امتصاص أيونات الكلوريد الديناميكي على ذرة إيريديوم مفردة يعزز تحفيز أكسدة مياه البحر Dynamic chloride ion adsorption on single iridium atom boosts seawater oxidation catalysis
تحليل مياه البحر بالكهرباء يوفر وسيلة متجددة وقابلة للتوسع واقتصادية لإنتاج الهيدروجين الأخضر. ومع ذلك، فإن تآكل الأنود بواسطةتطرح تحديات كبيرة لتسويقها. هنا، تختلف عن المحفزات التقليدية المصممة لطردالامتزاز، نقوم بتطوير محفز ذري من الإيريديوم على هيدروكسيد الحديد والكوبالت المزدوج الطبقات (Ir/CoFe-LDH) لتخصيص امتزاز الكلور وتعديل البنية الإلكترونية لمركز الإيريديوم النشط، وبالتالي إنشاء التنسيق لعملية التحليل الكهربائي لمياه البحر القلوية. تكشف التوصيفات العملية والحسابات النظرية عن الدور الحاسم لهذه الحالة التنسيقية في خفض طاقة تنشيط تفاعل تطور الأكسجين بمقدار 1.93. يظهر Ir/CoFe-LDH نشاطًا ملحوظًا في تفاعل تطور الأكسجين (فائض جهد 202 مللي فولت ومعدل دوران) ) في ، متفوق على إلكتروليت NaOH 6 M خالي من الكلور (فائض جهد 236 مللي فولت ومعدل دوران = )، مع الانتقائية التحفيزية والاستقرار عند كثافات تيار عالية لأكثر من .
التحليل الكهربائي للمياه على نطاق الشبكة يعد واعدًا لتخزين الكهرباء المتجددة في روابط الهيدروجين الجزيئية.استخدام مياه البحر الوفيرة كمادة خام بدلاً من المياه المحلاة يوفر استراتيجية أكثر استدامة لإنتاج الهيدروجين المتجدد.الذي ينتج NaCl كناتج ثانوي في نفس الوقت. ومع ذلك، فإن مياه البحر تحتوي على ملوحة من ، حيث يكون معظم الملحتواجه تقنية التحليل الكهربائي لمياه البحر العديد من التحديات، خاصة على جانب الأنود حيث تحدث تآكل شديد في المحفز وتفاعل أكسدة الكلوريد التنافسي (ClOR) في الوقت نفسه.، مما يعيق بشكل كبير تسويقه. تتطلب عملية التحليل الكهربائي للمياه المالحة بكفاءة واستدامة وجود أنود نشط للغاية واختياري قادر على العمل في وجود تركيز عالٍ من . كانت المحاولات السابقة لتحليل مياه البحر بالكهرباء تركز في الغالب على حظر الامتزاز على محفز الأنود لمنع ClOR. على سبيل المثال، كوبر وآخرون.مترسب كهربائيًا غير مفعللتحسين انتقائية تفاعل تطور الأكسجين (OER) في التحليل الكهربائي للمياه البحرية الحمضية بسبب الارتباط الضعيف للكلوريد علىسطح. كبريتيدات قائمة على النيكل والكوبالت والحديدالفوسفيدسيلينيداتوهيدروكسيدات الحديد والكوبالت المزدوجة الطبقات المعدلة بالبورنأظهر قدرة على مقاومة التآكل في التحليل الكهربائي للمياه البحرية القلوية بسبب التكوين في الموقعطبقة الأنيون الطارد أو البوليمر الأنيوني. للأسف، هذا أيضًا أضعف امتصاص الوسائط الوسيطة للأكسجين في عملية الأكسدة، مما قد يقلل من نشاط الأكسدة. بالإضافة إلى ذلك، عند الجهد الزائد العالي (أي، كثافة تيار عالية)،تصبح استراتيجية الردع أقل فعالية بسبب القوة الدافعة المعززة لـالامتزاز.
هنا، نصمم محفزًا ذريًا من الإيريديوم على هيدروكسيد الحديد والكوبالت المزدوج الطبقات (Ir/CoFe-LDH) لأكسدة مياه البحر الكهروكيميائية. يختلف عن المحفزات التقليدية المصممة لطرد بالكاملالامتصاص لتحليل مياه البحر بالكهرباء، المواقع الذرية للذهب الإريديوم على CoFe-LDH تسمحالامتزاز لتعديل البنية الإلكترونية لـ
مركز نشط. نتيجة لذلك،يظهر أداءً ملحوظًا في تفاعل الأكسدة الكهربائية في مياه البحر القلوية مع جهد زائد منخفض يصل إلى 202 مللي فولت عند كثافة تيارأقل من ذلك في إلكتروليت NaOH. علاوة على ذلك،يوفر نشاطًا ملحوظًا عند كثافات تيار ذات صلة بالصناعة ) مع قرب من كفاءة فارداي للأكسجين لأكثر من 1000 ساعة. في الوقت نفسه، يمكن أن تعمل أيضًا بشكل مستقر في مياه البحر الحقيقية، مما يصل إلىمع جهد زائد قدره 208 مللي فولت، ويحافظ على تشغيل مستقر لأكثر من 2000 ساعة عند كثافة تيارتظهر كل من التجارب في الموقع والتحليلات النظرية أن امتصاص أيونات الكلوريد الديناميكي على ذرة إيريديوم واحدة أثناء عملية أكسدة الماء يمكن أن يقلل بشكل فعال من حاجز الطاقة لتكوين *OOH (الخطوة المحددة للسرعة في أكسدة الماء) وبالتالي يعزز تحفيز أكسدة الماء، بينما في نفس الوقت يحافظ على حاجز طاقة مرتفع لتفاعل الكلوريد.
النتائج والمناقشة
تركيب وتوصيف هيكلي لـ Ir/CoFe-LDH
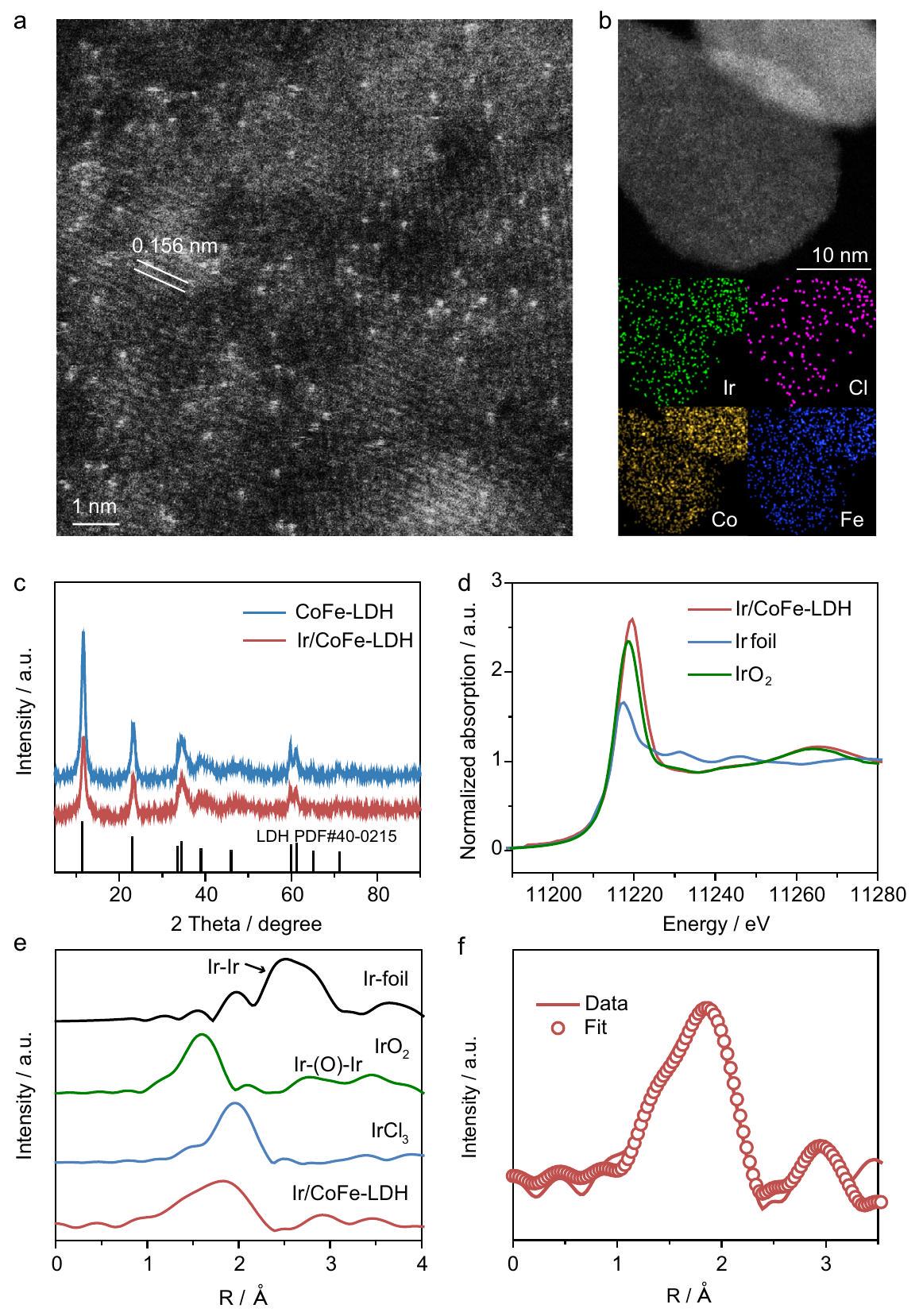
لقد تم استخدام المحفزات القائمة على الإريديوم على نطاق واسع في تحفيز تحليل الماء كهربائيًا.وعملية الكلور القلويحيث تحدد قوة رابطة الكلوريد الإيراني انتقائيتهقد تظهر تركيبات LDH العنصرية ذات الكهربية السالبة المنخفضة ارتباطًا إلكترونيًا معزّزًا مع ذرات المعادن الفردية.. استنادًا إلى قيم الكهربية السالبة ( )، من المتوقع أن تظهر ذرات المعادن النبيلة الفردية على LDHs من CoFe أداءً متفوقًا في تفاعل الأكسدة. في هذا العمل، نقوم بتطوير عملية تخليق من خطوتين لتحضير محفزات تحتوي على ذرة واحدة من الإيريديوم مع هيكل تنسيق ذري فردي قابل للتعديل على هيدروكسيد مزدوج الطبقات (LDH)، كما هو موضح في الشكل التكميلي 1. في الخطوة الأولى، تمت تخليقه من خلال طريقة الترسيب المشترك. بعد ذلك، تم تحضير محاليل مخففة من وتم إضافة NaOH إلى المستحلب المتجانس LDH لتثبيت الذرات الذرية من Ir على CoFe-LDH بالإضافة إلى ضبط الروابط الكلورية على مواقع Ir الذرية الفردية. تكشف صور SEM أن كل من CoFe-LDH و Ir/CoFe-LDH تظهر هياكل نانوية متجانسة، دون تغييرات ملحوظة في شكل السطح عند تحميل Ir (الشكل التكميلي 2). إن Ir في التحضير الأولي تم تحديد المحفز على أنه بواسطة مطيافية الكتلة مع البلازما المقترنة بالحث (ICP-MS). تظهر خصائص المجهر الإلكتروني الناقل (TEM) سطحًا نظيفًا لـدون تشكيل نانو جزيئات / نانو تجمعات (الشكل التكميلي 3). تظهر صور المجهر الإلكتروني الناقل الماسح ذو الزاوية العالية (HAADF-STEM) بوضوح بقعًا ساطعة، يمكن أن تُنسب إلى ذرات إيريديوم مفصولة على سطح CoFe-LDH (الشكل 1a). في الوقت نفسه، لا تزال ذرات إيريديوم تظهر بقعًا ساطعة معزولة في نفس الموضع في صور HAADF-STEM مع ميل (الشكل التوضيحي التكميلي 4)، مما يؤكد التشتت الذري الفردي للـ Ir. يبدو أن هناك تجمعًا في الدائرة الصفراء في الشكل التوضيحي التكميلي 5a وفي الدائرة الحمراء في الشكل التوضيحي التكميلي 5b، ومع ذلك، كلاهما يظهر ذرات فردية من زوايا مشاهدة أخرى، مما يشير إلى أن “تجمع” ذرات Ir الفردية قد يكون ناتجًا فقط عن زوايا المشاهدة. برنامج محاكاة STEM للدكتور بروبتم استخدامه لتناسب صورة HAADF-STEM لـ Ir/CoFe-LDH، مما يشير أيضًا إلى تشتت ذري فردي لـ Ir مع سطوع مختلف (الأشكال التكميلية 6 و 7). تُظهر تحليل خريطة العناصر (الشكل 1b) توزيعًا متجانسًا لـ Ir مع Cl على سطحعلاوة على ذلك، تظهر أنماط حيود الأشعة السينية (XRD) لـ CoFe-LDH المحضر و Ir/CoFe-LDH نفس انعكاسات براج بتوافق جيد مع LDH من المرحلة السداسية (الخط الأسود، PDF#40-0215، كما هو موضح في الشكل 1c)، بما يتماشى مع أنماط حيود الإلكترون في المنطقة المختارة (SAED) (الشكل التكميلية 8).
لتأكيد البنية الذرية للروثينيوم، تم إجراء قياسات DRIFTs في الموقع لـ و تمت (الشكل التكميلي 9). بعد امتصاص CO علىفرقتان (2080 ويمكن رؤيته بوضوح فيمنطقة تردد الاهتزاز (الشكل التوضيحي 9a). يمكن تعيين هذين النطاقين إلى الاهتزاز المتماثل (vs) والاهتزاز غير المتماثل (vas) أوضاع إير جيم-دايكاربونيل،على التوالي، استنادًا إلى تقارير عن توزيع ذري متفرقدعم الجيم-دايكربونيل على و من الجدير بالذكر أن الأنواع ثنائية الكربونيل لا يمكن أن تتشكل إلا على الأنواع المعزولة.، مما يؤكد التشتت الذري للروثينيوم في Ir/ CoFe-LDH. على النقيض، -LDH يظهر قمم IR متعددة متداخلة بين 2000 و (الشكل التوضيحي 9ب). القمة الأقوى عند يُعزى إلى الربط الخطي لـ CO على تجمعات Ir، مما يشير إلى وجود تجمعات Ir. ومع ذلك، لم يُلاحظ هذا الذروة العلوية فيتقدم هذه النتائج مزيدًا من التحقق التجريبي لتشتت الذرات من Ir في Ir/CoFe-LDH (الشكل التكميلي 9c). تشير هذه النتائج إلى غياب تجمعات أو جزيئات نانوية من Ir في Ir/CoFe-LDH، وهو ما يتماشى مع نتائج HAADF-STEM.
تم إجراء مطيافية الإلكترونات السينية (XPS) لفحص التركيب الإلكتروني السطحي والتأثير بين CoFe-LDH و Ir الذري المفرد (الشكل التكميلي 10). أظهر طيف XPS عالي الدقة لـ Ir 4f قمتين متميزتين تنتميان إلى Ir.و إيروفقًا لطاقة الربط للعينات القياسية لـ و (الشكل التوضيحي 11)، Ir 4f عند 61.7 إلكترون فولت و62.4 إلكترون فولت تم التعرف عليها كـ و على التوالي (الأقمار الصناعية لـ و تم أخذها في الاعتبار عند ملاءمة طيف XPS). يمكن تحليل طيف XPS عالي الدقة لـ Cl 2p إلى 199.1 eV و 198.45 eV، والتي تنشأ من و علىمقارنة حالات التكافؤ لكوبالت والحديد في و لوحظ أن Co و Fe لديهما تحول واضح في طاقة الربط، مما يدل على تفاعل إلكتروني قوي بين ذرة Ir المفردة و CoFe-LDH.
لتحديد التركيب الإلكتروني والبيئة التنسيقية المحلية للذرات الإيريديوم، تم إجراء تحليل الامتصاص بالأشعة السينية بالقرب من الحافة (XANES) وتحليل الامتصاص بالأشعة السينية الممتد (EXAFS).. الإيرطيف XANES عند حافة (الشكل 1d) يشير إلى حالة التكافؤ للـ Ir فيأعلى قليلاً من +4. في طيف EXAFS (الشكل 1e) كما هو المشار إليه في ورقة إيريديوم.“، و IrO2، يظهر Ir/CoFeLDH ذروتين عند 1.62 وå، التي يمكن تعيينها إلى IrO و، على التوالي. علاوة على ذلك، فإن غياب و الروابط تشير إلى توزيع ذري للـ Ir. أظهرت بيانات تركيب EXAFS (الشكل 1f، الشكل التكميلي 12 والجدول التكميلي 1) روابط Ir-O مع عدد تنسيقمن 3.1 وسنداتمع CN قدره 2.5 بدون أو المساهمة في Ir/CoFe-LDH. بالإضافة إلى ذلك، هناك مساهمات من الانعكاس الخلفي لـ Ir و M (Co و Fe) فيمع CN قدره 2.0 في القشرة الثانية.
الأداء الكهروكيميائي لـ
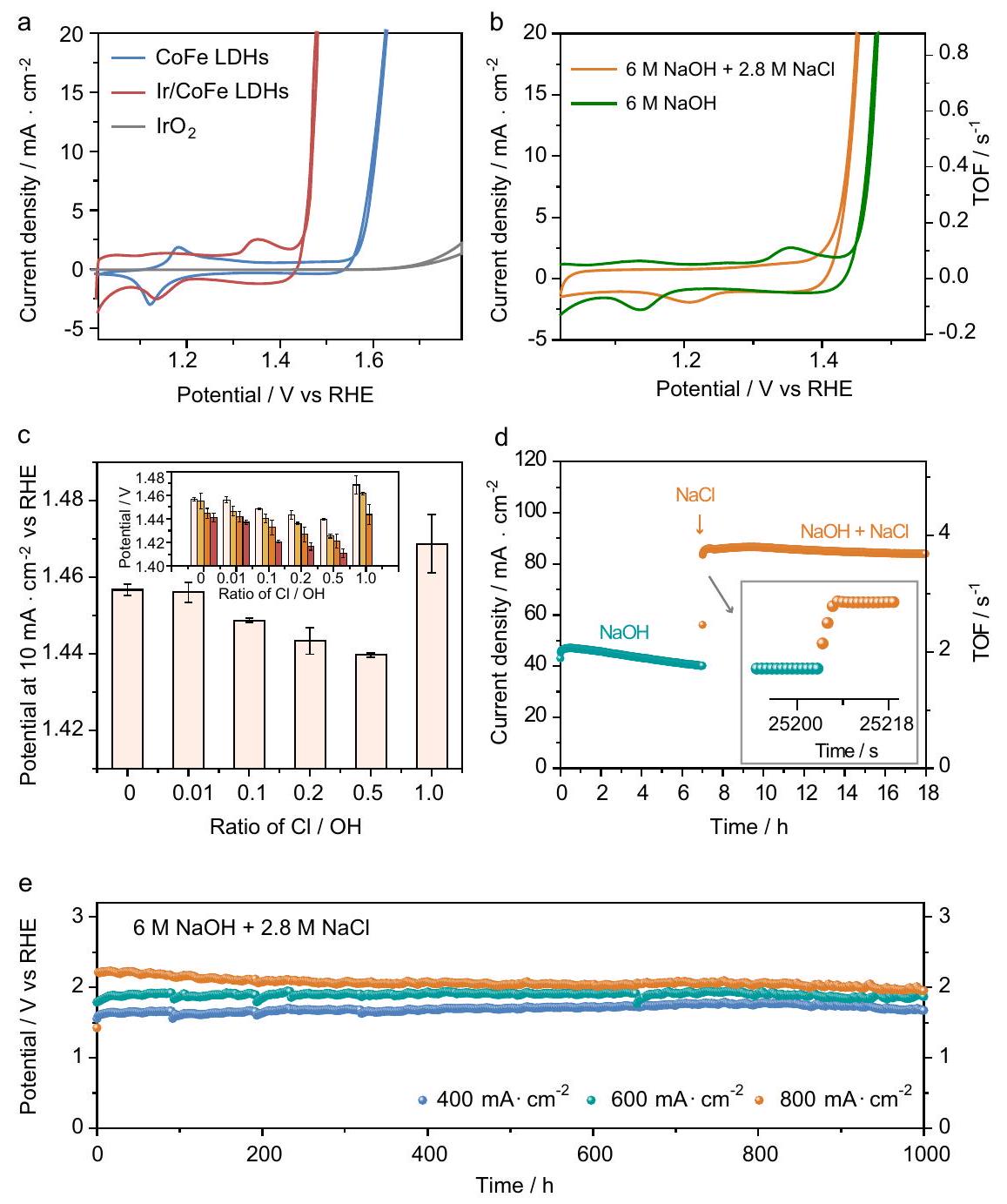
تم قياس أداء OER لـ Ir/CoFe-LDH المُعد في 6.0 م من هيدروكسيد الصوديوممحلول مائي باستخدام تكوين ثلاثي الأقطاب. تعرض الشكل التكميلي 13 صور المجهر الإلكتروني الماسح (SEM) للقطب العامل. تم قياس القطب المرجعي (SCE) (الأشكال التكميلية 14 و15) مباشرةً بواسطة إعداد ثلاثي الأقطاب، يتكون من سلكين من البلاتين وقطب مرجعي واحد ليتم معايرته عند التشبع. (الشكل التوضيحي الإضافي 16). يكشف منحنى الفولتمترية الدورية (الشكل 2a) أن Ir/CoFe-LDH يحتاج فقط إلى جهد زائد قدره 236 مللي فولت للوصول إلى كثافة تيار قدرها ، وهو أقل بمقدار 141 مللي فولت و 535 مللي فولت من CoFe-LDH والتجاري، على التوالي. يختلف عن CoFe -LDH، تم تقليل كبير في الـتُلاحظ قمم الأكسدة والاختزال بعد تحميل ذرات أحادية من الإيريديوم (الشكل التكميلي 17). وهذا يشير إلى أن قمة الأكسدة عند 1.24 فولت مقابل RHE وقمة الاختزال عند 1.17 فولت مقابل RHE هي زوج أكسدة واختزال، والذي يمكن أن يُعزى إلى أكسدة واختزال الإيريديوم. وقمة الأكسدة عند 1.33 فولت مقابل RHE وقمة الاختزال عند 1.07 فولت مقابل RHE هي زوج أكسدة واختزال، والذي يمكن أن يُعزى إلى أكسدة واختزال الكوبالت. ومن المثير للاهتمام، عندما يتم إضافة NaCl إلى الإلكتروليت لمحاكاة تراكم مياه البحر المشبعة.، يظهر انخفاض بمقدار 34 مللي فولت في الجهد الزائد من 236 مللي فولت إلى 202 مللي فولت للوصول إلى كثافة تيار قدرها (الشكل 2ب)، مما يشير إلى أن
الشكل 1 | التوصيفات الهيكلية لـ Ir/CoFe-LDH. أ صورة HAADF-STEM لـ Ir/CoFe-LDH تظهر تشتت ذرات Ir المفردة على CoFe-LDH. ب صورة TEM في مجال مظلم لـ Ir/CoFe-LDH والخرائط العنصرية المقابلة التي تظهر توزيع، وعناصر Ir. أنماط XRD لعينة CoFe المحضرة حديثًا –
LDH و Ir/CoFe-LDH. طيف XANES (d) و (e) تحويل فورييه لـ Ir-طيف EXAFS عند حافةورق الألمنيوم و -طيف EXAFS الموزون مع التناسب (الخط المنقط). وجود NaCl يحسن الأداء التحفيزي لـ. بالإضافة إلى ذلك، فإن تردد الدوران (TOF) لكل موقع Ir على Ir/CoFe-LDH (0.76 ) عند جهد 1.45 فولت (مقابل RHE) في إلكتروليت مياه البحر المشبعة هو 6.3 مرات أكبر من ذلك في إلكتروليت NaOH ( ) (الشكل 2ب). مع الأخذ في الاعتبار المشكلة المحتملة للتحميل المفرط للعامل المساعد في خمسة نقاط بيانات بتحميلات مختلفة للعوامل المساعدة في نطاقتم اختيارها لدراسة العلاقة بين تحميل المحفز والنشاط (الشكل التكميلي 18 أ، ب). تشير النتائج إلى وجود علاقة خطية بين تحميل المحفز والأداء ضمننطاق. ما وراء، يُظهر تغيير تحميل المحفز تأثيرًا ضئيلًا على الأداء التحفيزي (يصل إلى منطقة الاستقرار). وبالتالي، ضمن المنطقة الخطية، ترتبط بيانات CV بتحميل المحفز لـتم استخدامه لتحديد قيم TOF بدقة. النتائج أظهر أنه عند جهد 1.5 فولت (مقابل RHE)، فإن قيمة TOF لكل موقع Ir علىفيأعلى بـ 7 مرات من ذلك فيالشكل التكميلي 18c). في الوقت نفسه، يجب ملاحظة أن كمية التحميل من Ir يجب أن تكون مصممة بعناية؛ التحميل غير الكافي أو الزائد سيؤدي إلى كفاءة أقل في التحفيز (الأشكال التكميلية 19-21 والجداول التكميلية 2 و3)، وهو ما يتوافق مع خصائص المحفز أحادي الذرة المبلغ عنه. بالنسبة للاختيارية، حتى في إلكتروليت مياه البحر المشبعة، يظهر Ir/CoFe-LDH قريباً منكفاءة فاراداي في OER (الشكل التوضيحي التكميلي 22).
تظهر أداء OER لـ Ir/CoFe-LDH حساسية لتكوين الإلكتروليت مع نسب متغيرة من NaOH و NaCl. ومن الجدير بالذكر أن هناك تحسينًاتظهر نسبة OER كاعتبار مهم (الشكل 2c). من خلال تغيير تركيز NaOH إلى3 م و 6 م ، جهد الفائض في تفاعل الأكسدة عند كثافة تيار
الشكل 2 | أداء OER. أ منحنيات CV لـ Ir/CoFe-LDH و CoFe-LDH و IrOمسجل في 6 م NaOH (المقاومة المستخدمة لمعايرة iR هي، و على التوالي).مقارنة بين منحنيات CV و TOF لـ Ir/CoFe-LDH المسجلة في 6 M NaOH و (المقاومة المستخدمة لمعايرة iR هي 1.55 ، و، على التوالي). ج مقارنة جهد الفائض في OER عند كثافة تيار من-LDH في الإلكتروليت مع اختلافنسب في 1 م NaOH. يظهر الرسم الصغير جهد الفائض لتفاعل الأكسدة (OER) لـ Ir/CoFe-LDH عند كثافة تيار مع مختلفنسب في 1 م (أبيض)، 2 م (أصفر)، 3 م (برتقالي)، و6 م NaOH (أحمر). تم إجراء ثلاث قياسات لكل نقطة بيانات مع وجود أشرطة الخطأ التي تتوافق مع الانحراف المعياري.التغيير في كثافة التيار و TOF عند تبديل الإلكتروليت من 6 م NaOH إلى 6 مسُجلت عند 1.48 فولت مقابل القطب الهيدروجيني القابل للعكس (RHE). تم إجراء اختبار الاستقرار في. (كانت جميع تحميل الكتلة للعوامل المساعدة ، ودرجة حموضة الإلكتروليت تقيس 14.78.) تشهد انخفاضًا أوليًا يتبعه زيادة، تتماشى مع الارتفاع فيالتركيز. من المRemarkably، يظهر الأداء الأمثل لتفاعل الأكسدة عندنسبة (الشكل التوضيحي 23)، مما يبرز التأثير الحاسم لـعن أداء الموارد التعليمية المفتوحة.
بالإضافة إلى ذلك، عند الفحص في إلكتروليت NaOH الخالص عند جهد 1.49 فولت (مقابل RHE)، انخفض كثافة التيار لـ Ir/CoFe-LDH منإلىفي أقل من 7 ساعات (الشكل 2د). من المثير للاهتمام أن إدخال NaCl في محلول NaOH الكهربائي رفع كثافة التيار على الفور منإلىفي أقل من 6 ثوانٍ. بالإضافة إلى ذلك، لوحظ أنأظهرت قدرة مماثلة على تعزيز نشاط OER لـ Ir/CoFe-LDH، بينما على العكس، تم العثور على أنه يقلل من النشاط التحفيزي (الشكل التكميلي 24). على النقيض من ذلك، لم تظهر ذرات المعادن النبيلة الأخرى المفردة التي تم إدخالها على CoFe-LDH (مثل Ru/CoFe-LDH أو Rh/CoFe-LDH) مثل هذا التعزيز الملحوظ في نشاط OER من خلال تعديل الهالوجين (الشكل التكميلي 25)، مما يبرز الدور المميز الذي تلعبه Ir. قد يُعزى هذا التميز إلى القوة أو Fالتفاعل، كما يتضح من طيف الأشعة فوق البنفسجية والمرئية (الشكل التكميلية 26).
في الوقت نفسه، لم يمكن ملاحظة أي تعزيز كبير في نشاط OER عند دمج NaOH مع ملح آخر (على سبيل المثال، ) الكهارل ( الشكل التوضيحي 27).
للتحقيق فيما إذا كانسيؤثر على تنسيق Ir-Cl، قمنا بإجراء اختبارات الفولتمترية الدورية على Ir/CoFe-LDH في محلول من. بعد ذلك، قدمنا تدريجياً (تركيز أيون الكبريتات في مياه البحر ) واستمررنا في اختبارات CV. تشير النتائج إلى أداء مستدام تقريباً (ضمن 3 مللي فولت)، مما يؤكد أن لا يؤثر على نشاط المحفز (الشكل التكميلي 28). لإظهار ما إذا كان تحت ظروف دورية سيزيح المنسق ، أجرينا اختبار CV، وأظهرت النتائج عدم وجود تغيير ملحوظ في الأداء قبل وبعد دورة CV (الشكل التكميلي 29). بعد ذلك، تم إدخال المزيد من في الإلكتروليت، حتى الوصول إلى تركيز (زيادة عشرة أضعاف في تركيز الكبريتات في مياه البحر الحقيقية). تشير منحنيات CV إلى أن تقلب أداء المحفز ضئيل ( ) ضمن نطاق تركيز من (الشكل التكميلي 28). وهذا يشير إلى أنه بعد تشكيل حالة تنسيق مستقرة من Ir-Cl، فإن
وجود لا يمارس أي تأثير ملحوظ على الأداء.
بالإضافة إلى النشاط والانتقائية، تعتبر الاستقرار اعتباراً حاسماً آخر للتطبيق العملي . يمكن أن يُعزى تعطيل LDH إلى انخفاض pH المحلي، وتحمض الطبقات، وانحلال الكاتيونات، أو أكسدة مراكز المعادن إلى كاتيونات عالية التكافؤ مما يؤدي إلى تسربها وفي بيئة مياه البحر، قد يؤدي وجود أيونات الكلوريد إلى تفاقم هذه المخاطر. تم تقييم استقرار LDH بشكل أكبر في محلول عند كثافات تيار تتراوح من 400 إلى لتلبية متطلبات الصناعة. أظهرت الجهود المطبقة زيادة ضئيلة بعد 1000 ساعة من التفاعل المستمر (الشكل 2e). في الوقت نفسه، يحمل الأداء في مياه البحر الحقيقية دلالات كبيرة لتطبيق تكنولوجيا التحليل الكهربائي لمياه البحر. تطلب Ir/CoFeLDH جهدًا زائدًا قدره 208 مللي فولت فقط لتحقيق كثافة تيار قدرها في إلكتروليت مياه البحر (الشكل التكميلي 30)، مما يشير إلى قابليته للتطبيق تحت ظروف صناعية حقيقية. بالإضافة إلى ذلك، تؤكد اختبارات CV التي أجريت بعد اختبارات الاستقرار عند 24,200 و 400 ساعة استقرار Ir/CoFe-LDH تحت ظروف تشغيل واقعية (الشكل التكميلي 31). تم اختبار جهاز تحليل كهربائي مع -LDH كأنود وNiCoFeP ككاثود عند كثافة تيار قدرها في بيئة مياه البحر . توضح النتائج أن جهاز التحليل الكهربائي يمكنه الحفاظ على أداء مستقر تحت كثافة تيار عالية لأكثر من 2000 ساعة (الشكل التكميلي 32)، مما يؤكد جدوى محفز Ir/CoFe-LDH للتحليل الكهربائي لمياه البحر الحقيقية.
علاوة على ذلك، تم إجراء مزيد من التوصيفات لمحفزات ما بعد التفاعل لتأكيد استقرار Ir/CoFe-LDH. كشفت SEM بعد اختبار الاستقرار طويل الأمد عن عدم وجود تغييرات كبيرة في شكل النانو شيتس (الشكل التكميلي 33)، بينما أظهرت TEM بعد اختبار الاستقرار طويل الأمد غياب الكتل أو الجسيمات (الشكل التكميلي 34). تظهر توصيفات HADDF-STEM (الشكل التكميلي 35) أن توزيع ذرات Ir الذرية على سطح CoFe-LDH بعد اختبار الاستقرار طويل الأمد لم يتغير بشكل واضح. لا يزال لا يوجد إشارة Ir-Ir أو Ir-O-Ir في بيانات EXAFS لمحفز ما بعد OER، مما يكشف أيضًا عن تشتت معزول لذرات Ir بعد اختبارات استقرار OER طويلة الأمد. يعرض نمط XRD لمحفز Ir/CoFe-LDH بعد OER (الشكل التكميلي 36) الذروة المميزة لـ LDH عند حوالي . كشفت قياسات XPS لـ Ir/CoFe-LDH بعد اختبار استقرار OER طويل الأمد (الشكل التكميلي 37) أن حالة أكسدة Co وFe لم تظهر أي تغيير كبير، مما يؤكد أن تنسيق Ir-Cl يمكن أن يثبت CoFe-LDH، مما يمنع أكسدة وانحلال CoFe-LDH، وبالتالي يضمن استقرار ركيزة CoFe-LDH. علاوة على ذلك، أشارت التحليلات الكمية لـ XPS إلى تغيير ضئيل في تركيز Ir السطحي قبل وبعد OER. تم أيضًا قياس Ir المذاب في الإلكتروليت بعد اختبار استقرار OER بواسطة مطيافية انبعاث البلازما المقترنة بالتحريض (ICP-MS، 9.558 ppb)، وهو أقل بنحو تسع مرات من المزيج الفيزيائي لـ وCoFe -LDH (82.308 ppb)، كما هو موضح في الجدول التكميلي 4. تدعم هذه الأدلة التجريبية استقرار تحميل Ir على CoFeLDH. يثبت Ir استقرار CoFe من خلال تنسيق Ir-Cl القابل للتحكم، مما يثبط أكسدة CoFe، وفي الوقت نفسه، يثبت CoFe أيضًا Ir من خلال تفاعل إلكتروني قوي. تشير جميع التوصيفات المذكورة أعلاه مع البيانات الكهروكيميائية إلى استقرار جيد لـ Ir/CoFe-LDH في التحليل الكهربائي لمياه البحر. بالإضافة إلى ذلك، يجب ملاحظة أنه عند كثافات تيار تبلغ 600 و، كان الجهد الزائد مرتفعًا من الناحية الديناميكية الحرارية بما يكفي لتحفيز تفاعل أكسدة الكلوريد (الجهد الديناميكي الحراري: مقابل مقابل RHE عند 2.8 M NaCl)، ولكن لم يتم الكشف عن أي كلور أو هيبوكلوريت (مع كفاءة فاراداي للأكسجين > 99.9935% كما هو موضح في الأشكال التكملية 38-40)، مما يشير إلى انتقائية عالية لـ OER لـ في إلكتروليت NaCl عالي التركيز.
توصيف XAS وRaman في الموقع لـ Ir/CoFe-LDH
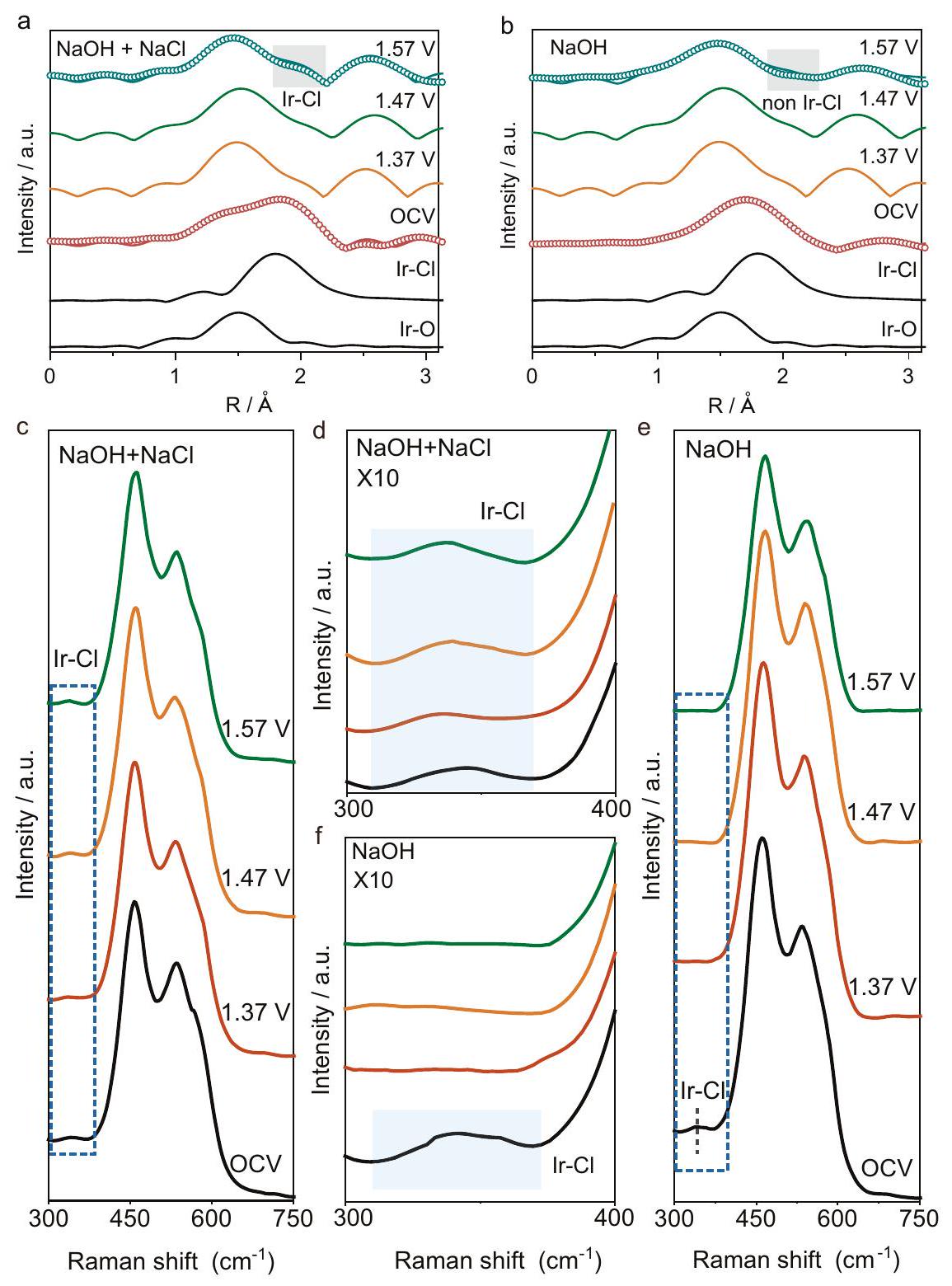
لاستكشاف لماذا وكيف يمكن أن يعزز نشاط OER لـ Ir/CoFe-LDH، تم إجراء EXAFS operando عند حافة Ir – وتمت مقارنته في (الشكل 3a) وإلكتروليت NaOH (الشكل 3b). التنسيق المحلي لعينتي Ir/CoFe-LDH المستخدمتين في EXAFS operando متشابه تقريبًا، مما يعني أن الاختلافات في طيف EXAFS في وإلكتروليت NaOH تنشأ من تطور هيكل المحفز أثناء OER (الشكل التكميلي 41 والجدول التكميلي 5). بعد ذلك، تحت جهد الدائرة المفتوحة (OCV)، يبقى تنسيق كلا المحفزين تقريبًا دون تغيير (الشكل التكميلي 42 والجدول التكميلي 6). ومع ذلك، عند جهد قدره 1.57 فولت مقابل RHE، كان تنسيق Ir-O واضحًا فقط في طيف EXAFS لـ Ir/CoFe-LDH المغمور في NaOH (الشكل التكميلي 43 والجدول التكميلي 7)، مما يشير إلى أن شكلت امتصاصًا تنافسيًا لتنسيق . في المقابل، يمكن ملاحظة كل من و عند نفس الجهد المطبق في إلكتروليت . يمكن أن يُعزى هذا الملاحظة إلى التركيز الكبير لـ ، مما يسمح باستمرار تنسيق . تشير النتائج أعلاه إلى وجود ارتباط تنافسي بين و مع مواقع Ir الذرية الفردية على CoFe-LDH أثناء OER في بيئة مياه البحر، وقد يكون الاستقرار الديناميكي لتنسيق في مياه البحر عاملاً رئيسيًا يدفع تحسين أداء OER. في الوقت نفسه، تم الكشف عن ذروة مميزة عند في أطياف EXAFS، والتي يمكن أن تُعزى إلى القرب الوثيق من Ir و أو Fe بسبب أكسدة سطح CoFe-LDH إلى MOOH أثناء عملية OER.
تم إجراء مطيافية Raman في الموقع أيضًا على LDH للحصول على رؤى آلية حول تأثير امتصاص Cl على موقع Ir الذري الفردي على نشاط OER. مقارنةً بـ CoFe-LDH (الشكل التكميلي 44)، أظهرت أطياف Raman لـ Ir/CoFe-LDH عند OCV ذروة جديدة عند حوالي (الشكل 3c-f). يمكن أن تُعزى هذه الذروة إلى اهتزاز وفقًا للعينة القياسية ، كما هو موضح في الشكل التكميلي 45.
علاوة على ذلك، في بيئة (الشكل 3c، d)، تم ملاحظة وجود تنسيق Ir-Cl عند بشكل متسق. في المقابل، في الإلكتروليتات القلوية بدون إضافة Cl، تم ملاحظة تنسيق فقط تحت حالة جهد الدائرة المفتوحة (OCV)، وتوقف وجوده عند تطبيق الجهد (الشكل 3e، f)، مما يشير إلى طبيعته الديناميكية. تشير هذه الظاهرة إلى أنه تحت تركيز عالٍ من ، هناك تنافس ملحوظ على الامتصاص بين و ، مما يتطلب تركيزًا عاليًا من NaCl للحفاظ على استقرار التنسيق. تتماشى هذه الملاحظة مع بيانات XAS لدينا، حيث يبقى التنسيق بين Ir وCl سليمًا في بيئة عالية التركيز من الملح، بينما يتبدد في بيئة غنية بـ . وبالتالي، يصبح من الضروري الحفاظ على بيئة ذات تركيز ملح مرتفع نسبيًا للحفاظ على تنسيق ، مما يسهل حركية OER.
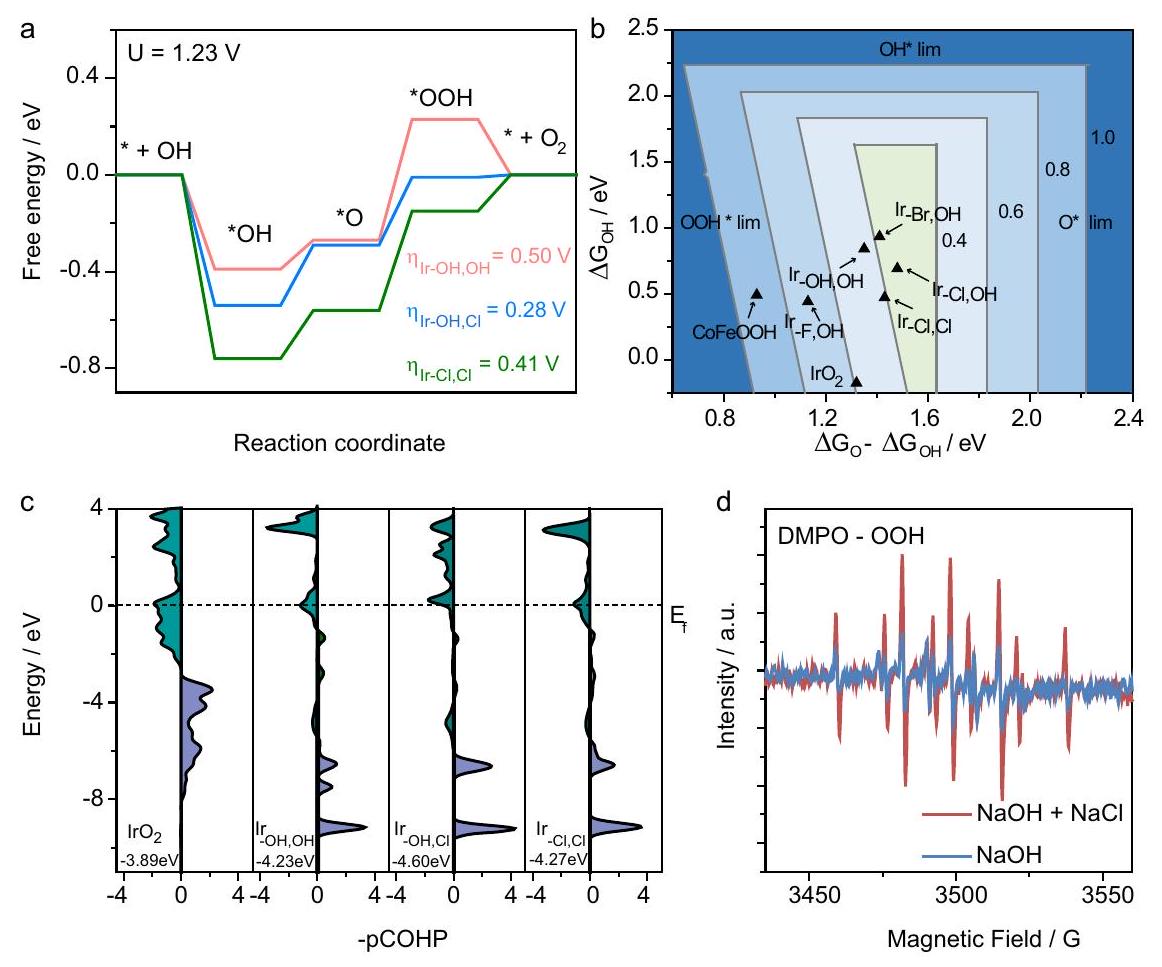
التحقيق النظري. أجرينا دراسات حسابية تهدف إلى تحديد تأثيرات تعديل بيئة التنسيق المحلي للإيريديوم والتأثيرات الناتجة على الهيكل الإلكتروني لمركز الإيريديوم وكذلك الطاقة الناتجة عن . بناءً على نتائج تركيب EXAFS وخصائص المحفز بعد OER، وُجد أن المحفز undergo إعادة بناء أثناء OER وأن الهيكل النشط الحقيقي هو ذرة الإيريديوم المفردة المرتبطة بسطح الهيدروكسيد الأكسجيني (أي، )، وقد تم ملاحظة إعادة بناء الهيدروكسيد إلى الهيدروكسيد الأكسجيني أثناء OER في الأدبيات . لذلك، قمنا ببناء نماذج المحاكاة الخاصة بنا كما هو موضح في الشكل التكميلي 46. تم بناء الإيريديوم الخماسي التنسيق مع ثلاث حالات: الإيريديوم المنسق مع اثنين من ، مع كلور واحد وواحد من ، ومع اثنين من الكلور ( ) المثبت على سطح LDH الدوري ذو الطبقتين، كنماذج المحاكاة. بالنسبة لـ CoFeOOH النقي، تم استخدام مواقع الحديد غير المشبعة في السطح (100) كمواقع نشطة. تشير التحليلات الديناميكية الحرارية (الشكل 4a والشكل التكميلي 47) إلى أن تشكيل وسيط * OOH هو الخطوة المحددة للسرعة
الشكل 3 | قياس XAS وRaman في الموقع. أ طيف EXAFS المحول فوريًا في الموقع للإيريديوم/CoFe-LDH في (أ) و(ب) NaOH. أطياف Raman في الموقع للإيريديوم/ المسجلة في (ج)، (د) ، و(هـ)، (و) NaOH. الأجزاء المميزة باللون الأزرق تتوافق مع تنسيق Ir-Cl.
(RDS) لـ OER على مواقع الحديد في CoFeOOH ومراكز الإيريديوم في Ir/CoFeOOH. يمكن أن تقلل ذرة الإيريديوم المفردة مع 2 OHs ( ) من الطاقة الحرة للأكسدة * من 2.24 eV إلى 1.73 eV. يمكن أن يقلل التنسيق أحادي الكلور مع ) من هذه الطاقة الحرة إلى 1.51 eV تفضيلًا لـ وفي الوقت نفسه يعزز امتصاص *OH، وهو الخطوة الرئيسية التي تتنافس مع امتصاص الكلور والأكسدة. ومع ذلك، فإن التنسيق الإضافي مع أيون كلوريد آخر لتشكيل رابطتين سيزيد من الطاقة الحرة للأكسدة إلى 1.64 eV. تتبع تسلسل النشاط للأنواع المنسقة من الإيريديوم . يعرض الشكل 4b الخريطة ثنائية الأبعاد (2D) للجهد الزائد النظري لبدء OER ( ) للإيريديوم الذري المفرد المثبت على CoFeOOH مع حالات تنسيق مختلفة من خلال افتراض العلاقة النسبية لـ (الشكل التكميلي 47c). يمكن أن تؤثر الهالوجينات الأخرى مثل و أيضًا على طاقة الامتصاص لـ *O و *OH بالإضافة إلى الجهد الزائد لـ OER، لكنها لم تكن فعالة مثل (الشكل التكميلي 47b) وفقًا للنتائج التجريبية.
لفهم آلية OER الأساسية ودور Cl بشكل أفضل، تم حساب تحليل كثافة الأوربيتال البلوري المتوقع (pCOHP)
بين مراكز الإيريديوم وذرات الأكسجين في *OOH الممتصة للحصول على معلومات حول امتصاص وسائط OOH . قمنا بحساب COHP المتكامل (ICOHP) حتى أعلى الحزم المشغولة (أسفل )، مما أعطى معلومات كمية مباشرة عن حالات الربط. بشكل عام، تشير -COHP السلبية إلى حالة مضادة للربط وتشير -COHP الإيجابية إلى حالة ربط . كما هو موضح في الشكل 4c، فإن قيمة ICOHP المنخفضة ( -4.60 eV ) توضح أن الأوربيتال الجزيئي لـ OOH الممتص يتفاعل بشكل أقوى بكثير مع حزم الإيريديوم في مقارنة بـ و و ، مما يشير إلى امتصاص أقوى لـ OOH على . تشير هذه النتائج إلى أن تشكيل *OOH، الذي هو RDS لـ OER، من الأسهل أن يحدث على مركز ، مما يعزز حركية OER.
للحصول على دليل تجريبي حول تشكيل وسيط *OOH، تم إجراء اختبار طيف الرنين المغناطيسي الإلكتروني (EPR) باستخدام 5،5-ثنائي ميثيل-1-بيرولين N-أكسيد (DMPO) كعامل حبس جذر حر *OOH . كما هو موضح في الشكل 4d، فإن الإشارة الناتجة عن *OOH المسجلة في الإلكتروليت هي تقريبًا ضعف تلك المسجلة في الإلكتروليت NaOH، مما يتطابق جيدًا مع
الشكل 4 | التحقيق النظري. أ الطاقة التنشيطية للإيريديوم/CoFe-LDH مع ثلاث حالات تنسيق. ب خريطة 2D للجهد الزائد النظري لبدء OER ( )، تم بناء خريطة الكنتور من خلال افتراض العلاقة النسبية لـ 3.17. ج الكثافة السكانية للأوربيتال البلوري المتوقع (pCOHP) بين مركز الإيريديوم وذرة الأكسجين في *OOH. د الكشف عن EPR لـ DMPO-OOH. تم أخذ العينة بعد 10 دقائق من التحليل الكهربائي عند 1.42 فولت مقابل RHE.
بيانات COHP. يمكن استنتاج الامتصاص المعزز لوسائط OER بشكل أكبر من الميل الأصغر لجهد الدائرة المفتوحة (OCP) المتجمع في الإلكتروليت مقارنةً بتلك التي تم جمعها في الإلكتروليت NaOH النقي، كما هو موضح في الشكل التكميلي 48.
للمزيد من التحقق من الدور المحدد لـ ، أجرينا حسابات لكثافة الحالات (DOS) وكثافة الحالات المتوقعة (PDOS) لـ Ir-Cl/CoFe-LDH وCl/CoFe-LDH. تشير النتائج إلى أن Cl تتداخل مع حزم التوصيل لـ Co وFe، ولكن لا يوجد تداخل مع حزم التكافؤ (الشكل التكميلي 49b). عند تحميل ذرة إيريديوم مفردة، يظهر Ir-Cl تداخلًا أقوى بكثير مع كل من حزم التكافؤ والتوصيل (الشكل التكميلي 49a)، مما يبرز تعزيز تداخل الأوربيتال وقدرة الربط لـ Ir-Cl. تتماشى بيانات الكثافة الكلية للحالات (TDOS) مع بيانات الكثافة الجزئية للحالات (PDOS)، مما يظهر زيادة ملحوظة في تداخل حزمة التكافؤ في وجود الإيريديوم السطحي، على عكس الحالة حيث لا تظهر حزم التكافؤ أي تفاعل (الشكل التكميلي 50).
لتبرير تحسين أداء OER واستكشاف دور أيون الكلوريد، تم دراسة الخصائص الحركية لـ في إلكتروليت NaCl وNaOH بشكل منهجي . تم تقدير الطاقة التنشيطية لـ OER على المحفز Ir/CoFe-LDH في الإلكتروليت لتكون باستخدام معادلة أرهينيوس، وهي أقل بكثير من تلك الموجودة في الإلكتروليت NaOH ( )، مما يثبت تحسين حركية OER بشكل كبير بواسطة الامتصاص (الأشكال التكملية 51 و52). من ناحية أخرى، تعرض CIOR جهدًا زائدًا نظريًا مرتفعًا بشكل ملحوظ (الشكل التكميلي 53)، مما يوفر مبررًا مقنعًا للاختيار المستمر القريب من 100% لـ OER لـ Ir/CoFe-LDH عند كثافات التيار الصناعية ( ) في التحليل الكهربائي للمياه المالحة.
لإظهار استقرار Ir/CoFe-LDH بشكل أكبر، اخترنا مواقع امتصاص Cl على Co أو Fe، على التوالي. تشير النتائج إلى أنه، في وجود لا يمكن أن تمتص بشكل فعال على Co (فيلم تكميلي 1) أو Fe (فيلم تكميلي 2)، على التوالي
(الشكل التكميلي 54). كشفت عملية التحسين عن امتصاص مباشر للكلور على مواقع الإيريديوم، مما يلغي وجود تكوينات مستقرة مثل أو (الأشكال التكملية 55 و56). وبالتالي، لا توجد هاتان الموقعان النشطان في وجود الإيريديوم. تؤكد هذه النتيجة بشكل أكبر على التحسين الكبير في الاستقرار الكهروكيميائي لـ CoFe LDH عند إدخال الإيريديوم. يثبت التنسيق القوي بين Ir-Cl مواقع CoFe، مما يمنع تنسيق الكلور مع كلا الموقعين المعدنيين، وبالتالي يضمن استقرار النظام في التحليل الكهربائي للمياه المالحة.
باختصار، لقد وضعنا استراتيجية بارعة تتضمن ترسيب الإيريديوم الذري المفرد على CoFe-LDH واستغلال أيونات الوفيرة في بيئة المياه المالحة لتنظيم حالة التنسيق لذرة الإيريديوم المفردة ديناميكيًا. لقد منح هذا التنسيق محفز Ir/CoFe-LDH نشاطًا متزايدًا بشكل كبير في OER في المياه المالحة، متجاوزًا أدائه في البيئة القلوية. تدعم التوصيفات العملية وحسابات DFT أن الأداء المعزز لـ OER لـ Ir/CoFe-LDH في المياه المالحة ينشأ من التنظيم الديناميكي لحالات تنسيق Cl وOH على الإيريديوم. يسهل هذا التعزيز في التنسيق الذي يتوسطه الكلور امتصاصًا قويًا لوسائط *OOH في OER، مما يقلل من حاجز الطاقة التنشيطية لتشكيل بمقدار 1.93 ويزيد بشكل كبير من معدل نقل الإلكترون بين الواجهة الكاثودية (CIER). بشكل مثير للإعجاب، يظهر Ir/CoFe-LDH استقرارًا تشغيليًا ملحوظًا عند كثافات التيار الصناعية ( ) في المياه المالحة، مع الحفاظ على نشاط مستمر لأكثر من 1000 ساعة مع الاحتفاظ باختيار ملحوظ لـ . لا تكشف هذه الدراسة فقط عن الإمكانات الهائلة لـ Ir/CoFe-LDH في التحليل الكهربائي للمياه المالحة، ولكنها تقدم أيضًا استراتيجية جديدة لتنظيم تنسيق الكلوريد الذري المفرد في بيئات المياه المالحة، وهو اختراق يعد بتقليل استهلاك الطاقة والتكاليف المرتبطة بالتحليل الكهربائي للمياه المالحة، مما يدفع النشر العملي لهذه التكنولوجيا.
طرق
مواد
نترات الحديد (III) تسعة هيدرات ، نترات الكوبالت (II) سداسية الهيدرات كلوريد الإريديوم المائي (كلوريد البوتاسيوم (تم شراءها من سيغما-ألدريتش. هيدروكسيد الصوديوم ( ) ، كلوريد الصوديوم ( ) كربونات الصوديوم ( ) تم شراؤها من شركة فوشين للمواد الكيميائية المحدودة. الإيثانول ( تم شراء ) من شركة تيانجين فوي للتكنولوجيا الكيميائية المحدودة. تم شراء 5,5-دايميثيل-1-بيرولين N-أكسيد (DMPO) من شركة دوجيندو الصين بكين المحدودة. تم شراء ورق ألياف الكربون (توراي 060) من شركة سوتشو سينيرو للتكنولوجيا المحدودة. تم أخذ مياه البحر من البحر الأصفر، الصين. تم استخدام مياه مؤينة (DI) (المقاومية: ) تم استخدامه لتحضير جميع المحاليل المائية.
تحضير CoFe-LDH
تم تخليق CoFe-LDH من خلال طريقة الترسيب المشترك. باختصار، 40 مل من المحلول A: و و 40 مل من المحلول ب: و تم تحضيرها أولاً. تم إضافة المحلول A و B بالتنقيط في نفس الوقت إلى دورق يحتوي على 40 مل من الماء المقطر. تم ضبط قيمة pH للمعلق النهائي إلى 8.5 تحت التحريك المغناطيسي، كما تم مراقبتها باستخدام مقياس pH في درجة حرارة الغرفة.تم التحكم في درجة الحموضة بشكل جيد حيث تم إضافة محلول الملح A ومحلول القلوي B في نفس الوقت، ويمكن تعديل نسبة هذين المحلولين لتلبية درجة الحموضة المستهدفة. تم ترك التعليق الناتج ليشيخ لمدة 12 ساعة في. بعد ذلك، تم جمع الراسب عن طريق الطرد المركزي، وغسله 3 مرات باستخدام الماء المقطر، تلاه غسل بالإيثانول 3 مرات. تم تجفيف الراسب النهائي في فرن مفرغ من الهواء عندبين عشية وضحاها.
تحضير /CoFe-LDH
المُحضَّر كما هوتمت إضافته إلى 50 مل من الماء المقطر تحت التحريك المغناطيسي لتشكيل تعليق غروي. ثم، 10 مل من المحلول المائي الطازج الذي تم تحضيرهتم إضافة محلول (5 ملغ) يحتوي على 0.02 م NaOH بالتنقيط إلى تعليق كولوييد CoFe-LDH المذكور أعلاه. تم تحريك التعليق مع التسخين المتزامن عندلمدة 6 ساعات. بعد ذلك، تم جمع الراسب بواسطة الطرد المركزي، وغسله بالماء المقطر والإيثانول، كل منهما 3 مرات. ثم، تم تجفيف الراسب النهائي في فرن مفرغ من الهواء عندبين عشية وضحاها.
تحضير-LDH
المُحضَّر كما هوتمت إضافته إلى 50 مل من الماء المقطر تحت التحريك المغناطيسي لتكوين تعليق غروي. ثم، 10 مل من المحلول المائي الطازج الذي تم تحضيرهتم إضافة محلول (10 ملغ) يحتوي على 0.02 م NaOH بالتنقيط إلى تعليق كولوييد CoFe-LDH المذكور أعلاه. تم تحريك التعليق مع تسخينه في نفس الوقت عندلمدة 6 ساعات. بعد ذلك، تم جمع الراسب عن طريق الطرد المركزي، وغسله بالماء المقطر والإيثانول كل منهما ثلاث مرات. ثم، تم تجفيف الراسب النهائي في فرن مفرغ من الهواء عندبين عشية وضحاها.
توصيف ومحاكاة HAADF-STEM
قبل التصوير، تم إضافة المحفزات المُعدة مسبقًا إلى الإيثانول اللامائي باستخدام جهاز الموجات فوق الصوتية لتشكيل تعليق غروي مخفف جدًا، ثمتمت إضافة التعليق على مصفوفات نحاسية بشبكة 230 مغطاة بالكربون فائق الرقة. تم الحصول على صورة HAADF-STEM عالية الدقة باستخدام مجهر Thermo Fisher Spectra 300 المزود بمصحح تشوهات لعدسة تشكيل الشعاع، وعمل عند 300 كيلوفولت. كانت شدة الشعاع أقل من 40 بيكو أمبير وزاوية التقارب في STEM كانتالذي قدم حجم مسبارعند 300 كيلو فولت. صور HADDF-STEM معتم أخذ ميل العينة عن طريق إمالة حامل الميل المزدوجالتي يمكن نقلها في و التوجيهات. تم إجراء محاكاة صور HADDF-STEM باستخدام برنامج Dr. Probeتم تعيين المعلمات على نفسه كما في حالة التجربة. كانت دقة نتائج المحاكاة.
أدوات التوصيف
تم إجراء قياس المجهر الإلكتروني الناقل على جهاز JEOLJEM 2100. تم تسجيل أنماط حيود الأشعة السينية (XRD) على جهاز حيود الأشعة السينية (Rigaku D/max 2500) معإشعاع ) بمعدل مسح فيتتراوح من 3 إلىتم تسجيل أطياف مطياف الإلكترونات الضوئية (XPS) على جهاز ESCALAB 250 (ثيرمو فيشر ساينتيفيك الولايات المتحدة الأمريكية) باستخدام أشعة كهرومغناطيسية أحادية اللون من نوع Al K.أشعة X. تم الإشارة إلى جميع طاقات الربط بالنسبة لقمة C 1s (284.8 eV). بشكل محدد، من خلال تقييم انحراف موضع طاقة الربط المرتبطة بقمة C 1s عند 284.8 eV، يتم استخدام الفرق الناتج لإجراء معايرة شاملة لمجموعة البيانات بأكملها. تم إجراء قياس ICP-MS على نظام Thermo X Series II ICP/MS رباعي القطب، Thermo Fisher Scientific، لت quantifying التركيب الكيميائي للمحفز. تم إجراء قياس طيف EPR على جهاز Magnettech MS-5000X باستخدام 5,5-dimethyl-1-pyrroline N-oxide (DMPO) كفخ للجذور الحرة.
طيف امتصاص الأشعة السينية
تمت القياسات في إعداد ثلاثي الأقطاب نموذجي في حاوية مصنوعة من التفلون مصممة خصيصًا مع نافذة مختومة بشريط كيبون. كانت ظروف الاختبار هي نفسها كما في حالة التوصيف الكهروكيميائي. تمت القياسات في BL-17C في مركز أبحاث الإشعاع المتزامن الوطني (NSRRC، هسينتشو، تايوان). تم جمع البيانات في الموقع في وضع الفلورة الكلية باستخدام كاشف انزلاق السيليكون.
تحليل بيانات XANES و EXAFS
تم تحليل بيانات هيكل الامتصاص القريب من حافة الأشعة السينية (XANES) وهيكل الامتصاص الدقيق الممتد للأشعة السينية (EXAFS) بواسطة برنامج أثينا من حزمة IFEFFIT. تم تحليل طيف EXAFS من خلال طرح الخلفية بعد الحافة من الامتصاص الكلي وتم تطبيعه بالنسبة لخطوة قفزة الحافة بواسطة برنامج أرتيميس وفقًا لمنهجية ملائمة البيانات المبلغ عنها سابقًا..
اختبار رامان في الموقع
تم جمع طيف رامان في الموقع باستخدام جهاز LabRAM ARAMIS (هوربا جوبين يفون، فرنسا) مع مصدر ليزر PSU-H-FDA بطول موجي 532 نانومتر (شركة تشانغتشون للصناعات الجديدة للتكنولوجيا البصرية المحدودة، الصين). تم استخدام عدسة LMPlanFLNتم استخدام عدسة هدف المجهر مع فتحة عددية تبلغ 0.5 (أوليمبوس، اليابان) لميكروسكوبية رامان. تم معايرة تردد رامان بواسطة شريحة سيليكون. ) خلال كل تجربة. تم إجراء تجارب رامان الكهروكيميائية في الموقع في خلية رامان في الموقع (شركة تيانجين أيدا هينغشينغ لتطوير التكنولوجيا المحدودة، الصين) والمح catalyst المعد. تم استخدام سلك البلاتين كإلكترود عمل، وإلكترود مرجعي، وإلكترود مضاد، على التوالي. تم استخدام جهاز العمل الكهربائي CHI 660e (شركة شنغهاي تشينهوا للأجهزة، الصين) للتحكم في الجهد حيث تم زيادة الجهود المطبقة خطوة بخطوة من جهد الدائرة المفتوحة (OCP) إلى 1.57 فولت مقابل RHE. تم الحصول على كل طيف على الأقل ثلاث مرات مع وقت تعرض قدره 30 ثانية.
تجارب DRIFTS في الموقع
تم استخدام DRIFTS في الموقع لتوصيف تفاعل المحفز مع CO. تم إجراء تجارب DRIFTS في الموقع على جهاز مطياف الأشعة تحت الحمراء بتحويل فورييه Bruker INVENIO R مزود بكاشف MCT/A مبرد بالنيتروجين السائل وملحق انعكاس منتشر من Harrick. تم تحميل حوالي 50 ملغ من المحفز في غرفة التفاعل عالية الحرارة Harrick Praying Mantis المزودة بنوافذ KBr (HVC-DRP-5). تم إغلاق الغرفة وتم تمرير الغازات من خلالها عند ضغط جوي. أر عالي النقاء. تم استخدامه كغاز تطهير، وCO المخفف (تم استخدام غاز الاستكشاف (درجة معتمدة من CO) كغاز استكشاف. تم التحكم في درجة الحرارة بواسطة ثيرموكوبل في اتصال مباشر مع العينة. تم استخدام الماء المتداول لتبريد جسم غرفة التفاعل (درجة الحرارة المحددة: ). جميع التوصيفات في الموقع اتبعت نفس إجراء المعالجة المسبقة، كل طيف تم الإبلاغ عنه هو متوسط 64 مسحًا. دقة طيفية من تم استخدامه لجمع الأطياف، التي تم الإبلاغ عنها بوحدات كوبيلكا-مونك. بالنسبة لـ Ir/CoFe-LDH، تم تسخين العينة في الأرجون (“معدل الزيادة” فيمعدل الزيادة) لمدة 30 دقيقة لإزالة المواد الماصة جسديًا. ثم تم تبريد درجة الحرارة في الأرجون إلى مع وجود الماء المتداول وتم تسجيل الطيف الخلفي. ثم تم وضع علامة على العينة فيدرجة معتمدة من CO“معدل الزيادة” فيلمدة 0.5 ساعة. بعد ذلك،، معدل التسارع) تم تطهيره عندلمدة 30 دقيقة لإزالة ثاني أكسيد الكربون الممتص فيزيائيًا حتى لم يُلاحظ أي تغيير في الطيف. تم تسجيل طيف DRIFTS في نطاق الأعداد الموجية من 1000 إلىعند 298 كلفن مع إجمالي 1000 قياس تم أخذها، كل منها مفصول بـفترة.
القياسات الكهروكيميائية
تم إجراء جميع القياسات الكهروكيميائية على إعداد ثلاثي الأقطاب باستخدام محطة العمل الكهروكيميائية CHI 660 e. تم استخدام سلك من البلاتين كقطب مضاد وإلكترود جسر الملح المزدوج SCE كإلكترود مرجعي. يتكون جسر الملح المزدوج SCE، الذي تم شراؤه من شركة تيانجين أيدا هينغشينغ لتطوير التكنولوجيا المحدودة، من KCl المشبع. تم إعداد القطب العامل من خلال الخطوات التالية: (1) تحضير حبر المحفز. 2 ملغ من المحفز الذي تم تصنيعه، 1 ملغ من الكربون الأسود، وتم dispersing محلول نافيون في الإيثانولوالماء منزوع الأيوناتتحت الموجات فوق الصوتية لمدة لا تقل عن ساعة واحدة لتكوين حبر محفز متجانس. (2) طلاء حبر المحفز على ورق ألياف الكربون (توراي، TGP-H-060): تم إسقاط حبر المحفز على ورق ألياف الكربونتوراي، تي جي بي-كل مرة مع التجفيف تحت مصباح الأشعة تحت الحمراء، يتبعه إسقاط آخرالحبر حتى وصلت كتلة التحفيز إلىتم قياس الفولتمترية الدورية (CV) من 0 إلى 1.0 فولت مقابل SCE بمعدل مسحتم قياس طيف الامتياز الكهربائي (EIS) عن طريق تطبيق جهد متردد قدره 5 مللي فولت عند جهد زائد قدره 10 مللي فولت بتردد يتراوح من 100 كيلو هرتز إلى 0.1 هرتز. تم تصحيح جميع منحنيات الاستقطاب لتعويض انخفاض أوم باستخدام المقاومة الأومية التي تم الحصول عليها من EIS. تم قياس قطب المرجع SCE مقابل مقياس RHE مباشرةً بواسطة إعداد ثلاثي الأقطاب، يتكون من سلكين من البلاتين وقطب مرجع واحد ليتم معايرته. تم قياس جهد الدائرة المفتوحة (OCP) تحت شرط تطهير عالي النقاء.الغاز في الإلكتروليت. تم تطبيق جهد مفتوح بعد التشبع بـالغاز، والحصول على جهد مستقر. هذا الجهد هو فرق الجهد بين RHE والقطب المرجعي. في اختيار الإلكتروليت للاختبار، فإن استخدام 6 م NaOH يعكس مستويات القلوية التي تُستخدم عادة في التحليل الكهربائي للمياه القلوية الصناعية. علاوة على ذلك، وفقًا لعملنا السابقمن الجدير بالذكر أن الحد الأقصى للذوبانية لـ NaCl في 6 M NaOH هو 2.8 M، حيث أن تركيز NaCl الزائد عن هذا الحد يؤدي إلى ترسيب بلوري. من المهم أن نلاحظ أنه في السيناريوهات العملية، من غير المحتمل أن تتجاوز تركيزات أملاح الإلكتروليت هذا الحد. من خلال إجراء اختبارات تحت هذه الظروف، نهدف إلى عرض أداء القطب عند أقصى طاقته.
قياس طاقة التنشيط ( )
طاقة التنشيط لتفاعل الأكسدة (OER) هي مؤشر على حركية التفاعل، والتي تعتمد فقط على المادة. المعادلة (1) هي معادلة أرهينيوس، حيثهو عامل ما قبل الأسية، هو ثابت الغاز المثالي، هي درجة الحرارة، و هو ثابت معدل، يمكن تطبيقه لتحديد طاقة التنشيط لتفاعل الأكسدة على المحفزات. عند جهد زائد ثابت، التيار الحركيلها علاقة خطية مع معدل ثابت ( )، كما هو موضح في المعادلة (2)، حيث هو ثابت فاراداي، هو عدد الإلكترونات المنقولة، هو مساحة القطب الكهربائي و هو التركيز. إذا افترضنا أن تأثيرات نقل الكتلة غير ملحوظة على تيارات OER، فإنيمكن التعبير عنها بالمعادلة (3)، حيثهو حاجز التنشيط عند جهد ثابت. يمكن التعبير عن طاقة التنشيط الحركية عند جهد معين كما هو موضح في المعادلة (4). وبالتالي، يمكن تعريف طاقة التنشيط لتفاعل الأكسدة (OER) بواسطة المعادلة (5):
طرق نظرية
تم إجراء جميع الحسابات النظرية باستخدام طريقة الموجة المعززة بالمش projectoraugmented wave ومجموعة أساس الموجة المستوية كما هو مطبق في حزمة المحاكاة الأولية في فيينا (VASP). تم تحسين خصائص الكتلة والسطح لـ CoFeOOH ضمن GGA-PBE. تم إجراء تحسين كامل لمواقع جميع الذرات في الكتلة من خلال إجراء عملية تحسين تدرج مترافق. مجموعة مونكهورست-باكتم تعيين عينات النقاط كـلتحسين الهندسة، ولحساب الهيكل الإلكتروني. وتم تحسين الثوابت الكثيفة باستخداممونكهورست-باك-نقطة أخذ العينات. تم تعيين طاقة القطع لوظائف الأساس ذات الموجة المستوية إلى 600 إلكترون فولت مع معيار تقارب تغيير الطاقة لـتم السماح لمواقع الذرات بالاسترخاء حتى وصل مجموع القوى المطلقة إلىتم تطبيق طريقة تصحيح هوبارد-يو لتحسين وصف الكوبالت المحلي.-الإلكترونات في CoFeOOH مع و لـ Fe و Co، على التوالي. تم أيضًا تضمين استقطاب الدوران، والتفاعل طويل المدى فان دير فالز (IVDW = 11)، وتصحيحات المذيب في حسابات السطح. تم اعتبار تأثير المذيب على المواد الممتصة باستخدام نموذج الذوبان الضمني بواسون-بولتزمان مع ثابت عازل قدره 78.4. تم حساب طاقات غيبس الحرة بواسطة:
حيث تمثل الرموز طاقة الربط ( )، التغير في طاقة النقطة الصفرية ( درجة الحرارة )، وتغير الإنتروبيا للنظام، على التوالي. تم حساب كثافة هاملتونيان المدارات البلورية (COHP) بواسطة LOBSTER، وتم تعيين عتبة التقارب للتكرار في المجال الذاتي المتسق عند .
توفر البيانات
تم توفير بيانات المصدر مع هذه الورقة.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294-303 (2012).
Zhu, J. et al. Hydrogen evolution using nanoparticles. Chem. Rev. 120, 851-918 (2020).
Bockris, J. O. A hydrogen economy. Science 176, 1323 (1972).
Mitchell, S. et al. Nanoscale engineering of catalytic materials for sustainable technologies. Nat. Nanotechnol. 16, 129-139 (2021).
Peter, S. et al. Electrolysis of low-grade and saline surface water. Nat. Energy 5, 367-377 (2020).
Wang, C. et al. Advances in hydrogen production from electrocatalytic seawater splitting. Nanoscale 17, 7897-7912 (2021).
Li, P. et al. Common-ion effect triggered highly sustained seawater electrolysis with additional NaCl production. Research 2020, 2872141 (2020).
Peter, S. et al. Design criteria, operating conditions, and nickel-iron hydroxide catalyst materials for selective seawater electrolysis. ChemSusChem 9, 962-972 (2016).
Rasmus, K. & Ann, C. Selectivity between oxygen and chlorine evolution in the chlor-alkali and chlorate processes. Chem. Rev. 116, 2982-3028 (2016).
Shao, H. et al. An earth-abundant catalyst-based seawater photoelectrolysis system with 17.9% solar-to-hydrogen efficiency. Adv. Mater. 30, 1707261 (2018).
Xie, H. et al. A membrane-based seawater electrolyser for hydrogen generation. Nature 612, 673-678 (2022).
Kuang, Y. et al. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl Acad. Sci. 116, 6624-6629 (2019).
Wu, L. Heterogeneous bimetallic phosphide as an efficient bifunctional catalyst for water/seawater splitting. Adv. Funct. Mater. 31, 2006484 (2021).
Marc, K. et al. as selective oxygen evolution electrocatalyst in acidic chloride solution. J. Am. Chem. Soc. 140, 10270-10281 (2018).
Yu, L. et al. Ultrafast room-temperature synthesis of porous S-doped Ni/Fe (oxy)hydroxide electrodes for oxygen evolution catalysis in seawater splitting. Energy Environ. Sci. 13, 3439 (2020).
Song, Z. et al. High-performance ammonium cobalt phosphate nanosheet electrocatalyst for alkaline saline water oxidation. Adv. Sci. 8, 2100498 (2021).
Zhao, Y. et al. Charge state manipulation of cobalt selenide catalyst for overall seawater electrolysis. Adv. Energy Mater. 29, 1801926 (2018).
Wu, L. et al. Boron-modified cobalt iron layered double hydroxides for high efficiency seawater oxidation. Nano Energy 83, 105838 (2021).
Mohammad, M. et al. Manganese compounds as water-oxidizing catalysts: from the natural water-oxidizing complex to nanosized manganese oxide structures. Chem. Rev. 116, 2886-2936 (2016).
Zenta, K. et al. Durability enhancement and degradation of oxygen evolution anodes in seawater electrolysis for hydrogen production. Appl. Surf. Sci. 257, 8230-8236 (2011).
Takuya, O. et al. A bilayer structure composed of deposited on a film to realize selective oxygen evolution from chloride-containing water. Langmuir 36, 5227-5235 (2020).
Oscar, D. et al. Iridium-based double perovskites for efficient water oxidation in acid media. Nat. Commun. 7, 12363 (2016).
Kim, Y. et al. Balancing activity, stability and conductivity of nanoporous core-shell iridium/iridium oxide oxygen evolution catalysts. Nat. Commun. 8, 1449 (2017).
Zhang, Y. et al. Atomic iridium incorporated in cobalt hydroxide for efficient oxygen evolution catalysis in neutral electrolyte. Adv. Mater. 30, 1707522 (2018).
Cai, C. et al. Ultrahigh oxygen evolution reaction activity achieved using Ir single atoms on amorphous nanosheets. ACS Catal. 11, 123-130 (2021).
Maximilian, M. et al. HCl oxidation on -based catalysts: from fundamentals to scale-up. ACS Catal. 3, 2813-2822 (2013).
Ricardo, E. et al. Heterogeneous model to distinguish the activity of electrogenerated chlorine species from soluble chlorine in an
electrochemical reactor. Ind. Eng. Chem. Res. 58, 22399-22407 (2019).
Zeradjanin, A. et al. On the Faradaic selectivity and the role of surface inhomogeneity during the chlorine evolution reaction on ternary Ti-Ru-Ir mixed metal oxide electrocatalysts. Phys. Chem. Chem. Phys. 16, 13741 (2014).
Johannes, G. et al. Selectivity trends between oxygen evolution and chlorine evolution on iridium-based double perovskites in acidic media. ACS Catal. 9, 8561-8574 (2019).
Rossmeisl, J. et al. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83-89 (2007).
Hansen, A. et al. Electrochemical chlorine evolution at rutile oxide (110) surfaces. Phys. Chem. Chem. Phys. 12, 283-290 (2010).
Li, P. et al. Boosting oxygen evolution of single-atomic ruthenium through electronic coupling with cobalt-iron layered double hydroxides. Nat. Commun. 10, 1711 (2019).
Barthel, J. Dr. Probe: a software for high-resolution STEM image simulation. Ultramicroscopy 193, 1-11 (2018).
Hernández-Cristóbal, O. et al. Effect of the reduction temperature on the activity and selectivity of titania-supported iridium nanoparticles for methylcyclopentane reaction. Ind. Eng. Chem. Res. 53, 10097-10104 (2014).
Shan, J. et al. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 551, 605-608 (2017).
Freakley, S. J. et al. The X-ray photoelectron spectra of Ir, IrO2 and IrCl3 revisited. Surf. Interface Anal. 49, 794-799 (2017).
Morgan, D. J. Resolving ruthenium: XPS studies of common ruthenium materials. Surf. Interface Anal. 47, 1072-1079 (2015).
Jiang, K. et al. Dynamic active-site generation of atomic iridium stabilized on nanoporous metal phosphides for water oxidation. Nat. Commun. 11, 2701 (2020).
Li, Z. et al. Iridium single-atom catalyst on nitrogen-doped carbon for formic acid oxidation synthesized using a general host-guest strategy. Nat. Chem. 12,, 764-772 (2020).
Sören, D. et al. Direct electrolytic splitting of seawater: opportunities and challenges. ACS Energy Lett. 4, 933-942 (2019).
Wang, G. et al. SC-IrO2NR-carbon hybrid: a catalyst with high electrochemical stability for oxygen reduction. Sci. China Mater. 56, 131-136 (2013).
Dao, J. et al. Layered double hydroxide-based electrocatalysts for the oxygen evolution reaction: identification and tailoring of active sites, and superaerophobic nanoarray electrode assembly. Chem. Soc. Rev. 50, 8790-8817 (2021).
Rossmeisl, J. et al. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 319, 178-184 (2005).
Michal, B. et al. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521-13530 (2013).
Zhang, B. et al. Homogeneously dispersed multimetal oxygenevolving catalysts. Science 352, 333-337 (2016).
Mefford, J. T. Correlative operando microscopy of oxygen evolution electrocatalysts. Nature 593, 67-73 (2021).
Zhang, J. Single-atom layered double hydroxide electrocatalyst: probing the origin of activity for oxygen evolution reaction. J. Am. Chem. Soc. 140, 3876-3879 (2018).
Richard, D. et al. Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 8617-8624 (1993).
Hoffman, R. Solids and Surfaces: A Chemist’s View of Bonding in Extended Structures (Wiley-VCH, 1988).
Liu, X. et al. Building up a picture of the electrocatalytic nitrogen reduction activity of transition metal single-atom catalysts. J. Am. Chem. Soc. 141, 9664-9672 (2019).
Lin, L. et al. A highly CO-tolerant atomically dispersed Pt catalyst for chemoselective hydrogenation. Nat. Nanotechnol. 14, 354-361 (2019).
Williamson, N. et al. Equilibrating (L) and species in hydrocarbon oxidations by bio-inspired nonheme iron catalysts using and AcOH. J. Am. Chem. Soc. 139, 17313-17326 (2017).
Song, H. et al. Direct and selective photocatalytic oxidation of to oxygenates with on cocatalysts/ ZnO at room temperature in water. J. Am. Chem. Soc. 141, 20507-20515 (2019).
Wang, N. et al. Hydration-effect-promoting Ni-Fe oxyhydroxide catalysts for neutral water oxidation. Adv. Mater. 32, 1906806 (2020).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537-541 (2005).
Liu, R. et al. Design of aligned porous carbon films with single-atom Co-N-C sites for high-current-density hydrogen generation. Adv. Mater. 33, 210533 (2021).
Zhao, S. et al. Structural transformation of highly active metal-organic framework electrocatalysts during the oxygen evolution reaction. Nat. Energy 5, 881-890 (2020).
شكر وتقدير
يشكر المؤلفون الدكتور سيجون هو على المساعدة في التوصيف. يعترف X.S. و Y.K. بالدعم المالي من مشروع البحث والتطوير الوطني الرئيسي (2021YFA1502200)، ومؤسسة العلوم الطبيعية الوطنية في الصين (21935001)، ومؤسسة العلوم الطبيعية في بكين (Z210016)، ومنحة طويلة الأجل من وزارة المالية الصينية ووزارة التعليم. يعترف B.L. بالدعم المالي من صندوق بدء التشغيل لجامعة مدينة هونغ كونغ (9020003) ومنحة ITF-RTH-Global STEM Professorship (9446006).
مساهمات المؤلفين
X.S. و B.L. و Y.K. أشرفوا على المشروع. X.D. ابتكر الفكرة وأجرى التجارب. X.D. و Q.S. و P.L. و D.Z. قاموا بتخليق المواد وإجراء القياسات الكهروكيميائية. Q.S. أجرى قياسات DRIFTs في الموقع وقياسات رامان في الموقع. T.L. و G.Y. و W.L. ساعدوا في توصيف المواد. W.C. و Y.C. و L.Z. أجروا قياسات XAS في الموقع. X.D. و Y.L. و E.Y. نفذوا حسابات DFT. H.Y. و G.Z. و W.L. ساعدوا في تحليل البيانات. X.S. و B.L. و Y.K. و X.D. و Q.S. كتبوا الورقة. S.H. أجرى قياسات XAS في الموقع في النسخة المعدلة. J.L. و J.D. حللوا بيانات
XAS في النسخة المعدلة. ساعد Z.W. و H.X. في محاكاة DFT. قام J.F. و Z.Z. بإجراء قياسات ATR-SEIRAS في النسخة المعدلة. قام C.J. و J.L. و L.B. بإجراء قياسات ومحاكاة HADDF-STEM. ناقش جميع المؤلفين النتائج وساهموا في إعداد المخطوطة.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح ما إذا تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/رخصة/بواسطة/4.0/. (ج) المؤلف(ون) 2024 مختبر الدولة الرئيسي للهندسة الكيميائية، مركز بكين المتقدم للابتكار في علوم المواد اللينة والهندسة، كلية الكيمياء، جامعة بكين للتكنولوجيا الكيميائية، بكين 100029، جمهورية الصين الشعبية.كلية الكيمياء، الهندسة الكيميائية والتكنولوجيا الحيوية، جامعة نانيانغ التكنولوجية، سنغافورة 637459، سنغافورة.المختبر الوطني في بكين للعلوم الجزيئية، مختبر كاس الرئيسي للغرويات، الواجهة والديناميكا الحرارية الكيميائية، معهد الكيمياء، الأكاديمية الصينية للعلوم، بكين 100190، جمهورية الصين الشعبية.مركز أبحاث طاقة الهيدروجين البحرية، معهد أبحاث جامعة تسينغhua في شنتشن، شنتشن 518057، جمهورية الصين الشعبية.مختبر الدولة الرئيسي للمركبات العضوية وغير العضوية، كلية الهندسة الكيميائية، جامعة بكين للتكنولوجيا الكيميائية، 100029 بكين، جمهورية الصين الشعبية.مركز الطاقة والتحفيز، كلية علوم المواد والهندسة، معهد بكين للتكنولوجيا، بكين 100081، جمهورية الصين الشعبية.المركز الوطني للبحوث في العلوم الفيزيائية على المقياس المجهري، جامعة العلوم والتكنولوجيا الصينية، هيفي 230026، جمهورية الصين الشعبية.منشأة الإشعاع السنكروتروني في بكين، معهد الفيزياء العالية الطاقة، الأكاديمية الصينية للعلوم، 100049 بكين، جمهورية الصين الشعبية.قسم الكيمياء، جامعة تسينغhua، 100084 بكين، جمهورية الصين الشعبية.قسم علوم المواد والهندسة، جامعة مدينة هونغ كونغ، منطقة هونغ كونغ الإدارية الخاصة 999077، جمهورية الصين الشعبية.كلية الطاقة، جامعة شاندونغ للعلوم والتكنولوجيا، تشينغداو 266590، جمهورية الصين الشعبية.مختبر بكين الرئيسي لتحفيز الطاقة البيئية، جامعة بكين للتكنولوجيا الكيميائية، 100029 بكين، جمهورية الصين الشعبية.قسم الكيمياء التطبيقية ومركز علوم المواد الوظيفية الناشئة، جامعة يانغ مينغ تشياو تونغ الوطنية، هسينتشو 300، تايوان.كلية علوم المواد والهندسة، مختبر تيانجين الرئيسي للمواد والأجهزة الكهروضوئية، جامعة تيانجين للتكنولوجيا، تيانجين 300384، جمهورية الصين الشعبية.مختبر شينسي، معهد شنتشن للدراسات المتقدمة، جامعة العلوم والتكنولوجيا الإلكترونية الصينية، شنتشن 518110، جمهورية الصين الشعبية.مختبر كاس الرئيسي للمعايير والقياسات لتكنولوجيا النانو، المركز الوطني لعلوم وتكنولوجيا النانو، 100190 بكين، جمهورية الصين الشعبية.قسم الكيمياء ومركز الماس الفائق والأفلام المتقدمة (COSDAF)، جامعة مدينة هونغ كونغ، منطقة هونغ كونغ الإدارية الخاصة 999077، جمهورية الصين الشعبية.ساهم هؤلاء المؤلفون بالتساوي: شينشوان دوآن، تشيهاو شا.البريد الإلكتروني:kuangy@tsinghua-sz.org;bliu48@cityu.edu.hk;sunxm@mail.buct.edu.cn
Seawater electrolysis offers a renewable, scalable, and economic means for green hydrogen production. However, anode corrosion by pose great challenges for its commercialization. Herein, different from conventional catalysts designed to repel adsorption, we develop an atomic Ir catalyst on cobalt iron layered double hydroxide (Ir/CoFe-LDH) to tailor Cl adsorption and modulate the electronic structure of the Ir active center, thereby establishing a unique coordination for alkaline seawater electrolysis. Operando characterizations and theoretical calculations unveil the pivotal role of this coordination state to lower OER activation energy by a factor of 1.93. The Ir/CoFe-LDH exhibits a remarkable oxygen evolution reaction activity ( 202 mV overpotential and TOF ) in , superior over Cl-free 6 M NaOH electrolyte ( 236 mV overpotential and TOF = ), with catalytic selectivity and stability at high current densities ( ) for more than .
Grid-scale water electrolysis is promising for storing renewable electricity into molecular hydrogen bond . Using earth-abundant seawater as feedstock instead of desalinated water provides a more sustainable strategy for renewable hydrogen production , which yields NaCl as a byproduct at the same time . However, seawater has a salinity of , in which most of the salt is . Implementation of seawater electrolysis technology confronts many challenges, especially at the anode side where severe catalyst corrosion and competitive chloride oxidation reaction (ClOR) occur simultaneously , which significantly hinder its commercialization. Efficient and sustained seawater electrolysis demands a highly active and selective anode that is able to work in the presence of concentrated . Previous attempts of seawater electrolysis mostly focused on the prohibition of adsorption on the anode catalyst so as to prevent ClOR. For instance, Koper et al. electrodeposited
on to improve the oxygen evolution reaction (OER) selectivity in acidic seawater electrolysis because of the weak chloride binding on surface. Ni, Co, Fe-based sulfides , phosphides , selenides , and boron-modified cobalt iron layered double hydroxides showed anticorrosion ability in alkaline seawater electrolysis due to the in situ formed repelling anion or polyanion layer. Unfortunately, this also weakened the adsorption of O-intermediates of OER, which might decrease the OER activity. Additionally, at high overpotential (i.e., high current density), the repelling strategy becomes less effective due to the enhanced driving force of adsorption .
Herein, we design an atomic Ir catalyst on cobalt iron layered double hydroxide (Ir/CoFe-LDH) for electrochemical seawater oxidation. Different from conventional catalysts designed to completely repel adsorption for seawater electrolysis, the atomic Ir sites on CoFe-LDH allow adsorption to modulate the electronic structure of
Ir active center. As a result, the exhibits a remarkable OER performance in alkaline seawater electrolysis with an overpotential as low as 202 mV at the current density of lower than that in NaOH electrolyte. Moreover, the affords a remarkable activity at industrial-relevant current densities ( ) with close to oxygen Faradaic efficiency for more than 1000 h . Meanwhile, it can also operate stably in real seawater, reaching with an overpotential of 208 mV , and maintains stable operation for more than 2000 h at a current density of . Both in situ experiments and theoretical analyses show that the dynamic chloride ion adsorption on single Ir atom during OER can effectively reduce the energy barrier to form *OOH (the ratedetermining step of OER) and thus boost water oxidation catalysis, while at the same time maintaining a high energy barrier for the competitive ClOR.
Results and discussion
Synthesis and structural characterization of Ir/CoFe-LDH
Iridium-based catalyst has been widely used in catalyzing water electrolysis and chloralkaline process , in which the Ir-chloride bond strength determines its selectivity . LDH elemental combinations with lower electronegativity may exhibit enhanced electron coupling with metal single atoms . Based on electronegativity values ( ), it is anticipated that noble metal single atoms on CoFe LDHs may demonstrate superior OER performance. In this work, we develop a two-step synthesis to prepare single Ir atom catalysts with tunable single atomic Ir coordination structure on layered double hydroxide (LDH), as shown in Supplementary Fig. 1. In the first step, was synthesized through a co-precipitation method. Subsequently, dilute solutions of and NaOH were added to the homogeneous LDH colloid to anchor atomic Ir onto CoFe-LDH as well as tune the chloride bonding on the single atomic Ir sites. SEM images reveal that both CoFe-LDH and Ir/CoFe-LDH exhibit uniform nanosheet structures, with no noticeable changes in surface morphology upon Ir loading (Supplementary Fig. 2). The Ir in the asprepared catalyst was determined to be by inductively coupled plasma mass spectrometry (ICP-MS). Transmission electron microscopy (TEM) characterization shows clean surface of without formation of nanoparticles/nanoclusters (Supplementary Fig. 3). The high angle annular dark field scanning transmission electron microscope (HADDF-STEM) images clearly show bright spots, which can be assigned to single Ir atoms anchored on the surface of CoFe-LDH (Fig. 1a). Meanwhile, Ir atoms still show isolated bright spots at the same position in the HAADF-STEM images with a tilt (Supplementary Fig. 4), confirming single atomic dispersion of Ir. There seems to be a cluster in the yellow circle in Supplementary Fig. 5a and in the red circle in Supplementary Fig. 5b, however, they both display single atoms at other viewing angles, indicating that the “agglomeration” of Ir single atoms may just be caused by viewing angles. Dr. probe STEM simulation software was used to fit the HAADF-STEM image of Ir/CoFe-LDH, which also suggests single atomic dispersion of Ir with different brightness (Supplementary Figs. 6 and 7). The elemental mapping (Fig. 1b) analysis shows homogeneous distribution of Ir along with Cl on the surface of . Furthermore, X-ray diffraction (XRD) patterns of the as-prepared CoFe-LDH and Ir/CoFe-LDH display the same Bragg reflections in good agreement with the hexagonal-phase LDH (black line, PDF#40-0215, as shown in Fig. 1c), in accordance with the selected area electron diffraction (SAED) patterns (Supplementary Fig. 8).
To further confirm the atomic structure of Ir, in situ DRIFTs measurements of and were performed (Supplementary Fig. 9). After adsorption of CO on , two bands (2080 and ) can be clearly seen in the vibrational frequency region (Supplementary Fig. 9a). These two bands can be assigned to the symmetric (vs) and the anti-symmetric (vas) vibrational
modes of Ir gem-dicarbonyl, , respectively, on the basis of reports of atomically dispersed gem-dicarbonyl supported on and . Of note, dicarbonyl species can only be formed on isolated species , thus confirming the atomic dispersion of Ir in Ir/ CoFe-LDH. In contrast, -LDH exhibits multiple overlapped IR peaks between 2000 and (Supplementary Fig. 9b). The strongest peak at is assigned to the linear binding of CO on Ir clusters, which indicates the existence of Ir clusters. However, this atop peak is not observed in , further providing experimental validation for the atomic dispersion of Ir in Ir/CoFe-LDH (Supplementary Fig. 9c). These results suggest the absence of Ir clusters or nanoparticles in Ir/CoFe-LDH, consistent with the HAADF-STEM results.
X-ray photoelectron spectroscopy (XPS) was performed to examine the surface electronic structure and the influence between the CoFe-LDH and the single atomic Ir (Supplementary Fig. 10). The high-resolution Ir 4f XPS spectrum showed two distinct peaks belonging to Ir and Ir : according to the binding energy of the standard samples of and (Supplementary Fig. 11), the Ir 4f at 61.7 eV and 62.4 eV were identified as the and , respectively (the satellites of and were taken into consideration in fitting the XPS spectrum). The high-resolution Cl 2p XPS spectrum can be deconvoluted into 199.1 eV and 198.45 eV , originating from and on . Comparing the valence states of Co and Fe in and , it was noticed that Co and Fe have obvious shift of binding energy, indicating strong electronic interaction between single atomic Ir and CoFe-LDH.
To further determine the electronic structure and local coordination environment of atomic Ir, X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were performed . The Ir -edge XANES spectrum (Fig. 1d) indicates that the valence state of Ir in is slightly higher than +4 . In the EXAFS spectra (Fig. 1e) as referred to Ir foil, , and IrO2, the Ir/CoFeLDH displays two peaks at 1.62 and å, which can be assigned to IrO and , respectively. Furthermore, the absence of and bonds suggested atomically distributed Ir. The EXAFS fitting data (Fig. 1f, Supplementary Fig. 12 and Supplementary Table 1) showed Ir-O bonds with a coordination number of 3.1 and bonds with a CN of 2.5 without or contribution in Ir/CoFe-LDH. Additionally, there are backscatter contributions of Ir and M (Co and Fe) at with a CN of 2.0 at the second shell.
Electrochemical performance of
The OER performance of the as-prepared Ir/CoFe-LDH was measured in 6.0 M sodium hydroxide aqueous solution using a threeelectrode configuration. Supplementary Fig. 13 displays the scanning electron microscopy (SEM) images of the working electrode. The saturated calomel electrode (SCE) (Supplementary Figs. 14 and 15) reference electrode against RHE scale was directly measured by a three-electrode setup, consisting of 2 Pt wires and 1 reference electrode to be calibrated at saturation of (Supplementary Fig. 16). The cyclic voltammetry (CV) curve (Fig. 2a) reveals that the Ir/CoFe-LDH just requires an overpotential of 236 mV to reach a current density of , which is 141 mV and 535 mV lower than that of CoFe-LDH and commercial , respectively. Different from CoFe -LDH, significant reduction in the redox peaks are observed after loading single atomic Ir (Supplementary Fig. 17). It suggests that the oxidation peak at 1.24 V versus RHE and the reduction peak at 1.17 versus RHE is a redox couple, which could be assigned to the redox of Ir. And the oxidation peak at 1.33 V versus RHE and the reduction peak at 1.07 vs . RHE is a redox couple, which could be assigned to the redox of Co . Interestingly, when NaCl is added into the electrolyte to mimic the accumulate-to-saturated seawater , there shows a 34 mV decrease in overpotential from 236 mV to 202 mV to reach a current density of (Fig. 2b), suggesting that
Fig. 1 | Structural characterizations of Ir/CoFe-LDH. a HAADF-STEM image of Ir/ CoFe-LDH showing single atomic Ir dispersion on CoFe-LDH. b Dark-field TEM image of Ir/CoFe-LDH and the corresponding elemental mappings showing the distribution of , and Ir elements. c XRD patterns of the as-prepared CoFe –
LDH and Ir/CoFe-LDH. d XANES spectra and (e) Fourier-transformed Ir -edge EXAFS spectra of , Ir foil, and -weighted EXAFS spectrum with fitting (dashed line).
presence of NaCl improve the catalytic performance of . In addition, the turnover frequency (TOF) per Ir-site on Ir/CoFe-LDH ( 0.76 ) at the potential of 1.45 V (vs. RHE) in saturated seawater electrolyte is 6.3 times greater than that in NaOH electrolyte ( ) (Fig. 2b). Taking into consideration the potential issue of excessive catalyst loading at , five data points with different catalyst’s loading in the range of were selected to examine the relationship between catalyst’s loading and activity (Supplementary Fig. 18a, b). The findings indicate a linear correlation between catalyst’s loading and performance within range. Beyond , the change in catalyst loading shows minimal effect on catalytic performance (reaching a plateau region). Thus, within the linear region, the CV data associated with a catalyst’s loading of was employed for the precise determination of TOF values. The results
demonstrate that, at a voltage of 1.5 V (vs. RHE), the TOF value per Ir site on in is 7 times higher than that in Supplementary Fig. 18c). Meanwhile, it should be noted that the loading amount of Ir should be carefully tailored; insufficient or over-loading would cause less efficient catalysis (Supplementary Figs. 19-21 and Supplementary Tables 2 and 3), which matches to the characteristic of the reported monatomic catalyst. For selectivity, even in saturated seawater electrolyte, Ir/CoFe-LDH displays a close to OER Faradaic efficiency (Supplementary Fig. 22).
The OER performance of Ir/CoFe-LDH exhibits sensitivity to the electrolyte composition with varying NaOH and NaCl ratios. Notably, an optimized ratio for OER emerges as a significant consideration (Fig. 2c). By altering the NaOH concentration to , 3 M , and 6 M , the OER overpotential at a current density of
Fig. 2 | OER performance. a CV curves of Ir/CoFe-LDH, CoFe-LDH, and IrO recorded in 6 M NaOH (the resistance used for iR calibration is , and , respectively). Comparison of CV curves and TOF of Ir/CoFe-LDH recorded in 6 M NaOH and (the resistance used for iR calibration is 1.55 , and , respectively). c Comparison of OER overpotential at a current density of of -LDH in electrolyte with different ratios in 1 M NaOH . The inset shows the OER overpotential of Ir/CoFe-LDH at a current density of with different ratios in 1 M (white), 2 M (yellow), 3 M (orange), and 6 M NaOH (red). Three measurements were conducted for each data point with the error bars corresponding to the standard deviation. The change in current density and TOF when the electrolyte was switched from 6 M NaOH to 6 M recorded at 1.48 V versus reversible hydrogen electrode (RHE). e The stability test performed in . (All the catalyst massloading was , and the PH of the electrolyte measures 14.78.).
experiences an initial decrease followed by an increase, correlating with the rise in concentration. Remarkably, the optimal OER performance consistently appears at the ratio of (Supplementary Fig. 23), highlighting the critical impact of on OER performance.
In addition, when examined in sole NaOH electrolyte at a voltage of 1.49 V (vs. RHE), the current density of Ir/CoFe-LDH decreased from to in less than 7 h (Fig. 2d). Intriguingly, the incorporation of NaCl into the NaOH electrolyte promptly elevated the current density from to within less than 6 s . Besides, it was noted that exhibited a similar capability to enhance the OER activity of Ir/CoFe-LDH, while conversely, was found to diminish the catalytic activity (Supplementary Fig. 24). In contrast, other noble metal single atoms introduced onto CoFe-LDH (such as Ru/CoFe-LDH or Rh/CoFe-LDH) did not exhibit such pronounced OER activity enhancement through halogen modification (Supplementary Fig. 25), underscoring the distinctive role played by Ir. This distinctiveness might be attributed to the robust or F interaction, as evident from the UV-visible spectra (Supplementary Fig. 26).
At the same time, no significant OER activity enhancement could be observed in NaOH combined with other salt (e.g., ) electrolyte (Supplementary Fig. 27).
To investigate whether would impact the Ir- Cl coordination, we conducted cyclic voltammetry tests on Ir/CoFe-LDH in a solution of . Subsequently, we gradually introduced (the sulfate ion concentration in seawater ) and continued the CV tests. The results indicate virtually sustained performance (within 3 mV ), affirming that does not affect the catalyst’s activity (Supplementary Fig. 28). To demonstrate whether under cyclic conditions would displace coordinated , we conducted a CV test, the results showed no discernible change in performance before and after the CV cycling (Supplementary Fig. 29). Subsequently, more was introduced into the electrolyte, eventually reaching a concentration of (tenfold enrichment of sulfate concentration in real seawater). CV curves indicate that the catalyst’s performance fluctuation is negligible ( ) within the range concentration of (Supplementary Fig. 28). This suggests that after the formation of a stable Ir-Cl coordination state, the
presence of does not exert any discernible influence on the performance.
Besides activity and selectivity, stability is another crucial consideration for practical application . The deactivation of LDH can be attributed to local pH reduction, interlayer acidification, cation dissolution, or oxidation of metal centers to high-valent cations leading to their leaching and in seawater environment, the presence of chloride ions may exacerbate these risks. The stability of LDH was further accessed in solution at current densities ranging from 400 to to meet the industry requirements. The applied potentials showed a negligible increase after 1000 h of continuous reaction (Fig. 2e). Meanwhile, performance in real seawater holds significant implications for the application of seawater electrolysis technology. Ir/CoFeLDH required an overpotential of only 208 mV to achieve a current density of in a seawater electrolyte (Supplementary Fig. 30), indicating its practicability under real industrial conditions. Additionally, CV tests conducted after stability tests at 24,200 , and 400 h confirm the stability of Ir/CoFe-LDH under realistic operating conditions (Supplementary Fig. 31). An electrolyzer with -LDH as an anode and NiCoFeP as a cathode at a current density of in a seawater environment was tested. The results illustrate that the electrolyzer can maintain stable performance under high current density for over 2000 h (Supplementary Fig. 32), confirming the viability of Ir/CoFe-LDH catalyst for real seawater electrolysis.
Furthermore, additional characterizations of post-reaction catalysts were performed to confirm the stability of Ir/CoFe-LDH. SEM after long-term stability test revealed no significant changes in the morphology of the nanosheets (Supplementary Fig. 33), while TEM after long-term stability test showed absence of clusters or particles (Supplementary Fig. 34). HADDF-STEM characterizations (Supplementary Fig. 35) show that the distribution of atomic Ir atoms on CoFe-LDH surface after long-term stability test has no obvious change. There is still no Ir-Ir or Ir-O-Ir signal in the EXAFS data of post-OER catalyst, also revealing isolated dispersion of Ir atoms after long-term OER stability tests. The XRD pattern of the post-OER Ir/ CoFe-LDH (Supplementary Fig. 36) displays the characteristic peak of LDH at about . The XPS measurement of Ir/CoFe-LDH after longterm OER stability test (Supplementary Fig. 37) revealed that the oxidation state of Co and Fe showed no significant change, confirming that Ir-Cl coordination could stabilize CoFe-LDH, preventing oxidation and dissolution of CoFe-LDH, thereby ensuring the stability of CoFe-LDH substrate. Furthermore, XPS quantitative analysis indicated a negligible change of the surface Ir concentration before and after OER. The dissolved Ir in the electrolyte after OER stability test was also quantified by inductively coupled plasma optical emission spectroscopy (ICP-MS, 9.558 ppb ), which is nearly nine times less than the physical mixture of and CoFe -LDH ( 82.308 ppb ), as shown in Supplementary Table 4. This experimental evidence further substantiates the stability of Ir loading on CoFeLDH. Ir stabilizes CoFe through controllable Ir-Cl coordination, suppressing CoFe oxidation, in the meanwhile CoFe also stabilizes Ir through strong electronic interaction. All of the above characterizations together with the electrochemical data suggest good stability of Ir/CoFe-LDH in seawater electrolysis. Additionally, it should be noted that at current densities of 600 and , the overpotential was thermodynamically high enough to trigger the chloride oxidation reaction (thermodynamic potential: versus versus RHE at 2.8 M NaCl ), but no chlorine or hypochlorite was detected (with oxygen Faradaic efficiency > 99.9935% as shown in Supplementary Figs. 38-40), suggesting a high OER selectivity for in high concentration NaCl electrolyte.
In situ XAS and Raman characterization of Ir/CoFe-LDH
To explore why and how can boost the OER activity of Ir/CoFe-LDH, operando EXAFS at the Ir -edge was performed and compared in (Fig. 3a) and NaOH (Fig. 3b) electrolyte. The local coordination of the two Ir/CoFe-LDH samples used for operando EXAFS are nearly the same, implying that the differences of EXAFS spectra in and NaOH electrolyte arise from the catalyst’s structure evolution during OER (Supplementary Fig. 41 and Supplementary Table 5). Subsequently, under open circuit voltage (OCV), the coordination of both catalysts remains nearly unaltered (Supplementary Fig. 42 and Supplementary Table 6). However, at a potential of 1.57 V versus RHE, only Ir-O coordination was evident in the EXAFS spectrum of Ir/CoFe-LDH immersed in NaOH (Supplementary Fig. 43 and Supplementary Table 7), suggesting that formed a competitive adsorption for coordination. In contrast, both and could be observed at the same applied potential in electrolyte. This observation can be attributed to the substantial concentration, allowing for the persistence of coordination. The above results suggest a competitive binding between and with single atomic Ir sites on CoFe-LDH during OER in the seawater environment, and the dynamic stability of coordination in seawater may stand as a key factor driving the OER performance enhancement. Concurrently, a distinctive peak at was detected in the EXAFS spectra, which could be attributed to the close proximity of Ir and or Fe due to the oxidation of CoFe-LDH surface to MOOH during the OER process.
In situ Raman spectroscopy was further performed on LDH to obtain mechanistic insight into the influence of Cl adsorption over single Ir atomic site on the OER activity. Compared to CoFe-LDH (Supplementary Fig. 44), the Raman spectra of Ir/CoFe-LDH at OCV showed a new peak at around (Fig. 3c-f). This peak can be assigned to vibration according to the standard sample , as shown in Supplementary Fig. 45.
Moreover, In environment (Fig. 3c, d), the presence of Ir-Cl coordination at was consistently observed. In contrast, in alkaline electrolytes without Cl addition, coordination was only observed under open circuit voltage (OCV) condition, and its presence ceased upon application of voltage (Fig. 3e, f), indicating its dynamic nature. This phenomenon suggests that under high concentration, there is a pronounced competition for adsorption between and , necessitating a high NaCl concentration to uphold the stability of the coordination. This observation is consistent with our XAS data, where the coordination between Ir and Cl remains intact in high salt concentration environment, whereas it dissipates in an -rich environment. Consequently, it becomes essential to maintain a relatively high salt concentration environment to uphold the coordination, thereby facilitating the kinetics of OER.
Theoretical investigation. We performed computational studies aimed at identifying the effects of modulation of the local iridium coordination environment and the resulting impacts on the electronic structure of iridium center as well as the energetics of . Based on the EXAFS fitting results and the characterization of catalyst after OER, the catalyst was found to undergo reconstruction during OER and the real active structure is the single-atom iridium bound on the surface of oxyhydroxide (i.e., ), and the reconstruction of hydroxide to oxyhydroxide during OER has been observed in the literature . Hence, we built our simulation models as shown in Supplementary Fig. 46. Penta-coordinated Ir with three states: Ir coordinated with two , with one Cl and one , and with two Cl ( ) anchored on the surface of two-layer periodic LDH, were constructed as the simulation models. For pure CoFeOOH , unsaturated Fe sites in the (100) surface were used as the active sites. Thermodynamic analyses (Fig. 4a and Supplementary Fig. 47) suggest that the formation of * OOH intermediate is the rate-determining step
Fig. 3 | In situ XAS and Raman measurement. a In situ Fourier-transformed EXAFS spectra of Ir/CoFe-LDH in (a) and (b) NaOH . In situ Raman spectra of Ir/ recorded in (c), (d) , and (e), (f) NaOH . The blue-highlighted portions correspond to the Ir-Cl coordination.
(RDS) of OER on Fe sites in CoFeOOH and Ir centers in Ir/CoFeOOH. The single Ir atom with 2 OHs ( ) could lower the * oxidation free energy from 2.24 eV to 1.73 eV . Mono chloride coordination with ) could further lower this free energy to 1.51 eV in preference to and meanwhile enhance the *OH adsorption, which is the key step competing with chloride adsorption and oxidation. However, further coordination with another chloride ion to form two bonds would increase the free energy of oxidation to 1.64 eV . The activity sequence of the coordinated Ir species follows . Figure 4b displays the two-dimensional (2D) map of theoretical OER onset overpotential ( ) for single atomic Ir anchored on CoFeOOH with different coordination states by assuming the scaling relationship of (Supplementary Fig. 47c). Other halogens like and could also affect the adsorption energy of *O and *OH as well as the overpotential of OER, but was not as efficient as (Supplementary Fig. 47b) in accordance with the experimental results.
To further understand the underlying OER mechanism and the role of Cl , projected crystal orbital Hamiltonian population (pCOHP)
analysis between the Ir centers and the O atoms in the adsorbed *OOH were calculated to give information on OOH intermediates adsorption . We calculated the integrated COHP (ICOHP) up to the highest occupied bands (below the ), which directly gave quantitative information on the bonding states. In general, a negative -COHP indicates an antibonding state and a positive -COHP indicates a bonding state . As shown in Fig. 4c, the lower ICOHP value ( -4.60 eV ) demonstrates that the molecular orbitals of adsorbed OOH interact much stronger with the Ir bands in than in , and , indicating stronger OOH adsorption on . These results suggest that the *OOH formation, which is the RDS of OER, is easier to take place on the center, thereby promoting the OER kinetics.
To get experimental evidence on the formation of *OOH intermediate, electron paramagnetic resonance (EPR) spectroscopy test was performed using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as an *OOH free radical trapping agent . As shown in Fig. 4d, the signal originating from *OOH recorded in electrolyte is nearly twice as much as that recorded in NaOH electrolyte, matching well with
Fig. 4 | Theoretical investigation. a The activation energy of Ir/CoFe-LDH with three coordination states. b 2D map of theoretical OER onset overpotential ( ), the contour map is constructed by assuming the scaling relationship of 3.17. c Projected crystal orbital Hamiltonian population (pCOHP) between the Ir center and the O atom in *OOH. d EPR detection of DMPO-OOH. The sample was taken after 10 min of electrolysis at 1.42 V versus RHE.
the COHP data. The enhanced adsorption of OER intermediates can be further deduced from the smaller slope of the open circuit potential (OCP) decay collected in electrolyte as compared to that collected in pure NaOH electrolyte, as shown in Supplementary Fig. 48.
To further verify the specific role of , we conducted calculations of the Density of States (DOS) and Projected Density of States (PDOS) for Ir-Cl/CoFe-LDH and Cl/CoFe-LDH. The results indicate that Cl hybridizes with the conduction bands of Co and Fe , but there is no hybridization with the valence bands (Supplementary Fig. 49b). Upon loading a Ir single-atom, Ir-Cl exhibits significantly stronger hybridization with both the valence and conduction bands (Supplementary Fig. 49a), highlighting the enhanced orbital hybridization and bonding capability of Ir-Cl. The total density of states (TDOS) data aligns with the partial density of states (PDOS) data, demonstrating a pronounced increase in valence band hybridization in the presence of surface Ir, in contrast to case where the valence bands show no interaction (Supplementary Fig. 50).
To rationalize the improved OER performance and explore the role of chloridion, kinetic characteristics of in NaCl and NaOH electrolyte were systematically studied . The activation energy for OER over the Ir/CoFe-LDH catalyst in electrolyte was estimated to be using the Arrhenius equation, much lower than that in NaOH electrolyte ( ), verifying the much-improved OER kinetics by adsorption (Supplementary Figs. 51 and 52). On the other hand, CIOR displays remarkably high theoretical overpotentials (Supplementary Fig. 53), offering a compelling rationale for the enduring near 100% OER selectivity of Ir/ CoFe-LDH at industrial current densities ( ) in seawater electrolysis.
To further demonstrate the stability of Ir/CoFe-LDH, we selected Cl adsorption sites on Co or Fe , respectively. The results indicate that, in the presence of cannot effectively adsorb onto Co (Supplementary Movie 1) or Fe (Supplementary Movie 2), respectively
(Supplementary Fig. 54). The optimization process revealed direct Cl adsorption onto Ir sites, eliminating the existence of stable configurations like or (Supplementary Figs. 55 and 56). Consequently, these two active sites do not exist at presence of Ir. This finding further confirms the significant improvement in the electrochemical stability of CoFe LDH upon the introduction of Ir. The strong Ir-Cl coordination stabilizes the CoFe sites, inhibiting Cl coordination with both metal sites, thus ensuring system stability in seawater electrolysis.
In summary, we have devised an ingenious strategy involving the deposition of single-atom Ir onto CoFe-LDH and leveraging abundant ions within the seawater milieu to dynamically regulate the coordination state of Ir single-atom catalyst. This orchestration has endowed Ir/CoFe-LDH catalyst with greatly enhanced OER reactivity in seawater, surpassing its performance in alkaline environment. The confluence of operando characterizations and DFT calculations substantiates that the amplified OER performance of Ir/CoFe-LDH in seawater originates from the dynamic regulation of the Cl and OH coordination states on Ir. This chloride-mediated coordination augmentation facilitates robust adsorption of *OOH intermediates in OER, thereby reducing the activation energy barrier for the ratedetermining formation by a factor of 1.93 and significantly increasing the cathodic interfacial electron transfer rate (CIER). Impressively, Ir/CoFe-LDH exhibits a remarkable operational stability at industrial current densities ( ) in seawater, maintaining uninterrupted activity for over 1000 h while retaining a remarkable selectivity. This study not only unveils the immense potential of Ir/CoFe-LDH in seawater electrolysis, but also presents a novel strategy for regulating single-atom Cl coordination in seawater environments, a breakthrough that holds the promise of minimizing energy consumption and costs associated with seawater electrolysis, thereby propelling the practical deployment of this technology.
Methods
Materials
Iron(III) nitrate nine-hydrate , cobalt(II) nitrate hexahydrate , iridium chloride hydrate ( ), potassium chloride ( ) were purchased from Sigma-Aldrich. Sodium hydroxide ( ), sodium chloride ( ), sodium carbonate ( ) were purchased from Fuchen Chemical Reagent Co., Ltd. Ethanol ( ) was purchased from Tianjin Fuyu Fine Chemical Co., Ltd. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was purchased from Dojindo China Beijing Co., Ltd. Carbon fiber paper (Toray 060) was purchased from Suzhou Sinero Technology Co., Ltd. Seawater was taken from the Yellow Sea, China. Deionized (DI) water (resistivity: ) was used for the preparation of all aqueous solutions.
Preparation of CoFe-LDH
CoFe-LDH was synthesized via a co-precipitation method. In brief, 40 ml solution A: and and 40 ml solution B: and were firstly prepared. Solution A and B were added dropwisely at the same time into a beaker filled with 40 ml of deionized water. The pH value of the final suspension was adjusted to 8.5 under magnetic stirring, as monitored using a pH meter at room temperature . The pH is well controlled since salt solution A and alkali solution B was added at the same time, and the ratio of these two solutions could be adjusted to meet the targeting pH . The resulted suspension was aged for 12 h at . Subsequently, the precipitate was collected by centrifugation, and washed 3 times using DI water, followed by ethanol wash for 3 times. The final precipitates were dried in a vacuum oven at overnight.
Preparation of /CoFe-LDH
The as-prepared was added to 50 ml DI water under magnetic stirring to form a colloid suspension. Then, 10 ml freshly made aqueous solution ( 5 mg ) containing 0.02 M NaOH was dropwisely added to the above CoFe-LDH colloid suspension. The suspension was stirred with simultaneous heating at for 6 h . Afterwards, the precipitate was collected by centrifugation, washed by DI water and ethanol, each for 3 times. Then, the final precipitates were dried in a vacuum oven at overnight.
Preparation of -LDH
The as-prepared was added to 50 ml DI water under magnetic stirring to form a colloid suspension. Then, 10 ml freshly made aqueous solution ( 10 mg ) containing 0.02 M NaOH was dropwisely added to the above CoFe-LDH colloid suspension. The suspension was stirred with simultaneous heating at for 6 h . Afterwards, the precipitate was collected by centrifugation, washed by DI water and ethanol each for 3 times. Then, the final precipitates were dried in a vacuum oven at overnight.
HAADF-STEM characterization and simulation
Before imaging, the as-prepared catalysts were added into anhydrous ethanol by using an ultrasonator to form a very dilute colloid suspension, then suspension was dripped onto 230 mesh Cu grids coated with ultrathin carbon. The high-resolution HAADF-STEM image was acquired using a Thermo Fisher Spectra 300 microscope equipped with an aberration corrector for the probe-forming lens, operated at 300 kV . The beam current was lower than 40 pA and the STEM convergence semi-angle was , which provided a probe size of at 300 kV . The HADDF-STEM images with a tilt of the sample were taken by tilting double tilt holder , which can be moved in and directions. The HADDF-STEM image simulations were carried out using Dr. Probe software , the parameters were set as
same as the experimental condition. The accuracy of simulation results was .
Characterization instruments
Transmission electron microscopy measurement was carried out on a JEOLJEM 2100. X-ray powder diffraction (XRD) patterns were recorded on an X-ray diffractometer (Rigaku D/max 2500) with radiation ( ) at a scan rate of in the range from 3 to . X-ray photoelectron spectroscopy (XPS) spectra were recorded on an ESCALAB 250 (Thermo Fisher Scientific USA) photoelectron spectrometer using monochromate Al K W X-ray beam. All binding energies were referenced to the C 1s peak ( 284.8 eV ). Specifically, by evaluating the deviation of the binding energy position corresponding to the C 1s peak at 284.8 eV , the obtained difference is then utilized to carry out a comprehensive calibration of the entire dataset. ICP-MS measurement was performed on a Thermo X Series II ICP/MS quadrupole system, Thermo Fisher Scientific to quantify the chemical composition of the catalyst EPR spectroscopy measurement was performed on a Magnettech MS-5000X using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the radical trap.
X-ray absorption spectroscopy
The measurements were performed in a typical three-electrode setup in a specially designed Teflon container with a window sealed by Kepton tape. The testing conditions were the same as in the electrochemical characterization case. The measurements were performed at BL-17C at the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan). In situ data were collected in totalfluorescence mode using a silicon drift detector.
XANES and EXAFS data analysis
The X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) data were analyzed by the software Athena of the IFEFFIT package. The EXAFS spectra were analyzed through post-edge background subtraction from the overall absorption and normalized with respect to the edge-jump step by the software Artemis following previously reported data fitting methodology .
In situ Raman test
In situ Raman spectra were collected on LabRAM ARAMIS (Horiba Jobin Yvon, France) with a PSU-H-FDA 532 nm laser source (Changchun New Industries Optoelectronics Tech. Co. Ltd, China). An LMPlanFLN microscope objective lens with a numerical aperture of 0.5 (OLYMPUS, Japan) was used for Raman microscopy. Raman frequency was calibrated by a Si wafer ( ) during each experiment. In situ electrochemical Raman experiments were performed in an in situ Raman cell (Tianjin Aida Hengsheng Technology Development Co., Ltd, China) and the prepared catalyst, , and Pt wire were employed as the working, reference, and counter electrode, respectively. A CHI 660e (Shanghai Chenhua Instrument Co., Ltd, China) electrochemical workstation was used to control the potential where the applied potentials were increased step by step from open circuit potential (OCP) to 1.57 V vs. RHE. Each spectrum was obtained at least three times with an exposure time of 30 s .
In situ DRIFTS experiments
In situ DRIFTS was used to characterize the interaction of the catalyst with CO. The in situ DRIFTS experiments were performed on a Bruker INVENIO R Fourier transform infrared spectrometer equipped with an MCT/A detector cooled by liquid nitrogen and a Harrick diffusereflectance attachment. Approximately 50 mg catalyst was loaded in the Harrick Praying Mantis high-temperature reaction chamber equipped with KBr windows (HVC-DRP-5). The chamber was sealed and gases were flown through at atmospheric pressure. High-purity Ar
was used as the purging gas, and dilute CO ( CO-certified grade) was used as the probe gas. The temperature was controlled by a thermocouple in direct contact with the sample. Circulating water was used to cool the body of the reaction chamber (set temperature: ). All of the in situ characterizations followed the same pretreatment procedure, each reported spectrum is an average of 64 scans. A spectral resolution of was used to collect the spectra, which are reported in Kubelka-Munk units. For Ir/CoFe-LDH, the sample was calcined in Ar ( ramp rate) at ramp rate) for 30 min to remove physically adsorbed . The temperature was then cooled in Ar to with circulating water and the background spectrum was recorded. The sample was then labeled in CO-certified grade, ramp rate) at for 0.5 h . Afterwards, , ramp rate) was purged at for 30 min to remove the physical adsorbed CO until there was no change observed in the spectra. The DRIFTS spectra were recorded in the wavenumber range from 1000 to at 298 K with a total of 1000 measurements taken, each separated by a interval.
Electrochemical measurements
All electrochemical measurements were performed on a threeelectrode setup using a CHI 660 e electrochemical workstation. A platinum wire and a SCE double salt bridge electrode were served as the counter electrode and the reference electrode, respectively. The SCE double salt bridge, which was purchased from Tianjin Aida Hengsheng Technology Development Co., Ltd, consists of saturated KCl . The working electrode was prepared by the following steps: (1) Preparation of catalyst ink. 2 mg of the as-synthesized catalyst, 1 mg of carbon black, and of Nafion solution were dispersed in ethanol and deionized water under ultrasonication for at least 1 h to form a homogeneous catalyst ink. (2) Coating the catalyst ink onto carbon fiber paper ( , Toray, TGP-H-060): the catalyst ink was drop-casted onto carbon fiber paper ( , Toray, TGP- each time with drying under an infrared lamp, followed by dropping another ink until the catalyst mass-loading of the catalyst reached . Cyclic voltammetry (CV) was measured from 0 to 1.0 V versus SCE at a scan rate of . The electrochemical impedance spectroscopy (EIS) was measured by applying an AC voltage of 5 mV at the overpotential of 10 mV with a frequency from 100 kHz to 0.1 Hz . All polarization curves were corrected for Ohmicdrop compensation with Ohmic resistance obtained by the EIS. The SCE reference electrode against RHE scale was directly measured by a three-electrode setup, consisting of 2 Pt wires and 1 reference electrode to be calibrated. The open circuit potential (OCP) was measured under the condition of purging high-purity gas into the electrolyte. An OCP was applied after saturation of gas, obtaining a stable potential. This potential is the potential difference between RHE and the reference electrode. In the choice of electrolyte for testing, the use of 6 M NaOH mirrors the alkalinity levels typically employed in industrial alkaline water electrolysis. Moreover, according to our previous work , it is notable that the maximal solubility of NaCl in 6 M NaOH is 2.8 M , as excess NaCl concentration beyond this threshold results in crystalline precipitation. Importantly, in practical scenarios, electrolyte salt concentrations are unlikely to exceed this limit. By conducting tests under these conditions, we aim to showcase the electrode’s performance at its utmost capacity.
Measurement of activation energy ( )
The activation energy of OER is an indicator of reaction kinetics, which is only dependent on the material. The Eq. (1) is Arrhenius equation, where is the preexponential factor, is the ideal gas constant, is the temperature, and is a rate constant, which can be applied to determine the activation energy of OER on the catalysts. At a fixed overpotential, the kinetic current has a linear relationship with a rate
constant ( ), as shown in Eq. (2), where is Faraday constant, is the number of transferred electrons, is the electrode area and is concentration. If we assume that mass transport effects are negligible on the OER currents, the can be expressed as Eq. (3), where is the activation barrier at a constant potential. The kinetic activation energy at a given voltage can be expressed as Eq. (4). Hence, the activation energy for OER can be defined by Eq. (5):
Theoretical methods
All theoretical calculations were performed using the projectoraugmented wave method and a plane-wave basis set as implementation in the Vienna Ab Initio Simulation Package (VASP). The bulk and surface properties of CoFeOOH were optimized within GGA-PBE. Full optimization of all atom positions in the bulk was performed via the action of a conjugated gradient optimization procedure. The Monkhorst-Pack -point samplings were set as for the geometry optimization, and for the computation of electronic structure. And the bulk constants were optimized using the Monkhorst-Pack -point sampling. The cutoff energy for plane-wave basis functions was set to 600 eV with the energy change convergence criterion of . Atomic positions were allowed to relax until the sum of the absolute forces reached down to . Hubbard-U correction method was applied to improve the description of localized Co and -electrons in the CoFeOOH with and for Fe and Co , respectively. The spin polarization, long-range van der Waals interaction (IVDW = 11), and solvent corrections were also included in surface calculations. The solvent effect on adsorbates was considered using the Poisson-Boltzmann implicit solvation model with a dielectric constant of 78.4. The Gibb’s free energies were calculated by:
where the symbols represent the binding energy ( ), the change in zero-point energy ( ), temperature ( ), and the entropy change of the system, respectively. The crystal orbital Hamiltonian population (COHP) was calculated by LOBSTER, and the convergence threshold for the iteration in self-consistent field was set at .
Data availability
Source data are provided with this paper.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294-303 (2012).
Zhu, J. et al. Hydrogen evolution using nanoparticles. Chem. Rev. 120, 851-918 (2020).
Bockris, J. O. A hydrogen economy. Science 176, 1323 (1972).
Mitchell, S. et al. Nanoscale engineering of catalytic materials for sustainable technologies. Nat. Nanotechnol. 16, 129-139 (2021).
Peter, S. et al. Electrolysis of low-grade and saline surface water. Nat. Energy 5, 367-377 (2020).
Wang, C. et al. Advances in hydrogen production from electrocatalytic seawater splitting. Nanoscale 17, 7897-7912 (2021).
Li, P. et al. Common-ion effect triggered highly sustained seawater electrolysis with additional NaCl production. Research 2020, 2872141 (2020).
Peter, S. et al. Design criteria, operating conditions, and nickel-iron hydroxide catalyst materials for selective seawater electrolysis. ChemSusChem 9, 962-972 (2016).
Rasmus, K. & Ann, C. Selectivity between oxygen and chlorine evolution in the chlor-alkali and chlorate processes. Chem. Rev. 116, 2982-3028 (2016).
Shao, H. et al. An earth-abundant catalyst-based seawater photoelectrolysis system with 17.9% solar-to-hydrogen efficiency. Adv. Mater. 30, 1707261 (2018).
Xie, H. et al. A membrane-based seawater electrolyser for hydrogen generation. Nature 612, 673-678 (2022).
Kuang, Y. et al. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl Acad. Sci. 116, 6624-6629 (2019).
Wu, L. Heterogeneous bimetallic phosphide as an efficient bifunctional catalyst for water/seawater splitting. Adv. Funct. Mater. 31, 2006484 (2021).
Marc, K. et al. as selective oxygen evolution electrocatalyst in acidic chloride solution. J. Am. Chem. Soc. 140, 10270-10281 (2018).
Yu, L. et al. Ultrafast room-temperature synthesis of porous S-doped Ni/Fe (oxy)hydroxide electrodes for oxygen evolution catalysis in seawater splitting. Energy Environ. Sci. 13, 3439 (2020).
Song, Z. et al. High-performance ammonium cobalt phosphate nanosheet electrocatalyst for alkaline saline water oxidation. Adv. Sci. 8, 2100498 (2021).
Zhao, Y. et al. Charge state manipulation of cobalt selenide catalyst for overall seawater electrolysis. Adv. Energy Mater. 29, 1801926 (2018).
Wu, L. et al. Boron-modified cobalt iron layered double hydroxides for high efficiency seawater oxidation. Nano Energy 83, 105838 (2021).
Mohammad, M. et al. Manganese compounds as water-oxidizing catalysts: from the natural water-oxidizing complex to nanosized manganese oxide structures. Chem. Rev. 116, 2886-2936 (2016).
Zenta, K. et al. Durability enhancement and degradation of oxygen evolution anodes in seawater electrolysis for hydrogen production. Appl. Surf. Sci. 257, 8230-8236 (2011).
Takuya, O. et al. A bilayer structure composed of deposited on a film to realize selective oxygen evolution from chloride-containing water. Langmuir 36, 5227-5235 (2020).
Oscar, D. et al. Iridium-based double perovskites for efficient water oxidation in acid media. Nat. Commun. 7, 12363 (2016).
Kim, Y. et al. Balancing activity, stability and conductivity of nanoporous core-shell iridium/iridium oxide oxygen evolution catalysts. Nat. Commun. 8, 1449 (2017).
Zhang, Y. et al. Atomic iridium incorporated in cobalt hydroxide for efficient oxygen evolution catalysis in neutral electrolyte. Adv. Mater. 30, 1707522 (2018).
Cai, C. et al. Ultrahigh oxygen evolution reaction activity achieved using Ir single atoms on amorphous nanosheets. ACS Catal. 11, 123-130 (2021).
Maximilian, M. et al. HCl oxidation on -based catalysts: from fundamentals to scale-up. ACS Catal. 3, 2813-2822 (2013).
Ricardo, E. et al. Heterogeneous model to distinguish the activity of electrogenerated chlorine species from soluble chlorine in an
electrochemical reactor. Ind. Eng. Chem. Res. 58, 22399-22407 (2019).
Zeradjanin, A. et al. On the Faradaic selectivity and the role of surface inhomogeneity during the chlorine evolution reaction on ternary Ti-Ru-Ir mixed metal oxide electrocatalysts. Phys. Chem. Chem. Phys. 16, 13741 (2014).
Johannes, G. et al. Selectivity trends between oxygen evolution and chlorine evolution on iridium-based double perovskites in acidic media. ACS Catal. 9, 8561-8574 (2019).
Rossmeisl, J. et al. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83-89 (2007).
Hansen, A. et al. Electrochemical chlorine evolution at rutile oxide (110) surfaces. Phys. Chem. Chem. Phys. 12, 283-290 (2010).
Li, P. et al. Boosting oxygen evolution of single-atomic ruthenium through electronic coupling with cobalt-iron layered double hydroxides. Nat. Commun. 10, 1711 (2019).
Barthel, J. Dr. Probe: a software for high-resolution STEM image simulation. Ultramicroscopy 193, 1-11 (2018).
Hernández-Cristóbal, O. et al. Effect of the reduction temperature on the activity and selectivity of titania-supported iridium nanoparticles for methylcyclopentane reaction. Ind. Eng. Chem. Res. 53, 10097-10104 (2014).
Shan, J. et al. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 551, 605-608 (2017).
Freakley, S. J. et al. The X-ray photoelectron spectra of Ir, IrO2 and IrCl3 revisited. Surf. Interface Anal. 49, 794-799 (2017).
Morgan, D. J. Resolving ruthenium: XPS studies of common ruthenium materials. Surf. Interface Anal. 47, 1072-1079 (2015).
Jiang, K. et al. Dynamic active-site generation of atomic iridium stabilized on nanoporous metal phosphides for water oxidation. Nat. Commun. 11, 2701 (2020).
Li, Z. et al. Iridium single-atom catalyst on nitrogen-doped carbon for formic acid oxidation synthesized using a general host-guest strategy. Nat. Chem. 12,, 764-772 (2020).
Sören, D. et al. Direct electrolytic splitting of seawater: opportunities and challenges. ACS Energy Lett. 4, 933-942 (2019).
Wang, G. et al. SC-IrO2NR-carbon hybrid: a catalyst with high electrochemical stability for oxygen reduction. Sci. China Mater. 56, 131-136 (2013).
Dao, J. et al. Layered double hydroxide-based electrocatalysts for the oxygen evolution reaction: identification and tailoring of active sites, and superaerophobic nanoarray electrode assembly. Chem. Soc. Rev. 50, 8790-8817 (2021).
Rossmeisl, J. et al. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 319, 178-184 (2005).
Michal, B. et al. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521-13530 (2013).
Zhang, B. et al. Homogeneously dispersed multimetal oxygenevolving catalysts. Science 352, 333-337 (2016).
Mefford, J. T. Correlative operando microscopy of oxygen evolution electrocatalysts. Nature 593, 67-73 (2021).
Zhang, J. Single-atom layered double hydroxide electrocatalyst: probing the origin of activity for oxygen evolution reaction. J. Am. Chem. Soc. 140, 3876-3879 (2018).
Richard, D. et al. Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 8617-8624 (1993).
Hoffman, R. Solids and Surfaces: A Chemist’s View of Bonding in Extended Structures (Wiley-VCH, 1988).
Liu, X. et al. Building up a picture of the electrocatalytic nitrogen reduction activity of transition metal single-atom catalysts. J. Am. Chem. Soc. 141, 9664-9672 (2019).
Lin, L. et al. A highly CO-tolerant atomically dispersed Pt catalyst for chemoselective hydrogenation. Nat. Nanotechnol. 14, 354-361 (2019).
Williamson, N. et al. Equilibrating (L) and species in hydrocarbon oxidations by bio-inspired nonheme iron catalysts using and AcOH. J. Am. Chem. Soc. 139, 17313-17326 (2017).
Song, H. et al. Direct and selective photocatalytic oxidation of to oxygenates with on cocatalysts/ ZnO at room temperature in water. J. Am. Chem. Soc. 141, 20507-20515 (2019).
Wang, N. et al. Hydration-effect-promoting Ni-Fe oxyhydroxide catalysts for neutral water oxidation. Adv. Mater. 32, 1906806 (2020).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537-541 (2005).
Liu, R. et al. Design of aligned porous carbon films with single-atom Co-N-C sites for high-current-density hydrogen generation. Adv. Mater. 33, 210533 (2021).
Zhao, S. et al. Structural transformation of highly active metal-organic framework electrocatalysts during the oxygen evolution reaction. Nat. Energy 5, 881-890 (2020).
Acknowledgements
The authors thank the help from Dr. Cejun Hu for the help on characterization. X.S. and Y.K. acknowledge financial support from the National Key Research and Development Project (2021YFA1502200), the National Natural Science Foundation of China (21935001), Beijing Natural Science Foundation (Z210016), a long-term subsidy from China’s Ministry of Finance and the Ministry of Education. B.L. acknowledges financial support from the City University of Hong Kong start up fund (9020003) and ITF-RTH-Global STEM Professorship (9446006).
Author contributions
X.S., B.L., and Y.K. supervised the project. X.D. conceived the idea and carried out the experiments. X.D., Q.S., P.L., and D.Z. conducted material synthesis and electrochemical measurements. Q.S. conducted the in situ DRIFTs and in situ Raman measurements. T.L., G.Y., and W.L. help with the material characterization. W.C., Y.C., and L.Z. conducted the in situ XAS measurements. X.D., Y.L. and E.Y. performed the DFT calculations. H.Y., G.Z., and W.L. assisted with the data analysis. X.S., B.L., Y.K., X.D., and Q.S. wrote the paper. S.H. conducted the in situ XAS measurements in the revised version. J.L. and J.D. analyzed the data of
XAS in the revised version. Z.W. and H.X. help with the DFT simulation. J.F. and Z.Z. conducted the ATR-SEIRAS measurements in the revised version. C.J., J.L., and L.B. conducted the HADDF-STEM measurements and simulations. All authors discussed the results and assisted the manuscript preparation.
Correspondence and requests for materials should be addressed to Yun Kuang, Bin Liu or Xiaoming Sun.
Peer review information Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024 State Key Laboratory of Chemical Resource Engineering, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, College of Chemistry, Beijing University of Chemical Technology, Beijing 100029, PR China. School of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, Singapore 637459, Singapore. Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, PR China. Ocean Hydrogen Energy R&D Center, Research Institute of Tsinghua University in Shenzhen, Shenzhen 518057, PR China. State Key Lab of Organic-Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology, 100029 Beijing, PR China. Energy & Catalysis Center, School of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, PR China. Hefei National Research Center for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei 230026, PR China. Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, 100049 Beijing, PR China. Department of Chemistry, Tsinghua University, 100084 Beijing, PR China. Department of Materials Science and Engineering, City University of Hong Kong, Hong Kong SAR 999077, PR China. College of Energy, Shandong University of Science and Technology, Tsingtao 266590, PR China. Beijing Key Laboratory of Energy Environmental Catalysis, Beijing University of Chemical Technology, 100029 Beijing, PR China. Department of Applied Chemistry and Center for Emergent Functional Matter Science, National Yang Ming Chiao Tung University, Hsinchu 300, Taiwan. School of Materials Science and Engineering, Tianjin Key Lab of Photoelectric Materials & Devices, Tianjin University of Technology, Tianjin 300384, PR China. ShenSi Lab, Shenzhen Institute for Advanced Study, University of Electronic Science and Technology of China, Shenzhen 518110, PR China. CAS Key Laboratory of Standardization and Measurement for Nanotechnology, National Center for Nanoscience and Technology, 100190 Beijing, PR China. Department of Chemistry & Center of Super-Diamond and Advanced Films (COSDAF), City University of Hong Kong, Hong Kong SAR 999077, PR China. These authors contributed equally: Xinxuan Duan, Qihao Sha. e-mail: kuangy@tsinghua-sz.org; bliu48@cityu.edu.hk; sunxm@mail.buct.edu.cn