DOI: https://doi.org/10.1038/s41586-024-07231-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38538783
تاريخ النشر: 2024-03-27
الهندسة الجينية المعقدة للبوليبلويد في قصب السكر
تاريخ الاستلام: 24 فبراير 2023
تم القبول: 23 فبراير 2024
نُشر على الإنترنت: 27 مارس 2024
الوصول المفتوح
الملخص
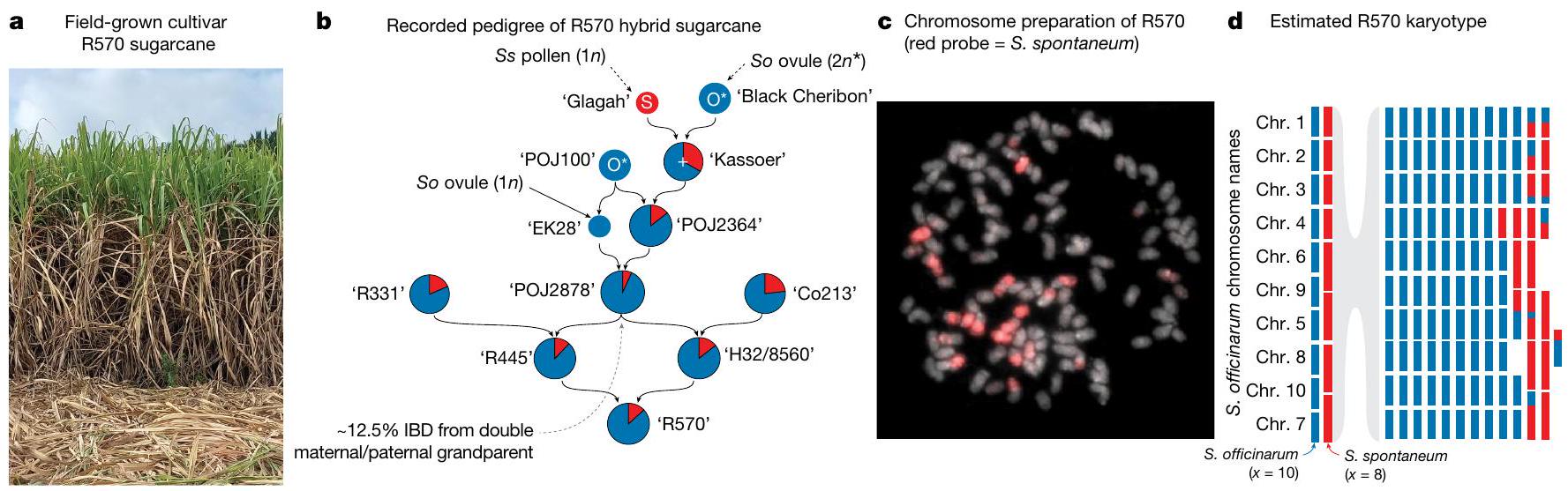
قصب السكر، المحصول الأكثر حصادًا في العالم من حيث الوزن، قد شكل التاريخ العالمي والتجارة والجغرافيا السياسية، وهو مسؤول حاليًا عن
في الأجيال الأولى من السلالات المعادة التهجين (BC1) كانت تحتوي على
مقدمة بين علامات اقتباس مفردة.
بين المتجانسات) ووراثة ثنائية الصيغة (مع ارتباط منهجي بين زوج من المتجانسات)
الطريق إلى جينوم تمثيلي
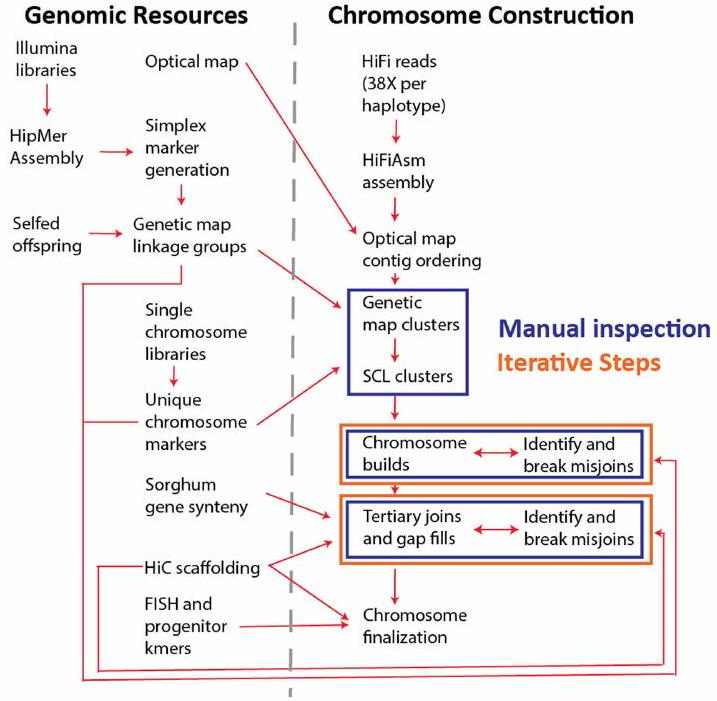
يمثل جينوم مثل R570 العديد من التحديات التقنية في التجميع وتمثيل الجينوم، حيث يحتوي R570 على جميع تعقيدات الجينومات المختلطة والمرباة. نظرًا للاختلافات في تفضيلات الاقتران بين كروموسومات R570، قد يكون من المناسب بيولوجيًا اتباع تمثيل الجينوم المختلط القياسي حيث يتم بناء تجميع لكل نظير ميوزي. ومع ذلك، نظرًا لسلالة التهجين الخلفي الخاصة به،
السابجينومات. لذلك، اخترنا تجميع جينوم جزئي قياسي لـ R570، حيث يمثل التجميع ‘الرئيسي’ تمثيلاً كاملاً للأنماط الوراثية الفريدة في R570 بينما يمثل ‘البديل’ أنماطًا وراثية إضافية متطابقة تقريبًا. بينما لا يحمل ‘البديل’ هنا نفس المعنى مقارنةً بالكائنات الحية ذات الاقتران الثنائي الصارم، قمنا بترتيب جينوم R570 بطريقة مماثلة لتحسين الفائدة للمجتمع.
مقالة
| متوقع
|
أخبار الأيام الثاني 5 | كر. 2 | أخبار الأيام الثالث | كر 10 | كر. 7 | كر. 8 | كر. 9 | أخبار الأيام الثاني 6 | كر. 4 | |
| كر. 1 // !! | ||||||||||
| تجمع أولي متطابق من الأنماط الوراثية انهار | كر. 2 | |||||||| ج I I I I I I |
|
|||||||
| سورغم بicolor v.3.1 | |||||| ج | |||||||||
|
||||||||||
|
|
 |
1 | ||||||||
| |''''!! | ||||||||||
|
||||||||||
| كر. 8 | 1 |
|
|
|||||||
|
جي، لي لي إي | |||||||||
| كر. 10 |
|
|||||||||
كشف المتغيرات. لدعم هذه الجهود، استخدمنا تجانس الجينات وأدلة تسلسل RNA (RNA-seq) لوصف مجموعة كاملة من تسلسلات البروتين المشفرة وتوضيح الجينات في التجميع الأساسي R570. التوضيح الأساسي مكتمل للغاية (BUSCO
المساهمة ضمن R570.c، خريطة التماثل الناتجة عن GENESPACE بين (من الأسفل إلى الأعلى) سورغوم بيكولور (الإصدار 3.1)، S. spontaneum (النوع الجيني AP85-441)، تجميعات الجينوم الأولية لـ R570 وR570 أحادي الصيغة الصبغية. تشير القطاعات الأفقية إلى الكروموسومات؛ الألوان (الأحمر-الأرجواني) تشير إلى الكروموسومات المتجانسة لسورغوم بيكولور (1-10) و”الضفائر” تمثل الكتل التماثلية بين كل زوج من الجينومات.
هندسة جينوم R570
| بشكل عام | س. أوفيشيناروم | س. سبونتانيوم | |
| حجم الجينوم الأساسي (N50 للكونتيغ) |
|
3.66 جيجابايت | 1.37 جيجابايت |
| حجم الجينوم البديل (N50 للكونتيغ) |
|
3.01 جيجابايت | 0.32 جيجابايت |
| حجم الجينوم مع الأخذ في الاعتبار الأنماط الوراثية المنهارة | 9.32 جيجابايت | – | – |
| الأنماط الوراثية المنهارة في التجميع
|
2.31 جيجابايت | 2.18 جيجابايت | 0.116 جيجابايت |
| عدد الجينات (عدد المجموعات المتجانسة المتجانسة) في التوصيف الأساسي
|
194,593
|
١٣٢٬٦١٨ | ٦١,١٩٧ |
| متوسط عدد الصبغيات للتجميع الأساسي (تغطية الكتل المتناظرة)
|
|
4.60x | 2.16x |
| متوسط هوية الببتيد بين الأليلات
|
– | 86% | 83% |
| الجينات المتأثرة بالمتغيرات الهيكلية
|
٥٣٦٢ | ٥٠٩٠ | ٢٦٠ |
وجدنا أن
استكشاف الأهداف للتربية
توصيف نظائر جينات المقاومة (RGAs)
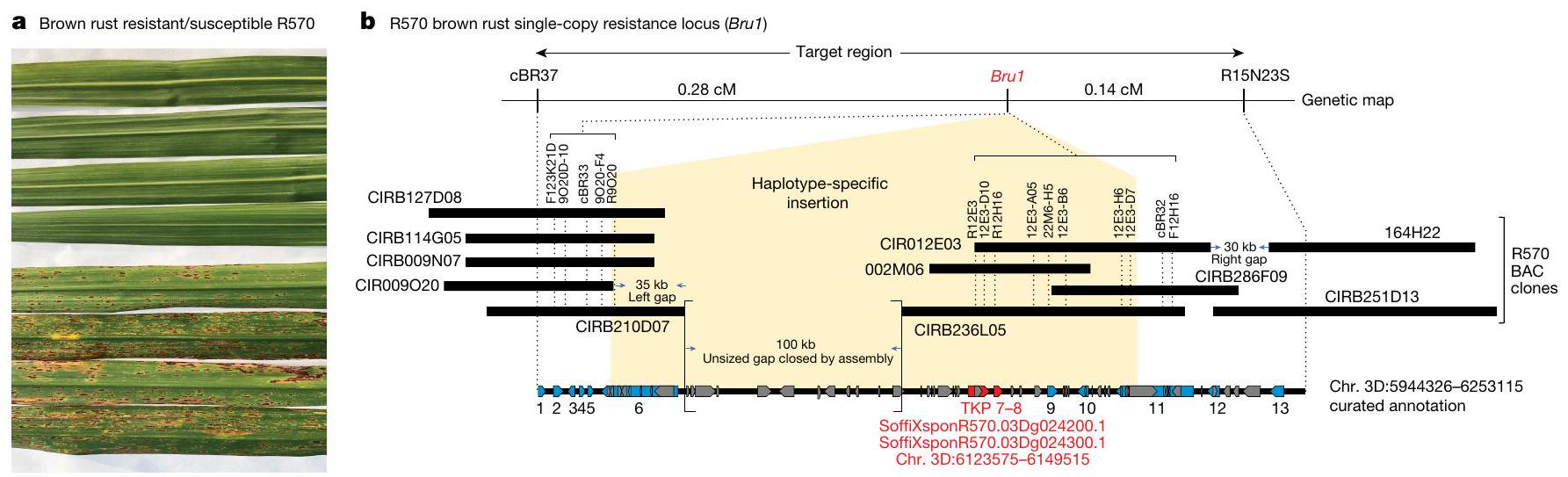
الأمراض التي تؤثر على جميع مناطق زراعة قصب السكر حول العالم هي الصدأ البني، الذي يسببه الفطر، Puccinia melanocephala. كان في السابق مسببا رئيسيا للأمراض في قصب السكر الذي تسبب في خسائر في المحصول تصل إلى 50%، وقد نجح المربون في التخفيف من خسائر P. melancocephala من خلال اختيار مقاومة الأمراض. تم تحديد موضع رئيسي (Bru1) يمنح مقاومة دائمة لهذه المرض (الشكل 3أ) في الصنف R570 (المراجع 43،44). لكشف الأليل المسبب الكامن وراء Bru1، استخدمت الدراسات السابقة نهج استنساخ قائم على الخريطة واسع النطاق الذي قام بفحص حوالي 2400 نسل ذاتي التلقيح من R570، مما قيد Bru1 إلى مجموعة من تسلسلات BAC التي امتدت لحوالي 209 كيلوبايت (المراجع 27،44) (الطرق). على الرغم من أن المنطقة احتوت على 13 نموذج جيني (الشكل 3ب والجدول التكميلي 13)، إلا أنها احتوت أيضًا على فجوة غير محددة وإدخال كبير من الهبلاي، وكلاهما يمنع المزيد من رسم الخرائط الدقيقة واكتشاف الجينات المرشحة بشكل شامل
المتغيرات (على سبيل المثال، الكينازات المتسلسلة وما إلى ذلك)) يصعب العثور عليها باستخدام التماثل التسلسلي فقط. تم التنبؤ بهيكلها عبر نطاق الحياة النباتية، ولكن تم التحقق من خمسة أمثلة فقط وظيفيًا في أحاديات الفلقة، جميعها منحت مقاومة لمسببات الأمراض الفطرية
الاستنتاجات
المحتوى عبر الإنترنت
- دينش بابو، ك. س. وآخرون. مراجعة قصيرة عن قصب السكر: استئناسه، التلاعبات الجزيئية وآفاق المستقبل. موارد جينية. تطور المحاصيل 69، 2623-2643 (2022).
- ياداف، س. وآخرون. تسريع المكاسب الجينية في تربية قصب السكر باستخدام الاختيار الجينومي. الزراعة 10، 585 (2020).
- غريفيت، ل.، غلازمان، ج.-سي. ودونت، أ. في حصاد داروين (تحرير موتلي، ت. ج. وآخرون) 49-66 (جامعة كولومبيا، 2006).
- بريمر، ج. مشاكل في تربية وعلم الخلايا لقصب السكر. يوفتيكا 10، 59-78 (1961).
- برنر، د. م. ولجندر، ب. ل. نقل الكروموسومات واستقرار الانقسام الميوزي لمشتقات قصب السكر (Saccharum spp.) الهجينة. علوم المحاصيل 33، 600-606 (1993).
- فييرا، م. ل. ج. وآخرون. إعادة النظر في الانقسام الميوزي في قصب السكر: عدم انتظامات الكروموسومات وانتشار التكوينات الثنائية. الجينات الأمامية 9، 213 (2018).
- أوليفيرا، ج. ك. وآخرون. الشذوذ الميوزي في قصب السكر (Saccharum spp.) والأنواع الأبوية: دليل على الانعكاسات المحيطية والبارامركزية. أن. أبليد. بيول.https://doi.org/ 10.1111/aab. 12855 (2023).
- هوار، ج.-ي. وآخرون. التحليل الجيني لنوع حديث من قصب السكر (Saccharum spp.). I. رسم الخرائط الجينومية باستخدام علامات AFLP. نظرية. تطبيق. علم الوراثة 103، 84-97 (2001).
- أيتكن، ك. س.، جاكسون، ب. أ. و مكينتاير، س. ل. توفر مجموعة من علامات AFLP و SSR تغطية شاملة للخريطة وتحديد المجموعات المرتبطة المتجانسة (أو المتشابهة) في صنف قصب السكر. نظرية. تطبيق. علم الوراثة. 110، 789-801 (2005).
- جانّو، ن.، غريفet، ل.، ديفيد، ج.، دُهُونت، أ. وغلازمان، ج.-سي. تباين في تفضيلات اقتران الكروموسومات أثناء الانقسام الاختزالي في قصب السكر متعدد الصيغ الصبغية كما كشفت عنه العلامات الجزيئية. الوراثة 93، 460-467 (2004).
- دومونت، ت. وآخرون. تربية قصب السكر في رينيون: التحديات والإنجازات وآفاق المستقبل. تكنولوجيا السكر 24، 181-192 (2022).
- دونت، أ. وآخرون. توصيف الهيكل الجيني المزدوج لأسلاف قصب السكر الحديثة (Saccharum spp.) بواسطة علم الوراثة الجزيئي. مول. جين. جينات. 250، 405-413 (1996).
- بايبيريديس، ن. ودي هونت، أ. تم فك شفرة بنية جينوم قصب السكر باستخدام مجسات أوليغو محددة الكروموسومات. مجلة النبات 103، 2039-2051 (2020).
- كوست، ل. وآخرون. هيكل الهبلايوت حول Bru1 يكشف عن أساس جيني ضيق لمقاومة الصدأ البني في أصناف قصب السكر الحديثة. نظرية. تطبيق. علم الوراثة. 125، 825-836 (2012).
- باركو، أ. س. وآخرون. توزيع وتكرار جين مقاومة الصدأ البني الرئيسي Bru1 في مجموعة قصب السكر العالمية. تربية النباتات. 136، 637-651 (2017).
- غارسميور، أ. وآخرون. تسلسل مرجعي أحادي الصيغة الجينية لمجموعة الجينوم المعقدة للغاية لقصب السكر. نات. كوم. 9، 2638 (2018).
- سوزا، ج. م. وآخرون. تجميع مساحة الجين 373k من جينوم قصب السكر متعدد الصيغ الصبغية يكشف عن خزانات من التنوع الوظيفي في المحصول الرائد في العالم للكتلة الحيوية. جيغا ساينس 8، giz129 (2019).
- شيرمان، ج. ر. وآخرون. مسودة تجميع الجينوم على مستوى الكروموسوم لقصب السكر التجاري. ساينس ريبورت. 12، 20474 (2022).
- نرك، س. وآخرون. التسلسل الكامل لجينوم الإنسان. ساينس 376، 44-53 (2022).
- سون، هـ. وآخرون. تجميع الجينوم على مستوى الكروموسوم وحل الهجينة لزراعة البطاطس رباعية الصيغة الصبغية. نات. جينت. 54، 342-348 (2022).
- أو، س.، تشين، ج. وجيانغ، ن. تقييم جودة تجميع الجينوم باستخدام مؤشر تجميع LTR (LAI). أبحاث الأحماض النووية 46، e126 (2018).
- سيمão، ف. أ.، ووترهاوس، ر. م.، إيوانيديس، ب.، كريفنتسيفا، إ. ف. وزدوبنوف، إ. م. BUSCO: تقييم اكتمال تجميع الجينوم والتعليق باستخدام الأورثولوجات ذات النسخة الواحدة. المعلوماتية الحيوية 31، 3210-3212 (2015).
- لوفيل، ج. ت. وآخرون. جين سبيس تتبع مناطق الاهتمام وتنوع عدد نسخ الجينات عبر عدة جينومات. إي لايف 11، e78526 (2022).
- كوادرا، أ.، أسيفيدو، ر.، مورينو دياث دي لا إسبينا، س.، جوف، ن. ودي لا توري، س. إعادة تشكيل الجينوم في ثلاثة أنواع حديثة من S. officinarum
أصناف قصب السكر S. spontaneum. ج. إكسب. بوت. 55، 847-854 (2004). - بايبيريديس، ج.، بايبيريديس، ن. ودي هونت، أ. تحقيق سيتوجينيتيكي جزيئي لتكوين الكروموسومات وانتقالها في قصب السكر. مول. جينت. جينومكس 284، 65-73 (2010).
- أيتكن، ك. س. وآخرون. خريطة جينية شاملة للقصب توفر تغطية محسّنة للخريطة وتدمج علامات تقنية مصفوفة التنوع عالية الإنتاجية (DArT). BMC جينوم. 15، 152 (2014).
- غارسميور، أ. وآخرون. الحفاظ العالي على الجينات المتجانسة على الرغم من الازدواجية الشديدة في الأوتوبوليبلويد في قصب السكر. نيو فيتولوجي. 189، 629-642 (2011).
- فيليلا، م. وآخرون. تحليل ثلاثة مناطق متجانسة/متجانسة من قصب السكر يقترح أحداث تعدد الصبغيات المستقلة لسكروم أوفيسيناروم وسكروم سبونتانيوم. علم الجينوم والتطور 9، 266-278 (2017).
- بومبيدور، ن. وآخرون. ثلاثة جينومات سلفية مؤسِّسة متورطة في أصل قصب السكر. آن. بوت. 127، 827-840 (2021).
- جانّو، ن. وآخرون. مقارنة الأشكال المتماثلة في منطقة غنية بالجينات بين الأعشاب تكشف عن الاستقرار في الجينوم متعدد الصيغ للسكر. مجلة النبات 50، 574-585 (2007).
- تشانغ، ق. وآخرون. رؤى جينومية حول الانخفاض الأخير في الكروموسومات في قصب السكر التلقائي متعدد الصيغ الصبغية Saccharum spontaneum. نات. جينت. 54، 885-896 (2022).
- Zhang، ج. وآخرون. الجينوم المحدد بالأليل من قصب السكر التلقائي Saccharum spontaneum L. نات. جينت. 50، 1565-1573 (2018).
- كمال، ن. وآخرون. جينوم الشوفان الموزاييك يقدم رؤى حول محصول حبوب صحي بشكل فريد. الطبيعةhttps://doi.org/10.1038/s41586-022-04732-y (2022).
- سونغ، ك.، لو، ب.، تانغ، ك. وأوزبورن، ت. سي. التغير السريع في الجينوم في البوليبloid الاصطناعي من براسكا وآثاره على تطور البوليبloid. وقائع الأكاديمية الوطنية للعلوم في الولايات المتحدة الأمريكية 92، 7719-7723 (1995).
- ليو، ب. وآخرون. تغييرات جينية سريعة في القمح متعدد الصيغ الوراثية والأنواع ذات الصلة: تداعيات على تطور الجينوم والتحسين الوراثي. مجلة الوراثة والجينوم 36، 519-528 (2009).
- لو، إكس. وآخرون. تحليل الفصل بين علامات الميكروساتلايت (SSR) في بوليبلود قصب السكر. جينات. مول. ريس. 14، 18384-18395 (2015).
- بايبيريديس، ن. وآخرون. الجينات المقارنة في قصب السكر تمكن من تحسين الخريطة الهيكلية والتحقق من ارتباطات العلامات والسمات. مول. بريد. 21، 233-247 (2008).
- ستورناغل، ب. وآخرون. أداة NLR-Annotator تمكّن من توضيح مجموعة مستقبلات المناعة داخل الخلوية. فيزيولوجيا النبات. 183، 468-482 (2020).
مقالة
- أيتكن، ك. س.، جاكسون، ب. أ. و مكينتاير، س. ل. تم تحديد مواقع الصفات الكمية للصفات المتعلقة بالسكر في صنف من قصب السكر (Saccharum spp.)
سكان سكروم أوفيسيناروم. نظرية. تطبيق. علم الوراثة 112، 1306-1317 (2006). - هوار، ج.-ي. وآخرون. التحليل الجيني لنوع حديث من قصب السكر (Saccharum spp.). II. الكشف عن QTLs لمكونات الإنتاج. نظرية. تطبيق. علم الوراثة 105، 1027-1037 (2002).
- مينغ، ر. وآخرون. التحليل الجزيئي للسمات المعقدة في الأوتوبوليبلويد: رسم خرائط QTLs التي تؤثر على إنتاج السكر والسمات ذات الصلة في قصب السكر. نظرية. تطبيق. علم الوراثة. 105، 332-345 (2002).
- كوبر، إ. أ. وآخرون. جينوم مرجعي جديد لسورغم بيكولور يكشف عن مستويات عالية من التشابه التسلسلي بين الأنماط الجينية الحلوة والحبوب: تداعيات على وراثيات استقلاب السكر. BMC Genom. 20، 420 (2019).
- داوغروس، ج. هـ. وآخرون. جين رئيسي محتمل لمقاومة الصدأ مرتبط بعلامة RFLP في صنف قصب السكر ‘R570’. نظرية. تطبيق. علم الوراثة. 92، 1059-1064 (1996).
- Le Cunff، L. وآخرون. رسم خرائط النقل المتناظر ثنائي الصيغة الصبغية/متعدد الصيغة الصبغية والمشي الكروموسومي المحدد بالنمط الوراثي نحو جين مقاومة الصدأ (Bru1) في قصب السكر عالي التعدد الصبغي.
علم الوراثة 180، 649-660 (2008). - جيش، ل. أ. وكلارك، س. إ. عائلة كينازات RLK/Pelle. مجلة النبات 66، 117-127 (2011).
- بروغمان، ر. وآخرون. جين مقاومة صدأ الساق في الشعير Rpg1 هو جين جديد لمقاومة الأمراض له تشابه مع كينازات المستقبلات. وقائع الأكاديمية الوطنية للعلوم في الولايات المتحدة الأمريكية 99، 9328-9333 (2002).
- كليميك، ف. وآخرون. استنساخ جين المقاومة Yr15 من القمح يسلط الضوء على عائلة كيناز-زائف الكيناز المت tandem في النباتات. نات. كوم. 9، 3735 (2018).
- لويس، ج. د.، لو، ت.، باستيدو، ب.، غوتمان، د. س. و ديسفيو، د. صعود الأموات الأحياء: الكينازات الزائفة كوسائط للمناعة المحفزة بواسطة المؤثرات. سلوك إشارة النبات. 9، e27563 (2014).
- كليميك، ف.، كوكير، ج.، فهيمة، ت. وبوزنياك، ج. ج. تظهر كينازات البروتين المتتالية كمنظمات جديدة لمناعة النباتات. تفاعل النبات والميكروبات الجزيئي 34، 1094-1102 (2021).
(ج) المؤلف(ون) 2024
طرق
تسلسل الجينوم
مكتبات الكروموسومات المفروزة بتدفق فردي. تم بناء مكتبات التسلسل باستخدام مجموعة مكتبة Illumina TruSeq DNA Nano وفقًا للبروتوكولات القياسية. تم تسلسل المكتبات إما على جهاز Illumina HiSeq2500 أو NovaSeq 6000 باستخدام نهايات متزاوجة وطول قراءة يبلغ 150 قاعدة.
المكتبات المتبقية من إلومينا. تم تقطيع شظايا الحمض النووي (DNA) بحجم 400 نقطة أساس (bp) إلى 2 ميكروغرام باستخدام جهاز كوفاريس LE220 وتم اختيار الحجم باستخدام جهاز بيبين (Sage Science). تم معالجة الشظايا بإصلاح الأطراف، وإضافة ذيول A، وربط المحولات المتوافقة مع إلومينا (IDT) باستخدام مجموعة إنشاء مكتبات كابا-إلومينا (KAPA Biosystems). تم قياس المكتبات المعدة باستخدام مجموعة qPCR لمكتبات تسلسل الجيل التالي من كابا بيوسيستمز (Roche) وتم تشغيلها على جهاز PCR الحقيقي من Roche LightCycler 480. ثم تم إعداد المكتبات المقاسة للتسلسل على منصة تسلسل إلومينا HiSeq باستخدام مجموعة TruSeq Rapid للعنقود المزدوج، الإصدار 2، مع جهاز تسلسل HiSeq 2500 لإنشاء خلية تدفق عنقودية للتسلسل. تم إجراء تسلسل خلية التدفق على جهاز تسلسل إلومينا HiSeq 2500 باستخدام مجموعات تسلسل HiSeq Rapid SBS، الإصدار 2، بعد
v.7989. تم إجراء تجميعين منفصلين باستخدام معلمات optArguments_nonhaplotype_noES_BG_DLE1_saphyr.xml. تم إجراء التجميع الأول بدون قطع المناطق متعددة المسارات المعقدة (CMPR) وأنتج 570 خريطة بطول N50 يبلغ 36.444 ميجاباز. والطول الإجمالي للخريطة هو 7,654.039 ميجاباز. تم إجراء تجميع إضافي باستخدام خيار قطع CMPR، الذي يقدم قطعًا في الخرائط عند التكرارات المحتملة لتقليل التبديل المحتمل بين الهومولوج والطور. التجميع الممكّن بقطع CMPR أنتج 1,512 خريطة بطول N50 يبلغ 9.546 ميجاباز والطول الإجمالي للخريطة هو
نطاقات التغطية
عبر GENESPACE (الإصدار 0.9.4)
كشفت أنهما ينتميان إلى عائلة RLK-PELLE-DSLV
ملخص التقرير
توفر البيانات
توفر الشيفرة
50. هوانغ، ي. وآخرون. العناصر الراجعة الوفيرة المحددة للأنواع توضح التركيب الجيني لأسلاف قصب السكر الحديثة. كروموسوما 129، 45-55 (2020).
51. ميتكالف، سي. جي. وآخرون. التوصيف باستخدام تقنية تحليل تدفق الخلايا للجينوم المعقد متعدد الصيغ الصبغية لقصب السكر (Saccharum officinarum) وسلالات قصب السكر الحديثة. تقارير العلوم 9، 19362 (2019).
52. ميتكالف، سي. جي. وآخرون. عزل وتسلسل نسخة واحدة من كروموسوم مدخل من جينوم معقد لتحديد الجينات والـ SNP. نظرية. تطبيق. علم الوراثة 135، 1279-1292 (2022).
53. فرانا، ج. وآخرون. فرز الكروموسومات باستخدام تقنية تحليل تدفق الخلايا في النباتات: الجيل القادم. طرق 57، 331-337 (2012).
54. فولجر، م. ر. وآخرون. تسلسل طويل القراءة وتجميع التكرارات القطاعية. نات. ميثودز 16، 88-94 (2019).
55. هافورد، م. ب. وآخرون. تجميع جديد، توضيح وتحليل مقارن لـ 26 جينوم متنوع من الذرة. ساينس 373، 655-662 (2021).
56. وانغ، م. وكونغ، ل. pblat: خوارزمية blat متعددة الخيوط تسرع محاذاة التسلسلات إلى الجينومات. BMC Bioinformatics 20، 28 (2019).
57. لي، هـ. ميني ماب 2: المحاذاة الزوجية لتسلسلات النوكليوتيدات. المعلوماتية الحيوية 34، 3094-3100 (2018).
58. لي، هـ. وآخرون. تنسيق المحاذاة/الخريطة وSAMtools. المعلوماتية الحيوية 25، 2078-2079 (2009).
59. كوينلان، أ. ر. BEDTools: الأداة متعددة الاستخدامات لتحليل ميزات الجينوم. بروتوكولات حالية. المعلوماتية الحيوية 47، 11.12.1-34 (2014).
60. لوفيل، ج. ت. وآخرون. المشهد الجينومي للاستجابات الجزيئية للإجهاد الناتج عن الجفاف الطبيعي في بانكوم هالي. نات. كوم. 9، 5213 (2018).
61. وو، ت. د. وناكو، س. الكشف السريع والمتسامح مع SNP عن المتغيرات المعقدة والتقطيع في القراءات القصيرة. المعلوماتية الحيوية 26، 873-881 (2010).
62. هاس، ب. ج. وآخرون. تحسين توضيح جينوم الأرابيدوبسيس باستخدام تجميعات محاذاة النسخ القصوى. أبحاث الأحماض النووية 31، 5654-5666 (2003).
63. سلاموف، أ. أ. وسولوفيف، ف. ف. العثور على الجينات من البداية في الحمض النووي الجينومي لذباب الفاكهة. أبحاث الجينوم 10، 516-522 (2000).
64. سلاتر، ج. س. س. و بيرني، إ. التوليد الآلي للحدس لمقارنة التسلسلات البيولوجية. BMC المعلوماتية الحيوية 6، 31 (2005).
65. ستانك، م. وآخرون. AUGUSTUS: التنبؤ من البداية بالنُسخ البديلة. أبحاث الأحماض النووية. 34، W435-9 (2006).
66. سميت، أ. ف. أ. & هوبلي، ر. نموذج التكرار Open-1.0.http://www.repeatmasker.org/ (2010).
67. مكورميك، ر. ف. وآخرون. الجينوم المرجعي للذرة الرفيعة (Sorghum bicolor): تحسين التجميع، تعليقات الجينات، أطلس النسخ، وتوقيعات تنظيم الجينوم. مجلة النبات 93، 338-354 (2018).
68. ماميتي، س. وآخرون. مورد جينومي للذرة الرفيعة الخضراء Setaria viridis يمكّن من اكتشاف المواقع ذات القيمة الزراعية. نات. بيولوجيا حيوية. 38، 1203-1210 (2020).
69. وانغ، ي. وآخرون. MCScanX: مجموعة أدوات للكشف والتحليل التطوري لتوافق الجينات والتوازي. أبحاث الأحماض النووية. 40، e49 (2012).
70. إيمس، د. م. وكيللي، س. أورثوفايندر: استنتاج التماثل النشوي لعلم الجينوم المقارن. جينوم بيو. 20، 238 (2019).
71. غويل، م.، صن، هـ.، جياو، و. ب. وشنيبرغر، ك. SyRI: العثور على إعادة ترتيب الجينوم والاختلافات التسلسلية المحلية من تجميعات الجينوم الكامل. علم الجينوم 20، 1-13 (2019).
72. بيجز، إتش.، أبويون، ب.، جنتلمان، ر. وديبروي، س. بيوسترينغز: كائنات سلسلة تمثل التسلسلات البيولوجية، وخوارزميات المطابقة (2008). إصدار حزمة R 2.0 (2015).
73. كاتوه، ك. وستاندلي، د. م. برنامج محاذاة التسلسلات المتعددة MAFFT الإصدار 7: تحسينات في الأداء وسهولة الاستخدام. مول. بيول. إيفول. 30، 772-780 (2013).
74. زو، ل. وآخرون. ggmsa: أداة استكشاف بصرية لمحاذاة التسلسل المتعدد والبيانات المرتبطة. بريف. بايوإنفورم. 23، bbac222 (2022).
75. لين، ز. وآخرون. التنبؤ بمستوى الذرة لهيكل البروتين على نطاق تطوري باستخدام نموذج لغوي. ساينس 379، 1123-1130 (2023).
76. سوياما، م.، تورنتس، د. وبورك، ب. PAL2NAL: تحويل موثوق لمحاذاة تسلسلات البروتين إلى المحاذاة المقابلة للكودونات. أبحاث الأحماض النووية 34، W609-12 (2006).
77. شريف، د. ولوبري، ج. ر. في الأساليب الهيكلية لتطور التسلسل: الجزيئات، الشبكات، السكان (محررون باستولا، أ.، بورتو، م.، رومان، هـ. إ. وفيندروسكولو، م.) 207-232 (سبرينجر برلين هايدلبرغ، 2007).
78. هوانغ، سي.-سي. وآخرون. معدلات التطور للعلامات النووية والعضوية المستخدمة بشكل شائع لأقارب الأرابيدوبسيس (Brassicaceae). جين 499، 194-201 (2012).
79. أسناغي، سي. وآخرون. تطبيق التماثل عبر عائلة الحشائش لتحديد موقع خريطة جين مقاومة صدأ قصب السكر. نظرية. تطبيق. علم الوراثة. 101، 962-969 (2000).
80. مينيك، ز. الأدوار الفسيولوجية للغليكوسيد هيدرازات النباتات. بلانتا 227، 723-740 (2008).
81. ليفي، إ.، شاني، ز. وشوسييف، أ. تعديل البوليسكاريدات وجدار الخلية النباتية بواسطة إنزيم الإندو-1،4-بيتا-غلوكاناز ومجالات ربط السليلوز. هندسة الجزيئات الحيوية 19، 17-30 (2002).
معلومات إضافية
يجب توجيه المراسلات والطلبات للحصول على المواد إلى A. L. Healey أو J. Schmutz أو A. D’Hont.
تُعرب مجلة Nature عن شكرها لإليزابيث كوبر والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل.
معلومات إعادة الطباعة والتصاريح متاحة علىhttp://www.nature.com/reprints.
مقارنة تسلسل البديل الأساسي C
| عمق الهبلايوتوب | الأسس المحسوبة (جيجابايت) | الأسس الممثلة (جيجابايت) | تمثيل الجينوم (%) |
| 0X | 1.47 | 0.74 | ٧.٩ |
| 1X | ٤.٦٣ | ٤.٦٣ | ٤٩.٦ |
| 2X | 1.15 | ٢.٣٠ | ٢٤.٦ |
| 3X | 0.35 | 1.05 | 11.3 |
| 4X | 0.15 | 0.60 | 6.6 |
| حجم الجينوم الممثل | 9.32 جيجابايت | ||
مقالة
سلسلتان طول كل منهما أكبر من 15 كيلوباس. يتم تلوين كل نقطة بناءً على نسبة تطابق المحاذاة. د) ملخص انهيار النمط الوراثي لـ R570. لتحديد مناطق انهيار النمط الوراثي داخل جينوم R570، تم محاذاة قراءات PacBio HiFi مع التجميع، باستخدام عمق القراءة لتحديد عدد نسخ النمط الوراثي. تمثل مناطق التغطية الفريدة 0 X كتل جينومية حيث لم يكن بالإمكان تحديد مواقع القراءة بشكل فريد بين التجميع الأساسي والبديل. تمثل القواعد المحسوبة تسلسلات الجينوم حيث يمكن حساب العمق بشكل موثوق.
مقالة
لكل متماثل متجانس بالنسبة لـ S.bicolor (BTx623). تم تقاطع أعداد النسخ (CN) لعمق قراءة PacBio HiFi (الذي يمثل الأنماط المتماثلة المتطابقة المنهارة) مع موضع الجين لتوفير مؤشر على الأليلات المنهارة الإضافية (مثل عدد النسخ
محفظة الطبيعة
آخر تحديث من المؤلفين: 13 فبراير 2024
ملخص التقرير
الإحصائيات
يجب أن تُوصف الاختبارات الشائعة فقط بالاسم؛ واصفًا التقنيات الأكثر تعقيدًا في قسم الطرق.
وصف كامل للمعلمات الإحصائية بما في ذلك الاتجاه المركزي (مثل المتوسطات) أو تقديرات أساسية أخرى (مثل معامل الانحدار) وَالتباين (مثل الانحراف المعياري) أو تقديرات مرتبطة بعدم اليقين (مثل فترات الثقة)
تحتوي مجموعتنا على الإنترنت حول الإحصائيات لعلماء الأحياء على مقالات تتناول العديد من النقاط المذكورة أعلاه.
البرمجيات والشيفرة
جمع البيانات
لم يتم استخدام أي برنامج لجمع البيانات لهذه المخطوطة.
تجميع الجينوم: RACON (الإصدار 1.4.10)، HiFiAsm (الإصدار 0.13-r308)، Juicebox (الإصدار 1.11.08)
بناء الخريطة الجينية: BWA-MEM (الإصدار 0.7.12)، JoinMap (الإصدار 4.0)، R/QTL (الإصدار 1.42-8)، DMwR (الإصدار 0.4.1)
بناء الخريطة البصرية: أدوات بيو نانو (الإصدار 1.3.8041.8044)، بيو نانو سولف (الإصدار 3.3_10252018)، برنامج بيو نانو جينومكس أكسس (الأدوات 1.3)
تجميع الجينوم: pblat (الإصدار 2.5) GENESPACE (الإصدار 0.9.4)، minimap2 (الإصدار 2.20-r1061)
توصيف الجينوم: GSNAP (الإصدار 2013-09-30)؛ PASA (الإصدار 2.0.2)؛ EXONERATE (الإصدار 2.4.0)؛ RepeatModeler (الإصدار open1.0.11)؛ FGENESH+ (الإصدار 3.1.0)؛
أغسطس (v3.1.0)
الجينوميات المقارنة: GENESPACE (الإصدار 0.9.4)، Orthofinder (الإصدار 2.5.4)، MCScanX (الإصدار 2)، SyRI (الإصدار 1.6)، Biostrings (الإصدار 2.70.2)، MAFFT (الإصدار 7.487)، seqinr (الإصدار 4.2-16)، ESMfold (الإصدار 2.0.1)، MAFFT (الإصدار 7.487)، NLR-Annotator (الإصدار 2)، pal2nal (الإصدار 13)، برنامج Olympus Cellsens (الإصدار)، FACSDiva (الإصدار 6.1.3)، Summit (الإصدار 6.2.2)، برنامج Olympus Cellsens (الإصدار 3.2)
بيانات
يجب أن تتضمن جميع المخطوطات بيانًا عن توفر البيانات. يجب أن يوفر هذا البيان المعلومات التالية، حيثما ينطبق:
- رموز الانضمام، معرفات فريدة، أو روابط ويب لمجموعات البيانات المتاحة للجمهور
- وصف لأي قيود على توفر البيانات
- بالنسبة لمجموعات البيانات السريرية أو بيانات الطرف الثالث، يرجى التأكد من أن البيان يتماشى مع سياستنا
البحث الذي يتضمن مشاركين بشريين، بياناتهم، أو مواد بيولوجية
التقارير عن العرق أو الإثنية، أو غير قابل للتطبيق
اجتماعي ذو صلة
تجمعات
خصائص السكان
غير قابل للتطبيق
غير قابل للتطبيق
الإشراف الأخلاقي
غير قابل للتطبيق
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
التقارير المتخصصة في المجال
علوم الحياة
لنسخة مرجعية من الوثيقة بجميع الأقسام، انظرnature.com/documents/nr-reporting-summary-flat.pdf
تصميم دراسة العلوم الحياتية
| حجم العينة | لم يتم تحديد حجم العينة مسبقًا قبل التحليل. تم تحديد جينات النسل الذاتي لإنشاء الخريطة الجينية.
|
| استثناءات البيانات | لم يتم استبعاد أي بيانات من التحليلات. |
| التكرار | تم استخراج الحمض النووي/الحمض النووي الريبي من نسخ موثوقة من نفس النمط الجيني (R570). لضمان التكرار، يمكن اختيار نفس النسخة (المحتفظ بها في البيوت الزجاجية) واختبارها. |
| التوزيع العشوائي | تم إجراء تجميع وتحليل الجينوم على جينوتيب هجين واحد من قصب السكر ‘R570’، لذا فإن العشوائية ليست ضرورية. |
| مُعَمي | تم إجراء تجميع وتحليل الجينوم على جينوتيب هجين واحد من قصب السكر ‘R570’، لذا فإن التعمية ليست ضرورية. |
التقارير عن مواد وأنظمة وطرق محددة

البحث ذو الاستخدام المزدوج الذي يثير القلق
المخاطر
تجارب مثيرة للقلق
النباتات
| مخزونات البذور | تم الحصول على المواد النباتية المستخدمة في التسلسل من صنف الهجين من قصب السكر R570، الموجود في البيوت الزجاجية في محطة أبحاث CIRAD في مونبلييه، فرنسا. |
| أنماط جينية نباتية جديدة | لا توجد أنماط جينية جديدة مستخدمة في هذه الدراسة. |
| المصادقة | لا توجد إجراءات مصادقة مطلوبة. |
تدفق الخلايا
المؤامرات
أكد أن:
المقاييس على المحاور مرئية بوضوح. قم بتضمين الأرقام على المحاور فقط للرسم البياني في الأسفل الأيسر من المجموعة (المجموعة هي تحليل للعلامات المتطابقة).
جميع الرسوم البيانية هي رسوم بيانية متساوية الارتفاع مع نقاط شاذة أو رسوم بيانية بالألوان الزائفة.
تم توفير قيمة عددية لعدد الخلايا أو النسبة المئوية (مع الإحصائيات).
المنهجية
| تحضير العينة | تم حصاد الجذور لفرز الكروموسومات الفردية من نباتات R570، المزروعة في أصص. تم معالجة الجذور بـ
|
| آلة | تم التقاط الصور باستخدام كاميرا CCD متصلة بمجهر BX53 من أوليمبوس. |
| برمجيات | برنامج أوليمبوس سيلسينس (الإصدار 3.2) |
| وفرة تجمع الخلايا | لم تُستخدم الخلايا الفردية، بل الكروموسومات التي تم إيقافها في الطور الاستوائي وتم صبغها. وبالتالي، فإن القيمة العددية للخلايا ليست ذات صلة. |
| استراتيجية البوابة | تم استخدام شدة الفلورة النسبية لفصل قمم الكاريوتيب التدريجي (I-V) وعزل الكروموسومات الفردية للتسلسل. |
مركز تسلسل الجينوم، معهد هودسون ألفا للتكنولوجيا الحيوية، هانتسفيل، ألاباما، الولايات المتحدة الأمريكية. سيراد، UMR AGAP المعهد، مونبلييه، فرنسا. معهد UMR AGAP، جامعة مونبلييه، CIRAD، INRAE، معهد أغرو، مونبلييه، فرنسا. معهد الجينوم المشترك التابع لوزارة الطاقة، مختبر لورانس بيركلي الوطني، بيركلي، كاليفورنيا، الولايات المتحدة الأمريكية. بحث السكر أستراليا، تي كواي، كوينزلاند، أستراليا. كورتيفا أجرساينس، جونستون، آيوا، الولايات المتحدة الأمريكية. CSIRO الزراعة والغذاء، منطقة علوم الحياة في كوينزلاند، سانت لوسيا، كوينزلاند، أستراليا. معهد علم النبات التجريبي بالأكاديمية التشيكية للعلوم، مركز الجينوم الهيكلي والوظيفي للنباتات، أولوموك، جمهورية التشيك. ERCANE، سانت كلوتيلد، لا ريونيون، فرنسا. معهد جينوم أريزونا، جامعة أريزونا، توكسون، أريزونا، الولايات المتحدة الأمريكية. CSIRO الزراعة والغذاء، أوربراي، جنوب أستراليا، أستراليا. تحالف كوينزلاند للابتكار في الزراعة والغذاء، جامعة كوينزلاند، بريسبان، كوينزلاند، أستراليا. المعهد المشترك للطاقة الحيوية، مختبر لورانس بيركلي الوطني، إيميريفيل، كاليفورنيا، الولايات المتحدة الأمريكية. مركز ARC للتميز في نجاح النباتات في الطبيعة والزراعة، جامعة كوينزلاند، بريسبان، كوينزلاند، أستراليا. البريد الإلكتروني:ahealey@hudsonalpha.org;jschmutz@hudsonalpha.org;dhont@cirad.fr
DOI: https://doi.org/10.1038/s41586-024-07231-4
PMID: https://pubmed.ncbi.nlm.nih.gov/38538783
Publication Date: 2024-03-27
The complex polyploid genome architecture of sugarcane
Received: 24 February 2023
Accepted: 23 February 2024
Published online: 27 March 2024
Open access
Abstract
Sugarcane, the world’s most harvested crop by tonnage, has shaped global history, trade and geopolitics, and is currently responsible for
in the first two generations so backcrossed (BC1) cultivars contained
are provided in single quotes.
between homologues) and disomic inheritance (with systematic association between a pair of homologues)
The road to a representative genome
A genome such as R570 poses many technical assembly and genome representation challenges, as R570 has all the complexities of both outbred and inbred genomes. Given variable pairing affinities among R570 chromosomes, it could potentially be biologically appropriate to follow the standard outbred genome representation where an assembly is built for each meiotic homologue. However, given its backcrossed pedigree,
subgenomes. Therefore, we opted for a standard partial-inbred genome assembly for R570, where the ‘primary’ assembly is a complete representation of unique haplotypes in R570 whereas the ‘alternate’ represents nearly identical, additional haplotypes. While ‘alternate’ here does not have the same meaning as compared to organisms with strict disomic pairing, we structured the R570 genome in a similar manner to improve utility for the community.
Article
| Expected
|
Chr. 5 | Chr. 2 | Chr. 3 | Chr 10 | Chr. 7 | Chr. 8 | Chr. 9 | Chr. 6 | Chr. 4 | |
| Chr. 1 //!!, | ||||||||||
| Primary assembly identical haplotypes collapsed | Chr. 2 | |||||||| j I I I I I I |
|
|||||||
| Sorghum bicolor v.3.1 | |||||| j | |||||||||
|
||||||||||
|
|
|
1 | ||||||||
| |''''!! | ||||||||||
|
||||||||||
| Chr. 8 | 1 |
|
|
|||||||
|
J,li li i'i | |||||||||
| Chr. 10 |
|
|||||||||
variant detection.To support these efforts,we used gene homology and RNA sequencing(RNA-seq)transcript evidence to describe the full suite of protein coding sequences and annotate genes in the primary R570 assembly.The primary annotation is highly complete(BUSCO
contribution within R570.c,GENESPACE-generated synteny map among (bottom to top)Sorghum bicolor(v.3.1),S.spontaneum(genotype AP85-441), R570 primary and R570 monoploid genome assemblies.Horizontal segments indicate chromosomes;colours(red-purple)indicate the orthologous Sorghum bicolor chromosomes(1-10)and'braids'represent syntenic blocks between each pair of genomes.
The architecture of the R570 genome
| Overall | S. officinarum | S. spontaneum | |
| Primary genome size (contig N50) |
|
3.66 Gb | 1.37 Gb |
| Alternate genome size (contig N50) |
|
3.01 Gb | 0.32 Gb |
| Genome size accounting for collapsed haplotypes | 9.32 Gb | – | – |
| Collapsed haplotypes in assembly
|
2.31 Gb | 2.18 Gb | 0.116 Gb |
| No. of genes (no. of syntenic orthogroups) in primary annotation
|
194,593
|
132,618 | 61,197 |
| Mean ploidy of primary assembly (coverage of syntenic blocks)
|
|
4.60x | 2.16x |
| Mean pairwise peptide identity among alleles
|
– | 86% | 83% |
| Genes impacted by structural variants
|
5,362 | 5,090 | 260 |
we found that
Exploration of targets for breeding
annotation of resistance gene analogues (RGAs)
important diseases that affects all sugarcane growing regions around the world is brown rust, caused by the fungus, Puccinia melanocephala. Once a major pathogen of sugarcane that caused yield losses of up to 50%, breeders have successfully mitigated P. melancocephala-derived losses by selecting for disease resistance. A major locus (Bru1) that confers durable resistance to this disease (Fig. 3a) was identified in cultivar R570 (refs. 43,44). To uncover the causative allele underlying Bru1, previous studies used an extensive map-based cloning approach that screened approximately 2,400 self-pollinated R570 progeny, constraining Bru1 to a set of BAC sequences that spanned approximately 209 kb (refs. 27,44) (Methods). Although the region contained 13 gene models (Fig. 3b and Supplementary Table 13), it also contained an unsized gap and large haplotype insertion, both of which prevent further fine-scale mapping and exhaustive candidate gene discovery
variants (for example, tandem kinase-kinases and so on)) are difficult to find using only sequence homology. Their structure has been predicted across the plant domain of life, but only five examples have been functionally validated in monocots, all of which conferred resistance to fungal pathogens
Conclusions
Online content
- Dinesh Babu, K. S. et al. A short review on sugarcane: its domestication, molecular manipulations and future perspectives. Genet. Resour. Crop Evol. 69, 2623-2643 (2022).
- Yadav, S. et al. Accelerating genetic gain in sugarcane breeding using genomic selection. Agronomy 10, 585 (2020).
- Grivet, L., Glaszmann, J.-C. & D’Hont, A. in Darwin’s Harvest (eds Motley, T. J. et al.) 49-66 (Columbia Univ. Press, 2006).
- Bremer, G. Problems in breeding and cytology of sugar cane. Euphytica 10, 59-78 (1961).
- Burner, D. M. & Legendre, B. L. Chromosome transmission and meiotic stability of sugarcane (Saccharum spp.) hybrid derivatives. Crop Sci. 33, 600-606 (1993).
- Vieira, M. L. C. et al. Revisiting meiosis in sugarcane: chromosomal irregularities and the prevalence of bivalent configurations. Front. Genet. 9, 213 (2018).
- Oliveira, G. K. et al. Meiotic abnormalities in sugarcane (Saccharum spp.) and parental species: evidence for peri- and paracentric inversions. Ann. Appl. Biol. https://doi.org/ 10.1111/aab. 12855 (2023).
- Hoarau, J.-Y. et al. Genetic dissection of a modern sugarcane cultivar (Saccharum spp.). I. Genome mapping with AFLP markers. Theor. Appl. Genet. 103, 84-97 (2001).
- Aitken, K. S., Jackson, P. A. & McIntyre, C. L. A combination of AFLP and SSR markers provides extensive map coverage and identification of homo(eo)logous linkage groups in a sugarcane cultivar. Theor. Appl. Genet. 110, 789-801 (2005).
- Jannoo, N., Grivet, L., David, J., D’Hont, A & Glaszmann, J.-C. Differential chromosome pairing affinities at meiosis in polyploid sugarcane revealed by molecular markers. Heredity 93, 460-467 (2004).
- Dumont, T. et al. Sugarcane breeding in reunion: challenges, achievements and future prospects. Sugar Tech 24, 181-192 (2022).
- D’Hont, A. et al. Characterisation of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Mol. Gen. Genet. 250, 405-413 (1996).
- Piperidis, N. & D’Hont, A. Sugarcane genome architecture decrypted with chromosome-specific oligo probes. Plant J. 103, 2039-2051 (2020).
- Costet, L. et al. Haplotype structure around Bru1 reveals a narrow genetic basis for brown rust resistance in modern sugarcane cultivars. Theor. Appl. Genet. 125, 825-836 (2012).
- Parco, A. S. et al. Distribution and frequency of Bru1, a major brown rust resistance gene, in the sugarcane world collection. Plant Breed. 136, 637-651 (2017).
- Garsmeur, O. et al. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat. Commun. 9, 2638 (2018).
- Souza, G. M. et al. Assembly of the 373k gene space of the polyploid sugarcane genome reveals reservoirs of functional diversity in the world’s leading biomass crop. GigaScience 8, giz129 (2019).
- Shearman, J. R. et al. A draft chromosome-scale genome assembly of a commercial sugarcane. Sci. Rep. 12, 20474 (2022).
- Nurk, S. et al. The complete sequence of a human genome. Science 376, 44-53 (2022).
- Sun, H. et al. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat. Genet. 54, 342-348 (2022).
- Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
- Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210-3212 (2015).
- Lovell, J. T. et al. GENESPACE tracks regions of interest and gene copy number variation across multiple genomes. eLife 11, e78526 (2022).
- Cuadrado, A., Acevedo, R., Moreno Díaz de la Espina, S., Jouve, N. & De La Torre, C. Genome remodelling in three modern S. officinarum
S. spontaneum sugarcane cultivars. J. Exp. Bot. 55, 847-854 (2004). - Piperidis, G., Piperidis, N. & D’Hont, A. Molecular cytogenetic investigation of chromosome composition and transmission in sugarcane. Mol. Genet. Genomics 284, 65-73 (2010).
- Aitken, K. S. et al. A comprehensive genetic map of sugarcane that provides enhanced map coverage and integrates high-throughput Diversity Array Technology (DArT) markers. BMC Genom. 15, 152 (2014).
- Garsmeur, O. et al. High homologous gene conservation despite extreme autopolyploid redundancy in sugarcane. New Phytol. 189, 629-642 (2011).
- Vilela, M. et al. Analysis of three sugarcane homo/homeologous regions suggests independent polyploidization events of Saccharum officinarum and Saccharum spontaneum. Genome Biol. Evol. 9, 266-278 (2017).
- Pompidor, N. et al. Three founding ancestral genomes involved in the origin of sugarcane. Ann. Bot. 127, 827-840 (2021).
- Jannoo, N. et al. Orthologous comparison in a gene-rich region among grasses reveals stability in the sugarcane polyploid genome. Plant J. 50, 574-585 (2007).
- Zhang, Q. et al. Genomic insights into the recent chromosome reduction of autopolyploid sugarcane Saccharum spontaneum. Nat. Genet. 54, 885-896 (2022).
- Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50, 1565-1573 (2018).
- Kamal, N. et al. The mosaic oat genome gives insights into a uniquely healthy cereal crop. Nature https://doi.org/10.1038/s41586-022-04732-y (2022).
- Song, K., Lu, P., Tang, K. & Osborn, T. C. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc. Natl Acad. Sci. USA 92, 7719-7723 (1995).
- Liu, B. et al. Rapid genomic changes in polyploid wheat and related species: implications for genome evolution and genetic improvement. J. Genet. Genom. 36, 519-528 (2009).
- Lu, X. et al. Segregation analysis of microsatellite (SSR) markers in sugarcane polyploids. Genet. Mol. Res. 14, 18384-18395 (2015).
- Piperidis, N. et al. Comparative genetics in sugarcane enables structured map enhancement and validation of marker-trait associations. Mol. Breed. 21, 233-247 (2008).
- Steuernagel, B. et al. The NLR-Annotator tool enables annotation of the intracellular immune receptor repertoire. Plant Physiol. 183, 468-482 (2020).
Article
- Aitken, K. S., Jackson, P. A. & McIntyre, C. L. Quantitative trait loci identified for sugar related traits in a sugarcane (Saccharum spp.) cultivar
Saccharum officinarum population. Theor. Appl. Genet. 112, 1306-1317 (2006). - Hoarau, J.-Y. et al. Genetic dissection of a modern sugarcane cultivar (Saccharum spp.).II. Detection of QTLs for yield components. Theor. Appl. Genet. 105, 1027-1037 (2002).
- Ming, R. et al. Molecular dissection of complex traits in autopolyploids: mapping QTLs affecting sugar yield and related traits in sugarcane. Theor. Appl. Genet. 105, 332-345 (2002).
- Cooper, E. A. et al. A new reference genome for Sorghum bicolor reveals high levels of sequence similarity between sweet and grain genotypes: implications for the genetics of sugar metabolism. BMC Genom. 20, 420 (2019).
- Daugrois, J. H. et al. A putative major gene for rust resistance linked with a RFLP marker in sugarcane cultivar ‘R570’. Theor. Appl. Genet. 92, 1059-1064 (1996).
- Le Cunff, L. et al. Diploid/polyploid syntenic shuttle mapping and haplotype-specific chromosome walking toward a rust resistance gene (Bru1) in highly polyploid sugarcane (
). Genetics 180, 649-660 (2008). - Gish, L. A. & Clark, S. E. The RLK/Pelle family of kinases. Plant J. 66, 117-127 (2011).
- Brueggeman, R. et al. The barley stem rust-resistance gene Rpg1 is a novel diseaseresistance gene with homology to receptor kinases. Proc. Natl Acad. Sci. USA 99, 9328-9333 (2002).
- Klymiuk, V. et al. Cloning of the wheat Yr15 resistance gene sheds light on the plant tandem kinase-pseudokinase family. Nat. Commun. 9, 3735 (2018).
- Lewis, J. D., Lo, T., Bastedo, P., Guttman, D. S. & Desveaux, D. The rise of the undead: pseudokinases as mediators of effector-triggered immunity. Plant Signal. Behav. 9, e27563 (2014).
- Klymiuk, V., Coaker, G., Fahima, T. & Pozniak, C. J. Tandem protein kinases emerge as new regulators of plant immunity. Mol. Plant Microbe Interact. 34, 1094-1102 (2021).
(c) The Author(s) 2024
Methods
Genome sequencing
Single flow-sorted chromosome libraries. Sequencing libraries were constructed using an Illumina TruSeq DNA Nano library kit using standard protocols. Libraries were sequenced on either the Illumina HiSeq2500 or NovaSeq 6000 instrument using paired ends and a read length of 150 base pairs.
Remaining Illumina libraries. Illumina Tight Insert Fragment, 400 bp-2 ug of DNA was sheared to 400 bp using the Covaris LE220 and size selected using the Pippin (Sage Science). The fragments were treated with end-repair, A-tailing and ligation of Illumina compatible adaptors (IDT) using the KAPA-Illumina library creation kit (KAPA Biosystems). The prepared libraries were quantified using KAPA Biosystems’ next-generation sequencing library qPCR kit (Roche) and run on a Roche LightCycler 480 real-time PCR instrument. The quantified libraries were then prepared for sequencing on the Illumina HiSeq sequencing platform using a TruSeq Rapid paired-end cluster kit, v.2, with the HiSeq 2500 sequencer instrument to generate a clustered flowcell for sequencing. Sequencing of the flowcell was performed on the Illumina HiSeq 2500 sequencer using HiSeq Rapid SBS sequencing kits, v.2, following a
v.7989. Two separated assemblies were performed using the optArguments_nonhaplotype_noES_BG_DLE1_saphyr.xml parameters. The initial assembly was performed without complex multi-path region (CMPR) cuts and produced 570 maps with a N50 length of 36.444 Mbp and total map length of 7,654.039 Mbp. One additional assembly was performed using the CMPR cut option, which introduces map cuts at potential duplications to reduce potential homeolog and phase switching. CMPR-cut-enabled assembly generated 1,512 maps with N50 length of 9.546 Mbp and total map length of
coverage ranges
via GENESPACE (v.0.9.4)
revealed they both belong to the RLK-PELLE-DSLV family
Reporting summary
Data availability
Code availability
50. Huang, Y. et al. Species-specific abundant retrotransposons elucidate the genomic composition of modern sugarcane cultivars. Chromosoma 129, 45-55 (2020).
51. Metcalfe, C. J. et al. Flow cytometric characterisation of the complex polyploid genome of Saccharum officinarum and modern sugarcane cultivars. Sci. Rep. 9, 19362 (2019).
52. Metcalfe, C. J. et al. Isolation and sequencing of a single copy of an introgressed chromosome from a complex genome for gene and SNP identification. Theor. Appl. Genet. 135, 1279-1292 (2022).
53. Vrána, J. et al. Flow cytometric chromosome sorting in plants: the next generation. Methods 57, 331-337 (2012).
54. Vollger, M. R. et al. Long-read sequence and assembly of segmental duplications. Nat. Methods 16, 88-94 (2019).
55. Hufford, M. B. et al. De novo assembly annotation and comparative analysis of 26 diverse maize genomes. Science 373, 655-662 (2021).
56. Wang, M. & Kong, L. pblat: a multithread blat algorithm speeding up aligning sequences to genomes. BMC Bioinformatics 20, 28 (2019).
57. Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094-3100 (2018).
58. Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078-2079 (2009).
59. Quinlan, A. R. BEDTools: the Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinformatics 47, 11.12.1-34 (2014).
60. Lovell, J. T. et al. The genomic landscape of molecular responses to natural drought stress in Panicum hallii. Nat. Commun. 9, 5213 (2018).
61. Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873-881 (2010).
62. Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654-5666 (2003).
63. Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516-522 (2000).
64. Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
65. Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435-9 (2006).
66. Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0. http://www.repeatmasker.org/ (2010).
67. McCormick, R. F. et al. The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 93, 338-354 (2018).
68. Mamidi, S. et al. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat. Biotechnol. 38, 1203-1210 (2020).
69. Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
70. Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
71. Goel, M., Sun, H., Jiao, W. B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 1-13 (2019).
72. Pages, H., Aboyoun, P., Gentleman, R. & DebRoy, S. Biostrings: string objects representing biological sequences, and matching algorithms (2008). R package version 2.0 (2015).
73. Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772-780 (2013).
74. Zhou, L. et al. ggmsa: a visual exploration tool for multiple sequence alignment and associated data. Brief. Bioinform. 23, bbac222 (2022).
75. Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123-1130 (2023).
76. Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609-12 (2006).
77. Charif, D. & Lobry, J. R. in Structural Approaches to Sequence Evolution: Molecules, Networks, Populations (eds Bastolla, U., Porto, M., Roman, H. E. & Vendruscolo, M.) 207-232 (Springer Berlin Heidelberg, 2007).
78. Huang, C.-C. et al. Evolutionary rates of commonly used nuclear and organelle markers of Arabidopsis relatives (Brassicaceae). Gene 499, 194-201 (2012).
79. Asnaghi, C. et al. Application of synteny across Poaceae to determine the map location of a sugarcane rust resistance gene. Theor. Appl. Genet. 101, 962-969 (2000).
80. Minic, Z. Physiological roles of plant glycoside hydrolases. Planta 227, 723-740 (2008).
81. Levy, I., Shani, Z. & Shoseyov, O. Modification of polysaccharides and plant cell wall by endo-1,4-beta-glucanase and cellulose-binding domains. Biomol. Eng. 19, 17-30 (2002).
Additional information
Correspondence and requests for materials should be addressed to A. L. Healey, J. Schmutz or A. D’Hont.
Peer review information Nature thanks Elizabeth Cooper and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
C Primary-alternate sequence comparison
| Haplotype Depth | Counted Bases (Gb) | Represented Bases (Gb) | Genome Representation (%) |
| 0X | 1.47 | 0.74 | 7.9 |
| 1X | 4.63 | 4.63 | 49.6 |
| 2X | 1.15 | 2.30 | 24.6 |
| 3X | 0.35 | 1.05 | 11.3 |
| 4X | 0.15 | 0.60 | 6.6 |
| Represented genome size | 9.32 Gb | ||
Article
two sequences that is greater than 15 kilobases in length. Each point is coloured based on alignment percent identity. D) Haplotype collapse summary for R570. To quantify regions of haplotype collapse within the R570 genome, PacBio HiFi reads were aligned to the assembly, using read depth to determine haplotype copy number. 0 X unique coverage regions represent genomic blocks where reads could not uniquely map between the primary and alternate assembly. Counted bases represent genome sequences where depth could be reliably calculated (
Article
for each syntenic ortholog relative to the S.bicolor(BTx623). Copy number (CN)-PacBio HiFi read depths (representing collapsed identical haplotypes) ranges were intersected with gene position to provide an indication of additional collapsed alleles (eg. copy number
natureportfolio
Last updated by author(s): Feb 13, 2024
Reporting Summary
Statistics
Only common tests should be described solely by name; describe more complex techniques in the Methods section.
A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals)
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Data collection
No software was used for data collection for this manuscript.
Genome Assembly: RACON(v1.4.10), HiFiAsm (v0.13-r308), Juicebox (v 1.11.08)
Genetic Map Construction: BWA-MEM (v0.7.12), JoinMap (v4.0), R/QTL (v1.42-8), DMwR (v0.4.1)
Optical Map Construction: Bionano tools (v1.3.8041.8044), Bionano Solve (v3.3_10252018), Bionano Genomics Access software (Tools 1.3)
Genome Assembly: pblat (v2.5) GENESPACE (v0.9.4), minimap2 (v 2.20-r1061)
Genome Annotation: GSNAP (v2013-09-30); PASA (v2.0.2);EXONERATE (v2.4.0) ; RepeatModeler (v.open1.0.11); FGENESH+(v3.1.0);
AUGUSTUS (v3.1.0)
Comparative Genomics: GENESPACE (v0.9.4), Orthofinder (v2.5.4), MCScanX (v2), SyRI (v1.6), Biostrings (v2.70.2), MAFFT (v7.487), seqinr (v4.2-16), ESMfold (v2.0.1), MAFFT (v7.487), NLR-Annotator (v2), pal2nal (v13), Olympus Cellsens software (v), FACSDiva (v6.1.3), Summit (v 6.2.2 ),Olympus Cellsens software (v3.2)
Data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
- Accession codes, unique identifiers, or web links for publicly available datasets
- A description of any restrictions on data availability
- For clinical datasets or third party data, please ensure that the statement adheres to our policy
Research involving human participants, their data, or biological material
Reporting on race, ethnicity, or Not applicable
other socially relevant
groupings
Population characteristics
Not applicable
Not applicable
Ethics oversight
Not applicable
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Field-specific reporting
Life sciences
For a reference copy of the document with all sections, see nature.com/documents/nr-reporting-summary-flat.pdf
Life sciences study design
| Sample size | Sample size was not predetermined prior to analysis. Selfed offspring genotyped for the genetic map (
|
| Data exclusions | No data was excluded from analyses. |
| Replication | DNA/RNA was extracted from verified clones of the same genotype (R570). For repeatability, the same clone (held in greenhouses) could be selected and tested. |
| Randomization | Genome assembly and analysis were conducted on a single sugarcane hybrid genotype ‘R570’, thus randomization is not necessary. |
| Blinding | Genome assembly and analysis were conducted on a single sugarcane hybrid genotype ‘R570’, thus blinding is not necessary. |
Reporting for specific materials, systems and methods
Dual use research of concern
Hazards
Experiments of concern
Plants
| Seed stocks | Plant material used for sequenced was derived from sugarcane hybrid cultivar R570, held in greenhouses at CIRAD research station in Montpellier, France |
| Novel plant genotypes | No novel genotypes used in this study. |
| Authentication | No authentication procedures required. |
Flow Cytometry
Plots
Confirm that:
The axis scales are clearly visible. Include numbers along axes only for bottom left plot of group (a ‘group’ is an analysis of identical markers).
All plots are contour plots with outliers or pseudocolor plots.
A numerical value for number of cells or percentage (with statistics) is provided.
Methodology
| Sample preparation | Roots for single chromosome sorting were harvested from R570 plants, grown in pots. Roots were treated with
|
| Instrument | Images were captured with a CCD camera attached to a BX53 Olympus microscope. |
| Software | Olympus Cellsens software (v3.2) |
| Cell population abundance | Individual cells were not used, rather chromosomes that were arrested in metaphase and stained. Thus, a numerical value for cells is not relevant. |
| Gating strategy | Relative fluorescence intensity was used to separate flow karyotype peaks (I-V) and isolate single chromosomes for sequencing. |
Genome Sequencing Center, HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA. CIRAD, UMR AGAP Institut, Montpellier, France. UMR AGAP Institut, Univ Montpellier, CIRAD, INRAE, Institut Agro, Montpellier, France. Department of Energy Joint Genome Institute, Lawrence Berkeley National Laboratory, Berkeley, CA, USA. Sugar Research Australia, Te Kowai, Queensland, Australia. Corteva Agriscience, Johnston, IA, USA. CSIRO Agriculture and Food, Queensland Bioscience Precinct, St Lucia, Queensland, Australia. Institute of Experimental Botany of the Czech Academy of Sciences, Centre of Plant Structural and Functional Genomics, Olomouc, Czech Republic. ERCANE, Sainte-Clotilde, La Réunion, France. Arizona Genomics Institute, University of Arizona, Tucson, AZ, USA. CSIRO Agriculture and Food, Urrbrae, South Australia, Australia. Queensland Alliance for Agriculture and Food Innovation, University of Queensland, Brisbane, Queensland, Australia. Joint BioEnergy Institute, Lawrence Berkeley National Laboratory, Emeryville, CA, USA. ARC Centre of Excellence for Plant Success in Nature and Agriculture, University of Queensland, Brisbane, Queensland, Australia. e-mail: ahealey@hudsonalpha.org; jschmutz@hudsonalpha.org; dhont@cirad.fr