الخلايا التائية المنقولة بالتبني والعوامل المصممة لحجب CD47-SIRPمحاور هي علاجات واعدة للسرطان تنشط أذرعًا مميزة من جهاز المناعة. هنا قمنا بإعطاء أجسام مضادة مضادة لـ CD47 بالاشتراك مع خلايا T المنقولة بالتبني بهدف تعزيز الفعالية المضادة للورم، ولكننا لاحظنا تراجع الفائدة العلاجية بسبب الإزالة السريعة لخلايا T التي تعبر عن مستقبلات المستضدات الكيميرية (CARs) أو مستقبلات خلايا T المهندسة بواسطة البلعميات. كانت إزالة خلايا CAR T بواسطة الأجسام المضادة المضادة لـ CD47 فعالة وسريعة بما يكفي لتكون بمثابة مفتاح أمان فعال. للتغلب على هذه التحديات، قمنا بتصميم متغير CD47 CD47(Q31P) (47E)، الذي يتفاعل مع SIRP.ويقدم إشارة ‘لا تأكلني’ التي لا يتم حجبها بواسطة الأجسام المضادة المضادة لـ CD47. خلايا TCR أو خلايا CAR T التي تعبر عنتكون مقاومة للإزالة بواسطة البلعميات بعد العلاج بالأجسام المضادة المضادة لـ CD47، وتساهم في جذب كبير ومستدام للبلعميات إلى بيئة الورم الدقيقة. على الرغم من أن العديد من البلعميات المجندة أظهرت نمطًا شبيهًا بـ M2.لقد عززت العلاج المشترك بشكل تآزري فعالية مضادة للأورام. تحدد دراستنا البلعميات كمنظمين رئيسيين لاستمرارية خلايا T وتوضح التحدي الأساسي في دمج العلاجات الموجهة نحو خلايا T مع تلك المصممة لتنشيط البلعميات. تقدم نهجًا علاجيًا قادرًا على استغلال التأثيرات المضادة للأورام لخلايا T والبلعميات في الوقت نفسه، مما يوفر قوة معززة ضد الأورام الصلبة.
تعتبر الخلايا النخاعية أكثر خلايا المناعة وفرة داخل بيئة الورم الدقيقة (TME) وقد كان هناك اهتمام كبير في استهدافها علاجياً لتحقيق تأثيرات مضادة للورم.ترتبط المستويات المرتفعة من البلعميات المرتبطة بالأورام (TAMs) بنتائج أسوأ في العديد من الدراسات، وتظهر بعض البيانات قبل السريرية أن تقليل أو القضاء على البلعميات المرتبطة بالأورام يعزز الاستجابة للعلاج الكيميائي والعلاج المناعي.. ومع ذلك، على الرغم من العشرات من الدراسات السريرية التي تختبر عوامل مثل مثبطات CSF1R و CCR2 لتقليل الخلايا المناعية المرتبطة بالورم والخلايا المايلويدية المرتبطة بالورم، لم يتم تحقيق فائدة سريرية واضحة. أظهربدلاً من ذلك، يرتبط زيادة كثافة TAM بتحسين النتائج السريرية في بعض أنواع السرطان.وزيادة نشاط البلعمة لدى TAM عن طريق حجب CD47-SIRPالمحور يتوسط التأثيرات المضادة للورم في عدة نماذج قبل السريريةالتجارب السريرية لـ CD47-SIRPأظهرت مثبطات المحور نشاطًا مضادًا للورم في بعض الأورام السائلة عند دمجها مع عوامل إضافية، لكن الأدلة السريرية على نشاطها كعوامل منفردة أو نشاطها في السرطانات الصلبة غير متوفرة.لذا، على الرغم من الجهود الكبيرة، فإن الأساليب العلاجية لاستهداف TAMs لتحقيق فائدة سريرية لا تزال مفقودة.
الشكل 1 | الأجسام المضادة المضادة لـ CD47 تلغي فعالية خلايا T المنقولة بالتبني من خلال الوساطة بواسطة البلعمياتنقص الخلايا. أ،نمو ورم الساركوما العظمية بعد العلاج بخلايا CAR T من نوع HER2-BBZ مع أو بدون B6H12.b، نمو ورم الساركوما العظمية MG63.3 بعد العلاج بـ أو خلايا T مع أو بدونالخلايا من الدم والورم في نموذج MG63.3 في اليوم 30. البيانات هي المتوسطانحراف معياري لـالفئران. د، نمو ورم الميلانوما A375 بعد العلاج بخلايا TCR NY-ESO-1 مع أو بدون B6H12. هـ، خلايا T في دم الفئران في نموذج A375 في اليوم 17. و، نمو ورم Nalm6 بعد العلاج بخلايا CAR T CD19-BBZ مع أو بدون CV-1 (موت Fc). ز، خلايا T في دم الفئران في نموذج Nalm6-CV-1 في اليوم 11. البيانات هي المتوسطانحراف معياري لـ (CD19-BBZ) أو فئران (CD19-BBZ+ CV-1) .نمو ورم اللوكيميا Nalm6 بعد العلاج مع حذف CD47خلايا CAR T (CD19-28). i، تصوير الخلايا التائية في Nalm6-47نموذج خلايا CAR T في اليوم 11.j، نمو ورم اللوكيميا Nalm6 بعد العلاج بـ
مضاد CD47 يلغي فعالية خلايا CAR T وخلايا TCR T
لاختبار الفرضية القائلة بأن تعزيز البلعمة بواسطة البلعميات من خلال حجب CD47 يمكن أن يحسن فعالية علاج خلايا CAR T،
المفرط التعبير عن CD47 ) خلايا CAR T CD19-28 مليار. ك، خلايا T في اليوم 45 بعد علاج CAR في دم الفئران في Nalm6-47نموذج خلايا CAR T. البيانات هي المتوسطانحراف معياري لـ أو (جميع الآخرين) الفئران.1، تصوير الخلايا التائية بعد استنفاد البلعميات (يسار). البيانات هي المتوسطانحراف معياري لـ (مستنفد) أو فئران (غير المستنفدة). اليمين، التغير النسبي في BLI لخلايا T، مع أو بدون B6H12، بعد استنفاد البلعميات. البيانات هي المتوسطانحراف معياري لـ (مستنفد + PBS) أو (جميع الآخرين) الفئران.تخطيط خلايا T قبل وبعد B6H12، بعد استنفاد البلعميات. و البيانات تعنيالانحراف المعياري للخطأالفئران لكل ذراع لنمو الورم. بالنسبة لـ e و i، البيانات هي المتوسطانحراف معياري لـالفئران. تم إجراء التحليل الإحصائي باستخدام تحليل التباين ثنائي الاتجاه (ANOVA) مع اختبار المقارنات المتعددة لتوكاي (a,b,d,f,h,j و l (يمينًا))، واختبار الطالب غير المقترن ذو الذيلين.-اختبارات ( (يمين) و (يسار) واختبار مان-ويتني ذو الذيلين-اختبارات (، اليسار)؛ NS، غير مهم. قمنا بإعطاء خلايا CAR T من نوع HER2-BB7 مع أو بدون الأجسام المضادة أحادية النسيلة المضادة لـ CD47 B6H12 للفئران التي تحمل زراعة ورم العظام 143B. أدت خلايا CAR T بمفردها إلى تأثيرات مضادة للورم، ولكن إضافة الأجسام المضادة المضادة لـ CD47 ألغت فعالية خلايا CAR T.
الشكل البياني الممتد 1a). لوحظت معارضة مماثلة مع سرطان العظام MG63.3 وورم الدماغ D425 (الشكل 1b والشكل البياني الممتد 1b-d). للتحقيق في سبب فشل العلاج مع العلاج المزدوج، قمنا بت quantifying خلايا T البشرية في الفئران الحاملة للأورام بعد العلاج معخلاياكانت خلايا T البشرية غائبة تمامًا في الأورام والدم لدى الفئران الحاملة لـ MG63.3 التي تلقت خلايا CAR T من نوع B7H3-BBZ بالإضافة إلى B6H12، ولكنها كانت موجودة في الفئران المعالجة بخلايا CAR T فقط والحيوانات المعالجة بعلاج التحكم المتماثل (الشكل 1c والشكل الإضافي 1e-g)، وتمت ملاحظة نتائج مماثلة في نموذج 143B (الشكل الإضافي 1h). كما أن B6H12 أزال تمامًا خلايا T المنقولة بالتبني التي تعبر عن TCR مستهدف لـ NY-ESO-1 في الفئران الحاملة لزراعة الميلانوما A375 وألغى تأثيراتها المضادة للأورام (الشكل 1d، e والشكل الإضافي 1i-k).
لتوصيف حركية استنفاد خلايا CAR T في متلقيات B6H12، استخدمنا تصوير الإضاءة الحيوية (BLI) لمراقبة خلايا CAR T CD19-28Z-nLuc التي تعبر عن نانولوسيفيراز في الفئران التي تحمل سرطان الدم Nalm6 الذي يعبر عن لوسيفيراز اليراعة. لم تتأثر الفعالية المتواضعة للدواء B6H12 كعامل وحيد في هذا النظام بالإعطاء المشترك لخلايا CAR T CD19-28Z. ومع ذلك، أزال B6H12 تمامًا فعالية خلايا CAR T CD19-28 وأظهر BLI فقدانًا كبيرًا لإشارة خلايا CAR T بعد علاج B6H12، مع غياب خلايا T من طحال الفئران في نهاية التجربة. معًا، تُظهر هذه البيانات أن الأجسام المضادة المضادة لـ CD47 تحفز استنفادًا سريعًا للخلايا T المنقولة بالتبني، بما في ذلك تلك المهندسة للتعبير عن TCR ترانسجيني أو CARs ذات مجالات استهداف وتحفيز مختلفة.
CD47 ضروري لاستمرار خلايا CAR T
لتحديد ما إذا كان استئصال خلايا T في هذه النماذج يحدث من خلال البلعمة المعتمدة على الأجسام المضادة بواسطة FcRقمنا بإدارة CV-1بروتين اندماجي يرتبط بـ CD47 ولكنه لا يرتبط بـ FcRs. مشابهة للنتائج مع B6H12، فإن العلاج المشترك مع CV-1 وخلايا CAR T المستهدفة لـ CD19 ألغى الفعالية المضادة للورم في الفئران الحاملة لـ Nalm6-fLuc، وأدى إلى استنفاد شبه كامل لخلايا T (الشكل 1f، g والشكل التمديدي 2a-f). بعد ذلك، اختبرنا ما إذا كان التعبير عن CD47 مطلوبًا لبقاء خلايا T المنقولة بالتبني من خلال استخدام CRISPR-Cas9 لإسكات CD47. ) في خلايا T البشرية الأولية، ثم استعادة تعبير بروتين CD47 في خلايا ( الشكل 2g من البيانات الموسعة).تم توسيع خلايا CAR T في فئران Nalm6-fLuc وأدت إلى السيطرة القوية على الورم وزيادة ملحوظة في البقاء على قيد الحياة، بينماتم استنفاد خلايا CAR T ولم تقدم أي نشاط مضاد للورم (الشكل 1h,i والشكل البياني الممتد 2h,i). حتى في غياب الورم،توسعت خلايا CAR T بشكل قوي، بينماتم استنفاد خلايا CAR T في الجسم الحي (الشكل التمديدي 2j-m).
نظرًا للدور الأساسي لتعبير CD47 في بقاء خلايا T في الجسم الحي، تساءلنا عما إذا كان الإفراط في التعبير عن CD47 )، والذي تم الإبلاغ عنه لمنع رفض المناعة بواسطة الخلايا المتبرعة يمكن أن يعزز بقاء خلايا CAR T وفعاليتها في فئران NSG، حيث لا يحدث رفض مناعي بسبب التثبيط المناعي العميق. لم يؤثر تعديل تعبير CD47 (سواء من خلال الإزالة أو الإفراط في التعبير) أو إضافة مضاد CD47 على وظيفة خلايا CAR T في المختبر (الشكل البياني الممتد 3a-1). ومع ذلك،أظهرت خلايا CAR T التي تعبر عن CD19-28 فعالية مضادة للأورام على المدى الطويل بشكل أفضل بشكل ملحوظ وتحسنت بقاء خلايا T مقارنةً بالتحكم في الجسم الحي (الشكل 1j، k والشكل الإضافي 3m-o). معًا، توضح هذه البيانات أن بقاء خلايا T المنقولة بالتبني يتطلب تعبير CD47 وSIRP.إن الارتباط وارتفاع تعبير CD47 يعززان بقاء وفعالية خلايا CAR T، حتى في غياب عوامل حجب CD47 وغياب الرفض المناعي..
استنادًا إلى الأدلة التي تشير إلى أن حجب CD47 يعزز ابتلاع البلعميات لخلايا الورمقمنا بفحص ما إذا كانت البلعميات تتوسط نقص خلايا T الناتج عن مضاد CD47 من خلال استنزاف البلعميات (الشكل البياني الموسع 3p،q) ثم معالجة الفئران بـ
خلايا CAR T من نوع CD19-28 7 -nLuc مع أو بدون B6H12. في اليوم التالي لعملية النقل التبني ولكن قبل إعطاء B6H12، أظهر تحليل BLI أعدادًا أعلى بشكل ملحوظ من خلايا CAR T في الفئران التي تم إزالة الماكروفاج منها، مما يتماشى مع نموذج حيث تقوم الماكروفاجات بوساطة نقص خلايا T حتى في غياب حجب CD47 (الشكل 11، م والشكل الإضافي 3ر). بعد إعطاء B6H12، لم نلاحظ فقدان إشارة BLI لخلايا CAR T في الفئران التي تم إزالة الماكروفاج منها، ولكن لوحظ انخفاض كبير في إشارة BLI لخلايا CAR T في الفئران التي تحتوي على قسم ماكروفاج سليم (الشكل 11، م والشكل الإضافي 3ر). معًا، تحدد هذه النتائج الماكروفاجات كحواجز أمام زراعة وفعالية خلايا T المضادة للورم التي تم نقلها بالتبني وتظهر الحاجة الأساسية لمستويات كافية من CD47 على خلايا T للتفاعل مع SIRP.حتى في المضيفين الذين لا يستطيعون التعرف على الفروق المستضدية. كما يشرحون عبثية دمج العلاج المناعي المضاد لـ CD47 مع العلاج بالخلايا التائية المتبناة، ويشيرون إلى البلعمة التي تتوسطها البلعميات كمنظم مهم لاستمرار الخلايا التائية في الجسم الحي.
تقوم البلعميات البشرية بابتلاع خلايا T
قمنا بعد ذلك بالتحقيق في إمكانية البلعمة من قبل البلعميات البشرية الأولية لخلايا T البشرية الأولية في المختبر. لاحظنا بلعمة البلعميات لخلايا T المعالجة بشكل وهمي في الأساس؛ ومع ذلك، تم بلعمة خلايا T المعالجة للتعبير عن CARs بمستويات أعلى بشكل ملحوظ، والتي زادت بشكل أكبر مع B6H12 (الشكل 2a، الشكل التمديدي 4a، b والفيديو التكميلي 1). يتم تنظيم بلعمة البلعميات بواسطة توازن إشارات ‘كلني’، مثل الكالريتولين.“، وإشارات “لا تأكلني” مثل CD47. أظهرت تقنية قياس التدفق الخلوي أن خلايا CAR T تعبر عن عدد أقل من جزيئات CD47 مقارنة بخطوط الورم المستخدمة في هذه الدراسة (الشكل 4c من البيانات الموسعة)، مما يتماشى مع نموذج حيث تقدم خلايا CAR T إشارات “لا تأكلني” المحدودة للبلاعم. كان تعبير CD47 متجانسًا نسبيًا بين CD4وتقلص تعبير CD47 مع مرور الوقت في الثقافة بينما زاد تعبير الكالريتكولين خلال نفس الفترة (الشكل 2ب والشكل الإضافي 4د-هـ)، مما يتماشى مع نموذج يُظهر أن خلايا CAR T المتقدمة في العمر أكثر عرضة للبلعمة.
خلال سير هذه التجارب، كشفت التحليلات الخلوية الروتينية لسائل الدماغ الشوكي (CSF) المأخوذ من مريض تم علاجه بعلاج خلايا CAR T التجارية axicabtagene ciloleucel (axi-cel) عن وجود الخلايا النسجية التي تبتلع اللمفاويات (الشكل 2c)، مما يتماشى مع البلعمة التي تتوسطها البلعميات. لمعالجة هذه الإمكانية بشكل أكثر منهجية، قمنا بتحليل بيانات تسلسل RNA على مستوى الخلية الواحدة (scRNA-seq) التي تم جمعها من دراستين سريريتين حديثتين لـ axi-cel.للكبيرلمفوما الخلايا البائية الكبيرة (LBCL) وخلايا CAR T GD2-BBZلورم الدبقي المنتشر في الخط الأوسط (DMG). أظهرت كلا المجموعتين من البيانات وجود mRNA لـ CAR في الخلايا النخاعية، مما يتماشى مع البلعمة التي تتوسطها البلعميات لخلايا CAR T في البشر (الشكل 2d والشكل الإضافي 4i). توفر هذه البيانات مزيدًا من الأدلة لدعم نموذج يتم فيه بلعمة الخلايا النخاعية لخلايا CAR T، وبالتالي قد تحد من زراعة الخلايا المنقولة بالتبني أو بقاء خلايا T المنشطة في البيئات السريرية.
يمكن أن يعمل مضاد CD47 كمفتاح أمان
افترضنا أن استنفاد خلايا T بواسطة مضاد CD47 يمكن أن يُستخدم كزر أمان جاهز للتخفيف من سمية خلايا CAR T. لاختبار ذلك، استخدمنا CAR مرتبط بببتيد ربط متعدد الخصائص (PIP)، الذي يعبر عن مجال ربط ببتيد على شكل عقدة سيستين تستهدف الإنتغرينات المعبر عنها على مجموعة واسعة من الأورام الخبيثة. (الشكل البياني الممتد 5a، b). قامت خلايا CAR T من نوع PIP بتحقيق نشاط قوي في المختبر، لكن الفئران المصابة بأورام العظام التي تم علاجها بخلايا CAR T من نوع PIP أظهرت سمية حادة (الشكل 3a والشكل البياني الممتد 5c). حتى في الفئران غير الحاملة للأورام، أدى العلاج بخلايا CAR T من نوع PIP-28 بسرعة إلى حدوث سمية (الشكل 3b والشكل البياني الممتد 5d-i). بالمقابل، لم يظهر متلقو خلايا CAR T من نوع PIP-28Z الذين تم علاجهم بـ B6H12
الشكل 2 | البلعمة بواسطة البلعميات لخلايا CAR T في المختبر وفي المرضى. أ، البلعمة لخلايا CFSE المزروعة معًاخلايا CAR T بواسطة البلعميات البشرية الأولية بواسطة تحليل تدفق الخلايا. البيانات هي المتوسطانحراف معياري لـآبار ثلاثية. قابلة للتكرار عبرمتبرعو الماكروفاجتغير الطي في تعبير CD47 وكالريتولين على خلايا CAR T بين اليومين 25 و16 من الثقافة بواسطة تحليل تدفق الخلايا. البيانات هي متوسطالانحراف المعياري للتغير النسبي للقيم (اليوم 25/اليوم 16) المستمد منالمتبرعين. MFI، شدة الفلورة المتوسطة. ج، صور مجهرية لخلايا هيستيوسايت ملونة بصبغة رايت-جيما تستوعب الخلايا اللمفاوية المجمعة من السائل الدماغي الشوكي لمريض مصاب بـ LBCL تم علاجه بخلايا CAR T من نوع CD19-28. تمثيلي من عينة تم جمعها من مريض واحد. قضبان المقياس،مناظر تسلسل RNA أحادي الخلية، مع عرض mRNA CAR باللون الأحمر. اليسار،الخلايا المفروزة من دمتم جمع بيانات المرضى الذين تم علاجهم بـ axi-cel CD19-28 مع LBCL في اليوم السابع بعد حقن خلايا CAR T.الخلايا من السائل الدماغي الشوكيGD2. المرضى المعالجون بـ BB7 الذين يعانون من DMG. تم أخذ عينات من 500 خلية لكل عينة مريض. تم إجراء التحليل الإحصائي باستخدام اختبار Student ذو الطرفين غير المتزاوج.-اختبارات (أ و ب)؛ بالنسبة لـ ب، المقارنة تكون بين قيم التعبير للمجموعة المحددة في اليوم 16 مقابل اليوم 25. لا توجد أي سمية ظاهرة أو فقدان للوزن، بما في ذلك عدم وجود سيتوكينات بشرية قابلة للاكتشاف في الدم (الشكل 3c-f). يمكن أن تؤدي استمرارية خلايا CAR T لفترة طويلة إلى مرض الطعم ضد المضيف (GvHD) في نماذج الزرع. لتحديد ما إذا كان علاج anti-CD47 يمكن أن يمنع GvHD بشكل دائم، قمنا بمراقبة الفئران الحاملة لنموذج Nalm6 المعالجة بخلايا CAR T من نوع CD19-BB7 مع أو بدون B6H12 لتطور GvHD بعد إزالة الورم بنجاح. بعد 48 يومًا، لاحظنا GvHD في الفئران المعالجة بخلايا CAR T من نوع CD19-BBZ، كما يتضح من تساقط الشعر وفقدان الوزن، بينما الفئران التي تم علاجها بخلايا CAR T بالإضافة إلى B6H12 لم تتطور لديها GvHD (الشكل التمديدي 5j-1). تشير هذه البيانات إلى أن حجب CD47 قد يحمل وعدًا كزر أمان جاهز للتخلص من السميات المرتبطة بخلايا CAR-T، كما يتضح من الإنقاذ في نموذج سمية حادة صارمة وكذلك نموذج سمية مزمنة لخلايا CAR T.
CD47 المهندسة لا ترتبط بمضاد CD47
لتحفيز البلعمة الانتقائية للأورام من خلال حجب CD47 مع حماية خلايا T من البلعمة، سعينا لتصميم متغير لـ CD47 يلغي ارتباط مضاد CD47 ولكنه يحتفظ بـ SIRP.التفاعل (الشكل 6a من البيانات الموسعة). عرضنا أولاً مجال CD47 الشبيه بالأجسام المضادة على سطح الخميرة واكتشفنا ارتباطًا قويًا بـ B6H12، ولكن ليس بـ SIRP. (الشكل البياني الموسعبسبب عدم وجود طرف N حر على CD47لذلك استخدمنا SIRP المعدلالمتغير CV-1كوكيل لـ SIRPربطت وخضعت مكتبة من متغيرات الطفرات المعروضة على الخميرة CD47 لستة فرزات متتالية باستخدام فرز الخلايا المعتمد على الفلورية (FACS)، بالتناوب بين الفرز السلبي ضد B6H12 والفرز الإيجابي نحو CV-1 (الشكل التمديدي 6f-h). احتوت جميع المتغيرات في الفرز النهائي على طفرة نقطية واحدة A30P (CD47(A30P)) أو Q31P (CD47(Q31P)) (الشكل 4a)، والتي أكدناها، عندما تم عرضها كمتغيرات CD47 فردية على الخميرة مع طرف N حر لـ CD47.لم يظهر أي ارتباط بـ B 6 H 12 ولكنه احتفظ بخصائص مشابهة أو ارتباط معزز بـ CV-1 و SIRP (الشكل 4ب والشكل الإضافي 6i-k). هذه النتائج تتماشى مع الفهم الهيكلي لـ CD47-SIRPالتفاعلات، حيث SIRPيتواصل بشكل أساسي مع CD47 من خلال حلقة FG الخاصة بـ CD47 والنهاية Nتشكيل اتصالات ثانوية أكثر مع حلقة CD47 BC، التي تشمل Thr26-Gln31 (الشكل 4c والشكل الإضافي 61,m). نظرًا لأن حلقة CD47 BC تقع بالقرب من حلقة CD47 FG الحرجة، يمكن أن تعمل كنقطة تثبيت للأجسام المضادة الأحادية النسيلة المضادة لـ CD47 مثل ، حيث يبدو أن بقايا Gln 31 مهمة بشكل خاص لارتباط الأجسام المضادة (الشكل 4c والشكل الإضافي 6m).
لتحديد ما إذا كانت متغيرات CD47 ترتبط بأجسام مضادة أحادية النسيلة أخرى مانعة لـ CD47، قمنا بتحليل ارتباطها بـ TJC4 (ليمزوبارليماب) و Hu5F9 (ماجروليماب) في اختبار عرض الخميرة. أظهر مسح الألانين للحلقة BC بالكامل (Thr26-Gln31) أن معظم الطفرات سمحت ببعض SIRPالارتباط، مع الطفرات في Ala30 أو Gln31 التي تظهر أقل تأثير (الشكل 4d والشكل الإضافي 6n). Hu5F9، الذي له بصمة ارتباط تتداخل إلى حد كبير مع SIRP“، أظهر فقدانًا طفيفًا في الارتباط بأي من متغيرات حلقة BC. ومع ذلك، فإن TJC4، الذي يرتبط هيكليًا بـ CD47 بطريقة مشابهة لـ B6H12لم يعد مرتبطًا بـ CD47(A30P) وCD47(Q31P) وCD47(A30P/Q31A) ولا بـ CD47(E29A)، وهو متغير إضافي لم يؤثر على ارتباط B6H12 (الشكل 4d). بعد ذلك، قمنا بتقييم ارتباط SIRP، B6H12 و TJC4 إلى CD47 البري الكامل الطول، CD47(A30P)، CD47(Q31P) و CD47(A30P/Q31A) المعبر عنها على خلايا T البشرية الأولية. ارتباط SIRP البشري والفأريلم تتأثر بشكل كبير بأي من المتغيرات الثلاثة ولم نكتشف أي ارتباط لـ B6H12 بأي من المتغيرات الثلاثة، بينما تم إلغاء ارتباط TJC4 تمامًا بواسطة الطفرة المزدوجة CD47(A30P/Q31A) (الشكل 4e والشكل التمديدي 7a). هذه البيانات توضح أن طفرات CD47 في حلقة BC، وخاصة Ala30 وGln31، تنتج بروتينات تحتفظ بـ SIRP.ترتبط ولكنها معفاة من الارتباط بعدة أجسام مضادة أحادية النسيلة ضد CD47، مما يوفر دليلاً على المفهوم لقدرة الهندسة على إشارات ‘لا تأكلني’
الشكل 3 | يمكن استخدام علاج مضاد CD47 كزر أمان. أ، بقاء أورام 143B بعد العلاج مع CD19-BB (غير مستهدف)، HER2-BB (الاستهداف)، خلايا CAR T PIP-28Z أو PIP-BBZ.فئران لكل ذراع. ب، إنترفيرون بشريفي دم الفئران التي تحتوي على أورام 143B-CD19 أو بدونها، المعالجة بخلايا CAR T من نوع CD19-BB7 أو PIP-28 7 أو PIP-BB 7 في اليوم الرابع، كما تم تحديده باستخدام قياس التدفق الكمي LEGENDPlex. البيانات هي متوسطانحراف معياري لـ (PIP-28 أو PIP-BB“مع ورم 143B-CD19″، (CD19-BB7 مع أورام 143B-CD19)، (تجربة مع ورم 143B-CD19 و CD19-BB7، PIP-287 أو PIP-BB7 بدون أورام) و (فئران بدون ورم) . ج، أوزان الفئران بعد علاج خلايا CAR T بـ PIP-28 مع أو بدون B6H12. البيانات هي متوسطانحراف معياري لـفئران لكل ذراع.
تمثيل لتجربتين مستقلتين. د، بقاء الفئران المعالجة بخلايا CAR T PIP-287 مع أو بدون B6H12. n=5 فئران لكل مجموعة. تمثيل لتجربتين مستقلتين. هـ، تصوير ضوئي لخلايا T بعد المعالجة بخلايا CAR T PIP-287 مع أو بدون B6H12. البيانات هي المتوسط.الانحراف المعياري للخطأفئران لكل ذراع. ف، IL-2 (يسار) و IFN (يمين) في دم الفئران المعالجة بـ PIP-28؟ خلايا CAR T مع أو بدون B6H12 في اليوم الرابع، كما تم تحديده باستخدام قياس التدفق الكمي LEGENDPlex. البيانات هي متوسطانحراف معياري لـالفئران. تم إجراء التحليل الإحصائي باستخدام اختبارات مانتل-كوكس ذات الرتبة اللوغاريتمية (أ و د)، واختبار الطالب ذو الذيلين غير المقترن.-اختبارات (ب)، تحليل التباين ثنائي الاتجاه (ج و هـ) وتحليل التباين أحادي الاتجاه مع اختبار المقارنات المتعددة توكي (و).
متغيرات CD47 التي لن يتم حجبها بواسطة الأجسام المضادة أحادية النسيلة المضادة لـ CD47، والتي توقعنا أنها ستدفع البلعمة المحددة للأورام مع الحفاظ على خلايا T في البيئة المجهرية للورم.
يمنع البلعمة المتوسطة بواسطة مضاد CD47
قمنا بعد ذلك بقياس البلعمة لـتم تعديل خلايا جوركات للتعبير عن إما CD47 WT أو CD47(A30P) أو CD47(Q31P) بواسطة البلعميات البشرية المتبرعة. أظهرت متغيرات CD47 المعبر عنها على خلايا جوركات خصائص ارتباط مشابهة للأجسام المضادة أحادية النسيلة المضادة لـ CD47 و SIRP.كما لوحظ في خلايا T الأولية، حيث أدى CD47(Q31P) إلى أكبر فقدان لارتباط B6H12 (الشكل البياني الممتد 7b). عبر عدة متبرعين، قامت البلعميات بتقليل كبير في البلعمة بعد حاضنة B6H12 مع أي من المتغيرين، لكن CD47(A30P) قدم حماية أقل مقارنة بـ CD47(Q31P)، الذي منع تمامًا البلعمة الإضافية بعد الحاضنة مع B6H12 (الشكل 4f والشكل البياني الممتد 7c). بناءً على هذا الملف الواعد، اخترنا المضي قدمًا مع CD47(Q31P)، والذي سيشار إليه فيما بعد بـ (CD47 المهندسة) لمزيد من الدراسة. لدراسة آثارفي الإنسانالخلايا، قمنا بإزالة CD47 الداخلي، ثم قدمنا CAR وTCR و/أو عبر الفيروسات الراجعةأو CD47 WT (ثم قمنا بقياس البلعمة في المختبر وفي الجسم الحي مع أو بدون B6H12. عبر عدة متبرعين من خلايا T والبلعميات، لاحظنا أن علاج B6H12 لم يعزز البلعمة لـCD19-28خلايا CAR T في المختبر، على عكس السيطرة خلايا CAR T CD19-28 (الشكل 7d من البيانات الموسعة). بالمثل، بينما أو CD19-28-نُسخ CAR T الخلوية المُعطاة للفئران غير الحاملة للأورام أظهرت مستويات BLI مشابهة قبل إعطاء B6H12، وقد أزال B6H12خلايا CAR T ولكن لم تؤثر على مستوياتخلايا CAR T (الشكل 4 ج والبيانات الموسعة الشكل) ). هذه البيانات توضح أن يعمل كإشارة ‘لا تأكلني’ في المختبر وفي الجسم الحي، لكنه لا يجعل خلايا T عرضة للبلع بعد حجب CD47 بواسطة B6H12.
تقوم خلايا CAR T بجذب البلعميات إلى الأورام
قمنا بعد ذلك بتحديد كميات البلعمة التي تقوم بها الخلايا التائية بواسطة البلعميات في نموذج 143B الصارم (الشكل 1a). تم حقن خلايا CAR T الموسومة بـ CFSE المضادة لـ HER2-BBZ داخل الأورام الموجودة، مع أو بدون علاج B6H12. أظهرت عينات الورم تسللًا قويًا للبلعميات، واتجاهات نحو انخفاض خلايا CAR T وزيادة CFSE.البلاعم بعد علاج B6H12، متسقة مع البلعمة لخلايا CAR T في الجسم الحي (الشكل التوضيحي الممتد 7h-1). بعد ذلك، عالجنا الفئران الحاملة لورم 143B بدون خلايا T، أو علاج وهمي، أو أو خلايا CAR T من نوع HER2-BB7 مع أو بدون B6H12 وتمت ملاحظة زيادات كبيرة في البلعميات لدى الفئران التي تلقت خلايا CAR T مقارنة بالحيوانات الضابطة أو غير المعالجة (الشكل 5a). لم يؤثر B6H12 على
الشكل 4 | متغير مُهندَس من CD47 يحتفظ بالارتباط بـ SIRP، ولكن لم يعد يرتبط بأجسام مضادة مضادة لـ CD47. أ، الطفرات المتوافقة التي تم تحديدها في الخميرة التي تم تسلسلها بعد الفرز 4 و5 و6. ترددات الطفرات المحددة من و 12 نسخة متسلسلة للأنواع 4 و 5 و 6، على التوالي. ب، الربط المنظم لـ B6H12، CV-1، SIRP البشري وفأر SIRP إلى CD47 WT المعروض على الخميرة، CD47(A30P) وCD47(Q31P). ج، الهياكل البلورية لـ CD47 (باللون الأحمر) المرتبطة بـ SIRP (وردي داكن، يسار) و B6H12 (أزرق فاتح، يمين)؛ يتم الإشارة إلى بقايا Ala30 (ذهبي) و Gln31 (أزرق) بواسطة صناديق. د، الربط المنظم لـ SIRP البشريو Hu5F9 إلى CD47 WT المعروض على الخميرة، CD47(A30P)، CD47(Q31P)، CD47(A30P/Q31A) و CD47(E29A). هـ، الربط العادي لـ B6H12، TJC4، SIRP البشري وفأر SIRP إلى CD47 WT كامل الطول,
CD47(A30P) و CD47(Q31P) المعبر عنهما على خلايا T البشرية الأولية. البيانات هي متوسطانحراف معياري لـ المتبرعين، تم تطبيعهم بالنسبة للجزء المرتبط بـ CD47WT. ف، البلعمة لخلايا جوركات مع CD47 KO الداخلي الذي يعبر عن CD47 WT، CD47(A30P) أو CD47(Q31P)، بواسطة البلعميات البشرية الأولية. البيانات هي متوسط الانحراف المعياري للآبار الثلاثية ( ). قابل للتكرار عبر متبرعو الماكروفاجالخلايا في الدم في اليوم السادس بعد العلاج بـخلايا CAR T مع أو بدون B 6 H 12. البيانات هي المتوسطانحراف معياري لـفئران. من أجل و البيانات تعنيانحراف معياري لـسلالات الخميرة الفردية، تم تطبيعها إلى MFI من الارتباط بـ CD47 WT. تم إجراء التحليل الإحصائي باستخدام ANOVA ثنائية الاتجاه مع اختبار المقارنات المتعددة لتوكاي (b و d-g). بالنسبة لـ d و e، فإن المقارنات تتم بين المجموعات المحددة والارتباط بالخلايا التي تعبر عن CD47 WT. مستويات البلعميات في البيئة الورمية لدى المتلقين لـخلايا CAR T، ولكنها قللت بشكل كبير من مستويات البلعميات في المتلقين لـتزامنت خلايا CAR T مع استنفاد خلايا CAR T (الشكل 5 أ والشكل الإضافي 8 أ، ب). تم التأكيد من خلال تحليل تسلسل RNA أحادي الخلية، حيث كانت تسلل خلايا CAR T والماكروفاج إلى الورم مرتبطين بشكل كبير، مما يتماشى مع نموذج يتم فيه استقطاب خلايا CAR T للماكروفاج إلى الورم، وتعتمد استمرارية الماكروفاج على استمرارية خلايا CAR T في الورم (الشكل 5 ب والأشكال الإضافية 8 ج-و و9 أ-ج). توصيف تسلسل RNA أحادي الخلية و أظهر مستلمو خلايا CAR T تعبيرًا قويًا عن الخلايا التائية لـ TNF وIFNG وCCL3 وCCL4 وCCL5 وCSF1 (المعروف أيضًا باسم M-SCF) وCSF2 (المعروف أيضًا باسم GM-CSF) (الشكل 9d من البيانات الموسعة)، والتي تجذب وتفعّل بشكل جماعي وحيدات النوى والبلاعم وقد تم الإشارة إليها في استقطاب البلاعم بواسطة الخلايا التائية إلى الأورام.تعبير الجينات في المتلقين لـكانت خلايا CAR T غير متغيرة أساسًا مع أو بدون علاج B6H12، في حين أن خلايا T في بيئة الورم المجهرية لمتلقي B6H12 الذين تم علاجهم معًا معأظهرت خلايا CAR T 595 جينًا معبرًا بشكل مختلف (DEGs) بالمقارنة مع أولئك الذين تم علاجهم معخلايا CAR T (الشكل التوضيحي الممتد 9e والجدول التكميلي 1)، بما في ذلك زيادة مجموعات الجينات المسببة للالتهابات مثل IL-12 وCD40/CD40L وإشارات NF-кB (الشكل 5c). توفر هذه البيانات دليلًا على وجود تواصل كبير بين الخلايا النخاعية وخلايا T داخل الـخلايا CAR T في البيئة المجهرية للورم، والتي تفتقر إلى البيئة المجهرية للورم لـمتلقي خلايا CAR T بعد استنفاد خلايا T الناتج عن B6H12. أظهرت تحليلات DEG ضمن مجموعات البلعميات الرئيسية عبر العلاجات أن العلاج معأدت خلايا CAR T وحدها إلى تحفيز 621 جينًا معبرًا مختلفًا في البلعميات مقارنةً بالحالة غير المعالجة، وقد تم تضخيم هذا التأثير بعدعلاج خلايا CAR T مع B6H12، مع 718 جينًا مختلفًا (الشكل 9f من البيانات الموسعة والجدول التكميلي 2). من الجدير بالذكر أن تأثير علاج B6H12 على تعبير الجينات في البلعميات عند إعطائه كعامل منفرد كان ضئيلاً (46 جينًا مختلفًا)، بينما كان للإعطاء المشترك لـ B6H12 معقلوب CAR T خفضت بشكل كبير من DEGs البلعميات، ربما بسبب استنفاد خلايا CAR T (الشكل 9f من البيانات الموسعة). بالمقابل، تحليل المسار للجينات التي تم تنظيمها لأعلى بواسطةتفعيل البلعميات المميز بواسطة خلايا CAR T بالإضافة إلى B6H12
شكل.علاج خلايا T بالإضافة إلى علاج مضاد CD47 يؤدي إلى تعزيز الفعالية المضادة للورم من خلال استقطاب ماكروفاجات متميزة
السكان. أ، تم تحديد البلعميات الكبيرة بواسطة تحليل تدفق الخلايا (تدفق؛ المحور الأيسر) والكيماويات المناعية (IHC؛ المحور الأيمن) لأورام الساركوما العظمية 143B المستأصلة التي تم علاجها بدون خلايا T، أو العلاج الوهمي، أو خلايا CAR T من نوع HER2-BBZ مع أو بدون B6H12. البيانات هي متوسط (تدفق: ) أو تعني انحراف معياري لـ (جميع الآخرين) الفئران.تركيب الخلايا المحددة باستخدام تسلسل RNA أحادي الخلية من الأورام المعالجة كما في أ. خلايا من 8 ظروف تجريبية. DC، خلايا دندريتية. ج، تحليل مسار Enrichr لأعلى 100 جين تم تنظيمه بشكل مرتفع في خلايا CAR T من الأورام المعالجة بخلايا EAR T 47 + B6H12 مقابل خلايا CAR T + B6H12. تظهر النتائج القيمة (اختبار فيشر الدقيق ذو الجانبين) لكل مسار. د، تكوين تجمعات البلعميات (c0-c6) بعد التقسيم وإعادة التجميع، ملون حسب التجمع، عبر التجربة الشروط الموضحة في خلايا من 8 ظروف تجريبية. ماك، بلاعم; مو-دي سي، خلية دندريتية مشتقة من وحيدة النواة; مو، وحيدة النواة. هـ، تصوير ضوئي لخلايا T في فئران 143B الحاملة للورم المعالجة بـ أو خلايا CAR T من نوع Antares HER2-BB7 مع أو بدون B6H12 في اليوم الثالث عشر. تم زراعة الأورام في الساق اليمنى. خلايا T في دم الفئران الحاملة لورم 143B، تم علاجها كما هو موصوف في، في اليوم الرابع عشر. البيانات هي المتوسطانحراف معياري لـفئران لكل ذراع.نمو الورم بعد العلاج كما هو موصوف في e. البيانات هي المتوسطالخطأ المعياري للمتوسطفئران لكل ذراع. هـ، خلايا T في دم الفئران الحاملة لورم A375، المعالجة بـخلايا T TCR NY-ESO-1 مع أو بدون B6H12، في اليوم 15. البيانات هي المتوسطانحراف معياري لـفئران لكل ذراع. أنا، نمو ورم الميلانوما A375 المعالج كما في هـ. البيانات هي المتوسطالانحراف المعياري للخطأفئران لكل ذراع. تم إجراء التحليل الإحصائي باستخدام تحليل التباين ثنائي الاتجاه مع اختبار المقارنات المتعددة توكي (أ، ب، ف، ج، و ي) واختبار الطالب غير المقترن ذو الذيلين.-اختبارات ( ). من خلال إثراء الليزوزوم، والمكمل، وعرض المستضد، ومسارات البلعمة (الشكل 9g من البيانات الموسعة). للمزيد من توصيف التغيرات في حجرة البلعميات التي تسببهاخلايا CAR T، قمنا بإعادة تجميع مجموعة البلعميات/وحيدات النواة (الشكل التمديدي 9h، i). كما لوحظ في البيانات السريرية (الشكل 2d)، حددنا العديد من البلعميات التي تحتوي على mRNA البشري CD3E ضمن مجموعات فرعية متعددة من البلعميات المعالجة بخلايا CAR-T (الشكل التمديدي 9j)، مما يتماشى مع البلعمة التي تتوسطها البلعميات من
خلايا CAR T. كما لاحظنا توسع تجمعات البلعميات c0، التي زادت بعد علاج خلايا CAR T وتوسعت أكثر بعد العلاج معCAR T بالإضافة إلى B6H12، ولكن تم تقليله بشكل كبير بعد العلاج مع خلايا CAR T بالإضافة إلى B6H12 (الشكل 5d)، مما يشير إلى أن هذه البلعميات تعتمد على تراكم خلايا CAR T داخل بيئة الورم. كانت الجينات المعبر عنها بشكل مختلف الرئيسية في المجموعة الموسعة، مثل Arg1 وMrc1 وChil3 وTlr1، مرتبطة بالبلعميات الشبيهة بـ M2c. (الشكل 9k من البيانات الموسعة والجدول التكميلي 3). بينما تعتبر البلعميات M2 بشكل عام يُفهم أنها تعزز الأورام، وقد تم إثبات أنها تظهر قدرة بلع قوية، خاصة تلك الموجودة في الفئة الفرعية M2c..
معًا، تُظهر هذه النتائج حلقة تغذية راجعة حيث تقوم خلايا CAR T بتحفيز تجنيد وتنشيط البلعميات داخل بيئة الورم، وفي الوقت نفسه، تعزز البلعميات المسارات المناعية المنشطة فيخلايا CAR T في البيئة المجهرية للورم. لم تحدث هذه التأثيرات داخلبيئة الورم المحيط بخلايا CAR T، حيث يؤدي حجب CD47 إلى استنفاد خلايا CAR T مما يلغي الدورة. وتظهر البيانات أيضًا تواصلًا قويًا بين خلايا CAR T والبلعميات في بيئة الورم، مما يؤدي إلى تحفيز عدة برامج تعبير جيني مؤيدة للالتهابات من المتوقع أن تعزز التأثيرات المضادة للورم.
مضاد CD47 يعزز فعاليةخلايا TCR T
قمنا بعد ذلك بتقييم التأثيرات المضادة للورم لـ ضد الخلايا بالإضافة إلى علاج مضاد CD47 في نماذج أورام متعددة. في 143B، حيث يكون لكل من خلايا CART وعلاج مضاد CD47 تأثير ضئيل كعلاجات فردية (الشكل 1a)، لاحظنا تجنيدًا ملحوظًا للخلايا التائية التي تتوسطها البلعميات إلى الأورام (الشكل 5a,b). بعد العلاج بـ B6H12،تم استنفاد خلايا CAR T تمامًا، بينمااستمرت خلايا CAR T لعدة أسابيع (الشكل 5e، f والبيانات الموسعة الشكل 10a-e). لا أو خلايا CAR T وحدها، ولا خلايا T الوهمية أوتمتاز خلايا CAR T المرتبطة بـ B6H12 بتأثيرات مضادة للأورام كبيرة، في حين أن B6H12 بالإضافة إلىسيارةأدت الخلايا إلى تأخير كبير في نمو الورم وتحسين في البقاء العام (الشكل 5g والشكل الإضافي 10f-h). تم ملاحظة نتائج مماثلة مع جرعات أقل من مضاد CD47، والتي قد تكون مرتبطة بتقليل السمية في الدراسات السريرية. (الشكل 10i,j من البيانات الموسعة).
قمنا أيضًا بدمج B 6 H 12 مع ضد خلايا CART في الورم العصبي النخاعي النقيلي وتمت ملاحظة زيادة في الاستمرارية وتحسين الفعالية المضادة للورم معخلايا CAR T بالإضافة إلى B6H12 (الشكل 11a-d من البيانات الموسعة) وزيادة الفعالية المضادة للورم مع ضد CD19-28 (خلايا CAR T بالإضافة إلى B6H12 في الفئران التي تحمل سرطان الدم Nalm6-fLuc (الشكل 11e، f). أخيرًا، في نموذج الميلانوما A375، قامت B6H12 بتقليل خلايا NY-ESO-1T التي تعبر عن CD47 الداخلي (الشكل 1e)، بينما استمرت خلايا NY-ESO-1T (الشكل 5h والشكل الإضافي 11g،h). تم إبطاء نمو ورم A375 بشكل طفيف بواسطة B6H12 بالإضافة إلى خلايا T الوهمية والعلاج بـتسببت خلايا T الخاصة بـ NY-ESO-1 وحدها في السيطرة الأولية على الورم، ولكنها أدت في النهاية إلى نمو الورم. بالمقابل، الفئران التي تم علاجها بـأظهرت خلايا T الخاصة بـ NY-ESO-1 و B6H12 السيطرة الكاملة على الورم والعلاج في جميع الفئران المعالجة (الشكل Si والشكل الإضافي 11i، j).
معًا، تُظهر هذه البيانات أن حجب CD47 مقترن بـالتعبير عن خلايا T العلاجية يحمي خلايا T من البلعمة التي تتوسطها البلعميات، مما يؤدي إلى تدفق كبير ومستدام من البلعميات داخل بيئة الورم، مرتبط بتواصل خلايا T والبلعميات. النتيجة هي تآزر قوي مضاد للورم في الأورام الصلبة والسائلة والمنتشرة، حتى عند الجرعات المنخفضة وفي الظروف التي لا تظهر فيها العلاجات الفردية أي نشاط.
نقاش
أفادت الدراسات السابقة أن CD47 ضروري لمنع رفض المناعةوأن زيادة التعبير عن CD47 مع حذف MHC تمنح خلايا CAR T مقاومة للاحتقار المناعي الأجنبي.تظهر نتائجنا أنه، حتى في غياب الرفض المناعي، فإن CD47 مطلوب لبقاء خلايا T المنقولة بالتبني، وأن زيادة التعبير عن CD47 تحسن من بقاء وفعالية خلايا CAR T (الشكل 1j، k). أدى علاج مضاد CD47 في مضيفي الفئران NSG إلى استنفاد سريع وكامل لخلايا T المنقولة بالتبني بواسطة البلعميات (الشكل 1)، وكان ذلك سريعًا وقويًا بما يكفي لتوفير حماية كاملة في نموذج سمية خلايا CAR T القاتلة (الشكل 3)، مما يوفر دليلًا لدعم اختبار CD47-SIRP.عوائق للتخفيف السموم الناتجة عن العلاجات الخلوية التبنيّة، والتي يمكن أن تحقق فوائد سريرية فورية. تتماشى هذه النتائج مع البيانات التي تظهر أن خلايا CAR T المعدلة جينياً لإزالة CD47 تظهر استمرارية محدودة في نماذج الزرع الغريب.وملاحظات حول انخفاض إجمالي اللمفاويات و اللمفاويات المحددة للمستضد، وزيادة القابلية للإصابة بالعدوى وانخفاض القابلية للاعتلال الذاتي في الفئران التي تم حذف جين Cd47 و Sirpa.تُوفر الصلة بالإعداد السريري من خلال ملاحظة نقص اللمفاويات في دراسات الماجروليماب والإيفورباست، وكلاهما يمنع ارتباط CD47 بـ SIRP..
على الرغم من أن استنفاد خلايا T قد زاد بشكل كبير من خلال حجب CD47 في دراساتنا، إلا أن بياناتنا تظهر أن بلع البلعميات يحد من بقاء خلايا T المنقولة بالتبني حتى في غياب CD47-SIRP.الحصار (الشكل 11 والأشكال البيانية الموسعة 7h-1 و9j). كما حددت بيانات تسلسل RNA أحادي الخلية السريرية بشكل متسق نسخ CAR داخل الخلايا النخاعية (الشكل 2d). معًا، تتماشى هذه النتائج مع نموذج يلعب فيه ابتلاع البلعميات دورًا مهمًا في تنظيم توازن خلايا T. من المعروف جيدًا أن حجب CD47 يسبب نقصًا انتقائيًا في خلايا الدم الحمراء القديمة.لقد لاحظنا زيادة في تعبير إشارات ‘كلني’ وانخفاض في تعبير CD47 على خلايا T بعد زراعة طويلة الأمد (الشكل 2ب والبيانات الموسعة الشكل). )، مما يثير احتمال أن CD47-SIRP قد ينظم المحور بشكل مشابه إزالة خلايا T القديمة. هناك حاجة إلى مزيد من البحث لتحديد العلاقة بشكل أفضل بينتنشيط الخلايا والتمايز، والتعبير عن مستقبلات مؤيدة للبلعمة ومناهضة للبلعمة، وقابلية البلعمة.
لقد لاحظنا أن خلايا CAR T تحفز تدفقًا سريعًا للخلايا البالعة إلى الأورام (الشكل 5 أ، ب)، وأن بقاء الخلايا البالعة يعتمد على بقاء خلايا CAR T. ونتيجة لذلك،أظهر مستلمو خلايا CAR T تواصلًا واسع النطاق بين الخلايا النخاعية وخلايا T في بيئة الورم، بما في ذلك تحفيز IL-12 وCD40L وإشارات NF-кB في خلايا T (الشكل 5c)، والتي تم إثبات أنها تعزز التأثيرات المضادة للورم.. وبالتالي، حمت خلايا T من البلعمة التي تتوسطها الأجسام المضادة CD47 (الشكل 4f، g والشكل الإضافي 7)، بينما عززت البلعمة الورمية، وعرض المستضدات، وأدت إلى بيئة ورمية مؤيدة للالتهاب. هناك حاجة إلى دراسات إضافية للاختبارفي أنظمة المناعة الكاملة لفهم التأثير على أنواع خلايا المناعة الأخرى، بما في ذلك خلايا T الذاتية وخلايا القاتل الطبيعي.. بينما يعتبر B 6 H 12 جسمًا مضادًا من الدرجة البحثية، فإن المتغيرات CD47 التي تم إنشاؤها في هذه الدراسة قد ألغت أيضًا الارتباط بجسم مضاد من الدرجة السريرية TJC4 (ليمزوبارلي (الشكل 4د،هـ)، مما يقودنا إلى التنبؤ بأنيمكن أن يسمح بدمج العلاجات التبنيّة مع الأجسام المضادة المضادة لـ CD47 من الدرجة السريرية. يمكن تطوير تحسينات إضافية لهذه الطريقة.بما في ذلك الاقتران مع أجسام مضادة إضافيةوإحداثإفراز الخلايا لـ CD47-SIRP” blockers ” يمكن ترجمتها إلى “موانع” أو “حواجز” حسب السياق..
تُعتبر الخلايا المناعية المرتبطة بالورم (TAMs) من بين أكثر الخلايا وفرة في بيئة الورم، وقد كان هناك اهتمام كبير في استغلال خصائصها المضادة للورم، ولكن تفتقر العلاجات المعدلة للماكروفاجات الفعالة لعلاج السرطان.لا مثبطات CSF1R ولا مثبطات CCR2، التي تمنع تجنيد وحيدات النوى إلى البيئة المجهرية للورم وتقلل أو تقضي على TAMsولا الاقترابات لتعديل حالات البلعميات من تلك ذات الملفات المناعية المثبطة، بما في ذلك مجموعة M2-like، نحو تلك ذات الملف الالتهابي الأكثر وضوحًا قد حققت فائدة علاجية واضحة.. وبالمثل، على الرغم من الحصار الجهازي لـ CD47-SIRPتأثيرات مضادة للورم بوساطة المحور في عدة نماذج قبل السريريةتفتقر الفائدة السريرية كعوامل فردية وفي السرطانات الصلبةتشير البيانات المقدمة هنا إلى أن هذا اللغز قد يُفسر بالسيف ذي الحدين الذي تمثله خلايا المناعة المرتبطة بالورم (TAMs) داخل بيئة الورم (TME)، حيث يتم تعويض التأثيرات المضادة للورم للخطط المصممة لزيادة بلع البلعميات من خلال بلع خلايا T التفاعلية للورم. وعلى العكس، قد يؤدي القضاء على خلايا TAMs أو تقليلها إلى تقليل الإضعاف المناعي وبلع خلايا الورم المتسللة.الخلايا، ولكن هذه الفوائد تعوضها فقدان البلعمة الورمية بواسطة البلعميات. تظهر بياناتنا أن الجمع بينسيارةالعلاج باستخدام مضاد CD47 هو احتمال مثير يمكن أن يعزز فعالية العلاج بالتبنيعلاجات الخلايا، خاصة في السرطانات الصلبة.
المحتوى عبر الإنترنت
أي طرق، مراجع إضافية، ملخصات تقارير Nature Portfolio، بيانات المصدر، بيانات موسعة، معلومات تكميلية، شكر وتقدير، معلومات مراجعة الأقران؛ تفاصيل مساهمات المؤلفين والمصالح المتنافسة؛ وبيانات توفر البيانات والرموز متاحة علىhttps://doi.org/10.1038/s41586-024-07443-8.
لبانية، ل. وماكال، س. ل. خلايا المناعة CAR: مبادئ التصميم، المقاومة والجيل القادم. ناتشر 614، 635-648 (2023).
كلوسترمين، د. ج. وأكاري، ل. البلعميات عند واجهة نظام السرطان المتطور بشكل مشترك. خلية 186، 1627-1651 (2023).
زيزو، ج.، هيلارد، ب. أ.، مونيستير، م. وكوهين، ب. ل. يتطلب التخلص الفعال من الخلايا المبرمجة للموت المبكر بواسطة البلعميات البشرية استقطاب M2c وتحفيز MerTK. مجلة المناعة 189، 3508-3520 (2012).
زو، ي. وآخرون. حجب CSF1/CSF1R يعيد برمجة البلعميات المتسللة إلى الورم ويحسن الاستجابة للعلاج المناعي عن طريق نقاط التفتيش T-cell في نماذج سرطان البنكرياس. أبحاث السرطان. 74، 5057-5069 (2014).
نويل، م. وآخرون. دراسة المرحلة 1ب لمضاد جزيئي صغير لمستقبل كيموكين الإنسان (C-C motif) 2 (PF-04136309) بالاشتراك مع ناب-باكليتاكسيل/جمسيتابين في العلاج الأولي لسرطان البنكرياس القنوي النقيلي. استثمار. N. أدوية 38، 800-811 (2020).
تشاو، م. ب. وآخرون. الكالريتولكين هو الإشارة الرئيسية المعززة للبلعمة في عدة أنواع من السرطان البشري ويتم موازنتها بواسطة CD47. علوم. ترانسلات. ميد. 2، 63ra94 (2010).
ويزكوف، ك. وآخرون. العلاجات المناعية التي تعيق CD47 تحفز تدمير سرطان الرئة ذو الخلايا الصغيرة بواسطة البلعميات. ج. استثمار سريري. 126، 2610-2620 (2016).
لاكاني، ن. ج. وآخرون. إيفورباستبت بمفرده وبالاشتراك مع بيمبروليزوماب أو تراستوزوماب في المرضى الذين يعانون من أورام صلبة متقدمة (ASPEN-01): دراسة أولية على البشر، مفتوحة، متعددة المراكز، من المرحلة 1 لتصعيد الجرعة وتوسيع الجرعة. لانسيت أونكول. 22، 1740-1751 (2021).
سيكيك، ب. آي. وآخرون. تجربة المرحلة الأولى الأولى في البشر، الأولى من نوعها، للأجسام المضادة المضادة لـ CD47 Hu5F9-G4 في مرضى السرطان المتقدم. ج. كلين. أونكول. 37، 946-946 (2019).
هو، إكس. وآخرون. خلايا T المعدلة وراثياً المضادة لـ CD19 ذات المناعة المنخفضة توفر تحكماً دائماً في الأورام في الفئران البشرية المتوافقة مناعياً. نات. كوميونيك. 14، 2020 (2023).
جيد، ز. وآخرون. بعد الحقن CARتحدد الخلايا المرضى المقاومين لعلاج CD19-CAR. نات. ميد. 28، 1860-1871 (2022).
ماجنزر، ر. ج. وآخرون. علاج خلايا T CAR GD2 للأورام الدبقية المنتشرة المتوسطة المتحورة H3K27M. ناتشر 603، 934-941 (2022).
هاذرلي، د. وآخرون. تم شرح خصوصية المستقبلات المزدوجة من خلال هياكل بروتينات تنظيم الإشارة بمفردها ومعقدة مع CD47. مول. خلية 31، 266-277 (2008).
بيتش، إي. سي. وآخرون. النشاط المضاد لسرطان الدم وقابلية التحمل للأجسام المضادة أحادية النسيلة المضادة للبشر CD47. مجلة سرطان الدم 7، e536 (2017).
مينغ، ز.، وانغ، ز.، قوه، ب.، كاو، و. وشين، هـ. TJC4، جسم مضاد مضاد لـ CD47 متميز بخصائص جديدة وموفرة لكريات الدم الحمراء. بلود 134، 4063-4063 (2019).
كيرستن، ك. وآخرون. الاعتماد المشترك الزماني والمكاني بين البلعميات والمستنفدينخلايا T في السرطان. خلية السرطان 40، 624-638 (2022).
بلازار، ب. ر. وآخرون. يتطلب تفاعل CD47 (بروتين مرتبط بالإنتيجرين) مع مستقبلات الخلايا التغصنية والبلعمية لمنع إزالة خلايا الدم المانحة. ج. التجارب الطبية. 194، 541-549 (2001).
بيكيت، أ. ن. وآخرون. تعبير CD47 ضروري لبقاء خلايا CAR T في الجسم الحي. ج. مناعة السرطان 11، e005857 (2023).
لي، ل. إكس.، عاطف، س. م.، شمييل، س. إ.، لي، س. ج. ومك سورلي، س. ج. زيادة القابلية للإصابة بسالمونيلا في الفئران التي تفتقر إلى بروتين الإشارة التنظيمية أ. مجلة المناعة 189، 2537-2544 (2012).
أوتيو، أ. وآخرون. محور SIRPa-CD47 ينظم تفاعلات الخلايا التغصنية مع خلايا T وتنشيط TCR خلال تحفيز خلايا T في الطحال. PLoS ONE 17، e0266566 (2022).
Deuse، T. وآخرون. نقطة التفتيش المناعية SIRPa-CD47 في خلايا NK. J. Exp. Med. 218، e20200839 (2021).
داشيك، م. م. وآخرون. تعزيز قتل السرطانات المعتمد على الأجسام المضادة باستخدام خلايا CAR T التي تفرز مثبط نقطة التفتيش CD47-SIRPa. بلود 141، 2003-2015 (2023).
تشن، هـ. وآخرون. توصيل مثبط CD47 SIRPa-Fc بواسطة خلايا CAR-T يعزز الفعالية المضادة للورم. ج. مناعة السرطان 10، e003737 (2022).
هونغ، د. س. وآخرون. إيغانيليسِب، مثبط PI3Ky الأول من نوعه، في المرضى الذين يعانون من أورام صلبة متقدمة: نتائج تجربة المرحلة 1/1ب MARIO-1. أبحاث السرطان السريرية 29، 2210-2219 (2023).
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا ما تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فسيتعين عليك الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/licenses/by/4.0/. (ج) المؤلف(ون) 2024
طرق
خطوط الخلايا
تم توفير خط خلايا Nalm6 B-ALL بواسطة D. Barrett وتم نقله فيروسياً للتعبير عن GFP و firefly luciferase. تم الحصول على خلايا 143B من مجموعة الثقافة الأمريكية (ATCC) ثم تم نقلها فيروسياً مع CD19 البشري. تم الحصول على خط خلايا CHLA-255 العصبية من R. Seeger وتم نقله فيروسياً مع GFP و firefly luciferase. تم توفير خلايا MG63.3 بواسطة C. Khanna وتم نقلها فيروسياً مع GFP و firefly luciferase. تم توفير خلايا D425 بواسطة S. Chesier وتم نقلها فيروسياً للتعبير عن GFP و firefly luciferase. تم الحصول على خلايا Nalm6 و MG63.3 في الأصل من ATCC. تم الحصول على خلايا D425 في الأصل من Sigma-Aldrich. تم الحصول على خلايا الميلانوما A375 وخلايا Jurkat (النسخة E6-1) من ATCC. تم توفير خط التعبئة الفيروسية 293GP بواسطة فرع الجراحة (المعهد الوطني للسرطان، المعاهد الوطنية للصحة). تم الحصول على خلايا التعبئة الفيروسية HEK293T من ATCC. تم الحصول على خلايا إنتاج البروتين Expi293 من ATCC.
تم الحفاظ على خلايا D425 في وسط خالٍ من المصل معزز بـ B27 (ثيرمو فيشر ساينتيفيك)، EGF، FGF (شيناندواه بيوتيكنولوجي)، LIF البشري المعاد تركيبه (ميلبورو) والهيبارين (ستيمسيل تكنولوجيز). تم زراعة خلايا Nalm6 و143B وA375 وMG63.3 وCHLA-255 وJurkat في RPMI-1640 (جيبكو). تم زراعة خلايا 293GP وHEK293T في DMEM (جيبكو). تم زراعة خلايا Expi293 في وسط Expi293 (ثيرمو فيشر ساينتيفيك). تم تعزيز وسط زراعة الخلايا بـ FBS، 10 مللي مولار HEPES، 2 مللي مولار l-glutamine، البنسلين وستربتوميسين (جيبكو)، باستثناء وسط Expi293. تم إجراء تحليل الحمض النووي باستخدام تكرارات قصيرة متتالية لجميع خطوط الخلايا مرة واحدة في السنة (اختبار خطوط الخلايا جينتيكا). تم اختبار جميع خطوط الخلايا بانتظام للكشف عن الميكوبلازما. تم زراعة خطوط الخلايا فيفيالبيئة.
مصدر خلايا T البشرية الأولية والبلاعم
تم شراء أكياس البوفي من متبرعين أصحاء من مركز ستانفورد للدم بموجب بروتوكول معفى من IRB. تم شراء كريات الدم البيضاء من متبرعين أصحاء من شركة StemCell Technologies. تم تنقية خلايا T البشرية الأولية عن طريق الاختيار السلبي باستخدام مجموعة RosetteSep لتغنيت خلايا T البشرية (StemCell Technologies) وأنابيب SepMate-50. تم حفظ خلايا T بالتبريد عندخلايا لكل مل في وسط التجميد CryoStor CS10 (تقنيات StemCell) حتى الاستخدام. تم تنقية المونوسيتات المحيطية الأولية من خلال تدرجات الكثافة المتعاقبة باستخدام فيكول (سيغما-ألدريتش) وبيركول (GE Healthcare). ثم تم تحويل المونوسيتات إلى بلاعم من خلال 7-9 أيام من الثقافة في IMDM.مصل بشري AB (تقنيات الحياة).
بناء الناقل الفيروسي
تمت رؤية جميع التركيبات الجينية باستخدام برنامج SnapGene (الإصدار 6.0.2؛ Dotmatics). تم استنساخ جميع التركيبات الفيروسية العكسية في المتجه الفيروسي العكسي MSGV1.تم وصف تراكيب CAR و TCR المستخدمة في هذه الدراسة سابقًا:HER2-BBو NY-ESO-“. تم إنتاج B7H3-BB7 سابقًا عن طريق دمج، من الطرف N إلى الطرف C، تسلسل قائد GM-CSF البشري، scFv المشتق من MGA271 في اتجاه VH-VL و(GGGS)سلسلة الربط، CD8الوصلة والتسلسل عبر الغشاء، و4-1BB وCD3 البشرية، مجالات الإشارة داخل الخلايا. تم إنتاج GD2-BB7 وHER2-BB7 وCD19-BB7 سابقًا عن طريق استنساخ scFvs المشتقة من الأجسام المضادة 14G2A و4D5 وFMC63، على التوالي، في متجه B7H3-BB7 بدلاً من scFv MGA271. تم إنتاج CD19-28 7 سابقًا عن طريق استبدال مجال 4-1BB في CD19-BB7 بمجال الإشارة داخل الخلايا للبشر CD28. تم إنتاج HA-287 سابقًا عن طريق استبدال scFv FMC63 بـ scFv 14G2a الذي يحتوي على طفرة نقطية (E101K) تليها منطقة فاصلة مشتقة من مجالات CH 2 CH 3 من IgG1. تم إنتاج PIP-28 وPIP-BB 3 عن طريق استبدال scFv FMC63 بـ 2.5 F knottin.تبع عن طريق تسلسل علامة العلم (DYKDDDDK) في ناقلات CD19-28 و CD19-BB على التوالي. تم بناء جهاز تقارير تنشيط خلايا T في الجسم الحي عن طريق استنساخ تسلسل يحتوي على لوسيفيراز اليراعة في ناقل فيروس اللينت pGreenFire1-NF-кB (نظام علوم الحياة) تحت المحفز المستجيب لـ NF-кB.تم إنشاء ناقلات CD47 عن طريق إدخال تسلسلات CD47 المحسّنة (المتغيرة و WT) بدلاً من تسلسل CD19-BB7. من أجل تتبع في الجسم الحي، تم إنشاء بلازميدات CAR-nLuc عن طريق استبدال كودون التوقف في CD37 بتسلسل يحتوي على تسلسل تخطي الريبوسوم من فيروس تيسش 12A (P2A)، متبوعًا بالنانو لوسيفيراز.تم إنشاء بلازميدات أنتايرس عن طريق إدخال تسلسل أنتايرسبدلاً من تسلسل CD19-BB7. تم إنشاء بناء TCR الخاص بـ NY-ESO-1 عن طريق إدخال NY-ESO-1 سلسلة، تليها تسلسل P2A، تليها سلسلة بدلاً من CD19-BB7.
إنتاج الفيروس
تم تعبئة السائل الفائق للفيروسات الرجعية باستخدام خلايا 293GP و بلازميد غلاف RD114. باختصار،RD114 وتم تسليم بلازميد نقل MSGV1 المقابل إلى خلايا 293GP المزروعة على أطباق بولي-D-لايسين بحجم 150 مم (كورنينغ) حتى تصل إلى 80% من التلاصق عن طريق التحويل المؤقت باستخدام Lipofectamine 2000 (ثيرمو فيشر ساينتيفيك). تم تجديد الوسط كل 24 ساعة. تم إنتاج الفيروس جنبًا إلى جنب من أجل مقارنات بين تراكيب CAR و TCR و CD47. تم جمع السائل الفيروسي بعد 48 و72 ساعة من التحويل. تم تجميع السوبرناتانت من الأطباق المكررة، وتم الطرد المركزي لإزالة بقايا الخلايا وتخزينه فيحتى الاستخدام. تم إنتاج سوائل فيروسية لنتيفيرال ذاتية التوقف من الجيل الثالث بطريقة مماثلة باستخدام خلايا HEK293T.غلاف pMD2.G (VSVg) pMDLg/pRRE ( ) ، pRSV-Rev و للنقل البلازميدات المقابلة. كانت جميع التركيبات فيروسية عكسية، باستثناء خلايا Tبناء التنشيط، الذي كان فيروسًا لينيًا.
تصنيع خلايا CAR T وخلايا TCR T
في اليوم 0، الإنسان الأساسيتم إذابة الخلايا وتنشيطها باستخدام كريات دينابيد المضادة لـ CD3/CD28 (ثيرمو فيشر ساينتيفيك) في أو نسبة الخرز إلى الخلية. في اليوم الثاني، تم تحضير أطباق الثقافة المغلفة بالفيروس على أطباق 12 بئر غير معالجة للزراعة النسيجية التي تم تغليفها مسبقًا بـ RetroNectin (تاكارا بيو) وفقًا لتعليمات الشركة المصنعة، عن طريق الحضانة مع 1 مل من السائل الفيروسي الرجعي. ) والطرد المركزي عند و لمدة ساعتين. ثم تم شفط السائل العلوي من الآبار وتم إضافة خلايا في 1 مل من وسط خلايا T الذي يتكون من AIM V (ثيرمو فيشر ساينتيفيك)،FBS،البنسلين (جيبكو)ستربتوميسين (جيبكو)، 2 مللي مول l-جلوتامين (جيبكو)، 10 مللي مول HEPES (جيبكو) و rhIL-2 (Peprotech). بعد إضافة خلايا T، تم تدوير الأطباق بلطف في جهاز الطرد المركزي عندلمدة دقيقتين ثم حضنت لمدة 24 ساعة عندتحت. تم تكرار عملية التحويل هذه في اليوم الثالث واليوم الرابع (إذا لزم الأمر). تم إزالة كرات الدينابيد في اليوم الرابع أو اليوم الخامس عن طريق الفصل المغناطيسي. تم الحفاظ على الخلايا بين و خلايا لكل مل وتم توسيعها حتى اليومعادةً، تم نقل خلايا T باستخدام CAR أو TCR وAntares (إذا تم استخدامه) في اليوم الثاني، ثم تم استخدام متغيرات CD47 في اليومين الثالث والرابع.
CRISPR-Cas9 KO لجين CD47 و AAVS1
تم تحضير ريبونوكليوبروتين (RNP) باستخدام sgRNA الاصطناعي مع تعديل الفوسفات الثيوي 2′-O-methyl (Synthego) مخفف في محلول TE عندإجماليتم تحضين sgRNA معذاكرة مؤقتة مزدوجة (IDT) وإنزيم نوكلياز كاس 9 من ستربتوكوكوس بيوجينيس V3 (IDT) لمدة 30 دقيقة في درجة حرارة الغرفة. التفاعلات (تم تجميعها مع 5 ملايين خلية T أو خلايا جوركات،عازل P3 (لونزا) وRNP. تم تنشيط الخلايا باستخدام بروتوكول EO115 باستخدام مجموعة P3 للخلايا الأولية 4D-Nucleofector ونظام 4D-Nucleofector (لونزا). تم استعادة الخلايا على الفور باستخدام وسط دافئ لمدة 6 ساعات قبل النقل باستخدام CAR أو TCR. تم إجراء التحفيز الكهربائي للخلايا باستخدام RNP على اليوم الثاني بعد الذوبان وتم نقله في نفس اليوم. كانت تسلسلات الدليل كما يلي: CD47، 5′-AUGCUUUGUUACUAAUAUGG-3′; AAVS1،-GGGGCCACUAGGGACAGGAU-.
تحليل تدفق الخلايا باستخدام تقنية تحليل تدفق الخلايا للمخلوقات الثديية
تم غسل الخلايا بمحلول FACS (2% FBS في PBS) قبل التلوين. تم إجراء التلوين في محلول FACS لمدة 30 دقيقة عند“. في تجارب معينة، تم تلوين الخلايا أولاً بصبغة الحياة القابلة للإصلاح eFluor 780 (eBioscience، ) في PBS لمدة 10 دقائق في درجة حرارة الغرفة قبل الغسيل بمحلول FACS وصبغها بأجسام مضادة أخرى. بعد الصبغ، تم غسل الخلايا مرة واحدة بمحلول FACS وتم تحليلها على نظام BD Fortessa. تم استخدام برنامج FACSDiva (الإصدار 8.0.1؛ BD) لجمع البيانات وبرنامج FlowJo (الإصدار 10.8.1؛ BD) لتحليل البيانات (استراتيجيات التصفية موضحة في الشكل التكميلية 2).
تم استخدام B7H3-Fc و HER2-Fc المعاد تركيبها (كلاهما من أنظمة R&D، بتخفيف 1:400) للكشف عن سطح CAR الخاص بـ B7H3 و HER2، على التوالي. وبالمثل، تم استخدام جسم مضاد مضاد لـ FMC63 (Genscript، 1:400) للكشف عن CARs CD19، بينما تم استخدام جسم مضاد مضاد لـ 14G2a (المعهد الوطني للسرطان، 1:400) للكشف عن CARs GD2 و HA. تم وسم كواشف الكشف عن CAR بالأجسام المضادة الفلورية باستخدام مجموعة وسم الأجسام المضادة DyLight 650 Microscale (Thermo Fisher Scientific). تم استخدام جسم مضاد لعلامة DYKDDDDK (علامة Flag، APC، L5، BioLegend، 1:400) للكشف عن CAR PIP. تم الكشف عن TCR NY-ESO-1 باستخدام أجسام مضادة محددة لـ V. (APC، H131، BioLegend، 1:100)، سلسلة البيتا من TCR NY-ESO-1. تم الكشف عن CD47 باستخدام B6H12 (BV711 و PE، B6H12، BD، 1:100؛ APC، B6H12، Invitrogen، 1:100؛ أو غير موسوم، Bio X Cell، التركيزات مدرجة في الأشكال)، TJC4 (غير معنون، تم إنتاجه داخليًا، التركيزات مدرجة في الأشكال)، (غير معنون، تم إنتاجه داخليًا، التركيزات مدرجة في الأشكال)، CV-1-Fc (غير معنون، ALX Oncology، التركيزات مدرجة في الأشكال)، (غير مُعَلَّم، Sino Biological، التركيزات مُدَرَجَة في الأشكال) أو hSIRP “-Fc (غير موسوم، Sino Biological، التركيزات مدرجة في الأشكال)، تلاها الكشف باستخدام أجسام مضادة متعددة النسائل ضد IgG الفأري أو البشري (AF488 و AF647، متعددة النسائل، Invitrogen، 1:500). تم استخدام أجسام مضادة للتحكم من نوع mIgG1 (غير موسومة، MPOC-21، Bio X Cell، 1:100 و PE، B11/6، Abcam، 1:100) كضوابط لتلوين B6H12. تم استخدام الأجسام المضادة التالية للكشف عن بروتينات سطح الخلية: الكالريتولين (PE، FMC 75، Abcam، 1:100)؛ CD4 البشري (BUV 395، SK3، BD، 1:200)؛ CD8 البشري (BUV 805، SK1، BD، 1:400)؛ CD45 البشري (Per-CP-Cy5.5، HI30، Invitrogen، 1:50)؛ CD69 البشري (BV421، FN50، BioLegend، 1:100)؛ CD39 البشري (BV605، A1، BioLegend، 1:100)؛ TIM3 البشري (BV510، F38-2E2، BioLegend، 1:100)؛ LAG3 البشري (PE، 3DS223H، Invitrogen، 1:100)؛ PD1 البشري (PE-Cy7، J105، Invitrogen، 1:100)؛ CD45RA البشري (BV785، HI100، BioLegend، 1:100)؛ CD62L البشري (BV605، DREG-56، BD، 1:100)؛ CD3 البشري (BUV 737، SK7، BD، 1:100)؛ CD45 الفأري (BUV 805، I3/2.3، BD، 1:100)؛ F4/80 (BV605، BM8، BioLegend، 1:100)؛ CD11b (APC، M1/70، BioLegend، 1:50 و BUV 395، M1/70، BD، 1:100)؛ CD19 الب
تحليل BLI
تم إعطاء الفئرانمندي-لوكسيفيرين لتصوير لوكفيراز اليراعات أو تخفيف 1:40 من ركيزة نانو-غلو (بروماجا، مخفف في DPBS) لتصوير أنتايرس ونانولوكفيراز عن طريق الحقن داخل الصفاق. تم التقاط الصور على نظام التصوير IVIS (بيركن إلمر) أو لاجو (سبكترا إنسترومنتس إيميجينغ) بعد 4 دقائق من الحقن لـ fluc و8 دقائق بعد الحقن لـ nLuc/Antares باستخدام تعريضات لمدة 30 ثانية وتجميع متوسط. إذا تم اكتشاف بكسلات مشبعة في الصورة، تم التقاط صورة إضافية باستخدام إعداد التعريض التلقائي. تم قياس التدفق الكلي باستخدام برنامج Living Image (الإصدار 4.7.3؛ بيركن إلمر) أو Aura (الإصدار 4.0.7؛ سبكترا إنسترومنتس إيميجينغ) مع منطقة اهتمام حول جسم كل فأر. تم استخدام الصور غير المشبعة فقط لتقدير BLI. تم عشوائية الفئران قبل إدارة خلايا T. لضمان توزيع موحد لحمولة الورم بين المجموعات. في نهاية التجربة، تم جمع جميع الصور في تسلسل واحد على أورا وضبطها على نفس مقياس اللمعان.
استنساخ وإنتاج البروتينات المؤتلفة
تم استخدام متجه gWIZ مع ببتيد إشارة BM40 للتعبير عن البروتين. يحتوي الحمض النووي على شفرة Hu5F9 (ماجروليماب)سلسلة ثقيلة مع مجال Fc من hIgG1، سلسلة خفيفة Hu5F9، TJC4 (ليمزوبارليماب)تم طلب سلسلة ثقيلة مع مجال Fc من hIgG1 وسلسلة خفيفة TJC4 من IDT. تم استنساخ السلاسل الثقيلة والخفيفة بشكل فردي في متجه gWIZ المنقوص بواسطة إنزيمات AscI و BamHI باستخدام تجميع جيبسون. تم نقل البلازميدات إلى خلايا Expi293F (Thermo Fisher Scientific) بنسبة 1:1 من السلسلة الثقيلة إلى السلسلة الخفيفة باستخدام ExpiFectamine وفقًا لتعليمات الشركة المصنعة. ثم، بعد 5 أيام من النقل، تم جمع السائل الفائق، وضبطه إلى pH 8.0 وتم تصفيته بشكل معقم. ثم تم تنقية Hu 5 F 9 و TJC4 باستخدام بروتين A-Sepharose 4 B (Thermo Fisher Scientific) وتم تبادل العازلة إلى PBS وتركيزه باستخدام مرشحات الطرد المركزي Amicon (Millipore Sigma). لتقييم ارتباط CD47، تم صبغ الخلايا بـ Hu5F9 أو TJC4 ثم تم صبغها بأجسام مضادة ثانوية مضادة للبشر موسومة (AF488 أو AF647، Invitrogen، 1:500).وتم الحصول على التحكم في النظير mIgG1 (MOPC-21) من Bio X Cell. وتم الحصول على متغيرات CV-1 (ALX-222، CV-1-hIgG1 Fc؛ وALX-90، CV-1-hIgG1 dead Fc) من ALX Oncology. SIRP البشري وفأر SIRP تم الحصول عليها من Sino Biologic.
نماذج حيوانية
فئران NSG (NOD.Cg-Prkdcإل2رغتتم شراء فئران SzJ من مختبر جاكسون وتربيتها داخليًا وفقًا لبروتوكولات معتمدة من APLAC بجامعة ستانفورد. كانت الفئران الذكور والإناث الصحية المستخدمة في التجارب الحية تتراوح أعمارها بين 6 و 10 أسابيع عند زراعة الورم أو خلايا T، وكانت خالية من الأدوية، ولم تشارك في إجراءات سابقة. تم إيواء الفئران في أقفاص معقمة في منشأة حواجز بجامعة ستانفورد في و الرطوبة تحتدورة الضوء والظلام. قام موظفو مركز خدمات الطب البيطري (VSC) في جامعة ستانفورد بمراقبة الفئران يوميًا. تم euthanize الفئران عندما أظهرت وضعية منحنية مستمرة، ومعطف غير مرتب مستمر، وشلل، وضعف في الحركة، أكثر منفقدان الوزن، إذا كانت الأورام تتداخل بشكل كبير مع الوظائف الجسدية الطبيعية أو إذا تجاوزت الحدود المحددة في بروتوكولات معتمدة من APLAC والتي تبلغ 1.70 سم في أي اتجاه. وفقًا لتوصيات موظفي VSC، تم دعم الفئران التي تعاني من الأمراض بـمحلول ملحي تحت الجلد، هلام الحمية (DietGel 76A، ClearH2O) وطعام رطب. بالنسبة لجميع التجارب، لم يتم إجراء حسابات لحجم العينة، ولكن تم تحديد أحجام المجموعات بناءً على الخبرة مع نماذج مثبتة ومعروفة تم نشرها سابقًا.تم تخصيص أقفاص الفئران التي تم زراعتها سابقًا بورم بشكل عشوائي لظروف خلايا CAR T و anti-CD47 للحقن، مع ضمان توزيع متساوٍ تقريبًا لحجم الورم بين المجموعات قبل العلاج. تم إجراء زراعة الورم وحقن خلايا T بواسطة فني كان غير مطلع على العلاجات والنتائج المتوقعة.
نموذج ورم العظام 143B
أو أو خلايا 143B-CD19 (خلايا 143B المعدلة للتعبير المفرط عن CD19؛ خلايا 143B لا تعبر بشكل طبيعي عن CD19) فيتم حقن DPBS في السمحاق الظنبوبي لذكور أو إناث الفئران من نوع NSG التي تتراوح أعمارها بين ستة إلى عشرة أسابيع (جرعة الزرع موضحة أدناه لكل دراسة محددة)بشكل عام، بعد 5 أيام من زراعة الورم وبعد التأكيد البصري على تكوين الورم، تم علاج الفئران بخلايا CAR T من نوع HER2-BB7، تلاها جرعتان من B6H12. تم مراقبة تقدم الورم من خلال القياس باستخدام الكالبر. تم euthanizing الفئران وفقًا للمعايير الموضحة في قسم ‘نماذج الحيوانات’. التفاصيل الخاصة بالنسخ المختلفة من النموذج المعروض هي كما يلي:
دراسات خلايا CAR T + B6H12 (الشكل 1أ، الشكل التوضيحي 1أ والشكل البياني الموسع 1أ، هـ): الفئران المزروعة بـ-خلايا CD19 تمت معالجتهم بـ-تم حقن خلايا CAR T من نوع BB 7 عبر الوريد الذيل في اليوم الخامس. ثم تم علاج الفئران مرتين باستخدام B6H12 ( ) أو PBS عن طريق الحقن داخل الصفاق في اليوم السادس واليوم العاشر. تم قياس عدد خلايا T في الدم باستخدام تحليل تدفق الخلايا في اليوم الثاني عشر.
دراسة بقاء خلايا CAR T في نموذج PIP (الشكل 3أ): الفئران التي تم زراعتها بـتم علاج 143B بـCD19-BB7 (تحكم غير مستهدف للأورام)، HER2-BB (التحكم في استهداف الورم)، PIP-28 أو PIP-BBخلايا CAR T في اليوم الخامس عن طريق حقن الوريد الذيل.
دراسة السيتوكينات في مصل خلايا CAR T (الشكل 3ب): فئران غير حاملة للأورام أو فئران مزروعة بـ-تم معالجة CD19 بـتم حقن خلايا CAR T CD19-BB (تحكم محدد للورم)، PIP-28Z أو PIP-BBZ، أو خلايا T وهمية عن طريق الحقن في الوريد الذيل في اليوم الرابع. تم جمع الدم لتحليل السيتوكينات في المصل في اليوم الثامن (4 أيام بعد إعطاء خلايا CAR T). دراسات خلايا CAR T مع جرعة عالية من B6H12 (الشكل 5e-g، الشكل التوضيحي 1p والشكل الممتد 10a-h): الفئران المزروعة بـتم معالجة خلايا 143B-CD19 بـخلايا CAR T من نوع HER2-BBZ Antares مع حذف CD47 الداخلي والتعبير المفرط عن CD47WT ) أو CD47(Q31P) ( )، أو عدد مكافئ من خلايا T المزيفة من نوع أنطارس عن طريق الحقن الوريدي في الوريد الذيل في اليوم الخامس. ثم تم علاج الفئران مرتين باستخدام B6H12 ( ) أو PBS عن طريق الحقن داخل البطن في اليوم السابع واليوم الحادي عشر. تم قياس خلايا T بواسطة BLI باستخدام النانو لوسيفيراز قبل (اليوم السابع) وبعد (اليوم الثالث عشر) علاج anti-CD47 وفي الدم بواسطة تحليل تدفق الخلايا في اليوم الرابع عشر. دراسات خلايا CAR T مع جرعة منخفضة من B6H12 (الشكل التوضيحي التكميلي 1q والشكل البياني الموسع 10i، j): الفئران المزروعة بـتم معالجة خلايا -CD19 بـخلايا CAR T HER2-BB7 مع CD47KO داخلي ومرتفع التعبير عن أي منهما أو ، أو عدد مكافئ من خلايا T المزيفة عن طريق الحقن الوريدي في الوريد الذيل في اليوم الخامس. ثم تم علاج الفئران مرتين بـ B6H12 ( أو ) أو PBS عن طريق الحقن داخل البطن في اليوم السادس واليوم العاشر. تم قياس عدد خلايا T في الدم باستخدام تحليل تدفق الخلايا في اليوم الثاني عشر. فقط تلك الفئران التي تم علاجها بـتم تقييم خلايا CAR T لفعاليتها المضادة للورم بالاشتراك مع B6H12.
نموذج ورم الميلانوما A375
إجماليخلايا A375 فيتم حقن I DPBS في جوانب ذكور أو إناث الفئران من نوع NSG الذين تتراوح أعمارهم بينأسابيعبشكل عام، بعد 7 إلى 14 يومًا من زراعة الورم وبعد التأكيد البصري على تكوين الورم، تم علاج الفئران بخلايا T TCR الخاصة بـ NY-ESO-1، تلاها جرعتان أو ثلاث جرعات من B6H12. تم مراقبة تقدم الورم من خلال القياس باستخدام الكالبر. تم euthanizing الفئران وفقًا للمعايير الموضحة في قسم نماذج الحيوانات. التفاصيل الخاصة بالنسخ المختلفة من النموذج المعروض هي كما يلي:
دراسات خلايا T TCR NY-ESO-1 بجرعة منخفضة + B6H12 (الشكل 1d، e، الشكل التوضيحي 1d والشكل البياني الموسع 1i): تم علاج الفئران بـتم حقن خلايا T من نوع NY-ESO-1TCR أو عدد مكافئ من خلايا T وهمية عن طريق الحقن الوريدي في الوريد الذيل في اليوم التاسع بعد زراعة الورم. ثم تم علاج الفئران مرتين باستخدام B6H12 ( ) أو PBS عن طريق الحقن داخل الصفاق في اليوم 10 و 15. تم قياس خلايا T في الدم بواسطة تحليل تدفق الخلايا في اليوم 17.
دراسات خلايا T TCR NY-ESO-1 بجرعات عالية + B6H12 (الشكل التوضيحي التكميلي 1d والشكل البياني الموسع 1j، k): تم علاج الفئران بـتم حقن خلايا NY-ESO-1TCRT أو عدد مكافئ من خلايا T الوهمية عن طريق الحقن في الوريد في اليوم السابع بعد زراعة الورم. ثم تم علاج الفئران مرتين باستخدام B6H12 ( ) أو PBS عن طريق الحقن داخل الصفاق في اليوم التاسع والثالث عشر. تم قياس عدد خلايا T في الدم باستخدام تحليل تدفق الخلايا في اليوم السادس عشر. دراسات قياس خلايا T TCR لـ NY-ESO-1 (الشكل 5h، الشكل التوضيحي 1t والشكل البياني الممتد 11g،h): بعد 7 أيام من زراعة الورم، تم علاج الفئران بـخلايا T TCR NY-ESO-1-Antares مع ذاتية وزيادة التعبير عن ، أو عدد مكافئ من خلايا T المزيفة من نوع أنطارس عن طريق الحقن الوريدي في الوريد الذيل. ثم تم علاج الفئران ثلاث مرات بـ B6H12 ( ) أو PBS عن طريق الحقن داخل البطن في الأيام 9 و 11 و 14. تم قياس خلايا T بواسطة النانولوسيفيراز
مؤشر BLI قبل (اليوم 9) وبعد (اليوم 14) علاج anti-CD47 وفي الدم بواسطة تحليل تدفق الخلايا في اليوم 15. دراسات فعالية خلايا T المناعية المضادة للورم لـ NY-ESO-1 TCR (الشكل Si، الشكل التوضيحي 1u والشكل البياني الممتد 11i، j): بعد 7 أيام (تجربة متبرع خلايا T 1؛ الشكل البياني الممتد 11j) أو 14 يومًا (تجربة متبرع خلايا T 2؛ الشكل 5i والشكل البياني الممتد 11i) بعد زراعة الورم، تم علاج الفئران بـتم حقن خلايا T من نوع NY-ESO-1-Antares TCR مع CD47KO داخليًا وذو تعبير مفرط لـ 47E، أو عدد مكافئ من خلايا T من نوع mock-Antares عن طريق الحقن الوريدي في الوريد الذيل. ثم تم علاج الفئران إما: ثلاث مرات باستخدام B6H12 ( ) أو PBS عن طريق الحقن داخل الصفاق في الأيام 9 و 11 و 14 (التجربة 1)؛ أو مرتين مع B6H12 ( ) أو PBS عن طريق الحقن داخل الصفاق في اليومين 15 و 19 (التجربة 2).
نموذج ورم العظام MG63.3
إجماليخلايا MG63.3 فيتم حقن DPBS في السمحاق الظنبوبي لفئران NSG الذكور أو الإناث في سنأسابيعابتداءً من 15 يومًا بعد زراعة الورم وبعد التأكيد البصري على تكوين الورم، تم علاج الفئران بـبـ B6H12 أو PBS ثلاث مرات في الأسبوع عن طريق الحقن داخل البطن. في اليوم 21، تم علاج الفئران عن طريق الوريد بـأو خلايا CAR T من نوع B7H3-BB7 أو بدون خلايا T. تم قياس تقدم الورم باستخدام الكالبر الرقمي مرتين في الأسبوع. تم euthanizing الفئران وفقًا للمعايير الموضحة في قسم ‘نماذج الحيوانات’ (الشكل التوضيحي التكميلي 1b). بالنسبة لتجارب تحديد كمية خلايا T، تم زراعة الفئران بشكل موضعي معتم معالجة الخلايا عن طريق الوريد بـخلايا CAR T B7H3-BB7 في اليوم 15 مع أو بدون 3 جرعات من علاج B6H12 (لكل جرعة؛ عن طريق البطن). تم جمع الدم والأورام في اليوم 30 بعد زراعة الورم.
نموذج ورم الدبقيات المتوسطة D425
تم تخدير الفئران (التي تتراوح أعمارها بين 6-10 أسابيع) بـالإيزوفلوران (مينراد إنترناشيونال) في غرفة التحريضتم الحفاظ على التخدير على إطار ستيريوتاكتيك (أدوات ديفيد كوف) عندتم توصيل الإيزوفلورين من خلال موصل أنفي. تم حقن خلايا د425 ميدولوبلاستوما عند الإحداثيات 2 مم خلف لامدا على الخط الأوسط و2 مم عمق باستخدام إبرة ذات طرف غير حاد.مقياس S / 2 بوصة / نمط النقطة“; هاملتون). باستخدام مضخة حقن دقيقة (UMP-3؛ أدوات الدقة العالمية)،تم حقن خلايا D425-GFP-fLuc في حجم منفيبعد ترك الإبرة في مكانها لمدة دقيقة، تم سحبها. ثم، بعد 4 أيام من زراعة الورم وبعد تأكيد تكوين الورم بواسطة البيولومينسنس، تم توزيع الفئران عشوائيًا وعولجت بدون خلايا T (مجموعة B6H12 فقط)، أو ج خلايا T أو عدد مكافئ من خلايا CAR CD19-BBZ غير المستهدفة للأورامالخلايا التائية عن طريق الحقن في الوريد الذيل. بدءًا من اليوم الرابع، تم أيضًا علاج الفئران بـ من B6H12 أو PBS ثلاث مرات في الأسبوع عن طريق الحقن داخل الصفاق. تم مراقبة تقدم الورم بواسطة بيوإضاءة اليراعة (BLI) (الشكل التوضيحي التكميلي 1c). في الشكل التوضيحي التكميلي 1c,d، تعتبر علاجات CD19-BBZ و B7H3-BBZ تكرارًا للبيانات المنشورة سابقًا.، مدرج للمقارنة مع، حيث تم تشغيل هذه الأذرع جميعها في نفس التجربة في وقت واحد.
نماذج أورام اللوكيميا Nalm6
إجماليخلايا Nalm6-GFP-fLuc فيتم زرع DPBS عن طريق حقن الوريد الذيل في ذكور أو إناث من الفئران NSG التي تتراوح أعمارهاأسابيعبشكل عام، بعد أربعة أيام من زراعة الورم وبعد تأكيد تكوين الورم بواسطة BLI، تم علاج الفئران بخلايا CAR T CD19-BB7 أو CD19-287، تلتها جرعات من مضاد CD47. تم مراقبة تقدم الورم من خلال قياس fLuc BLI. تم euthanize الفئران وفقًا للمعايير الموضحة في قسم ‘نماذج الحيوانات’. التفاصيل الخاصة بالنسخ المختلفة من النموذج المعروض هي كما يلي:
دراسات خلايا CAR T بجرعات عالية + B6H12 (الشكل التوضيحي التكميلي 1e والشكل البياني الموسع 11,n,p,s): الفئران المزروعة بـتم معالجة خلايا Nalm6-GFP-fLuc بـ B6H12 ) أو PBS عن طريق البطن حقن ثلاث مرات في الأسبوع، بدءًا من اليوم الثالث. ثم تم علاج الفئران بـتم حقن خلايا CAR T CD19-287-nLuc عن طريق الوريد الذيل في اليوم الرابع. وتم قياس خلايا T والأورام بواسطة التصوير الضوئي الأسبوعي.
دراسات خلايا CAR T بجرعة منخفضة + B6H12 (الشكل التوضيحي التكميلي 1f وبيانات موسعة الشكل) ): الفئران المزروعة بـ تم معالجة خلايا Nalm6-GFP-fLuc بـتم حقن خلايا CAR T CD19-287-nLuc عن طريق الوريد الذيل في اليوم الرابع. تم علاج الفئران مرتين باستخدام B6H12.أو PBS عن طريق الحقن داخل الصفاق في اليوم الخامس والسابع. تم قياس خلايا T والأورام بواسطة BLI أسبوعيًا.
دراسات خلايا CAR T بجرعات عالية + CV-1 (الشكل 1f,g، الشكل التوضيحي 1g والشكل البياني الموسع 2a,b,e): الفئران المزروعة بـتم معالجة خلايا Nalm6-GFP-fLuc بـتم حقن خلايا CAR T CD19-BB7-nLuc عن طريق الوريد الذيل في اليوم الرابع. تم علاج الفئران بـ CV-1-Fc (ALX-90؛ Fc ميت؛ ) أو PBS عن طريق الحقن داخل البطن ثلاث مرات في الأسبوع بدءًا من اليوم تم قياس الخلايا والأورام بواسطة BLI أسبوعياً.
دراسات خلايا CAR T بجرعة منخفضة + CV-1 (الشكل التوضيحي التكميلي 1h والشكل البياني الموسع 2c-f): الفئران المزروعة بـتم معالجة خلايا Nalm6-GFP-fLuc بـتم حقن خلايا CAR T CD19-287-nLuc عن طريق الوريد الذيل في اليوم الرابع. تم علاج الفئران بـ CV-1-Fc (ALX-90؛ Fc ميت؛أو PBS عن طريق الحقن داخل الصفاق ثلاث مرات في الأيام 5 و 7 و 10. تم قياس خلايا T والأورام بواسطة BLI مرتين أسبوعياً.
جرعة منخفضةدراسات خلايا CAR T (الشكل 1h,i والشكل البياني الممتد 2h,i): الفئران المزروعة بـتم معالجة خلايا Nalm6-GFP-fLuc بـ خلايا CAR T CD19-287-nLuc مع حذف جين CD47 الداخلي نقص CD47 مع فرط التعبير عن CD47 WT )، أو عدد مكافئ من خلايا T المزيفة عن طريق حقن الوريد الذيل في اليوم الرابع. تم علاج الفئران مرتين بـ B6H12 ( ) أو PBS عن طريق الحقن داخل الصفاق في اليومين 5 و 7. تم قياس الأورام بواسطة BLI أسبوعيًا. تم قياس خلايا T بواسطة BLI في اليوم 11.
جرعة منخفضةدراسات خلايا CAR T + B6H12 (الشكل التوضيحي التكميلي 1s والشكل البياني الموسع 11e، f): الفئران المزروعة بـتم معالجة خلايا Nalm6-GFP-fLuc بـ خلايا CAR T CD19-287-nLuc مع CD47KO داخلي ومرتفع التعبير عن إما أو ، أو عدد مكافئ من خلايا T المزيفة عن طريق حقن الوريد الذيل في اليوم الرابع. تم علاج الفئران مرتين بـ B6H12 ( ) أو PBS عن طريق الحقن داخل البطن في اليومين 5 و 7. تم قياس الأورام بواسطة BLI أسبوعياً.
نموذج استنفاد خلايا T
فئران NSG ذكور أو إناث (بعمرأسابيع) تم زراعتها بـ أو خلايا CAR T CD19-287-nLuc مع حذف CD47 الداخلي وارتفاع التعبير عن إما أو لا بروتين إضافي ( ) عن طريق حقن الوريد الذيل (اليوم 0). ثم تم علاج الفئران مرتين بـ B 6 H 12 أو PBS عن طريق الحقن داخل الصفاق في اليومين 3 و 5. تم قياس خلايا T بواسطة BLI باستخدام النانو لوسيفيراز قبل (جرعة، يوم الجرعة، اليوم 3 ) وبعد ( جرعة، يومالجرعة، اليوم السابع) علاج مضاد-CD47 وفي الدم بواسطة قياس التدفق الخلوي (جرعة، يوم الجرعة، اليوم 6). للدراسات الخاصة بالتحكم في النوع (الشكل 1f، g من البيانات الموسعة)، تم زرع الفئران بـتم حقن خلايا CAR T CD19-287 عن طريق الوريد الذيل (اليوم 0) ثم تم علاجها بـتحكم من نوع mIgG1 ) أو PBS عن طريق الحقن داخل البطن في اليوم الأول. تم قياس عدد خلايا T في الدم باستخدام تحليل تدفق الخلايا في اليوم الخامس. تم euthanizing الفئران وفقًا للمعايير الموضحة في قسم ‘نماذج الحيوانات’ في نهاية التجربة (الشكل التوضيحي 1i,m).
نموذج سمية PIP CAR
تم تصنيع متجهات PIP CAR كما هو موضح في قسم بناء المتجهات الفيروسية. ذكور أو إناث من فئران NSG (بعمرتم علاج (أسابيع) مع خلايا PIP CAR T أو خلايا التحكم CD19-BB7 أو HER2-BB7 أو خلايا وهمية بالجرعة المشار إليها في الشكل ( أو تم حقن خلايا CAR T عن طريق الحقن في الوريد الذيل. عانت الفئران في مجموعتي PIP-28 7 و PIP-BB 7 من ظهور سريع للتسمم (خلال 1-5 أيام، حسب الجرعة) تم ملاحظته سريرياً كوضعيّة منحنية، وفراء غير مرتب، وحركة بطيئة، وجفاف، وفقدان الوزن. تم مراقبة التسمم المرتبط بالعلاج من خلال تغيير الوزن، الذي تم قياسه قبل إعطاء خلايا T و في الأسبوع التالي. تم حساب نسبة تغيير الوزن على النحو التالي: نسبة تغيير الوزنالوزن في الوقتالوزن الابتدائي) -1توفيت الفئران بسبب السمية أو تم euthanized إذا وصلت إلى فقدان وزن بنسبة 20% أو أظهرت علامات سريرية على سمية شديدة، كما هو موضح في قسم ‘نماذج الحيوانات’. لتقييم توطين وتفعيل خلايا T، تم نقل خلايا CAR T CD19-287-nLuc أو PIP-287-nLuc مع تقرير لوسيفراز اليراعة تحت سيطرة محفز قابل للتحفيز بواسطة NF-кB. تم زراعة الفئران بـتمت معالجة خلايا CAR T وتصويرها يوميًا باستخدام ركيزة Nano-GLO (nLuc؛ إجمالي CART) واللوكسيفيرين (fluc؛ CART النشط)، مع فصل كل جرعة ركيزة بفترة 12 ساعة. لتحليل BLI للأعضاء، بعد 4 أيام من العلاج معتم حقن الفئران بـ CD19-287-nLuc أو PIP-287-nLuc باستخدام إما ركيزة Nano-GLO (nLuc؛ CAR T الكلي) أو لوكفيرين (fLuc؛ CAR T النشط) وتم euthanized بعد 10 دقائق من الحقن. تم جمع الأعضاء وفقًا للإجراءات القياسية وتم تصويرها باستخدام BLI على آلة IVIS (Perkin Elmer) في أطباق بستة آبار. بالنسبة لنماذج مفتاح الأمان، تم إعطاء الفئران الجرعة بـتم إعطاء خلايا CAR T PIP-287 أو CD19-287 عن طريق الوريد. تم علاج الفئران بخلايا CAR T PIP-28.تم إعطاء B6H12 أو PBS على مدى ثلاثة أيام متتالية (الأيام 2 و3 و4)، بعد يومين من إعطاء CAR (اليوم 0). تم جمع الدم لتحليل السيتوكينات في المصل في اليوم 4 بعد إعطاء CAR (الشكل التكميلي 1k).
نموذج ورم العصبي الصماوي النقيلي CHLA-255
تم زراعة فئران NSG الذكور أو الإناث التي تتراوح أعمارها بين ستة إلى عشرة أسابيع بـخلايا CHLA-255-GFP-fLuc عن طريق حقن الوريد الذيلبعد 7 أيام من زراعة الورم وبعد تأكيد تكوين الورم بواسطة BLI، تم توزيع الفئران عشوائيًا وعلاجها بـ-نخاع CAR T خلايا مع KO ذاتي لـ CD47 و تعبير مفرط أو أو عدد مكافئ من خلايا T غير المعدلة (غير المنقولة) عن طريق الحقن الوريدي في الوريد الذيل. ثم تم علاج الفئران ثلاث مرات باستخدام B6H12أو PBS عن طريق الحقن داخل الصفاق في الأيام 7 و 9 و 13. تم مراقبة تقدم الورم بواسطة تصوير بيوفوتونيك باستخدام لوسيفيراز اليراعة. تم قياس خلايا T بواسطة تصوير بيوفوتونيك باستخدام نانو لوسيفيراز بعد علاج مضاد CD47 في اليوم 14 وفي الدم بواسطة تحليل تدفق الخلايا في اليوم 15. تم euthanizing الفئران وفقًا للمعايير الموضحة في قسم ‘نماذج الحيوانات’ (الشكل التوضيحي التكميلي 1r).
نموذج GvHD لخلايا CART
فئران NSG ذكور أو إناث (بعمرأسابيع) تم زراعتها بـخلايا Nalm6-GFP-fLuc عن طريق حقن الوريد الذيل. ثم تم علاج الفئران بـخلايا CAR T CD19-BB7 في اليوم الرابع. تلقت نصف مجموعة الفئران ثلاث جرعات من B6H12 (250 ميكروغرام) على مدى 3 أيام بعد إعطاء خلايا CAR T. تم مراقبة الفئران لنمو الورم بواسطة التصوير الضوئي للانبعاثات الحيوية (BLI) وعلامات مرض الطعم ضد المضيف (GvHD)، مثل تساقط الشعر، وخلل التقرن، وفقدان الوزن.تم استخراج الطحال والجلد جراحيًا. تم تحضير مقاطع الجلد كشرائح وصبغها بصبغة هيماتوكسيلين وإيوزين باستخدام الطريقة القياسية (الشكل التوضيحي 11).
عزل خلايا T من الطحال والأورام
تم جمع الطحال والأورام وتفكيكها ميكانيكياً باستخدام جهاز التفكيك gentleMACS (ميلتينيي). تم إعداد تعليقات خلوية مفردة عن طريق تمرير الطحال والأورام من خلالمنخل الخلايا (ثيرمو فيشر ساينتيفيك)، إزالة كريات الدم الحمراء بواسطة تحليل ACK (كوالتي بيو لوجيكال)، وتصفية إضافية من خلال أنابيب فلتر تحليل التدفق معأغطية مصفاة الخلايا (فالكون). ثم تم تجميد التعليقات الفردية للخلايا في محلول CryoStor (تقنيات ستيمسيل) في النيتروجين السائل، أو تم صبغها وتشغيلها مباشرة على نظام تحليل تدفق الخلايا.
تحديد كمية خلايا T والسيتوكينات من الدم
تم جمع دم الفئران من الجيب خلف العين في أنابيب جمع الدم Microvette مع EDTA (ثيرمو فيشر ساينتيفيك). تم إزالة كريات الدم الحمراء بواسطة تحلل ACK (كوالتي بايولوجيكال)، تلا ذلك غسلتين مع محلول FACS (PBS + 2% FBS). تم صبغ العينات وخلطها مع كريات العد CountBright Absolute Counting (ثيرمو فيشر).
علمي) قبل تحليل تدفق الخلايا. IL-2 و IFNتم قياس مستويات السيتوكينات في الدم باستخدام اختبارات المناعة LEGENDPlex (BioLegend) وفقًا لتعليمات الشركة المصنعة من مصل تم جمعه بعد طرد عينات الدم.لمدة 10 دقائق. تم تعيين قيم السيتوكينات السلبية إلى 0.
اختبارات قتل الورم باستخدام إنكو سايت، تحليل السيتوكينات واكتشاف علامات تنشيط خلايا T
إجماليتم زراعة خلايا الورم المعلّمة بـ GFP معخلايا CART في RPMI المدعوم بـ FBS، 10 مللي مولار هيبس، 2 مللي مولار -جلوتامين، البنسلين وستربتوميسين. للحالات التي تحتوي على B6H12، تركيز منتم استخدامه. تم زراعة آبار ثلاثية في صفائح مسطحة القاع مكونة من 96 بئرًا لكل حالة. تم مراقبة فلورية الورم كلمعالهدف باستخدام نظام تحليل الخلايا الحية IncuCyte S3 (Sartorius)، الموجود في حاضنة زراعة الخلايا في و تم إعدادها لالتقاط 4 صور لكل بئر في كل نقطة زمنية. تم قياس إجمالي كثافة GFP المتكاملة باستخدام ميزة برنامج محلل IncuCyte الأساسي (IncuCyte S3 v.2019B Rev2؛ Sartorius). تم تطبيع البيانات إلى نقطة الزمن الأولى ورسمها كتغير مضاعف في فلورية الورم مع مرور الوقت. بالنسبة لتحليل إفراز السيتوكينات وعلامات خلايا T، تم إعداد الثقافات المشتركة كما هو موضح أعلاه باستثناء في أطباق ذات 96 بئرًا ذات قاع دائري. بعد حوالي 24 ساعة، تم طرد الأطباق لتجميع الخلايا وتم جمع السائل العلوي وتخزينه فيحتى التحليل، بينما تم معالجة كريات الخلايا على الفور لقياس التدفق الخلوي. IFNتم قياس مستويات IL-2 في سوائل الثقافة المشتركة بواسطة ELISA (Human ELISA MAX Deluxe، BioLegend) وفقًا لتعليمات الشركة المصنعة. تم تعيين قيم السيتوكين السلبية إلى 0. تم قياس قيم الامتصاص باستخدام جهاز Synergy H1 Hybrid Multi-Mode Reader مع برنامج Gen5 (الإصدار 2.00.18؛ BioTek). لتحليل علامات خلايا T بعد تنشيطها بواسطة خلايا الورم، تم تجميع الكريات من الأطباق التي تم طردها مركزيًا معًا لآبار ثلاثية، وتم صبغها وتحليلها باستخدام تقنية تدفق الخلايا. تم إعداد تجارب الثقافة المشتركة باستخدام خلايا T في اليوم العاشر.
نقص الماكروفاج وغسل الصفاق
تمت معالجة ذكور أو إناث الفئران من نوع NSG (التي تتراوح أعمارها بين 6-10 أسابيع) مسبقًا بحقن وريدية بـمن ليفوسومات الكلوردونات (Liposoma)، تليهامن مضاد مستقبل CSF1R الفأري (AFS98؛ بايو إكس سيل) عن طريق الحقن داخل الصفاقتم معالجة الفئران بـمضاد CSF1R ثلاث مرات في الأسبوع طوال مدة التجربة. ثم، بعد 6 أيام من علاج الكلودرونات، تم إعطاء الفئرانCD19-28-نقل خلايا CAR T عن طريق الوريد، تليهاتم إعطاء B6H12 عن طريق البطن في اليوم السابع. تم قياس خلايا T بواسطة BLI باستخدام النانو لوسيفيراز قبل (اليوم السابع) وبعد (اليوم التاسع) علاج anti-CD47. تم إجراء غسيل البطن في اليوم الثالث عشر باستخدام 10 مل من محلول FACS وإبرة قياس 25. تم جمع خلايا البطن، وغسلها بمحلول FACS وصبغها، قبل أن يتم تشغيلها على نظام قياس التدفق (الشكل التوضيحي 1j).
اختبار البلعمة
في جميع اختبارات البلعمة المعتمدة على التدفق في المختبر، تم زراعة خلايا T والبلاعم البشرية معًا بنسبة 2:1 (على سبيل المثال، 100,000 خلية T: 50,000 بلاعم) في أطباق 96 بئرًا ذات قاع U منخفض الالتصاق (كورنينج) في RPMI خالي من المصل (ثيرمو فيشر ساينتيفيك). تم وسم خلايا T بـ CFSE (إنفيتروجين) عن طريق تعليق الخلايا في PBS (حل عملي) وفقًا لتعليمات الشركة المصنعة لمدة 20 دقيقة فيمحمي من الضوء وغسل مرتين بـ 20 مل من وسط يحتوي على FBS قبل الزراعة المشتركة. ثم تم حضن الخلايا إما بمفردها أو بحضور مضاد CD47 (B6H12؛ Bio X Cell) أو التحكم في نوع IgG1 (MOPC-21؛ Bio X Cell) بتركيزتم تحضين خلايا T والأجسام المضادة لمدة 30 دقيقة في بيئة رطبةحاضنة فيتم غسل الأطباق مرتين؛ وتم إضافة البلعميات البشرية إلى الطبق؛ وتم حضن الأطباق لمدةفيتم إيقاف البلعمة عن طريق الغسل بـPBS والطرد المركزي فيقبل أن يتم صبغ الخلايا بصبغة الحياة/الموت و anti-CD11b (APC، M1/70، BioLegend، 1:50). تم تحليل التجارب بواسطة قياس التدفق الخلوي، وتم قياس البلعمة كعدد منوالبلاعم، مقاسة كنسبة مئوية من إجمالي CD11bالبلاعم وتم تطبيعها مع حالة التحكم.
لإجراء اختبارات البلعمة في المختبر المعتمدة على IncuCyte، تم زراعة خلايا T والماكروفاجات البشرية معًا بنسبة (على سبيل المثال، 100,000 خلية T: 50,000 بلعوم) في أطباق مسطحة القاع سعة 96 بئر (كورنينغ) في وسط RPMI مدعوم بـ FBS، 10 مللي مولار هيبس، 2 مللي مولار -جلوتامين، البنسلين وستربتوميسين. تم وسم خلايا T بصبغة pHrodo Red (إنفيتروجن) عن طريق حضن خلايا T فيخلايا لكل مل بتركيز عمل من pHrodo Red قدرهفي PBS لمدة ساعة واحدة عندفي الظلام في مكان رطبالحاضنة. تم إيقاف تفاعل التوسيم وتم غسل الصبغة الزائدة عن طريق الغسل مرتين بوسيط كامل. ثم تم حضن الخلايا إما بمفردها أو بحضور مضاد CD47 (B6H12؛ Bio X Cell) بتركيزفي RPMI خالي من المصل. تم حضن خلايا T والأجسام المضادة لمدة 30 دقيقة في بيئة رطبةحاضنة في، قبل أن يتم غسلها مرتين بوسط كامل. تم إضافة البلعميات إلى كل بئر وسمح لها بالالتصاق لمدة ساعتين في بيئة رطبةحاضنة فيبعد ساعتين، تم إضافة خلايا T الموسومة إلى الصفيحة بنسبة 2:1 من خلايا T إلى البلعميات. تم مراقبة فلورية pHrodo Red الناتجة عن البلعمة بعد 3 ساعات معالهدف باستخدام نظام تحليل الخلايا الحية IncuCyte S3 (Sartorius)، الموجود في حاضنة زراعة الخلايا في و تم تعيينه لالتقاط أربع صور لكل بئر في كل نقطة زمنية. تم قياس إجمالي شدة الفلورسنت الحمراء المتكاملة باستخدام ميزة برنامج محلل IncuCyte الأساسي (IncuCyte S3 v.2019B Rev2؛ سارتوريوس).
المجهر الضوئي التداخلي لتفاعلات خلايا T والبلعميات
تم وسم خلايا CAR T CD19-28 بصبغة pHrodo Red كما هو موضح أعلاه. ثم تم وسم خلايا T الموصومة بـ pHrodo-Red بصبغة DiO Vybrant المحبة للدهون (Invitrogen) وفقًا لتعليمات الشركة المصنعة في PBS لمدة دقيقتين عندفي الظلام. تم وسم البلعميات بصبغة DiD Vybrant (Invitrogen) وفقًا لتعليمات الشركة المصنعة في PBS لمدة 15 دقيقة عندفي الظلام. بعد صبغ الخلايا، تم غسل الخلايا ثلاث مرات بوسط كامل لإزالة الصبغة الزائدة. ثم تم حضانة خلايا T مع B6H12 فيلمدة 20 دقيقة عندفي PBS، تليها غسلتان بوسط كامل. تم تضمين خلايا T الموسومة والبلعميات في مصفوفة الكولاجين (نوع I-A من Cellmatrix، FUJIFILM Wako chemicals) بنسبة 2:1 (على سبيل المثال، 1,000,000 خلية T: 500,000 بلعمية) في طبق زراعة زجاجي ذو 24 بئر (Mattek). أربعة أبعاد (تم إجراء تصوير حي باستخدام المجهر الضوئي الماسح بالليزر (Zeiss LSM900). تم تحليل الصور باستخدام برنامج Imaris (الإصدار 10.0؛ أكسفورد إنسترومنتس).
تحديد كمية تعبير CD47 على خلايا الورم وخلايا T باستخدام QuantiBrite
تم قياس تعبير CD47 باستخدام جسم مضاد مضاد لـ CD47-PE (B6H12، BD، 1:20) ومجموعة قياس QuantiBrite PE (BD) وفقًا لتعليمات الشركة المصنعة.تم إنتاج خلايا CAR T من CD19-28 كما هو موضح أعلاه، باستثناء أنه تم الاحتفاظ بالخلايا في الثقافة لمدة يوم واحد بعد إذابتها قبل تنشيطها باستخدام كريات مضادة لـ CD3/CD28. تم تحليل خلايا T بواسطة تحليل التدفق الخلوي في اليوم 0 (قبل التنشيط؛ يوم واحد بعد الإذابة)، اليوم 4 (على الفور بعد الإزالة من تنشيط الكريات)، اليوم 7 واليوم 11 (متوسط وقت النقل في الجسم الحي). تم صبغ خلايا T باستخدام مضاد hCD4 (BUV 395، SK3، BD، 1:200)، مضاد hCD8 (BUV 805، SK1، BD،المضادات الحيوية المضادة لـ hCD47 أو التحكم في نوع المصل mIgG1 (PE، B11/6، Abcam، 1:20)، المضاد لـ hCD45RA (BV785، HI100، BioLegend، 1:100) والمضاد لـ hCD62L (BV605، DREG-56، BD، 1:100). تم تعريف أنواع تمايز خلايا T كما يلي: T naive (CD45RA“), ذاكرة مركزية T (CD45RA), خلايا الذاكرة التائية الفعالة (CD45RA) و T الذاكرة الفعالة التي تعيد التعبير عن CD45RA (CD45RAتم صبغ خلايا الورم باستخدام مضاد hCD47 فقط أو التحكم في النظير المجهري mIgG1. تم حساب جزيئات CD47 وفقًا لتعليمات مجموعة QuantiBrite. باستخدام الاستقراء من إشارات MFI لخرز BD QuantiBrite-PE مع كميات معروفة من PE. تم تحديد درجة التوسيم لمضاد CD47-PE (BD، 2040745) تجريبيًا على أنها 0.842 جزيء من الصبغة لكل جسم مضاد، باستخدام الامتصاص الأقصى عند 566 نانومتر، معامل الانقراض لـ PE. ) وتركيز الأجسام المضادة المدرجة.
تصوير عينات السائل الدماغي الشوكي للمريض
تم جمع تحضير السائل الدماغي الشوكي باستخدام تقنية السيتوسبين من مريض تم علاجه بعلاج خلايا CAR T CD19-287 axicabtagene ciloleucel (axi-cel)، وتم صبغه بصبغة رايت-جيما وتم تصويره بواسطة المجهر فيتكبير، التقاط الخلايا النسجية مع الخلايا اللمفاوية المبتلعة (تظهر الصور الخام في الشكل التكميلية 3).
تحليل الخلايا المفردة لعينات المرضى
تم إعادة تحليل مجموعتين من البيانات: المرجع 14 (GSE168940)، بما في ذلك بيانات scRNA-seq التي تم جمعها من تسعة مرضى مصابين بـ LBCL تم علاجهم بعلاج خلايا CAR T من نوع axi-cel CD19287، حيث تم استخدام 50,000-70,000 خلية CAR T (حية واحدةأوتم فرز الأحداث باستخدام FACS إلىالنقاء؛ ومرجع 15 (GSE186802)، بما في ذلك بيانات scRNA-seq المجمعة من أربعة مرضى مصابين بـ DMG تم علاجهم بعلاج خلايا CAR T GD2.BBZ، مع خلايا مستمدة من السائل الدماغي الشوكي. تم تحليل كلا المجموعتين من البيانات على منصة 10x Genomics.حيثما تم الإشارة، تم استخدام خلايا تعبر عن CAR-mRNA تم توثيقها سابقًا.
علم الأنسجة لعينات الأنسجة
تشمل الأنسجة التي تم تقييمها الجلد والرئة. تم جمع الأنسجة وتثبيتها في محلول الفورمالين المحايد بنسبة 10%. بعد التثبيت، تم معالجة الأنسجة بشكل روتيني، وتضمينها في الشمع، وتقطيعها عند وتم صبغ الأنسجة بانتظام باستخدام الهيماتوكسيلين والإيوزين (H&E). تم تصور الأنسجة باستخدام ميكروسكوب أوليمبوس BX43 العمودي ذو المجال الساطع، وتم التقاط الصور باستخدام كاميرا أوليمبوس DP27 وبرنامج cellSens (الإصدار 3.2؛ أوليمبوس لعلوم الحياة).
ناقلات عرض سطح الخميرة
تم استنساخ تسلسل الحمض النووي الذي يشفر مجال CD47 الشبيه بالأجسام المضادة (Gln19-Ser135) في متجه عرض سطح الخميرة pCTCON2 (Addgene) باستخدام مواقع Nhel و BamHI. كان متجه pFreeNTerm (pFNT) مستندًا إلى هيكل pCL.، تصميم موقع قطع Nhel جوهري في تسلسل إشارة Aga2p كـ موقع الاستنساخ واستخدام موقع قطع MluI قبلرابط جلايسين 4 سيرين كموقع استنساخ 3′. تم استنساخ متغيرات مجال CD47 الشبيه بالغلوبولين المناعي (Gln19-Ser135) في متجه عرض سطح الخميرة pFNT باستخدام مواقع Nhel وMlul هذه.
تحليل تدفق الخلايا لعرض سطح الخميرة
تم تحويل خميرة Saccharomyces cerevisiae (السلالة، EBY100؛ ATCC) باستخدام بلازميدات pCTCON2 أو pFNT وتم اختيارها على أطباق SD-CAA-Agar. تم زراعة الخميرة (~100,000 لكل عينة) وتحفيزها في SG-CAA، وتم إعداد الربط على مدى مجموعة من تركيزات الليغاند القابلة للذوبان في PBS المحتوي على BSA (BPBS)، مع الأخذ في الاعتبار استنفاد الربائط ووقت التوازن. بعد الحضانة مع شريك الربط، تم غسل خلايا الخميرة مرة واحدة بمحلول BPBS، ثم تم الحضانة مع تخفيف 1:5000 من جسم مضاد الدجاج المضاد لـ MYC (وحيد النسيلة، إنفيتروجين) للبروتينات المعروضة على pCTCON2، وتم الحضانة لمدة 30 دقيقة في في الظلام. بعد الإضافة الأولية، تم غسل العينات مرة واحدة باستخدام BPBS، وتم إضافة الأجسام المضادة الثانوية. تم الكشف عن التعبير باستخدام تخفيف 1:500 من الألكسا فلور 488 المضاد للدجاج من الماعز (متعدد النسائل، إنفيتروجن) أو الألكسا فلور 647 (متعدد النسائل، أبكام). بالنسبة للبروتينات المعروضة pFNT، تم استخدام GFP المعروض معًا لمراقبة التعبير. ارتباط البروتينات مع مجالات Fc الفأر (تم الكشف عنه بتخفيف 1:500 من مضاد الماوس من الماعز أليكسا فلور 488 أو أليكسا فلور 647 (متعدد النسائل، إنفيتروجن). ارتباط البروتينات ذات مجال Fc البشري (CV-1(ALX-222)، mSIRPتم الكشف عن Hu5F9، TJC4) باستخدام تخفيف 1:500 من الأجسام المضادة الثانوية من الماعز المضادة للبشر أليكسافلور 488 أو أليكسافلور 647 (وحيدة النسيلة، إنفيتروجن). تم تحضين الأجسام المضادة الثانوية لمدة 15 دقيقة فيفي الظلام. بعد الحضانة الثانوية، تم غسل العينات مرة واحدة بمحلول BPBS، ثم تم تجميعها وتركها مجمعة على الثلج حتى التحليل. تم تحليل العينات عن طريق إعادة تعليقها فيمن BPBS وتشغيل تحليل تدفق الخلايا على نظام BD Accuri C6 (BD Biosciences). تم استخدام برنامج Accuri C6 (الإصدار 1.0.264.21؛ BD) لجمع البيانات، وتم استخدام برنامج FlowJo (الإصدار 10.8.1؛ BD) لتحليل البيانات (استراتيجيات التصفية موضحة في الشكل التكميلية 2). تم تصفية العينات لخلايا الخميرة الكثيفة (التشتت الأمامي (FSC) مقابل التشتت الجانبي (SSC)) ثم للخلايا الفردية (ارتفاع FSC مقابل مساحة FSC). تم تحديد الخمائر المعبرة وتصفيتها من خلال علامة MYC الطرفية C أو الكشف عن GFP. تم قياس المتوسط الهندسي لإشارة الفلورسنت المرتبطة من المجموعة المعبرة واستخدامه كقيمة ربط خام. عند مقارنة إشارات الربط، تم قياس متوسط إشارة الفلورسنت التعبيرية لمتغيرات البروتين المختلفة واستخدامها لتطبيع إشارة الربط. لتحديد ‘الجزء المرتبط’، تم قسمة إشارات الربط على الإشارة المستمدة من أعلى تركيز لشريك الربط المستخدم، أو تلك المستمدة من الربط بـ WT CD47. لحسابتم تحليل القيم والبيانات في برنامج بريزم (الإصدار 9.5.1، غراف باد) باستخدام نموذج انحدار غير خطي.
تم إنشاء مكتبة عرض سطح الخميرة، وتصنيفها، وتسلسلها. تم التعبير عن CD47 في S. cerevisiae (السلالة، EBY100؛ ATCC) كدمج جيني مع بروتين التزاوج الأجلوكتين Aga2p في متجه pCTCON2. تم إنشاء مكتبة PCR عرضة للأخطاء باستخدام مجال CD47 الشبيه بالأجسام المضادة (Gln19 إلى Ser135) كقالب وتم إدخال الطفرات باستخدام مجموعة التحوير العشوائي Gene Morph II (Agilent) وفقًا لتعليمات الشركة المصنعة. تم إجراء PCRs منفصلة باستخدام تركيزات مختلفة من إنزيم Mutazyme II. تم تنقية منتجات هذه التفاعلات بواسطة الرحلان الكهربائي في الهلام، وتم تجميعها وتكبيرها باستخدام PCR القياسي مع بوليميراز Phusion (New England BioLabs). تم إدخال الحمض النووي الطافري المنقى والمتجه pCTCON2 المفرغ كهربائيًا إلى خميرة EBY100، حيث تم تجميعها في الجسم الحي من خلال إعادة التركيب المتجانس. قدرنامتغيرات المكتبة، التي تم تحديدها من خلال تخفيف الطلاء وعدّ المستعمرات. تم زراعة الخميرة في وسط SD-CAA وتم تحفيز التعبير عن بروتين CD47 من خلال النمو في وسط يحتوي على 90% SG-CAA و10% SD-CAA طوال الليل.تم عزل الخميرة التي تعرض متغيرات CD47 بواسطة FACS باستخدام جهاز فرز الخلايا SONY SH800S (SONY؛ برنامج فرز الخلايا SH800S الإصدار 2.1.5) وتم تحليلها باستخدام مقياس التدفق BD Accuri C6 (BD Biosciences؛ برنامج BD Accuri الإصدار 1.0.264.21). استراتيجيات التصفية موضحة في الشكل التوضيحي التكميلي 2. تم تحليل البيانات باستخدام برنامج FlowJo (الإصدار 10.8.1، BD). تم إجراء الفحوصات باستخدام ظروف ربط متوازنة حيث تم حضانة الخميرة في درجة حرارة الغرفة في BPBS مع التركيزات التالية من B6H12 أو CV-1 (ALX-222) لمدة ساعتين. بالنسبة للتصنيفات السلبية لـ B6H12، تم جمع تجمعات الخميرة التي تعبر عن CD47 ولكنها غير مرتبطة. بالنسبة للتصنيفات الإيجابية لـ CV-1، تم جمع تجمعات الخميرة التي تعبر عن CD47 وترتبط (الشكل التوضيحي الموسع 6g). التصنيف 1، تصنيف سلبي، 500 pM B6H12؛ التصنيف 2، تصنيف سلبي، 5 nM B6H12؛ التصنيف 3، تصنيف إيجابي، 20 nM CV-1؛ التصنيف 4، تصنيف سلبي، 20 nM B6H12؛ التصنيف 5، تصنيف سلبي، 50 nM B6H12؛ التصنيف 6، تصنيف إيجابي،بعد الحضانة مع B6H12 أو CV-1، تم ترسيب الخميرة، وغسلها، ووضع علامة عليها باستخدام الأجسام المضادة الفلورية كما هو موضح أعلاه قبل الفرز. تم تكاثر سلالات الخميرة المفرزة، وتحفيزها للتعبير عن CD47، وخضعت لجولات متكررة من FACS كما هو موضح أعلاه. بعد كل جولة من الفحص، تم استرداد الحمض النووي البلازميدي باستخدام مجموعة Zymoprep لعمل الميني بريب للخميرة (Zymo Research)، وتحويله إلى خلايا DH10B الكهربائية (Thermo Fisher Scientific) وعزله باستخدام مجموعة GeneJET لعمل الميني بريب (Thermo Fisher Scientific). تم إجراء التسلسل بواسطة ELIM Biopharmaceuticals.
نمذجة هيكل CD47
تم تنزيل هياكل CD47 من بنك بيانات البروتين (PDB) وتحليلها باستخدام PyMol (الإصدار 2.5.8؛ شرويدنجر). الهيكل المستخدم لـ CD47-hSIRPa كان PDB 2JJS (المرجع 18). الهيكل المستخدم لـ CD47-B6H12 كان PDB 5TZU (المرجع 19).
نموذج البلعمة الحية 143B
إجماليالخلايا فيتم حقن DPBS في السمحاق الظنبوبي لفئران NSG (التي تتراوح أعمارهاأسابيع). ثم، بعد 20 يومًا من زراعة الورم، تم علاج الفئران بـتم وسم خلايا CART HER2-BB7 بـ CFSE داخل الورم. قبل الإعطاء، تم وسم خلايا T بـ CFSE.حل عملي)، وفقًا لتعليمات الشركة المصنعة، مع خلايا موسومة بتركيز خلايا لكل مل في PBS لمدة 5 دقائق. تم غسل الخلايا في وسط كامل، ثم تم حضنها مع أو بدونPBS لمدة 20 دقيقة عندتم إعادة تعليق الخلايا في من PBS/ورم للحقن داخل الورم. مباشرة بعد إعطاء خلايا T، تم علاج الفئران بعد ذلك بـ B6H12 ( ) أو PBS عن طريق الحقن داخل الصفاق. تم جمع الأورام بعد 16 ساعة في اليوم 21 بعد زراعة الورم. تم تفكيك الأورام ميكانيكياً باستخدام جهاز التفكيك gentleMACS (Miltenyi). تم إعداد تعليقات الخلايا المفردة عن طريق تمرير الأورام عبر منخل الخلايا (ثيرمو فيشر ساينتيفيك)، إزالة كريات الدم الحمراء بواسطة تحلل ACK (كوالتي بيو لوجيك) وفلترة إضافية من خلال أنابيب فلترة تدفق الخلايا معأغطية مصفاة الخلايا (فالكون). تم صبغ التعليق الخلوي المفرد لاحقًا لتحليل التدفق الخلوي للكشف عن CFSE.خلايا T والبلعميات من الأورام المفككة (انظر أدناه) (الشكل التوضيحي التكميلي 1n).
143B دراسة ارتباطية وتفكك الورم
إجماليالخلايا فيتم حقن DPBS في السمحاق الظنبوبي لفئران NSG (التي تتراوح أعمارهاأسابيع). ثم، بعد 13 يومًا من زراعة الورم وبعد التأكيد البصري على تكوين الورم، تم علاج الفئران بـخلايا CAR T HER2-BB7 مع ذاتية وزيادة التعبير عن أي منهما أو ، عدد مكافئ من خلايا T المزيفة عن طريق الحقن في الوريد عبر ذيل الفأر، أو عدم استخدام خلايا T. ثم تم علاج الفئران مرتين بـ B6H12 ( ) أو PBS عن طريق الحقن داخل الصفاق في اليوم 15 واليوم 19. تم جمع الأورام في اليوم 21 بعد زراعة الورم (اليوم 8 بعد علاج خلايا CAR T). تم تقسيم الأورام باستخدام شفرة حلاقة، حيث تم تثبيت قسم واحد في تمت إزالة البارافورمالدهيد من الورم المثبت بالفورمالدهيد بعد 24 ساعة واستبداله بـالإيثانول للتخزين على المدى الطويل. ثم تم تثبيت مقاطع الورم في الفورمالين وتضمينها في البارافين وفقًا للبروتوكول القياسي (الشكل التوضيحي التكميلي 1o).
تحليل تدفق الخلايا المناعية وتحليل الأنسجة المناعية للأورام المفككة
لتحليل تدفق الخلايا، تم جمع الأورام كما هو موضح أعلاه. تم صبغ تعليقات الخلايا المفردة للأورام المفككة باستخدام CAR (APC، HER2-Fc، R&D، 1:400)، hCD19 (BUV 496، SJ25C1، BD، 1:100)، CD11b (BUV 395، M1/70، BD، 1:100)، F4/80 (BV605، BM8، BioLegend، 1:100)، hCD45 (PerCP-Cy5.5، HI30، Invitrogen، 1:50)، hCD3 (BUV 737، SK7، BD، 1:100)، mCD45 (BUV 805، I3/2.3، BD، 1:100) و hCD47 (BV711، B6H12، BD، 1:100)، ومع صبغة حيوية قابلة للإصلاح (eFluor 780، Invitrogen، 1:2000) لمدة 30 دقيقة في PBS + 2% FBS (محلول فاس) قبل أن يتم تحليلها بواسطة تحليل تدفق الخلايا.
لتحليل IHC، تم جمع الأورام كما هو موضح أعلاه. تم استخدام مقاطع أورام زراعة الأنسجة المثبتة بالفورمالين والمحفوظة في البارافين. تم إجراء صبغة F4/80 (D2S9R، تكنولوجيا الإشارات الخلوية، 1:200) يدويًا، وتم إجراء صبغة hCD3 (SP7، أبكام، 1:100) وARG1 (D4E3M، تكنولوجيا الإشارات الخلوية، 1:250) باستخدام منصة Ventana Discovery. باختصار، تم تحضين مقاطع الأنسجة في إما محلول سترات 6 مليمول (F4/80، تخفيف 1:200) أو محلول Tris EDTA (CD3/ARG1، تخفيف 1:100 و1:250 على التوالي؛ معيار تكييف الخلايا 1) لمدة 25 دقيقة (F4/80) أو لمدة ساعة واحدة (CD3/ARG1) لاسترجاع المستضدية، تلتها حضانة مع الأجسام المضادة الأولية المعنية لمدة ساعة واحدة. تم حضانة الأجسام المضادة الأولية المرتبطة مع الأجسام المضادة الثانوية المعنية (ماعز مضاد للأرنب، متعددة النسائل، F4/80: مختبرات فيكتور، غير مخفف أو CD3/ARG1: جاكسون إيمونوريسيرش، 1:500)، تلتها UltraMap HRP (روش، F4/80) or الكشف عن ChromoMap DAB (روش، CD3/ARG1). لتحليل IHC، تم تحديد مناطق الورم بناءً على علم الأنسجة. تم تحليل إيجابية F4/80 وCD3 وARG1 لكل منطقة ورم. تم قياس درجات إيجابية IHC لـ F4/80 وCD3 وARG1 تلقائيًا في مناطق الاهتمام باستخدام برنامج Aperio ImageScope (الإصدار 12.3.2.8013). تم اختيار مناطق الاهتمام عشوائيًا داخل الورم لاستبعاد البلعميات الموجودة في الأنسجة الطبيعية المحيطة بالورم.
تحليل الخلايا المفردة للأورام المفصولة 143B
تم فرز الأورام المفصولة من نموذج الساركوما العظمية 143B الموضح أعلاه لخلايا حية باستخدام صبغة Live/Dead (Invitrogen) في منشأة FACS المشتركة في جامعة ستانفورد (استراتيجية التصفية موضحة في الشكل التكميلية 2). تم إعداد مكتبات scRNA-seq باستخدام منصة Chromium Next GEM Single Cell 5′ v2 (10x GENOMICS). تم إرسال المكتبات إلى Novogene للتسلسل على مسار NovoSeq S4 (PE150) مع حوالي 30,000 قراءة متوسطة لكل خلية. تم محاذاة القراءات وقياسها باستخدام CellRanger (الإصدار 6.0؛ 10x GENOMICS) باستخدام سير العمل القياسي، مع مرجع النسخ GRCh38 للبشر وmm 10 للفئران. تم استيراد مخرجات CellRanger إلى R (الإصدار 4.2.2) باستخدام Seurat (الإصدار 4.2.0). تم تطبيق الفلاتر التالية باستخدام وظيفة subset لاختيار الخلايا الحية: nFeature_RNA > 200 و nFeature_RNA < 5000؛ نسبة قراءات الميتوكوندريا < 5%. بعد التصفية، تراوحت العينات البيولوجية الثمانية من 7,658-9,327 معرفات جزيئية فريدة متوسطة لكل خلية. تم تطبيع مصفوفة البيانات باستخدام NormalizeData وتم قياسها باستخدام Seurat. تم تعيين أنواع الخلايا باستخدام التعرف التلقائي على نوع الخلية SingleR. تم إجراء تحليل التعبير التفاضلي، والتجميع، وتحليل تقليل الأبعاد UMAP على مصفوفة البيانات الناتجة باستخدام Seurat.تم إجراء تحليل المسار باستخدام Enrichr، مع مجموعة جينات NCI-Nature 2016 التي تم الاستعلام عنها لخلايا T البشرية ومجموعة مسارات KEGG البشرية التي تم الاستعلام عنها باستخدام معرفات الجينات المحولة للفئران لخلايا البلعميات الفئران.
التحليلات الإحصائية
تم الإشارة إلى الاختبارات الإحصائية المحددة في أساطير الأشكال. تم إجراء التحليلات الإحصائية باستخدام R (الإصدار 4.2.2) وExcel (الإصدار 16.64؛ مايكروسوفت) أو Prism (الإصدار 9.3.1، GraphPad). بالنسبة للمقارنات بين مجموعتين، تم تقييم الدلالة الإحصائية باستخدام اختبار Student غير المقترن ذو الطرفين.-اختبارات أو مان-ويتني-اختبارات. للمقارنة ضمن الدراسات الحية وبين الدراسات المجمعة، تم إجراء تحليل التباين الثنائي مع اختبار توكي للمقارنات المتعددة للتحليل بعد التجربة. تم حساب الأهمية لبيانات البقاء باستخدام اختبار مانتل-كوكس ذو الرتبة اللوغاريتمية. تم تحديد أحجام العينات بناءً على تباين نماذج الأورام المستخدمة، والذي تم تحديده من خلال الخبرة مع النماذج المنشورة مسبقًا والمثبتة جيدًا.تم توزيع الحيوانات الحاملة للأورام على مجموعات العلاج عشوائيًا لضمان توزيع متساوٍ لأحجام الأورام بين المجموعات. يتم تمثيل البيانات كمتوسطانحراف معياري (دراسات في المختبر) أو متوسط س.م. (بعض الدراسات الحية). لجميع التحليلات الإحصائية، تُشير القيم إلى كل لوحة من اللوحات.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
جميع البيانات المرتبطة بهذه الورقة مدرجة في المقالة والمعلومات التكميلية. تم إيداع مجموعة بيانات scRNA-seq في مستودع التعبير الجيني NCBI (GEO) تحت رقم الوصول GSE261475. تم الحصول على البيانات المستخدمة لإنشاء مخططات UMAP لـ scRNA-seq من بيانات المرضى (الشكل 2d) من مجموعات بيانات متاحة للجمهور باستخدام أرقام الوصول لسلسلة GEO GSE168940 (المرجع 14) وGSE186802 (المرجع 15). بالنسبة لنمذجة بنية بلورات البروتين، تم استخدام ملفات PDB المتاحة للجمهور التالية: 2JJS (hCD47-hSIRPa) و5TZU (hCD47-B6H12). يتم توفير بيانات المصدر مع هذه الورقة. 30. هيوز، م. س. وآخرون. نقل جين TCR مستمد من مريض لديه استجابة مضادة للأورام ملحوظة ينقل وظائف فعالة للغاية لخلايا T. العلاج الجيني البشري 16، 457-472 (2005). 31. ماجنزر، ر. ج. وآخرون. خلايا CAR T المستهدفة لبروتين B7-H3، وهو مستضد شامل للسرطان، تظهر نشاطًا قويًا في الدراسات السريرية ضد الأورام الصلبة لدى الأطفال وأورام الدماغ. أبحاث السرطان السريرية 25، 2560-2574 (2019). 32. لبانيه، ل. وآخرون. تعزيز السلامة والفعالية لمستقبلات خلايا CAR-T المنظمة بواسطة البروتياز. خلية 185، 1745-1763 (2022). 33. لين، ر. س. وآخرون. الإفراط في التعبير عن c-Jun في خلايا CAR T يؤدي إلى مقاومة الإرهاق. ناتشر 576، 293-300 (2019). 34. فريتاس، ك. أ. وآخرون. تعزيز نشاط خلايا T الفعالة من خلال استهداف وحدة كيناز الوسيط. ساينس 378، eabn5647 (2022). 35. كيمورا، ر. هـ.، ليفين، أ. م.، كوكرا، ف. ف. وكوكرا، ج. ر. ببتيدات عقد السيستين المهندسة التي ترتبط، ، و الإنتغرينات ذات الألفة المنخفضة النانومولارية. البروتينات 77، 359-369 (2009). 36. تشو، ج. وآخرون. بروتين فلوري برتقالي مشع يمكن تحفيزه باللون السماوي يسهل المجهر ثنائي الانبعاث ويعزز التصوير البيولومينسنتي في الكائنات الحية. نات. بيولوجيا تقنية. 34، 760-767 (2016). 37. غريشام، إتش. دي، جودوين، ج. إل، ألين، ب. م، أندرسون، د. سي & براون، إ. ج. عضو جديد من عائلة مستقبلات الإنتغرين يساهم في بلع الكريات البيضاء المحفز بواسطة الأرجينين-غليسين-أسبارتيك. مجلة البيولوجيا الخلوية 108، 1935-1943 (1989). 38. ثيروفاث، ج. وآخرون. مضاد GD2 يتعاون مع حجب CD47 لتحقيق القضاء على الورم. نات. ميد. 28، 333-344 (2022). 39. علي، ن. وآخرون. مرض الطعم ضد المضيف الخيفي في NOD-scid IL-2Ryتظهر الفئران نمط ذاكرة T-الفعالة. PLoS ONE 7، e44219 (2012). 40. فيرارا، ج.، غيليين، ف. ج.، سليكمان، ب.، بوراكوفسكي، س. ج. ومورفي، ج. ف. مرض الطعم ضد المضيف الحاد الجلدي تجاه مستضدات التوافق النسيجي الثانوية في نموذج فئري: تحليل نسيجي وارتباط بالمرض السريري. مجلة أبحاث الأمراض الجلدية 86، 371-375 (1986). 41. رادوسيفيتش، م. ت. وآخرون. قياس كثافة المستضدات لأهداف المناعية على سطح الخلايا بواسطة تحليل تدفق الخلايا: اختبار متعدد المستضدات لنقائل الورم العصبي في نخاع العظام. بروتوكولات ستار. 4، 102709 (2023). 42. شاه، ن. ن. & هارينجتون، أ. م. البلعمة الدموية في السائل الدماغي الشوكي بعد علاج خلايا CAR T. بلود 139، 1116 (2022). 43. هاو، ي. وآخرون. تحليل متكامل لبيانات الخلايا الفردية متعددة الأنماط. خلية 184، 3573-3587 (2021). 44. ليم، س.، غلاسكو، ج. إ.، فيلسينجر إنتيررانتي، م.، ستورم، إ. م. وكوكران، ج. ر. العرض المزدوج للبروتينات على سطح خلايا الخميرة يبسط قياس تفاعلات الارتباط وتفاعلات البيوكوجوجين الإنزيمية. مجلة التكنولوجيا الحيوية 12، 1600696 (2017). 45. هانتر، س. أ. وكوكران، ج. ر. اختبارات ارتباط الخلايا لتحديد ألفة تفاعلات البروتين-بروتين: التقنيات والاعتبارات. طرق الإنزيمات. 580، 21-44 (2016). 46. هافيمستر، سي. وساتيجا، آر. تطبيع وتثبيت التباين لبيانات تسلسل RNA أحادي الخلية باستخدام الانحدار الثنائي السالب المنتظم. جينوم بيو. 20، 296 (2019). 47. تشين، إ. واي. وآخرون. إنريش: أداة تحليل إثراء قوائم الجينات التفاعلية والتعاونية بتنسيق HTML5. BMC Bioinform. 14، 128 (2013).
الشكر والتقدير تم دعم هذا العمل من قبل منح المعاهد الوطنية للصحة 1R01CA263500-01 (C.L.M. وM.M.); ومنحة EPICC للبحث الانتقالي (مؤسسة سانت بالدرِك، C.L.M.); وصندوق فيرجينيا وD.K. لودفيغ لأبحاث السرطان (C.L.M.). C.L.M. وS.A.Y.-H. وL.L. وZ.G. هم أعضاء في معهد باركر لعلاج السرطان المناعي، الذي يدعم برنامج علاج السرطان المناعي في جامعة ستانفورد. تم دعم B.J.M. من خلال منحة زمالة الدراسات العليا متعددة التخصصات في ستانفورد؛ وK.A.F. من قبل زمالة أبحاث الدراسات العليا من مؤسسة العلوم الوطنية تحت المنحة DGE-1656518؛ وF.L. من خلال منحة مشروع Capstone من M-TRAM في ستانفورد؛ وA.L. من قبل مؤسسة Nuovo-Soldati ومن قبل ITMO Cancer AVIESAN (التحالف الوطني لعلوم الحياة والصحة) في إطار خطة السرطان الفرنسية؛ وR.P. من قبل مركز الأميرة ماكسيما لعلم الأورام pediatrics وصندوق أكاديمية تير مولن من الأكاديمية الملكية الهولندية للفنون والعلوم؛ وS.H. من خلال منحة U54 CA232568-01؛ وO.K. من خلال زمالة بول وديزي سورو للأمريكيين الجدد. حصل J.B. على دعم من منحة تدريب علوم الحياة في ستانفورد 5T32-GM11999505 من خلال معهد بيولوجيا الخلايا الجذعية والطب التجديدي، ومن برنامج زمالة أبحاث الدراسات العليا من مؤسسة العلوم الوطنية تحت رقم الجائزة 2146755؛ وT.M. من منحة برنامج تدريب العلماء الطبيين في ستانفورد T32GM007365، ومن NCI تحت رقم الجائزة F30CA271797، ومن زمالة الدراسات العليا متعددة التخصصات في ستانفورد، ومن واجهة الكيمياء/البيولوجيا ChEM-H في ستانفورد.
برنامج التدريب قبل الدكتوراه وزمالة O’Leary-Thiry للدراسات العليا في Stanford ChEM-H. تم إجراء الفرز على جهاز في مرفق Stanford Shared FACS تم الحصول عليه باستخدام منحة الأدوات المشتركة NIH S10 Shared Instrument Grant S10RR025518-01. نشكر موظفي مرفق Stanford Shared FACS على فرز عينات الأورام، ومركز خدمات علم الأمراض/الهستولوجيا البشرية في ستانفورد على صبغ المناعية، ومركز الخدمات البيطرية في ستانفورد على الدعم العام لأعمال الفئران ودراسات التشريح، وALX Oncology على مساهمتها في مادة CV-1. تم إنشاء الرسوم التوضيحية في الشكل 5a و6a من البيانات الموسعة والشكل 1 من الملحق باستخدام BioRender.
مساهمات المؤلفين: S.A.Y.-H. وJ.T. وC.L.M. اقترحوا فكرة الدراسة. S.A.Y.-H. صمم التجارب، واستنسخ التركيبات، وأجرى التوصيفات في المختبر، بما في ذلك اختبارات البلعمة، والتجارب في الجسم الحي، وهندسة البروتين، وتجارب توصيف ارتباط الخميرة، وتجارب scRNA-seq، بما في ذلك تحليل وتفسير البيانات. J.T. صمم التجارب، واستنسخ التركيبات، وأجرى التوصيفات في المختبر، بما في ذلك اختبارات البلعمة، والتجارب في الجسم الحي، بما في ذلك تحليل وتفسير البيانات. B.J.M. أجرى تجارب هندسة البروتين وتوصيف ارتباط الخميرة، وحلل البيانات وفسر النتائج. K.A.F. أجرى التجارب، وحلل البيانات وفسر النتائج المتعلقة بـ scRNA-seq. F.L. وM.T.R. وS.D. استنسخوا التركيبات، وأجروا التوصيفات في المختبر وأجروا دراسات في الجسم الحي. A.L. صمم وميز نموذج سمية PIP CAR، بما في ذلك إجراء التجارب، وتحليل البيانات وتفسير النتائج. N.M.-V. صمم التجارب في الجسم الحي، وساعد في دراسات تفكيك الورم، وصمم، وحلل وفسر تجارب تدفق الخلايا. P.X. وJ.H. نظما وأجريا دراسات في الجسم الحي. A.D. أجرى تجارب IHC وحلل وفسر البيانات. M.H.D. أجرى تجارب scRNA-seq. Z.G. وY.C. حلالا بيانات scRNA-seq وفسرا النتائج. R.P. صمم وأجرى تجارب مجهرية، بما في ذلك تحليل البيانات وتفسير النتائج. A.M. أجرى اختبارات البلعمة. L.L. استنسخ التركيبات وصمم التجارب. J.B. أجرى التوصيفات في المختبر وتجارب تدفق الخلايا. T.M. وZ.E. حلالا بيانات تدفق الخلايا وفسرا النتائج. C.W.M. أجرى دراسات في الجسم الحي وتحليل الأنسجة وتفسير البيانات. S.H. أجرى تجارب التوصيف في المختبر وساعد في الدراسات في الجسم الحي. K.D.M. أجرى التجارب في الجسم الحي. A.B. وO.K. قدما وميزتا البلعميات البشرية الأولية لاختبارات البلعمة. S.L.W. استنسخ التركيبات وأجرى اختبارات التوصيف في المختبر. J.Y.S. وS.F.-P. قدما وحللا صور السائل الدماغي الشوكي للمرضى. R.G.M. صمم وأجرى التجارب في الجسم الحي، وحلل البيانات وفسر النتائج. E.S. وJ.R.C. وC.L.M. صمموا التجارب، وحللوا البيانات وفسروا النتائج. C.J.K. وP.H.S. وM.M. وI.L.W. وB.S. وE.S. وJ.R.C. وC.L.M. أشرفوا على العمل. S.A.Y.-H. وE.S. وC.L.M. كتبوا المخطوطة مع ملاحظات من جميع المؤلفين.
المصالح المتنافسة S.A.Y.-H.، J.T.، B.J.M.، J.R.C. وC.L.M. مدرجون كمخترعين مشاركين في براءة اختراع تتعلق بهذا العمل (PCT/US2O24/013209، المقدمة من مجلس أمناء جامعة ليلاند ستانفورد جونيور). C.L.M. يمتلك أسهماً في CARGO Therapeutics وLink Cell Therapies وEnsoma، التي تطور علاجات تعتمد على CAR؛ ويستشير CARGO وLink وImmatics وEnsoma وRed Tree Capital؛ ويتلقى تمويلًا بحثيًا من Lyell Immunopharma وTune Therapeutics. S.A.Y.-H. هو مستشار لشركة Quince Therapeutics. J.T. هو مستشار لشركة Dorian Therapeutics. L.L. وE.S. هما مستشاران ويمتلكان أسهماً في Lyell Immunopharma. L.L. هو مؤسس مشارك، ويستشير ويمتلك أسهماً في CARGO Therapeutics. O.K. هو زميل أول في ARTIS Ventures. C.J.K. هو مؤسس وعضو في المجلس الاستشاري العلمي لشركات NextVivo وSurrozen وMozart Therapeutics. R.G.M. هو مؤسس مشارك ويمتلك أسهماً في Link Cell Therapies؛ وهو مستشار لشركات NKarta وArovella Pharmaceuticals وInnervate Radiopharmaceuticals وGammaDelta Therapeutics وAptorum Group وZai Labs وImmunai وGadeta وFATE Therapeutics (DSMB) وWaypoint Bio. I.L.W. هو مدير ومساهم ومستشار لشركة Forty Seven (لكن ليس Gilead)؛ ومؤسس مشارك ومدير ومستشار لشركتي Bitterroot Bio وPHeast، ومؤسس مشارك لشركة 48. I.L.W. هو أيضًا عضو في المجلس الاستشاري العلمي لشركة Appia. E.S. يستشير لشركتي Lepton Pharmaceuticals وGalaria. J.R.C. هو مؤسس مشارك ومالك أسهم في Trapeze Therapeutics وCombangio وVirsti Therapeutics؛ لديه مصالح مالية في Aravive وXyence Therapeutics وCARGO Therapeutics؛ وهو عضو في مجلس إدارة Ligand Pharmaceuticals وRevel Pharmaceuticals. المؤلفون الآخرون يعلنون عدم وجود مصالح متنافسة.
معلومات إضافية
معلومات إضافية النسخة الإلكترونية تحتوي على مواد إضافية متاحة فيhttps://doi.org/10.1038/s41586-024-07443-8. يجب توجيه المراسلات والطلبات للحصول على المواد إلى كريستال ل. ماكال. تشكر مجلة نيتشر سميتا تشاندرا والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة الأقران لهذا العمل. معلومات إعادة الطباعة والتصاريح متاحة علىhttp://www.nature.com/reprints.
الشكل البياني الممتد 1 | انظر الصفحة التالية للتعليق.
الشكل البياني الممتد 1 | علاج مضاد CD47 يخفف من فعالية خلايا CAR وTCR T عن طريق استنفاد خلايا T المنقولة بالتبني. (أ)عولجت بقاء ورم 143B.فئران/ذراع. (ب) B7H3.BB7- أو GD2.BB7نجاة ورم MG63.3 المعالج.فئران/ذراع. (ج)تم معالجة نمو الورم D 425 بواسطة BLI. تم تضمين CD19.BB7-CAR كعنصر تحكم غير مستهدف للورم. المتوسطSEM لـ أو (جميع الآخرين) الفئران/الذراع. ممثل لتجربتين مستقلتين. (د) B7H3.BBCتم علاج بقاء الورم D425. تم تضمين CD19.BB7-CAR كعنصر تحكم غير مستهدف للورم. (B6H12) أو (جميع الآخرين) الفئران لكل ذراع علاج. تمثيلي من تجربتين مستقلتين. (هـ) مخططات تدفق خلوية تمثيلية لـالخلايا المحددة في الدم والورم في B7H3.BB7-CARنموذج MG63.3 المعالج بـ B6H12 في اليوم 30 بعد زراعة الورم. (f) مخططات تمثيلية لتدفق السيتومتريةالخلايا المحددة في دم الفئران غير الحاملة للأورام التي تم علاجها مع خلايا CD19.287CAR T وخلايا PBS أو B6H12 أو التحكم في النظير mIgG1. تمثل تجربتين مستقلتين. (ز) hCD8(الأعلى) و hCD4(أسفل) خلايا T في دم الفئران في اليوم الخامس في نموذج التحكم في النوع، تم علاجها كما في (f). المتوسطانحراف معياري لـالفئران. تمثيل لتجربتين مستقلتين. (ح) خلايا T (و) في دم الفئران في اليوم الثاني عشر في هير2.بي بي فئران معالجة في نموذج 143B. المتوسطانحراف معياري لـالفئران. (ط) جرعة منخفضة من NY-ESO-1عولجت بقاء ورم 375.فئران/ذراع. (ج) جرعة عالية من NY-تم علاج نمو ورم 375. المتوسطمنفئران/ذراع. (ك) خلايا T (hCD4و) في الدم بواسطة تحليل تدفق الخلايا في اليوم 16 من الفئران المعالجة بجرعة عالية من NY-ESO-1-TCR B6H12 في نموذج A375. المتوسطانحراف معياري لـفئران. (1) جرعة عالية من CD19.28-سيارةتم علاج نمو ورم Nalm6 بواسطة B6H12 باستخدام التصوير الضوئي بالأشعة تحت الحمراء. المتوسطSEM لـفئران/ذراع. (م) جرعة منخفضة CD19.28-سيارةتم علاج نمو ورم Nalm6 بواسطة B6H12 باستخدام التصوير الضوئي للانبعاثات.SEM لـفئران/ذراع. (اسم) صور لورم نالم6 BLI في نموذج CAR-T بجرعة عالية (يسار) وجرعة منخفضة (يمين) في اليوم 17. (o) CD19.28 7 -CAR بجرعة منخفضةنجاة Nalm6 المعالجة بـ B6H12.فئران/ذراع. (ب) خلايا T BLI في نموذج CAR-T عالي الجرعة – Nalm6، تم معالجتها كما في (ل). المتوسطSEM لـفئران/ذراع. (ق) خلايا T BLI في نموذج CAR-T منخفض الجرعة – Nalm6، تم علاجها كما في (م). المتوسطSEM لـفئران/ذراع. (ر) صور لخلايا CAR T CD19.28-nLuc-BLI في نموذج Nalm6 بجرعة منخفضة، تم علاجها كما في (م)، في اليوم 11. (س) قياس خلايا T بواسطة تحليل التدفق من الطحال في نموذج CART-Nalm6 بجرعة عالية، تم علاجها كما في (I). المتوسطانحراف معياري لـالفئران. [(أ)، (ب)، (د)، (ي)، (ع)] اختبار لوج-رانك مانتل-كوكس. [(ج)، (ي)، (ل)، (م)، (ب)، (ق)] اختبار تحليل التباين ثنائي الاتجاه (ANOVA) مع اختبار المقارنة المتعددة لتوكاي.اختبار ستودنت ذو الذيلين غير المتزاوجاختبار. للجميع: ns = غير ذي دلالة.
الشكل البياني الممتد 2 | انظر الصفحة التالية للتعليق.
الشكل 2 من البيانات الموسعة | العلاج باستخدام CV-1 أو حذف CD47 يؤدي إلى استنفاد خلايا T وإلغاء الفعالية المضادة للورم. (أ) CD19.BBC-CARالسيرة الذاتية-1
(Fc-dead) عالج بقاء ورم نالم6.فئران/ذراع. (ب) صور لنالم6 BLI في اليوم 11 من جرعة عالية من CD19.BB7-CARالفئران المعالجة بـ CV-1، تم معالجتها كما في (أ). (ج) جرعة منخفضة من CD19.28 7 -CARنمو ورم نالم6 المعالج بـ CV-1 (موت Fc). المتوسطSEM لـفئران/ذراع. (د) صور لنظام التصوير الضوئي في نالم6 بجرعة منخفضة من CD19.28 CARفئران معالجة بـ CV-1، تم معالجتها كما في (ج). (هـ-و) صور (هـ) وقياس (و) لتصوير الخلايا التائية بعد معالجة CV-1 لفئران تحمل ورم نالم6. الأعلى: جرعة عالية من CD19.BB7-CAR CV-1، تم معالجته كما في (أ)، في اليوم 9 (بعد أربعة أيام من علاج CV-1). الأسفل: جرعة منخفضة من CD19.28て-CAR CV-1، تم معالجته كما في (ج)، اليوم 11 (ستة أيام بعد علاج CV-1). بالنسبة لـ (و): المتوسطانحراف معياري لـفئران لكل حالة جرعة. (ز) حذف CD47 بواسطة تقنية كريسبر/كاس9 (CD47الكفاءة في خلايا T البشرية الأولية بواسطة تحليل تدفق الخلايا.الخلايا هيمع التعبير الخارجي عن نوع البرية CD47. تمثيلي لـالمتبرعين. (ح) صور لـ
تقدم ورم نالم6 (أعلى خمسة صفوف) وخلايا T في اليوم 11 (الصف السفلي) بواسطة BLI في نالم6-47-نموذج CAR T-CD19.287. (ط) بقاء الفئران الحاملة لنموذج Nalm6 موضح في (ح).فئران لكل ذراع علاج. (ج) مخططات تمثيلية لتدفق السيتومترية لـالخلايا المحددة في دمأوCD19.28 7 -nLuc-CARفئران غير مصابة بالأورام المعالجة بـ B6H12. (ك) صور لتصوير الخلايا التائية بعد العلاج بـأو-CD19.28-نقطة-CAR T خلايا قبل وبعد علاج B6H12. (I) تصوير بيوفوتوني لتلك الخلايا التائية في الفئران الموضحة في (k) المعالجة بـأو-CD19.28-nLuc-CART، قبل وبعد معالجة B6H12. الخط المتقطع يشير إلى حد الكشف. المتوسطمنفئران.وخلايا T في الدم في اليوم السادس أو السابع بواسطة تحليل تدفق الخلايا بعد العلاجأو-نلوك-كارتفي تجارب منفصلة، تم التعامل معها كما في (ك). المتوسطانحراف معياري لـالفئران. [(ج)، (ل)، (م)] اختبار تحليل التباين ثنائي الاتجاه (ANOVA) مع اختبار المقارنة المتعددة لتوكاي. ns = غير دال. [(ف – الأعلى)] اختبار مان-ويتني ثنائي الطرف. [(ف – الأسفل)] اختبار الطالب غير المقترن ثنائي الطرف.اختبار.
الشكل البياني الممتد 3 | انظر الصفحة التالية للتعليق.
الشكل البياني الموسع 3 | الإفراط في التعبير عن CD47 ) لا يغير وظيفة خلايا T في المختبر، ولكنه يعزز الفعالية في الجسم الحي. (أ) توسيع CD19.28-, ، أو خلايا CAR-T على مدى أيام في الثقافة. المتوسطانحراف معياري لـمتبرعو الخلايا. (ب) CD19.28CD19.28CD19.28، أو -CD19.28حيوية خلايا CAR-T على مدار الأيام في الثقافة. المتوسطSD منمتبرعو الخلايا. (ج) قتل ورم Nalm6-GFP بواسطة الموك، CD19.28CD19.28، -CD19.28-, أو -CD19.28-خلايا CAR-T عند نسبة T المقاسة عبر اختبار إنكو سايت. المتوسطانحراف معياري لـمتبرعي الخلايا، مع اشتقاق كل نقطة بيانات من المتوسط لـآبار ثلاثية لكل متبرع. وهمي، CD19.28%، و-CD19.287 هي حالات مشتركة مكررة في (د). (د) قتل ورم Nalm6GFP بواسطة الموك، CD19.28، أو CD19.28-خلايا CAR T B6H12 بنسبة 1:1E:T تم قياسها عبر اختبار IncuCyte. المتوسطانحراف معياري لـمتبرعي الخلايا، مع اشتقاق كل نقطة بيانات من المتوسط لـآبار ثلاثية لكل متبرع. الشروط بدون B6H12 هي شروط مشتركة مكررة في (ج). (هـ-و) IFN- (e) وإفراز IL-2 (f) عند الزراعة المشتركة لمدة 24 ساعة مع وبدون خلايا ورم Nalm6 بنسبة 1:1 من E:T من العينة الوهمية، CD19.28て-,47 CD19.28، CD19.28 – أو CD19.28-CAR-T تم قياسه عبر ELISA. نموذج، CD19.28-, و تم تقييم خلايا (-CAR-T) أيضًا في وجود B6H12 و Nalm6. المتوسطانحراف معياري لـمتبرعي الخلايا، مع اشتقاق كل نقطة بيانات من المتوسط لـآبار ثلاثية لكل متبرع. (ج-ل) أنكسين V (ج)، CD69 (ح)، CD39 (i) تعبير TIM3 (j) و LAG3 (k) و PD1 (l) عند زراعة مشتركة لمدة 24 ساعة مع وبدون خلايا ورمية Nalm6 بنسبة 1:1 E:T من نموذج التحكم، CD19.28-CD19.28 AAVS1 -CD19.28 – أو -CD19.28-CAR-T تم قياسه عبر تحليل تدفق الخلايا. نموذج، CD19.28، و -CD19.28تم تقييم خلايا CAR-T أيضًا في وجود B6H12 و Nalm6. المتوسطانحراف معياري لـمتبرعو الخلايا. (م) تعبير CD47 و CAR (الثاني من اليسار) على خلايا T بواسطة قياس التدفق الخلوي بعد الإفراط في التعبير عن CD47ممثل لـ المتبرعين. ( ن ) صور لـ Nalm6 BLI في اليوم 45 بعد الجرعة مع CD19.28 – أو -CD19.28-خلايا CAR-T في الفئران الحاملة لورم نالم6. (o) CD19.28 – أو -CD19.28-تم علاج نمو ورم نالم6 بواسطة CAR T باستخدام التصوير الضوئي بالأشعة تحت الحمراء. المتوسطSEM لـفئران/ذراع. (ص)تم جمع البلعميات عبر غسل الصفاق باستخدام تقنية تحليل تدفق الخلايا في اليوم الثالث عشر بعد علاج الكلودرونات. المتوسطانحراف معياري لـ (مستنفد من البلعميات) أو فئران (غير مستنفدة من البلعميات). (q) مخططات التدفق التمثيلية واستراتيجية التصفية لاكتشاف البلعميات في العينات المجمعة بعد غسل الصفاق. (r) CD19.287-nLuc-CAR T BLI قبل (اليوم 7) وبعد (اليوم 9) العلاج بـ B6H12 للفئران.نقص الماكروفاجات (بدأ في اليوم 0). المتوسطانحراف معياري لـ (مستنفدة من البلعميات، معالجة بمحلول فوسفات البفر) أو (جميع المجموعات الأخرى) الفئران. [(أ)، (ب)، (ج)، (د)، (هـ)، (و)، (ز)، (ح)، (ط)، (ي)، (ك)، (ل)، (م)، (ن)] اختبار تحليل التباين ثنائي الاتجاه (ANOVA) مع اختبار المقارنة المتعددة لتوكاي. ns = غير دال. [(ص)] اختبار ستودنت غير المقترن ذو الذيليناختبار.
الشكل البياني الممتد 4 | انظر الصفحة التالية للتعليق.
الشكل البياني الممتد 4 | تعبير CD47 موحد بين أنواع خلايا CAR T وينخفض مع مرور الوقت في الثقافة. (أ) البلعمة لخلايا CD19.28 (-CAR-T الموسومة بـ CFSE من ثلاثة متبرعين بواسطة البلعميات البشرية الأولية من ثلاثة متبرعين بواسطة قياس التدفق، بعد ساعة واحدة من الثقافة المشتركة. كانت خلايا CAR T إما غير معالجة أو معالجة بـ B6H12 أو التحكم في النظير المجهري IgG1 قبل إضافة البلعميات. المتوسطانحراف معياري لـمتبرعي الخلايا، مع اشتقاق كل نقطة بيانات من المتوسط لـآبار ثلاثية لكل متبرع. (ب) صور تمثيلية من المجهر الضوئي التداخلي لابتلاع خلايا CD19.287-CAR-T البشرية الأولية بواسطة ماكروفاج بشري أولي بعد المعالجة بـ B 6 H 12 في مصفوفة كولاجين ثلاثية الأبعاد. الصور من اليسار إلى اليمين تمثل 0 دقيقة (الصورة الأولية)، و 3 ساعات و 21 دقيقة من التعايش بين خلايا T – البلعميات. تم وسم خلايا CAR T (باللون الأخضر) والبلعميات (باللون الأحمر) بشكل منفصل باستخدام أصباغ دهنية. كما تم وسم خلايا CAR T أيضًا بصبغة pH حساسة pHrodo Red (المعروضة باللون الأزرق) قبل معالجة B6H12. تشير الأسهم البيضاء إلى حدث بلعمي. راجع الفيديو المرافق SI Video 1 لمشاهدة الفيديو الزمني. الصور تمثل عينة عبر التجارب. (ج) تعبير CD47 على خلايا الورم، خلايا T الوهمية، وخلايا CD19.287-CAR-T بواسطة قياس التدفق الكمي QuantiBrite. تم تقييم خلايا T في اليوم الرابع واليوم الحادي عشر من الثقافة. تم قياس الكميات في الرسوم البيانية الموضحة على اليسار لكل نوع من الخلايا. تمثيلي لـعينة ورم ومتبرعو الخلايا وقابل للتكرار عبرالتجارب. (د) تعبير CD47 علىوCD19.28خلايا CAR-T بواسطة قياس التدفق الكمي QuantiBrite. تم تقييم الخلايا بواسطة قياس التدفق في اليوم 0 (قبل التنشيط)، اليوم 4 (بعد إزالة حبات التنشيط)، اليوم 7، واليوم 11 (وقت النقل النموذجي في الجسم الحي). المتوسطانحراف معياري لـمتبرعو الخلايا. تظهر فقط الفروقات المهمة. جميع المجموعات الأخرى ليست مختلفة بشكل ملحوظ، عند المقارنة بين نفس النوع الفرعي على مر الزمن وبين الأنواع الفرعية. في نفس النقطة الزمنية. (هـ) تعبير CD47 على، و (يسار) و CD8 (اليمين) أنواع خلايا CD19.28-CAR-T في الأيام، و 11 بعد التفعيل بواسطة قياس التدفق الكمي QuantiBrite. المتوسطانحراف معياري لـمتبرعو الخلايا. تظهر فقط الفروقات المهمة. جميع المجموعات الأخرى ليست مختلفة بشكل ملحوظ، عند المقارنة بين نفس النوع الفرعي على مر الزمن وبين الأنواع الفرعية في نفس النقطة الزمنية. (f) مخططات تمثيلية لقياس التدفق لخلايا CD19.287-CAR-T قبل (اليوم 0) وبعد التنشيط (الأيام 4 و 7 و 11). العمود الأيسر: تصنيف لحالات تمايز خلايا T، T نايف (CD45RA/CD62Lذاكرة مركزية T (CD45RAذاكرة التأثير T (CD45RACD62L )، و T ذاكرة المؤثرين التي تعيد التعبير عن CD45RA (CD45RA/ CD62L . العمود الأيمن: هيستوجرامات تعبير CD47 عبر حالات تمايز خلايا T. (g) تعبير الكالريتولكين (يسار) و CD47 (يمين) على خلايا CAR-T بواسطة قياس التدفق في اليوم العاشر من الثقافة. الكمية من الهيستوجرامات الموضحة في الأسفل لكل نوع من خلايا CAR-T. تمثيلي لـ المتبرعين. (ح) قياس تعبير CD47 (أعلى) و الكالريتiculin (أسفل) على الخلايا في اليوم 16 واليوم 25 من الثقافة بواسطة تحليل تدفق الخلايا. المتوسطمنالمتبرعون. (ط) مناظر الخلايا الفردية ملونة حسب نوع الخلية. الأعلى: UMAP المستمد من الجوار الأقرب الموزون (wnn)الخلايا المفروزة من الدم الم collected في اليوم السابع بعد حقن CAR Tأكسي-سيل (CD19.28 ) تم معالجة مرضى LBCL، تم أخذ عينة من 500 خلية/عينة مريض. الأسفل: UMAP مستمد من الخلايا من السائل الدماغي الشوكي (مرضى الورم الدبقي المنتشر في الخط الأوسط (DMG) المعالجين، تم أخذ عينات من 500 خلية/عينة مريض. [(أ)، (د)، (هـ)] اختبار تحليل التباين ثنائي الاتجاه (ANOVA) مع اختبار المقارنة المتعددة لتوكاي. [(ح)] اختبار ستودنت غير المقترن ذو الذيليناختبار. للجميع: ns = غير ذي دلالة.
الشكل 5 من البيانات الموسعة | يمكن أن يحد مضاد CD47 من السميات الناتجة عن استهداف الانتيجرين الشامل للأورام PIP-CAR و GvHD. (أ) رسم توضيحي لتصميم PIP-CAR.
تم إنشاء الرسم البياني باستخدام BioRender. (ب) تعبير PIP.28 7 -CAR بواسطة تحليل تدفق الخلايا. تمثيلي لـ المتبرعين. (ج) قتل الورم 143B-GFP بواسطة PIP.28 –PIP.BB-, أو استهداف غير الورمCD19.BB-خلايا CAR T بنسبة 1:1E:T تم قياسها عبر اختبار IncuCyte. المتوسطانحراف معياري لـآبار ثلاثية وقابلة للتكرار في ثلاثة تجارب مستقلة مع متبرعين مختلفين. (د) أوزان الفئران بعد العلاج مع CD19.28 7 – أو PIP.28 7 -CAR-T. المتوسطSEM لـفئران/ذراع. (هـ) كارتون لتصميم تقرير ثنائي اللوكيفيراز لتتبع نشاط CAR-T في الجسم الحي. nLuc مرتبط بتعبير CAR، و fLuc مرتبط بتنشيط NF-KB. (و) تصوير ضوئي لتفاصيل T في الفئران المعالجة بـ CD19.287 أو PIP.28 [-nLuc_NFkB-fLuc-CART خلايا. اليسار: nLuc (إجمالي خلايا T). اليمين: fLuc (خلايا T النشطة). المتوسطSEM لـفئران/ذراع. (ج) صور لتصوير الخلايا التائية باستخدام BLI لفئران تم علاجها كما في (و)، بعد 3 أيام من الحقن. الأعلى: nLuc (إجمالي الخلايا التائية). الأسفل: fLuc (خلايا T المنشطة).فئران/ذراع. (ح) صور تمثيلية من BLI من الأعضاء المستخرجة من الفئران المعالجة كما في (و)، بعد أربعة أيام من إعطاء CAR T. اليسار: nLuc (خلايا T الكلية). اليمين: fluc (خلايا T النشطة). تمثيلي لـفئران/ذراع. (i) صور تمثيلية ملونة بصبغة الهيماتوكسيلين والإيوزين (H&E) لنسج الرئة المجمعة من الفئران المعالجة كما في (f)، بعد أربعة أيام من إعطاء CAR T. اللوحة اليمنى هي عرض مكبر لمنطقة الإطار في اللوحة الوسطى. الصور تمثلفئران/ذراع. (ج-1) صور تمثيلية لـ (ج) فئران تظهر تساقط الشعر الناتج عن مرض GvHD (اليوم 48 بعد حقن CART)، (ك) طحال من الفئرانفي اليوم 48 بعد حقن CAR T)، و(ل) صبغة H&E لشرائح الجلد (المجمعة في اليوم 48 بعد حقن CAR T) من الفئران المعالجة بـ CD19.BB7-CARTب6هـ12. ممثل لـفئران/ذراع. [(ج)، (د)، (و)] اختبار تحليل التباين ثنائي الاتجاه (ANOVA) مع اختبار المقارنة المتعددة لتوكاي.
الشكل البياني الممتد 6 | انظر الصفحة التالية للتعليق.
مقالة
الشكل 6 من البيانات الموسعة | تحتفظ المتغيرات المهندسة من CD47 بارتباط SIRPa وتظهر فقدان الارتباط ببعض، ولكن ليس جميع، الأجسام المضادة المضادة لـ CD47. (أ) CD47 المُهندَسآلية:تربط الأجسام المضادة CD 47 خلايا الورم، ولكن لا-تسبب خلايا T في تحفيز البلعمة المحددة للورم. تم إنشاء الرسم البياني باستخدام BioRender. (ب) رسم كاريكاتوري للخميرة يعرض مجال Ig-like CD47 باستخدام متجه pCTCON2. يتم عرض CD47 كإندماج في الطرف N. (ج) ربط بـلخميرة عرضت CD47 في pCTCON2 بواسطة تحليل تدفق الخلايا. تمثيلي لـتجارب مستقلة. (د) منحنى الارتباط لـ B6H12 مع الخميرة الذي عرض CD47 في pCTCON2، تم قياسه عبر تركيزات متعددة بواسطة قياس التدفق الخلوي. MFI منتجربة. (هـ) ارتباط 300 نانومول من SIRP البشري (أعلى) والفأري (أسفل)لخميرة عرضت CD47 في pCTCON2 بواسطة تحليل تدفق الخلايا. البيانات تمثلتجارب مستقلة. (و) ارتباطلخميرة عرضت CD47 في pCTCON2 بواسطة تحليل تدفق الخلايا. البيانات تمثلتجارب مستقلة. (ج) مخططات فرز تدفق السيتومتر لجميع الأنواع الستة من مكتبة CD47، تشير الأنواع السلبية إلى B6H12 والأنواع الإيجابية إلى CV-1. السكان المجمّع المشار إليه بالصندوق الأسود في كل مخطط. (ح) ارتباطتم جمع بيانات CV-1 من مجموعة مكتبة الخميرة المعروضة CD47 بعد الفرز 6 أو الخميرة المعروضة WT CD47. البيانات تمثلمستقل التجارب. (ط) ارتباطأو 100 نانومتر مكعب – 1 إلى متغيرات CD 47 المعروضة على الخميرة باستخدام متجه pCTCON2. المتوسطانحراف معياري لـسلالات الخميرة الفردية، تم تطبيعها إلى MFI من الارتباط بـ. (j) كارتون للخميرة يعرض مجال Ig-like لـ CD47 باستخدام متجه pFreeNTerm (pFNT). يتم عرض CD47 كإندماج في الطرف C، مع GFP لمراقبة تعبير البروتين. (k) ارتباط 100 نانومتر من SIRP البشري والفأريلخميرة عرضت CD47 في pFNT بواسطة تحليل تدفق الخلايا. تمثيلي لـتجارب مستقلة. (I) التركيب البلوري لـ CD47 (أصفر) المرتبط بـ SIRPa (برتقالي) [PDB: 2JJS]، مع تحديد حلقة BC لـ CD47 (أخضر)، التي تحتوي على بقايا CD47 T26-Q31. (m) الهياكل البلورية لـ CD47 (أحمر) المرتبطة بـ SIRP (وردي داكن، يسار) [PDB: 2JJS] و B6H12 (أزرق فاتح، يمين) [PDB: 5TZU]، مع تحديد البقايا A30 (ذهبي) و Q31 (أزرق)، وحلقات BC و FG من CD47. الهياكل هي تكبير للمناطق المربعة في الهياكل الكاملة الموضحة في الشكل 4c. (ن) ارتباط ، TJC4، Hu5F9، و hSIRP إلى الخميرة المعروضة، و متغيرات. تعنيانحراف معياري لـسلالات الخميرة الفردية، تم تطبيعها إلى MFI من الارتباط بـ [(i), (n)] اختبار تحليل التباين ثنائي الاتجاه (ANOVA) مع اختبار المقارنة المتعددة لتوكاي. ns = غير دال. المقارنة بين المجموعة المحددة والارتباط بخلايا تعبر عن CD47 WT.
الشكل البياني الممتد 7| انظر الصفحة التالية للتعليق.
الشكل البياني الموسع 7| تعبير عنعلى خلايا T يقلل من البلعمة المستحثة بواسطة B 6 H 12 بسبب عدم ارتباط الأجسام المضادة في المختبر وفي الجسم الحي. (أ) هيستوجرامات تدفق سيتومترية تمثيلية لـ، و ربط بـ، ، و مفرط التعبير على خلايا T البشرية الأولية. البيانات تمثلمتبرعون مختلفون من خلايا T في تجارب مستقلة. (ب) اليسار: مخططات هيستوجرام تدفق الخلايا التمثيلية لـ، و500 نانومتر من B 6 H 12 الارتباط بـ، و معبر عنها في Jurkats مع . اليمين: ارتباط B6H12، hSIRP، وmSIRPإلى الطول الكامل، و معبر عنها في خلايا جوركات مع وجودها الذاتييعنيانحراف معياري لـتجارب مستقلة، تم تطبيعها إلى نسبة الارتباط بـ. (ج) البلعمة بواسطة البلعميات البشرية الأولية من عدة متبرعين مع خلايا يوركات الموسومة بـ CFSE مع وجودها الذاتي، معبرًا عن، أو المتغيرات بعد ساعة واحدة من التعايشيعنيمنآبار ثلاثية. (د) البلعمة بواسطة البلعميات البشرية الأولية من متبرعين اثنين من خلايا CD19.287-CAR-T البشرية الأولية المعلّمة بـ pHrodo Red من ثلاثة متبرعين مع وجود داخلي.، معبرًا عن أو بعد ثلاث ساعات من الثقافة المشتركةيعنيمنآبار ثلاثية. (هـ) صور لتصوير الخلايا التائية بعد العلاج معأو-CD19.28-نيوك-سي آي آر-تي خلايا B6H12 في الفئران غير الحاملة للأورام. (f) تصوير الخلايا التائية قبل وبعدCD47 علاج، للفئران المعالجة كما في (هـ). المتوسطانحراف معياري لـفئران.والخلايا في الدم في اليوم السادس بواسطة تحليل تدفق الخلايا للفئران المعالجة كما في (هـ). المتوسطانحراف معياري لـالفئران. (ح) مخططات التدفق التمثيلية واستراتيجية التصفية لاكتشافالبلاعم والخلايا التائية في عينات ورم 143 B المفصولة في نموذج البلعمة داخل الجسم المعالج بخلايا Her2.BB7-CARTب6هـ12. ممثل لـ. (i) رسم بياني تمثيلي للتدفق واستراتيجية التصفية لاكتشاف CFSEخلايا ورم 143B المفكك من فأر لم يتلقَ خلايا CAR T.البلاعم الكبيرة المحددة بواسطة تحليل تدفق الخلايا للأورام المفككة المعالجة كما في (هـ). المتوسطانحراف معياري لـفئران. (ك) hCD3تم تحديد خلايا T البشرية بواسطة تحليل تدفق الخلايا للأورام المفككة المعالجة كما في (هـ). المتوسطانحراف معياري لـالفئران. (ل) نسبة خلايا T المبتلعة المحددة في كل ورم مفكك، تم معالجتها كما في (ح)، محسوبة كعددالبلاعم لكل 100,000 خلية حية لكل عينة مقسومة على العدد الإجمالي لـالبلاعم والخلايا التائية لكل 100,000 خلية حية لكل عينة، تم تحديدها بواسطة تحليل تدفق الخلايا. المتوسطانحراف معياري لـالفئران. [(ب)، (ج)، (د)، (ف)، (ج)] اختبار تحليل التباين ثنائي الاتجاه (ANOVA) مع اختبار المقارنة المتعددة لتوكاي. ns = غير دال. (ب) المقارنة بين المجموعة المحددة والارتباط بـ خلايا التعبير. [(ج)، (ك)] اختبار ستودنت ذو الذيلين غير المقترن اختبار. [(ل)] اختبار مان-ويتني ذو الذيلين.
الشكل البياني الممتد 8 | علاج CAR-T يزيد من تسلل الأورام للخلايا البلعمية. (أ) hCDتم تحديد خلايا T بواسطة تحليل التدفق الخلوي للأورام المنفصلة 143B في نموذج 143B التبادلي، المعالج بدون خلايا T، أو بشكل وهمي، أو -خلايا CAR-Tمتوسط أو يعني (جميع العينات الأخرى) الفئران. (ب) نسبة الإيجابية لخلايا CD 3 T التي تم تحديدها بواسطة IHC من مقاطع الورم المعالجة كما في (أ). المتوسط من ( ) أو تعني (جميع العينات الأخرى) الفئران. (ج) ارتباط hCD 3 وتلوين في مقاطع IHC للأورام المعالجة كما في (أ). نقاط البيانات تمثل أورام فردية، ملونة حسب مجموعة العلاج ( ). محسوب بواسطة الانحدار الخطي البسيط. (د) نسبة الإيجابية لـ hCD3 (خلايا T)، mArg1 (ماكروفاجات M2)، و mF4/80 (ماكروفاجات إجمالية) التي تم تحديدها بواسطة IHC من مقاطع الورم المعالجة كما في (أ). المتوسط لـ ( ) أو تعني من (جميع الآخرين) الفئران. (هـ) ارتباط نسبة إيجابية hCD3 و mArg1 بواسطة IHC لشرائح الورم المعالجة كما في (أ). نقاط البيانات تمثل أورام فردية، ملونة حسب مجموعة العلاج ( ). تم حسابها بواسطة الانحدار الخطي البسيط. (و) صور تمثيلية لشرائح الأورام الملطخة بـ mF4/80 و mArg1 و hCD3، تم معالجتها كما في (أ). تمثيلية لشرائح الأورام المجمعة منفئران/ذراع. [(أ)، (ب)] اختبار ANOVA ثنائي الاتجاه مع اختبار المقارنات المتعددة توكي. ns = غير ذي دلالة.
الشكل البياني الموسع 9 | انظر الصفحة التالية للتعليق.
الشكل 9 من البيانات الموسعة | يزيد علاج CAR-T من تسلل الورم لمجموعات الماكروفاج المتميزة. (أ و ب) ملف scRNA-seq للخلايا المناعية المنفصلة عن الورم والخلايا المناعية المتسللة في نموذج 143B التبادلي، المعالج بدون خلايا T، نموذج زائف، – أو -خلايا CAR-T BBZ-Her2 B6H12. النقاط تمثل خلايا فردية. خلايا من 8 ظروف تجريبية مع ثلاثة فئران لكل مجموعة معالجة، ملونة بـ (أ) نوع الخلية (الرسوم الثمانية على اليسار؛ تمثل UMAPs ظروف المعالجة المتميزة)، النوع (أقصى اليمين)، أو (ب) مستوى تعبير الجين. (ج) تركيب الخلايا الفأرية التي تم تحديدها عبر scRNA-seq من أورام 143B المعالجة كما في (أ). البيانات مستمدة من تم تجميع الخلايا من 8 ظروف تجريبية مع ثلاثة فئران لكل مجموعة معالجة. تم تعيين أنواع الخلايا باستخدام التعرف التلقائي على نوع الخلية من SingleR. (د) رسم نقطي يوضح تعبير scRNA-seq لمؤشرات مجموعة خلايا T المختارة، السيتوكينات، والكيموكينات.خلايا T المتسللة من الأورام البشرية من العلاجات الموصوفة في (أ). (هـ) مقارنة بين الجينات المعبر عنها بشكل مختلف بين خلايا CAR-T من مجموعات العلاج المختلفة الموصوفة في (أ). تم تحديد الأهمية الإحصائية باستخدام Seurat؛ *Padj <0.05. (و) مقارنة بين الجينات المعبر عنها بشكل مختلف بين البلعميات من مجموعات العلاج المختلفة الموصوفة في (أ). تم تحديد الأهمية الإحصائية باستخدام Seurat؛ صفة. (g) تحليل مسار Enrichr لأعلى 100 جين مرتفع التعبير في البلعميات المتسللة للأورام [سكان البلعميات/وحيدات النواة في (أ)] فيالأورام المعالجة مقارنةً بالضوابط غير المعالجة. تظهر النتائج قيمة (اختبار فيشر الدقيق ذو الجانبين) لكل مسار. (ح) UMAP للسكان المحددين من البلعميات/وحيدات النواة في (أ)، مقسمة ومُعاد تصنيفها، ملونة حسب الكتلة (هويات الكتل على اليسار). النقاط تمثل خلايا فردية. خلايا من 8 ظروف تجريبية. (ط) خرائط UMAP للسكان المحددين من البلعميات/وحيدات النواة، مقسمة ومعاد تصنيفها، ملونة حسب الكتلة (هويات الكتل في (ح)). تمثل خرائط UMAP ظروف العلاج المميزة. النقاط تمثل خلايا فردية.الخلايا من 8 ظروف تجريبية موصوفة في (أ). (ي) UMAP للسكان المعاد تصنيفهم من البلعميات/وحيدات النواة من (م)، ملونة بالأحمر إذا كان hCDتعبير mRNAالنقاط تمثل خلايا فردية.خلايا من 8 ظروف تجريبية، معإجمالي الخلايا المحددة على أنها. تمثل UMAPs ظروف العلاج المميزة الموضحة في (أ). (ك) رسم نقطي يوضح تعبير scRNA-seq لجينات محددة تعرف الكتل داخل تجمعات البلعميات المحددة في (ح). Padj.<0.0001 لكل جين مختار.
الشكل 10 من البيانات الموسعة | اقتران – تعزز علاج CAR T والعلاج المضاد لـ CD47 السيطرة على الورم في نموذج ساركوما العظام العدوانية، حتى عند الجرعات المنخفضة.
( ) (يسار، أ)(يمين، خلايا T من النوعين a و b) مشتقة من متبرعين مستقلين (ممثلة بالألواح a و b) في الدم في اليوم 14 بواسطة تحليل تدفق الخلايا بعد علاج B6H12 في فئران 143B الحاملة للورم، المعالجة بـ – أو -أنتاريس-سيارة-تييعنيمنفئران. (ج)والخلايا في الدم في اليوم 14 واليوم 27 من نمو الورم في الفئران الحاملة لورم 143B، المعالجة بـأنتاريس-CAR-T +B 6 H 12. تمثل نقاط البيانات الفئران الفردية، مع القيم من نفس الفأر متصلة بواسطة خطوط بين نقاط الوقت. (د) تصوير الخلايا التائية قبل علاج B6H12 في الفئران المعالجة كما في (أ). المتوسطانحراف معياري لـالفئران. (هـ) خلايا T BLI بعد علاج B6H12 في اليوم 13 في الفئران المعالجة كما في (أ). المتوسطانحراف معياري لـفئران أو -أنتاريس-سيارة-تيب6H12 عالج بقاء ورم 143B. فئران/ذراع. (ج و ح)أو-أنتاريس-سيارة-تيتم علاج نمو ورم 143B (ج) و(ح) البقاء على قيد الحياة، باستخدام خلايا T المأخوذة من متبرع مختلف عن (ف). المتوسطSEM (ج) أو ممثل (ح) لـفئران. (ط) (يسار) و خلايا T (اليمين) المستمدة من الدم في اليوم الثاني عشر بواسطة تحليل تدفق الخلايا بعد العلاج بـأو.بي بي سي – سيارة – تجرعات منخفضة من أو علاج B6H12 في الفئران الحاملة لورم 143B. المتوسطانحراف معياري لـفئران. (ج)-هير2.بي بي زد-كار-تيب6H12 بجرعة منخفضة أو عالجت نمو ورم 143B، باستخدامالخلايا المشتقة من متبرعين مختلفين (اللوحات اليسرى واليمنى، على التوالي). تم معالجة الفئران بخلايا T وهمية وتمت معالجتها معاً ( ) B6H12 (اللوحة اليسرى) أو B6H12 (اللوحة اليمنى). المتوسط SEM لـالفئران. [(أ)، (ب)، (د)، (هـ)، (ز)، (ي)، (ج)] اختبار ANOVA ثنائي الاتجاه مع اختبار المقارنة المتعددة لتوكاي. غير ذي دلالة = ns. [(ت)، (ح)] اختبار مانتل-كوكس لوج-رانك. غير ذي دلالة = ns. ج
جرعة:ب7هـ3.بي بي سي
بروتين T الخلوي NY-ESO-1
ج
قبل-علاج CD47 (اليوم 9) الجرعة:NY-ESO-1
الشكل البياني الممتد 11 | تعبير عنعلى استهداف الورمتسمح الخلايا بالتزاوج مع علاج مضاد لـ CD47 وتؤدي إلى تحسين السيطرة على الورم. (أ)(يسار) و خلايا T (اليمين) بواسطة تحليل التدفق الخلوي من دم الفئران المعالجة بـ CAR-T في اليوم الخامس عشر في الفئران الحاملة لورم CHLA-255، المعالجة بـ – أو .بي.بي.سي-نلوك-سي.أيه.آر-تييعنيمنالفئران. (ب) تصوير الخلايا التائية باستخدام تقنية BLI في اليوم الرابع عشر في الفئران الحاملة لورم CHLA-255، المعالجة كما في (أ). المتوسطانحراف معياري لـالفئران. (ج) صور لتقدم ورم CHLA-255 (الأربعة صفوف العليا) وخلايا T في اليوم 14 (السفلي) بواسطة BLI، تم علاجها كما في (أ). (د)أو-B7H3.BB7-nLuc-CAR-Tتم علاج نمو ورم CHLA-255 بواسطة B6H12 باستخدام التصوير الضوئي الحيوي. المتوسطSEM لـفئران/ذراع. (هـ)أو-CD19.28-CAR-Tتم علاج نمو ورم نالم6 بواسطة B6H12 باستخدام التصوير الضوئي بالليزر. المتوسطSEM لـفأر/ذراع.أو تم علاج بقاء Nalm6.فئران/ذراع. (ج) خلايا T بواسطة BLI في فئران A375 الحاملة للورم، المعالجة بـخلايا T من نوع أنطارس-TCRقبل (اليوم 9، يسار) وبعد (اليوم 14، يمين)علاج CD 47. المتوسط الانحراف المعياري لـ ن فئران. (ح) hCD4 (يسار) و خلايا T (اليمين) بواسطة تحليل التدفق من الدم في الفئران الحاملة لورم A375، المعالجة كما في (g). المتوسطانحراف معياري لـفئران. (ط)-NY-ESO-1-TCR-Tنمو ورم A375 المعالج بـ B6H12. البيانات هي مسارات نمو الورم الفردية لـفئران/ذراع. (ج)-NY-ESO-1-TCR-T نمو ورم A375 المعالج بـ B6H12، مع خلايا T مشتقة من متبرع مختلف عن الموضح في (i). المتوسط SEM من الفئران/الذراع. [(أ)، (ب)، (د)، (هـ)، (ز)، (ح)، (ط)] اختبار ANOVA ثنائي الاتجاه مع اختبار المقارنة المتعددة لتوكاي. ns = غير دال. [(و)] اختبار لوغ-رانك مانتل-كوكس. ns = غير دال.
natureportfolio
المؤلف(المؤلفون) المراسلون:
كريستال ماكال
آخر تحديث من قبل المؤلف(المؤلفين): 26 مارس 2024
ملخص التقرير
تسعى Nature Portfolio لتحسين قابلية إعادة إنتاج العمل الذي ننشره. يوفر هذا النموذج هيكلًا للاتساق والشفافية في التقرير. لمزيد من المعلومات حول سياسات Nature Portfolio، انظر سياسات التحرير وقائمة مراجعة سياسة التحرير.
الإحصائيات
لجميع التحليلات الإحصائية، تأكد من أن العناصر التالية موجودة في أسطورة الشكل، أسطورة الجدول، النص الرئيسي، أو قسم الطرق.
تم التأكيد
حجم العينة الدقيقة ( ) لكل مجموعة/شرط تجريبي، معطاة كرقم منفصل ووحدة قياس بيان حول ما إذا كانت القياسات مأخوذة من عينات متميزة أو ما إذا كانت نفس العينة قد تم قياسها عدة مرات X
اختبار(اختبارات) الإحصائية المستخدمة وما إذا كانت أحادية أو ثنائية الجانب
يجب وصف الاختبارات الشائعة فقط بالاسم؛ وصف تقنيات أكثر تعقيدًا في قسم الطرق. X
وصف لجميع المتغيرات التي تم اختبارها X
وصف لأي افتراضات أو تصحيحات، مثل اختبارات الطبيعية والتعديل للمقارنات المتعددة X
وصف كامل للمعلمات الإحصائية بما في ذلك الاتجاه المركزي (مثل المتوسطات) أو تقديرات أساسية أخرى (مثل معامل الانحدار) و X
لإجراء اختبار الفرضية الصفرية، إحصائية الاختبار (مثل ) مع فترات الثقة، أحجام التأثير، درجات الحرية و القيمة المذكورة أعطِ القيم كقيم دقيقة كلما كان ذلك مناسبًا. لمعلومات التحليل البايزي، معلومات حول اختيار الأولويات وإعدادات سلسلة ماركوف مونت كارلو للتصاميم الهرمية والمعقدة، تحديد المستوى المناسب للاختبارات والتقارير الكاملة للنتائج تقديرات أحجام التأثير (مثل حجم كوهين ، حجم بيرسون )، مشيرًا إلى كيفية حسابها
تحتوي مجموعتنا على الويب حول الإحصائيات لعلماء الأحياء على مقالات حول العديد من النقاط أعلاه.
البرمجيات والرمز
معلومات السياسة حول توفر كود الكمبيوتر
جمع البيانات
FACSDiva الإصدار 8.0.1 (BD Biosciences): جمع بيانات تحليل الخلايا
برنامج BD Accuri C6 الإصدار 1.0.264.21 (BD Biosciences): جمع بيانات تحليل الخلايا
برنامج IncuCyte S3 الإصدار 2019B Rev2 (Sartorius): جمع بيانات السمية الخلوية
LivingImage الإصدار 4.7.3 (Perkin Elmer): جمع بيانات البيولومينسنس في الجسم الحي
Aura الإصدار 4.0.7 (Spectral Instruments Imaging): جمع بيانات البيولومينسنس في الجسم الحي
برنامج فرز الخلايا SH800S الإصدار 2.1.5 (SONY): جمع بيانات فرز الخلايا
cellSens الإصدار 3.2 (Olympus Life Science): المجهر النسيجي
تحليل البيانات
FlowJo الإصدار 10.8.1 (BD): تحليل بيانات تحليل الخلايا
Excel الإصدار 16.64 (Microsoft): تحليل البيانات الضخمة
Prism الإصدار 9.5.1 (GraphPad): إنشاء الرسوم البيانية والتحليل الإحصائي
SnapGene الإصدار 6.0.2 (Dotmatics): تصميم ناقلات DNA والاستنساخ الجزيئي
Aura الإصدار 4.0.7 (Spectral Instruments Imaging): تقدير بيانات البيولومينسنس في الجسم الحي
Imaris الإصدار 10.0 (Oxford Instruments): المجهر الضوئي التداخلي
PyMol الإصدار 2.5.8 (Schrödinger, LLC): نمذجة بنية البروتين
Aperio ImageScope الإصدار 12.3.2.8013: تحليل درجة إيجابية IHC
CellRanger الإصدار 6.0: فصل ومواءمة قراءات التسلسل إلى جينومات مرجعية مضيفة
Seurat الإصدار 4.2.0: تحديد نوع الخلايا، وتحليلات التعبير الجيني التفاضلي، والتصورات الإصدار 4.2.2: تحليل البيانات الإحصائية وscRNA-seq
بالنسبة للمخطوطات التي تستخدم خوارزميات أو برمجيات مخصصة تكون مركزية للبحث ولكن لم يتم وصفها بعد في الأدبيات المنشورة، يجب أن تكون البرمجيات متاحة للمحررين والمراجعين. نشجع بشدة على إيداع الكود في مستودع مجتمعي (مثل GitHub). انظر إرشادات Nature Portfolio لتقديم الكود والبرمجيات لمزيد من المعلومات.
البيانات
معلومات السياسة حول توفر البيانات
يجب أن تتضمن جميع المخطوطات بيانًا حول توفر البيانات. يجب أن يوفر هذا البيان المعلومات التالية، حيثما ينطبق:
رموز الوصول، معرفات فريدة، أو روابط ويب لمجموعات البيانات المتاحة للجمهور
وصف لأي قيود على توفر البيانات
بالنسبة لمجموعات البيانات السريرية أو بيانات الطرف الثالث، يرجى التأكد من أن البيان يتماشى مع سياستنا
جميع البيانات المرتبطة بهذه الورقة مدرجة في المخطوطة والمواد التكميلية. جميع البيانات الخام متاحة في ملفات البيانات المصدر. تم إيداع مجموعة بيانات scRNAseq في NCBI Gene Expression Omnibus (GEO) ويمكن الوصول إليها من خلال رقم الوصول لسلسلة GEO GSE261475. البيانات المستخدمة لإنشاء مخططات UMAP من بيانات المرضى (الشكل 2د) تم الحصول عليها من مجموعات بيانات متاحة للجمهور باستخدام أرقام الوصول لسلسلة GEO GSE168940 (Good Z. وآخرون، Nature Medicine، 2022) وGSE186802 (Majzner، R.G. وآخرون، Nature، 2022). لنمذجة بنية بلورات البروتين، تم استخدام ملفات PDB المتاحة للجمهور التالية: 2JJS (hCD47-hSIRPa) و5TZU (hCD47 – B6H12).
المشاركون في البحث البشري
معلومات السياسة حول الدراسات التي تشمل المشاركين في البحث البشري والجنس والنوع في البحث.
التقرير عن الجنس والنوع
يمكن العثور على معلومات الجنس للمرضى في المنشورات السابقة حيث تم نشر مجموعات البيانات التي أعيد تحليلها في الأصل: (Good، Z. وآخرون. Nature Medicine، 2022 وMajzner، R.G. وآخرون، Nature، 2022). لم يتم جمع معلومات الجنس. تم عزل خلايا T البشرية من أكياس الدم أو لوكوباك من متبرعين أصحاء مجهولين (ذكور وإناث؛ لم يتم جمع معلومات الجنس) تم شراؤها من مركز ستانفورد للدم أو تقنيات STEMCELL، على التوالي.
خصائص السكان
يمكن العثور على خصائص المرضى في المنشورات السابقة حيث تم نشر مجموعات البيانات التي أعيد تحليلها في الأصل: (Good، Z. وآخرون. Nature Medicine، 2022 وMajzner، R.G. وآخرون، Nature، 2022). تم عزل خلايا T البشرية من أكياس الدم أو لوكوباك من متبرعين أصحاء مجهولين (ذكور وإناث، تحت 45) تم شراؤها من مركز ستانفورد للدم أو تقنيات STEMCELL، على التوالي.
التجنيد
يمكن العثور على معلومات تجنيد المرضى في المنشورات السابقة حيث تم نشر مجموعات البيانات التي أعيد تحليلها في الأصل: (Good، Z. وآخرون. Nature Medicine، 2022 وMajzner، R.G. وآخرون، Nature، 2022). تم الحصول على موافقة خطية مستنيرة من جميع متبرعي خلايا T الأصحاء من مركز ستانفورد للدم وتقنيات STEMCELL.
الإشراف الأخلاقي
تمت الموافقة على كلا الدراستين اللتين أعيد تحليل البيانات منهما (Good، Z. وآخرون. Nature Medicine، 2022 وMajzner، R.G. وآخرون، Nature، 2022) من قبل لجنة الأخلاقيات بجامعة ستانفورد. تم الحصول على الموافقة الأخلاقية المتعلقة بمتبرعي خلايا T من مركز ستانفورد للدم وتقنيات STEMCELL. كانت خلايا الدم المستخدمة معفاة من موافقة لجنة الأخلاقيات حيث لم يتم تقديم معلومات قابلة للتحديد حول المتبرعين بالدم.
لاحظ أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
التقرير الخاص بالمجال
يرجى اختيار الخيار أدناه الذي يناسب بحثك بشكل أفضل. إذا لم تكن متأكدًا، اقرأ الأقسام المناسبة قبل اتخاذ اختيارك.
علوم الحياة العلوم السلوكية والاجتماعية العلوم البيئية والتطورية والإيكولوجية
لنسخة مرجعية من الوثيقة مع جميع الأقسام، انظر nature.com/documents/nr-reporting-summary-flat.pdf
تصميم دراسة علوم الحياة
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبيًا.
حجم العينة لم يتم إجراء حسابات حجم العينة. تم تحديد أحجام المجموعات بناءً على الخبرة مع نماذج مثبتة جيدًا ومنشورة سابقًا
حجم العينة
1. Labanieh، L. وآخرون، السلامة المعززة والفعالية لمستقبلات CAR-T المنظمة بواسطة البروتياز. Cell، 2022. 185(10): ص. 1745-1763.e22.
2. Theruvath، J. وآخرون، Anti-GD2 يتعاون مع حجب CD47 لتحقيق القضاء على الورم. Nature Medicine 2022 28:2، 2022. 28(2): ص. 333-344.
استبعاد البيانات
في التجارب الموصوفة في الشكل 1I,m، توفيت فأرة واحدة في مجموعة استنفاد البلعميات مبكرًا وتم استبعادها من الدراسة. في تحليل IHC والتدفق الموصوف في الشكل 5a والشكل الإضافي 8a-e، تم استبعاد بعض البيانات بسبب عدم وجود ورم قابل للاكتشاف بعد التفكيك، وخاصة في مجموعة المعالجة بـ 47E-CAR.
التكرار
تم استخدام خلايا T متبرعة مختلفة على الأقل في كل تجربة مع خلايا T الأولية (باستثناء تلك المذكورة أدناه) وتم إجراء كل تجربة بشكل مستقل. بالنسبة للتجارب التي تم فيها عرض متبرع واحد تمثيلي، كانت البيانات تمثيلية لجميع المتبرعين. تم إجراء التجارب مع خلايا Jurkat بشكل مستقل على الأقل 3 مرات ومع ماكروفاجات مشتقة من متبرعين مختلفين. تم إجراء التجارب مع الخميرة باستخدام ثلاثة مستنسخات مختلفة وتم تنفيذها بشكل مستقل. كانت جميع محاولات التكرار ناجحة. تم استخدام متبرع واحد فقط لنموذج CHLA-255 النقيلي الموصوف في الشكل التمديدي 11a-d، والدراسة التوافقية 143B الموصوفة في الشكل 5a-d والأشكال التمديدية 8 و9، ونموذج البلعمة في الجسم الحي الموصوف في الشكل التمديدي 7h-I، ونموذج CD19 CAR-Nalm6 بجرعة منخفضة الموصوف في الشكل التمديدي 1، الشكل 1h,i، والأشكال التمديدية 11e,f. تم استخدام متبرع واحد في هذه الحالات بسبب القيود العملية التجريبية (مثل تجارب تسلسل RNA للخلايا المفردة)، وتجارب سابقة مماثلة مع متبرعين مختلفين وجرعات مختلفة، ولكن نتائج مماثلة (مثل تجارب Nalm6)، أو تأكيد التكرار من خلال دراسات تجريبية سابقة أصغر (مثل CHLA-255، البلعمة في الجسم الحي، وتجارب تفكيك ورم 143B).
العشوائية
للتجارب الحية، تم تخصيص أقفاص الفئران التي تم زراعتها مسبقًا بالورم بشكل عشوائي لظروف خلايا CAR T وanti-CD47 للحقن، مما يضمن توزيعًا متساويًا تقريبًا لحجم الورم بين المجموعات قبل العلاج. تم إجراء التجارب باستخدام خلايا T البشرية الأولية باستخدام خلايا T المعزولة من متبرعين أصحاء (تحت سن 45) تم اختيارهم عشوائيًا. تم اختيار سلالات الخميرة المتحولة بشكل عشوائي لكل حالة. بخلاف ذلك، لم تكن جميع التجارب الأخرى عشوائية، بل تضمنت ضوابط لأخذ المتغيرات المشتركة في الاعتبار (مثل خلايا T المعالجة بشكل وهمي المأخوذة من نفس المتبرع مثل خلايا CAR T أو خلايا الخميرة الملطخة بالأجسام المضادة الثانوية فقط).
مُعَمي
تمت زراعة الأورام في الجسم الحي وحقن خلايا T بواسطة فني كان غير مطلع على العلاجات والنتائج المتوقعة. بخلاف ذلك، تم إجراء تحليل البيانات بشكل كامل ومستنير. لم تكن التجارب المزدوجة التعمية ممكنة بسبب توفر الأفراد لاستيعاب مثل هذه الحالات.
التقارير عن مواد وأنظمة وطرق محددة
نحتاج إلى معلومات من المؤلفين حول بعض أنواع المواد والأنظمة التجريبية والأساليب المستخدمة في العديد من الدراسات. هنا، يرجى الإشارة إلى ما إذا كانت كل مادة أو نظام أو طريقة مدرجة ذات صلة بدراستك. إذا لم تكن متأكدًا مما إذا كان عنصر القائمة ينطبق على بحثك، يرجى قراءة القسم المناسب قبل اختيار رد.
المواد والأنظمة التجريبية
طرق
غير متوفر
مشارك في الدراسة
غير متوفر
مشارك في الدراسة
الأجسام المضادة
خطوط خلايا حقيقية النواة X
تدفق الخلايا X
علم الحفريات وعلم الآثار
X الحيوانات وغيرها من الكائنات الحية
الأجسام المضادة
الأجسام المضادة المستخدمة
من أبكام:
جلوبيولين مضاد للدجاج من نوع الماعز أليكسا فلور 647 (متعدد النسائل؛ الكتالوج: NC0928213)
من مختبرات فيكتور: جلوبيولين مضاد للأرانب (متعدد النسائل؛ كتالوج: BP-9100-50)
من إنفيتروجين: مضاد hCD45-PerCP-Cyanine5.5 (نسخة: HI30، كتالوج: 45-0459-42) دجاج مضاد c-myc (مُتعدد النسائل؛ كتالوج: A21281) جلوبيولين مضاد للدجاج – أليكسا فلور 488 (متعدد النسائل؛ كتالوج: A11039) جلوبيولين مضاد للفأر أليكسا فلور 488 (متعدد النسائل؛ كتالوج: A11029) جلوبيولين مضاد للفأر من نوع جوت (ألكسا فلور 647؛ متعدد النسائل؛ كتالوج: A32728) جلوبيولين مضاد للبشر من نوع الماعز أليكسا فلور 488 (وحيد النسيلة؛ كتالوج: A11013) جلوبيولين مضاد للبشر من نوع الماعز أليكسافلور 647 (متعدد النسائل؛ كتالوج: A21445) مضاد CD47-APC (النسخة: B6H12؛ الكتالوج: 17-0479-42) صبغة القابلية للإصلاح eFluor 780 (الكتالوج: 65-0865-14) مضاد hLAG3-PE (نسخة: 3DS223H؛ كتالوج: 12-2239-42) مضاد hPD1-PE-Cy7 (النسخة: J105؛ الكتالوج: 25-2799-41) مجموعة كشف موت الخلايا المبرمج أنكسين V – APC (الرقم التسلسلي: A35110) الأجسام المضادة والبروتينات المخصصة: مستمد من المعهد الوطني للسرطان: CAR مضاد 14G2a (كشف GD2 و HA CARs؛ النسخة 1A7، مرتبطة بـ Dylight 650 باستخدام مجموعة تمييز Dylight 650 من ثيرمو فيشر؛ الكتالوج: 84535) مستخرج من جينسكيب عبر تحضير مخصص: CAR مضاد لـ CD19 (نسخة FMC63، مرتبطة بـ Dylight 650 باستخدام مجموعة تمييز Dylight 650 من ثيرمو ساينتيفيك؛ كتالوج 84535) مستمد من البحث والتطوير: hB7H3-Fc (رقم الكتالوج: 1027-B3-100) و hHer2-Fc (رقم الكتالوج: 1129-ER-050) مستمد من Sino Biological: hSIRPa-mFc (رقم الكتالوج: 11612-H38H) و mSIRPa-hFc (رقم الكتالوج: 50956-M02H) مأخوذ من مختبر كوكرا: Hu5F9 و TJC4 مستمد من ALX Oncology: CV-1-Fc التحقق تم التحقق من البروتينات المنتجة حسب الطلب (anti-14G2a-CAR، anti-CD19-CAR، Hu5F9، TJC4، وCV-1) من خلال ارتباطها بالبروتين المستهدف على خلايا T وعرضها على الخميرة (لربط CD47)، من خلال مقارنة تلوين الأجسام المضادة المحددة مع النوع، والثانوي فقط، والشهادات غير الملونة.
معلومات السياسة حول خطوط الخلايا والجنس والنوع في البحث
مصدر(s) خط الخلايا
المصادقة
تلوث الميكوبلازما
خطوط يتم التعرف عليها بشكل خاطئ بشكل شائع (انظر سجل ICLAC)
تم توفير خط خلايا Nalm6 B-ALL من قبل ديفيد بارنت (مستشفى الأطفال في فيلادلفيا) وتم نقله بواسطة الفيروسات الراجعة للتعبير عن GFP و luciferase اليراع. تم الحصول على خلايا الساركوما العظمية 143B من مجموعة الثقافة الأمريكية (ATCC، ماناساس) ثم تم نقلها بواسطة الفيروسات الراجعة مع CD19 البشري. تم الحصول على خط خلايا CHLA-255 العصبية من روبرت سيجر (مستشفى الأطفال في لوس أنجلوس) وتم نقله بواسطة الفيروسات الراجعة مع GFP و luciferase اليراع. تم توفير MG63.3 من قبل تشاند خانا (المعهد الوطني للسرطان، المعاهد الوطنية للصحة) وتم نقله بواسطة الفيروسات الراجعة مع GFP و luciferase اليراع. تم توفير D425 من قبل س. تشيسير (جامعة ستانفورد، ستانفورد، كاليفورنيا) وتم نقله بواسطة الفيروسات الراجعة للتعبير عن GFP و luciferase اليراع. تم الحصول على Nalm6 و MG63.3 في الأصل من ATCC. تم الحصول على D425 في الأصل من سيغما ألدريش. تم الحصول على خلايا الميلانوما A375 وخلايا Jurkat (النسخة E6-1) من ATCC. تم توفير خط التعبئة الفيروسية الراجعة 293GP من قبل فرع الجراحة (المعهد الوطني للسرطان، المعاهد الوطنية للصحة). تم الحصول على خلايا التعبئة الفيروسية اللينتية 293T من ATCC. تم الحصول على خلايا إنتاج البروتين Expi293 من ATCC.
تم التحقق من خطوط الخلايا المهندسة للتعبير عن البروتينات ذات الاهتمام عبر قياس التدفق الخلوي. تم التحقق من جميع خطوط الخلايا مسبقًا عبر بصمة STR قبل استخدامها في هذه المخطوطة. كما تم التحقق من خطوط الخلايا من قبل البائع التجاري، يمكن العثور على تقنيات التحقق على موقع البائع:https://www.atcc.org/ و https:// www.emdmillipore.com/.
تم اختبار الخلايا بشكل متكرر للكشف عن الميكوبلازما باستخدام مجموعة الكشف عن الميكوبلازما MycoAlert من Lonza. جميع التجارب المبلغ عنها في هذه الدراسة استخدمت خلايا اختبرت سلبية للميكوبلازما.
لم يتم استخدام خطوط خلوية يتم التعرف عليها بشكل خاطئ بشكل شائع.
الحيوانات وغيرها من الكائنات البحثية
معلومات السياسة حول الدراسات التي تشمل الحيوانات؛ توجيهات ARRIVE الموصى بها للإبلاغ عن أبحاث الحيوانات، والجنس والنوع في البحث
الحيوانات المخبرية تم استخدام ذكور وإناث الفئران NOD/SCID/IL2Ry-/- (NSG؛ NOD.Cg-Prkdcscid II2rgtm1WjI/SzJ) في جميع التجارب الحية. كانت الفئران
الحيوانات المخبرية
6-10 أسابيع من العمر في وقت زراعة الورم أو خلايا T. تم إيواء الفئران في و رطوبة مع إضاءة لمدة 12 ساعةدورة الظلام لمدة ساعة.
الحيوانات البرية
لم يتم استخدام حيوانات برية في هذه الدراسة
التقارير عن الجنس
تنطبق النتائج على كلا الجنسين
عينات تم جمعها من الميدان
لم يتم استخدام عينات ميدانية.
رقابة الأخلاقيات
تم إجراء جميع الدراسات الحيوانية وفقًا لبروتوكولات معتمدة من APLAC بجامعة ستانفورد. تم إيواء الحيوانات في مركز خدمات الطب البيطري بجامعة ستانفورد (VSC) في منشأة الحواجز في و رطوبة مع إضاءة لمدة 12 ساعةدورة مظلمة لمدة ساعة. تم مراقبة الفئران يوميًا من قبل موظفي VSC وتم euthanized إذا تم استيفاء معايير النهاية.
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
تدفق الخلايا
المؤامرات
أكد أن:
توضح تسميات المحاور العلامة والفلوكروم المستخدم (مثل CD4-FITC). المقاييس على المحاور مرئية بوضوح. قم بتضمين الأرقام على المحاور فقط للرسم البياني في أسفل اليسار من المجموعة (المجموعة هي تحليل للعلامات المتطابقة). جميع الرسوم البيانية هي رسوم بيانية متساوية الارتفاع مع نقاط شاذة أو رسوم بيانية بالألوان الزائفة. تم توفير قيمة عددية لعدد الخلايا أو النسبة المئوية (مع الإحصائيات).
المنهجية
تحضير العينة
للتلوين السطحي: وفقًا للطرق، تم غسل الخلايا بمحلول FACS (PBS + 2% FBS)، وتم تلوينها بأجسام مضادة مرتبطة بالفلوروفور في محلول FACS (100 ميكرولتر إجمالي حجم التلوين لكل عينة) لمدة 30 دقيقة على الثلج، ثم تم غسلها مرة أخرى بمحلول FACS، ثم تم تحليلها، أو تم تلوينها بجسم مضاد ثانوي (في 100 ميكرولتر إجمالي حجم التلوين) لمدة 30 دقيقة على الثلج، قبل غسلها مرة أخرى، ثم تحليلها.
آلة
بي دي فورتيسا إكس-20، بي دي إل إس آر II، بي دي أكوري، سوني SH800S
برمجيات
برنامج FACSDiva الإصدار 8.0.1 (BD Biosciences)، برنامج BD Accuri (BD Biosciences)، برنامج فرز الخلايا SH800S الإصدار 2.1.5 (SONY)، وبرنامج FlowJo الإصدار 10.8.1 (BD Biosciences)
وفرة تجمع الخلايا
تم تحديد نقاء خلايا الخميرة المفروزة بواسطة قياس التدفق الخلوي. تم زراعة الخلايا المفروزة، وتم تشغيلها عبر قياس التدفق الخلوي لقياس الارتباط. تم تقدير نقاء المجموعات المفروزة عندفي جميع العينات.
استراتيجية البوابة
تم تحديد العينات على اللمفاويات (FSC-A/SSC-A)، الخلايا المفردة (FSC-H/FSC-A)، والعلامات ذات الصلة (CD47، CAR، CD4، CD8، إلخ كما هو محدد في نص المخطوطة الرئيسي). بالنسبة للخلايا التي تم صبغها بصبغة الحياة/الموت، تم أيضًا تحديد الخلايا الحية ضمن مجموعة الاهتمام (تم إجراء صبغة الحياة/الموت باستخدام مجموعة صلاحية قابلة للإصلاح eFluor 780 [كatalog eBioscience #65-0865-14]). تم استخدام التحكم في الفلورسنت بدون واحد (FMO) والتحكم في النوع لتحديد تحديد البوابات عند الضرورة.
تم تحديد كريات QuantiBrite-PE (BD Biosciences؛ الكتالوج 340495) وLegendPLEX Th1 Panel (BioLegend؛ الكتالوج: 741035) وفقًا لتعليمات الشركة المصنعة.
لتحليل الدم في الفئران الحية، تم تصنيف العينات على اللمفاويات والخلايا المفردة كما هو موضح أعلاه، مع تصنيف إضافي على خلايا CD45hi البشرية. تم استخدام كرات العد CountBright للتحقق من الأعداد المطلقة للخلايا.
تم تصنيف الخمائر أولاً بناءً على التشتت الأمامي مقابل التشتت الجانبي (FSC-A مقابل SSC-A)، ثم تم تصنيفها للعينات الفردية باستخدام التشتت الأمامي (FSC-H مقابل FSC-A). أخيرًا، تم تحديد الخمائر المعبرة من خلال صبغة c-myc-tag أو تعبير GFP، حيث تم تعريف السكان الإيجابيين لـ c-myc أو GFP من خلال تشغيل ضوابط غير المحفزة وبدون ثانوية. في جميع الحالات، لوحظ وجود سكان سلبي وإيجابي واضح في الخمائر المحفزة.
قم بتحديد هذا المربع لتأكيد أنه تم تقديم رقم يوضح استراتيجية البوابة في المعلومات التكميلية.
مركز علاج خلايا السرطان، معهد ستانفورد للسرطان، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.معهد باركر لعلاج السرطان المناعي، سان فرانسيسكو، كاليفورنيا، الولايات المتحدة الأمريكية.برنامج بيولوجيا السرطان، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.برنامج الدراسات العليا في علم المناعة، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.ماجستير في البحث الانتقالي والطب التطبيقي، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.وكالة سرطان كولومبيا البريطانية، فانكوفر، كولومبيا البريطانية، كندا.قسم علوم البيانات الطبية الحيوية، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.قسم الطب، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.مركز الأميرة ماكسيما لسرطان الأطفال، أوتريخت، هولندا.قسم الهندسة الحيوية، جامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.معهد بيولوجيا الخلايا الجذعية والطب التجديدي، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.برنامج في الفيزياء الحيوية، جامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.برنامج تدريب العلماء الطبيين، جامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.قسم الأعصاب، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.برنامج علوم الأعصاب، جامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.معهد ستانفورد للسرطان، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.مركز سيلفستر الشامل للسرطان، جامعة ميامي، ميامي، فلوريدا، الولايات المتحدة الأمريكية.قسم علم الأمراض، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.قسم طب الأطفال، كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.مركز لودفيغ لأبحاث وعلاج خلايا السرطان الجذعية، ستانفورد جوهانا ثيروفاث.البريد الإلكتروني: cmackall@stanford.edu
Sean A. Yamada-Hunter , Johanna Theruvath , Brianna J. McIntosh , Katherine A. Freitas , Frank Lin , Molly T. Radosevich¹, Amaury Leruste¹, Shaurya Dhingra’, Naiara Martinez-Velez¹, Peng Xu , Jing Huang , Alberto Delaidelli , Moksha H. Desai , Zinaida Good , Roel Polak , Audre May , Louai Labanieh , Jeremy Bjelajac , Tara Murty , Zach Ehlinger , Christopher W. Mount , Yiyun Chen , Sabine Heitzeneder , Kristopher D. Marjon , Allison Banuelos , Omair Khan , Savannah L. Wasserman , Jay Y. Spiegel , Sebastian Fernandez-Pol , Calvin J. Kuo , Poul H. Sorensen , Michelle Monje , Robbie G. Majzner , Irving L. Weissman , Bita Sahaf , Elena Sotillo , Jennifer R. Cochran Crystal L. Mackall
Abstract
Adoptively transferred T cells and agents designed to block the CD47-SIRP axis are promising cancer therapeutics that activate distinct arms of the immune system . Here we administered anti-CD47 antibodies in combination with adoptively transferred T cells with the goal of enhancing antitumour efficacy but observed abrogated therapeutic benefit due to rapid macrophage-mediated clearance of T cells expressing chimeric antigen receptors (CARs) or engineered T cell receptors. Anti-CD47-antibody-mediated CAR T cell clearance was potent and rapid enough to serve as an effective safety switch. To overcome this challenge, we engineered the CD47 variant CD47(Q31P) (47E), which engages SIRP and provides a ‘don’t eat me’ signal that is not blocked by anti-CD47 antibodies. TCR or CAR T cells expressing are resistant to clearance by macrophages after treatment with anti-CD47 antibodies, and mediate substantial, sustained macrophage recruitment to the tumour microenvironment. Although many of the recruited macrophages manifested an M2-like profile , the combined therapy synergistically enhanced antitumour efficacy. Our study identifies macrophages as major regulators of T cell persistence and illustrates the fundamental challenge of combining T-cell-directed therapeutics with those designed to activate macrophages. It delivers a therapeutic approach that is capable of simultaneously harnessing the antitumour effects of T cells and macrophages, offering enhanced potency against solid tumours.
Myeloid cells are the most plentiful immune cells within the tumour microenvironment (TME) and there has been great interest in therapeutically targeting them for antitumour effects . Increased levels of tumour-associated macrophages (TAMs) associate with poorer outcomes in numerous studies, and some preclinical data demonstrate that reducing or eliminating TAMs enhances responses to chemotherapy and immunotherapy . However, despite dozens of clinical studies testing agents such as CSF1R and CCR2 inhibitors to deplete TAMs and tumour-associated myeloid cells, a clear clinical benefit has not been
demonstrated . Alternatively, increased TAM density is correlated with improved clinical outcomes in some cancers , and augmenting TAM phagocytic activity by blocking the CD47-SIRP axis mediates antitumour effects in several preclinical models . Clinical trials of CD47-SIRP axis blockers demonstrated antitumour activity in some liquid tumours when combined with additional agents, but clinical evidence for single-agent activity or activity in solid cancers is lacking . Thus, despite extensive effort, therapeutic approaches to target TAMs for clinical benefit are lacking.
Fig. 1 | Anti-CD47 antibodies abrogate adoptively transferred T cell efficacy through macrophage-mediated cell depletion. a, osteosarcoma tumour growth after treatment with HER2-BBZ CAR T cells with or without B6H12.b, MG63.3 osteosarcoma tumour growth after treatment with or T cells with or without cells from blood and tumour in the MG63.3 model at day 30. Data are mean s.d. of mice. d, A375 melanoma tumour growth after treatment with NY-ESO-1 TCR cells with or without B6H12.e, T cells in the blood of mice in the A375 model at day 17.f, Nalm6 tumour growth after treatment with CD19-BBZ CAR T cells with or without CV-1 (Fc-dead). g, T cells in the blood of mice in the Nalm6-CV-1 model at day11. Data are mean s.d. of (CD19-BBZ) or (CD19-BBZ+ CV-1) mice.h, Nalm6 leukaemia tumour growth after treatment with CD47 knock out ( ) CD19-28 ( CAR T cells. i, T cell BLI in the Nalm6-47 CAR T cell model at day 11.j, Nalm6 leukaemia tumour growth after treatment with
Anti-CD47 abrogates CAR T and TCR T cell efficacy
To test the hypothesis that augmenting macrophage phagocytosis through CD47 blockade could improve efficacy of CAR T cell therapy,
CD47-overexpressing ( ) CD19-28 亿 CAR T cells. k, T cells on day 45 after CAR treatment in the blood of mice in the Nalm6-47 CAR T cell model. Data are mean s.d. of or (all others) mice.1, T cell BLI after macrophage depletion (left). Data are mean s.d. of (depleted) or (non-depleted) mice. Right, the fold change in T cell BLI, with or without B6H12, after macrophage depletion. Data are mean s.d. of (depleted + PBS) or (all others) mice. , T cell BLI before and after B6H12, after macrophage depletion. For and , data are mean s.e.m. of mice per arm for tumour growth. For eand i, data are mean s.d. of mice. Statistical analysis was performed using two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test (a,b,d,f,h,jandl(right)), unpaired two-tailed Student’s -tests ( (right) and (left)) and two-tailed Mann-Whitney -tests ( , left); NS, not significant.
we administered HER2-BB7 CAR T cells with or without the anti-CD47 monoclonal antibody B6H12 to mice bearing 143B osteosarcoma xenografts. CAR T cells alone induced antitumour effects, but the addition of anti-CD47 antibodies ablated CAR T cell efficacy (Fig. 1a and
Extended Data Fig. 1a). Similar antagonism was observed with MG63.3 osteosarcoma and D425 medulloblastoma (Fig. 1b and Extended Data Fig. 1b-d). To investigate the cause of therapeutic failure with dual treatment, we quantified human T cells in tumour-bearing mice after treatment with cells . Human T cells were completely absent in the tumours and blood of MG63.3-bearing mouse recipients of B7H3-BBZ CAR T cells plus B6H12 but present in mice treated with CAR T cells only and isotype-control-treated animals (Fig. 1c and Extended Data Fig. 1e-g), and similar results were observed in the 143B model (Extended Data Fig. 1h). B6H12 also completely depleted adoptively transferred T cells expressing an NY-ESO-1-targeting TCR in mice bearing A375 melanoma xenografts and ablated their antitumour effects (Fig. 1d, e and Extended Data Fig. li-k).
To characterize the kinetics of CAR T cell depletion in B6H12 recipients, we used bioluminescence imaging (BLI) to monitor CD19-28て CAR T cells expressing nanoluciferase (CD19-28Z-nLuc) in mice bearing Nalm6 leukaemia expressing firefly luciferase (Nalm6-fLuc). The modest single-agent efficacy of B6H12 in this system was not affected by co-administration of CD19-28Z CAR T cells. However, B6H12 completely ablated CD19-28 CAR T cell efficacy and BLI demonstrated a significant loss of CAR T cell signal after B6H12 treatment, with T cells absent from the spleens of mice at the conclusion of the experiment (Extended Data Fig. 11-s). Together, these data demonstrate that anti-CD47 antibodies induce rapid depletion of adoptively transferred T cells, including those engineered to express a transgenic TCR or CARs with differing targeting and costimulatory domains.
CD47 is essential for CAR T cell persistence
To determine whether T cell ablation in these models occurs through FcR-mediated antibody-dependent phagocytosis , we administered CV-1 , a fusion protein that binds to CD47 but does not bind to FcRs. Similar to the results with B6H12, CV-1 co-treatment with CD19 CAR T cells abrogated antitumour efficacy in Nalm6-fLuc-bearing mice, and induced near total T cell depletion (Fig. 1f,g and Extended Data Fig. 2a-f). We next tested whether CD47 expression was required for the survival of adoptively transferred T cells by using CRISPR-Cas9 to knock out CD47 ( ) in primary human T cells, and then restoring CD47 protein expression in cells ( , Extended Data Fig. 2g). CAR T cells expanded in Nalm6-fLuc mice and mediated robust tumour control and significantly prolonged survival, whereas CAR T cells were depleted and delivered no anti-tumour activity (Fig. 1h,i and Extended Data Fig. 2h,i). Even in the absence of tumour, CAR T cells robustly expanded, while CAR T cells were depleted in vivo (Extended Data Fig. 2j-m).
Given the essential role for CD47 expression for T cell persistence in vivo, we wondered whether overexpression of CD47 ( ), which has been reported to prevent immune rejection by allogeneic cells , could enhance CAR T cell persistence and efficacy in NSG mice, in which immune rejection does not occur due to profound immune suppression. Modulation of CD47 expression (either through knock out or overexpression) or addition of anti-CD47 did not alter CAR T cell function in vitro (Extended Data Fig. 3a-1). However, CD19-28て CAR T cells mediated significantly better long-term antitumour efficacy and improved T cell persistence compared with the controls in vivo (Fig. 1j,k and Extended Data Fig. 3m-o). Together, these data demonstrate that the survival of adoptively transferred T cells requires CD47 expression and SIRP engagement and that CD47 overexpression enhances CAR T cell persistence and efficacy, even in the absence of CD47 blocking agents and in the absence of immune rejection .
On the basis of evidence that CD47 blockade enhances macrophage phagocytosis of tumour cells , we examined whether macrophages mediated T cell depletion induced by anti-CD47 by depleting macrophages (Extended Data Fig. 3p,q) and then treating the mice with
CD19-28 7 -nLuc CAR T cells with or without B6H12. The day after adoptive transfer but before B6H12 administration, BLI analysis revealed significantly higher CAR T cell numbers in macrophage-depleted mice, consistent with a model in which macrophages mediate T cell depletion even in the absence of CD47 blockade (Fig. 11,m and Extended Data Fig. 3r).After B6H12 administration, we observed no loss of CAR T cell BLI signal in macrophage-depleted mice, but a significantly reduced CAR T cell BLI signal in mice with an intact macrophage compartment (Fig. 11,m and Extended Data Fig. 3r). Together, these results identify macrophages as barriers to engraftment and antitumour efficacy of adoptively transferred T cells and demonstrate an essential requirement for adequate levels of CD47 on T cells to engage SIRP , even in hosts that are incapable of recognizing allogeneic disparities. They further explain the futility of combining anti-CD47 with adoptive T cell therapy and implicate macrophage-mediated phagocytosis as an important regulator of T cell persistence in vivo.
Human macrophages phagocytose T cells
We next investigated the potential for primary human macrophages to phagocytose primary human T cells in vitro. We observed macrophage phagocytosis of mock-transduced T cells at baseline; however, T cells transduced to express CARs were phagocytosed at significantly higher levels, which was further increased with B6H12 (Fig. 2a, Extended Data Fig. 4a,b and Supplementary Video 1). Macrophage phagocytosis is regulated by a balance of ‘eat me’ signals, such as calreticulin , and ‘don’t eat me’ signals, such as CD47. Flow cytometry revealed that CAR T cells expressed fewer CD47 molecules than the tumour lines used in this study (Extended Data Fig. 4c), consistent with a model in which CAR T cells present limiting ‘don’t eat me’ signals to macrophages. CD47 expression was relatively uniform between CD4 and T cells and among T cell differentiation states; however, CD47 expression decreased over time in culture while calreticulin expression increased during the same period (Fig. 2b and Extended Data Fig. 4d-h), consistent with a model in which aged CAR T cells are more susceptible to phagocytosis.
During the course of these experiments, routine cytologic analysis of cerebrospinal fluid (CSF) collected from a patient treated with axicabtagene ciloleucel (axi-cel), a commercial CD19-28Z CAR T cell therapy, revealed histiocytes engulfing lymphocytes (Fig. 2c), consistent with macrophage-mediated phagocytosis. To address this possibility more systematically, we analysed single-cell RNA-sequencing (scRNA-seq) data collected from two recent clinical studies of axi-cel for large cell lymphoma (LBCL) and GD2-BBZ CAR T cells for diffuse midline glioma (DMG). Both datasets demonstrated CAR mRNA in myeloid cells, consistent with macrophage-mediated phagocytosis of CAR T cells in humans (Fig. 2d and Extended Data Fig. 4i). These data provide further evidence in support of a model in which myeloid cells phagocytose CAR T cells, and may therefore limit durable engraftment of adoptively transferred cells or the survival of activated T cells in clinical settings.
Anti-CD47 can function as a safety switch
We reasoned that anti-CD47-mediated T cell depletion could be harnessed as an off-the-shelf safety switch to mitigate CAR T cell toxicity. To test this, we used a polyspecific integrin-binding peptide (PIP) CAR, which expresses a cystine-knot peptide binding domain that targets integrins expressed on a wide array of malignancies (Extended Data Fig. 5a,b). PIP CAR T cells mediated potent activity in vitro, but osteosarcoma-bearing mice that were treated with PIP CAR T cells demonstrated acute toxicity (Fig. 3a and Extended Data Fig. 5c). Even in non-tumour-bearing mice, treatment with PIP-28, CAR T cells quickly resulted in toxicity (Fig. 3b and Extended Data Fig. 5d-i). By contrast, PIP-28Z CAR T cell recipients treated with B6H12 did not demonstrate
Fig. 2 | Macrophages phagocytose CAR T cells in vitro and in patients. a, Phagocytosis of co-cultured CFSE CAR T cells by primary human macrophages by flow cytometry. Data are mean s.d. of triplicate wells. Reproducible across macrophage donors. , The fold change in CD47 and calreticulin expression on CAR T cells between days 25 and 16 of culture by flow cytometry. Data are mean s.d. of fold change of values (day 25/day 16) derived from donors. MFI, mean fluorescence intensity. c, Microscopy images of Wright-Giemsa-stained histiocytes engulfing lymphocytes collected from the CSF of a patient with LBCL who was treated with CD19-28 (CAR T cells. Representative
of a sample collected from a single patient. Scale bars, , scRNA-seq landscapes, with CAR mRNA shown in red. Left, cells sorted from blood of axi-cel CD19-28 -treated patients with LBCL collected on day 7 after CAR T cell infusion. Right, cells from the CSF of GD2. BB7-treated patients with DMG. Both sampled to 500 cells per patient sample. Statistical analysis was performed using unpaired two-tailed Student’s -tests (a and b); for b, comparison is between expression values of the indicated group on day 16 versus day 25 .
any overt toxicity or weight loss, including no detectable human cytokines in the blood (Fig. 3c-f). Prolonged CAR T cell persistence can lead to graft versus host disease (GvHD) in xenograft models. To determine whether anti-CD47 treatment could durably prevent GvHD, we monitored Nalm6-bearing mice treated with CD19-BB7 CAR T cells with or without B6H12 for the development of GvHD after successful tumour clearance. After 48 days, we observed GvHD in mice treated with CD19-BBZ CAR T cells, evidenced by alopecia and weight loss, whereas mice that were treated with CAR T cells plus B6H12 did not develop GvHD (Extended Data Fig. 5j-1). These data suggest that CD47 blockade could hold promise as an off-the-shelf safety switch to eliminate CAR-T-cell-associated toxicities, as evidenced by rescue in a stringent acute toxicity model as well as a model of chronic CAR T cell toxicity.
Engineered CD47 does not bind to anti-CD47
To induce selective tumour phagocytosis through CD47 blockade while protecting T cells from phagocytosis, we sought to engineer a CD47 variant that ablates anti-CD47 binding but retains SIRP interaction (Extended Data Fig. 6a). We first displayed the CD47 Ig-like domain on the surface of yeast and detected strong binding to B6H12, but not SIRP (Extended Data Fig. ), due to the lack of a free N terminus on CD47 . We therefore used the engineered SIRP variant CV-1 as a proxy for SIRP binding and subjected a library of yeast-displayed CD47 mutant variants to six successive sorts using fluorescence-activated cell sorting (FACS), alternating between negative sorts against B6H12 and positive sorts towards CV-1 (Extended Data Fig. 6f-h). All of the variants in the final sort contained a single A30P (CD47(A30P)) or Q31P (CD47(Q31P)) point mutation (Fig. 4a), which we confirmed, when displayed as individual CD47 variants on yeast with a free CD47 N terminus , manifested no binding to B 6 H 12 but retained similar
or enhanced binding to CV-1 and SIRP (Fig. 4b and Extended Data Fig. 6i-k). These results are consistent with structural understanding of CD47-SIRP interactions, whereby SIRP predominantly contacts CD47 through the CD47 FG loop and N-terminus , forming more minor contacts with the CD47 BC loop, which encompasses Thr26-Gln31 (Fig. 4c and Extended Data Fig. 61,m). As the CD47 BC loop lies near the critical CD47 FG loop, it can serve as an anchoring point for anti-CD47 blocking monoclonal antibodies like , with the Gln 31 residue appearing to be particularly important for antibody binding (Fig. 4c and Extended Data Fig. 6m).
To determine whether the CD47 variants bound to other CD47 blocking monoclonal antibodies, we analysed their binding to TJC4 (lemzoparlimab) and Hu5F9 (magrolimab) in a yeast display assay. An alanine scan of the entire BC loop (Thr26-Gln31) revealed that most mutations allowed for some SIRP binding, with mutations to Ala30 or Gln31 manifesting the most minimal impact (Fig. 4d and Extended Data Fig. 6n). Hu5F9, which has a binding footprint that largely overlaps with SIRP , demonstrated minimal loss of binding to any of the BC loop variants. However, TJC4, which structurally binds to CD47 similarly to B6H12 , no longer bound to CD47(A30P), CD47(Q31P) and CD47(A30P/Q31A) nor to CD47(E29A), an additional variant that did not affect B6H12 binding (Fig. 4d). We next profiled the binding of SIRP , B6H12 and TJC4 to full-length WT CD47, CD47(A30P), CD47(Q31P) and CD47(A30P/Q31A) expressed on primary human T cells. Binding of human and mouse SIRP was largely unaffected by any of the three variants and we detected no B6H12 binding to any of the three variants, while TJC4 binding was completely ablated by the CD47(A30P/ Q31A) double mutant (Fig. 4e and Extended Data Fig. 7a). These data demonstrate that CD47 mutations in the BC loop, and Ala30 and Gln31 specifically, generate proteins that retain SIRP binding but are exempt from binding to multiple anti-CD47 monoclonal antibodies, providing a proof of concept for the ability to engineer ‘don’t eat me’ signalling
Fig. 3 | Anti-CD47 therapy can be used as a safety switch. a, The survival of 143B tumours after treatment with CD19-BB (non-targeting), HER2-BB (targeting), PIP-28Z or PIP-BBZ CAR T cells. mice per arm. b, Human IFN in the blood of mice with or without 143B-CD19 tumours, treated with CD19-BB7, PIP-28 7 or PIP-BB 7 CAR T cells on day 4, as determined using LEGENDPlex quantitative flow cytometry. Data are mean s.d. of (PIP-28 or PIP-BB with 143B-CD19 tumour), (CD19-BB7 with 143B-CD19 tumours), (mock with 143B-CD19 tumour and CD19-BB7, PIP-287 or PIP-BB7 without tumours) and (mock without tumour) mice. c, Mouse weights after PIP-28 CAR T cell treatment with or without B6H12. Data are mean s.d. of mice per arm.
Representative of two independent experiments. d, The survival of mice treated with PIP-287 CAR T cells with or without B6H12.n=5 mice per arm. Representative of two independent experiments. e, T cell BLI after treatment with PIP-287 CAR T cells with or without B6H12. Data are mean s.e.m. of mice per arm. f, IL-2 (left) and IFN (right) in the blood of mice treated with PIP-28? CAR T cells with or without B6H12 on day 4, as determined using LEGENDPlex quantitative flow cytometry. Data are mean s.d. of mice. Statistical analysis was performed using log-rank Mantel-Cox tests (a and d), unpaired two-tailed Student’s -tests (b), two-way ANOVA (c and e) and one-way ANOVA with Tukey’s multiple-comparison test (f).
CD47 variants that will not be blocked by anti-CD47 monoclonal antibodies, which we predicted would drive tumour-specific phagocytosis while sparing T cells in the TME.
prevents anti-CD47-mediated phagocytosis
We next measured phagocytosis of Jurkat cells engineered to express either CD47 WT, CD47(A30P) or CD47(Q31P) by human donor macrophages. CD47 variants expressed on Jurkat cells demonstrated similar binding properties to anti-CD47 monoclonal antibodies and SIRP as observed on primary T cells, with CD47(Q31P) leading to the greatest loss of B6H12 binding (Extended Data Fig. 7b). Across multiple donors, macrophages mediated significantly reduced phagocytosis after B6H12 incubation with either variant, but CD47(A30P) provided less protection compared with CD47(Q31P), which completely prevented additional phagocytosis after incubation with B6H12 (Fig. 4f and Extended Data Fig. 7c). On the basis of this promising profile, we chose to move forward with CD47(Q31P), hereafter (engineered CD47) for further study.
To study the effects of in human cells, we knocked out endogenous CD47, then retrovirally introduced CAR, TCR, and/or or CD47 WT ( ), then measured phagocytosis in vitro and in vivo with or without B6H12. Across multiple T cell and macrophage donors, we observed
that B6H12 treatment did not enhance phagocytosis of CD19-28 CAR T cells in vitro, in contrast to control CD19-28 CAR T cells (Extended Data Fig. 7d). Similarly, while or CD19-28 -nLuc CAR T cells administered to non-tumour-bearing mice manifested similar BLI levels before B6H12 administration, B6H12 depleted CAR T cells but did not affect the levels of CAR T cell (Fig. 4 g and Extended Data Fig. ). These data demonstrate that functions as a ‘don’t eat me’ signal in vitro and in vivo but does not render T cells susceptible to phagocytosis after B6H12-mediated CD47 blockade.
CAR T cells recruit macrophages into tumours
We next quantified macrophage-mediated T cell phagocytosis in the stringent 143B model (Fig. 1a). CFSE-labelled HER2-BBZ CAR T cells were injected intratumourally into established orthotopic tumours, with or without B6H12 treatment. Tumour samples revealed robust macrophage infiltration, and trends of decreased CAR T cells and increased CFSE macrophages after B6H12 treatment, consistent with phagocytosis of CAR T cells in vivo (Extended Data Fig. 7h-1). We next treated 143B-tumour-bearing mice with no T cells, mock, or or HER2-BB7 CAR T cells with or without B6H12 and observed significant increases in mouse macrophages in CAR T cell recipients compared with in the mock or untreated animals (Fig. 5a). B6H12 did not affect
Fig. 4 | An engineered variant of CD47 retains binding to SIRP , but no longer binds to anti-CD47 antibodies. a, Consensus mutations identified in yeast sequenced after sorts 4,5 and 6 . Frequencies of identified mutations out of and 12 sequenced clones for sorts 4,5 and 6 , respectively. b, Normalized binding of B6H12, CV-1, human SIRP and mouse SIRP to yeast-displayed CD47 WT, CD47(A30P) and CD47(Q31P). c, Crystal structures of CD47 (red) binding to SIRP (dark pink, left) and B6H12 (light blue, right); residues Ala30 (gold) and Gln31 (blue) are indicated by boxes. d, Normalized binding of human SIRP and Hu5F9 to yeast-displayed CD47 WT, CD47(A30P), CD47(Q31P), CD47(A30P/Q31A) and CD47(E29A). e, Normalized binding of B6H12, TJC4, human SIRP and mouse SIRP to full-length CD47 WT,
CD47(A30P) and CD47(Q31P) expressed on primary human T cells. Data are mean s.d. of donors, normalized to the fraction binding to CD47WT. f, Phagocytosis of Jurkat cells with endogenous CD47 KO expressing CD47 WT, CD47(A30P) or CD47(Q31P), by primary human macrophages. Data are mean s.d. of triplicate wells ( ). Reproducible across macrophage donors. cells in the blood on day 6 after treatment with CAR T cells with or without B 6 H 12 . Data are mean s.d. of mice. For and , data are mean s.d. of individual yeast clones, normalized to MFI from binding to CD47 WT. Statistical analysis was performed using two-way ANOVA with Tukey’s multiple-comparison test (b and d-g). For d and e, the comparisons are between the indicated groups and binding to cells expressing CD47 WT.
the TME macrophage levels in the recipients of CAR T cells, but significantly reduced the macrophage levels in the recipients of CAR T cells coincident with CAR T cell depletion (Fig. 5a and Extended Data Fig. 8a,b). Confirmed through scRNA-seq analysis, CAR T cell and macrophage tumour infiltration were highly correlated, consistent with a model in which CAR T cells recruit macrophages into the tumour, and macrophage persistence is dependent on CAR T cell persistence in the tumour (Fig. 5b and Extended Data Figs. 8c-f and 9a-c).
scRNA-seq profiling of and CAR T cell recipients demonstrated robust T cell expression of TNF, IFNG, CCL3, CCL4, CCL5, CSF1 (also known as M-SCF) and CSF2 (also known as GM-CSF) (Extended Data Fig. 9d), which collectively attract and activate monocytes and macrophages and have been implicated in T-cell-mediated recruitment of macrophages into tumours . Gene expression in recipients of CAR T cells was essentially unchanged with or without B6H12 therapy, whereas T cells in the TME of B 6 H 12 recipients co-treated with CAR T cells showed 595 differentially expressed genes (DEGs) compared
with those co-treated with CAR T cells (Extended Data Fig. 9e and Supplementary Table 1), including increased proinflammatory genes sets such as IL-12, CD40/CD40L and NF-кB signalling (Fig. 5c). These data provide evidence for substantial cross-talk between myeloid cells and T cells within the CAR T cell TME, which is lacking in the TME of CAR T cell recipients after B6H12-induced T cell depletion. DEG analyses within the major macrophage clusters across treatments showed that treatment with CAR T cells alone induced 621 DEGs in macrophages compared with the untreated condition, and this effect was magnified after CAR T cell co-treatment with B6H12, with 718 DEGs (Extended Data Fig. 9f and Supplementary Table 2). Notably, the effect of B6H12 therapy on macrophage gene expression when administered as a single agent was minimal (46 DEGs), while B6H12 co-administration with CAR T cells substantially reduced macrophage DEGs, probably due to CAR T cell depletion (Extended Data Fig. 9f). By contrast, pathway analysis of genes upregulated by CAR T cells plus B6H12 highlighted macrophage activation indicated
Fig. T cell therapy plus anti-CD47 treatment leads to enhanced antitumour efficacy through recruitment of distinct macrophage
populations.a, Macrophages identified by flow cytometry (flow; left axis) and immunohistochemistry (IHC; right axis) of excised 143B osteosarcoma tumours treated with no T cells, mock, or HER2-BBZ CAR T cells with or without B6H12. Data are the mean of (flow: ) or mean s.d. of (all others) mice. , The composition of cells identified using scRNA-seq from tumours treated as in a. cells from 8 experimental conditions. DC, dendritic cells. c, Enrichr pathway analysis of the top 100 upregulated genes in CAR T cells from tumours treated with 47 EAR T cells + B6H12 versus CAR T cells + B6H12. The results show the value (two-sided Fisher’s exact test) for each pathway. d, The composition of macrophage clusters (c0-c6) after subsetting and reclustering, coloured by cluster, across the experimental
conditions described in cells from 8 experimental conditions. Mac, macrophage; Mo-DC, monocyte-derived dendritic cell; Mo, monocyte. e, T cell BLI in 143B-tumour-bearing mice treated with or Antares HER2-BB7 CAR T cells with or without B6H12 on day 13. Tumours were engrafted in the right leg.f, T cells in the blood of 143B-tumour-bearing mice, treated as described in , on day 14 . Data are mean s.d. of mice per arm. tumour growth after treatment as described in e. Data are mean s.e.m. of mice per arm. h, T cells in the blood of A375-tumour-bearing mice, treated with NY-ESO-1 TCR T cells with or without B6H12, on day 15. Data are mean s.d. of mice per arm. i, A375 melanoma tumour growth treated as in h. Data are mean s.e.m. of mice per arm. Statistical analysis was performed using two-way ANOVA with Tukey’s multiple-comparison test (a,b,f,g and i) and unpaired two-tailed Student’s -tests ( ).
by enrichment of lysosome, complement, antigen presentation and phagosome pathways (Extended Data Fig. 9g).
To further characterize changes in the macrophage compartment induced by CAR T cells, we reclustered the macrophage/monocyte cluster (Extended Data Fig. 9h,i). As observed in clinical data (Fig. 2d), we identified numerous macrophages that contained human CD3E mRNA within multiple CAR-T-cell-treated macrophage subclusters (Extended Data Fig. 9j), consistent with macrophage-mediated phagocytosis of
CAR T cells. We also observed expansion of macrophage cluster c0, which was enriched after CAR T cell treatment and further expanded after treatment with CAR T plus B6H12, but substantially reduced after treatment with CAR T cells plus B6H12 (Fig. 5d), suggesting that these macrophages are dependent on CAR T cell accumulation within the TME. Key DEGs in the expanded cluster, such as Arg1, Mrc1, Chil3 and Tlr1, were associated with M2c-like macrophages (Extended Data Fig. 9k and Supplementary Table 3). While M2 macrophages are
generally understood to be protumorigenic, they have also been demonstrated to manifest strong phagocytic potential, especially those in the M2c subclass .
Together, these results demonstrate a feedforward loop in which CAR T cells drive the recruitment and activation of macrophages within the TME and, simultaneously, macrophages enhance immune-activating pathways in CAR T cells in the TME. These effects did not occur within the CAR T cell TME, in which CD47-blockade-mediated CAR T cell depletion abrogates the cycle. The data further demonstrate robust cross-talk between CAR T cells and macrophages in the TME, resulting in the induction of several proinflammatory gene expression programs that are predicted to enhance antitumour effects.
Anti-CD47 enhances efficacy of /TCR T cells
We next assessed the antitumour effects of versus cells plus anti-CD47 therapy in multiple tumour models. In 143B, in which both CART cell and anti-CD47 therapy have minimal effect as monotherapies (Fig. 1a), we observed marked T-cell-mediated recruitment of macrophages into tumours (Fig. 5a,b). After treatment with B6H12, CAR T cells were completely depleted, while CAR T cells persisted for multiple weeks (Fig.5e,fand Extended Data Fig.10a-e). Neither or CAR T cells alone, nor mock T cells or CAR T cells paired with B6H12, mediated significant antitumour effects, whereas B6H12 plus CAR cells induced a significant delay in tumour growth and an improvement in overall survival (Fig. 5g and Extended Data Fig. 10f-h). Similar results were seen with lower doses of anti-CD47, which may be associated with reduced toxicity in clinical studies (Extended Data Fig. 10i,j).
We also paired B 6 H 12 with versus CART cells in metastatic neuroblastoma and observed enhanced persistence and improved antitumour efficacy with CAR T cells plus B6H12 (Extended Data Fig. 11a-d) and enhanced antitumour efficacy with versus CD19-28 (CAR T cells plus B6H12 in mice bearing Nalm6-fLuc leukaemia (Extended Data Fig. 11e,f). Finally, in the A375 melanoma model, B6H12 depleted NY-ESO-1T cells expressing endogenous CD47 (Fig. 1e), while NY-ESO-1T cells persisted (Fig. 5h and Extended Data Fig. 11g,h). A375 tumour growth was minimally slowed by B6H12 plus mock T cells and treatment with NY-ESO-1 T cells alone led to initial tumour control, but ultimate tumour outgrowth. By contrast, mice treated with NY-ESO-1 T cells and B6H12 demonstrated complete tumour control and cure in all of the mice treated (Fig. Si and Extended Data Fig. 11i, j).
Together, these data demonstrate that CD47 blockade paired with expressed in therapeutic T cells protects T cells from macrophage-mediated phagocytosis and results in a considerable and sustained influx of macrophages within the TME, associated with T cell-macrophage cross-talk. The outcome is a strong antitumour synergy in solid, liquid and metastatic tumours, even at low doses and in conditions in which both single-agent therapies manifest no activity.
Discussion
Previous studies reported that CD47 is necessary to prevent immune rejection and that CD47 overexpression combined with MHC knockout imparts resistance of CAR T cells to allogeneic immune rejection . Our findings demonstrate that, even in the absence of immune rejection, CD47 is required for the survival of adoptively transferred T cells and CD47 overexpression improves CAR T cell persistence and efficacy (Fig. 1j,k). Anti-CD47 treatment in NSG mouse hosts induced rapid and complete macrophage-mediated depletion of adoptively transferred T cells (Fig.1), which was sufficiently rapid and potent to mediate complete protection in a lethal CAR T cell toxicity model (Fig. 3), providing evidence to support testing of CD47-SIRP blockers to mitigate
toxicities resulting from adoptive T cell therapeutics, which could have immediate clinical benefits. These results align with data demonstrating that CD47knockout CAR T cells show limited persistence in xenograft models and observations of reduced total and antigen specific lymphocytes, increased susceptibility to infection and reduced susceptibility to autoimmunity in Cd47 and Sirpa knockout mice . Relevance to the clinical setting is provided by the observation of lymphopaenia in studies of magrolimab and evorpacept, both of which block CD47 engagement of SIRP .
Although T cell depletion was greatly enhanced by CD47 blockade in our studies, our data demonstrate that macrophage phagocytosis limits the persistence of adoptively transferred T cells even in the absence of CD47-SIRP blockade (Fig. 11 and Extended Data Figs. 7h-1 and 9j). Clinical scRNA-seq data also consistently identified CAR transcripts within myeloid cells (Fig. 2d). Together, these findings are consistent with a model in which macrophage phagocytosis has an important role in regulating T cell homeostasis. It is well recognized that CD47 blockade selectively depletes aged red blood cells . We observed increased expression of ‘eat me’ signals and decreased expression of CD47 on T cells after prolonged culture (Fig. 2b and Extended Data Fig. ), raising the prospect that the CD47-SIRP axis may similarly regulate the clearance of aged T cells. Future research is needed to better define the relationship between cell activation and differentiation, and expression of pro-phagocytic and anti-phagocytic receptors, and phagocytosis susceptibility.
We observed that CAR T cells induce a rapid influx of macrophages into tumours (Fig. 5a,b), and that macrophage persistence is dependent on CAR T cell persistence. As a result, CAR T cell recipients manifested extensive cross-talk between myeloid cells and T cells in the TME, including induction of IL-12, CD40L and NF-кB signalling in T cells (Fig. 5c), which have been demonstrated to augment antitumour effects . Thus, protected T cells from anti-CD47-mediated phagocytosis (Fig. 4f,g and Extended Data Fig. 7), while enhancing tumour phagocytosis, antigen presentation and inducing a pro-inflammatory TME. Additional studies are needed to test in fully immunocompetent systems to understand the impact on other immune cell types, including endogenous T and natural killer cells . While B 6 H 12 is a research-grade antibody, the CD47 variants generated in this study also ablated binding to the clinical grade antibody TJC4 (lemzoparli (Fig. 4d,e), leading us to predict that could allow pairing of adoptive therapies with clinical grade anti-CD47 antibodies. Further enhancements could be developed for this approach , including pairing with additional antibodies and inducing cell secretion of CD47-SIRP blockers .
TAMs are among the most plentiful cells in the TME, and there has been great interest in harnessing their antitumour properties, but effective macrophage-modulating therapeutics for cancer are lack. Neither CSF1R and CCR2 inhibitors, which inhibit recruitment of monocytes to the TME and reduce or eliminate TAMs , nor approaches to modulate macrophage states from those with immunosuppressive profiles, including the M2-like subset, toward those with more inflammatory profile have mediated clear therapeutic benefit . Similarly, although systemic blockade of the CD47-SIRP axis mediated antitumour effects in several preclinical models , clinical benefit as single agents and in solid cancers is lacking . The data presented here suggest that this conundrum may be explained by the double-edged sword that TAMs represent within the TME, whereby antitumour effects of manoeuvres designed to augment macrophage phagocytosis are offset by phagocytosis of tumour reactive T cells. Conversely, eliminating or reducing TAMs may diminish immunosuppression and phagocytosis of tumour infiltrating cells, but these benefits are offset by loss of tumour phagocytosis by macrophages. Our data demonstrate that pairing CAR therapy with anti-CD47 is an exciting prospect that could enhance the potency of adoptive cell therapies, especially in solid cancers.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-024-07443-8.
Labanieh, L. & Mackall, C. L. CAR immune cells: design principles, resistance and the next generation. Nature 614, 635-648 (2023).
Kloosterman, D. J. & Akkari, L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell 186, 1627-1651 (2023).
Zizzo, G., Hilliard, B. A., Monestier, M. & Cohen, P. L. Efficient clearance of early apoptotic cells by human macrophages requires M 2 c polarization and MerTK induction. J. Immunol. 189, 3508-3520 (2012).
Zhu, Y. et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057-5069 (2014).
Noel, M. et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest. N. Drugs 38, 800-811 (2020).
Osorio, J. C., Smith, P., Knorr, D. A. & Ravetch, J. V. The antitumor activities of anti-CD47 antibodies require Fc-FcүR interactions. Cancer Cell https://doi.org/10.1016/j. ccell.2023.10.007 (2023).
Chao, M. P. et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2, 63ra94 (2010).
Weiskopf, K. et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Invest. 126, 2610-2620 (2016).
Weiskopf, K. et al. Engineered SIRPa variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88-91 (2013).
Lakhani, N. J. et al. Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): a first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 22, 1740-1751 (2021).
Jiang, Z., Sun, H., Yu, J., Tian, W. & Song, Y. Targeting CD47 for cancer immunotherapy. J. Hematol. Oncol. 14, 180 (2021).
Sikic, B. I. et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 37, 946-946 (2019).
Hu, X. et al. Hypoimmune anti-CD19 chimeric antigen receptor T cells provide lasting tumor control in fully immunocompetent allogeneic humanized mice. Nat. Commun. 14, 2020 (2023).
Good, Z. et al. Post-infusion CAR cells identify patients resistant to CD19-CAR therapy. Nat. Med. 28, 1860-1871 (2022).
Majzner, R. G. et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 603, 934-941 (2022).
Miller, C. L. et al. Systemic delivery of a targeted synthetic immunostimulant transforms the immune landscape for effective tumor regression. Cell Chem. Biol. 29, 451-462 (2022).
Ho, C. C. M. et al. “Velcro” engineering of high affinity CD47 ectodomain as signal regulatory protein a (SIRPa) antagonists that enhance antibody-dependent cellular phagocytosis. J. Biol. Chem. 290, 12650-12663 (2015).
Hatherley, D. et al. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol. Cell 31, 266-277 (2008).
Pietsch, E. C. et al. Anti-leukemic activity and tolerability of anti-human CD47 monoclonal antibodies. Blood Cancer J. 7, e536 (2017).
Meng, Z., Wang, Z., Guo, B., Cao, W. & Shen, H. TJC4, a differentiated anti-CD47 antibody with novel epitope and RBC sparing properties. Blood 134, 4063-4063 (2019).
Kersten, K. et al. Spatiotemporal co-dependency between macrophages and exhausted T cells in cancer. Cancer Cell 40, 624-638 (2022).
Blazar, B. R. et al. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J. Exp. Med. 194, 541-549 (2001).
Beckett, A. N. et al. CD47 expression is critical for CAR T-cell survival in vivo. J. Immunother. Cancer 11, e005857 (2023).
Li, L. X., Atif, S. M., Schmiel, S. E., Lee, S. J. & McSorley, S. J. Increased susceptibility to Salmonella infection in signal regulatory protein a-deficient mice. J. Immunol. 189, 2537-2544 (2012).
Autio, A. et al. SIRPa-CD47 axis regulates dendritic cell-T cell interactions and TCR activation during T cell priming in spleen. PLoS ONE 17, e0266566 (2022).
Deuse, T. et al. The SIRPa-CD47 immune checkpoint in NK cells. J. Exp. Med. 218, e20200839 (2021).
Dacek, M. M. et al. Potentiating antibody-dependent killing of cancers with CAR T cells secreting CD47-SIRPa checkpoint blocker. Blood 141, 2003-2015 (2023).
Chen, H. et al. Delivery of CD47 blocker SIRPa-Fc by CAR-T cells enhances antitumor efficacy. J. Immunother. Cancer 10, e003737 (2022).
Hong, D. S. et al. Eganelisib, a first-in-class PI3Ky inhibitor, in patients with advanced solid tumors: results of the phase 1/1b MARIO-1 trial. Clin. Cancer Res. 29, 2210-2219 (2023).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
(c) The Author(s) 2024
Methods
Cell lines
The Nalm6 B-ALL cell line was provided by D. Barrett and retrovirally transduced to express GFP and firefly luciferase. 143B osteosarcoma cells were acquired from the American Type Culture Collection (ATCC) and then retrovirally transduced with human CD19. The CHLA-255 neuroblastoma line was obtained and provided by R. Seeger and retrovirally transduced with GFP and firefly luciferase. MG63.3 cells were provided by C. Khanna and retrovirally transduced with GFP and firefly luciferase. D425 cells were provided by S. Chesier and retrovirally transduced to express GFP and firefly luciferase. Nalm6 and MG63.3 cells were originally obtained from ATCC. D425 cells were originally obtained from Sigma-Aldrich. A375 melanoma cells and Jurkat cells (clone E6-1) were obtained from ATCC. The 293GP retroviral packaging line was provided by the Surgery Branch (National Cancer Institute, National Institutes of Health). HEK293T lentiviral packaging cells were obtained from ATCC. Expi293 protein production cells were obtained from ATCC.
D425 cells were maintained in serum-free medium supplemented with B27 (Thermo Fisher Scientific), EGF, FGF (Shenandoah Biotechnology), human recombinant LIF (Millipore) and heparin (StemCell Technologies). Nalm6, 143B, A375, MG63.3, CHLA-255 and Jurkat cells were cultured in RPMI-1640 (Gibco). 293GP and HEK293T cells were cultured in DMEM (Gibco). Expi293 cells were cultured in Expi293 medium (Thermo Fisher Scientific). Cell line culture medium was supplemented with FBS, 10 mM HEPES, 2 mM l -glutamine, penicillin and streptomycin (Gibco), except for the Expi293 medium. Short tandem repeat DNA profiling of all cell lines was conducted once per year (Genetica Cell Line testing). All cell lines were routinely tested for mycoplasma. Cell lines were cultured at in a environment.
Source of primary human T cells and macrophages
Buffy coats from healthy donors were purchased from the Stanford Blood Center under an IRB-exempt-protocol. Leukopaks from healthy donors were purchased from StemCell Technologies. Primary human T cells were purified by negative selection using the RosetteSep Human T cell Enrichment kit (StemCell Technologies) and SepMate-50 tubes. T cells were cryopreserved at cells per ml in CryoStor CS10 cryopreservation medium (StemCell Technologies) until use. Primary peripheral monocytes were purified through successive density gradients using Ficoll (Sigma-Aldrich) and Percoll (GE Healthcare). Monocytes were then differentiated into macrophages by 7-9 days of culture in IMDM AB human serum (Life Technologies).
Viral vector construction
All DNA constructs were visualized using SnapGene software(v.6.0.2; Dotmatics). All retroviral constructs were cloned into the MSGV1 retroviral vector . The following CAR and TCR constructs used in this study were previously described: , HER2-BB and NY-ESO- . B7H3-BB7 was previously generated by fusing, from N to C terminus, a human GM-CSF leader sequence, scFv derived from MGA271 in the VH-VL orientation and (GGGS) linker sequence, CD8 hinge and transmembrane sequence, and human 4-1BB and CD3, intracellular signalling domains. GD2-BB7,HER2-BB7 and CD19-BB7 were generated previously by cloning scFvs derived from 14G2A, 4D5 and FMC63 antibodies, respectively, into the B7H3-BB7 vector in place of the MGA271 scFv. CD19-28 7 was generated previously by replacing the 4-1BB domain in CD19-BB7 with the intracellular signalling domain of human CD28. HA-287 was generated previously by replacing the FMC63 scFv with the 14G2a scFv containing a point mutation (E101K) followed by a spacer region derived from the CH 2 CH 3 domains of IgG1. PIP-28 and PIP-BB 3 were generated by replacing the FMC63 scFv with the 2.5 F knottin followed
by a Flag-tag sequence (DYKDDDDK) in the CD19-28 and CD19-BB 7 vectors, respectively. The in vivo T cell activation reporter was constructed by cloning a sequence containing firefly luciferase into the pGreenFire1-NF-кB lentiviral vector (System Biosciences) under the NF-кB responsive promoter . CD47 vectors were generated by inserting codon-optimized CD47 sequences (variant and WT) in place of the CD19-BB7 sequence. For in vivo tracking, CAR-nLuc plasmids were generated by replacing the stop codon in the CD37 with a sequence containing a porcine teschovirus-12A (P2A) ribosomal skipping sequence, followed by nanoluciferase . Antares plasmids were generated by inserting the Antares sequence in place of the CD19-BB7 sequence. The NY-ESO-1 TCR construct was generated by inserting the NY-ESO-1 chain, followed by a P2A sequence, followed by the chain in place of CD19-BB7.
Virus production
Retroviral supernatant was packaged using 293GP cells and the RD114 envelope plasmid. In brief, RD114 and of the corresponding MSGV1 transfer plasmid were delivered to 293GP cells grown on 150 mm poly-D-lysine dishes (Corning) to 80% confluency by transient transfection with Lipofectamine 2000 (Thermo Fisher Scientific). The medium was replenished every 24 h . Virus production was performed side by side for comparable CAR, TCR and CD47 constructs. The retroviral supernatant was collected 48 and 72 h after transfection. The supernatants from replicate dishes were pooled, centrifuged to deplete cell debris and stored at until use. Third-generation, self-inactivating lentiviral supernatant was similarly produced with HEK293T cells using pMD2.G (VSVg) envelope, pMDLg/pRRE ( ), pRSV-Rev and of the corresponding transfer plasmids. All of the constructs were retroviral, except for the T cell activation construct, which was lentiviral.
CAR T and TCR T cell manufacturing
At day 0 , primary human cells were thawed and activated with anti-CD3/CD28 human T-Expander Dynabeads (Thermo Fisher Scientific) at a or bead to cell ratio. On day 2 , virus-coated culture plates were prepared on non-tissue-culture-treated 12-well plates that had been precoated with RetroNectin (Takara Bio) according to the manufacturer’s instructions, by incubating with 1 ml of retroviral supernatant ( ) and centrifugation at and for 2 h . The supernatant was subsequently aspirated off of the wells and cells were added in 1 ml of T cell medium comprising AIM V (Thermo Fisher Scientific), FBS, penicillin (Gibco), streptomycin (Gibco), 2 mM l-glutamine (Gibco), 10 mM HEPES (Gibco) and rhIL-2 (Peprotech). After addition of the T cells, the plates were gently centrifuged at for 2 min then incubated for 24 h at under . This transduction process was repeated on day 3 and day 4 (if necessary). Dynabeads were removed on day 4 or day 5 by magnetic separation. Cells were maintained between and cells per ml and expanded until day . Typically, T cells were transduced with CAR or TCR and Antares (if used) on day 2 , and then CD47 variants on days 3 and 4.
CRISPR-Cas9 KO of CD47 and AAVS1
Ribonucleoprotein (RNP) was prepared using synthetic sgRNA with 2′-O-methyl phosphorothioate modification (Synthego) diluted in TE buffer at . A total of sgRNA was incubated with duplex buffer (IDT) and Alt-R Streptococcus pyogenes Cas 9 Nuclease V3 (IDT) for 30 min at room temperature. Reactions ( ) were assembled with 5 million T cells or Jurkat cells, P3 buffer (Lonza) and RNP. Cells were pulsed with protocol EO115 using the P3 Primary Cell 4D-Nucleofector Kit and 4D-Nucleofector System (Lonza). Cells were recovered immediately with warm medium for 6 h before transduction with CAR or TCR. Cells were electroporated with RNP on
day 2 after thaw and transduced later the same day. Guide sequences were as follows: CD47, 5′-AUGCUUUGUUACUAAUAUGG-3′; AAVS1, -GGGGCCACUAGGGACAGGAU- .
Flow cytometry analysis of mammalian cells
Cells were washed with FACS buffer (2% FBS in PBS) before staining. Staining was performed in FACS buffer for 30 min at . In certain experiments, cells were first stained with Fixable Viability Dye eFluor 780 (eBioscience, ) in PBS for 10 min at room temperature before washing with FACS buffer and staining with other antibodies. After staining, cells were then washed once with FACS buffer and analysed on the BD Fortessa system. FACSDiva (v.8.0.1; BD) software was used for data collection and FlowJo software (v.10.8.1; BD) was used for data analysis (gating strategies are shown in Supplementary Fig. 2).
Recombinant B7H3-Fc and HER2-Fc (both R&D systems, 1:400 dilution) were used to detect B7H3 and HER2 surface CAR, respectively. Likewise, anti-FMC63 idiotype antibody (Genscript, 1:400) was used to detect CD19 CARs, while anti-14G2a idiotype antibody (National Cancer Institute, 1:400) was used to detect GD2 and HA CARs. CAR detection reagents were fluorescently labelled using the DyLight 650 Microscale Antibody Labelling Kit (Thermo Fisher Scientific). Anti-DYKDDDDK tag (Flag tag, APC, L5, BioLegend, 1:400) was used to detect the PIP CAR. NY-ESO-1 TCR was detected with antibodies specific for V (APC, H131, BioLegend, 1:100), the beta chain of the NY-ESO-1 TCR. CD47 was detected with B6H12 (BV711 and PE, B6H12, BD, 1:100; APC, B6H12, Invitrogen, 1:100; or unlabelled, Bio X Cell, concentrations are listed in the figures), TJC4 (unlabelled, produced in-house, concentrations are listed in the figures), (unlabelled, produced in-house, concentrations are listed in the figures), CV-1-Fc (unlabelled, ALX Oncology, concentrations are listed in the figures), (unlabelled, Sino Biological, concentrations are listed in the figures) or hSIRP -Fc (unlabelled, Sino Biological, concentrations are listed in the figures), followed by detection with polyclonal anti-mouse or anti-human IgG antibodies (AF488 and AF647, polyclonal, Invitrogen, 1:500). mIgG1 isotype control antibodies (unlabelled, MPOC-21, Bio X Cell, 1:100 and PE, B11/6, Abcam, 1:100) were used as controls for B6H12 staining. The following antibodies were used for detection of cell surface proteins: calreticulin (PE, FMC 75, Abcam, 1:100); human CD4 (BUV 395, SK3, BD, 1:200); human CD8 (BUV 805, SK1, BD, 1:400); human CD45 (Per-CP-Cy5.5, HI30, Invitrogen, 1:50); human CD69 (BV421, FN50, BioLegend, 1:100); human CD39 (BV605, A1, BioLegend, 1:100); human TIM3 (BV510, F38-2E2, BioLegend, 1:100); human LAG3 (PE, 3DS223H, Invitrogen, 1:100); human PD1 (PE-Cy7, J105, Invitrogen, 1:100); human CD45RA (BV785, HI100, BioLegend, 1:100); human CD62L (BV605, DREG-56, BD, 1:100); human CD3 (BUV 737, SK7, BD, 1:100); mouse CD45 (BUV 805, I3/2.3, BD, 1:100); F4/80 (BV605, BM8, BioLegend, 1:100); CD11b (APC, M1/70, BioLegend, 1:50 and BUV 395, M1/70, BD, 1:100); human CD19 (BUV 496, SJ25C1, BD, 1:100). Annexin V was detected using the eBioscience Annexin V Apoptosis Detection Kit (Invitrogen) according to the manufacturer’s instructions.
BLI analysis
Mice were administered of D-luciferin for firefly luciferase imaging or a 1:40 dilution of Nano-Glo substrate (Promega, diluted in DPBS) for Antares and nanoluciferase imaging by intraperitoneal injection. Images were acquired on the IVIS (Perkin Elmer) or Lago (Spectral Instruments Imaging) imaging system 4 min after injection for fluc and 8 min after injection for nLuc/Antares using 30 s exposures and medium binning. If saturated pixels were detected in the image, an additional image was acquired using the auto-expose setting. Total flux was measured using Living Image (v.4.7.3; Perkin Elmer) or Aura (v.4.0.7; Spectral Instruments Imaging) software with a region of interest around the body of each mouse. Only non-saturated images were used for quantification of BLI. Mice were randomized before T cell administration
to ensure uniform distribution of tumour burden between groups. At the end of the experiment, all of the images were collected into a single sequence on Aura and set to the same luminescence scale.
Recombinant protein cloning and production
The gWIZ vector with a BM40 signal peptide was used for protein expression. DNA encoding the Hu5F9 (magrolimab ) heavy chain with an hIgG1 Fc domain, Hu5F9 light chain, TJC4 (lemzoparlimab ) heavy chain with an hIgG1 Fc domain and TJC4 light chain were ordered from IDT. Heavy and light chains were individually cloned into an AscI/ BamHI-digested gWIZ vector using Gibson assembly. Plasmids were transfected into Expi293F cells (Thermo Fisher Scientific) at a 1:1 ratio of heavy chain:light chain using ExpiFectamine according to the manufacturer’s instructions. Then, 5 days after transfection, the supernatant was collected, adjusted to pH 8.0 and sterile-filtered. Hu 5 F 9 and TJC4 were then purified using recombinant Protein A-Sepharose 4 B (Thermo Fisher Scientific) buffer-exchanged into PBS and concentrated using Amicon Centrifugal Filters (Millipore Sigma). To assess CD47 binding, cells were stained with Hu5F9 or TJC4 and then stained with labelled anti-human secondary antibodies (AF488 or AF647, Invitrogen, 1:500). and mIgG1 isotype control (MOPC-21) were acquired from Bio X Cell. CV-1 variants (ALX-222, CV-1-hIgG1 Fc; and ALX-90, CV-1-hIgG1 dead Fc) were acquired from ALX Oncology. Human SIRP and mouse SIRP were acquired from Sino Biologic.
Animal models
NSG mice (NOD.Cg-Prkdc Il2rgt SzJ ) were purchased from the Jackson Laboratory and bred in-house under Stanford University APLAC-approved protocols. Healthy male and female mice that were used for in vivo experiments were aged between 6 and 10 weeks at tumour or T cell engraftment and were drug naive, and not involved in previous procedures. The mice were housed in sterile cages in a barrier facility at Stanford University at and humidity under a light-dark cycle. Veterinary Services Center (VSC) staff at Stanford University monitored the mice daily. Mice were euthanized when they manifested persistent hunched posture, persistent scruffy coat, paralysis, impaired mobility, greater than weight loss, if tumours significantly interfered with normal bodily functions or if they exceeded limits designated in APLAC-approved protocols of 1.70 cm in any direction. According to the recommendations of VSC staff, mice with morbidities were supported with subcutaneous saline, diet gel (DietGel 76A, ClearH2O) and wet chow. For all experiments, no sample size calculations were performed, but group sizes were determined by experience with well-established, previously published models . Cages of mice that were previously engrafted with tumour were randomly assigned CAR T cell and anti-CD47 conditions for infusion, ensuring approximately equal distributions of tumour size between groups before treatment. Tumour engraftments and T cell infusions were performed by a technician who was blinded to treatments and expected outcomes.
143B osteosarcoma tumour model
or or 143B-CD19 cells (143B cells engineered to over-express CD19; 143B cells do not naturally express CD19) in DPBS were injected into the tibial periosteum of six- to ten-week-old NSG male or female mice (engraftment dose indicated below for each specific study) . Generally, 5 days after tumour implantation and after visual confirmation of tumour formation, mice were treated with HER2-BB7 CAR T cells, followed by two doses of B6H12. Tumour progression was monitored by measurement using callipers. Mice were euthanized according to the criteria described in the ‘Animal models’ section. Specifics for different iterations of the model presented are as follows:
CAR T cell + B6H12 studies (Fig. 1a, Supplementary Fig. 1a and Extended Data Fig. 1a,h): mice engrafted with -CD19 cells
were treated with -BB 7 CAR T cells by tail-vein injection on day 5. Mice were then treated twice with B6H12 ( ) or PBS by intraperitoneal injection on day 6 and day 10 . T cells were quantified in the blood by flow cytometry on day 12.
PIP CAR T cell survival study (Fig. 3a): mice engrafted with 143B were treated with CD19-BB7 (non-tumour-targeting control), HER2-BB (tumour targeting control), PIP-28 or PIP-BB CAR T cells on day 5 by tail-vein injection.
PIP CAR T cell serum cytokine study (Fig. 3b): non-tumour-bearing mice or mice engrafted with -CD19 were treated with CD19-BB (tumour-specific control), PIP-28Z or PIP-BBZ CAR T cells, or mock T cells by tail-vein injection on day 4. Blood was collected for serum cytokine analysis on day 8 ( 4 days after CAR T cell administration). CAR T cell studies with high-dose B6H12 (Fig. 5e-g, Supplementary Fig. 1p and Extended Data Fig. 10a-h): mice engrafted with 143B-CD19 cells were treated with HER2-BBZ Antares CAR T cells with endogenous CD47KO and overexpressing either CD47WT ( ) or CD47(Q31P) ( ), or an equivalent number of mock-Antares T cells intravenously by tail-vein injection on day 5. Mice were then treated twice with B6H12 ( ) or PBS by intraperitoneal injection on day 7 and day 11. T cells were quantified by nanoluciferase BLI before (day 7) and after (day 13) anti-CD47 treatment and in the blood by flow cytometry on day 14. CAR T cell studies with low-dose B6H12 (Supplementary Fig. 1q and Extended Data Fig. 10i, j): mice engrafted with -CD19 cells were treated with HER2-BB7 CAR T cells with endogenous CD47KO and overexpressing either or , or an equivalent number of mock T cells intravenously by tail-vein injection on day 5 . The mice were then treated twice with B6H12 ( or ) or PBS by intraperitoneal injection on day 6 and day 10 . T cells were quantified in the blood by flow cytometry on day 12 . Only those mice that were treated with CAR T cells were evaluated for antitumour efficacy in combination with B6H12.
A375 melanoma tumour model
A total of A375 cells in I DPBS was injected into the flanks of NSG male or female mice aged weeks . Generally, 7 to 14 days after tumour implantation and after visual confirmation of tumour formation, mice were treated with NY-ESO-1 TCR T cells, followed by two or three doses of B6H12. Tumour progression was monitored by measurement using callipers. Mice were euthanized according to the criteria described in the Animal Models section. Specifics for different iterations of the model presented are as follows:
Low-dose NY-ESO-1TCR T cell + B6H12 studies (Fig. 1d,e, Supplementary Fig. 1d and Extended Data Fig. 1i): mice were treated with NY-ESO-1TCR T cells or an equivalent number of mock T cells intravenously by tail-vein injection on day 9 after tumour implantation. Mice were then treated twice with B6H12 ( ) or PBS by intraperitoneal injection on day 10 and 15 . T cells were quantified in the blood by flow cytometry on day 17.
High-dose NY-ESO-1 TCR T cell + B6H12 studies (Supplementary Fig. 1d and Extended Data Fig. 1j,k): mice were treated with NY-ESO-1TCRT cells or an equivalent number of mock T cells intravenously by tail-vein injection on day 7 after tumour implantation. Mice were then treated twice with B6H12 ( ) or PBS by intraperitoneal injection on day 9 and 13. T cells were quantified in the blood by flow cytometry on day 16. NY-ESO-1 TCR T cell quantification studies (Fig. 5h, Supplementary Fig. 1t and Extended Data Fig. 11g,h):7 days after tumour implantation, mice were treated with NY-ESO-1-Antares TCR T cells with endogenous and overexpressing , or an equivalent number of mock-Antares T cells intravenously by tail-vein injection. Mice were then treated three times with B6H12 ( ) or PBS by intraperitoneal injection on days 9,11 and 14 . T cells were quantified by nanoluciferase
BLI before (day 9) and after (day 14) anti-CD47 treatment and in the blood by flow cytometry on day 15. NY-ESO-1 TCR T cell antitumour efficacy studies (Fig. Si, Supplementary Fig. 1u and Extended Data Fig. 11i, j): 7 days ( T cell donor experiment 1; Extended Data Fig. 11j) or 14 days (T cell donor experiment 2; Fig. 5i and Extended Data Fig. 11i) after tumour implantation, mice were treated with NY-ESO-1-Antares TCR T cells with endogenous CD47KO and overexpressing 47E, or an equivalent number of mock-Antares T cells intravenously by tail-vein injection. Mice were then treated either: three times with B6H12 ( ) or PBS by intraperitoneal injection on days 9, 11 and 14 (experiment 1); or twice with B6H12 ( ) or PBS by intraperitoneal injection on days 15 and 19 (experiment 2).
MG63.3 osteosarcoma tumour model
A total of MG63.3 cells in DPBS was injected into the tibia periostea of NSG male or female mice aged weeks . Starting 15 days after tumour implantation and after visual confirmation of tumour formation, the mice were treated with of B6H12 or PBS three times per week by intraperitoneal injection. On day 21, the mice were treated intravenously with or B7H3-BB7 CAR T cells or no T cells. Tumour progression was measured using digital callipers twice per week. Mice were euthanized according to the criteria described in the ‘Animal models’ section (Supplementary Fig. 1b). For T cell quantification experiments, mice engrafted orthotopically with cells were treated intravenously with B7H3-BB7 CAR T cells on day 15 with or without 3 doses of B6H12 treatment ( per dose; intraperitoneal). Blood and tumours were collected on day 30 after tumour engraftment.
D425 medulloblastoma tumour model
Mice (aged 6-10 weeks) were anaesthetized with isoflurane (Minrad International) in an induction chamber . Anaesthesia on a stereotactic frame (David Kopf Instruments) was maintained at isoflurane delivered through a nose adaptor. D425 medulloblastoma cells were injected at coordinates 2 mm posterior to lambda on midline and 2 mm deep using a blunt-ended needle ( s gauge/ 2 inch/point style ; Hamilton). Using a microinjection pump (UMP-3; World Precision Instruments), D425-GFP-fLuc cells were injected in a volume of at . After leaving the needle in place for 1 min , it was retracted at . Then, 4 days after tumour implantation and after confirmation of tumour formation by bioluminescence, mice were randomized and treated with no T cells (B6H12 only group), or C T cells or an equivalent number of non-tumour targeting CD19-BBZ CAR T cells intravenously by tail-vein injection. Starting on day 4, the mice were also treated with of B6H12 or PBS three times per week by intraperitoneal injection. Tumour progression was monitored by firefly luciferase BLI (Supplementary Fig. 1c). In Extended Data Fig. 1c,d, CD19-BBZ and B7H3-BBZ treatments are reproductions of previously published data , included for comparison with , as these arms were all run simultaneously in the same experiment.
Nalm6 leukaemia tumour models
A total of Nalm6-GFP-fLuc cells in DPBS was implanted by tail-vein injection into NSG male or female mice aged weeks . Generally, four days after tumour implantation and after confirmation of tumour formation by BLI, mice were treated with CD19-BB7 or CD19-287 CAR T cells, followed by doses of anti-CD47. Tumour progression was monitored by fLuc BLI measurement. Mice were euthanized according to the criteria described in the ‘Animal models’ section. Specifics for different iterations of the model presented are as follows:
High-dose CAR T cell + B6H12 studies (Supplementary Fig. 1e and Extended Data Fig. 11,n,p,s): mice engrafted with Nalm6-GFP-fLuc cells were treated with B6H12 ( ) or PBS by intraperitoneal
injection three times per week, starting on day 3. Mice were then treated with CD19-287-nLuc CAR T cells by tail-vein injection on day 4 . T cells and tumours were quantified by BLI weekly.
Low-dose CAR T cell + B6H12 studies (Supplementary Fig. 1f and Extended Data Fig. ): mice engrafted with Nalm6-GFP-fLuc cells were treated with CD19-287-nLuc CAR T cells by tail-vein injection on day 4. Mice were treated twice with B6H12 or PBS by intraperitoneal injection on day 5 and 7. T cells and tumours were quantified by BLI weekly.
High-dose CAR T cell + CV-1 studies (Fig. 1f,g, Supplementary Fig. 1g and Extended Data Fig. 2a,b,e): mice engrafted with Nalm6-GFP-fLuc cells were treated with CD19-BB7-nLuc CAR T cells by tail-vein injection on day 4. Mice were treated with CV-1-Fc (ALX-90; dead Fc; ) or PBS by intraperitoneal injection three times per week starting on day cells and tumours were quantified by BLI weekly.
Low-dose CAR T cell + CV-1 studies (Supplementary Fig. 1h and Extended Data Fig. 2c-f): mice engrafted with Nalm6-GFP-fLuc cells were treated with CD19-287-nLuc CAR T cells by tail-vein injection on day 4. Mice were treated with CV-1-Fc (ALX-90; dead Fc; or PBS by intraperitoneal injection three times on days 5, 7 and 10. T cells and tumours were quantified by BLI twice weekly.
Low-dose CAR T cell studies (Fig. 1h,i and Extended Data Fig. 2h,i): mice engrafted with Nalm6-GFP-fLuc cells were treated with CD19-287-nLuc CAR T cells with endogenous CD47 KO ( ), CD47 KO with overexpression of CD47 WT ( ), or an equivalent number of mock T cells by tail-vein injection on day 4. Mice were treated twice with B6H12 ( ) or PBS by intraperitoneal injection on days 5 and 7. Tumours were quantified by BLI weekly. T cells were quantified by BLI on day 11.
Low-dose CAR T cell + B6H12 studies (Supplementary Fig. 1s and Extended Data Fig. 11e,f): mice engrafted with Nalm6-GFP-fLuc cells were treated with CD19-287-nLuc CAR T cells with endogenous CD47KO and overexpressing either or , or an equivalent number of mock T cells by tail-vein injection on day 4 . Mice were treated twice with B6H12 ( ) or PBS by intraperitoneal injection on days 5 and 7. Tumours were quantified by BLI weekly.
T cell depletion model
NSG male or female mice (aged weeks) were implanted with or CD19-287-nLuc CAR T cells with endogenous CD47 KO and overexpressing either or no additional protein ( ) by tail-vein injection (day 0 ). Mice were then treated twice with B 6 H 12 or PBS by intraperitoneal injection on days 3 and 5. T cells were quantified by nanoluciferase BLI before ( dose, day dose, day 3 ) and after ( dose, day dose, day 7 ) anti-CD47 treatment and in the blood by flow cytometry ( dose, day dose, day 6). For isotype control studies (Extended Data Fig.1f,g), mice were implanted with CD19-287 CAR T cells by tail-vein injection (day 0 ) and were then treated with , mIgG1 isotype control ( ) or PBS by intraperitoneal injection on day 1 . T cells were quantified in the blood by flow cytometry on day 5 . Mice were euthanized according to the criteria described in the ‘Animal models’ section at the conclusion of the experiment (Supplementary Fig.1i,m).
PIP CAR toxicity model
PIP CAR vectors were made as described in the viral vector construction section. NSG male or female mice (aged weeks) were treated with the PIP CAR T or control CD19-BB7, HER2-BB7 or mock T cells at the dose indicated in the figure ( or CAR T cells) by tail-vein injection. Mice in the PIP-28 7 and PIP-BB 7 groups experienced rapid onset of toxicity (within 1-5 days, depending on dose) observed clinically as a hunched posture, scruffy coat, slow movement, dehydration and weight loss. Treatment-related toxicity was monitored by weight change, which was measured before T cell administration and per week thereafter. The percentage weight change was calculated as follows: percentage weight change weight at time initial weight) -1. Mice died from toxicity or were euthanized if they reached 20% weight loss or showed clinical signs of severe toxicity, as described in the ‘Animal models’ section. For assessment of T cell localization and activation, CD19-287-nLuc or PIP-287-nLuc CAR T cells were transduced with a firefly luciferase reporter under control of an NF-кB-inducible promoter. Mice were implanted with CAR T cells and imaged daily with Nano-GLO substrate (nLuc; total CART) and luciferin (fluc; active CART), with each substrate dose separated by 12 h . For organ BLI analysis, 4 days after treatment with CD19-287-nLuc or PIP-287-nLuc, the mice were injected with either Nano-GLO substrate (nLuc; total CAR T) or luciferin (fLuc; active CAR T) and euthanized 10 min after injection. Organs were collected according to standard procedures and imaged using BLI on the IVIS machine (Perkin Elmer) in six-well plates. For safety-switch models, mice were dosed with PIP-287 or CD19-287 CAR T cells intravenously. Mice treated with PIP-28 CAR T cells were administered B6H12 or PBS over three consecutive days (days 2, 3 and 4), 2 days after CAR administration (day 0 ). Blood was collected for serum cytokine analysis on day 4 after CAR administration (Supplementary Fig. 1k).
CHLA-255 neuroblastoma metastatic tumour model
Six- to ten-week-old NSG male or female mice were implanted with CHLA-255-GFP-fLuc cells by tail-vein injection . The, 7 days after tumour implantation and after confirmation of tumour formation by BLI, mice were randomized and treated with -nLuc CAR T cells with endogenous CD47 KO and overexpressing or or an equivalent number of mock (non-transduced) T cells intravenously by tail-vein injection. Mice were then treated three times with B6H12 or PBS by intraperitoneal injection on days 7, 9 and 13. Tumour progression was monitored by firefly luciferase BLI. T cells were quantified by nanoluciferase BLI after anti-CD47 treatment on day 14 and in the blood by flow cytometry on day 15. Mice were euthanized according to the criteria described in the ‘Animal models’ section (Supplementary Fig.1r).
CART cell GvHD model
NSG male or female mice (aged weeks) were implanted with Nalm6-GFP-fLuc cells by tail-vein injection. The mice were then treated with CD19-BB7 CAR T cells on day 4. Half of the cohort of mice received three doses of B6H12 (250ug) over 3 days after CAR T cell administration. Mice were monitored for tumour growth by BLI and signs of GvHD, such as alopecia, dyskeratosis and weight loss . Spleens and skin were extracted surgically. Skin sections were prepared as slides and stained with H&E using the standard method (Supplementary Fig. 11).
Isolation of T cells from spleens and tumours
Spleens and tumours were collected and mechanically dissociated using the gentleMACS dissociator (Miltenyi). Single-cell suspensions were made by passing spleens and tumours through a cell strainer (Thermo Fisher Scientific), depleting red blood cells by ACK lysis (Quality Biological), and further filtration through flow cytometry filter tubes with cell strainer caps (Falcon). Single-cell suspensions were then frozen in CryoStor buffer (StemCell Technologies) in liquid nitrogen, or stained and run directly on the flow cytometry system.
Quantification of T cells and cytokines from the blood
Mouse blood was collected from the retro-orbital sinus into Microvette blood collection tubes with EDTA (Thermo Fisher Scientific). Red blood cells were depleted by ACK lysis (Quality Biological), followed by two washes with FACS buffer (PBS + 2% FBS). The samples were stained and mixed with CountBright Absolute Counting beads (Thermo Fisher
Scientific) before flow cytometry analysis. IL-2 and IFN cytokine levels in blood were quantified using LEGENDPlex immunoassays (BioLegend) according to the manufacturer’s instructions from serum collected after centrifuging blood samples at for 10 min . Negative cytokine values were set to 0 .
IncuCyte tumour killing assays, cytokine analysis and T cell activation marker detection
A total of GFP-labelled tumour cells was cocultured with CART cells in RPMI supplemented with FBS, 10 mM HEPES, 2 mMl -glutamine, penicillin and streptomycin. For conditions with B6H12, a concentration of was used. Triplicate wells were plated in 96-well flat-bottom plates for each condition. Tumour fluorescence was monitored every with a objective using the IncuCyte S3 Live-Cell Analysis System (Sartorius), housed in a cell culture incubator at and , set to take 4 images per well at each timepoint. The total integrated GFP intensity was quantified using the IncuCyte basic analyzer software feature (IncuCyte S3 v.2019B Rev2;Sartorius). Data were normalized to the first timepoint and plotted as the fold change in tumour fluorescence over time. For cytokine secretion and T cell marker analysis, cocultures were set up as described above except in 96-well round-bottom plates. After approximately 24 h , the plates were centrifuged to pellet cells and of supernatant was collected and stored at until analysis, while the cell pellets were immediately processed for flow cytometry. IFN and IL-2 levels in the coculture supernatants were quantified by ELISA (Human ELISA MAX Deluxe,BioLegend) according to the manufacturer’s instructions. Negative cytokine values were set to 0 . Absorbance values were measured using the Synergy H1 Hybrid Multi-Mode Reader with Gen5 software (v.2.00.18; BioTek). For analysis of T cell markers after activation by tumour cells, pellets from centrifuged plates were pooled together for triplicate wells, stained and analysed using flow cytometry. Coculture experiments were set-up using day 10 T cells.
Macrophage depletion and peritoneal lavage
NSG male or female mice (aged 6-10 weeks) were pretreated with intravenous injection with of clodronate liposomes (Liposoma), followed by of anti-mouse-CSF1R (AFS98; Bio X Cell) by intraperitoneal injection . Mice were treated with of anti-CSF1R three times per week for the duration of the experiment. Then, 6 days after clodronate treatment, the mice were administered with CD19-28 -nLuc CAR T cells intravenously, followed by B6H12 intraperitoneally on day 7. T cells were quantified by nanoluciferase BLI before (day 7) and after (day 9) anti-CD47 treatment. Peritoneal lavage was performed on day 13 with 10 ml of FACSbuffer and a 25 gauge needle. Peritoneal cells were collected, washed with FACS buffer and stained, before being run on the flow cytometry system (Supplementary Fig. 1j).
Phagocytosis assay
For all flow-based in vitro phagocytosis assays, T cells and human macrophages were co-cultured at a ratio of 2:1 (for example, 100,000 T cells:50,000 macrophages) in ultra-low-attachment 96-wellU-bottom plates (Corning) in serum-free RPMI (Thermo Fisher Scientific). T cells were labelled with CFSE (Invitrogen) by suspending cells in PBS ( working solution) according to the manufacturer’s instructions for 20 min at protected from light and washed twice with 20 ml of FBS-containing medium before co-culture. Cells were then either incubated alone or in the presence of anti-CD47 (B6H12;Bio X Cell) or mIgG1 isotype control (MOPC-21; Bio X Cell) at a concentration of . T cells and antibodies were incubated for 30 min in a humidified incubator at . Plates were washed twice; human macrophages were added to the plate; and plates were incubated for at . Phagocytosis was stopped by washing with PBS and centrifugation at before the cells were stained with Live/Dead stain
and anti-CD11b (APC, M1/70, BioLegend, 1:50). Assays were analysed by flow cytometry, and phagocytosis was measured as the number of and macrophages, quantified as a percentage of the total CD11b macrophages and normalized to the control condition.
For IncuCyte-based in vitro phagocytosis assays, T cells and human macrophages were co-cultured at a ratio of (for example, 100,000 T cells:50,000 macrophages) in 96-well flat-bottom plates (Corning) in RPMI supplemented with FBS, 10 mM HEPES, 2 mMl -glutamine, penicillin and streptomycin. T cells were labelled with pHrodo Red dye (Invitrogen) by incubating T cells at cells per ml with a working concentration of pHrodo Red of in PBS for 1 h at in the dark in a humidified incubator. The labelling reaction was quenched and excess dye was washed away by washing twice with complete medium. Cells were then either incubated alone or in the presence of anti-CD47 (B6H12; Bio X Cell) at a concentration of in serum-free RPMI. T cells and antibodies were incubated for 30 min in a humidified incubator at , before being washed twice with complete medium. Macrophages were added to each well and allowed to adhere for 2 h in a humidified incubator at . After 2 h , labelled T cells were added to the plate at a 2:1 T cell:macrophage ratio. pHrodo Red fluorescence due to phagocytosis was monitored after 3 h with a objective using the IncuCyte S3 Live-Cell Analysis System (Sartorius), housed in a cell culture incubator at and , set to take four images per well at each timepoint. Total integrated red fluorescence intensity was quantified using the IncuCyte basic analyzer software feature (IncuCyte S3 v.2019B Rev2; Sartorius).
Confocal microscopy of T cell-macrophage interactions
CD19-28 CAR T cells were labelled with pHrodo Red dye as described above. pHrodo-Red-labelled T cells were then labelled with DiO Vybrant lipophilic dye (Invitrogen) according to the manufacturer’s instructions in PBS for 2 min at in the dark. Macrophages were labelled with DiD Vybrant dye (Invitrogen) according to the manufacturer’s instructions in PBS for 15 min at in the dark. After dye labelling, cells were washed three times with complete medium to remove excess dye. T cells were then incubated with B6H12 at for 20 min at in PBS, followed by two washes with complete medium. Labelled T cells and macrophages embedded in a collagen matrix (Cellmatrix type I-A, FUJIFILM Wako chemicals) at a ratio of 2:1 (for example, 1,000,000 T cells:500,000 macrophages) in a 24 -well glass-bottom culture plate (Mattek). Four-dimensional ( ) live confocal imaging was performed on a confocal laser-scanning microscope (Zeiss LSM900). Images were analysed using Imaris software (v.10.0; Oxford Instruments).
Quantification of CD47 expression on tumour and T cells using QuantiBrite
CD47 expression was quantified using an anti-CD47-PE antibody (B6H12, BD, 1:20) and a QuantiBrite PE Quantitation Kit (BD) according to the manufacturer’s instructions . CD19-28て CAR T cells were produced as described above, except that cells were kept in culture 1 day after thawing before activation with anti-CD3/CD28 beads. T cells were analysed by flow cytometry on day 0 (before activation; 1 day after thaw), day 4 (immediately after removal from bead activation), day 7 and day 11 (average time of transfer in vivo). T cells were stained with anti-hCD4 (BUV 395, SK3, BD, 1:200), anti-hCD8 (BUV 805, SK1, BD, ), anti-hCD47 or mIgG1 isotype control (PE, B11/6, Abcam, 1:20), anti-hCD45RA (BV785, HI100, BioLegend, 1:100) and anti-hCD62L (BV605, DREG-56, BD, 1:100) antibodies. T cell differentiation subtypes were defined as follows: T naive (CD45RA ), T central memory (CD45RA ), T effector memory (CD45RA ) and T effector memory re-expressing CD45RA (CD45RA ). Tumour cells were stained with only anti-hCD47 or mIgG1 isotype control. Molecules of CD47 were calculated according to the QuantiBrite kit instructions
using extrapolation from MFI signals of BD QuantiBrite-PE beads with known quantities of PE. The degree of labelling for anti-CD47-PE (BD, 2040745) was determined experimentally as 0.842 molecules of dye per antibody, using the maximum absorbance at 566 nm , the extinction coefficient for PE ( ) and the listed antibody concentration.
Imaging of patient CSF samples
A CSF cytospin preparation was collected from a patient treated with axicabtagene ciloleucel (axi-cel) CD19-287 CAR T cell therapy, stained with Wright-Giemsa and imaged by microscopy at magnification, capturing histiocytes with engulfed lymphocytes (raw images are shown in Supplementary Fig. 3).
Single-cell analysis of patient samples
Two datasets were reanalysed: ref. 14 (GSE168940), including scRNA-seq data collected from nine patients with LBCL treated with axi-cel CD19287 CAR T cell therapy, where 50,000-70,000 CAR T cells (single live or events) were FACS sorted to purity; and ref. 15 (GSE186802), including scRNA-seq data collected from four patients with DMG treated with GD2.BBZ CAR T cell therapy, with cells derived from CSF. Both datasets were analysed on the 10x Genomics platform . Where indicated, previously annotated CAR-mRNA-expressing cells were used.
Histology of tissue samples
The tissues assessed include skin and lung. Tissues were collected and immersion-fixed in 10% neutral-buffered formalin. After fixation, tissues were routinely processed, embedded in paraffin, sectioned at and routinely stained with haematoxylin and eosin (H&E). Tissues were visualized using the Olympus BX43 upright bright-field microscope, and images were captured using the Olympus DP27 camera and cellSens software (v.3.2; Olympus Life Science).
Yeast surface display vectors
A DNA sequence encoding the CD47 Ig-like domain (Gln19-Ser135) was cloned into the pCTCON2 yeast-surface display vector (Addgene) using the Nhel and BamHI sites. The pFreeNTerm (pFNT) vector was based on the pCL backbone , designing an intrinsic Nhel cutsite into the Aga2p signal sequence as the cloning site and using a MluI cutsite prior to a Gly 4 Ser linker as the 3 ‘ cloning site. Variants of the CD47 Ig-like domain (Gln19-Ser135) were cloned into the pFNT yeast-surface display vector using these Nhel and Mlul sites.
Flow cytometry analysis of yeast surface display constructs
Saccharomyces cerevisiae (strain, EBY100; ATCC) yeast were transformed with pCTCON2 or pFNT plasmids and selected on SD-CAA-Agar plates. Yeast (~100,000 per sample) were grown and induced in SG-CAA, and binding was set up over a range of soluble ligand concentrations in PBS containing BSA (BPBS), taking into account ligand depletion and equilibrium time . After incubation with binding partner, yeast cells were washed once with BPBS, then incubated with a 1:5,000 dilution of chicken anti-MYC antibody (polyclonal, Invitrogen) for pCTCON2 displayed proteins, and incubated for 30 min at in the dark. After primary addition, the samples were washed once with BPBS, and secondary antibodies were added. Expression was detected with a 1:500 dilution of goat anti-chicken Alexa Fluor 488 (polyclonal, Invitrogen) or Alexa Fluor 647 (polyclonal, Abcam). For pFNT displayed proteins, co-displayed GFP was used to monitor expression. Binding of proteins with mouse Fc domains ( ) was detected with a 1:500 dilution of goat anti-mouse Alexa Fluor 488 or Alexa Fluor 647 (polyclonal, Invitrogen). Binding of proteins with a human Fc domain (CV-1(ALX-222), mSIRP , Hu5F9, TJC4) was detected using a 1:500 dilution of goat anti-human Alexa Fluor 488 or Alexa Fluor 647 (polyclonal, Invitrogen). Secondary antibodies were incubated for 15 min at in
the dark. After secondary incubation, the samples were washed once with BPBS, pelleted and left pelleted on ice until analysis. The samples were analysed by resuspending them in of BPBS and running flow cytometry on the BD Accuri C6 (BD Biosciences) system. Accuri C6 software (v.1.0.264.21; BD) was used for data collection and FlowJo software (v.10.8.1; BD) was used for data analysis (gating strategies are shown in Supplementary Fig. 2). The samples were gated for bulk yeast cells (forward scatter (FSC) versus side scatter (SSC)) and then for single cells (FSC-height versus FSC-area). Expressing yeast were determined and gated through the C-terminal MYC tag or GFP detection. The geometric mean of the binding fluorescence signal was quantified from the expressing population and used as a raw binding value. When comparing binding signals, the average fluorescence expression signal was quantified for different protein variants and used to normalize binding signal. To determine the ‘fraction bound’, binding signals were divided by the signal derived from the highest concentration of binding partner used, or that derived from binding to WT CD47. To calculate values, data were analysed in Prism (v.9.5.1, GraphPad) using a nonlinear regression curve fit.
Yeast surface display library generation, sorting and sequencing CD47 was expressed in S. cerevisiae (strain, EBY100; ATCC) as a genetic fusion to the agglutinin mating protein Aga2p in the pCTCON2 vector. An error-prone PCR library was created using the CD47 Ig-like domain (Gln19 to Ser135) as a template and mutations were introduced using the Gene Morph II random mutagenesis kit (Agilent) according to the manufacturer’s instructions. Separate PCRs were performed using various concentrations of Mutazyme II enzyme. Products from these reactions were purified by gel electrophoresis, pooled and amplified with standard PCR using Phusion polymerase (New England BioLabs). Purified mutant DNA and linearized pCTCON2 plasmid were electroporated into EBY100 yeast, where they were assembled in vivo through homologous recombination. We estimated variants for the library, determined by dilution plating and colony counting. Yeast were grown in SD-CAA medium and induced for CD47 protein expression by growth in medium containing 90% SG-CAA and 10% SD-CAA overnight . Yeast displaying CD47 variants were isolated by FACS using the SONY SH800S cell sorter (SONY; SH800S cell sorter software v.2.1.5) and analysed using the BD Accuri C6 flow cytometer (BD Biosciences; BD Accuri software v.1.0.264.21). Gating strategies are shown in Supplementary Fig. 2. Data were analysed using FlowJo software (v.10.8.1, BD). Screens were performed using equilibrium binding conditions where yeast were incubated at room temperature in BPBS with the following concentrations of B6H12 or CV-1 (ALX-222) for 2 h . For negative sorts to B6H12, the CD47-expressing, but non-binding populations of yeast were collected. For positive sorts to CV-1, the CD47-expressing and binding populations of yeast were collected (Extended Data Fig. 6g). Sort 1, negative sort, 500 pM B6H12; sort 2, negative sort, 5 nM B6H12; sort 3, positive sort, 20 nM CV-1; sort 4, negative sort, 20 nM B6H12; sort 5, negative sort, 50 nM B6H12; sort 6, positive sort, . After incubation with B6H12 or CV-1, yeast was pelleted, washed and labelled with fluorescent antibodies as described above before sorting. Sorted yeast clones were propagated, induced for CD47 expression and subjected to iterative rounds of FACS as described above. After each round of screening, plasmid DNA was recovered using the Zymoprep yeast plasmid miniprep I kit (Zymo Research), transformed into DH10B electrocompetent cells (Thermo Fisher Scientific) and isolated using the GeneJET plasmid miniprep kit (Thermo Fisher Scientific). Sequencing was performed by ELIM Biopharmaceuticals.
CD47 structure modelling
CD47 structures were downloaded from the Protein Data Bank (PDB) and analysed using PyMol (v.2.5.8; Schrödinger). The CD47-hSIRPa structure used was PDB 2JJS (ref. 18). The CD47-B6H12 structure used was PDB 5TZU (ref. 19).
143B in vivo phagocytosis model
A total of cells in DPBS was injected into the tibia periosteum of NSG mice (aged weeks). Then, 20 days after tumour implantation, mice were treated with CFSE-labelled HER2-BB7 CART cells intratumourally. Before administration, T cells were labelled with CFSE ( working solution), according to the manufacturer instructions, with cells labelled at a concentration of cells per ml in PBS for 5 min . Cells were washed in complete medium, and then incubated with or without PBS for 20 min at . Cells were resuspended in of PBS/tumour for intratumoural injection. Immediately after T cell administration, the mice were then treated with B6H12 ( ) or PBS by intraperitoneal injection. Tumours were collected 16 h later on day 21 after tumour implantation. Tumours were mechanically dissociated using the gentleMACS dissociator (Miltenyi). Single-cell suspensions were made by passing tumours through a cell strainer (Thermo Fisher Scientific), depleting red blood cells by ACK lysis (Quality Biological) and further filtration through flow cytometry filter tubes with cell strainer caps (Falcon). Single-cell suspensions were subsequently stained for flow cytometry to detect CFSE T cells and macrophages from dissociated tumours (see below) (Supplementary Fig. 1n).
143B correlative study and tumour dissociation
A total of cells in DPBS was injected into the tibia periosteum of NSG mice (aged weeks). Then, 13 days after tumour implantation and after visual confirmation of tumour formation, the mice were treated with HER2-BB7 CAR T cells with endogenous and overexpressing either or , an equivalent number of mock T cells intravenously by tail-vein injection, or no T cells. The mice were then treated twice with B6H12 ( ) or PBS by intraperitoneal injection on day 15 and day 19 . Tumours were collected at day 21 after tumour implantation (day 8 after CAR T cell treatment). Tumours were split with a razor, with one section being fixed in paraformaldehyde, and the other mechanically dissociated as described above, before being stained for flow cytometry and FACS. Formaldehyde-fixed tumour had paraformaldehyde removed after 24 h and replaced with ethanol for long-term storage. Tumour sections were then formalin-fixed and paraffin-embedded according to the standard protocol (Supplementary Fig. 1o).
Flow cytometry and IHC analysis of dissociated tumours
For flow cytometry, tumours were collected as described above. Single-cell suspensions of dissociated tumours were stained for CAR (APC, HER2-Fc, R&D, 1:400), hCD19 (BUV 496, SJ25C1, BD, 1:100), CD11b (BUV 395, M1/70, BD, 1:100), F4/80 (BV605, BM8, BioLegend, 1:100), hCD45 (PerCP-Cy5.5, HI30, Invitrogen, 1:50), hCD3 (BUV 737, SK7, BD, 1:100), mCD45 (BUV 805, I3/2.3, BD, 1:100) and hCD47 (BV711, B6H12, BD, 1:100), and with Fixable Viability Dye (eFluor 780, Invitrogen, 1:2000) for 30 min in PBS + 2% FBS (FACS Buffer) before being analysed by flow cytometry.
For IHC analysis, tumours were collected as described above. Formalin-fixed, paraffin-embedded xenograft tumour sections were used. F4/80 (D2S9R, Cell Signaling Technology, 1:200) staining was performed manually, and hCD3 (SP7, Abcam, 1:100) and ARG1 (D4E3M, Cell Signaling Technology, 1:250) staining was performed using the Ventana Discovery platform. In brief, tissue sections were incubated in either 6 mM citrate buffer (F4/80,1:200 dilution) or Tris EDTA buffer (CD3/ARG1, 1:100 and 1:250 dilution respectively; cell conditioning 1 standard) at for 25 min (F4/80) or for 1 h (CD3/ARG1) to retrieve antigenicity, followed by incubation with the respective primary antibody for 1 h . Bound primary antibodies were incubated with the respective secondary antibodies (goat anti-rabbit, polyclonal, F4/80: Vector Laboratories, undiluted or CD3/ARG1: Jackson ImmunoResearch, 1:500), followed by UltraMap HRP (Roche, F4/80)
or ChromoMap DAB (Roche, CD3/ARG1) detection. For IHC analysis, tumour regions were identified on the basis of histology. F4/80, CD3 and ARG1 positivity were analysed for each tumour region. F4/80, CD3 and ARG1 IHC positivity scores were automatically quantified in the regions of interest using Aperio ImageScope software (v.12.3.2.8013). Regions of interest were randomly selected within the tumour to exclude macrophages present in the normal tissue around the tumour.
Single-cell analysis of dissociated 143B tumours
Dissociated tumours from the 143B osteosarcoma model described above were sorted for live cells using a Live/Dead stain (Invitrogen) at the Stanford Shared FACS facility (the gating strategy is shown in Supplementary Fig. 2). scRNA-seq libraries were prepared using the Chromium Next GEM Single Cell 5′ v2 platform (10x GENOMICS). Libraries were sent to Novogene for sequencing on a NovoSeq S4 lane (PE150) with approximately 30,000 mean reads per cell. Reads were aligned and quantified using CellRanger (v.6.0;10x GENOMICS) using the standard workflow, with the reference transcriptomes GRCh38 for human and mm 10 for mouse. The CellRanger output was imported into R (v.4.2.2) using Seurat (v.4.2.0). The following filters were applied using the subset function to select for live cells: nFeature_RNA > 200 and nFeature_RNA < 5000; percent mitochondrial reads < 5%. After filtering, the eight biological samples ranged from 7,658-9,327 mean unique molecular identifiers per cell. The data matrix was normalized with NormalizeData and scaled with Seurat. Cell types were assigned using SingleR automated cell type recognition. Differential expression analysis, clustering and UMAP dimensionality reduction analysis were performed on the resulting data matrix using Seurat . Pathway analysis was performed using Enrichr , with the NCI-Nature 2016 gene set collection queried for human T cells and the KEGG Human Pathway collection queried with converted mouse gene IDs for mouse macrophages.
Statistical analyses
The specific statistical tests used are indicated in the figure legends. Statistical analyses were performed using R (v.4.2.2), Excel (v.16.64; Microsoft) or Prism (v.9.3.1, GraphPad). For comparisons between two groups, statistical significance was assayed using two-tailed unpaired Student’s -tests or Mann-Whitney -tests. For comparison within in vivo studies and between grouped studies, two-way ANOVA combined with Tukey’s multiple-comparison test for post hoc analysis was performed. Significance for survival data was calculated using the log-rank Mantel-Cox test. Sample sizes were determined on the basis of the variability of tumour models used, determined by experience with well-established, previously published models . Tumour-bearing animals were assigned to the treatment groups randomly to ensure an equal distribution of tumour sizes between groups. Data are represented as mean s.d. (in vitro studies) or mean s.e.m. (some in vivo studies). For all statistical analyses, values are indicated in each figure panel.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data associated with this paper are included in the Article and the Supplementary Information. The scRNA-seq dataset has been deposited at the NCBI Gene Expression Omnibus (GEO) under accession number GSE261475. Data used to generate scRNA-seq UMAP plots from patient data (Fig. 2d) were obtained from publicly available datasets using the GEO series accession numbers GSE168940 (ref. 14) and GSE186802 (ref. 15). For protein crystal structure modelling, the following publicly availably PDB files were used: 2JJS (hCD47-hSIRPa) and 5TZU (hCD47-B6H12). Source data are provided with this paper.
30. Hughes, M. S. et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum. Gene Ther. 16, 457-472 (2005).
31. Majzner, R. G. et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res. 25, 2560-2574 (2019).
32. Labanieh, L. et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 185, 1745-1763 (2022).
33. Lynn, R. C. et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293-300 (2019).
34. Freitas, K. A. et al. Enhanced T cell effector activity by targeting the Mediator kinase module. Science 378, eabn5647 (2022).
35. Kimura, R. H., Levin, A. M., Cochran, F. V. & Cochran, J. R. Engineered cystine knot peptides that bind , , and integrins with low-nanomolar affinity. Proteins 77, 359-369 (2009).
36. Chu, J. et al. A bright cyan-excitable orange fluorescent protein facilitates dual-emission microscopy and enhances bioluminescence imaging in vivo. Nat. Biotechnol. 34, 760-767 (2016).
37. Gresham, H. D., Goodwin, J. L., Allen, P. M., Anderson, D. C. & Brown, E. J. A novel member of the integrin receptor family mediates Arg-Gly-Asp-stimulated neutrophil phagocytosis. J. Cell Biol. 108, 1935-1943 (1989).
38. Theruvath, J. et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med. 28, 333-344 (2022).
39. Ali, N. et al. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Ry mice display a T-effector memory phenotype. PLoS ONE 7, e44219 (2012).
40. Ferrara, J., Guillen, F. J., Sleckman, B., Burakoff, S. J. & Murphy, G. F. Cutaneous acute graft-versus-host disease to minor histocompatibility antigens in a murine model: histologic analysis and correlation to clinical disease. J. Invest. Dermatol. 86, 371-375 (1986).
41. Radosevich, M. T. et al. Antigen density quantification of cell-surface immunotherapy targets by flow cytometry: Multi-antigen assay of neuroblastoma bone marrow metastasis. STAR Protoc. 4, 102709 (2023).
42. Shah, N. N. & Harrington, A. M. Hemophagocytosis in cerebrospinal fluid after CAR T-cell therapy. Blood 139, 1116 (2022).
43. Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587 (2021).
44. Lim, S., Glasgow, J. E., Filsinger Interrante, M., Storm, E. M. & Cochran, J. R. Dual display of proteins on the yeast cell surface simplifies quantification of binding interactions and enzymatic bioconjugation reactions. Biotechnol. J. 12, 1600696 (2017).
45. Hunter, S. A. & Cochran, J. R. Cell-binding assays for determining the affinity of proteinprotein interactions: technologies and considerations. Methods Enzymol. 580, 21-44 (2016).
46. Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
47. Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128 (2013).
Acknowledgements This work was supported by National Institutes of Health grants 1R01CA263500-01 (C.L.M. and M.M.); an EPICC Translational Research Grant (St Baldrick’s Foundation, C.L.M.); and the Virginia and D.K. Ludwig Fund for Cancer Research (C.L.M.). C.L.M., S.A.Y.-H., L.L. and Z.G. are members of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program. B.J.M. was supported by a Stanford Interdisciplinary Graduate Fellowship; K.A.F. by the National Science Foundation Graduate Research Fellowship under grant DGE-1656518; F.L. by a Stanford M-TRAM Capstone project grant; A.L. by the Nuovo-Soldati Foundation and by ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé/National Alliance for the Life Sciences and Health) within the framework of the French Cancer Plan; R.P. by the Princess Máxima Center for Pediatric Oncology and Academy Ter Meulen Fund of the Royal Netherlands Academy of Arts & Sciences; S.H. by a U54 CA232568-01 grant; and O.K. by a Paul and Daisy Soros Fellowship for New Americans. J.B. received support from the Stanford Biosciences Training grant 5T32-GM11999505 through the Institute for Stem Cell Biology & Regenerative Medicine, and the National Science Foundation Graduate Research Fellowship Program under Award Number 2146755; T.M. from the Stanford Medical Scientist Training Program grant T32GM007365, the NCI under Award Number F30CA271797, the Stanford Interdisciplinary Graduate Fellowship, the Stanford ChEM-H Chemistry/Biology Interface
Predoctoral Training Program and the Stanford ChEM-H O’Leary-Thiry Graduate Fellowship. Sorting was performed on an instrument at the Stanford Shared FACS Facility obtained using NIH S10 Shared Instrument Grant S10RR025518-01. We acknowledge the staff at the Stanford Shared FACS facility for sorting tumour samples, the Stanford Human Pathology/Histology Service Center for immunohistochemistry staining, the Stanford Veterinary Service Center for general support of mouse work and necropsy studies, and ALX Oncology for contributing the CV-1 reagent. Illustrations in Extended Data Fig. 5a and 6a and Supplementary Fig. 1 were created using BioRender.
Author contributions S.A.Y.-H., J.T. and C.L.M. conceived the idea of the study. S.A.Y.-H. designed experiments, cloned constructs, performed in vitro characterizations, including phagocytosis assays, in vivo experiments, protein engineering and yeast binding characterization experiments, and scRNA-seq experiments, including analysing and interpreting data. J.T. designed experiments, cloned constructs and performed in vitro characterizations, including phagocytosis assays, and in vivo experiments, including analysing and interpreting data. B.J.M. conducted protein engineering and yeast binding characterization experiments, analysed data and interpreted results. K.A.F. performed experiments, analysed data and interpreted results relating to scRNA-seq. F.L., M.T.R. and S.D. cloned constructs, performed in vitro characterizations and conducted in vivo studies. A.L. designed and characterized the PIP CAR toxicity model, including performing experiments, analysing data and interpreting results. N.M.-V. designed in vivo experiments, assisted with tumour dissociation studies, and designed, analysed and interpreted flow cytometry experiments. P.X. and J.H. organized and conducted in vivo studies. A.D. performed IHC experiments and analysed and interpreted data. M.H.D. performed scRNA-seq experiments. Z.G. and Y.C. analysed scRNA-seq data and interpreted results. R.P. designed and performed confocal experiments, including analysing data and interpreting results. A.M. performed phagocytosis assays. L.L. cloned constructs and designed experiments. J.B. performed in vitro characterizations and flow cytometry experiments. T.M. and Z.E. analysed flow cytometry data and interpreted results. C.W.M. performed in vivo studies and histology analysis and data interpretation. S.H. performed in vitro characterization experiments and assisted with in vivo studies. K.D.M. performed in vivo experiments. A.B. and O.K. provided and characterized primary human macrophages for phagocytosis assays. S.L.W. cloned constructs and performed in vitro characterization assays. J.Y.S. and S.F.-P. provided and analysed images of patient CSF. R.G.M. designed and performed in vivo experiments, analysed data and interpreted results. E.S., J.R.C. and C.L.M. designed experiments, analysed data and interpreted results. C.J.K., P.H.S., M.M., I.L.W., B.S., E.S., J.R.C. and C.L.M. supervised the work. S.A.Y.-H., E.S. and C.L.M. wrote the manuscript with feedback from all of the authors.
Competing interests S.A.Y.-H., J.T., B.J.M., J.R.C. and C.L.M. are listed as coinventors on a patent related to this work (PCT/US2O24/013209, submitted by the board of trustees of the Leland Stanford Junior University). C.L.M. holds equity in CARGO Therapeutics, Link Cell Therapies and Ensoma, which are developing CAR-based therapies; consults for CARGO, Link, Immatics, Ensoma and Red Tree Capital; and receives research funding from Lyell Immunopharma and Tune Therapeutics. S.A.Y.-H. is a consultant for Quince Therapeutics. J.T. is a consultant for Dorian Therapeutics. L.L. and E.S. are consultants for and hold equity in Lyell Immunopharma. L.L. is a cofounder of, consults for and holds equity in CARGO Therapeutics. O.K. is a senior fellow with ARTIS Ventures. C.J.K. is founder and scientific advisory board member for NextVivo, Surrozen and Mozart Therapeutics. R.G.M. is a co-founder of and holds equity in Link Cell Therapies; and is a consultant for NKarta, Arovella Pharmaceuticals, Innervate Radiopharmaceuticals, GammaDelta Therapeutics, Aptorum Group, Zai Labs, Immunai, Gadeta, FATE Therapeutics (DSMB) and Waypoint Bio. I.L.W. is a director, stockholder in and consultant for Forty Seven (but not Gilead); a co-founder of and director and consultant for Bitterroot Bio and PHeast, and a co-founder of 48. I.L.W. is also on the scientific advisory board of Appia. E.S consults for Lepton Pharmaceuticals and Galaria. J.R.C. is a cofounder and equity holder of Trapeze Therapeutics, Combangio and Virsti Therapeutics; has financial interests in Aravive, Xyence Therapeutics and CARGO Therapeutics; and is a member of the board of directors of Ligand Pharmaceuticals and Revel Pharmaceuticals. The other authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-024-07443-8.
Correspondence and requests for materials should be addressed to Crystal L. Mackall. Peer review information Nature thanks Smita Chandran and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
Extended Data Fig.1 | See next page for caption.
Extended Data Fig. 1 | Anti-CD47 therapy blunts CAR and TCR T cell efficacy by depleting adoptively transferred T cells. (a) treated 143B tumour survival. mice/arm. (b) B7H3.BB7- or GD2.BB7 treated MG63.3 tumour survival. mice/arm. (c) treated D 425 tumour growth by BLI. CD19.BB7-CAR is included as a non-tumour targeting control. Mean SEM of or (all others) mice/arm. Representative of two independent experiments. (d) B7H3.BBC treated D425 tumour survival. CD19.BB7-CAR is included as a nontumour targeting control. (B6H12) or (all others) mice per treatment arm. Representative of two independent experiments. (e) Representative flow cytometry plots of cells identified in the blood and tumour in the B7H3.BB7-CAR B6H12 treated MG63.3 model on day 30 post tumour engraftment. (f) Representative flow cytometry plots of cells identified in the blood of non-tumour bearing mice co-treated with CD19.287CAR T cells and either PBS, B6H12, or mIgG1 isotype control. Representative of two independent experiments. (g) hCD8 (top) and hCD4 (bottom) T cells in the blood of mice on day 5 in the isotype control model, treated as in (f). Mean SD of mice. Representative of two independent experiments. (h) T cells ( and ) in the blood of mice on day 12 in Her2.BB
treated mice in the 143B model. Mean SD of mice. (i) Low-dose NY-ESO-1 treated A 375 tumour survival. mice/arm. (j) High-dose NY- treated A 375 tumour growth. Mean of mice/arm. (k) T cells (hCD4 and ) in the blood by flow cytometry on day 16 of mice treated with high-dose NY-ESO-1-TCR B6H12 in the A375 model. Mean SD of mice. (1) High-dose CD19.28 -CAR B6H12 treated Nalm6 tumour growth by BLI. Mean SEM of mice/arm. (m) Low-dose CD19.28 -CAR B6H12 treated Nalm6 tumour growth by BLI. Mean SEM of mice/arm. (n) Images of Nalm6 tumour BLI in the high-dose (left) and low-dose (right) CAR-T model on day 17. (o) Low-dose CD19.28 7 -CAR B6H12 treated Nalm6 survival. mice/arm. (p) T cell BLI in the high-dose CAR-T – Nalm6 model, treated as in (l). Mean SEM of mice/arm. (q) T cell BLI in the low-dose CAR-T – Nalm6 model, treated as in (m). Mean SEM of mice/arm. (r) Images of CD19.28-nLuc-CAR T cell BLI in the low-dose CAR T – Nalm6 model, treated as in (m), on day 11. (s) Quantification of T cells by flow cytometry from the spleen in the high-dose CART-Nalm6 model, treated as in (I). Mean SD of mice. [(a), (b), (d), (i), (o)] Log-rank Mantel-Cox test. [(c), (j), (l), (m), (p), (q)] Two-way analysis of variance (ANOVA) test with Tukey’s multiple comparison test. Unpaired two-tailed Student’s test. For all: ns = not significant.
Extended Data Fig. 2 | See next page for caption.
Extended Data Fig. 2 | Treatment with CV-1 or CD47 knock-out leads to T cell depletion and ablation of antitumour efficacy. (a) CD19.BBC-CAR CV-1
(Fc-dead) treated Nalm6 tumour survival. mice/arm. (b) Images of Nalm6 BLI on day 11 of high-dose CD19.BB7-CAR CV-1 treated mice, treated as in (a). (c) Low-dose CD19.28 7 -CAR CV-1 (Fc-dead) treated Nalm6 tumour growth. Mean SEM of mice/arm. (d) Images of Nalm6 BLI of low-dose CD19.28 CAR CV-1 treated mice, treated as in (c). (e-f) Images (e) and quantification (f) of T cell BLI post CV-1 treatment of Nalm6 tumour-bearing mice. Top: high-dose CD19.BB7-CAR CV-1, treated as in (a), on day 9 (four days post CV-1 treatment). Bottom: low-dose CD19.28て-CAR CV-1, treated as in (c), day 11 (six days post CV-1 treatment). For ( f ): mean SD of mice for each dose condition.
(g) CRISPR/Cas9 mediated CD47 knock-out (CD47 ) efficiency in primary human T cells by flow cytometry. cells are with exogenous expression of wild-type CD47. Representative of donors. (h) Images of
Nalm6 tumour progression (top five rows) and T cells on day 11 (bottom row) by BLI in the Nalm6-47 -CD19.287-CAR T model. (i) Survival of Nalm6 bearing mice shown in (h). mice per treatment arm. (j) Representative flow cytometry plots of cells identified in the blood of or CD19.28 7 -nLuc-CAR B6H12 treated, non-tumour bearing mice. (k) Images of T cell BLI following treatment with or -CD19.28 -nLuc-CAR T cells before and after B6H12 treatment. (I) T cell BLI of mice shown in (k) treated with or -CD19.28 -nLuc-CART, before and after B6H12 treatment. Dashed line indicates limit of detection. Mean of mice. and T cells in the blood on day 6 or 7 by flow cytometry following treatment or -nLuc-CART in separate experiments, treated as in ( k ). Mean SD of mice. [(c), (l), (m)] Two-way analysis of variance (ANOVA) test with Tukey’s multiple comparison test. ns = not significant. [(f – top)] Two -tailed Mann-Whitney test. [(f-bottom)] Unpaired two-tailed Student’s test.
Extended Data Fig. 3 | See next page for caption.
Extended Data Fig. 3 | CD47 over-expression ( ) does not alter T cell function in vitro, but enhances efficacy in vivo. (a) Expansion of CD19.28 -, , or CAR-T cells over days in culture. Mean SD of cell donors. (b) CD19.28 CD19.28 CD19.28 , or -CD19.28 CAR-T cell viability over days in culture. Mean SD of cell donors. (c) Nalm6-GFP tumour killing by mock, CD19.28 CD19.28 , -CD19.28 -, or -CD19.28 -CAR-T cells at a T ratio measured via IncuCyte assay. Mean SD of cell donors, with each datapoint derived from the average of triplicate wells per donor. Mock, CD19.28%, and -CD19.287 are shared conditions duplicated in (d). (d) Nalm6GFP tumour killing by mock, CD19.28 , or CD19.28 -CAR T cells B6H12 at a 1:1E:T ratio measured via IncuCyte assay. Mean SD of cell donors, with each datapoint derived from the average of triplicate wells per donor. Conditions without B6H12 are shared conditions duplicated in (c). (e-f) IFN- (e) and IL-2 (f) secretion upon co-culture for 24 h with and without Nalm6 tumour cells at a1:1E:T ratio of mock, CD19.28て-,47 CD19.28 , CD19.28 – or CD19.28 -CAR-T measured via ELISA. Mock, CD19.28 -, and (-CAR-T cells were also assessed in the presence of B6H12 and Nalm6. Mean SD of cell donors, with each datapoint derived from the average of triplicate wells per donor. (g-l) Annexin V(g), CD69 (h), CD39
(i), TIM3 (j), LAG3 (k), and PD1 (l) expression upon co-culture for 24 h with and without Nalm6 tumour cells at a 1:1E:T ratio of mock, CD19.28 -CD19.28 , AAVS1 -CD19.28 – or -CD19.28 -CAR-T measured via flow cytometry. Mock, CD19.28 , and -CD19.28 -CAR-T cells were also assessed in the presence of B6H12 and Nalm6. Mean SD of cell donors. (m) CD47 and CAR (second from left) expression on T cells by flow cytometry after CD47 over-expression . Representative of donors. ( n ) Images of Nalm6 BLI on day 45 after dosing with CD19.28 – or -CD19.28 -CAR-T cells in Nalm6 tumour-bearing mice. (o) CD19.28 – or -CD19.28 -CAR T treated Nalm6 tumour growth by BLI. Mean SEM of mice/arm. (p) macrophages collected via peritoneal lavage by flow cytometry on day 13 after clodronate treatment. Mean SD of (macrophage depleted) or (macrophage non-depleted) mice. (q) Representative flow plots and gating strategy for detection of macrophages in samples collected following peritoneal lavage. (r) CD19.287-nLuc-CAR T BLI before (day 7) and after (day 9) treatment with B6H12 of mice macrophage depletion (started on day 0 ). Mean SD of (macrophage depleted, PBS treated) or (all other groups) mice. [(a), (b), (c), (d), (e), (f), (g), (h), (i), (j), (k), (l), (o), (r)] Two-way analysis of variance (ANOVA) test with Tukey’s multiple comparison test. ns = not significant. [(p)] Unpaired two-tailed Student’s test.
Extended Data Fig. 4 | See next page for caption.
Extended Data Fig. 4 | CD47 expression is uniform among CAR T cell subtypes and decreases over time in culture. (a) Phagocytosis of CFSE labelled CD19.28 (-CAR-T cells from three donors by primary human macrophages from three donors by flow cytometry, following one hour of co-culture. CAR T cells were either untreated or treated with B6H12 or mIgG1 isotype control prior to addition of macrophages. Mean SD of cell donors, with each datapoint derived from the average of triplicate wells per donor. (b) Representative confocal images of primary human CD19.287-CAR-T cell phagocytosis by a primary human macrophage after treating with B 6 H 12 in a three-dimensional collagen matrix. Images from left to right represent 0 min (initial image), and 3 h 21 min of T cell – macrophage co-incubation. CAR T cells (green) and macrophages (red) were labelled separately with lipophilic dyes. CAR T cells were also labelled with pH sensitive pHrodo Red dye (shown in blue) prior to B6H12 treatment. White arrows indicate a phagocytic event. See SI Video 1 for accompanying time-lapse video. Images are representative across experiments. (c) CD47 expression on tumour cells, mock T cells, and CD19.287-CAR-T cells by QuantiBrite quantitative flow cytometry. T cells were assessed on day 4 and day 11 of culture. Quantification of histograms shown on the left for each cell type. Representative of tumour sample and cell donors and reproducible across experiments. (d) CD47 expression on and CD19.28 CAR-T cells by QuantiBrite quantitative flow cytometry. Cells were assessed by flow cytometry on day 0 (prior to activation), day 4 (after activation bead removal), day 7, and day 11 (time of typical transfer in vivo). Mean SD of cell donors. Only significant differences are shown. All other groups are not significantly different, comparing between the same subtype over time and between subtypes
at the same timepoint. (e) CD47 expression on , and (left) and CD8 (right) CD19.28-CAR-T cell subtypes on days , and 11 post-activation by QuantiBrite quantitative flow cytometry. Mean SD of cell donors. Only significant differences are shown. All other groups are not significantly different, comparing between the same subtype over time and between subtypes at the same timepoint. (f) Representative flow cytometry plots of CD19.287-CAR-T cells pre- (day 0 ) and post-activation (days 4, 7, and11). Left column: gating for T cell differentiation states, T naïve (CD45RA /CD62L ), T central memory (CD45RA ), T effector memory (CD45RA CD62L ), and T effector memory re-expressing CD45RA (CD45RA / CD62L . Right column: histograms of CD47 expression across T cell differentiation states. (g) Calreticulin (left) and CD47 (right) expression on CAR-T cells by flow cytometry on day 10 of culture. Quantification of histograms shown on the bottom for each type of CAR-T cell. Representative of donors. (h) Quantification of CD47 (top) and calreticulin (bottom) expression on cells on day 16 and day 25 of culture by flow cytometry. Mean of donors. (i) Single cell landscapes colour coded for cell type. Top: Weighted nearest neighbour (wnn) UMAP derived from cells sorted from the blood collected on day 7 after CAR T infusion of axi-cel (CD19.28 ) treated LBCL patients, sampled to 500 cells/patient sample. Bottom: UMAP derived from cells from the CSF of (treated diffuse midline glioma (DMG) patients, sampled to 500 cells/patient sample. [(a), (d), (e)] Two-way analysis of variance (ANOVA) test with Tukey’s multiple comparison test. [(h)] Unpaired two-tailed Student’s test. For all: ns = not significant.
Extended Data Fig. 5 | Anti-CD47 can limit toxicities from a pan-tumour integrin targeting PIP-CAR and GvHD. (a) Cartoon of the PIP-CAR design.
The diagram was created using BioRender. (b) PIP.28 7 -CAR expression by flow cytometry. Representative of donors. (c) 143B-GFP tumour killing by PIP.28 -,PIP.BB-, or non-tumour targeting CD19.BB-CAR T cells at a 1:1E:T ratio measured via IncuCyte assay. Mean SD of triplicate wells and reproducible in three independent experiments with different donors. (d) Weights of mice following treatment with CD19.28 7 – or PIP.28 7 -CAR-T. Mean SEM of mice/arm. (e) Cartoon of the dual luciferase reporter design for tracking CAR-T activity in vivo. nLuc is linked to CAR expression, fLuc is linked to NF-KB activation. (f) T cell BLI of mice treated with CD19.287or PIP.28 [-nLuc_NFkB-fLuc-CART cells. Left: nLuc (total T cells). Right: fLuc (active T cells). Mean SEM of mice/arm. (g) Images of T cell BLI of mice treated as in (f), 3 days after infusion. Top: nLuc (total T cells). Bottom: fLuc
(activated T cells). mice/arm. (h) Representative images of BLI from organs extracted from mice treated as in (f), four days after CAR T administration. Left: nLuc (total T cells). Right: fluc (active T cells). Representative of mice/arm. (i) Representative hematoxylin and eosin (H&E) stained images of lung tissue collected from mice treated as in (f), four days after CAR T administration. Right panel is a higher magnification view of the boxed region in the middle panel. Images are representative of mice/arm. (j-1) Representative images of (j) mice demonstrating GvHD derived alopecia (day 48 post-CART infusion), (k) spleens from mice on day 48 post-CAR T infusion), and (l) H&E staining of skin sections (collected on day 48 post-CAR T infusion) from mice treated with CD19.BB7-CART B6H12. Representative of mice/arm. [(c), (d), (f)] Two-way analysis of variance (ANOVA) test with Tukey’s multiple comparison test.
Extended Data Fig. 6 | See next page for caption.
Article
Extended Data Fig. 6 | Engineered variants of CD47 retain SIRPa binding and demonstrate a loss of binding to some, but not all, anti-CD47 antibodies.
(a) Engineered CD47 mechanism: CD 47 antibodies bind tumour cells, but not -T cells, triggering tumour-specific phagocytosis. The diagram was created using BioRender. (b) Cartoon of yeast displayed CD47 Ig-like domain using the pCTCON2 vector. CD47 is displayed as an N-terminal fusion.
(c) Binding of to yeast displayed CD47 in pCTCON2 by flow cytometry. Representative of independent experiments. (d) Binding curve of B6H12 to yeast displayed CD47 in pCTCON2, measured over multiple concentrations by flow cytometry. MFI of experiment. (e) Binding of 300 nM human (top) and mouse (bottom) SIRP to yeast displayed CD47 in pCTCON2 by flow cytometry. Data are representative of independent experiments. (f) Binding of to yeast displayed CD47 in pCTCON2 by flow cytometry. Data are representative of independent experiments. (g) Flow cytometry sorting plots of all six sorts of the CD47 library, indicating negative sorts to B6H12 and positive sorts to CV-1. Collected population indicated by the black box in each plot. (h) Binding of CV-1 to the yeast displayed CD47 library population collected after sort 6 or yeast displayed WT CD47. Data are representative of independent
experiments. (i) Binding of or 100 nMCV – 1 to CD 47 variants displayed on yeast using the pCTCON2 vector. Mean SD of individual yeast clones, normalized to MFI from binding to . (j) Cartoon of yeastdisplayed CD47 Ig-like domain using the pFreeNTerm (pFNT) vector. CD47 is displayed as a C-terminal fusion, along with GFP to monitor protein expression. (k) Binding of 100 nM human and mouse SIRP to yeast displayed CD47 in pFNT by flow cytometry. Representative of independent experiments. (I) Crystal structure of CD47 (yellow) binding SIRPa (orange) [PDB: 2JJS], identifying the CD47 BC loop (green), containing CD47 residues T26-Q31. (m) Crystal structures of CD47 (red) binding SIRP (dark pink, left) [PDB: 2JJS] and B6H12 (light blue, right) [PDB: 5TZU], identifying residues A30 (gold) and Q31 (blue), and the BC and FG loops of CD47. Structures are enlargements of the boxed regions in the full structures shown in Fig. 4c. (n) Binding of , TJC4, Hu5F9, and hSIRP to yeast displayed , and variants. Mean SD of individual yeast clones, normalized to MFI from binding to [(i), (n)] Two-way analysis of variance (ANOVA) test with Tukey’s multiple comparison test. ns = not significant. Comparison is between indicated group and binding to CD47 WT expressing cells.
Extended Data Fig. 7| See next page for caption.
Extended Data Fig. 7| Expression of on T cells mitigates B 6 H 12 induced phagocytosis due to lack of antibody binding in vitro and in vivo.
(a) Representative flow cytometry histograms of , and binding to , , and over-expressed on primary human T cells. Data are representative of different T cell donors in independent experiments.
(b) Left: Representative flow cytometry histograms of , and 500 nM B 6 H 12 binding to , and expressed on Jurkats with endogenous . Right: Binding of B6H12, hSIRP , and mSIRP to full-length , and expressed on Jurkat cells with endogenous . Mean SD of independent experiments, normalized to fraction binding to . (c) Phagocytosis by primary human macrophages from multiple donors of CFSE labelled Jurkats with endogenous , expressing , or variants after one hour of co-culture . Mean of triplicate wells. (d) Phagocytosis by primary human macrophages from two donors of pHrodo Red labelled primary human CD19.287-CAR-T cells from three donors with endogenous , expressing or after three hours of co-culture . Mean of triplicate wells. (e) Images of T cell BLI following treatment with or -CD19.28 -nLuc-CAR-T cells B6H12 in non-tumour bearing mice. (f) T cell BLI before and after CD47
treatment, of mice treated as in (e). Mean SD of mice. and cells in the blood on day 6 by flow cytometry of mice treated as in(e). Mean SD of mice. (h) Representative flow plots and gating strategy for detection of macrophages and T cells in dissociated 143 B tumour samples in the in vivo phagocytosis model treated with Her2.BB7-CART cells B6H12. Representative of . (i) Representative flow plot and gating strategy for detection of CFSE cells of a dissociated 143B tumour from a mouse that did not receive CAR T cells. (j) macrophages identified by flow cytometry of dissociated tumours treated as in (h). Mean SD of mice. (k) hCD3 human T cells identified by flow cytometry of dissociated tumours treated as in (h). Mean SD of mice. (l) Percentage of phagocytosed T cells identified in each dissociated tumour, treated as in (h), calculated as the number of macrophages per 100,000 lives cells per sample divided by the total number of macrophages and T cells per 100,000 live cells per sample, identified by flow cytometry. Mean SD of mice. [(b), (c), (d), (f), (g)] Two-way analysis of variance (ANOVA) test with Tukey’s multiple comparison test. ns = not significant. (b) comparison is between indicated group and binding to expressing cells. [(j), (k)] Unpaired two-tailed Student’s test. [(l)] Two-tailed Mann-Whitney Test.
Extended Data Fig. 8 | CAR-T treatment increases tumour infiltration of macrophages. (a) hCD T cells identified by flow cytometry of dissociated 143 B tumours in the 143B correlative model, treated with no T cells, mock, or -CAR-T cells . Mean of or mean (all other samples) mice. (b) Percent positivity for CD 3 T cells identified by IHC from tumour sections treated as in (a). Mean of ( ) or mean (all other samples) mice. (c) Correlation of hCD 3 and staining in IHC sections of tumours treated as in (a). Data points are representative of individual tumours, coloured by treatment group ( ). calculated by simple linear regression. (d) Percent positivity
for hCD3 (T cells), mArg1 (M2 macrophages), and mF4/80 (total macrophages) cells identified by IHC from tumour sections treated as in (a). Mean of ( ) or mean of (all others) mice. (e) Correlation of hCD3 and mArg1 percent positivity by IHC of tumour sections treated as in (a). Datapoints are representative of individual tumours, coloured by treatment group ( ). calculated by simple linear regression. (f) Representative images of IHC tumour sections stained for mF4/80, mArg1, and hCD3, treated as in (a). Representative of tumour sections collected from mice/arm. [(a), (b)] Two-way ANOVA test with Tukey’s multiple comparison test. ns = not significant.
Extended Data Fig. 9 | See next page for caption.
Extended Data Fig. 9 | CAR-T treatment increases tumour infiltration of distinct macrophage populations. (a and b) scRNA-seq profile of dissociated tumour and infiltrating immune cells in the 143B correlative model, treated with no T cells, mock, – or -Her2.BBZ-CAR-T cells B6H12. Dots represent individual cells. cells from 8 experimental conditions with three mice per treatment group, coloured by (a) cell type (left eight plots; UMAPs represent distinct treatment conditions), species (far right), or (b) gene expression level. (c) Composition of murine cells identified via scRNA-seq from 143B tumours treated as in (a). Data are derived from cells pooled from 8 experimental conditions with three mice per treatment group. Cell types assigned using SingleR automated cell type recognition. (d) Dot Plot depicting scRNA-seq expression of selected T cell subset markers, cytokines, and chemokines. human tumour infiltrating T cells from treatments described in (a). (e) Comparison of differentially expressed genes between CAR-T cells of different treatment groups described in (a). Statistical significance was determined with Seurat;*Padj <0.05. (f) Comparison of differentially expressed genes between macrophages of different treatment groups described in (a). Statistical significance was determined with Seurat; adj . (g) Enrichr pathway analysis of the top 100 upregulated genes in tumour infiltrating macrophages [macrophage/monocyte population in (a)] in treated tumours compared with untreated controls. Results depict the value (two-sided Fisher’s exact test) of each pathway. (h) UMAP of the identified macrophage/monocyte population in (a), subsetted and re-clustered, coloured by cluster (cluster identities on the left). Dots represent individual cells. cells from 8 experimental conditions. (i) UMAPs of the identified macrophage/monocyte population, subsetted and re-clustered, coloured by cluster (cluster identities in (h)). UMAPs represent distinct treatment conditions. Dots represent individual cells. cells from 8 experimental conditions described in (a). (j) UMAP of the re-clustered macrophage/monocyte population from (m), coloured for red if hCD mRNA expression . Dots represent individual cells. cells from 8 experimental conditions, with total cells identified as . UMAPs represent distinct treatment conditions described in (a). (k) Dot plot depicting scRNA-seq expression of selected cluster-defining genes within the macrophage populations identified in (h).Padj.<0.0001 for each selected gene.
Extended Data Fig. 10 | Pairing of – CAR T and anti-CD47 therapy enhances tumour control in an aggressive osteosarcoma model, even at low doses.
( ) (left, a ) and (right, a & b ) T cells derived from two independent donors (represented by panels a & b) in the blood on day 14 by flow cytometry after B6H12 treatment in 143B tumour bearing mice, treated with – or -Antares-CAR-T . Mean of mice. (c) and cells in the blood on day 14 and day 27 of tumour growth in 143B tumour bearing mice, treated with Antares-CAR-T +B 6 H 12 . Datapoints represent individual mice, with values from the same mouse connected by lines between time points. (d) T cell BLI prior to B6H12 treatment in mice treated as in (a). Mean SD of mice. (e) T cell BLI after B6H12 treatment on day 13 in mice treated as in (a). Mean SD of mice. (f) or -Antares-CAR-T B6H12 treated 143B tumour survival. mice/arm. (g and h) or -Antares-CAR-T treated 143B tumour (g) growth and (h) survival, using T cells derived from a different donor than (f). Mean SEM (g) or representative (h) of mice. (i) (left) and (right) T cells derived from in the blood on day 12 by flow cytometry after treatment with or .BBC-CAR-T low-doses of or B6H12 treatment in 143B tumour bearing mice. Mean SD of mice. (j) -Her2.BBZ-CAR-T low-dose B6H12 ( or ) treated 143B tumour growth, using cells derived from two different donors (left and right panels, respectively). Mice treated with mock T cells were co-treated ( ) B6H12 (left panel) or B6H12 (right panel). Mean SEM of mice. [(a), (b), (d), (e), (g), (i), (j)] Two-way ANOVA test with Tukey’s multiple comparison test. ns = not significant. [(f), (h)] Log-rank Mantel-Cox test. ns = not significant. c
Dose: B7H3.BBC
NY-ESO-1 T cell BLI
g
Pre- CD47 treatment (Day 9) Dose: NY-ESO-1
Extended Data Fig. 11 | Expression of on tumour-targeting cells permits pairing with anti-CD47 therapy and results in improved tumour control.
(a) (left) and (right) T cells by flow cytometry from the blood of CAR-T treated mice on day 15 in CHLA-255 tumour bearing mice, treated with – or .BBC-nLuc-CAR-T . Mean of mice. (b) T cell BLI on day 14 in CHLA-255 tumour bearing mice, treated as in (a). Mean SD of mice. (c) Images of CHLA-255 tumour progression (top four rows) and T cells on day 14 (bottom) by BLI, treated as in (a). (d) or -B7H3.BB7-nLuc-CAR-T B6H12 treated CHLA-255 tumour growth by BLI. Mean SEM of mice/arm. (e) or -CD19.28 -CAR-T B6H12 treated Nalm6 tumour growth by BLI. Mean SEM of mice/arm. (f) or
treated Nalm6 survival. mice/arm. (g) T cell by BLI in A375 tumour bearing mice, treated with Antares-TCR-T cells , before (day 9, left) and after (day 14, right) CD 47 treatment. Mean SD of n mice. (h) hCD4 (left) and (right) T cells by flow cytometry from the blood in the A375 tumour bearing mice, treated as in (g). Mean SD of mice. (i) -NY-ESO-1-TCR-T B6H12 treated A375 tumour growth. Data are individual tumour growth traces of mice/arm. (j) -NY-ESO-1-TCR-T B6H12 treated A375 tumour growth, with T cells derived from a different donor than shown in (i). Mean SEM of mice/arm. [(a), (b), (d), (e), (g), (h), (j)] Two-way ANOVA test with Tukey’s multiple comparison test. ns = not significant. [(f)] Log-rank Mantel-Cox test. ns = not significant.
natureportfolio
Corresponding author(s):
Crystal Mackall
Last updated by author(s): Mar 26, 2024
Reporting Summary
Nature Portfolio wishes to improve the reproducibility of the work that we publish. This form provides structure for consistency and transparency in reporting. For further information on Nature Portfolio policies, see our Editorial Policies and the Editorial Policy Checklist.
Statistics
For all statistical analyses, confirm that the following items are present in the figure legend, table legend, main text, or Methods section.
Confirmed
The exact sample size ( ) for each experimental group/condition, given as a discrete number and unit of measurement A statement on whether measurements were taken from distinct samples or whether the same sample was measured repeatedly X
The statistical test(s) used AND whether they are one- or two-sided
Only common tests should be described solely by name; describe more complex techniques in the Methods section. X
A description of all covariates tested X
A description of any assumptions or corrections, such as tests of normality and adjustment for multiple comparisons X
A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals) X
For null hypothesis testing, the test statistic (e.g. ) with confidence intervals, effect sizes, degrees of freedom and value noted Give values as exact values whenever suitable. For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes Estimates of effect sizes (e.g. Cohen’s , Pearson’s ), indicating how they were calculated
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Policy information about availability of computer code
Data collection
FACSDiva ver 8.0.1 (BD Biosciences): Flow cytometry data acquisition
BD Accuri C6 software ver 1.0.264.21 (BD Biosciences): Flow cytometry data acquisition
IncuCyte S3 ver 2019B Rev2 Software (Sartorius): Cytotoxicity data acquisition
LivingImage ver 4.7.3 (Perkin Elmer): In Vivo Bioluminescence data acquisition
Aura ver 4.0.7 (Spectral Instruments Imaging): In Vivo Bioluminescence data acquisition
SH800S cell sorter software ver 2.1.5 (SONY): Cell sorting data acquisition
Gen5 ver 2.00.18 (BioTek): ELISA quantification
Imaris ver 10.0 (Oxford Instruments): confocal microscopy
cellSens ver 3.2 (Olympus Life Science): histology microscopy
Data analysis
FlowJo ver 10.8.1 (BD): Flow cytometry data analysis
Excel ver 16.64 (Microsoft): Analysis of bulk data
Prism ver 9.5.1 (GraphPad): Generation of graphs and statistical analysis
SnapGene ver 6.0.2 (Dotmatics): DNA vector design and molecular cloning
Aura ver 4.0.7 (Spectral Instruments Imaging): In Vivo Bioluminescence data quantification
Imaris ver 10.0 (Oxford Instruments): confocal microscopy
PyMol ver 2.5.8 (Schrödinger, LLC): protein structure modeling
Aperio ImageScope ver 12.3.2.8013: IHC positivity score analysis
CellRanger ver 6.0: demultiplexing and alignment of sequencing reads to host reference genomes
Seurat ver 4.2.0: cell type identification, differential gene expression analyses, and visualizations ver 4.2.2: statistical and scRNA-seq data analysis
For manuscripts utilizing custom algorithms or software that are central to the research but not yet described in published literature, software must be made available to editors and reviewers. We strongly encourage code deposition in a community repository (e.g. GitHub). See the Nature Portfolio guidelines for submitting code & software for further information.
Data
Policy information about availability of data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
Accession codes, unique identifiers, or web links for publicly available datasets
A description of any restrictions on data availability
For clinical datasets or third party data, please ensure that the statement adheres to our policy
All data associated with this paper are included in the manuscript and the supplementary materials. All raw data are provided in the source data files. The scRNAseq dataset has been deposited in the NCBI Gene Expression Omnibus (GEO) and is accessible through the GEO series accession number GSE261475. Data used to generate scRNA-seq UMAP plots from patient data (Fig. 2d) were obtained from publicly available data sets using the GEO series accession numbers GSE168940 (Good Z., et. al., Nature Medicine, 2022) and GSE186802 (Majzner, R.G., et. al., Nature, 2022). For protein crystal structure modeling, the following publicly availably PDB files were used: 2JJS (hCD47-hSIRPa) and 5TZU (hCD47 – B6H12).
Human research participants
Policy information about studies involving human research participants and Sex and Gender in Research.
Reporting on sex and gender
Sex information for patients can be found in previous publications where the datasets we re-analyzed were originally published: (Good, Z. et. al. Nature Medicine, 2022 and Majzner, R.G., et. al., Nature, 2022). Gender information was not collected. Human T cells were isolated from buffy coats or leuokopaks from anonymous healthy donors (male and female; gender information was not collected) purchased from the Stanford Blood Center or STEMCELL Technologies, respectively.
Population characteristics
Patient characteristics can be found in previous publications where the datasets we re-analyzed were originally published: (Good, Z. et. al. Nature Medicine, 2022 and Majzner, R.G., et. al., Nature, 2022). Human T cells were isolated from buffy coats or leuokopaks from anonymous healthy donors (male and female, under 45) purchased from the Stanford Blood Center or STEMCELL Technologies, respectively.
Recruitment
Patient recruitment information can be found in previous publications where the datasets we re-analyzed were originally published: (Good, Z. et. al. Nature Medicine, 2022 and Majzner, R.G., et. al., Nature, 2022). Written informed consent was obtained from all healthy T cell donors by the Stanford Blood Center and STEMCELL Technologies.
Ethics oversight
Both studies we re-analyzed data from (Good, Z. et. al. Nature Medicine, 2022 and Majzner, R.G., et. al., Nature, 2022) were approved by the Stanford University IRB. Ethical approval pertaining to T cell donors was obtained by the Stanford Blood Center and STEMCELL Technologies. Blood cells used were exempt from IRB approval as there was no identifiable information provided about blood donors.
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Field-specific reporting
Please select the one below that is the best fit for your research. If you are not sure, read the appropriate sections before making your selection.
Life sciences Behavioural & social sciences Ecological, evolutionary & environmental sciences
For a reference copy of the document with all sections, see nature.com/documents/nr-reporting-summary-flat.pdf
Life sciences study design
All studies must disclose on these points even when the disclosure is negative.
Sample size No sample size calculations were performed. Group sizes were determined by experience with well-established, previously published models
Sample size
1. Labanieh, L., et al., Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell, 2022. 185(10): p. 1745-1763.e22.
2. Theruvath, J., et al., Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nature Medicine 2022 28:2, 2022. 28(2): p. 333-344.
Data exclusions
In the experiments described in Figure 1I,m, one mouse in the macrophage depletion group died prematurely and was excluded from the study. In the IHC and flow analysis described in Figure 5a and Extended Data Figure 8a-e some data were excluded due to lack of detectable tumor after dissociation, notably in the 47E-CAR treated group.
Replication
At least 2 different donor T cells were used for every experiment with primary T cells (save for those noted below) and each experiment was performed independently. For experiments where one representative donor was shown, data were representative of all donors. Experiments with Jurkat cells were performed independently at least 3 times and with different donor derived macrophages. Experiments with yeast were performed with three different clones and were performed independently. All attempts at replication were successful. Only one T cell donor was used for the CHLA-255 metastatic model described in Extended Data Figure 11a-d, the 143B correlative study described in Figure 5a-d and Extended Data Figures 8 and 9, the in vivo phagocytosis model described in Extended Data Figure 7h-I, and the low-dose CD19 CAR-Nalm6 model described in Extended Data Figure 1, Figure 1h,i, and Extended Data Figure 11e,f. A single donor was used in these cases due to practical experimental limitations (such as for single cell RNA sequencing experiments), similar prior experiments with different donors and different doses, but similar results (such as Nalm6 experiments), or confirmation of replication though smaller scale prior pilot studies (such as for CHLA-255, in vivo phagocytosis, and 143B tumor dissociation experiments).
Randomization
For in vivo experiments, cages of mice that were previously engrafted with tumor were randomly assigned CAR T cell and anti-CD47 conditions for infusion, ensuring roughly equal distributions of tumor size between groups prior to treatment. Experiments with primary human T cells were performed using T cells isolated from healthy T cell donors (under age 45 ) chosen at random. Transformed yeast clones were chosen at random for each condition. Otherwise, all other experiments were not randomized, but instead included controls to account for covariates (such as mock transduced T cells derived from the same donor as CAR T cells or yeast cells stained with only secondary antibodies).
Blinding
In vivo tumor engraftment and T cell infusion were performed by a technician who was blinded to treatments and expected outcomes. Otherwise, fully informed data analysis was performed. Fully blinded experiments were not possible due to personnel availability to accommodate such situations.
Reporting for specific materials, systems and methods
We require information from authors about some types of materials, experimental systems and methods used in many studies. Here, indicate whether each material, system or method listed is relevant to your study. If you are not sure if a list item applies to your research, read the appropriate section before selecting a response.
Policy information about cell lines and Sex and Gender in Research
Cell line source(s)
Authentication
Mycoplasma contamination
Commonly misidentified lines
(See ICLAC register)
The Nalm6 B-ALL cell line was provided by David Barrett (Children’s Hospital of Philadelphia) and retrovirally transduced to express GFP and firefly luciferase. 143B osteosarcoma cells were acquired from the American Type Culture Collection (ATCC, Manassas) and then retrovirally transduced with human CD19. CHLA-255 neuroblastoma line was obtained and provided by Robert Seeger (Children’s Hospital Los Angeles) and retrovirally transduced with GFP and firefly luciferase. MG63.3 was provided by Chand Khanna (National Cancer Institute, National Institutes of Health) and retrovirally transduced with GFP and firefly luciferase. D425 was provided by S. Chesier (Stanford University, Stanford, CA) and retrovirally transduced to express GFP and firefly luciferase. Nalm6 and MG63.3 were originally obtained from ATCC. D425 was originally obtained from Sigma Aldrich. A375 melanoma cells and Jurkat cells (clone E6-1) were obtained from ATCC. The 293GP retroviral packaging line was provided by the Surgery Branch (National Cancer Institute, National Institutes of Health). 293T lentiviral packaging cells were obtained from ATCC. Expi293 protein production cells were obtained from ATCC.
Engineered cell lines were verified to express proteins of interest via flow cytometry. All cell lines were previously authenticated via STR fingerprinting prior to use in this manuscript. Cell lines were also authenticated by the commercial vendor, authentication techniques can be found on the vendor website: https://www.atcc.org/ and https:// www.emdmillipore.com/.
Cells were frequently tested for mycoplasma using the Lonza MycoAlert Mycoplasma Detection kit. All experiments reported in this study used cells that tested negative for Mycoplasma.
No commonly misidentified cell lines were used.
Animals and other research organisms
Policy information about studies involving animals; ARRIVE guidelines recommended for reporting animal research, and Sex and Gender in Research
Laboratory animals
NOD/SCID/IL2Ry-/- (NSG; NOD.Cg-Prkdcscid II2rgtm1WjI/SzJ) male and female mice were used for all in vivo experiments. Mice were
Laboratory animals
6-10 weeks old at the time of tumor or T cell engraftment. Mice were housed at and humidity with a 12 hour light hour dark cycle.
Wild animals
No wild animals were used in this study
Reporting on sex
Findings apply to both sexes
Field-collected samples
No field samples were used.
Ethics oversight
All animal studies were undertaken under Stanford University APLAC-approved protocols. Animals were housed in the Stanford Veterinary Service Center (VSC) Barrier Facility at and humidity with a 12 -hour light hour dark cycle. Mice were monitored daily by VSC staff and euthanized if endpoint criteria were met.
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Flow Cytometry
Plots
Confirm that:
The axis labels state the marker and fluorochrome used (e.g. CD4-FITC).
The axis scales are clearly visible. Include numbers along axes only for bottom left plot of group (a ‘group’ is an analysis of identical markers).
All plots are contour plots with outliers or pseudocolor plots.
A numerical value for number of cells or percentage (with statistics) is provided.
Methodology
Sample preparation
For surface staining: as per methods, cells were washed with FACS buffer (PBS + 2% FBS), stained with fluorophoreconjugated antibodies in FACS buffer (100uL total staining volume per sample) for 30 minutes on ice, washed again with FACS buffer, and then analyzed, or stained with secondary antibody (in 100uL total staining volume) for 30 minutes on ice, before washing again, and then analyzing.
Instrument
BD Fortessa X-20, BD LSR II, BD Accuri, SONY SH800S
Software
FACSDiva ver 8.0.1 (BD Biosciences), BD Accuri software (BD Biosciences), SH800S cell sorter software ver 2.1.5 (SONY), and FlowJo ver 10.8.1 (BD Biosciences)
Cell population abundance
Purity of sorted yeast cells was determined by flow cytometry. Sorted cells were grown up, and run via flow cytometry for binding. Purity of sorted populations was estimated at in all samples.
Gating strategy
Samples were gated on lymphocytes (FSC-A/SSC-A), single cells (FSC-H/FSC-A), and relevant markers (CD47, CAR, CD4, CD8, etc. as specified in the manuscript main text). For cells that were stained with live/dead staining, live cells were also gated into the population of interest (live/dead staining was performed using Fixable Viability Kit eFluor 780 [eBioscience catalog #65-0865-14]). Fluorescence minus one (FMO) and isotype controls were used to determine gating where necessary.
QuantiBrite-PE (BD Biosciences; catalog 340495) and LegendPLEX Th1 Panel (BioLegend; catalog: 741035) beads were gated following the manufacturer’s instructions.
For in vivo murine blood analysis, samples were gated on lymphocytes and single cells as above, with further gating on human CD45hi cells. CountBright absolute counting beads were used to validate absolute cell numbers.
Yeast were first gated for forward scatter vs side scatter (FSC-A vs SSC-A), then gated for singlets using forward scatter (FSC-H vs FSC-A). Finally, expressing yeast were identified via c-myc-tag staining or GFP expression, with the c-myc or GFP positive population defined by running uninduced and no-secondary controls. In all cases a clear negative and positive population were observed in induced yeast.
Tick this box to confirm that a figure exemplifying the gating strategy is provided in the Supplementary Information.
Center for Cancer Cell Therapy, Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA, USA. Parker Institute for Cancer Immunotherapy, San Francisco, CA, USA. Cancer Biology Program, Stanford University School of Medicine, Stanford, CA, USA. Immunology Graduate Program, Stanford University School of Medicine, Stanford, CA, USA. Masters in Translational Research and Applied Medicine Program, Stanford University School of Medicine, Stanford, CA, USA. British Columbia Cancer Agency, Vancouver, British Columbia, Canada. Department of Biomedical Data Science, Stanford University School of Medicine, Stanford, CA, USA. Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA. Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands. Department of Bioengineering, Stanford University, Stanford, CA, USA. Institute for Stem Cell Biology and Regenerative Medicine, Stanford, CA, USA. Program in Biophysics, Stanford University, Stanford, CA, USA. Medical Scientist Training Program, Stanford University, Stanford, CA, USA. Department of Neurology, Stanford University School of Medicine, Stanford, CA, USA. Neurosciences Program, Stanford University, Stanford, CA, USA. Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA, USA. Sylvester Comprehensive Cancer Center, University of Miami, Miami, FL, USA. Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA. Department of Pediatrics, Stanford University School of Medicine, Stanford, CA, USA. Ludwig Center for Cancer Stem Cell Research and Medicine, Stanford Johanna Theruvath. e-mail: cmackall@stanford.edu
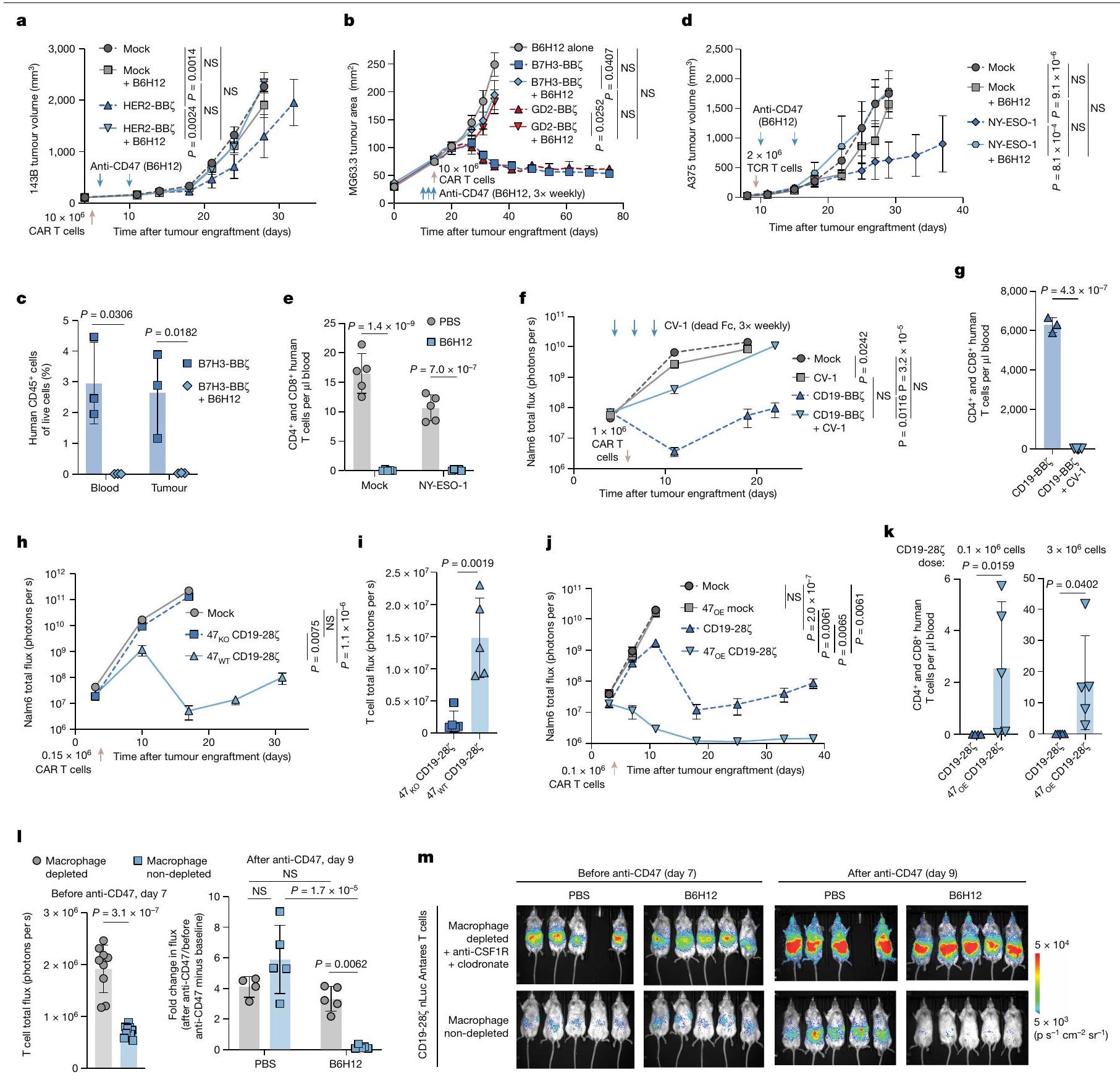
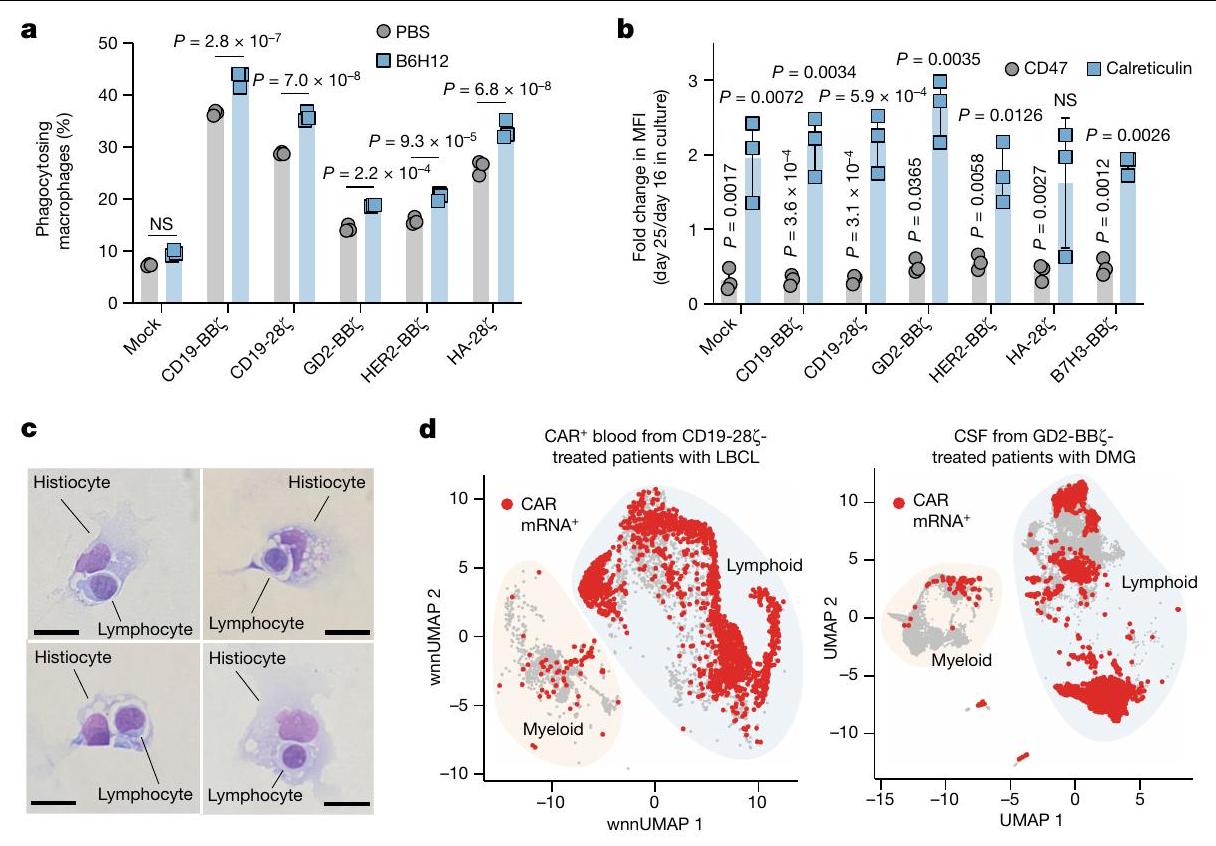
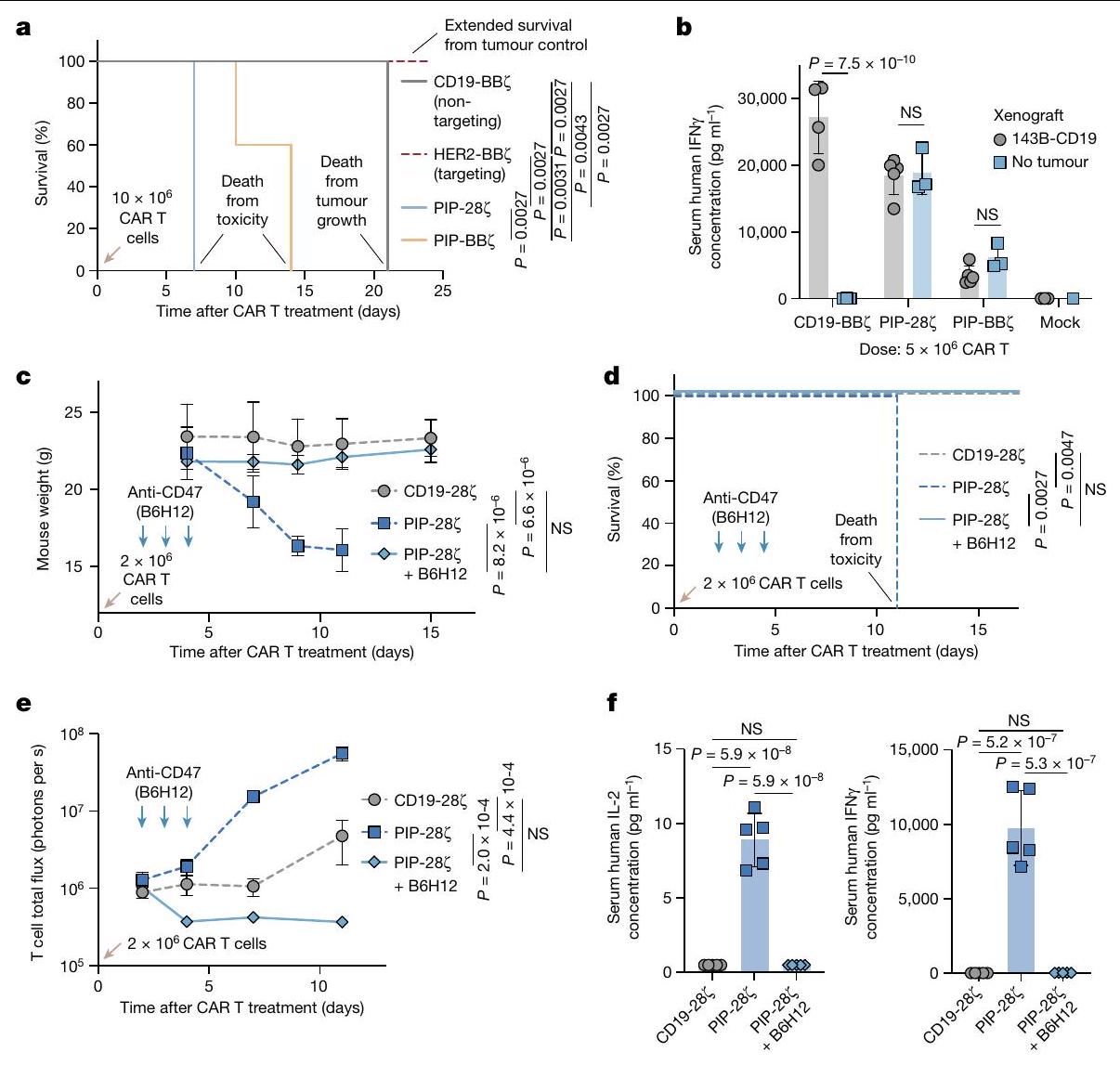
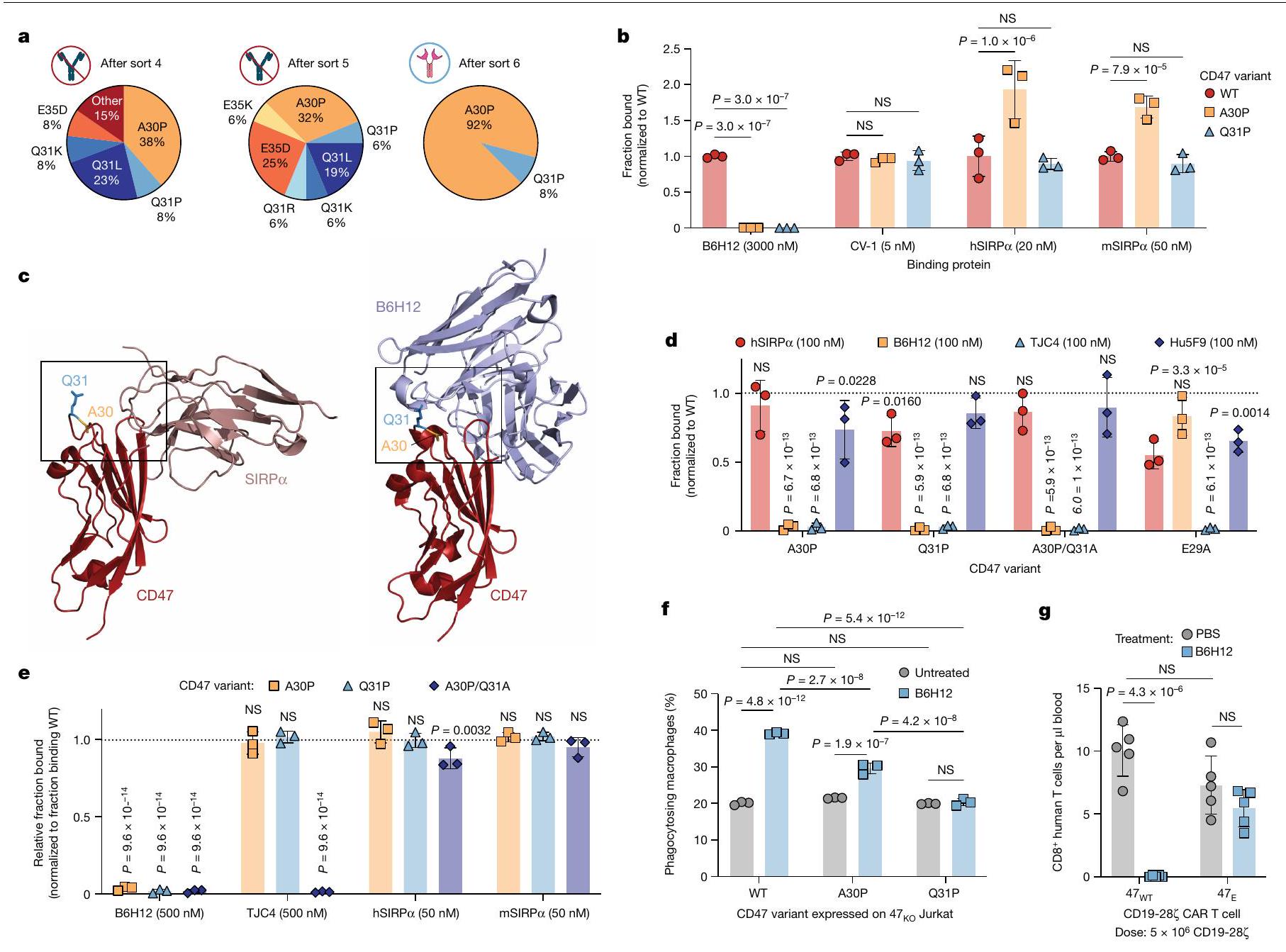
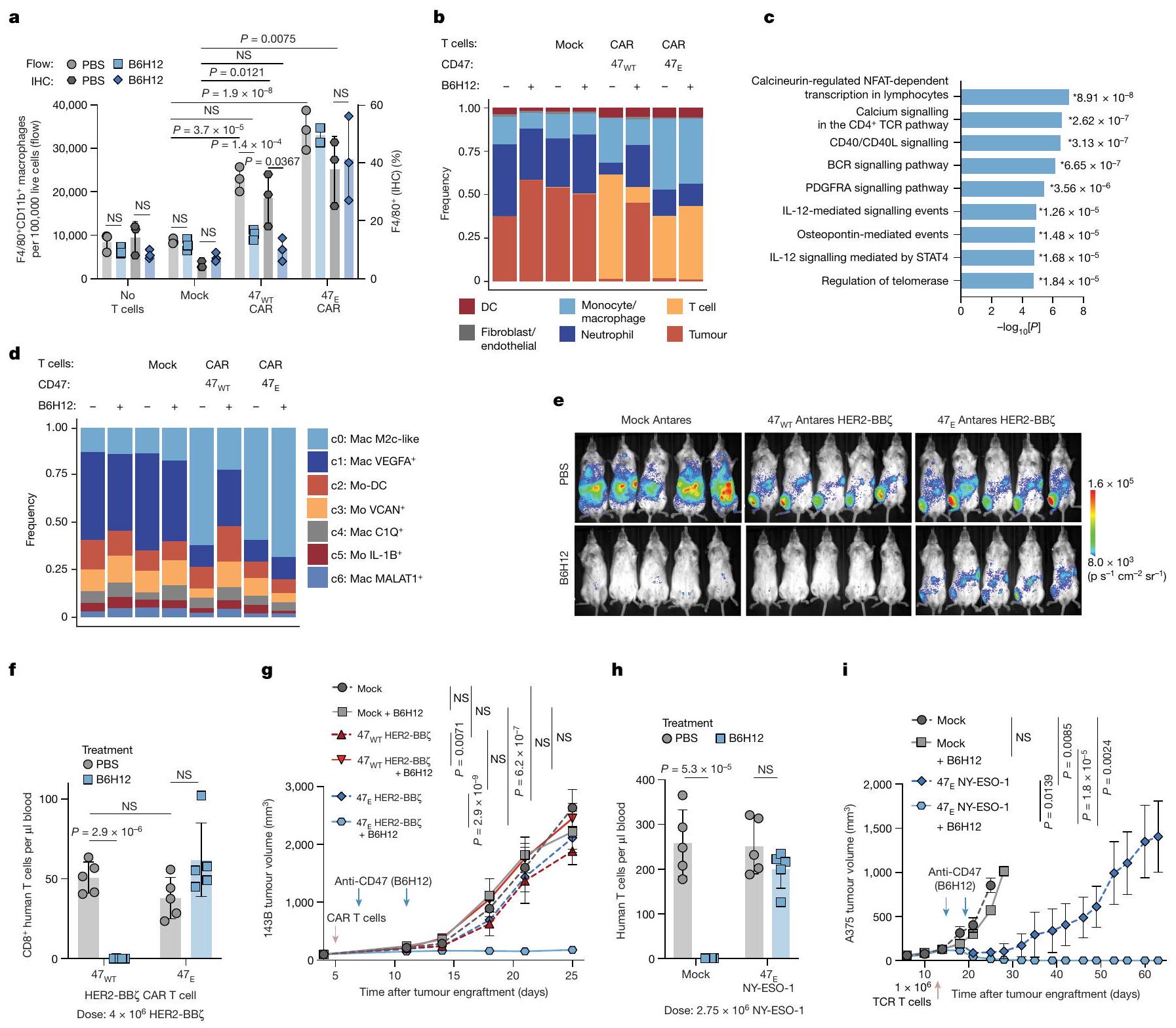
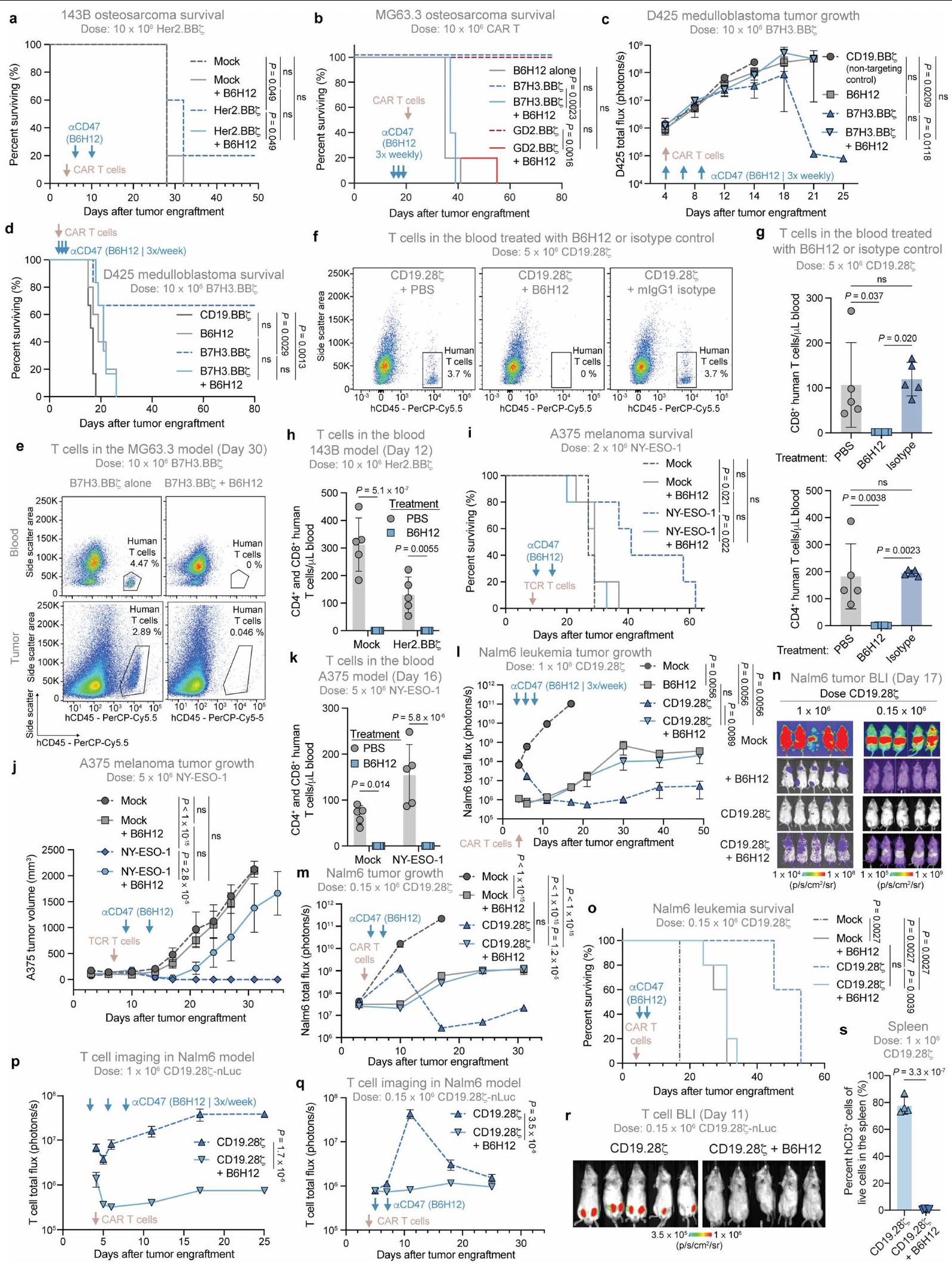
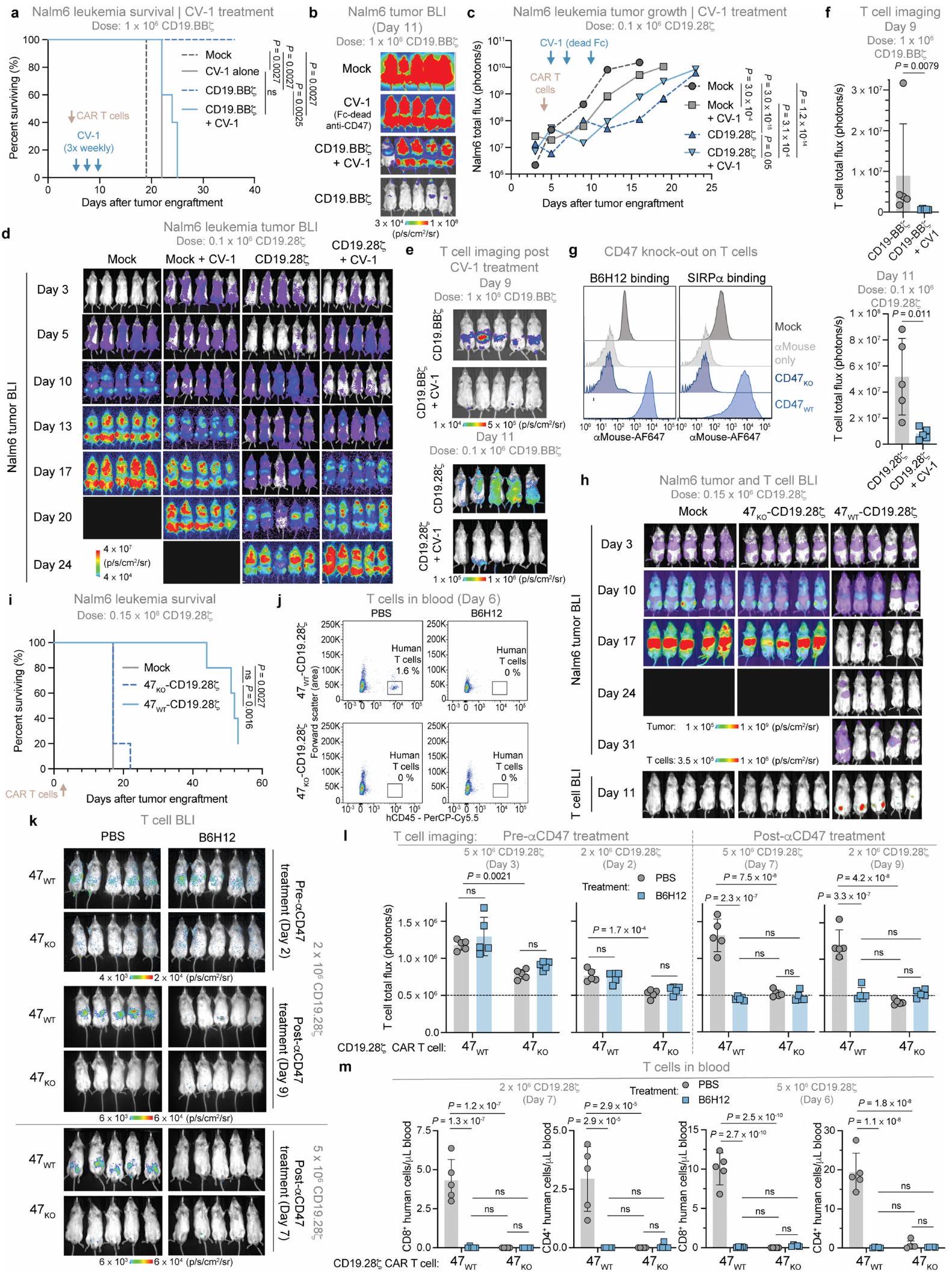
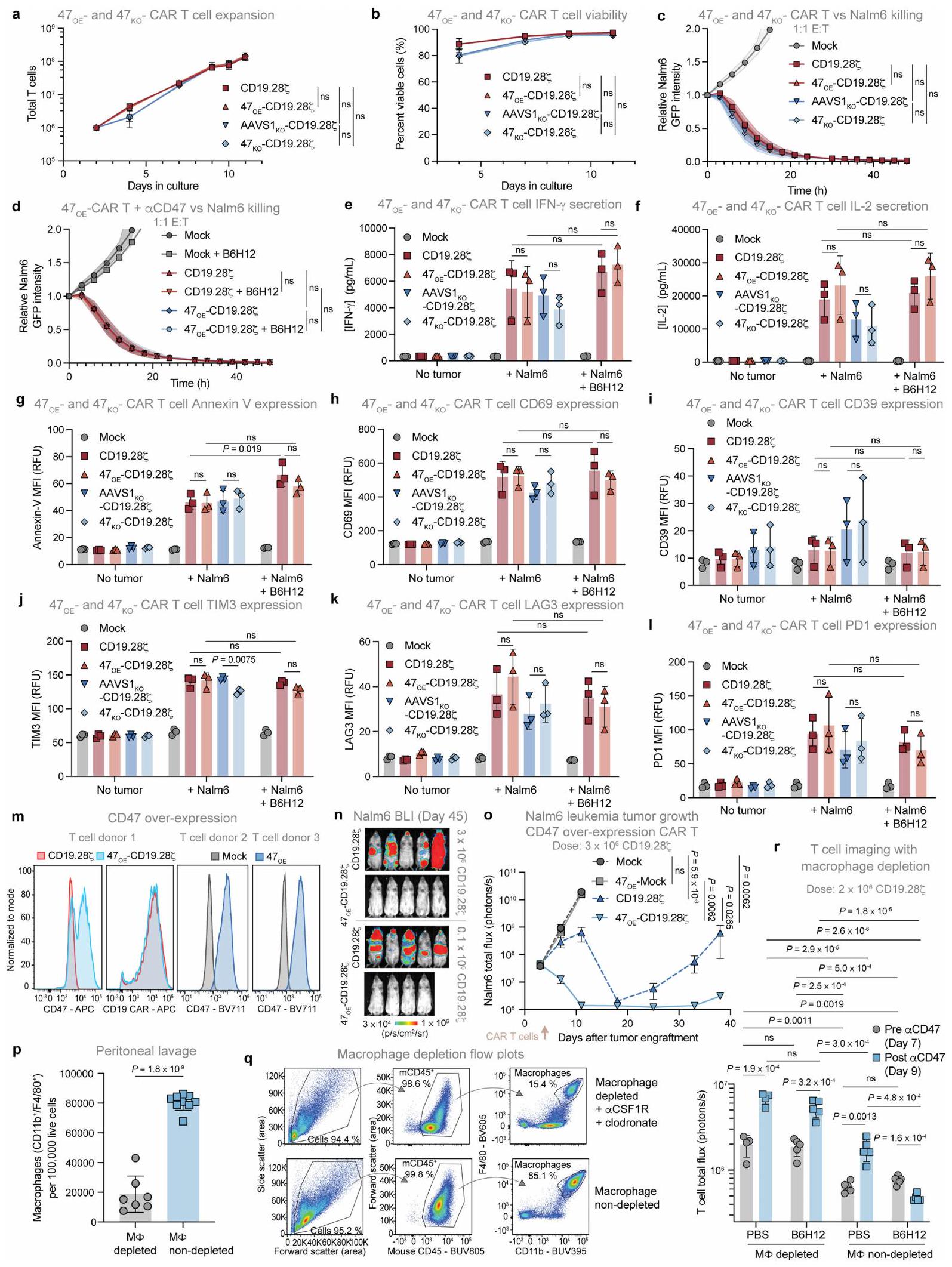
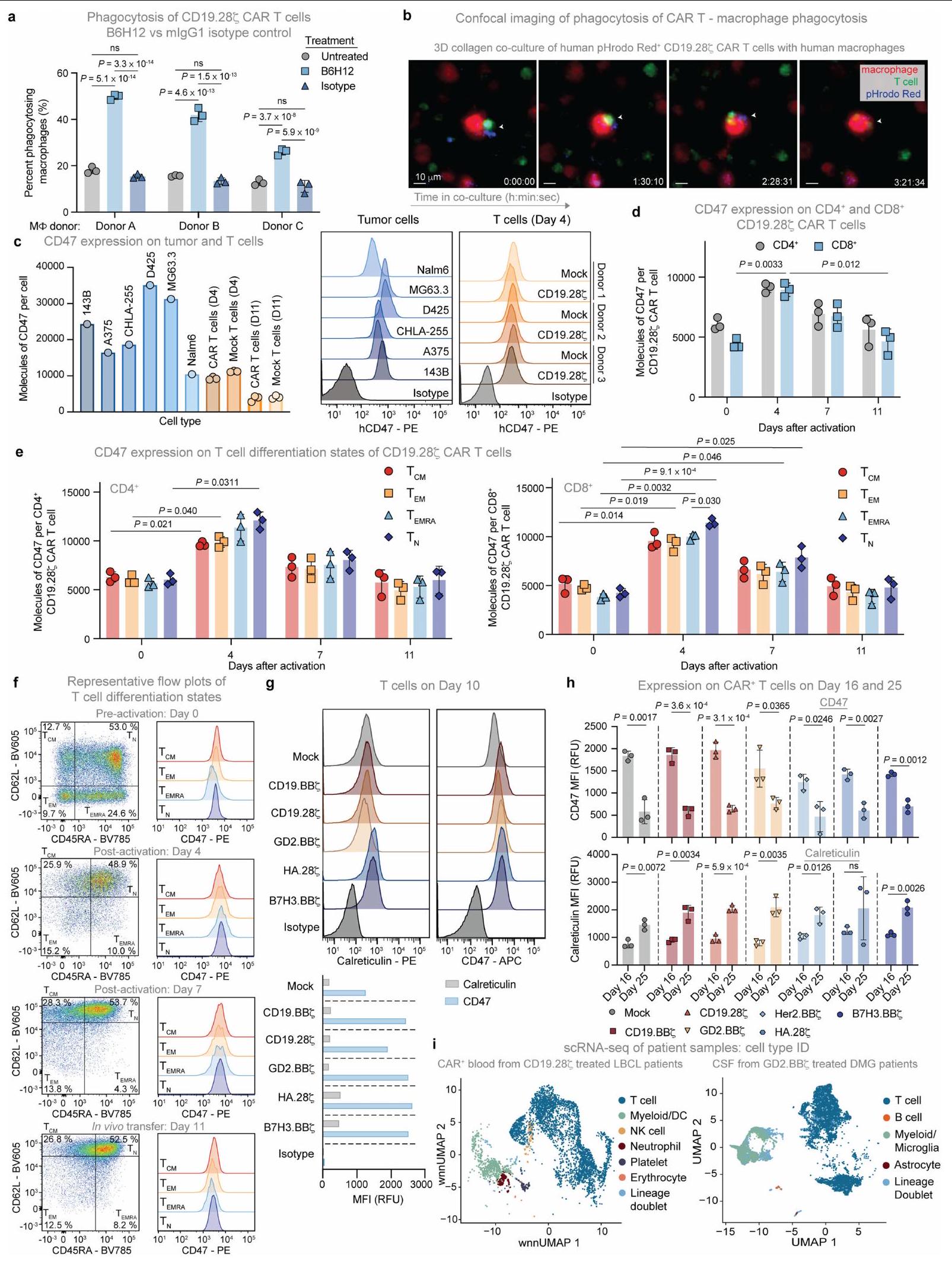
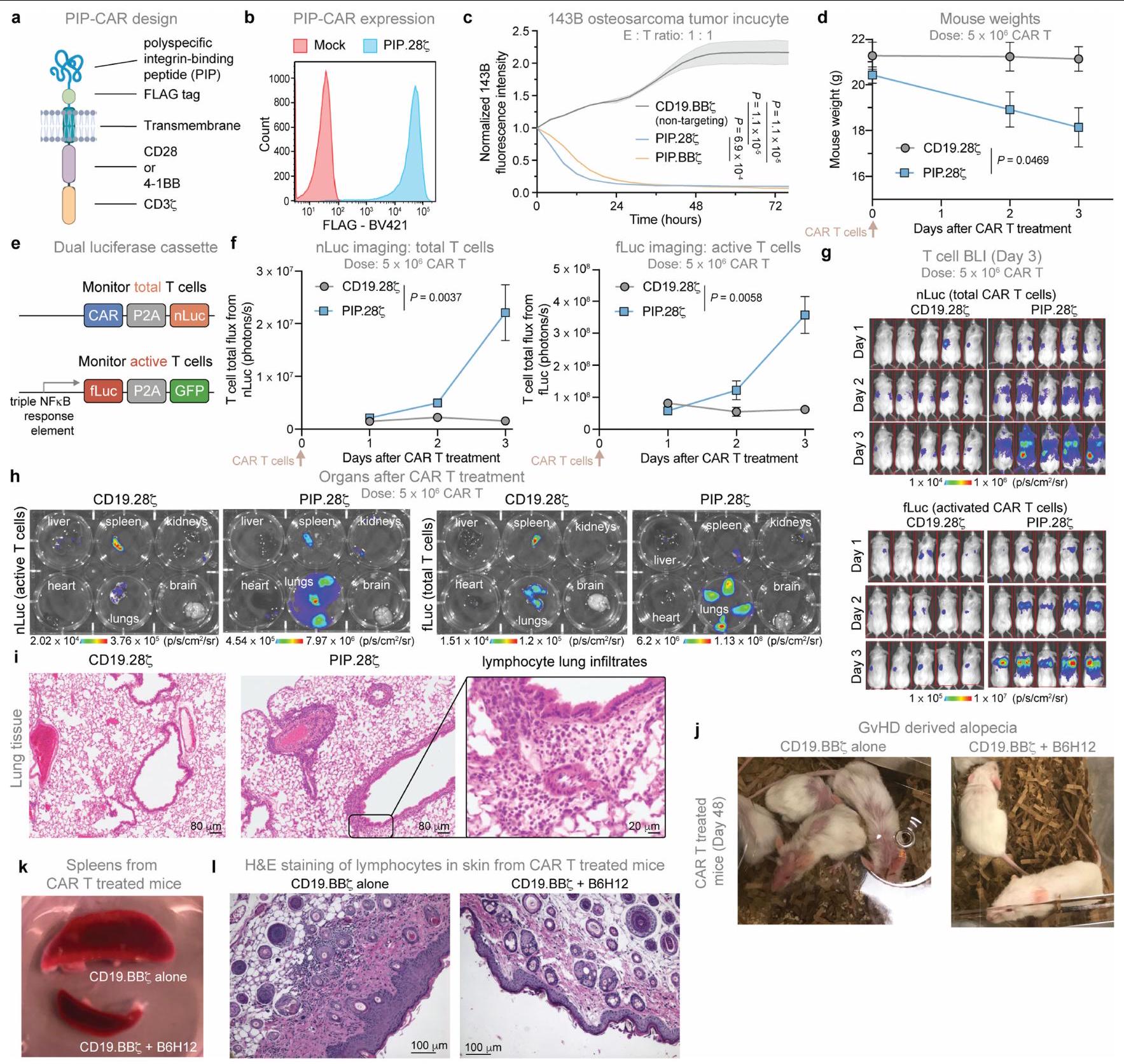
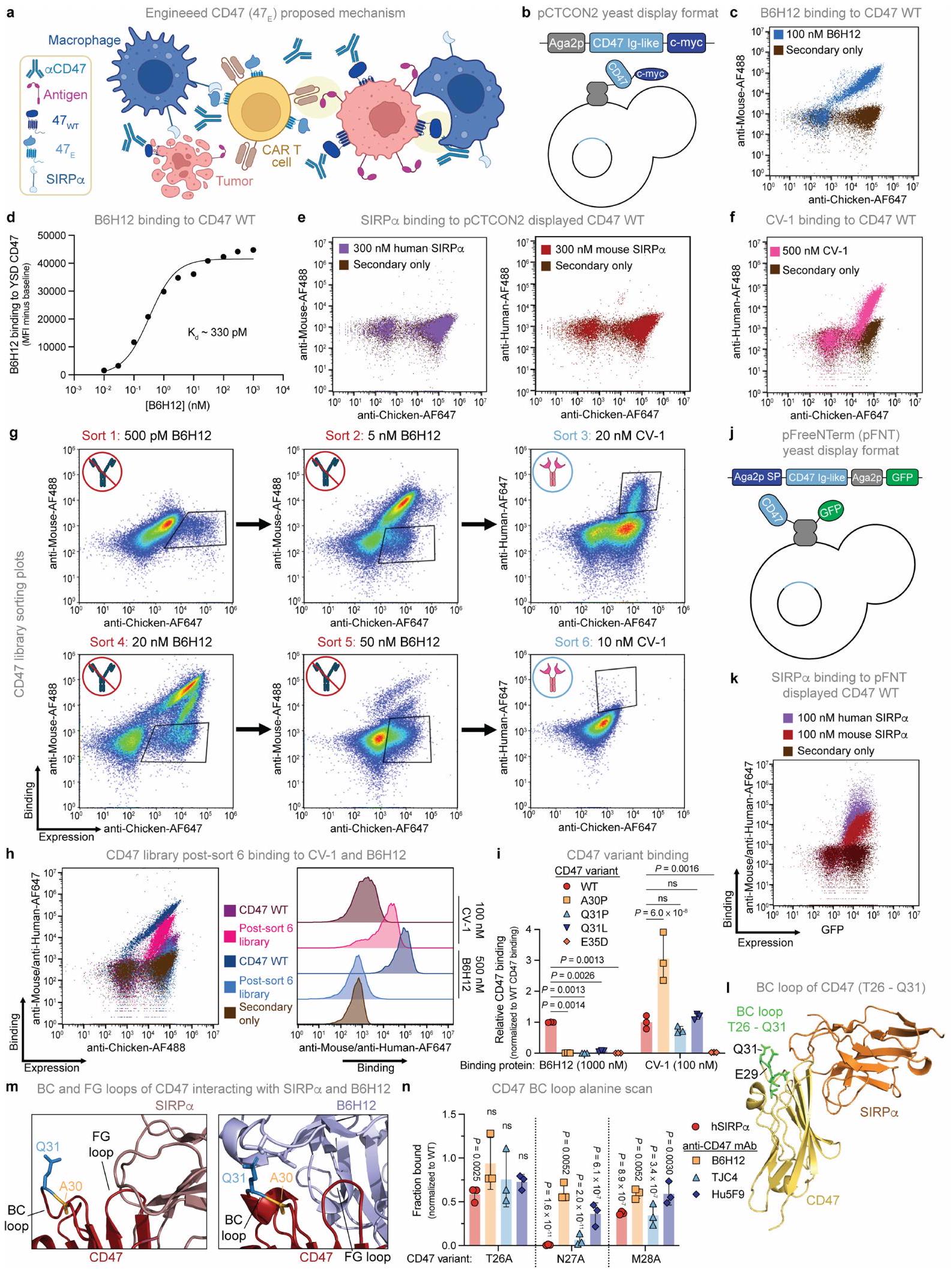
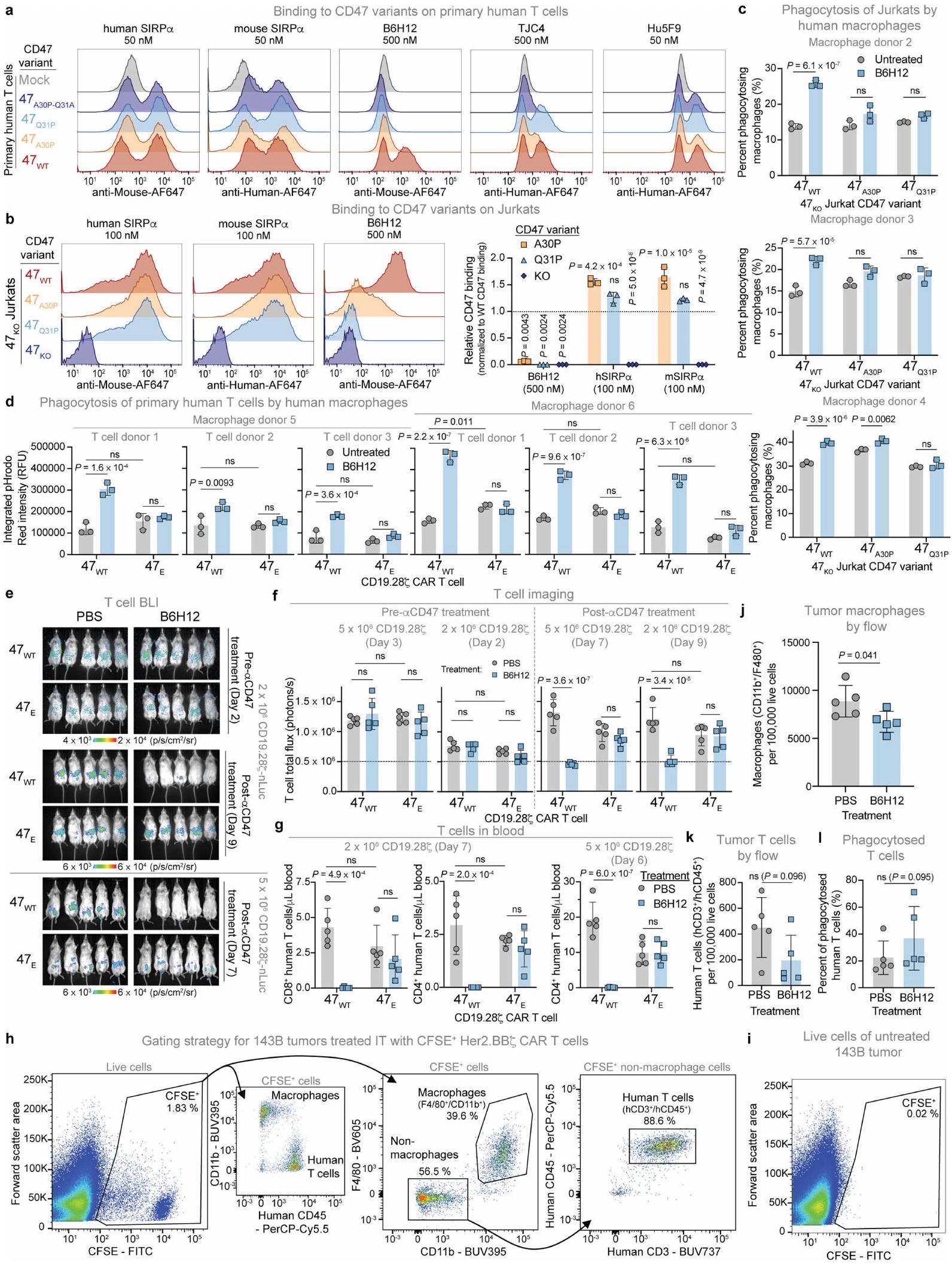
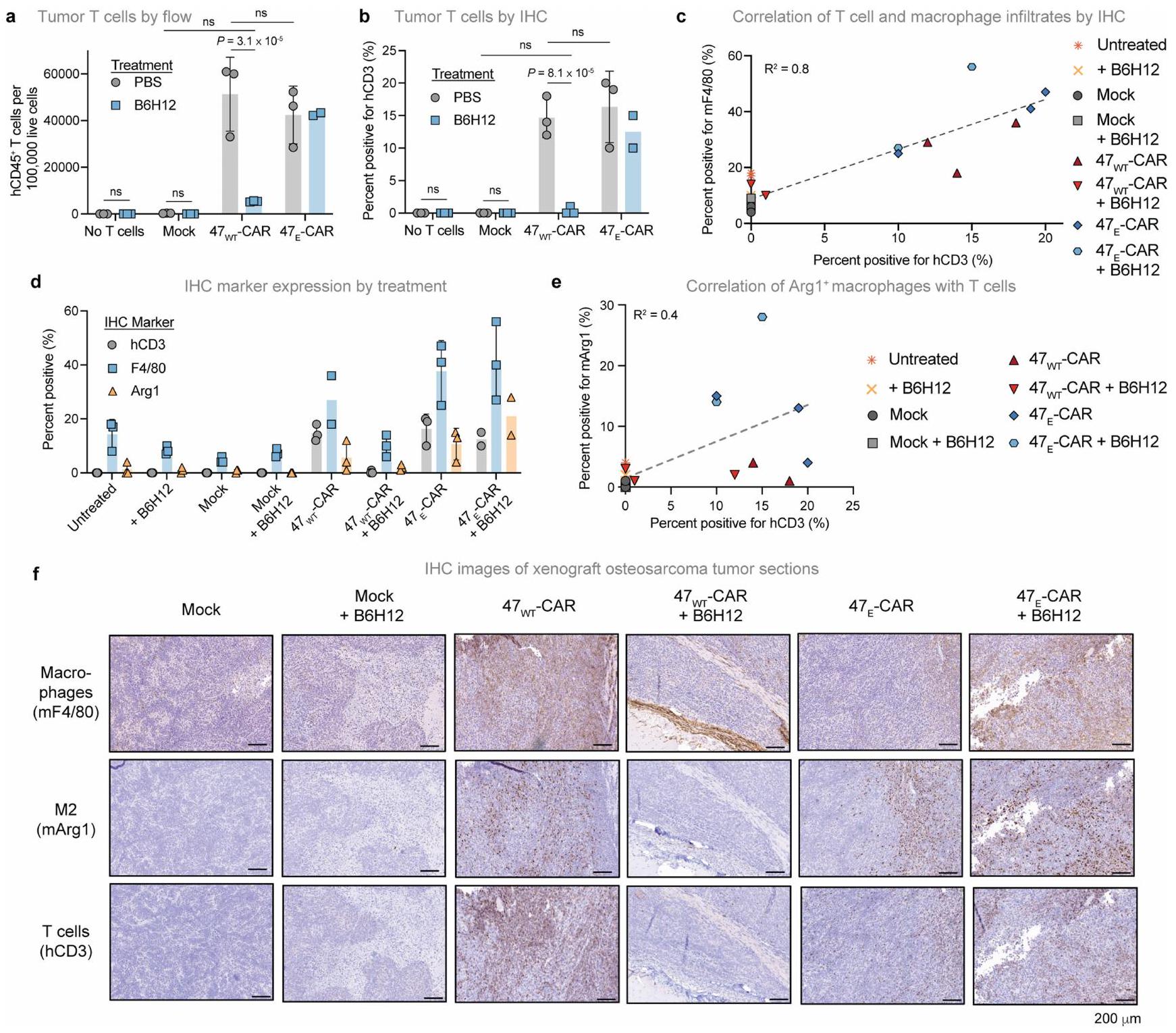
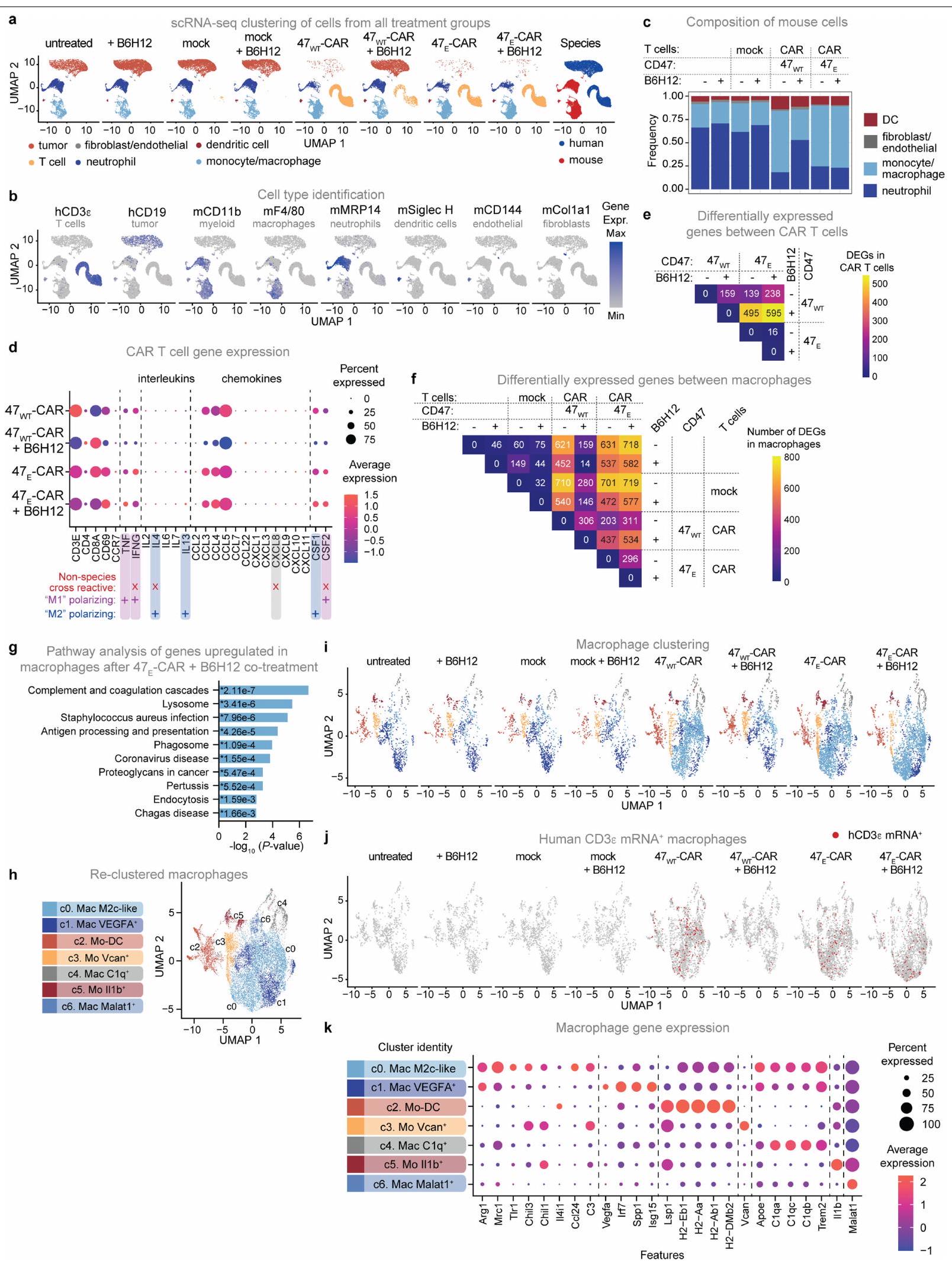
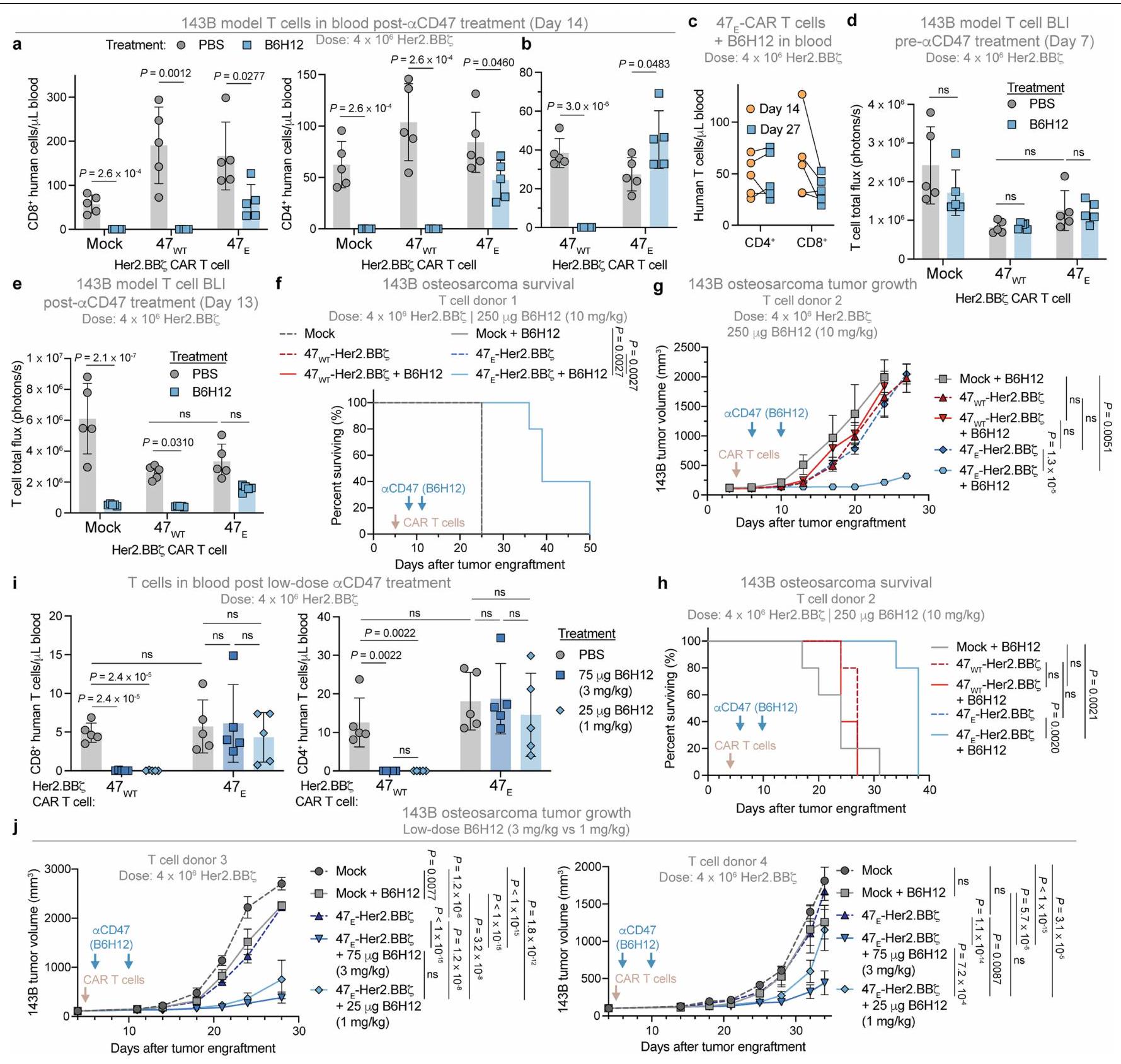
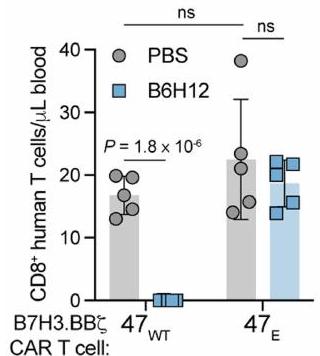 ج
ج
 جوهانا ثيروفاث.
جوهانا ثيروفاث.