DOI: https://doi.org/10.1038/s41467-024-47402-5
PMID: https://pubmed.ncbi.nlm.nih.gov/38609380
تاريخ النشر: 2024-04-12
الهيدروجين الانتقائي الخالي من المذيبات للنيتروأرومات إلى مركبات الأزوكسي على ذرات الكوبالت المفروشة على شبكات نانوية من Nb2O5
تم القبول: 1 أبريل 2024
نُشر على الإنترنت: 12 أبريل 2024
(د) التحقق من التحديثات
الملخص
الهيدروجين الانتقائي الخالي من المذيبات للنيتروأرومات إلى مركبات الأزوكسي هو أمر مهم للغاية، ولكنه يمثل تحديًا. هنا، نبلغ عن استراتيجية فعالة لبناء ذرات الكوبالت الموزعة بشكل فردي والمزينة على نانوميشات أكسيد النيوبيوم الخماسية مع خصائص هندسية وإلكترونية فريدة. إن استخدام هذه المحفزات أحادية الذرة المدعومة من الكوبالت في الهيدروجين الانتقائي للنيتروبنزين إلى الأزوكسي بنزين يؤدي إلى نشاط تحفيزي عالي وانتقائية، مع
مثبطات البلمرة، وإضافات الطعام
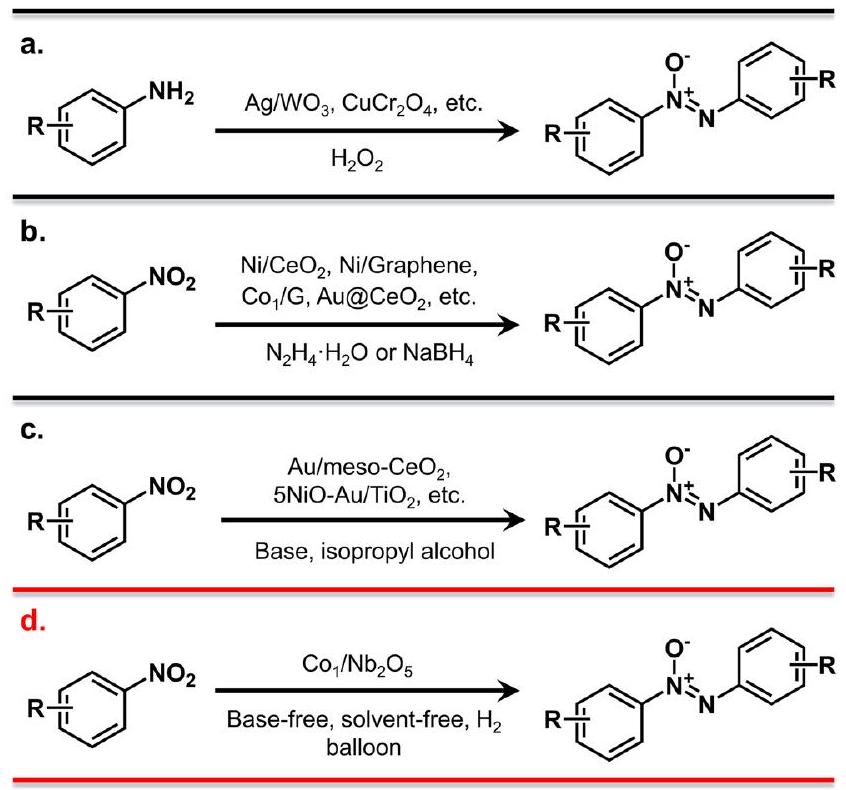
أكسدة الأنيلينات إلى مركبات الأزوكسيد العطرية.
النتائج
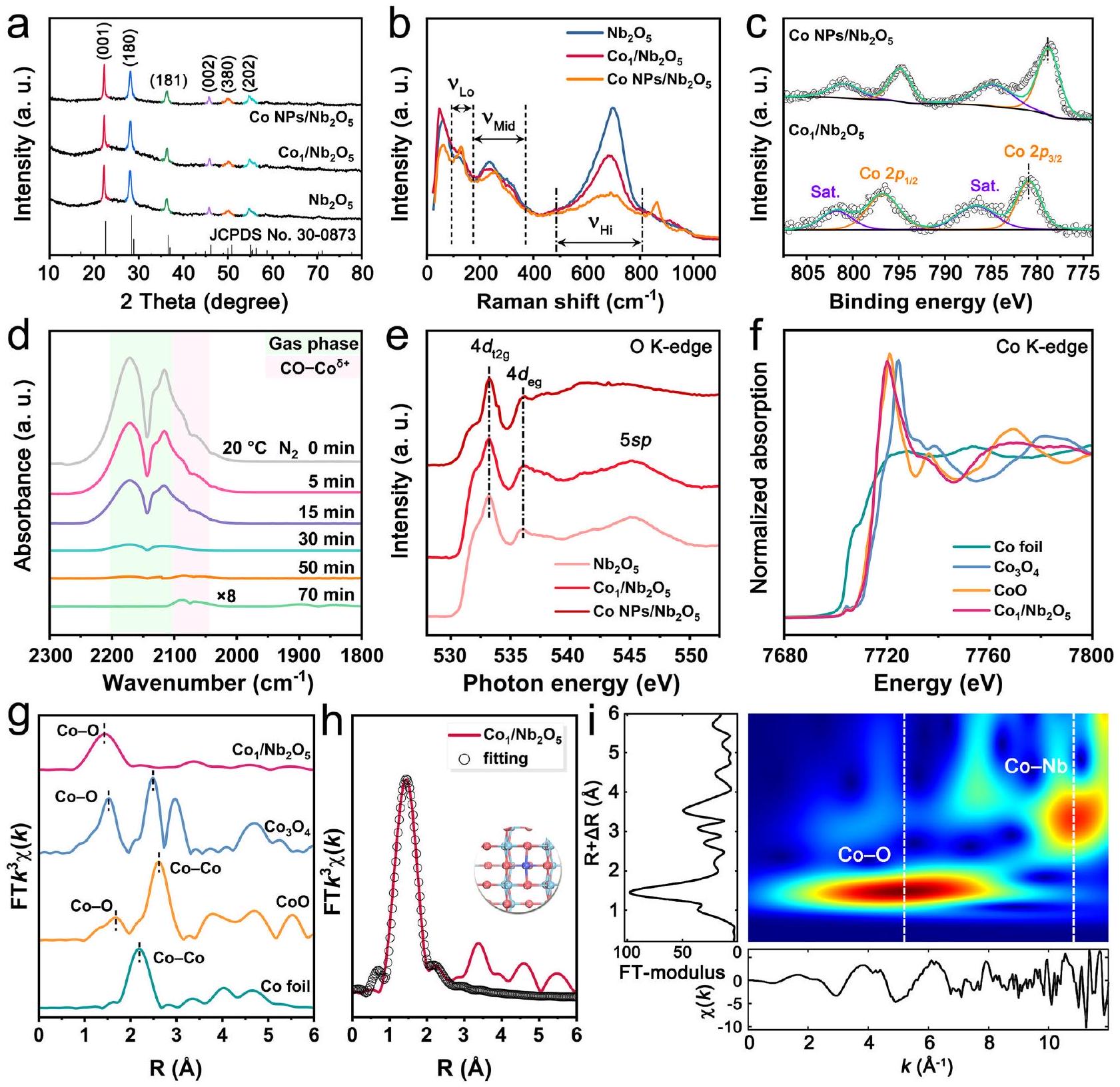
تركيب وتوصيف محفز الكوبالت الموزع ذريًا
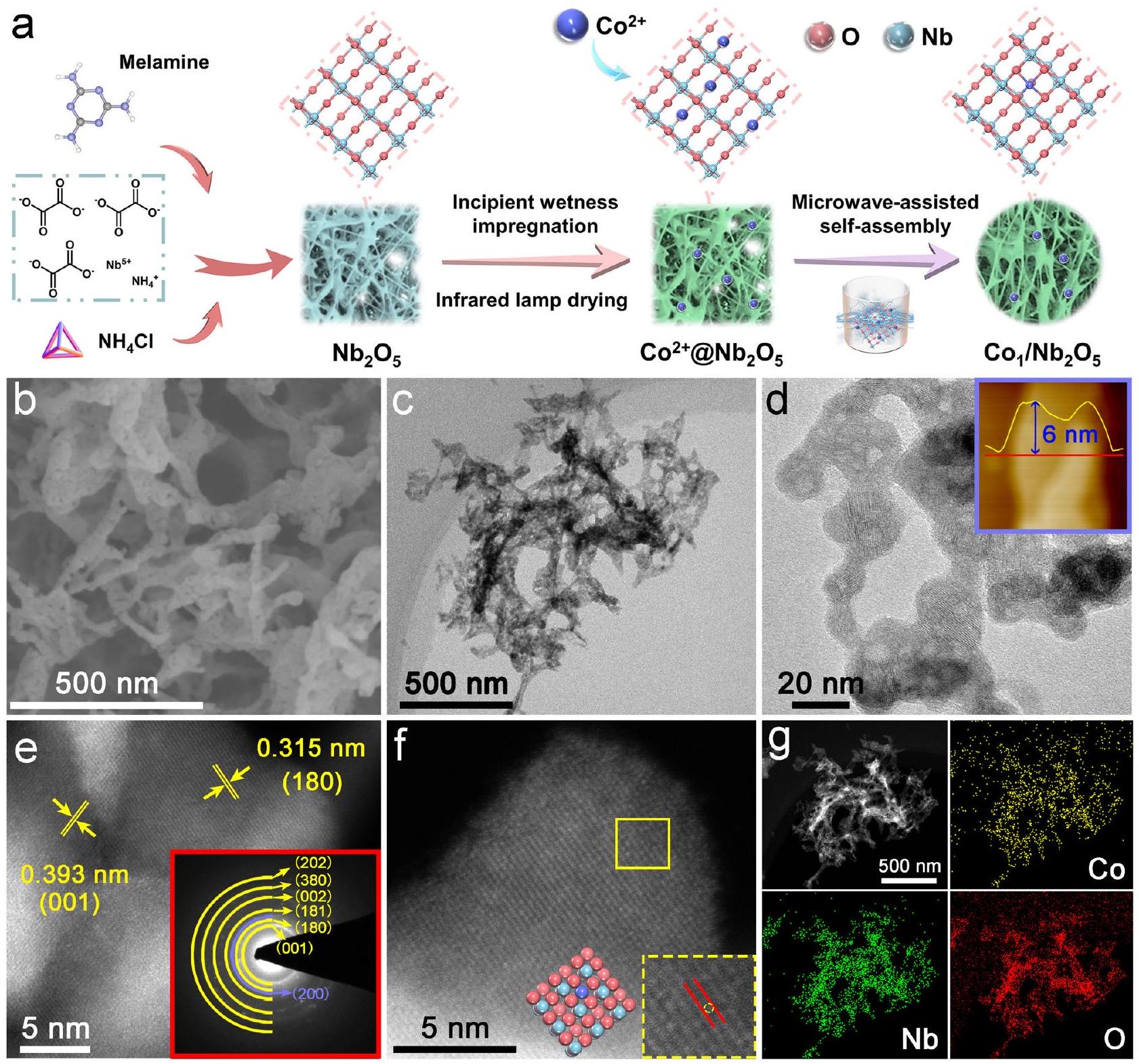
(النمط المدعوم هو نمط SAED).
و الشكل التوضيحي الإضافي 5). بالمقارنة مع
تقييم الأداء التحفيزي
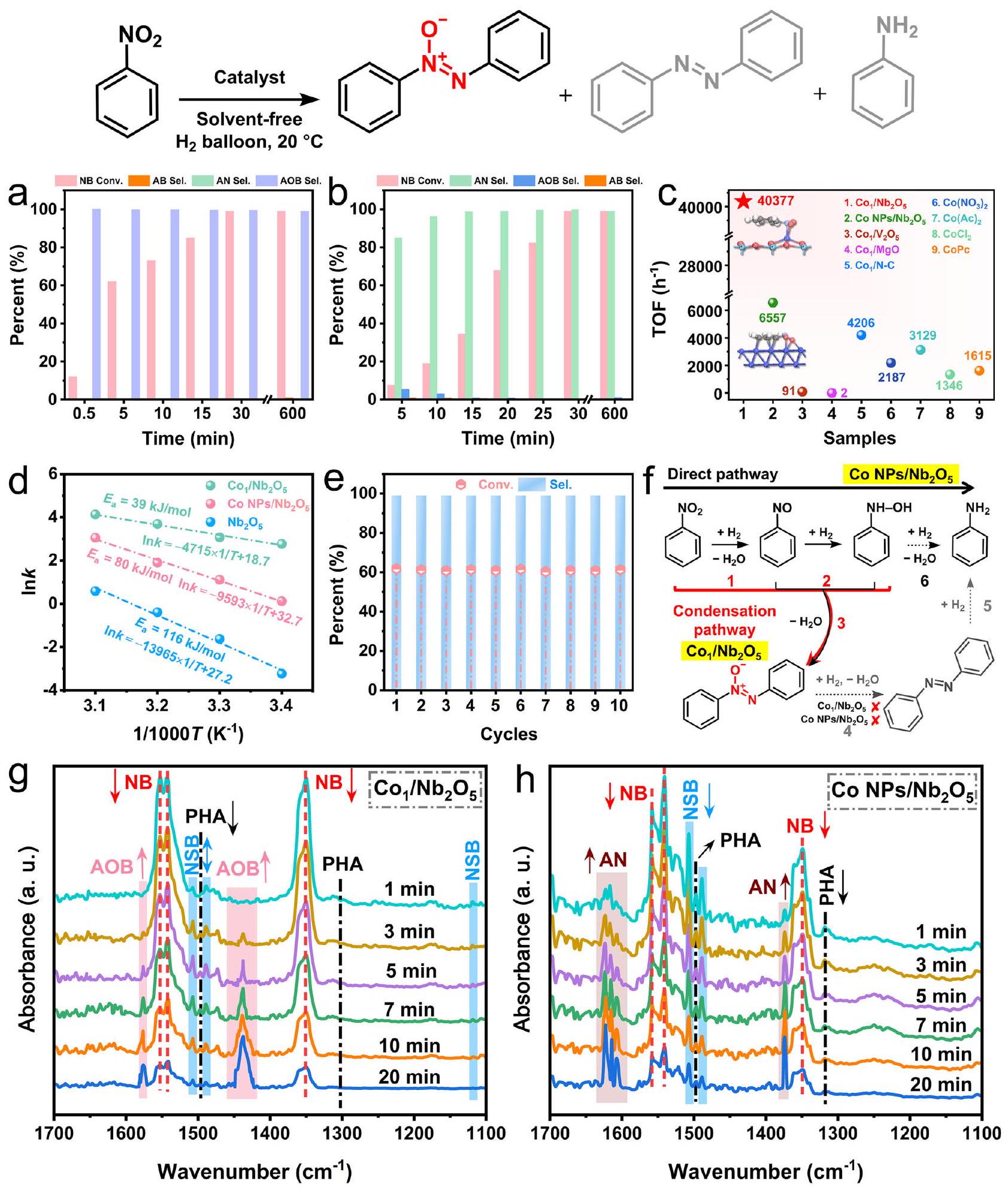
معدل المعلومات أكثر من
يؤثر على الأداء التحفيزي العام للعامل الحفاز. يمكن العثور على مزيد من المناقشة حول أصل اختلاف الانتقائية في محاكاة الحركية للخطوات الرئيسية للهدرجة في قسم التحقيق في الآلية.
خطوات من 1-2-6 تحت ظروف التفاعل (الشكل 4f). لتأكيد هذه النقطة، تم فحص الهدرجة الانتقائية للنيتروبنزين على
تحقيق في الآلية
في الأشكال التكميلية 53 و 54. طاقات امتصاص النيتروبنزين (
نقاش
في
طرق
تركيب
تركيب
تركيب
تقييم التحفيز
توفر البيانات
References
- Doherty, S. et al. Highly selective and solvent-dependent reduction of nitrobenzene to N-phenylhydroxylamine, azoxybenzene, and aniline catalyzed by phosphino-modified polymer immobilized ionic liquid-stabilized AuNPs. ACS Catal. 9, 4777-4791 (2019).
- Formenti, D., Ferretti, F., Scharnagl, F. K. & Beller, M. Reduction of nitro compounds using 3d-non-noble metal catalysts. Chem. Rev. 119, 2611-2680 (2019).
- Dai, Y. et al. Light-tuned selective photosynthesis of azo- and azoxyaromatics using graphitic
. Nat. Commun. 9, 60 (2018). - Yan, H. et al. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 9, 3197 (2018).
- Zhao, J.-X. et al. Selectivity regulation in Au-catalyzed nitroaromatic hydrogenation by anchoring single-site metal oxide promoters. ACS Catal. 10, 2837-2844 (2020).
- Lou, Y. et al. Pocketlike active site of
single-atom catalyst for selective crotonaldehyde hydrogenation. J. Am. Chem. Soc. 141, 19289-19295 (2019). - Ma, Y. et al. High-density and thermally stable palladium singleatom catalysts for chemoselective hydrogenations. Angew. Chem. Int. Ed. 59, 21613-21619 (2020).
- Jin, H. et al. Sabatier phenomenon in hydrogenation reactions induced by single-atom density. J. Am. Chem. Soc. 145, 12023-12032 (2023).
- Wu, B. et al. Ru single atoms for efficient chemoselective hydrogenation of nitrobenzene to azoxybenzene. Green Chem. 23, 4753-4761 (2021).
- Chen, Z., Liu, J., Koh, M. J. & Loh, K. P. Single-atom catalysis: from simple reactions to the synthesis of complex molecules. Adv. Mater. 34, 2103882 (2022).
- Guo, J. et al. Metal-organic frameworks as catalytic selectivity regulators for organic transformations. Chem. Soc. Rev. 50, 5366-5396 (2021).
- Kaden, W. E., Wu, T., Kunkel, W. A. & Anderson, S. L. Electronic structure controls reactivity of size-selected Pd clusters adsorbed on
surfaces. Science 326, 826-829 (2009). - Zaera, F. Designing sites in heterogeneous catalysis: are we reaching selectivities competitive with those of homogeneous catalysts? Chem. Rev. 122, 8594-8757 (2022).
- Giannakakis, G., Mitchell, S. & Pérez-Ramírez, J. Single-atom heterogeneous catalysts for sustainable organic synthesis. Trends Chem. 4, 264-276 (2022).
- Clarke, C. J., Tu, W.-C., Levers, O., Bröhl, A. & Hallett, J. P. Green and sustainable solvents in chemical processes. Chem. Rev. 118, 747-800 (2018).
- Ni, S. et al. Mechanochemical solvent-free catalytic C-H methylation. Angew. Chem. Int. Ed. 60, 6660-6666 (2021).
- Mi, R. et al. Solvent-free heterogeneous catalytic hydrogenation of polyesters to diols. Angew. Chem. Int. Ed. 62, e202304219 (2023).
- Miao, Y. et al. Photothermal recycling of waste polyolefin plastics into liquid fuels with high selectivity under solvent-free conditions. Nat. Commun. 14, 4242 (2023).
- Shi, X. et al. High performance and active sites of a ceria-supported palladium catalyst for solvent-free chemoselective hydrogenation of nitroarenes. Chem. Cat. Chem. 9, 3743-3751 (2017).
- Sun, Z. et al. The solvent-free selective hydrogenation of nitrobenzene to aniline: an unexpected catalytic activity of ultrafine Pt nanoparticles deposited on carbon nanotubes. Green Chem. 12, 1007-1011 (2010).
- Di Liberto, G., Tosoni, S., Cipriano, L. A. & Pacchioni, G. A few questions about single-atom catalysts: When modeling helps. Acc. Mater. Res. 3, 986-995 (2022).
- Liang, X., Fu, N., Yao, S., Li, Z. & Li, Y. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 144, 18155-18174 (2022).
- Li, H., Li, R., Liu, G., Zhai, M. & Yu, J. Noble-metal-free single- and dual-atom catalysts for artificial photosynthesis. Adv. Mater. 13, 2301307 (2023).
- Liu, L. & Corma, A. Isolated metal atoms and clusters for alkane activation: translating knowledge from enzymatic and homogeneous to heterogeneous systems. Chem 7, 2347-2384 (2021).
- Li, Z., Wang, D., Wu, Y. & Li, Y. Recent advances in the precise control of isolated single-site catalysts by chemical methods. Natl. Sci. Rev. 5, 673-689 (2018).
- Swain, S., Altaee, A., Saxena, M. & Samal, A. K. A comprehensive study on heterogeneous single atom catalysis: current progress, and challenges. Coord. Chem. Rev. 470, 214710 (2022).
- Ji, S. et al. Construction of a single-atom palladium catalyst by electronic metal-support interaction and interface confinement
effect with remarkable performance in Suzuki coupling reaction. Chem. Eng. J. 452, 139205 (2023). - Li, Z. et al. Engineering the electronic structure of single-atom iron sites with boosted oxygen bifunctional activity for zinc-air batteries. Adv. Mater. 35, 2209644 (2023).
- Chen, C. et al. Zero-valent palladium single-atoms catalysts confined in black phosphorus for efficient semi-hydrogenation. Adv. Mater. 33, 2008471 (2021).
- Yang, H. et al. Catalytically active atomically thin cuprate with periodic Cu single sites. Natl. Sci. Rev. 10, https://doi.org/10.1093/ nsr/nwac100 (2023).
- Li, Z. et al. A heterogeneous single atom cobalt catalyst for highly efficient acceptorless dehydrogenative coupling reactions. Small 19, 2207941 (2023).
- Dong, X. et al. Molten salt-induction of geometrically deformed ruthenium single atom catalysts with high performance for aerobic oxidation of alcohols. Chem. Eng. J. 451, 138660 (2023).
- Guo, Y., Jing, Y., Xia, Q. & Wang, Y. NbO
-based catalysts for the activation of and bonds in the valorization of waste carbon resources. Acc. Chem. Res. 55, 1301-1312 (2022). - Jing, Y., Xin, Y., Guo, Y., Liu, X. & Wang, Y. Highly efficient
catalyst for aldol condensation of biomass-derived carbonyl molecules to fuel precursors. Chin. J. Catal. 40, 1168-1177 (2019). - Jing, Y. et al. Towards the circular economy: converting aromatic plastic waste back to arenes over a
catalyst. Angew. Chem. Int. Ed. 60, 5527-5535 (2021). - Shao, Y. et al. Selective production of arenes via direct lignin upgrading over a niobium-based catalyst. Nat. Commun. 8, 16104 (2017).
- Xia, J. et al. Identifying the activity origin of a single-atom
catalyst for hydrodeoxygenation of methylcatechol: A stable substitutional site. ACS Catal. 13, 6093-6103 (2023). - Lim, E. et al. Facile synthesis of
@carbon core-shell nanocrystals with controlled crystalline structure for high-power anodes in hybrid supercapacitors. ACS Nano 9, 7497-7505 (2015). - Zheng, Y. et al. Defect-concentration-mediated T-
anodes for durable and fast-charging Li-ion batteries. Adv. Funct. Mater. 32, 2107060 (2022). - Chen, D. et al. Unraveling the nature of anomalously fast energy storage in
. J. Am. Chem. Soc. 139, 7071-7081 (2017). - Zheng, Y. et al. Triple conductive wiring by electron doping, chelation coating and electrochemical conversion in fluffy
anodes for fast-charging Li-ion batteries. Adv. Sci. 9, 2202201 (2022). - Zhang, Y., Song, T., Zhou, X. & Yang, Y. Oxygen-vacancy-boosted visible light driven photocatalytic oxidative dehydrogenation of saturated N -heterocycles over
nanorods. Appl. Catal. B Environ. 316, 121622 (2022). - Liu, R. et al. Design of aligned porous carbon films with single-atom Co-N-C sites for high-current-density hydrogen generation. Adv. Mater. 33, 2103533 (2021).
- Liu, Y. et al. Improving
photoconversion with ionic liquid and Co single atoms. Nat. Commun. 14, 1457 (2023). - Li, L., Dong, L., Liu, X., Guo, Y. & Wang, Y. Selective production of ethylbenzene from lignin oil over
modified catalyst. Appl. Catal. B Environ. 260, 118143 (2020). - Su, K. et al. Tuning the Pt species on
by support-induced modification in the photocatalytic transfer hydrogenation of phenylacetylene. Appl. Catal. B Environ. 298, 120554 (2021). - Li, Y. et al. A single site ruthenium catalyst for robust soot oxidation without platinum or palladium. Nat. Commun. 14, 7149 (2023).
- Shi, X . et al. Protruding Pt single-sites on hexagonal
to accelerate photocatalytic hydrogen evolution. Nat. Commun. 13, 1287 (2022). - Singh, J. A. et al. Understanding the active sites of CO hydrogenation on Pt-Co catalysts prepared using atomic layer deposition. J. Phys. Chem. C 122, 2184-2194 (2018).
- Kumar, N., Jothimurugesan, K., Stanley, G. G., Schwartz, V. & Spivey, J. J. In situ FT-IR study on the effect of cobalt precursors on CO adsorption behavior. J. Phys. Chem. C 115, 990-998 (2011).
- Song, D., Li, J. & Cai, Q. In situ diffuse reflectance FTIR study of CO adsorbed on a cobalt catalyst supported by silica with different pore sizes. J. Phys. Chem. C 111, 18970-18979 (2007).
- Kumar, P. et al. High-density cobalt single-atom catalysts for enhanced oxygen evolution reaction. J. Am. Chem. Soc. 145, 8052-8063 (2023).
- Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829-7836 (2023).
- Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the
catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022). - Chen, X. et al. Structure-dependence and metal-dependence on atomically dispersed Ir catalysts for efficient n-butane dehydrogenation. Nat. Commun. 14, 2588 (2023).
- Feng, Z. et al. Asymmetric sites on the
catalyst for promoting formate formation and transformation in hydrogenation. J. Am. Chem. Soc. 145, 12663-12672 (2023). - Lee, J. D. et al. Facilitating hydrogen dissociation over dilute nanoporous Ti-Cu catalysts. J. Am. Chem. Soc. 144, 16778-16791 (2022).
- Zhao, H. et al. The role of
species in single-atom catalyst for hydrogenation. Nat. Catal. 5, 818-831 (2022). - Kyriakou, G. et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 335, 1209-1212 (2012).
- Liu, W. et al. Highly-efficient RuNi single-atom alloy catalysts toward chemoselective hydrogenation of nitroarenes. Nat. Commun. 13, 3188 (2022).
- Richner, G., van Bokhoven, J. A., Neuhold, Y.-M., Makosch, M. & Hungerbühler, K. In situ infrared monitoring of the solid/liquid catalyst interface during the three-phase hydrogenation of nitrobenzene over nanosized Au on
. Phys. Chem. Chem. Phys. 13, 12463-12471 (2011). - Liu, L., Concepción, P. & Corma, A. Modulating the catalytic behavior of non-noble metal nanoparticles by inter-particle interaction for chemoselective hydrogenation of nitroarenes into corresponding azoxy or azo compounds. J. Catal. 369, 312-323 (2019).
- Zhou, H. et al. Hydrogenolysis cleavage of the
bond over a metal-free catalyst. ACS Catal. 12, 4806-4812 (2022). - Yu, M. et al. High coverage
adsorption and dissociation on fcc Co surfaces from DFT and thermodynamics. Int. J. Hydrog. Energy 43, 5576-5590 (2018).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-47402-5.
(ج) المؤلف(ون) 2024
المختبر الوطني الرئيسي لزيت الصخر الزيتي القاري، كلية الكيمياء والهندسة الكيميائية، جامعة النفط الشمالية، داتشينغ، جمهورية الصين الشعبية.
مركز الابتكار التعاوني لمواد الوظائف الحيوية في جيانغسو، كلية الكيمياء وعلوم المواد، جامعة نانجينغ العادية، نانجينغ، جمهورية الصين الشعبية. مختبر الإشعاع السنكروتروني الوطني، جامعة العلوم والتكنولوجيا في الصين، هيفي، جمهورية الصين الشعبية. المركز الوطني للبحوث الدولية في تكنولوجيا التحفيز، كلية الكيمياء وعلوم المواد، جامعة هيلونغجيانغ، هاربين، جمهورية الصين الشعبية. قسم الكيمياء، جامعة كوينز، كينغستون، كندا. ساهم هؤلاء المؤلفون بالتساوي: شياو وين لو، كونغ قوه. البريد الإلكتروني: zhijun.li@queensu.ca; yu.wang@njnu.edu.cn
DOI: https://doi.org/10.1038/s41467-024-47402-5
PMID: https://pubmed.ncbi.nlm.nih.gov/38609380
Publication Date: 2024-04-12
Solvent-free selective hydrogenation of nitroaromatics to azoxy compounds over Co single atoms decorated on
Accepted: 1 April 2024
Published online: 12 April 2024
(D) Check for updates
Abstract
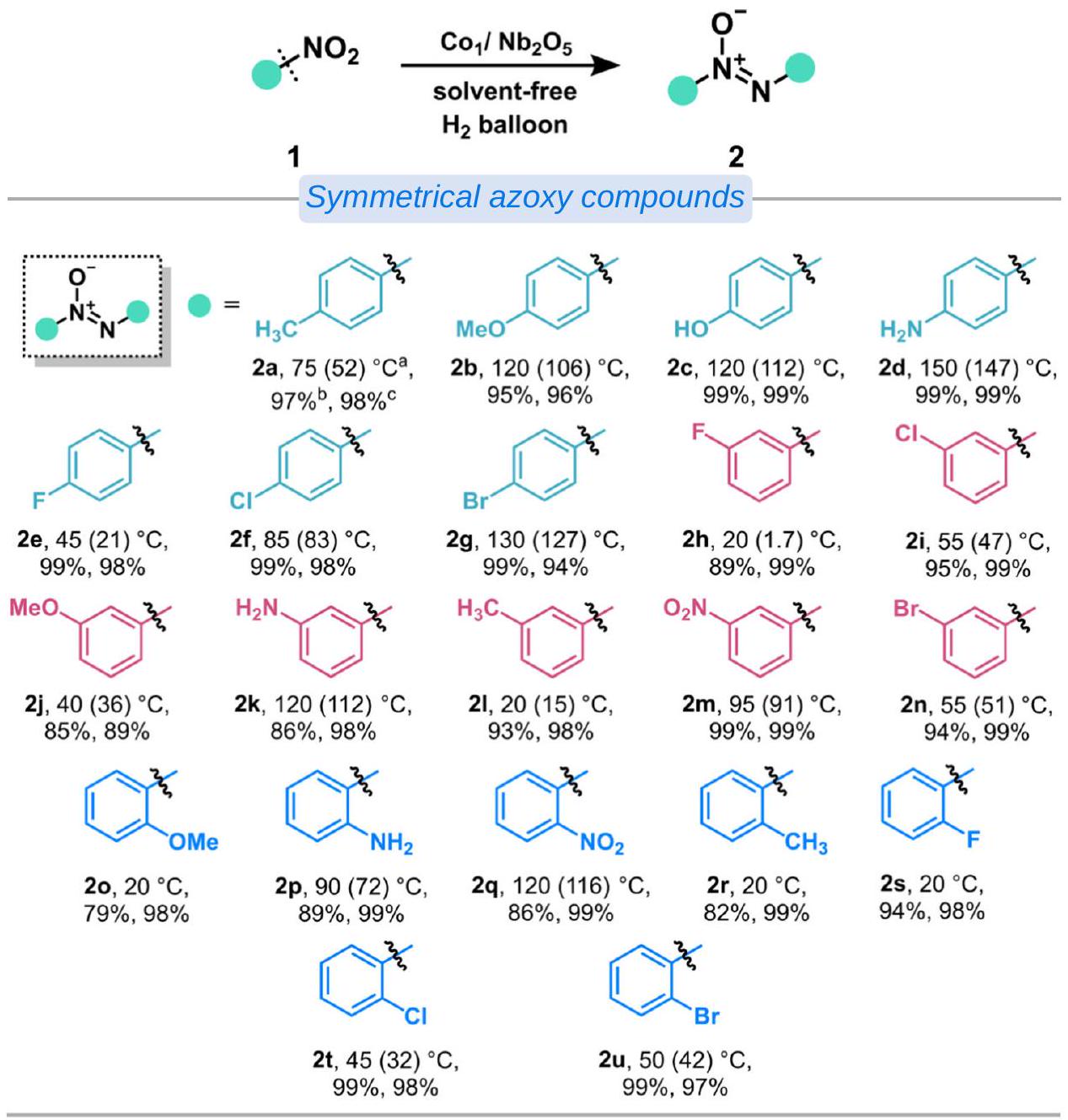
The solvent-free selective hydrogenation of nitroaromatics to azoxy compounds is highly important, yet challenging. Herein, we report an efficient strategy to construct individually dispersed Co atoms decorated on niobium pentaoxide nanomeshes with unique geometric and electronic properties. The use of this supported Co single atom catalysts in the selective hydrogenation of nitrobenzene to azoxybenzene results in high catalytic activity and selectivity, with
polymerization inhibitors, and food additives
a Oxidation of anilines into aromatic azoxy compounds.
Results
Synthesis and characterization of atomically dispersed Co catalyst
(the inset is SAED pattern).
and Supplementary Fig. 5). Compared with the
Evaluation of catalytic performance
formation rate than those of
affecting the overall catalytic performance of the catalyst. More discussion on the origin of the selectivity difference can be found in the kinetics simulations of key hydrogenation steps in the Mechanism investigation section.
steps from 1-2-6 under the reaction conditions (Fig. 4f). To confirm this point, the selective hydrogenation of nitrobenzene was examined on
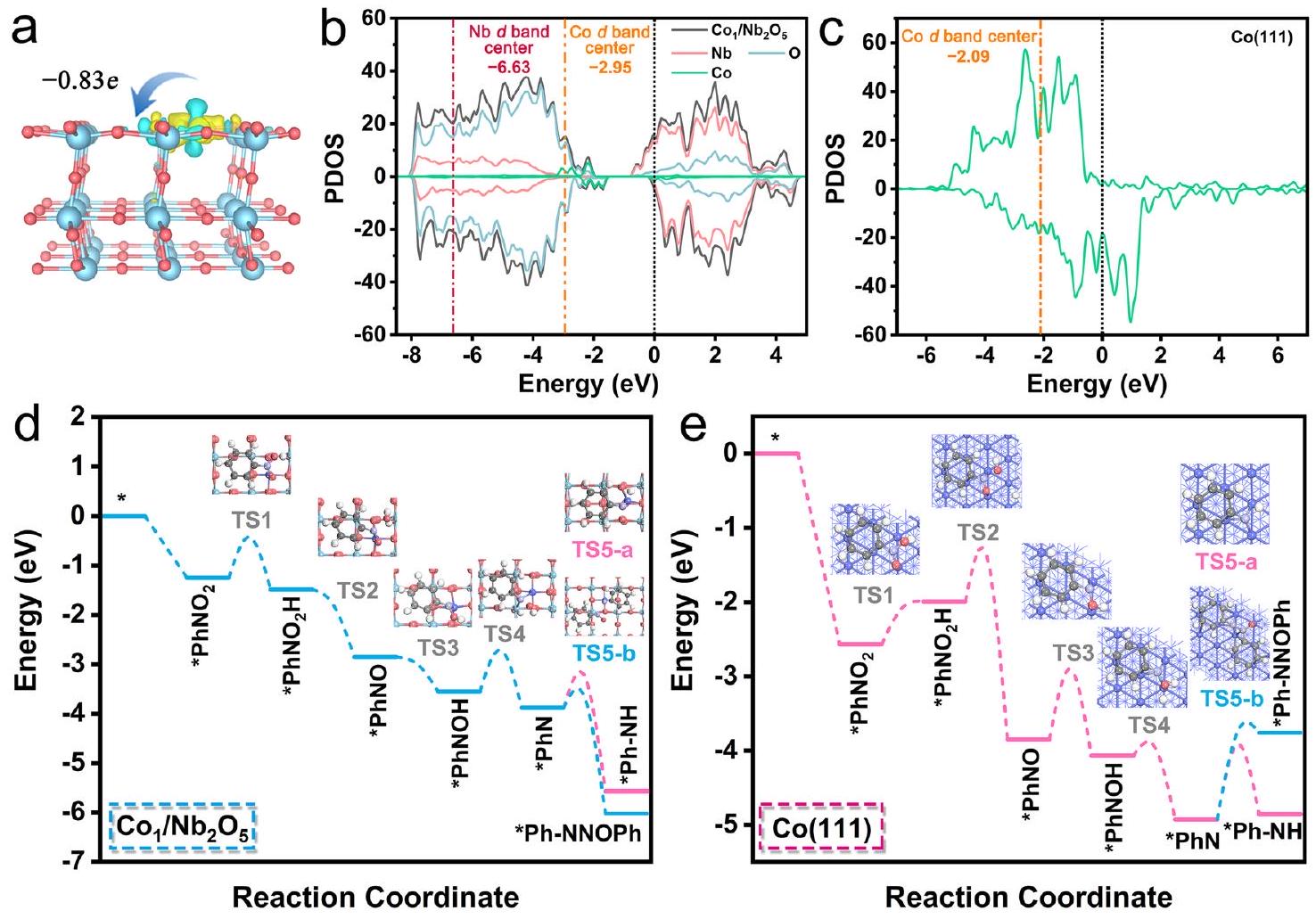
Mechanism investigation
in Supplementary Figs. 53 and 54. The nitrobenzene adsorption energies (
Discussion
in
Methods
Synthesis of
Synthesis of
Synthesis of
Catalytic evaluation
Data availability
References
- Doherty, S. et al. Highly selective and solvent-dependent reduction of nitrobenzene to N-phenylhydroxylamine, azoxybenzene, and aniline catalyzed by phosphino-modified polymer immobilized ionic liquid-stabilized AuNPs. ACS Catal. 9, 4777-4791 (2019).
- Formenti, D., Ferretti, F., Scharnagl, F. K. & Beller, M. Reduction of nitro compounds using 3d-non-noble metal catalysts. Chem. Rev. 119, 2611-2680 (2019).
- Dai, Y. et al. Light-tuned selective photosynthesis of azo- and azoxyaromatics using graphitic
. Nat. Commun. 9, 60 (2018). - Yan, H. et al. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 9, 3197 (2018).
- Zhao, J.-X. et al. Selectivity regulation in Au-catalyzed nitroaromatic hydrogenation by anchoring single-site metal oxide promoters. ACS Catal. 10, 2837-2844 (2020).
- Lou, Y. et al. Pocketlike active site of
single-atom catalyst for selective crotonaldehyde hydrogenation. J. Am. Chem. Soc. 141, 19289-19295 (2019). - Ma, Y. et al. High-density and thermally stable palladium singleatom catalysts for chemoselective hydrogenations. Angew. Chem. Int. Ed. 59, 21613-21619 (2020).
- Jin, H. et al. Sabatier phenomenon in hydrogenation reactions induced by single-atom density. J. Am. Chem. Soc. 145, 12023-12032 (2023).
- Wu, B. et al. Ru single atoms for efficient chemoselective hydrogenation of nitrobenzene to azoxybenzene. Green Chem. 23, 4753-4761 (2021).
- Chen, Z., Liu, J., Koh, M. J. & Loh, K. P. Single-atom catalysis: from simple reactions to the synthesis of complex molecules. Adv. Mater. 34, 2103882 (2022).
- Guo, J. et al. Metal-organic frameworks as catalytic selectivity regulators for organic transformations. Chem. Soc. Rev. 50, 5366-5396 (2021).
- Kaden, W. E., Wu, T., Kunkel, W. A. & Anderson, S. L. Electronic structure controls reactivity of size-selected Pd clusters adsorbed on
surfaces. Science 326, 826-829 (2009). - Zaera, F. Designing sites in heterogeneous catalysis: are we reaching selectivities competitive with those of homogeneous catalysts? Chem. Rev. 122, 8594-8757 (2022).
- Giannakakis, G., Mitchell, S. & Pérez-Ramírez, J. Single-atom heterogeneous catalysts for sustainable organic synthesis. Trends Chem. 4, 264-276 (2022).
- Clarke, C. J., Tu, W.-C., Levers, O., Bröhl, A. & Hallett, J. P. Green and sustainable solvents in chemical processes. Chem. Rev. 118, 747-800 (2018).
- Ni, S. et al. Mechanochemical solvent-free catalytic C-H methylation. Angew. Chem. Int. Ed. 60, 6660-6666 (2021).
- Mi, R. et al. Solvent-free heterogeneous catalytic hydrogenation of polyesters to diols. Angew. Chem. Int. Ed. 62, e202304219 (2023).
- Miao, Y. et al. Photothermal recycling of waste polyolefin plastics into liquid fuels with high selectivity under solvent-free conditions. Nat. Commun. 14, 4242 (2023).
- Shi, X. et al. High performance and active sites of a ceria-supported palladium catalyst for solvent-free chemoselective hydrogenation of nitroarenes. Chem. Cat. Chem. 9, 3743-3751 (2017).
- Sun, Z. et al. The solvent-free selective hydrogenation of nitrobenzene to aniline: an unexpected catalytic activity of ultrafine Pt nanoparticles deposited on carbon nanotubes. Green Chem. 12, 1007-1011 (2010).
- Di Liberto, G., Tosoni, S., Cipriano, L. A. & Pacchioni, G. A few questions about single-atom catalysts: When modeling helps. Acc. Mater. Res. 3, 986-995 (2022).
- Liang, X., Fu, N., Yao, S., Li, Z. & Li, Y. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 144, 18155-18174 (2022).
- Li, H., Li, R., Liu, G., Zhai, M. & Yu, J. Noble-metal-free single- and dual-atom catalysts for artificial photosynthesis. Adv. Mater. 13, 2301307 (2023).
- Liu, L. & Corma, A. Isolated metal atoms and clusters for alkane activation: translating knowledge from enzymatic and homogeneous to heterogeneous systems. Chem 7, 2347-2384 (2021).
- Li, Z., Wang, D., Wu, Y. & Li, Y. Recent advances in the precise control of isolated single-site catalysts by chemical methods. Natl. Sci. Rev. 5, 673-689 (2018).
- Swain, S., Altaee, A., Saxena, M. & Samal, A. K. A comprehensive study on heterogeneous single atom catalysis: current progress, and challenges. Coord. Chem. Rev. 470, 214710 (2022).
- Ji, S. et al. Construction of a single-atom palladium catalyst by electronic metal-support interaction and interface confinement
effect with remarkable performance in Suzuki coupling reaction. Chem. Eng. J. 452, 139205 (2023). - Li, Z. et al. Engineering the electronic structure of single-atom iron sites with boosted oxygen bifunctional activity for zinc-air batteries. Adv. Mater. 35, 2209644 (2023).
- Chen, C. et al. Zero-valent palladium single-atoms catalysts confined in black phosphorus for efficient semi-hydrogenation. Adv. Mater. 33, 2008471 (2021).
- Yang, H. et al. Catalytically active atomically thin cuprate with periodic Cu single sites. Natl. Sci. Rev. 10, https://doi.org/10.1093/ nsr/nwac100 (2023).
- Li, Z. et al. A heterogeneous single atom cobalt catalyst for highly efficient acceptorless dehydrogenative coupling reactions. Small 19, 2207941 (2023).
- Dong, X. et al. Molten salt-induction of geometrically deformed ruthenium single atom catalysts with high performance for aerobic oxidation of alcohols. Chem. Eng. J. 451, 138660 (2023).
- Guo, Y., Jing, Y., Xia, Q. & Wang, Y. NbO
-based catalysts for the activation of and bonds in the valorization of waste carbon resources. Acc. Chem. Res. 55, 1301-1312 (2022). - Jing, Y., Xin, Y., Guo, Y., Liu, X. & Wang, Y. Highly efficient
catalyst for aldol condensation of biomass-derived carbonyl molecules to fuel precursors. Chin. J. Catal. 40, 1168-1177 (2019). - Jing, Y. et al. Towards the circular economy: converting aromatic plastic waste back to arenes over a
catalyst. Angew. Chem. Int. Ed. 60, 5527-5535 (2021). - Shao, Y. et al. Selective production of arenes via direct lignin upgrading over a niobium-based catalyst. Nat. Commun. 8, 16104 (2017).
- Xia, J. et al. Identifying the activity origin of a single-atom
catalyst for hydrodeoxygenation of methylcatechol: A stable substitutional site. ACS Catal. 13, 6093-6103 (2023). - Lim, E. et al. Facile synthesis of
@carbon core-shell nanocrystals with controlled crystalline structure for high-power anodes in hybrid supercapacitors. ACS Nano 9, 7497-7505 (2015). - Zheng, Y. et al. Defect-concentration-mediated T-
anodes for durable and fast-charging Li-ion batteries. Adv. Funct. Mater. 32, 2107060 (2022). - Chen, D. et al. Unraveling the nature of anomalously fast energy storage in
. J. Am. Chem. Soc. 139, 7071-7081 (2017). - Zheng, Y. et al. Triple conductive wiring by electron doping, chelation coating and electrochemical conversion in fluffy
anodes for fast-charging Li-ion batteries. Adv. Sci. 9, 2202201 (2022). - Zhang, Y., Song, T., Zhou, X. & Yang, Y. Oxygen-vacancy-boosted visible light driven photocatalytic oxidative dehydrogenation of saturated N -heterocycles over
nanorods. Appl. Catal. B Environ. 316, 121622 (2022). - Liu, R. et al. Design of aligned porous carbon films with single-atom Co-N-C sites for high-current-density hydrogen generation. Adv. Mater. 33, 2103533 (2021).
- Liu, Y. et al. Improving
photoconversion with ionic liquid and Co single atoms. Nat. Commun. 14, 1457 (2023). - Li, L., Dong, L., Liu, X., Guo, Y. & Wang, Y. Selective production of ethylbenzene from lignin oil over
modified catalyst. Appl. Catal. B Environ. 260, 118143 (2020). - Su, K. et al. Tuning the Pt species on
by support-induced modification in the photocatalytic transfer hydrogenation of phenylacetylene. Appl. Catal. B Environ. 298, 120554 (2021). - Li, Y. et al. A single site ruthenium catalyst for robust soot oxidation without platinum or palladium. Nat. Commun. 14, 7149 (2023).
- Shi, X . et al. Protruding Pt single-sites on hexagonal
to accelerate photocatalytic hydrogen evolution. Nat. Commun. 13, 1287 (2022). - Singh, J. A. et al. Understanding the active sites of CO hydrogenation on Pt-Co catalysts prepared using atomic layer deposition. J. Phys. Chem. C 122, 2184-2194 (2018).
- Kumar, N., Jothimurugesan, K., Stanley, G. G., Schwartz, V. & Spivey, J. J. In situ FT-IR study on the effect of cobalt precursors on CO adsorption behavior. J. Phys. Chem. C 115, 990-998 (2011).
- Song, D., Li, J. & Cai, Q. In situ diffuse reflectance FTIR study of CO adsorbed on a cobalt catalyst supported by silica with different pore sizes. J. Phys. Chem. C 111, 18970-18979 (2007).
- Kumar, P. et al. High-density cobalt single-atom catalysts for enhanced oxygen evolution reaction. J. Am. Chem. Soc. 145, 8052-8063 (2023).
- Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829-7836 (2023).
- Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the
catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022). - Chen, X. et al. Structure-dependence and metal-dependence on atomically dispersed Ir catalysts for efficient n-butane dehydrogenation. Nat. Commun. 14, 2588 (2023).
- Feng, Z. et al. Asymmetric sites on the
catalyst for promoting formate formation and transformation in hydrogenation. J. Am. Chem. Soc. 145, 12663-12672 (2023). - Lee, J. D. et al. Facilitating hydrogen dissociation over dilute nanoporous Ti-Cu catalysts. J. Am. Chem. Soc. 144, 16778-16791 (2022).
- Zhao, H. et al. The role of
species in single-atom catalyst for hydrogenation. Nat. Catal. 5, 818-831 (2022). - Kyriakou, G. et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 335, 1209-1212 (2012).
- Liu, W. et al. Highly-efficient RuNi single-atom alloy catalysts toward chemoselective hydrogenation of nitroarenes. Nat. Commun. 13, 3188 (2022).
- Richner, G., van Bokhoven, J. A., Neuhold, Y.-M., Makosch, M. & Hungerbühler, K. In situ infrared monitoring of the solid/liquid catalyst interface during the three-phase hydrogenation of nitrobenzene over nanosized Au on
. Phys. Chem. Chem. Phys. 13, 12463-12471 (2011). - Liu, L., Concepción, P. & Corma, A. Modulating the catalytic behavior of non-noble metal nanoparticles by inter-particle interaction for chemoselective hydrogenation of nitroarenes into corresponding azoxy or azo compounds. J. Catal. 369, 312-323 (2019).
- Zhou, H. et al. Hydrogenolysis cleavage of the
bond over a metal-free catalyst. ACS Catal. 12, 4806-4812 (2022). - Yu, M. et al. High coverage
adsorption and dissociation on fcc Co surfaces from DFT and thermodynamics. Int. J. Hydrog. Energy 43, 5576-5590 (2018).
Acknowledgements
Author contributions
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-47402-5.
(c) The Author(s) 2024
National Key Laboratory of Continental Shale Oil, College of Chemistry and Chemical Engineering, Northeast Petroleum University, Daqing, PR China.
Jiangsu Collaborative Innovation Centre of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing, PR China. National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, PR China. National Center for International Research on Catalytic Technology, School of Chemistry and Material Sciences, Heilongjiang University, Harbin, PR China. Department of Chemistry, Queen’s University, Kingston, Canada. These authors contributed equally: Xiaowen Lu, Cong Guo. e-mail: zhijun.li@queensu.ca; yu.wang@njnu.edu.cn