بمبرووليزوماب والعلاج الكيميائي في سرطان الثدي المبكر عالي المخاطر، ER+/HER2−: تجربة عشوائية من المرحلة الثالثة Pembrolizumab and chemotherapy in high-risk, early-stage, ER+/HER2− breast cancer: a randomized phase 3 trial
بمبرووليزوماب والعلاج الكيميائي في سرطان الثدي المبكر عالي المخاطر، ER+/HER2−: تجربة عشوائية من المرحلة الثالثة
تاريخ الاستلام: 17 يوليو 2024 تم القبول: 12 نوفمبر 2024 نُشر على الإنترنت: 21 يناير 2025 تحقق من التحديثات
تظهر قائمة بالمؤلفين وانتماءاتهم في نهاية الورقة.
أدى إضافة بيمبروليزوماب إلى العلاج الكيميائي المساعد يليه بيمبروليزوماب المساعد إلى تحسين النتائج لدى المرضى الذين يعانون من سرطان الثدي الثلاثي السلبي في مراحله المبكرة وعالي المخاطر. ومع ذلك، فإن ما إذا كانت إضافة بيمبروليزوماب المساعد إلى العلاج الكيميائي ستؤدي إلى تحسين النتائج في حالات سرطان الثدي الإيجابي لمستقبلات الاستروجين/سلبي لمستقبلات عامل نمو البشرة البشري 2 (ERيبقى سبب سرطان الثدي غير واضح. أجرينا دراسة مزدوجة التعمية، خاضعة للرقابة باستخدام الدواء الوهمي في المرحلة الثالثة (KEYNOTE-756) حيث تم تضمين مرضى لم يتلقوا علاجًا سابقًا لـ ERسرطان الثدي الغازي عالي المخاطر من الدرجة الثالثة (T1c-2 ( )، cN1-2 أو T3-4، cNO-2) تم تعيينهم عشوائيًا (1:1) لتلقي البمبروليزوماب 200 ملغ أو الدواء الوهمي كل 3 أسابيع مع الباكليتاكسيل أسبوعيًا لمدة 12 أسبوعًا، تليها أربع دورات من الدوكسوروبيسين أو الإبيروبسين بالإضافة إلى السيكلوفوسفاميد كل أسبوعين أو كل 3 أسابيع. بعد الجراحة (مع/بدون العلاج الإشعاعي المساعد)، تلقى المرضى البمبروليزوماب المساعد أو الدواء الوهمي لتسع دورات بالإضافة إلى العلاج الهرموني المساعد. كانت الأهداف الأولية المزدوجة هي الاستجابة الكاملة المرضية والبقاء خاليًا من الأحداث في مجموعة النية للعلاج. في المجموع، تم تعيين 635 مريضًا إلى ذراع البمبروليزوماب-العلاج الكيميائي و643 إلى ذراع الدواء الوهمي-العلاج الكيميائي. في التحليل المؤقت الأول المحدد مسبقًا للدراسة، كانت نسبة الاستجابة الكاملة المرضية ( فترة الثقة (CI)، 21.0-27.8%) في مجموعة البمبروليزوماب-العلاج الكيميائي و ( ) في ذراع العلاج الوهمي-العلاج الكيميائي (فرق العلاج المقدر، 8.5 نقطة مئوية؛ 95% CI، 4.2-12.8؛ ). لم تكن فترة البقاء خالية من الأحداث ناضجة في هذا التحليل. خلال المرحلة المساعدة قبل الجراحة، كانت الأحداث السلبية المتعلقة بالعلاج من الدرجة تم الإبلاغ عنها في و للمرضى في ذراعي العلاج الكيميائي مع البمبروليزوماب والعلاج الكيميائي مع الدواء الوهمي، على التوالي. باختصار، فإن إضافة البمبروليزوماب إلى العلاج الكيميائي المساعد قد حسنت بشكل كبير من معدل الاستجابة الكاملة المرضية لدى المرضى ذوي المخاطر العالية في المراحل المبكرة من ER.سرطان الثدي. كانت السلامة متوافقة مع الملفات المعروفة لكل علاج دراسي. يستمر المتابعة من أجل البقاء خالياً من الأحداث.ClinicalTrials.govمعرف: NCT03725059.
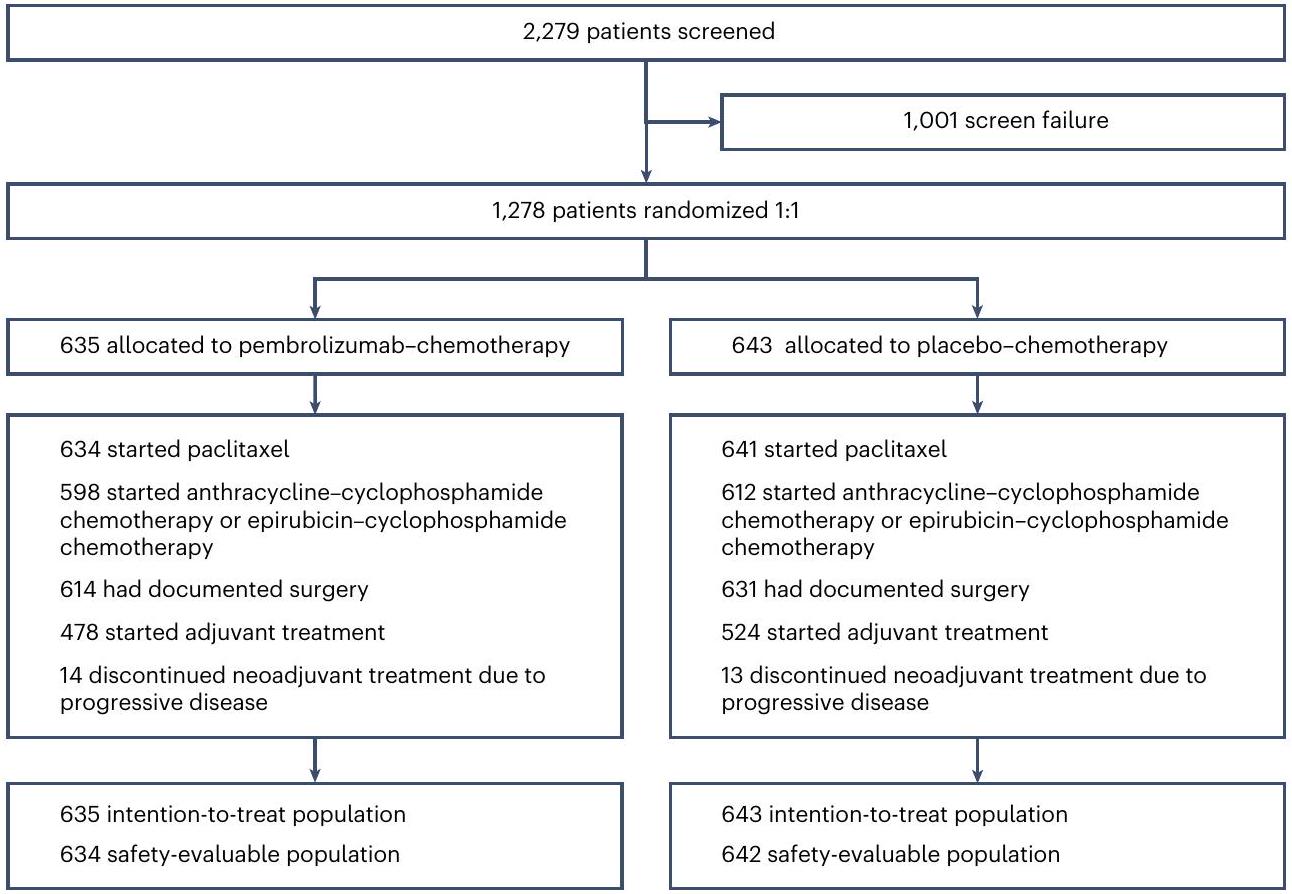
الشكل 1 | توزيع المرضى في الدراسة. تضمنت المرضى المتقدمة المرض المتقدم بالأشعة. لم يكن من الضروري أن يكمل المرضى جميع العلاج المساعد للخضوع للجراحة.
مستقبلات الإستروجين الإيجابية / مستقبلات عامل نمو البشرة البشري 2 السلبيةسرطان الثدي هو مرض غير متجانس يتضمن مجموعة فرعية من المرضى (بما في ذلك أولئك الذين يعانون من أورام عالية الدرجة ووجود الغدد اللمفاوية) في خطر مرتفع من الانتكاس والذين لديهم نتائج طويلة الأمد سيئة على الرغم من العلاج الكيميائي المساعد (النيودي) والعلاج الهرموني المساعد.. بالنسبة لهؤلاء المرضى، تتراوح معدلات الاستجابة الكاملة المرضية المبلغ عنها (pCR) من إلىومعدلات البقاء بدون أحداث (EFS) مشابهة لتلك الخاصة بالمرضى الذين يعانون من سرطان الثدي الثلاثي السلبي (TNBC). على الرغم من المرض متنوع، في المرضى الذين يعانون منسرطان الثدي، أظهرت دراسة تحليلية شاملة للدراسات السابقة للعلاج المساعد وجود علاقة إيجابية بين الاستجابة المرضية الكاملة (pCR) وكلاً من فترة البقاء الخالية من الأحداث (EFS) والبقاء العام (OS).تدعم الإرشادات التنظيمية استخدام pCR كنقطة نهاية مناسبة لتقييم فعالية العلاج المساعد..
العلاج المركب مع مثبطات نقاط التفتيش المناعية بالإضافة إلى العلاج الكيميائي يحدث تغييرات في بيئة الورم الدقيقة قد تعزز المناعة المضادة للسرطان الذاتية، وتقلل من حجم الورم وتزيد من معدل الاستجابة مقارنة بالعلاج الكيميائي وحده.يدعم هذه الفرضية، حيث أظهرت البيانات السريرية أن الأجسام المضادة وحيدة النسيلة المضادة لبروتين الموت الخلوي المبرمج 1 (anti-PD-1) بيمبروليزوماب، عند دمجه مع العلاج الكيميائي المساعد في التجربة السريرية المرحلة الثانية I-SPY2، قد زاد من معدلات الاستجابة الكاملة المحتملة (pCR) المقدرة لأكثر من الضعف للمرضى ذوي المخاطر العالية (المحددة بواسطة درجة ماما برينت)./HER2الأورام مقارنة بالمرضى الذين يتلقون العلاج الكيميائي المساعد فقط ضد على التوالي. بالإضافة إلى ذلك، أظهر استخدام بيمبروليزوماب مع العلاج الكيميائي المساعد تحسينًا في معدل الاستجابة الكاملة المرضية (pCR) ومدة البقاء الخالية من الأحداث (EFS) لدى المرضى الذين يعانون من سرطان الثدي الثلاثي السلبي في مراحله المبكرة..
استنادًا إلى هذه النتائج، تم تصميم KEYNOTE-756 (NCT03725059) كدراسة عشوائية مزدوجة التعمية من المرحلة 3 لتقييم فعالية وسلامة العلاج المساعد باستخدام بيمبروليزوماب بالإضافة إلى العلاج الكيميائي، تليها بيمبروليزوماب المساعد بالإضافة إلى العلاج الهرموني، مقابل العلاج الوهمي المساعد بالإضافة إلى العلاج الكيميائي، تليها العلاج الوهمي المساعد بالإضافة إلى العلاج الهرموني في المرضى ذوي المخاطر العالية، في المراحل المبكرة.سرطان الثدي.
النتائج
المرضى والعلاج
من 27 ديسمبر 2018 إلى 5 أغسطس 2022، تم تخصيص 1,278 مريضًا من 222 موقعًا عالميًا عشوائيًا إلى مجموعة العلاج بالبمبروليزوماب-العلاج الكيميائي (635 مريضًا) أو إلى مجموعة العلاج الوهمي-العلاج الكيميائي (643 مريضًا؛ الشكل 1). كانت الخصائص الديموغرافية وخصائص المرض الأساسية متوازنة بين مجموعات العلاج (الجدول 1).
في أول تحليل مؤقت (تاريخ قطع البيانات، 25 مايو 2023؛ متوسط مدة المتابعة، 33.2 (النطاق = 9.7-51.8) شهراً)، تلقى 1,275 مريضاً العلاج النيوأدجوانت الأول، وبدأ 1,210 مرضى العلاج النيوأدجوانت الثاني، وجرى توثيق إجراء الجراحة لـ 1,245 مريضاً وبدأ 1,002 مريض العلاج المساعد (الشكل 1). كانت مدة العلاج في المرحلة النيوأدجوانت 4.9 شهراً (النطاقالشهور) في مجموعة العلاج الكيميائي مع بيمبروليزوماب و 4.9 شهور (النطاقالشهور) في مجموعة العلاج الوهمي-العلاج الكيميائي (الجدول البياني الموسع 1). تلقت المجموعتان عددًا مشابهًا من دورات العلاج الكيميائي.
فعالية
تمت ملاحظة pCR (ypTO/Tis ypNO) في 154 من 635 مريضًا. ) في ذراع البمبروليزوماب-العلاج الكيميائي و 100 من 643 مريضًا في مجموعة العلاج الوهمي-العلاج الكيميائي. كانت الفجوة المقدرة في معدل الاستجابة الكاملة المرضية 8.5 نقطة مئوية (فترة الثقة (CI); الجدول 2). كانت معايير الدلالة الإحصائية المحددة مسبقًا لهذه التحليل هي ؛ وبالتالي، كانت نسبة المرضى الذين حققوا استجابة كاملة (pCR) أعلى بشكل ملحوظ في مجموعة العلاج الكيميائي مع بيمبروليزوماب مقارنة بمجموعة العلاج الكيميائي مع الدواء الوهمي. تم ملاحظة نتائج مماثلة فيما يتعلق بنسبة المرضى الذين حققوا استجابة كاملة (pCR) وفقًا للنقاط النهائية الثانوية لـ ypTO و ypNO و ypTO/Tis (الجدول 2).
فوائد البمبروليزوماب-العلاج الكيميائي مقارنة بالعلاج الكيميائي الوهمي بالنسبة لـ pCRكانت النتائج عمومًا متسقة عبر المجموعات الفرعية المحددة من خلال الخصائص السكانية والسريرية الأساسية (الشكل 2). ومن الجدير بالذكر أنه تم ملاحظة معدل أعلى عدديًا من فرق الاستجابة الكاملة (pCR) مع زيادة تعبير PD-L1 في الورم. كانت الفروقات المقدرة في العلاج في المجموعات الفرعية المحددة مسبقًا بناءً على
الجدول 1 | الخصائص السكانية وخصائص المرض الأساسية
خاصية
بمبروليزوماب الكيميائي )
العلاج الكيميائي الوهمي )
العمر (سنة)
الوسيط (النطاق)
٤٩ (٢٤-٨٢)
٤٩ (١٩-٧٨)
. (%)
89 (14.0)
76 (11.8)
الدولة/المنطقة – الرقم (%)
الصين
٨٨ (١٣.٩)
91 (14.2)
أوروبا الشرقية
١٣٩ (٢١.٩)
130 (20.2)
آخر
٤٠٨ (٦٤.٣)
422 (65.6)
PD-L1 CPS-لا. (%)
<1
153 (24.1)
154 (24.0)
٤٨٢ (٧٥.٩)
489 (76.0)
253 (39.8)
259 (40.3)
125 (19.7)
١٢٩ (٢٠.١)
حالة أداء ECOG – العدد (%)
0
570 (89.8)
٥٨٨ (٩١.٤)
1
65 (10.2)
55 (8.6)
جدول الأنثراسيكلين – لا. (%)
كل 3 أسابيع
415 (65.4)
425 (66.1)
كل أسبوعين
183 (28.8)
187 (29.1)
لم يبدأ
٣٧ (٥.٨)
31 (4.8)
تصنيف الورم – العدد (%)
T1-T2
402 (63.3)
413 (64.2)
T3-T4
233 (36.7)
230 (35.8)
تأثير العقدة – لا. (%)
إيجابي
570 (89.8)
582 (90.5)
سلبي
65 (10.2)
61 (9.5)
المرحلة العامة للمرض – العدد (%)
المرحلة الثانية
٣٩٩ (٦٢.٨)
٤٠٨ (٦٣.٥)
المرحلة الثالثة
236 (37.2)
235 (36.5)
درجة الورم – العدد (%)
الصف الثالث
635 (100)
642 (99.8)
الصف الثاني
0
إيجابية مستقبلات هرمون الاستروجين – لا. (%)
٦٠١ (٩٤.٦)
600 (93.3)
<10%
٣٤ (٥.٤)
٤٣ (٦.٧)
حالة انقطاع الطمث – لا. (%)
قبل انقطاع الطمث
٣٥٤ (٥٥.٧)
353 (54.9)
ما بعد انقطاع الطمث
278 (43.8)
287 (44.6)
غير قابل للتطبيق
3 (0.5)
3 (0.5)
البيانات من مجموعة النية للعلاج. جميع المرضى كانوا غير معالجين سابقًا، وتم تأكيدهم مركزيًا.، مرض.إجمالي ستة رجال معوتم تضمين حالات سرطان الثدي في الدراسة (ثلاث حالات في كل مجموعة علاج).تم تعريف CPS لـ PD-L1 على أنه عدد خلايا الورم الإيجابية لـ PD-L1، واللمفاويات، والبلاعم مقسومًا على العدد الإجمالي لخلايا الورم مضروبًا في 100.تتراوح حالة الأداء وفقًا لمقياس ECOG من 0 إلى 5، حيث تشير الدرجات الأعلى إلى إعاقة أكبر.انتهاك البروتوكول. درجة الجمع الإيجابية غير PD-L1 (CPS) لـ و كانواإلى 10.1 و نقاط مئوية، على التوالي. كانت الفروق المقدرة في العلاج 17.4 (، نقاط النسبة المئوية
الجدول 2 | نسبة الاستجابة الكاملة المرضية في التحليل المؤقت الأول وفقًا للمرحلة المرضية
نقطة النهاية
بمبروليزوماب الكيميائي )
العلاج الكيميائي الوهمي )
فرق العلاج المقدرنقاط النسبة المئويةسي آي)
قيمة
المرحلة المرضية ypTO/Tis ypNO
عدد المرضى
154
100
–
–
نسبة المرضى الذين استجابوا (95% فاصل ثقة)
٢٤.٣ (٢١.٠-٢٧.٨)
15.6 (12.8-18.6)
8.5 (4.2-12.8)
0.00005
المرحلة المرضية ypTO ypNO
عدد المرضى
135
82
–
–
نسبة المرضى الذين استجابوا (95% فترة الثقة)
21.3 (18.1-24.7)
12.8 (10.3-15.6)
8.3 (4.2-12.4)
–
المرحلة المرضية ypTO/Tis
عدد المرضى
187
١١٧
–
–
نسبة المرضى الذين استجابوا (95% فترة الثقة)
٢٩.٤ (٢٥.٩-٣٣.٢)
18.2 (15.3-21.4)
11.0 (6.5-15.7)
–
اعتُبر المشاركون غير مستجيبين إذا لم يتلقوا دواء الدراسة، أو أوقفوا علاج الدراسة واستمروا في العلاج المساعد باستخدام فئات الأدوية غير المحددة من قبل الدراسة قبل الجراحة (بغض النظر عن نتيجة الجراحة)، أو أوقفوا علاج الدراسة لأسباب تمنع إجراء الجراحة، أو كانت لديهم بيانات مفقودة عن pCR لأي سبب. تم تقييم pCR من قبل أخصائي الأمراض المحلي في وقت الجراحة وفقًا لمعايير تصنيف سرطان الثدي الحالية من AJCC. تم تعريف المرحلة المرضية ypTO/Tis ypNO على أنها غياب سرطان غازي متبقي في عينة الثدي المزالة بالكامل وجميع العقد اللمفاوية الإقليمية المأخوذة. تم تعريف المرحلة المرضية ypTO ypNO على أنها غياب سرطان غازي ومتواجد في الموقع متبقي في عينة الثدي المزالة بالكامل وجميع العقد اللمفاوية الإقليمية المأخوذة. تم تعريف المرحلة المرضية ypTO/Tis على أنها غياب سرطان غازي ومتواجد في الموقع متبقي في عينة الثدي المزالة بالكامل (بغض النظر عن تورط العقد اللمفاوية) وجميع العقد اللمفاوية الإقليمية المأخوذة.تم حساب الفرق المقدر في العلاج باستخدام طريقة ميتينين-نورمين المنهجية. في تحليل المجموعات الفرعية بعد الحدث لدرجة الاستجابة الكاملة (pCR) بناءً على مقياس PD-L1 CPS لـ ( ). بالإضافة إلى ذلك، تم ملاحظة فائدة pCR لعلاج البمبروليزوماب مع العلاج الكيميائي في المرضى الذين لديهم إيجابية لمستقبلات هرمون الاستروجين و على الرغم من ذلك، مع وجود حجم أكبر بين أولئك الذين لديهم إيجابية لمستقبلات هرمون الاستروجينبين المرضى الذين لديهم إيجابية لمستقبلات هرمون الاستروجين، كانت نسبة pCR (19 من 34 مريضًا) في مجموعة العلاج بالبمبروليزوماب-العلاج الكيميائي مقابل 30.2% (13 من 43 مريضًا) في مجموعة العلاج الوهمي-العلاج الكيميائي (فرق العلاج المقدر، 25.6 نقطة مئوية (بينما بين المرضى الذين لديهم إيجابية لمستقبلات هرمون الاستروجينكان الـ pCR ( 135 من 601 مريض) مقابل 14.5% (87 من 600 مريض؛ الفرق المقدر في العلاج، 8.0 نقاط مئوية; الشكل 2).
في تحليلات استكشافية لاحقة، تم تحسينتمت ملاحظة (Tis ypNO) بناءً على كل من CPSs PD-L1 وإيجابية ER، مع أكبر فرق عددي لوحظ في المرضى الذين لديهم CPS PD-L1.وإيجابية مستقبلات هرمون الاستروجين ( ; فرق العلاج، 24.2 نقطة مئوية ( ); الشكل البياني الممتد 1).
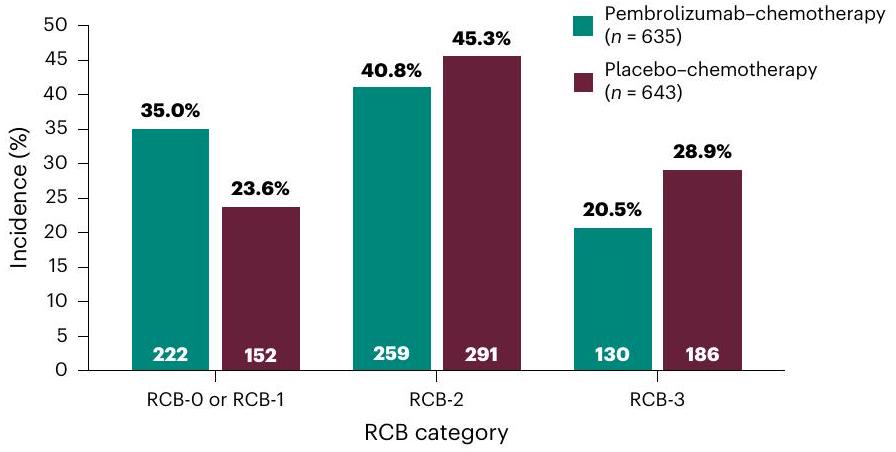
أظهر تحليل النقطة النهائية الاستكشافية لحمل السرطان المتبقي (RCB) أن إضافة بيمبروليزوماب إلى العلاج الكيميائي المساعد قد حولت المزيد من المرضى إلى فئات RCB أقل (RCB-0 أو RCB-1، 35.0% مقابل 23.6%; RCB-2، 40.8% مقابل 45.3%; RCB-3، 20.5% مقابل 28.9%; الشكل 3).
لم يكن البقاء الخالي من الأحداث ناضجًا في التحليل الوسيط الأول؛ لا يزال يتم تقييم هذه النقطة النهائية من خلال تحليلات وسيطة لاحقة محددة مسبقًا وتحليل نهائي.
السلامة
خلال مرحلة العلاج المساعد، حدثت أحداث سلبية مرتبطة بالعلاج (AEs) من أي درجة في 624 من 634 مريضًا. ) في ذراع العلاج الكيميائي مع بيمبروليزوماب و 633 من 642 مريضًا ( ) في ذراع العلاج الوهمي الكيميائي (الجدول 3). درجة متعلقة بالعلاج حدثت أحداث سلبية في 333 ) و 298 مريضاً ( )، على التوالي. جاد
بمبروليزوماب
العلاج الكيميائي الوهمي
عدد المرضى الذين استجابوا/عدد المرضى (%)
الفرق في الاستجابة الكاملة المرضية (95% CI)
نقاط النسبة المئوية
بشكل عام
154/635 (24.3)
100/643 (15.6)
– –
–
8.5 (4.2 إلى 12.8)
مرحلة المرض
المرحلة الثانية
١٠٣/٣٩٩ (٢٥.٨)
68/408 (16.7)
9.1 (3.5 إلى 14.8)
المرحلة الثالثة
51/236 (21.6)
32/235 (13.6)
8.0 (1.1 إلى 14.9)
حالة العقدة
إيجابي
143/570 (25.1)
92/582 (15.8)
9.3 (4.6 إلى 13.9)
سلبي
11/65 (16.9)
8/61 (13.1)
3.8 (-9.2 إلى 16.7)
حجم الورم
T1-T2
111/402 (27.6)
71/413 (17.2)
10.4 (4.7 إلى 16.1)
تي3-تي4
٤٣/٢٣٣ (١٨.٥)
٢٩/٢٣٠ (١٢.٦)
5.8 (-0.8 إلى 12.5)
جدول الأنثراسيكلين
كل 3 أسابيع
97/415 (23.4)
55/425 (12.9)
10.4 (5.3 إلى 15.7)
كل أسبوعين
54/183 (29.5)
44/187 (23.5)
6.0 (-3.0 إلى 15.0)
لم يبدأ
3/37 (8.1)
1/31 (3.2)
4.9 (-9.2 إلى 18.8)
مؤشر PD-L1
<1
11/153 (7.2)
4/154 (2.6)
4.5 (-0.4 إلى 10.1)
143/482 (29.7)
96/489 (19.6)
9.8 (4.4 إلى 15.2)
107/253 (42.3)
75/259 (29.0)
13.2 (4.9 إلى 21.4)
67/125 (53.6)
47/129 (36.4)
17.4 (5.1 إلى 29.1)
إيجابية مستقبلات هرمون الاستروجين
135/601 (22.5)
87/600 (14.5)
8.0 (3.6 إلى 12.4)
<10
19/34 (55.9)
13/43 (30.2)
25.6 (3.3 إلى 45.8)
عمر
<65 سنة
135/546 (24.7)
89/567 (15.7)
9.0 (4.3 إلى 13.7)
19/89 (21.3)
11/76 (14.5)
□
6.9 (-5.2 إلى 18.6)
حالة أداء ECOG
0
142/570 (24.9)
91/588 (15.5)
9.4 (4.8 إلى 14.1)
1
12/65 (18.5)
9/55 (16.4)
2.1 (-12.2 إلى 15.8)
المنطقة الجغرافية
الصين
11/88 (12.5)
9/91 (9.9)
□
2.6 (-7.0 إلى 12.5)
أوروبا الشرقية
41/139 (29.5)
21/130 (16.2)
13.3 (3.3 إلى 23.2)
آخر
102/408 (25.0)
70/422 (16.6)
8.4 (2.9 إلى 13.9)
حالة انقطاع الطمث
قبل انقطاع الطمث
83/354 (23.4)
57/353 (16.1)
م
ما بعد انقطاع الطمث
69/278 (24.8)
42/287 (14.6)
10.2 (3.7 إلى 16.8)
العلاج الكيميائي الوهمي أفضل
الشكل 2 | تحليل الفئات الفرعية للاختلاف في النسب المئوية للمرضى الذين حققوا استجابة كاملة جزئية (pCR) في التحليل المؤقت الأول. تم عرض البيانات من الفئات الفرعية الرئيسية. بالنسبة للسكان الإجماليين، تم حساب الفرق المتوسط المقدر في العلاج مع فترة الثقة 95% باستخدام طريقة ميتينين-نورمين المنهجية. كان التحليل لفئات PD-L1CPS الفرعية مصنفًا. كانت جميع التحليلات الأخرى غير مصنفة.
حاشيةتشير ‘ إلى أن تحليلات المجموعات الفرعية لـ pCR بناءً على حد PD-L1CPS البالغ 20 لم تكن محددة مسبقًا. تم تعريف PD-L1CPS على أنه عدد خلايا الورم الإيجابية لـ PD-L1، واللمفاويات، والبلاعم مقسومًا على العدد الإجمالي لخلايا الورم مضروبًا في 100. تتراوح حالة الأداء وفقًا لـ ECOG من 0 إلى 5، مع الإشارة إلى أن الدرجات الأعلى تدل على إعاقة أكبر. تم الإبلاغ عن الآثار الجانبية المرتبطة بالعلاج في 18.5% و 10.3% من المرضى، على التوالي. مريض واحد ( ) في مجموعة البمبروليزوماب توفي بسبب مضاعفات حادة غير مرتبطة بالعلاج – احتشاء عضلة القلب الموجي. حدثت إيقاف أي علاج دراسي بسبب الآثار الجانبية المرتبطة بالعلاج في 121 مريضًا ( ) في مجموعة العلاج الكيميائي مع بيمبروليزوماب و 65 مريضًا ( ) في مجموعة العلاج الوهمي-العلاج الكيميائي. كانت الآثار الجانبية المرتبطة بالعلاج الأكثر شيوعًا هي تساقط الشعر ( و ، على التوالي)، الغثيان ( و ، على التوالي)، التعب ( و ، على التوالي) وفقر الدم ( و على التوالي؛ الجدول 3).
تمت ملاحظة آثار جانبية ناتجة عن المناعة، باستثناء ردود الفعل الناتجة عن التسريب، من أي درجة في 208 مرضى. ) في ذراع العلاج الكيميائي مع البمبروليزوماب و 45 مريضًا ( ) في ذراع العلاج الكيميائي الوهمي (الجدول 3). الدرجة تم الإبلاغ عن آثار جانبية ناتجة عن المناعة فيو 8 مرضى (” )، على التوالي. كانت الآثار الجانبية المناعية الأكثر شيوعًا هي قصور الغدة الدرقية ( و فرط نشاط الغدة الدرقية و على التوالي) والتهاب الرئة (2.8% و1.4%، على التوالي). لم تُنسب أي وفيات إلى الآثار الجانبية الناتجة عن المناعة.
نقاش
في هذه التجربة العشوائية من المرحلة الثالثة التي تشمل مرضى لم يتلقوا علاجًا سابقًا، ذوي المخاطر العالية، في المرحلة المبكرة،سرطان الثدي، كانت النسبة المئوية للمرضى في مجموعة العلاج الكيميائي مع البمبروليزوماب أعلى بشكل ملحوظ من تلك في مجموعة العلاج الكيميائي مع الدواء الوهمي عند وقت الجراحة. كانت الفروق بين المجموعتين في نسبة الاستجابة الكاملة (pCR) لصالح العلاج الكيميائي مع البمبروليزوماب عبر جميع الفئات الفرعية المحددة مسبقًا، على الرغم من اختلاف حجم الفائدة ووجود فترات ثقة واسعة بنسبة 95% في بعض الحالات.
الجدول 3 | الأحداث الضائرة خلال المرحلة المساعدة قبل الجراحة في التحليل المؤقت الأول
حدث سلبي
بمبروليزوماب الكيميائي )
العلاج الكيميائي الوهمي )
أي درجة
درجة
أي درجة
درجة
عدد المرضى (%)
أي AE
634 (100.0)
٣٨١ (٦٠.١)
638 (99.4)
٣٥٠ (٥٤.٥)
الأحداث الضائرة المرتبطة بالعلاج
624 (98.4)
٣٣٣ (٥٢.٥)
٦٣٣ (٩٨.٦)
298 (46.4)
الثعلبة
٤٠٦ (٦٤.٠)
0
٣٩١ (٦٠.٩)
0
غثيان
306 (48.3)
8 (1.3)
321 (50.0)
7 (1.1)
فقر الدم
205 (32.3)
21 (3.3)
164 (25.5)
18 (2.8)
إرهاق
١٩٠ (٣٠.٠)
17 (2.7)
180 (28.0)
9 (1.4)
إسهال
172 (27.1)
11 (1.7)
130 (20.2)
10 (1.6)
زيادة في إنزيم الألانين أمينوترانسفيراز
158 (24.9)
٢٤ (٣.٨)
147 (22.9)
18 (2.8)
نقص العدلات
١٤٦ (٢٣.٠)
85 (13.4)
158 (24.6)
١٠١ (١٥.٧)
زيادة في إنزيم الأسبارتات أمينوترانسفيراز
137 (21.6)
10 (1.6)
١٠٧ (١٦.٧)
6 (0.9)
انخفاض عدد العدلات
137 (21.6)
89 (14.0)
153 (23.8)
١٠٣ (١٦.٠)
وهن
134 (21.1)
10 (1.6)
١١٦ (١٨.١)
4 (0.6)
التقيؤ
127 (20.0)
9 (1.4)
١٠٨ (١٦.٨)
9 (1.4)
اعتلال الأعصاب المحيطية
111 (17.5)
4 (0.6)
130 (20.2)
5 (0.8)
مُعَزَّز بالمناعة
٢٠٨ (٣٢.٨)
٤٥ (٧.١)
٤٥ (٧.٠)
8 (1.2)
قصور الغدة الدرقية
111 (17.5)
1 (0.2)
11 (1.7)
0
فرط نشاط الغدة الدرقية
57 (9.0)
1 (0.2)
3 (0.5)
0
التهاب الرئة
18 (2.8)
9 (1.4)
9 (1.4)
2 (0.3)
قصور الغدة الكظرية
16 (2.5)
5 (0.8)
0
0
تفاعلات جلدية شديدة
14 (2.2)
8 (1.3)
3 (0.5)
2 (0.3)
التهاب الغدة النخامية
12 (1.9)
6 (0.9)
1 (0.2)
0
التهاب الغدة الدرقية
11 (1.7)
1 (0.2)
2 (0.3)
0
التهاب الكبد
8 (1.3)
7 (1.1)
3 (0.5)
0
التهاب القولون
6 (0.9)
3 (0.5)
5 (0.8)
1 (0.2)
التهاب الأوعية الدموية
5 (0.8)
1 (0.2)
4 (0.6)
0
تم جمع الأحداث الضائرة حتى 30 يومًا بعد التوقف عن العلاج (90 يومًا للأحداث الضائرة الخطيرة). تم سرد الأحداث بترتيب تنازلي حسب التكرار في مجموعة العلاج بالبمبروليزوماب والعلاج الكيميائي. شملت مجموعة الأمان القابلة للتقييم المرضى الذين تلقوا على الأقل دواءً واحدًا من التجربة، أو خضعوا لعملية جراحية، أو كليهما. تم تصنيف شدة الأحداث الضائرة وفقًا لمعايير المصطلحات الشائعة للمعهد الوطني للسرطان (الإصدار 4.0).كانت الأحداث السلبية المرتبطة بالعلاج هي الأحداث التي اعتبرها الباحث مرتبطة بعلاج الدراسة. الأحداث السلبية المرتبطة بالعلاج التي حدثت في على الأقليتم الإبلاغ عن المرضى.تمت معالجة الآثار الجانبية المعتمدة على المناعة، باستثناء ردود الفعل الناتجة عن التسريب، بناءً على قائمة من المصطلحات المفضلة التي تهدف إلى التقاط المخاطر المعروفة للبينبروليزوماب، وتم اعتبارها بغض النظر عن نسبتها إلى علاج الدراسة من قبل الباحث. الآثار الجانبية المعتمدة على المناعة التي حدثت فيتم الإبلاغ عن المرضى. المجموعات الفرعية. من الجدير بالذكر أن نسبة الاستجابة الكاملة (pCR) كانت أعلى عددياً في المجموعات الفرعية من المرضى الذين لديهم تعبير أعلى عن PD-L1 وفي مجموعة المرضى الذين لديهم إيجابية لمستقبلات هرمون الاستروجين (ER). (الذين تم الإبلاغ عنهم سابقًا بأن لديهم خصائص بيولوجية ونتائج سريرية مشابهة لتلك الخاصة بالمرضى الذين يعانون من سرطان الثدي الثلاثي السلبي ). ومع ذلك، يجب تفسير هذه النتائج بحذر حيث أن هذه المجموعات الفرعية غير قوية، والهدف الوحيد من تحليلات المجموعات الفرعية هو استكشاف الصلاحية المتقاربة. تحليلات المجموعات الفرعية الاستكشافية بعد الحدث لـ pCR بناءً على PD-L1CPS من وتم الإبلاغ عن ذلك استنادًا إلى كل من PD-L1CPSs وإيجابية ER. من المهم توخي الحذر عند تفسير هذه الملاحظات.
الشكل 3 | RCB في التحليل المؤقت الأول. كان RCB نقطة نهاية استكشافية وتم تقييمه بواسطة أخصائي علم الأمراض المحلي في وقت الجراحة. تشير RCB-0 و RCB-1 و RCB-2 و RCB-3 إلى أمراض متبقية تزداد حجمًا. كانت بيانات RCB مفقودة لـ و 14 مريضاً ( ) في ذراعي العلاج الكيميائي مع البمبروليزوماب والعلاج الكيميائي مع الدواء الوهمي، على التوالي.
خصوصًا مع أحجام العينات الصغيرة جدًا. لإقامة الثقة، فإن التكرار في تجارب أخرى أمر ضروري تمامًا. كانت الفعالية المرتبطة بالعلاج الكيميائي مع البمبروليزوماب في المرحلة السابقة للجراحة مدعومة بالنتائج المماثلة التي لوحظت عند تعريفها وفقًا للنقاط الثانوية (أي، ypTO و ypNO و ). إن إضافة بيمبروليزوماب إلى العلاج الكيميائي المساعد قد حولت أيضًا المزيد من المرضى إلى فئات RCB الأقل (RCB-0 و RCB-1)، مما يظهر قدرة التركيبة على تقليل نسيج الورم المتبقي بعد الجراحة بين أولئك الذين ليس لديهم pCR. وقد ارتبط انخفاض RCB بتحسن .
تتوافق نتائج دراستنا مع تلك من دراسة المرحلة الثانية I-SPY2 التي أظهرت أن إضافة بيمبروليزوماب إلى العلاج الكيميائي حسنت من معدل الاستجابة الكاملة (pCR) لدى المرضى الذين لم يتلقوا علاجًا مسبقًا، والذين يعانون من سرطان الثدي في مراحله المبكرة، عالي المخاطر، وHER2-.. في دراسة CheckMate 7FL المرحلة 3، تم الإبلاغ عن تحسين مماثل في نتائج pCR بين المرضى ذوي المخاطر العالية، في المرحلة المبكرة، من الدرجة 2/الدرجةسرطان الثدي الذين تلقوا نيفولوماب بالإضافة إلى العلاج الكيميائي المساعد مقارنةً بالذين تلقوا دواءً وهمياً بالإضافة إلى العلاج الكيميائي المساعد مقابل 13.8%; نسبة الأرجحية .
في المرحلة المساعدة من دراستنا، تلقى المرضى بيمبروليزوماب أو دواء وهمي بالإضافة إلى العلاج الهرموني القياسي. أظهرت دراسة monarchE أن إضافة مثبط كيناز السيكلين المعتمد على السيكلين 4/كيناز السيكلين المعتمد على السيكلين 6، أباستيكليب، إلى العلاج الهرموني المساعد حسنت من البقاء بدون انتكاسة لدى المرضى الذين لديهم مستقبلات هرمونية إيجابية، HER2.سرطان الثدي في مراحله المبكرة، إيجابي العقدة، عالي المخاطر. ومع ذلك، تم تصميم الدراسة الحالية قبل الإبلاغ عن هذه النتائج، ولم يُسمح باستخدام الأبيماسيكلب وفقًا للبروتوكول. نظرًا لأن الاستجابة الكاملة المرضية تم تقييمها قبل المرحلة المساعدة، لم يكن من الممكن أن يؤثر استخدام الأبيماسيكلب على هذه النتيجة. تم الإبلاغ عن مخاطر السلامة المحتملة في الدراسات التي تحقق في العلاج المشترك لمثبطات كيناز السيكلين المعتمد على 4/6 مع مثبطات نقاط التفتيش المناعية في المرضى الذين يعانون من مستقبلات هرمونية إيجابية، HER2سرطان الثدي النقيليلا توصي الإرشادات الحالية أيضًا بمثل هذه العلاجات المركبة..
البيانات المتعلقة بالنقطة الأساسية الأخرى للدراسة وهي فترة البقاء خالية من الأحداث (EFS) ليست ناضجة ولا تزال قيد التقييم. ستعكس نتيجة EFS تأثير نظام العلاج الشامل، بما في ذلك كل من مراحل العلاج المساعد والعلاج المساعد. لم يتم تصميم هذه الدراسة لتمييز المساهمة النسبية لكل مرحلة؛ سيكون من الضروري إجراء تجربة مستقبلية لمعالجة هذا السؤال.
كانت الأحداث الضائرة في مجموعة العلاج بالبمبروليزوماب-العلاج الكيميائي متوافقة مع ملفات السلامة المعروفة للعوامل الفردية. مشابهة للنتائج من دراسة KEYNOTE-522 (التي قيمت البمبروليزوماب بالاشتراك مع العلاج الكيميائي المساعد بين المرضى الذين يعانون من سرطان الثدي الثلاثي السلبي).لم يكن هناك دليل على أن إضافة أدى استخدام بيمبروليزوماب مع العلاج الكيميائي المساعد القياسي إلى تفاقم السمية المرتبطة بالعلاج الكيميائي، وكانت الأحداث الأكثر شيوعًا في كلا الذراعين هي تلك المرتبطة عادة بالعلاج الكيميائي السام، مثل تساقط الشعر والغثيان وفقر الدم. كانت حالات الدرجة المرتبطة بالعلاجكانت الآثار الجانبية والأعراض المرتبطة بالعلاج التي أدت إلى توقف العلاج أعلى بشكل معتدل مع العلاج الكيميائي مع بيمبروليزوماب مقارنة بالعلاج الكيميائي مع الدواء الوهمي. كما كان متوقعًا استنادًا إلى الدراسات السابقة التي تقيم الأجسام المضادة وحيدة النسيلة المضادة لـ PD-1 و PD-L1.حدثت الآثار الجانبية ذات الآلية المحتملة المناعية بشكل أكثر تكرارًا بين المرضى في مجموعة العلاج بالبمبروليزوماب-العلاج الكيميائي مقارنةً بمجموعة العلاج الوهمي-العلاج الكيميائي. بشكل عام، كان ملف الأمان في هذه الدراسة متسقًا مع ذلك في دراسة KEYNOTE-522..
في الختام، أدى العلاج الكيميائي مع البمبروليزوماب كعلاج مساعد إلى تحسين معدل الاستجابة الكاملة المرضية مقارنة بالعلاج الكيميائي وحده في المرضى ذوي المخاطر العالية في المراحل المبكرة.سرطان الثدي. تقييم فترة البقاء الخالية من الأحداث جارٍ.
المحتوى عبر الإنترنت
أي طرق، مراجع إضافية، ملخصات تقارير Nature Portfolio، بيانات المصدر، بيانات موسعة، معلومات إضافية، شكر وتقدير، معلومات مراجعة الأقران؛ تفاصيل مساهمات المؤلفين والمصالح المتنافسة؛ وبيانات توفر البيانات والرموز متاحة فيhttps://doi.org/10.1038/s41591-024-03415-7.
References
Jatoi, I., Anderson, W. F., Jeong, J. H. & Redmond, C. K. Breast cancer adjuvant therapy: time to consider its time-dependent effects. J. Clin. Oncol. 29, 2301-2304 (2011).
Torrisi, R. et al. Neoadjuvant chemotherapy in hormone receptor-positive/HER2-negative early breast cancer: when, why and what? Crit. Rev. Oncol. Hematol. 160, 103280 (2021).
Cortazar, P. et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164-172 (2014).
U.S. Department of Health and Human Services, FDA, Oncology Center of Excellence, CDER & CBER. Pathological complete response in neoadjuvant treatment of high-risk early-stage breast cancer: use as an endpoint to support accelerated approval guidance for industry. www.fda.gov/media/83507/download (accessed 21 December 2023).
Emens, L. A. & Middleton, G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol. Res. 3, 436-443 (2015).
Xue, Y. et al. Platinum-based chemotherapy in combination with PD-1/PD-L1 inhibitors: preclinical and clinical studies and mechanism of action. Expert Opin. Drug Deliv. 18, 187-203 (2021).
Nanda, R. et al. Effect of pembrolizumab plus neoadjuvant chemotherapy on pathologic complete response in women with early-stage breast cancer: an analysis of the ongoing phase 2 adaptively randomized I-SPY2 trial. JAMA Oncol. 6, 676-684 (2020).
Schmid, P. et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 382, 810-821 (2020).
Schmid, P. et al. Event-free survival with pembrolizumab in early triple-negative breast cancer. N. Engl. J. Med. 386, 556-567 (2022).
Schrodi, S. et al. Outcome of breast cancer patients with low hormone receptor positivity: analysis of a 15-year populationbased cohort. Ann. Oncol. 32, 1410-1424 (2021).
Fredriksson, I., Acs, B., Hartman, J., Sönmez, D. & Lindman, H. 241MO patient characteristics and real-world outcomes in HER2 negative/ER zero and ER low patients treated as triple-negative breast cancer in Sweden 2008-2020. Ann. Oncol. 34, S278-S324 (2023).
Iwamoto, T. et al. Estrogen receptor (ER) mRNA and ER-related gene expression in breast cancers that are 1% to 10% ER-positive by immunohistochemistry. J. Clin. Oncol. 30, 729-734 (2012).
Yoder, R. et al. Impact of low versus negative estrogen/ progesterone receptor status on clinico-pathologic characteristics and survival outcomes in HER2-negative breast cancer. NPJ Breast Cancer 8, 80 (2022).
Yau, C. et al. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: a multicentre pooled analysis of 5161 patients. Lancet Oncol. 23, 149-160 (2022).
Loi, S. et al. LBA20 A randomized, double-blind trial of nivolumab (NIVO) vs placebo (PBO) with neoadjuvant chemotherapy (NACT) followed by adjuvant endocrine therapy (ET) ± NIVO in patients (pts) with high-risk, ER HER2 primary breast cancer (BC). Ann. Oncol. 34, S1259-S1260 (2023).
Johnston, S. R. D. et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. 24, 77-90 (2023).
Rugo, H. S. et al. Abemaciclib in combination with pembrolizumab for , metastatic breast cancer: phase 1b study. NPJ Breast Cancer 8, 118 (2022).
Masuda, J. et al. Efficacy, safety, and biomarker analysis of nivolumab in combination with abemaciclib plus endocrine therapy in patients with HR-positive HER2-negative metastatic breast cancer: a phase II study (WJOG11418B NEWFLAME trial). J. Immunother. Cancer 11, e007126 (2023).
Jerusalem, G. et al. Neoadjuvant nivolumab + palbociclib + anastrozole for oestrogen receptor-positive/human epidermal growth factor receptor 2-negative primary breast cancer: results from CheckMate 7A8. Breast 72, 103580 (2023).
NCCN clinical practice guidelines in oncology (NCCN guidelines). Breast cancer, version 4.2023. www.nccn.org/ (accessed 15 February 2023).
Brahmer, J. R. et al. Society for immunotherapy of cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 9, e002435 (2021).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons. org/licenses/by/4.0/.
¹وحدة الثدي، مركز شامباليماود السريري/مؤسسة شامباليماود، لشبونة، البرتغال. ²مركز بايلور الطبي، أونكولوجيا تكساس، شبكة أونكولوجيا الولايات المتحدة، دالاس، تكساس، الولايات المتحدة الأمريكية.قسم أمراض الثدي، مركز سرطان الثدي في هنان، المستشفى السرطاني التابع لجامعة تشنغتشو ومستشفى هنان للسرطان، تشنغتشو، الصين.مركز جامعة تكساس Southwestern الطبي، دالاس، تكساس، الولايات المتحدة الأمريكية.مركز أبحاث السرطان التجريبية، معهد بارتس للسرطان، جامعة كوين ماري لندن، لندن، المملكة المتحدة.مؤسسة ميديكا للعلوم والابتكار، ريدج وود، نيو جيرسي، الولايات المتحدة الأمريكية.مؤسسة ميديكا للعلوم والابتكار، برشلونة، إسبانيا.مركز سرطان الثدي الدولي (IBCC)، بانجيا أونكولوجي، مجموعة كويرون، برشلونة، إسبانيا.الجامعة الأوروبية في مدريد، كلية العلوم الطبية والصحية، قسم الطب، مدريد، إسبانيا.معهد الأورام، مستشفى بياتا ماريا آنا، مدريد، إسبانيا.مركز الثدي، قسم النساء والتوليد، مستشفى جامعة LMU، ميونيخ، ألمانيا.كلية الطب بجامعة ستانفورد، ستانفورد، كاليفورنيا، الولايات المتحدة الأمريكية.مركز الأميرة مارجريت للسرطان، شبكة الصحة الجامعية، تورونتو، أونتاريو، كندا.مستشفى جامعة إيرلانغن، مركز السرطان الشامل إيرلانغن-EMN، مركز أبحاث السرطان البافاري (BZKF)، إيرلانغن، ألمانيا.قسم جراحة الثدي، مركز فودان للسرطان في شنغهاي، قسم الأورام، كلية الطب بجامعة فودان، شنغهاي، الصين.معهد كوري، باريس، فرنسا.قسم الطب في سامسونغ، كلية الطب بجامعة سونغكيونكوان، سيول، جمهورية كوريا.أونكولوجي الدم، IMAT أونكوميديك، مونتيريا، كولومبيا.المعهد الوطني للأورام، بودابست، المجر.مستشفى ماساتشوستس العام وكلية الطب بجامعة هارفارد، بوسطن، ماساتشوستس، الولايات المتحدة الأمريكية.مستشفى جامعة سيول، معهد أبحاث السرطان، كلية الطب بجامعة سيول، جامعة سيول، سيول، جمهورية كوريا.معهد سرطان الثدي ويست ميد، مستشفى ويست ميد وجامعة سيدني، سيدني، نيو ساوث ويلز، أستراليا.مركز الطب السرطاني، جامعة هونغ كونغ، بوكفولام، هونغ كونغ.مستشفى معهد السرطان JFCR، طوكيو، اليابان.كلية الطب، جامعة باريس ساكلاي، غاستاف روسي، فيليجويف، فرنسا.مستشفى أوساكا الوطني NHO، أوساكا، اليابان.ميرك وشركاه، رحواي، نيو جيرسي، الولايات المتحدة الأمريكية.جامعة كاليفورنيا لوس أنجلوس، مركز جونسن الشامل للسرطان، لوس أنجلوس، كاليفورنيا، الولايات المتحدة الأمريكية. البريد الإلكتروني:fcardoso@abcglobalalliance.org
طرق
المرضى
كان المرضى المؤهلون عامًا من العمر مع تأكيد مركزي لـ HER2 سرطان الثدي القنوي الغازي من الدرجة 3 (تم تقييمه وفقًا لأحدث إرشادات الجمعية الأمريكية للأورام السريرية – كلية أطباء الأمراض الأمريكية )، تم تشخيصهم حديثًا، غير معالجين سابقًا، مرض غير نقيل (T1c-2 ( )، cN1-2 أو T3-4، cNO-2، وفقًا لأحدث معايير التصنيف للجنة الأمريكية المشتركة للسرطان (AJCC))، كما حددها الباحث في التقييم الإشعاعي، التقييم السريري أو كليهما؛ درجة أداء مجموعة التعاون الشرقي للأورام (ECOG) ; ووظيفة الأعضاء الكافية. كان المرضى الذين لديهم أورام أولية متعددة البؤر، سرطان الثدي الالتهابي وأولئك الذين لديهم إيجابية منخفضة لمستقبلات هرمون الاستروجين مؤهلين.
شملت معايير الاستبعاد سرطان الثدي النشط الثنائي، متعدد المراكز، اللوبولي أو النقيل؛ الأمراض المناعية الذاتية التي تم علاج المريض لها بالعلاج النظامي خلال العامين الماضيين؛ تشخيص نقص المناعة أو استخدام العلاج المثبط للمناعة خلال أسبوع واحد؛ تاريخ التهاب رئوي غير معدي تم علاجه بالستيرويدات؛ التهاب رئوي؛ السل النشط؛ عدوى نشطة كان المريض يتلقى لها علاجًا نظاميًا؛ أو مرض قلبي وعائي ذو دلالة سريرية. تم إدراج معايير الأهلية الكاملة في المعلومات التكميلية.
تصميم التجربة والعلاج
في هذه التجربة العشوائية المزدوجة التعمية (ClinicalTrials.gov المعرف، NCT03725059)، تلقى المرضى العلاج في مراحل ما قبل الجراحة وما بعدها. لم يُسمح بالتداخل بين أذرع العلاج بين المراحل. بناءً على البيانات السريرية الناشئة في المرضى الذين يعانون من سرطان الثدي في مراحله المبكرة والتي تظهر نتائج سريرية طويلة الأمد غير متجانسة عبر البلدان/المناطق على الرغم من تشابه العلاجات القياسية، تم تصنيف المرضى عند العشوائية حسب المنطقة (أوروبا الشرقية مقابل الصين مقابل جميع البلدان الأخرى) لضمان التوازن عبر أذرع العلاج. تم تصنيف المرضى من أوروبا الشرقية بشكل إضافي حسب حالة PD-L1 للورم (CPS مقابل ). لم يتم تصنيف المرضى من الصين بشكل إضافي. تم تصنيف المرضى من جميع البلدان الأخرى (أي، باستثناء أوروبا الشرقية والصين) بشكل إضافي حسب حالة PD-L1 للورم (CPS مقابل )، ووجود العقد اللمفاوية (إيجابية مقابل سلبية)، وجدول جرعات الأنثراسيكلين (Q2W مقابل Q3W) وإيجابية مستقبلات هرمون الاستروجين ( مقابل ). تم تعيين المرضى عشوائيًا (بنسبة ) إلى ذراع العلاج بالبمبروليزوماب-العلاج الكيميائي أو ذراع العلاج الوهمي-العلاج الكيميائي باستخدام نظام استجابة صوتية تفاعلية مركزي مع نظام استجابة ويب مدمج. في مرحلة ما قبل الجراحة، تلقى المرضى أربع دورات من البمبروليزوماب الوريدي أو العلاج الوهمي مرة واحدة كل 3 أسابيع بالإضافة إلى باكليتاكسيل كل أسبوع ; أول علاج ما قبل الجراحة)، تليها أربع دورات من البمبروليزوماب أو العلاج الوهمي بالاشتراك مع إما دوكسوروبيسين أو إبيروبيسين ( ) بالإضافة إلى سيكلوفوسفاميد ( ) يتم إعطاؤها إما كل أسبوعين أو كل ثلاثة أسابيع (ثاني علاج ما قبل الجراحة). يمكن للمرضى الذين أكملوا أو أوقفوا أول علاج ما قبل الجراحة بدء العلاج الثاني ما قبل الجراحة أو الخضوع للجراحة، وأولئك الذين أكملوا أو أوقفوا العلاج الثاني ما قبل الجراحة يمكنهم الخضوع للجراحة. خضع المرضى للجراحة (حفظ الثدي أو استئصال الثدي مع/بدون خزعة العقدة اللمفاوية الحارسة أو استئصال الإبط) في موعد لا يتجاوز 6 أسابيع بعد آخر جرعة من العلاج ما قبل الجراحة. في مرحلة ما بعد الجراحة، تلقى المرضى (بدءًا خلال 60 يومًا بعد الجراحة) البمبروليزوماب أو العلاج الوهمي كل 3 أسابيع لمدة تصل إلى تسع دورات (حتى 6 أشهر)، بالإضافة إلى اختيار الباحث للعلاج الهرموني (وفقًا لإرشادات المؤسسة) لمدة تصل إلى 10 سنوات. لم يُسمح بالعلاج الهرموني المساعد مع أباستيكليب. كان العلاج الإشعاعي المساعد (وفقًا لإرشادات المؤسسة) مسموحًا به، كما هو موضح، إما قبل بدء العلاج المساعد أو بالتزامن. تم إيقاف علاج التجربة في المرضى الذين يعانون من تقدم المرض أو تكراره أو آثار سامة غير مقبولة.
تم تطوير الدراسة بواسطة لجنة استشارية علمية وموظفي الراعي (ميرك شارب ودوهيم LLC، شركة تابعة لميرك وشركاه، رحواي، نيو جيرسي، الولايات المتحدة الأمريكية). أشرفت لجنة مراقبة البيانات المستقلة الخارجية على الدراسة، وقامت بتقييم السلامة بشكل دوري، وقامت بتقييم الفعالية في التحليلات المؤقتة المحددة مسبقًا. تمت الموافقة على البروتوكول من قبل هيئة أخلاقية في كل موقع دراسي (انظر المعلومات التكميلية للحصول على قائمة بمواقع الدراسة). قدم المرضى موافقة خطية مستنيرة
التقييمات
عند الانتهاء من العلاج ما قبل الجراحة، تم تقييم pCR وفقًا لمعايير تصنيف AJCC (ypTO/Tis ypNO، ypTO ypNO وypTO/Tis) بواسطة طبيب أمراض محلي غير مطلع على تخصيص العلاج. تم تقييم EFS (المعرف على أنه الوقت من العشوائية إلى تقدم المرض الذي يمنع الجراحة، أو تكرار محلي أو بعيد، أو سرطان أولي ثانٍ أو وفاة لأي سبب، أيهما يحدث أولاً) بشكل أعمى بواسطة الباحث. تم تقييم تعبير PD-L1 في عينات خزعة الإبرة الأساسية الجديدة أو الحديثة في مختبر مركزي باستخدام PD-L1IHC 22 C 3 pharmDx (Agilent Technologies). تم تقييم الآثار الجانبية طوال التجربة ولمدة 30 يومًا بعد إيقاف العلاج (90 يومًا للآثار الجانبية الخطيرة) وتم تصنيفها وفقًا لمعايير المصطلحات الشائعة للآثار الجانبية (v4.0) . كانت الآثار الجانبية المناعية تعتمد على قائمة من المصطلحات المفضلة التي تهدف إلى التقاط المخاطر المعروفة للبمبروليزوماب وتم اعتبارها بغض النظر عن نسبتها إلى علاج الدراسة من قبل الباحث.
النقاط النهائية
كانت النقاط النهائية الأساسية للدراسة هي pCR، المحددة على أنها ypNO في وقت الجراحة، وEFS في مجموعة النية للعلاج. شملت النقاط النهائية الثانوية pCR وفقًا لتعريفات ypTO ypNO و Tis في جميع المرضى، pCR وفقًا لجميع التعريفات في المرضى الذين لديهم PD-L1 CPS ورم، EFS بين المرضى الذين لديهم PD-L1 CPS ورم وOS بين جميع المرضى والمرضى الذين لديهم PD-L1CPS ورم. تم تقييم السلامة خلال مراحل ما قبل الجراحة وما بعدها في جميع المرضى الذين تلقوا دواء التجربة، أو خضعوا للجراحة أو كليهما. كانت تقييم RCB (المرض المتبقي في الثدي أو العقد اللمفاوية في وقت الجراحة) نقطة نهائية استكشافية.
التحليل الإحصائي
تم تقييم الفعالية في مجموعة النية للعلاج، والتي شملت جميع المرضى الذين خضعوا للعشوائية. تم تقييم السلامة في مجموعة المعالجة، والتي شملت جميع المرضى الذين خضعوا للعشوائية وتلقوا دواء التجربة، أو خضعوا للجراحة أو كليهما. استخدمنا طريقة ميتينين-نورمين المنهجية المصنفة, مع أوزان تتناسب مع حجم عينة الطبقة، لمقارنة الفروق بين الذراعين في نسب المرضى الذين لديهم pCR. اعتُبر المرضى الذين لم تكن لديهم نتائج pCR لأي سبب أو الذين تلقوا علاجًا مسبقًا غير محدد في البروتوكول على أنهم لم يكن لديهم استجابة.
الـ لم يتم تعديل فترات الثقة المرتبطة بالفروق بين الذراعين في نسب المرضى الذين لديهم pCR لمقارنات متعددة، وبالتالي لا يمكن استخدامها لاستنتاج التأثيرات. تم استخدام عوامل التصنيف المستخدمة في التوزيع العشوائي مع استراتيجية التجميع المحددة مسبقًا في جميع التحليلات المصنفة.
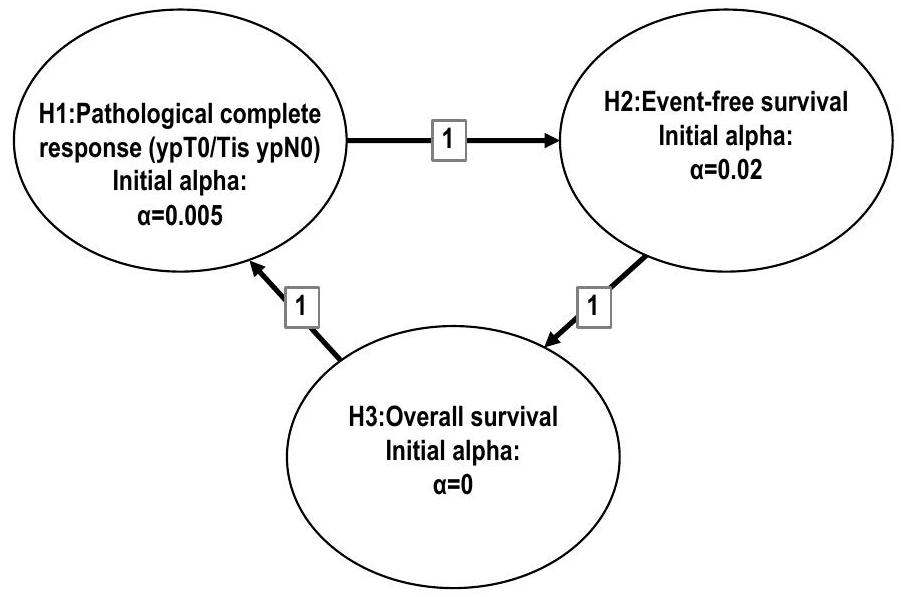
تم تطبيق الطريقة الرسومية التي نوقشت في المرجع 27 للتحكم في معدل الخطأ من النوع الأول عند مستوى أحادي الجانب 0.025 عبر كلا نقطتي النهاية الرئيسيتين وجميع التحليلات المؤقتة والنهائية (الشكل البياني الممتد 2). تم استخدام دالة إنفاق لان-دي ميتس أوبراين-فليمنغ للتحكم في الخطأ من النوع الأول في التحليلات المؤقتة والنهائية. كان الهدف الأساسي من التحليل المؤقت الأول هو تقييم تفوق العلاج الكيميائي مع بيمبروليزوماب على العلاج الكيميائي الوهمي فيما يتعلق بنسبة المرضى الذين لديهم pCR (المرحلة ypT0/Tis، ypNO)؛ كان من المقرر أن يحدث هذا التحليل بعد الانتهاء من التسجيل وأن يكون جميع المرضى العشوائيين قد خضعوا للجراحة بعد حوالي 6 أشهر من العلاج المسبق.
مع تسجيل حوالي 1,240 مريضًا، كان لدى التجربة القدرة على اكتشاف فرق حقيقي قدره 15 نقطة مئوية لـ
مقارنة معدل pCR (المرحلة ypT0/Tis ypNO) بين أذرع العلاج عند مستوى أحادي الجانب 0.005. كان من الممكن أن تكون لديها قدرة على لاكتشاف نسبة المخاطر لـ EFS قدرها 0.73 عند مستوى أحادي الجانب 0.02 في التحليل النهائي.
تم إجراء التحليلات الإحصائية باستخدام SAS v9.4 (معهد SAS).
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقرير Nature Portfolio المرتبط بهذه المقالة.
توفر البيانات
تلتزم شركة ميرك شارب ودوهيم LLC، وهي شركة تابعة لشركة ميرك وشركاه، رحواي، نيو جيرسي، الولايات المتحدة الأمريكية (MSD)، بتوفير الوصول إلى البيانات المجهولة وتقارير الدراسات السريرية من التجارب السريرية للشركة للباحثين العلميين المؤهلين بغرض إجراء أبحاث علمية مشروعة. كما تلتزم MSD بحماية حقوق وخصوصية المشاركين في التجارب، وبالتالي لديها إجراء لتقييم وتلبية الطلبات لمشاركة بيانات التجارب السريرية للشركة مع الباحثين العلميين الخارجيين المؤهلين. يوضح موقع مشاركة بيانات MSD (المتاح على engagezone.msd.com/ds_documentation.php) العملية والمتطلبات لتقديم طلب بيانات. سيتم تقييم الطلبات بسرعة من حيث الاكتمال والامتثال للسياسات. ستتم مراجعة الطلبات الممكنة من قبل لجنة من خبراء الموضوع في MSD لتقييم الصلاحية العلمية للطلب ومؤهلات مقدمي الطلبات. وفقًا لتشريعات خصوصية البيانات، يجب على مقدمي الطلبات المعتمدة الدخول في اتفاقية مشاركة بيانات قياسية مع MSD قبل منح الوصول إلى البيانات. ستتوفر البيانات للطلب بعد الموافقة على المنتج في الولايات المتحدة والاتحاد الأوروبي أو بعد توقف تطوير المنتج. هناك ظروف قد تمنع MSD من مشاركة البيانات المطلوبة، بما في ذلك اللوائح الخاصة بالبلد أو المنطقة. إذا تم رفض الطلب، سيتم إبلاغ المحقق. يتطلب الوصول إلى بيانات العلامات الحيوية الجينية أو الاستكشافية خطة تحليل إحصائي مفصلة مدفوعة بالفرضيات يتم تطويرها بالتعاون بين مقدم الطلب وخبراء الموضوع في MSD؛ بعد الموافقة على خطة التحليل الإحصائي وتنفيذ اتفاقية مشاركة البيانات، ستقوم MSD إما بإجراء التحليلات المقترحة ومشاركة النتائج مع مقدم الطلب أو ستقوم بإنشاء متغيرات العلامات الحيوية وإضافتها إلى ملف مع البيانات السريرية التي يتم تحميلها إلى بوابة التحليل بحيث يمكن لمقدم الطلب إجراء التحليلات المقترحة.
References
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer: ASCO-College of American Pathologists guideline update. J. Clin. Oncol. 41, 3867-3872 (2023).
Allison, K. H. et al. Estrogen and progesterone receptor testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists guideline update. Arch. Pathol. Lab. Med. 144, 545-563 (2020).
National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. https://ctep.cancer.gov/ protocoldevelopment/electronic_applications/ctc.htm (2017).
Miettinen, O. & Nurminen, M. Comparative analysis of two rates. Stat. Med. 4, 213-226 (1985).
Maurer, W. & Bretz, F. Memory and other properties of multiple test procedures generated by entangled graphs. Stat. Med. 32, 1739-1753 (2013).
الشكر والتقدير
تم تمويل هذا البحث من قبل ميرك شارب ودوهيم LLC، وهي شركة تابعة لشركة ميرك وشركاه، رحواي، نيو جيرسي، الولايات المتحدة الأمريكية. نشكر المرضى وعائلاتهم ومقدمي الرعاية على مشاركتهم في هذه الدراسة، بالإضافة إلى جميع المحققين وموظفي الموقع. بالإضافة إلى ذلك، يشكر المؤلفون الموظفين التاليين من ميرك شارب ودوهيم LLC، وهي شركة تابعة لشركة ميرك وشركاه، رحواي، نيو جيرسي، الولايات المتحدة الأمريكية، الذين دعموا الدراسة: ج. أكتان (إشراف الدراسة)؛ ك. ماري هيرشفيلد (قيادة الدراسة)؛ ج. زهاو، إكس. بينغ وج. وي (خبرة إحصائية)؛ ج. تشابمان، ج. كيميل، س. كروز، د. فالك، ل. مونيز، إ. رامزاني، أ.ت. أدورنو، أ. غارسيا وك. بونادر (جمع البيانات، إشراف البحث ومراجعة الوثائق)؛ م.ر. باباساني، ج. باي ول.و. بانغ (تحليل وتقرير البيانات). تم تقديم المساعدة في الكتابة الطبية من قبل أ. حسن وأ. لي من ICON plc. تم تمويل هذه المساعدة من قبل ميرك شارب ودوهيم LLC، وهي شركة تابعة لشركة ميرك وشركاه، رحواي، نيو جيرسي، الولايات المتحدة الأمريكية.
مساهمات المؤلفين
كان لدى جميع المؤلفين الوصول إلى البيانات المستخدمة لإعداد المخطوطة وشاركوا في كتابة أو مراجعة نقدية وتحرير المخطوطة. تم كتابة المسودة الأولى من قبل المؤلف الأول وكبير المؤلفين مع المساعدة التحريرية المقدمة من كاتب طبي تم تمويله من قبل الراعي. قام أ.ب، ف.س، د.و.س، ج.س، ي.د، ن.هـ، س.-أ.ي، ل.ج، ف.ك، هـ.م، ب.س، م.ل.ت وك.ت بتصميم وتخطيط الدراسة. قام أ.ب، د.و.س، ب.أ.ف، م.ج.ف، ن.هـ، ر.هـ، س.-أ.ي، ز.ل، د.ل، ي.هـ.ب، ج.ر، ب.س، ز.س، ل.س، ت.ت، م.ل.ت، ك.ت و هـ.ي بجمع البيانات. قام أ.ب، ف.س، ي.د، ل.ج، ف.ك، ب.س، م.ل.ت وك.ت بتحليل البيانات. قام أ.ب، ف.س، د.و.س، ي.د، ب.أ.ف، ن.هـ، ر.هـ، س.-أ.ي، ل.ج، ف.ك، د.ل، هـ.م، ج.أ.س، ي.هـ.ب، ب.س، م.ل.ت، ك.ت و هـ.ي بتفسير النتائج. قام أ.ب، ف.س، ي.د، ل.ج، ي.هـ.ب، ب.س، ك.ت و هـ.ي بصياغة المخطوطة. قام أ.ب، ف.س، د.و.س، ج.س، ي.د، ب.أ.ف، م.ج.ف، ن.هـ، ر.هـ، س.-أ.ي، ل.ج، ف.ك، ز.ل، د.ل، هـ.م، ج.أ.س، ي.هـ.ب، ج.ر، ب.س، ز.س، ل.س، ت.ت، م.ل.ت وك.ت بمراجعة أو تعديل المخطوطة لمحتوى فكري مهم. قام ف.س، د.و.س، ج.س، ب.أ.ف، م.ج.ف، ن.هـ، ر.هـ، س.-أ.ي، ز.ل، هـ.م و ت.ت بتوفير مواد الدراسة/المرضى. قدم ج.ر الدعم الإداري أو اللوجستي أو الفني. قرر أ.ب، ف.س، د.و.س، ج.س، ي.د، ب.أ.ف، م.ج.ف، ن.هـ، ر.هـ، س.-أ.ي، ل.ج، ف.ك، ز.ل، د.ل، هـ.م، ج.أ.س، ي.هـ.ب، ج.ر، ب.س، ز.س، ل.س، ت.ت، م.ل.ت، ك.ت و هـ.ي تقديم المخطوطة للنشر. وافق جميع المؤلفين على المسودة النهائية للمخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى فاطمة كاردوسو.
معلومات مراجعة الأقران تشكر مجلة ناتشر ميديسين كارين جيلمون، بينغه شيو، والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل. المحرر الرئيسي: أولريكه هارجيس، بالتعاون مع فريق ناتشر ميديسين.
الشكل 1 من البيانات الموسعة | تحليل استكشافي لاحق للاختلاف في نسب المرضى الذين حققوا استجابة كاملة مرضية حسب إيجابية مستقبلات الاستروجين ودرجة الإيجابية المجمعة لـ PD-L1 في التحليل المؤقت الأول. تم تعريف درجة الإيجابية المجمعة لمستضد موت الخلايا المبرمج 1 (PD-L1) (CPS) على أنها عدد خلايا الورم الإيجابية لـ PD-L1، اللمفاويات والبلعميات مقسومًا على العدد الإجمالي لخلايا الورم مضروبًا في 100. لم تحدث استجابة كاملة مرضية في المرضى الذين كانت درجة PD-L1CPS لديهم <1. مع إيجابية مستقبلات الإستروجين (ER)ذراع بيمبروليزوماب،؛ دواء وهمي ). بالنسبة للسكان بشكل عام، تم حساب الفرق في العلاج المقدر باستخدام طريقة ميتينين-نورمينين المصنفة حسب المنطقة (الصين مقابل أوروبا الشرقية مقابل جميع البلدان الأخرى) وحالة PD-L1 (CPS مقابل تم تقدير الفروق في العلاج للنقاط النهائية الأخرى بناءً على طريقة ميتينين-نورمينين (غير مصنفة).
الشكل البياني الممتد 2 | رسم بياني لتعددية التحكم في خطأ النوع الأول لفرضيات الدراسة. يسمح خطة التحليل المحددة مسبقًا بانتقال ألفا من نقطة النهاية الناجحة إلى نقاط أخرى. H، فرضية.
البيانات الموسعة الجدول 1 | ملخص تعرض الدواء خلال المرحلة المساعدة
بمبروليزوماب العلاج الكيميائي )
العلاج الكيميائي الوهمي )
جميع أدوية الدراسة
ن
634
641
الشهور المتوسطة (النطاق)
4.9 (0.0-6.9)
4.9 (0.0-7.8)
بمبروليزوماب/دواء وهمي (كل 3 أسابيع)
ن
634
641
الشهور المتوسطة (النطاق)
4.9 (0.0-6.9)
4.9 (0.0-7.8)
عدد الإدارات الوسيطة (النطاق)
8.0 (1.0-8.0)
8.0 (1.0-8.0)
باكليتاكسيل (مرة في الأسبوع)
ن
634
641
الشهور المتوسطة (النطاق)
2.6 (0.0-4.7)
2.6 (0.0-4.7)
عدد الإدارات الوسيطة (النطاق)
12.0 (1.0-12.0)
12.0 (1.0-12.0)
دوكسوروبيسين (كل 3 أسابيع)
ن
164
165
الشهور المتوسطة (النطاق)
2.1 (0.0-3.1)
2.1 (0.0-2.8)
عدد الإدارات الوسيطة (النطاق)
4.0 (1.0-4.0)
4.0 (1.0-4.0)
دوكسوروبيسين (كل أسبوعين)
ن
١٣٦
١٣٣
الشهور المتوسطة (النطاق)
1.4 (0.5-2.8)
1.4 (0.0-1.9)
عدد الإدارات الوسيطة (النطاق)
4.0 (2.0-4.0)
4.0 (1.0-4.0)
إبيروبيسين (كل 3 أسابيع)
ن
٢٥١
261
الشهور المتوسطة (النطاق)
2.1 (0.0-4.2)
2.1 (0.0-3.9)
عدد الإدارات الوسيط (النطاق)
4.0 (1.0-4.0)
4.0 (1.0-4.0)
إبيروبيسين (كل أسبوعين)
ن
٤٧
٥٤
الشهور المتوسطة (النطاق)
1.4 (1.0-2.9)
1.4 (0.5-2.3)
عدد الإدارات الوسيط (النطاق)
4.0 (2.0-4.0)
4.0 (2.0-4.0)
سيكلوفوسفاميد (كل 3 أسابيع)
ن
415
425
الشهور المتوسطة (النطاق)
2.1 (0.0-4.2)
2.1 (0.0-3.9)
عدد الإدارات الوسيطة (النطاق)
4.0 (1.0-4.0)
4.0 (1.0-4.0)
سيكلوفوسفاميد (كل أسبوعين)
ن
183
187
الشهور المتوسطة (النطاق)
1.4 (0.5-2.9)
1.4 (0.0-2.3)
عدد الإدارات الوسيطة (النطاق)
4.0 (2.0-4.0)
4.0 (1.0-4.0)
شملت مجموعة الأمان القابلة للتقييم المرضى الذين تلقوا على الأقل دواءً واحدًا من التجربة، أو خضعوا لعملية جراحية، أو كليهما.
محفظة الطبيعة
المؤلف(المؤلفون) المراسلون:
فاطمة كاردوسو
آخر تحديث بواسطة المؤلفين: 24 أكتوبر 2024
ملخص التقرير
تتمنى Nature Portfolio تحسين قابلية إعادة إنتاج الأعمال التي ننشرها. يوفر هذا النموذج هيكلًا للاتساق والشفافية في التقرير. لمزيد من المعلومات حول سياسات Nature Portfolio، يرجى الاطلاع على سياسات التحرير وقائمة مراجعة سياسة التحرير.
الإحصائيات
لجميع التحليلات الإحصائية، تأكد من أن العناصر التالية موجودة في أسطورة الشكل، أسطورة الجدول، النص الرئيسي، أو قسم الطرق.
مؤكد □ □ حجم العينة الدقيقلكل مجموعة/شرط تجريبي، معطاة كرقم منفصل ووحدة قياس □ بيان حول ما إذا كانت القياسات قد أُخذت من عينات متميزة أو ما إذا كانت نفس العينة قد تم قياسها عدة مرات □
اختبار(ات) الإحصاء المستخدمة وما إذا كانت أحادية الجانب أو ثنائية الجانب □ X يجب أن تُوصف الاختبارات الشائعة فقط بالاسم؛ واصفًا التقنيات الأكثر تعقيدًا في قسم الطرق. □【 وصف لأي افتراضات أو تصحيحات، مثل اختبارات الطبيعية والتعديل للمقارنات المتعددة □
وصف كامل للمعلمات الإحصائية بما في ذلك الاتجاه المركزي (مثل المتوسطات) أو تقديرات أساسية أخرى (مثل معامل الانحدار) و التباين (مثل الانحراف المعياري) أو تقديرات مرتبطة بعدم اليقين (مثل فترات الثقة) □ X لاختبار الفرضية الصفرية، فإن إحصائية الاختبار (على سبيل المثال، ) مع فترات الثقة، أحجام التأثير، درجات الحرية وقيمة ملحوظة أعطِالقيم كقيم دقيقة كلما كان ذلك مناسبًا. □ لتحليل بايزي، معلومات حول اختيار القيم الأولية وإعدادات سلسلة ماركوف مونت كارلو □ لتصميمات هرمية ومعقدة، تحديد المستوى المناسب للاختبارات والتقارير الكاملة عن النتائج □【 تقديرات أحجام التأثير (مثل حجم تأثير كوهينبيرسون )، مما يشير إلى كيفية حسابها تحتوي مجموعتنا على الإنترنت حول الإحصائيات لعلماء الأحياء على مقالات تتناول العديد من النقاط المذكورة أعلاه.
البرمجيات والشيفرة
معلومات السياسة حول توفر كود الكمبيوتر جمع البيانات تم جمع البيانات السريرية في قاعدة بيانات الدراسة السريرية.
تحليل البيانات تم إجراء التحليلات الإحصائية باستخدام برنامج SAS الإصدار 9.4 (معهد SAS، كاري، نورث كارولينا). بالنسبة للمخطوطات التي تستخدم خوارزميات أو برامج مخصصة تكون مركزية في البحث ولكن لم يتم وصفها بعد في الأدبيات المنشورة، يجب أن تكون البرمجيات متاحة للمحررين والمراجعين. نحن نشجع بشدة على إيداع الشيفرة في مستودع مجتمعي (مثل GitHub). راجع إرشادات مجموعة Nature لتقديم الشيفرة والبرمجيات لمزيد من المعلومات.
بيانات
معلومات السياسة حول توفر البيانات يجب أن تتضمن جميع المخطوطات بيانًا حول توفر البيانات. يجب أن يوفر هذا البيان المعلومات التالية، حيثما ينطبق:
رموز الانضمام، معرفات فريدة، أو روابط ويب لمجموعات البيانات المتاحة للجمهور
وصف لأي قيود على توفر البيانات
بالنسبة لمجموعات البيانات السريرية أو بيانات الطرف الثالث، يرجى التأكد من أن البيان يتماشى مع سياستنا
ميرك شارب ودوهيم ذ.م.م، وهي شركة تابعة لميرك وشركاه، راهووي، نيو جيرسي، الولايات المتحدة الأمريكية (MSD) ملتزمة بتوفير الوصول إلى البيانات المجهولة وتقارير الدراسات السريرية من تجارب الشركة السريرية للباحثين العلميين المؤهلين بغرض إجراء أبحاث علمية مشروعة. كما أن MSD ملزمة بحماية حقوق وخصوصية المشاركين في التجارب، وبالتالي لديها إجراء لتقييم وتلبية الطلبات المتعلقة بمشاركة بيانات التجارب السريرية للشركة مع الباحثين العلميين الخارجيين المؤهلين. موقع مشاركة بيانات MSD (متاح على: http://engagezone.msd.com/ds_documentation.php) يحدد العملية و متطلبات تقديم طلب بيانات. سيتم تقييم الطلبات بسرعة من حيث الاكتمال والامتثال للسياسات. سيتم مراجعة الطلبات القابلة للتنفيذ من قبل لجنة من خبراء MSD لتقييم الصلاحية العلمية للطلب ومؤهلات مقدمي الطلبات. وفقًا لتشريعات خصوصية البيانات، يجب على مقدمي الطلبات المعتمدة الدخول في اتفاقية مشاركة بيانات قياسية مع MSD قبل منح الوصول إلى البيانات. ستتوفر البيانات للطلب بعد الموافقة على المنتج في الولايات المتحدة والاتحاد الأوروبي أو بعد توقف تطوير المنتج. هناك ظروف قد تمنع MSD من مشاركة البيانات المطلوبة، بما في ذلك اللوائح الخاصة بالبلدان أو المناطق. إذا تم رفض الطلب، سيتم إبلاغ الباحث بذلك. يتطلب الوصول إلى بيانات العلامات الحيوية الجينية أو الاستكشافية خطة تحليل إحصائي مفصلة مدفوعة بالفرضيات يتم تطويرها بالتعاون بين مقدم الطلب وخبراء MSD؛ بعد الموافقة على خطة التحليل الإحصائي وتنفيذ اتفاقية مشاركة البيانات، ستقوم MSD إما بإجراء التحليلات المقترحة ومشاركة النتائج مع مقدم الطلب أو ستقوم بإنشاء متغيرات العلامات الحيوية وإضافتها إلى ملف مع بيانات سريرية يتم تحميلها إلى بوابة التحليل حتى يتمكن مقدم الطلب من إجراء التحليلات المقترحة.
البحث الذي يتضمن مشاركين بشريين، بياناتهم، أو مواد بيولوجية
معلومات السياسة حول الدراسات التي تشمل مشاركين بشريين أو بيانات بشرية. انظر أيضًا معلومات السياسة حول الجنس، الهوية/التقديم الجنسي، والتوجه الجنسي والعرق، والعرقية والعنصرية.
التقارير عن الجنس والنوع الاجتماعي
التقارير عن العرق أو الإثنية أو غيرها من المجموعات الاجتماعية ذات الصلة
خصائص السكان
التوظيف
رقابة الأخلاقيات
تقرير المخطوطة عن مرضى سرطان الثدي.
تم تصنيف المرضى عند التوزيع العشوائي حسب المنطقة (أوروبا الشرقية مقابل الصين مقابل جميع الدول الأخرى). يتم الإبلاغ عن الخصائص الأساسية ذات الصلة في الجدول 1.
تم تقديم هذه المعلومات في الجدول 1.
تم تسجيل المرضى من 222 موقعًا عالميًا. تم إجراء التوظيف بواسطة المحققين وكان محدودًا بالمرضى الذين استوفوا معايير الأهلية المحددة في البروتوكول. يتم تقديم تفاصيل هذه المواقع في المواد التكميلية.
تمت الموافقة على البروتوكول من قبل هيئة أخلاقية في كل مؤسسة. قدم المرضى موافقة مستنيرة مكتوبة. أشرف لجنة مستقلة لمراقبة البيانات على الدراسة، وقامت بتقييم السلامة بشكل دوري، وقامت بتقييم الفعالية في تحليلات مؤقتة محددة مسبقًا (انظر المواد التكميلية للتفاصيل).
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
التقارير المتخصصة في المجال
يرجى اختيار الخيار أدناه الذي يناسب بحثك بشكل أفضل. إذا لم تكن متأكدًا، اقرأ الأقسام المناسبة قبل اتخاذ قرارك. علوم الحياة العلوم السلوكية والاجتماعية العلوم البيئية والتطورية والبيئية لنسخة مرجعية من الوثيقة بجميع الأقسام، انظرnature.com/documents/nr-reporting-summary-flat.pdf
تصميم دراسة العلوم الحياتية
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبياً. حجم العينة مع تسجيل حوالي 1240 مريضًا، كانت التجربة قدالقدرة على اكتشاف فرق حقيقي قدره 15 نقطة مئوية لمقارنة معدل pCR (المرحلة ypT0/Tis ypNO) بين مجموعات العلاج، عند مستوى ألفا أحادي الجانب قدره 0.005. سيكون لديها قدرة علىلكشف نسبة المخاطر لـ EFS تبلغ 0.73، عند مستوى ألفا أحادي الجانب قدره 0.02 في التحليل النهائي. تم تقديم خطة التحليل الإحصائي الكاملة في البروتوكول.
استثناءات البيانات تم تقييم الفعالية في مجموعة النية للعلاج، والتي شملت جميع المرضى الذين خضعوا للتوزيع العشوائي. وتم تقييم السلامة في مجموعة المعالجة، والتي شملت جميع المرضى الذين خضعوا للتوزيع العشوائي وتلقوادواء تجريبي، خضع لعملية جراحية، أو كليهما.
استنساخ كانت هذه تجربة سريرية. لم يكن من المناسب التكرار.
العشوائية تم تعيين المرضى عشوائيًا (بنسبة 1:1) إلى مجموعة العلاج الكيميائي مع البمبروليزوماب أو مجموعة العلاج الكيميائي مع الدواء الوهمي باستخدام نظام استجابة صوتية تفاعلية مركزي مع نظام استجابة ويب مدمج.
مُعَمي كانت هذه دراسة مزدوجة التعمية.
التقارير عن مواد وأنظمة وطرق محددة
نحتاج إلى معلومات من المؤلفين حول بعض أنواع المواد والأنظمة التجريبية والأساليب المستخدمة في العديد من الدراسات. هنا، يرجى الإشارة إلى ما إذا كانت كل مادة أو نظام أو طريقة مدرجة ذات صلة بدراستك. إذا لم تكن متأكدًا مما إذا كان عنصر القائمة ينطبق على بحثك، يرجى قراءة القسم المناسب قبل اختيار رد.
المواد والأنظمة التجريبية
طرق
غير متوفر
مشارك في الدراسة
غير متوفر
إكس
□ الأجسام المضادة
إكس
إكس
□
إكس
إكس
□
إكس
إكس
□
□
إكس
□
إكس
□
البيانات السريرية
معلومات السياسة حول الدراسات السريرية يجب أن تتوافق جميع المخطوطات مع إرشادات ICMJE لنشر الأبحاث السريرية ويجب أن تتضمن جميع التقديمات قائمة مراجعة CONSORT مكتملة.
تسجيل التجارب السريرية
ClinicalTrials.gov، NCT03725059
بروتوكول الدراسة
بروتوكول الدراسة متاح عند الطلب (وفقًا لبيان توفر البيانات)
جمع البيانات
من 27 ديسمبر 2018 حتى 5 أغسطس 2022، تم تسجيل ما مجموعه 1278 مريضًا من 222 موقعًا عالميًا في الدراسة.
النتائج
النقاط النهائية الرئيسية للدراسة كانت pCR، والتي تُعرف بـ ypNO في وقت الجراحة، وEFS في مجموعة النية للعلاج. تضمنت النقاط الثانوية pCR وفقًا لتعريفات ypTO ypNO وypTO/Tis في جميع المرضى، وpCR وفقًا لجميع التعريفات في المرضى الذين لديهم PD-L1 CPSالأورام، EFS بين المرضى الذين لديهم PD-L1 CPSالأورام، وOS بين جميع المرضى والمرضى الذين لديهم PD-L1 CPSأورام.
النباتات
مخزونات البذور
تقرير عن مصدر جميع مخزونات البذور أو المواد النباتية الأخرى المستخدمة. إذا كان ذلك مناسبًا، يرجى ذكر مركز مخزون البذور ورقم الفهرس. إذا تم جمع عينات نباتية من الحقل، يرجى وصف موقع الجمع، التاريخ وإجراءات أخذ العينات.
أنماط جينية نباتية جديدة
وصف الطرق التي تم من خلالها إنتاج جميع الأنماط الجينية النباتية الجديدة. يشمل ذلك تلك التي تم إنشاؤها من خلال الأساليب الجينية المتحولة، وتحرير الجينات، والطفرات المعتمدة على المواد الكيميائية/الإشعاع، والتهجين. بالنسبة لخطوط الجينات المتحولة، وصف طريقة التحويل، وعدد الخطوط المستقلة التي تم تحليلها، والجيل الذي أجريت عليه التجارب. بالنسبة لخطوط تحرير الجينات، وصف المحرر المستخدم، والتسلسل الداخلي المستهدف للتحرير، وتسلسل RNA الدليل المستهدف (إذا كان ذلك مناسبًا) وكيفية عمل المحرر.
المصادقة
تم تطبيقه.
وصف أي إجراءات تحقق لكل مخزون بذور مستخدم أو جينوتيب جديد تم إنشاؤه. وصف أي تجارب تم استخدامها لتقييم تأثير الطفرة، وحيثما ينطبق، كيف تم فحص التأثيرات الثانوية المحتملة (مثل إدخالات T-DNA في موقع ثانٍ، التباين، تحرير الجينات خارج الهدف).
Pembrolizumab and chemotherapy in high-risk, early-stage,ER HER2 breast cancer: a randomized phase 3trial
Received: 17 July 2024
Accepted: 12 November 2024
Published online: 21 January 2025
Check for updates
A list of authors and their affiliations appears at the end of the paper
Addition of pembrolizumab to neoadjuvant chemotherapy followed by adjuvant pembrolizumab improved outcomes in patients with high-risk, early-stage, triple-negative breast cancer. However, whether the addition of neoadjuvant pembrolizumab to chemotherapy would improve outcomes in high-risk, early-stage, estrogen receptor-positive/human epidermal growth factor receptor 2-negative (ER ) breast cancer remains unclear. We conducted a double-blind, placebo-controlled phase 3 study (KEYNOTE-756) in which patients with previously untreated ER grade 3 high-risk invasive breast cancer (T1c-2 ( ), cN1-2 or T3-4, cNO-2) were randomly assigned (1:1) to neoadjuvant pembrolizumab 200 mg or placebo Q3W given with paclitaxel QW for 12 weeks, followed by four cycles of doxorubicin or epirubicin plus cyclophosphamide Q2W or Q3W. After surgery (with/without adjuvant radiation therapy), patients received adjuvant pembrolizumab or placebo for nine cycles plus adjuvant endocrine therapy. Dual primary endpoints were pathological complete response and event-free survival in the intention-to-treat population. In total, 635 patients were assigned to the pembrolizumab-chemotherapy arm and 643 to the placebo-chemotherapy arm. At the study’s prespecified first interim analysis, the pathological complete response rate was ( confidence interval (CI), 21.0-27.8%) in the pembrolizumab-chemotherapy arm and ( ) in the placebo-chemotherapy arm (estimated treatment difference, 8.5 percentage points; 95% CI, 4.2-12.8; ). Event-free survival was not mature in this analysis. During the neoadjuvant phase, treatment-related adverse events of grade were reported in and of patients in the pembrolizumabchemotherapy and placebo-chemotherapy arms, respectively. In summary, the addition of pembrolizumab to neoadjuvant chemotherapy significantly improved the pathological complete response rate in patients with high-risk, early-stage ER breast cancer. Safety was consistent with the known profiles of each study treatment. Follow-up continues for event-free survival. ClinicalTrials.gov identifier: NCT03725059.
Fig.1|Disposition of patients in the study. Progressive disease included radiographic progressive disease. Patients did not have to complete all neoadjuvant therapy to undergo surgery.
Estrogen receptor-positive/human epidermal growth factor receptor 2-negative ( ) breast cancer is a heterogeneous disease that includes a subpopulation of patients (including those with high-grade tumors and lymph node involvement) at high risk of recurrence who have poor long-term outcomes despite (neo) adjuvant chemotherapy and adjuvant endocrine therapy . For these patients, reported pathological complete response (pCR) rates range from to and event-free survival (EFS) rates are similar to those of patients with triple-negative breast cancer (TNBC) . Although disease is heterogeneous, in patients with breast cancer, a meta-analysis of neoadjuvant studies has demonstrated a positive correlation between pCR and both EFS and overall survival (OS) . Regulatory guidance supports pCR as an appropriate endpoint for the evaluation of the efficacy of neoadjuvant treatment .
Combination therapy with immune checkpoint inhibitors plus chemotherapy induces changes to the tumor microenvironment that may enhance endogenous anticancer immunity, reduce tumor volume and increase response rate compared with chemotherapy alone . Supporting this hypothesis, clinical data have demonstrated that the antiprogrammed cell death protein 1 (anti-PD-1) monoclonal antibody pembrolizumab combined with neoadjuvant chemotherapy in the phase 2 I-SPY2 trial more than doubled the estimated pCR rates for patients with high-risk (defined by MammaPrint score) ER /HER2 tumors compared with patients receiving neoadjuvant chemotherapy alone ( versus , respectively) . Additionally, pembrolizumab combined with neoadjuvant chemotherapy has been shown to improve pCR and EFS in patients with earlystage TNBC .
Building on these results, KEYNOTE-756 (NCT03725059) was designed as a randomized, double-blind, phase 3 study to evaluate the efficacy and safety of neoadjuvant pembrolizumab plus chemotherapy followed by adjuvant pembrolizumab plus endocrine therapy versus neoadjuvant placebo plus chemotherapy followed by adjuvant placebo plus endocrine therapy in patients with high-risk, early-stage, breast cancer.
Results
Patients and treatment
From 27 December 2018 to 5 August 2022, a total of 1,278 patients from 222 global sites were randomly assigned to the pembrolizumab-chemotherapy arm (635 patients) or to the placebo-chemotherapy arm ( 643 patients; Fig. 1). Demographics and baseline disease characteristics were balanced between treatment arms (Table 1).
At the first interim analysis (data cutoff, 25 May 2023; median duration of follow-up, 33.2 (range = 9.7-51.8) months), 1,275 patients had received the first neoadjuvant treatment, 1,210 patients had started the second neoadjuvant treatment, 1,245 patients had documented surgery and 1,002 patients had started adjuvant treatment (Fig. 1). Median duration of treatment in the neoadjuvant phase was 4.9 months (range months) in the pembrolizumab-chemotherapy arm and 4.9 months (range months) in the placebo-chemotherapy arm (Extended Data Table 1). Both groups received a similar median number of chemotherapy cycles.
Efficacy
A pCR (ypTO/Tis ypNO) was observed in 154 of 635 patients ( ) in the pembrolizumab-chemotherapy arm and 100 of 643 patients in the placebo-chemotherapy arm. The estimated treatment difference in the rate of pCR was 8.5 percentage points ( confidence interval (CI), ; Table 2). The prespecified statistical significance criterion for this analysis was ; thus, the percentage of patients who had a pCR was significantly higher in the pem-brolizumab-chemotherapy arm than in the placebo-chemotherapy arm. Similar results were observed with respect to the percentage of patients who had a pCR defined per the secondary endpoints of ypTO ypNO and ypTO/Tis (Table 2).
Benefits of pembrolizumab-chemotherapy as compared to pla-cebo-chemotherapy with respect to pCR ( ypNO) were generally consistent across subgroups defined by demographics and baseline clinical characteristics (Fig. 2). Notably, a numerically higher rate of pCR difference was observed with higher tumor PD-L1 expression. The estimated treatment differences in the prespecified subgroups based
Table 1 | Demographics and baseline disease characteristics
Characteristic
Pembrolizumabchemotherapy ( )
Placebochemotherapy ( )
Age (year)
Median (range)
49 (24-82)
49 (19-78)
. (%)
89 (14.0)
76 (11.8)
Country/region-no. (%)
China
88 (13.9)
91 (14.2)
Eastern Europe
139 (21.9)
130 (20.2)
Other
408 (64.3)
422 (65.6)
PD-L1 CPS-no. (%)
<1
153 (24.1)
154 (24.0)
482 (75.9)
489 (76.0)
253 (39.8)
259 (40.3)
125 (19.7)
129 (20.1)
ECOG performance status-no. (%)
0
570 (89.8)
588 (91.4)
1
65 (10.2)
55 (8.6)
Anthracycline schedule-no. (%)
Every 3weeks
415 (65.4)
425 (66.1)
Every 2weeks
183 (28.8)
187 (29.1)
Not started
37 (5.8)
31 (4.8)
Tumor classification-no. (%)
T1-T2
402 (63.3)
413 (64.2)
T3-T4
233 (36.7)
230 (35.8)
Nodal involvement-no. (%)
Positive
570 (89.8)
582 (90.5)
Negative
65 (10.2)
61 (9.5)
Overall disease stage-no. (%)
Stage II
399 (62.8)
408 (63.5)
Stage III
236 (37.2)
235 (36.5)
Tumor grade-no. (%)
Grade 3
635 (100)
642 (99.8)
Grade 2
0
ER positivity-no. (%)
601 (94.6)
600 (93.3)
<10%
34 (5.4)
43 (6.7)
Menopausal status-no. (%)
Premenopausal
354 (55.7)
353 (54.9)
Postmenopausal
278 (43.8)
287 (44.6)
Not applicable
3 (0.5)
3 (0.5)
Data are from the intention-to-treat population. All patients had previously untreated, centrally confirmed , disease. A total of six men with and breast cancer were included in the study (three in each treatment arm). PD-L1 CPS was defined as the number of PD-L1-positive tumor cells, lymphocytes and macrophages divided by the total number of tumor cells multiplied by 100 . ECOG performance status ranges from 0 to 5 , with higher scores indicating greater disability. Protocol violation.
on PD-L1 combined positive score (CPS) of and were to 10.1 and percentage points, respectively. The estimated treatment difference was 17.4 ( , ) percentage points
Table 2 | pCR at the first interim analysis according to pathological stage
Participants were considered non-responders if they did not receive the study medication, discontinued study treatment and continued neoadjuvant treatment with drug categories not specified by the study before surgery (regardless of surgical outcome), discontinued study treatment for reasons that precluded surgery or had missing data for pCR for any reason. pCR was assessed by the local pathologist at the time of surgery per the current AJCC breast cancer staging criteria. Pathological stage ypTO/Tis ypNO was defined as the absence of residual invasive cancer in the complete resected breast specimen and all sampled regional lymph nodes. Pathological stage ypTO ypNO was defined as the absence of residual invasive and in situ cancer in the complete resected breast specimen and all sampled regional lymph nodes. Pathological stage ypTO/Tis was defined as the absence of residual invasive and in situ cancer in the complete resected breast specimen (independent of lymph node involvement) and all sampled regional lymph nodes. Estimated treatment difference was calculated using the stratified Miettinen-Nurminen method.
in the post hoc subgroup analysis of pCR based on PD-L1 CPS of ( ). Additionally, pCR benefit for pembrolizumab-chemotherapy was observed both in patients with ER positivity and , albeit with a greater magnitude among those with ER positivity . Among patients with ER positivity , the pCR rate was (19 of 34 patients) in the pembrolizumab-chemotherapy arm versus 30.2% ( 13 of 43 patients) in the placebo-chemotherapy arm (estimated treatment difference, 25.6 percentage points ( )), whereas among patients with ER positivity , the pCR was ( 135 of 601 patients) versus 14.5% (87 of 600 patients; estimated treatment difference, 8.0 percentage points ; Fig. 2).
In post hoc exploratory analyses, an improvement in Tis ypNO) was observed based on both PD-L1 CPSs and ER positivity, with the largest numerical difference observed in patients with PD-L1 CPS and ER positivity ( ; treatment difference, 24.2 percentage points ( ); Extended Data Fig. 1).
Analysis of the exploratory endpoint of residual cancer burden (RCB) showed that the addition of pembrolizumab to neoadjuvant chemotherapy shifted more patients to lower RCB categories (RCB-0 or RCB-1,35.0% versus 23.6%; RCB-2,40.8% versus 45.3%; RCB-3,20.5% versus 28.9%; Fig. 3).
EFS was not mature at the first interim analysis; this endpoint continues to be evaluated by prespecified subsequent interim analyses and a final analysis.
Safety
During the neoadjuvant phase, treatment-related adverse events (AEs) of any grade occurred in 624 of 634 patients ( ) in the pembroli-zumab-chemotherapy arm and 633 of 642 patients ( ) in the pla-cebo-chemotherapy arm (Table 3). Treatment-related grade AEs occurred in 333 ( ) and 298 patients ( ), respectively. Serious
Pembrolizumab-
Placebochemotherapy chemotherapy
No. of patients with response/no. of patients (%)
Difference in pathological complete response (95% CI)
Percentage points
Overall
154/635 (24.3)
100/643 (15.6)
– –
–
8.5 (4.2 to 12.8)
Disease stage
Stage II
103/399 (25.8)
68/408 (16.7)
9.1 (3.5 to 14.8)
Stage III
51/236 (21.6)
32/235 (13.6)
8.0 (1.1 to 14.9)
Nodal status
Positive
143/570 (25.1)
92/582 (15.8)
9.3 (4.6 to 13.9)
Negative
11/65 (16.9)
8/61 (13.1)
3.8 (-9.2 to 16.7)
Tumor size
T1-T2
111/402 (27.6)
71/413 (17.2)
10.4 (4.7 to 16.1)
T3-T4
43/233 (18.5)
29/230 (12.6)
5.8 (-0.8 to 12.5)
Anthracycline schedule
Every 3 week
97/415 (23.4)
55/425 (12.9)
10.4 (5.3 to 15.7)
Every 2 week
54/183 (29.5)
44/187 (23.5)
6.0 (-3.0 to 15.0)
Not started
3/37 (8.1)
1/31 (3.2)
4.9 (-9.2 to 18.8)
PD-L1 CPS
<1
11/153 (7.2)
4/154 (2.6)
4.5 (-0.4 to 10.1)
143/482 (29.7)
96/489 (19.6)
9.8 (4.4 to 15.2)
107/253 (42.3)
75/259 (29.0)
13.2 (4.9 to 21.4)
67/125 (53.6)
47/129 (36.4)
17.4 (5.1 to 29.1)
ER positivity
135/601 (22.5)
87/600 (14.5)
8.0 (3.6 to 12.4)
<10
19/34 (55.9)
13/43 (30.2)
25.6 (3.3 to 45.8)
Age
<65 yr
135/546 (24.7)
89/567 (15.7)
9.0 (4.3 to 13.7)
19/89 (21.3)
11/76 (14.5)
□
6.9 (-5.2 to 18.6)
ECOG performance status
0
142/570 (24.9)
91/588 (15.5)
9.4 (4.8 to 14.1)
1
12/65 (18.5)
9/55 (16.4)
2.1 (-12.2 to 15.8)
Geographic region
China
11/88 (12.5)
9/91 (9.9)
□
2.6 (-7.0 to 12.5)
Eastern Europe
41/139 (29.5)
21/130 (16.2)
13.3 (3.3 to 23.2)
Other
102/408 (25.0)
70/422 (16.6)
8.4 (2.9 to 13.9)
Menopausal status
Premenopausal
83/354 (23.4)
57/353 (16.1)
m
Postmenopausal
69/278 (24.8)
42/287 (14.6)
10.2 (3.7 to 16.8)
Placebochemotherapy better
Fig. 2 | Subgroup analysis of the difference in percentages of patients with a pCR at the first interim analysis. Data from key subgroups are shown. For the overall population, the estimated mean treatment difference with 95% CI was calculated using the stratified Miettinen-Nurminen method. The analysis for PD-L1CPS subgroups was stratified. All other analyses were unstratified. The
footnote ‘ ‘ indicates the subgroup analyses of pCR based on a PD-L1CPS cutoff of 20 were not prespecified. PD-L1CPS was defined as the number of PD-L1positive tumor cells, lymphocytes and macrophages divided by the total number of tumor cells multiplied by 100 . ECOG performance status ranges from 0 to 5 , with higher scores indicating greater disability.
treatment-related AEs were reported in 18.5% and 10.3% of patients, respectively. One patient ( ) in the pembrolizumab arm died due to treatment-related acute non- wave myocardial infarction. Discontinuation of any study treatment due to treatment-related AEs occurred in 121 patients ( ) in the pembrolizumab-chemotherapy arm and 65 patients ( ) in the placebo-chemotherapy arm. The most frequently occurring treatment-related AEs were alopecia ( and , respectively), nausea ( and , respectively), fatigue ( and , respectively) and anemia ( and , respectively; Table 3).
Immune-mediated AEs, excluding infusion reactions, of any grade were observed in 208 patients ( ) in the pembrolizumabchemotherapy arm and 45 patients ( ) in the placebo-chemotherapy arm (Table 3). Grade immune-mediated AEs were reported
in and 8 patients ( ), respectively. The most frequently occurring immune-mediated AEs were hypothyroidism ( and , respectively), hyperthyroidism ( and , respectively) and pneumonitis (2.8% and 1.4%, respectively). No deaths were attributed to immune-mediated AEs.
Discussion
In this randomized phase 3 trial involving patients with previously untreated, high-risk, early-stage, breast cancer, a significantly higher percentage of patients in the pembrolizumab-chemotherapy arm than in the placebo-chemotherapy arm had a pCR at the time of surgery. The between-group difference in pCR favored pembrolizumab-chemotherapy across all prespecified subgroups, albeit with a differing magnitude of benefit and wide 95% CIs in some
Table 3 | AEs during the neoadjuvant phase at the first interim analysis
Adverse event
Pembrolizumabchemotherapy ( )
Placebochemotherapy ( )
Any grade
Grade
Any grade
Grade
Number of patients (%)
Any AE
634 (100.0)
381 (60.1)
638 (99.4)
350 (54.5)
Treatment-related AE
624 (98.4)
333 (52.5)
633 (98.6)
298 (46.4)
Alopecia
406 (64.0)
0
391 (60.9)
0
Nausea
306 (48.3)
8 (1.3)
321 (50.0)
7 (1.1)
Anemia
205 (32.3)
21 (3.3)
164 (25.5)
18 (2.8)
Fatigue
190 (30.0)
17 (2.7)
180 (28.0)
9 (1.4)
Diarrhea
172 (27.1)
11 (1.7)
130 (20.2)
10 (1.6)
Alanine aminotransferase increased
158 (24.9)
24 (3.8)
147 (22.9)
18 (2.8)
Neutropenia
146 (23.0)
85 (13.4)
158 (24.6)
101 (15.7)
Aspartate aminotransferase increased
137 (21.6)
10 (1.6)
107 (16.7)
6 (0.9)
Neutrophil count decreased
137 (21.6)
89 (14.0)
153 (23.8)
103 (16.0)
Asthenia
134 (21.1)
10 (1.6)
116 (18.1)
4 (0.6)
Vomiting
127 (20.0)
9 (1.4)
108 (16.8)
9 (1.4)
Peripheral neuropathy
111 (17.5)
4 (0.6)
130 (20.2)
5 (0.8)
Immune-mediated
208 (32.8)
45 (7.1)
45 (7.0)
8 (1.2)
Hypothyroidism
111 (17.5)
1 (0.2)
11 (1.7)
0
Hyperthyroidism
57 (9.0)
1 (0.2)
3 (0.5)
0
Pneumonitis
18 (2.8)
9 (1.4)
9 (1.4)
2 (0.3)
Adrenal insufficiency
16 (2.5)
5 (0.8)
0
0
Severe skin reactions
14 (2.2)
8 (1.3)
3 (0.5)
2 (0.3)
Hypophysitis
12 (1.9)
6 (0.9)
1 (0.2)
0
Thyroiditis
11 (1.7)
1 (0.2)
2 (0.3)
0
Hepatitis
8 (1.3)
7 (1.1)
3 (0.5)
0
Colitis
6 (0.9)
3 (0.5)
5 (0.8)
1 (0.2)
Vasculitis
5 (0.8)
1 (0.2)
4 (0.6)
0
AEs were collected up to 30 days after discontinuation of treatment ( 90 days for serious AEs). Events are listed in descending order of frequency in the pembrolizumab-chemotherapy arm. The safety-evaluable population included patients who received at least one trial drug, underwent surgery, or both. The severity of AEs was graded per the National Cancer Institute Common Terminology Criteria for AEs (v4.0). Treatment-related AEs were events that were considered related to a study treatment by the investigator. Treatment-related AEs that occurred in at least of patients are reported. Immune-mediated AEs, excluding infusion reactions, were based on a list of preferred terms intended to capture known risks of pembrolizumab and were considered regardless of attribution to study treatment by the investigator. Immune-mediated AEs that occurred in patients are reported.
subgroups. Notably, pCR was numerically higher in subgroups of patients with higher tumor PD-L1 expression and in the subgroup of patients with ER positivity (who have previously been reported to have biological characteristics and clinical outcomes similar to those of patients with TNBC ). However, these results should be interpreted with caution as these subgroups are underpowered, and the only objective of subgroup analyses is to explore convergent validity. Post hoc exploratory subgroup analyses of pCR based on PD-L1CPS of and based on both PD-L1CPSs and ER positivity were reported. It is important to exercise caution when interpreting these observations,
Fig. 3 | RCB at the first interim analysis. RCB was an exploratory endpoint and was assessed by a local pathologist at the time of surgery. RCB-0, RCB-1, RCB-2 and RCB-3 denote increasingly larger residual diseases. RCB data were missing for and 14 patients ( ) in the pembrolizumab-chemotherapy and placebo-chemotherapy arms, respectively.
especially with very small sample sizes. To establish confidence, replication in other trials is absolutely essential. The efficacy associated with neoadjuvant pembrolizumab-chemotherapy was supported by the similar results observed when defined per secondary endpoints (that is, ypTO ypNO and ). The addition of pembrolizumab to neoadjuvant chemotherapy also shifted more patients to lower RCB categories (RCB-0 and RCB-1), demonstrating the ability of the combination to reduce tumor tissue remaining after surgery among those without pCR. Reduced RCB has been associated with improved .
Results from our study are consistent with those from the phase 2 I-SPY2 study in which the addition of pembrolizumab to chemotherapy improved pCR in patients with previously untreated, high-risk, early-stage,HER2- breast cancer . In the phase 3 CheckMate 7FL study, a similar improvement in pCR outcomes was reported among patients with high-risk, early-stage, grade 2/grade breast cancer who received nivolumab plus neoadjuvant chemotherapy compared to those who received placebo plus neoadjuvant chemotherapy ( versus 13.8%; odds ratio .
In the adjuvant phase of our study, patients received pembrolizumab or placebo plus standard endocrine therapy. The monarchE study demonstrated that the addition of the cyclin-dependent kinase 4/cyclin-dependent kinase 6 inhibitor abemaciclib to adjuvant endocrine therapy improved relapse-free survival in patients with hormone receptor-positive, HER2 , node-positive, high-risk, early-stage breast cancer . However, the present study was designed before these results were reported, and the use of adjuvant abemaciclib was not permitted per protocol. Given that pCR was assessed before the adjuvant phase, the use of abemaciclib could not have influenced this outcome. Potential safety risks have been reported in studies investigating cyclin-dependent kinase 4/6 inhibitor plus immune checkpoint inhibitor combination therapy in patients with hormone receptor-positive, HER2 metastatic breast cancer . Current guidelines also do not recommend such combination therapy .
Data for the study’s other primary endpoint of EFS are not mature and continue to be evaluated. The EFS outcome will reflect the overall treatment regimen effect, including both the neoadjuvant and adjuvant treatment phases. This study was not designed to discriminate the relative contribution of each phase; a prospective trial would be needed to address this question.
AEs in the pembrolizumab-chemotherapy arm were consistent with the known safety profiles of the individual agents. Similar to results from the KEYNOTE-522 study (which evaluated pembrolizumab in combination with neoadjuvant chemotherapy among patients with TNBC) , there was no evidence that the addition of
pembrolizumab to standard neoadjuvant chemotherapy exacerbated chemotherapy-associated toxicity, and the most frequently occurring events in both arms were those typically associated with cytotoxic chemotherapy, such as alopecia, nausea and anemia. Incidences of treatment-related grade AEs and treatment-related AEs leading to treatment discontinuation were moderately higher with pem-brolizumab-chemotherapy than with placebo-chemotherapy. As anticipated based on prior studies evaluating anti-PD-1 and anti-PD-L1 monoclonal antibodies , AEs with a potentially immune-mediated mechanism occurred more frequently among patients in the pem-brolizumab-chemotherapy arm than in the placebo-chemotherapy arm. Broadly, the safety profile in this study was consistent with that in the KEYNOTE-522 study .
In conclusion, neoadjuvant pembrolizumab-chemotherapy resulted in an improved pCR rate compared with chemotherapy alone in patients with high-risk, early-stage, breast cancer. Evaluation of EFS is ongoing.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, Supplementary Information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-024-03415-7.
References
Jatoi, I., Anderson, W. F., Jeong, J. H. & Redmond, C. K. Breast cancer adjuvant therapy: time to consider its time-dependent effects. J. Clin. Oncol. 29, 2301-2304 (2011).
Torrisi, R. et al. Neoadjuvant chemotherapy in hormone receptor-positive/HER2-negative early breast cancer: when, why and what? Crit. Rev. Oncol. Hematol. 160, 103280 (2021).
Cortazar, P. et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164-172 (2014).
U.S. Department of Health and Human Services, FDA, Oncology Center of Excellence, CDER & CBER. Pathological complete response in neoadjuvant treatment of high-risk early-stage breast cancer: use as an endpoint to support accelerated approval guidance for industry. www.fda.gov/media/83507/download (accessed 21 December 2023).
Emens, L. A. & Middleton, G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol. Res. 3, 436-443 (2015).
Xue, Y. et al. Platinum-based chemotherapy in combination with PD-1/PD-L1 inhibitors: preclinical and clinical studies and mechanism of action. Expert Opin. Drug Deliv. 18, 187-203 (2021).
Nanda, R. et al. Effect of pembrolizumab plus neoadjuvant chemotherapy on pathologic complete response in women with early-stage breast cancer: an analysis of the ongoing phase 2 adaptively randomized I-SPY2 trial. JAMA Oncol. 6, 676-684 (2020).
Schmid, P. et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 382, 810-821 (2020).
Schmid, P. et al. Event-free survival with pembrolizumab in early triple-negative breast cancer. N. Engl. J. Med. 386, 556-567 (2022).
Schrodi, S. et al. Outcome of breast cancer patients with low hormone receptor positivity: analysis of a 15-year populationbased cohort. Ann. Oncol. 32, 1410-1424 (2021).
Fredriksson, I., Acs, B., Hartman, J., Sönmez, D. & Lindman, H. 241MO patient characteristics and real-world outcomes in HER2 negative/ER zero and ER low patients treated as triple-negative breast cancer in Sweden 2008-2020. Ann. Oncol. 34, S278-S324 (2023).
Iwamoto, T. et al. Estrogen receptor (ER) mRNA and ER-related gene expression in breast cancers that are 1% to 10% ER-positive by immunohistochemistry. J. Clin. Oncol. 30, 729-734 (2012).
Yoder, R. et al. Impact of low versus negative estrogen/ progesterone receptor status on clinico-pathologic characteristics and survival outcomes in HER2-negative breast cancer. NPJ Breast Cancer 8, 80 (2022).
Yau, C. et al. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: a multicentre pooled analysis of 5161 patients. Lancet Oncol. 23, 149-160 (2022).
Loi, S. et al. LBA20 A randomized, double-blind trial of nivolumab (NIVO) vs placebo (PBO) with neoadjuvant chemotherapy (NACT) followed by adjuvant endocrine therapy (ET) ± NIVO in patients (pts) with high-risk, ER HER2 primary breast cancer (BC). Ann. Oncol. 34, S1259-S1260 (2023).
Johnston, S. R. D. et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. 24, 77-90 (2023).
Rugo, H. S. et al. Abemaciclib in combination with pembrolizumab for , metastatic breast cancer: phase 1b study. NPJ Breast Cancer 8, 118 (2022).
Masuda, J. et al. Efficacy, safety, and biomarker analysis of nivolumab in combination with abemaciclib plus endocrine therapy in patients with HR-positive HER2-negative metastatic breast cancer: a phase II study (WJOG11418B NEWFLAME trial). J. Immunother. Cancer 11, e007126 (2023).
Jerusalem, G. et al. Neoadjuvant nivolumab + palbociclib + anastrozole for oestrogen receptor-positive/human epidermal growth factor receptor 2-negative primary breast cancer: results from CheckMate 7A8. Breast 72, 103580 (2023).
NCCN clinical practice guidelines in oncology (NCCN guidelines). Breast cancer, version 4.2023. www.nccn.org/ (accessed 15 February 2023).
Brahmer, J. R. et al. Society for immunotherapy of cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 9, e002435 (2021).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons. org/licenses/by/4.0/.
¹Breast Unit, Champalimaud Clinical Centre/Champalimaud Foundation, Lisbon, Portugal. ²Baylor University Medical Center, Texas Oncology, US Oncology Network, Dallas, TX, USA. Department of Breast Disease, Henan Breast Cancer Center, Affiliated Cancer Hospital of Zhengzhou University & Henan Cancer Hospital, Zhengzhou, China. University of Texas Southwestern Medical Center, Dallas, TX, USA. Centre for Experimental Cancer Medicine, Barts Cancer Institute, Queen Mary University London, London, UK. Medica Scientia Innovation Research, Ridgewood, NJ, USA. Medica Scientia Innovation Research, Barcelona, Spain. International Breast Cancer Center (IBCC), Pangaea Oncology, Quiron Group, Barcelona, Spain. Universidad Europea de Madrid Faculty of Biomedical and Health Sciences, Department of Medicine, Madrid, Spain. IOB Madrid, Institute of Oncology, Hospital Beata María Ana, Madrid, Spain. Breast Center, Department of OB&GYN, LMU University Hospital, Munich, Germany. Stanford University School of Medicine, Stanford, CA, USA. Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, Canada. University Hospital Erlangen, Comprehensive Cancer Center Erlangen-EMN, Bavarian Cancer Research Center (BZKF), Erlangen, Germany. Department of Breast Surgery, Fudan University Shanghai Cancer Center, Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China. Institute Curie, Paris, France. Samsung Medical Division, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea. Hemato Oncólogo, IMAT Oncomedica, Montería, Colombia. National Institute of Oncology, Budapest, Hungary. Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA. Seoul National University Hospital, Cancer Research Institute, Seoul National University College of Medicine, Seoul National University, Seoul, Republic of Korea. Westmead Breast Cancer Institute, Westmead Hospital and the University of Sydney, Sydney, New South Wales, Australia. Centre of Cancer Medicine, University of Hong Kong, Pokfulam, Hong Kong. The Cancer Institute Hospital of JFCR, Tokyo, Japan. Faculté de Médecine, Paris Saclay University, Gustave Roussy, Villejuif, France. NHO Osaka National Hospital, Osaka, Japan. Merck & Co., Inc., Rahway, NJ, USA. University of California Los Angeles, Jonsson Comprehensive Cancer Center, Los Angeles, CA, USA. e-mail: fcardoso@abcglobalalliance.org
Methods
Patients
Eligible patients were years of age with centrally confirmed HER2 grade 3 invasive ductal breast carcinoma (evaluated according to the most recent guidelines of the American Society of Clinical Oncology-College of American Pathologists ); newly diagnosed, previously untreated, nonmetastatic disease (T1c-2 ( ), cN1-2 or T3-4, cNO-2, per the most current staging criteria of the American Joint Committee on Cancer (AJCC)), as determined by the investigator in radiologic assessment, clinical assessment or both; an Eastern Cooperative Oncology Group (ECOG) performance-status score ; and adequate organ function. Patients with multifocal primary tumors, inflammatory breast cancer and those with low ER positivity were eligible.
Exclusion criteria included active bilateral, multicentric, lobular or metastatic breast cancer; autoimmune disease for which the patient was treated with systemic therapy within 2 years; diagnosis of immunodeficiency or use of immunosuppressive therapy within 1 week; history of noninfectious pneumonitis treated with steroids; pneumonitis; active tuberculosis; active infection for which the patient was receiving systemic therapy; or clinically significant cardiovascular disease. Full eligibility criteria are listed in the Supplementary Information.
Trial design and treatment
In this randomized, double-blind trial (ClinicalTrials.gov identifier, NCT03725059), patients received treatment in neoadjuvant and adjuvant phases. No crossover between treatment arms was permitted between the phases. Based on emerging clinical data in patients with early-stage breast cancer demonstrating heterogeneous long-term clinical outcomes across countries/regions despite similar standard of care treatments, patients were stratified at randomization by region (Eastern Europe versus China versus all other countries) to ensure balance across treatment arms. Patients from Eastern Europe were further stratified by tumor PD-L1 status (CPS versus ). Patients from China were not further stratified. Patients from all other countries (that is, excluding Eastern Europe and China) were further stratified by tumor PD-L1 status (CPS versus ), lymph node involvement (positive versus negative), anthracycline dosing schedule (Q2W versus Q3W) and ER positivity ( versus ). Patients were randomly assigned (in a ratio) to the pembrolizumab-chemotherapy arm or the pla-cebo-chemotherapy arm using a central interactive voice-response system with an integrated web-response system. In the neoadjuvant phase, patients received four cycles of intravenous pembrolizumab or placebo once Q3W plus paclitaxel QW ; first neoadjuvant treatment), followed by four cycles of pembrolizumab or placebo in combination with either doxorubicin or epirubicin ( ) plus cyclophosphamide ( ) administered either Q2W or Q3W (second neoadjuvant treatment). Patients who either completed or discontinued the first neoadjuvant treatment could start the second neoadjuvant treatment or undergo surgery, and those who completed or discontinued the second neoadjuvant treatment could undergo surgery. Patients underwent surgery (breast conservation or mastectomy with/without sentinel lymph node biopsy or axillary dissection) no later than 6 weeks after the last dose of the neoadjuvant treatment. In the adjuvant phase, patients received (beginning within 60 days after surgery) pembrolizumab or placebo Q3W for up to nine cycles (up to 6 months), plus the investigator’s choice of endocrine therapy (per institutional guidelines) for up to 10 years. Adjuvant therapy with abemaciclib was not permitted. Adjuvant radiation therapy (per institutional guidelines) was permitted, as indicated, either before initiation of adjuvant therapy or concurrently. Trial treatment was discontinued in patients with disease progression or recurrence or unacceptable toxic effects.
The study was developed by a scientific advisory committee and employees of the sponsor (Merck Sharp & Dohme LLC, a subsidiary
of Merck & Co., Inc., Rahway, NJ, USA). An external independent data monitoring committee oversaw the study, periodically assessed safety, and assessed efficacy at prespecified interim analyses. The protocol was approved by an ethics body at each study site (see Supplementary Information for a list of study sites). Patients provided written informed consent
Assessments
Upon completion of neoadjuvant therapy, pCR was assessed according to the AJCC staging criteria (ypTO/Tis ypNO, ypTO ypNO and ypTO/Tis) by a local pathologist blinded to treatment assignment. EFS (defined as the time from randomization to disease progression that precluded surgery, local or distant recurrence, second primary cancer or death due to any cause, whichever occurred first) was evaluated in ablinded fashion by the investigator.PD-L1 expression in new or recent core needle biopsy samples was assessed at a central laboratory using PD-L1IHC 22 C 3 pharmDx (Agilent Technologies). AEs were assessed throughout the trial and for 30 days after discontinuation of treatment ( 90 days for serious AEs) and graded according to Common Terminology Criteria for AEs (v4.0) . Immune-mediated AEs were based on a list of preferred terms intended to capture known risks of pembrolizumab and were considered regardless of attribution to study treatment by the investigator.
Endpoints
The study’s primary endpoints were pCR , defined as ypNO at the time of surgery, and EFS in the intention-to-treat population. Secondary endpoints included pCR according to the definitions of ypTO ypNO and Tis in all patients, pCR according to all definitions in patients with PD-L1 CPS tumor, EFS among patients with PD-L1 CPS tumor and OS among all patients and patients with PD-L1CPS tumor. Safety during the neoadjuvant and adjuvant phases was evaluated in all patients who received trial drug, underwent surgery or both. Evaluation of RCB (residual disease in either the breast or lymph node at the time of surgery) was an exploratory endpoint.
Statistical analysis
Efficacy was evaluated in the intention-to-treat population, which included all patients who had undergone randomization. Safety was evaluated in the as-treated population, which included all patients who had undergone randomization and received trial drug, underwent surgery or both. We used the stratified Miettinen-Nurminen method , with weights proportional to the stratum sample size, to compare between-arm differences in percentages of patients with a pCR. Patients who did not have pCR results for any reason or who received neoadjuvant treatment not specified in the protocol were considered as not having a response.
The CIs associated with the between-arm differences in the percentages of patients with a pCR were not adjusted for multiple comparisons and therefore cannot be used to infer effects. The stratification factors used at randomization with the prespecified pooling strategy were used in all stratified analyses.
The graphical method discussed in ref. 27 was applied to control the type I error rate at a one-sided level of 0.025 across both primary endpoints and all interim and final analyses (Extended Data Fig. 2). The Lan-DeMets O’Brien-Fleming spending function was used to control the type I error in the interim and final analyses. The primary purpose of the first interim analysis was to evaluate the superiority of pembrolizumab-chemotherapy over placebo-chemotherapy with respect to the percentage of patients with a pCR (stage ypT0/Tis, ypNO ); this analysis was to occur after enrollment was completed and all randomized patients would have had surgery after approximately 6 months of neoadjuvant treatment.
With an enrollment of approximately 1,240 patients, the trial had power to detect a true difference of 15 percentage points for the
comparison of the rate of pCR (stage ypT0/Tis ypNO) between the treatment arms at a one-sided level of 0.005 . It would have a power of to detect a hazard ratio for EFS of 0.73 at a one-sided level of 0.02 at the final analysis.
Statistical analyses were conducted using SAS v9.4 (SAS Institute).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD), is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data-sharing website (available at engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the United States and European Union or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country- or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
References
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer: ASCO-College of American Pathologists guideline update. J. Clin. Oncol. 41, 3867-3872 (2023).
Allison, K. H. et al. Estrogen and progesterone receptor testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists guideline update. Arch. Pathol. Lab. Med. 144, 545-563 (2020).
National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. https://ctep.cancer.gov/ protocoldevelopment/electronic_applications/ctc.htm (2017).
Miettinen, O. & Nurminen, M. Comparative analysis of two rates. Stat. Med. 4, 213-226 (1985).
Maurer, W. & Bretz, F. Memory and other properties of multiple test procedures generated by entangled graphs. Stat. Med. 32, 1739-1753 (2013).
Acknowledgements
Funding for this research was provided by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. We thank the patients and their families and caregivers for participating in this study, along with all investigators and site personnel. Additionally, the authors thank the following employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, who supported
the study: G. Aktan (study oversight); K. Marie Hirshfield (study leadership); J. Zhao, X. Peng and J. Wei (statistical expertise); J. Chapman, J. Kimmel, S. Cruz, D. Falk, L. Muniz, E. Ramezani, A.T. Adorno, A. Garcia and K. Ponader (collection of data, supervision of research and document review); M.R. Papasani, C. Bai and L.W. Pang (analysis and reporting of data). Medical writing assistance was provided by A. Hassan and A. Lee of ICON plc. This assistance was funded by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Author contributions
All authors had access to the data used to prepare the manuscript and participated in the writing or critical review and editing of the manuscript. The first draft was written by the first and senior author with editorial assistance provided by a medical writer funded by the sponsor. A.B., F.C., D.W.C., J.C., Y.D., N.H., S.-A.I., L.J., V.K., H.M., P.S., M.L.T. and K.T. conceptualized, designed and planned the study. A.B., D.W.C., P.A.F., M.G.F., N.H., R.H., S.-A.I., Z.L., D.L., Y.H.P., G.R., P.S., Z.S., L.S., T.T., M.L.T., K.T. and H.Y. did the acquisition of the data. A.B., F.C., Y.D., L.J., V.K., P.S., M.L.T. and K.T. analyzed the data. A.B., F.C., D.W.C., Y.D., P.A.F., N.H., R.H., S.-A.I., L.J., V.K., D.L., H.M., J.O’.S., Y.H.P., P.S., M.L.T., K.T. and H.Y. interpreted the results. A.B., F.C., Y.D., L.J., Y.H.P., P.S., K.T. and H.Y. drafted the manuscript. A.B., F.C., D.W.C., J.C., Y.D., P.A.F., M.G.F., N.H., R.H., S.-A.I., L.J., V.K., Z.L., D.L., H.M., J.O’.S., Y.H.P., G.R., P.S., Z.S., L.S., T.T., M.L.T. and K.T. did the reviewing or revising the manuscript for important intellectual content. F.C., D.W.C., J.C., P.A.F., M.G.F., N.H., R.H., S.-A.I., Z.L., H.M. and T.T. did the provision of study materials/patients. G.R. provided administrative, logistical or technical support. A.B., F.C., D.W.C., J.C., Y.D., P.A.F., M.G.F., N.H., R.H., S.-A.I., L.J., V.K., Z.L., D.L., H.M., J.Ó.S., Y.H.P., G.R., P.S., Z.S., L.S., T.T., M.L.T., K.T. and H.Y. decided to submit the manuscript for publication. All authors approved the final draft of the manuscript.
Competing interests
F.C. receives consultancy honorarium from Amgen, Astellas/ Medivation, AstraZeneca, Celgene, Daiichi Sankyo, Eisai, GE Oncology, Genentech, Gilead, GlaxoSmithKline, Iqvia, Macrogenics, Medscape, Merck Sharp & Dohme, Merus BV, Mylan, Mundipharma, Novartis, Pfizer, Pierre Fabre, prIME Oncology, Roche, Sanofi, Samsung, Bioepis, Seagen, Teva, and Touchime. J.O’.S. receives honoraria for consulting and/or advisory boards from AbbVie, Agendia, Amgen, Aptitude Health, AstraZeneca, BioNTech, Byondis, Carrick Therapeutics, Daiichi Sankyo Company, DAVA Oncology, Eisai, Eli Lilly, Fishawack Health, G1 Therapeutics, Genzyme, GlaxoSmithKline, Genentech, Gilead, Loxo Oncology, Merck Sharp & Dohme, Novartis, Ontada, Pfizer, Pierre Fabre Pharmaceuticals, Puma Biotechnology, Roche, Samsung Bioepis, Sanofi, Seagen, Stemline Therapeutics, Taiho Oncology and Veru. H.M. receives consultancy fees from Amgen, AstraZeneca, Bristol Myers Squibb, Calithera, Celgene, Crown Bioscience, Daiichi Sankyo, Eli Lilly, Genentech/Roche, Gilead, Immunomedics, Merck Sharp & Dohme, OBI Pharma, Peregrine, Pfizer, Puma, Seattle Genetics, Spectrum Pharmaceuticals, Syndax Pharmaceuticals and Taplmmune and research support from Bristol Myers Squibb, BTG, MedImmune/AstraZeneca and Merck Sharp & DohmeD. P.S. receives consultant fees from or received honoraria from AstraZeneca, Bayer, Boehringer Ingelheim, Merck, Novartis, Pfizer, Puma, Roche, Eisai and Celgene; and receives grant funding (to institution) from Astellas, AstraZeneca, Genentech, Novartis, Oncogenex, Roche and Medivation. J.C. is a consultant/advisor for Roche, AstraZeneca, Seattle Genetics, Daiichi Sankyo, Lilly, Merck Sharp & Dohme, Leuko, Bioasis, Clovis Oncology, Boehringer Ingelheim, Ellipses, Hibercell, BioInvent, Gemoab, Gilead, Menarini, Zymeworks, Reveal Genomics, Scorpion Therapeutics, Expres2ion Biotechnologies, Jazz Pharmaceuticals, Abbvie, BridgeBio, Biontech and Biocon; receives honoraria from Roche, Novartis, Eisai, Pfizer,
Lilly, Merck Sharp & Dohme, Daiichi Sankyo, AstraZeneca, Gilead and Stemline Therapeutics; receives research funding (to institution) from Roche, Ariad Pharmaceuticals, AstraZeneca, Baxalta GMBH/Servier Affaires, Bayer Healthcare, Eisai, F. Hoffman-La Roche, Guardant Health, Merck Sharp & Dohme, Pfizer, Piqur Therapeutics, Iqvia and Queen Mary University of London; holds stock in MAJ3 Capital, Leuko (relative); has receives travel, accommodation and expenses from Roche, Novartis, Eisai, Pfizer, Daiichi Sankyo, AstraZeneca, Gilead, Merck Sharp & Dohme and Stemline Therapeutics; listed on patents for pharmaceutical combinations of a Pi3k inhibitor and a microtubule destabilizing agent (J.C. Castán, A.P. Giménez, V.S. Elizalde; WO 2014/199294 A; ISSUED) and for Her2 as a predictor of response to dual HER2 blockade in the absence of cytotoxic therapy (A. Prat, A. Llombart, J.C.; US 2019/O338368 A1; LICENSED). N.H. is a consultant for Gilead, Hoffmann-La Roche and Seagen; holds business ownership in the West German Study Group; and receives honoraria for lectures from AstraZeneca, Daiichi Sankyo, F. Gilead, Hoffmann-La Roche, Merck Sharp & Dohme, Lilly, Novartis, Pfizer Pharma GmbH, Pierre Fabre Pharmaceuticals, Seagen, Viatris and Zuelligpharma. M.L.T. is a consultant for AstraZeneca, Blueprint Medicines, Daiichi Sankyo, Genentech, Gilead (DSMC), GlaxoSmithKline, G1 Therapeutics (DSMC), Guardant, Menarini Stemline, Merck, Natera, Novartis, Pfizer, RefleXion, Replicate and Sanofi Aventis; receives grant/research support (institutional) from Arvinas, AstraZeneca, Bayer, Genentech/ Roche, GlaxoSmithKline, Hummingbird Biosciences, Merck, OncoSec Medical and Pfizer. D.W.C. is a consultant/advisor for AstraZeneca, Exact Sciences, Eisai, Gilead, GlaxoSmithKline, Inflex, Inivata/ Neogenomics, Lilly, Merck Sharp & Dohme, Novartis, Pfizer, Roche and SAGA; receives research support to institutions from AstraZeneca, GlaxoSmithKline, Inivata, Knight, Merck Sharp & Dohme, Pfizer and Roche; is a member of a trial steering committee for AstraZeneca, Merck Sharp & Dohme and GlaxoSmithKline; holds a patent (US62/675,228) for methods of treating cancers characterized by a high expression level of spindle and kinetochore-associated complex subunit 3 (ska3) gene. P.A.F. receives consulting fees from Agendia, AstraZeneca, Daiichi Sankyo, Eisai, Hexal, Lilly, Merck Sharp & Dohme, a subsidiary of Merck & Co., Novartis, Pfizer, Pierre Fabre, Roche and Seagen; receives fees for non-CME services received directly from commercial interests or their agents (Daiichi Sankyo, Lilly, Novartis and Seagen); and receives research grants from Biontech and Cepheid. D.L. is a consultant/advisor for AstraZeneca and Merck Sharp & Dohme and receives honoraria from Novartis, Pfizer, Roche, Lilly, Gilead, Merck Sharp & Dohme and AstraZeneca. Y.H.P. receives consultancy or advisory fees from AstraZeneca, Eisai, Eli Lilly Export S.A. Puerto Rico Branch, Novartis, Pfizer and Roche; and receives research funding from AstraZeneca, Merck Sharp & Dohme, Novartis,
Pfizer and Roche. G.R. receives honoraria from AstraZeneca, Gilead, Med-Gen Sol, Merck Sharp & Dohme, Roche, Novartis, Lilly, Swixx and Pfizer. L.S. is a consultant/advisor for Novartis, Daiichi Pharma, AstraZeneca, Eli Lilly, Precede and Seagen; and received institutional research support from Merck, Genentech, Gilead and Eli Lilly. S.-A.I. is a consultant/advisor for AstraZeneca, Novartis, Roche/Genentech, Pfizer, Amgen, Hanmi, Lilly, GlaxoSmithKline, Merck Sharp & Dohme, Daiichi Sankyo, Idience and Bertis; receives research funding to institutions from AstraZeneca, Pfizer, Roche/Genentech, Daewoong Pharmaceutical, Eisai Korea and Boryung Pharm. R.H. is a consultant/ advisor for Amgen, AstraZeneca, Bristol Myers Squibb, Eisai, Eli Lilly and Company, Janssen, Merck KGaA, Merck Sharp & Dohme, Novartis, OncoSec Medical Incorporated, Pfizer Australia, Roche Products Pty, Seagen, Takeda and Zai Lab; receives honorarium from AstraZeneca, Eli Lilly, Janssen, Merck Sharp & Dohme and Novartis; and receives research funding (to institutions) from AstraZeneca, BMS, Corvus, Eisai, Eli Lilly, Janssen, Merck Sharp & Dohme, Novartis, Oncosec, Roche and Seagen. T.T. receives honoraria from Daiichi Sankyo, Chugai and Eli Lilly. F.A. receives research grants (to institutions) from AstraZeneca, Daiichi Sankyo, Lilly, Novartis, Pfizer and Roche. Y.D., L.J., V.K. and K.T. are employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and stockholders in Merck & Co., Inc. A.B. is a consultant/advisor for Pfizer, Novartis, Genentech, Merck & Co., Radius Health, Immunomedics/Gilead, Sanofi, Daiichi Pharma/AstraZeneca, Phillips, Eli Lilly and Foundation Medicine and research/grant (to institution) from Genentech, Novartis, Pfizer, Merck & Co., Sanofi, Radius Health, Immunomedics/Gilead, Daiichi Pharma/ AstraZeneca and Eli Lilly.
Correspondence and requests for materials should be addressed to Fatima Cardoso.
Peer review information Nature Medicine thanks Karen Gelmon, Binghe Xu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ulrike Harjes, in collaboration with the Nature Medicine team.
Difference in Pathological Complete Response (95% CI)
no. of patients with response/no. of patients (%)
percentage points
Overall
154/635 (24.3)
100/643 (15.6)
→-
8.5 (4.2 to 12.8)
PD-L1 CPS/ER positivity
CPS , ER positivity <10
19/33 (57.6)
13/39 (33.3)
24.2 (1.0 to 45.1)
CPS , ER positivity
124/449 (27.6)
83/450 (18.4)
→ –
9.2 (3.7 to 14.6)
CPS <1, ER positivity
11/152 (7.2)
4/150 (2.7)
→
4.6 (-0.4 to 10.2)
-20-10
0
PlaceboChemotherapy
Better
10
304050
PembrolizumabChemotherapy
Better
Extended Data Fig. 1| Post hoc exploratory analysis of the difference in percentages of patients with a pathological complete response by estrogen receptor positivity and PD-L1 combined positive score at the first interim analysis. Programmed cell death ligand1 (PD-L1) combined positive score (CPS) was defined as the number of PD-L1-positive tumor cells, lymphocytes and macrophages divided by the total number of tumor cells multiplied by 100 . No pathological complete response occurred in patients with tumor PD-L1CPS<1
with estrogen receptor (ER) positivity (pembrolizumab arm, ; placebo ). For the overall population, the estimated treatment difference was calculated using the Miettinen-Nurminen method stratified by region (China vs Eastern Europe vs all other countries) and PD-L1 status (CPS vs ). Estimated treatment differences for other endpoints were based on the MiettinenNurminen method (unstratified).
Extended Data Fig. 2 | Multiplicity graph for type I error control of study hypotheses. The prespecified analysis plan allows alpha passing from successful endpoint(s) to other(s). H, hypothesis.
Extended Data Table 1| Summary of drug exposure during the neoadjuvant phase
PembrolizumabChemotherapy ( )
PlaceboChemotherapy ( )
All study drugs
n
634
641
Median months (range)
4.9 (0.0-6.9)
4.9 (0.0-7.8)
Pembrolizumab/placebo (Q3W)
n
634
641
Median months (range)
4.9 (0.0-6.9)
4.9 (0.0-7.8)
Median no. administrations (range)
8.0 (1.0-8.0)
8.0 (1.0-8.0)
Paclitaxel (QW)
n
634
641
Median months (range)
2.6 (0.0-4.7)
2.6 (0.0-4.7)
Median no. administrations (range)
12.0 (1.0-12.0)
12.0 (1.0-12.0)
Doxorubicin (Q3W)
n
164
165
Median months (range)
2.1 (0.0-3.1)
2.1 (0.0-2.8)
Median no. administrations (range)
4.0 (1.0-4.0)
4.0 (1.0-4.0)
Doxorubicin (Q2W)
n
136
133
Median months (range)
1.4 (0.5-2.8)
1.4 (0.0-1.9)
Median no. administrations (range)
4.0 (2.0-4.0)
4.0 (1.0-4.0)
Epirubicin (Q3W)
n
251
261
Median months (range)
2.1 (0.0-4.2)
2.1 (0.0-3.9)
Median no. administrations (range)
4.0 (1.0-4.0)
4.0 (1.0-4.0)
Epirubicin (Q2W)
n
47
54
Median months (range)
1.4 (1.0-2.9)
1.4 (0.5-2.3)
Median no. administrations (range)
4.0 (2.0-4.0)
4.0 (2.0-4.0)
Cyclophosphamide (Q3W)
n
415
425
Median months (range)
2.1 (0.0-4.2)
2.1 (0.0-3.9)
Median no. administrations (range)
4.0 (1.0-4.0)
4.0 (1.0-4.0)
Cyclophosphamide (Q2W)
n
183
187
Median months (range)
1.4 (0.5-2.9)
1.4 (0.0-2.3)
Median no. administrations (range)
4.0 (2.0-4.0)
4.0 (1.0-4.0)
The safety-evaluable population included patients who received at least one trial drug, underwent surgery, or both.
natureportfolio
Corresponding author(s):
Fatima Cardoso
Last updated by author(s): Oct 24, 2024
Reporting Summary
Nature Portfolio wishes to improve the reproducibility of the work that we publish. This form provides structure for consistency and transparency in reporting. For further information on Nature Portfolio policies, see our Editorial Policies and the Editorial Policy Checklist.
Statistics
For all statistical analyses, confirm that the following items are present in the figure legend, table legend, main text, or Methods section.
Confirmed
□
□ The exact sample size for each experimental group/condition, given as a discrete number and unit of measurement
□ A statement on whether measurements were taken from distinct samples or whether the same sample was measured repeatedly
□
The statistical test(s) used AND whether they are one- or two-sided
□ X
Only common tests should be described solely by name; describe more complex techniques in the Methods section.
□【 A description of any assumptions or corrections, such as tests of normality and adjustment for multiple comparisons
□
A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals)
□ X
For null hypothesis testing, the test statistic (e.g. ) with confidence intervals, effect sizes, degrees of freedom and value noted Give values as exact values whenever suitable.
□ For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings
□ For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes
□【 Estimates of effect sizes (e.g. Cohen’s , Pearson’s ), indicating how they were calculated
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Policy information about availability of computer code
Data collection Clinical data were captured in the study clinical database.
Data analysis
Statistical analyses were conducted using SAS version 9.4 (SAS Institute, Cary, NC).
For manuscripts utilizing custom algorithms or software that are central to the research but not yet described in published literature, software must be made available to editors and reviewers. We strongly encourage code deposition in a community repository (e.g. GitHub). See the Nature Portfolio guidelines for submitting code & software for further information.
Data
Policy information about availability of data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
Accession codes, unique identifiers, or web links for publicly available datasets
A description of any restrictions on data availability
For clinical datasets or third party data, please ensure that the statement adheres to our policy
Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD) is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and
requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the US and EU or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country- or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
Research involving human participants, their data, or biological material
Policy information about studies with human participants or human data. See also policy information about sex, gender (identity/presentation), and sexual orientation and race, ethnicity and racism.
Reporting on sex and gender
Reporting on race, ethnicity, or other socially relevant groupings
Population characteristics
Recruitment
Ethics oversight
The manuscript reports on patients with breast cancer.
Patients were stratified at randomization by region (Eastern Europe vs China vs all other countries). Relevant baseline characteristics are reported in Table 1.
This information is provided in Table 1.
Patients were enrolled from 222 global sites. Recruitment was done by investigators and was limited to patients who met protocol-specified criteria for eligibility. Details of these sites are provided in the Supplemental Material.
The protocol was approved by an ethics body at each institution. Patients provided written informed consent. An external independent data monitoring committee oversaw the study, periodically assessed safety, and assessed efficacy at prespecified interim analyses (see Supplementary Material for details).
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Field-specific reporting
Please select the one below that is the best fit for your research. If you are not sure, read the appropriate sections before making your selection.
Life sciences
Behavioural & social sciences
Ecological, evolutionary & environmental sciences
For a reference copy of the document with all sections, see nature.com/documents/nr-reporting-summary-flat.pdf
Life sciences study design
All studies must disclose on these points even when the disclosure is negative.
Sample size
With enrollment of approximately 1240 patients, the trial had power to detect a true difference of 15 percentage points for the comparison of the rate of pCR (stage ypT0/Tis ypNO) between the treatment arms, at a one-sided alpha level of 0.005 . It would have a power of to detect a hazard ratio for EFS of 0.73 , at a one-sided alpha level of 0.02 at the final analysis. The full statistical analysis plan is provided in the protocol.
Data exclusions
Efficacy was evaluated in the intention-to-treat population, which included all patients who had undergone randomization. Safety was evaluated in the as-treated population, which included all patients who had undergone randomization and received trial drug, underwent surgery, or both.
Replication
This was a clinical trial. Replication was not appropriate.
Randomization
Patients were randomly assigned (in a 1:1 ratio) to the pembrolizumab-chemotherapy arm or the placebo-chemotherapy arm using a central interactive voice-response system with an integrated Web-response system.
Blinding
This was a double-blind study.
Reporting for specific materials, systems and methods
We require information from authors about some types of materials, experimental systems and methods used in many studies. Here, indicate whether each material, system or method listed is relevant to your study. If you are not sure if a list item applies to your research, read the appropriate section before selecting a response.
Materials & experimental systems
Methods
n/a
Involved in the study
n/a
X
□ Antibodies
X
X
□
X
X
□
X
X
□
□
X
□
X
□
Clinical data
Policy information about clinical studies
All manuscripts should comply with the ICMJE guidelines for publication of clinical research and a completed CONSORT checklist must be included with all submissions.
Clinical trial registration
ClinicalTrials.gov, NCT03725059
Study protocol
The study protocol is available upon request (per data availability statement)
Data collection
From December 27, 2018 through August 5, 2022, a total of 1278 patients from 222 global sites were enrolled in the study.
Outcomes
The study’s primary endpoints were pCR , defined as ypNO at the time of surgery, and EFS in the intention-to-treat population. Secondary endpoints included pCR according to the definitions of ypTO ypNO and ypTO/Tis in all patients, pCR according to all definitions in patients with PD-L1 CPS tumors, EFS among patients with PD-L1 CPS tumors, and OS among all patients and patients with PD-L1 CPS tumors.
Plants
Seed stocks
Report on the source of all seed stocks or other plant material used. If applicable, state the seed stock centre and catalogue number. If plant specimens were collected from the field, describe the collection location, date and sampling procedures.
Novel plant genotypes
Describe the methods by which all novel plant genotypes were produced. This includes those generated by transgenic approaches, gene editing, chemical/radiation-based mutagenesis and hybridization. For transgenic lines, describe the transformation method, the number of independent lines analyzed and the generation upon which experiments were performed. For gene-edited lines, describe the editor used, the endogenous sequence targeted for editing, the targeting guide RNA sequence (if applicable) and how the editor
Authentication
was applied.
Describe any authentication procedures for each seed stock used or novel genotype generated. Describe any experiments used to assess the effect of a mutation and, where applicable, how potential secondary effects (e.g. second site T-DNA insertions, mosiacism, off-target gene editing) were examined.