بناء دعائم قابلة للتنظيم من خلال الهندسة غير الستيوكيومترية لاستقرار جزيئات النانو من الروثينيوم لتحسين انقسام الماء الشامل على مستوى الرقم الهيدروجيني Constructing regulable supports via non-stoichiometric engineering to stabilize ruthenium nanoparticles for enhanced pH-universal water splitting
بناء دعائم قابلة للتنظيم من خلال الهندسة غير الستيوكيومترية لاستقرار جزيئات النانو من الروثينيوم لتحسين انقسام الماء الشامل على مستوى الرقم الهيدروجيني
تاريخ الاستلام: 7 نوفمبر 2023 تم القبول: 6 مارس 2024 نُشر على الإنترنت: 29 مارس 2024 (د) التحقق من التحديثات
إنشاء تفاعلات مناسبة بين المعدن والداعم أمر ضروري للحصول على محفزات فعالة ومقاومة للتآكل لتفكيك الماء. هنا، آلية التفاعل بين جزيئات نانو الروثينيوم وسلسلة من أكاسيد التيتانيوم، بما في ذلك و تمت دراسة التصميم عبر الهندسة غير الستيوكيومترية السهلة بشكل منهجي.، مع الهيكل الفريد للفرقة، والتوصيل العالي، والاستقرار الكيميائي، يمنح تفاعل دعم المعادن المبتكر من خلال الواجهة الوحدات، التي تثبت أنواع الروثينيوم خلال عملية تطوير الأكسجين وتؤدي إلى تسرب الهيدروجين لتسريع حركية تطوير الهيدروجين. كما هو متوقع، يعرض جهدًا زائدًا منخفضًا للغاية قدره 8 مللي فولت و150 مللي فولت لتفاعل تقليل الهيدروجين وتفاعل أكسدة الأكسجين مع فترة تشغيل طويلة تبلغ 500 ساعة فيفي الوسائط الحمضية، التي تتوسع في بيئات الرقم الهيدروجيني الشامل. مستفيدًا من الأداء الثنائي الممتاز، تم تجميع خلايا التحليل الكهربائي لغشاء تبادل البروتون وغشاء تبادل الأنيون معيحقق أداءً متفوقًا وتشغيلًا قويًا. تمهد هذه الأعمال الطريق لأجهزة تحويل الطاقة الفعالة.
لقد حظي تحليل الماء باهتمام واسع باعتباره أكثر الطرق الواعدة للحصول على الهيدروجين الأخضر.تسمح تجميع الأقطاب الكهربائية الغشائية (MEA) بتحليل الماء كهربائيًا لإنتاج الهيدروجين الأخضر النظيف والفعال.تعتبر أغشية تبادل الأنيونات (AEM) وأغشية تبادل البروتونات (PEM) أجهزة واعدة لتحليل الماء، والتي تعتمد على المحفزات القائمة على البلاتين (Pt) لتفاعل تطور الهيدروجين (HER).علاوة على ذلك، تتطلب خلايا التحليل الكهربائي من نوع PEM أيضًا مواد قائمة على الإريديوم (Ir) المقاومة للتآكل بسبب الظروف الحمضية القاسية لتفاعل تطور الأكسجين (OER).لقد أدى الاستهلاك الواسع النطاق للمعادن الثمينة إلى ارتفاع التكاليف لتقنية التحليل الكهربائي للماء باستخدام أغشية البوليمر، مما أعاق تسويق هذه التقنية.. لذلك، من الضروري تطوير محفزات منخفضة التكلفة مع أقل أو بدونالمحفزات القائمة على الروثينيوم. الروثينيوم هو بديل فعال لـبسبب حوالي من تكلفةالأهم من ذلك، تظهر جزيئات نانو الروثينيوم مزايا ثنائية الوظيفة الفريدة في تفاعل تقليل الهيدروجين وتفاعل أكسدة الأكسجين في تحليل الماء لتقليل تكلفة المحفز.. ومع ذلك، فإن الألفة الأقوى للروثينيوم تجاه الوسط *H في تفاعل تقليل البروتونات تؤثر بشكل كبير على حركية التفاعل. في عملية OER، يتشكل Ru بسهولة في صورة قابلة للذوباننوع ) تحت إمكانيات الأكسدة، مما يظهر عمرًا قصيرًا للغاية تعزيز نشاط واستقرار جزيئات نانو الروثينيوم في كل من تفاعل الهيدروجين (HER) وتفاعل الأكسدة (OER) هو تحدٍ. من بين العديد من استراتيجيات تصميم المحفزات، حظيت المحفزات المحملة باهتمام واسع بسبب عملية التخليق البسيطة، وانخفاض استخدام المعادن الثمينة، والتعرض الواسع للمواقع النشطة، وكفاءة استخدام الذرات العالية.الأهم من ذلك، يمكن أن يؤدي الاتصال الكافي بين الدعم والمرحلة النشطة إلى بناء تفاعلات معدنية-دعم مناسبة ووحدات واجهة مستقرة لتنظيم التنسيق.
تكوين وبنية الواجهة الإلكترونية، التي تعزز الأداء التحفيزي واستقرار المراكز النشطةبشكل عام، يحتاج الدعم المثالي للكهروكاتاليست إلى الحفاظ على الاستقرار مع موصلية كهربائية عالية لتقليل استهلاك الطاقة المفرط وانحلال الكاتاليست تحت الفولتية التشغيلية.. من بين الدعائم المستقرة الشائعة، مثل ، ، إلخ، لقد جذبت الكثير من الانتباه بسبب مقاومتها الممتازة للتآكل وقابلية تعديل التكافؤ.. ومع ذلك، عادةً ما يظهر فجوة طاقة واسعة كمواد شبه موصلة من النوع n، مما يمنحه موصلية ضعيفة ويؤدي إلى تطبيقات محدودة في التحفيز الكهربائي.إدخال فراغات الأكسجين المناسبة فيمن خلال هندسة العيوب يمكن إنشاء مستويات طاقة العيوب لتحسين الموصلية. ومع ذلك، فإن توزيع فراغات الأكسجين العشوائية يجعل من الصعب ضبط حالات العيوب بدقة ويسبب اختلالات هيكلية محتملة في أشباه الموصلات. لذلك، فإن بناء دعائم أكسيد غير متكافئة مستقرة مع عيوب موزعة بشكل دوري يمكن أن يقدم عيوب شبكية بهياكل مستقرة. أكاسيد التيتانيوم غير المتكافئة ( )، مثل ، و تمتلك موصلية كهربائية أعلى لعدة أوامر من حيث الحجم من. على وجه الخصوص،، كأكسيد التيتانيوم غير الستويكيometric النموذجي، له موصلية مشابهة لتلك الخاصة بالجرافين، والتي تُعزى إلى الكثافة الوفيرة لتوزيع حالات الطاقة عند مستوى فيرمي.. بالإضافة إلى ذلك،يعرض هيكلًا أكثر استقرارًا مقارنة بأكاسيد التيتانيوم ذات القيمة المنخفضة مثل TiO. والأهم من ذلك، أن التصميم غير المتكافئ الدقيق للدعم يمكن أن يحصل على الهيكل البلوري والإلكتروني المناسب لتحقيق التكيف مع الأنواع النشطة، مما يفيد في تحسين النشاط الجوهري واستقرار المواقع النشطة.
هنا، آلية التفاعل بين جزيئات النانو من الروثينيوم وسلسلة من أكاسيد التيتانيوم، بما في ذلك و تم استكشافه بشكل منهجي. التفاعل المناسب بين المعدن والدعم بين الروثينيوم ويتم تحقيقه، مما يحفز بشكل معتدل إثراء الإلكترونات لمواقع الروثينيوم (Ru) لمنع آلية الأكسدة الكهربائية للأكسجين (LOM) ويسهل إزالة البروتون من خلال استقراروحدات، التي توازن بين النشاط والاستقرار لـ OER. علاوة على ذلك، فإن المقاومة المنخفضة عند الواجهة بينويبدأ الروثينيوم آلية تسرب الهيدروجين لتفاعل تقليل الهيدروجين (HER) لتسريع حركية التفاعل. يتم تأكيد النشاط الثنائي الاستثنائي في بيئات ذات درجة حموضة شاملة. بشكل خاص، في الظروف الحمضية،يظهر جهدًا زائدًا فائق الانخفاض قدره 8 مللي فولت و150 مللي فولت عندلها و OER، على التوالي، وتحافظ على تشغيل مستدام لمدة 500 ساعة. تم تجميع أجهزة PEM معتظهر فولتية خلايا أقل واستقرارًا أطول من تلك الموجودة في المنتجات التجارية. يوفر هذا العمل رؤى جديدة حول تطوير المحفزات المدعومة بشكل عقلاني ويمهد الطريق لتصميم أجهزة تحويل الطاقة.
النتائج
مبادئ تصميم الحامل
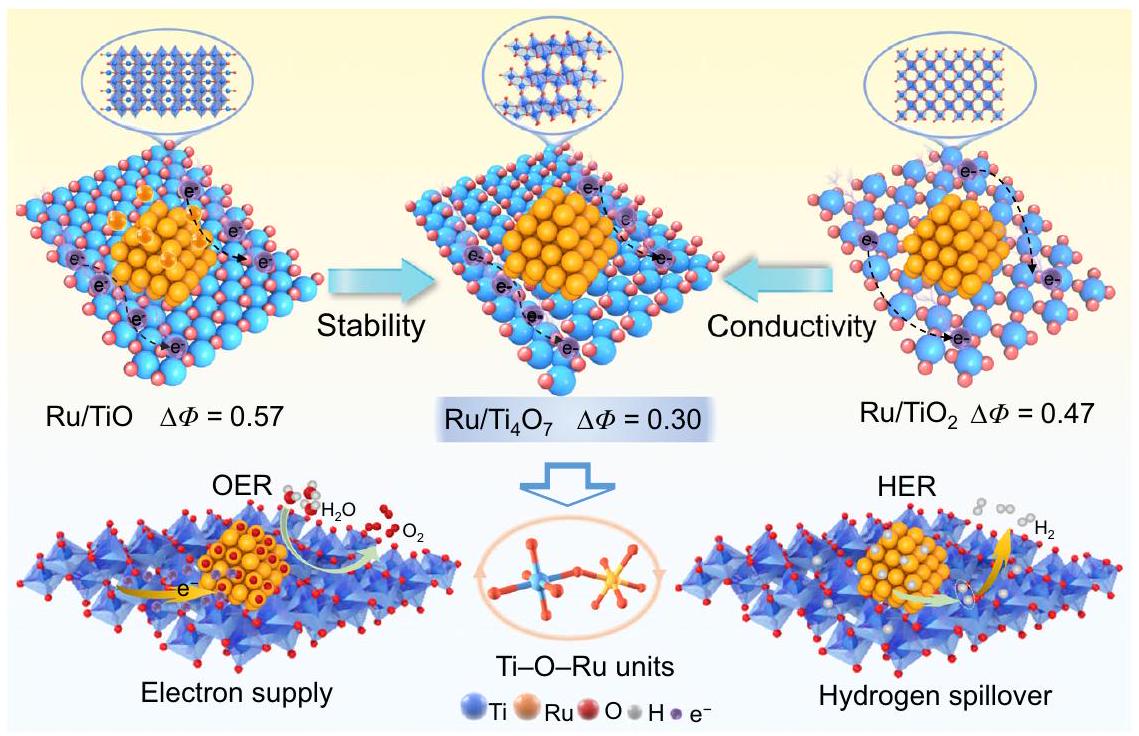
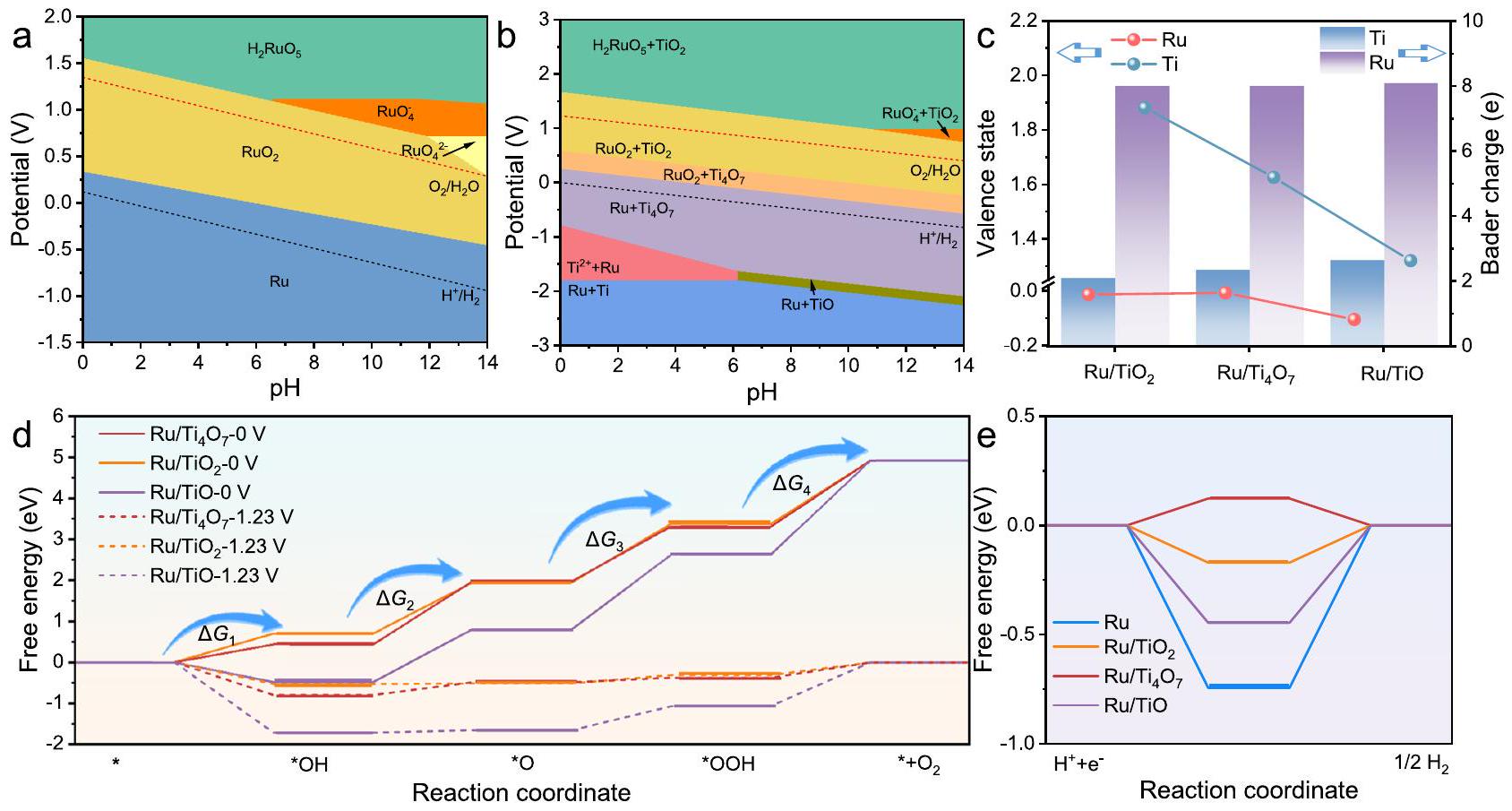
كما هو موضح في الشكل 1، تم استخدام الهندسة غير الستيوكيومترية لتخصيص دعائم أكسيد التيتانيوم بدقة.، و ) مع فراغات الأكسجين ذات الترتيبات الدورية. ، مع عيوب مرتبة بانتظام، يظهر كمنافس محتمل يتمتع باستقرار هيكلي متفوق على TiO متعدد العيوب وموصلية أعلى بكثير منالأهم من ذلك،يعرض تفاعل دعم المعادن المناسب، الذي يؤسس لنشاط الوظيفة المزدوجة لـفي كل من HER وOER. من ناحية، فإن وظيفة العمل الأعلى مقارنة بجزيئات Ru النانوية تعزز غنى الإلكترونات في Ru، مما يمكن أن يخفف من ذوبان Ru في OER. من ناحية أخرى، مقارنة بـ TiO والحد الأدنى من فرق دالة العمل بين و جزيئات Ru ( يمكن أن يقلل من حاجز شوتكي للواجهة (الشكل التكميلي 1)، مما يعزز نقل الإلكترونات في المحفز المركب ويحفز تسرب الهيدروجين أثناء تفاعل تطور الهيدروجين.
علم الشكل والبنية البلورية
تم ترسيب جزيئات نانوية من الروثينيوم (NPs) على دعائم أكسيد التيتانيوم بنسب ستوكيومترية مختلفة.و TiO) عبر طريقة كيميائية رطبة (الشكل 2أ).لديه مزايا محتملة في التحفيز الكهربائي لدعم جزيئات نانو الروثينيوم بسبب الاستقرار الكيميائي المتفوق على TiO والموصلية الكهربائية المتفوقة بشكل كبير علىتظهر صور المجهر الإلكتروني الماسح (SEM) والمجهر الإلكتروني الناقل (TEM) الطلاء المتجانس لدعائم أكسيد التيتانيوم بواسطة جزيئات نانو الروثينيوم (Ru NPs) بحجم (الأشكال 2ب و 1ج). الفراغات المناسبة للأكسجين في تعزيزالامتزاز على، مما يمنح جزيئات النانو من الروديوم توزيعًا أكثر انتظامًا علىمقارنة بـمع شغور منخفض (الأشكال التكميلية 2-6)في التصوير المجهري الإلكتروني عالي الدقة المصحح للانحرافات (AC-HRTEM)يمكن ملاحظة الربط المحكم والتطابق المناسب على النانو بين المراحل المختلفة، مصحوبًا بانتقال سلس عند الواجهة (الشكل 2d). تم الحصول على صور مجهر الإلكترون الناقل المظلم ذو الزاوية العالية (HAADF-STEM) المحاكية من خلال التركيب البلوري لـ وتظهر الروبيديوم اتفاقًا ملحوظًا مع النتائج التجريبية (الأشكال 2e و 1f). استنادًا إلى هذه الصور الذرية، يمكن أن يُعزى تباعد الشبكة البالغ 0.377 نانومتر إلى المستوى (102) من (JCPDS 50-0787)، بينما يتوافق تباعد الشبكة البالغ 0.234 نانومتر مع المستوى (100) من الروثينيوم (JCPDS 89-4903، الأشكال 2g، 1h، والشكل التكميلي 7). بالإضافة إلى ذلك، ، وعناصر O موزعة بشكل موحد في رسم الطيف الكمي للطاقة (EDS) لـ (الشكل 2i)، والقمم الانكسارية المميزة المعينة للروثينيوم وتتواجد في نمط حيود الأشعة السينية (XRD) (الشكل 2j). تشير النتائج أعلاه إلى أنيمكن أن تحقق تلامس شبكي واجهي مناسب مع جزيئات نانو الروثينيوم، مما يوفر إمكانية للتفاعل التكيفي بين المعدن والدعم وهجرة سريعة للإلكترونات عبر الواجهة..
تحليل الهيكل الإلكتروني والتنسيقي
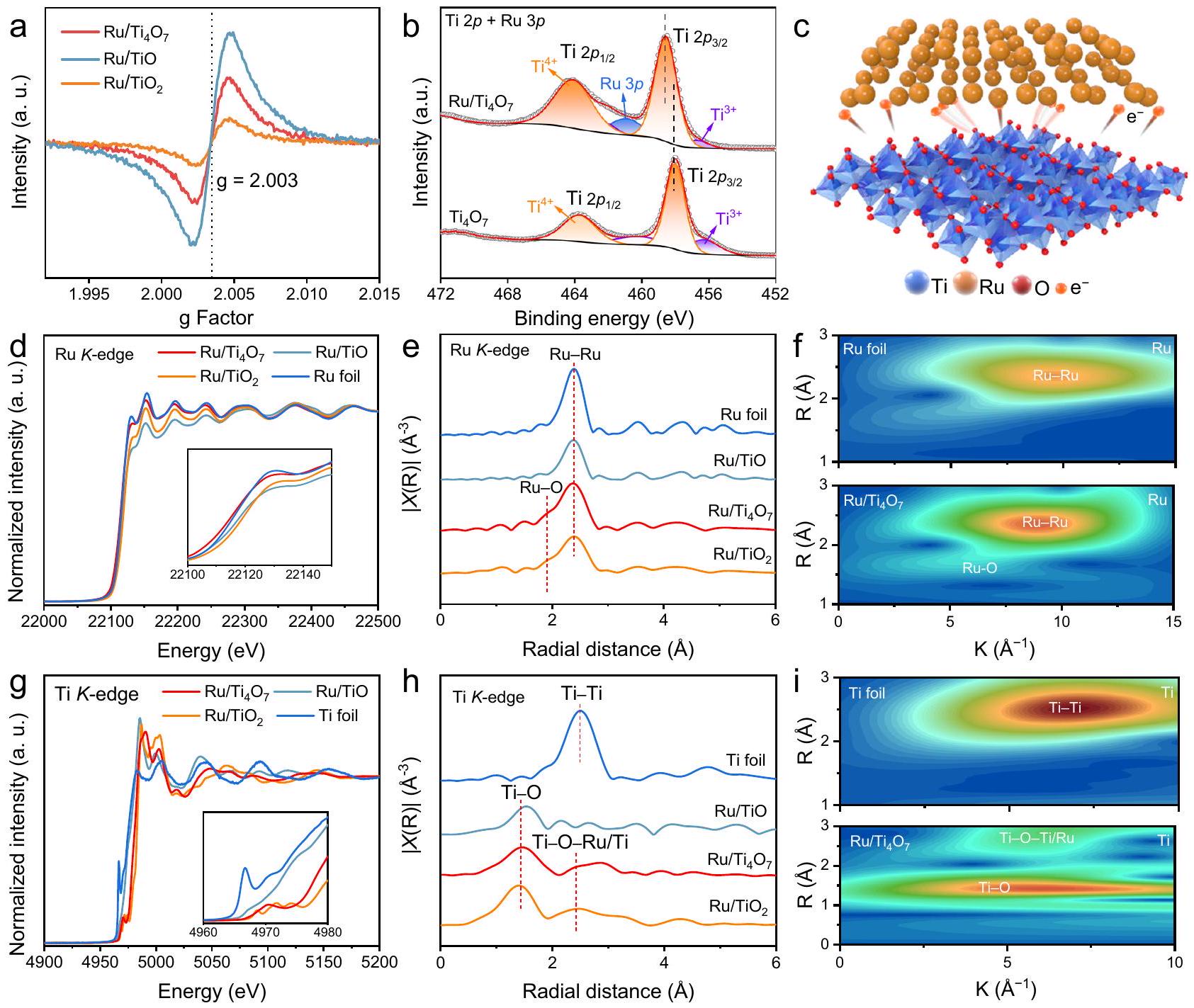
تركيز فراغات الأكسجين في، و يظهر اتجاهًا متزايدًا بسبب النسب الستيوكيومترية المختلفة من Ti و O (الشكل 3a). إن الفراغات الأكسجينية المرتبة بشكل دوري لا تحسن فقط من موصلية الدعم ولكنها توفر أيضًا إمكانية تشكيل الواجهات الجسرية بينوجزيئات النانو من الروديوم. في طيف الأشعة السينية للالكترونيات (XPS) الدقيق لـ، و ، الـ و تظهر المناطق تداخلًا وثيقًا (الشكل 3ب). طاقة الربط لـ Tiفي مواد أكسيد التيتانيوم المحملة بشكل مختلف تظهر درجات مختلفة من التحول نحو طاقة الربط العالية (الشكل 3ب، والأشكال التكميلية 8-10)، مما يشير إلى أن أكسيد التيتانيوم يمكن أن يعمل كمانح للإلكترونات لتعزيز غنى الإلكترونات في Ru (الشكل 3ج). كما هو موضح في الشكل التكميلية 8ج، تظهر جزيئات Ru فقط القمم المميزة للروتينيوم المعدني. في منطقة Ru 3d منالإشارات المنسوبة إلىويمكن ملاحظة Ru-Ru، مما يتوافق مع الواجهةوحداتيمكن ملاحظة تغييرات إلكترونية محلية مماثلة في البنية الدقيقة لامتصاص الأشعة السينية (XAFS). في بنية امتصاص الأشعة السينية بالقرب من الحافة (XANES)، يتم رودي-edge يظهر أيضًا كثافة ذروة خط أبيض مختلفة بعد التلامس مع دعائم أكسيد التيتانيوم المختلفة (الشكل 3d). من المهم أن يكون الروديوم فيلديه أقرب حالة تكافؤ إلى رقائق الروثينيوم مقارنةً بتلك في و ، والذي يُعزى إلى انخفاض فرق وظيفة العملوتم الحصول على Ru من قياسات طيف الانبعاث الضوئي فوق البنفسجي (UPS) (الشكل التكميلي 1). في الوقت نفسه، تم تحضير Ti--حافة Ru/يظهر تحولًا إلى طاقة أعلى مقارنةً بتلك الخاصة بـ، مما يشير إلى أن الحالة المتزايدة من الفالنس لتيتانيوم في (الشكل التوضيحي 11). تشير النتائج أعلاه إلى أن الروديوم يعمل كقابل للإلكترونات لتشكيل اتصال شوتكي مع دعم أكسيد التيتانيوم، ويمتلك أفضل قدرة على التكيف مع الهيكل الإلكتروني لجزيئات Ru لتقليل حاجز شوتكي عند الواجهة. طيف الامتصاص بالأشعة السينية الممتد (EXAFS) لجزيئات Ru المقابلة-تظهر طيف تحويل فورييه EXAFS الموزون إشارة ضعيفة لـ Ru-O تقع عندبعد التواصل مع أو
الشكل 1 | تفعيل ثنائي الوظيفة استنادًا إلى دعائم مختلفة. مخطط توضيحي لآلية التفاعل بين دعائم أكسيد التيتانيوم المختلفة وجزيئات Ru النانوية لتفعيل تفاعل الأكسدة (OER) وتفاعل الاختزال (HER).
على التوالي.تباعد مستوى البلورة ورسم تخطيطي لواجهة الاتصال بين و تخطيط EDS، و O في على التوالي.نمط حيود الأشعة السينية.
الشكل 2 | التخليق والتوصيف الهيكلي. أ صورة مجهر إلكتروني مسحّي لـصور TEM و AC-HRTEM لـ. د، هـ الهيكل الذري التخطيطي والصور المحاكاة المقابلة بتقنية HAADF-STEM لـورو في،
الشكل 3 | توصيف هيكل الإلكترون والتنسيق. أ طيف EPR لـ، و طيف XPS الدقيق في و منطقة، و رسم تخطيطي يوضح التفاعل الإلكتروني بينو Ru. د مُعَيار-edge XANES من رقائق التيتانيوم،، و
TiO. و المعادل-تحويلات فورييه الموزونة و(تحويل المويجات-إشارات EXAFS الموزونة. ج مُعَيارَة-edge XANES من رقائق الروثينيوم،، ، و المقابل-تحويلات فورييه الموزونة وتحويل الموجات لـ-إشارات EXAFS الموزونة. (الشكل 3e). يجب أن ينشأ هذا الظاهرة من الواجهةوحدات بين الروثينيوم وأكسيد التيتانيوم، والتي تُلاحظ في تحويل الموجات بشكل أوضح (الشكل 3f). المستقرتعمل الوحدات على استقرار الأنواع النشطة بفعالية وتعمل كقناة إلكترونية لتقليل مقاومة نقل الإلكترونات عند الواجهة.كما هو موضح في الشكل 3 ج، فإن انتقال التيتانيوم-edge يعكس حالات التكافؤ المتغيرة للأكسيد للتيتانيوم في دعائم أكسيد التيتانيوم المختلفة من حيث النسبة المولية. علاوة على ذلك، كما هو موضح في -تحويلات فورييه EXAFS الموزونة لـ-حافة و (الشكل 3 ح) والتحويل الموجي المقابل (الشكل 3ي)، الهيكل الذي ينتمي إلىيؤكد بشكل أكبر تشكيل الواجهةوحدات.
تقييم النشاط الكهروتحفيزي والاستقرار
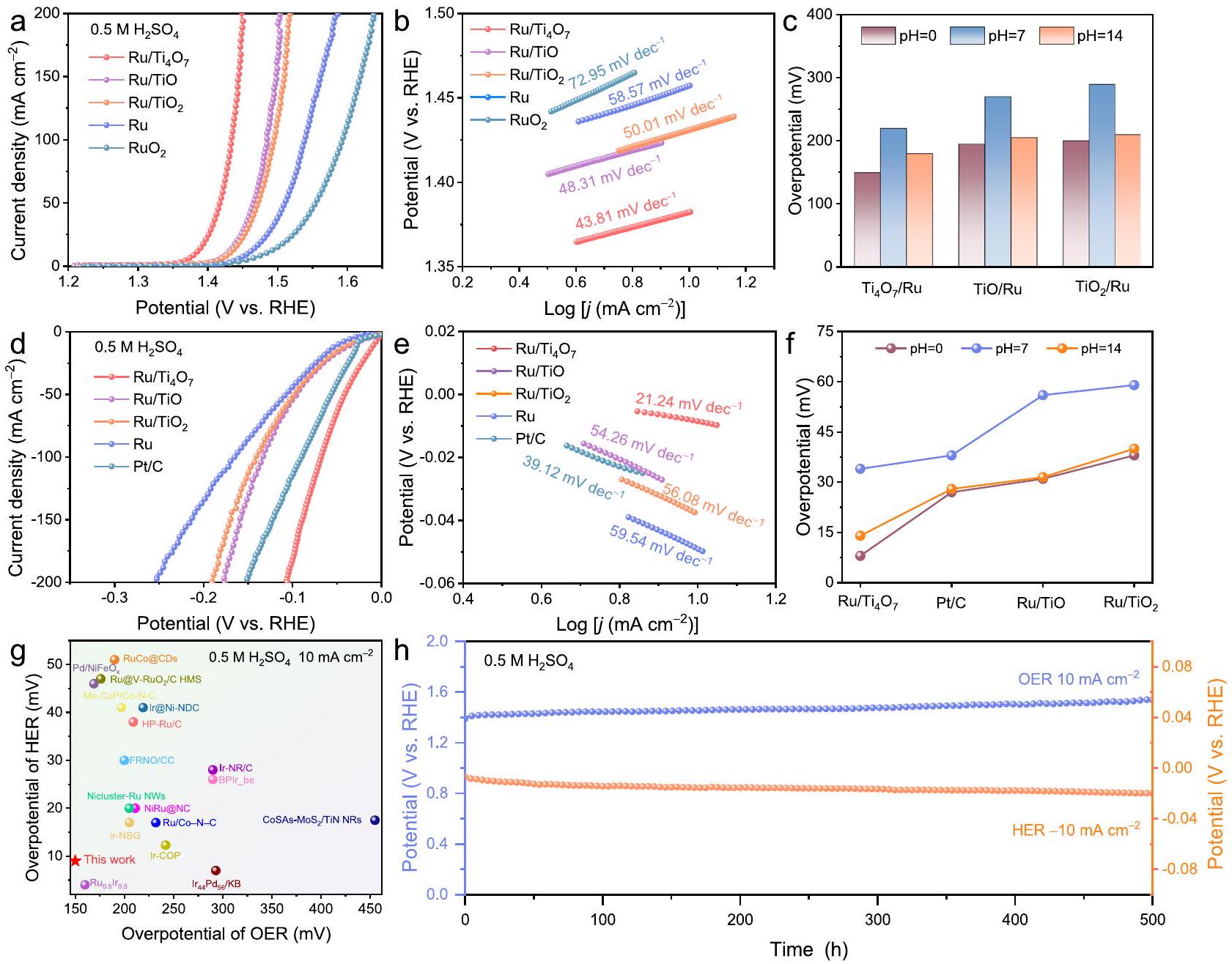
تم تقييم الأنشطة الكهروكيميائية لأكاسيد التيتانيوم المحملة بالروثينيوم في نظام ثلاثي الأقطاب قياسي للتحقق من المزايا الفريدة لـالدعم. في الوسط الحمضي،مع تحميل رو المحسن ودرجة حرارة التلدين يظهر أقل جهد زائد لتفاعل الأكسدة (150 مللي فولت عند، الشكل 4 أ والشكل التكميلي 12) وانحدار تافل (الشكل 4ب، ج). بالنسبة لتفاعل تقليل الهيدروجين،كما يظهر جهدًا منخفضًا للغاية قدره 8 مللي فولت عندفي وسط حمضي البيئات، التي تتفوق على التجاريةوأكسيد التيتانيوم المدعوم من جزيئات نانو الروثينيوم (الشكل 4d). ومن المRemarkably، كما هو موضح في الشكل 4e، فإن ميل تافل لـمع فرق في وظيفة العمل الأدنى فقط، وهو أقل بكثير من قيمة آلية فولمر-هيدروفسكي/تافل التقليدية (30 مللي فولتالفرق المنخفض في وظيفة العمل بين المعدن والدعم عرضة لتحفيز آلية تسرب الهيدروجين خلال تفاعل تقليل الهيدروجين وتوفير حاجز شوتكي بيني منخفض وهجرة سريعة للإلكترونات على الواجهة.تؤكد طيفية الامتصاص الكهربائي الكيميائي (EIS) أيضًا أدنى مقاومة لنقل الإلكترونات لـ (الشكل التكميلي 13). من الجدير بالذكر أن النشاط الثنائي الوظيفي المتفوق لـكما أظهرت قابلية التوسع في البيئات المحايدة والأساسية (الشكل 4c و f، والأشكال التكميلية 14-17). بالإضافة إلى ذلك،يعرض أعلى سعة مزدوجة الطبقات ( ) القيمة ( 68.95 م ف ) بنفس تحميل الروثينيوم، مما يشير إلى أن المزيد من مواقع النشاط النشطة للروثينيوم المكشوفة مقارنة بـ و (الأشكال التكميلية 18 و19). منحنيات استقطاب LSV المعيارية بواسطة وكتلة الروثينيوم تشير إلى أن مواقع الروثينيوم النشطة في تمتلك أعلى أنشطة نوعية (الشكل التكميلي 20) ونشاط كتلي (الشكل التكميلي 21). الاستثنائي
الشكل 4 | تقييم النشاط الكهروكيميائي. أ منحنيات الاستقطاب لقياس الجهد الخطي (LSV) لتفاعل الأكسدة.رسوم بيانية لجدول Ru ، تجاري، و (تحميل المبلغ: ) في . ج الجهد الزائد المقابل لتفاعل تطور الأكسجين عند في بيئات pH مختلفة. منحنيات استقطاب LSV لـ HER و (e) مخططات Tafel لـ Ru، Pt/C التجاري، Ru/
، و (تحميل المبلغ: ) في ). ف الجهد الزائد المقابل لتفاعل تقليل الهيدروجين عند في بيئات pH مختلفة. ج مقارنة الجهد الزائد مع جهد المحفزات الثنائية الوظيفة الأخرى لتفكيك الماء فيتم الإبلاغ عنه مؤخرًا.منحنيات الكرونوپوتنشيومتريةفي، و في. الأداء الثنائي الوظائف لـيتجاوز ذلك معظم المحفزات الثنائية الوظائف الممتازة في التقارير الحديثة (الجداول التكميلية 2، 3)، خاصة في البيئات الحمضية (الشكل 4g).
استقرارتم التحقق منه عبر الكرونو بوتنشيومترية. في البيئات الحمضية الشديدة،يظهر استقرارًا مفاجئًا لتفاعل الأكسدة (OER) وتفاعل الاختزال (HER) لمدة 500 ساعة مع تدهور نشاط ضئيل (الشكل 4 ح) وانهيار هيكلي طفيف (الشكل التوضيحي 22)، مما يتجاوز عمر التشغيل لـ (الشكل التوضيحي 23). يتطلب أيضًا إمكانيات أقل لتحفيز OER مقارنةً بـمساهمًا في تقليل استهلاك الطاقة لأجهزة التحليل الكهربائي للمياه على مدى فترة طويلة (الشكل التكميلي 23). والأهم من ذلك،يمكن أن تحافظ على تشغيل مستقر لمدة 300 ساعة عند كثافة تيار عالية منمع تدهور طفيف في النشاط بعد 3000 دورة من الفولتمترية الدورانية (CV) (الأشكال التكميلية 24 و 25). الاستجابة الحالية المنخفضة في الفولتمترية الدورانية المرتبطة بأكسدة-اختزال Ru في مقارنةً بـ Ru و يشير إلى الأكسدة المثبطة للروثينيوم بواسطة (الشكل التوضيحي الإضافي 26). الشكل التوضيحي الإضافي 27 يظهر أن فقدان الروثينيوم والتيتانيومتتباطأ تدريجياً خلال عملية التحليل الكهربائي الطويلة الأمد. يذوب التيتانيوم تدريجياً في أول 50 ساعة من الدورة، وهو ما يُعزى إلى التفاعل بين السطح غير المستقر للقطب الكهربائي. بعد 50 ساعة، يتباطأ معدل ذوبان التيتانيوم بسبب استقرار سطح القطب الكهربائي. علاوة على ذلك، قمم الانكسار المميزة التي تنتمي إلى الروثينيوم ولا يزال يمكن ملاحظته في نمط XRD لـبعد OER دون وجود قمم مميزة جديدة مقارنة بالذوبان الظاهر لـ (الشكل التوضيحي 28). كما هو موضح في طيف الرنين المغناطيسي الإلكتروني (EPR) (الشكل التوضيحي 29) وطيف XPS للأكسجينقبل وبعد اختبارات الاستقرار (الشكل التوضيحي 30أ)، كان هناك انخفاض طفيف في فراغات الأكسجين فييتم ملاحظة الدعم، مما يدل على الاستقرار الهيكلي لـالدعم تحت كثافات تيار عالية. بالإضافة إلى ذلك، فإن إشارات XPS غير المتغيرة تقريبًا لـ Ti و Ru مع انزياح طفيف نحو طاقة ربط أعلى بعد OER توفر مزيدًا من الأدلة على الصيانة الهيكلية الجيدة لـخلال التفاعل (الشكل التكميلي 30ب). الاستقرار الممتاز لـ يُعزى ذلك إلى عملية الأكسدة المثبطة، والتي يجب أن تُشتق من إثراء الإلكترونات للروثينيوم من خلال الاستقرار.وحدات.
أصل النشاط الثنائي المحسن
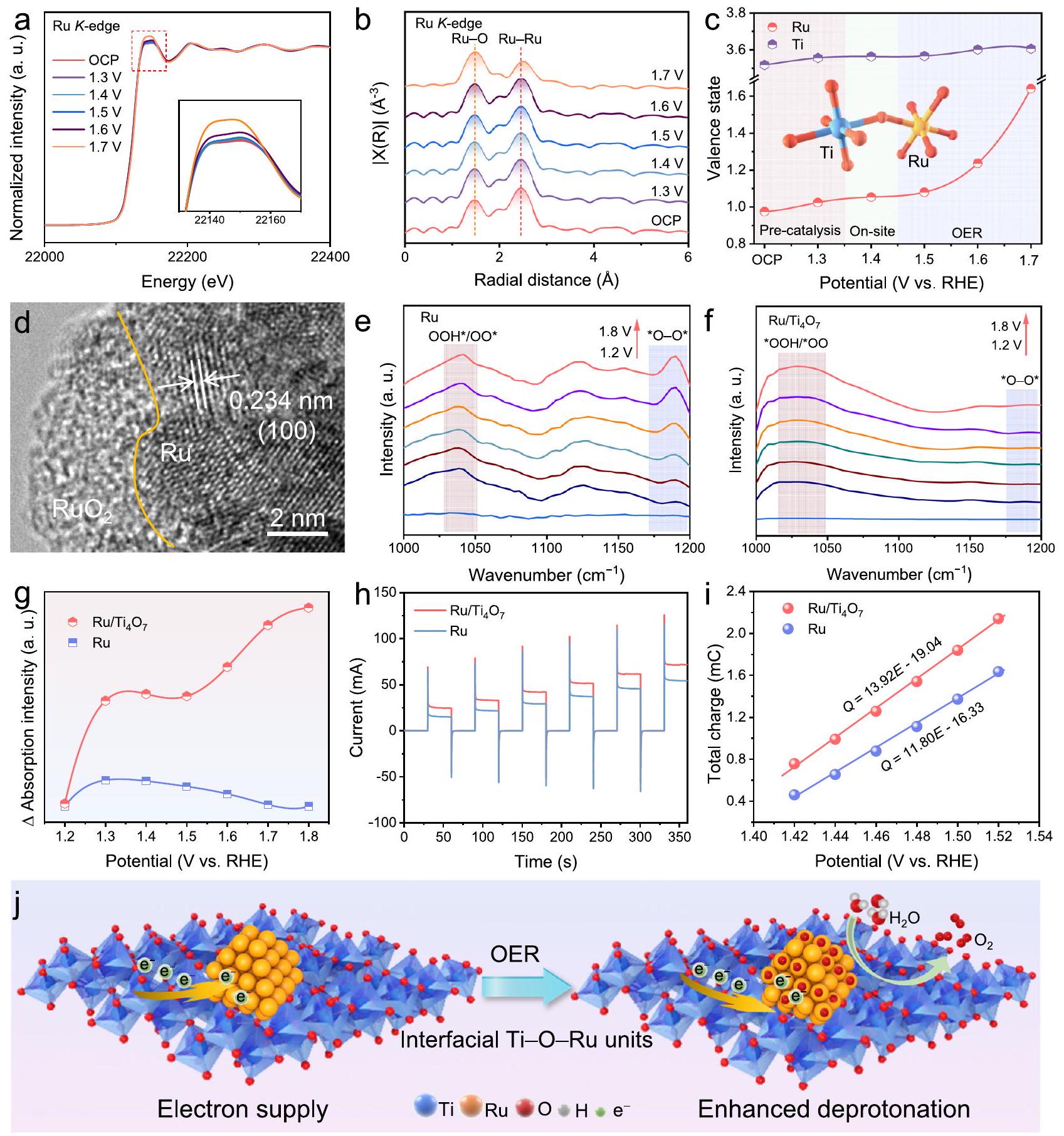
تم إجراء سلسلة من الاختبارات في الموقع في بيئات حمضية لتتبع أصل النشاط والاستقرار لـاستكشفت تقنية XAFS في الموقع تطور الهيكلخلال OER.خضعت لـ 20 دورة CV للحصول على السطح المستقر قبل تسجيل طيف XAFS في الموقع. في أكثر البيئات الحمضية شدة،
شكل. | توصيف في الموقع وآلية التفاعل. أ Ru في الموقع المعياري-edge XANES من. المقابل-تحويلات فورييه الموزونة. ج حالات التكافؤ للروثينيوم والتيتانيوم فيتم الحصول عليه عبر-الحافة تحت إمكانيات مختلفة. d HRTEM منبعد OER. طيف ATR-SEIRAS في الموقع لـ (e) Ru ولـ OER فيتحت إمكانيات مختلفة مقابل RHE. فرق الشدة للإشارات تحت الحمراء عند 1038 والاستجابات الحالية للقياس الطيفي بالنبض لعنصر الروثينيومالعلاقة بين تخزين الشحنة والجهد لـ Ru ورسم تخطيطي لعملية OER المحسّنة الناتجة عندعم. شدة الخط الأبيض للرو-edge يزيد مع الجهود المطبقة لتفاعل الأكسدة (OER) (الشكل 5a)، مما يشير إلى أكسدة جزيئات نانو الروثينيوم (Ru NPs). علاوة على ذلك، في تحويل فورييه-الفضاء (الشكل 5ب)، الزيادة التدريجية في شدة الإشارة لمسار Ru-O تؤكد تشكيلمن الجدير بالذكر أن القمم المنسوبة إلى معدن الروثينيوم (Ru-Ru) يمكن ملاحظتها دائمًا حتى تحت الجهود العالية لتفاعل تطور الأكسجين (OER)، وهو ما يتوافق مع المرحلة المعدنية للروثينيوم الموضحة في نمط حيود الأشعة السينية (XRD).بعد OER. تشير النتائج أعلاه إلى أنالأنواع التي تشكلت محليًا على يمكن أن تعمل كطبقة حاجزة لتقليل الأكسدة الإضافية وذوبان جزيئات نانو الروثينيوم. تظهر طيفيات XAFS في الموقع أن التيتانيومحافةيتداخل تقريبًا تمامًا تحت إمكانيات OER دون خصائص أكسدة بارزة، مما يستفيد من الهيكل المستقر لـ (الشكل التكميلي 31a). -فضاء Ti يؤكد أنرابطة فيلا يتغير بشكل كبير خلال OER (الشكل التكميلي 31b). استقراريمكن التحقق منها بشكل أكبر في حالات التكافؤ الصريحة حالة التكافؤ المصححة بواسطة شدة الخط الأبيض المعيارية المرجعية المقابلة لـ Ti فييتغير قليلاً بين 3.52 و 3.61 مع زيادة إمكانيات التطبيق (الشكل التكميلي 32). في الوقت نفسه، يزيد متوسط حالة التكافؤ للروثينيوم من 0.98 إلى 1.64. لا يظهر التغيير الكبير حتى يصل الجهد إلى 1.7 فولت مقابل RHE (الشكل 5c). تؤكد هذه الظاهرة مرة أخرى أن جزيئات الروثينيوم النانوية لا تزال تحافظ على الاستقرار الهيكلي تحت الجهود العالية لعملية الأكسدة الكهربائية. إن الأكسدة غير المكتملة لجزيئات الروثينيوم النانوية مفيدة للحفاظ على الموصلية الممتازة لمادة المركب خلال عملية التحفيز الكهربائي. في صورة HRTEM لـبعد OER، الاتصال السطحي الوثيق بين جزيئات Ru NPs ويمكن أن تظل محفوظة (الشكل التوضيحي 33). بالإضافة إلى ذلك، يمكن ملاحظة بنية غير متبلورة محليًا في الطبقة الخارجية لجزيئات نانو الروثينيوم.بعد OER، الذي يجب أن يتوافق معالأنواع (الشكل 5د).
علاوة على ذلك، تم الكشف عن آلية تفاعل OER من خلال طيف الامتصاص تحت السطح المعزز بالأشعة تحت الحمراء بتقنية الانعكاس الكلي المخفف في الموقع (ATR-SEIRAS). توضح الشكل 5e أنه مع زيادة الجهد إلى 1.4 فولت مقابل RHE، تظهر أطياف ATR-SEIRAS في الموقع لـ Ru نطاقات امتصاص حول تردد الاهتزاز لـ، مما يتوافق مع * المتوسطات. في الوقت نفسه، إشارة امتصاص مميزة عند تردد الاهتزاز لـتقوى تدريجياً مع تطبيق إمكانيات OER، مما يتوافق مع *في LOM. طيف ATR-SEIRAS في الموقع لـتظهر في مواقع مشابهة لـ Ru، مما يعزى إلى نطاقات الامتصاص لـ *OOH/*OO (الشكل 5f). قد تنشأ توسيع وتحويل قمم الامتصاص من انتقال عملية إزالة البروتون. علاوة على ذلك، لا يوجد تقريبًا أي إشارة تتوافق مع *يلاحظ حول تردد الاهتزاز لـ. بالإضافة إلى ذلك، فإن الفرق في الكثافة المعايرة لطرز ATR-SEIRAS في الموقع المرتبطة بـ *OOH/*OO وأوه-أوهتُعرض الإشارات في الشكل 5g لتحديد النسبة التي تشغلها آلية تطور الامتزاز وLOM في عملية OER الحمضية.إن فرق كثافة الامتزاز الأعلى يعني نسبة أعلى من آلية تطور الامتزاز في عملية التفاعل. في نطاق الجهد العالي، يخضع الروديوم لإعادة بناء السطح لتشكيلالأنواع، مما يمكّن كل من آلية تطور الامتصاص وLOM لدفع OER. في الوقت نفسه، طيف ATR-SEIRAS في الموقع لـتظهر باستمرار فرق كثافة امتصاص أعلى من الروديوم خلال عملية الأكسدة الكهربائية للماء. تشير هذه الظاهرة إلى أنيميل إلى دفع تفاعل الأكسدة الكهربية للأكسجين من خلال آلية تطور الامتصاص، مما يمكن أن يحقق تفاعل OER أكثر استقرارًا مقارنةً بـ LOM مع إمكانية ذوبان المحفز. في اختبارات الاعتماد على الرقم الهيدروجيني، مقارنةً بجزيئات نانو الروثينيوم،أظهرت ارتباطًا أقل مع الحموضة، مما أكد أنها كانت أكثر ميلًا لدفع تفاعل الأكسدة الكهربائي مع آلية تطور الامتزاز (الشكل التكميلي 34). هذه النتائج توضح أنيميل إلى دفع تفاعل الأكسدة الكهربية للأكسجين من خلال آلية تطور الامتزاز السطحي، مما يمكن أن يحقق تفاعل أكسدة كهربية للأكسجين أكثر استقرارًا مقارنةً بآلية التحلل المحتمل للمحفز..
تم استخدام اختبار الفولتمترية النبضية لتقييم قدرة المحفزات على إزالة البروتون، مما يمكن أن يؤكد مصدر النشاط المعزز لـفيتحت نبضات جهد مختلفة (الشكل التوضيحي 35)، روديوم وتظهر نبضات تيار كاثودي وأنودي متناوبة (الشكل 5h). تم قياس سعة تخزين الشحنة المؤكسدة لمختلف المحفزات من خلال تكامل استجابة التيار الأنودي لنبضات الجهد. كما هو موضح في الشكل 5i،تظهر سعة تخزين شحنة الأكسدة أعلى مقارنة بجزيئات نانو الروثينيوم، مما يعني أنتخضع لعملية إزالة بروتون أسرع لتكوين وسيط تفاعل *. تشير هذه النتائج إلى أن التفاعلات المناسبة بين المعدن والدعم ويمكن لرو تفعيل مواقع رو من خلال تعزيز عملية إزالة البروتون في تفاعل الأكسدة الكهربائي. علاوة على ذلك، في مخططات بود EIS، فإن عملية ما قبل تفاعل الأكسدة الكهربائييؤدي إلى توزيع غير متساوٍ للشحنات السطحية (الشكل التكميلي 36)، والذي يتجلى في انخفاض في قمم التردد ضمن النطاقمقارنةً بـ RHE وتحول نحو ترددات أعلى مقارنةً بقمم مرحلة الانتقال الأوسع لجزيئات نانو الروثينيوم ( مقابل RHE). تشير هذه الظاهرة إلى أن يعرض تفريغ شحنات أسرع لتسريع إزالة البروتون أثناء تفاعل الأكسدة-الاختزال.باختصار، التفاعلات المعتدلة بين المعادن والدعم بين جزيئات نانو الروثينيوم ومن خلاليمكن أن تقلل الوحدات من حواجز شوتكي على الواجهة لتسريع نقل الإلكترونات وتحقيق إثراء الإلكترونات في مواقع الروثينيوم لإبطاء تآكل الروثينيوم أثناء. بالإضافة إلى ذلك، فإن التعديل الإلكتروني لمواقع الروثينيوم النشطة يسهل عملية إزالة البروتون في تفاعل الأكسدة (OER) (الشكل 5j). خلال عملية تفاعل الهيدروجين (HER)، تم تسجيل مخططات نايكويست (EIS) عند كثافات تيار مختلفة وتم ملاءمتها بواسطة الدائرة المعادلة الموضحة في الشكل التكميلي 37a.يعكس مقاومة امتصاص الهيدروجين على سطح المادة في الدائرة المعادلةتم قياس حركيات امتصاص الهيدروجين من خلال رسم مقابل الجهد الزائد وحساب انحدارات تافل المستمدة من EIS. كما هو موضح في الشكل التكميلية 37، فإن الانحدار المخفض لـ21.3 مللي فولت مقارنةً مع و تشير إلى الديناميات المعجلة لامتصاص الهيدروجين، والتي تتعلق بتسرب الهيدروجين (الشكل التكميلي 38، والجدول التكميلي 4). النتائج المذكورة أعلاه توضح بشكل أكبر أنيحقق ديناميات سريعة لتفاعل الهيدروجين من خلال آلية تسرب الهيدروجين المحتملة.
الحساب النظري للنشاط الجوهري وتحليل الاستقرار
تكشف حسابات نظرية الكثافة الوظيفية (DFT) عن تأثير قابلية دعمعلى تعديل الهيكل الإلكتروني والتفاعل الثنائي الوظيفة لمواقع الروثينيوم. يظهر هيكل نطاق الطاقة أنوتمتلك TiO توزيع كثافة حالات غني عند مستوى فيرمي، مما يرمز إلى توصيلها الفائق مقارنة بفجوة نطاق ملحوظة من (الشكل التكميلي 39). تُظهر كثافة الشحنة التفاضلية منطقة صفراء كبيرة بالقرب من ذرات الروثينيوم عند الواجهة بين الروثينيوم والذي يؤكد غنى الإلكترونات في ذرات Ruatoms (الأشكال التكميلية 40 و 36c). في الملف الشخصي المحتمل المتوسط (الشكل التكميلي 41d)، فرق الجهد الكهروستاتيكي بين الروثينيوم ويمكن ملاحظته بشكل أكثر وضوحًا. بعد ذلك، تم استخدام مخططات بورباكس للتحقق من استقرار الروثينيوم بناءً على وحدات Ti-O-Ru بين الروثينيوم ودعائم أكسيد التيتانيوم. وفقًا لمخطط بورباكس للروثينيوم-أكسجين (الشكل 6أ)، فإن الروثينيوم عرضة للاكسدة المفرطة لتكوين حالة عالية من الفلزات. عند الجهود العالية لتفاعل تطور الأكسجين، خاصة في البيئات الحمضية. علاوة على ذلك، فإن الواجهة القوية الوحدات تزيد بشكل فعال من الإمكانية النظرية للذوبان للروثينيوم في الأكسدة الكهروكيميائية كما هو موضح في نظام بوربايكس لـ Ti-O-Ru (الشكل 6ب). في الشكل 6ج، تظهر حسابات الحالة الأساسية والحالة التكافؤية عند الواجهة أن مواقع الروثينيوم في الواجهة لديها أعلى حالة تكافؤ (-0.007)، والتي هي الأقرب إلى حالة التكافؤ الأولية للروثينيوم ومتوافقة مع نتائج XAFS. هذه القابلية للتكيف في الهيكل الإلكتروني تعطيحاجز شوتكي بواجهة أدنى مقارنة بـ و لتسريع هجرة الإلكترونات في الواجهة وتحفيز آلية تسرب الهيدروجين لـ.
النموذج الهيكلي المقابل لجزيئات نانو الروثينيوم المؤكسدة جزئيًا المحملة علىتم بناؤه لحساب مسارات OER استنادًا إلى آلية تطور الامتصاص (الأشكال التكميلية 41-43). إن الأكسدة المحلية جنبًا إلى جنب مع إعادة توزيع الإلكترونات بين Ru ودعائم أكسيد التيتانيوم المختلفة تمنح مواقع Ru أنشطة جوهرية متباينة من خلال-تعديل مركز النطاق. الانخفاض في-مركز النطاق يقلل من طاقة المدارات المضادة للرابطة التي تتكون من وسائط تفاعل الامتصاص و-مدارات الروثينيوم، مما يعني الامتصاص الضعيف لوسائط التفاعل. لذلك، في الأكسيد السطحي و ، الـ-مراكز النطاقات لمواقع الروثينيوم هيو -2.07 إلكترون فولت، على التوالي (الشكل التكميلية 44 والجدول التكميلية 5)، مما يمنح طاقات الامتزاز الحرة لـ *OH المقابلة بـ، و 0.703 إلكترون فولت عند، على التوالي (الشكل 6د). بعد ذلك، يتم أيضًا تحسين عملية إزالة البروتون.يظهر أقل إزالة بروتون
الشكل 6 | الحساب النظري وعلاقة الهيكل بالنشاط. أ Ru-O، و (ب)رسوم بوربايكس التي تم إنشاؤها مع تركيز أيونات مائية منمات. شحن باد في واجهة ، و وحالات التكافؤ المحسوبة المقابلة لـ Ru و Ti. د ملفات الطاقة الحرة لـ
وسائط OER المختلفة عند 0 فولت و 1.23 فولت لموقع Ru في، و . ملفات الطاقة الحرة للوسطاء في تفاعل الهيدروجين (HER) لموقع Ru في Ru، Ru/TiO، ، و . الطاقة الحرة وأدنى جهد نظري لـالجدول التكميلي 6). تتماشى عملية إزالة البروتون المعززة مع النتائج التجريبية. تحفز المحفزات تفاعل تقليل الهيدروجين في هيكلها الأولي عند إمكانيات الاختزال (الشكل التكميلي 45)يوضح مخطط خطوة الطاقة الحرة أن النشاط الضعيف لتفاعل الهيدروجين (HER) لمواقع الروديوم (Ru) في جزيئات نانوية من الروديوم (Ru NPs) ناتج عن الامتصاص القوي للوسائط الوسيطة لتفاعل الهيدروجين (HER) (الشكل 6e). على وجه التحديد،-مركز نطاق الروديوم عند الواجهة يظهر تباينات واضحة مع هجرة الإلكترونات عند الواجهة الناتجة عن الاتصال بين الروديوم وأكاسيد التيتانيوم المختلفة.-مركز نطاق Ru هو -1.72 eV. بعد الاتصال مع، و المقابل-تتحول مراكز نطاقات الروثينيوم إلى، و -1.98 إلكترون فولت، على التوالي (الشكل التكميلية 46). بسبب الانخفاض مقارنةً بجزيئات Ru الأصلية الناتج عن تفاعل المعدن والدعم، فإن مواقع Ru في Ru، Ru/TiO، Ru/TiO، و Ru/ لديطاقة الامتزاز الحرة لـ0.123 إلكترون فولت، على التوالي (الشكل 6e، الجدول التكميلي 7). لذلك، يمتلك أقل جهد تفاعل نظري يبلغ 0.123 إلكترون فولت لتفاعل الهيدروجين. وقد أكدت الحسابات النظرية أن الدعم المناسب يمكن أن يعزز النشاط الجوهري لمواقع الروثينيوم لكل من تفاعل الأكسدة وتفاعل الهيدروجين.
أداء جهاز التحليل الكهربائي للماء
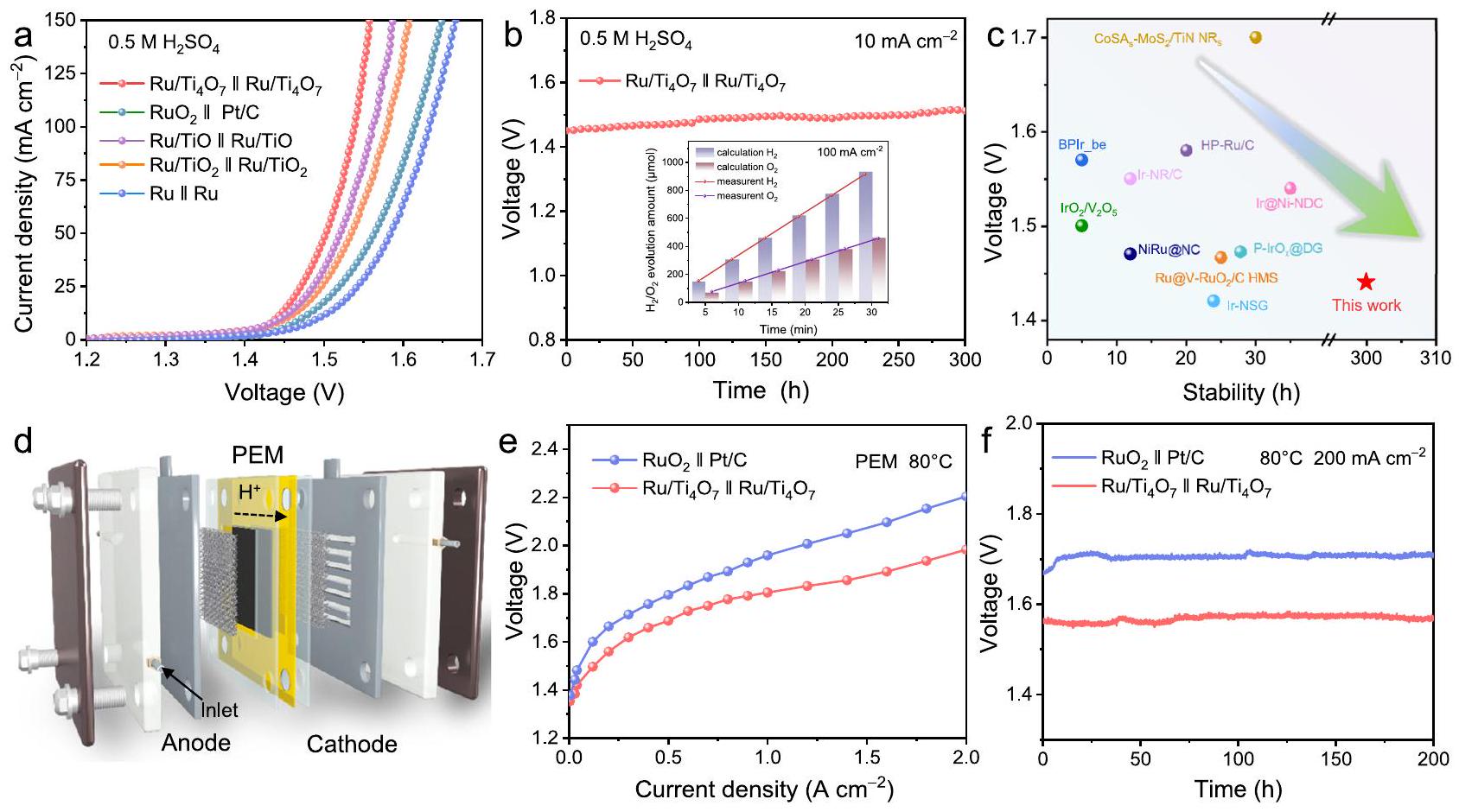
أولاً، تم التحقق من المزايا الفريدة لتصميم الدعم المناسب من خلال اختبارات التحليل الكهربائي للماء بدون غشاء. كما هو موضح في الشكل 7a، فإن التحليل الكهربائي للماء المدفوع بـمع نشاط ثنائي الوظيفة الممتاز يتطلب فقط جهد خلية يبلغ 1.44 فولت للوصول إلى كثافة تيارمع تشغيل مستقر لمدة 300 ساعة (الشكل 7ب) في البيئات الحمضية. ومن الجدير بالذكر أن الكفاءة فاراداي المقابلة قريبة أيضًا منإن أداء فصل الماء الممتاز هذا يتجاوز أداء المحفزات الثنائية الوظيفة المتطورة التي تم الإبلاغ عنها مؤخرًا في الوسط الحمضي (الشكل 7c). تفوق الـتم التحقق أيضًا من دعم تحليل الماء في البيئات ذات الرقم الهيدروجيني الشامل (الأشكال التكميلية 47، 48 والجدول التكميلية 8). مستفيدًا من مزايا الموصلية والاستقرار،تم استخدامه كعوامل حفازة لكل من الكاثود والأنود في إلكتروليزرات MEA (الشكل 7د). بالنسبة لتحليل الماء باستخدام غشاء تبادل البروتون (الشكل 7هـ)،يعرض جهد خلية أقل بكثير من ذلك لـويحافظ على التشغيل طويل الأمد لمدة 200 ساعة عندو 300 ساعة عند (الشكل 7f والشكل التكميلي 49)، والذي يتفوق على معظم المحفزات المعتمدة على الروثينيوم التي تم الإبلاغ عنها مؤخرًا (الجدول التكميلي 9). المزايا الهيكلية الفريدة لمحول الإلكتروليت البوليمري وخصائص التشغيل عند درجات حرارة عالية، جنبًا إلى جنب مع الاستقرار الممتاز لـتحقيق التحليل الكهربائي للماء باستخدام غشاء تبادل البروتون القويلقد أكدت منحنيات نايكويست الخاصة بـ EIS لمجموعة MEA ميزة التوصيل.، مما يقلل بشكل كبير من استهلاك الطاقة للخلية الكهربية تحت كثافات التيار العالية (الشكل التكميلي 50). علاوة على ذلك، يمتلك أيضًا نشاطًا عاليًا وقابلية للتوسع واستقرارًا في AEM (الشكل التكميلي 51). المزايا الظاهرة للثنائي الوظيفةفي تجميع الأقطاب الغشائية تظهر آفاق تطبيق صناعي مثيرة للإعجاب.
نقاش
في هذه الدراسة، تم استخدام أكسيد التيتانيوم المصمم من خلال الهندسة غير المتوازنة لتثبيت المعدن رو (Ru) من أجل تقسيم الماء بشكل ثنائي الوظيفة عالي النشاط والثبات. كان التصميم غير المتوازن لـيسهل تحقيق التفاعل المناسب لدعم المعادن مع الروثينيوم. إن إثراء الإلكترونات لجزيئات الروثينيوم النانوية من خلال الاستقرارتعزز الوحدات مقاومة التآكل للروثينيوم للحصول على نشاط جزئيالنوع، الذي يثبط LOM ويسرع إزالة البروتون للتفاعل الأكسدة-اختزال. الموصلية الكهربائية العالية لـيسمحلتحفيز OER مع استهلاك منخفض للطاقة. علاوة على ذلك، فإن التكيف في وظيفة العمل بينوتجعل رو HER تهتم بآلية تسرب الهيدروجين لتحقيق حركية تفاعل سريعة. كما هو متوقع،يعرض جهدًا زائدًا منخفضًا للغاية قدره 8 مللي فولت و150 مللي فولت عندلـ HER و OER في البيئات الحمضية، على التوالي، مع تشغيل مستقر لمدة 500 ساعة. الأداء الممتاز ثنائي الوظيفة يمكّنلتحفيز انقسام الماء عندعند جهد 1.44 فولت، والذي تم التحقق منه أيضًا في بيئات pH العالمية. تم تجميع أجهزة PEM معتظهر فولتية خلايا أقل واستقرارًا أعلى من تلك الموجودة في المنتجات التجارية
الشكل 7 | أداء تحليل الماء. أ منحنيات استقطاب LSV لـ، ، و (تحميل المبلغ: ) في لتفكيك الماء الحمضي. ب اختبارات الاستقرار لـفي، ويعرض الإطار الداخلي الكفاءة الفارادية المقابلة في. ج مقارنة جهد الخلية واستقرارها مع تلك التي تم الإبلاغ عنها مؤخرًا ثنائية الوظيفة محفزات لتفكيك الماء الحمضي. نموذج مفاهيمي لخلايا تدفق إلكتروليزر الماء PEM باستخدام الماء النقي كمواد خام. منحنيات الاستقطاب الثابتة لـ Ru/ و (تحميل المبلغ: ) لتحليل الماء باستخدام كهرباء PEM في الماء النقي في. اختبارات الاستقرارلإلكتروليزر الماء PEM في. وعملية قوية فيلمدة 300 ساعة. تؤكد هذه الأداء المتميز على تقدم تصميم الدعم الدقيق لأجهزة التحليل الكهربائي للماء وتفتح آفاقًا جديدة للبناء العقلاني لمحفزات تفاعل الأكسدة.
طرق
تركيب المواد
إعدادأولاً، تم طحن 1 جرام من TiO2 و40 ملغ من الكربون الأسود لمدة 10 ساعات لتشكيل خليط متجانس. بعد ذلك، تم تسخين الخليط عندلمدة 3.5 ساعة للحصول علىمسحوق.
إعداد. محملة بـ Ruتم تحضيره بواسطة اختزال رطب بسيط. أولاً، و تم توزيعها بالتساوي في 50 مل من الماء المنزوع الأيونات. بعد ذلك، 7 مل منتم إضافة المحلول ببطء قطرة قطرة إلى الخليط مع التحريك بقوة. بعد التحريك لمدة 4 ساعات، تم جمع المنتج عن طريق الطرد المركزي وتجفيفه تحت الفراغ عندلمدة 12 ساعة. تم تسخين المسحوق الناتج مرة أخرى عند، و لمدة ساعة واحدة في جو الأرغون. تم وضع علامة على المنتجوتم تخزينه في البيئة المملوءة بالغاز الأرجون. كتلةتم الحفاظ على 60 ملغ، وجرعةتم تغييره إلىو 60 ملغ لتحقيق تحميل مختلف من الروثينيوم.
إعداد و طرق التحضير والتخزين و كانت هي نفسها التي لـ. وتم استبدال TiOللحصول على و ، على التوالي.
خصائص المواد
تم إجراء اختبارات XRD باستخدام جهاز Bruker D8 advance (Billerica) مع معدل مسح مضمون لـتم التقاط صور SEM باستخدام جهاز Regulus 8100 (هيتاشي). AC-TEM (JEM-ARM200F، جيول) تم استخدامه للتحقيق في البنية الدقيقة للمواد. تم الحصول على صور TEM و HRTEM باستخدام Tecnai G2 F20 (FEI). استخدم XPS أشعة X من نوع Al Ka كمصدر للإثارة (Escalab 250Xi، Thermo Fisher Scientific). تم الحصول على جميع طيف XPS الضيق تحت ظروف طاقة تمرير تبلغ 20 eV وخطوة طاقة تبلغ 0.05 eV، وتم معايرتها بواسطة قمة الكربون.تقع عند 284.8 إلكترون فولت. تم إجراء قياسات UPS باستخدام محلل VG Scienta R4000 (مصدر ضوء هيليوم أحادي اللون بقدرة 20.2 إلكترون فولت) مع انحياز قدره 10 إلكترون فولت. تم إجراء قياسات XAFS في خط شعاع 44 A في مصدر الفوتون التايواني باستخدام مطيافية امتصاص الأشعة السينية السريعة (هسينتشو). تم جمع طيف XAFS في درجة حرارة الغرفة وتحليله باستخدام برنامج أثينا. تم إجراء قياسات الطيف الكتلي بالتحليل الطيفي للبلز المتصل بالتحريض (ICP-MS) باستخدام جهاز Optima 7300 DV (PerkinElmer).
القياسات الكهروكيميائية
تم تنفيذ الاختبارات الكهروكيميائية باستخدام محطة عمل كهروكيميائية (Autolab PGSTAT302، Metrohm). تم الإشارة إلى الجهود الكهربائية باستخدام قطب هيدروجين قابل للعكس (RHE). تم إجراء القياسات الكهروكيميائية في خلية كهروكيميائية ذات ثلاثة أقطاب مع غاز الأرجون المشبع.محلول مائي، ومحلول عازل فوسفات بتركيز 1 م (PBS). تم خلط المحفزات النشطة والرباط (فلوريد بولي فينيليدين، PVDF) بنسبة وزن 7:1 مع N-methyl-2-pyrrolidone (NMP) كمذيب. ثم تم تجفيف حبر المحفز بالتنقيط على ورق الكربون وتجفيفه لتحضير القطب العامل حتى كان تحميل المحفز. Pt/C تجاري (20 وزن. %) وتم تحضيرها كأقطاب عمل باستخدام نفس الطريقة للمقارنة. تم استخدام رقائق البلاتين وقضيب الكربون كأقطاب مضادة لتفاعل الأكسدة (OER) وتفاعل الاختزال (HER) على التوالي.، و تم استخدام الأقطاب الكهربائية كأقطاب مرجعية في الأوساط الحمضية والقلوية والمحايدة، على التوالي. تم الحصول على اختبارات CV بمعدلات مسح مختلفة ضمننطاق RHE مقابل لتحديد التم توليد منحنيات LSV بمعدل مسح قدرهمعالتعويض. تم إجراء EIS عند جهد 1.5 فولت مقابل RHE لتفاعل الأكسدة (OER) و-0.5 فولت لتفاعل الاختزال (HER)، مع تغطية ترددات من 100 كيلوهرتز إلى 0.1 هرتز مع سعة 10 مللي فولت. في قياس الاعتماد على الرقم الهيدروجيني لتفاعل الأكسدة (OER)، تم تحضير المحلول الكهربائي عن طريق إضافة مكونات مخزن بريتون-روبنسون (0.4 م لكل من الفوسفات، البورات، والأسيتات) إلىمحلول، ثم تم تعديل الرقم الهيدروجيني إلى القيمة المرغوبة عن طريق إضافة. تم تنشيط جميع الزجاجيات في ماء نقي للغاية مباشرة قبل المعالجة الكهروكيميائية. تم إجراء الفولتمترية النبضية مع متابعة التيار على مر الزمن. تم الحفاظ على الجهد عند جهد منخفض ( مقابل RHE)، ثم تم التبديل والاحتفاظ بجهد أعلى ( ) قبل العودة إلى . تم تكرار هذه الدورة مع زيادةمن 1.42 فولت إلى 1.50 فولت فيخطوة والحفاظ علىغير متغير. تم حساب الشحنات المتعلقة بالخطوة المحتملة من خلال تكامل نبضة التيار على مدى الزمن، مع الأخذ في الاعتبار إشارة التيار الخلفية.
الخصائص الكهروكيميائية في الموقع
قياسات XAFS في الموقع. تشمل مطيافية امتصاص الأشعة السينية في الموقع، XANES وEXAFS عند كل من Ru-حافة و-الحافة، تم جمعه في وضع العائد الكلي للفلوريسنس باستخدام كاشف انزلاق السيليكون في مركز أبحاث الإشعاع المتناظر الوطني (NSRRC) في اليابان وتايوان. تمت القياسات، تحت نفس الظروف كما في حالة التوصيف الكهروكيميائي في إعداد ثلاثي الأقطاب النموذجي، في حاوية مصممة خصيصًا من التفلون مع نافذة مختومة بشريط كيمبتون. تم الحفاظ على نطاقات المسح في نطاق طاقة من و لـ رو-edge (BL-12B2 في SPring-8، NSRRC) و Ti-الحافة (17C في مصدر الضوء في تايوان، NSRRC)، على التوالي. تم الحصول على الأطياف عن طريق طرح خط الأساس من الحافة السابقة وتطبيع تلك الخاصة بالحافة اللاحقة. تحويل فورييه علىتم استخدام تذبذبات EXAFS الموزونة لتحليل EXAFS. جميع طيفيات EXAFS مقدمة بدون تصحيح الطور.
قياسات ATR-SEIRAS في الموقع. تم إجراء قياسات ATR-SEIRAS بواسطة مطياف الأشعة تحت الحمراء بتقنية تحويل فورييه (FT-IR) من نوع نيكوليت iS50 مزود بكاشف MCT مبرد بالنيتروجين السائل ومسار بصري ثابت بزاوية. كانت دقة الطيف للقياسات هيوتم إضافة 32 تداخلاً لكل طيف.
قياسات تجميع الأقطاب الغشائية (MEA)
قياسات إلكتروليزر PEM. تم تحضير مجموعة الأقطاب الغشائية باستخدام نافيون 117 بطريقة الغشاء المغطى بالمواد الحفازة بمساحة هندسية قدرهاتم توزيع المحفزات النشطة ثنائية الوظيفة في الإيزوبروبانول، والماء المقطر، ومحلول الإيثانول من نافيون. ) لتشكيل حبر. ثم تم رش الحبر على كلا جانبي غشاء تبادل البروتون (PEM) بواسطة نقل بولي تترافلوروإيثيلين (PTFE) بمساحة فعالة من حتى كانت تحميل المحفزلتجنب التآكل عند جهد الأكسدة، تم استخدام شعر التيتانيوم كطبقة انتشار غاز (GDL) للأنود. أخيرًا، تم ضغط الغشاء مع المحفزات الكهربائية المطلية، وGDL الأنود، وGDL الكاثود (ورق الكربون) معًا تحت الحرارة لتأسيس MEA تحتبضغط 10 ميغاباسكال لمدة 5 دقائق. المنتج التجاري و تم استخدامهما بنفس الطريقة مثل الكاثود والأنود على التوالي للمقارنة. تم إرسال الماء النقي إلى الكاثود والأنود بواسطة مضخة بيرستالتية بسرعة 40 دورة في الدقيقة. قبل الاختبار، تم تنشيط MEA المعدة في وضع الجهد الثابت عند جهد خلية قدره 2 فولت. بعد ذلك، تم تشغيل الإلكتروليزر عندتمت إزالة الهيدروجين والأكسجين في المحلول الكهربائي باستخدام الأرجون لتجنب التأثير المحتمل للفقاعات المتكونة على الاستقرار طويل الأمد للأقطاب. تم الحصول على منحنيات الاستقطاب في حالة الاستقرار بواسطة مصدر تيار مباشر (ITECH، IT-M3223). تم تسجيل جهد الخلية عند كثافات تيار مختلفة.
قياسات إلكتروليزر AEM. تم استخدام PiperION-A60 لبناء إلكتروليزر AEM. كانت حبر المحفز لـ AEM هو نفسه المستخدم في PEM، باستثناء أن محلول الإيثانول Nafion تم استبداله بمحلول الإيثانول PiperION A. ). تم دعم الحبر على ورق الكربون، وشعر تي بالمساحة الفعالة لـ حتى كانت تحميل المحفزلتحسين الاتصال السطحي بين طبقة المحفز وAEM، تم رش كمية صغيرة من محلول الإيثانول لآيونومر PiperION A على سطح طبقات المحفز للأنود والكاثود ثم تم تجفيفها في درجة حرارة الغرفة لمدة 48 ساعة. أخيرًا، تم ضغط AEM وGDLs مع تحميل المحفز معًا تحت الحرارة لتأسيس MEA تحتبضغط 10 ميغاباسكال لمدة 5 دقائق. المنتج التجاري و تم استخدامهما أيضًا بنفس الطريقة كالكاثود والأنود على التوالي للمقارنة. تم إرسال محلول مائي من KOH بتركيز 1 مول إلى الكاثود والأنود بواسطة مضخة بيرستالتية بسرعة 40 دورة في الدقيقة. تم تشغيل الإلكتروليزر عند.
طرق حسابية
تم إجراء جميع حسابات DFT من خلال حزمة المحاكاة الأولية فيينا (VASP). استخدمت الحسابات طريقة الموجة المعززة بواسطة المشروع (PAW).الزيف المحتمل مع PBEتقريب التدرج العام (GGA) لوظيفة التبادل-الارتباط. كانت مجموعة الأساس الموجي المسطح لها طاقة قطع تبلغ 500 إلكترون فولت. استخدم أخذ العينات K في حساب طاقة الامتزاز شبكة مونكهورست-باك منبينما في حساب DOS، تم استخدام الشبكة. تم تطبيق استقطاب الدوران على جميع الهياكل، وتم ضمان الاسترخاء الكامل لجميع الذرات مع تحمل تقارب الطاقةلكل ذرة. تم الحفاظ على القوة النهائية على كل ذرة تحتتم حساب مخططات بورباي باستخدام بيئة المحاكاة الذرية (ASE)مع طاقة تشكيل المدخل بواسطة حسابات DFT لنماذج الكتلة والسطح.
في بناء الهجائن، يتم استخدام الحد الأدنى من عدم التطابق الشبكي كمعيار لبناء نموذج الاتصال بين Ru وأكاسيد التيتانيوم المختلفة. يتصل وجه (001) من TiO بوجه (010) من Ru.وجه من يتصل بوجه ( 210 ) من Ru. الـ ( وجه منيتصل بوجه (001) من الروثينيوم.
يمكن حساب طاقة الامتزاز للوسطاء التفاعليين باستخدام المعادلة التالية (1):
أين الإعلانات، و ( ) هي طاقة الربط، هو تغيير طاقة النقطة الصفرية، هو تغيير الإنتروبيا. في هذا العمل، قيم و تم الحصول عليها من خلال حساب تردد الاهتزاز.
يمكن حساب الطاقة الحرة لجيبس لخطوات التفاعل الخمس من خلال المعادلات التالية (2-7):
في هذا العمل،تم حسابها عند.
توفر البيانات
البيانات التي تم توليدها في هذه الدراسة متوفرة في ملف البيانات المصدرية.
References
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609-616 (2023).
Zhang, Y. et al. Hydrogen-bond regulation of the microenvironment of Ni(II)-porphyrin bifunctional electrocatalysts for efficient overall water splitting. Adv. Mater. 35, 2210727 (2023).
Hao, S. et al. Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotechnol. 16, 1371-1377 (2021).
Zheng, X. et al. Tailoring a local acid-like microenvironment for efficient neutral hydrogen evolution. Nat. Commun. 14, 4209 (2023).
Chen, Z. et al. Stabilizing Pt single atoms through Pt-Se electron bridges on vacancy-enriched nickel selenide for efficient electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 62, e202308686 (2023).
Zhang, X.-L. et al. Efficient acidic hydrogen evolution in proton exchange membrane electrolyzers over a sulfur-doped marcasitetype electrocatalyst. Sci. Adv. 9, eadh2885 (2023).
Hao, Y. et al. Methanol upgrading coupled with hydrogen product at large current density promoted by strong interfacial interactions. Energy Environ. Sci. 16, 1100-1110 (2023).
Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022).
Ruiz Esquius, J. et al. Lithium-directed transformation of amorphous iridium (oxy)hydroxides to produce active water oxidation catalysts. J. Am. Chem. Soc. 145, 6398-6409 (2023).
Chen, M. et al. Recent progress in transition-metal-oxide-based electrocatalysts for the oxygen evolution reaction in natural seawater splitting: a critical review. eScience 3, 100111 (2023).
Yang, X. et al. IrPd nanoalloy-structured bifunctional electrocatalyst for efficient and pH-universal water splitting. Small 19, 2208261 (2023).
Li, Y. et al. Arming Ru with oxygen-vacancy-enriched subnanometer skin activates superior bifunctionality for pH-universal overall water splitting. Adv. Mater. 35, 2206351 (2023).
Li, J. et al. Boosting electrocatalytic activity of Ru for acidic hydrogen evolution through hydrogen spillover strategy. ACS Energy Lett. 7, 1330-1337 (2022).
Ling, C., Shi, L., Ouyang, Y., Zeng, X. C. & Wang, J. Nanosheet supported single-metal atom bifunctional catalyst for overall water splitting. Nano Lett. 17, 5133-5139 (2017).
Chen, J. et al. Reversible hydrogen spillover in Ru-WO enhances hydrogen evolution activity in neutral pH water splitting. Nat. Commun. 13, 5382 (2022).
Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829-7836 (2023).
Zheng, X. et al. Ru-Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Wei, J. et al. Site-specific metal-support interaction to switch the activity of Ir single atoms for oxygen evolution reaction. Nat. Commun. 15, 559 (2024).
Monai, M. et al. Restructuring of titanium oxide overlayers over nickel nanoparticles during catalysis. Science 380, 644-651 (2023).
Wu, Z. et al. Microwave synthesis of Pt clusters on black with abundant oxygen vacancies for efficient acidic electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 62, e202300406 (2023).
Deng, L. et al. Electronic modulation caused by interfacial Ni-O-M ( ) bonding for accelerating hydrogen evolution kinetics. Angew. Chem. Int. Ed. 60, 22276-22282 (2021).
Wang, W., Wang, Z., Hu, Y., Liu, Y. & Chen, S. A potential-driven switch of activity promotion mode for the oxygen evolution reaction at interface. eScience 2, 438-444 (2022).
Park, J. E. et al. Three-dimensional unified electrode design using a NiFeOOH catalyst for superior performance and durable anionexchange membrane water electrolyzers. ACS Catal. 12, 135-145 (2022).
Song, W. et al. Upscaled production of an ultramicroporous anionexchange membrane enables long-term operation in electrochemical energy devices. Nat. Commun. 14, 2732 (2023).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212-222 (2021).
Kosmala, T. et al. Operando visualization of the hydrogen evolution reaction with atomic-scale precision at different metal-graphene interfaces. Nat. Catal. 4, 850-859 (2021).
Wang, X. et al. Electronic structure modulation of by enriched with oxygen vacancies to boost acidic evolution. ACS Catal. 12, 9437-9445 (2022).
Lin, C. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021).
Shi, X. et al. Efficient and stable acidic water oxidation enabled by low-concentration, high-valence iridium sites. ACS Energy Lett. 7, 2228-2235 (2022).
Liu, X. et al. Restructuring highly electron-deficient metal-metal oxides for boosting stability in acidic oxygen evolution reaction. Nat. Commun. 12, 5676 (2021).
Mahdavi-Shakib, A. et al. The role of surface hydroxyls in the entropy-driven adsorption and spillover of on catalysts. Nat. Catal. 6, 710-719 (2023).
Han, H. G., Choi, J. W., Son, M. & Kim, K. C. Unlocking power of neighboring vacancies in boosting hydrogen evolution reactions on two-dimensional NiPS monolayer. eScience, 100204 https://doi. org/10.1016/j.esci.2023.100204 (2023).
Xia, J. et al. hierarchical bifunctional catalyst anchored on graphene aerogel toward flexible and high-energy Li-S batteries. ACS Nano 16, 19133-19144 (2022).
Yang, B. et al. Flatband towards extraordinary solar steam generation. Nature 622, 499-506 (2023).
Dai, J. et al. Single-phase perovskite oxide with super-exchange induced atomic-scale synergistic active centers enables ultrafast hydrogen evolution. Nat. Commun. 11, 5657 (2020).
Ioroi, T., Siroma, Z., Yamazaki S.-i. & Yasuda K. Electrocatalysts for PEM fuel cells. Adv. Energy Mater. 9, 1801284 (2019).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659-23669 (2023).
He, Q. et al. Confining high-valence iridium single sites onto nickel oxyhydroxide for robust oxygen evolution. Nano Lett. 22, 3832-3839 (2022).
Hu, F. et al. Lattice-matching formed mesoporous transition metal oxide heterostructures advance water splitting by active bridges. Adv. Energy Mater. 12, 2200067 (2022).
Zhang, B. et al. Enhanced spatial charge separation in a niobium and tantalum nitride core-shell photoanode: In situ interface bonding for efficient solar water splitting. Angew. Chem. Int. Ed. 62, e202305123 (2023).
Chen, R. et al. Promoting the efficiency and selectivity of -to reduction on active sites via preferential glycol oxidation with holes. P. Natl Acad. Sci. USA 120, e2312550120 (2023).
Li, M. et al. Revealing the regulation mechanism of interfacial chemical bonding for improving hydrogen oxidation reaction. ACS Catal. 11, 14932-14940 (2021).
Dai, J. et al. Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis. Nat. Commun. 13, 1189 (2022).
Fu, H. Q. et al. Hydrogen spillover-bridged volmer/tafel processes enabling ampere-Level current density alkaline Hydrogen evolution reaction under low overpotential. J. Am. Chem. Soc. 144, 6028-6039 (2022).
Zheng, T. et al. Intercalated iridium diselenide electrocatalysts for efficient pH-universal water splitting. Angew. Chem. Int. Ed. 58, 14764-14769 (2019).
Li, J. et al. A fundamental viewpoint on the hydrogen spillover phenomenon of electrocatalytic hydrogen evolution. Nat. Commun. 12, 3502 (2021).
Hou, L. et al. Electronic and lattice engineering of ruthenium oxide towards highly active and stable water splitting. Adv. Energy Mater. 13, 2300177 (2023).
Nong, H. N. et al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 587, 408-413 (2020).
Yang, L. et al. A highly active, long-lived oxygen evolution electrocatalyst derived from open-framework iridates. Adv. Mater. 35, 2208539 (2023).
Hao, Y. et al. Switchingsed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659-23669 (2023).
Du, K. et al. Interface engineering breaks both stability and activity limits of for sustainable water oxidation. Nat. Commun. 13, 5448 (2022).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304-313 (2019).
Zhang, Q. et al. Solid-solution alloy nanoparticles of a combination of immiscible Au and Ru with a large gap of reduction potential and their enhanced oxygen evolution reaction performance. Chem. Sci. 10, 5133-5137 (2019).
Yang, Y. et al. Hierarchical nano assembly of as a highly efficient electrocatalyst for overall water splitting in a wide pH range. J. Am. Chem. Soc. 141, 10417-10430 (2019).
Zheng, X. et al. Enriched d-band holes enabling fast oxygen evolution kinetics on atomic-layered defect-rich lithium cobalt oxide nanosheets. Adv. Funct. Mater. 32, 2200663 (2022).
Tan, H. et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 13, 2024 (2022).
Hammer, B. & Norskov, J. K. Why gold is the noblest of all the metals. Nature 376, 238-240 (1995).
Ruban, A., Hammer, B., Stoltze, P., Skriver, H. L. & Nørskov, J. K. Surface electronic structure and reactivity of transition and noble metals. J. Mol. Catal. A: Chem. 115, 421-429 (1997).
Bornet, A. et al. Influence of temperature on the performance of carbon- and ATO-supported oxygen evolution reaction catalysts in a gas diffusion electrode setup. ACS Catal. 13, 7568-7577 (2023).
Kwon, J. et al. Tailored electronic structure of Ir in high entropy alloy for highly active and durable bifunctional electrocatalyst for water splitting under an acidic environment. Adv. Mater. 35, 2300091 (2023).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Hjorth Larsen, A. et al. The atomic simulation environment-a Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017).
شكر وتقدير
تم دعم هذا العمل من قبل المؤسسة الوطنية للعلوم الطبيعية في الصين (22075141)، وصندوق الابتكار العلمي والتكنولوجي الخاص بقمم الكربون والحياد الكربوني في مقاطعة جيانغسو (BK20220039)، ومؤسسة العلوم الطبيعية في مقاطعة جيانغسو (BK20210311). يشكر المؤلفون مركز أبحاث الإشعاع المتزامن الوطني (NSRRC) في هسينتشو، تايوان.
مساهمات المؤلفين
قام S.Z. بإجراء التجارب، وجمع البيانات، وتحليل البيانات. كتب S.Z. و L.D. المخطوطة. قام T.X. بإجراء التجارب. قام S.H. و W.Z. بإجراء اختبار XAFS في الموقع. قام S.L. و F.H. بتحليل البيانات وكتابة المخطوطة. قام C.K. و H.C. بإجراء قياس XAFS. صمم S.P. الدراسة، وحلل البيانات، وراجع المخطوطة.
¹كلية علوم المواد والتكنولوجيا، جامعة نانجينغ للطيران والفضاء، نانجينغ 210016، الصين.قسم الكيمياء التطبيقية، جامعة يانغ مينغ تشياو تونغ الوطنية، هسينتشو 300، تايوان.قسم علوم المواد والهندسة، جامعة تشينغ هوا الوطنية، هسينتشو 30013، تايوان.البريد الإلكتروني: pengshengjie@nuaa.edu.cn
Establishing appropriate metal-support interactions is imperative for acquiring efficient and corrosion-resistant catalysts for water splitting. Herein, the interaction mechanism between Ru nanoparticles and a series of titanium oxides, including and , designed via facile non-stoichiometric engineering is systematically studied. , with the unique band structure, high conductivity and chemical stability, endows with ingenious metalsupport interaction through interfacial units, which stabilizes Ru species during OER and triggers hydrogen spillover to accelerate HER kinetics. As expected, displays ultralow overpotentials of 8 mV and 150 mV for HER and OER with a long operation of 500 h at in acidic media, which is expanded in pH -universal environments. Benefitting from the excellent bifunctional performance, the proton exchange membrane and anion exchange membrane electrolyzer assembled with achieves superior performance and robust operation. The work paves the way for efficient energy conversion devices.
Water splitting has received extensive concern as the most promising pathway to obtain green hydrogen . Membrane electrode assembly (MEA) water electrolysis allows for clean and efficient green hydrogen production . Anion exchange membrane (AEM) and proton exchange membrane (PEM) electrolyzers are promising devices for water electrolysis, which rely on platinum (Pt)-based catalysts for hydrogen evolution reaction (HER) . Furthermore, PEM electrolyzers also require corrosion-resistant iridium (Ir)-based materials due to the harsh acidic conditions for oxygen evolution reaction (OER) . The large-scale consumption of precious metals has resulted in high costs for PEM, hindering the commercialization of PEM water electrolysis . Therefore, it is necessary to develop low-cost catalysts with less or non based catalysts. Ru is an effective alternative to due to about of the cost of . More importantly, Ru NPs exhibit unique HER and OER bifunctionality advantages in water splitting to reduce the catalyst cost . However, the stronger affinity of Ru for the *H intermediate in HER significantly affects reaction kinetics . In the OER process, Ru easily forms soluble species ( ) under the oxidation potentials, thus exhibiting an extremely short lifetime . Enhancing the activity and stability of Ru NPs in both HER and OER is a challenge. Among many strategies of catalyst design, loaded catalysts have garnered widespread attention due to the simple synthesis process, low usage of precious metals, ample exposure of active sites, and high atomic utilization efficiency . More importantly, sufficient contact between the support and active phase can construct suitable metal-support interactions and stable interface units to regulate the coordination
configuration and electronic structure of interface sites, which optimizes the catalytic performance and stability of the active centers . In general, ideal electrocatalyst support needs to maintain stability with high electroconductibility to minimize excessive energy consumption and catalyst dissolution under the operating voltages . Among the common stabilized supports, such as , , etc., has attracted much attention due to its excellent corrosion resistance and adjustable valence . However, usually displays a broad energy band gap as a typical n-type semiconductor material, which gives it poor conductivity and results in limited application in electrocatalysis . Introducing appropriate oxygen vacancies into through defect engineering can create defect energy levels to improve the conductivity . Nevertheless, random oxygen vacancy distribution makes it difficult to adjust defect states accurately and causes potential structural imbalances in semiconductors. Therefore, constructing non-stoichiometrically stable oxide supports with periodically distributed defects can introduce lattice defects with stable structures. Non-stoichiometric titanium oxides ( ), such as , and , possess higher electrical conductivity for several orders of magnitude than . In particular, , as a typical non-stoichiometric titanium oxide, has a similar conductivity to that of graphene, which is attributed to the abundant density of state distribution at the Fermi level . In addition, exhibits a more stable structure compared to lower-valence titanium oxides such as TiO . More importantly, the precise nonstoichiometric design of the support can obtain the suitable crystal and electronic structure to achieve adaptability with the active species, which is beneficial to improve the intrinsic activity and stability of the active sites.
Herein, the interaction mechanism between Ru nanoparticles and a series of titanium oxides, including and , was systematically explored. The appropriate metal-support interaction between Ru and is achieved, which moderately induces electron enrichment of the Ru sites to inhibit the lattice oxygen mechanism (LOM) of OER and facilitate deprotonation through stable units, which balances the activity and stability of OER. Furthermore, the low interface resistance between and Ru initiates the hydrogen spillover mechanism for HER to accelerate reaction kinetics. The exceptional bifunctional activity is confirmed in pH -universal environments. Specifically, in acidic conditions, demonstrates ultra-low overpotentials of 8 mV and 150 mV at for HER and OER, respectively, and maintains durable operation for 500 h . PEM devices assembled with show lower cell voltages and longer stability than those of commercial . This work provides new insight into the development of rationally supported catalysts and paves the way for the design of energy conversion devices.
Results
Principles of carrier design
As shown in Fig. 1, non-stoichiometric engineering was employed to finely customize titanium oxide supports ( , and ) with oxygen vacancies of periodic arrangements. , with regularly arranged defects, emerges as a potential competitor with superior structural stability over multi-defect TiO and significantly higher conductivity than . More importantly, exhibits a suitable metalsupport interaction, which lays the foundation for the dual-function activity of in both HER and OER. On the one hand, the higher work function compared to Ru NPs promotes the electron richness of Ru, which can alleviate the dissolution of Ru in OER. On the other hand, compared to TiO and , the minimum work function difference between and Ru NPs ( ) can reduce the interface Schottky barrier (Supplementary Fig. 1), which promotes electron transport in the composite catalyst and trigger hydrogen spillover during HER.
Morphology and crystal structure
Ru nanoparticles (NPs) were deposited on titanium oxide supports with different stoichiometric ratios ( , and TiO ) via a wet chemical method (Fig. 2a). has potential electrocatalytic advantages in support of Ru NPs due to the chemical stability superior to TiO and electrical conductivity far superior to . Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images display the uniform coating of the titanium oxide supports by Ru NPs with a size of (Figs. 2b and 1c). The suitable oxygen vacancies in promote the adsorption on , which endows the Ru NPs with a more uniform distribution on compared to with low vacancies (Supplementary Figs. 2-6) . In aberration-corrected high-resolution (AC-HRTEM) of , tight binding and appropriate matching at the nanoscale between the different phases can be observed, accompanied by a smooth transition at the interface (Fig. 2d). The simulated High Angle toroidal dark field image-scanning transmission electron microscope (HAADF-STEM) images obtained through the crystal structure of and Ru show remarkable agreement with the experimental results (Figs. 2e and 1f). Based on these atomic images, the lattice spacing of 0.377 nm can be attributed to the (102) plane of (JCPDS 50-0787), while the lattice spacing of 0.234 nm corresponds to the ( 100 ) plane of Ru (JCPDS 89-4903, Figs. 2g, 1h, and Supplementary Fig. 7). In addition, , and O elements are uniformly distributed in the energydispersive spectroscopy (EDS)-mapping of (Fig. 2i), and the characteristic diffraction peaks assigned to Ru and coexist in the X-ray diffraction (XRD) pattern (Fig. 2j). The above results indicate that can achieve a suitable interfacial lattice contact with Ru NPs, which provides the possibility for adaptive metal-support interaction and rapid interface electron migration .
Electronic and coordination structure analysis
The oxygen vacancy concentration in , and shows an increasing trend due to the different stoichiometric ratios of Ti and O (Fig. 3a). The periodically arranged oxygen vacancy not only improves the conductivity of the support but also provides the possibility to form the bridging interfaces between the and the Ru NPs. In the X-ray photoelectron spectroscopy (XPS) fine spectra of , and , the and regions display close overlap (Fig. 3b). The binding energy of Ti in different Ruloaded titanium oxide materials shows various degrees of shift to the high binding energy (Fig. 3b, and Supplementary Figs. 8-10), which indicates that titanium oxide can serve as an electron donor to promote electron enrichment of Ru (Fig. 3c). As shown in Supplementary Fig. 8c, Ru NPs exhibit only the characteristic peaks of metallic Ru. In the Ru 3 d region of , the signals attributed to and Ru-Ru can be observed, corresponding to the interface units . Similar local electronic changes can be further observed in the fine structure of X-ray absorption (XAFS). In the X-ray absorption near-edge structure (XANES), the Ru -edge also exhibits different white line peak intensity after contacting with various titanium oxide supports (Fig. 3d). Importantly, Ru in has the closest valence state to Ru foil compared with that in and , which is attributed to the low work function difference of and Ru obtained by the ultraviolet photoemission spectroscopy (UPS) measurements (Supplementary Fig. 1). Meanwhile, Ti pre- -edge of Ru/ shows a shift to higher energy compared with that of , suggesting that the increased valence state of Ti in (Supplementary Fig. 11). The above results indicate that Ru serves as an electron acceptor to form a Schottky contact with the titanium oxide support, and possesses the best adaptability of the electronic structure with Ru NPs to reduce the interface Schottky barrier. Extended X-ray absorption fine structure (EXAFS) spectra of Ru corresponding -weighted EXAFS Fourier transform spectra display a weak Ru-O signal located at after contacting with or
Fig. 1 | Bifunctional activation based on different supports. Schematic diagram of the interaction mechanism between different titanium oxide supports and Ru NPs to activate OER and HER.
respectively. Crystal plane spacing of and Ru. g Schematic diagram of interface contact between and EDS mapping of , and O in , respectively. XRD pattern of .
Fig. 2 | Synthesis and structural characterization. a SEM of . b TEM and c AC-HRTEM images of . d, e The schematic atom structure and the corresponding simulated HAADF-STEM images of and Ru in ,
Fig. 3 | Electron and coordination structure characterization. a EPR spectra of , and XPS fine spectra in and region of , and . c Schematic illustration of the electronic interaction between and Ru. d Normalized -edge XANES of Ti foil, , and
TiO. e The corresponding -weighted Fourier transforms and ( ) Wavelet transform of -weighted EXAFS signals. g Normalized -edge XANES of Ru foil, , , and The corresponding -weighted Fourier transforms and i Wavelet transform of -weighted EXAFS signals.
(Fig. 3e). This phenomenon should originate from the interface units between Ru and titanium oxide, which is observed in the wavelet transform more clearly (Fig. 3f). The stable units effectively stabilize the active species and serve as an electron channel to further reduce the interface electron transfer resistance . As shown in Fig. 3 g , the shift of Ti pre- -edge reflects the changed oxide valence states of Ti in different stoichiometric titanium oxide supports. Furthermore, As illustrated in the -weighted EXAFS Fourier transforms of -edge of and (Fig. 3 h ) and the corresponding wavelet transform (Fig. 3i), the structure belonging to further verifies the formation of the interface units.
Electrocatalytic activity and stability evaluation
The electrocatalytic activities of the Ru-loaded titanium oxides were evaluated in a standard three-electrode system to verify the unique advantages of the support. In the acidic media, with optimized Ru loading and annealing temperature exhibits the lowest OER overpotential ( 150 mV at , Fig. 4a and Supplementary Fig. 12) and Tafel slope ( , Fig. 4b, c). For HER, also displays an ultra-low overpotential of 8 mV at in acidic
environments, which is superior to commercial and other titanium oxide supported Ru NPs (Fig. 4d). Remarkably, as shown in Fig. 4e, the Tafel slope of with minimum work function difference is only , which is significantly lower than the value of the conventional Volmer-Heyrovsky/Tafel mechanism ( 30 mV . The low work function difference between metal and support is prone to trigger the hydrogen spillover mechanism during HER and provide a low interfacial Schottky barrier and fast interfacial electron migration . Electrochemical impedance spectroscopy (EIS) further verifies the lowest electron-transfer resistance of (Supplementary Fig. 13). Notably, the superior difunctional activity of also exhibited scalability in the neutral and basic environments (Fig. 4c, f, and Supplementary Figs. 14-17). In addition, exhibits the highest double-layer capacitance ( ) value ( 68.95 mF ) with the same Ru loading, suggesting that more exposed Ru active sites of the compared to and (Supplementary Figs. 18 and 19). The LSV polarization curves normalized by and the mass of Ru indicate that the active Ru sites in have the highest specific activities (Supplementary Fig. 20) and mass activity (Supplementary Fig. 21). The outstanding
Fig. 4 | Electrocatalytic activity evaluation. a Linear sweep voltammetry (LSV) polarization curves of OER and Tafel plots of Ru , commercial , and (loading amount: ) in . c The corresponding OER overpotential at in different pH environments. d LSV polarization curves of HER and (e) Tafel plots of Ru, commercial Pt/C, Ru/
, and (loading amount: ) in ). f The corresponding HER overpotential at in different pH environments. g Comparison of overpotential with that of other bifunctional catalysts for water splitting in recently reported. Chronopotentiometry curves of at , and in .
bifunctional performance of exceeds that of most of the excellent bifunctional catalysts in the recent reports (Supplementary Tables 2, 3), especially in acidic environments (Fig. 4g).
The stability of was validated via chronopotentiometry. In severe acidic environments, demonstrates surprising stability of OER and HER for 500 h with a negligible activity decay (Fig. 4 h ) and slight structural collapse (Supplementary Fig. 22), which exceeds the operating life of (Supplementary Fig. 23). also requires lower potentials to drive OER compared to , contributing to the lower energy consumption of the water electrolysis devices over a long period (Supplementary Fig. 23). More importantly, can maintain stable operation for 300 h at a high current density of with only slight activity decay after 3000 cyclic voltammetry (CV) cycles (Supplementary Figs. 24 and 25). The lower current response in the CV corresponding to the oxidation-reduction of Ru in compared to Ru and implies the inhibited oxidation of Ru by (Supplementary Fig. 26). Supplementary Fig. 27 manifests that the Ru and Ti loss of gradually slows down during long-term OER. Ti gradually dissolves in the first 50 h of cycling, which is attributed to the reaction between the unstable surface of the electrode. After 50 h , the dissolution rate of Ti slows down due to the stabilization of the surface of the electrode. Furthermore,
the characteristic diffraction peaks belonging to Ru and can still be observed in the XRD pattern of after OER without new characteristic peaks compared to the apparent dissolution of (Supplementary Fig. 28). As shown in the electron paramagnetic resonance (EPR) spectra (Supplementary Fig. 29) and XPS spectra of O before and after stability tests (Supplementary Fig. 30a), a slight decrease in oxygen vacancies in the support is observed, indicating the structural stability of the support under high current densities. In addition, the almost unchanged XPS signals of Ti and Ru with slight shift towards high binding energy after OER further provide evidence for the good structural maintenance of during the reaction (Supplementary Fig. 30b). The excellent stability of is attributed to inhibited oxidation process, which should be derived from the electron enrichment of Ru through the stabilized units.
Origin of enhanced difunctional activity of
A series of in-situ tests were performed in acid environments to trace the origin of the activity and stability of . In-situ XAFS explored the structural evolution of during OER. underwent 20 CV cycles to obtain the stable surface before recording the in-situ XAFS spectra. In the most severe acidic environments, the
Fig. | In-situ characterization and reaction mechanism. a Normalized in-situ Ru -edge XANES of . The corresponding -weighted Fourier transforms. c The valence states of Ru and Ti in obtained via -edge under different potentials. d HRTEM of after OER. In-situ ATR-SEIRAS spectra of (e) Ru and for OER in under different potentials vs. RHE. Intensity difference of the infrared signals at 1038 and Current responses to pulse voltammetry for Ru and . i Relationship between charge storage and potential of Ru and Schematic illustration of the optimized OER process induced by the support.
intensity of the white line of the Ru -edge increases with the applied potentials of OER (Fig. 5a), indicating the oxidation of Ru NPs. Furthermore, in the Fourier transform -space (Fig. 5b), the gradually increased signal intensity of the Ru-O path verifies the formation of . Notably, the peaks attributed to metallic Ru-Ru can always be observed even under the high potentials of OER, which corresponds to the Ru metallic phase shown in the XRD pattern of after OER. The above results indicate that the species locally formed on the can act as a barrier layer to slow down the further oxidation and dissolution of Ru NPs. The in-situ XAFS spectra show that the Ti edge of almost completely overlaps under the OER potentials without prominent oxidation characteristics, which benefits from the stable structure of (Supplementary Fig. 31a). The -space of Ti further verifies that the bond in does not change significantly during OER (Supplementary Fig. 31b). The stability of can be more verified in the explicit valence states
of Ti, and Ru. The valence state corrected by the normalized white line intensity of the corresponding reference of Ti in changes slightly between 3.52 and 3.61 with the application potentials growing (Supplementary Fig. 32). Meanwhile, the average valence state of Ru increases from 0.98 to 1.64 . The significant change does not appear until the potential reaches 1.7 V vs. RHE (Fig. 5c). This phenomenon further verifies that the Ru NPs still maintain structural stability under high potentials of OER. The incomplete oxidation of Ru NPs is beneficial to maintaining the outstanding conductivity of the composite material during the electrocatalytic process. In the HRTEM image of after OER, the tight interfacial contact between Ru NPs and can still be maintained (Supplementary Fig. 33). In addition, a locally amorphous structure can be observed in the outer layer of Ru NPs in after OER, which should correspond to the species (Fig. 5d).
Moreover, the reaction mechanism of OER was revealed by in-situ attenuated total reflection-surface enhanced infrared absorption spectra (ATR-SEIRAS). Figure 5e shows that, as the bias increases to 1.4 V vs. RHE, in-situ ATR-SEIRAS spectra of Ru exhibit absorption bands around the vibration frequency of , corresponding to the * intermediates. Simultaneously, a distinct absorption signal at the vibration frequency of gradually strengthens with the application of OER potential, corresponding to * in the LOM. In-situ ATR-SEIRAS spectra of appear at similar positions as Ru, attributing to the absorption bands of *OOH/*OO (Fig. 5f). The broadening and shifting of absorption peaks may originate from the transition of the deprotonation process. Furthermore, almost no signal corresponding to * is observed around the vibration frequency of . Additionally, the normalized density difference of in-situ ATR-SEIRAS spectra corresponding to *OOH/*OO and O-O signals is presented in Fig. 5g to determine the proportion occupied by adsorption evolution mechanism and LOM in the acidic OER process . The higher adsorption density difference implies a higher proportion of adsorption evolution mechanism in the reaction process. In the high potential range, Ru undergoes surface reconstruction to form species, enabling both the adsorption evolution mechanism and LOM to drive OER. Meanwhile, in-situ ATR-SEIRAS spectra of consistently exhibit a higher adsorption density difference than Ru during OER. This phenomenon indicates that tends to drive OER through the adsorption evolution mechanism, which can realize more stable OER compared with the LOM with potential catalyst dissolution. In the pH-dependence tests, compared with Ru NPs, showed less correlation with acidity, which verified that it was more inclined to drive OER with adsorption evolution mechanism (Supplementary Fig. 34). These results demonstrate that tends to drive OER through the surface adsorption evolution mechanism, which can realize more stable OER compared with the LOM with potential catalyst dissolution .
Pulse voltammetry test was employed to assess the deprotonation capability of catalysts, which can confirm the source of the enhanced activity of in . Under different voltage pulses (Supplementary Fig. 35), Ru and exhibit alternating cathodic and anodic current pulses (Fig. 5h). The oxidation charge storage capacity of different catalysts was further measured by integrating the anodic current response to voltage pulses. As shown in Fig. 5i, demonstrates a higher oxidation charge storage capacity compared to Ru NPs, implying that undergoes a faster deprotonation process to form reaction intermediate * . These findings suggest that the appropriate metal-support interactions between and Ru can activate Ru sites by promoting the deprotonation process in OER. Furthermore, in the EIS bode plots, the pre-OER process of results in an uneven distribution of surface charges (Supplementary Fig. 36), which is manifested by a reduction in the frequency peaks within the range of vs. RHE and a shift towards higher frequencies compared to the broader transition phase peaks of Ru NPs
( vs. RHE). This phenomenon suggests that exhibits a faster charge dissipation to accelerate deprotonation during OER to reaction kinetics . In summary, the moderate metal-support interactions between Ru NPs and through units can reduce interface Schottky barriers to accelerate electron transfer and achieve electron enrichment at Ru sites to slow down the corrosion of Ru during . In addition, electronic modulation of active Ru sites facilitates the deprotonation process of OER (Fig. 5j). During the HER process, EIS Nyquist plots at different current densities were recorded and fitted by the equivalent circuit inset of Supplementary Fig. 37a. reflects the hydrogen adsorption resistance on the material surface in the equivalent circuit . Hydrogen adsorption kinetics were quantified by plotting versus overpotential and calculating the EISderived Tafel slopes. As shown in Supplementary Fig. 37, the reduced slope of of 21.3 mv dec compared with that of and indicates the accelerated hydrogen adsorption kinetics, which is related to the hydrogen spillover (Supplementary Fig. 38, and Supplementary Table 4). The above results further demonstrate that achieves fast HER kinetics through a potential hydrogen spillover mechanism.
Theoretical calculation for intrinsic activity and stability analysis
Density functional theory (DFT) calculations reveal the impact of the support adaptability of on the electronic structure modulation and the bifunctional reactivity of the Ru sites. The energy band structure manifests that and TiO have a rich density of state distribution at the Fermi level, symbolizing their superior conductivity compared to a noticeable band gap of (Supplementary Fig. 39). The differential charge density shows a significant yellow area near the Ru atoms at the interface between Ru and , which verifies the electron enrichment of Ruatoms (Supplementary Figs. 40 and 36c) . In the average potential profile (Supplementary Fig. 41d), the electrostatic potential difference between Ru and can be more explicitly observed. Subsequently, Pourbaix diagrams were utilized to verify the stabilization of Ru based on Ti-O-Ru units between Ru and titanium oxide supports. According to the Pourbaix diagram of Ru-O (Fig. 6a), Ru is prone to over-oxidation to form high-valent at high potentials for OER, especially in acidic environments. Furthermore, the robust interface units effectively increases the theoretical dissolution potential of Ru in electrochemical oxidation as shown in the Pourbaix system of Ti-O-Ru (Fig. 6b). In Fig. 6c, the Bade and valence state calculations at the interface show that the Ru sites at the interface have the highest valence state ( -0.007 ), which is closest to the initial valence state of Ru and consistent with the XAFS results. This electronic structure adaptability gives a lower interface Schottky barrier compared to and to accelerate interface electron migration and trigger the hydrogen spillover mechanism for .
The structural model corresponding to the partially oxidized Ru NPs loaded on was constructed to calculate the OER pathways based on the adsorption evolution mechanism (Supplementary Figs. 41-43). The local oxidation combined with the electron redistribution between Ru and different titanium oxide supports endow the Ru sites with varying intrinsic activities through the -band center modulation. The downward shift of the -band center reduces the energy of the antibonding orbitals formed by the adsorption reaction intermediates and the -orbitals of Ru, which implies the weak adsorption of reaction intermediates . Therefore, in the surface oxidized and , the -band centers of the Ru sites are and -2.07 eV , respectively (Supplementary Fig. 44 and Supplementary Table 5), which endow the corresponding *OH adsorption free energies with , and 0.703 eV at , respectively (Fig. 6d). Subsequently, the deprotonation process is also optimized. exhibits the smallest deprotonation
Fig. 6 | Theoretical calculation and structure-activity relationship. a Ru-O, and (b) Pourbaix diagrams generated with aqueous ions concentration of M at . c Bade charge at the interface of , and and corresponding calculated valence states of Ru and Ti. d Free energy profiles of
different OER intermediates at 0 V and 1.23 V for Ru site in , and . e Free energy profiles of HER intermediates for Ru site in Ru, Ru/TiO , , and .
free energy and the lowest theoretical potential for , Supplementary Table 6). The enhanced deprotonation process is consistent with experimental results. The catalysts drive HER in its initial structure at the reduction potentials (Supplementary Fig. 45) . The free energy step diagram verifies that the weak HER activity of Ru sites in Ru NPs originated from the strong adsorption of HER intermediates (Fig. 6e). Specifically, the -band center of Ru at the interface exhibits distinct variations with the interface electron migration induced by the contact between Ru and different titanium oxides. The -band center of Ru is -1.72 eV . After contact with , and , the corresponding -band centers of Ru shift to , and -1.98 eV , respectively (Supplementary Fig. 46). Owing to the downward shift compared to the original Ru NPs induced by the metalsupport interaction, the Ru sites in Ru, Ru/TiO, Ru/TiO , and Ru/ have adsorption free energies of , and 0.123 eV , respectively (Fig. 6e, Supplementary Table 7) . Therefore, has the lowest theoretical reaction potential of 0.123 eV for HER. The theoretical calculations further verified that the appropriate support can promote the intrinsic activity of the Ru sites for both OER and HER.
Performance of the water electrolysis device
Firstly, the unique advantages of suitable support design were verified through membrane-free water electrolysis tests. As shown in Fig. 7a, the water electrolysis driven by with excellent bifunctional activity only requires a cell voltage of only 1.44 V to reach a current density of with stable operation for 300 h (Fig. 7b) in the acidic environments. Noteworthily, the corresponding Faraday efficiency is also close to . This excellent water splitting performance exceeds that of the state-of-the-art difunctional catalysts recently reported in the acidic media (Fig. 7c). The superiority of the support for water splitting in the pH -universal environments has also been verified (Supplementary Figs. 47, 48 and Supplementary Table 8). Benefitting from the advantages of conductivity and stability, was used as both cathode and anode catalysts in the MEA electrolyzers
(Fig. 7d). For the PEM water electrolysis (Fig. 7e), exhibits significantly lower cell voltage than that of and maintains long-term operation of 200 h at and 300 h at (Fig. 7f and Supplementary Fig. 49), which is superior to most recently reported Ru-based catalysts (Supplementary Table 9). The unique structural advantages of the PEM electrolyzer and the characteristics of high-temperature operation combined with the excellent stability of achieve robust PEM water electrolysis . The EIS Nyquist curves of the MEA further verified the conductivity advantage of , which significantly reduces the energy consumption of the electrolyzer under high current densities (Supplementary Fig. 50) . Furthermore, also possesses extensible high activity and stability in AEM (Supplementary Fig. 51). The apparent advantages of bifunctional in membrane electrode assembly show impressive industrialized application prospects.
Discussion
In this study, titanium oxide designed through rational nonstoichiometric engineering was used to stabilize metal Ru for highly active and stable bifunctional water splitting. The non-stoichiometric design of facilitates the achievement of appropriate metalsupport interaction with Ru. The electron enrichment of Ru NPs through stable units enhances the corrosion resistance of Ru to obtain partially active species, which inhibits the LOM and accelerates deprotonation for OER. The high electrical conductivity of allows to drive OER with low energy consumption. Furthermore, the work function adaptation between and Ru makes HER tend to the hydrogen spillover mechanism to achieve fast reaction kinetics. As expected, displays ultralow overpotentials of 8 mV and 150 mV at for HER and OER in acidic environments, respectively, with steady operation for 500 h . The excellent difunctional performance enables to drive water splitting at at a voltage of 1.44 V , which is also validated in pH-universal environments. PEM devices assembled with show lower cell voltages and higher stability than those of commercial
Fig. 7 | The performance of water splitting. a LSV polarization curves of , , and (loading amount: ) in for the acidic water splitting. b Stability tests of at , and the inset display the corresponding Faraday efficiency at . c Comparison of the cell voltage and stability of with those of recently reported bifunctional
catalysts for acidic water splitting. d Conceptual model of the PEM water electrolyzer flow cell using pure water as feedstock. e Steady polarization curves of Ru/ and (loading amount: ) for PEM water electrolyzer in pure water at . f Stability tests of for PEM water electrolyzer at . and robust operation at for 300 h . This outstanding performance validates the advancement of delicate support design for water electrolysis devices and opens new avenues for the rational construction of OER catalysts.
Methods
Material synthesis
Preparation of . Firstly, 1 g TiO 2 and 40 mg carbon black were ball-milled for 10 h to form a uniform mixture. Subsequently, the mixture was annealed at for 3.5 h to obtain powder.
Preparation of . Ru -loaded was prepared by simple wet reduction. First, and were evenly dispersed in 50 mL of deionized water. Subsequently, 7 mL of solution was slowly added dropwise into the mixture with vigorous stirring. After stirring for 4 h , the product was collected by centrifugation and dried under vacuum at for 12 h . The obtained powder was further annealed at , and for 1 h in the Ar atmosphere. The product was labeled and stored in the Ar-filled environment. The mass of was maintained at 60 mg , and the dosage of was changed to , and 60 mg to achieve different Ru loading.
Preparation of and . The preparation and storage methods of and were the same as those of . and TiO replaced to obtain and , respectively.
Material characterizations
XRD tests were carried out utilizing the Bruker D8 advance (Billerica) with a guaranteed scanning rate of . SEM images were captured using the Regulus 8100 (Hitachi). AC-TEM (JEM-ARM200F, JEOL)
was employed to investigate the fine structure of the materials. The TEM and HRTEM images were acquired using the Tecnai G2 F20 (FEI). XPS utilized Al Ka X-ray as the excitation source (Escalab 250Xi, Thermo Fisher Scientific). All narrow XPS spectra were obtained under the conditions of 20 eV pass energy and 0.05 eV energy step, calibrated by the peak of C located at 284.8 eV . UPS was performed using a VG Scienta R4000 analyzer (monochromatic He I light source of 20.2 eV ) with a 10 eV bias. XAFS measurements were conducted at the Taiwan Photon Source 44 A beamline quick-scanning X-ray absorption spectroscopy (Hsinchu). The XAFS spectra were collected at room temperature and analyzed using the Athena program. Inductively coupled plasma-mass spectrometry (ICP-MS) was carried out using the Optima 7300 DV instrument (PerkinElmer).
Electrochemical measurements
The electrochemical tests were executed using an electrochemical workstation (Autolab PGSTAT302, Metrohm). The potentials were referenced to a reversible hydrogen electrode (RHE). The electrochemical measurements were performed in a three-electrode electrochemical cell with Ar-saturated aqueous solution, and 1 M phosphate buffered solution (PBS). The active catalysts and binder (Polyvinylidene fluoride, PVDF) were mixed in a weight ratio of 7:1 with N-methyl-2-pyrrolidone (NMP) as the solvent. The catalyst ink was then drop-dried onto the carbon paper and dried for preparation of the working electrode until the catalyst loading was . Commercial Pt/C (20 wt. %) and were prepared as the working electrodes using the same method for comparison. A platinum foil and a carbon rod were used as the counter electrodes for OER and HER, respectively. The , and electrodes were employed as the reference electrode in acidic, alkaline, and neutral media, respectively. The CV tests at different scan rates were acquired within the vs. RHE range to determine
the . LSV curves were generated at a scan rate of with compensation. EIS was performed at the potentials of 1.5 V vs. RHE for OER and -0.5 V for HER, covering frequencies from 100 kHz to 0.1 Hz with an amplitude of 10 mV . In the pH -dependence measurement of OER, the electrolyte was prepared by adding the components of Britton-Robinson buffer ( 0.4 M each of phosphate, borate, and acetate) to a solution, and the pH was then adjusted to the desired value by addition of . All glassware was sonicated in ultrapure water directly before electrochemical treatment. Pulse voltammetry was performed while following the current over time. The potential was kept at a low potential ( vs. RHE), then switched and kept at a higher potential ( ) before returning to . This cycle was repeated while increasing from 1.42 V to 1.50 V in step and keeping unchanged. Charges related to the potential step were calculated by integrating the current pulse over time, accounting for the background current signal.
In-situ electrochemical characterizations
In-situ XAFS Measurements. In-situ X-ray absorption spectroscopy, encompassing XANES and EXAFS at both Ru -edge and -edge, was gathered in total-fluorescence-yield mode utilizing a silicon drift detector at the National Synchrotron Radiation Research Center (NSRRC) in Japan and Taiwan. The measurement, under the same conditions as the electrochemical characterization case in a typical three-electrode setup, took place in a specially designed Teflon container with a window sealed by Kempton tape. The scan ranges were kept in an energy range of and for Ru -edge (BL-12B2 at SPring-8, NSRRC) and Ti -edge (17C at Taiwan Light Source, NSRRC), respectively. The spectra were obtained by subtracting the baseline of the pre-edge and normalizing that of the post-edge. Fourier transform on -weighted EXAFS oscillations was employed for EXAFS analysis. All EXAFS spectra are presented without phase correction.
In-situ ATR-SEIRAS Measurements. ATR-SEIRAS measurements were performed by a Nicolet iS50 Fourier transform infrared spectrometer (FT-IR) spectrometer with a liquid nitrogen-cooled MCT detector and a fixed angle IR optical path. The spectral resolution of the measurements was , and 32 interferograms were added for each spectrum.
Membrane Electrode Assembly (MEA) measurements
PEM electrolyzer measurements. The membrane electrode assembly was prepared using Nafion 117 by the catalyst-coated membrane method with a geometric area of . The bifunctional active catalysts were dispersed in isopropanol, deionized water, and a Nafion ethanol solution ( ) to form an ink. Then, the ink was sprayed on both sides of PEM by the polytetrafluoroethylene (PTFE) transfer with an effective area of until the catalyst loading was . To avoid corrosion at oxidation potential, Ti felt was used as a gas diffusion layer (GDL) for the anode. Finally, the membrane with electrocatalysts coated, the anode GDL, and the cathode GDL (carbon paper) were hot pressed together to establish the MEA under with a pressure of 10 MPa for 5 min . The commercial and were used in the same way as the cathode and anode respectively for comparison. Pure water was sent to the cathode and anode by a peristaltic pump at a speed of 40 rpm . Before testing, the prepared MEA was activated in potentiostatic mode at a cell voltage of 2 V . Subsequently, the electrolyzer was operated at , and the hydrogen and oxygen in the electrolyte were removed with Ar to avoid the possible influence of the bubbles formed on the long-term stability of the electrodes. Steadystate polarization curves were obtained by a direct-current power (ITECH, IT-M3223). The cell voltages at different current densities were recorded.
AEM electrolyzer measurements. PiperION-A60 was utilized to construct the AEM electrolyzer. The catalyst ink of AEM was the same as that of PEM, except the Nafion ethanol solution was replaced by PiperION A ionomer ethanol ( ). The ink was supported on carbon paper, and Ti felt with the effective area of until the catalyst loading was . To improve the interfacial contact between the catalyst layer and AEM, a small amount of PiperION A ionomer ethanol solution was sprayed on the surface of the anode and cathode catalyst layers and then dried at room temperature for 48 h . Finally, the AEM and the GDLs with catalyst loading were hot pressed together to establish the MEA under with a pressure of 10 MPa for 5 min . The commercial and were also used in the same way as the cathode and anode respectively for comparison. One M KOH aqueous solution was sent to the cathode and anode by a peristaltic pump at a speed of 40 rpm . The electrolyzer was operated at .
Computational methods
All DFT calculations were conducted through the Vienna Ab initio Simulation Package (VASP). The computations utilized the projector augmented wave (PAW) pseudopotential with the PBE generalized gradient approximation (GGA) exchange-correlation function. The plane wave basis set had a cutoff energy of 500 eV . K-sampling in the calculation of adsorption energy used a Monkhorst-Pack mesh of , while in the calculation of DOS, a mesh was employed. Spin polarization was applied to all structures, and complete relaxation of all atoms was ensured with an energy convergence tolerance of per atom. The final force on each atom was maintained below . Porbaix diagrams were calculated using atomic simulation environment (ASE) with input formation energy by DFT calculations of bulk and surface models.
In the construction of heterojunctions, the minimum lattice mismatch is used as a criterion to construct the interface contact model between Ru and different titanium oxides. The ( 001 ) facet of TiO contacts the ( 010 ) facet of Ru . The ( ) facet of contacts the ( 210 ) facet of Ru. The ( ) facet of contacts the ( 001 ) facet of Ru.
The adsorption energy of reaction intermediates can be computed using the following Eq. (1):
Where ads , and ( ) is the binding energy, is the zero-point energy change, is the entropy change. In this work, the values of and were obtained by vibration frequency calculation.
The Gibbs free energy of the five reaction steps can be calculated by the following Eqs. (2-7):
In this work, were calculated at .
Data availability
The data generated in this study are provided in the Source Data file.
References
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609-616 (2023).
Zhang, Y. et al. Hydrogen-bond regulation of the microenvironment of Ni(II)-porphyrin bifunctional electrocatalysts for efficient overall water splitting. Adv. Mater. 35, 2210727 (2023).
Hao, S. et al. Torsion strained iridium oxide for efficient acidic water oxidation in proton exchange membrane electrolyzers. Nat. Nanotechnol. 16, 1371-1377 (2021).
Zheng, X. et al. Tailoring a local acid-like microenvironment for efficient neutral hydrogen evolution. Nat. Commun. 14, 4209 (2023).
Chen, Z. et al. Stabilizing Pt single atoms through Pt-Se electron bridges on vacancy-enriched nickel selenide for efficient electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 62, e202308686 (2023).
Zhang, X.-L. et al. Efficient acidic hydrogen evolution in proton exchange membrane electrolyzers over a sulfur-doped marcasitetype electrocatalyst. Sci. Adv. 9, eadh2885 (2023).
Hao, Y. et al. Methanol upgrading coupled with hydrogen product at large current density promoted by strong interfacial interactions. Energy Environ. Sci. 16, 1100-1110 (2023).
Shi, Z. et al. Enhanced acidic water oxidation by dynamic migration of oxygen species at the catalyst/support interfaces. Angew. Chem. Int. Ed. 61, e202212341 (2022).
Ruiz Esquius, J. et al. Lithium-directed transformation of amorphous iridium (oxy)hydroxides to produce active water oxidation catalysts. J. Am. Chem. Soc. 145, 6398-6409 (2023).
Chen, M. et al. Recent progress in transition-metal-oxide-based electrocatalysts for the oxygen evolution reaction in natural seawater splitting: a critical review. eScience 3, 100111 (2023).
Yang, X. et al. IrPd nanoalloy-structured bifunctional electrocatalyst for efficient and pH-universal water splitting. Small 19, 2208261 (2023).
Li, Y. et al. Arming Ru with oxygen-vacancy-enriched subnanometer skin activates superior bifunctionality for pH-universal overall water splitting. Adv. Mater. 35, 2206351 (2023).
Li, J. et al. Boosting electrocatalytic activity of Ru for acidic hydrogen evolution through hydrogen spillover strategy. ACS Energy Lett. 7, 1330-1337 (2022).
Ling, C., Shi, L., Ouyang, Y., Zeng, X. C. & Wang, J. Nanosheet supported single-metal atom bifunctional catalyst for overall water splitting. Nano Lett. 17, 5133-5139 (2017).
Chen, J. et al. Reversible hydrogen spillover in Ru-WO enhances hydrogen evolution activity in neutral pH water splitting. Nat. Commun. 13, 5382 (2022).
Wang, N. et al. Doping shortens the metal/metal distance and promotes OH coverage in non-noble acidic oxygen evolution reaction catalysts. J. Am. Chem. Soc. 145, 7829-7836 (2023).
Zheng, X. et al. Ru-Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Wei, J. et al. Site-specific metal-support interaction to switch the activity of Ir single atoms for oxygen evolution reaction. Nat. Commun. 15, 559 (2024).
Monai, M. et al. Restructuring of titanium oxide overlayers over nickel nanoparticles during catalysis. Science 380, 644-651 (2023).
Wu, Z. et al. Microwave synthesis of Pt clusters on black with abundant oxygen vacancies for efficient acidic electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 62, e202300406 (2023).
Deng, L. et al. Electronic modulation caused by interfacial Ni-O-M ( ) bonding for accelerating hydrogen evolution kinetics. Angew. Chem. Int. Ed. 60, 22276-22282 (2021).
Wang, W., Wang, Z., Hu, Y., Liu, Y. & Chen, S. A potential-driven switch of activity promotion mode for the oxygen evolution reaction at interface. eScience 2, 438-444 (2022).
Park, J. E. et al. Three-dimensional unified electrode design using a NiFeOOH catalyst for superior performance and durable anionexchange membrane water electrolyzers. ACS Catal. 12, 135-145 (2022).
Song, W. et al. Upscaled production of an ultramicroporous anionexchange membrane enables long-term operation in electrochemical energy devices. Nat. Commun. 14, 2732 (2023).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212-222 (2021).
Kosmala, T. et al. Operando visualization of the hydrogen evolution reaction with atomic-scale precision at different metal-graphene interfaces. Nat. Catal. 4, 850-859 (2021).
Wang, X. et al. Electronic structure modulation of by enriched with oxygen vacancies to boost acidic evolution. ACS Catal. 12, 9437-9445 (2022).
Lin, C. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021).
Shi, X. et al. Efficient and stable acidic water oxidation enabled by low-concentration, high-valence iridium sites. ACS Energy Lett. 7, 2228-2235 (2022).
Liu, X. et al. Restructuring highly electron-deficient metal-metal oxides for boosting stability in acidic oxygen evolution reaction. Nat. Commun. 12, 5676 (2021).
Mahdavi-Shakib, A. et al. The role of surface hydroxyls in the entropy-driven adsorption and spillover of on catalysts. Nat. Catal. 6, 710-719 (2023).
Han, H. G., Choi, J. W., Son, M. & Kim, K. C. Unlocking power of neighboring vacancies in boosting hydrogen evolution reactions on two-dimensional NiPS monolayer. eScience, 100204 https://doi. org/10.1016/j.esci.2023.100204 (2023).
Xia, J. et al. hierarchical bifunctional catalyst anchored on graphene aerogel toward flexible and high-energy Li-S batteries. ACS Nano 16, 19133-19144 (2022).
Yang, B. et al. Flatband towards extraordinary solar steam generation. Nature 622, 499-506 (2023).
Dai, J. et al. Single-phase perovskite oxide with super-exchange induced atomic-scale synergistic active centers enables ultrafast hydrogen evolution. Nat. Commun. 11, 5657 (2020).
Ioroi, T., Siroma, Z., Yamazaki S.-i. & Yasuda K. Electrocatalysts for PEM fuel cells. Adv. Energy Mater. 9, 1801284 (2019).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659-23669 (2023).
He, Q. et al. Confining high-valence iridium single sites onto nickel oxyhydroxide for robust oxygen evolution. Nano Lett. 22, 3832-3839 (2022).
Hu, F. et al. Lattice-matching formed mesoporous transition metal oxide heterostructures advance water splitting by active bridges. Adv. Energy Mater. 12, 2200067 (2022).
Zhang, B. et al. Enhanced spatial charge separation in a niobium and tantalum nitride core-shell photoanode: In situ interface bonding for efficient solar water splitting. Angew. Chem. Int. Ed. 62, e202305123 (2023).
Chen, R. et al. Promoting the efficiency and selectivity of -to reduction on active sites via preferential glycol oxidation with holes. P. Natl Acad. Sci. USA 120, e2312550120 (2023).
Li, M. et al. Revealing the regulation mechanism of interfacial chemical bonding for improving hydrogen oxidation reaction. ACS Catal. 11, 14932-14940 (2021).
Dai, J. et al. Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis. Nat. Commun. 13, 1189 (2022).
Fu, H. Q. et al. Hydrogen spillover-bridged volmer/tafel processes enabling ampere-Level current density alkaline Hydrogen evolution reaction under low overpotential. J. Am. Chem. Soc. 144, 6028-6039 (2022).
Zheng, T. et al. Intercalated iridium diselenide electrocatalysts for efficient pH-universal water splitting. Angew. Chem. Int. Ed. 58, 14764-14769 (2019).
Li, J. et al. A fundamental viewpoint on the hydrogen spillover phenomenon of electrocatalytic hydrogen evolution. Nat. Commun. 12, 3502 (2021).
Hou, L. et al. Electronic and lattice engineering of ruthenium oxide towards highly active and stable water splitting. Adv. Energy Mater. 13, 2300177 (2023).
Nong, H. N. et al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 587, 408-413 (2020).
Yang, L. et al. A highly active, long-lived oxygen evolution electrocatalyst derived from open-framework iridates. Adv. Mater. 35, 2208539 (2023).
Hao, Y. et al. Switchingsed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659-23669 (2023).
Du, K. et al. Interface engineering breaks both stability and activity limits of for sustainable water oxidation. Nat. Commun. 13, 5448 (2022).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304-313 (2019).
Zhang, Q. et al. Solid-solution alloy nanoparticles of a combination of immiscible Au and Ru with a large gap of reduction potential and their enhanced oxygen evolution reaction performance. Chem. Sci. 10, 5133-5137 (2019).
Yang, Y. et al. Hierarchical nano assembly of as a highly efficient electrocatalyst for overall water splitting in a wide pH range. J. Am. Chem. Soc. 141, 10417-10430 (2019).
Zheng, X. et al. Enriched d-band holes enabling fast oxygen evolution kinetics on atomic-layered defect-rich lithium cobalt oxide nanosheets. Adv. Funct. Mater. 32, 2200663 (2022).
Tan, H. et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 13, 2024 (2022).
Hammer, B. & Norskov, J. K. Why gold is the noblest of all the metals. Nature 376, 238-240 (1995).
Ruban, A., Hammer, B., Stoltze, P., Skriver, H. L. & Nørskov, J. K. Surface electronic structure and reactivity of transition and noble metals. J. Mol. Catal. A: Chem. 115, 421-429 (1997).
Bornet, A. et al. Influence of temperature on the performance of carbon- and ATO-supported oxygen evolution reaction catalysts in a gas diffusion electrode setup. ACS Catal. 13, 7568-7577 (2023).
Kwon, J. et al. Tailored electronic structure of Ir in high entropy alloy for highly active and durable bifunctional electrocatalyst for water splitting under an acidic environment. Adv. Mater. 35, 2300091 (2023).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Hjorth Larsen, A. et al. The atomic simulation environment-a Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075141), Scientific and Technological Innovation Special Fund for Carbon Peak and Carbon Neutrality of Jiangsu Province (BK20220039), and Natural Science Foundation of Jiangsu Province (BK20210311). The authors thank the National Synchrotron Radiation Research Center (NSRRC), Hsinchu, Taiwan.
Author contributions
S.Z. performed the experiments, collected the data, and analyzed the data. S.Z. and L.D. wrote the manuscript. T.X. performed the experiments. S.H. and W.Z. performed the in-situ XAFS test. S.L. and F.H. analyzed the data and wrote the manuscript. C.K. and H.C. performed the XAFS measurement. S.P. designed the study, analyzed the data, and revised the manuscript.
Correspondence and requests for materials should be addressed to Shengjie Peng.
Peer review information Nature Communications thanks Xue Yong, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
¹College of Materials Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing 210016, China. Department of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu 300, Taiwan. Department of Materials Science and Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan. e-mail: pengshengjie@nuaa.edu.cn