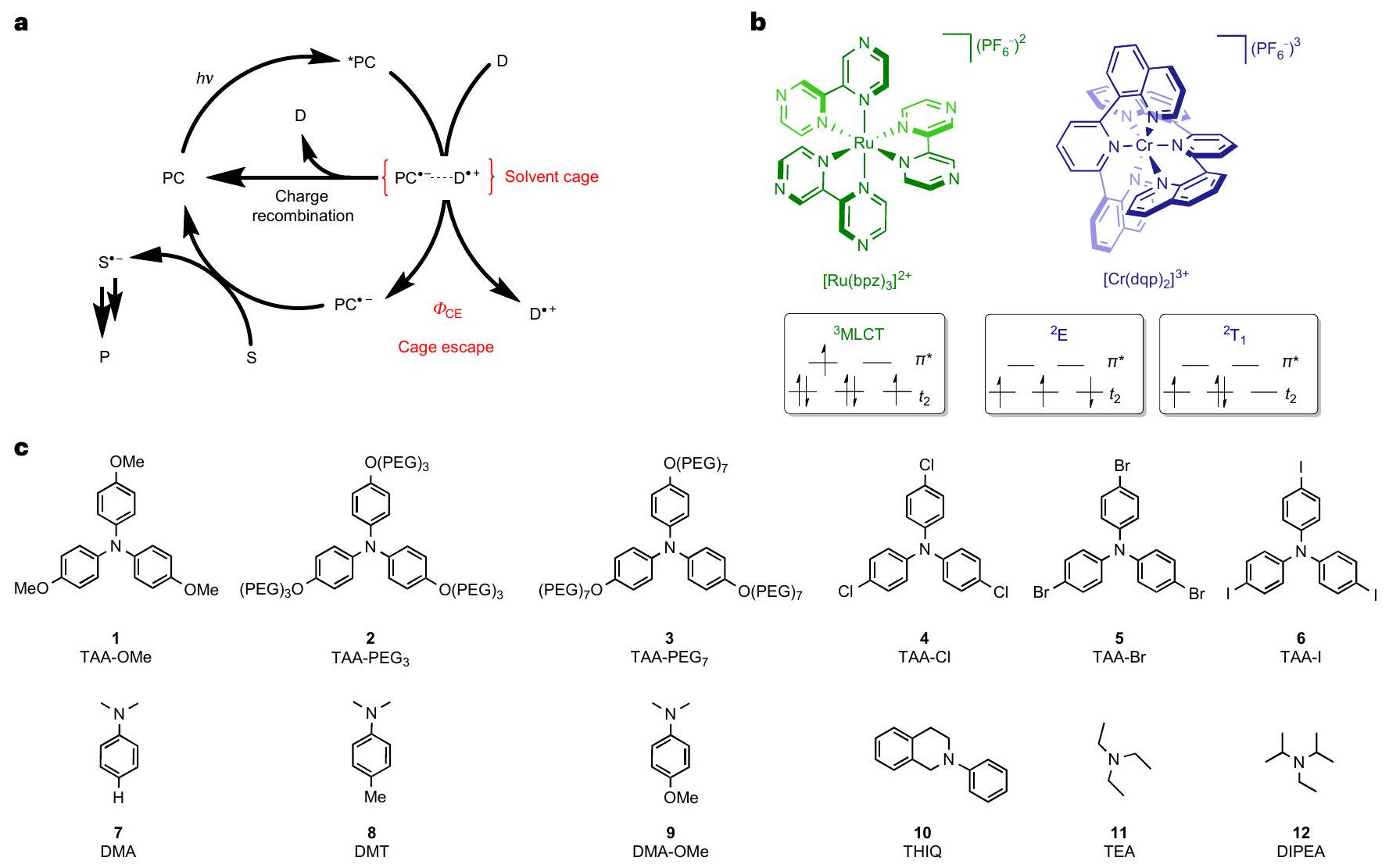
(2) تأثير حجم المانح (TAA-OMe، TAA-PEG
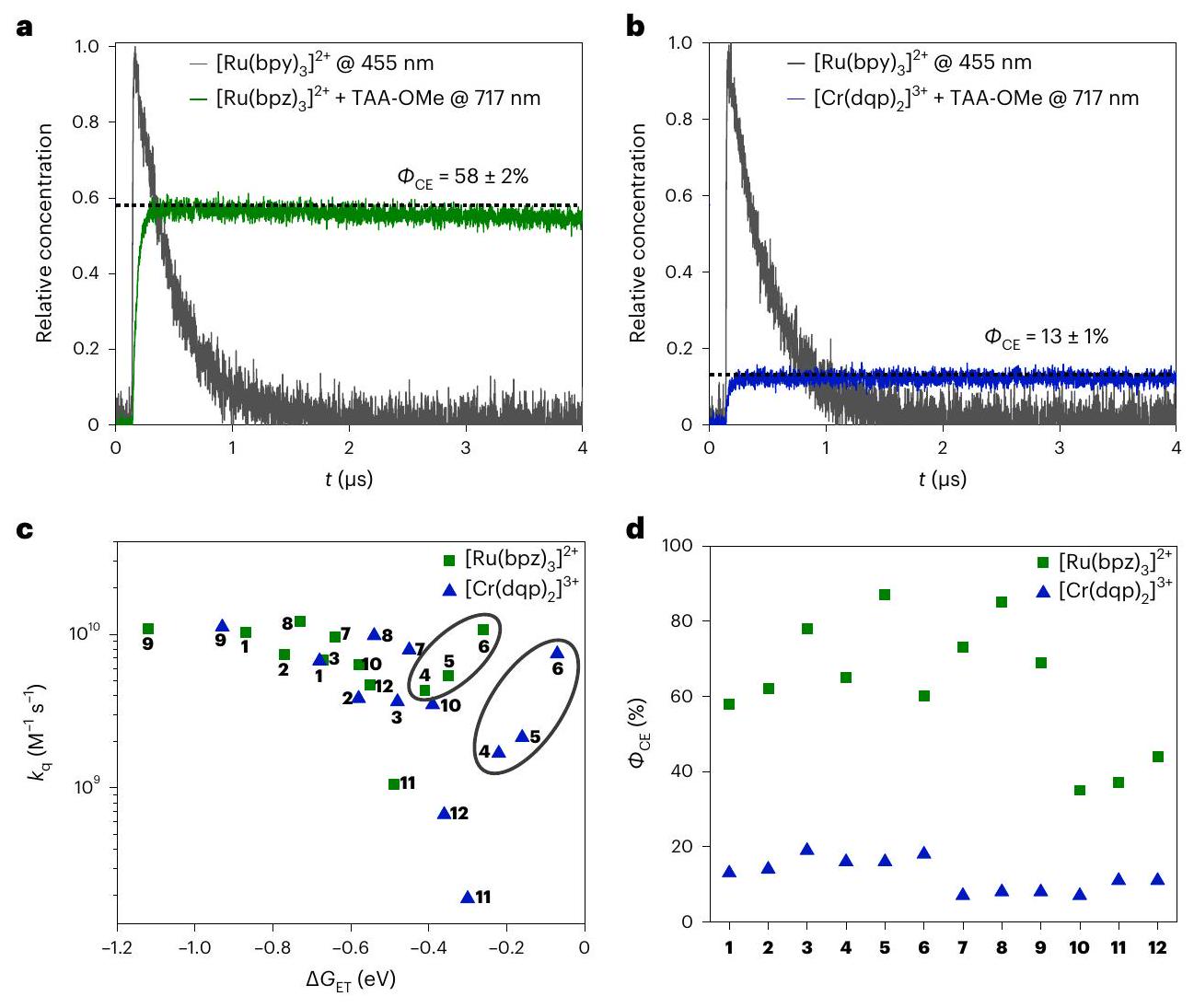
اتجاهات في عوائد الهروب من القفص الكمي
تم تحديد وجود المتبرعين 1-12 (الشكل 1c) في الأسيتونيتريل المؤكسد بواسطة القياس النسبي باستخدام التحليل الضوئي بالليزر (الأقسام التكميلية 6 و 7). المبدأ الأساسي لهذه الطريقة هو مقارنة تركيزات المنتجات الضوئية التي تتكون بعد إثارة عينة وحل مرجعي بشكل نبضي. المرجع، هنا، هو محلول مائي مؤكسد من
أ, زوال الامتصاص العابر لمركب المرجع
فائض كبير بالنسبة لمركبات المعادن (تركيزات ملليمولارية مقابل ميكرومولارية) لضمان نقل الإلكترونات المحفز بالضوء بسرعة وكفاءة قصوى (كمية)، ومع ذلك، ظل من الممكن تحفيز مركبات المعادن بشكل انتقائي في جميع الحالات بسبب معامل الانكسار الضئيل للمانحين عند الأطوال الموجية ذات الصلة. TAA-OMe
نقل الإلكترون، في حين أن مانحي الإلكترون الآخرين كانوا مانحين غير قابلين للعكس بسبب إزالة البروتون بعد الأكسدة الأولية لالكترون واحد
هروب مختلف من القفص لمركبات Ru و Cr
التكوينات (دوران منخفض
تم تحديده بالمعادلة (2). تعتمد طاقة إعادة التنظيم لعملية نقل الإلكترون العكسي داخل القفص على زوج المحفز الضوئي والمانح الفردي؛ ومع ذلك، في تحليلنا، تم السماح لطاقة إعادة التنظيم مع مانحين مختلفين بالتغير، ولكن بالنسبة لمانح إلكترون معين، كانت متطابقة.
ترابط الأداء الضوئي المحفز مع هروب القفص
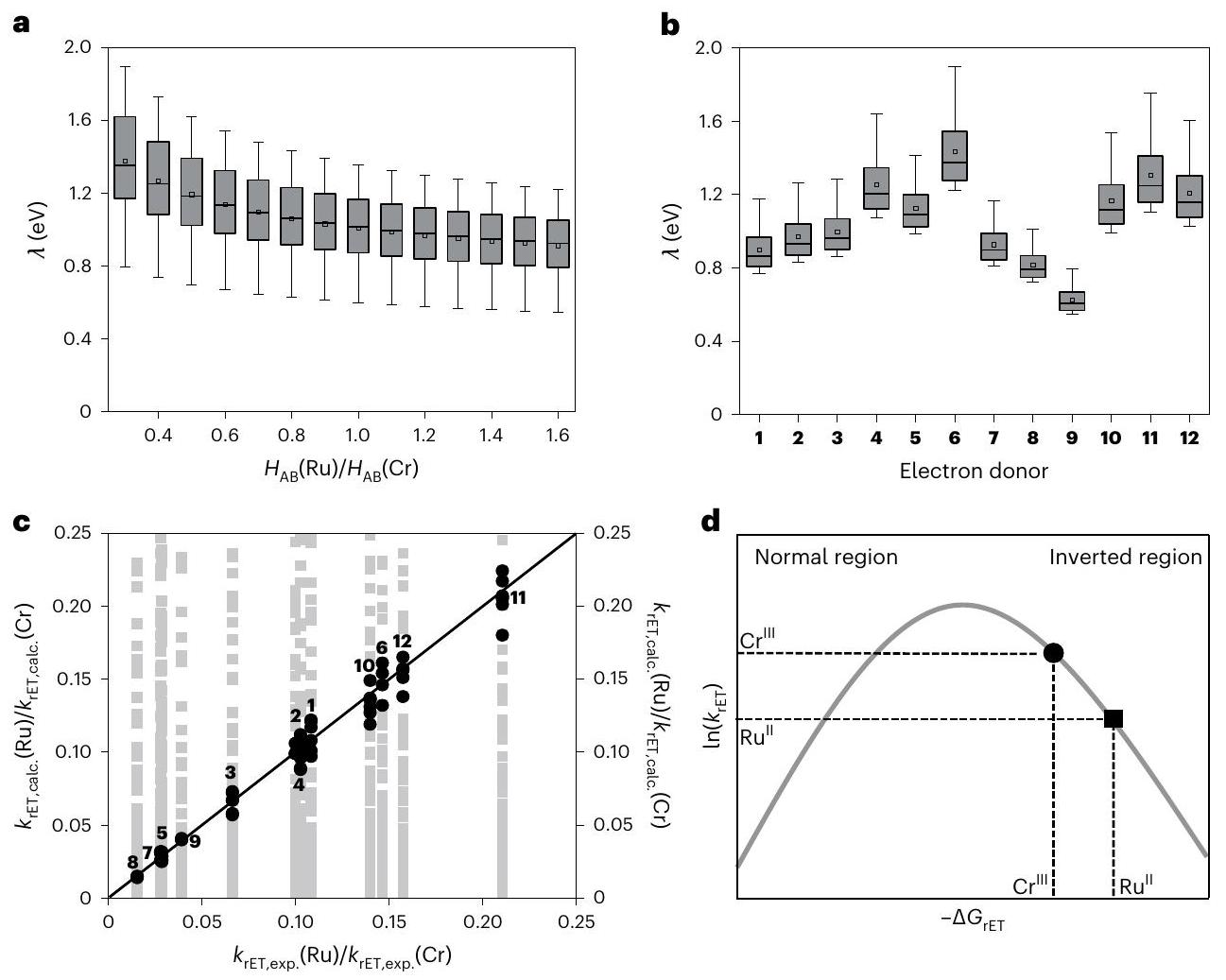
طاقة إعادة التنظيم لنسب معينة من التوصيلات الإلكترونية القادرة على إنتاج
و (3) تفاعل أزا-هنري الضوئي الحفاز، حيث يعمل THIQ كمانح للإلكترون وركيزة في نفس الوقت (الشكل 4 ج والملحق القسم 8.3). تم إجراء كل تفاعل مع كلا
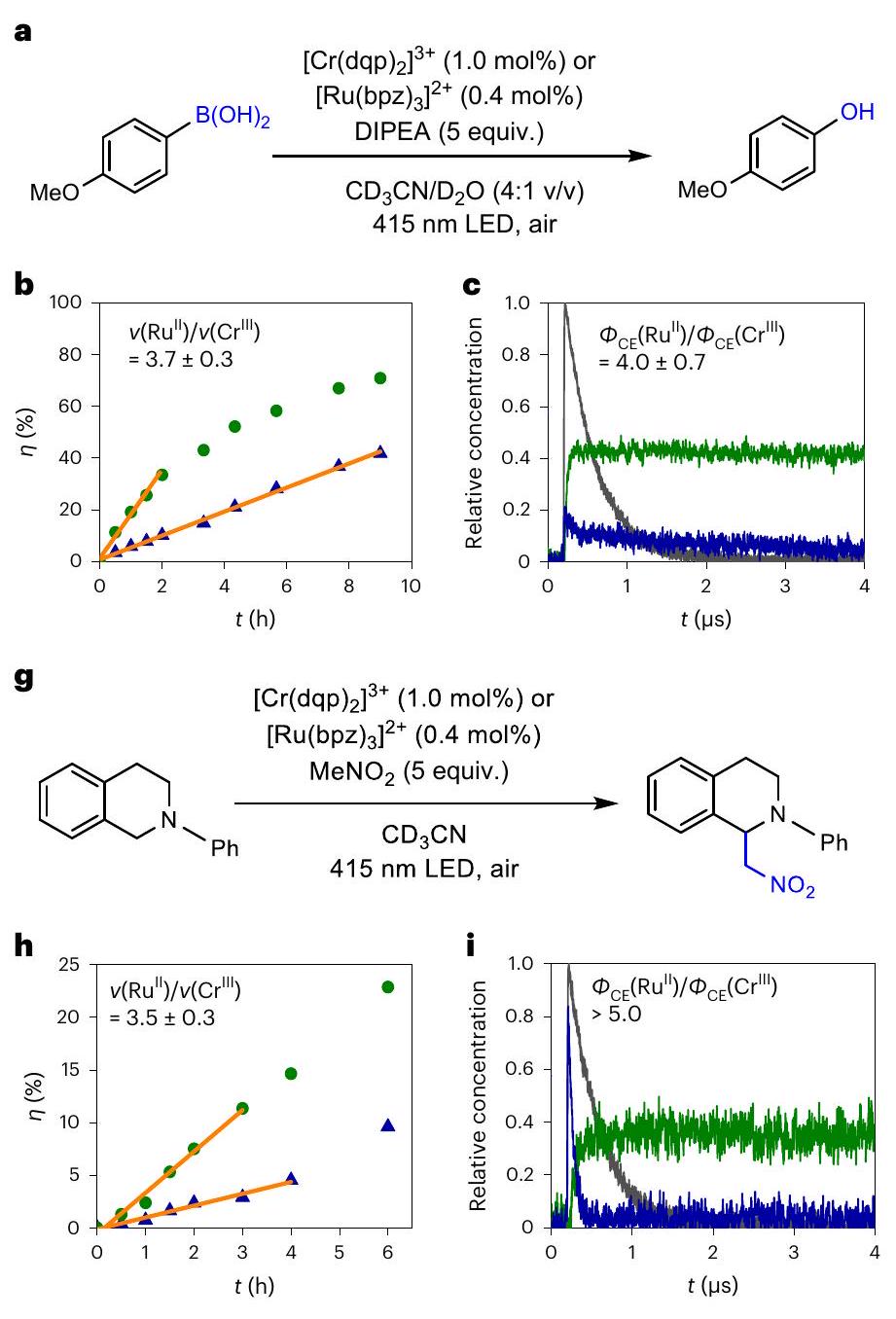
أ، د، ج، الهيدروكسيل الهوائي لحمض 4-ميثوكسي فينيل بورونيك (أ)، إزالة البروم الاختزالية لـ 2-برومو أسيتوفينون (د) وتفاعل آزا-هنري (ج). تم تعديل تركيزات
حدود التجارب المنفذة (القسم التكميلي 8.2 والشكل التكميلي 107).
الاستنتاجات
المحتوى عبر الإنترنت
References
- Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075-10166 (2016).
- Karkas, M. D., Porco, J. A. Jr & Stephenson, C. R. Photochemical approaches to complex chemotypes: applications in natural product synthesis. Chem. Rev. 116, 9683-9747 (2016).
- Yoon, T. P., Ischay, M. A. & Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2, 527-532 (2010).
- Marzo, L., Pagire, S. K., Reiser, O. & König, B. Visible-light photocatalysis: does it make a difference in organic synthesis? Angew. Chem. Int. Ed. 57, 10034-10072 (2018).
- Großkopf, J., Kratz, T., Rigotti, T. & Bach, T. Enantioselective photochemical reactions enabled by triplet energy transfer. Chem. Rev. 122, 1626-1653 (2022).
- Twilton, J. et al. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 1, 0052 (2017).
- Arias-Rotondo, D. M. & McCusker, J. K. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 45, 5803-5820 (2016).
- Stevenson, B. G. et al. Mechanistic investigations of an
-aminoarylation photoredox reaction. J. Am. Chem. Soc. 143, 8878-8885 (2021). - Buzzetti, L., Crisenza, G. E. M. & Melchiorre, P. Mechanistic studies in photocatalysis. Angew. Chem. Int. Ed. 58, 3730-3747 (2019).
- Balzani, V., Bergamini, G. & Ceroni, P. Light: a very peculiar reactant and product. Angew. Chem. Int. Ed. 54, 11320-11337 (2015).
- Aydogan, A. et al. Accessing photoredox transformations with an iron(III) photosensitizer and green light. J. Am. Chem. Soc. 143, 15661-15673 (2021).
- Kjær, K. S. et al. Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 363, 249-253 (2019).
- Hoffman, M. Z. Cage escape yields from the quenching of *Ru(bpy)
by methylviologen in aqueous solution. J. Phys. Chem. 92, 3458-3464 (1988). - Olmsted, J. III & Meyer, T. J. Factors affecting cage escape yields following electron-transfer quenching. J. Phys. Chem. 91, 1649-1655 (1987).
- Ripak, A. et al. Photosensitized activation of diazonium derivatives for C-B bond formation. Chem. Catal. 3, 100490 (2023).
- DiLuzio, S., Connell, T. U., Mdluli, V., Kowalewski, J. F. & Bernhard, S. Understanding Ir(III) photocatalyst structure-activity relationships: a highly parallelized study of light-driven metal reduction processes. J. Am. Chem. Soc. 144, 1431-1444 (2022).
- Sittel, S. et al. Visible-light induced fixation of
into organic molecules with polypyridine chromium(III) complexes. ChemCatChem 15, e202201562 (2023). - Sun, H., Neshvad, G. & Hoffman, M. Z. Energy gap dependence of the efficiency of charge separation upon the sacrificial reductive quenching of the excited states of Ru(II)-diimine photosensitizers in aqueous solution. Mol. Cryst. Liq. Cryst. 194, 141-150 (2006).
- Gould, I. R. et al. Electron-transfer reactions in the Marcus inverted region. Charge recombination versus charge shift reactions. J. Am. Chem. Soc. 111, 1917-1919 (1989).
- Wolff, H.-J., Bürßner, D. & Steiner, U. E. Spin-orbit coupling controlled spin chemistry of Ru(bpy)
photooxidation: detection of strong viscosity dependence of in-cage backward electron transfer rate. Pure Appl. Chem. 67, 167-174 (1995). - Gibbons, D. J., Farawar, A., Mazzella, P., Leroy-Lhez, S. & Williams, R. M. Making triplets from photo-generated charges: observations, mechanisms and theory. Photochem. Photobiol. Sci. 19, 136-158 (2020).
- Kikuchi, K., Hoshi, M., Niwa, T., Takahashi, Y. & Miyashi, T. Heavy-atom effects on the excited singlet-state electron-transfer reaction. J. Phys. Chem. 95, 38-42 (1991).
- Jayanthi, S. & Ramamurthy, P. Photoinduced electron transfer reactions of 2,4,6-triphenylpyrylium: solvent effect and charge-shift type of systems. Phys. Chem. Chem. Phys. 1, 4751-4757 (1999).
- Meidlar, K. & Das, P. K. Tris(2,2′-bipyridine)ruthenium(II)-sensitized photooxidation of phenols. Environmental effects on electron transfer yields and kinetics. J. Am. Chem. Soc. 104, 7462-7469 (1982).
- Ohno, T. & Lichtin, N. N. Electron transfer in the quenching of triplet methylene blue by complexes of iron(II). J. Am. Chem. Soc. 102, 4636-4643 (1980).
- Gould, I. R., Ege, D., Moser, J. E. & Farid, S. Efficiencies of photoinduced electron-transfer reactions: role of the Marcus inverted region in return electron transfer within geminate radical-ion pairs. J. Am. Chem. Soc. 112, 4290-4301 (1990).
- Kalyanasundaram, K. & Neumann-Spallart, M. Influence of added salts on the cage escape yields in the photoredox quenching of Ru(bpy)
excited states. Chem. Phys. Lett. 88, 7-12 (1982). - Delaire, J. A., Sanquer-Barrie, M. & Webber, S. E. Role of electrostatic interaction in light-induced charge separation in polyelectrolyte bound vinyldiphenylanthracene. J. Phys. Chem. 92, 1252-1257 (1988).
- Das, P. K., Encinas, V. & Scaiano, J. C. Laser flash photolysis study of the reactions of carbonyl triplets with phenols and photochemistry of p-hydroxypropiophenone. J. Am. Chem. Soc. 103, 4154-4162 (1981).
- Bürgin, T. H., Glaser, F. & Wenger, O. S. Shedding light on the oxidizing properties of spin-flip excited states in a
polypyridine complex and their use in photoredox catalysis. J. Am. Chem. Soc. 144, 14181-14194 (2022). - Rehm, D. & Weller, A. Kinetik und Mechanismus der Elektronübertragung bei der Fluoreszenzlöschung in Acetonitril. Ber. Bunsen-Ges. Phys. Chem. 73, 834-839 (1969).
- Ballardini, R., Varani, G., Indelli, M. T., Scandola, F. & Balzani, V. Free energy correlation of rate constants for electron transfer quenching of excited transition metal complexes. J. Am. Chem. Soc. 100, 7219-7223 (1978).
- Neumann, S., Kerzig, C. & Wenger, O. S. Quantitative insights into charge-separated states from one- and two-pulse laser experiments relevant for artificial photosynthesis. Chem. Sci. 10, 5624-5633 (2019).
- DeLaive, P. J., Foreman, T. K., Giannotti, C. & Whitten, D. G. Photoinduced electron transfer reactions of transition-metal complexes with amines. Mechanistic studies of alternate pathways to back electron transfer. J. Am. Chem. Soc. 102, 5627-5631 (1980).
- Kitzmann, W. R. & Heinze, K. Charge-transfer and spin-flip states: thriving as complements. Angew. Chem. Int. Ed. 62, e202213207 (2022).
- Gould, I. R., Moser, J. E., Ege, D. & Farid, S. Effect of molecular dimension on the rate of return electron transfer within photoproduced geminate radical ion pairs. J. Am. Chem. Soc. 110, 1991-1993 (1988).
- Pitre, S. P., McTiernan, C. D., Ismaili, H. & Scaiano, J. C. Mechanistic insights and kinetic analysis for the oxidative hydroxylation of arylboronic acids by visible light photoredox catalysis: a metal-free alternative. J. Am. Chem. Soc. 135, 13286-13289 (2013).
- Zou, Y.-Q. et al. Highly efficient aerobic oxidative hydroxylation of arylboronic acids: photoredox catalysis using visible light. Angew. Chem. Int. Ed. 51, 784-788 (2012).
- Maji, T., Karmakar, A. & Reiser, O. Visible-light photoredox catalysis: dehalogenation of vicinal dibromo-,
-halo-, and -dibromocarbonyl compounds. J. Org. Chem. 76, 736-739 (2011). - Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322-5363 (2013).
- Nicewicz, D. A. & MacMillan, D. W. C. Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes. Science 322, 77-80 (2008).
- Glaser, F., Kerzig, C. & Wenger, O. S. Sensitization-initiated electron transfer via upconversion: mechanism and photocatalytic applications. Chem. Sci. 12, 9922-9933 (2021).
- Bartling, H., Eisenhofer, A., König, B. & Gschwind, R. M. The photocatalyzed aza-Henry reaction of
-aryltetrahydroisoquinolines: comprehensive mechanism, – versus -abstraction, and background reactions. J. Am. Chem. Soc. 138, 11860-11871 (2016). - Condie, A. G., González-Gómez, J. C. & Stephenson, C. R. J. Visible-light photoredox catalysis: aza-Henry reactions via C-H functionalization. J. Am. Chem. Soc. 132, 1464-1465 (2010).
- Scaiano, J. C. T. Photochemistry Essentials (American Chemical Society, 2022).
- Talbott, E. D., Burnett, N. L. & Swierk, J. R. Mechanistic and kinetic studies of visible light photoredox reactions. Chem. Phys. Rev. 4, 031312 (2023).
- Marchini, M., Bergamini, G., Cozzi, P. G., Ceroni, P. & Balzani, V. Photoredox catalysis: the need to elucidate the photochemical mechanism. Angew. Chem. Int. Ed. 56, 12820-12821 (2017).
- Ghosh, I., Bardagi, J. I. & König, B. Reply to “Photoredox Catalysis: The Need to Elucidate the Photochemical Mechanism”. Angew. Chem. Int. Ed. 56, 12822-12824 (2017).
- Coles, M. S., Quach, G., Beves, J. E. & Moore, E. G. A photophysical study of sensitization-initiated electron transfer: insights into the mechanism of photoredox activity. Angew. Chem. Int. Ed. 59, 9522-9526 (2020).
- Tlili, A. & Lakhdar, S. Acridinium salts and cyanoarenes as powerful photocatalysts: opportunities in organic synthesis. Angew. Chem. Int. Ed. 60, 19526-19549 (2021).
- Speckmeier, E., Fischer, T. G. & Zeitler, K. A toolbox approach to construct broadly applicable metal-free catalysts for photoredox chemistry: deliberate tuning of redox potentials and importance of halogens in donor-acceptor cyanoarenes. J. Am. Chem. Soc. 140, 15353-15365 (2018).
- Kim, D. & Teets, T. S. Strategies for accessing photosensitizers with extreme redox potentials. Chem. Phys. Rev. 3, 021302 (2022).
- Krzywinski, M. & Altman, N. Visualizing samples with box plots. Nat. Methods 11, 119-120 (2014).
- Wang, C., Li, H., Bürgin, T. H. & Wenger, O. S. Data of figures and tables contained in the paper titled ‘Cage escape governs photoredox reaction rates and quantum yields’. figshare https:// doi.org/10.6084/m9.figshare. 22294531 (2024).
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
توفر البيانات
شكر وتقدير
مساهمات المؤلفين
التمويل
المصالح المتنافسة
معلومات إضافية
المواد الإضافية المتاحة على
https://doi.org/10.1038/s41557-024-01482-4.
يجب توجيه المراسلات وطلبات المواد إلى أوليفر س. وينجر.
- ¹قسم الكيمياء، جامعة بازل، بازل، سويسرا.
العنوان الحالي: قسم البيولوجيا والكيمياء، جامعة أوسنابروك، أوسنابروك، ألمانيا. البريد الإلكتروني: oliver.wenger@unibas.ch
DOI: https://doi.org/10.1038/s41557-024-01482-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38499849
Publication Date: 2024-03-18
Cage escape governs photoredox reaction rates and quantum yields
Accepted: 20 February 2024
Published online: 18 March 2024
(D) Check for updates
Abstract
Photoredox catalysis relies on light-induced electron transfer leading to a radical pair comprising an oxidized donor and a reduced acceptor in a solvent cage. For productive onward reaction to occur, the oxidized donor and the reduced acceptor must escape from that solvent cage before they undergo spontaneous reverse electron transfer. Here we show the decisive role that cage escape plays in three benchmark photocatalytic reactions, namely, an aerobic hydroxylation, a reductive debromination and an aza-Henry reaction. Using ruthenium(II)- and chromium(III)-based photocatalysts, which provide inherently different cage escape quantum yields, we determined quantitative correlations between the rates of photoredox product formation and the cage escape quantum yields. These findings can be largely rationalized within the framework of Marcus theory for electron transfer.
interactions
Results and discussion
Photoinduced electron transfer of Ru and Cr complexes
(2) the effect of donor size (TAA-OMe, TAA-PEG
Trends in cage escape quantum yields
the presence of the donors 1-12 (Fig. 1c) in aerated acetonitrile were determined by relative actinometry using laser flash photolysis (Supplementary Sections 6 and 7). The basic principle of this method is to compare the concentrations of the photoproducts formed after pulsed excitation of a sample and a reference solution. The reference, here an aqueous aerated solution of
a, Transient absorption decays of the reference complex
large excess relative to the metal complexes (millimolar versus micromolar concentrations) to ensure rapid and maximally efficient (quantitative) photoinduced electron transfer, yet selective excitation of the metal complexes remained possible in all cases due to the negligible extinction coefficient of the donors at the relevant wavelengths. The TAA-OMe
electron transfer, whereas the other electron donors were irreversible donors due to deprotonation after the initial one-electron oxidation
Different cage escape for Ru and Cr complexes
configurations (low-spin
determined with equation (2). The reorganization energy for in-cage reverse electron transfer depends on the individual photocatalystdonor pair; however, in our analysis, the reorganization energy with different donors was allowed to vary, but for a given electron donor, identical
Correlation of photocatalytic performance with cage escape
a, Reorganization energies for given ratios of electronic couplings able to produce
and (3) a photocatalytic aza-Henry reaction, where THIQ acts both as the electron donor and substrate (Fig. 4 g and Supplementary Section 8.3). Each reaction was performed with both
a,d,g, Aerobic hydroxylation of 4-methoxyphenylboronic acid (a), reductive debromination of 2-bromoacetophenone (d) and aza-Henry reaction (g). The concentrations of
limits of the experiments performed (Supplementary Section 8.2 and Supplementary Fig. 107).
Conclusions
Online content
References
- Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075-10166 (2016).
- Karkas, M. D., Porco, J. A. Jr & Stephenson, C. R. Photochemical approaches to complex chemotypes: applications in natural product synthesis. Chem. Rev. 116, 9683-9747 (2016).
- Yoon, T. P., Ischay, M. A. & Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2, 527-532 (2010).
- Marzo, L., Pagire, S. K., Reiser, O. & König, B. Visible-light photocatalysis: does it make a difference in organic synthesis? Angew. Chem. Int. Ed. 57, 10034-10072 (2018).
- Großkopf, J., Kratz, T., Rigotti, T. & Bach, T. Enantioselective photochemical reactions enabled by triplet energy transfer. Chem. Rev. 122, 1626-1653 (2022).
- Twilton, J. et al. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 1, 0052 (2017).
- Arias-Rotondo, D. M. & McCusker, J. K. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 45, 5803-5820 (2016).
- Stevenson, B. G. et al. Mechanistic investigations of an
-aminoarylation photoredox reaction. J. Am. Chem. Soc. 143, 8878-8885 (2021). - Buzzetti, L., Crisenza, G. E. M. & Melchiorre, P. Mechanistic studies in photocatalysis. Angew. Chem. Int. Ed. 58, 3730-3747 (2019).
- Balzani, V., Bergamini, G. & Ceroni, P. Light: a very peculiar reactant and product. Angew. Chem. Int. Ed. 54, 11320-11337 (2015).
- Aydogan, A. et al. Accessing photoredox transformations with an iron(III) photosensitizer and green light. J. Am. Chem. Soc. 143, 15661-15673 (2021).
- Kjær, K. S. et al. Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 363, 249-253 (2019).
- Hoffman, M. Z. Cage escape yields from the quenching of *Ru(bpy)
by methylviologen in aqueous solution. J. Phys. Chem. 92, 3458-3464 (1988). - Olmsted, J. III & Meyer, T. J. Factors affecting cage escape yields following electron-transfer quenching. J. Phys. Chem. 91, 1649-1655 (1987).
- Ripak, A. et al. Photosensitized activation of diazonium derivatives for C-B bond formation. Chem. Catal. 3, 100490 (2023).
- DiLuzio, S., Connell, T. U., Mdluli, V., Kowalewski, J. F. & Bernhard, S. Understanding Ir(III) photocatalyst structure-activity relationships: a highly parallelized study of light-driven metal reduction processes. J. Am. Chem. Soc. 144, 1431-1444 (2022).
- Sittel, S. et al. Visible-light induced fixation of
into organic molecules with polypyridine chromium(III) complexes. ChemCatChem 15, e202201562 (2023). - Sun, H., Neshvad, G. & Hoffman, M. Z. Energy gap dependence of the efficiency of charge separation upon the sacrificial reductive quenching of the excited states of Ru(II)-diimine photosensitizers in aqueous solution. Mol. Cryst. Liq. Cryst. 194, 141-150 (2006).
- Gould, I. R. et al. Electron-transfer reactions in the Marcus inverted region. Charge recombination versus charge shift reactions. J. Am. Chem. Soc. 111, 1917-1919 (1989).
- Wolff, H.-J., Bürßner, D. & Steiner, U. E. Spin-orbit coupling controlled spin chemistry of Ru(bpy)
photooxidation: detection of strong viscosity dependence of in-cage backward electron transfer rate. Pure Appl. Chem. 67, 167-174 (1995). - Gibbons, D. J., Farawar, A., Mazzella, P., Leroy-Lhez, S. & Williams, R. M. Making triplets from photo-generated charges: observations, mechanisms and theory. Photochem. Photobiol. Sci. 19, 136-158 (2020).
- Kikuchi, K., Hoshi, M., Niwa, T., Takahashi, Y. & Miyashi, T. Heavy-atom effects on the excited singlet-state electron-transfer reaction. J. Phys. Chem. 95, 38-42 (1991).
- Jayanthi, S. & Ramamurthy, P. Photoinduced electron transfer reactions of 2,4,6-triphenylpyrylium: solvent effect and charge-shift type of systems. Phys. Chem. Chem. Phys. 1, 4751-4757 (1999).
- Meidlar, K. & Das, P. K. Tris(2,2′-bipyridine)ruthenium(II)-sensitized photooxidation of phenols. Environmental effects on electron transfer yields and kinetics. J. Am. Chem. Soc. 104, 7462-7469 (1982).
- Ohno, T. & Lichtin, N. N. Electron transfer in the quenching of triplet methylene blue by complexes of iron(II). J. Am. Chem. Soc. 102, 4636-4643 (1980).
- Gould, I. R., Ege, D., Moser, J. E. & Farid, S. Efficiencies of photoinduced electron-transfer reactions: role of the Marcus inverted region in return electron transfer within geminate radical-ion pairs. J. Am. Chem. Soc. 112, 4290-4301 (1990).
- Kalyanasundaram, K. & Neumann-Spallart, M. Influence of added salts on the cage escape yields in the photoredox quenching of Ru(bpy)
excited states. Chem. Phys. Lett. 88, 7-12 (1982). - Delaire, J. A., Sanquer-Barrie, M. & Webber, S. E. Role of electrostatic interaction in light-induced charge separation in polyelectrolyte bound vinyldiphenylanthracene. J. Phys. Chem. 92, 1252-1257 (1988).
- Das, P. K., Encinas, V. & Scaiano, J. C. Laser flash photolysis study of the reactions of carbonyl triplets with phenols and photochemistry of p-hydroxypropiophenone. J. Am. Chem. Soc. 103, 4154-4162 (1981).
- Bürgin, T. H., Glaser, F. & Wenger, O. S. Shedding light on the oxidizing properties of spin-flip excited states in a
polypyridine complex and their use in photoredox catalysis. J. Am. Chem. Soc. 144, 14181-14194 (2022). - Rehm, D. & Weller, A. Kinetik und Mechanismus der Elektronübertragung bei der Fluoreszenzlöschung in Acetonitril. Ber. Bunsen-Ges. Phys. Chem. 73, 834-839 (1969).
- Ballardini, R., Varani, G., Indelli, M. T., Scandola, F. & Balzani, V. Free energy correlation of rate constants for electron transfer quenching of excited transition metal complexes. J. Am. Chem. Soc. 100, 7219-7223 (1978).
- Neumann, S., Kerzig, C. & Wenger, O. S. Quantitative insights into charge-separated states from one- and two-pulse laser experiments relevant for artificial photosynthesis. Chem. Sci. 10, 5624-5633 (2019).
- DeLaive, P. J., Foreman, T. K., Giannotti, C. & Whitten, D. G. Photoinduced electron transfer reactions of transition-metal complexes with amines. Mechanistic studies of alternate pathways to back electron transfer. J. Am. Chem. Soc. 102, 5627-5631 (1980).
- Kitzmann, W. R. & Heinze, K. Charge-transfer and spin-flip states: thriving as complements. Angew. Chem. Int. Ed. 62, e202213207 (2022).
- Gould, I. R., Moser, J. E., Ege, D. & Farid, S. Effect of molecular dimension on the rate of return electron transfer within photoproduced geminate radical ion pairs. J. Am. Chem. Soc. 110, 1991-1993 (1988).
- Pitre, S. P., McTiernan, C. D., Ismaili, H. & Scaiano, J. C. Mechanistic insights and kinetic analysis for the oxidative hydroxylation of arylboronic acids by visible light photoredox catalysis: a metal-free alternative. J. Am. Chem. Soc. 135, 13286-13289 (2013).
- Zou, Y.-Q. et al. Highly efficient aerobic oxidative hydroxylation of arylboronic acids: photoredox catalysis using visible light. Angew. Chem. Int. Ed. 51, 784-788 (2012).
- Maji, T., Karmakar, A. & Reiser, O. Visible-light photoredox catalysis: dehalogenation of vicinal dibromo-,
-halo-, and -dibromocarbonyl compounds. J. Org. Chem. 76, 736-739 (2011). - Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322-5363 (2013).
- Nicewicz, D. A. & MacMillan, D. W. C. Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes. Science 322, 77-80 (2008).
- Glaser, F., Kerzig, C. & Wenger, O. S. Sensitization-initiated electron transfer via upconversion: mechanism and photocatalytic applications. Chem. Sci. 12, 9922-9933 (2021).
- Bartling, H., Eisenhofer, A., König, B. & Gschwind, R. M. The photocatalyzed aza-Henry reaction of
-aryltetrahydroisoquinolines: comprehensive mechanism, – versus -abstraction, and background reactions. J. Am. Chem. Soc. 138, 11860-11871 (2016). - Condie, A. G., González-Gómez, J. C. & Stephenson, C. R. J. Visible-light photoredox catalysis: aza-Henry reactions via C-H functionalization. J. Am. Chem. Soc. 132, 1464-1465 (2010).
- Scaiano, J. C. T. Photochemistry Essentials (American Chemical Society, 2022).
- Talbott, E. D., Burnett, N. L. & Swierk, J. R. Mechanistic and kinetic studies of visible light photoredox reactions. Chem. Phys. Rev. 4, 031312 (2023).
- Marchini, M., Bergamini, G., Cozzi, P. G., Ceroni, P. & Balzani, V. Photoredox catalysis: the need to elucidate the photochemical mechanism. Angew. Chem. Int. Ed. 56, 12820-12821 (2017).
- Ghosh, I., Bardagi, J. I. & König, B. Reply to “Photoredox Catalysis: The Need to Elucidate the Photochemical Mechanism”. Angew. Chem. Int. Ed. 56, 12822-12824 (2017).
- Coles, M. S., Quach, G., Beves, J. E. & Moore, E. G. A photophysical study of sensitization-initiated electron transfer: insights into the mechanism of photoredox activity. Angew. Chem. Int. Ed. 59, 9522-9526 (2020).
- Tlili, A. & Lakhdar, S. Acridinium salts and cyanoarenes as powerful photocatalysts: opportunities in organic synthesis. Angew. Chem. Int. Ed. 60, 19526-19549 (2021).
- Speckmeier, E., Fischer, T. G. & Zeitler, K. A toolbox approach to construct broadly applicable metal-free catalysts for photoredox chemistry: deliberate tuning of redox potentials and importance of halogens in donor-acceptor cyanoarenes. J. Am. Chem. Soc. 140, 15353-15365 (2018).
- Kim, D. & Teets, T. S. Strategies for accessing photosensitizers with extreme redox potentials. Chem. Phys. Rev. 3, 021302 (2022).
- Krzywinski, M. & Altman, N. Visualizing samples with box plots. Nat. Methods 11, 119-120 (2014).
- Wang, C., Li, H., Bürgin, T. H. & Wenger, O. S. Data of figures and tables contained in the paper titled ‘Cage escape governs photoredox reaction rates and quantum yields’. figshare https:// doi.org/10.6084/m9.figshare. 22294531 (2024).
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
Data availability
Acknowledgements
Author contributions
Funding
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41557-024-01482-4.
Correspondence and requests for materials should be addressed to Oliver S. Wenger.
- ¹Department of Chemistry, University of Basel, Basel, Switzerland.
Present address: Department of Biology and Chemistry, Osnabrück University, Osnabrück, Germany. e-mail: oliver.wenger@unibas.ch