الهندسة المتسارعة للإنزيمات بواسطة التعبير الخالي من الخلايا الموجه بتعلم الآلة Accelerated enzyme engineering by machine-learning guided cell-free expression
الهندسة المتسارعة للإنزيمات بواسطة التعبير الخالي من الخلايا الموجه بتعلم الآلة
تاريخ الاستلام: 30 يوليو 2024 تم القبول: 9 ديسمبر 2024 نُشر على الإنترنت: 20 يناير 2025
Grant م. لاندويرجوناثان و. بوغارتكارول ماغالهايسإريك جي. هامرلوندأشتى س. كريم و مايكل سي. جويت □
هندسة الإنزيمات محدودة بتحدي توليد واستخدام مجموعات بيانات كبيرة من علاقات التسلسل والوظيفة للتصميم التنبؤي بسرعة. لمعالجة هذا التحدي، نقوم بتطوير منصة موجهة بواسطة التعلم الآلي (ML) تدمج تجميع الحمض النووي الخالي من الخلايا، والتعبير الجيني الخالي من الخلايا، والاختبارات الوظيفية لرسم خرائط سريعة لبيئات اللياقة عبر مساحة تسلسل البروتين وتحسين الإنزيمات لعدة تفاعلات كيميائية متميزة. نطبق هذه المنصة لتصميم مركبات الأميد من خلال تقييم تفضيل الركيزة لـ 1217 نوعًا من الإنزيمات في 10,953 تفاعلًا فريدًا. نستخدم هذه البيانات لبناء نماذج تعلم آلي معززة باستخدام الانحدار ال Ridge للتنبؤ بأنواع إنزيمات الأميد القادرة على إنتاج 9 أدوية جزيئية صغيرة. على مدار هذه المركبات التسعة، تظهر أنواع الإنزيمات المتنبأ بها بواسطة التعلم الآلي نشاطًا محسّنًا يتراوح بين 1.6 إلى 42 مرة مقارنة بالنموذج الأصلي. تعد إطار العمل الخالي من الخلايا والموجه بواسطة التعلم الآلي بوعد تسريع هندسة الإنزيمات من خلال تمكين الاستكشاف التكراري لمساحة تسلسل البروتين لبناء محفزات حيوية متخصصة بشكل متوازي.
الإنزيمات المصممة جاهزة لتحقيق تأثيرات تحويلية عبر التطبيقات في الطاقةالموادوالطبلإنشاء مثل هذه الإنزيمات، يتم تغيير تسلسل الأحماض الأمينية للبروتين لتعزيز الوظيفة الأصلية أو تسهيل تفاعلات كيميائية جديدة. تتضمن هذه العملية عادةً تحديد الإنزيمات التي تتمتع بمرونة طبيعية وتنوع في التفاعل المطلوب، تليها استخدام التطور الموجه.لسوء الحظ، فإن الأساليب الحالية للتطور الموجه محدودة لأنها غالبًا ما تستطيع فقط رسم علاقات التسلسل-الوظيفة في منطقة ضيقة من فضاء التسلسل. على سبيل المثال، فإن استراتيجيات الفحص عمومًا ذات إنتاجية منخفضة، مما يقيد إعادة أخذ عينات الطفرات في حملات الطفرات المشبعة بالموقع التكرارية ويمكن أن تفوت التفاعلات التآزرية التي تلتقط التآزرات المفيدة الثنائية (أو الأكبر) عندما تكون الطفرات الفردية محايدة أو حتى ضارة.. بالإضافة إلى ذلك، تركز طرق الاختيار للتطور الموجه على الإنزيمات “الناجحة” لتحويل واحد، مما يحد من القدرة على جمع العلاقات الإيجابية والسلبية بين التسلسل والوظيفة لهندسة ردود فعل مماثلة..
ظهرت التقنيات الحاسوبية لتسريع أساليب التطور الموجه الحالية. يمكن أن يؤدي تصميم البروتين من الصفر إلى إنشاء إنزيمات جديدة على الطبيعة، ولكن تنوع الكيميائيات و تظل التطبيقات محدودةتم استخدام نماذج التعلم الآلي (ML) لاكتشاف الإنزيمات من خلال استنتاج اللياقة بناءً على المتجانسات ذات الصلة و/أو تسلسلات البروتين من جميع الكائنات الحية (ما يُعرف بتوقع عدم وجود بيانات سابقة) وكذلك للتنقل في مشاهد اللياقة البروتينية بناءً على بيانات اللياقة المختبرة (مثل الانحدار غير الخطي باستخدام ترميزات واحدة ساخنة محددة للموقع).بينما تُظهر طرق هندسة الإنزيم المدعومة بالتعلم الآلي وعودًا، لا يزال بناء مجموعات بيانات بسرعة لاستكشاف الفضاء التسلسلي الواسع يمثل تحديًا.، خاصةً بالنظر إلى أن معظم الروابط بين النمط الجيني والنمط الظاهري تُفقد في حملات هندسة الإنزيمات عالية الإنتاجية .
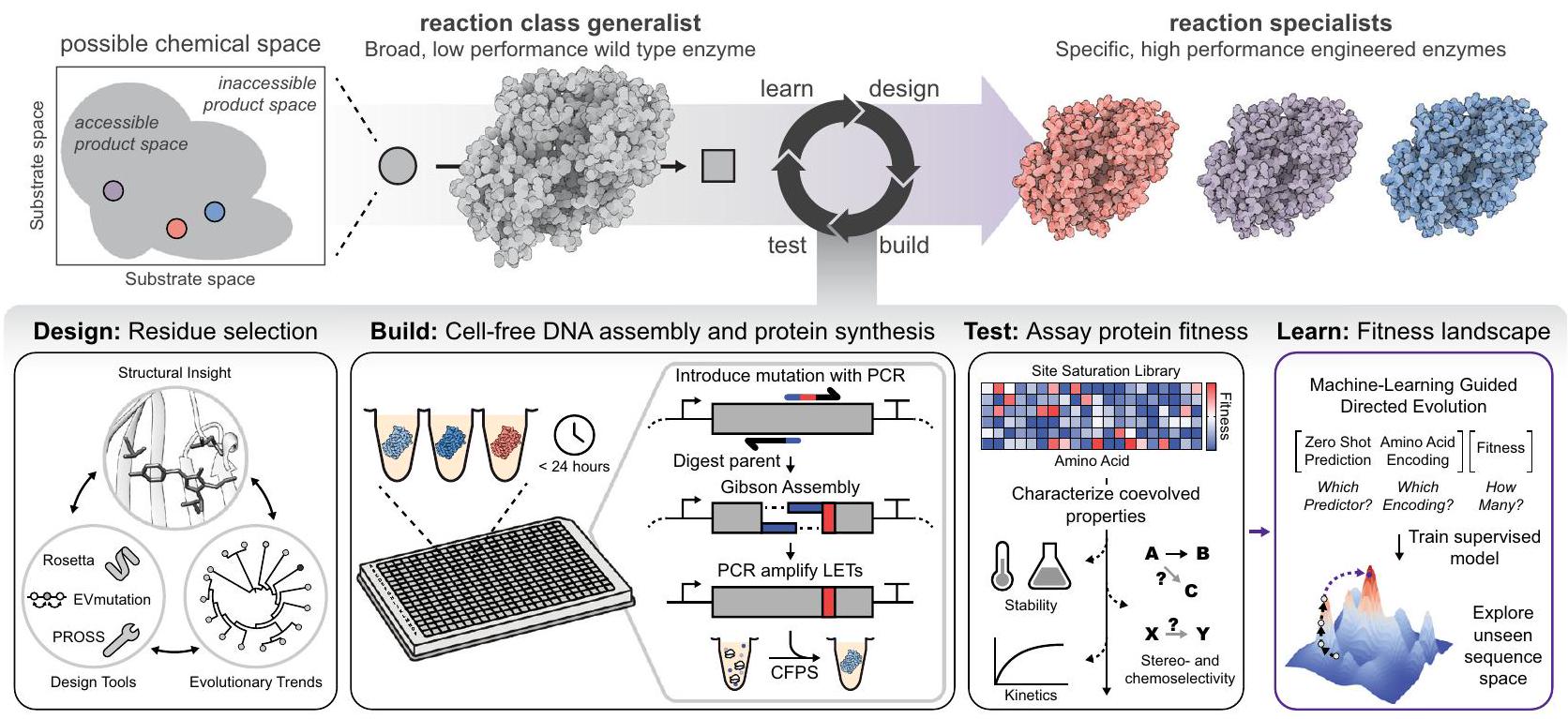
هنا، قمنا بتطوير نهج عالي الإنتاجية وموجه بواسطة التعلم الآلي لتمكين استكشاف المناظر الطبيعية للملاءمة عبر مناطق متعددة من الفضاء الكيميائي لتصميم المحفزات الحيوية (الشكل 1). ميزة رئيسية في نهجنا هي استخدام أنظمة التعبير الجيني الخالية من الخلايا (CFE) للسماح بالتخليق السريع والاختبار الوظيفي للبروتينات.في سير عمل التصميم-البناء-الاختبار-التعلم (DBTL). يقوم هذا الإطار أولاً برسم علاقات التسلسل-الوظيفة لمتغيرات الإنزيم مع طفرات من رتبة واحدة لتحويل كيميائي محدد تم تحديده من خلال تقييم تعددية الركيزة الإنزيمية. ثم تُستخدم هذه البيانات لتناسب نماذج التعلم الآلي المعززة بالانحدار المتدرج الخاضع للإشراف مع
الشكل 1 | منصة هندسة إنزيم خالية من الخلايا موجهة بواسطة التعلم الآلي. يوضح المخطط كيفية تطبيق سير عمل التصميم-البناء-الاختبار-التعلم لرسم خرائط سريعة لمشاهد التسلسل-الوظيفة. يتم اختيار البقايا المحتملة التي توجه تحفيز الإنزيم بشكل منطقي استنادًا إلى رؤى هيكلية، واتجاهات تطورية، وأدوات حسابية (مثل ROSETTA).تحور EVبروسيتطفير التشبع في الموقع وخلايا- تتم التعبيرات الجينية الحرة في أقل من 24 ساعة لإنشاء مكتبات محددة التسلسل (بناء). يمكن بعد ذلك فحص المكتبات للحصول على مقاييس ملاءمة البروتين المرغوبة (اختبار). تُستخدم المعلومات من مرحلة الاختبار، بما في ذلك الفشل، لتحديد بقايا الأحماض الأمينية المهمة وظيفيًا التي تعود بالتغذية الراجعة على التصاميم التكرارية، بالإضافة إلى ملاءمتها لنماذج التعلم الآلي (تعلم). مُتنبئ لياقة صفرية تطورية واستنباط طفرات من مرتبة أعلى مع نشاط متزايد. من المهم أن نماذج التعلم الآلي يمكن تشغيلها على وحدة المعالجة المركزية لجهاز كمبيوتر عادي مما يجعل نهجنا بالكامل سهل الاستخدام ومتاحة. تقدم طريقتنا تكملة لأدوات استراتيجيات التطور الموجه المتزايدة، مثل تلك التي تتنبأ بالميزات الحفازة للإنزيمات..
قمنا بتطبيق إطار عملنا لتنفيذ التطور المتباين، محولين إنزيمًا عامًا يشكل روابط الأميد إلى عدة إنزيمات متخصصة ومتميزة. التكوين البيوكاتاليتيكي لروابط الأميد – وهو نمط موجود بشكل شائع في الأدوية، والكيماويات الزراعية، والبوليمرات، والعطور، والنكهات، وغيرها من المنتجات عالية القيمة.-يمكن أن تقدم مزايا فريدة على نظيراتها الاصطناعية (مثل، ظروف تفاعل معتدلة والاختيارات الكيميائية، والستيريو، والإقليمية) وتسهيل التصنيع الحيوي المستداممcbA من ماريناكتينوسبورا ثيرموترولارنسهو إنزيم تمثيل الأمييد المعتمد على ATP والذي يشارك في تخليق المستقلبات الثانوية لمارينكاربولين.. McbA، وقرينها القريب ShABSلقد أظهرت أنها تتمتع بنطاق ركيزة مريح، حيث تقبل العديد من الأحماض والأمينات البسيطة الموجودة عادة في الأدوية.. تشير هذه الخلفية إلى أن McbA يمكن أن يكون نقطة انطلاق مرنة لتطوير إنزيم عام إلى متخصصين في تفاعلات متعددة، كل منهم قادر على تنفيذ تفاعل كيميائي مختلف. كانت طريقتنا الموجهة بواسطة التعلم الآلي قادرة على تحسين نشاط إنزيم McbA مقارنةً بالإنزيم البري بنسبة تتراوح بين 1.6 و 42 مرة لإنتاج تسعة مركبات.
النتائج
استكشاف مشهد التخليق البيوكاتاليتيكي لـ McbA
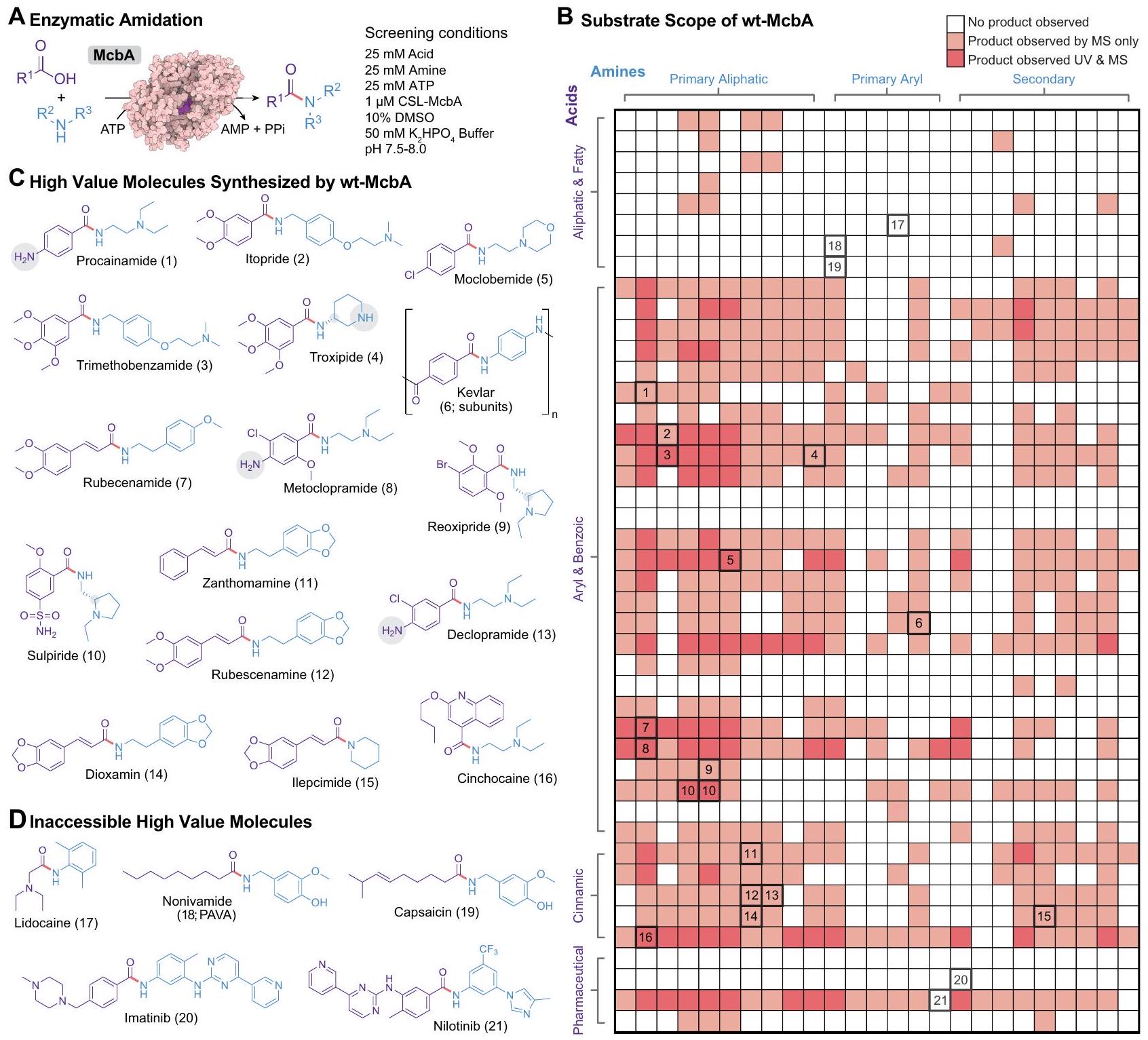
كان الهدف من هذا العمل هو تطوير سير عمل موجه بواسطة التعلم الآلي (ML) لتسريع حملات التطور الموجه المتزامنة للتفاعل البيوكاتاليتيكي من خلال تقليل عبء الفحص. تطلب هذا الهدف توليد بيانات تسلسل-ملاءمة لتحولات كيميائية فريدة، والتي يمكن من خلالها إنشاء نماذج تعلم آلي تنبؤية. لتحديد التفاعلات ذات الاهتمام، استكشفنا أولاً مساحة تفاعل الأميدية الممكنة للنوع البري من McbA (wt-McbA) من خلال تقييم تعددية الركيزة الإنزيمية (الشكل 2A). درسنا مجموعة واسعة من الركائز التي انحرفت عن الأحماض الحلقية غير المتجانسة والأمينات الأولية أو العطرية المفضلة من قبل wt-McbA. شملت هذه الركائز الأمينات الأولية والثانوية والألكيلية، مركبات عطرية، هيكل دوائي معقد، أنواع فقيرة أو غنية بالإلكترونات، وركائز تحتوي على عناصر غير متجانسة أخرى، هالوجينات، و”نيوكليوفيلات” أو “إلكتروفيلات” غير محمية. تم تضمين ركائز أكثر تحديًا (مثل الأحماض والأمينات المعقدة غير المتجانسة، الإنزيمات المتناظرة، وركائز تحتوي على كل من الأحماض والأمينات أو أحماض وأمينات متعددة) لتحديد القيود والتفضيلات الفطرية لـ wt-McbA. قمنا بإجراء 1100 تفاعل فريد مع تركيز إنزيم منخفض. ) وتركيز عالٍ من الركيزة ( 25 مللي مول )، مما يغطي العديد من الجزيئات ذات القيمة المعروفة بما في ذلك الأدوية والعطور والبوليمرات (الشكل 2B).
من المثير للاهتمام أن wt-McbA أظهر تحملًا لمجموعات وظيفية وهندسية “غير محمية” متعددة. بشكل عام، كانت الأحماض الأليفاتية تتحمل بشكل ضعيف بينما كانت الأحماض الأريلية والحمض البنزويكي والأحماض السينامكية تعتبر ركائز مقبولة بسهولة. كانت الأحماض الأريلية المشحونة استثناءً فريدًا وعادة ما كانت مرتبطة بعدد قليل جدًا من الأمينات. على العكس من ذلك، كان wt-McbA يرتبط بسهولة بالأمينات الأليفاتية الأولية والثانوية لكنه واجه صعوبة مع الأمينات الأريلية. لاحظنا أن McbA كان قادرًا على تخليق 11 مركبًا صيدلانيًا بالإضافة إلى العشرات من الجزيئات الهجينة (الشكل 2C)، تتراوح من كميات ضئيلة يمكن اكتشافها فقط بواسطة مطيافية الكتلة (MS) إلى التحويل. في هذه التفاعلات، اكتشفنا كل من الانتقائية الاستيريو (على سبيل المثال، تفضيل قوي لتخليق S-sulpiride على Rsulpiride) وتفضيلات صارمة للانتقائية الكيميائية والإقليمية (على سبيل المثال، الركائز التي تحتوي على كل من الأحماض والأمينات لا تتبلمر). نظرًا لأن آلية تفاعل McbA تبدأ أولاً مع أدينيل الحمض الكربوكسيلي، لاحظنا أيضًا عدة حالات حيث تم ملاحظة فقط وسيط acyl-AMP. العديد من الجزيئات المهمة لا يتم تخليقها بواسطة McbA من النوع البري (الشكل 2D). تشير هذه المنتجات غير القابلة للوصول إلى أن McbA قد لا يكون قادرًا على التفاعل مع الأحماض الأليفاتية والدهنية (على سبيل المثال، nonivamide، capsaicin) أو ركائز كبيرة معينة (على سبيل المثال، imatinib، nilotinib). يمكن أن يساعد فهم أفضل لنطاق الركيزة في توجيه أعمال هندسة الإنزيم المستقبلية.
هندسة البروتين الخالي من الخلايا لفحص مكتبات البروتين المعرفة بالتسلسل بسرعة
مع تحديد التحولات الكيميائية المحددة من تقييمنا لتنوع الركيزة الإنزيمية، أردنا بعد ذلك توليد كميات كبيرة من بيانات علاقة التسلسل-الوظيفة لإنزيمات McbA الطافرة لتدريب نماذج ML للتنبؤ بالمتغيرات عالية النشاط.
الشكل 2 | يشير الفضاء الكيميائي المتنوع القابل للوصول لـ McbA إلى محفز حيوي قادر على تخليق عدة جزيئات ذات قيمة عالية. مخطط التفاعل وظروف الفحص لاستكشاف نطاق الركيزة لـ McbA لتخليق الأميدات الإنزيمي. تم التعبير عن McbA باستخدام CFE وتم بدء التفاعل بإضافة تركيبات مختلفة من الأحماض والأمينات.فحص الركيزة الشامل لـ McbA، تم تحليله باستخدام HPLC (RP) المعكوسة ( ). اللون الأحمر الداكن يتوافق مع منتج يمكن ملاحظته بواسطة الأشعة فوق البنفسجية (UV)
الامتصاص بينما يتوافق اللون الأحمر الفاتح مع كميات ضئيلة يمكن اكتشافها فقط بواسطة مطيافية الكتلة. يمكن العثور على قائمة كاملة من الركائز في الشكل S1. ج من بين 21 جزيءًا ذا قيمة عالية كان ممكنًا في نطاق الركيزة، لاحظنا أن McbA كان قادرًا على تخليق 16 (11 منها أدوية جزيئية صغيرة). د مثال على الجزيئات ذات القيمة العالية التي لم يكن McbA قادرًا على تخليقها تحت ظروف التفاعل المختبرة. تم توفير بيانات المصدر كملف بيانات مصدر.
للقيام بذلك، قمنا بتنفيذ نهج تخليق البروتين الخالي من الخلايا الذي لا يتطلب خطوات تحويل واستنساخ شاقة (الشكل 1). اعتمد نهجنا على تجميع الحمض النووي الخالي من الخلايا و لبناء مكتبات بروتين معرفة بالتسلسل مشبعة بالمواقع. كان هذا سير العمل يتكون من خمس خطوات: (i) تمهيد الحمض النووي يحتوي على عدم تطابق نوكليوتيد يقدم طفرة مرغوبة من خلال PCR، (ii) يقوم DpnI بهضم البلازميد الأصلي، (iii) يشكل تجميع جيبسون الجزيئي بلازميدًا متحورًا، (iv) يقوم PCR الثاني بتكبير قوالب التعبير الحمض النووي الخطية (LETs)، و(v) يتم التعبير عن البروتين المتحور من خلال CFE. بهذه الطريقة، يمكن بناء مئات إلى آلاف من الطفرات البروتينية المعرفة بالتسلسل في تفاعلات فردية خلال يوم واحد، ويمكن تراكم الطفرات من خلال تكرارات سريعة لسير العمل. يتجنب نهجنا أي تحيزات محتملة في مكتبات التشبع بالمواقع النموذجية التي تنشأ من استخدام تمهيدات متدهورة.
قمنا بالتحقق من صحة سير العمل الخاص بنا باستخدام بروتين الفلورسنت الأخضر الفائق الاستقرار المعروف جيدًا (muGFP) من خلال استهداف أربعة بقايا معروفة بأنها مهمة للاستقرار والفلورية (الشكل S2). عند بناء مكتبتنا المشبعة بالمواقع التي تستهدف هذه البقايا الأربعة (77 متغيرًا)، وجدنا تحملًا عاليًا لانحرافات تصميم التمهيد (على سبيل المثال، التداخل المتجانس، درجات حرارة الانصهار) (الشكل S3، S4) وأن LETs من متغيرات muGFP منحت جميع الطفرات المرغوبة (الشكل S5). أشارت البروتينات القابلة للذوبان بطول كامل إلى أن التغيرات في الفلورية لم تكن ناتجة عن تغييرات في التعبير أو الذوبانية (الشكل S6). إن رسم خريطة المنظر المشبع بالمواقع للبروتين لا يسلط الضوء فقط على البقايا التي تعتبر حاسمة للملاءمة (على سبيل المثال، البقايا التي تشكل الفلورسنت وتؤثر على تعبئة النواة الكارهة للماء كانت غير قابلة للتحمل للطفرات ) ولكن أيضًا يوفر رؤى حول القابلية العامة للطفرات في المواقع.
بعد التحقق من الصحة، طبقنا سير العمل الخاص بنا على McbA لتوليد بيانات علاقة التسلسل-الوظيفة التي يمكن أن تدرب نماذج ML لتسريع حملات الهندسة لدينا. قمنا في البداية بهندسة McbA لتخليق ثلاث جزيئات ذات قيمة عالية تم تحديدها من خلال تقييم نطاق الركيزة لدينا: (i) مثبط مونوامين أوكسيداز A، moclobemide، بسبب تنوع McbA العالي تجاه هذا التفاعل (الشكل 2C (5); تحويل wt); (ii) ميتوكلوبراميد، بسبب التحدي الفريد الذي تطرحه مكون الحمض الذي يحتوي على أمين حر يمكن أن يتنافس مع الأمين المقصود (الشكل 2C (8); 3% تحويل wt); و(iii) سينشوكائين، الذي يحتوي على مكون حمضي فريد ولكنه يشترك في نفس جزء الأمين مثل ميتوكلوبراميد (الشكل 2C (16); تحويل wt). من خلال إجراء هذه الحملات الهندسية بالتوازي، كنا نأمل في استنتاج الطفرات التي تؤثر على خصوصية الركيزة للأمين (الطفرات المشتركة) والحمض (الطفرات الفريدة) التي قد تؤدي إلى مبادئ تصميم عامة لـ McbA.
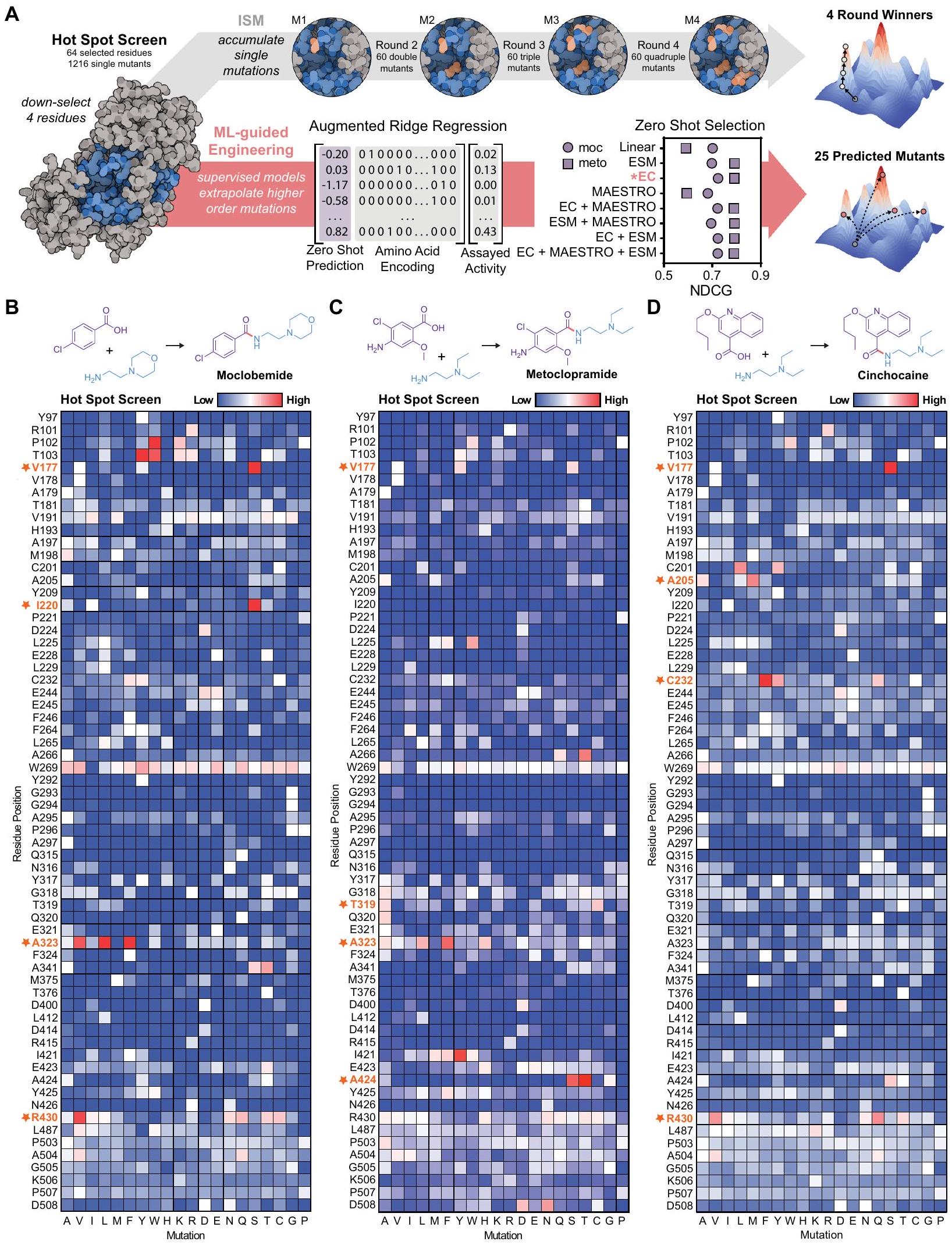
باستخدام تركيزات ركيزة مرتفعة نسبيًا وتحميل إنزيم منخفض كخطوة نحو ظروف تفاعل أكثر صلة بالصناعة (الشكل S7)، قمنا بإجراء فحص النقاط الساخنة (HSS) لكل جزيء يتكون من طفرات التشبع بالمواقع على مساحة تسلسل واسعة لتحديد مواقع البقايا التي، عند تحويرها، تؤثر إيجابيًا على الملاءمة (الشكل 3A). مسترشدين بالهيكل البلوري لـ McbA (PDB: 6SQ8)، اخترنا 64 بقايا تغلق تمامًا الموقع النشط وأنفاق الركيزة المحتملة (على سبيل المثال، البقايا داخل من الركائز الأصلية المربوطة). كشفت HSS الخاصة بنا لهذه البقايا (64 بقايا أحماض أمينية طفرات فردية إجمالية) عن عدة بقايا كان لها تأثير إيجابي على تخليق moclobemide (الشكل 3B)، ميتوكلوبراميد (الشكل 3C)، وسينشوكائين (الشكل 3D) عند تحويرها مقارنة بـ wtMcbA كما تم قياسه بواسطة الكروماتوغرافيا السائلة-مطيافية الكتلة (LC-MS).
تعبير خالي من الخلايا موجه بواسطة ML لهندسة البروتين
مع وجود مجموعة بيانات كبيرة في متناول اليد لعدة طفرات فردية متميزة من McbA، بدأنا في الاستفادة من نماذج ML لتسريع هندسة McbA لإنتاج جزيئات صغيرة عبر مناطق متنوعة من الفضاء الكيميائي. كانت الفكرة الرئيسية هي استخدام بيانات الطفرات الفردية من HSS لاستنتاج طفرات من مرتبة أعلى مع نشاط متزايد (الشكل 3A). لتحقيق هذا الهدف، اخترنا ملاءمة نماذج الانحدار المتزايد مع بياناتنا – حيث أن هذه النماذج بسيطة وقد أظهرت سابقًا أنها تتفوق على النماذج الأكثر تعقيدًا في هندسة البروتين – مما يسمح لنا بالتنبؤ بطفرات من مرتبة أعلى مع نشاط متزايد.
قمنا أولاً باختيار هيكل نموذج تنبؤي. كانت تمثيلات ميزات متغيرات McbA تتكون من ترميزات أحماض أمينية محددة بالموقع متصلة بتنبؤ ملاءمة بدون تجارب . اعتبرنا عدة ترميزات لأحماض الأمينية، تتراوح من الترميزات البسيطة ذات النقطة الواحدة إلى أوصاف أكثر تعقيدًا تحاول دمج الخصائص الفيزيائية الكيميائية للأحماض الأمينية . كما استكشفنا مؤشرات ملاءمة متغيرات البروتين المرجعية لدمج تنبؤات صفرية قائمة على عالمية، تطورية، وبنية. اختبرنا ثلاثة مؤشرات ملاءمة محددة: نموذج التحويل ESM-1b المدرب على قاعدة بيانات UniRef50 (عالمي)، نموذج كثافة الاحتمال EVmutation (EC) المدرب على MSA من تسلسلات مرتبطة تطوريًا (تطوري)، وMAESTRO لتقدير التغيرات المستندة إلى الهيكل في طاقة الانحلال الحرة (الهيكلية). تم إجراء التدريب وضبط المعلمات الفائقة باستخدام بيانات الطفرات الفردية. ) من HSS (أعلى أربع نقاط ساخنة؛ الشكل 3B، C).
بالتوازي، قمنا بإجراء حملة تطور موجهة أكثر تقليدية على كل منتج أميد (موكلوبيميد، ميتوكلوبراميد، وسينشوكائين) من خلال الطفرات المشبعة التكرارية (ISM). سيوفر ذلك طفرات أعلى قيمة للتحقق من صحة أداء النموذج وتقييمه نظرًا لهدفنا المتمثل في استقراء الطفرات من الطفرات الفردية إلى الطفرات الأعلى.
بالنسبة للموكلابيمايد، اخترنا ستة بقايا من HSS فوق عتبة نشاط تبلغ 1.5 ضعف النشاط مقارنة بالنمط البري لإجراء طفرات من خلال ثلاث جولات من ISM (الشكل S7). قمنا أولاً بتثبيت الطفرة الأعلى من HSS. (V177S) وأجرينا طفرات تشبع الموقع على خمسة بقايا إضافية. من خلال إعادة إدخال الطفرات الثابتة سابقًا في الجولات اللاحقة، استكشفنا التفاعلات المحتملة بين الطفرات (على سبيل المثال، تم تشبع S177 في الجولة الثانية من ISM، نظرًا لأن V177S تم دمجه قبل A323F). بالإضافة إلى ذلك، استكشفنا تمامًا جميع الطفرات المزدوجة التوافقية لأعلى بقايا اثنتين، والتي أظهرت تأثيرات إضافية على تخليق الموكلوباميد (الشكل S7). بعد ثلاث جولات من ISM، حيث اخترنا الطفرة الأكثر نشاطًا في كل جولة لبدء الجولة التالية، حددنا طفرة رباعية. ) مع زيادة النشاط من لـ wt-McbA إلىالتحويل – لتخليق موكلوبيميد (الشكل S7). قمنا بتوصيف المعلمات الحركية الظاهرة في الحالة المستقرة واستقرار هذه الطفرات الإنزيمية من كل جولة من ISM. على وجه التحديد، قمنا بتعبير وتنقية وتقييم كل متغير من McbA، حيث لاحظنا زيادة بمقدار 42 مرة في الكفاءة التحفيزية من wt-McbA إلى qm-Mcba.ارتفع من 18.2 إلى ) للأمين (الأشكال S8، S9). لم يتغير نقطة الانصهار بشكل ملحوظ بين wt-McbA و qm-McbA لكن الطفرة الثانية (A323F) زادتبواسطةعند إضافته إلى الطفرة الأولى (V177S) (الشكل S10). بالإضافة إلى ذلك، أظهرنا أننا نستطيع إنتاج كميات من الميليجرام من موكلوبيميد فيرد فعل (العائد المعزول) وأكدت التركيب بواسطة الرنين المغناطيسي النووي (الأشكال S11، S12).
بالنسبة للميتوكلوبراميد، اخترنا 10 بقايا لحملة ISM، والتي كانت جميعها فوق عتبة نشاط تبلغ 1.25 ضعف النشاط مقارنة بالنمط البري. أسفرت ثلاث جولات من ISM للميتوكلوبراميد عن طفرات رباعية أظهرت نشاطًا محسّنًا يقارب 30 ضعفًا مقارنة بـ wt-McbA (الشكل S13).
كانت الحملة من أجل السينشوكائين أكثر صعوبة في التنقل، وفشلنا في ملاحظة الطفرات المفيدة بخلاف الطفرة المزدوجة، على الرغم من اتخاذ مسارات ISM متعددة (الشكل S14). هذه النتيجة (أي، مواجهة طرق مسدودة خلال ISM) دعمت الحاجة إلى تضمين نماذج التعلم الآلي في إطار عملنا التي قد تلتقط التفاعلات الإيبيستاتية. استخدمنا بيانات ISM لموكلاوبيميد وميتوكلوبراميد التي تحتوي على طفرات مزدوجة وثلاثية ورباعية.للموكليبيميد ولتقييم أداء كل نموذج، بينما سيوفر السينشوكائين اختبار ضغط فريد لنموذجنا الأعلى أداءً الذي تم تحديده.
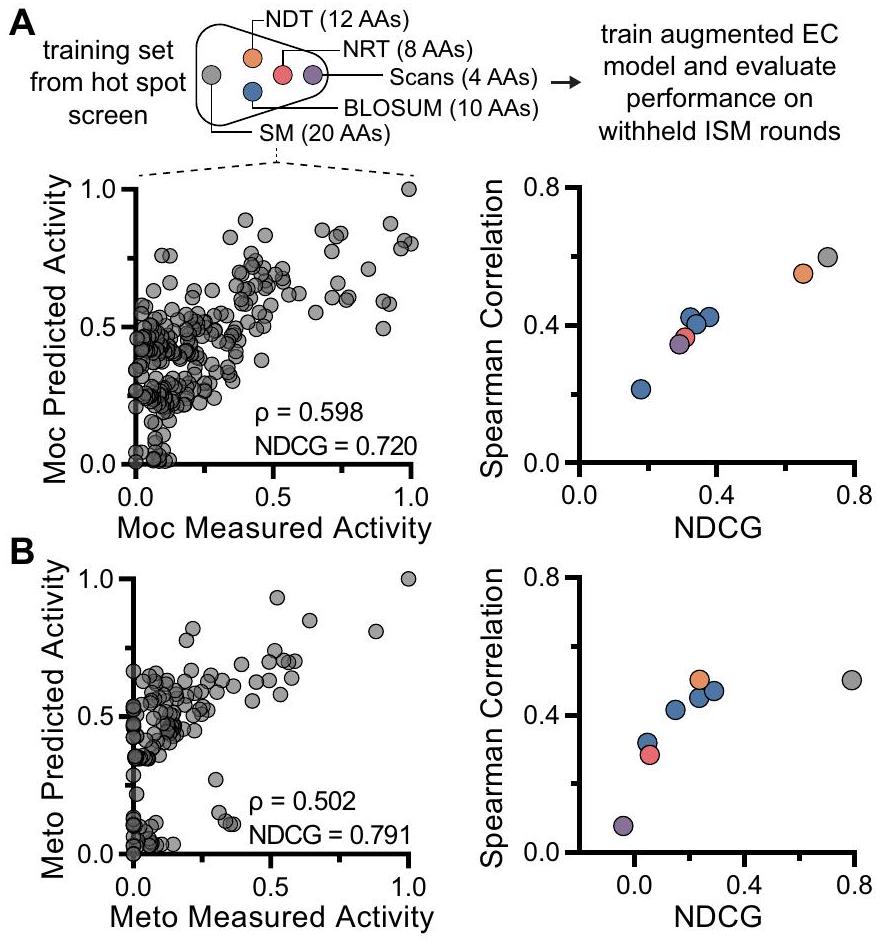
تم تقييم أداء توقع النموذج أولاً باستخدام المكسب التراكمي المخفض المنظم (NDCG)مقياس تقييم يقيم النماذج بناءً على قدرتها على تصنيف المتغيرات عالية اللياقة بشكل صحيح (متماشياً مع هدفنا التجريبي في اكتشاف المتغيرات عالية اللياقة مع الحد الأدنى من عبء الفحص)، والذي تطابق بشكل عام مع النتائج من معامل ارتباط سبيرمان (الشكل S15). تفوقت النماذج المعززة على نموذج الانحدار المتدرج وحده عند تقييمها باستخدام NDCG. كما حاولنا دمج المتنبئين في ميزات المتغيرات لدينا (مثل التنبؤات من كل من ESM-1b و EVmutation)، لكن لم يُلاحظ أي زيادة في أداء النموذج. أخيرًا، اختبرنا ضرورة مجموعة بيانات تشبع الموقع بالكامل ( ) لتدريب النماذج لتحقيق أداء تنبؤي عالٍ ولتقييم ما إذا كانت مجموعات البيانات الأصغر المستخدمة عادة في هندسة البروتين ستكون كافية. لقد احتفظنا بالمتغيرات في مجموعة التدريب لتعكس استراتيجيات هندسة البروتين الشائعة التي لا تبحث بشكل شامل في فضاء التسلسل، بما في ذلك مكتبات الكودون المخفضة (NDTو NRTمسحات الأحماض الأمينية الفردية (هنا، نجمع بين المسحات الشائعة لاستخدام الجلايسين، الألانين، البرولين، والسيستين)، والأبجديات المختصرة التي تجمع بشكل طبيعي الأحماض الأمينية حسب الخصائص الفيزيائية والكيميائية (BLOSUMعند تدريب نفس نموذج الانحدار ال Ridge المعزز باستخدام ترميزات جورجييف، أشار هذا التحليل إلى أن استخدام جميع البيانات المجمعة من مجموعة بيانات تشبع الموقع يوفر قوة تنبؤية أكبر (الشكل 4A، B). يمكن أن يُعزى ذلك على الأرجح إلى طبيعة مجموعات البيانات الغنية التي تحتوي في الغالب على طفرات ذات نشاط غير صفري (64/77 للموكلاوبيميد و62/77 للميتوكلوبراميد)، مما يمنع وجود “ثغرات” في مجموعات التدريب.المضي قدمًا، قررنا استخدام مجموعة بيانات تشبع الموقع ونموذج EVmutation المعزز مع ترميزات جورجييف نظرًا للأداء التنبؤي القوي بين كلاهما.
الشكل 3 | توليد سريع لبيانات مشهد ملاءمة التسلسل لتوجيه التطور الموجه لـ McbA باستخدام التعلم الآلي. مخطط سير العمل: يتم تدريب نموذج الانحدار الخطي المراقب على بيانات نسبة التحويل لأربعة مواقع قابلة للتغيير تم اختيارها من HSS، مع ميزات التسلسل التي تتكون من ترميز الأحماض الأمينية معززة بتوقع بدون تدريب لملاءمة الإنزيم. من مجموعة تدريب مننستنتج من الطفرات الفردية الطفرات ذات الرتبة الأعلى ونختبر أعلى 25 توقعًا. تم مقارنة اختيار الصفر مع جميع استراتيجيات الترميز (يظهر جورجييف) و مرتبة حسب NDCG. شاشة النقاط الساخنة لـ 64 بقايا محددة في McbA تظهر نسبة التحويل المئوية للتغيرموكلوبيميدميتوكلوبراميد، ودي سينشوكائين تم تطبيعهما إلى WT; يشير اللون الأزرق إلى نسبة تحويل منخفضة، بينما يشير اللون الأحمر إلى نسبة تحويل عالية). تم تمييز أعلى أربعة مواقع قابلة للتغيير، أو النقاط الساخنة، باللون البرتقالي وعليها نجوم. تُستخدم هذه المواقع كمجموعة تدريب للتحقق من النموذج. تم تقديم نتائج التحقق الكاملة في الشكل S15. تم توفير بيانات المصدر كملف بيانات مصدر.
الشكل 4 | التطور الموجه بواسطة التعلم الآلي يتنبأ بوجود طفرات عالية النشاط مع عبء فحص أقل من الطفرات الناتجة عن الإشباع الموضعي التكراري. تحليل A، B لوفاء النموذج مع مجموعات التدريب المبنية بمكتبات أصغر من الطفرات المشبعة، بما في ذلك مجموعات الكودونات المخفضة (NDT، NRT) وأبجديات الأحماض الأمينية المخفضة بناءً على BLOSUM50، مقاسة بواسطة ارتباط سبيرمان. ) و NDCG. يتم مقارنة النشاط المقاس مقابل المتوقع على جولات ISM المحتجزة للطرز المدربة على مجموعة بيانات الطفرات الكاملة. كلا من موكليبيميد (موك) (A) وميتوكلوبراميد (ميتو) (B). النسبة المئوية للتحويل التي تم التحقق منها تجريبياً (; تشير أشرطة الخطأ إلى SD) من توقعات ML لموكليبيميد (C) ، ميتوكلوبراميد (D) ، وسينشوكائين (E) مع الطفرة الرباعية من ISM (M4) الملونة باللون الرمادي. بالنسبة لسينشوكائين، لم تتضمن توقعات نموذج ML الطفرة الأكثر أداءً من ISM. النوع البري McbA ملون باللون الرمادي الداكن. تم قياس نسبة التحويل بواسطة RP-HPLC في تجارب مستقلة. تم توفير بيانات المصدر كملف بيانات مصدر. المركبات وحقيقة أن نموذج كثافة الاحتمال المدرب مسبقًا سهل تطبيقه على مركبات أخرى (الشكل 3A). كما أن EVmutation يتطلب موارد حاسوبية ووقت أقل من ESM.
باستخدام نماذج التعلم الآلي المدربة لدينا (نموذج واحد ذو هدف واحد لكل مركب)، قمنا بفرزمتغيرات إنزيمية تركيبية لتخليق الموكلاوبيميد، الميتوكلوبراميد، والسينشوكائين في السليكو وتم اختيار أفضل 25 توقعًا للبناء والاختبار لاحقًا. وجدنا أن نموذج التعلم الآلي المعزز كان قادرًا على توقع متغيرات McbA الغنية بالنشاط العالي لكل منتج أميد عند اختبارها تجريبيًا، حيث تجاوز بعضها qm-McbA من حملات الموكلاوبيميد والميتوكلوبراميد (الشكل 4C-E والجدول S1). ومن الجدير بالذكر أن أفضل طفرات متوقعة للميتوكلوبراميد احتوت على طفرة (A424S) تم استبدالها في HSS بطفرة أكثر نشاطًا (A424T) تم نقلها إلى ISM، مما يشير إلى أن النموذج وجد طفرة متفوقة كانت ستُهمل باستخدام ISM فقط. كان أفضل متغير متوقع للسينشوكائين يتمتع بنشاط أعلى بكثير من أفضل طفرة فردية بمفردها، ومن المدهش أنه احتوى على طفرة (A205L) التي قللت النشاط مقارنة بـ wt-McbA في HSS (الأشكال S16، 17 والجدول S2)؛ لم نتمكن من اختيار ودمج الطفرات من HSS بشكل منطقي للوصول إلى نفس النتائج. تظهر هذه النتائج أن استراتيجيتنا الموجهة بواسطة التعلم الآلي يمكن أن تكتشف متغيرات ذات ملاءمة عالية لمجموعة متنوعة من الجزيئات باستخدام نفس الإنزيم الأساسي. بينما من غير الواضح ما إذا كان النموذج يمكنه استنتاج التفاعلات غير الخطية مباشرة، وجدنا أنه قادر على تجنب الاعتماد على المسارات وتقليل عبء الفحص. ومع ذلك، فإن ترتيب التوقعات التي تم اختبارها تجريبيًا يرتبط بشكل ضعيف بالترتيب المتوقع (متوسط ارتباط ترتيب سبيرمان )، مما يشير إلى أن النماذج قد لا تلتقط بدقة كامل مشهد تسلسل اللياقة.
تنويع البيوكاتاليست المدعوم بالذكاء الاصطناعي للأدوية عالية القيمة
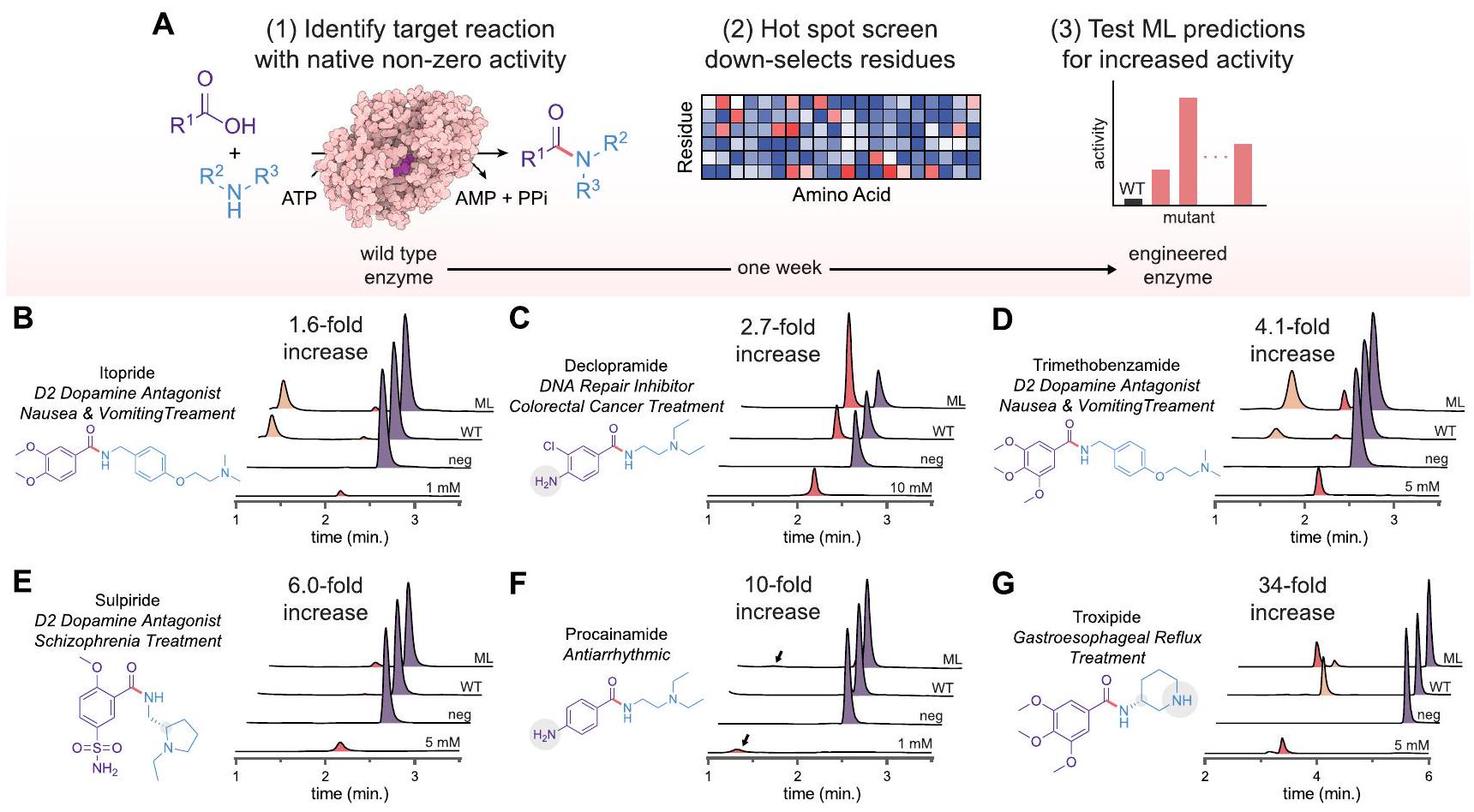
لتقييم قوة نهجنا، قمنا بعد ذلك بتطبيق إطار عملنا المدعوم بالتعلم الآلي للتنبؤ بمتغيرات McbA المميزة من أجل تخليق ستة مركبات صيدلانية إضافية. بدءًا من تفاعل الهدف المحدد من فحص نطاق الركيزة لدينا (الشكل 2)، استخدمنا نفس مجموعة مكتبة المتغيرات الفردية لمتغير McbA المكونة من 1,216 متغيرًا من أعلاه لإجراء HSS (7,302 تفاعل فريد إجمالاً)، واختيار أربع نقاط ساخنة، وتدريب نموذج التعلم الآلي لدينا للتنبؤ بمتغيرات أعلى ترتيب مع زيادة النشاط (الشكل 5A؛ الأشكال S18-23). تم اختبار أفضل 24 تنبؤًا لكل تفاعل، وتم التعبير عن أفضل متغير وتنقيته ومقارنته بنشاط wt-McbA. لاحظنا زيادات في العوائد تتراوح من 1.6 مرة إلى 34 مرة مقارنة بـ wt-McbA للستة مركبات التي اختبرناها (الشكل 5B-G والأشكال S18-23). بالنسبة لكل مركب، كان أفضل متغير متوقع دائمًا يتفوق على أفضل تصميم عقلاني (أي، دمج أفضل أربع طفرات من HSS دون استخدام نموذج التعلم الآلي؛ الجدول S3-5). أعطت بعض المتغيرات تحسينات متواضعة فقط، والتي قد تكون نتيجة لإشارة منخفضة إلى ضوضاء في فحوصات النقاط الساخنة لبعض المركبات المستهدفة التي كانت قابلة للاكتشاف فقط بواسطة MS. يمكن أن يؤدي ذلك إلى مشاهد لياقة مسطحة يصعب نمذجتها. ومع ذلك، أنتج إطار عملنا متغيرات إنزيمية ذات نشاط متزايد لعدة منتجات كانت في البداية ملحوظة فقط بكميات ضئيلة.
قمنا أيضًا بمقارنة مدى كفاءة بعض متغيرات الإنزيم في أداء كل خطوة من خطوات التفاعل (الشكل S24). على سبيل المثال، يبدو أن wt-McbA بارع في خطوة الأدينيلation لدواء التروكسيبيد (الأدينيلating 3,4,5-trimethoxybenzoic acid)، لكنه غير قادر على تحفيز تكوين رابطة الأميد (الشكل 5G). يمكن لمتغير الإنزيم المهندَس بعد ذلك قبول الأمين، مما يؤدي إلى انخفاض كبير في الوسط الملحوظ. بشكل غير متوقع، كانت متغيرات McbA المهندَسة لكل منتج مستهدف
الشكل 5 | الهندسة الموجهة بواسطة التعلم الآلي لإنزيمات الأميد المتميزة من أجل تخليق مجموعة واسعة من الأدوية الصغيرة. أ. الاستراتيجية التي استخدمناها لهندسة البروتين الموجهة بواسطة التعلم الآلي لـ McbA موضحة. أولاً، حددنا التفاعلات غير الأصلية التي يمكن أن يحفزها wt-McbA وأعطينا الأولوية لتلك التي تنتج أدوية صغيرة قيمة. ثانياً، يتم استخدام مجموعة فرعية من 64 بقايا لاختيار البقايا التي تؤثر إيجابياً على النشاط. ثالثاً، يتم تدريب نموذج الانحدار المتزايد على بيانات من مجموعة الفرعية، وتتم التنبؤات بواسطة التعلم الآلي. تم اختباره تجريبيًا. مقارنة B-G لأعلى نشاط متوقع للمتغير لمجموعة من الأدوية الصغيرة مقارنةً بـ wt-McbA ومعيار أصلي. تم تطبيع تركيز الإنزيم إلى وتم تحليل المنتجات بواسطة RP-HPLC. الزيادة المضاعفة في العائد الملحوظة تقارن بين wt-McbA و ML-McbA ( ). تظهر آثار HPLC التمثيلية للمنتج (بالأحمر)، الركيزة الحمضية (بالأرجواني)، والحمض المؤدي (بالبرتقالي) لكل تفاعل. تم أخذ الآثار من ثلاثة تجارب مستقلة على الأقل ( ). كما أظهرت انتقائية إقليمية صارمة على الرغم من عدم وجود أي ضغط انتقائي للحفاظ عليها. يتجلى ذلك في الطفرة الرباعية لـ troxipide التي تظهر زيادة بمقدار 34 مرة في النشاط دون أي تضحيات في التخصص. وبالمثل، يتم الحفاظ على التفضيلات الاستيريوselective مع S-sulpiride. مجتمعة، يسمح لنا إطار العمل الموجه بواسطة التعلم الآلي باستخدام بيانات وظيفية من متغيرات إنزيم الطفرات الفردية للتنبؤ بسرعة وفعالية بالطفرات العليا ذات الترتيب الأعلى.
نقاش
في هذا العمل، أنشأنا إطار عمل لهندسة البروتينات مدعوم بتقنيات التعلم الآلي، يتيح التصميم التنبؤي دون الحاجة إلى موارد حاسوبية متخصصة. هذا الإطار دمج بشكل فريد طريقة CFE وطرق الطفرات، والتعلم الآلي لتسريع حملات التطور الموجه، والتطور المتباين لتحويل إنزيم عام إلى عدة إنزيمات متخصصة. عرضنا هذا الإطار من خلال التنقل السريع في تسع حملات لهندسة البروتينات لإنزيم الأميد سينثيتاز McbA، حيث تم تنفيذ ست من هذه الحملات في وقت واحد.
من خلال الجهود المبذولة لبناء نماذج التعلم الآلي وجميع جولات ISM، استكشفنا مشهد التسلسل-الوظيفة لـ McbA من خلال تقييم 2856 متغيرًا من McbA (1217 متغيرًا منها كانت لنماذج التعلم الآلي)، و1100 منتج أميد محتمل، و12,584 تفاعل زوج الركيزة-المتحور. حددنا 19 موضعًا فريدًا من البقايا داخل McbA تؤثر بشكل كبير على التحفيز الحيوي، حيث ينتج عن كل تفاعل مجموعة فريدة من هذه البقايا الساخنة. عبر جميع المتغيرات التسعة النهائية لـ McbA المهندسة، أجرينا إجمالي 21 طفرة مختلفة تحدث عبر 14 بقايا مختلفة (الشكل S25). بينما تحتوي العديد من أزواج الركيزة على نفس الحمض أو الأمين، فإنه من الصعب تبرير سبب ظهور بعض الطفرات حيث إنها ليست محفوظة بين العديد من هذه الإنزيمات. على سبيل المثال، تحتوي منتجات الأميد مثل الميتوكلوبراميد، والسينشوكائين، والبروكاييناميد، والدكلوبراميد جميعها على N,N-ثنائي إيثيل إيثيلين ديامين ولكن مع أحماض مختلفة. يمكن اقتراح أن V177S هو مفيد. طفرة لهذه التفاعل، لكنها مجرد تعميم وليست عالمية حيث أن البروكاييناميد لم يحتوي على هذه الطفرة. ومع ذلك، يبدو أن بياناتنا تشير إلى أن اختيار البقايا (نقاط ساخنة) مرتبط بشكل أقوى بالتفاعل العام وليس بالأحماض أو الأحماض الأمينية بمفردها. في جميع الحالات، أظهرت المتغيرات الإنزيمية الجديدة نشاطًا محسنًا مقارنةً بمتغيرات wt-McbA (تحسين من 1.6 إلى 42 مرة). في أحد الأمثلة، حقق متغير إنزيمي لتخليق الموكلوبيميدتحويل (زيادة بمقدار 42 مرة في الكفاءة التحفيزية مقارنة بـ wt-McbA) وتمت زيادته إلى كميات ملليغرام.
كانت ميزة مهمة في عملنا هي استخدام نماذج التعلم الآلي المدربة على طفرات بقايا فردية للتنبؤ بالطفرات ذات الرتبة الأعلى مع تحسين اللياقة. اخترنا نموذج الانحدار المتزايد لأنه قد أظهر سابقًا أداءً جيدًا مقارنةً بالأساليب الأكثر تعقيدًا وكان قادرًا على الاستقراء من الطفرات الفردية إلى الطفرات ذات الرتبة الأعلى، وهو ما يتماشى مع بياناتنا.على سبيل المثال، في كل من حالات الاختبار التسع، كانت المتغيرات الإنزيمية المتوقعة بواسطة التعلم الآلي التي تحتوي على 4 طفرات أكثر نشاطًا من مجموعة الطفرات الفردية الأربعة الأكثر نشاطًا بمفردها. هذه الملاحظة صحيحة في الحالات التي تكون فيها جودة البيانات عالية (مثل، موكلوباميد)، حيث يكون مشهد اللياقة مسطحًا (أي، العديد من الطفرات ذات النشاط الصفري وليس العديد من الطفرات التي تحسن النشاط)، ونسبة الإشارة إلى الضوضاء عالية (مثل، ديكلوبراميد)، مما يبرز قوة نهجنا. قد تكون هناك حالات حيث ستكون النماذج الأكثر تعقيدًا (مثل، الغابات العشوائية، آلات الدعم الشعاعي، الشبكات العصبية، إلخ) ضرورية للتعميم بشكل أفضل على مساحة التسلسل بأكملها للتنقل في مشاهد اللياقة المعقدة. على الرغم من ذلك، ساعد الإطار الموجه بواسطة التعلم الآلي المستخدم هنا في البحث حول المسارات الافتراضية لـ ISM، مما قلل من الجهد وزاد من معدلات النجاح في عدة حملات هندسة إنزيم.
يمكن تطبيق نهجنا، من الناحية النظرية، على أي إنزيم ولكن سيتطلب ضبطًا دقيقًا خاصًا بالتفاعل حول جمع البيانات وتوليد نموذج التعلم الآلي. فيما يتعلق بجمع البيانات، تظل طرق الفحص التجريبية للتفاعلات البيوكاتاليتيكية عقبة. هنا، لأن المركبات الناتجة من McbA كانت مستقرة في وجود مستخلص التعبير الخالي من الخلايا وكانت الطرق الكروماتوغرافية فعالة (على سبيل المثال، لكل عينة)، قدمت تقنية LC-MS حلاً قابلاً للإدارة، كما تم العثور عليه في أمثلة أخرى كملحق لعملية الفحص، ستكون هناك تطبيقات لهندسة الإنزيم حيث تكون استراتيجيات الاختيار مفيدة (على سبيل المثال، عندما توجد طريقة اختيار قابلة للتطبيق، يمكن إجراء قفزات أكبر في فضاء التسلسل). قد تتطلب حملات الهندسة مع بروتينات مختلفة استكشاف نماذج ومعلمات تعلم الآلة المختلفة. بينما لاحظنا أداءً ممتازًا مع نموذج EVmutation المعزز، قد تتطلب أهداف اللياقة البدنية البديلة أيضًا متنبئين بديلين للياقة البدنية. على سبيل المثال، إذا كان الهدف هو هندسة الاستقرار، فمن المنطقي أن يكون المتنبئ باللياقة البدنية القائم على الهيكل أفضل.هناك العديد من المتنبئين بتأثيرات المتغيرات البروتينية الآخرين الذين يدفعون باستمرار حدود المعرفة إلى الأمام والذين يمكن أن يحسنوا توقعاتنا.قد تتفوق ترميزات التسلسل الأكثر تعقيدًا المستندة إلى معالجة اللغة الطبيعية أيضًا على الترميزات المعززة.أخيرًا، نلاحظ أن تدريب نماذج التعلم الآلي على مزيد من المتبقيات، وبيانات متعددة الطفرات (بما في ذلك البيانات من جولات متعددة من الطفرات أو التركيبات العشوائية للطفرات التي تنوع الأحماض الأمينية عبر البروتين بالكامل المعني)، أو القياسات الحركية قد يكون مفيدًا في هندسة محفزات أفضل.
باختصار، إطار عملنا القائم على التعلم الآلي القابل للوصول والذي لا يعتمد على الخلايا يتغلب على التحديات التقليدية للتطور الموجه من خلال تجاوز الاعتماد على المسارات التي تقيد مساحة البحث عن التسلسل في الأساليب المتطورة. بالإضافة إلى ذلك، فإن التعبير الخالي من الخلايا من قوالب التعبير الخطية يسرع عملية استكشاف المناظر الطبيعية للتسلسل-الوظيفة، حيث يمكن إجراء العملية الكاملة لتعبير البروتين وتقييمه دون خلايا، مما يسمح بتجنب خطوات الاستنساخ المرهقة، مما يستغرق ساعات بدلاً من أيام إلى أسابيع. هذه الميزات تسرع وتيرة الهندسة مقارنةً بـ ISM وحده؛ حيث مكن إطار عملنا من إكمال ست حملات هندسة إنزيمات في وقت واحد في أسبوع واحد فقط لكل مركب. علاوة على ذلك، نلاحظ أيضًا التكلفة المنخفضة (أي، سنتات لكل…رد الفعل) وقابلية التوسع العالية لـ CFE التي تمكّن سير العمل لدينابالإضافة إلى فوائد الإطار الخالي من الخلايا، يبرز عملنا أيضًا تعددية إنزيم الأميد McbA الذي يمكن توجيهه لتحفيز العديد من التفاعلات الفريدة ذات الأهمية، بما في ذلك تلك المستخدمة في إنتاج الأدوية الجزيئية الصغيرة. نتطلع إلى أن تكون الطريقة الموصوفة هنا، خاصة عندما يتم تعزيزها بتصميم البروتين من الصفر.ستسرع حملات هندسة الإنزيمات لفتح إنزيمات متخصصة ذات وظائف وخصائص متنوعة.
طرق
تجميع الحمض النووي الخالي من الخلايا والتعبير الجيني
تم إنشاء مكتبات الحمض النووي لكل من wt-McbA و muGFP. تم تحسين الكودون لـ wt-McbA من Marinactinospora thermotolerans (UniProt: R4R1U5) ليكون مناسبًا لبكتيريا E. coli وتم استنساخه في بلازميد pJL1 (Addgene, 69496) مع علامة CSL في الطرف N. (اندماج CAT-Strep-Linker الذي يحتوي على علامة سترب II). تم تحسين كودون muGFP ليكون مناسبًا لبكتيريا الإشريكية القولونية وتم استنساخه في بلازميد pJL1 دون تنقية..
تم إجراء توليد مكتبة الحمض النووي الخالي من الخلايا على النحو التالي: (1) تم إجراء تفاعل البوليميراز المتسلسل الأول في تفاعل مع 1 نانوغرام من قالب البلازميد المضاف، (2)تم إضافة DpnI وتم حضنه فيلمدة ساعتين، (3) تم تخفيف الـ PCR بنسبة 1:4 عن طريق إضافةمن ماء خالٍ من النوكلياز (NF)، (4)تم إضافة DNA مخفف إلىتفاعل تجميع جيبسون (صنع ذاتي) وتم حضنه لمدة لمدة ساعة واحدة، تم تخفيف تفاعل التجميع بنسبة 1:10 عن طريق إضافةمن مياه NF، (6)تم إضافة رد فعل التجميع المخفف إلىتفاعل PCR. تم إعداد جميع خطوات الاستنساخ باستخدام روبوت معالجة السوائل Integra VIAFLO في ألواح PCR ذات 384 بئر (Bio-Rad). تم تصميم البرايمرات باستخدام Benchling مع حساب درجة انصهارها بواسطة الإعداد الافتراضي.
خوارزمية سانتا لوسيا 1998. لقد لاحظنا أن درجات حرارة الانصهار لأدوات تصميم البرايمر البديلة أحيانًا تنحرف عن تلك المحسوبة في بنشلينغ، لذا يجب على المستخدمين أخذ ذلك في الاعتبار عند تصميم البرايمرات. كانت القواعد العامة التي اتبعناها لتصميم البرايمرات هي برايمر عكسي منبادئ أمامي منوتداخل متماثل منتم طلب جميع البرايمرات من شركة Integrated DNA Technologies (IDT)؛ وتم تصنيع البرايمرات الأمامية واستلامها في أطباق تحتوي على 384 بئرًا وتم تعديلها إلىلتسهيل إعداد التفاعلات. يمكن العثور على معلومات إضافية حول تصميم البرايمر والأكواد التي استخدمناها لجميع الأحماض الأمينية العشرين في الشكل S4 والجدول S7. جميع تفاعلات PCR استخدمت إنزيم Q5 Hot Start DNA Polymerase (NEB). يمكن العثور على معلومات إضافية حول معلمات جهاز التدوير الحراري في الجدول S8.
لتجميع الطفرات من أجل ISM،من “الفائز” من لوحة تجميع جيبسون المخففة تم تحويله إلىمن الكفاءة الكيميائية. كولاي (خلايا NEB 5-alpha). تم زراعة الخلايا على أطباق LB تحتوي علىكاناميسين (LB-Kan). تم استخدام مستعمرة واحدة لزراعة 50 مل من LB-Kan، وتم نموها طوال الليل فيمع اهتزاز بسرعة 250 دورة في الدقيقة. تم تنقية البلازميد باستخدام مجموعات ZymoPURE II Midiprep وتم تأكيد التسلسل. يمكن بعد ذلك دمج الطفرات المتعاقبة من خلال طريقة توليد مكتبة الحمض النووي الخالي من الخلايا المذكورة أعلاه.
تم إنشاء مكتبة الطفرات المزدوجة الشاملة McbA (المستخدمة في الشكل S7e) من خلال جولتين متتاليتين من التحوير المشبع دون استهداف أي بقايا معينة أولاً. بعد التحوير المشبع الأول، تم إعداد البلازميدات التي تحتوي على كل طفرة وفقًا للبروتوكول المذكور أعلاه باستثناء أنه تم استخدام 5 مل من الثقافات الليلية في LB-Kan لتنقية البلازميدات باستخدام مجموعات ZymoPURE II Miniprep. تم استخدام هذه البلازميدات العشرين كقوالب للجولة التالية من التحوير المشبع لتجميع جميع الطفرات المزدوجة الـ 400.
تم طلب جميع المتغيرات McbA المتوقعة بواسطة ML ككتل جينية من IDT تحتوي على زوائد جيبسون 5′ و3′ من pJL1. تم إعادة تعليق الحمض النووي بتركيزتم طلب هيكل بلازميد pJL1 المبسط ككتلة جينية من IDT، وتم تضخيمه بواسطة تفاعل البوليميراز المتسلسل (PCR)، وتنقيته باستخدام مجموعة تنظيف وتركيز الحمض النووي (Zymo Research)، وتم تخفيفه إلى تركيز قدرهتم استخدام تجميع جيبسون لتجميع الحمض النووي الذي يشفر متغيرات McbA مع هيكل pJL1. تم دمج 10 نانوجرام من هيكل pJL1 المنقى والمستقيم و10 نانوجرام من إدخال gblock فيتفاعل تجميع جيبسون وتم حضنه فيلـتم تخفيف تفاعلات التجميع غير المنقاة فيمن مياه NF وتم استخدام رد الفعل المخفف كقالب لـتفاعل PCR (باستخدام بوليميراز DNA Q5 Hot Start) لإنتاج LETs لعملية CFPS.
تم إعداد مستخلصات الخلايا الخام كما هو موصوف سابقًا باستخدام خلايا E. coli BL21 Star (DE3) (إنفيتروجن).تمت إجراء تفاعلات CFE استنادًا إلى نظام PANOx-SPوتم تنفيذها في أطباق PCR بسعة 384 بئر (بايو راد) كـتفاعلات معمن LET التي تعمل كقالب DNA. تم تحضين التفاعلات فيلمدة 16 ساعة.
تعبير وتنقية البروتينات المؤتلفة
تم تحويل بلازميد pJL1-McbA إلى خلايا E. coli BL21 Star (DE3) المعالجة كيميائيًا (Invitrogen) وفقًا لتعليمات الشركة المصنعة. تم زراعة الخلايا على وسط LB-Kan وتم حضنها طوال الليل فيتم استخدام مستعمرة واحدة لتلقيحثقافة الليل في LB-Kan، نمت في مع اهتزاز بسرعة 250 دورة في الدقيقة. تم تحضير 1 لتر من وسط TB Express Overnight (Millipore) وفقًا لتعليمات الشركة المصنعة وتمت إضافته بـ كاناميسين. تم تلقيح وسط السل في اليوم التالي باستخدام 5 مل من الثقافة الليلية وتم زراعته فيمع اهتزاز 250 دورة في الدقيقة حتى التشبعتم جمع الخلايا عن طريق الطرد المركزي (بيكمان كولتر أفانتي J-26) عندلمدة 10 دقائق عندتم تجميد كريات الخلايا بسرعة باستخدام النيتروجين السائل وتخزينها فيحتى الاستخدام المستقبلي أو إعادة التعليق في 25 مل من محلول الغسيلتريس- HCl pH 8.0EDTAالجلسرين). تم تحلل الخلايا المعاد تعليقها بواسطة الموجات فوق الصوتية (جهاز QSonica Q700) باستخدام ست دورات من 10 ثوانٍ تشغيل و10 ثوانٍ إيقاف.السعة، وتم إزالة الكسر غير القابل للذوبان عن طريق الطرد المركزي عندلمدة 20 دقيقة عند. أوضح تم تحضين المستخلصات مع 2 مل من راتنج Strep-Tactin XT Superflow (IBA Lifesciences) المعادل مسبقًا مع التحريك لمدة 30 دقيقة فيتم تحميل الراتنج على عمود تدفق الجاذبية وغسله ثلاث مرات بـ 20 مل من محلول الغسيل. تم استرجاع بروتين McbA باستخدام 10 مل من محلول الإلخاء.تريس-EDTA، 50 مللي مولار البيوتين،الجلسرين) وتركيزها معفلتر الطرد المركزي Amicon Ultra (Millipore Sigma؛ بحد قطع 30 كيلودالتون). تم تبادل McbA المنقى إلى محلول التخزين.HEPES pH 7.5الجليسرول) باستخدام عمود إزالة الملح PD-10 المعادل مسبقًا (Cytiva). تم تخزين McbA في للاستخدام الفوري ( ) أو للتخزين على المدى الطويل. تم قياس تركيز البروتين عن طريق قياس A280 على جهاز NanoDrop 2000c (ثيرمو ساينتيفيك)، مع حساب معامل انقراض McbA والوزن الجزيئي بواسطة Expasy ProtParam. تم تنقية wt-McbA وستة متغيرات McbA المهندسة الموجودة في الشكل 5 بهذه الطريقة.
اختبار نشاط muGFP
تم قياس أداء متغيرات muGFP من خلال قياس الفلورية باستخدام جهاز قراءة الألواح (BioTek Synergy H1) مع تحفيز عند 485 نانومتر وانبعاث لـتم نقل 100 ميكرولتر من تفاعل CFPS الخام الذي يحتوي على متغير muGFP المعبر عنه إلى لوحة 384 بئر ذات قاع دائري سوداء (Nunc) قبل القياسات.
اختبار نشاط أميد سينثيتاز
تم تجميع جميع الفحوصات عالية الإنتاجية (فحص النقاط الساخنة، التطفير المتكرر لمواقع الإشباع، نطاق الركيزة، التحقق من توقعات التعلم الآلي، واستكشاف توقعات التعلم الآلي) في ألواح مكونة من 384 بئرًا (بايو راد) باستخدام روبوت معالجة السوائل إنتيغرا VIAFLO.تم توزيع خليط التفاعل الذي يحتوي على الركائز (ATP، حمض، أمين، وDMSO) مع حجم زائد مملوء بمحلول فوسفات البوتاسيوم 50 مللي مولار pH 7.5 كـعينات في لوحة مكونة من 384 بئر. تم بدء اختبار الأميد بواسطة إضافةمن تفاعل CFPS الخام الذي يحتوي على متغير McbA المعبر عنه، مع تركيزات نهائية تبلغ 25 مللي مول من ATP، 25 مللي مول من الحمض، 25 مللي مول من الأمين،دي ميثيل سلفوكسيد، والإنزيم (المحدد بواسطةدمج -ليوسين باستخدام البروتوكولات الموصوفة سابقًاتم تحضير محاليل مخزنة من الأحماض في DMSO وتم أخذ ذلك في الاعتبار للوصول إلىدي إم إس أو. للتفاعلات التي تم تنفيذها في ثلاث نسخ،من نفستم استخدام تفاعل CFPS لثلاثة اختبارات منفصلة. تم حضن التفاعل فيلمدة 16 ساعة ثم تم تبريده بـالميثانول. تم تخزين الألواح فيحتى يتم التحضير للتحليل عن طريق LC-MS.
تم إعداد تجارب الأميدation للمتغيرات المنقاة من McbA الموجودة في الشكل 5 بطريقة مشابهة كما هو موصوف أعلاه في ألواح 384 بئر.تم تجميع التفاعلات في ثلاث نسخ، تحتوي على 25 مللي مول من ATP، 25 مللي مول من الحمض، 25 مللي مول من الأمين،بيوروفوسفاتاز (سيغما I5907)، DMSO، وحجم التعبئة من 50 مليمول من فوسفات البوتاسيوم pH 7.5. لاختبار إنتاج السينشوكائين والبروكائيناميد، تم تقليل الركائز بكميات ستوكيومترية إلى 20 مليمول و 10 مليمول، على التوالي. كان ذلك لتعويض عن ضعف الذوبانية الملحوظ لهذين الحمضين (حمض 2-بيوتوكسي كوينولين-4-كربوكسيليك وحمض 4-أمينوبنزويك) في التفاعل المنقى عنددي إم إس أو. تم تحضين التفاعلات فيلمدة 16 ساعة ثم تم تبريده بـالميثانول. تم تخزين العينات فيحتى يتم التحضير للتحليل عبر LC-MS. يمكن العثور على أرقام CAS لجميع المواد الكيميائية المستخدمة في شاشات النقاط الساخنة، بالإضافة إلى معايير الأميد التي قمنا بشرائها، في الجدول S13.
اختبار تخليق الأميد وتجديد ATP
تم استنساخ كيناز البوليفوسفات، PPK12 من عائلة إريسيبيلوتريشاسي غير المصنفة (يونيبروت: A0A847P5F2_9FIRM)، وتم التعبير عنه وتنقيته إلى نقاء كامل كما هو موصوف سابقًا.تم تجميع التفاعلات في ثلاث نسخ، تحتوي على 25 مللي مول من الأمين، 25 مللي مول من الحمض،بوليفوسفات (سيغما 1.06529)بيروفوسفاتاز (سيغما I5907)،مكبAPPK12، 10% حجماً/حجماً
DMSO، وحجم التعبئة من 50 مليمول من فوسفات البوتاسيوم pH 7.5. تم إعداد تخفيف تسلسلي بمعدل 2 وتمت إضافته إلى مزيج التفاعل لتركيزات نهائية تتراوح من 25 مليمول إلى 0.02 مليمول. تم حضن التفاعلات عندلمدة 16 ساعة ثم تم تبريده بـمن الميثانول وتم تحليله بواسطة LC-MS.
التحضير على نطاق واسع لتخليق الموكليبيميد
تم إعداد اختبارات التفاعل المقياس لإعداد الموكلابيميد الإنزيمي بطريقة مشابهة لما تم وصفه أعلاه.تفاعل يحتوي على 25 مللي مول من ATP، 25 مللي مول من الحمض، 25 مللي مول من الأمين،، بيوروفوسفاتاز (سيغما I5907)،دي إم إس أو، وحجم التعبئة من 50 مللي مولار من فوسفات البوتاسيوم pH 7.5. بعد 16 ساعة، تم إيقاف التفاعل وتم استخراج المنتج بإضافة 30 مل من الإيثيل أسيتات.تم جمع المراحل العضوية، وغسلها بـومحلول ملحيمجفف فوقتمت تصفيته، وتم تبخير المذيب تحت ضغط منخفض للحصول على المنتج المطلوب على شكل مسحوق أبيض.العائد المعزول) دون أي تنقية إضافية. الـ و تتوافق نتائج الرنين المغناطيسي النووي (الموجودة أدناه وفي الشكل S12) بشكل جيد مع تلك التي تم الإبلاغ عنها سابقًا.. طيف لـ و تم تسجيل طيف الرنين المغناطيسي النووي عند درجة حرارة الغرفة باستخدام نظام Bruker Avance III بتردد 500 ميجاهرتز. يتم الإبلاغ عن الانزياحات الكيميائية فيوحدات نسبية لقمم المذيبات المتبقية DMSO-d6 (2.50 جزء في المليون لـ 1H و39.5 جزء في المليون لـ 13C). يتم تعيين أنماط الانقسام كالتالي: s (أحادي)، d (ثنائي)، t (ثلاثي)، q (رباعي)، quint (خماسي)، m (متعدد). يتم الإبلاغ عن ثوابت الاقتران بوحدات هرتز، تليها التكامل. NMR. ( 500 ميغاهرتز ، DMSO-d6) ، “، . C NMR. ( 126 ميغاهرتز، DMSO) 165.51، 136.38، 133.69، 129.55، 128.85، 66.66، 57.77، 53.76، 37.06.
تحليلات LC-MS
تم تحليل منتجات الأميد (بالإضافة إلى الركائز الحمضية وبعض الوسائط الحمضية المؤيدة) باستخدام نظام Agilent G6125B LC/MSD أحادي الرباعي المزود بمصدر تأين بالرش الكهربائي مضبوط على وضع التأين الإيجابي. تم طرد العينات المروقة في جهاز الطرد المركزي لمدة 10 دقائق عندلإزالة البروتينات المترسبة. تم إعداد لوحة منفصلة تحتوي على 384 بئرًا لحقن العينة في جهاز HPLC-MS عن طريق التخفيفمن العينات المطفأة معالميثانول باستخدام جهاز Integra VIAFLO. تم الكشف عن كميات ضئيلة من المركبات باستخدام مطياف الكتلة، بينما كانت العديد من المركبات موجودة بتركيز عالٍ بما يكفي للت quantification بواسطة كاشف مصفوفة الصمام الثنائي (DAD) عند 254 نانومتر. تم فصل المركبات على عمود Luna C18 (Phenomenex 00D-4251-BO) باستخدام مراحل متنقلة (A)معحمض الفورميك و (ب) الأسيتونيتريل. تم تنفيذ الطريقة العامة للفصل الكروماتوغرافي باستخدام التدرجات التالية عند معدل تدفق ثابت من : . بالنسبة لشاشات النقاط الساخنة، تم استخدام طريقة مسرعة مع التدرجات التالية عند معدل تدفق ثابت من : ; بالنسبة لجهاز الطيف الكتلي، تم ضبط جهد الشعيرات على 3 كيلو فولت، وتم استخدام غاز النيتروجين للتبخير (35 رطل لكل بوصة مربعة) والتجفيف.تم معايرة جهاز الطيف الكتلي باستخدام خليط الضبط (Agilent G2421-60001) قبل إجراء القياسات. تم الحصول على بيانات الطيف الكتلي بنطاق مسح منمع بطاقات SIM متنوعةوفقًا للمركب الذي كنا نقوم بفحصه. تم جمع بيانات LC-MS وتحليلها باستخدام برنامج Agilent OpenLab CDS ChemStation. تم تقدير عائد المنتج عن طريق قسمة مساحة ذروة DAD لمنتج الأميد على مجموع مساحات الذروات لكل من الأميد والركيزة الحمضية. تم تسجيل عائد كمي دقيق لموكليبيميد بعد تخليقها على نطاق تحضيري وعزلها.
تحديد درجة انصهار
تم تحديد درجة انصهار البروتين باستخدام مطياف الدائرة الضوئية Jasco J-810 مع قنينة بطول مسار 10 مم تم رصدها عند 222 نانومتر. تم تبادل عينات McbA أولاً إلى محلول ملحي معزز بالفوسفات 1X، pH 7.4، وتم تخفيفها إلى.
حركيات الإنزيمات
تم تحديد الحركيات الظاهرة لـ McbA لزوج الأمين من موكلوبيمايد (4-(2 أمينو إيثيل) مرفولين) من خلال ربط تكوين رابطة الأميد إنزيميًا (والإفراج المتزامن لـ AMP من وسيط الأسيل-AMP عن طريق استبداله بالأمين) مع أكسدة NADH (الشكل S9). احتوت التفاعلات على 100 مليمول من MOPS-KOH بدرجة حموضة 7.8،فوسفوإنول البيروفات NADH، 50 مللي مولار حمض 4-كلوروبنزويك، خليط إنزيمات كيناز البيروفات وديهيدروجيناز اللاكتات (سيغما-ألدريتش P0294)مايوكيناز (سيغما-ألدريتش 475941)، وتركيزات مختلفةمن المتغير المدروس McbA. حيث أن الحمض هنا (حمض 4-كلوروبنزويك) له قابلية ذوبان ضعيفة في الماء وتم إذابته في DMSO، كانت التفاعلات النهائية تحتوي علىدي إم إس أو (ما يعادل شاشات الأميدation لدينا).تم تحقيق التوازن في التفاعلات أولاً عندلمدة 3 دقائق ثم بدأ بإضافةتم تحديد السرعة الابتدائية لتركيزات مختلفة من الأمين.عن طريق قياس امتصاص NADH عند 340 نانومتر على جهاز Cary 60 UV-Vis (Agilent). تم جمع البيانات وتحليلها باستخدام برنامج Cary WinUV Kinetics Application (Agilent). تم رسم مخططات مايكلس-منتن في برنامج GraphPad Prism وتناسبها باستخدام أداة تحليل الانحدار غير الخطي الافتراضية لمعادلة مايكلس-منتن.
تم قياس الحركيات لزوج الحمض من موكلوبيمايد (حمض 4-كلوروبنزويك) بطريقة مشابهة لما تم وصفه أعلاه، باستثناء أنه تم تثبيت الأمين عند 50 مللي مول، وتم بدء التفاعل بإضافة كميات مختلفة من الحمض. وتم الحفاظ على تركيز DMSO النهائي ثابتًا عند. لقد لاحظنا سلوكًا غير ميكاليز-مينتين عند محاولة تحديد الحركيات للحامض، فيما بدا أنه تثبيط تحت تأثير الركيزة بواسطة الحامض (البيانات غير معروضة). كما حاولنا قياس خطوة أدينيلت الحامض مباشرة من خلال الربط الإنزيمي لتكوين أسيل-AMP (مع الإفراج المتزامن عن ) مع أكسدة NADH لاستكشاف آلية التفاعل بشكل أعمق. تم استخدام مجموعة اختبار البيروفوسفات Piper™ (Fisher Scientific P22062)، ولكن إضافة تركيزات صغيرة من DMSO أدت إلى ترسيب الإنزيمات الموجودة في المجموعة.
ترميزات الأحماض الأمينية
تمت دراسة خمس استراتيجيات مختلفة لترميز الأحماض الأمينية هنا بناءً على عمل ويتمن وآخرون وفورن هولت وآخرون. : ترميز واحد حار، جورجييف، VHSE، مقاييس z، ووصف الخصائص الفيزيائية. بالإضافة إلى الترميزات ذات الواحد الحار (التي لا تحتوي على أي معلومات حول طبيعة الحمض الأميني في كل موضع)، أردنا أيضًا تضمين الترميزات التي تحاول تجسيد الخصائص الفيزيائية الكيميائية للأحماض الأمينية. نشرح هذه الترميزات بإيجاز أدناه (ترتيبًا من الأكثر إلى الأقل عددًا من المعلمات) ونشجع القراء على زيارة هذه المصادر لمزيد من المعلومات. لجعل تمثيلات عددية معلوماتية لخصائص الأحماض الأمينية، تقوم هذه الاستراتيجيات بإجراء تحليل المكونات الرئيسية لمجموعات مختلفة تم تنسيقها يدويًا إما من خصائص تم قياسها تجريبيًا أو تم التنبؤ بها/تقديرها حسابيًا. جورجييفالميزات (19 معلمة) هي المكونات الرئيسية لأكثر من 500 مؤشر للأحماض الأمينية مأخوذة من قاعدة بيانات AAindex. VHSEالميزات (8 معلمات) هي المكونات الرئيسية لـ 50 متغيرًا، تركز على الخصائص الكارهة للماء، والستيريكية، والإلكترونية. مقاييس Zتتميز الخصائص (5 معلمات) بأنها المكونات الرئيسية لـ 26 متغيرًا، تركز على المحبة للدهون، والحجم، والقطبية. الوصف الفيزيائيتُشتق الميزات (3-معلمات) من تعديل عقلاني غير رسمي للمكونات الرئيسية للخصائص الكارهة للماء والخصائص الاستيركية للببتيدات. بالنسبة لجميع الاستراتيجيات، قمنا أولاً بإنشاء ترميزات لمكتبة التركيب الكاملة التي تم اختبارها (المخزنة في موتر من “متغيرات فريدة” × 4 حمض أميني الأحماض” × ” -المعلمات”، حيث -المعلمات تساوي عدد الأحماض الأمينية لنظام one-hot). ثم تم تسطيح البعدين الأخيرين من التنسور لإنشاء مصفوفة. وبشكل خاص بالنسبة للتشفيرات الفيزيائية الكيميائية (باستثناء one-hot)، تم توحيد كل عمود من المصفوفة (مركزيًا حول المتوسط ومقاسًا بوحدة واحدة).
تنبؤات بدون تدريب مسبق
تطوري. الطفرات الكهربائيةتم تدريب نموذج كثافة الاحتمال باستخدام خادم الويب EVcouplings (https://evcouplings.org/) مع المعلمات الافتراضية، مع تسلسل الإدخال لـ McbA مأخوذ من UniProt (R4R1U5). كان لدى النموذج الذي اخترناه عتبة تضمين نقاط البت تبلغ 0.7. النموذج والرمز لتكرار التنبؤات بدون تدريب متاحة في مستودع GitHub الخاص بنا. تم توفير رمز توقع تأثيرات الطفرات في مستودع GitHub الخاص بـ EVcouplings (https://github.com/debbiemarkslab/EVcouplings/blob/develop/تم استخدام (notebooks/model_parameters_mutation_effects.ipynb) كقالب. تم اشتقاق الميزات للنماذج المعززة من الطاقة الإحصائية للتسلسل بالنسبة لنوع البرية.
عالمي. التنبؤات باستخدام ESM-1bتم إنشاء نموذج لغة محول مدرب مسبقًا باستخدام الشيفرة المقدمة من العمل الممتاز لـ ويتمن وآخرون حول التطور الموجه المدعوم بالذكاء الاصطناعي (https:// github.com/fhalab/MLDE) مع نموذج ESM-1b المقدم في مستودع ESM على GitHub (https://github.com/facebookresearch/esm). باختصار، تم استخدام بروتوكول ملء القناع للتنبؤ باحتمالية الطفرات المختلفة من خلال تقديم النموذج مع التسلسل الكامل و”إخفاء” موضع معين. استخدمنا نهج ملء القناع البسيط، الذي يعتبر كل موضع متغير مستقلاً عن الآخر. تم استخدام هذا النهج لأنه أقل تكلفة حسابية وقدم تنبؤات أفضل قليلاً من النهج الشرطي (الذي لا يفترض استقلالية المواضع المتغيرة) في هذا العمل السابق. يمكن العثور على وصف كامل للكود في المنشور الأصلي ومستودع GitHub المرتبط. تم اشتقاق الميزات للنماذج المعززة من احتمال السجل التسلسلي بالنسبة لنوع البرية.
هيكلية. تم إجراء التنبؤات المعتمدة على الهيكل باستخدام MAESTROأداة سطر الأوامر لنظام ويندوز (الإصدار 1.2.35). استخدمنا هيكل بنك البيانات البروتينية (PDB) لـ McbA (6SQ8) كمدخل وحسبنا التغيرات في الاستقرار (طاقة الانحلال الحرة) باستخدام أمر “evalmut”. تم اشتقاق الميزات للنماذج المعززة باستخدام ناتج ‘energy’.
التطور الموجه المدعوم بتعلم الآلة
تم تعزيز نماذج الانحدار Ridge وفقًا للكود المرافق للعمل الأنيق لـ Hsu وآخرين. (https://github.com/تم تنفيذ ميزات تسلسل متغير McbA من خلال دمج التنبؤات بدون تدريب مع ترميزات الأحماض الأمينية المحددة للموقع. تم أولاً توحيد التنبؤات بدون تدريب وتنظيمها بواسطة قوة تنظيم شائعة.قوة تنظيم L2 لرجوع ريدجتم تحديده خلال ضبط المعلمات الفائقة باستخدام التحقق المتقاطع. للحصول على الكود الكامل المستخدم في هذا العمل، يرجى الاطلاع على مستودع GitHub المرافق لدينا فيhttps://github.com/grantlandwehr/accelerated-enzyme-engineering. نظرًا لبعض التغييرات التي تم إجراؤها بين تطوير النموذج الأولي وإعادة تنفيذ الشيفرة للنشر (على سبيل المثال، ضبط المعلمات الفائقة، مخطط التحقق المتقاطع، نطاق البحث عن معامل الانتظام، إلخ.) هناك اختلافات طفيفة في التوقعات المرتبة 23-25 لمتوكلوبراميد وموكليبيميد كما هو موضح في الشكل 4.
تم إجراء تقييم واختيار النموذج أولاً بشكل رجعي باستخدام مجموعات البيانات المعلمة بالتحليل من حملات هندسة الموكلابيمايد والميتوكلوبراميد الخاصة بنا. تم تدريب نماذج معززة (باستخدام تركيبات من المتنبئين بدون تدريب المذكورين أعلاه وترميزات الأحماض الأمينية) على مكتبات تشبع الموقع الفردي لأربعة المخلفات ( ) وتم اختباره على الطفرات العليا المحتجزة من الجولات الإضافية من الطفرات المشبعة ( تم تحديد الأربعة بقايا (نقاط ساخنة) من خلال اختيار الأربعة بقايا التي تحتوي على طفرات مع أعلى تحسين في العائد من بين 64 بقايا تم اختبارها. ضبط المعلمات الفائقة لـتم تنفيذ ذلك باستخدام التحقق المتقاطع المتكرر 5 مرات (مع 20 تكرارًا) عن طريق أخذ عينات عشوائيةمن بيانات التدريب والاختبار على 20% المحتجزة؛ تم تقييم أداء النموذج لتعديل المعلمات الفائقة باستخدام متوسط الخطأ التربيعي (MSE). مع المعلمات الفائقة المحسّنة، تم استخدام جميع النماذج المدربة لإجراء التنبؤات على مجموعة الاختبار المحتجزة. تم استخدام معامل ارتباط سبيرمان وNDCG لاختيار أفضل متنبئ بدون تدريب واستراتيجية الترميز، مع تفضيل NDCG.
بعد تحديد أفضل نموذج (الذي كان في حالتنا هو تعزيز نموذج كثافة احتمال الطفرة EV باستخدام ترميزات جورجييف)، قمنا بعمل توقعات على مجموعة البيانات التوافقية بأكملها.تم اختبار أفضل 25 توقعًا لموكلاوبيميد وميتوكلوبراميد تجريبيًا (الشكل 4). تم إجراء تدريب النموذج والتوقعات للسبعة منتجات الأميد المتبقية بطريقة مشابهة لما سبق. نظرًا لنموذج كثافة احتمال EVmutation المدرب مسبقًا والأبعاد المنخفضة للترميزات، يمكن إجراء تدريب النموذج والتوقعات لمجموعة البيانات التوافقية بالكامل في دقائق على معالج Intel Core i7 من الجيل الثاني عشر مع 32 جيجابايت من الذاكرة العشوائية دون تسريع وحدة معالجة الرسومات.
جمع البيانات وتحليلها
جميع المعلومات الإحصائية المقدمة في هذه المخطوطة مستمدة منتجارب مستقلة ما لم يُذكر خلاف ذلك في النص أو أساطير الأشكال. تمثل أشرطة الخطأ انحرافًا معياريًا واحدًا عن المتوسط المستمد من هذه التجارب. تم إجراء تحليل البيانات وتوليد الأشكال باستخدام Excel الإصدار 2304، ChimeraX الإصدارتم قياس فلورية muGFP باستخدام جهاز قراءة الألواح الدقيقة BioTek Synergy H1 وتم تحليلها باستخدام Gen5 الإصدار 2.09.2. تم إجراء الصور الشعاعية كما هو موصوف سابقًا وتم مسحها باستخدام جهاز التصوير Typhoon FLA 7000 الإصدار 1.2..
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
جميع البيانات المقدمة في هذه المخطوطة متاحة في ملف البيانات المصدر أو مودعة في GitHub المرتبط (https://github.com/تسلسل البروتينات والحمض النووي لجميع الإنزيمات المعبر عنها في هذا العمل متاحة في المعلومات التكميلية. تم توفير بيانات المصدر مع هذه الورقة.
توفر الشيفرة
الكود لإعادة إنتاج النتائج متاح علىhttps://github.com/grantlandwehr/هندسة الإنزيمات المعجلة.
References
Schwander, T., von Borzyskowski, L. S., Burgener, S., Cortina, N. S. & Erb, T. J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 354, 900-904 (2016).
Sarai, N. S. et al. Directed evolution of enzymatic silicon-carbon bond cleavage in siloxanes. Science 383, 438-443 (2024).
Fryszkowska, A. et al. A chemoenzymatic strategy for site-selective functionalization of native peptides and proteins. Science 376, 1321-1327 (2022).
Arnold, F. H. Design by directed evolution. Acc. Chem. Res 31, 125-131 (1998).
Packer, M. S. & Liu, D. R. Methods for the directed evolution of proteins. Nat. Rev. Genet 16, 379-394 (2015).
Miton, C. M. & Tokuriki, N. How mutational epistasis impairs predictability in protein evolution and design. Protein Sci. 25, 1260-1272 (2016).
Rix, G. et al. Scalable continuous evolution for the generation of diverse enzyme variants encompassing promiscuous activities. Nat. Commun. 11, 5644 (2020).
Kalvet, I. et al. Design of heme enzymes with a tunable substrate binding pocket adjacent to an open metal coordination site. J. Am. Chem. Soc. 145, 14307-14315 (2023).
Chu, A. E., Lu, T. & Huang, P.-S. Sparks of function by de novo protein design. Nat. Biotechnol. 42, 203-215 (2024).
Yeh, A. H.-W. et al. De novo design of luciferases using deep learning. Nature 614, 774-780 (2023).
Yang, K. K., Wu, Z. & Arnold, F. H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687-694 (2019).
Freschlin, C. R., Fahlberg, S. A. & Romero, P. A. Machine learning to navigate fitness landscapes for protein engineering. Curr. Opin. Biotech. 75, 102713 (2022).
Zhang, S. et al. EvoAI enables extreme compression and reconstruction of the protein sequence space. Res. Sq. rs.3.rs-3930833. https://doi.org/10.21203/rs.3.rs-3930833/v1 (2024).
Wittmann, B. J., Yue, Y. & Arnold, F. H. Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Syst. 12, 1026-1045.e7 (2021).
Bell, E. L. et al. Directed evolution of an efficient and thermostable PET depolymerase. Nat. Catal. 5, 673-681 (2022).
Silverman, A. D., Karim, A. S. & Jewett, M. C. Cell-free gene expression: an expanded repertoire of applications. Nat. Rev. Genet. 21, 151-170 (2020).
Karim, A. S. et al. In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat. Chem. Biol. 16, 912-919 (2020).
Hunt, A. C. et al. A rapid cell-free expression and screening platform for antibody discovery. Nat. Commun. 14, 3897 (2023).
Hunt, A. C. et al. Multivalent designed proteins neutralize SARS-CoV-2 variants of concern and confer protection against infection in mice. Sci. Transl. Med. 14, eabn1252-eabn1252 (2022).
Rapp, J. T., Bremer, B. J. & Romero, P. A. Self-driving laboratories to autonomously navigate the protein fitness landscape. Nat. Chem. Eng. 1, 97-107 (2024).
Fallah-Araghi, A., Baret, J.-C., Ryckelynck, M. & Griffiths, A. D. A completely in vitro ultrahigh-throughput droplet-based microfluidic screening system for protein engineering and directed evolution. Lab Chip 12, 882-891 (2012).
Ao, Y. et al. Structure- and data-driven protein engineering of transaminases for improving activity and stereoselectivity. Angew. Chem. Int. Ed. 62, e202301660 (2023).
Yu, T. et al. Enzyme function prediction using contrastive learning. Science 379, 1358-1363 (2023).
Wang, X. et al. Multi-modal deep learning enables efficient and accurate annotation of enzymatic active sites. Nat. Commun. 15, 7348 (2024).
Pattabiraman, V. R. & Bode, J. W. Rethinking amide bond synthesis. Nature 480, 471-479 (2011).
Bryan, M. C. et al. Key Green Chemistry research areas from a pharmaceutical manufacturers’ perspective revisited. Green. Chem. 20, 5082-5103 (2018).
Boström, J., Brown, D. G., Young, R. J. & Keserü, G. M. Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discov. 17, 709-727 (2018).
Sabatini, M. T., Boulton, Lee, T., Sneddon, H. F. & Sheppard, T. D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2, 10-17 (2019).
Wu, S., Snajdrova, R., Moore, J. C., Baldenius, K. & Bornscheuer, U. T. Biocatalysis: enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 60, 88-119 (2021).
Winn, M. et al. Discovery, characterization and engineering of ligases for amide synthesis. Nature 593, 391-398 (2021).
Schnepel, C. et al. Thioester-mediated biocatalytic amide bond synthesis with in situ thiol recycling. Nat. Catal. 6, 89-99 (2023).
Petchey, M. et al. The broad aryl acid specificity of the amide bond synthetase mcba suggests potential for the biocatalytic synthesis of amides. Angew. Chem. Int. Ed. 57, 11584-11588 (2018).
Chen, Q. et al. Discovery of McbB, an enzyme catalyzing the carboline skeleton construction in the marinacarboline biosynthetic Pathway. Angew. Chem. Int. Ed. 52, 9980-9984 (2013).
Tang, Q. et al. Broad spectrum enantioselective amide bond synthetase from Streptoalloteichus hindustanus. ACS Catal. 14, 1021-1029 (2024).
Petchey, M. R., Rowlinson, B., Lloyd, R. C., Fairlamb, I. J. S. & Grogan, G. Biocatalytic synthesis of moclobemide using the amide bond synthetase McbA coupled with an ATP recycling system. ACS Catal. 10, 4659-4663 (2020).
Jewett, M. C. & Swartz, J. R. Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng. 86, 19-26 (2004).
Scott, D. J. et al. A novel ultra-stable, monomeric green fluorescent protein for direct volumetric imaging of whole organs using CLARITY. Sci. Rep.-uk 8, 667 (2018).
Yong, K. J. & Scott, D. J. Rapid directed evolution of stabilized proteins with cellular high-throughput encapsulation solubilization and screening (CHESS). Biotechnol. Bioeng. 112, 438-446 (2015).
Pédelacq, J.-D., Cabantous, S., Tran, T., Terwilliger, T. C. & Waldo, G. S . Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79-88 (2006).
Hsu, C., Nisonoff, H., Fannjiang, C. & Listgarten, J. Learning protein fitness models from evolutionary and assay-labeled data. Nat. Biotechnol. 1-9. https://doi.org/10.1038/s41587-021-01146-5 (2022).
Georgiev, A. G. Interpretable numerical descriptors of amino acid space. J. Comput Biol. 16, 703-723 (2009).
Mei, H., Liao, Z. H., Zhou, Y. & Li, S. Z. A new set of amino acid descriptors and its application in peptide QSARs. Pept. Sci. 80, 775-786 (2005).
Sandberg, M., Eriksson, L., Jonsson, J., Sjöström, M. & Wold, S. New chemical descriptors relevant for the design of biologically active peptides. a multivariate characterization of 87 amino acids. J. Med Chem. 41, 2481-2491 (1998).
Barley, M. H., Turner, N. J. & Goodacre, R. Improved descriptors for the quantitative structure-activity relationship modeling of peptides and proteins. J. Chem. Inf. Model 58, 234-243 (2018).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 118, e2016239118 (2021).
Hopf, T. A. et al. Mutation effects predicted from sequence covariation. Nat. Biotechnol. 35, 128-135 (2017).
Laimer, J., Hofer, H., Fritz, M., Wegenkittl, S. & Lackner, P. MAESTRO -multi agent stability prediction upon point mutations. BMC Bioinform. 16, 116 (2015).
Järvelin, K. & Kekäläinen, J. Cumulated gain-based evaluation of IR techniques. ACM Trans. Inf. Syst. (TOIS) 20, 422-446 (2002).
Reetz, M. T., Kahakeaw, D. & Lohmer, R. Addressing the numbers problem in directed evolution. Chembiochem 9, 1797-1804 (2008).
Aslan, A. S., Birmingham, W. R., Karagüler, N. G., Turner, N. J. & Binay, B. Semi-rational design of Geobacillus stearothermophilus L-lactate dehydrogenase to access various chiral -hydroxy acids. Appl. Biochem. Biotech. 179, 474-484 (2016).
Gray, V. E., Hause, R. J. & Fowler, D. M. Analysis of large-scale mutagenesis data to assess the impact of single amino acid substitutions. Genetics 207, 53-61 (2017).
Murphy, L. R., Wallqvist, A. & Levy, R. M. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein Eng. Des. Sel. 13, 149-152 (2000).
O’Kane, P. T., Dudley, Q. M., McMillan, A. K., Jewett, M. C. & Mrksich, M. High-throughput mapping of CoA metabolites by SAMDI-MS to optimize the cell-free biosynthesis of HMG-CoA. Sci. Adv. 5, eaaw9180 (2019).
Goldenzweig, A. et al. Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell 63, 337-346 (2016).
Mansoor, S., Baek, M., Juergens, D., Watson, J. L. & Baker, D. Zeroshot mutation effect prediction on protein stability and function using RoseTTAFold. Protein Science. 32, e4780 (2023).
Biswas, S., Khimulya, G., Alley, E. C., Esvelt, K. M. & Church, G. M. Low-N protein engineering with data-efficient deep learning. Nat. Methods 18, 389-396 (2021).
Lauko, A. et al. Computational design of serine hydrolases. bioRxiv 2024.08.29.610411. https://doi.org/10.1101/2024.08.29. 610411 (2024).
Kightlinger, W. et al. Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases. Nat. Chem. Biol. 14, 627-635 (2018).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343-345 (2009).
Kwon, Y.-C. & Jewett, M. C. High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep. 5, 8663 (2015).
Jewett, M. C., Calhoun, K. A., Voloshin, A., Wuu, J. J. & Swartz, J. R. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol. 4, 220-220 (2008).
Jewett, M. C. & Swartz, J. R. Substrate replenishment extends protein synthesis with an in vitro translation system designed to mimic the cytoplasm. Biotechnol. Bioeng. 87, 465-471 (2004).
Tavanti, M., Hosford, J., Lloyd, R. C. & Brown, M. J. B. ATP regeneration by a single polyphosphate kinase powers multigram-scale aldehyde synthesis in vitro. Green Chem. 23, 828-837 (2020).
Lavayssiere, M. & Lamaty, F. Amidation by reactive extrusion for the synthesis of active pharmaceutical ingredients teriflunomide and moclobemide. Chem. Commun. 59, 3439-3442 (2023).
Vornholt, T. et al. Systematic engineering of artificial metalloenzymes for new-to-nature reactions. Sci. Adv. 7, eabe4208 (2021).
Hellberg, S., Sjoestroem, M., Skagerberg, B. & Wold, S. Peptide quantitative structure-activity relationships, a multivariate approach. J. Med Chem. 30, 1126-1135 (1987).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70-82 (2021).
Zubi, Y. S. et al. Metal-responsive regulation of enzyme catalysis using genetically encoded chemical switches. Nat. Commun. 13, 1864 (2022).
Landwehr, G. et al. Accelerated enzyme engineering by machinelearning guided cell-free expression. accelerated-enzymeengineering. https://doi.org/10.5281/zenodo. 14262332 (2024).
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031-3048 (2017).
شكر وتقدير
نشكر كوسوكي سيكي، أندرو سي. هانت، وستيف ر. فليمنغ على المحادثات المتعلقة بهذا العمل. نعترف باستخدام مرفق كيك للبيوفيزياء، وهو مورد مشترك لمؤسسة روبرت إتش لوري.
مركز السرطان الشامل بجامعة نورث وسترن مدعوم جزئيًا من قبل منحة دعم مركز السرطان NCI #P30 CA060553. بالإضافة إلى ذلك، نقر باستخدام الموارد الحاسوبية ومساهمات الموظفين المقدمة لمرافق الحوسبة عالية الأداء Quest في جامعة نورث وسترن، والتي تدعمها بشكل مشترك مكتب نائب الرئيس، ومكتب البحث، وتقنية المعلومات بجامعة نورث وسترن. كما استخدمت هذه العمل مرافق NMR IMSERC في جامعة نورث وسترن، التي تلقت دعمًا من مورد تكنولوجيا النانو الناعمة والهجينة (SHyNE) (NSF ECCS-2025633)، والمعهد الدولي لتكنولوجيا النانو، وجامعة نورث وسترن. تم إجراء الرسوم البيانية الجزيئية والتحليلات باستخدام UCSF ChimeraX، التي تم تطويرها من قبل مورد الحوسبة الحيوية، والتصور، والمعلوماتية في جامعة كاليفورنيا، سان فرانسيسكو، بدعم من المعاهد الوطنية للصحة R01-GM129325 ومكتب البنية التحتية السيبرانية وعلم الأحياء الحاسوبي، المعهد الوطني للحساسية والأمراض المعدية. يقر M.C.J. بالدعم من منحة وزارة الطاقة DE-SCOO23278، ومنحة وكالة خفض التهديدات الدفاعية DTRA1-21-1-0038، ومنحة المعاهد الوطنية للصحة 1U19AI142780-01، وبرنامج LDRD في مختبرات سانديا الوطنية منحة DE-NAOOO3525.
قد قدم كل من ج.م.ل.، ج.و.ب.، أ.س.ك.، و م.س.ج. إقراراً بالاختراع استناداً إلى العمل المقدم. لدى م.س.ج. مصلحة مالية في National Resilience و Gauntlet Bio و Pearl Bio, Inc. و Stemloop Inc. يتم مراجعة وإدارة مصالح م.س.ج. من قبل جامعة نورث وسترن وجامعة ستانفورد وفقاً لسياساتهما المتعلقة بالمصالح المتنافسة. جميع المؤلفين الآخرين يعلنون عدم وجود مصالح متنافسة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى أشتى س. كريم أو مايكل سي. جويت.
معلومات مراجعة الأقران تشكر مجلة Nature Communications جيمس كارثيرز، سونغ هي والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب-غير التجارية-عدم الاشتقاق 4.0 الدولية، التي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع، واستنساخ في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/. (ج) المؤلف(ون) 2025
¹قسم الهندسة الكيميائية والبيولوجية، جامعة نورث وسترن، إيفانستون، إلينوي، الولايات المتحدة الأمريكية. ²مركز البيولوجيا التركيبية، جامعة نورث وسترن، إيفانستون، إلينوي، الولايات المتحدة الأمريكية.قسم الهندسة الحيوية، جامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.ساهم هؤلاء المؤلفون بالتساوي: غرانت م. لاندوير، جوناثان و. بوغارت. البريد الإلكتروني:ashty.karim@northwestern.edu; mjewett@stanford.edu
Accelerated enzyme engineering by machine-learning guided cell-free expression
Received: 30 July 2024
Accepted: 9 December 2024
Published online: 20 January 2025
Grant M. Landwehr , Jonathan W. Bogart , Carol Magalhaes , Eric G. Hammarlund , Ashty S. Karim & Michael C. Jewett □
Enzyme engineering is limited by the challenge of rapidly generating and using large datasets of sequence-function relationships for predictive design. To address this challenge, we develop a machine learning (ML)-guided platform that integrates cell-free DNA assembly, cell-free gene expression, and functional assays to rapidly map fitness landscapes across protein sequence space and optimize enzymes for multiple, distinct chemical reactions. We apply this platform to engineer amide synthetases by evaluating substrate preference for 1217 enzyme variants in 10,953 unique reactions. We use these data to build augmented ridge regression ML models for predicting amide synthetase variants capable of making 9 small molecule pharmaceuticals. Over these nine compounds, ML-predicted enzyme variants demonstrate 1.6 – to 42 -fold improved activity relative to the parent. Our ML-guided, cell-free framework promises to accelerate enzyme engineering by enabling iterative exploration of protein sequence space to build specialized biocatalysts in parallel.
Engineered enzymes are poised to have transformative impacts across applications in energy , materials , and medicine . To create such enzymes, a protein’s amino acid sequence is changed to enhance native function or facilitate new chemical reactions. This process typically involves identifying enzymes with natural plasticity and promiscuity for the reaction of interest, followed by using directed evolution . Unfortunately, current approaches to directed evolution are limited because they can often only map sequence-function relationships in a narrow region of sequence space. For example, screening strategies are generally low throughput, which constrains resampling mutations in iterative site saturation mutagenesis campaigns and can miss epistatic interactions that capture beneficial pairwise (or greater) synergies when the single mutations are neutral or even detrimental . Additionally, selection methods for directed evolution focus on “winning” enzymes for a single transformation, which limits the ability to collect positive and negative sequence-function relationships for forward engineering of similar reactions .
Computational technologies have emerged to accelerate existing directed evolution approaches. De novo protein design can create new-to-nature enzymes, but the diversity of chemistries and
applications remains limited . Machine learning (ML) models have been used to discover enzymes by inferring fitness based on related homologs and/or protein sequences from all organisms (a so-called zero-shot prediction) as well as to navigate protein-fitness landscapes based on assayed fitness data (e.g., nonlinear regression using sitespecific one-hot encodings) . While ML-assisted enzyme engineering methods show promise, rapidly building datasets to navigate vast sequence space remains a challenge , especially considering most genotype-phenotype links are lost in high-throughput enzyme engineering campaigns .
Here, we developed a high-throughput, ML-guided approach to enable exploration of fitness landscapes across multiple regions of chemical space for forward design of biocatalysts (Fig. 1). A key feature of our approach is the use of cell-free gene expression (CFE) systems to allow for the rapid synthesis and functional testing of proteins in a design-build-test-learn (DBTL) workflow. This framework first maps sequence-function relationships for enzyme variants with single-order mutations for a specific chemical transformation identified from an evaluation of enzymatic substrate promiscuity. Then, these data are used to fit supervised ridge regression ML models augmented with an
Fig. 1 | An ML-guided, cell-free enzyme engineering platform. Schematic shows how a design-build-test-learn workflow is applied to rapidly map sequence-function landscapes. Putative residues directing enzyme catalysis are rationally selected based on structural insights, evolutionary trends, and computational tools (e.g., ROSETTA , EVmutation , PROSS ) (design). Site saturation mutagenesis and cell-
free gene expression are carried out in less than 24 h to generate sequence-defined libraries (build). The libraries can then be screened for desirable protein fitness metrics (test). Information from the test phase, including failures, is used to identify functionally important amino acid residues that feedback on iterative designs, as well as fit ML models (learn).
evolutionary zero-shot fitness predictor and extrapolate higher-order mutants with increased activity. Importantly, the ML models can be run on the central processing unit of a typical computer making our entire approach user-friendly and accessible. Our method offers a compliment to the growing toolbox of directed evolution strategies, such as those that predict catalytic features of enzymes .
We applied our framework to carry out divergent evolution, converting an amide bond-forming generalist enzyme into multiple, distinct specialist enzymes. The biocatalytic formation of amide bonds-a motif ubiquitously found in pharmaceuticals, agrochemicals, polymers, fragrances, flavors, and other high-value products -could offer unique advantages over synthetic counterparts (e.g., mild reaction conditions and chemo-, stereo-, and regioselectivities) and facilitate sustainable biomanufacturing . McbA from Marinactinospora thermotolerans is one representative ATP-dependent amide bond synthetase involved in the biosynthesis of marinacarboline secondary metabolites . McbA, and its close homolog ShABS , have been shown to have a relaxed substrate scope, accepting several simple acids and amines commonly found in pharmaceuticals . This backdrop suggests that McbA could serve as a flexible starting point for engineering a generalist enzyme into multiple reaction specialists each capable of carrying out a different chemical reaction. Our MLguided approach was able to improve the McbA enzyme activity relative to the wild type enzyme between 1.6 – and 42 -fold for producing nine compounds.
Results
Exploring the biocatalytic synthesis landscape of McbA
The goal of this work was to develop an ML-guided, DBTL workflow that expedites simultaneous directed evolution campaigns for biocatalysis by reducing screening burden. This goal required generating sequence-fitness data for unique chemical transformations, from which to create predictive ML models. To identify reactions of interest, we first explored the possible amidation reaction space of wild-type McbA (wt-McbA) by evaluating enzymatic substrate promiscuity (Fig. 2A). We studied an extensive array of substrates that deviated from the heterocyclic acids and primary or aromatic amines preferred by wt-McbA. These substrates included primary, secondary, alkyl,
aromatic, complex pharmacophore, electron poor or rich species, and substrates containing other heteroatoms, halogens, and “unprotected” nucleophiles or electrophiles. More challenging substrates (e.g., complex heterocyclic acids and amines, enantiomers, and substrates containing both acids and amines or multiple acids and amines) were also included to determine the innate limitations and preferences of wt-McbA. We carried out 1100 unique reactions with low enzyme concentration ( ) and high substrate concentration ( 25 mM ), covering numerous molecules of known value including pharmaceuticals, fragrances, and polymers (Fig. 2B).
Interestingly, wt-McbA displayed a tolerance to multiple “unprotected” functional groups and geometries. Generally, aliphatic acids were poorly tolerated while aryl, benzoic, and cinnamic acids were readily accepted substrates. Charged aryl acids were a unique exception and usually coupled to very few amines. Conversely, wt-McbA readily coupled primary and secondary aliphatic amines but struggled with aryl amines. We observed that McbA was able to synthesize 11 pharmaceutical compounds as well as dozens of hybrid molecules (Fig. 2C), ranging from trace amounts detectable only by mass spectrometry (MS) to conversion. In these reactions, we uncovered both stereoselectivity (e.g., strongly favoring the synthesis of S-sulpiride over Rsulpiride) and strict chemo- and regioselectivity preferences (e.g., substrates containing both acids and amines not polymerizing). Given that the reaction mechanism of McbA first begins with the adenylation of the carboxylic acid, we also noticed several instances where only the acyl-AMP intermediate was observed. Several important molecules are not synthesized by wild-type McbA (Fig. 2D). These inaccessible products suggest that McbA may not be able to react with aliphatic and fatty acids (e.g., nonivamide, capsaicin) or particular large substrates (e.g., imatinib, nilotinib). A better understanding of substrate scope can guide future enzyme engineering work.
Cell-free protein engineering to rapidly screen sequence-defined protein libraries
With specific chemical transformations identified from our evaluation of enzymatic substrate promiscuity, we next wanted to quickly generate large amounts of sequence-function relationship data of mutant McbA enzymes for training ML models to predict high-activity variants.
Fig. 2 | The diverse accessible chemical space of McbA suggests a biocatalyst capable of synthesizing several high value molecules. A Reaction scheme and screening conditions for exploring the substrate scope of McbA for the enzymatic synthesis of amides. McbA was expressed using CFE and the reaction was initiated by the addition of different combinations of acid and amine substrates. The all-by-all substrate screen for McbA, analyzed with reversed phase (RP)-HPLC ( ). Darker red corresponds to a product that was observable by ultraviolet (UV)
absorbance while lighter red corresponds to trace amounts only detectable by mass spectrometry. A complete list of substrates can be found in Fig. S1. C Among the 21 high value molecules that were possible in the substrate scope, we observed that McbA was able to synthesize 16 ( 11 of which are small-molecule pharmaceuticals). D Example high value molecules that McbA was unable to synthesize under the tested reaction conditions. Source data are provided as a Source Data file.
To do this, we implemented a cell-free protein synthesis approach that does not require laborious transformation and cloning steps (Fig. 1). Our approach relied on cell-free DNA-assembly and to build site-saturated, sequence-defined protein libraries. This workflow had five steps: (i) a DNA primer containing a nucleotide mismatch introduces a desired mutation through PCR, (ii) DpnI digests the parent plasmid, (iii) an intramolecular Gibson assembly forms a mutated plasmid, (iv) a second PCR amplifies linear DNA expression templates (LETs), and (v) the mutated protein is expressed through CFE. In this way, hundreds to thousands of sequence-defined protein mutants can be built in individual reactions within a day, and mutations can be accumulated through rapid iterations of the workflow. Our approach avoids any potential biases in typical site-saturation libraries that arise from the use of degenerate primers.
We validated our workflow using the well-characterized, monomeric ultra-stable green fluorescent protein (muGFP) by targeting four residues that are known to be important for stability and fluorescence (Fig. S2). When building our site-saturated library targeting these four residues ( 77 variants), we found a high tolerance to primer design deviations (e.g., homologous overlaps, melting temperatures) (Fig. S3, S4) and that LETs of muGFP variants conferred all desired mutations (Fig. S5). Full-length soluble proteins indicated that changes in fluorescence were not due to changes in expression or solubility (Fig. S6). Mapping the protein site-saturated landscape not only highlights residues that are crucial for fitness (e.g., residues composing the fluorophore and impacting hydrophobic core packing were intolerable to mutations ) but also provides insight into the general mutability of sites.
After validation, we applied our workflow to McbA to generate sequence-function relationship data that could train ML models to expedite our engineering campaigns. We initially engineered McbA to synthesize three high-value molecules identified by our substrate scope evaluation: (i) the monoamine oxidase A inhibitor, moclobemide, due to McbA’s high promiscuity towards this reaction (Fig. 2C (5); wt conversion); (ii) metoclopramide, due to the unique challenge posed by the acid component containing a free amine that could potentially compete with the intended amine (Fig. 2C (8); 3% wt conversion); and (iii) cinchocaine, which contains a unique acid component but shares the same amine fragment as metoclopramide (Fig. 2C (16); wt conversion). By performing these engineering campaigns in parallel we hoped to infer mutations that influence substrate specificity for the amine (shared mutations) and the acid (unique mutations) that may lead to general design principles for McbA.
Using relatively high substrate concentrations and low enzyme loading as a step towards more industrially relevant reaction conditions (Fig. S7), we performed a hot spot screen (HSS) for each molecule consisting of site-saturation mutagenesis on a wide sequence space to identify residue positions that, when mutated, positively impact fitness (Fig. 3A). Guided by the crystal structure of McbA (PDB: 6SQ8), we selected 64 residues that completely enclosed the active site and putative substrate tunnels (e.g., residues within of the docked native substrates). Our HSS of these residues ( 64 residues amino acids total single mutants) revealed multiple residues that had a positive impact on moclobemide (Fig. 3B), metoclopramide (Fig. 3C), and cinchocaine (Fig. 3D) synthesis when mutated compared to wtMcbA as measured by liquid chromatography-mass spectrometry (LC-MS).
ML-guided, cell-free expression for protein engineering
With a large data set at hand for multiple, distinct single McbA mutants, we set out to leverage ML models to accelerate the engineering of McbA for the production of small molecules across diverse regions of chemical space. The key idea was to use single mutant data from the HSS to extrapolate higher order mutants with increased activity (Fig. 3A). To achieve this goal, we chose to fit augmented ridge regression models with our data-as such models are simple and have been previously shown to outperform more sophisticated models for protein engineering -allowing us to predict higher-order mutants with increased activity.
We first selected a predictive model architecture. McbA variant feature representations consisted of site-specific amino acid encodings concatenated with a zero-shot fitness prediction . We considered several amino acid encodings, ranging from simple one-hot encodings to more complex descriptors that attempt to incorporate amino acid physiochemical properties . We also explored benchmark protein variant fitness predictors to incorporate universal, evolutionary, and structural based zero-shot predictions. We tested three specific fitness predictors: the Evolutionary Scale Modeling (ESM)-1b transformer trained on the UniRef50 database (universal), an EVmutation (EC) probability density model trained on an MSA of evolutionarily related sequences (evolutionary), and MAESTRO to estimate structure-based changes in unfolding free energy (structural). Training and hyperparameter tuning were performed using single mutant data ( ) from the HSS (top four hot spots; Fig. 3B, C).
In parallel, we conducted a more traditional directed evolution campaign on each amide product (moclobemide, metoclopramide, and cinchocaine) via iterative saturation mutagenesis (ISM). This would provide valuable higher order mutations to validate and benchmark model performance given our objective of extrapolating from single to higher order mutations.
For moclobemide, we selected six residues from the HSS above a threshold of 1.5 -fold activity over wild type to mutate through three rounds of ISM (Fig. S7). We first fixed the top mutation from the HSS
(V177S) and performed site-saturation mutagenesis on the five additional residues. By reintroducing previously fixed mutations in subsequent rounds, we explored potential epistatic interactions (e.g., S177 was saturated in ISM round 2, given V177S was incorporated before A323F). In addition, we completely explored all combinatorial double mutants of the top two residues, which showed additive impacts for moclobemide synthesis (Fig. S7). After three rounds of ISM, whereby we selected the most highly active mutant in each round to start the next round, we identified a quadruple mutant ( ) with increased activity-from for wt-McbA to conversion-for the synthesis of moclobemide (Fig. S7). We characterized the apparent steady-state kinetic parameters and stability of these enzyme mutants from each round of ISM. Specifically, we expressed, purified, and evaluated each McbA variant observing a 42 -fold increase in catalytic efficiency from wt-McbA to qm-Mcba increased from 18.2 to ) for the amine (Figs. S8, S9). The melting point did not significantly change between wt-McbA and qm-McbA , but the second mutation (A323F) increased by when added to the first mutation (V177S) (Fig. S10). Additionally, we showed that we could make milligram quantities of moclobemide in a reaction ( isolated yield) and confirmed the structure by NMR (Figs. S11, S12).
For metoclopramide, we selected 10 residues for the ISM campaign, which were all above a threshold of 1.25-fold activity over wild type. Three rounds of ISM for metoclopramide yielded a quadruple mutant that displayed nearly 30 -fold improved activity over wt-McbA (Fig. S13).
The campaign for cinchocaine was more difficult to navigate and we failed to observe beneficial mutations beyond a double mutant, despite taking multiple ISM paths (Fig. S14). This result (i.e., running into dead ends during ISM) supported the need to include ML models in our framework that might capture epistatic interactions. We used the ISM data for moclobemide and metoclopramide containing double, triple, and quadruple mutants ( for moclobemide and for metoclopramide) to evaluate each model’s performance, while cinchocaine would provide a unique pressure-test for our identified top-performing model.
Model prediction performance was first evaluated using the normalized discounted cumulative gain (NDCG) , an evaluation metric that scores models on their ability to correctly rank highfitness variants (aligning with our experimental goal of discovering high-fitness variants with minimal screening burden), which generally matched results from the Spearman rank correlation coefficient (Fig. S15). The augmented models outperformed the ridge regression model alone when evaluated with NDCG. We also tried combining predictors in our variant features (e.g., predictions from both ESM-1b and EVmutation), but no increase in model performance was observed. Lastly, we tested the necessity of the entire site saturation dataset ( ) for training models to achieve high predictive performance and to evaluate whether smaller datasets commonly used in protein engineering would be sufficient. We withheld variants in the training set to reflect common protein engineering strategies that do not exhaustively search the sequence space, including reduced codon libraries (NDT and NRT ), single amino acid scans (here, we combine the commonly used glycine, alanine, proline, and cysteine scans), and reduced alphabets that naturally group amino acids by physiochemical properties (BLOSUM ). When training the same augmented ridge regression model with Georgiev encodings, this analysis indicated that utilizing all the data gathered from the site saturation dataset provides more predictive power (Fig. 4A, B). This can likely be attributed to the nature of the rich datasets mostly containing mutants with non-zero activity (64/77 for moclobemide and 62/77 for metoclopramide), preventing “holes” in training sets . Moving forward, we decided to use the site saturation dataset and the augmented EVmutation model with Georgiev encodings given the strong predictive performance among both
Fig. 3 | Rapid generation of sequence-fitness landscape data for ML-guided directed evolution of McbA. A Workflow schematic: a supervised ridge regression model is trained on percent conversion data of four mutable sites selected from the HSS, with sequence features consisting of an amino acid encoding augmented with a zero-shot prediction of enzyme fitness. From a training set of single mutants, we extrapolate higher order mutations and test the top 25 predictions. Zero shot selection was compared against all encoding strategies (Georgiev is shown) and
ranked by NDCG. Hot spot screen of 64 identified residues in McbA showing foldchange percent conversion of moclobemide, metoclopramide, and D cinchocaine normalized to WT ( ; blue indicates low percent conversion, red indicates high percent conversion). The top four highly mutable sites, or hot spots, are highlighted in orange and starred. They are used as the training set for model validation. Full validation results are provided in Fig. S15. Source data are provided as a Source Data file.
Fig. 4 | ML-guided directed evolution predicts highly active mutants with a lower screening burden than iterative site saturation mutagenesis.
A, B Analysis of model fidelity with training sets built with smaller libraries than saturation mutagenesis, including reduced codon sets (NDT, NRT) and reduced amino acid alphabets based on BLOSUM50, quantified by spearman correlation ( ) and NDCG. Comparing measured versus predicted activity on withheld ISM rounds is shown for models trained on the complete saturation mutagenesis dataset for
both moclobemide (moc) (A), and metoclopramide (meto) (B). The experimentally validated percent conversion ( ; error bars indicate SD) of ML-predictions for moclobemide (C), metoclopramide (D), and cinchocaine (E) with the quadruple mutant from ISM (M4) colored gray. For cinchocaine, the ML model predictions did not include the highest performing mutant from ISM. Wild type McbA is colored dark gray. Percent conversion was measured by RP-HPLC in independent experiments. Source data are provided as a Source Data file.
compounds and the fact that the already-trained probability density model simplified application to other compounds (Fig. 3A). EVmutation is also less computationally resource and time intensive than ESM.
Using our trained ML models (one single-objective model per compound), we screened combinatorial enzyme variants for the synthesis of moclobemide, metoclopramide, and cinchocaine in silico and selected the top 25 predictions to subsequently build and test. We found that the augmented ML model was able to predict McbA variants enriched in high activity for each amide product when tested experimentally, some even surpassing qm-McbA from both moclobemide and metoclopramide ISM campaigns (Fig. 4C-E and Table S1). Notably, the best predicted mutant for metoclopramide contained a mutation (A424S) that was superseded in the HSS by a more active mutation (A424T) carried forward in ISM, indicating the model found a superior mutant that would have been overlooked using ISM alone. The best predicted variant for cinchocaine had significantly higher activity than the best single mutation on its own and surprisingly contained a mutation (A205L) that decreased activity compared to wt-McbA in the HSS (Figs. S16, 17 and Table S2); we could not rationally select and combine mutations from the HSS to reach the same results. These results show that our ML-guided strategy can discover high fitness variants for a variety of molecules using the same starting enzyme. While it is unclear if the model can directly infer nonlinear interactions, we found that it is able to avoid path dependencies and reduce the screening burden. However, the rank of experimentally tested predictions correlates poorly with predicted rank (average Spearman’s rank correlation of ), indicating the models may not accurately capture the entire sequence-fitness landscape.
ML-guided biocatalytic diversification for high-value pharmaceuticals
To assess the robustness of our approach, we next applied our MLguided framework to predict distinct McbA mutants for the synthesis of an additional six pharmaceutical compounds. Starting with an identified target reaction from our substrate scope screen (Fig. 2), we used the same instance of our 1,216 single mutant McbA variant library from above to perform an HSS (7,302 unique reactions total), select four hot spots, and train our ML model to predict higher order mutants with increased activity (Fig. 5A; Figs. S18-23). For each reaction, the top 24 predictions were tested, and the best variant was expressed, purified, and compared to wt-McbA activity. We observed increases in yields ranging from 1.6 -fold to 34 -fold over wt-McbA for the six compounds we tested (Fig. 5B-G and Figs. S18-23). For each compound, the best predicted mutant always outperformed the best rational design (i.e., combining the four best mutations from the HSS without using the ML model; Table S3-5). Some mutants gave only modest improvements, which may be an artifact of low signal-to-noise in the hot spot screens for some of the target compounds that were only detectable by MS. This can lead to flat fitness landscapes that are more difficult to model. Nonetheless, our framework yielded enzyme mutants with increased activity for multiple products that were initially only observed in trace amounts.
We also compared how efficiently some enzyme variants perform each reaction step (Fig. S24). For example, wt-McbA appears to be proficient at the adenylation step for troxipide (adenylating 3,4,5-trimethoxybenzoic acid), but unable to catalyze amide bond formation (Fig. 5G). The engineered enzyme variant can subsequently accept the amine, leading to a large decrease in the observed intermediate. Serendipitously, the engineered McbA variants for each target product
Fig. 5 | ML-guided engineering of distinct amide synthetases for the biosynthesis of a broad panel of small-molecule pharmaceuticals. A The strategy we used for machine learning-guided protein engineering of McbA is shown. First, we identified non-native reactions that wt-McbA can catalyze and prioritized those that produce valuable small-molecule pharmaceuticals. Second, an HSS of 64 residues is used to down-select residues that positively impact activity. Third, an augmented ridge regression model is trained on data from the HSS, and ML predictions are
experimentally tested. B-G Comparison of the highest activity predicted variant for a panel of small-molecule pharmaceuticals compared to wt-McbA and an authentic standard. Enzyme concentration was normalized to and products were analyzed by RP-HPLC. The fold-increase in yield observed compares wt-McbA to ML-McbA ( ). Representative HPLC traces of product (red), acid substrate (purple), and adenylated acid (orange) for each reaction are shown. Traces are taken from at least three independent experiments ( ).
also displayed strict regioselectivity despite the lack of any selective pressure to maintain it. This is exemplified by the quadruple mutant for troxipide that exhibits a 34 -fold increase in activity without any sacrifice in specificity. Similarly, stereoselective preferences with S -sulpiride are maintained. Taken together, our ML-guided framework allows us to use functional data from single mutant enzyme variants to predict superior higher-order mutants rapidly and effectively.
Discussion
In this work, we established a high-throughput, ML-guided protein engineering framework for predictive design that does not require specialized computational resources. This framework uniquely integrated a CFE and mutagenesis method, ML to expedite directed evolution campaigns, and divergent evolution to convert a generalist enzyme into multiple specialists. We showcased this framework by rapidly navigating nine protein engineering campaigns for the amide synthetase McbA, six of which were performed simultaneously.
Through efforts to build ML models and all ISM rounds, we explored the sequence-function landscape of McbA by assessing 2856 variants of McbA (1217 variants of which were for ML models), 1100 possible amide products, and 12,584 substrate pair-mutant reactions. We identified 19 unique residue positions within McbA that significantly impact biocatalysis, with each reaction yielding a unique set of these hot spot residues. Across all nine final engineered McbA variants, we made a total of 21 different mutations occurring across 14 different residues (Fig. S25). While many of the substrate pairs contain the same acid or amine, it is difficult to rationalize why certain mutations arise as they are not conserved among many of these enzymes. For example, the amide products metoclopramide, cinchocaine, procainamide, and declopramide all contain N,N-diethylethylenediamine but different acids. One could propose that V177S is a beneficial
mutation for this reaction, but it is only a generalization and not universal as procainamide did not contain this mutation. However, our data seems to indicate that residue selection (hot spots) are more strongly related to the overall reaction and not the acid or amine fragments on their own. In all cases, newly generated enzyme variants demonstrated improved activity relative to wt-McbA variants ( 1.6 -fold to 42 -fold improvement). In one example, an enzyme variant for moclobemide synthesis achieved conversion (a 42 -fold increase in catalytic efficiency over wt-McbA) and was scaled to milligram quantities.
An important feature of our work was the use of ML models trained on single-residue mutations to predict higher order mutants with improved fitness. We selected an augmented ridge regression ML model because it had previously been shown to perform well compared to more sophisticated approaches and was able to extrapolate from single to higher order mutations, an observation that is consistent with our data . For example, in each of the nine test cases, ML-predicted enzyme variants with 4 mutations had greater activity than the combination of the four most active single-residue mutants alone. This observation holds in instances where the data generation is high quality (e.g., moclobemide), the fitness landscape is flat (i.e., many zero activity mutants and not many mutants that improve activity), and the signal-to-noise ratio is high (e.g., declopramide), which highlights the robustness of our approach. There may be instances where more complex models (e.g., random forests, support vector machines, neural networks, etc.) that can generalize better to the entire sequence space will be necessary to navigate complex fitness landscapes. Despite this, the ML-guided framework used here aided in the search around hypothetical ISM trajectories, reducing effort and increasing success rates in multiple enzyme engineering campaigns.
Our approach can, in theory, be applied to any enzyme but will require reaction-specific fine-tuning around data collection and ML model generation. In terms of data collection, experimental screening methods for biocatalytic reactions remain a bottleneck. Here, because the product compounds of McbA were stable in the presence of the cell-free expression lysate and chromatographic methods were efficient (e.g., per sample), LC-MS provided a manageable solution, as has been found in other examples . As a complement to screening, there will be enzyme engineering applications where selection strategies are beneficial (e.g., when a tractable selection method exists, larger jumps in sequence space can be made). Engineering campaigns with different proteins may also warrant exploring various ML models and parameters. While we observed excellent performance with the augmented EVmutation model, alternative fitness goals may also require alternative fitness predictors. For example, if the goal is to engineer stability, it would reason that a structural-based fitness predictor may be superior . There are numerous other protein variant effect predictors continuously pushing the state-of-the-art forward that could improve our predictions . More complex sequence encodings based on natural language processing may also outperform augmented encodings . Finally, we note that training the ML models on more residues, multi-mutant data (including data from multiple rounds of mutagenesis or random combinations of mutants that diversify amino acids across the entire protein of interest), or kinetic measurements could be beneficial in engineering better catalysts.
In sum, our accessible ML-guided, cell-free framework overcomes traditional directed evolution challenges by circumventing path dependencies that constrain sequence search space in state-of-the-art methods. Additionally, cell-free expression from linear expression templates expedites the process of exploring sequence-function landscapes since the entire process for protein expression and assessment can be done without cells and we could avoid laborious cloning steps, taking hours instead of days to weeks. These features speed the pace of engineering relative to ISM alone; our framework enabled six enzyme engineering campaigns to be simultaneously completed in just 1 week per compound. Furthermore, we also note the low cost (i.e., cents per reaction) and high scalability of CFE enabling our workflow . Beyond the benefits of the cell-free framework, our work also highlights the versatility of the amide synthetase McbA to be directed to catalyze many unique reactions of interest, including those used in small-molecule pharmaceutical production. Looking forward, we anticipate that the approach described here, especially when augmented with de novo protein design , will accelerate enzyme engineering campaigns to unlock specialized enzymes with diverse functions and properties.
Methods
Cell-free DNA assembly and gene expression
DNA libraries were created for both wt-McbA and muGFP. wt-McbA from Marinactinospora thermotolerans (UniProt: R4R1U5) was codonoptimized for E. coli and cloned into the pJL1 plasmid (Addgene, 69496) with an N-terminal CSL-tag (CAT-Strep-Linker fusion containing Strep-tag II). muGFP was codon-optimized for E. coli and cloned into the pJL1 plasmid without a purification .
The cell-free DNA library generation was performed as follows: (1) the first PCR was performed in a reaction with 1 ng of plasmid template added, (2) of DpnI was added and incubated at for 2 h , (3) the PCR was diluted 1:4 by the addition of of nuclease-free (NF) water, (4) of diluted DNA was added to a Gibson assembly reaction (self-made) and incubated for for 1 h , (5) the assembly reaction was diluted 1:10 by the addition of of NF water, (6) of the diluted assembly reaction was added to a PCR reaction. All cloning steps were set up using an Integra VIAFLO liquid handling robot in 384-well PCR plates (Bio-Rad). Primers were designed using Benchling with melting temperature calculated by the default
SantaLucia 1998 algorithm. We have noticed that melting temperatures of alternative primer design tools sometimes deviate from those calculated in Benchling, so users should consider this when designing primers. The general heuristics we followed for primer design were a reverse primer of , a forward primer of , and a homologous overlap of . All primers were ordered from Integrated DNA Technologies (IDT); forward primers were synthesized and received in 384-well plates and normalized to for ease of setting up reactions. Additional information on primer design and the codons we used for all 20 amino acids can be found in Fig. S4 and Table S7. All PCR reactions used Q5 Hot Start DNA Polymerase (NEB). Additional information on thermocycler parameters can be found in Table S8.
To accumulate mutations for ISM, of the “winner” from the diluted Gibson assembly plate was transformed into of chemically competent . coli (NEB 5-alpha cells). Cells were plated onto LB plates containing kanamycin (LB-Kan). A single colony was used to inoculate 50 mL of LB-Kan, grown overnight at with 250 RPM shaking. The plasmid was purified using ZymoPURE II Midiprep kits and sequence confirmed. Successive mutations can then be incorporated via our cell-free DNA library generation method above.
The comprehensive combinatorial double mutant McbA library (used in Fig. S7e) was generated by two successive rounds of saturation mutagenesis with no particular residue targeted first. After the first site saturation, plasmids containing each mutation were prepared following the above protocol except 5 mL of overnight cultures in LB-Kan were used to purify plasmids using ZymoPURE II Miniprep kits. These 20 plasmids were used as templates for the next round of site saturation mutagenesis to accumulate all 400 double mutants.
All ML-predicted McbA variants were ordered as gblocks from IDT containing pJL1 5′ and 3′ Gibson assembly overhangs. DNA was resuspended at a concentration of . A linearized pJL1 plasmid backbone was ordered as a gblock from IDT, PCR amplified, purified using a DNA Clean and Concentrate Kit (Zymo Research), and diluted to a concentration of . Gibson assembly was used to assemble the DNA encoding McbA variants with the pJL1 backbone. 10 ng of purified, linearized pJL1 backbone and 10 ng of gblock insert were combined in a Gibson assembly reaction and incubated at for . The unpurified assembly reactions were diluted in of NF water and of the diluted reaction was used as the template for a PCR reaction (using Q5 Hot Start DNA polymerase) to generate LETs for CFPS.
Crude cell extracts were prepared as previously described using E. coli BL21 Star (DE3) cells (Invitrogen) . CFE reactions were performed based on the PANOx-SP system and carried out in 384-well PCR plates (Bio-Rad) as reactions with of LET serving as the DNA template. Reactions were incubated at for 16 h .
Expression and purification of recombinant proteins
pJL1-McbA plasmid was transformed into chemically competent E. coli BL21 Star (DE3) cells (Invitrogen) following the manufacturer’s instructions. Cells were plated onto LB-Kan and incubated overnight at . A single colony was used to inoculate a overnight culture in LB-Kan, grown at with 250 RPM shaking. 1 L of Overnight Express TB Medium (Millipore) was prepared following the manufacturer’s instructions and supplemented with kanamycin. The TB medium was inoculated the following day using the 5 mL overnight culture and grown at with 250 RPM shaking until saturation . Cells were harvested by centrifugation (Beckman Coulter Avanti J-26) at for 10 min at . Cell pellets were either flash frozen with liquid nitrogen and stored at until future use or resuspended in 25 mL of Wash Buffer Tris- HCl pH 8.0 , EDTA, glycerol). Resuspended cells were lysed by sonication (QSonica Q700 Sonicator) using six 10 s ON and 10 s OFF cycles at amplitude, and the insoluble fraction was removed by centrifugation at for 20 min at . Clarified
lysates were incubated with 2 mL of pre-equilibrated Strep-Tactin XT Superflow resin (IBA Lifesciences) with shaking for 30 min at . Resin was loaded onto a gravity-flow column and washed three times with 20 mL Wash Buffer. McbA protein was eluted with 10 mL of Elution Buffer Tris- EDTA, 50 mM biotin, glycerol) and concentrated with a Amicon Ultra Centrifugal filter (Millipore Sigma; 30 kDa cutoff). Purified McbA was buffer exchanged into Storage Buffer HEPES pH 7.5, glycerol) using a pre-equilibrated PD-10 desalting column (Cytiva). McbA was stored at for immediate use ( ) or for longer term storage. Protein concentration was quantified by measuring A280 on a NanoDrop 2000c (Thermo Scientific), with McbA extinction coefficient and molecular weight calculated by Expasy ProtParam. wt-McbA and the six engineered McbA variants found in Fig. 5 were purified in this manner.
muGFP activity assay
Performance of muGFP variants were quantified by measuring fluorescence on a plate reader (BioTek Synergy H1) using an excitation of 485 nm and emission of of crude CFPS reaction containing an expressed muGFP variant was transferred to a black, round bottom 384-well plate (Nunc) prior to measurements.
Amide synthetase activity assay
All high-throughput assays (hot spot screen, iterative site saturation mutagenesis, substrate scope, ML predictions validation, and ML prediction exploration) were assembled in 384-well plates (Bio-Rad) using an Integra VIAFLO liquid handling robot. A reaction mix containing the substrates (ATP, acid, amine, and DMSO) with excess volume filled with 50 mM potassium phosphate pH 7.5 was dispensed as aliquots in a 384 -well plate. The amidation assay was initiated by adding of crude CFPS reaction containing an expressed McbA variant, with final concentrations of 25 mM ATP, 25 mM acid, 25 mM amine, DMSO, and of enzyme (determined by -leucine incorporation using previously described protocols ). Stock solutions of the acids were prepared in DMSO and this was taken into account to reach DMSO. For reactions that were performed in triplicates, from the same CFPS reaction was used for three separate assays. The reaction was incubated at for 16 h and then quenched with of methanol. Plates were stored at until prepared for analysis via LC-MS.
Amidation assays for the purified McbA variants found in Fig. 5 were set up similarly as described above in 384 -well plates. reactions were assembled in triplicate, containing 25 mM ATP, 25 mM acid, 25 mM amine, pyrophosphatase (Sigma I5907), DMSO, and volume to fill of 50 mM potassium phosphate pH 7.5. For assaying the production of cinchocaine and procainamide, substrates were decreased in stoichiometric amounts to 20 mM and 10 mM , respectively. This was to compensate for an observed poor solubility of these two acids (2-butoxyquinoline-4-carboxylic acid and 4-aminobenzoic acid) in the purified reaction at DMSO. Reactions were incubated at for 16 h and then quenched with of methanol. Samples were stored at until prepared for analysis via LC-MS. The CAS numbers of all chemicals used in the hot spot screens, as well as the amide standards we purchased, can be found in Table S13.
Amide synthetase & ATP regeneration assay
Polyphosphate kinase, PPK12 from an unclassified Erysipelotrichaceae (Uniprot: A0A847P5F2_9FIRM), was cloned, expressed, and purified to homogeneity as previously described reactions were assembled in triplicate, containing 25 mM amine, 25 mM acid, polyphosphate (Sigma 1.06529 ), pyrophosphatase (Sigma I5907), McbA, PPK12, 10% v/v
DMSO, and volume to fill of 50 mM potassium phosphate pH 7.5. A 2-fold serial dilution of AMP was prepared and added to the reaction mix to final concentrations ranging from 25 mM to 0.02 mM . Reactions were incubated at for 16 h and then quenched with of methanol and analyzed by LC-MS.
Preparative scale biosynthesis of moclobemide
Scaled amidation assays for the enzymatic preparation of moclobemide were set up similarly as described above. A reaction containing 25 mM ATP, 25 mM acid, 25 mM amine, , pyrophosphatase (Sigma I5907), DMSO, and volume to fill of 50 mM potassium phosphate pH 7.5 . After 16 h , the reaction was quenched and product was extracted by the addition of 30 mL of ethyl acetate . The organic phases were collected, washed with , and brine , dried over , filtered, and the solvent was evaporated under reduced pressure to afford the desired product as a white powder ( isolated yield) without any further purification. The and NMR (found below and in Fig. S12) are in good agreement with those previously reported . Spectra for and NMR were recorded at room temperature with a Bruker Avance III 500 MHz system. Chemical shifts are reported in relative units to residual solvent peaks DMSO-d6 ( 2.50 ppm for 1 H and 39.5 ppm for 13 C ). Splitting patterns are assigned as s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), m (multiplet). Coupling constants are reported as Hz, followed by integration. NMR. ( 500 MHz , DMSO-d6) , , . C NMR. ( 126 MHz, DMSO) 165.51, 136.38, 133.69, 129.55, 128.85, 66.66, 57.77, 53.76, 37.06.
LC-MS analytics
Amide products (along with acid substrates and some adenylated acid intermediates) were analyzed using an Agilent G6125B Single Quadrupole LC/MSD system equipped with an electrospray ionization source set to positive ionization mode. The quenched samples were centrifuged for 10 min at to remove precipitated proteins. A separate 384 -well plate for sample injection into the HPLC-MS was prepared by diluting of the quenched samples with of methanol using the Integra VIAFLO. Trace amounts of compounds were detected using MS, while many compounds were present in high enough concentration to quantify by diode array detector (DAD) at 254 nm . Compounds were separated on a Luna C18 Column (Phenomenex 00D-4251-BO) using mobile phases (A) with formic acid and (B) Acetonitrile. The general method for chromatographic separation was carried out using the following gradients at a constant flow rate of : . For hot spot screens, an expediated method was used with the following gradients at a constant flow rate of : ; . For the MS, capillary voltage was set at 3 kV , and nitrogen gas was used for nebulizing ( 35 psig ) and drying . The MS was calibrated using Tuning Mix (Agilent G2421-60001) before measurements were taken. MS data were acquired with a scan range of with various SIM according to which compound we were screening for. LC-MS data were collected and analyzed using Agilent OpenLab CDS ChemStation software. The product yield was estimated by dividing the DAD peak area for the amide product by the sums of the peak areas of both the amide and the acid substrate. An exact quantitative yield for moclobemide was recorded after its preparative scale synthesis and isolation.
Melting temperature determination
Protein melting temperature was determined using a Jasco J-810 circular dichroism spectrophotometer with a 10 mm path length cuvette monitored at 222 nm . McbA samples were first buffer exchanged into a 1X phosphate buffered saline solution, pH 7.4 , and diluted to .
Enzyme kinetics
McbA apparent kinetics for the amine pair of moclobemide (4-(2aminoethyl)morpholine) were determined by enzymatically coupling amide bond formation (and the concomitant release of AMP from the acyl-AMP intermediate by its substitution with the amine) with the oxidation of NADH (Fig. S9). Reactions contained 100 mM MOPS-KOH pH 7.8, phosphoenolpyruvate, NADH, 50 mM 4-chlorobenzoic acid, pyruvate kinase and lactate dehydrogenase enzyme mix (Sigma-Aldrich P0294), myokinase (Sigma-Aldrich 475941), and various concentrations of the studied McbA variant. As the acid here (4chlorobenzoic acid) has poor solubility in water and was dissolved in DMSO, the final reactions contained DMSO (equivalent to our amidation screens). reactions were first equilibrated at for 3 min and then initiated by adding of amine. The initial velocity was determined for different concentrations of amine by measuring NADH absorbance at 340 nM on a Cary 60 UV-Vis (Agilent). Data was collected and analyzed using the Cary WinUV Kinetics Application software (Agilent). Michaelis-Menten graphs were plotted in GraphPad Prism and fit using the default Michaelis-Menten non-linear regression analysis tool.
Kinetics for the acid pair of moclobemide (4-chlorobenzoic acid) were measured similarly as described above, except the amine was held constant at 50 mM , and the reaction was initiated by addition of various amounts of the acid. The final DMSO concentration was still held constant at . We observed non-Michaelis-Menten behavior when attempting to determine the kinetics for the acid, in what appeared to be substrate inhibition by the acid (data not shown). We also attempted to measure the acid adenylation step directly by enzymatically coupling acyl-AMP formation (and the concomitant release of ) with the oxidation of NADH to further probe the reaction mechanism. The Piper™ pyrophosphate assay kit (Fisher Scientific P22062) was used, but the addition of small concentrations of DMSO resulted in the precipitation of enzymes found in the kit.
Amino acid encodings
Five different amino acid encoding strategies were studied here following the work of Wittman et al. and Vornholt et al. : one-hot, Georgiev, VHSE, z-scales, and physical descriptors. Beyond one-hot encodings (that contain no information about the nature of the amino acid at each position), we also wanted to include encodings that attempt to encapsulate physiochemical properties of amino acids. We briefly explain these encodings below (in order of most to least parameters) and encourage readers to visit these sources for further information. To make informative numerical representations of amino acid properties, these strategies perform principal component analysis of different manually curated sets of either experimentally measured or computationally predicted/estimated properties. Georgiev features (19-parameters) are principal components of the over 500 amino acid indices taken from the AAindex database. VHSE features (8parameters) are principal components of 50 variables, focused on hydrophobic, steric, and electronic properties. Z-scales (5-parameters) features are principal components of 26 variables, focused on lipophilicity, size, and polarity. Physical descriptors (3-parameters) features are derived from a rational ad hoc modification of principal components of hydrophobic and steric properties of peptides. For all strategies, we first generated encodings for the entire combinatorial library tested (stored in a tensor of ” unique variants” × 4 amino
acids” × ” -parameters”, where -parameters is equal to the number of amino acids for one-hot). The last two dimensions of the tensor were then flattened to generate a matrix. Specifically for the physiochemical encodings (excluding one-hot), each column of the matrix was standardized (mean-centered and unit-scaled).
Zero-shot predictions
Evolutionary. The EVmutations probability density model was trained using the EVcouplings webserver (https://evcouplings.org/) with default parameters, with the input sequence for McbA taken from UniProt (R4R1U5). The model we selected had a bitscore inclusion threshold of 0.7. The model and code for replicating zero-shot predictions are provided in our GitHub repository. The mutation effects prediction code provided in the EVcouplings GitHub repository (https://github.com/debbiemarkslab/EVcouplings/blob/develop/ notebooks/model_parameters_mutation_effects.ipynb) was used as a template. Features for the augmented models were derived from the sequence statistical energy relative to wild type.
Universal. Predictions using the ESM-1b pre-trained transformer language model were made using the code provided from the excellent work of Wittman et al. on ML-guided directed evolution (https:// github.com/fhalab/MLDE) with the ESM-1b model provided in the ESM GitHub repository (https://github.com/facebookresearch/esm). Briefly, a mask-filling protocol was used to predict the probability of different mutants by presenting the model with the entire sequence and “masking” a position of interest. We used a naïve mask-filing approach, which considers each variable position as independent from each other. This mask-filing approach was used as it is less computationally expensive and provided slightly superior predictions than a conditional approach (which does not assume independence of variable positions) in this previous work. A complete description of the code can be found in the original publication and the associated GitHub repository. Features for the augmented models were derived from the sequence log-probability relative to wild type.
Structural. Structural-based predictions were made using the MAESTRO command line tool for Windows (v1.2.35). We used the Protein Data Bank (PDB) structure for McbA (6SQ8) as the input and calculated changes in stability (unfolding free energy) with the “evalmut” command. Features for the augmented models were derived using the ‘energy’ output.
Machine learning-guided directed evolution
Ridge regression models were augmented following the code accompanying the elegant work of Hsu et al. (https://github.com/ chloechsu/combining-evolutionary-and-assay-labeled-data). McbA variant sequence featurization was performed by concatenating zeroshot predictions with site-specific amino acid encodings. Zero-shot predictions were first standardized and regularized by a common regularization strength . The L 2 regularization strength for ridge regression ( ) was determined during hyperparameter tuning using cross-validation. For our complete code used in this work, please see our accompanying GitHub repository at https://github.com/ grantlandwehr/accelerated-enzyme-engineering. Given some changes made between initial model development and reimplementation of the code for publication (e.g., hyperparameter tuning cross validation scheme, search range of the regularization coefficient , etc.) there are minor differences in predictions ranked 23-25 for metoclopramide and moclobemide found in Fig. 4.
Model evaluation and selection were first performed retrospectively by using the assay-labeled datasets from our moclobemide and metoclopramide engineering campaigns. Augmented models (using combinations of the above zero-shot predictors and amino acid encodings) were trained on the single site saturation libraries for four
residues ( ) and tested on the withheld higher-order mutants from the additional rounds of saturation mutagenesis ( ). The four residues (hot spots) were determined by selecting the four residues that contained mutations with highest improvement in yield among the 64 residues tested. Hyperparameter turning of was performed using repeated 5 -fold cross-validation (with 20 repeats) by randomly sampling of the training data and testing on the withheld 20%; model performance for hyperparameter tuning was scored using mean squared error (MSE). With the optimized hyperparameter, all trained models were used to make predictions on the withheld test set. Spearman correlation coefficient and NDCG were used to select the best zero-shot predictor and encoding strategy, with a preference given to NDCG.
After identifying the best model (which in our case was augmenting the EVmutation probability density model with Georgiev encodings), we made predictions on the entire combinatorial dataset ( ). The top 25 predictions for moclobemide and metoclopramide were then experimentally tested (Fig. 4). Model training and predictions for the remaining seven amide products was performed similarly as above. Given the already trained EVmutation probability density model and the low dimensionality of the encodings, model training and predictions for the entire combinatorial dataset could be made in minutes running on 12th Gen Intel Core i7 with 32 GB RAM without GPU acceleration.
Data collection and analysis
All statistical information provided in this manuscript is derived from independent experiments unless otherwise noted in the text or figure legends. Error bars represent 1 s.d. of the mean derived from these experiments. Data analysis and figure generation were conducted using Excel Version 2304, ChimeraX Version , GraphPad Prism Version 9.5.0, and Python 3.9 using custom scripts available on GitHub. muGFP fluorescence was measured on a BioTek Synergy H1 Microplate Reader and analyzed using Gen5 Version 2.09.2. Autoradiograms were performed as previously described and scanned using the Typhoon FLA 7000 Imager v1.2 .
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data presented in this manuscript are available in the Source Data file or deposited in the associated GitHub (https://github.com/ grantlandwehr/accelerated-enzyme-engineering). Protein and DNA sequences for all enzymes expressed in this work are available in the Supplementary Information. Source data are provided with this paper.
Code availability
The code to reproduce the results is available at https://github.com/ grantlandwehr/accelerated-enzyme-engineering .
References
Schwander, T., von Borzyskowski, L. S., Burgener, S., Cortina, N. S. & Erb, T. J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 354, 900-904 (2016).
Sarai, N. S. et al. Directed evolution of enzymatic silicon-carbon bond cleavage in siloxanes. Science 383, 438-443 (2024).
Fryszkowska, A. et al. A chemoenzymatic strategy for site-selective functionalization of native peptides and proteins. Science 376, 1321-1327 (2022).
Arnold, F. H. Design by directed evolution. Acc. Chem. Res 31, 125-131 (1998).
Packer, M. S. & Liu, D. R. Methods for the directed evolution of proteins. Nat. Rev. Genet 16, 379-394 (2015).
Miton, C. M. & Tokuriki, N. How mutational epistasis impairs predictability in protein evolution and design. Protein Sci. 25, 1260-1272 (2016).
Rix, G. et al. Scalable continuous evolution for the generation of diverse enzyme variants encompassing promiscuous activities. Nat. Commun. 11, 5644 (2020).
Kalvet, I. et al. Design of heme enzymes with a tunable substrate binding pocket adjacent to an open metal coordination site. J. Am. Chem. Soc. 145, 14307-14315 (2023).
Chu, A. E., Lu, T. & Huang, P.-S. Sparks of function by de novo protein design. Nat. Biotechnol. 42, 203-215 (2024).
Yeh, A. H.-W. et al. De novo design of luciferases using deep learning. Nature 614, 774-780 (2023).
Yang, K. K., Wu, Z. & Arnold, F. H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687-694 (2019).
Freschlin, C. R., Fahlberg, S. A. & Romero, P. A. Machine learning to navigate fitness landscapes for protein engineering. Curr. Opin. Biotech. 75, 102713 (2022).
Zhang, S. et al. EvoAI enables extreme compression and reconstruction of the protein sequence space. Res. Sq. rs.3.rs-3930833. https://doi.org/10.21203/rs.3.rs-3930833/v1 (2024).
Wittmann, B. J., Yue, Y. & Arnold, F. H. Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Syst. 12, 1026-1045.e7 (2021).
Bell, E. L. et al. Directed evolution of an efficient and thermostable PET depolymerase. Nat. Catal. 5, 673-681 (2022).
Silverman, A. D., Karim, A. S. & Jewett, M. C. Cell-free gene expression: an expanded repertoire of applications. Nat. Rev. Genet. 21, 151-170 (2020).
Karim, A. S. et al. In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat. Chem. Biol. 16, 912-919 (2020).
Hunt, A. C. et al. A rapid cell-free expression and screening platform for antibody discovery. Nat. Commun. 14, 3897 (2023).
Hunt, A. C. et al. Multivalent designed proteins neutralize SARS-CoV-2 variants of concern and confer protection against infection in mice. Sci. Transl. Med. 14, eabn1252-eabn1252 (2022).
Rapp, J. T., Bremer, B. J. & Romero, P. A. Self-driving laboratories to autonomously navigate the protein fitness landscape. Nat. Chem. Eng. 1, 97-107 (2024).
Fallah-Araghi, A., Baret, J.-C., Ryckelynck, M. & Griffiths, A. D. A completely in vitro ultrahigh-throughput droplet-based microfluidic screening system for protein engineering and directed evolution. Lab Chip 12, 882-891 (2012).
Ao, Y. et al. Structure- and data-driven protein engineering of transaminases for improving activity and stereoselectivity. Angew. Chem. Int. Ed. 62, e202301660 (2023).
Yu, T. et al. Enzyme function prediction using contrastive learning. Science 379, 1358-1363 (2023).
Wang, X. et al. Multi-modal deep learning enables efficient and accurate annotation of enzymatic active sites. Nat. Commun. 15, 7348 (2024).
Pattabiraman, V. R. & Bode, J. W. Rethinking amide bond synthesis. Nature 480, 471-479 (2011).
Bryan, M. C. et al. Key Green Chemistry research areas from a pharmaceutical manufacturers’ perspective revisited. Green. Chem. 20, 5082-5103 (2018).
Boström, J., Brown, D. G., Young, R. J. & Keserü, G. M. Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discov. 17, 709-727 (2018).
Sabatini, M. T., Boulton, Lee, T., Sneddon, H. F. & Sheppard, T. D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2, 10-17 (2019).
Wu, S., Snajdrova, R., Moore, J. C., Baldenius, K. & Bornscheuer, U. T. Biocatalysis: enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 60, 88-119 (2021).
Winn, M. et al. Discovery, characterization and engineering of ligases for amide synthesis. Nature 593, 391-398 (2021).
Schnepel, C. et al. Thioester-mediated biocatalytic amide bond synthesis with in situ thiol recycling. Nat. Catal. 6, 89-99 (2023).
Petchey, M. et al. The broad aryl acid specificity of the amide bond synthetase mcba suggests potential for the biocatalytic synthesis of amides. Angew. Chem. Int. Ed. 57, 11584-11588 (2018).
Chen, Q. et al. Discovery of McbB, an enzyme catalyzing the carboline skeleton construction in the marinacarboline biosynthetic Pathway. Angew. Chem. Int. Ed. 52, 9980-9984 (2013).
Tang, Q. et al. Broad spectrum enantioselective amide bond synthetase from Streptoalloteichus hindustanus. ACS Catal. 14, 1021-1029 (2024).
Petchey, M. R., Rowlinson, B., Lloyd, R. C., Fairlamb, I. J. S. & Grogan, G. Biocatalytic synthesis of moclobemide using the amide bond synthetase McbA coupled with an ATP recycling system. ACS Catal. 10, 4659-4663 (2020).
Jewett, M. C. & Swartz, J. R. Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng. 86, 19-26 (2004).
Scott, D. J. et al. A novel ultra-stable, monomeric green fluorescent protein for direct volumetric imaging of whole organs using CLARITY. Sci. Rep.-uk 8, 667 (2018).
Yong, K. J. & Scott, D. J. Rapid directed evolution of stabilized proteins with cellular high-throughput encapsulation solubilization and screening (CHESS). Biotechnol. Bioeng. 112, 438-446 (2015).
Pédelacq, J.-D., Cabantous, S., Tran, T., Terwilliger, T. C. & Waldo, G. S . Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79-88 (2006).
Hsu, C., Nisonoff, H., Fannjiang, C. & Listgarten, J. Learning protein fitness models from evolutionary and assay-labeled data. Nat. Biotechnol. 1-9. https://doi.org/10.1038/s41587-021-01146-5 (2022).
Georgiev, A. G. Interpretable numerical descriptors of amino acid space. J. Comput Biol. 16, 703-723 (2009).
Mei, H., Liao, Z. H., Zhou, Y. & Li, S. Z. A new set of amino acid descriptors and its application in peptide QSARs. Pept. Sci. 80, 775-786 (2005).
Sandberg, M., Eriksson, L., Jonsson, J., Sjöström, M. & Wold, S. New chemical descriptors relevant for the design of biologically active peptides. a multivariate characterization of 87 amino acids. J. Med Chem. 41, 2481-2491 (1998).
Barley, M. H., Turner, N. J. & Goodacre, R. Improved descriptors for the quantitative structure-activity relationship modeling of peptides and proteins. J. Chem. Inf. Model 58, 234-243 (2018).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 118, e2016239118 (2021).
Hopf, T. A. et al. Mutation effects predicted from sequence covariation. Nat. Biotechnol. 35, 128-135 (2017).
Laimer, J., Hofer, H., Fritz, M., Wegenkittl, S. & Lackner, P. MAESTRO -multi agent stability prediction upon point mutations. BMC Bioinform. 16, 116 (2015).
Järvelin, K. & Kekäläinen, J. Cumulated gain-based evaluation of IR techniques. ACM Trans. Inf. Syst. (TOIS) 20, 422-446 (2002).
Reetz, M. T., Kahakeaw, D. & Lohmer, R. Addressing the numbers problem in directed evolution. Chembiochem 9, 1797-1804 (2008).
Aslan, A. S., Birmingham, W. R., Karagüler, N. G., Turner, N. J. & Binay, B. Semi-rational design of Geobacillus stearothermophilus L-lactate dehydrogenase to access various chiral -hydroxy acids. Appl. Biochem. Biotech. 179, 474-484 (2016).
Gray, V. E., Hause, R. J. & Fowler, D. M. Analysis of large-scale mutagenesis data to assess the impact of single amino acid substitutions. Genetics 207, 53-61 (2017).
Murphy, L. R., Wallqvist, A. & Levy, R. M. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein Eng. Des. Sel. 13, 149-152 (2000).
O’Kane, P. T., Dudley, Q. M., McMillan, A. K., Jewett, M. C. & Mrksich, M. High-throughput mapping of CoA metabolites by SAMDI-MS to optimize the cell-free biosynthesis of HMG-CoA. Sci. Adv. 5, eaaw9180 (2019).
Goldenzweig, A. et al. Automated structure- and sequence-based design of proteins for high bacterial expression and stability. Mol. Cell 63, 337-346 (2016).
Mansoor, S., Baek, M., Juergens, D., Watson, J. L. & Baker, D. Zeroshot mutation effect prediction on protein stability and function using RoseTTAFold. Protein Science. 32, e4780 (2023).
Biswas, S., Khimulya, G., Alley, E. C., Esvelt, K. M. & Church, G. M. Low-N protein engineering with data-efficient deep learning. Nat. Methods 18, 389-396 (2021).
Lauko, A. et al. Computational design of serine hydrolases. bioRxiv 2024.08.29.610411. https://doi.org/10.1101/2024.08.29. 610411 (2024).
Kightlinger, W. et al. Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases. Nat. Chem. Biol. 14, 627-635 (2018).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343-345 (2009).
Kwon, Y.-C. & Jewett, M. C. High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep. 5, 8663 (2015).
Jewett, M. C., Calhoun, K. A., Voloshin, A., Wuu, J. J. & Swartz, J. R. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol. 4, 220-220 (2008).
Jewett, M. C. & Swartz, J. R. Substrate replenishment extends protein synthesis with an in vitro translation system designed to mimic the cytoplasm. Biotechnol. Bioeng. 87, 465-471 (2004).
Tavanti, M., Hosford, J., Lloyd, R. C. & Brown, M. J. B. ATP regeneration by a single polyphosphate kinase powers multigram-scale aldehyde synthesis in vitro. Green Chem. 23, 828-837 (2020).
Lavayssiere, M. & Lamaty, F. Amidation by reactive extrusion for the synthesis of active pharmaceutical ingredients teriflunomide and moclobemide. Chem. Commun. 59, 3439-3442 (2023).
Vornholt, T. et al. Systematic engineering of artificial metalloenzymes for new-to-nature reactions. Sci. Adv. 7, eabe4208 (2021).
Hellberg, S., Sjoestroem, M., Skagerberg, B. & Wold, S. Peptide quantitative structure-activity relationships, a multivariate approach. J. Med Chem. 30, 1126-1135 (1987).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70-82 (2021).
Zubi, Y. S. et al. Metal-responsive regulation of enzyme catalysis using genetically encoded chemical switches. Nat. Commun. 13, 1864 (2022).
Landwehr, G. et al. Accelerated enzyme engineering by machinelearning guided cell-free expression. accelerated-enzymeengineering. https://doi.org/10.5281/zenodo. 14262332 (2024).
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031-3048 (2017).
Acknowledgements
We thank Kosuke Seki, Andrew C. Hunt, and Steve R. Fleming for conversations regarding this work. We acknowledge the use of the Keck Biophysics Facility, a shared resource of the Robert H. Lurie
Comprehensive Cancer Center of Northwestern University supported in part by the NCI Cancer Center Support Grant #P30 CA060553. In addition, we acknowledge the use of the computational resources and staff contributions provided for the Quest high performance computing facility at Northwestern University which is jointly supported by the Office of the Provost, the Office for Research, and Northwestern University Information Technology. This work also made use of the IMSERC NMR facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), Int. Institute of Nanotechnology, and Northwestern University. Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. M.C.J. acknowledges support from the Department of Energy Grant DE-SCOO23278, the Defense Threat Reduction Agency Grant DTRA1-21-1-0038, the National Institutes of Health Grant 1U19AI142780-01, and the LDRD Program at Sandia National Laboratories Grant DE-NAOOO3525.
G.M.L., J.W.B., A.S.K., and M.C.J. have filed an invention disclosure based on the work presented. M.C.J. has a financial interest in National Resilience, Gauntlet Bio, Pearl Bio, Inc., and Stemloop Inc. M.C.J.’s interests are reviewed and managed by Northwestern University and Stanford University in accordance with their competing interest policies. All other authors declare no competing interests.
Correspondence and requests for materials should be addressed to Ashty S. Karim or Michael C. Jewett.
Peer review information Nature Communications thanks James Carothers, Song He and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2025
¹Department of Chemical and Biological Engineering, Northwestern University, Evanston, IL, USA. ²Center for Synthetic Biology, Northwestern University, Evanston, IL, USA. Department of Bioengineering, Stanford University, Stanford, CA, USA. These authors contributed equally: Grant M. Landwehr, Jonathan W. Bogart. e-mail: ashty.karim@northwestern.edu; mjewett@stanford.edu