حجم وعيوب المواد غير العضوية البلورية لها أهمية في العديد من التطبيقات، وخاصة في التحفيز، حيث تؤدي غالبًا إلى تحسين/ظهور خصائص جديدة. حتى الآن، لم تتمكن استراتيجية كيمياء التعديل من توفير بلورات نانوية عالية الجودة من الإطارات المعدنية العضوية (MOFs) بحجم مصغر مع عرض عيوب متزايدة. نحن نبلغ هنا عن استراتيجية عامة مستدامة لتصميم MOFs رباعية التكافؤ ذات العيوب العالية والصغيرة جدًا.بلورات (حواليرابط مفقود،تم إجراء توصيفات متقدمة لتسليط الضوء على العوامل الرئيسية التي تحكم آلية التبلور ولتحديد طبيعة العيوب. أظهرت النانوMOFs فائقة الصغر أداءً استثنائيًا في تفاعل تحلل الببتيد، بما في ذلك التفاعل العالي، الانتقائية، الانتشار، الاستقرار، وتظهر تفاعلية وانتقائية قابلة للتعديل تجاه تكوين روابط الببتيد ببساطة عن طريق تغيير مذيب التفاعل. لذلك، تفتح هذه الجسيمات فائقة الصغر ذات العيوب العالية من M(IV)-MOFs آفاقًا جديدة لتطوير محفزات MOF غير المتجانسة ذات الوظائف المزدوجة.
على مدى العقود القليلة الماضية، أدى تطوير البلورات النانوية الغروية إلى ثورة في علم المواد بسبب خصائصها الجذابة للغاية في التحفيز غير المتجانس، والبصريات، وعلم الأحياء، والهندسة.. في الواقع، تخضع معظم المواد النانوية لتغيرات دراماتيكية في خصائصها عندما يكون حجم جزيئاتها في النطاق الصغير جداً (على سبيل المثال، أقل منمثل تأثير حجم الكم في مواد أشباه الموصلاتخصائص تحفيزية للمعادن النبيلة الخاملةأو الموصلية الكهربائية للعوازلبلورات نانوية من الإطارات المعدنية العضوية (nanoMOFs) هي مواد مسامية تتكون من أيونات معدنية/كتل أكسيدية وروابط عضوية.لقد منح تقليل حجم MOFs إلى النانو مقياسًا لـ nanoMOFs خصائص محسّنة متنوعة (مثل: التحفيز، الاستشعار، الطب الحيوي…)وميزات جديدة (المرونة، الخصائص البصرية)ولكن على الرغم من التقدم، لا يزال تصميم جزيئات MOF النانوية فائقة الصغر يواجه صعوبات شديدة. يمكن أن يكون ذلك إما بسبب المعلمات الأكبر بكثير لوحدات الخلية لمواد الإطار العضوي المعدنية مقارنة بالمواد النانوية غير العضوية و/أو بسبب المحدود الاستقرار الكيميائي لـ nanoMOFs المستهدفة. ومع ذلك، في هذا الحجم الصغير للغاية، تقع الغالبية العظمى من ذرات MOFs بالقرب من السطح الخارجي، مزينة بأماكن أكبر من الأماكن الداخلية المكونة. هذا يزيد من واجهة التفاعل مع الركيزة إلى جانب تقليل طول مسار الانتشار/التحرر بشكل كبير.مما يؤدي بشكل طبيعي إلى تحسين الخصائص التحفيزية.
هندسة العيوب هي اهتمام طويل الأمد في المواد النانوية البلورية، وخاصة بسبب تأثير المواقع الشاغرة على التحفيز.من المثير للاهتمام أن العيوب الهيكلية في MOFs قد أظهرت تحسينات مماثلة تجاه الخصائص الحفازة و/أو فصل الغاز.. ومع ذلك، فإن هذا يرتبط في معظم الحالات بانخفاض الاستقرار الكيميائي بسبب تقليل الاتصال بين المعدن والليغاند و/أو وجود مواقع معدنية إضافية متاحة.. UiO-66(Zr) أو ( حمض البنزين-1,4-ديكربوكسيليك هو نموذج أولي لمادة الإطار العضوي المعدنية القائمة على الزركونيوم، ويتميز بثبات حراري وكيميائي ممتاز بسبب اتصاله العالي بين المعدن والليغاند (وضع متصل بـ 12) وزركونيوم قوي.
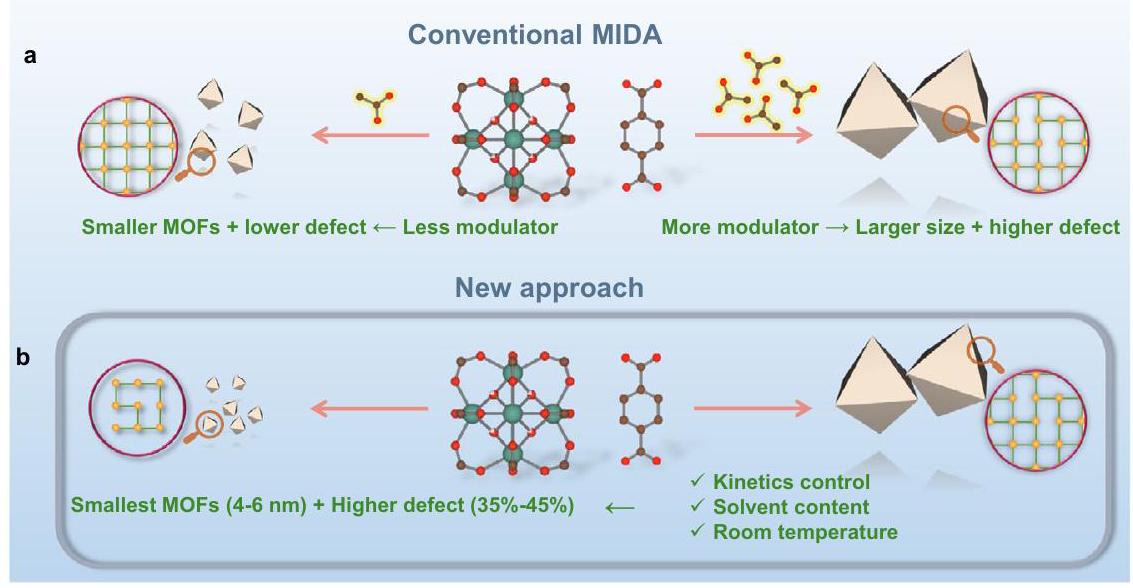
رابطة الكربوكسيلاتوبالتالي، كانت هندسة العيوب في MOFs حتى الآن تركز بشكل رئيسي على UiO-66(Zr) ومشتقاته.الطريقة الأكثر شيوعًا في هندسة العيوب في Zr -MOFs هي نهج العيب الناتج عن المنظم (MIDA)، حيث يتم إضافة منظم أحادي الكربوكسيلات (مثل الفورمات، الأسيتات…)، الذي يرتبط بشكل تفضيلي مع مراكز المعادن بدلاً من الوصلات، مما يؤدي إلى عيوب فراغات الرابطة المفقودة (الشكل 1أ). وبالتالي، يمكن التحكم في محتوى العيوب من خلال كمية المنظم المستخدم..
أثناء تخليق MOF، يرتبط المنظم بـعُقد لإنتاج بلورات ذات ترابط أقل، وبالتالي بحجم أكبر بسبب انخفاض معدل النواة وحركية نمو البلورات. على الرغم من أن هذه طريقة موثوقة لإنتاج جزيئات MOF بأحجام جزيئات قابلة للتعديل.، يكون ذلك على حساب السيطرة على عدد العيوب (الشكل 1أ)لذلك، تمنع استراتيجية MIDA تخليق النانوMOFs الفائقة الصغر ذات العيوب العالية، والتي تعتبر مرشحة مثالية للتفاعل الحفزي. ومن الجدير بالذكر أن العديد من التقارير قد أشارت إلى أهمية التغلب على حواجز الانتشار، حيث تحدث التفاعلات المحفزة بواسطة MOF بشكل رئيسي على السطح الخارجي للجزيئات.. هذه الحالة تكون خاصة عندما يكون حجم الركيزة قابلاً للمقارنة مع حجم فتحة مسام MOF. على الرغم من أن الركائز قد تنتشر جزئيًا داخل إطار MOF، إلا أن إزالة المنتجات الناتجة يمكن أن تعيقها القيود الحركية. الاعتماد على رقائق نانوية مسامية ثنائية الأبعاد ذات نسبة أبعاد عالية مقشرة هو استراتيجية بديلة جذابة للتغلب على هذه القيود.. ومع ذلك، فإن هذه المواد النانوية عادة ما تكون أكثر تحديًا في التحضير (أو التقشير) و/أو تظهر عادةً استقرارًا أقل مقارنةً بنظيراتها ثلاثية الأبعاد. لقد تم استكشاف نهج MIDA، الذي يتم عادةً في مادة N,N-dimethylformamide (DMF) السامة عند درجات حرارة وضغوط عالية، بشكل رئيسي مع UiO-66 بمحتوى عيب في الوصلات المفقودة يبلغ حوالي.. بالإضافة إلى ذلك، لم يتم توسيعه ليشمل المشتقات الوظيفية، وبالتالي لم يتم استغلال الإمكانيات الكاملة لعائلة هذه النانو MOFs المعيبة حتى الآن. لذلك، فإن تطوير طرق جديدة متعددة الاستخدامات لتقليل حجم النانو MOFs القوية، مع ضمان محتوى عيوب مرتفع والحفاظ على استقرار حراري/كيميائي معقول لجزيئات MOF النانوية فائقة الصغر لمواجهة التفاعلات الحفازة الصعبة هو تحدٍ رئيسي يجب التغلب عليه.
نقدم هنا طريقة مستدامة لإنتاج سلسلة من MOFs من نوع UiO فائقة الصغر ) بمحتوى عيوب مرتفع بشكل استثنائي. كشفت مجموعة من تقنيات التوصيف المتقدمة أن المحتوى الكبير من العيوب في جزيئات UiO-66 النانوية الصغيرة يعود إلى وجود عيوب في الروابط المفقودة وأن التبلور يهيمن عليه النمو. ومن الجدير بالذكر أن هذه الاستراتيجية متعددة الاستخدامات ويمكن تطبيقها على العديد من مشتقات UiO-66(Zr)-X ( )، إلى نظير Hf UiO-66(Hf) وأخيرًا إلى هياكل Zr-MOFs الأخرى مثل Zr fumarate MOF-801(Zr)، مما يؤدي إلى إنتاج nanoMOFs فائقة الصغر مع محتوى عيوب مرتفع جدًا. بالإضافة إلى ذلك، فإن ظروفنا الاصطناعية الخضراء المعتدلة أكثر استدامة بكثير من الطرق التقليدية المائية، مما يساهم في توفير الطاقة و/أو تقليل كمية النفايات الخطرة بشكل كبير. علاوة على ذلك، فإن nanoMOFs التي تم تصنيعها هنا تظهر أداءً تحفيزيًا ممتازًا في تحلل روابط الببتيد، حيث تُظهر تفاعلية، وانتشار كيميائي، وانتقائية، واستقرارًا أفضل بكثير من المحفزات القياسية. بشكل ملحوظ، تُظهر هذه nanoMOFs ثنائية الوظيفة حيث يمكن ببساطة تغيير المذيب التفاعلي، مما يسمح باستبدال تحلل روابط الببتيد بتفاعل التكثيف المعاكس، مما يؤدي إلى تكوين روابط الأميد. بالإضافة إلى ذلك، تُظهر هذه nanoMOFs أيضًا انتقائية قابلة للتعديل بسبب تأثير غربلة الجزيئات.
النتائج والمناقشة
تحضير المواد وخصائصها
لإعداد جزيئات نانوية فائقة الصغر عالية الجودة من UiO-66(Zr) بمحتوى عيوب مرتفع، قمنا أولاً بدراسة استراتيجيات الحالة الراهنة ذات الصلة بعناية. على سبيل المثال، أظهرت حموضة المحلول تأثيرًا كبيرًا على حركية التبلور بسبب التغيرات في حالة بروتنة الأحماض الكربوكسيلية التي تؤدي إلى حركية أسرع عند مستويات أعلى من الحموضة.. بالإضافة إلى ذلك، يبدو أن وجود الماء في خليط التفاعل كان عاملاً حاسماً في تحديد العيوب الناتجة عن التكوين أو الروابط بدلاً من اتصالات زركونيوم-الليغاند كما أظهر استخدام درجة حرارة تخليق منخفضة أنها مفيدة في هندسة العيوب.وتقليص الحجم بسبب التكوين المثبط للروابط التنسيقية ولتحديد نضوج أوستوالد (كما هو موضح في الشكل 1b). وبالتالي، لتحقيق هدفنا الطموح في تحضير نانوMOFs فائقة الصغر بمحتوى عيوب عالٍ، قمنا بتطوير استراتيجية خضراء بسيطة تتجنب: (i) استخدام أملاح الزركونيوم الحمضية جداً (مثل، ) وعوامل تعديل حمضية قليلاً (مثل حمض الفورميك) باستخدام مواد تم تصنيعها مسبقاً أوكسوكلوستر الأسيتات (الشكل التكميلي 1)؛ (ii) يتخلص من DMF ويستبدله بالماء والإيثانول لتجنب إطلاق الفورمات عند تحلل DMF (ويمكن أن يتيح نهجًا مستدامًا)؛ (iii) يضمن ذوبانًا أسرع للموصل العضوي عن طريق تخفيف وسط التفاعل في الإيثانول، مما يسرع من حركيات التخليق؛ و (iv) يعمل في درجة حرارة الغرفة، مما يوفر الطاقة.
تمت عملية التخليق الأولي لـ UiO-66(Zr) من خلال خلط الأسيتات المغلفة أولاًالأوكسيكلاسترات مع حمض الأسيتيك. تم إدخال الماء والإيثانول وحمض البنزين-1،4-ثنائي الكربوكسيليك (BDC) لاحقًا في محلول الأوكسيكلاستر (انظر التفاصيل في SI). بعد ساعتين في درجة حرارة الغرفة
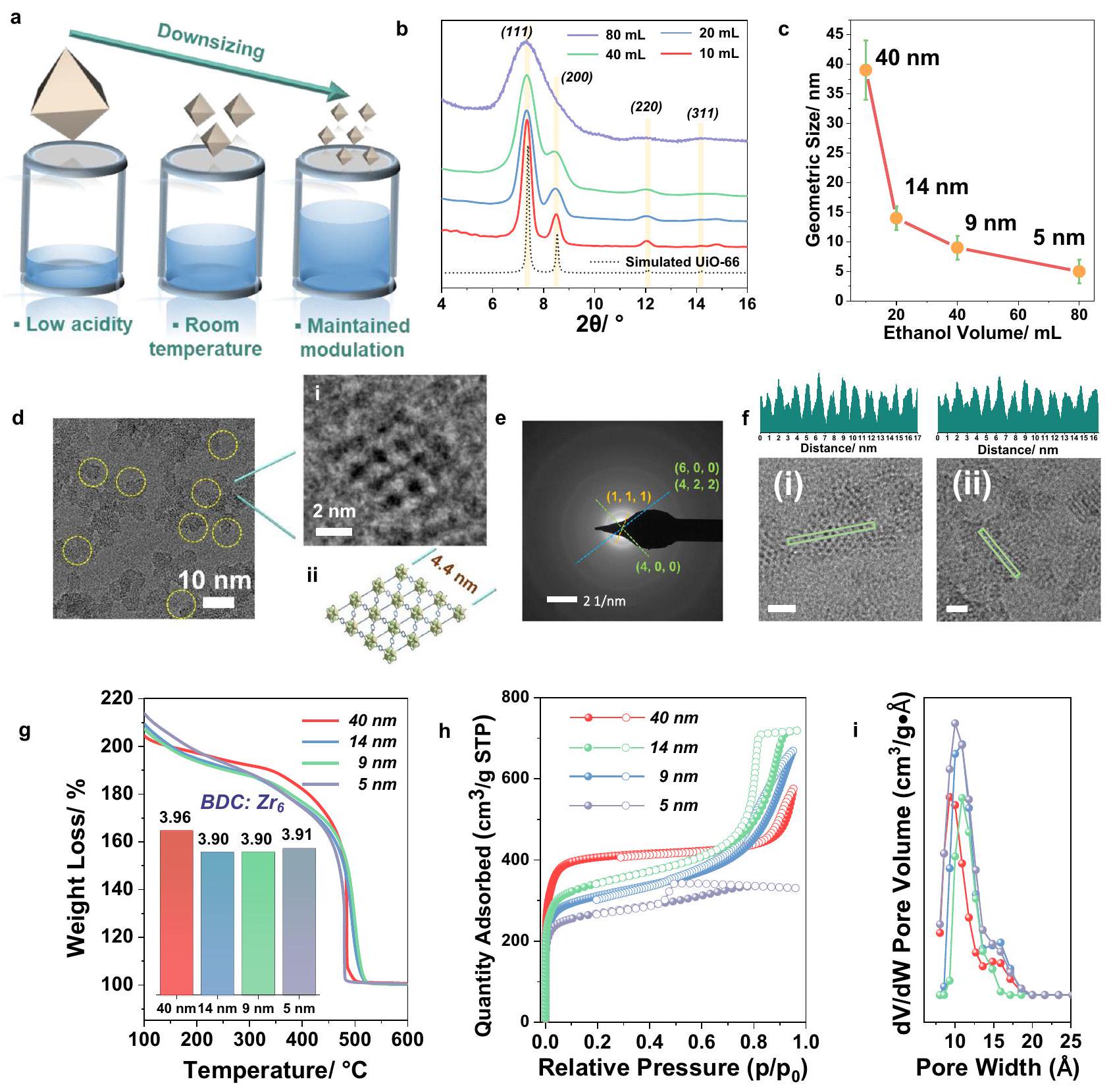
الشكل 1 | مقارنة طرق تخليق جزيئات MOF. مخطط (أ) نهج العيب الناتج عن المحفز التقليدي (MIDA) لضبط حجم العيوب في MOFs، (ب) نهجنا الذي ينتج جزيئات صغيرة جداً وعالية العيوب جزيئات النانو من MOFs رباعية التكافؤ، يمثل الأحمر والأخضر والبني ذرات الأكسجين والمعادن (IV) والكربون على التوالي؛ الكرة الصفراء والخطوط الخضراء داخل الدوائر البنفسجية تشير إلى العقد المعدنية والليغاند العضوي على التوالي. درجة الحرارة تحت التحريك، أظهر الصلب الناتج نمط PXRD (انظر الشكل التوضيحي 2) يتوافق مع نمط الانكسار النظري لـ UiO-66. أشار المجهر الإلكتروني الناقل (TEM) إلى حجم الجسيمات بـ (الشكل التكميلي 3). هذه القيمة قريبة من حجم الجسيمات (44 نانومتر) المحسوب من معادلة شيرر، مما يشير إلى أن الجسيمات هي في الغالب مجالات بلورية مفردة. أظهر الامتزاز (الشكل التوضيحي التكميلي 4) منحنى إيزوثيرم من النوع الأول مع ارتفاع شديدسعة (ج) ومساحة سطح محسوبة باستخدام طريقة بروناور-إيميت-تيلر (BET) (1617أكبر من مساحة سطح BET لـ UiO-66(Zr) الخالي من العيوبإن هذا التوسع الكبير في المساحة السطحية يدل على تكوين كمية كبيرة من العيوب.لا توجد قمم محظورة متناظرة عند الزاوية المنخفضةعند حوالي 4 وتمت ملاحظة ( ) في نمط PXRD للعينة، مما يشير إلى غياب عيوب الكتل المفقودة التي قد تؤدي إلى هيكل منظم بتوبولوجيا reo (الشكل التوضيحي 2).. وبالتالي، افترضنا وجود روابط مفقودة في موادنا. أظهرت مطيافية الأشعة تحت الحمراء بتحويل فورييه (FTIR) غياب مجموعات الأحماض الكربوكسيلية غير المنسقة في المواد المغسولة، بما يتماشى مع وجود عيوب في الروابط بدلاً من وجود أكوكلوسترات مفقودة (الشكل التكميلي 5). أظهرت التحليل الحراري الوزني (TGA) تحت جو من الأكسجين أن نانوMOF UiO-66 بحجم 40 نانومتر أظهر نسبة منخفضة جداً من الرابطة إلى المعدن (الرابط: ، الشكل التوضيحي التكميلي 6)، بما يتماشى مع محتوى الروابط المفقودة العالي. عدد الروابط المفقودة في عيّنتنا يتوافق مع واحدة من أكثر نماذج UiO-66 عيبًا التي تم الإبلاغ عنها حتى الآن، وفي الوقت نفسه، حسب علمنا، تمثل المساحة السطحية لـ 40 نانومتر من UiO-66 قيمة مرتفعة مقارنة بأحدث التقنيات..
توضح الطريقة العامة التي طورناها للتحكم في حجم الجسيمات في الشكل 2a. حيث أدت محاولتنا الأولى إلى الحصول على UiO-66 مع عيوب كبيرة بحجم 40 نانومتر عند استخدام 10 مل فقط من الإيثانول مع 50 ملغ من BDC، تم تخفيف وسط التفاعل بشكل تدريجي مع، على التوالي) مما يتيح ذوبانًا أفضل لـالليغاند. من المثير للاهتمام أن هذا أدى إلى تقليل كبير في حجم الجسيمات النانوية، كما يتضح من أنماط PXRD في الشكل 2ب. ومن الجدير بالذكر أنه عندما وصل حجم الإيثانول إلى 80 مل، لم يكن بالإمكان ملاحظة سوى غلاف واسع من قمم الانكسار الرئيسية المميزة لـ UiO-66 بسبب الفقد الكبير في النظام طويل المدى، وهو ما يتماشى مع معادلة شيرر. الصور المأخوذة بواسطة TEM (الأشكال التكميلية 3، 7، 8، والأشكال 2ج، د) أثبتت السيطرة الدقيقة على تقليل الحجم إلى ما بين 4 نانومتر و6 نانومتر. ومع ذلك، لا تزال مستويات الشبكة البلورية مرئية بواسطة TEM (الشكل 2د)، مما يؤكد جودة الجسيمات النانوية. نمط حيود الإلكترون في المنطقة المختارة (SAED) أظهر فقط الحلقات المميزة لجسيمات UiO-66 النانوية مع اتجاهات بلورية مختلفة (الشكل 2هـ)، مما يثبت أكثر جودة هذه النانوMOFs، فضلاً عن توزيع الحجم المتجانس. التوسيع الإضافي للشكل 2د(ط) يظهر بوضوح المسام والشبكة لجسيم نانوي بحجم 4.4 نانومتر، بما يتماشى جيدًا مع النموذج الهيكلي من الشكل 2د(ث). من تحليل الملف على صور HRTEM على طول الاتجاهين (220) و(011) (الشكل 2و)، المسافات بين تتميز الأوكسيكلوسترز بالتجانس العالي مع قيمة متوسطة تبلغ 1.1 نانومتر، وهي قريبة من القيمة النظرية (1.2 نانومتر)، مما يشير إلى غياب عيوب الأوكسيكلوستر في UiO-66 الفائق الصغر لدينا (HD-US-UiO-66). يجب ملاحظة أن مثل هذه الجسيمات الصغيرة تت correspond فقط إلى حوالي 2 أبعاد وحدة الخلية، أي 8 وحدات خلايا أو 12 قفصًا ثماني السطوح لكل جزيء نانوي. وبالتالي، يقع هذا النظام عند الحدود بين البلورات النانوية والبوليهيدرات العضوية المعدنية المنفصلة. ومع ذلك، على عكس جزيئاتنا الفائقة الصغر (انظر بعد ذلك لوصف الاستقرار الحراري والكيميائي)، غالبًا ما تعاني الأقفاص/البوليهيدرات العضوية المعدنية (MOCs/MOPs) بشدة من ضعف الاستقرار الكيميائي/الهيدروليكي والانهيار الهيكلي عند التنشيط، مما يمنع تطبيقاتها..
وفقًا لـ TGA، أظهرت جميع الجسيمات النانوية المختلفة المُعدة مسبقًا محتوى رابط مشابه جدًا، مع ما يقرب من رابطين مفقودين لكل صيغة (الشكل 2g). هذا، عند دمجه مع النواة في الطور السائل تحليل الرنين المغناطيسي النووي (NMR) (الشكل التوضيحي 9) وطيف الأشعة السينية المشتتة للطاقة (EDX) (الشكل التوضيحي 10) يؤديان إلى صيغة عامة لـيرجى ملاحظة أن ارتباط حجم البلورة بالعيوب في تخليقنا هو، حسب علمنا، الأول من نوعه ويسمح بتحقيقجزيئات MOF مع عيوب تصل إلى 4.2 روابط مفقودة لكل مجموعة أكسيد، متجاوزة بكثير القيم المبلغ عنها عادةً (الشكل التكميلي 11)أظهرت قياسات المسامية بالنيتروجين عند 77 كلفن على جزيئات HD-US-UiO-66 المنشطة في جميع الحالات سعة امتصاص عالية (منإلى، الشكل 2h) المرتبط بالهيسترسيس. انخفاض فيحدثت سعة الامتزاز عند تقليل الحجم، بما يتماشى مع الزيادة التدريجية في نسبة السطح الخارجي إلى السطح الداخلي. أشارت تحليلات توزيع حجم المسام (نموذج DFT) بشكل عام إلى الحفاظ على حجم المسام (الشكل 2i)، وهو ما يتماشى مرة أخرى مع المحتوى الثابت من الوصلات المفقودة. من أجل المقارنة، اتبعنا MIDA التقليدية، بالإضافة إلى المعلمات الاصطناعية لـ 40 نانومتر UiO-66 وأجرينا مجموعة من التجارب الاصطناعية عن طريق تقليل كمية حمض الأسيتيك. ومن الجدير بالذكر أن تحضير جزيئات أصغر، تصل إلى 5 نانومتر، والتي تُعرف باسم MI-US-UiO66 (UiO-66 الفائق الصغر الناتج عن المحفز)، مرتبط بعدد أقل من العيوب (2.6 وصلات مفقودة لكل)الأوكسيكلاسترات) تتماشى بشكل جيد مع النتائج السابقة حيث كان محتوى عيب الوصل المفقود يعتمد بشكل كبير على كمية المعدل (انظر الملحق للحصول على التفاصيل).
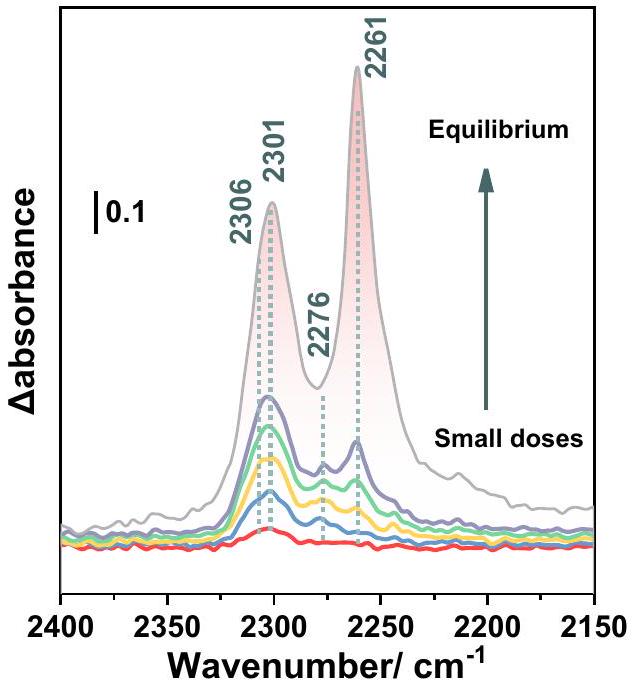
للمزيد من التحقيق في طبيعة العيوب المفقودة في الروابط على المستوى الذري، تم استخدام مطيافية FTIR في الموقع بوجود الأسيتونيتريل- ( تم إجراء التجارب على الأبخرة. يتم غالبًا نسب حموضة UiO-66(Zr) النقية إلى مواقع الحمض برونستيد الداخلية الخاصة بها (أربعة )، بينما، عند تفعيل درجة حرارة عالية، تكون هناك مواقع إضافية لحمض لويس مرتبطة بالعيوب (الرابط المفقود) موجودة. عند إدخال عند ضغط توازن قدره 10 تور، يمكن ملاحظة ثلاث نطاقات اهتزازية عند 2306 و2301 ومرتبط بامتصاص كيميائي لـعلى مواقع الأحماض لويس وبرونستيد المختلفة (الشكل 3).نطاقات عند 2306 وتسيطر في الغالب على الأطياف في الجرعات الثلاث الأولى، مما يشير إلى تفاعل قوي بين مواقع الحمض لويسمن MOF و. من المثير للاهتمام، أن هذهالأشرطة أظهرت انزياحًا أزرق طفيف، منإلى 2306 و، مقارنةً بموقع الذروة الثابت للـالممتصة في الأعمال المبلغ عنها الأخرى. وهذا يشير بوضوح إلى قوة حمضية أعلى للمواقع، ربما تعززت بتركيز كبير من العيوب. يمكن حساب تركيزها من خلال دمج الأشرطة المقابلة مقابل الكمية المولية منالمقدمة. القيمة الناتجة منتتوافق مع عدد أكبر بكثير من مواقع الحمض لويس مقارنةً بما تم الإبلاغ عنه عادةً لعيوب UiO-66 الأكبر حجمًا عبر MIDA، على سبيل المثال، عادةً حوالي، مما يوضح أكثر أن جزيئات UiO-66 النانوية الصغيرة جدًا لدينا تظهر درجة أعلى بكثير من العيوب، وبالتالي فهي مثيرة للاهتمام بشكل خاص للتفاعل الحفاز القائم على حمض لويس.
يمكن اقتراح عدة فرضيات لفهم تشكيل HD-US-UiO-66. أولاً، تقومأوكسوكلاستر الأسيتات المُشكلة مسبقًا بتحديد الرقم الهيدروجيني للمحلول بالقرب من 4، مما يفضل إزالة البروتون من مجموعات الكربوكسيل للرابطة وبالتالي يؤدي إلى نواة أسرع عند استبدال الأسيتات الطرفية من الأوكسوكلاستر في وجود وحدات الديكربوكسيليت. ثم، تؤدي التخفيف عند إضافة الإيثانول إلى إعاقة معدل التصادم الفعال، وعندما يتم دمجه مع محتوى المعدل العالي، قد يحد من نمو البلورات وكذلك نضوج أوستوالد. ومع ذلك، عند تقليل الحجم، تصبح حركية التفاعل أسرع، من 3 ساعات لأكبر الجزيئات إلى أقل من ساعة واحدة لجزيئات. وبالتالي، من المحتمل أن تكون هناك معلمات أخرى تلعب دورًا. على سبيل المثال، تكون قابلية ذوبان الرابطة محدودة عند درجة حرارة الغرفة في الإيثانول وبالتالي، تزداد نسبة الرابطة التي تذوب مع تخفيف الإيثانول، مما يفضل حركية أسرع للجزيئات الأصغر. للتحقق من هذه الفرضية، مع الأخذ في الاعتبار قابلية ذوبان BDC الأفضل بكثير في DMF، استبدلنامن EtOH بـ
الشكل 2 | خصائص جزيئات UiO-66 النانوية المُصنعة. أ رسم تخطيطي لاستراتيجيتنا، (ب) حيود الأشعة السينية البودرة (PXRD) () أنماط UiO-66 المُصنعة بأحجام مختلفة من EtOH، الخطوط الصفراء توضح قمم الحيود من النمط المحسوب، (ج) متوسط الحجم الإحصائي لـ UiO-66 المُصنعة، (د) صورة TEM لجزيئات UiO-66 بحجم 5 نانومتر (تم الحصول عليها باستخدام 80 مل من EtOH، مميزة باستخدام دوائر صفراء)، i) منطقة مختارة مكبرة، ii) هيكل UiO-66 من منظور محور (101)، (هـ) نمط SAED لجزيئات UiO-66 بحجم 5 نانومتر، (و) صور مجهر الإلكترون الناقل عالي الدقة (HRTEM) لـ HD-US-UiO-
66 وملفات كثافة التباين الخاصة بها، من منظور (i) (220) و(ii) (011) الاتجاهات، شريط القياس، المستطيلات الخضراء تمثل المناطق المختارة لتحليل التباين، (ز) TGA تحت جو الأكسجين (معدل المسح) لـ UiO-66 بأحجام مختلفة، الرسم البياني المدرج: نسبة الرابط إلىللعينات المختلفة؛ (ح)الامتصاص هو توازن UiO-66 بأحجام مختلفة (شريط)، يتم تمثيل الامتصاص والإزالة بواسطة كرات مملوءة، وكرات مفتوحة، على التوالي، i) توزيع حجم المسام لأحجام مختلفة من UiO-66 (نفس لون التسمية كما في (ح)).
DMF مع الحفاظ على جميع المعلمات الاصطناعية الأخرى ثابتة. وُجد أن الجزيئات النانوية الناتجة كانت حوالي 8 نانومتر فقط بدلاً من 40 نانومتر مقارنةً باستخدام EtOH (الشكل التكميلي 18)، مما يؤكد تأثير قابلية ذوبان الرابط على الحركية. تم ملاحظة مثل هذا الاختناق سابقًا من قبل بعضنا عند زيادة حجم الفاصل العضوي الحمضي الديكربوكسيلي أثناء التبلور تحت ظروف حل حراري لـ UiO-66(Zr) ونظائره الممتدة.
للحصول على فهم أعمق حول آلية تشكيل HD-US-UiO-66، تم إجراء تجارب تشتت الضوء الديناميكي المعتمد على الزمن (TD-DLS) في الموقع. الشكل 4 أ أظهر تطور حجم الجزيئات كدالة لزمن التبلور لـ HD-US-UiO-66. تم عزو عملية زيادة الحجم السريعة في الدقائق الخمس عشرة الأولى إلى تشكيل نوى MOF. تم ملاحظة نمو MOF في النطاق بين 15 دقيقة و130 دقيقة وبلغت ذروتها بعد 130 دقيقة بحجم هيدروديناميكي حوالي 18 نانومتر. أظهر الميل الواضح أن تبلور HD-US-UiO-66 يتبع عملية تهيمن عليها النمو، بما يتماشى مع دور ذوبان الرابط. يشير مؤشر التوزيع المتدني جدًا (Pdi) (الشكل التكميلي 19) إلى نواة متجانسة تليها نمو بلورات UiO-66 في المحلول. لتأكيد حركية النمو لـ HD-US-UiO-66، تم إجراء دراسة HRTEM/STEM خارج الموقع بعدو
180 دقيقة. الشكل 4 ب يظهر أنه تم ملاحظة فقطأوكسوكلاستر (حوالي 0.6 نانومتر) قبل إدخال BDC. تم تشكيل جزيئات نانوية صغيرة جدًا (حوالي 1.3 نانومتر) بسرعة كبيرة بمجرد إضافة الرابط (1 دقيقة)، مما يشير إلى نواة سريعة جدًا لجزيئات UiO-66 في المحلول، بما يتماشى مع المرحلة (i) الموضحة في الشكل 4 أ. أظهرت صور TEM عند 120 و180 دقيقة نمو نوى MOF الصغيرة وبلوغ ذروة نمو الجزيئات النانوية. هذه النتائج متوافقة تمامًا مع TD-DLS، مما يؤكد أن ذوبان الرابط يعمل كاختناق في التحكم في نمو MOF. من الجدير بالذكر أن TD-DLS في الموقع لا يسلط الضوء فقط على عملية التبلور ولكن أيضًا يبرز بشدة الاستقرار الكولودي الممتاز لـ HD-US-UiO-66. تم ملاحظة هذا الاستقرار، المرتبط بالشحنة الإيجابية العالية التي تم تقييمها من خلال تحليل جهد زتا، بغض النظر عن حجم البلورة النانوية (الأشكال S20، S21) وهو ميزة قوية لعملية استخدامها في التطبيقات مثل تصنيع الأفلام الرقيقة جدًا، توصيل الأدوية، الاستشعار والإلكترونيات، من بين أمور أخرى.
تعتبر إحدى الميزات الجذابة جدًا لـ MOFs هي تفعيل روابطها لتحقيق الخصائص المرغوبة. وبالتالي، قمنا بتوسيع الاستراتيجية الاصطناعية لإنتاج مشتقات أخرى من HD-US-UiO-66-X،
الشكل 3 | خصائص المواقع الحمضية المتاحة بواسطة FTIR في الموقع. طيف FTIR في الموقع عند 298 كلفن لـ(أحمر إلى رمادي، استكشاف جرعات صغيرة تصل إلى ضغط توازن 10 تور) الممتصة على HD-US-UiO-66 (5 نانومتر)، يتم عرض موضع القمم ومساحة القمة في خطوط منقطة ومنطقة حمراء، على التوالي، تشير الجرعات الصغيرة والتوازن إلى زيادةالجرعات من 0.2 تور إلى 6.4 تور مشبعة.
بما في ذلكو Br، بالإضافة إلى استبدالبـأوكسوكلاستر وموصلات مسطحة قائمة على BDC بواسطة الفومارات. جميع هذه النانوMOFs الصغيرة جدًا أظهرت أحجام جزيئات خلوية من 2-3 ووصلات مشابهة جدًا (حوالي 3.3-4 روابط لكل صيغة) مثل HD-US-UiO-66 الأصلي (انظر التحليل التفصيلي في SI).
التفاعل الحفاز غير المتجانس
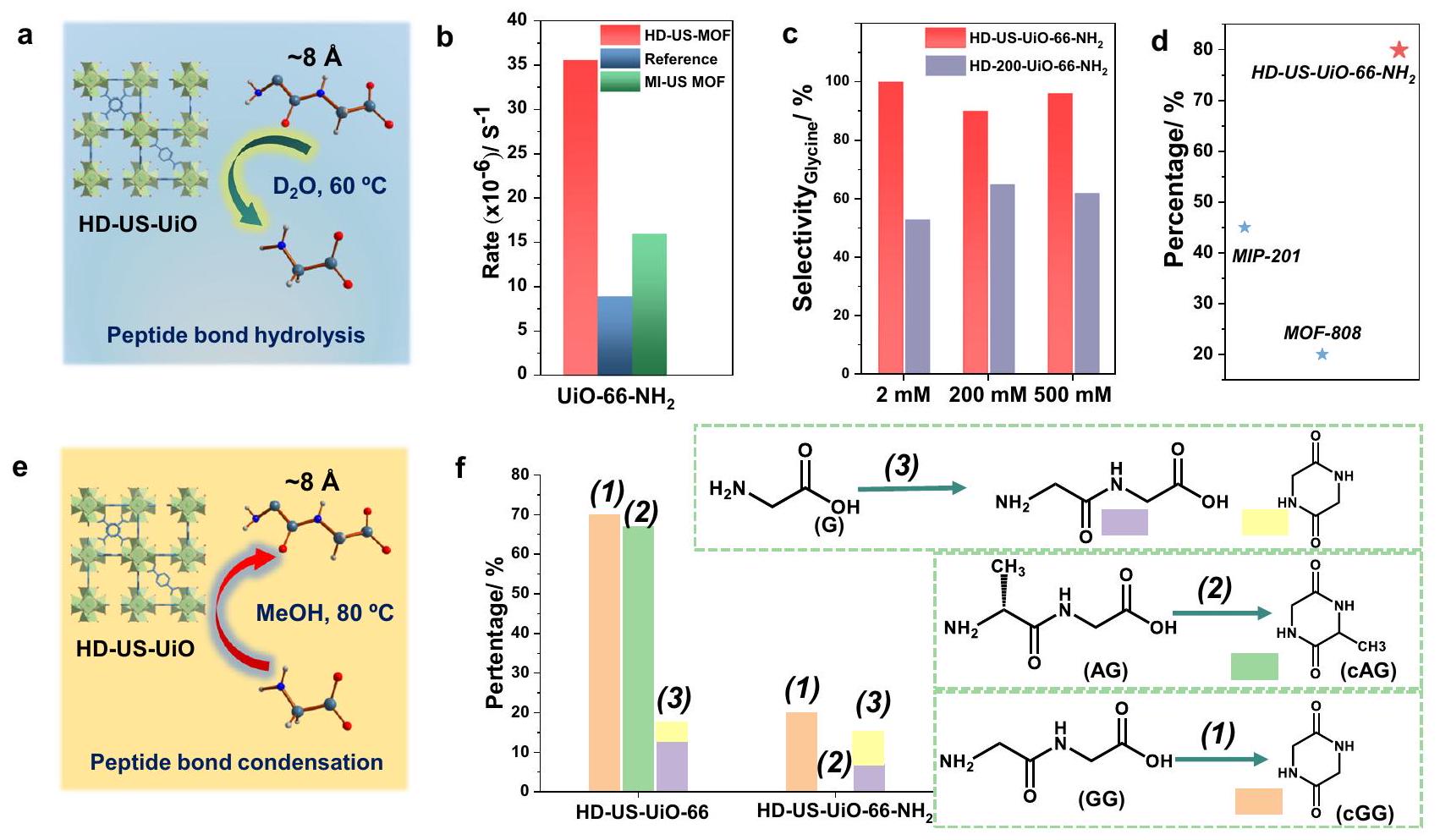
في ضوء خصائص HD-US-UiO-66-X، قررنا استكشاف استخدامها في التفاعل الحفاز غير المتجانس. تم اختيار التحلل المائي لرابطة الببتيد في الجلايسيل-جلايسين (GG) كالتفاعل النموذجي للتحقيق في أهمية تقليل الحجم/تشكيل العيوب لجزيئات MOF على الأداء الحفاز العام (الشكل 5أ). تم اختيار HD-US-UiO-66-NH2 نانوMOF لاحقًا للتفاعل الحفاز بسبب إمكانية حدوث روابط H – بين الركيزة الببتيدية و-مجموعة على الرابط، أو بين مجموعة –والنيوكليوفيل، مما قد يساهم في التفاعل الحفاز العام. كما هو متوقع، أدى استخدام HD-US-UiO-إلى زيادة النشاط بمقدار حوالي 3 مرات مقارنةً بجزيئات UiO-66 التقليدية(حوالي 200 نانومتر) التي تم الإبلاغ عنها سابقًا (الشكل 5ب). قد يكون هذا التحسين بسبب زيادة مساحة السطح الخارجية بشكل كبير و/أو محتوى العيوب الكبير. وبالتالي، قمنا بمقارنة أداء HD-US-UiO-66-NH2 وMI-US-UiO-66-NH2 نانوMOFs مع الدراسة السابقة. من الجدير بالذكر أنه على الرغم من أن MI-US-UiO-66أظهرت تفاعلية أفضل مقارنة بالدراسة السابقة، لكنها أدت بشكل أسوأ (حوالي مرتين) من HD-US-UiO-66-NH2، على الرغم من حجم الجسيمات المتشابه جداً. وهذا يشير إلى أن زيادة كمية العيوب في الروابط المفقودة أمر حاسم لتعزيز تفاعلية مشتقات UiO-66 تجاه تحلل الروابط الببتيدية، بالتزامن مع تقليل حجم الجسيمات. ومن الجدير بالذكر أن التفاعلية هنا قابلة للمقارنة مع المواد المرجعية مثل MOF-808 “فائق النشاط” (35 نانومتر)، الذي أظهر معدل تحلل GG قدره.
تم اختبار قابلية إعادة تدوير HD-US-UiO-66-NH2 nanoMOF على مدى خمس دورات تفاعل متتالية (الشكل التكميلي 35)، مع تسجيل فقدان طفيف في التفاعل فقط بعددورة، حيث انخفضت التفاعلية إلىمن الأداء الملحوظ في الدورة 1. ومع ذلك، لا يزال هذا قابلًا لإعادة التدوير بشكل ممتاز، خاصة عند مقارنته بالمواد المرجعية، مثل MIP-201 و MOF-808.التي عانت من انخفاض القابلية لإعادة التدوير (فقط و النشاط بعد 5 دورات، على التوالي، الشكل 5د)، ويبرز استقرار الماء لهذه النانوMOFs فائقة الصغر وعالية العيوب.
تمت دراسة خصوصية المحفز بشكل أكبر، حيث يمكن أن يتم تحلل GG إلى G، أو قد يحدث تفاعل حلقي (تكوين رابطة أميد)، مما يشكل GG حلقي (cGG) (الشكل التوضيحي التكميلي 32). من خلال استخدام HD-UiO-66تم فحص تأثير أحجام الجسيمات والمساحة السطحية الخارجية مقابل الداخلية على خصوصية التفاعل في nanoMOFs بحجمين مختلفين (4 نانومتر مقابل 200 نانومتر). تم دراسة UiO- بحجم 200 نانومتر.يتم تصنيفه على أنه HD-200-UiO-66-NH2، وقد تم توصيفه بالكامل قبل التفاعل الحفزي
الشكل 4 | التوصيفات في الموقع وخارج الموقع لنمو MOF. أ الحجم الهيدروديناميكي لكرات HD-US-UiO-66ج) محدد بواسطة قياس تشتت الضوء الديناميكي في الموقع (دقة الوقت ) ، و (ب) HAADF-STEM خارج الموقع ( ) و HRTEM ( صور لـ HD-US-UiO-66 في أوقات مختلفة.
الشكل 5 | تقييم أداء التحفيز لـ HD-US-UiO-66-X. توضيح لتحلل الببتيد باستخدام HD-US-UiO-66، (ب) معدل التحلل من الدرجة الأولى الزائفة لجلايسيل جلايسين (GG) إلى جلايسين (G) باستخدام HD-US-UiO-66- و MI-US-UiO-66- النانو MOFs، تشير الإشارة إلى القيمة المبلغ عنها في الدراسات السابقة تحت نفس الظروف، (ج) انتقائية التحلل المائي بواسطة HD-US-UiO-66-NH2 و HD-200-UiO-66-في إنتاج المنتج المطلوب G ، حيث التركيز الابتدائي لـ
يزداد GG من 2 مللي مول إلى 500 مللي مول، (د) قابلية إعادة الاستخدام لـ HD-US-UiO-66-NH2 على مدى 5 دورات تفاعل مقارنة بأفضل MOFs حتى الآن، النسبة مقارنة بعائد الدورة 1، (هـ) توضيح تكثيف رابطة الأميد باستخدام HD-US-UiO-66، في الميثانول، (و) عائد تكوين رابطة الأميد مع HD-US-UiO-66 و HD-US-UiO-66-بدءًا من، وAG. (الأشكال التكميلية 36-38). من المثير للاهتمام أن HD-US-UiO-66-NH2 وُجد أنه أكثر انتقائية، حيث كان يعطي باستمرار نسبة أكبر من منتج التحلل G، بدلاً من CGG، بغض النظر عن التركيز الابتدائي لركيزة GG (الشكل 5c). أنتج HD-200-UiO-66-NH2 كمية أكبر بكثير من cGG (للمنتج)، من المحتمل أن يكون ذلك بسبب زيادة المساحة السطحية الداخلية مقارنة بـ HD-US-UiO-الذي يعزز تكثف روابط الأميد في غياب الماء في المسام الأكثر كرهًا للماء، بدلاً من تفاعل التحلل المائي على السطح الخارجي للنانوMOF المعرض للماء.
نظرًا لأن cGG وُجد أنه منتج جانبي للتحلل المائي مع HD-200-UiO-66-NH2، تم فحص النانو MOFs بشكل إضافي لقدرتها على تحفيز تكوين الروابط الببتيدية بين الأحماض الأمينية والببتيدات الصغيرة. يمكن تعزيز تفاعل التكثيف بدلاً من التحلل المائي ببساطة عن طريق تغيير المذيب من الماء إلى الميثانول.أظهر كل من HD-US-UiO-66-NH2 و HD-US-UiO-66 nanoMOFs القدرة على تعزيز تكوين روابط الأميد في الميثانول (الشكل 5e، f)، لكن HD-US-UiO-66 أظهر تفاعلية أعلى بكثير من HD-US-UiO-66-NH2 المُوظف. مشابه للتقرير السابق.، بالنسبة لكلا MOFs، كانت عملية تكوين رابطة الببتيد داخل الجزيء مفضلة على التكثيف بين جزيئات الجلايسين الفردية (التفاعل 3، الشكل 5f). من المثير للاهتمام أن HD-US-UiO-66-NH2 المُوظف كان غير فعال في تكوين cGG عند البدء من GG (التفاعل 2)، ولكن حقيقة أن cGG تم ملاحظته في التفاعلين الآخرين الموضحين في الشكل 5f، تشير إلى أن كفاءة تكوين الرابطة داخل الجزيء تتأثر بتفاعل الركيزة مع MOF، بدلاً من أن تكون نتيجة لنشاط تحفيزي منخفض. قد تتأثر هذه الاختلافات في التفاعلات بوجود مجموعة مانحة للإلكترون – المجموعات على MOF، بالإضافة إلى أحجام مسام MOF، حيث أن MOF المُوظف يظهر مسامًا أصغر قليلاً بسبب العائق الفراغي لـمجموعة، التي تزيد من حاجز الانتشار للركيزة إلى داخل وخارج مسام MOF.
في هذا العمل، أبلغنا عن استراتيجية عامة لتحضير سلسلة من المواد ذات العيوب العالية (رابط مفقود) وصغير جداً (بلورات نانوية قائمة على UiO-66 تحت ظروف مستدامة بالكامل، مما يشير إلى إمكانية التوسع. يعتبر نمو البلورات عنق الزجاجة في التبلور، كما يتضح من TD-DLS في الموقع مع HRTEM/STEM خارج الموقع، ويمكن التلاعب به ببساطة باستخدام مذيب إضافي. تم تقييم عيوب الوصلات المفقودة بواسطة تقنيات متقدمة متعددة، بما في ذلك PXRD وFTIR وTGA وHRTEM وطيف FTIR في الموقع المدمج معاستكشاف، الذي كشف عن أهمية حموضة لويس في HD-US-UiO-66. أظهر HD-US-UiO-66-X الناتج أداءً تحفيزيًا ممتازًا في كل من تحلل وتكوين روابط الببتيد مع تفاعل تحفيزي، انتقائية، كفاءة استرداد المنتج، وإمكانية إعادة الاستخدام مقارنةً بالمواد الأخرى المبلغ عنها. قدم التحقيق المفصل في تأثير العيوب، حجم الجسيمات، والتوظيف على النشاط التحفيزي لـ nanoMOFs رؤى فريدة حول المعلمات الرئيسية التي تؤثر على التفاعل، وبالتالي توضح كيف يمكن ضبط nanoMOFs لإظهار تفاعل محدد وانتقائي من خلال التحكم الدقيق في خصائص المحفزات. لذلك، قد تعزز الاكتشافات المبلغ عنها هنا تطوير nanoMOFs كمواد تحفيزية غير متجانسة ذات وظائف مزدوجة وتحسينات في الأداء في جوانب متنوعة. علاوة على ذلك، قد تُستخدم المواد المقدمة هنا أيضًا في تطوير أجهزة استشعار/بصرية (نتائج أولية في المناقشة التكميلية، والشكل التكميلية 24)، الأغشية، تركيبات النانوميديسين، واستكشاف خصائص أخرى تعتمد على الحجم.
طرق
تركيبأوكسيكلوسترات
تم إضافته إلى خليط من 3 مل من حمض الأسيتيك الجليدي و5 مل من الإيزوبروبانول تحت التحريك بسرعة 500 دورة في الدقيقة وتم تسخينه عند لمدة 60 دقيقة. تم جمع المنتج إما من خلال الترشيح بالشفط أو الطرد المركزي في تم غسل المادة الصلبة البيضاء المجمعة مرتين بأسيتون وتجفيفها تحت الفراغ في درجة حرارة الغرفة. تم تصنيعيمكن تخزين الأوكسيكلوستر في ظروف محيطة لمدة 12 شهرًا على الأقل دون تغيير في الخصائص. تم إعدادتحتوي الأوكسيكلاسترات على الصيغة التالية:.
تركيبأوكسيكلوستر
تمت إضافة إلى خليط مكون من 4 مل من حمض الأسيتيك الجليدي و 18 مل من الإيزوبروبانول تحت التحريك بسرعة 500 دورة في الدقيقة وتم تسخينه عند لمدة 60 دقيقة. تم جمع المنتج إما من خلال الترشيح بالشفط أو الطرد المركزي في تم غسل المادة الصلبة البيضاء المجمعة بمذيب الأسيتون مرتين ثم تم تجفيفها تحت الفراغ عند درجة حرارة الغرفة.
تركيب
أوكسيكلوستر ) تم تفريقها في حمض الأسيتيك تحت التحريك فيثم أُضيف، وتم تحريك خليط التفاعل حتى أصبح شفافًا تمامًا. تم إدخال 10 مل من الإيثانول إلى المحلول تلاه الإضافة الفورية لحمض 1,4-بيزينديكربوكسيليك (BDC، )، وتم تحريك التفاعل لمدة 3 ساعات في درجة حرارة الغرفة. ثم تم طرد المحلول الناتج في جهاز الطرد المركزي عند لمدة 45 دقيقة ثم تم غسلها مرتين بالإيثانولتم تجفيف المادة الصلبة المجمعة تحت الفراغ لمدة 3 ساعات من أجل التحليلات والتطبيقات.
تركيب MI-US-UiO-66 ( )
أوكسيكلوسترتم تفريقها في حمض الأسيتيك ) تحت التحريك في ثم أُضيف، وتم تحريك خليط التفاعل حتى أصبح شفافًا تمامًا. تم إدخال 10 مل من الإيثانول إلى المحلول تلاه الإضافة الفورية لحمض 1,4-بيزينديكربوكسيليك (BDC، )، وتم تحريك التفاعل لمدة ساعة واحدة في درجة حرارة الغرفة. ثم تم طرد المحلول الناتج في جهاز الطرد المركزي عند لمدة 45 دقيقة ثم تم غسلها مرتين بالإيثانولتم تجفيف المادة الصلبة المجمعة تحت الفراغ لمدة 3 ساعات من أجل التحليلات والتطبيقات. يمكن تطبيق التحكم في الحجم الناتج عن المودولátor باستخدام كميات مختلفة من حمض الأسيتيك المستخدم في دفعة التخليق.
تركيب HD-US-UiO-66 ( )
أوكسيكلوسترتم تفريقها في حمض الأسيتيك ) تحت التحريك في ثم أُضيف، وتم تحريك خليط التفاعل حتى أصبح شفافًا تمامًا. تم إدخال 80 مل من الإيثانول إلى المحلول تلاه الإضافة الفورية لحمض 1,4-بيزينديكربوكسيليك (BDC، )، وتم تحريك التفاعل لمدة ساعتين في درجة حرارة الغرفة. تم تبخير المحلول الناتج بواسطة التبخير الدوار في درجة حرارة الغرفة حتى تم ترك الحجم. تم طرد التعليق الغروي في جهاز الطرد المركزي عندلمدة 60 دقيقة ثم تم غسلها مرتين بمزيج من 20 مل من الأسيتون و20 مل من الإيثانولتم تجفيف المادة الصلبة المجمعة تحت الفراغ لمدة 3 ساعات من أجل التحليلات والتطبيقات. يمكن تطبيق نهج التحكم في الحجم هنا مع كميات مختلفة من الإيثانول المستخدمة في دفعة التخليق.
تركيب HD-US-UiO-66-NH2 ( )
أوكسيكلوستراتتم تفريقها في حمض الأسيتيك ) تحت التحريك في تمت الإضافة بعد ذلك، وتم تحريك خليط التفاعل حتى أصبح واضحًا تمامًا. تم إدخال 80 مل من الإيثانول إلى المحلول تلاه الإضافة الفورية لحمض 2-أمينوبنزويك-1،4-ثنائي الكربوكسيليك (BDC-NH2، )، وكانت ردود الفعل تم التحريك لمدة ساعة واحدة في درجة حرارة الغرفة. تم تبخير المحلول الناتج بواسطة التبخير الدوار في درجة حرارة الغرفة حتىتم ترك حجم. تم طرد التعليق الغروي في جهاز الطرد المركزي عندلمدة 60 دقيقة ثم تم غسلها مرتين بمزيج من 20 مل من الأسيتون و 20 مل من الإيثانولتم تجفيف المادة الصلبة المجمعة تحت الفراغ لمدة 3 ساعات من أجل التحليلات والتطبيقات.
تحلل الجلايسيل جلايسين
قبل التحلل المائي، تم تنشيط MOFs عندلمدة 20 ساعة. أضيف إلى قنينة زجاجية سعة 1 ملمولات MOF و. التالي، محلول بتركيز 40 مللي مولار من جلايسيل جلايسين فيتمت إضافة، وتم ضبط pD للمزيج إلى 7.4 وتم حضنه فيمع التحريك. تم إعداد الوعاء وفقًا لنقطة الزمن في ثلاث نسخ. تم إيقاف التفاعلات عند، ، و48 ساعة وتم الطرد المركزي عندلمدة 10 دقائق.تم تحليل السائل العلوي باستخداميستخدم0.1 م TMSPمعيار داخلي. تم غسل MOFs في الأسيتون وتحليلها باستخدام PXRD بعد التفاعل للتحقق من سلامة هيكلها.
للتجارب المتعلقة بإعادة التدوير، بعد التفاعل، تم غسل المواد العضوية المعدنية.بين عشية وضحاها لإزالة الركيزة والمنتج الممتص، تليها غسلة في 10 مل من الأسيتون وتجفيف في الفرن لمدة 8 ساعات عند. بعد ذلك تم تكرار التفاعل.
تكوين الروابط الببتيدية
تم استخدام MOFs بعد التنشيط. أضيف إلى قنينة بسدادة قابلة للضغط سعة 10 مل 5مولات MOF ومولات الركيزة (جلايسيل جلايسين، ل-ألانيل جلايسين أو جلايسين) تليها 1 مل من الميثانول. تم إغلاق الخلطات وتحريكها، ثم تم حضنها فيلمدة 24 ساعة مع التحريك. بعد الحضانة، تم تخفيف العينات بـوتم تحريكها في درجة حرارة الغرفة لمدة ساعة واحدة لاستخراج الركائز والمنتجات من MOFs. بعد ذلك، تم جمع السائل العلوي عن طريق الطرد المركزي عندلـتم تخفيف السائل العلوي بـلإعطاء حجم نهائي منالذي تم تحليله باستخداميستخدم0.1 م TMSP-معيار داخلي.
توفر البيانات
جميع البيانات التي تدعم نتائج هذه الدراسة متاحة من المؤلفين المقابلين عند الطلب. يتم توفير بيانات المصدر مع هذه الورقة.
References
Roduner, E. Size matters: why nanomaterials are different. Chem. Soc. Rev. 35, 583-592 (2006).
Chen, Y. et al. Phase engineering of nanomaterials. Nat. Rev. Chem. 4, 243-256 (2020).
Wang, Y. & Herron, N. Nanometer-sized semiconductor clustersmaterials synthesis, quantum size effects, and photophysical properties. J. Phys. Chem. 95, 525-532 (1991).
Valden, M., Lai, X. & Goodman, D. W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 281, 1647-1650 (1998).
Zhang, P. et al. Electronic transport in nanometre-scale silicon-oninsulator membranes. Nature 439, 703-706 (2006).
Cai, X. C. et al. Nano-sized metal-organic frameworks: synthesis and applications. Coord. Chem. Rev. 417, 213366 (2020).
Protesescu, L. et al. Colloidal nano-MOFs nucleate and stabilize ultra-small quantum dots of lead bromide perovskites. Chem. Sci. 12, 6129-6135 (2021).
Sakata, Y. et al. Shape-memory nanopores induced in coordination frameworks by crystal downsizing. Science 339, 193-196 (2013).
Krause, S. et al. The effect of crystallite size on pressure amplification in switchable porous solids. Nat. Commun. 9, 1573 (2018).
Horcajada, P. et al. Porous metal-organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat. Mater. 9, 172-178 (2010).
Marshall, C. R. et al. Size-dependent properties of solutionprocessable conductive MOF nanocrystals. J. Am. Chem. Soc. 144, 5784-5794 (2022).
Fabrizio, K. & Brozek, C. K. Size-dependent thermal shifts to MOF nanocrystal optical gaps induced by dynamic bonding. Nano Lett. 23, 925-930 (2023).
Xia, H.-L. et al. Size- and emission-controlled synthesis of full-color luminescent metal-organic frameworks for tryptophan detection. Angew. Chem. Int. Ed. 62, e202308506 (2023).
Zhao, M. et al. Ultrathin 2D metal-organic framework nanosheets. Adv. Mater. 27, 7372-7378 (2015).
Nowotny, M. K., Sheppard, L. R., Bak, T. & Nowotny, J. Defect chemistry of titanium dioxide. Application of defect engineering in processing of -based photocatalysts. J. Phys. Chem. C 112, 5275-5300 (2008).
Busch, G. Early history of the physics and chemistry of semiconductors-from doubts to fact in a hundred years. Eur. J. Phys. 10, 254 (1989).
Furukawa, H., Müller, U. & Yaghi, O. M. Heterogeneity within order” in metal-organic frameworks. Angew. Chem. Int. Ed. 54, 3417-3430 (2015).
Dissegna, S., Epp, K., Heinz, W. R., Kieslich, G. & Fischer, R. A. Defective metal-organic frameworks. Adv. Mater. 30, 1704501 (2018).
Cavka, J. H. et al. A new zirconium inorganic building brick forming metal-organic frameworks with exceptional stability. J. Am. Chem. Soc. 130, 13850-13851 (2008).
Wu, H. et al. Unusual and highly tunable missing-linker defects in zirconium metal-organic framework UiO-66 and their important effects on gas adsorption. J. Am. Chem. Soc. 135, 10525-10532 (2013).
Vermoortele, F. et al. Synthesis modulation as a tool to increase the catalytic activity of metal-organic frameworks: the unique case of UiO-66 (Zr). J. Am. Chem. Soc. 135, 11465-11468 (2013).
Shearer, G. C. et al. Defect engineering: tuning the porosity and composition of the metal-organic framework UiO-66 via modulated synthesis. Chem. Mater. 28, 3749-3761 (2016).
Liang, W. et al. Defect engineering of UiO-66 for and uptake-a combined experimental and simulation study. Dalton Trans. 45, 4496-4500 (2016).
Schaate, A. et al. Modulated synthesis of Zr-based metal-organic frameworks: from nano to single crystals. Chem. Eur. J. 17, 6643-6651 (2011).
Forgan, R. S. Modulated self-assembly of metal-organic frameworks. Chem. Sci. 11, 4546-4562 (2020).
Fang, Z. et al. Structural complexity in metal-organic frameworks: simultaneous modification of open metal sites and hierarchical porosity by systematic doping with defective linkers. J. Am. Chem. Soc. 136, 9627-9636 (2014).
Taylor, J. M., Dekura, S., Ikeda, R. & Kitagawa, H. Defect control to enhance proton conductivity in a metal-organic framework. Chem. Mater. 27, 2286-2289 (2015).
Abánades Lázaro, I., Wells, C. J. & Forgan, R. S. Multivariate modulation of the Zr MOF UiO-66 for defect-controlled combination anticancer drug delivery. Angew. Chem. Int. Ed. 59, 5211-5217 (2020).
Wei, R. et al. Tuning the properties of nodes in the metal organic framework UiO-66 by selection of node-bound ligands and linkers. Chem. Mater. 31, 1655-1663 (2019).
Gao, W. Y., Cardenal, A. D., Wang, C. H. & Powers, D. C. In Operando analysis of diffusion in porous metal-organic framework catalysts. Chem. Eur. J. 25, 3465-3476 (2019).
Sharp, C. H. et al. Nanoconfinement and mass transport in metal-organic frameworks. Chem. Soc. Rev. 50, 11530-11558 (2021).
Wang, M., Dong, R. & Feng, X. Two-dimensional conjugated metal-organic frameworks (2D c-MOFs): chemistry and function for MOFtronics. Chem. Soc. Rev. 50, 2764-2793 (2021).
Decker, G. E., Stillman, Z., Attia, L., Fromen, C. A. & Bloch, E. D. Controlling size, defectiveness, and fluorescence in nanoparticle UiO-66 through water and ligand modulation. Chem. Mater. 31, 4831-4839 (2019).
DeStefano, M. R., Islamoglu, T., Garibay, S. J., Hupp, J. T. & Farha, O. K. Room-temperature synthesis of UiO-66 and thermal modulation of densities of defect sites. Chem. Mater. 29, 1357-1361 (2017).
Dai, S., Nouar, F., Zhang, S., Tissot, A. & Serre, C. One-step roomtemperature synthesis of metal (IV) carboxylate metal-organic frameworks. Angew. Chem. Int. Ed. 133, 4328-4334 (2021).
Dai et al. Monodispersed MOF-808 nanocrystals synthesized via a scalable room-temperature approach for efficient heterogeneous peptide bond hydrolysis. Chem. Mater. 33, 7057-7066 (2021).
Firth, F. C. et al. Engineering new defective phases of UiO family metal-organic frameworks with water. J. Mater. Chem. A 7, 7459-7469 (2019).
Liu, L. et al. Imaging defects and their evolution in a metal-organic framework at sub-unit-cell resolution. Nat. Chem. 11, 622-628 (2019).
Wang, X. et al. Robust ultrathin nanoporous MOF membrane with intra-crystalline defects for fast water transport. Nat. Commun. 13, 1-11 (2022).
Sánchez-González, E., Tsang, M. Y., Troyano, J., Craig, G. A. & Furukawa, S. Assembling metal-organic cages as porous materials. Chem. Soc. Rev. 51, 4876-4889 (2022).
Ragon, F. et al. Acid-functionalized UiO-66 (Zr) MOFs and their evolution after intra-framework cross-linking: structural features and sorption properties. J. Mater. Chem. A 3, 3294-3309 (2015).
Chakarova, K., Strauss, I., Mihaylov, M., Drenchev, N. & Hadjiivanov, K. Evolution of acid and basic sites in UiO-66 and UiO-66-NH2 metalorganic frameworks: FTIR study by probe molecules. Microporous Mesoporous Mater. 281, 110-122 (2019).
Ragon, F., Chevreau, H., Devic, T., Serre, C. & Horcajada, P. Impact of the Nature of the Organic Spacer on the Crystallization Kinetics of UiO-66(Zr)-Type MOFs. Chem. Eur. J. 21, 7135-7143 (2015).
Sindoro, M., Yanai, N., Jee, A.-Y. & Granick, S. Colloidal-sized metal-organic frameworks: synthesis and applications. Acc. Chem. Res. 47, 459-469 (2014).
Simms, C., Mullaliu, A., Swinnen, S., de Azambuja, F. & Parac-Vogt, T. N. MOF catalysis meets biochemistry: molecular insights from the hydrolytic activity of MOFs towards biomolecules. Mol. Syst. Des. Eng. 8, 270-288 (2023).
Ly, H. G. T., Fu, G., de Azambuja, F., De Vos, D. & Parac-Vogt, T. N. Nanozymatic activity of UiO-66 metal-organic frameworks: tuning the nanopore environment enhances hydrolytic activity toward peptide bonds. ACS Appl. Nano Mater. 3, 8931-8938 (2020).
Ly, H. G. T. et al. Superactivity of MOF-808 toward peptide bond hydrolysis. J. Am. Chem. Soc. 140, 6325-6335 (2018).
Wang et al. A zirconium metal-organic framework with SOC topological net for catalytic peptide bond hydrolysis. Nat. Commun. 13, 1-8 (2022).
de Azambuja, F. et al. En route to a heterogeneous catalytic direct peptide bond formation by Zr -based metal-organic framework catalysts. ACS Catal. 11, 7647-7658 (2021).
شكر وتقدير
يُعرب S.D. وC. Serre وM.D. عن شكرهم لبرنامج الأبحاث والابتكار التابع للاتحاد الأوروبي Horizon 2020 بموجب اتفاقية المنحة رقم 831975 (مشروع MOF4AIR) لتقديم الدعم المالي. كما يشكر C. Simms مؤسسة الأبحاث – فلاندرز (FWO) على منحة الزمالة (68090/11C9320N). يقدّر المؤلفون مساعدة الدكتور X. Xu في قياسات TEM. وقد أعرب S.D. وA.T. وG.P. وC. Serre عن شكرهم لباريس. المنطقة، من خلال مشروع DIM Respore ذو الأولوية، للوصول إلى HRTEM.
مساهمات المؤلفين
قام S.D. و A.T. و C. Serre بتصور البحث. صمم S.D. و C. Simms و T.V. التجارب وحللوا البيانات. أجرى G.P. تحليل STEM/HRTEM. أشرف M.D. على تحقيقات FTIR في الموقع. كتب S.D. المسودة الأصلية بمساعدة C. Simms. ساهم جميع المؤلفين في مناقشة الورقة ومراجعتها. أشرف A.T. و C. Serre على المشروع.
ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا ما تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/رخصة/بواسطة/4.0/. (ج) المؤلف(ون) 2024
معهد المواد المسامية في باريس، المدرسة العليا، ESPCI باريس، CNRS، جامعة PSL، 75005 باريس، فرنسا.جامعة نورماندي، ENSICAEN، UNICAEN، CNRS، مختبر التحفيز والطيفية، 14000 كاين، فرنسا.مختبر الكيمياء الحيوية غير العضوية، قسم الكيمياء، جامعة KU Leuven، شارع سيليستينينلاان 200F، 3001 لوفين، بلجيكا.جامعة باريس-ساكلاي، CNRS، مركز النانوسك sciences والنانوتكنولوجيات، 91120 باليزو، فرنسا.
The size and defects in crystalline inorganic materials are of importance in many applications, particularly catalysis, as it often results in enhanced/ emerging properties. So far, applying the strategy of modulation chemistry has been unable to afford high-quality functional Metal-Organic Frameworks (MOFs) nanocrystals with minimized size while exhibiting maximized defects. We report here a general sustainable strategy for the design of highly defective and ultra-small tetravalent MOFs ( ) crystals (ca. missing linker, ). Advanced characterizations have been performed to shed light on the main factors governing the crystallization mechanism and to identify the nature of the defects. The ultra-small nanoMOFs showed exceptional performance in peptide hydrolysis reaction, including high reactivity, selectivity, diffusion, stability, and show emerging tailorable reactivity and selectivity towards peptide bond formation simply by changing the reaction solvent. Therefore, these highly defective ultra-small M(IV)-MOFs particles open new perspectives for the development of heterogeneous MOF catalysts with dual functions.
Over the past few decades, the development of colloidal nanocrystals has led to a revolution in material science due to their very appealing properties in heterogeneous catalysis, optics, biology, and engineering . Indeed, most nanomaterials undergo dramatic changes in their properties when their particle size lies in the ultra-small scale (e.g., below ) such as the quantum size effect in semiconductor materials , catalytic properties for inert noble metals , or electrical conductivity for insulators . Metal-organic framework nanocrystals (nanoMOFs) are porous solids assembled from metal ions/oxoclusters and organic linkers . The reduction of the MOFs size to the nanoscale has imparted nanoMOFs with various enhanced properties (i.e., catalysis, sensing, biomedicine…) and novel features (flexibility, optical properties) , but despite advances, the design of ultra-small MOF nanoparticles still faces severe difficulties . This can be due either to the much larger unit-cell parameters of MOFs in comparison with inorganic nanomaterials and/or to the limited
chemical stability of targeted nanoMOFs. However, at this ultra-small size, the majority of the atoms of MOFs lie close to the external surface, decorated with larger cavities than the constitutive inner ones. This maximizes the interface for substrate interaction alongside largely decreased diffusion/desorption path length , naturally resulting in enhanced catalytic properties.
Defect engineering is a long term interest in crystalline nanomaterials, particularly due to the influence of vacant sites on catalysis . Interestingly, structural defects in MOFs have shown similar optimizations towards catalytic properties and/or gas separation . However, this is in most cases associated with a lower chemical stability due to the reduction of metal-ligand connectivity and/or the presence of additional accessible metal sites . UiO-66(Zr) or ( Benzene-1,4-dicarboxylic acid) is a prototypical zirconiumbased MOF with an excellent thermal and chemical stability due to its high metal-ligand connectivity ( 12 -connected mode) and robust Zr –
carboxylate bonds . Consequently, defect engineering in MOFs has been to date mainly focused on UiO-66(Zr) and its derivatives . The most typical method for the defect engineering in Zr -MOFs is the modulator-induced-defect approach (MIDA), where a monocarboxylate modulator (formate, acetate…) is added, binding preferentially with metal centers in place of the linker, leading to the missing ligand vacancy defects (Fig. 1a). As such, the defect content can be controlled by the amount of the modulator used .
During the MOF synthesis, the modulator binds to the nodes to produce crystals with lower connectivity, and consequently with larger size due to lower nucleation rate and crystal growth kinetics. Although this is a reliable way to produce MOF particles with tunable particle sizes , it is at the expense of control over the number of defects (Fig. 1a) . The MIDA strategy therefore prevents the synthesis of highly defective ultra-small nanoMOFs which are optimal candidates for catalysis. Notably, numerous reports have pointed out the importance of overcoming diffusion barriers, with MOF catalyzed reactions mainly taking place on the outer surface of the particles . This is the case particularly when the size of the substrate is comparable to the aperture size of the MOF’s pores. Although the substrates may partially diffuse inside the MOF framework, the desorption of the resulting products can be hampered by kinetic limitations. Relying on exfoliated high aspect ratio 2D porous nanosheets is an appealing alternative strategy to overcome these limitations . However, these nanomaterials are usually more challenging to prepare (or exfoliate) and/or exhibit usually a reduced stability compared with their related 3D counterparts. The MIDA approach, usually carried out in toxic N,Ndimethylformamide (DMF) at high temperature and pressure, has mainly been explored with UiO-66 with missing linker defect content of ca. . Additionally, it has not been extended to the functionalized derivatives and thus, the full potential of this defective nanoMOFs family has also not been exploited so far. Therefore, developing new versatile routes to both downsize robust nanoMOFs, whilst ensuring a high defect content and maintaining reasonable thermal/chemical stability of the ultra-small MOF nanoparticles to address challenging catalytic reactions is a key challenge to overcome.
We report here a sustainable route to produce a series of ultrasmall UiO-type MOFs ( ) with exceptionally high defect content. A set of advanced characterization techniques revealed that the large defect content on such small UiO-66 nanoparticles is attributed to the presence of missing linker defects and that the crystallization is growth-dominated. Noteworthy, this strategy is versatile and can be applied to many UiO-66(Zr)-X derivatives ( ), to
the Hf counterpart UiO-66( Hf ) and finally to other Zr -MOFs structures like the Zr fumarate MOF-801(Zr), resulting in ultra-small nanoMOFs with very high defect content. Additionally, our mild green synthetic conditions are far more sustainable than the traditional solvothermal routes, which is of interest to save energy and/or strongly reduce the quantity of hazardous wastes. Moreover, the nanoMOFs synthesized here present excellent catalytic performance in peptide bond hydrolysis showing much better reactivity, chemical diffusion, selectivity and stability than benchmark catalysts. Significantly, these nanoMOFs show bifunctionality as by simply changing the reaction solvent, the hydrolysis of peptide bonds can be replaced by the opposite condensation reaction, resulting in amide bond formation. Additionally, these nanoMOFs also show tailorable selectivity due to the molecularsieving effect.
Results and discussion
Materials synthesis and characterizations
To prepare high quality ultra-small nanoparticles of UiO-66(Zr) with high defect content, we first considered carefully the main relevant state-of-the-art strategies. For instance, the acidity of the solution was shown to significantly influence the kinetics of crystallization due to the changes in protonation state of the carboxylic acids that lead to faster kinetics at higher . In addition, the presence of water in the reaction mixture appeared to be a critical factor in determining the defects resulting from the formation of or bonds rather than Zr-ligand connections . Using a low synthesis temperature was also shown to be beneficial towards both the defect engineering and downsizing due to the inhibited formation of coordination bonds and to limited Ostwald ripening (illustrated in Fig. 1b) . Thus, to achieve our ambitious goal to prepare ultra-small nanoMOFs with a high defect content, we developed a simple green strategy that: (i) avoids the use of very acidic Zr salts (e.g., ) and slightly acidic modulators (e.g., formic acid) by using pre-synthesized acetate oxoclusters (Supplementary Fig. 1); (ii) discards DMF and replaces it by water and ethanol to avoid the release of formates upon DMF degradation (and enables a sustainable approach); (iii) ensures a faster dissolution of the organic linker by diluting the reaction media in ethanol, subsequently accelerating the synthesis kinetics; and (iv) is operated at room temperature, which is energy saving.
The initial synthesis of UiO-66(Zr) was performed by first mixing acetate capped oxoclusters with acetic acid. Water, ethanol, and benzene-1,4-dicarboxylic acid (BDC) were subsequently introduced in the oxocluster solution (see detail in SI). After 2 h at room
Fig. 1 | Comparison of MOF nanoparticles synthesis approaches. Scheme of (a) the conventional modulator-induced-defect approach (MIDA) for the size-defect tuning of MOFs, (b) our approach that produces ultra-small and highly defective
tetravalent-MOFs nanoparticles, Red, green, and brown represent O, Metal(IV), and C atoms, respectively; yellow sphere and green lines within the purple circles indicate the metal nodes and organic ligand, respectively.
temperature under stirring, the resulting solid showed a PXRD pattern (see Supplementary Fig. 2) in agreement with the theoretical diffraction pattern of UiO-66. Transmission Electron Microscope (TEM) indicated a particle size of (Supplementary Fig. 3). This value is close to the particle size ( 44 nm ) calculated from Scherrer equation, indicating that the particles are mainly single crystal domains. The adsorption (Supplementary Fig. 4) showed a type I isotherm with extremely high capacity ( g) and a calculated Brunauer-Emmett-Teller (BET) surface area ( 1617 , larger than the BET surface area of defect-free UiO-66(Zr) . Such a huge expansion in the surface area is indicative of the formation of a large amount of defects . No symmetryforbidden peaks at low angle ( at ca. 4 and ) were observed in the PXRD pattern of the sample, suggesting the absence of missing cluster defects that would lead to an ordered structure with reo topology (Supplementary Fig. 2) . Thus, we hypothesized the presence of missing linkers in our material. Fourier transform infrared spectroscopy (FTIR) demonstrated the absence of uncoordinated carboxylic acid residual groups in the washed materials, in agreement with the presence of linker defects rather than missing oxoclusters (Supplementary Fig. 5). Thermogravimetric analysis (TGA) under oxygen atmosphere evidenced that the 40 nm UiO-66 nanoMOF exhibited a very low ligand-to-metal ratio (linker: , Supplementary Fig. 6), in agreement with a high missing linker content. The number of missing ligands in our sample corresponds to one of the most defective UiO-66 reported so far and concomitantly, to the best of our knowledge, the surface area of 40 nm UiO-66 represents a comparably high value compared to the state-of-the-art .
The general method we developed to control the particle size is illustrated in Fig. 2a. As our first attempt led to highly defective 40 nm UiO-66 while using only 10 mL EtOH with 50 mg of BDC, the reaction media was further diluted stepwise with , respectively) enabling a better dissolution of the ligand. Interestingly, this led to a dramatic reduction of nanoparticle size, as evidenced by the PXRD patterns in Fig. 2b. Notably, when the volume of ethanol reached 80 mL , only a broad envelop of the main characteristic diffraction peaks of UiO-66 could be observed due to the considerable loss of long-range order, which is consistent with the Scherrer equation. The TEM images (Supplementary Figs. 3, 7, 8, and Figs. 2c, d) evidenced the precise control of downsizing down to between 4 nm and 6 nm . Nevertheless, the crystal lattice planes can still be observed by TEM (Fig. 2d), confirming the quality of the nanoparticles. The selected area electron diffraction (SAED) pattern only showed the characteristic rings of the UiO-66 nanoparticles with different crystal orientations (Fig. 2e), further proving the quality of these nanoMOFs, as well as homogeneous size distribution. Further enlargement of Fig. 2d(i) clearly shows the pores and lattice of a 4.4 nm nanoparticle, in good accordance with the structural model from Fig. 2d(ii). From the profile analysis on HRTEM images along (220) and (011) directions (Fig. 2f), the distances between two adjacent oxoclusters are highly homogeneous with an average value of 1.1 nm that is close to the theoretical one ( 1.2 nm ), suggesting the absence of oxocluster defects in our ultrasmall UiO-66 (HD-US-UiO-66). To be noted, such a small particle corresponds to only ca. 2 unit-cell dimensions, i.e., 8 unit-cells or 12 octahedral-cages per nanoparticle. This system therefore lies at the frontier between nanocrystals and discrete metal-organic polyhedra. However, in contrast with our ultrasmall particles (see after for the description of the thermal and chemical stability), Metal-Organic Cages/Polyhedra (MOCs/MOPs) often suffer strongly from poor chemical/hydrolytic stabilities and structural collapse upon activation, preventing their applications .
According to TGA, all the different as-prepared nanoparticles exhibited very similar linker content, with close to 2 missing linkers per formula (Fig. 2g). This, once combined with liquid phase nuclear
magnetic resonance (NMR) analysis (Supplementary Fig. 9) and Energy Dispersive X-ray spectroscopy (EDX) (Supplementary Fig. 10), leads to a general formula of . Note that the connection of crystal size to defectiveness in our syntheses is, to the best our knowledge, the first of its kind and allows for achieving MOF nanoparticles with defectiveness of up to 4.2 missing linkers per oxocluster, by far exceeding the commonly reported values (Supplementary Fig. 11) . Nitrogen porosimetry at 77 K on the activated HD-US-UiO-66 particles evidenced in all cases a high sorption capacity (from to , Fig. 2h) associated with a hysteresis. A decrease of adsorption capacity occurred upon downsizing, in line with a progressive increase of the external to internal surface ratio. Pore size distribution analyses (DFT model) indicated overall a preserved pore size (Fig. 2i), which is once again in line with the constant missing linker content. For the sake of comparison, we followed the conventional MIDA, as well as the synthetic parameters of 40 nm UiO-66 and carried out a set of synthetic experiments by reducing the amount of acetic acid. Noteworthy, the preparation of smaller particles, down to 5 nm , denoted as MI-US-UiO66 (modulator-induced ultrasmall UiO-66), is associated with lower number of defects ( 2.6 missing linkers per oxoclusters) in good accordance with the previous findings where the missing linker defect content strongly depended on the modulator quantity (see SI for details).
To further investigate the nature of the missing linker defects at atomic level, in situ FTIR spectroscopy in presence of acetonitrile- ( ) vapors was performed. The acidity of pristine UiO-66(Zr) is mostly assigned to its intrinsic Brønsted acid sites (four ), while, upon high temperature activation, additional Lewis acid sites associated to the defects (missing linker) are present. When introducing aliquots to 10 torr equilibrium pressure, three vibrational bands could be observed at 2306, 2301, and , associated with the chemisorption of on different Lewis and Brønsted acid sites (Fig. 3). The bands at 2306 and mostly dominate the spectra in the first three doses, indicating a strong interaction between the Lewis acid sites of the MOF and . Interestingly, these bands showed a slight blue shift, from to 2306 and , in comparison to the constant peak position of the physisorbed in other reported works . This clearly indicates a higher acidic strength of the sites, likely promoted by the large concentration of defects. Their concentration could be calculated by integrating the corresponding bands vs. the molar amount of introduced. The obtained value of corresponds to a much larger number of Lewis acid sites than commonly reported for much larger-sized defective UiO-66 via MIDA, e.g., typically around , which further demonstrates that our ultra-small UiO-66 nanoparticles exhibit a much higher degree of defects, being therefore particularly interesting for Lewis acid based catalysis.
Several hypotheses can be proposed to understand the formation of HD-US-UiO-66. First, the pre-formed acetate oxoclusters establish the pH of the solution near 4 , which favors ligand deprotonation of the carboxylic groups of the ligand and thus leads to a faster nucleation upon substitution of the terminal acetates from the oxoclusters in the presence of the dicarboxylate moieties. Then, the dilution upon addition of ethanol impairs the effective collision rate, and once combined with the high modulator content, might limit the crystal growth as well as Ostwald ripening. However, upon downsizing, the reaction kinetics becomes faster, from 3 h for the largest particles to less than 1 h for the particles. Thus, other parameters are likely in play. For instance, the solubility of the ligand is limited at RT in ethanol and therefore, the proportion of ligand that is solubilized increases with the ethanol dilution, which favors a faster kinetics for the smallest particles. To validate this hypothesis, considering the much better solubility of BDC in DMF, we replaced of EtOH by
Fig. 2 | Characterizations of the synthesized UiO-66 nanoparticles. a Schematic diagram of our strategy, (b) powder X-ray diffraction (PXRD) ( ) patterns of UiO-66 synthesized with different volumes of EtOH, the yellow lines evidence the diffraction peaks from the calculated pattern, (c) statistical mean size of the synthesized UiO-66, (d) TEM image of the 5 nm UiO-66 (obtained with 80 mL EtOH, highlighted using yellow circles), i) enlarged selected zone, ii) structure of UiO-66 viewed from (101) axis direction, (e) SAED pattern of the 5 nm UiO-66, (f) High-resolution Transmission Electron Microscope (HRTEM) images of HD-US-UiO-
66 and their contrast intensity profiles, viewed along (i) (220) and (ii) (011) directions, scale bar , the green rectangles represent the selected regions for contrast analysis, (g) TGA under oxygen atmosphere (scan rate of ) of the UiO-66 with different sizes, inserted bar chart: linker to ratio of the different samples; (h) sorption isotherms of UiO-66 with different sizes ( bar), adsorption and desorption are represented by filled spheres, and open spheres, respectively, i) pore size distribution for different sizes of UiO-66 (same color label as in (h)).
DMF whilst keeping all other synthetic parameters constant. The resulting nanoparticles were found to be only ca. 8 nm instead of 40 nm in comparison with the use of EtOH (Supplementary Fig. 18), which corroborates the influence of the linker’s solubility on the kinetics. Such a bottleneck was observed previously by some of us when increasing the size of the dicarboxylic acid organic spacer over the crystallization under solvothermal conditions of UiO-66(Zr) and its extended analogs .
To gain further understanding about the formation mechanism of the HD-US-UiO-66, in situ time-dependent dynamic light scattering experiments (TD-DLS) were conducted. Figure 4 a showed the
evolution of particle size as a function of crystallization time of HD-US-UiO-66. The fast size increase process in the first 15 min was attributed to the formation of MOF nuclei. The MOF’s growth was observed in the range between 15 min and 130 min and reached saturation after 130 min with a hydrodynamic size at ca. 18 nm . The clear slope revealed that the crystallization of HD-US-UiO-66 follows a growthdominated process, in good agreement with the role of linker dissolution. The very low polydispersity index (Pdi) (Supplementary Fig. 19) implied a homogeneous nucleation followed by crystal growth of UiO-66 in solution. To confirm the growth kinetics of HD-US-UiO-66, an ex-situ HRTEM/STEM study was carried out after , and
180 min . Figure 4 b shows that only oxoclusters (ca. 0.6 nm ) were observed before the introduction of BDC. Ultra-small nanoparticles (around 1.3 nm ) were formed very quickly as soon as the ligand was added ( 1 min ), which indicates the very fast nucleation of UiO-66 nanoparticles in solution, in agreement with the stage (i) shown in Fig. 4 a . The TEM images at 120 and 180 min demonstrated the growth of the small MOF nuclei and the saturation of nanoparticles growth. These results are fully consistent with TD-DLS, confirming that the ligand dissolution acts as the bottleneck in controlling the MOF growth. Notably, the in situ TD-DLS not only sheds light on the crystallization process but also strongly highlighted the excellent colloidal stability of the HD-US-UiO-66. This stability, correlated to the highly positive charge evaluated by Zeta potential analysis, was observed whatever the nanocrystal size (Figs. S20, S21) and is a strong asset for their solution processability in a view of applications such as ultrathin film fabrication, drug delivery, sensing and electronics, among others .
One very appealing feature of MOFs is their ligand functionalization to achieve desired properties. Thus, we extended the synthetic strategy to produce other functional HD-US-UiO-66-X derivatives,
Fig. 3 | Characterization of the accessible acid sites by in situ FTIR. In situ FTIR spectra at 298 K of (red to gray, probe small doses to up to 10 torr equilibrium pressure) adsorbed on HD-US-UiO-66 ( 5 nm ), the peaks position and peak area are shown in dotted lines and red area, respectively, the small doses and equilibrium indicate the increasing dosing from 0.2 torr to saturated 6.4 torr.
including , and Br , as well as replacing by oxoclusters and BDC-based planar linkers by fumarate. All these ultrasmall nanoMOFs exhibited 2-3 unit-cell particle sizes and very similar connectivities (ca. 3.3-4 linkers per formula) as the pristine HD-US-UiO-66 (see detailed analysis in SI).
Heterogeneous catalysis
In light of the properties of our HD-US-UiO-66-X, we decided to explore their use in heterogeneous catalysis . The challenging hydrolysis of the peptide bond in glycylglycine (GG) was selected as the model reaction to investigate the significance of downsizing/ defect formation of MOF nanoparticles on the overall catalytic performance (Fig. 5a). HD-US-UiO-66-NH2 nanoMOF was subsequently selected for catalysis due to the potential H -bonding that could occur between the peptide substrate and the – group on the ligand, or between the – group and nucleophile, which could contribute to the overall catalysis. As anticipated, the use of HD-US-UiO- led to ca. 3 times higher reactivity compared to conventional UiO-66 particles (ca. 200 nm ) reported previously (Fig. 5b) . Such an enhancement might be due to the significantly expanded external surface area and/or the large defect content. Thus, we have compared the performance of HD-US-UiO-66-NH2 and MI-US-UiO-66-NH2 nanoMOFs with the previous study. Noteworthy, although MI-US-UiO-66 showed better reactivity compared to the previous study, it performed worse (ca. 2 times) than HD-US-UiO-66-NH2, despite their very similar particle size. This suggests that maximizing the amount of missing linker defects is critical to enhance the reactivity of UiO-66 derivatives towards peptide bond hydrolysis, in combination with decreasing the particle size. Notably, the reactivity here is comparable to benchmarks materials such as the “superactive” MOF-808 ( 35 nm ), which exhibited a GG hydrolysis rate of .
The recyclability of HD-US-UiO-66-NH2 nanoMOF was tested over five subsequent reaction cycles (Supplementary Fig. 35), with the slight loss of reactivity being only recorded after the cycle, where the reactivity dropped to of the performance observed in cycle 1 . This is however still an excellent recyclability, especially when compared to benchmark materials, such as MIP-201 and MOF-808 , which suffered from lower recyclability (only and activity after 5 cycles, respectively, Fig. 5d), and highlights the water stability of these ultrasmall highly defective nanoMOFs.
The specificity of the catalyst was further studied, as GG may be hydrolyzed to G , or may undergo cyclisation (amide bond formation), forming cyclic GG (cGG) (Supplementary Fig. 32). By using HD-UiO-66 nanoMOFs of two different sizes ( 4 nm vs 200 nm ), the influence on particle size and external vs internal surface area on the reaction specificity was examined. The 200 nm UiO- is labeled as HD-200-UiO-66-NH2, and was fully characterized prior to catalytic reaction
Fig. 4 | In situ and ex-situ characterizations of the MOF growth. a Hydrodynamic size of HD-US-UiO- 66 colloids ( C) determined by in situ time-dependent DLS (time resolution ), and the (b) ex-situ HAADF-STEM ( ) and HRTEM ( ) images of HD-US-UiO-66 at different times.
Fig. 5 | Catalytic performance evaluation of the HD-US-UiO-66-X. a llustration of peptide hydrolysis using HD-US-UiO-66, (b) Pseudo first order hydrolysis rate of glycylglycine (GG) to glycine (G) using HD-US-UiO-66- and MI-US-UiO-66- nanoMOFs, reference refers to the value reported in the previous studies under the same conditions , (c) Selectivity of hydrolysis by HD-US-UiO-66-NH2 and HD-200-UiO-66- in producing the desired product G , as the starting concentration of
GG increases from 2 mM to 500 mM , (d) Recyclability of HD-US-UiO-66-NH2 over 5 reaction cycles in comparison to best-performing MOFs to date, percentage compared to yield of cycle 1, (e) illustration of amide bond condensation using HD-US-UiO-66, in MeOH, (f) Amide bond formation yield with HD-US-UiO-66 and HD-US-UiO-66- starting from , and AG.
(Supplementary Figs. 36-38). Interestingly, the HD-US-UiO-66-NH2 was found to be more selective, consistently giving a greater proportion of hydrolysis product G , rather than CGG , regardless of the starting concentration of GG substrate (Fig. 5c). The HD-200-UiO-66-NH2 produced considerably more cGG ( of product), likely due to the increased internal surface area compared to HD-US-UiO- , which promotes amide bond condensation in the absence of water in the more hydrophobic pores, rather than hydrolysis reaction at the water exposed external surface of the nanoMOF.
As cGG was found to be a side product of hydrolysis with HD-200-UiO-66-NH2, the nanoMOFs were further examined for their ability to catalyze the peptide bond formation between amino acids and small peptides. The condensation reaction can be promoted instead of hydrolysis simply by changing the solvent from water to methanol . Both HD-US-UiO-66-NH2 and HD-US-UiO-66 nanoMOFs showed the ability to promote amide bond formation in MeOH (Fig. 5e, f), but the HD-US-UiO-66 exhibited much higher reactivity than the functionalized HD-US-UiO-66-NH2. Similar to the previous report , for both MOFs the intramolecular peptide bond formation was favored over intermolecular condensation between individual glycine molecules (Reaction 3, Fig. 5f). Interestingly, the functionalized HD-US-UiO-66-NH2 was ineffective in forming cGG when starting from GG (Reaction 2), but the fact that cGG was observed in other two reactions shown in Fig. 5f, suggests that the efficiency of intramolecular bond formation is influenced by substrate interaction with the MOF, rather than by a reduced catalytic activity. These differences in the interactions could be influenced by the presence of the electron donating – groups on the MOF, as well as by the MOF pore sizes, as the functionalized MOF exhibits slightly smaller pores due to the steric hindrance of the group, which increases the diffusion barrier of the substrate into and out of the pores of the MOF.
In this work, we have reported a general strategy for the preparation of a series of highly defective ( missing linker) and ultra-small ( ) UiO-66-based nanocrystals under fully sustainable conditions, suggesting feasibility towards upscaling. Crystal growth acts as a bottleneck in crystallization, as evidenced by in situ TD-DLS with ex-situ HRTEM/STEM, and can be manipulated by simply using additional solvent. Missing linker defects have been assessed by multiple advanced techniques, including PXRD, FTIR, TGA, HRTEM and in situ FTIR spectroscopy coupled with probe, which revealed the importance of Lewis-acidity of the HD-US-UiO-66. The resulting HD-US-UiO-66-X showed excellent catalytic performance in both peptide bond hydrolysis and formation with catalytic reactivity, selectivity, product recovery efficiency, and recyclability compared to other reported materials. Detailed investigation of the influence of defects, particle size and functionalization on the catalytic activity of the nanoMOFs provided unique insights into the key parameters that influence the reactivity, and as such demonstrate how nanoMOFs can be tuned to show specific and selective reactivity through precise control of catalysts’ properties. Therefore, the discoveries reported here might further promote the development of nanoMOFs as heterogeneous catalysts having dual functions and performance enhancements in varying aspects. Furthermore, the materials presented here may be also used for the development of sensing/optical devices (preliminary results in Supplementary Discussion, and Supplementary Fig. 24), membranes, nanomedicine formulations and to explore other fundamental size-dependent properties.
Methods
Synthesis of oxoclusters
was added into a mixture of 3 mL of glacial acetic acid and 5 mL of isopropanol under stirring at 500 rpm and heated at for 60 min . The product was collected either through suction filtration or centrifugation at . The collected white solid was subsequently washed with acetone twice and dried under vacuum at RT. The synthesized oxoclusters could be stored in ambient condition for at least 12 months without properties change. The prepared oxoclusters have the following formula: .
Synthesis of oxoclusters
was added into a mixture of 4 mL of glacial acetic acid and 18 mL of isopropanol under stirring at 500 rpm and heated at for 60 min . The product was collected either through suction filtration or centrifugation at . The collected white solid was subsequently washed with acetone twice and dried under vacuum at RT.
Synthesis of
oxoclusters ( ) were dispersed in acetic acid under stirring at was subsequently added, and the reaction mixture was stirred until it became completely clear. 10 mL of ethanol was introduced into the solution followed by the immediate addition of 1,4-benzenedicarboxylic acid (BDC, ), and the reaction was stirred for 3 h at room temperature. The resulting solution was centrifuged at for 45 min and then washed twice with ethanol ( ). The collected solid was dried under vacuum for 3 h for characterizations and applications.
Synthesis of MI-US-UiO-66 ( )
oxoclusters ( ) were dispersed in acetic acid ( ) under stirring at was subsequently added, and the reaction mixture was stirred until it became completely clear. 10 mL of ethanol was introduced into the solution followed by the immediate addition of 1,4-benzenedicarboxylic acid (BDC, ), and the reaction was stirred for 1 h at room temperature. The resulting solution was centrifuged at for 45 min and then washed twice with ethanol ( ). The collected solid was dried under vacuum for 3 h for characterizations and applications. Modulator-induced size control can be applied with different amount of acetic acid used in the synthesis batch.
Synthesis of HD-US-UiO-66 ( )
oxoclusters ( ) were dispersed in acetic acid ( ) under stirring at was subsequently added, and the reaction mixture was stirred until it became completely clear. 80 mL of ethanol was introduced into the solution followed by the immediate addition of 1,4-benzenedicarboxylic acid (BDC, ), and the reaction was stirred for 2 h at room temperature. The resulting solution was evaporated by rotary evaporation at room temperature until volume was left. The colloidal suspension was centrifuged at for 60 min and then washed twice with the mixture of 20 mL of acetone and 20 mL of ethanol ( ). The collected solid was dried under vacuum for 3 h for characterizations and applications. The size control approach here can be applied with different amount of ethanol used in the synthesis batch.
Synthesis of HD-US-UiO-66-NH2 ( )
oxoclusters ( ) were dispersed in acetic acid ( ) under stirring at was subsequently added, and the reaction mixture was stirred until it became completely clear. 80 mL of ethanol was introduced into the solution followed by the immediate addition of 2 -aminobenzene-1,4dicarboxylic acid (BDC-NH2, ), and the reaction was
stirred for 1 h at room temperature. The resulting solution was evaporated by rotary evaporation at room temperature until volume was left. The colloidal suspension was centrifuged at for 60 min and then washed twice with the mixture of 20 mL of acetone and 20 mL of ethanol ( ). The collected solid was dried under vacuum for 3 h for characterizations and applications.
Glycylglycine hydrolysis
Prior to hydrolysis, MOFs were activated at for 20 h . To a 1 mL glass vial was added moles of MOF and . Next, of a 40 mM solution of Glycylglycine in was added, the mixture pD was adjusted to 7.4 and incubated at with stirring. Vessel was prepared per time point in triplicate. Reactions were stopped at , , and 48 h and centrifuged at for 10 minutes. of supernatant was analyzed with using of 0.1 M TMSP internal standard. MOFs were washed in acetone and analyzed with PXRD after reaction to check their structure integrity.
For recycling experiments, after reaction, the MOFs were washed in overnight to remove adsorbed substrate and product, followed by washing in 10 mL of acetone and drying in the oven for 8 h at . After which the reaction was repeated.
Peptide bond formation
MOFs were used after activation. To a 10 mL crimp cap vial was added 5 moles of MOF and moles of substrate (glycylglycine, L-alanylglycine or glycine) followed by 1 mL methanol. The mixtures were sealed and stirred, and then incubated at for 24 h with stirring. After incubation, samples were diluted with and stirred at room temperature for 1 h to elute substrates and products from the MOFs. After which the supernatant was collected via centrifugation at for of supernatant was then diluted with to give a final volume of , which was analyzed with using of 0.1 M TMSP- internal standard.
Data availability
All data supporting the finding of this study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Roduner, E. Size matters: why nanomaterials are different. Chem. Soc. Rev. 35, 583-592 (2006).
Chen, Y. et al. Phase engineering of nanomaterials. Nat. Rev. Chem. 4, 243-256 (2020).
Wang, Y. & Herron, N. Nanometer-sized semiconductor clustersmaterials synthesis, quantum size effects, and photophysical properties. J. Phys. Chem. 95, 525-532 (1991).
Valden, M., Lai, X. & Goodman, D. W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 281, 1647-1650 (1998).
Zhang, P. et al. Electronic transport in nanometre-scale silicon-oninsulator membranes. Nature 439, 703-706 (2006).
Cai, X. C. et al. Nano-sized metal-organic frameworks: synthesis and applications. Coord. Chem. Rev. 417, 213366 (2020).
Protesescu, L. et al. Colloidal nano-MOFs nucleate and stabilize ultra-small quantum dots of lead bromide perovskites. Chem. Sci. 12, 6129-6135 (2021).
Sakata, Y. et al. Shape-memory nanopores induced in coordination frameworks by crystal downsizing. Science 339, 193-196 (2013).
Krause, S. et al. The effect of crystallite size on pressure amplification in switchable porous solids. Nat. Commun. 9, 1573 (2018).
Horcajada, P. et al. Porous metal-organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat. Mater. 9, 172-178 (2010).
Marshall, C. R. et al. Size-dependent properties of solutionprocessable conductive MOF nanocrystals. J. Am. Chem. Soc. 144, 5784-5794 (2022).
Fabrizio, K. & Brozek, C. K. Size-dependent thermal shifts to MOF nanocrystal optical gaps induced by dynamic bonding. Nano Lett. 23, 925-930 (2023).
Xia, H.-L. et al. Size- and emission-controlled synthesis of full-color luminescent metal-organic frameworks for tryptophan detection. Angew. Chem. Int. Ed. 62, e202308506 (2023).
Zhao, M. et al. Ultrathin 2D metal-organic framework nanosheets. Adv. Mater. 27, 7372-7378 (2015).
Nowotny, M. K., Sheppard, L. R., Bak, T. & Nowotny, J. Defect chemistry of titanium dioxide. Application of defect engineering in processing of -based photocatalysts. J. Phys. Chem. C 112, 5275-5300 (2008).
Busch, G. Early history of the physics and chemistry of semiconductors-from doubts to fact in a hundred years. Eur. J. Phys. 10, 254 (1989).
Furukawa, H., Müller, U. & Yaghi, O. M. Heterogeneity within order” in metal-organic frameworks. Angew. Chem. Int. Ed. 54, 3417-3430 (2015).
Dissegna, S., Epp, K., Heinz, W. R., Kieslich, G. & Fischer, R. A. Defective metal-organic frameworks. Adv. Mater. 30, 1704501 (2018).
Cavka, J. H. et al. A new zirconium inorganic building brick forming metal-organic frameworks with exceptional stability. J. Am. Chem. Soc. 130, 13850-13851 (2008).
Wu, H. et al. Unusual and highly tunable missing-linker defects in zirconium metal-organic framework UiO-66 and their important effects on gas adsorption. J. Am. Chem. Soc. 135, 10525-10532 (2013).
Vermoortele, F. et al. Synthesis modulation as a tool to increase the catalytic activity of metal-organic frameworks: the unique case of UiO-66 (Zr). J. Am. Chem. Soc. 135, 11465-11468 (2013).
Shearer, G. C. et al. Defect engineering: tuning the porosity and composition of the metal-organic framework UiO-66 via modulated synthesis. Chem. Mater. 28, 3749-3761 (2016).
Liang, W. et al. Defect engineering of UiO-66 for and uptake-a combined experimental and simulation study. Dalton Trans. 45, 4496-4500 (2016).
Schaate, A. et al. Modulated synthesis of Zr-based metal-organic frameworks: from nano to single crystals. Chem. Eur. J. 17, 6643-6651 (2011).
Forgan, R. S. Modulated self-assembly of metal-organic frameworks. Chem. Sci. 11, 4546-4562 (2020).
Fang, Z. et al. Structural complexity in metal-organic frameworks: simultaneous modification of open metal sites and hierarchical porosity by systematic doping with defective linkers. J. Am. Chem. Soc. 136, 9627-9636 (2014).
Taylor, J. M., Dekura, S., Ikeda, R. & Kitagawa, H. Defect control to enhance proton conductivity in a metal-organic framework. Chem. Mater. 27, 2286-2289 (2015).
Abánades Lázaro, I., Wells, C. J. & Forgan, R. S. Multivariate modulation of the Zr MOF UiO-66 for defect-controlled combination anticancer drug delivery. Angew. Chem. Int. Ed. 59, 5211-5217 (2020).
Wei, R. et al. Tuning the properties of nodes in the metal organic framework UiO-66 by selection of node-bound ligands and linkers. Chem. Mater. 31, 1655-1663 (2019).
Gao, W. Y., Cardenal, A. D., Wang, C. H. & Powers, D. C. In Operando analysis of diffusion in porous metal-organic framework catalysts. Chem. Eur. J. 25, 3465-3476 (2019).
Sharp, C. H. et al. Nanoconfinement and mass transport in metal-organic frameworks. Chem. Soc. Rev. 50, 11530-11558 (2021).
Wang, M., Dong, R. & Feng, X. Two-dimensional conjugated metal-organic frameworks (2D c-MOFs): chemistry and function for MOFtronics. Chem. Soc. Rev. 50, 2764-2793 (2021).
Decker, G. E., Stillman, Z., Attia, L., Fromen, C. A. & Bloch, E. D. Controlling size, defectiveness, and fluorescence in nanoparticle UiO-66 through water and ligand modulation. Chem. Mater. 31, 4831-4839 (2019).
DeStefano, M. R., Islamoglu, T., Garibay, S. J., Hupp, J. T. & Farha, O. K. Room-temperature synthesis of UiO-66 and thermal modulation of densities of defect sites. Chem. Mater. 29, 1357-1361 (2017).
Dai, S., Nouar, F., Zhang, S., Tissot, A. & Serre, C. One-step roomtemperature synthesis of metal (IV) carboxylate metal-organic frameworks. Angew. Chem. Int. Ed. 133, 4328-4334 (2021).
Dai et al. Monodispersed MOF-808 nanocrystals synthesized via a scalable room-temperature approach for efficient heterogeneous peptide bond hydrolysis. Chem. Mater. 33, 7057-7066 (2021).
Firth, F. C. et al. Engineering new defective phases of UiO family metal-organic frameworks with water. J. Mater. Chem. A 7, 7459-7469 (2019).
Liu, L. et al. Imaging defects and their evolution in a metal-organic framework at sub-unit-cell resolution. Nat. Chem. 11, 622-628 (2019).
Wang, X. et al. Robust ultrathin nanoporous MOF membrane with intra-crystalline defects for fast water transport. Nat. Commun. 13, 1-11 (2022).
Sánchez-González, E., Tsang, M. Y., Troyano, J., Craig, G. A. & Furukawa, S. Assembling metal-organic cages as porous materials. Chem. Soc. Rev. 51, 4876-4889 (2022).
Ragon, F. et al. Acid-functionalized UiO-66 (Zr) MOFs and their evolution after intra-framework cross-linking: structural features and sorption properties. J. Mater. Chem. A 3, 3294-3309 (2015).
Chakarova, K., Strauss, I., Mihaylov, M., Drenchev, N. & Hadjiivanov, K. Evolution of acid and basic sites in UiO-66 and UiO-66-NH2 metalorganic frameworks: FTIR study by probe molecules. Microporous Mesoporous Mater. 281, 110-122 (2019).
Ragon, F., Chevreau, H., Devic, T., Serre, C. & Horcajada, P. Impact of the Nature of the Organic Spacer on the Crystallization Kinetics of UiO-66(Zr)-Type MOFs. Chem. Eur. J. 21, 7135-7143 (2015).
Sindoro, M., Yanai, N., Jee, A.-Y. & Granick, S. Colloidal-sized metal-organic frameworks: synthesis and applications. Acc. Chem. Res. 47, 459-469 (2014).
Simms, C., Mullaliu, A., Swinnen, S., de Azambuja, F. & Parac-Vogt, T. N. MOF catalysis meets biochemistry: molecular insights from the hydrolytic activity of MOFs towards biomolecules. Mol. Syst. Des. Eng. 8, 270-288 (2023).
Ly, H. G. T., Fu, G., de Azambuja, F., De Vos, D. & Parac-Vogt, T. N. Nanozymatic activity of UiO-66 metal-organic frameworks: tuning the nanopore environment enhances hydrolytic activity toward peptide bonds. ACS Appl. Nano Mater. 3, 8931-8938 (2020).
Ly, H. G. T. et al. Superactivity of MOF-808 toward peptide bond hydrolysis. J. Am. Chem. Soc. 140, 6325-6335 (2018).
Wang et al. A zirconium metal-organic framework with SOC topological net for catalytic peptide bond hydrolysis. Nat. Commun. 13, 1-8 (2022).
de Azambuja, F. et al. En route to a heterogeneous catalytic direct peptide bond formation by Zr -based metal-organic framework catalysts. ACS Catal. 11, 7647-7658 (2021).
Acknowledgements
S.D., C. Serre, and M.D. acknowledge the European Union’s Horizon 2020 research and innovation program under grant agreement No. 831975 (MOF4AIR project) for providing financial support. C. Simms thanks the Research Foundation-Flanders (FWO) for the fellowship grant (68090/11C9320N). The authors appreciate the help of Dr. X. Xu for TEM measurements. S.D., A.T., G.P., and C. Serre acknowledged the Paris
region, through the DIM Respore priority project, for the access to HRTEM.
Author contributions
S.D., A.T., and C. Serre conceived the research. S.D., C. Simms, and T.V. designed the experiments and analyzed the data. G.P. conducted the STEM/HRTEM analysis. M.D. supervised the in situ FTIR investigations. S.D. wrote the original draft with the help of C. Simms. All authors contributed in the paper discussion and revision. A.T. and C. Serre supervised the project.
Correspondence and requests for materials should be addressed to Antoine Tissot, Tatjana N. Parac-Vogt or Christian Serre.
Peer review information Nature Communications thanks Carol Hua, Stefano Canossa, Ravichandar Babarao, and the other, anonymous,
reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Reprints and permissions information is available at
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
Institut des Matériaux Poreux de Paris, Ecole Normale Supérieure, ESPCI Paris, CNRS, PSL University, 75005 Paris, France. Normandie Université, ENSICAEN, UNICAEN, CNRS, Laboratoire Catalyse et Spectrochimie, 14000 Caen, France. Laboratory of Bioinorganic Chemistry, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. Université Paris-Saclay, CNRS, Centre de Nanosciences et de Nanotechnologies, 91120 Palaiseau, France.