بناء محفزات كهربائية فائقة التنسيق من الكبريت والأكسجين لإنتاج H2O2 الانتقائي والتراكمي Constructing sulfur and oxygen super-coordinated main-group electrocatalysts for selective and cumulative H2O2 production
التحضير المباشر لبيروكسيد الهيدروجينتقدم تفاعل اختزال الأكسجين بواسطة إلكترونين بديلاً متزايدًا للعملية التقليدية المستهلكة للطاقة والمعروفة بعملية الأنثراكوينون للتطبيقات في الموقع. ومع ذلك، فإن اعتمادها معوق حاليًا بسبب ضعفالانتقائية والتقليلالعائد الناتج عن التتابعتقليل أو تفاعلات فينتون. هنا، مسترشدين بالحسابات النظرية، نسعى لتجاوز هذا التحدي من خلال تنشيط محفز ذرة واحدة من الرصاص من المجموعة الرئيسية عبر استراتيجية هندسة بيئة محلية باستخدام هيكل فائق التنسيق من الكبريت والأكسجين. يظهر المحفز من المجموعة الرئيسية، الذي تم تصنيعه باستخدام تقنية التحلل الحراري المدعومة بنقاط الكربون، كثافة تيار صناعية تصل إلىوالمتراكم المرتفعتركيزات بكفاءات فارادائية ملحوظة. توضح كل من النتائج التجريبية والمحاكاة النظرية أن التناسق الفائق للكبريت والأكسجين يوجه جزءًا من الإلكترونات من مواقع الرصاص الرئيسية إلى ذرات الأكسجين المنسقة، مما يؤدي إلى تحسين طاقة ارتباط *OOH وزيادة نشاط تقليل الأكسجين. يكشف هذا العمل عن طرق جديدة للتخفيف من تحدي إنتاج النقص فيالتحليل الكهربائي من خلال التصميم العقلاني لمحفزات المجموعات الرئيسية.
بيروكسيد الهيدروجينتحتل ( ) مرتبة بين المواد الكيميائية الأساسية والضرورية، حيث يتم استخدامها على نطاق واسع في الصناعات الكيميائية والطبية، بالإضافة إلى معالجة البيئة.نظرًا للنمو المستمر في الاحتياجات الصناعية المرتبطة مباشرة، من المتوقع أن السوق العالميةسيقترب الطلب في السوق من حوالي 6 ملايين طن بحلول. حاليًا، أكثر من من جميعيتم تصنيعه صناعيًا من خلال عملية دورة الأنثراكوينون، التي تعاني من استهلاك مكثف للطاقة، ونفايات عضوية كبيرة، ومخاوف تتعلق بالسلامة ناتجة عن عدم الاستقرار.أثناء النقل والتخزين. في تناقض واضح، التفاعل الكهروكيميائي ذو الإلكترونين (تفاعل اختزال الأكسجين (ORR) يوفر مسارًا أكثر مباشرة لـالإنتاج في الموقع تحت ظروف البيئة المحيطة. عادةً، العديد من الكاثود المواد تفضل المنافسةORR إلىبدلاً منأو، مما أدى إلى انخفاضي yieldsمحفزات سبائك المعادن النبيلة ذات المواقع التفاعلية المعزولة (على سبيل المثال،تُكتشف أنها مفيدة لامتصاص “نهاية إلى نهاية” (نوع بولينغ) لـ، الذي يقلل منكسر الروابط، تسهيلعملية ORR لـإنتاجومع ذلك، فإن التكلفة الباهظة وندرة الموارد من المعادن النبيلة تعيق بشدة تنفيذها على نطاق واسع. لذلك، من الضروري تطوير محفزات ذات انتقائية عالية وغير نبيلة.التحليل الكهربائي
لقد أظهرت المحفزات أحادية الذرة غير المتجانسة (SACs)، التي تتميز بمواقع نشطة معزولة وهياكل إلكترونية مميزة، وعدًا كبيرًا كمواد لتحويل الطاقة والتحول الكيميائي.. بشكل خاص، المعادن الانتقالية
SACs ذات التعريف الواضحتم التعرف على الأجزاء كعوامل تحفيز كهربائية استثنائية لـإنتاج. أخذ الـكمثال على ذلك، حيث إن ضبط هيكل التنسيق والتكوين الذري المحيط يعمل بشكل فعال على تحسين الوسائط الأكسجينية ويحدد الانتقائية تجاهتركيب. يمكن أن يُعزى ذلك بشكل أساسي إلى خصائصها الإلكترونية الفريدة، التي تظهر شغلًا جزئيًاالإلكترونات التي تمنح قدرة قوية على الامتصاص والتفعيلكما تم التنبؤ به وتوضيحه بواسطةنظرية مركز النطاقللأسف، فإن مواقع المعادن الانتقالية التفاعلية تظهر أيضًا تفاعلات قوية مع المركبات المصنعة.تحريض غير مرغوب فيهتفاعلات الاختزال ) ضمن نطاق جهد ORR. وبالتالي، فإن الناتج يتعرض للتحلل، مما يضعف بشكل كبيرمحصول. بالإضافة إلى ذلك، قد تسهم تفاعلات فنتون الناتجة عن المعادن الانتقالية في تقليلإنتاج وتحمل متانة الأقطاب الكهربائية. ولهذا الغرض، قد تستنفد القوة القوية لامتصاص مواقع المعادن الانتقالية للأنواع المحتوية على الأكسجين المتراكمة، مما يؤدي إلى تقليص الإنتاج الكلي. من المعقول أن نتوقع أن تحسين اختيارية ORR للمواد الحفازة الأقل تفاعلاً بطبيعتهاقد تؤدي التحلل إلى نتائج أعلىالكميات، على الرغم من أن هذا لا يزال تحديًا واعدًا ولكنه هائل لـالتحليل الكهربائي. متميز عنمعادن الانتقال من المجموعة -، والمعادن من المجموعة الرئيسية ذات الشواغر المملوءة بالكامل-مدارات ( تعتبر التكوينات الإلكترونية عمومًا أقل كفاءة تحفيزية لعملية نقل الإلكترون في التفاعل التحفيزي.يجب أن تتفوق المعادن الرئيسية بشكل طبيعي على المعادن الانتقالية من حيث النشاط الخامل لـوتفاعلات شبيهة بفنتون، بسبب نقص الجمع بين المدارات المضيفة الفارغة والمشغولة. ومن المثير للاهتمام، أن المحفزات من المجموعة الرئيسية قد أظهرت أنها مرشحة واعدة للتفاعلات الكهروكيميائية، حيث تتمتع بمتانة محسّنة من خلال التخفيف من تفاعلات فنتون الناتجة عن المعادن الانتقالية.. وبالتالي، يجب أن يكون من الممكن تحقيق الانتقاء بشكل متزامنORR لـالإنتاج وتخفيف التحلل المتتالي من خلال تطوير المجموعة الرئيسيةORR SACs.
مدفوعين بالتحليل أعلاه، توقعنا أولاً عملية ORR على SACs الرصاص (Pb) مع بيئات تنسيق محلية متنوعة باستخدام حسابات نظرية الكثافة الوظيفية (DFT). وبناءً عليه، تم وضع استراتيجية متعددة الاستخدامات لتنظيم الميكروبيئات لمواقع المعادن في المجموعة الرئيسية من خلال تعديل التنسيق الأول بدقة مع الكبريت.وأكسجين. ثم قمنا بتصنيع المحفزات الأحادية للمجموعة الرئيسية Pb SACs (Pb SA/OSC) باستخدام نهج التحلل الحراري المدعوم بنقاط الكربون، حيث تم التحكم بشكل منطقي في نسبة S/O لنقاط الكربون. أكدت التحليلات المجهرية المتقدمة وطيف الامتصاص الدقيق للأشعة السينية من السنكروترون التنظيم الناجح لتنسيق المحفزات الأحادية للمجموعة الرئيسية عبر التنسيق المزدوج للكبريت والأكسجين.تم تقييم أداء ORR لساعة Pb SA/OSC المصنوعة حديثًا باستخدام تقنية القطب الدائري الدوار. علاوة على ذلك، لاستكشاف إمكانية التطبيق العملي،تم تجميعه في جهاز تدفق وظيفي لـالإنتاج عند التيارات الصناعية. هذا المحفز المميز من المجموعة الرئيسية Pb قد يبشر بسبيل جديد لاستغلال عناصر المجموعة الرئيسية في تصميم المحفزات الكهروكيميائية لـتركيب.
النتائج
هندسة التنسيق لـ Pb SA/OSC
قمنا أولاً بدراسة امتصاص وسائط تفاعل اختزال الأكسجين على نماذج ذرات الرصاص الفردية مع بيئات تنسيق محلية متنوعة للحصول على رؤى حولاختيارية ORR باستخدام حسابات DFT. تم اختيار المحفزات القائمة على الرصاص كمحفزات نموذجية من المجموعة الرئيسية، حيث أظهرت مواد الرصاص نشاطًا عاليًا في تفاعلات التحفيز الكهربائي المتنوعة.تشير الأبحاث السابقة إلى أن التفاعلات الحفزية لمواقع المعادن تتأثر ببيئات التنسيق الخاصة بها بسبب التغيرات في الهيكل الإلكتروني.. بالإضافة إلى ذلك، تم الإبلاغ في الأدبيات أن أكثر أرقام التنسيق شيوعًا لمواقع الرصاص في المجموعة الرئيسية هي 4 وفي هذه الحالة،
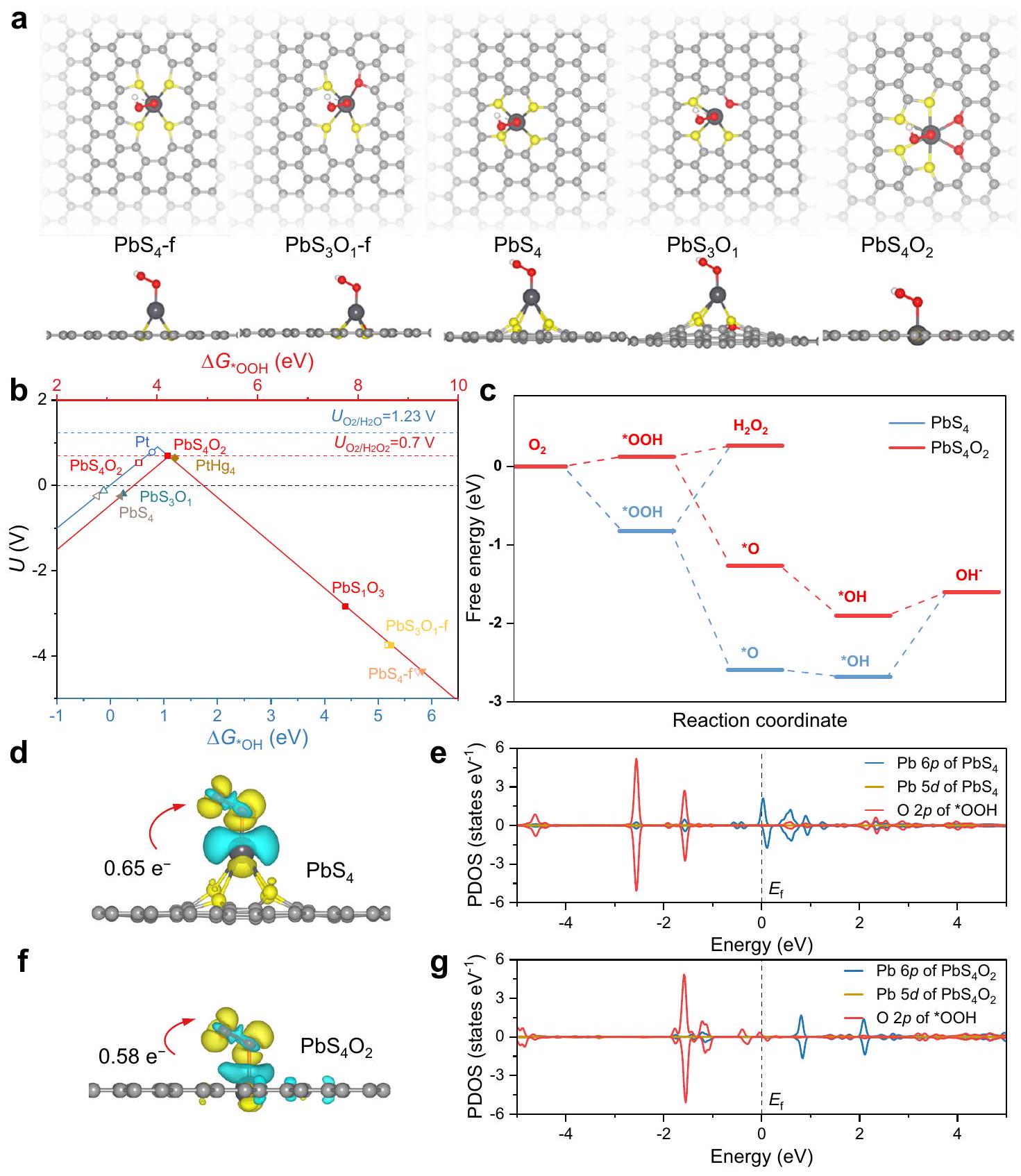
تم بناء نماذج هياكل Pb SA/OSC من خلال دعم ذرات Pb على الجرافين مع تركيبات مختلفة من ذرات S و O المنسقة (الشكل 1a والشكل التكميلي 1). تم تحسين هندسةيعرض هيكل تنسيق مستوي مع مسافات من ( ) و (الجدول التكميلي 1). على النقيض، فإن المسافات لـوفي الهياكل الأخرى لـ Pb SACs، تظهر المسافات بين الذرات أطول. وبناءً عليه، فإن طاقات الامتزاز لـوعلى المحددوتم حساب هياكل Pb SACs الأخرى. العلاقات بينوالإمكانات المحدودة لـتم إنشاء ORR لتصوير علاقات التدرج (الشكل 1ب)، مما يشير إلى أنيقع بالقرب من قمة البركان المحتمل المحدود، مشابه لـمحفز. وبالتالي،يوفر انتقائية عالية لـتكوين بجهد منخفض قدره 0.006 فولت. تقع مواقع الرصاص الأخرى إما على الجانب الأيسر أو الأيمن من مخططات البركان (الشكل التكميلي 2)، مما يشير إلى أن امتصاص *OOH قوي جدًا أو ضعيف جدًا، على التوالي. لتقديم وصف أكثر دقة لتفاعل ORR، يتم تحديد العلاقات بينوالإمكانات المحدودة لـتم فحص ORR. الجهد المحدد لـكان بعيدًا عن الجهد التوازني لـORR (1.23 فولت)، مما يوحي بأنغير نشط من أجل التخفيض لـإلىعلاوة على ذلك، يكشف مخطط الطاقة الحرة لتفاعل اختزال الأكسجين (ORR) أن امتصاص * OOH علىمناسب لتشكيل وإزالةبينما ذلك علىأقوى، مما يشير مرة أخرى إلى الانتقائية العالية لـORR على الـالهيكل (الشكل 1ج).
تُعتبر قوة امتصاص *OOH من العوامل الأساسية التي تحكم انتقائية تفاعل اختزال الأكسجين (ORR) في المحفزات. على وجه التحديد، يعتمد ارتباط *OOH على مستوى الطاقة الإلكترونية لموقع النشاط والنقل الشحني المقابل عند واجهة التحفيز. كشفت تحليل شحنة بادير أن *OOH تحمل شحنة سالبة مع عدد أقل من الإلكترونات المنقولة من ( ، الشكل 1د) مقارنة بـ ( ، الشكل 1f)، متوافق مع القوة المحسوبة لامتصاص *OOH. تُظهر خريطة الألوان لشحنة بادر على كل ذرة أن الإلكترونات المقدمة من موقع الرصاص تُنقل ليس فقط إلى المادة الممتصة ، ولكن أيضًا إلى ذرات الأكسجين المنسقة لـ (الشكل التكميلي 3). على النقيض من ذلك، فإن إلكترونات موقع الرصاص فيلا تُنقل إلى ذرات الكبريت المنسقة، مما يؤدي إلى اكتساب *OOH الم adsorb بقوة مزيد من الإلكترونات. تُظهر مخططات كثافة الحالة المتوقعة (PDOS) تداخل مستويات الطاقة لعنصر الرصاص.المدارات ومدارات * OOH (الشكل 1e)، مما يعني أن امتصاص * OOH يساهم فيهمدارات. للم يُلاحظ أي منطقة متداخلة بالقرب من مستوى فيرمي (الشكل 1g)، مما يشير إلى أنتم التحكم في المدارات بواسطة ذرة الأكسجين المنسقة. تأثير بيئات التنسيق على تحويل * OOH إلىيمكن فهمه ليس فقط من خلال ضعف الـالرابطة، ولكن أيضًا من خلال تشديد الـرابطة فيالممتص على موقع الرصاص (الجدول التكميلي 2). لذلك، كشفت حسابات DFT أن تشكيل وحدات الرصاص المتناسقة بشكل فائق من الكبريت والأكسجين هو مصدر الاستثنائيةانتقائية ORR لـ Pb SA/OSC، كتفاعل مناسب بينمداراتتؤدي المدارات إلى قوة امتصاص مثالية لمتوسطات * OOH.
تركيب وتوصيف Pb SA/OSC
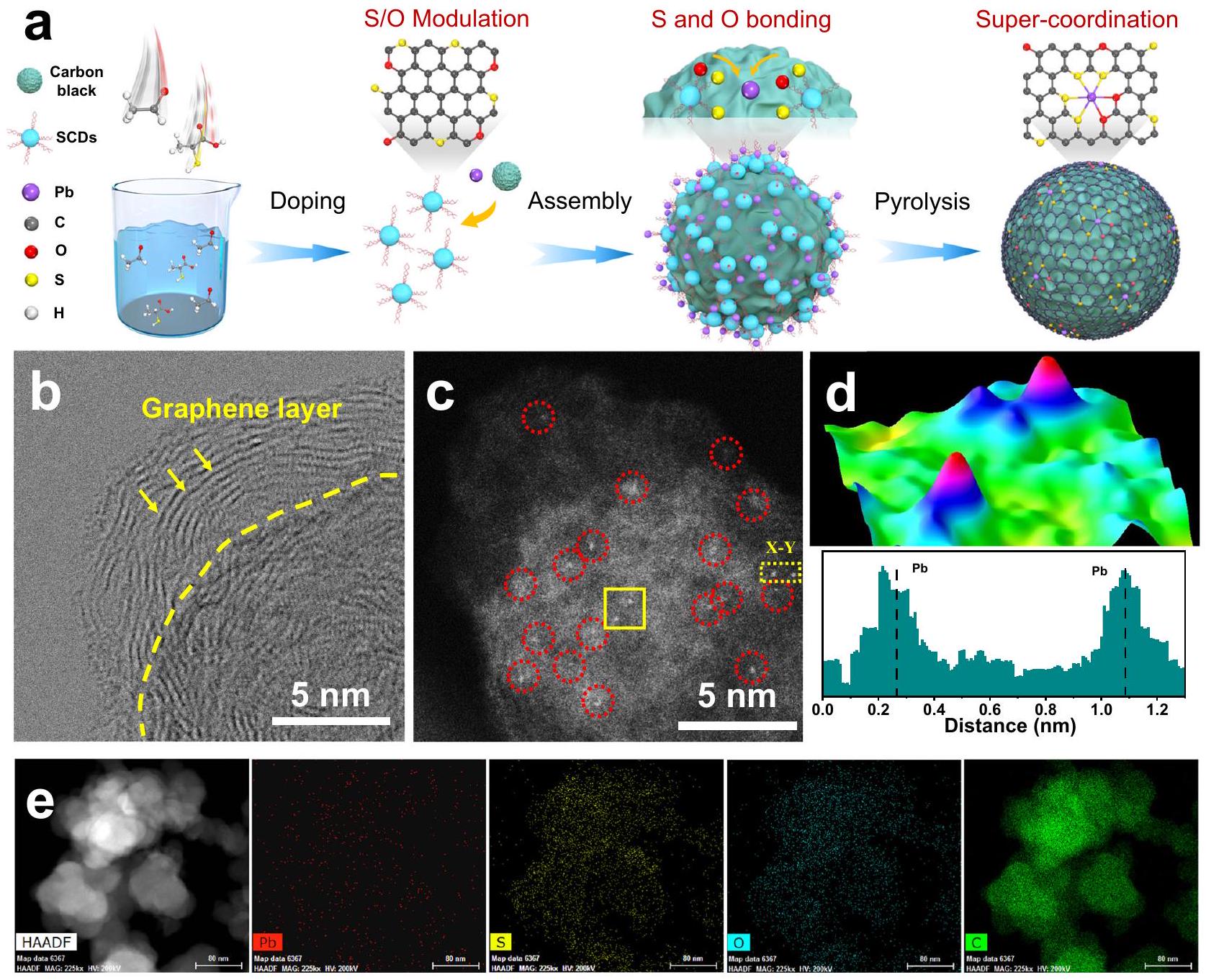
لتأكيد التنبؤ النظري الاستثنائيتم تثبيت الذرات الفردية من الرصاص (Pb SA/OSC) على دعائم كربونية باستخدام نهج التحلل الحراري المدعوم بنقاط كربونية. يتم توضيح عملية التحضير التخطيطية لـ Pb SA/OSC في الشكل 2a. تم أولاً تحضير نقاط الكربون الوظيفية بالكبريت والأكسجين (SCDs) باستخدام نسب متغيرة من المواد الأولية المحتوية على الكبريت والأكسجين، مما يوفر منصة متعددة الاستخدامات لتعديل البيئات الذرية المحلية لـ Pb SACs. ومن الجدير بالذكر أن التفاعل الكيميائي القوي بين المعادن وذرات الكبريت المدعومة في SCDs منح تشكيل روابط معدنية-كبريت قوية ومستقرة حرارياً.
الشكل 1 | النشاط النظري لتفاعل الأكسدة الاختزالية للأكسجين (ORR) لـ Pb SA/OSC. أ. وجهات نظر علوية وجانبية لعينات *OOH الممتصة على Pb SA/OSC. الأنماط الهيكلية التي تم اعتبارها في نموذج المحفز لـ Pb SA/OSC، بما في ذلك الهياكل في المستوى.والهياكل الرباعيةو. يتم الإشارة إلى التكوينات بـ نسبة الذرات والنوع، مع ” ” تشير إلى وجود الكبريت من نوع الثيوفين.مخططات البراكين لـوORR على مختلفو، مع رسم الإمكانية المحدودة كدالة لـ (المحور الأفقي الأزرق) و (أحمر
المحور الأفقي). إمكانيات التوازن لـوهي 1.23 فولت و 0.7 فولت، على التوالي. ج مخطط الطاقة الحرة لتفاعل اختزال الأكسجينوعند جهد مطبق يساوي صفر في وسط قلوي. د، ف مخططات كثافة الشحنة التفاضلية لأنواع * OOH الممتصة علىو، حيث تمثل المناطق الزرقاء والصفراء فقدان واكتساب الإلكترونات، على التوالي. تم أخذ الأسطح المتساوية عند كثافة شحنةبور. (على سبيل المثال) مخططات PDOS لـومحفزات مع *OOH. تخفيف تجمع مواقع المعادنتمت معالجة كاتيونات الرصاص من المجموعة الرئيسية بشكل كافٍ مع مجموعات الوظائف S و O في SCDs، مما أدى إلى تشكيل مركب تنسيق معدني، تم تجميعه على ركائز من الكربون الأسود عبرالتفاعلات. لذلك، فإن الترابط بين المعادن والكبريت عند الواجهة بين أيونات الرصاص (Pb) وSCDs قد ربط وثبت المواقع الرئيسية، مما قدم سلف معدني موزع على المستوى الذري. ثم تم الحصول على SACs الرصاص الرئيسية مع تنسيق مزدوج مميز من الكبريت والأكسجين من خلال معالجة التحلل الحراري تحت جو واقٍ من غاز الأرجون المتدفق. للمقارنة، تم إعداد سلسلة من عينات التحكم بتنسيقات متباينة. تمت أيضًا تخليق البيئات تحت ظروف متطابقة بخلاف ذلك ولكن باستخدام سوائل كربونية مختلفة. وتم الإشارة إليها على أنهاو، على التوالي.
كشفت صور المجهر الإلكتروني الناقل (TEM) أن Pb SA/OSC احتفظت إلى حد كبير بالتكوين الكروي للفحم الأسود، على الرغم من حجمها الأكبر قليلاً (الأشكال التكميلية 8a، b). كما هو موضح في الشكل 2b، كانت العينة المستخرجة تمتلك هيكل جرافين طبقي نما بشكل متجانس على ركائز الفحم الأسود، دون الكشف عن أي جزيئات نانوية تحتوي على الرصاص. وبالمثل، تم إجراء الملاحظات في صورة المجهر الإلكتروني الناقل بتقنية الحقل المظلم بزاوية عالية (HAADF-STEM).
الشكل 2 | الشكل والمظهر لـ Pb SA/OSC. أ رسم تخطيطي لتحضير Pb SA/OSC.صورة STEM وصورة AC HAADF-STEM لـتوافق دالة غاوسي المتداخلة مع الذرات لتخطيط المربع من (ج)
ملف الكثافة على طولفي (ج). خريطة العناصر EDS لـ Pb SA/OSC، مما يشير إلى توزيعات متجانسة لـوعناصر C. (الشكل التوضيحي 8c) أكد الاستنتاج المذكور أعلاه. بالإضافة إلى ذلك، لم تُلاحظ أي قمم بلورية مميزة للرصاص المعدني أو PbS في نمط حيود الأشعة السينية (الشكل التوضيحي 9) لـ Pb SA/OSC، مما يتماشى مع نتائج TEM السابقة. ومن الجدير بالذكر أن نتائج رامان (الشكل التوضيحي 10) كشفت عن الميزة المميزة “وأشرطة من المواد الكربونية الموصلّة )، مما يشير إلى وجود عيوب وفيرة في المواد الكربونية المستمدة من SCDs، مما يسهل تثبيت الذرات المعدنية المعزولة . تم دراسة الهيكل المسامي لـ Pb SA/OSC بواسطة طريقة بروناوير-إيميت-تيلر (BET)، حيث أظهر مساحة سطح محددة كبيرة وفقًا لطريقة BET (الشكل التكميلي 11). علاوة على ذلك، أظهرت عينات OSC و Pb SA/OC و Pb SA/OSC2.5 و Pb SA/OSC-7.5 أشكالًا متشابهة، وبلورات كربونية وعيوب، مما يدل على التحضير الناجح لمجموعات Pb SACs الرئيسية (الأشكال التكملية 10 و 13-15). يمكن مراقبة التوزيع الأحادي لمركبات Pb مباشرةً بواسطة HAADF-STEM المصحح للانحراف (AC HAADF-STEM). كما هو موضح في الشكل 2c والشكل التكميلي 16، يمكن تمييز النقاط الساطعة الفردية (المعلمة بدوائر حمراء) التي تتوافق مع ذرات Pb المعزولة بوضوح عن مصفوفة الكربون. تم تأكيد التوزيع الذري لـ Pb في Pb SA/OSC بشكل أكبر من خلال رسم خرائط تركيب دالة غاوسي المتداخلة وتحليل ملف الكثافة (الشكل 2d). أظهرت خرائط التحليل الطيفي بالأشعة السينية المشتتة للطاقة (EDS) من الشكل 2e أن و C على الدعامات كانت موزعة بشكل متساوٍ. علاوة على ذلك، تم قياس المحتوى الفعلي للرصاص على أنهعبر تحليل طيف الانبعاث الضوئي للبلازما المقترنة بالحث (ICP-MS).
تحليل البنية الذرية والإلكترونية
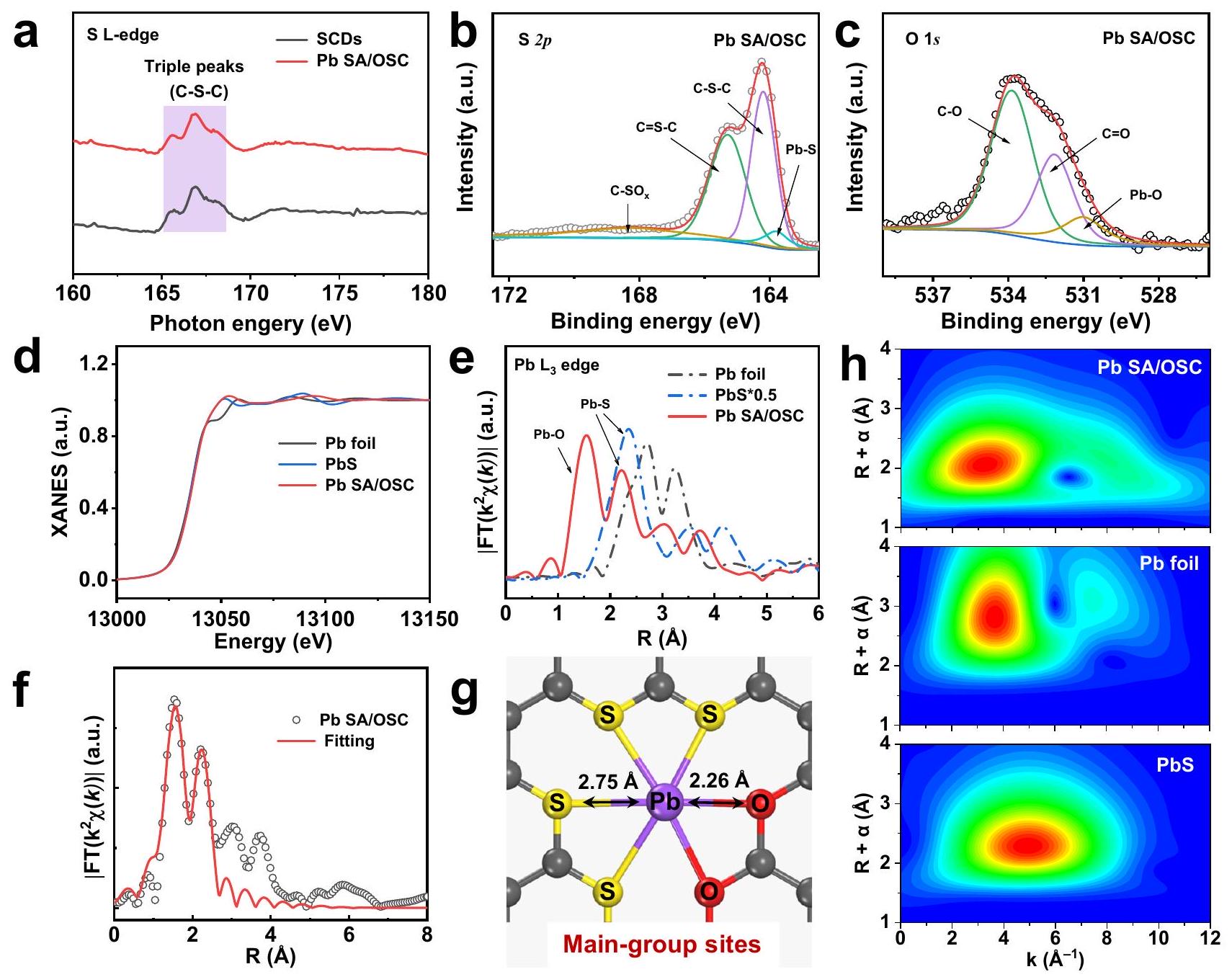
لتحليل الهيكل الإلكتروني والذري المحلي لمجموعات الرصاص SACs، تم إجراء امتصاص الأشعة السينية الناعمة القريبة من الحافة (XANES) باستخدام السنكروترون. كان طيف XANES عند حافة S L (الشكل 3a) لمجموعة Pb SA/OSC مهيمنًا بثلاثة قمم ضمن النطاقالمتوافقة مع أنواع التنسيق C-S-C، والتي تتوافق مع نتائج عينة SCDs. هذه الملاحظة اقترحت بشكل مقنع تثبيت الكبريت (S) في الإطار الكربوني، وهو ما تم التحقق منه من خلال نتائج مسح طيف الإلكترون السيني (XPS) (الشكل التكميلي 17). في الوقت نفسه، أظهر طيف حافة كربون (الشكل التكميلي 18) أربعة قمم مميزة تقع عند 286.2 إلكترون فولت (القمة A)، 288.1 إلكترون فولت (القمة289.3 إلكترون فولت (ذروة ) و 293.5 إلكترون فولت (ذروة C)، المنسوبة إلى انتقال الإلكترون الأساسي للكربون في، و “حالات غير الرابطة، على التواليتمت دراسة الكبريت بشكل أعمق بواسطة تقنية عالية الدقة الطيف. كما هو موضح في الشكل 3ب، تم نسب القمة عند 168.3 إلكترون فولت إلى أنواع الكبريتات بينما كانت القمتان عند 165.2 إلكترون فولت و 164.2 إلكترون فولت مرتبطتين بـ والسندات، على التوالييجدر بالذكر أن هناك قمة عند 163.8 إلكترون فولت تتوافق معالتنسيق كان
الشكل 3 | التركيب الذري والإلكتروني لمحفز Pb SA/OSC. أ طيف XANES لحافة S L لعناصر SCDs وعينات Pb SA/OSC.وطيف XPS لـ Pb SA/OSC.المُعَايَر-طيف XANES الحافة لعنصر الرصاص في محفز Pb SA/OSC والمراجع (رقائق الرصاص و PbS).-طيف FT-EXAFS عند حافة
وعينات مرجعية.منحنيات التوافق لتناسب EXAFS لـ Pb SA/OSC في فضاء R.نموذج واجهة ذرية تخطيطي لـ Pb SA/OSC.رسوم WT-EXAFS للرصاصرقائق و PbS، على التوالي. تمت ملاحظته. الدقة العاليةيمكن تحليل الطيف إلى، و “، مما يشير إلى تشكيل الروابط (الشكل 3ج). أشارت هذه النتائج إلى أن الرصاص الموزع على المستوى الذري أظهر خصائص نموذجية وبيئات تنسيق مزدوجة.
تم فحص هيكل الامتصاص الدقيق للأشعة السينية الممتدة (EXAFS) للحصول على معلومات هيكلية مفصلة عن SACs من المجموعة الرئيسية. كما هو موضح في الشكل 3d، فإن شدة الخط الأبيض عند الـ “حافةكان شكل OSC مشابهًا لشكل PbS، مما يدل على أن ذرة الرصاص كانت تتميز بحالة أيونية. تم توضيح طيف EXAFS المحول فوريًا (FT) لـ Pb SA/ OSC والمراجع في الشكل 3e. أظهر العينة قمة سائدة تقع عند حوالي، يُنسب في الغالب إلى تشتتالتنسيق. ومن الجدير بالذكر، وجود ذروة عند حواليتم نسب Pb SA/OSC إلىتشتت المسار، كما يتضح من طيف FT-EXAFS مقارنة بـ PbS. علاوة على ذلك، غياب الميزة المقابلة لـالرابطة أثبتت التشتت الذري للرصاص على الإطار الكربوني. كميًا، يمكن الحصول على تكوين التنسيق لذرة الرصاص في محفز Pb SA/OSC من خلال ملاءمة EXAFS، كما هو موضح في الشكل 3f. تشير أفضل نتيجة ملاءمة لبيانات EXAFS في الجدول التكميلي 6 إلى أن ذرة الرصاص المعزولة كانت متشتتة ذريًا علىمصفوفة الكربون المضافة معًا ومن المحتمل أن تكون منسقة ضمن مزيج منوأجزاء (يشار إليها بـ، الشكل 3g). من الجدير بالذكر أن المتوسط توافقت المسافات بين الذرات في نتائج تركيب EXAFS مع المسافات لـوفيالهيكل لكنه انحرف عن المسافات منوفي الهياكل الأخرى لـ Pb SACs، مما يدعم الهيكل الفائق التنسيق لـ Pb SA/OSC. تحويل الموجات (WT) لـتم إجراء تذبذبات EXAFS عند حافة لتأكيد أحادية الذرات للأنواع الرصاصية في Pb SA/OSC. من مخططات الكنتور WT لمعايير رقاقة الرصاص و PbS (الشكل 3h)، كانت ذروة الشدة عند 3.7 وكانت مرتبطة على الأرجح بـو، على التوالي. في المقابل، عرض مخطط الكنتور WT لـ Pb SA/OSC أقصى شدة عندناتج عنالترابط. ومن ثم، فإن النتائج المذكورة أعلاه تشير بشكل قاطع إلى الخصائص المعزولة والمفرطة التنسيق لأنواع الرصاص في Pb SA/OSC، مما يؤكد أن البيئات الذرية المحلية لمواقع الرصاص الفردية تم تعديلها من خلال هندسة التنسيق.
قياس أداء ORR
مختلفًا عن SACs المتنوعة المبلغ عنها في الأدبيات، أظهر مادة Pb SA/OSC المصنوعة خصيصًا مركزًا رئيسيًا مميزًا وتكوينًا فائق التنسيق للكبريت والأكسجين. مستلهمًا من البيئة المحلية المميزة لـ Pb SA/OSC، أداءتم تقييم ORR بعد ذلك باستخدام نظام قياسي مكون من ثلاثة أقطاب مع قطب دائري دوار (RRDE) في-مشبع 0.10 م
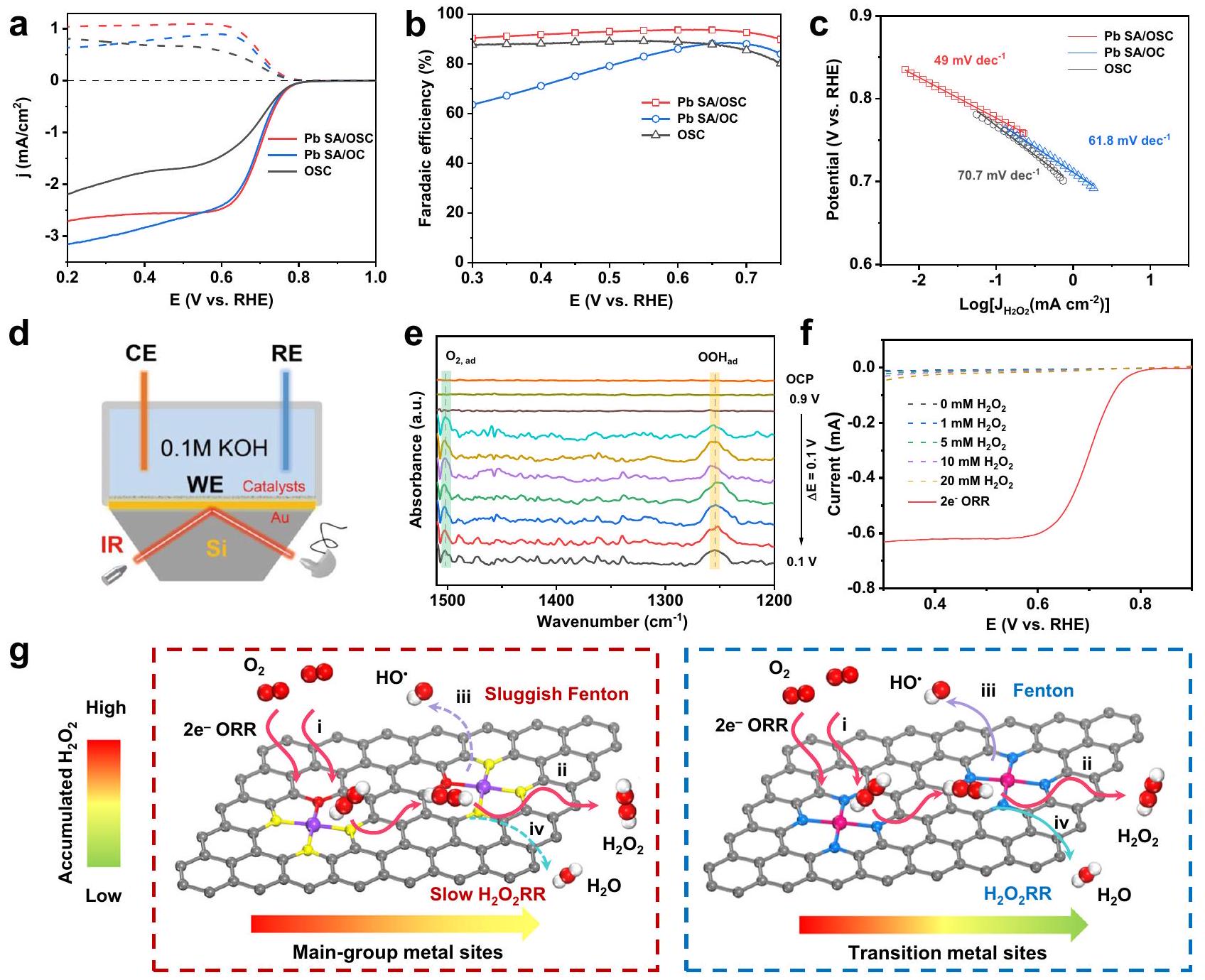
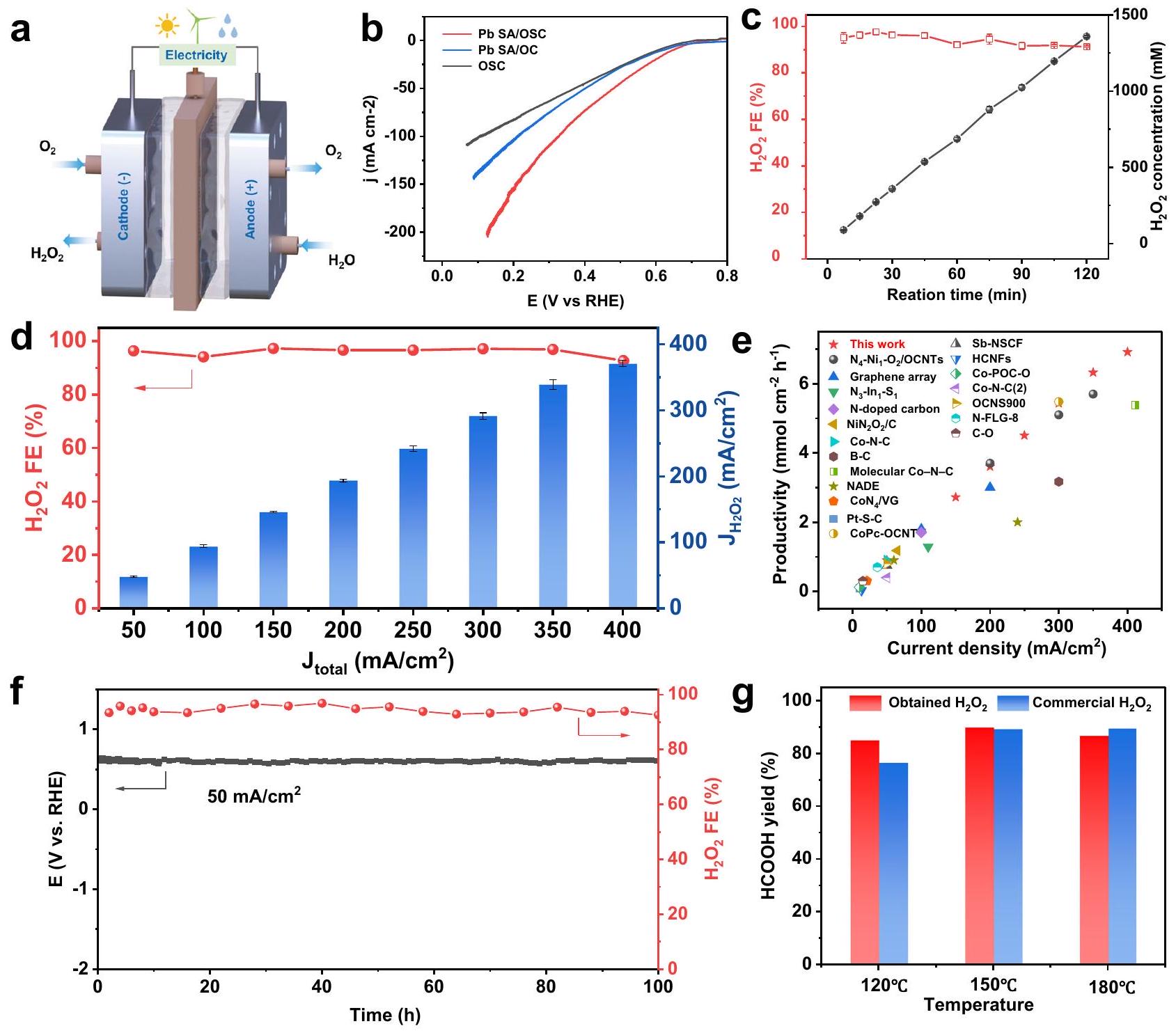
الشكل 4 | تقييم نشاط وانتقائية تفاعل اختزال الأكسجين ذو الإلكترونين لـ Pb SA/OSC باستخدام RRDE. أ منحنيات استقطاب اختزال الأكسجين الكهروكيميائية (الخطوط الصلبة) جنبًا إلى جنب معتيارات الكشف (الخطوط المتقطعة) لـ و OSC في -مشبع 0.10 م محلول كOH.الانتقائية المستندة إلى قياسات RRDE. ج. منحدرات تافل لمختلف المحفزات المستمدة من التيار الحركي لـالإنتاج. رسم توضيحي تخطيطي للإعداد التجريبي لقياسات ATR-SEIRAS في الموقع. WE القطب الكهربائي العامل، CE القطب الكهربائي المضاد، RE إلكترود مرجعي. تم جمع طيف ATR-SEIRAS في الموقع على محفز Pb SA/OSC في-مشبع 0.10 م KOH كاثوليت عند إمكانيات مختلفة تتراوح من 0.9 إلى 0.1 فولت مقابل RHE.LSV لـفيإلكتروليت KOH بتركيز 0.10 م مشبع بـ، أو “. مخططات التراكم الكهروكيميائيإنتاج المحفزات المعدنية للمجموعة الرئيسية (يسار) ومحفزات المعادن الانتقالية (يمين).
كOH. لدراسة أدوار مركز المجموعة الرئيسية وبنية التناسق الفائق في التأثير على النشاط التحفيزي لـتم أيضًا تحضير عينات تحكم من OSC خالية من المعادن و Pb SA/OC (مع O كذرة التنسيق الوحيدة). قبل تقييم الانتقائية لـتم تحديد معامل الجمع لقطب حلقة البلاتين ليكون حوالي 0.37، تم معايرته باستخدامتفاعل الأكسدة والاختزال (الشكل التوضيحي 23)تظهر الشكل 4a منحنيات الفولتمترية المسحية الخطية (LSV) لمختلف المحفزات، مقدمةً إشارات التيار لتقليل الأكسجين (الخطوط الصلبة) والتيار الجزئي المقابل لـ (الخطوط المتقطعة) المسجلة على القطب القرصي والقطب الحلقي، على التوالي. من الواضح أن Pb SA/OSC أظهر أداءً متفوقًانشاط ORR مقارنة بـ وOSC. أظهر أعلى تيار حلقي (0.204 مللي أمبير عند 0.5 فولت مقابل RHE)، مما يشير إلى توليد أعلى لـخلال عملية ORR.تم حساب الانتقائية ورسمها كدالة للجهد المطبق في الشكل 4b. في نطاق جهد واسع من 0.30 فولت إلى 0.70 فولت،انتقائية الـتجاوزت المحفز 90% (بحد أقصى 94%)، مما يبرز انتقائية عاليةمسار ORR. بالمقارنة،وأظهرت OSC أقل بكثيرالانتقائية وقيم عدد نقل الإلكترون الأعلى (n) (الشكل التوضيحي 24)، مما يؤكد أهمية هيكل التناسق الفائق في ضبط انتقائية تفاعل اختزال الأكسجين. بالإضافة إلى ذلك، قدم Pb SA/OSC أعلىكثافة التيار ) عند 0.65 فولت (الشكل التوضيحي 25) وأعلى جهد بداية إيجابي ( ) (المحدد كإمكانات كثافة التيار الحلقي لـ )، مقارنةً بعينات Pb SA/OC و OSC.
علاوة على ذلك، تم تحديد ميل تافل لـ Pb SA/OSC ليكون، أصغر بكثير من تلك الخاصة بـ Pb SA/OC () و OSC ( )، مما يشير إلى حركياته الأكثر ملاءمة لـ تم حساب المساحات السطحية النشطة كهربائيًا (ECSA) من خلال قياس سعات الطبقة المزدوجة ( ) من المحفزات. كما هو موضح في الشكل التوضيحي 26، أظهر عينة Pb SA/OSC حجمًا أكبر قيمة وتوافر أعلى للمواقع النشطة المكشوفة مقارنةً بالمواد المحفزة الأخرى المُعدة، مما يساهم بشكل كبير في أدائها المتفوق فيالإنتاج. الـنشاط ORR لـتمت دراسة المحفز بشكل أعمق في إلكتروليت محايد (0.10 م محلول ملحي مخفف بالفوسفات، PBS،كما هو موضح في الملحق
الشكل 27،أظهرت تيار استجابة الأكسدة العالي (0.56 مللي أمبير عند 0.1 فولت مقابل RHE) والانتقائية (ضمن النطاق المحتمل لـلنا. RHE. لتأكيد دور بيئة التنسيق في الانتقائية التحفيزية، أداء ORR لـتمت دراسة عينات بمحتويات مختلفة من الكبريت. جميع العينات قدمت نتائج مرضية.كثافة التيار ونافذة الجهد التشغيلية (الشكل التكميلي 28)، وأظهر محفز Pb SA/OSC بمحتوى S الأمثل أفضل انتقائية لـنشاط ORR.
لكشف عن الوسيط الرئيسي للأكسجين الممتص (*OOH) على Pb SA/OSC أثناء التحليل الكهربائيتم إجراء اختبارات الطيف الكهرومغناطيسي الممتد المعزز بالانعكاس الكلي المخفف في الموقع (ATRSEIRAS) (الشكل 4d). ومن المثير للإعجاب، تم رصد نطاق امتصاص جديد عند حواليظهر عند تطبيق جهد أقل من 0.8 فولت مقابل RHE (الشكل 4e). يمكن أن يُعزى هذا النطاق الامتصاصي المميز على Pb SA/OSC إلىاهتزاز الشد لـ * OOH ، وهو ما يتماشى مع الدراسات السابقة. بالإضافة إلى ذلك، فإن نطاق الامتصاص عند حوالينُسِبَ إلىوضع الشد للأكسجين الجزيئي الممتصتظهر النتائج التجريبية المؤشرة أن نطاق الاهتزاز لـ *OOH قد شهد انخفاضًا إلى (الشكل التكميلي 31)، مما يشير إلى مشاركة ذرة الهيدروجين ويؤكد أصل *OOH. بشكل عام، دعمت نتائج ATR-SEIRAS المعتمدة على الجهد الوساطة *OOHمسار ORR على المحفزات المعدة (الشكل التوضيحي التكميلي 32).
لتقييم المجموع المتراكممن الضروري تقييم التفاعل الجانبي المتتالي بشكل كميتفاعل اختزالعلى نفس المحفز. التجارب الكهروكيميائية لـتمت في-مشبع 0.10 م كOH بتركيزات متغيرة من. كما هو موضح في الشكل 4f، معدل أظهر الاختزال الكهربائي على Pb SA/OSC زيادة طفيفة فقط مع زيادة الجهد الزائد والكثافة الحالية لـعلى Pb SA/OSC كان أقل منعندما تجاوز الجهد 0.40 فولت مقابل RHE، مما يشير إلى نشاط محدود لمواقع المجموعة الرئيسية لـ Pb SA/OSC تجاهتمت ملاحظة نتائج مماثلة لمحفزات المجموعة الرئيسية خلالعملية ORR. لذلك، تم تحديد محفز Pb SA/OSC كمرشح واعد للغاية لـالإنتاج بسبب حداثتهنشاط ومرتفعالانتقائية خلال عملية تقليل الأكسجين. وبالتالي، فإن المعدل الصافي لـالإنتاج (أي، معدل الإنتاج ناقص معدل الاختزال الكهربائي لـيمكن الحفاظ على مستوى مرتفع بما فيه الكفاية من Pb SA/OSC حيث أن تركيزتراكم. بالإضافة إلى ذلك، قد تؤدي تفاعلات فنتون المحتملة التي تحفزها مواقع المعادن الانتقالية إلى استنفاد المتراكم.وتقوض متانة القطب الكهربائيعلى عكس مواقع المعادن الانتقالية، فإن مواقع المعادن من المجموعة الرئيسية تمتلك بشكل طبيعي نشاطًا أقل تجاه تفاعلات فينتون.كما هو موضح في الشكل التوضيحي 35، لم تُلاحظ أي إشارات مميزة لجذور الهيدروكسيل الناتجة عن تفاعلات فنتون في طيف EPR لـ Pb SACs خلالعملية ORR. وبالتالي، من المتوقع أن تكون المحفزات الخاصة بـ ORR المستندة إلى مواقع المعادن الرئيسية خيارًا جذابًا للتخفيف من تفاعلات فنتون خلالالإنتاج. بشكل عام، يُظهر محفز Pb SA/OSC إمكانيات كبيرة في تلبية المتطلبات للتراكم.الإنتاج من خلال القمعوتفاعلات فينتون (الشكل 4g).
مقياس عمليالتحليل الكهربائي
مستوحاة من الانتقائية الملحوظة لتفاعل اختزال الأكسجين 2e- والإمكانات لـتم استخدام إنتاج محفز Pb SA/OSC، وقطب غاز منتشر (GDE) وخلية كهربائية مخصصة للتدفق لأغراض عملية.التخليق. كما هو موضح في الشكل 5a، تم إجراء LSV في البداية في إعداد خلية تدفق في 1 م KOH مع يدويتعويض iRكما هو موضح في الشكل 5b، أظهر محفز Pb SA/OSC كثافة تيار أعلى بكثير مقارنة بـ وOSC، مما يشير إلى الدور الحاسم لبيئة التنسيق لـفيالتحليل الكهربائي. ثم تم إجراء تجارب التحليل الكهربائي على نطاق واسع. عند كثافات تيار مختلفة تتراوح من 50 إلىلتقييم الأسعارالإنتاج. التركيز المتراكم لـتم تحديدها في الكاثوليت باستخدام طريقة الكميّة اللونية (الشكل التكميلي 39). من المهم أن كفاءات فاراداي (FEs) لـلـالجيل ظل فوقعبر نطاق كثافة التيار المطبق (الشكل 5d). عندما تكون تركيز يتجمع في الإلكتروليت، كثافة عالية منيؤدي إلى التحلل الذاتي أوبشكل مثير للإعجاب، خلال التحليل الكهربائي بالجملة في (الشكل 5ج)، تركيز المتراكمزاد تقريبًا بشكل خطي وبلغ ) بعد ساعتين من التحليل الكهربائي. الأداء المتفوق لـ أشار محفز OSC إلى أن المواقع الرئيسية للمجموعة ذات بيئة تنسيق محلية منظمة منعت بشكل فعال التحلل الذاتي أو المزيد من الاختزال الكهروكيميائي الناتج.. تتماشى هذه النتيجة مع الملاحظات التي تم الحصول عليها من قياسات EPRتجارب تفاعل الاختزال، مما يؤكد الحد الأدنى من فنتون ونشاط الـ. علاوة على ذلك، تشير الحسابات إلى أن الجزيئاتيمكن أن يتم استقراره وتراكمه عند واجهة الصلب-السائل لـ OSC، مما يمنع بشكل فعال تحلل . من الجدير بالذكر أن “أظهرت FEs فقط اتجاه انخفاض طفيف (من 97% إلى 91%) خلال التحليل الكهربائي عند تيار ثابت في.
علاوة على ذلك، أظهر محفز Pb SA/OSC أداءً عاليًامعدل الإنتاج عند كثافات تيار مختلفة تتراوح من 50 إلى(الشكل 5e). عندمعدلالجيل وصل، مما يضعه ضمن أفضل أداء لمحفزات ORR المتطورة (الجدول التكميلي 7)لتحقيق استقرار Pb SA/OSC، تم إجراء التحليل الكهربائي على المدى الطويل عند كثافة تيار ثابتة من. كما هو موضح في الشكل 5f، فإن هناك ارتفاعاً FE (أعلىتم الحفاظ على ( ) دون تدهور ملحوظ لمدة لا تقل عن 100 ساعة، مما يتيح إنتاجًا مستمرًا ومستقرًا لـ، وهو أمر حاسم للتطبيقات الصناعية. الشكل، وتشتت الرصاص الذري، وبلورية الكربون لـتم الحفاظ على OSC بشكل جيد بعد اختبار الاستقرار (الأشكال التكميلية 44-46). وهذا يبرز الاستقرار الهيكلي للعامل المساعد من أجلORR. علاوة على ذلك، أظهرت النتائج التي تم الحصول عليها من ICP-MS تسربًا ضئيلًا لكل من Pb و S (حواليمن الرصاص ومن S في Pb SA/OSC) إلى الإلكتروليت الكتلي بعد 6 ساعات من التحليل الكهربائي المستمر.
أظهر محفز Pb SA/OSC أداءً مثيرًا للإعجاب في التخليق الكهربائي لـلتقييم تطبيقاته العملية بشكل أكبر، قمنا بدمج ما تم الحصول عليهحل مع تحويل الكربوهيدرات المشتقة من الكتلة الحيوية لإنتاج حمض الفورميك (HCOOH). وقد أفادت الدراسات السابقة أن الكربوهيدرات المشتقة من الكتلة الحيوية يمكن تحويلها إلى حمض الفورميك، وهو مهمحامل، من خلال الأكسدة الهيدروحرارية في وجودوقلويباستخدام تحويل الجلوكوز إلى HCOOH كمثال تمثيلي، تم الحصول على عائد مرتفع من HCOOH باستخدام القلويات كعامل حفاز وكعامل مؤكسد. لعب القلوي دورًا حاسمًا في تسهيل الأكسدة الانتقائية ومنع الأكسدة المفرطة لـ HCOOH المتكون (الشكل التكميلي 47). في هذا الصدد،الحل الذي تم الحصول عليه من خلال التخليق الكهربائي قدم منصة مثالية لتحويل الكربوهيدرات المشتقة من الكتلة الحيوية، حيث زودت كل من القلوياتوفي الوقت نفسه. المتراكممحلول (1358 مليمول) تم الحصول عليه عن طريق التحليل الكهربائي الكتليتم استخدامه لمدة ساعتين في الأكسدة الهيدروحرارية للجلوكوز عند درجات حرارة مختلفة. للمقارنة، تجاريتم استخدام كمية محسّنة من KOH تحت نفس الظروف للتفاعلات المذكورة أعلاه. كما هو موضح في الشكل 5g، تم إنتاج كلاهما كهربائيًاالحل والتجاريحقق عوائد استثنائية من HCOOH (أكثر من ) عند درجات حرارة تبلغ 150 وبالإضافة إلى ذلك، لوحظ انخفاض طفيف في إنتاج HCOOH عند درجة حرارة أقل.الذي تم إنتاجه بواسطة التحفيز الكهربائييمكن تطبيق الحل مباشرة على تحويل الكربوهيدرات المشتقة من الكتلة الحيوية دون الحاجة إلى معالجة إضافية.
الشكل 5 | أداء التخليق الكهربائي لـفي جهاز تحليل كهربائي بتدفق يعتمد على GDE. توضيح تخطيطي لجهاز الخلية المتدفقة.منحنيات LSV لـ PbSA / OSC و Pb SA/OC و OSC. المنحنيات تم رسمها يدويًاتعويض iR. ج المتراكمتركيزات وكفاءات فارادايفي.كفاءات فارادايمن عند كثافات تيار مختلفة. تشير أشرطة الخطأ في (ج، د) إلى الانحراف المعياري
انحراف ثلاث قياسات مستقلة. eمعدل الإنتاجOSC مقارنةً بالعوامل المحفزة الكهربائية المتطورة المبلغ عنها.اختبار الاستقرار وانتقائية محفز Pb SA/OSC تحت، مع تجديد الإلكتروليت كل 6 ساعات خلال الاختبار. غ إنتاج حمض الفورميك من أكسدة الجلوكوز بواسطة المنتج والتجاريعند درجات حرارة مختلفة.
نقاش
باختصار، قمنا بتطوير محفز كهربائي ذي ذرة واحدة من المجموعة الرئيسية يتميز بمواقع الرصاص (Pb) المتناسقة بشكل فائق مع الكبريت (S) والأكسجين (O) والمثبتة في هياكل كربونية من خلال نهج التحلل الحراري المدعوم بنقاط كربونية. من خلال الهندسة المدروسة لبيئات التنسيق لمواقع المجموعة الرئيسية، أظهرت محفزات Pb SA/OSC أداءً ملحوظًا فيORR نحومع انتقائية عالية تصل إلى. علاوة على ذلك، كثافات التيار الصناعية تصل إلىمعلـتم تحقيق الإنتاج بمعدل مثير للإعجابفي خلية تدفق كهربائية تعتمد على GDE في وسط قلوي، متجاوزًا أداء معظم التقنيات الحديثة.محفزات ORR. حافظ محفز Pb SA/OSC على استقرار كافٍ دون انخفاض واضح فيالإنتاج وكفاءة فاراداي خلال 100 ساعة من التحليل الكهربائي المستمر. ومن الجدير بالذكر أن النشاط الخامل لمجموعات الرصاص الرئيسية SACs هو لـوتفاعلات شبيهة بفينتون، تركيز المتراكمحل وصلت إلى 1358 مليمول مع كفاءة عالية خلال التحليل الكهربائي الكتلي. علاوة على ذلك، فإن الناتجتم استخدام الحل بنجاح في الأكسدة الهيدروحرارية للجلوكوز المشتق من الكتلة الحيوية المتجددة، مما أدى إلى تحقيق عائد استثنائي من HCOOH (أكثر منتشير الدراسات التجريبية والنظرية المجمعة إلى أن الأداء التحفيزي المتفوق لـ Pb SA/OSC فييمكن أن يُعزى ORR إلى “البيئات الدقيقة المنظمة بواسطة التنسيق الفائقالأجزاء. يقدم هذا العمل طريقًا واعدًا لتصميممحفزات ORR لمعالجة معضلة إنتاج الاستنفاد فيالتحليل الكهربائي وقد يفتح الباب للإنتاج الأخضر لحمض الفورميك من الكتلة الحيوية المتجددة.
طرق
المواد الكيميائية
أسيتالدهيد (محلول مائي 40%)، هيدروكسيد الصوديوم، هيدروكسيد البوتاسيوم، حمض الهيدروكلوريك، خلات الرصاص (II) ثلاثي الهيدرات ) ، تم توفير الجلوكوز والإيثانول اللامائي من شركة سينوفارم للمواد الكيميائية، الصين. تم شراء حمض 2-ميركابتوبروبيونيك (98%) وأكسيد التيتانيوم (IV) – هيدرات حمض الكبريتيك (93%) من شركة ماكلين، الصين. محلول نافيون (تم توفيره من قبل شركة سيغما-ألدريتش، الولايات المتحدة الأمريكية. تم شراء الكربون الأسود (فولكان XC-72) من شركة كابوت، الولايات المتحدة الأمريكية. تم استخدام جميع المواد الكيميائية والمركبات مباشرة كما تم استلامها دون مزيد من التنقية.
إعداد SCDs و OCDs
تم تحضير النقاط الكربونية المضافة بالكبريت (SCDs) باستخدام طريقة تكثيف الألدول عالية الكفاءة في درجة حرارة الغرفة.في إجراء تحضير نموذجي، تم إذابة 5 مل من حمض 2-ميركابتوبروبيونيك في 40 مل من الأسيتالديهيد. ثم، تم إضافة 15 جرام من NaOH ببطء وتم التحريك لمدة 2.5 ساعة. المنتج الذي تم الحصول عليه بعد انتهاء التفاعل تم تعريضه للموجات فوق الصوتية مع كمية معينة من الماء المنزوع الأيونات لمدة 30 دقيقة. تم طرد محلول مائي من المنتج الخام باستخدام جهاز الطرد المركزي.لمدة 10 دقائق لإزالة الجسيمات الكبيرة أو المتجمعة. تم جمع السائل العلوي ونقله إلى كيس غسيل الكلى (MWCO: 3500 دالتون) ليتم غسله لمدة يومين.
تم إعداد عينات SCDs بمحتويات مختلفة من الكبريت باستخدام إجراء مشابه باستثناء تغيير كمية حمض 2-ميركابتوبروبيونيك.، و 7.5 مل. يتم الإشارة إليها على أنها OCDs و SCDs-2.5 و SCDs-7.5، على التوالي.
تركيب Pb SA/OSC
في تخليق نموذجي لمحفز Pb SA/OSC، تم أولاً تحضير محلول سلفيد الرصاص عن طريق إذابةفيلتحقيقمحلول مخزون. بعد ذلك، تم توزيع 300 ملغ من SCDs المنقاة في 50 مل منتحت تأثير الموجات فوق الصوتية لمدة 10 دقائق للحصول على محلول SCDs. ثم، 1 مل منتم إضافة 300 ملغ من Vulcan XC-72 في الإيثانول (10 مل) إلى المحلول المذكور أعلاه، تلا ذلك عملية الصوتنة لمدة 15 دقيقة. بعد ذلك، تم تجفيف المحلول المختلط أعلاه أولاً عن طريق التبخر الدوراني ثم في فرن مفرغ.. تم تسخين المسحوق الأسود الناتج إلىمعدل التسخين، ) لمدة ساعتين في تدفق النيتروجين. بعد الغسل الشامل بالماء والإيثانول، تم تعريض المساحيق المجففة للتلدين عند درجات حرارة عالية في معدل التسخين، ) لمدة ساعتين في تدفق النيتروجين. بعد أن تم تبريده بشكل طبيعي إلى درجة حرارة الغرفة، تم الحصول على محفز Pb SA/OSC.
لـوبالنسبة لعينات Pb SA/OSC-7.5، فإن عملية التخليق هي نفسها كما في Pb SA/OSC، ولكن النقاط الكربونية المضافة لها الكبريت هي SCDs-2.5 وSCDs-7.5، على التوالي. علاوة على ذلك، تم تحضير عينة OSC بنفس الإجراء الموصوف لـ Pb SA/OSC ولكن دون إضافة أي سلف Pb.
تركيب Pb SA/OC
لتحضير محفز Pb SA/OC، تم توزيع 300 ملغ من OCDs المنقاة في 50 مل منتحت تأثير الموجات فوق الصوتية لمدة 10 دقائق للحصول على محلول OCDs. ثم، 1 مل منتم إضافة 300 ملغ من Vulcan XC-72 في الإيثانول (10 مل) إلى المحلول المذكور أعلاه، تلا ذلك عملية الصوتنة لمدة 15 دقيقة. بعد ذلك، تم تجفيف المحلول المختلط أعلاه أولاً عن طريق التبخر الدوراني ثم في فرن مفرغ.. تم تسخين المسحوق الأسود الناتج إلىمعدل التسخين، ) لمدة ساعتين في تدفق النيتروجين. بعد الغسل الشامل بالماء والإيثانول، تم تعريض المساحيق المجففة للتلدين عند درجات حرارة عالية في معدل التسخين، ) لمدة ساعتين في تدفق النيتروجين. بعد أن تم تبريده بشكل طبيعي إلى درجة حرارة الغرفة، تم الحصول عليه.
توصيفات
تمت دراسة مورفولوجيات المحفزات الكهربية باستخدام مجهر إلكتروني نافذ (TEM) مع مجهر (7700، شركة هيتاشي، اليابان) بجهد تسريع قدره 100 كيلو فولت. تم الحصول على صور مجهرية بتصحيح الانحراف من نوع HAADF-STEM ورسم خرائط العناصر باستخدام جهاز STEM (JEM-ARM200CF، شركة جيول، اليابان). تم دمجها مع مصححات انحراف كروي مزدوج تعمل عند 200 كيلو فولت. تم تسجيل قياسات حيود الأشعة السينية (XRD) على جهاز حيود Rigaku Miniflex-600 باستخدامالإشعاع ) بحجم خطوة وزمن العد 0.5 ثانية. تم جمع طيف الإلكترونات الضوئية بالأشعة السينية (XPS) باستخدام مجهر أشعة سينية مسح (PHI 5000 Verasa، ULAC-PHI Inc.، الولايات المتحدة الأمريكية) باستخدام إشعاع Al Ka وقمة C1s عند 284.8 eV كمعيار داخلي. تم أخذ الأطياف رامان باستخدام جهاز طيف رامان (LabRAM HR Evolution، شركة هوريبا، اليابان) مزودًا بالليزر الأخضر الذي ينبعث عند 532 نانومتر. تم إجراء تحليل عنصري للرصاص في العينات الصلبة التي تم تحليلها باستخدام مطياف الكتلة المتصل بالبلازما المحفزة (ICP-MS).
تم إجراء طيف امتصاص الأشعة السينية الناعمة (Soft-XAS) في محطة نهاية التحفيز وعلوم السطح في خط الأشعة BL11U ومحطة نهاية الانبعاث الضوئي في خط الأشعة BL10B في المختبر الوطني للأشعة السينية المتزامنة (NSRL) في هيفي، الصين. تم إجراء قياسات طيف امتصاص الأشعة السينية الدقيقة (XAFS) في الـ “-الحافة. تم جمع البيانات في خط الشعاع BL14W1 في منشأة الإشعاع السنكروتروني في شنغهاي (SSRF)، الصين. تم تشغيل حلقة التخزين عند 3.5 جيجا إلكترون فولت مع أقصى تيار إلكتروني قدرهتم استخدام الرقائق و PbS كمرجع.تم تسجيل بيانات طيف الامتصاص القريب من حافة الأشعة السينية (XANES) في وضع الفلورية. تم تلوين الأشعة السينية الصلبة باستخدام بلورات مزدوجة من السيليكون (111). تم استخراج ومعالجة بيانات EXAFS المكتسبة وفقًا للإجراءات القياسية باستخدام وحدة ATHENA المدمجة في حزمة برامج IFEFFIT.تم الحصول على طيف EXAFS الموزون عن طريق طرح الخلفية بعد الحافة من الامتصاص الكلي ثم تطبيعها بالنسبة لخطوة قفزة الحافة. بعد ذلك،-مرجحالبيانات في-مساحة تتراوح منتم تحويلها باستخدام تحويل فورييه إلى الفضاء الحقيقي (R) باستخدام نوافذ هانينغ ) لفصل مساهمات EXAFS من قذائف التنسيق المختلفة. تم إجراء قياسات طيف الامتصاص تحت السطح المعزز بالأشعة تحت الحمراء المنعكسة الكلية المخففة (ATRSEIRAS) على جهاز نيكوليتجهاز مطياف FT-IR iS50 (شركة ثيرمو ساينتيفيك، الولايات المتحدة الأمريكية) مزود بكاشف MCT.
القياسات الكهروكيميائية
تم إجراء التجارب الكهروكيميائية في نظام قياسي مكون من ثلاثة أقطاب يتم التحكم فيه بواسطة محطة عمل CHI 760e (شركة CH Instruments، الصين). يتكون القطب الدوار ذو الحلقة القرصية (RRDE، أدوات البحث باين، الولايات المتحدة الأمريكية) من قطب كربوني زجاجي (مساحة القرص: ) وإلكترود حلقة من البلاتين (مساحة الحلقة: ) تم استخدامه كالكاثود العامل، مع كفاءة جمع نظرية قدرها تم استخدام قطب (محلول KCl المشبع) وسلك Pt كأقطاب مرجعية وأقطاب مضادة، على التوالي. تم تطبيع جميع الجهود المسجلة عن طريق تحويلها إلى قطب الهيدروجين القابل للعكس (RHE) وفقًا للمعادلةتم اعتماد الجهد المحتمل مقابل RHE ما لم يُذكر خلاف ذلك.
لإعداد القطب العامل، 4 ملغ من المحفزات ومحلول نافيون (تم خلطها معمن 1:1 (محلول مختلط من الماء/الإيثانول ثم تم تفريقه بواسطة الموجات فوق الصوتية لمدة 30 دقيقة حتى تم الحصول على حبر محفز متجانس. بعد ذلك، تم تلميع RRDE ميكانيكياً باستخدام تعليق الألومينا،تم إسقاط حبر المحفز على القطب القرصي وتجفيفه لتشكيل فيلم رقيق موحد في درجة حرارة الغرفة. تم إجراء جميع القياسات الكهروكيميائية في أو -مشبع 0.10 م KOH و 0.10 م PBS محلول مائي. قبل قياس ORR، تم إجراء عملية التنشيط المسبق عن طريق مسح منحنيات CV بمعدلتم إجراء القياس من 0.05 إلى 1.0 فولت على جهاز RRDE حتى تم الوصول إلى حالة مستقرة. ثم تم الحصول على منحنيات استقطاب ORR من قياسات LSV تحتظروف مشبعة بين 0.05 و 1.0 فولت عند معدل مسحعند 1600 دورة في الدقيقة. بينما تم تثبيت جهد حلقة البلاتين عند 1.2 فولت لقياس كمية الناتج.على القطب القرصي. خاصة، تم تنظيف القطب الحلقي من البلاتين كهربائيًا من خلال مسح منحنيات الجهد المتناوب لمدة 20 دورة. (نطاق محتمل:معدل المسح: ) على حلقة Pt قبل قياس LSV.
تم معايرة كفاءة الجمع (N) على قطب RRDE فيالكهرباء السائلة عند سرعات دوران مختلفة. وبالتالي، تم تحديد كفاءة الجمع المقاسة على أنها، وهو قريب بشكل معقول من القيمة النظرية. الـ تفاعل اختزالتمت دراسة ) من خلال إجراء قياسات LSV لقطب القرص المغطى بالكاتاليس.محلول 0.1 م من هيدروكسيد البوتاسيوم المشبع، أو “عند 1600 دورة في الدقيقة.
انتقائية بيروكسيد الهيدروجين ) وعدد نقل الإلكترون ( n ) تم حسابه باستخدام المعادلة التالية:
أينهو التيار الحلقي،هو التيار القرصي وN هو كفاءة الجمع المحددة.
كثافة التيار الحركي (تم حساب ( ) باستخدام معادلة كوتيكي-ليفش:
أينهو كثافة التيار المقاسة وهو كثافة التيار المحدودة بالانتشار.يتم الحصول على القيمة من خلال قسمة التيار الحلقي على كفاءة الجمع (N) ومساحة القطب القرصي.تم أخذ القيمة من أعلى قيمة في الرسم البياني j المقاسة على مدى النطاق المحتمل الكامل الذي تم التحقيق فيه ).
تم حساب انحدارات تافل وفقًا لمعادلة تافل:
أين هو الثابت، هو الجهد المطبق، هو ميل تافل، وهو كثافة التيار الحركي لـالإنتاج.
تم إجراء قياسات السعة الكهروكيميائية من منحنيات شحن الطبقة المزدوجة باستخدام الفولتامترات عند نافذة الجهد غير الفارادي في محلول 0.10 م كOH. تم اختبار هذه العينات بمعدلات مسح تتراوح من 10 إلىأظهرت مخططات كثافة التيار (عند 1.05 فولت) كدالة لمعدل المسح سعة الطبقة المزدوجة من خلال الميل.
التحليل الكهربائي بالجملة لـ
تم تصنيع الكاثود عن طريق رش الطلاء على المادة التي تم تخليقها.محفز OSC على إلكترود انتشار الغاز (GDE، YLS30، شركة سوزو سينيرو للتكنولوجيا، الصين). في عملية نموذجية، تم خلط 8 ملغ من المحفز معمنمحلول نافيون و 4 مل منمذيب مختلط من الإيثانول/الماء (حجم/حجم). تم تعريض المحلول أعلاه للموجات فوق الصوتية لمدة لا تقل عن 60 دقيقة لتكوين حبر متجانس. ثم تم رش الحبر المتناثر بشكل جيد على GDE. ) بمبلغ تحميل قدره (الشكل التكميلي 41). تم تجفيف إلكترود انتشار الغاز (GDE) في غرفة فراغ طوال الليل وقطعه إلى حجم قطب كهربائي من التيتانيوم مغطى بـوتم استخدام القطب (KCl المشبع) كأنود وقطب مرجعي، على التوالي. تم تحضير الأنود عن طريق ترسيبعلى دعم من التيتانيوم بواسطة طريقة الطلاء بالغمر والتحلل الحراريباختصار، تم قطع شبكة التيتانيوم إلى حجمتم غسل المنطقة بأسيتون وماء منزوع الأيونات، ثم تم استخدام الموجات فوق الصوتية في محلول مائي من HCl بتركيز 6 م تم تسخينه إلى لمدة 45 دقيقة قبل الطلاء بالغمر. كانت محلول الطلاء يحتوي على 30 ملغ من (ألفا أيسر) مذاب في 10 مل من محلول الإيزوبروبانول مع حمض الهيدروكلوريك المركز. تم غمر شبكة التيتانيوم فيمحلول، يليه التجفيف في فرن مفرغ من الهواء عندلمدة 10 دقائق قبل التكليس في فرن فيلمدة 10 دقائق. تم تكرار عملية الغمر والتكلس حتىتم تحقيق التحميل.
عمليتم تنفيذ الإنتاج باستخدام نظام خلية تدفق محكم الغاز معحجرة الغاز وحجرتان سائل. كانت غرف الكاثوليت والأنوليت مفصولة بغشاء تبادل الكاتيونات (نافيون 117، شركة فويل سيل ستور، الولايات المتحدة الأمريكية). كانت خلية التحليل الكهربائي تتكون من لوحين خلفيين من التيتانيوم معحقل تدفق متعرج. خلال اختبارات التحفيز الكهربائي، تم إعادة تدوير الإلكتروليت (1 م كOH) بمعدل تدفق ثابت يبلغ حواليمع مضخة دافعة، وتم الحفاظ على تغذية الغاز عنديتدفق عبر الكاثود باستخدام جهاز التحكم في التدفق. تم توفير التيارات الثابتة بواسطة محطة عمل (PMC1000، PARSTAT Inc.، الولايات المتحدة الأمريكية). لاختبار الـمعدل الإنتاج، تم زيادة كثافة تيار الكاثود من 50 إلى. تم تجديد محلول الإلكتروليت 1 م KOH بشكل دوري كل 6 ساعات في الحالة المستقرةإنتاج يصل إلى 100 ساعة.تم تحديد التركيز كميًا بواسطة سلفونات التيتانيومطريقة المعايرة. بعد إضافة محلول أصفر منتم إنتاجه. علاقة معايرة خطية بين التركيز و تم الحصول على قمة الامتصاص عند 408 نانومتر باستخدام مطيافية الأشعة فوق البنفسجية والمرئية. هنا،فيتم تحضيره عن طريق التحريك طوال الليل حتى تم تشكيل محلول شفاف، والذي تم استخدامه لتحديد الكمية العالية بشكل طيفي.تم إنتاجه في خلية التدفق.
تم إجراء طيف الامتصاص الكهربائي الكيميائي (EIS) عند 0.75 فولت (مقابل RHE) من 100,000 إلى 0.1 هرتز لتحديد المقاومة غير المصححة في نطاق التردد العالي لتصحيح iR في الخلية الكهروكيميائية.
الكفاءة الفارادائية لـالإنتاجتم تحديده بواسطة المعادلة التالية:
أينهو المنتجتركيز ) في الإلكتروليت، يشير إلى حجم الإلكتروليت (لتر)، وF يدل على ثابت فاراداي ( )، وهي التيار التشغيلي (أ) ووقت الاختبار (ث)، على التوالي.
أكسدة الجلوكوز لتحليل حمض الفورميك والمنتجات
تمت دراسة الأكسدة الانتقائية للجلوكوز من خلال تجارب دفعة. في تجربة نموذجية، تم استخدام 20 مل من المحلول القلوي.تم تدوير المحلول في خلية التدفق لمدة ساعتين عند. ثم، 4 مل من القلوي المذكور أعلاهتم إدخال محلول و70 ملغ من الجلوكوز إلى المفاعل. ثم تم نقل المفاعل إلى فرن تم تسخينه مسبقًا إلى درجة الحرارة المطلوبة (120، 150 أوبعد 15 دقيقة، تم إزالة المفاعل من الفرن وغمره في حمام ماء بارد. بالمقارنة، تم استخدام نفس الكمية من المنتج التجاريمع التركيز المحسن لـتم استخدامه للتفاعلات المذكورة أعلاه تحت نفس الظروف. بعد التفاعل، تم ضبط الرقم الهيدروجيني للمحلول إلى 3 باستخدام HCl لتحليل الكروماتوغرافيا السائلة عالية الأداء (HPLC) (LC16، شركة شيمادزو، اليابان). تم إجراء تحليلات HPLC على أمينعمود HPX-87H مزود بكاشف معامل الانكسار.
طرق حسابية
تم إجراء حسابات نظرية الكثافة الوظيفية (DFT) بواسطة حزمة المحاكاة الأولية في فيينا (VASP) باستخدام طريقة الموجة المعززة بالمشروطة (PAW).تم التعامل مع طاقة تبادل الإلكترون والتداخل ضمن تقريب التدرج العام باستخدام طريقة بيردو-بورك-إرنزرهوف (GGA-PBE).تم أخذ استقطاب الدوران في الاعتبار في جميع الحسابات. تم تعيين أخذ عينات نقاط k لمنطقة بريليو علىتم تعيين معايير التقارب للهندسة (مجال ذاتي الاتساق) إلىللطاقة وللقوى. كانت طاقة القطع لطاقة الحركة للموجات المستوية
400 إلكترون فولت. التوزيع الإلكتروني للرصاص هووتم اعتبار جهد الإلكترون التكافؤ في الحسابات. تم تحديد الشغلات الجزئية باستخدام نظام تمويه غاوسي بعرض تمويه قدره 0.05 إلكترون فولت. تم استرخاء جميع الهندسات بالكامل إلى الحالة الأساسية. هياكل الـتم أخذ عينات من واجهة الماء من خلال التوزيع العشوائي لـ 36 جزيئًا من الماء في منطقة شريحة الماء، تلاها تحسين محلي بدأ بمجموعتين من التكوينات. لوصف الكهروستاتيكا والتشتت عند الواجهة، تم استخدام نموذج الاستمرارية القابل للاستقطاب المطبق في VASPsol مع السماحية النسبية 78.4 كما هو الحال في الماء.تم اعتبار تفاعلات فان der Waals باستخدام طريقة DFT-D3 التجريبية المقترحة من قبل غريمي وآخرين.. الهياكل في حالة الأرض للمواد الممتصة *وتم تحديد *OH من خلال البحث عن أدنى طاقة واحدة بين جميع التكوينات الممكنة على المواقع النشطة الممكنة.
تغير الطاقة الحرة لجيبس ( ) تم حسابه على أنه ، حيث هو فرق الطاقة الإلكترونية الذي يتم الحصول عليه مباشرة من حسابات DFT،هو فرق طاقة النقطة الصفرية،هي درجة حرارة الغرفة (298.15 كلفن) وهو تغيير الإنتروبيا.أين هو الجهد عند القطب، e هو الشحنة المنقولة وأين هو ثابت بولتزمان و تقليل إلىيتبع آلية تفاعل ذات إلكترونين كما يلي:
حيث يشير النجمة (*) إلى موقع الرصاص في المحفز و * OOH تشير إلى الوسيط الممتص على سطح المحفز. الجهد الزائد الذي يقيم أداء الـيتم الحصول على ORR كـ، حيث تمثل 0.7 الجهد التوازني. الجهد المحدد هو الجهد الذي تصبح عنده خطوة التفاعل طاقة حرة..
نفترضلوسط حمضي. تتكون تفاعل الإلكترونات الأربعة المرتبطة من الخطوات الأولية التالية:
الجهد الزائد لـيتم الحصول على ORR كـ، حيث يمثل 1.23 الجهد التوازني. يتم تحديد الجهد المحدد بواسطة ، e. يتم حساب طاقات الامتزاز الحرة لأي وسيط تفاعل i تحت الظروف القياسية بشكل مريح كـ ، حيث هي طاقة الامتزاز وهي مساهمات الطاقة الحرة للمواد الممتصة. القيم لـللممتصات في موقع الرصاصتُقدم المحفزات في الجدول التكميلي 5. ثم يتم الإشارة إلى جميع طاقات الامتزاز بشكل متسق بالنسبة للجزيئات المستقرة لـوفي الجدول التكميلي 6، مع التغير الطاقي التجريبي لعملية الاختزال لـإلىلبناء مخطط الطاقة الحرة. الطاقات الحرة لـوعند جهد معينبالنسبة إلى القطب الهيدروجيني القياسي (NHE)تعرف على النحو التالي:
نفترضلوسط قلوي. تتكون التفاعل الرباعي الإلكترون المرتبط المقابل من الخطوات الأولية التالية:
توفر البيانات
البيانات التي تدعم نتائج هذه الدراسة متاحة ضمن المقال وملفات المعلومات التكميلية الخاصة به. جميع بيانات المصدر الأخرى ذات الصلة متاحة من المؤلف المجيب عند الطلب. تم توفير بيانات المصدر الخاصة بالمقال مع هذه الورقة. تم توفير بيانات المصدر مع هذه الورقة.
References
Perry, S. C. et al. Electrochemical synthesis of hydrogen peroxide from water and oxygen. Nat. Rev. Chem. 3, 442-458 (2019).
Lane, B. S. & Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 103, 2457-2474 (2003).
Brillas, E., Sirés, I. & Oturan, M. A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 109, 6570-6631 (2009).
Bu, Y. et al. Carbon-based electrocatalysts for efficient hydrogen peroxide production. Adv. Mater. 33, 2103266 (2021).
Xia, C., Xia, Y., Zhu, P., Fan, L. & Wang, H. Direct electrosynthesis of pure aqueous solutions up to by weight using a solid electrolyte. Science 366, 226-231 (2019).
Xu , J. et al. Organic wastewater treatment by a single-atom catalyst and electrolytically produced . Nat. Sustain. 4, 233-241 (2021).
Lu, Z. et al. High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials. Nat. Catal. 1, 156-162 (2018).
Jiang, Y. et al. Selective electrochemical production through two-electron oxygen electrochemistry. Adv. Energy Mater. 8, 1801909 (2018).
Wang, Y., Waterhouse, G. I. N., Shang, L. & Zhang, T. Electrocatalytic oxygen reduction to hydrogen peroxide: from homogeneous to heterogeneous electrocatalysis. Adv. Energy Mater. 11, 2003323 (2021).
Siahrostami, S . et al. Enabling direct production through rational electrocatalyst design. Nat. Mater. 12, 1137-1143 (2013).
Verdaguer-Casadevall, A. et al. Trends in the electrochemical synthesis of : enhancing activity and selectivity by electrocatalytic site engineering. Nano Lett. 14, 1603-1608 (2014).
Jirkovský, J. S. et al. Single atom hot-spots at Au-Pd nanoalloys for electrocatalytic production. J. Am. Chem. Soc. 133, 19432-19441 (2011).
Kulkarni, A., Siahrostami, S., Patel, A. & Nørskov, J. K. Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 118, 2302-2312 (2018).
Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374-384 (2021).
Wang, A., Li, J. & Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2, 65-81 (2018).
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981-5079 (2018).
Gao, J. et al. Enabling direct production in acidic media through rational design of transition metal single atom catalyst. Chem 6, 658-674 (2020).
Sun, Y. et al. Activity-selectivity trends in the electrochemical production of hydrogen peroxide over single-site metal-nitrogencarbon catalysts. J. Am. Chem. Soc. 141, 12372-12381 (2019).
Xiao, C. et al. Super-coordinated nickel site single-atom catalyst for selective electrosynthesis at high current densities. Angew. Chem. Int. Ed. 61, e202206544 (2022).
Jung, E. et al. Atomic-level tuning of catalyst for highperformance electrochemical production. Nat. Mater. 19, 436-442 (2020).
Chen, S. et al. Identification of the highly active Co-N4 coordination motif for selective oxygen reduction to hydrogen peroxide. J. Am. Chem. Soc. 144, 14505-14516 (2022).
Hammer, B. & Nørskov, J. K. Theoretical surface science and catalysis-calculations and concepts. Adv. Catal. 45, 71-129 (2000).
Nørskov, J. K., Bligaard, T., Rossmeisl, J. & Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37-46 (2009).
Yang, . et al. Tuning two-electron oxygen-reduction pathways for electrosynthesis via engineering atomically dispersed single metal site catalysts. Adv. Mater. 34, 2107954 (2022).
Choi, C. H. et al. Hydrogen peroxide synthesis via enhanced twoelectron oxygen reduction pathway on carbon-coated Pt surface. J. Phys. Chem. C 118, 30063-30070 (2014).
Sheng, H. et al. Linear paired electrochemical valorization of glycerol enabled by the electro-Fenton process using a stable cathode. Nat. Catal. 5, 716-725 (2022).
Choi, C. H. et al. The Achilles’ heel of iron-based catalysts during oxygen reduction in an acidic medium. Energy Environ. Sci. 11, 3176-3182 (2018).
Liu, S. et al. Turning main-group element magnesium into a highly active electrocatalyst for oxygen reduction reaction. Nat. Commun. 11, 938 (2020).
Luo, F. et al. P-block single-metal-site tin/nitrogen-doped carbon fuel cell cathode catalyst for oxygen reduction reaction. Nat. Mater. 19, 1215-1223 (2020).
Yan, M. et al. -templated synthesis of sulfur-doped with hierarchical architecture and high metal loading for electrosynthesis. Nat. Commun. 14, 368 (2023).
Gu, Y., Xi, B. J., Zhang, H., Ma, Y. C. & Xiong, S. L. Activation of maingroup antimony atomic sites for oxygen reduction catalysis. Angew. Chem. Int. Ed. 61, e202202200 (2022).
Zhang, E. et al. Engineering the local atomic environments of indium single-atom catalysts for efficient electrochemical production of hydrogen peroxide. Angew. Chem. Int. Ed. 61, e202117347 (2022).
Zheng, T. et al. Copper-catalysed exclusive to pure formic acid conversion via single-atom alloying. Nat. Nanotechnol. 16, 1386-1393 (2021).
Ross, M. B. et al. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2, 648-658 (2019).
Tong, Y., Yan, X., Liang, J. & Dou, S. X. Metal-based electrocatalysts for methanol electro-oxidation: progress, opportunities, and challenges. Small 17, 1904126 (2021).
Li, X. et al. Microenvironment modulation of single-atom catalysts and their roles in electrochemical energy conversion. Sci. Adv. 6, eabb6833 (2020).
Magyar, J. S. et al. Reexamination of Lead(II) Coordination Preferences in Sulfur-Rich Sites: implications for a critical mechanism of lead poisoning. J. Am. Chem. Soc. 127, 9495-9505 (2005).
Shimoni-Livny, L., Glusker, J. P. & Bock, C. W. Lone pair functionality in divalent lead compounds. Inorg. Chem. 37, 1853-1867 (1998).
Yin, P. et al. Sulfur stabilizing metal nanoclusters on carbon at high temperatures. Nat. Commun. 12, 3135 (2021).
Wang, L. et al. A sulfur-tethering synthesis strategy toward highloading atomically dispersed noble metal catalysts. Sci. Adv. 5, eaax6322 (2019).
Jin, S. et al. A universal graphene quantum dot tethering design strategy to synthesize single-atom catalysts. Angew. Chem. Int. Ed. 59, 21885-21889 (2020).
Jiang, R. et al. Edge-site engineering of atomically dispersed by Selective bond cleavage for enhanced oxygen reduction reaction activities. J. Am. Chem. Soc. 140, 11594-11598 (2018).
Zhao, Y. et al. Stereodefined codoping of sp-N and S atoms in fewlayer graphdiyne for oxygen evolution reaction. J. Am. Chem. Soc. 141, 7240-7244 (2019).
Chen, P. et al. Interfacial engineering of cobalt sulfide/graphene hybrids for highly efficient ammonia electrosynthesis. Proc. Natl. Acad. Sci. USA. 116, 6635-6640 (2019).
Shang, H. et al. Engineering unsymmetrically coordinated single atom sites with enhanced oxygen reduction activity. Nat. Commun. 11, 3049 (2020).
Yang, H. B. et al. Atomically dispersed as the active site for electrochemical reduction. Nat. Energy 3, 140-147 (2018).
Yang, Q. et al. Atomically dispersed Lewis acid sites boost 2-electron oxygen reduction activity of carbon-based catalysts. Nat. Commun. 11, 5478 (2020).
Cao, P. et al. Metal single-site catalyst design for electrocatalytic production of hydrogen peroxide at industrial-relevant currents. Nat. Commun. 14, 172 (2023).
Li, H. et al. Scalable neutral electrosynthesis by platinum diphosphide nanocrystals by regulating oxygen reduction reaction pathways. Nat. Commun. 11, 3928 (2020).
Tang, C. et al. Tailoring acidic oxygen reduction selectivity on single-atom catalysts via modification of first and second coordination spheres. J. Am. Chem. Soc. 143, 7819-7827 (2021).
Martinez, U. et al. Progress in the development of Fe-based PGMfree electrocatalysts for the oxygen reduction reaction. Adv. Mater. 31, 1806545 (2019).
Lin, Z. et al. Tuning the p-orbital electron structure of s-block metal Ca enables a high-performance electrocatalyst for oxygen reduction. Adv. Mater. 33, 2107103 (2021).
Wang, X. et al. Efficient methane electrosynthesis enabled by tuning local availability. J. Am. Chem. Soc. 142, 3525-3531 (2020).
Zhao, Q. et al. Approaching a high-rate and sustainable production of hydrogen peroxide: oxygen reduction on single-atom electrocatalysts in simulated seawater. Energy Environ. Sci. 14, 5444-5456 (2021).
Xia, Y. et al. Highly active and selective oxygen reduction to on boron-doped carbon for high production rates. Nat. Commun. 12, 4225 (2021).
Liu, C. et al. Heterogeneous molecular Co-N-C catalysts for efficient electrochemical synthesis. Energy Environ. Sci. 16, 446-459 (2023).
Zhang, Q. et al. Highly efficient electrosynthesis of hydrogen peroxide on a superhydrophobic three-phase interface by natural air diffusion. Nat. Commun. 11, 1731 (2020).
Lin, Z. et al. Atomic Co decorated free-standing graphene electrode assembly for efficient hydrogen peroxide production in acid. Energy Environ. Sci. 15, 1172-1182 (2022).
Zhao, J. et al. Manipulating the oxygen reduction reaction pathway on Pt-coordinated motifs. Nat. Commun. 13, 685 (2022).
Dong, K. et al. Honeycomb carbon nanofibers: a superhydrophilic -entrapping electrocatalyst enables ultrahigh mass activity for the two-electron oxygen reduction reaction. Angew. Chem. Int. Ed. 60, 10583-10587 (2021).
Li, B.-Q., Zhao, C.-X., Liu, J.-N. & Zhang, Q. Electrosynthesis of hydrogen peroxide synergistically catalyzed by atomic sites and oxygen functional groups in noble-metal-free electrocatalysts. Adv. Mater. 31, 1808173 (2019).
Chen, S. et al. Chemical identification of catalytically active sites on oxygen-doped carbon nanosheet to decipher the high activity for electro-synthesis hydrogen peroxide. Angew. Chem. Int. Ed. 60, 16607-16614 (2021).
Li, L. et al. Tailoring selectivity of electrochemical hydrogen peroxide generation by tunable pyrrolic-nitrogen-carbon. Adv. Energy Mater. 10, 2000789 (2020).
Wang, Y. et al. Vertical graphene array for efficient electrocatalytic reduction of oxygen to hydrogen peroxide. Nano Energy 96, 107046 (2022).
Cao, P. et al. Durable and selective electrochemical synthesis under a large current enabled by the cathode with highly hydrophobic three-phase architecture. ACS Catal. 11, 13797-13808 (2021).
Wang, Y. et al. High-efficiency oxygen reduction to hydrogen peroxide catalyzed by nickel single-atom catalysts with tetradentate N2O2 coordination in a three-phase flow cell. Angew. Chem. Int. Ed. 59, 13057-13062 (2020).
Jin, F. et al. Hydrothermal conversion of carbohydrate biomass into formic acid at mild temperatures. Green Chem. 10, 612-615 (2008).
Yun, J. et al. Low-temperature and highly efficient conversion of saccharides into formic acid under hydrothermal conditions. AIChE J. 62, 3657-3663 (2016).
Li, L. et al. Kilogram-scale synthesis and functionalization of carbon dots for superior electrochemical potassium storage. ACS Nano 15, 6872-6885 (2021).
Gabardo, C. M. et al. Continuous carbon dioxide electroreduction to concentrated multi-carbon products using a membrane electrode assembly. Joule 3, 2777-2791 (2019).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15-50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Rossmeisl, J., Qu, Z. W., Zhu, H., Kroes, G. J. & Nørskov, J. K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83-89 (2007).
Rossmeisl, J., Logadottir, A. & Nørskov, J. K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 319, 178-184 (2005).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886-17892 (2004).
Kattel, S., Atanassov, P. & Kiefer, B. Catalytic activity of Co-N/C electrocatalysts for oxygen reduction reaction: a density functional theory study. Phys. Chem. Chem. Phys. 15, 148-153 (2013).
شكر وتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2021YFA1501003 إلى Y.W.)، ومؤسسة العلوم الطبيعية الوطنية في الصين (52100195 إلى X.Z.، 51821006 إلى H.Q.Y.، 52027815 إلى H.Q.Y.، 52192684 إلى H.Q.Y.، 92261105 إلى Y.W. و 22221003 إلى Y.W.)، ومؤسسة العلوم الطبيعية الإقليمية في آنهوي (2108085QB70 و 2108085UD06 إلى Y.W.)، وبرنامج البحث والتطوير للتقنيات الرئيسية في مقاطعة آنهوي (2022a05020053 إلى Y.W.)، ومؤسسة الابتكار وريادة الأعمال للطلاب في جامعة العلوم والتكنولوجيا في الصين (CY2022G42 إلى S.L.X.) وبرنامج الابتكار التعاوني لمركز هيفي العلمي، الأكاديمية الصينية للعلوم (2021HSC-CIP002 إلى Y.W.). نشكر محطات نهاية التصوير الضوئي BL1W1B في منشأة الإشعاع المتزامن في بكين (BSRF)، وBL14W1 في منشأة الإشعاع المتزامن في شنغهاي (SSRF)، وBL10B وBL11U في المختبر الوطني للإشعاع المتزامن (NSRL) على المساعدة في التوصيفات.
مساهمات المؤلفين
قام H.Q.Y. و Y.E.W. بتصميم الدراسة والإشراف على المشروع. قام X.Z. بإجراء التخليق، والتوصيف، والاختبارات الكهروكيميائية. قام Y.M. و J.J.C. بإجراء الدراسات النظرية. ساعد C.M.Z. و M.K.K. في تخليق المحفزات. ساعد C.C. و S.L.X. في قياسات XANES و EXAFS والنقاش. قام X.Z. و Y.M. و H.Q.Y. و Y.E.W. بتحليل النتائج وكتابة الورقة معًا. ساهم جميع المؤلفين في مناقشة النتائج والمخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى يوان وو أو هان-تشينغ يو.
تُعرب مجلة Nature Communications عن شكرها لتاج تشوكسي، وتشونيو دو، والمراجعين الآخرين المجهولين، على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
مختبر المفاتيح لعلوم تحويل الملوثات الحضرية، قسم علوم البيئة والهندسة، جامعة العلوم والتكنولوجيا في الصين، هيفي 230026، الصين.كلية الكيمياء وعلوم المواد، جامعة العلوم والتكنولوجيا في الصين، هيفي 230026، الصين.ساهم هؤلاء المؤلفون بالتساوي: شياو تشو، يوان مين، تشانغ مينغ تشاو. البريد الإلكتروني:yuenwu@ustc.edu.cn؛hqyu@ustc.edu.cn
Constructing sulfur and oxygen supercoordinated main-group electrocatalysts for selective and cumulative production
Received: 24 July 2023
Accepted: 20 December 2023
Published online: 02 January 2024
(A) Check for updates
Xiao Zhou , Yuan Min , Changming Zhao , Cai Chen , Ming-Kun Ke , Shi-Lin Xu , Jie-Jie Chen ¹, Yuen Wu® & Han-Qing Yu
Direct electrosynthesis of hydrogen peroxide via the two-electron oxygen reduction reaction presents a burgeoning alternative to the conventional energy-intensive anthraquinone process for on-site applications. Nevertheless, its adoption is currently hindered by inferior selectivity and diminished yield induced by consecutive reduction or Fenton reactions. Herein, guided by theoretical calculations, we endeavor to overcome this challenge by activating a main-group Pb single-atom catalyst via a local micro-environment engineering strategy employing a sulfur and oxygen super-coordinated structure. The main-group catalyst, synthesized using a carbon dot-assisted pyrolysis technique, displays an industrial current density reaching and elevated accumulated concentrations with remarkable Faradaic efficiencies. Both experimental results and theoretical simulations elucidate that S and O super-coordination directs a fraction of electrons from the main-group Pb sites to the coordinated oxygen atoms, consequently optimizing the *OOH binding energy and augmenting the oxygen reduction activity. This work unveils novel avenues for mitigating the production-depletion challenge in electrosynthesis through the rational design of main-group catalysts.
Hydrogen peroxide ( ) ranks among the most essential and fundamental chemicals, as it is extensively applied in chemical and medical industries, as well as environmental remediation . Owing to the persistent growth in directly correlated industrial needs, it is projected that the global market demand will approach approximately 6 million tons by . Presently, over of all is industrially synthesized via the anthraquinone cycling process, which is encumbered by intensive energy consumption, significant organic waste, and safety concerns stemming from the instability of during transport and storage . In apparent contrast, the electrochemical two-electron ( ) oxygen reduction reaction (ORR) offers a more direct pathway for production onsite under ambient conditions . Typically, numerous cathode
materials favor the competing ORR to rather than the ORR, resulting in diminished yields . Noble metal alloy catalysts with isolated reactive sites (e.g., are discovered to be advantageous for “end-on” adsorption (Paulingtype) of , which minimizes bond breaking, facilitating the ORR process for production . However, the exorbitant cost and resource scarcity of noble metals severely impede their largescale implementation. Therefore, it is the key to develop highselectivity and non-noble metal catalysts for electrosynthesis.
Heterogeneous single-atom catalysts (SACs), characterized by isolated active sites and distinctive electronic structures, have been shown to hold considerable promise as materials for energy conversion and chemical transformation . In particular, transition-metal
SACs with well-defined moieties have been recognized as exceptional electrocatalysts for production . Taking the moiety as an example, wherein tuning the coordination structure and surrounding atomic configuration effectively optimizes the oxygen intermediates and governs the selectivity towards synthesis . This can be primarily attributed to their unique electronic properties, which exhibit partially occupied electrons that confer a robust capacity for adsorbing and activating , as predicted and elucidated by the -band center theory . Regrettably, reactive transition-metal sites also exhibit strong interactions with synthesized , instigating undesired reduction reactions ( ) within the ORR potential range. Consequently, the generated undergoes decomposition, significantly undermining yield . In addition, transition metal-induced Fenton reactions may contribute to a reduction in production and compromise electrode durability . To this end, the potent adsorption strength of transition-metal sites to oxygencontaining species may exhaust accumulated , thereby curtailing overall production. It is reasonable to predict that optimizing ORR selectivity for intrinsically less reactive catalysts for decomposition may yield higher quantities, although this remains a promising yet formidable challenge for electrosynthesis. Distinct from -block transition metals, main-group metals with fully occupied -orbitals ( electronic configuration) are generally deemed catalytically inferior for the electron transfer process of catalytic reaction . Main-group metals should inherently surpass transition metals in terms of inert activity for and Fenton-like reactions, owning to the lack of a combination of empty and occupied host orbitals. Intriguingly, main-group catalysts have been demonstrated as promising candidates for electrocatalytic reactions, boasting enhanced durability by mitigating transition metal-induced Fenton reactions . Thus, it should be feasible to concurrently attain selective ORR for production and alleviate consecutive decomposition through the development of main-group ORR SACs.
Motivated by the above analysis, we first predicted the ORR process on lead (Pb) SACs with diverse local coordination environments using density functional theory (DFT) calculations. Accordingly, a versatile strategy was devised to regulate the microenvironments of main-group metal sites by meticulously modulating the first coordination with sulfur and oxygen . We then fabricated the maingroup Pb SACs (Pb SA/OSC) employing a carbon dot-assisted pyrolysis approach, wherein the S/O ratio of the carbon dots was rationally controlled. Advanced microscopy analysis and synchrotron X-ray absorption fine structure spectroscopy corroborated the successful regulation of the coordination configuration of the main-group SACs via S and O dual coordination. The ORR performance of the asfabricated Pb SA/OSC was subsequently assessed utilizing the rotating ring-disk electrode technique. Moreover, to explore the possibility for practical application, was assembled into a functional flowcell device for production at industrial currents. This distinctive main-group Pb catalyst potentially heralds a novel avenue for the exploitation of main-group elements in designing electrochemical catalysts for synthesis.
Results
Coordination engineering of Pb SA/OSC
We first examined the adsorption of ORR reaction intermediates on Pb single-atom models with varying local coordination environments to gain insights into the ORR selectivity employing DFT calculations. Pb -based catalysts were chosen as main-group model catalysts, as Pb materials have been demonstrated to exhibit high activity for diverse electrocatalytic reactions . Prior research has indicated that the catalytic reactivities of metal sites are influenced by their coordination environments due to electronic structure alterations . Additionally, it has been reported in the literature that the most common coordination numbers for main-group Pb sites are 4 and . In this instance,
Pb SA/OSC structure models were constructed by supporting Pb atoms on graphene with various combinations of coordinated S and O atoms (Fig. 1a and Supplementary Fig. 1). The optimized geometry of exhibits a planar coordination structure with distances of ( ) and (Supplementary Table 1). In contrast, the distances of and in the other Pb SACs structures display longer interatomic distances. Accordingly, the adsorption energies of and on the selected and other Pb SACs structures were calculated. Correlations between and the limiting potential of the ORR were established to depict scaling relations (Fig. 1b), suggesting that lies nearest to the apex of the limiting potential volcano, akin to the catalyst . Thus, affords high selectivity to for formation with a low overpotential of 0.006 V . Other Pb sites are situated on either the left or right side of the volcano plots (Supplementary Fig. 2), indicating that the adsorption of *OOH is excessively strong or weak, respectively. To provide a more precise description of the ORR reaction, correlations between the and the limiting potential of the ORR were examined. The limiting potential of was far away from the equilibrium potential of the ORR ( 1.23 V ), insinuating that is inactive for the reduction of to . Furthermore, the free energy diagram of the ORR reveals that the adsorption of * OOH on is suitable for the formation and desorption of , while that on is stronger, once again indicating the high selectivity of the ORR on the structure (Fig. 1c).
The adsorption strength of *OOH is recognized as a pivotal descriptors in governing the ORR selectivity of catalysts. Specifically, the binding of *OOH relies upon the electronic energy level of the active site and the corresponding charge transfer at the catalytic interface. Bader charge analysis disclosed that *OOH bears a negative charge with fewer electrons transferred from ( , Fig. 1d) compared to ( , Fig. 1f), consonant with the calculated adsorption strength of *OOH. Color mapping of the Bader charge on each atom shows that the electrons provided by the Pb site are transferred not only to the adsorbed , but also to the coordinated O atoms of (Supplementary Fig. 3). In contrast, electrons of Pb site in are not transferred to the coordinated S atoms, resulting in the strongly adsorbed *OOH gaining more electrons. Projected density of states (PDOS) plots exhibits an overlap of the energy levels of the Pb orbitals and the orbitals of * OOH (Fig. 1e), implying that the adsorption of * OOH is contributed by the orbitals of . For , no overlapped region was observed near the Fermi level (Fig. 1g), indicating that the orbitals were controlled by the coordinated O atom. The impact of the coordination environments on the conversion of * OOH to can be comprehended not only by the weakening of the bond, but also by the tightening of the bond in adsorbed on the Pb site of (Supplementary Table 2). Therefore, the DFT calculations unveiled that the formation of S and O super-coordinated Pb moieties is the origin of the exceptional ORR selectivity of Pb SA/OSC, as a suitable interaction between orbitals and orbitals gives rise to an optimal adsorption strength of * OOH intermediates.
Synthesis and characterizations of Pb SA/OSC
To corroborate the theoretical prediction of exceptional ORR selectivity of Pb SA/OSC, the main-group single atoms were in-situ anchored onto carbon supports employing a carbon dot-assisted pyrolysis approach. The schematic preparation process of Pb SA/OSC is illustrated in Fig. 2a. S, O-functionalized carbon dots (SCDs) were first prepared by utilizing varying ratios of S/O-containing precursors, furnishing a versatile platform for modulating of the local atomic environments of Pb SACs. Notably, the strong chemical interaction between metal and doped sulfur atoms in SCDs endowed the formation of strong and thermally stable metal-sulfur bonding, markedly
Fig. 1 | Theoretical ORR activity of Pb SA/OSC. a Top and side views of *OOH species adsorbed on Pb SA/OSC. Structural motifs considered in the catalyst model of Pb SA/OSC, including in-plane structures of and , tetragonal structures of and . Configurations are denoted by the atomic ratio and tpye, with ” ” indicating the presence of thiophene-type sulfur. Volcano plots for the and ORR on various and , with the limiting potential plotted as a function of (blue horizontal axis) and (red
horizontal axis). The equilibrium potentials of and are 1.23 V and 0.7 V , respectively. c Free energy diagram of ORR on and at an applied potential of zero in alkaline media. d, f Differential charge density plots for * OOH species adsorbed on and , with blue and yellow areas representing a loss and gain of electrons, respectively. The isosurfaces were taken at a charge density of bohr . (e.g.) PDOS plots of the and catalysts with *OOH.
mitigating the aggregation of metal sites . The main-group Pb cations were adequately chelated with the S and O -function groups of SCDs, forming a metal coordination composite, which was assembled on carbon black substrates via interactions . Therefore, the interfacial metal-sulfur bonding between the Pb cations and SCDs effectively anchored and stabilized the main-group sites, proffering an atomically dispersed metal precursor. Then, the main-group Pb SACs with a distinctive S and O dual coordination were obtained via pyrolysis treatment under a protective flowing Ar atmosphere. For comparison, a series of control samples with divergent coordination
environments were also synthesized under otherwise identical conditions but employing distinct carbon dot precursors. These were denoted as and , respectively.
Transmission electron microscopy (TEM) images revealed that Pb SA/OSC largely retained the spherical configuration of carbon black, albeit with a marginally larger size (Supplementary Figs. 8a, b). As shown in Fig. 2b, the obtained sample possessed a layered graphene structure epitaxially grown on carbon black substrates, with no discernible Pb -containing nanoparticles detected. Likewise, observations in the high-angle dark-field scanning TEM (HAADF-STEM) image
Fig. 2 | Morphology and structure of Pb SA/OSC. a Schematic illustration of the preparation of Pb SA/OSC. STEM image and (c) AC HAADF-STEM image of OSC. d Atom-overlapping Gaussian-function fitting mapping of the square from (c),
intensity profile along in (c). e EDS elemental mapping of Pb SA/OSC, suggesting uniform distributions of and C elements.
(Supplementary Fig. 8c) corroborated the aforementioned conclusion. Additionally, no characteristic crystal peaks of metallic Pb or PbS was observed in the XRD pattern (Supplementary Fig. 9) of Pb SA/OSC, harmonizing with the prior TEM results. It merits attention that Raman results (Supplementary Fig. 10) disclosed the characteristic and bands of conductive carbon materials ( ), implying the presence of copious defects in the SCDs-derived carbon materials, favorable for anchoring isolated metal atoms . The porous structure of Pb SA/OSC was studied by the Brunauer-Emmett-Teller (BET) method, displaying a large BET specific surface area of (Supplementary Fig. 11). Furthermore, the OSC, Pb SA/OC, Pb SA/OSC2.5, and Pb SA/OSC-7.5 samples exhibited analogous morphologies, carbon crystallinities and defects, indicating the successful preparation of the main-group Pb SACs (Supplementary Figs. 10, 13-15). The monodispersion of Pb species could be directly monitored by aberration-corrected HAADF-STEM (AC HAADF-STEM). As shown in Fig. 2c and Supplementary Fig. 16, the individual bright dots (labeled with red circles) corresponding to isolated Pb atoms can be unambiguously distinguished from the carbon matrix. The atomic dispersion of Pb in Pb SA/OSC was further substantiated by atomoverlapping Gaussian-function fitting mapping and analysis of intensity profile (Fig. 2d). The energy-dispersive X-ray spectroscopy (EDS) mapping from Fig. 2e indicated that and C on the supports
were uniformly distributed. Furthermore, the actual content of Pb was quantified as via inductively coupled plasma optical emission spectroscopy (ICP-MS) analysis.
Analysis of atomic and electronic structure
To scrutinize the local electronic and atomic structure of the maingroup Pb SACs, synchrotron soft X-ray absorption near-edge structure (XANES) was conducted. The S L-edge XANES spectrum (Fig. 3a) of Pb SA/OSC was dominated by three peaks within the range of corresponding to C-S-C coordination species, which concurred with the findings of the SCDs sample . This observation compellingly suggested the anchoring of S in the carbon framework, validated by the X-ray photoelectron spectroscopy (XPS) survey results (Supplementary Fig. 17). Meanwhile, the carbon K-edge spectrum (Supplementary Fig. 18) showed four characteristic peaks situated at 286.2 eV (peak A), 288.1 eV (peak ), 289.3 eV (peak ) and 293.5 eV (peak C), attributable to the transition of the core electron of carbon into the , and antibonding states, respectively . Sulfur was further probed by the high-resolution spectrum. As shown in Fig. 3b, the peak at 168.3 eV was ascribed to the sulfate species ( ), while the two peaks at 165.2 eV and 164.2 eV were associated with and bonds, respectively . It should be noted that a peak at 163.8 eV corresponding to coordination was
Fig. 3 | Atomic and electronic structure of the Pb SA/OSC catalyst. a S L-edge XANES spectra of SCDs and of Pb SA/OSC samples. and XPS spectra of Pb SA/OSC. The normalized -edge XANES spectra of the Pb SA/OSC catalyst and the references ( Pb foil and PbS ). e -edge FT-EXAFS spectra of
and reference samples. Corresponding EXAFS fitting curves of Pb SA/OSC in R space. Schematic atomic interface model of Pb SA/OSC. WT-EXAFS plots of Pb foil and PbS , respectively.
observed. The high-resolution spectrum can be deconvoluted into , and , indicating the formation of bonds (Fig. 3c). These findings insinuated that the atomically dispersed Pb exhibited typical and dual coordination environments.
Extended X-ray absorption fine structure (EXAFS) was examined to glean detailed structural information for the main-group SACs. As depicted in Fig. 3d, the white line intensity at the edge of OSC resembled that of PbS , signifying that the Pb atom was featured with an ionic state. The Fourier transform (FT) EXAFS spectra of Pb SA/ OSC and the references are illustrated in Fig. 3e. The sample displayed a dominant peak located at about , predominantly attributed to the scattering of coordination. Notably, a peak at about of Pb SA/OSC was ascribed to path scattering, as discerned by the FT-EXAFS spectra compared with PbS . Moreover, the absence of the corresponding feature of the bond substantiated the atomic dispersion of Pb on the carbon framework. Quantitatively, the coordination configuration of the Pb atom in the Pb SA/OSC catalyst could be acquired via EXAFS fitting, as shown in Fig. 3f. The best-fit result of the EXAFS data in Supplementary Table 6 indicates that the isolated Pb atom was atomically dispersed on the co-doped carbon matrix and likely coordinated within a mixture of and moieties (denoted as , Fig. 3g). It is worth noting that the averaged
interatomic distances of EXAFS fitting results concurred with the distances of and in the structure but diverged from the distances of and in the other Pb SACs structures, substantiating the super-coordinated structure of Pb SA/OSC. Wavelet transform (WT) of -edge EXAFS oscillations was conducted to further corroborate the atomic monodispersity of the Pb species in Pb SA/OSC. From the WT contour plots of Pb foil and PbS standards (Fig. 3h), the intensity maxima at 3.7 and were most likely associated with and , respectively. In contrast, the WT contour plot of Pb SA/OSC displayed an intensity maximum at , attributable to bonding. Hence, the above results compellingly suggested the isolated and super-coordinated feature of Pb species in Pb SA/OSC, confirming that the local atomic environments of Pb single sites were modulated by coordination engineering.
ORR performance measurement
Differing from the various SACs reported in the literature, the asfabricated Pb SA/OSC material exhibited a characteristic main-group center and a S, O super-coordinated configuration. Inspired by the distinctive local environment of Pb SA/OSC, the performance of ORR was subsequently assessed using a standard three-electrode system with rotating ring-disk electrode (RRDE) in -saturated 0.10 M
Fig. 4 | Evaluation of two-electron ORR activity and selectivity of Pb SA/OSC using RRDE. a Electrochemical oxygen reduction polarization curves (solid lines) along with detection currents (dashed lines) for and OSC in -saturated 0.10 M KOH electrolyte. selectivity based on the RRDE measurements. c Tafel slopes of different catalysts derived from kinetic current for production. d Schematic illustration of the experimental setup for in-situ ATR-SEIRAS measurements. WE working electrode, CE counter electrode, RE
reference electrode. e In-situ ATR-SEIRAS spectra collected on the Pb SA/OSC catalyst in -saturated 0.10 M KOH catholyte at various potentials ranging from 0.9 to 0.1 V vs RHE. LSV of in -saturated 0.10 M KOH electrolyte containing , or . g Schematics of the electrochemical cumulative production on the main-group metal catalysts (left) and transition metal catalysts (right).
KOH . To study the roles of the main-group center and supercoordination structure in affecting the catalytic reactivity of OSC, control samples of metal-free OSC and Pb SA/OC (with O as only the coordination atom) were also prepared. Before evaluating the selectivity of , the collection coefficient of the Pt ring electrode was determined to be approximately 0.37, calibrated using the redox reaction (Supplementary Fig. 23) . Figure 4a shows the linear sweep voltammetry (LSV) curves of the various catalysts, presenting the current signals for oxygen reduction (solid lines) and the corresponding partial current of (dashed lines) recorded on the disk electrode and ring electrode, respectively. It is obvious that Pb SA/OSC exhibited a superior ORR activity compared to and OSC. exhibited the highest ring current ( 0.204 mA at 0.5 V vs. RHE), suggesting a higher generation of during the ORR process. The selectivity was calculated and plotted as a function of applied potential in Fig. 4b. In a wide potential window from 0.30 V to 0.70 V , the selectivity of the catalyst exceeded 90% (with a maximum of 94%), highlighting a highly selective ORR pathway. In comparison, and OSC showed
much lower selectivity and higher electron transfer number (n) values (Supplementary Fig. 24), confirming the importance of the super-coordination structure in tuning the ORR selectivity. Besides, Pb SA/OSC delivered the highest current density ( ) at 0.65 V (Supplementary Fig. 25) and the most positive onset potential ( ) (defined as the potential at the ring current density of ), compared to the Pb SA/OC and OSC samples.
Furthermore, the Tafel slope for the Pb SA/OSC was determined to be , significantly smaller than those of Pb SA/OC ( ) and OSC ( ), indicating its more favorable kinetics for generation (Fig. 4c). The electrochemically active surface areas (ECSA) were calculated by measuring the double-layer capacitances ( ) of the catalysts. As depicted in Supplementary Fig. 26, the Pb SA/OSC sample exhibited a larger value and a higher availability of exposed active sites compared to the other as-prepared catalysts, making a significant contribution to its superior performance in production. The ORR activity of the catalyst was further investigated in a neutral electrolyte ( 0.10 M phosphate-buffered saline, PBS, ). As shown in Supplementary
Fig. 27, demonstrated a high ORR current ( 0.56 mA at 0.1 V vs. RHE) and selectivity ( ) within the potential range of us. RHE. To confirm the role of the coordination environment in the catalytic selectivity, the ORR performance of samples with different S contents was also studied. All samples delivered satisfactory current density and working potential window (Supplementary Fig. 28), and the Pb SA/OSC catalyst with optimal S content exhibited the best selectivity for ORR activity.
To detect the key adsorbed oxygen intermediate (*OOH) on Pb SA/OSC during electrolytic synthesis, in-situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATRSEIRAS) tests were performed (Fig. 4d). Impressively, a new absorption band at about emerged when applying a potential lower than 0.8 V vs RHE (Fig. 4e). This featured absorption band on Pb SA/OSC can be assigned to the stretching vibration of * OOH , which is consistent with previous studies . Additionally, the absorption band at around was attributed to the stretching mode of adsorbed molecular oxygen ( , ad). The deuterated experimental results show that the vibration band of *OOH underwent a downshift to (Supplementary Fig. 31), indicating the involvement of hydrogen atom and confirming the origin of *OOH. Overall, the potential-dependent ATR-SEIRAS results supported the *OOH mediated ORR pathway on the prepared catalysts (Supplementtary Fig. 32).
To evaluate the cumulated , it is imperative to quantitatively assess the side reaction of the consecutive reduction reaction on the same catalyst . The electrochemical experiments for were conducted in -saturated 0.10 M KOH with varying concentrations of . As shown in Fig. 4f, the rate of electroreduction on Pb SA/OSC exhibited only marginal increase with higher overpotential and concentration. The current density for on Pb SA/OSC was less than when the potential exceeded 0.40 V vs. RHE, indicating limited activity of the main-group sites of Pb SA/OSC towards RR. Similar results were observed for main-group catalysts during the ORR process . Therefore, the Pb SA/OSC catalyst was identified as a highly promising candidates for production owing to its minimal activity and high selectivity during ORR process. Consequently, the net rate of production (i.e., production rate minus electroreduction rate of ) on Pb SA/OSC can be maintained at a sufficiently high level as the concentration of built up. Additionally, the potential Fenton reactions catalyzed by transition-metal sites may deplete the accumulated and compromise the durability of the electrode . In contrast to transition-metal sites, the main-group metal sites inherently possess lower activity towards Fenton reactions . As shown in Supplementary Fig. 35, no characteristic signals of hydroxyl radical from Fenton reactions were observed in the EPR spectra of Pb SACs during the ORR process. Hence, ORR catalysts based on maingroup metal sites are expected to be an attractive option for mitigating Fenton reactions during production. Overall, the Pb SA/OSC catalyst demonstrates significant potential in meeting the requirements for accumulative production by suppressing and Fenton reactions (Fig. 4g).
Practical scale electrosynthesis
Inspired by the remarkable 2e- ORR selectivity and potential for production of the Pb SA/OSC catalyst, a gas diffusion electrode (GDE) and a customized flow-cell electrolyzer were employed for practical synthesis. As illustrated in Fig. 5a, LSV was initially performed in a flow-cell setup in 1 M KOH with manual iR compensation . As shown in Fig. 5b, the Pb SA/OSC catalyst exhibited significantly higher current density compared to the and OSC , suggesting the crucial role of the coordination environment of the in electrosynthesis. Bulk electrolysis experiments were then conducted
at different current densities ranging from 50 to to evaluate the rates of production. The accumulated concentration of in the catholyte was determined using a colorimetric quantification method (Supplementary Fig. 39). Importantly, the Faradaic efficiencies (FEs) of for generation remained above across the applied current density range (Fig. 5d). When the concentration of builds up in the electrolyte, a high density of leads to self-decomposition or . Impressively, during bulk electrolysis at (Fig. 5c), the concentration of the accumulated increased almost linearly and reached ) after 2 h of electrolysis. The superior performance of the OSC catalyst indicated that the main-group sites with a regulated local coordination environment effectively prevented the selfdecomposition or further electrochemical reduction of generated . This finding aligns with the observations obtained from the EPR measurements and reduction reaction experiments, thereby confirming the minimal Fenton and activity of the . Furthermore, the calculations indicate that molecular could be stabilized and accumulated at the solid-liquid interface of the OSC, effectively inhibiting the decomposition of . It is noteworthy that the FEs only exhibited a slight decrease trend (97% to 91%) during constant current electrolysis at .
Furthermore, the Pb SA/OSC catalyst exhibited a high production rate at different current densities ranging from 50 to (Fig. 5e). At , the rate of generation reached , ranking among the top performances of the state-of-the-art 2e- ORR catalysts (Supplementary Table 7) . To investigate the stability of Pb SA/OSC, long-term electrolysis was conducted at a constant current density of . As shown in Fig. 5f, a high FE (above ) was maintained without noticeable degradation for at least 100 h , enabling continuous and stable production of , which is critical for industrial applications. The morphology, atomic Pb dispersion, and carbon crystallinity of the OSC were well-preserved after stability testing (Supplementary Figs. 44-46). This underscores the structural stability of the catalyst for the ORR. Moreover, the results obtained from ICP-MS showed negligible leaching of both Pb and S (around of Pb and of S in Pb SA/OSC) into the bulk electrolyte after 6 h of continuous electrolysis.
The Pb SA/OSC catalyst exhibited impressive performance in electrosynthesis of . To further evaluated its practical applications, we coupled the obtained solution with biomass-derived carbohydrate conversion for the production of formic acid ( HCOOH ). Previous studies have reported that biomass-derived carbohydrates can be transformed to formic acid, an important carrier, through hydrothermal oxidation in the presence of and alkali . Taking glucose to HCOOH conversion as a representative example, a high HCOOH yield was obtained using alkali as a catalyst and as an oxidant. The alkali played a crucial role in facilitating selective oxidation and inhibiting overoxidation of the formed HCOOH (Supplementary Fig. 47). In this regard, the solution obtained by electrosynthesis provided an ideal platform for biomass-derived carbohydrate conversion, as it supplied both alkali and simultaneously. The accumulated solution ( 1358 mM ) obtained by bulk electrolysis ( for 2 h ) was used for the hydrothermal oxidation of glucose at different temperatures. For comparison, commercial with an optimized amount of KOH was used under the same conditions for the above reactions. As shown in Fig. 5g, both the electrochemically produced solution and the commercial delivered extraordinary yields of HCOOH (over ) at temperatures of 150 and . Additionally, a slight decrease in the yield of HCOOH was observed at a lower temperature . The electrocatalytically produced solution can be directly applied for biomass-derived carbohydrate conversion without further treatment.
Fig. 5 | Electrosynthesis performance of in a flow-cell electrolyzer based on the GDE. a Schematic illustration of the flow-cell device. LSV curves for PbSA / OSC, Pb SA/OC, and OSC. The curves are manually iR-compensated . c The accumulated concentrations and Faradaic efficiencies of at . Faradaic efficiencies and of at different current densities. Error bars in (c, d) correspond to the standard
deviation of three independent measurements. e production rate of OSC compared to the reported state-of-the-art electrocatalysts. Stability test and selectivity of the Pb SA/OSC catalyst under , with the electrolyte refreshed every 6 h during the test. g Yield of formic acid from the oxidation of glucose by the produced and commercial at different temperatures.
Discussion
In summary, we developed a main-group single-atom electrocatalyst featuring S , O super-coordinated Pb sites anchored in carbon frameworks through a carbon dot-assisted pyrolysis approach. Through rational engineering of the coordination environments of the maingroup sites, the Pb SA/OSC catalysts exhibited remarkable performances in the ORR toward with a high selectivity of up to . Furthermore, industrial current densities up to with for production were achieved at an impressive rate of in a GDE-based flow-cell electrolyzer in an alkaline medium, surpassing the performance of most of the state-of-the-art ORR catalysts. The Pb SA/OSC catalyst maintained adequate stability without an obvious decrease in production and Faradaic efficiency during 100 -h continuous electrolysis. Notably, due to the inert activity of the main-group Pb SACs for the and Fentonlike reactions, the concentration of the accumulated solution
reached 1358 mM with high FE during bulk electrolysis. Moreover, the obtained solution was successfully utilized for the hydrothermal oxidation of renewable biomass-derived glucose, delivering an extraordinary yield of HCOOH (over ). Combined experimental and theoretical studies suggest that the superior catalytic performance of Pb SA/OSC in the ORR can be attributed to the supercoordination regulated microenvironments of the moieties. This work presents a promising avenue for the design of ORR catalysts to address the production-depletion dilemma in electrosynthesis and could open the door for green production of formic acid from renewable biomass.
Methods
Chemicals
Acetaldehyde (40% water solution), sodium hydroxide, potassium hydroxide, hydrochloric acid, lead (II) acetate trihydrate ( ),
glucose and anhydrous ethanol were supplied by Sinopharm Chemical Reagent Co., China. 2-Mercaptopropionic acid (98%) and titanium (IV) oxysulfate-sulfuric acid hydrate (93%) were purchased from Macklin Co., China. Nafion solution ( ) was provided by Sigma-Aldrich Co., USA. Carbon black (Vulcan XC-72) was bought from The Cabot Co., USA. All chemicals and reagents were directly used as received without further purification.
Preparation of SCDs and OCDs
The sulfur-doped carbon dots (SCDs) were synthesized by using a high-efficiency aldol condensation method at room temperature . In a typical preparation procedure, 5 mL of 2-mercaptopropionic acid was dissolved in 40 mL of acetaldehyde. Then, 15 g of NaOH was slowly added and stirred for 2.5 h . The product obtained after the completion of the reaction was sonicated with a certain amount of deionized water for 30 min . An aqueous solution of the crude product was centrifuged ( for 10 min to remove large or agglomerated particles. The supernatant was collected and transferred into a dialysis bag (MWCO: 3500 Da) to dialyze for two days.
The SCDs samples with different S contents were prepared by using a similar procedure except for changing the amount of 2 -mercaptopropionic acid to , and 7.5 mL . They are denoted as OCDs, SCDs-2.5 and SCDs-7.5, respectively.
Synthesis of Pb SA/OSC
In a typical synthesis of the Pb SA/OSC catalyst, first lead precursor solution was prepared by dissolving in to achieve a stock solution. Subsequently, 300 mg of purified SCDs was dispersed in 50 mL of under sonication for 10 min to obtain the SCDs solution. Then, 1 mL of solution and 300 mg of Vulcan XC-72 in ethanol ( 10 mL ) were added to the above solution, followed by sonication for 15 min . Afterward, the abovemixed solution was dried first by rotary evaporation and then in a vacuum oven at . The resulting black powder was heated to (heating rate, ) for 2 h in a nitrogen flow. After thorough washing with water and ethanol, the dried powders were subjected to high-temperature annealing at (heating rate, ) for 2 h in a nitrogen flow. After being naturally cooled to room temperature, the Pb SA/OSC catalyst was obtained.
For the and Pb SA/OSC-7.5 samples, the synthesis process is the same as that of Pb SA/OSC, but the S-doped carbon dots are SCDs-2.5 and SCDs-7.5, respectively. Moreover, the OSC sample was prepared by the same procedure as described for Pb SA/OSC but without any Pb precursor addition.
Synthesis of Pb SA/OC
For the Pb SA/OC catalyst preparation, 300 mg of purified OCDs was dispersed in 50 mL of under sonication for 10 min to obtain the OCDs solution. Then, 1 mL of solution and 300 mg of Vulcan XC-72 in ethanol ( 10 mL ) were added to the above solution, followed by sonication for 15 min . Afterward, the above-mixed solution was dried first by rotary evaporation and then in a vacuum oven at . The resulting black powder was heated to (heating rate, ) for 2 h in a nitrogen flow. After thorough washing with water and ethanol, the dried powders were subjected to high-temperature annealing at (heating rate, ) for 2 h in a nitrogen flow. After being naturally cooled to room temperature, was obtained.
Characterizations
The morphologies of the electrocatalysts were characterized by transmission electron microscopy (TEM) using a microscope (7700, Hitachi Co., Japan) with an accelerating voltage of 100 kV . Aberrationcorrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and element mapping were obtained on a STEM instrument (JEM-ARM200CF, JEOL Co., Japan)
incorporated with double spherical aberration correctors operated at 200 kV . X-ray diffraction (XRD) measurements were recorded on a Rigaku Miniflex-600 diffractometer using radiation ( ) with a step size of and a counting time of 0.5 s . X-ray photoelectron spectroscopy (XPS) was collected on a scanning X-ray microprobe (PHI 5000 Verasa, ULAC-PHI Inc., USA) using Al Ka radiation and the C1s peak at 284.8 eV as an internal standard. Raman spectra were taken using a Raman spectroscopy instrument (LabRAM HR Evolution, Horiba Co., Japan) equipped with a green laser emitting at 532 nm . Elemental analysis of Pb in the solid samples analyzed with inductively coupled plasma-mass spectrometry (ICP-MS).
Soft X-ray absorption spectra (Soft-XAS) were carried out at the Catalysis and Surface Science endstation at the BL11U Beamline and Photoemission Endstation at the BL10B beamline in the National Synchrotron Radiation Laboratory (NSRL) in Hefei, China. X-ray absorption fine structure (XAFS) spectroscopy measurements were performed at the -edge. Data were collected at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF), China. The storage ring was operated at 3.5 GeV with a maximum electron current of foil and PbS were used as references. The edge X-ray absorption near-edge spectroscopy (XANES) data were recorded in fluorescence mode. The hard X-ray was monochromatized with Si (111) double-crystals. The acquired EXAFS data were extracted and processed according to standard procedures using the ATHENA module implemented in the IFEFFIT software package. The -weighted EXAFS spectra were obtained by subtracting the post-edge background from the overall absorption and then normalizing with respect to the edge-jump step. Subsequently, -weighted data in the -space ranging from were Fourier transformed to real (R) space using hanning windows ( ) to separate the EXAFS contributions from different coordination shells. In situ attenuated total reflection surface-enhanced infrared absorption spectra (ATRSEIRAS) measurements were performed on a Nicolet iS50 FT-IR spectrometer (Thermo Scientific Co., USA) with an MCT detector.
Electrochemical measurements
The electrochemical experiments were performed in a standard three electrode system controlled by a CHI 760e workstation (CH Instruments Co., China). A rotating ring disk electrode (RRDE, Pine Research Instrumentation, USA) consisting of a glassy carbon electrode (disk area: ) and a platinum ring electrode (ring area: ) was used as the working electrode, with a theoretical collection efficiency of electrode (saturated KCl solution) and a Pt wire were used as the reference and counter electrodes, respectively. All the recorded potentials were normalized by converting to reversible hydrogen electrode (RHE) according to the equation . The potential versus RHE was adopted unless otherwise specified.
To prepare the working electrode, 4 mg of the catalysts and of Nafion solution ( ) were mixed with of 1:1 ( ) water/ethanol mixed solution and then dispersed by ultrasonication for 30 min until a homogeneous catalyst ink was obtained. After polishing the RRDE mechanically with an alumina suspension, of catalyst ink was dropped on the disk electrode and dried to form a uniform thin film at room temperature. All electrochemical measurements were carried out in or -saturated 0.10 M KOH and 0.10 M PBS aqueous solution. Prior to the ORR measurement, the pre-activation process by scanning CV scans at a rate of from 0.05 to 1.0 V was performed on the RRDE until a stable state was reached. Then, the ORR polarization curves were acquired from LSV measurements under -saturated conditions between 0.05 and 1.0 V at a scan rate of at 1600 rpm . While the Pt ring potential was held at 1.2 V to quantify the amount of generated at the disc electrode. Especially, the Pt ring electrode was electrochemically cleaned through scanning CV curves for 20 cycles
(potential range: , scanning rate: ) on the Pt ring before LSV measurement.
The collection efficiency ( N ) on the RRDE electrode was calibrated in electrolyte at different rotation rates. Thus, the measured collection efficiency was determined to be , which is reasonably close to the theoretical value. The reduction reaction ( ) was studied by conducting LSV measurements of the catalyst-coated disk electrode in -saturated 0.1 M KOH solution containing , or at 1600 rpm .
The selectivity of hydrogen peroxide ( ) and electron transfer number ( n ) were calculated with the following equation:
where is the ring current, is the disk current and N is the determined collection efficiency.
The kinetic current density ( ) was calculated using the Koutecký-Levich equation:
where is the measured current density and is the diffusion-limited current density. The value is acquired by dividing the ring current by the collection efficiency ( N ) and the disk electrode area. The value was taken from the highest value in the j plot measured over the entire potential range investigated ( ).
Tafel slopes were calculated according to the Tafel equation:
where is the constant, is the applied potential, is the Tafel slope, and is the kinetic current density for production.
Electrochemical capacitance measurements were conducted from double-layer charging curves using the CVs at the non-Faradic potential window in 0.10 M KOH solution. These samples were tested with scan rates ranging from 10 to . The plots of current density (at 1.05 V ) as a function of the scan rate showed the double-layer capacitance by the slopes.
Bulk electrosynthesis of
The cathode was fabricated by spray-coating the as-synthesized OSC catalyst on a gas diffusion electrode (GDE, YLS30, Suzhou Sinero Technology Co., China). In a typical process, 8 mg of catalyst was mixed with of Nafion solution and 4 ml of (vol/vol) ethanol/water mixed solvent. The above solution was sonicated for at least 60 min to form a homogeneous ink. The well-dispersed ink was then airbrushed onto a GDE ( ) with a loading amount of (Supplementary Fig. 41). The gas diffusion electrode (GDE) was dried in a vacuum chamber overnight and cut to a size of . A Ti-mesh electrode coated with and an electrode (saturated KCl ) were employed as the anode and reference electrode, respectively. The anode was prepared by depositing on a titanium support by a dip coating and thermal decomposition method . Briefly, the Ti mesh was cut to a size of area and washed with acetone and DI water, and then ultrasonicated in a 6 M HCl aqueous solution heated to for 45 min before dip coating. The coating stock solution consisted of 30 mg of (Alfa Aesar) dissolved in 10 mL of an isopropanol solution with concentrated HCl . The Ti mesh was dipped into the solution, followed by drying in a vacuum oven at for 10 min before calcination in a
furnace at for 10 min . The dipping and calcination process was repeated until a loading was achieved.
Practical production was carried out using a gas-tight flow cell system with an gas compartment and two liquid compartments. The catholyte and anolyte chambers were separated by a cation exchange membrane (Nafion 117, Fuel Cell Store Co., USA). The electrolyzer cell consisted of two titanium backplates with a serpentine flow field. During the electrocatalytic tests, the electrolyte ( 1 M KOH ) was recycled at a constant flow rate of approximately with a peristaltic pump, and the gas feed was maintained at flowing through the cathode using a flow controller. The constant currents were provided by a workstation (PMC1000 , PARSTAT Inc., USA). For the test of the production rate, the cathode current density was increased from 50 to . The 1 M KOH electrolyte was refreshed periodically every 6 h in the stable production up to 100 h . The concentration was quantitatively determined by the titanium sulfonate titration method . After the addition of , a yellow solution of was produced. A linear calibration relationship between the concentration and the absorbance peak at 408 nm was acquired using UVvis spectroscopy. Here, in was prepared by stirring overnight until a transparent solution was formed, which was used to spectrophotometrically quantify the high concentration of produced in the flow cell.
Electrochemical impedance spectroscopy (EIS) was conducted at 0.75 V (vs. RHE) from 100,000 to 0.1 Hz to determine the uncompensated resistance in a high-frequency range for iR-correction in the electrochemical cell.
The faradaic efficiency of production ( ) was determined by the following equation:
where is the produced concentration ( ) in the electrolyte, refers to the volume of electrolyte ( L ), F indicates the Faraday constant ( ), and are the operating current (A) and test time (s), respectively.
Glucose oxidation for formic acid and product analysis
The selective oxidation of glucose was studied by batch experiments. In a typical experiment, 20 mL of the alkaline solution in the flow cell was cycled for 2 h at . Then, 4 mL of the above alkaline solution and 70 mg of glucose were introduced into the reactor. Then, the reactor was transferred to an oven preheated to the required temperature ( 120,150 or ). After 15 min , the reactor was removed from the oven and immersed in a cold-water bath. By comparison, the same amount of commercial with the optimized concentration of were used for the above reactions under the same conditions. After the reaction, the pH of the solution was adjusted to 3 with HCl for high-performance liquid chromatography (HPLC) analysis (LC16, Shimadzu Co., Japan). HPLC analyses were performed on an Amine HPX-87H column with a refractive index detector.
Computational methods
Density functional theory (DFT) calculations were performed by Vienna Ab initio Simulation Package (VASP) with the projector augment wave (PAW) method . The electron exchange and correlation energy were treated within the generalized gradient approximation with the Perdew-Burke-Ernzerhof (GGA-PBE) . Spin polarization was taken into account in all calculations. The k -points sampling for Brillioun zone was set as . The convergence criteria for geometry (self-consistent field) were set to for energy and for forces. The cutoff energy for the kinetic energy of the plane waves was
400 eV . The electronic configuration of lead is and a valence electron potential was considered in the calculations. Partial occupancies were determined using a Gaussian smearing scheme with a smearing width of 0.05 eV . All geometries were fully relaxed to the ground state. The structures of the -water interface were sampled through the random placement of 36 water molecules in the water-slab region followed by local optimization started with two sets of configurations. To describe the electrostatics and dispersion at the interface, the polarizable continuum model implemented in VASPsol with relative permittivity of 78.4 as water was employed . The van der Waals interactions were considered using the empirical DFT-D3 method proposed by Grimme et al. . The ground state structures of adsorbed * and *OH were determined by searching the lowest energy one among all the possible configurations on possible active sites.
Gibbs free energy change ( ) was calculated as , where is the electronic energy difference directly obtained from DFT calculations, is the zero-point energy difference, is the room temperature ( 298.15 K ) and is the entropy change. where is potential at the electrode, e is charge transferred and where is the Boltzmann constant and reduction to follows a two-electron reaction mechanism as below:
where asterisk (*) denotes the Pb site of catalyst and * OOH denotes the intermediate adsorbed on the catalyst surface. The overpotential that evaluates the performance of the ORR is obtained as , where 0.7 represents the equilibrium potential. The limiting potential is the potential at which the reaction step becomes exergonic, .
We assume for acidic medium. The associative fourelectron reaction is composed of the following elementary steps:
The overpotential of the ORR is obtained as , , where 1.23 represents the equilibrium potential. The limiting potential is determined by , e. Adsorption free energies of any reaction intermediate i under standard conditions are conveniently calculated as , where is the adsorption energy and is free energy contributions to the free adsorbate. The values for for adsorbates at the Pb site of catalysts are provided in Supplementary Table 5. All adsorption energies are then consistently referenced relative to the stable molecules of and in Supplementary Table 6, together with experimental energy change of reduction of to to construct the free energy diagram. The free energies of and at a given potential relative to normal hydrogen electrode (NHE) are defined as follows:
We assume for alkaline medium. The corresponding associative four-electron reaction is composed of the following elementary steps:
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information files. All other relevant source data are available from the responding author upon request. Source data of the article are provided with this paper. Source data are provided with this paper.
References
Perry, S. C. et al. Electrochemical synthesis of hydrogen peroxide from water and oxygen. Nat. Rev. Chem. 3, 442-458 (2019).
Lane, B. S. & Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 103, 2457-2474 (2003).
Brillas, E., Sirés, I. & Oturan, M. A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 109, 6570-6631 (2009).
Bu, Y. et al. Carbon-based electrocatalysts for efficient hydrogen peroxide production. Adv. Mater. 33, 2103266 (2021).
Xia, C., Xia, Y., Zhu, P., Fan, L. & Wang, H. Direct electrosynthesis of pure aqueous solutions up to by weight using a solid electrolyte. Science 366, 226-231 (2019).
Xu , J. et al. Organic wastewater treatment by a single-atom catalyst and electrolytically produced . Nat. Sustain. 4, 233-241 (2021).
Lu, Z. et al. High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials. Nat. Catal. 1, 156-162 (2018).
Jiang, Y. et al. Selective electrochemical production through two-electron oxygen electrochemistry. Adv. Energy Mater. 8, 1801909 (2018).
Wang, Y., Waterhouse, G. I. N., Shang, L. & Zhang, T. Electrocatalytic oxygen reduction to hydrogen peroxide: from homogeneous to heterogeneous electrocatalysis. Adv. Energy Mater. 11, 2003323 (2021).
Siahrostami, S . et al. Enabling direct production through rational electrocatalyst design. Nat. Mater. 12, 1137-1143 (2013).
Verdaguer-Casadevall, A. et al. Trends in the electrochemical synthesis of : enhancing activity and selectivity by electrocatalytic site engineering. Nano Lett. 14, 1603-1608 (2014).
Jirkovský, J. S. et al. Single atom hot-spots at Au-Pd nanoalloys for electrocatalytic production. J. Am. Chem. Soc. 133, 19432-19441 (2011).
Kulkarni, A., Siahrostami, S., Patel, A. & Nørskov, J. K. Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 118, 2302-2312 (2018).
Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374-384 (2021).
Wang, A., Li, J. & Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2, 65-81 (2018).
Liu, L. & Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 118, 4981-5079 (2018).
Gao, J. et al. Enabling direct production in acidic media through rational design of transition metal single atom catalyst. Chem 6, 658-674 (2020).
Sun, Y. et al. Activity-selectivity trends in the electrochemical production of hydrogen peroxide over single-site metal-nitrogencarbon catalysts. J. Am. Chem. Soc. 141, 12372-12381 (2019).
Xiao, C. et al. Super-coordinated nickel site single-atom catalyst for selective electrosynthesis at high current densities. Angew. Chem. Int. Ed. 61, e202206544 (2022).
Jung, E. et al. Atomic-level tuning of catalyst for highperformance electrochemical production. Nat. Mater. 19, 436-442 (2020).
Chen, S. et al. Identification of the highly active Co-N4 coordination motif for selective oxygen reduction to hydrogen peroxide. J. Am. Chem. Soc. 144, 14505-14516 (2022).
Hammer, B. & Nørskov, J. K. Theoretical surface science and catalysis-calculations and concepts. Adv. Catal. 45, 71-129 (2000).
Nørskov, J. K., Bligaard, T., Rossmeisl, J. & Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37-46 (2009).
Yang, . et al. Tuning two-electron oxygen-reduction pathways for electrosynthesis via engineering atomically dispersed single metal site catalysts. Adv. Mater. 34, 2107954 (2022).
Choi, C. H. et al. Hydrogen peroxide synthesis via enhanced twoelectron oxygen reduction pathway on carbon-coated Pt surface. J. Phys. Chem. C 118, 30063-30070 (2014).
Sheng, H. et al. Linear paired electrochemical valorization of glycerol enabled by the electro-Fenton process using a stable cathode. Nat. Catal. 5, 716-725 (2022).
Choi, C. H. et al. The Achilles’ heel of iron-based catalysts during oxygen reduction in an acidic medium. Energy Environ. Sci. 11, 3176-3182 (2018).
Liu, S. et al. Turning main-group element magnesium into a highly active electrocatalyst for oxygen reduction reaction. Nat. Commun. 11, 938 (2020).
Luo, F. et al. P-block single-metal-site tin/nitrogen-doped carbon fuel cell cathode catalyst for oxygen reduction reaction. Nat. Mater. 19, 1215-1223 (2020).
Yan, M. et al. -templated synthesis of sulfur-doped with hierarchical architecture and high metal loading for electrosynthesis. Nat. Commun. 14, 368 (2023).
Gu, Y., Xi, B. J., Zhang, H., Ma, Y. C. & Xiong, S. L. Activation of maingroup antimony atomic sites for oxygen reduction catalysis. Angew. Chem. Int. Ed. 61, e202202200 (2022).
Zhang, E. et al. Engineering the local atomic environments of indium single-atom catalysts for efficient electrochemical production of hydrogen peroxide. Angew. Chem. Int. Ed. 61, e202117347 (2022).
Zheng, T. et al. Copper-catalysed exclusive to pure formic acid conversion via single-atom alloying. Nat. Nanotechnol. 16, 1386-1393 (2021).
Ross, M. B. et al. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2, 648-658 (2019).
Tong, Y., Yan, X., Liang, J. & Dou, S. X. Metal-based electrocatalysts for methanol electro-oxidation: progress, opportunities, and challenges. Small 17, 1904126 (2021).
Li, X. et al. Microenvironment modulation of single-atom catalysts and their roles in electrochemical energy conversion. Sci. Adv. 6, eabb6833 (2020).
Magyar, J. S. et al. Reexamination of Lead(II) Coordination Preferences in Sulfur-Rich Sites: implications for a critical mechanism of lead poisoning. J. Am. Chem. Soc. 127, 9495-9505 (2005).
Shimoni-Livny, L., Glusker, J. P. & Bock, C. W. Lone pair functionality in divalent lead compounds. Inorg. Chem. 37, 1853-1867 (1998).
Yin, P. et al. Sulfur stabilizing metal nanoclusters on carbon at high temperatures. Nat. Commun. 12, 3135 (2021).
Wang, L. et al. A sulfur-tethering synthesis strategy toward highloading atomically dispersed noble metal catalysts. Sci. Adv. 5, eaax6322 (2019).
Jin, S. et al. A universal graphene quantum dot tethering design strategy to synthesize single-atom catalysts. Angew. Chem. Int. Ed. 59, 21885-21889 (2020).
Jiang, R. et al. Edge-site engineering of atomically dispersed by Selective bond cleavage for enhanced oxygen reduction reaction activities. J. Am. Chem. Soc. 140, 11594-11598 (2018).
Zhao, Y. et al. Stereodefined codoping of sp-N and S atoms in fewlayer graphdiyne for oxygen evolution reaction. J. Am. Chem. Soc. 141, 7240-7244 (2019).
Chen, P. et al. Interfacial engineering of cobalt sulfide/graphene hybrids for highly efficient ammonia electrosynthesis. Proc. Natl. Acad. Sci. USA. 116, 6635-6640 (2019).
Shang, H. et al. Engineering unsymmetrically coordinated single atom sites with enhanced oxygen reduction activity. Nat. Commun. 11, 3049 (2020).
Yang, H. B. et al. Atomically dispersed as the active site for electrochemical reduction. Nat. Energy 3, 140-147 (2018).
Yang, Q. et al. Atomically dispersed Lewis acid sites boost 2-electron oxygen reduction activity of carbon-based catalysts. Nat. Commun. 11, 5478 (2020).
Cao, P. et al. Metal single-site catalyst design for electrocatalytic production of hydrogen peroxide at industrial-relevant currents. Nat. Commun. 14, 172 (2023).
Li, H. et al. Scalable neutral electrosynthesis by platinum diphosphide nanocrystals by regulating oxygen reduction reaction pathways. Nat. Commun. 11, 3928 (2020).
Tang, C. et al. Tailoring acidic oxygen reduction selectivity on single-atom catalysts via modification of first and second coordination spheres. J. Am. Chem. Soc. 143, 7819-7827 (2021).
Martinez, U. et al. Progress in the development of Fe-based PGMfree electrocatalysts for the oxygen reduction reaction. Adv. Mater. 31, 1806545 (2019).
Lin, Z. et al. Tuning the p-orbital electron structure of s-block metal Ca enables a high-performance electrocatalyst for oxygen reduction. Adv. Mater. 33, 2107103 (2021).
Wang, X. et al. Efficient methane electrosynthesis enabled by tuning local availability. J. Am. Chem. Soc. 142, 3525-3531 (2020).
Zhao, Q. et al. Approaching a high-rate and sustainable production of hydrogen peroxide: oxygen reduction on single-atom electrocatalysts in simulated seawater. Energy Environ. Sci. 14, 5444-5456 (2021).
Xia, Y. et al. Highly active and selective oxygen reduction to on boron-doped carbon for high production rates. Nat. Commun. 12, 4225 (2021).
Liu, C. et al. Heterogeneous molecular Co-N-C catalysts for efficient electrochemical synthesis. Energy Environ. Sci. 16, 446-459 (2023).
Zhang, Q. et al. Highly efficient electrosynthesis of hydrogen peroxide on a superhydrophobic three-phase interface by natural air diffusion. Nat. Commun. 11, 1731 (2020).
Lin, Z. et al. Atomic Co decorated free-standing graphene electrode assembly for efficient hydrogen peroxide production in acid. Energy Environ. Sci. 15, 1172-1182 (2022).
Zhao, J. et al. Manipulating the oxygen reduction reaction pathway on Pt-coordinated motifs. Nat. Commun. 13, 685 (2022).
Dong, K. et al. Honeycomb carbon nanofibers: a superhydrophilic -entrapping electrocatalyst enables ultrahigh mass activity for the two-electron oxygen reduction reaction. Angew. Chem. Int. Ed. 60, 10583-10587 (2021).
Li, B.-Q., Zhao, C.-X., Liu, J.-N. & Zhang, Q. Electrosynthesis of hydrogen peroxide synergistically catalyzed by atomic sites and oxygen functional groups in noble-metal-free electrocatalysts. Adv. Mater. 31, 1808173 (2019).
Chen, S. et al. Chemical identification of catalytically active sites on oxygen-doped carbon nanosheet to decipher the high activity for electro-synthesis hydrogen peroxide. Angew. Chem. Int. Ed. 60, 16607-16614 (2021).
Li, L. et al. Tailoring selectivity of electrochemical hydrogen peroxide generation by tunable pyrrolic-nitrogen-carbon. Adv. Energy Mater. 10, 2000789 (2020).
Wang, Y. et al. Vertical graphene array for efficient electrocatalytic reduction of oxygen to hydrogen peroxide. Nano Energy 96, 107046 (2022).
Cao, P. et al. Durable and selective electrochemical synthesis under a large current enabled by the cathode with highly hydrophobic three-phase architecture. ACS Catal. 11, 13797-13808 (2021).
Wang, Y. et al. High-efficiency oxygen reduction to hydrogen peroxide catalyzed by nickel single-atom catalysts with tetradentate N2O2 coordination in a three-phase flow cell. Angew. Chem. Int. Ed. 59, 13057-13062 (2020).
Jin, F. et al. Hydrothermal conversion of carbohydrate biomass into formic acid at mild temperatures. Green Chem. 10, 612-615 (2008).
Yun, J. et al. Low-temperature and highly efficient conversion of saccharides into formic acid under hydrothermal conditions. AIChE J. 62, 3657-3663 (2016).
Li, L. et al. Kilogram-scale synthesis and functionalization of carbon dots for superior electrochemical potassium storage. ACS Nano 15, 6872-6885 (2021).
Gabardo, C. M. et al. Continuous carbon dioxide electroreduction to concentrated multi-carbon products using a membrane electrode assembly. Joule 3, 2777-2791 (2019).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15-50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865-3868 (1996).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Rossmeisl, J., Qu, Z. W., Zhu, H., Kroes, G. J. & Nørskov, J. K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83-89 (2007).
Rossmeisl, J., Logadottir, A. & Nørskov, J. K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 319, 178-184 (2005).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886-17892 (2004).
Kattel, S., Atanassov, P. & Kiefer, B. Catalytic activity of Co-N/C electrocatalysts for oxygen reduction reaction: a density functional theory study. Phys. Chem. Chem. Phys. 15, 148-153 (2013).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA1501003 to Y.W.), the National Natural Science Foundation of China (52100195 to X.Z., 51821006 to H.Q.Y., 52027815 to H.Q.Y., 52192684 to H.Q.Y., 92261105 to Y.W. and 22221003 to Y.W.), the Anhui Provincial Natural Science Foundation (2108085QB70 and 2108085UD06 to Y.W.), the Key Technologies R&D Program of Anhui Province (2022a05020053 to Y.W.), the Students’ Innovation and Entrepreneurship Foundation of University of Science and Technology of China (CY2022G42 to S.L.X.) and the Collaborative Innovation Program of Hefei Science Center, CAS (2021HSC-CIP002 to Y.W.). We thank the photoemission endstations BL1W1B in Beijing Synchrotron Radiation Facility (BSRF), BL14W1 in Shanghai Synchrotron Radiation Facility (SSRF), BL10B and BL11U in National Synchrotron Radiation Laboratory (NSRL) for the help in characterizations.
Author contributions
H.Q.Y. and Y.E.W. conceived and designed the study and supervised the project. X.Z. carried out the synthesis, characterization, and electrochemical tests. Y.M. and J.J.C. conducted the theoretical studies. C.M.Z. and M.K.K. helped to synthesize the catalysts. C.C. and S.L.X. helped with XANES and EXAFS measurements and discussion. X.Z., Y.M., H.Q.Y., and Y.E.W. analysed the results and co-wrote the paper. All authors contributed to discussion of the results and the manuscript.
Correspondence and requests for materials should be addressed to Yuen Wu or Han-Qing Yu.
Peer review information Nature Communications thanks Tej Choksi, Chunyu Du and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
CAS Key Laboratory of Urban Pollutant Conversion, Department of Environmental Science and Engineering, University of Science and Technology of China, Hefei 230026, China. School of Chemistry and Materials Science, University of Science and Technology of China, Hefei 230026, China. These authors contributed equally: Xiao Zhou, Yuan Min, Changming Zhao. e-mail: yuenwu@ustc.edu.cn; hqyu@ustc.edu.cn