DOI: https://doi.org/10.1038/s41557-023-01428-2
PMID: https://pubmed.ncbi.nlm.nih.gov/38238464
تاريخ النشر: 2024-01-18
تحرير الهيكل العظمي للبيريدينات من خلال تبديل أزواج الذرات من CN إلى CC
تم القبول: 15 ديسمبر 2023
نُشر على الإنترنت: 18 يناير 2024
(د) التحقق من التحديثات
الملخص
تحرير الهيكل العظمي هو استراتيجية تركيبية بسيطة لاستبدال دقيق أو إعادة ترتيب الذرات في هياكل الحلقات الأساسية للجزيئات المعقدة؛ حيث يتيح تنويع المركبات بسرعة لا يمكن تحقيقها من خلال تطبيق استراتيجيات التحرير المحيطي. يعتمد تحرير الهيكل العظمي المبلغ عنه سابقًا للأرينات الشائعة بشكل رئيسي على تفاعلات إدخال من نوع الكاربين أو النيتريين أو إعادة الترتيب. على الرغم من كونها قوية وفعالة وقابلة للتطبيق على تعديل هياكل الهتروأرين الأساسية في المراحل المتأخرة، إلا أن هذه الاستراتيجيات لا يمكن استخدامها في تحرير الهيكل العظمي للبيريدينات. هنا نبلغ عن تحرير الهيكل العظمي المباشر للبيريدينات من خلال تبديل أزواج الذرات من CN إلى CC لتوليد البنزين والنفتالين بطريقة معيارية. على وجه التحديد، نستخدم تفاعلات إزالة الأروماتية المتسلسلة، والإضافة الحلقية، وإعادة الأروماتية من خلال تفاعلات الإضافة العكسية في تسلسل وعاء واحد لتحويل البيريدينات الأصلية إلى بنزين ونفتالين يحملان بدائل متنوعة في مواقع محددة، كما تحددها مكونات تفاعل الإضافة الحلقية. يتم عرض التطبيقات على تنويع الهيكل العظمي في المراحل المتأخرة لأسس البيريدين في عدة أدوية.
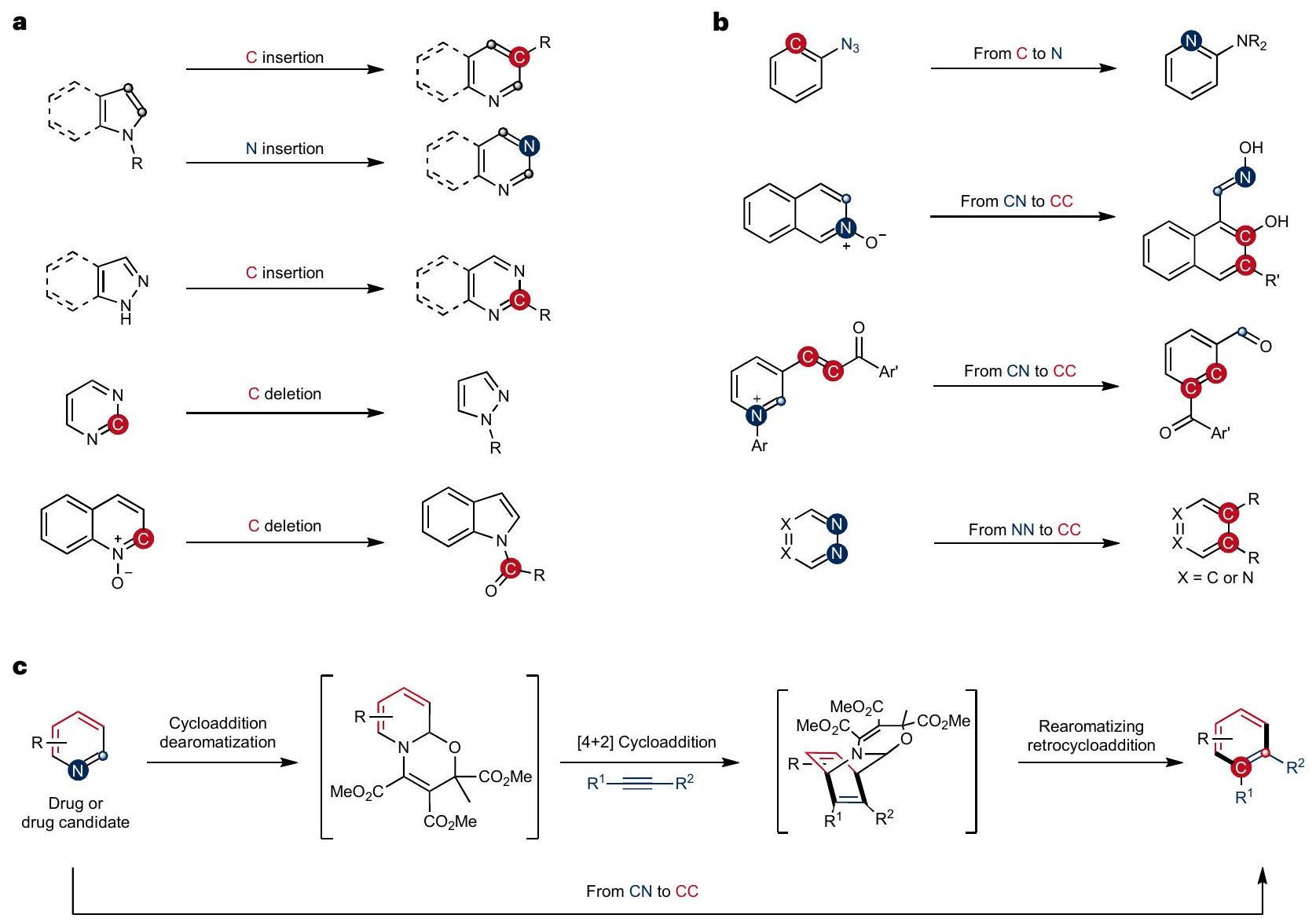
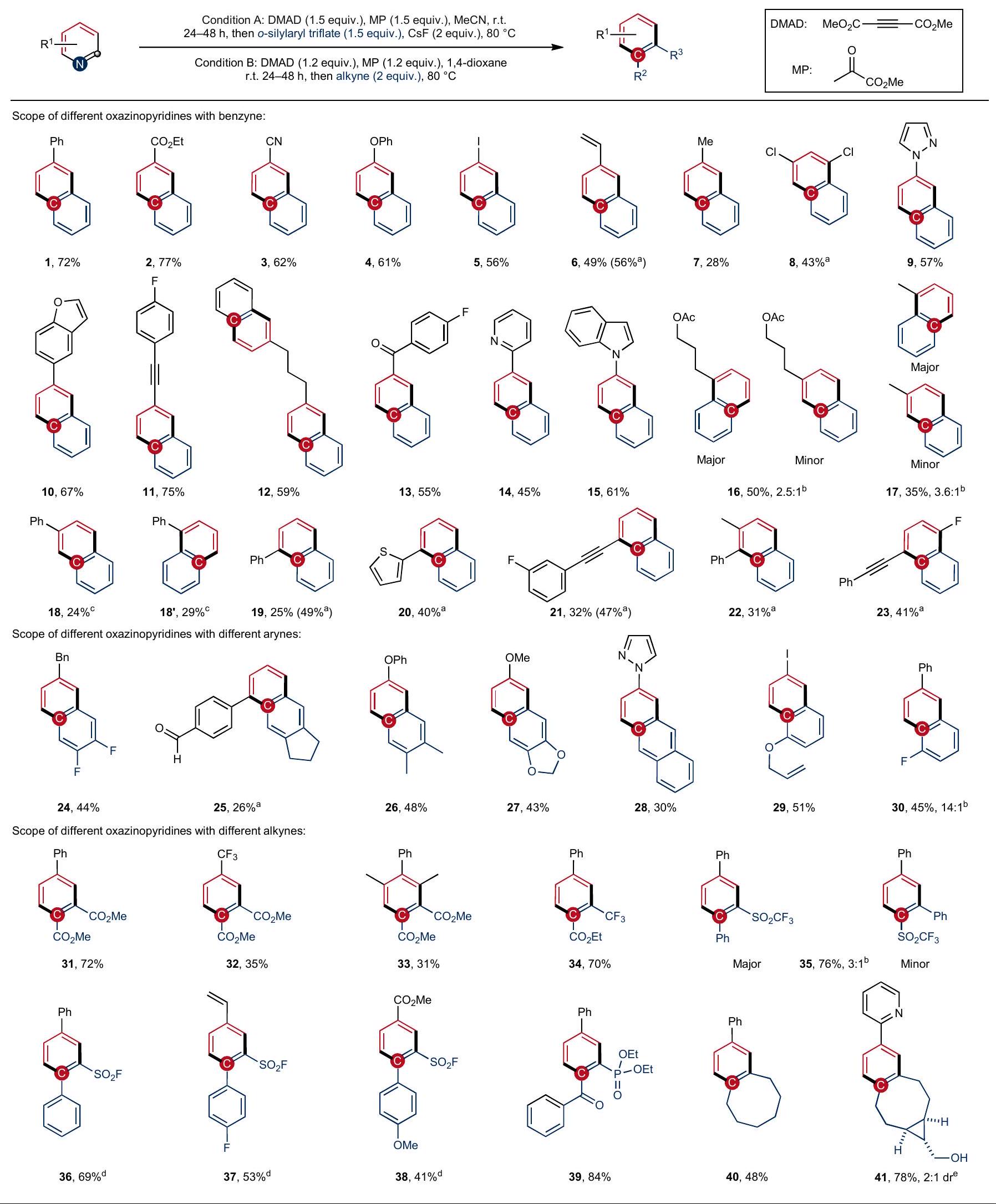
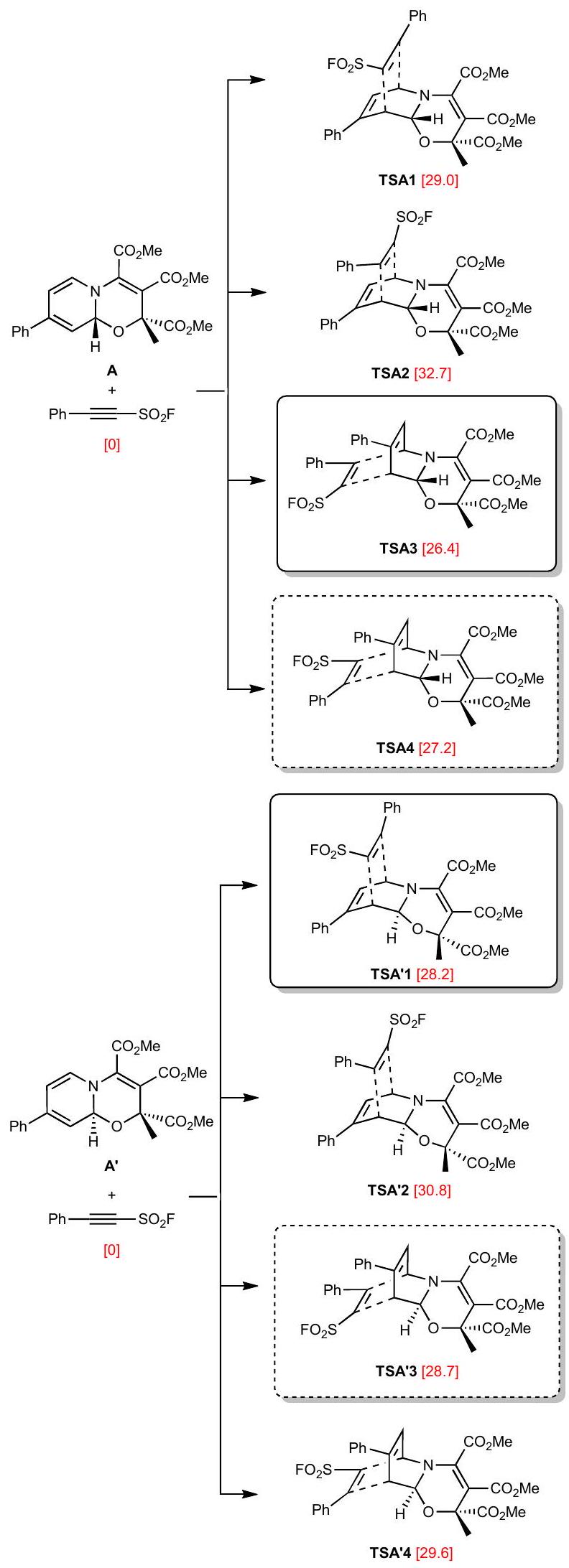
تم تطوير استراتيجية من خلال إزالة العطرية المتسلسلة، والإضافة الدائرية، وإعادة العطرية من خلال إعادة الدورة، مما يتيح تبادل أزواج الذرات من CN إلى CC في البيريدينات. الطريقة هي نهج موحد في وعاء واحد لتعديل البيريدين مع نطاق واسع من الركائز القابلة للتطبيق على التعديل في المراحل المتأخرة.
أقل استكشافًا، ربما لأن نطاق الركيزة محدود لمعظم الطرق المبلغ عنها، مما يجعل من الصعب تطبيق هذه التفاعلات على تنويع المراحل المتأخرة للأنظمة الأكثر تعقيدًا (الشكل 1ب)
نواة البيريدين إلى
النتائج والمناقشة
للمركبات الصيدلانية البنزينية
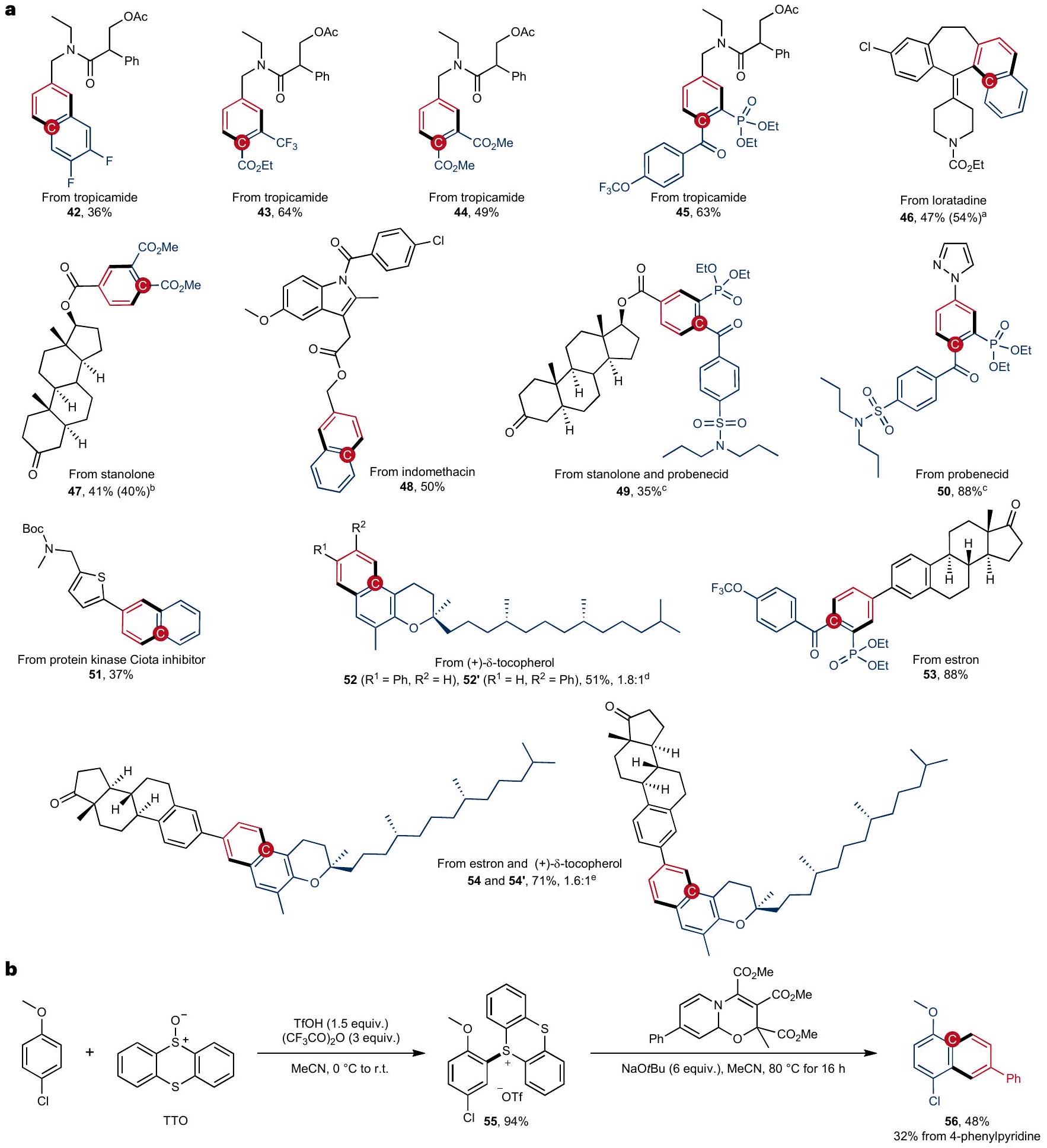
الشرط
من حمض التريفليك وأنهيدريد التريفلوأسيتيك أعطى ملح ثيانثرين 55. تم تفاعل هذا الملح القابل للعزل بسهولة مع الأوكازينو بيريدين المشتق من 4-فينيلبيريدين في الأسيتونيتريل باستخدام
من 4-فينيلبيريدين و 55 قدم أيضًا المنتج 56، على الرغم من أنه بعائد أقل (32%).
الخاتمة
المحتوى عبر الإنترنت
References
- Baumann, M. & Baxendale, I. R. An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem. 9, 2265-2319 (2013).
- Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257-10274 (2014).
- Kallitsis, J. K., Geormezi, M. & Neophytides, S. G. Polymer electrolyte membranes for high-temperature fuel cells based on aromatic polyethers bearing pyridine units. Polym. Int. 58, 1226-1233 (2009).
- Zhou, F.-Y. & Jiao, L. Recent developments in transition-metal-free functionalization and derivatization reactions of pyridines. Synlett 32, 159-178 (2021).
- Murakami, K., Yamada, S., Kaneda, T. & Itami, K. C-H functionalization of azines. Chem. Rev. 117, 9302-9332 (2017).
- Dolewski, R. D., Hilton, M. C. & McNally, A. 4-Selective pyridine functionalization reactions via heterocyclic phosphonium salts. Synlett 29, 8-14 (2018).
- Proctor, R. S. J. & Phipps, R. J. Recent advances in Minisci-type reactions. Angew. Chem. Int. Ed. 58, 13666-13699 (2019).
- Cao, H., Cheng, Q. & Studer, A. Radical and ionic meta-C-H functionalization of pyridines, quinolines, and isoquinolines. Science 378, 779-785 (2022).
- Boyle, B. T., Levy, J. N., Lescure, L. D., Paton, R. S. & McNally, A. Halogenation of the 3-position of pyridines through Zincke imine intermediates. Science 378, 773-779 (2022).
- Liu, Z. et al. Borane-catalyzed C3-alkylation of pyridines with imines, aldehydes, or ketones as electrophiles. J. Am. Chem. Soc. 144, 4810-4818 (2022).
- Zhou, X.-Y., Zhang, M., Liu, Z., He, J.-H. & Wang, X.-C. C3-selective trifluoromethylthiolation and difluoromethylthiolation of pyridines and pyridine drugs via dihydropyridine intermediates. J. Am. Chem. Soc. 144, 14463-14470 (2022).
- Zhang, M. et al. C3-cyanation of pyridines: constraints on electrophiles and determinants of regioselectivity. Angew. Chem. Int. Ed. 62, e202216894 (2022).
- Fan, Z. et al. Molecular editing of aza-arene
bonds by distance, geometry and chirality. Nature 610, 87-93 (2022). - Jurczyk, J. et al. Single-atom logic for heterocycle editing. Nat. Synth. 1, 352-364 (2022).
- Joynson, B. W. & Ball, L. T. Skeletal editing: interconversion of arenes and heteroarenes. Helv. Chim. Acta 106, e2022001 (2023).
- Hu, Y., Stumpfe, D. & Bajorath, J. Recent advances in scaffold hopping. J. Med. Chem. 60, 1238-1246 (2017).
- Buchner, E. & Curtius, T. H. Ueber die einwirkung von diazoessigäther auf aromatische kohlenwasserstoffe. Ber. Dtsch. Chem. Ges. 18, 2377-2379 (1885).
- Ciamician, G. L. & Dennstedt, M. Ueber die einwirkung des chloroforms auf die kaliumverbindung pyrrols. Ber. Dtsch. Chem. Ges. 14, 1153-1163 (1881).
- Dherange, B. D., Kelly, P. Q., Liles, J. P., Sigman, M. S. & Levin, M. D. Carbon atom insertion into pyrroles and indoles promoted by chlorodiazirines. J. Am. Chem. Soc. 143, 11337-11344 (2021).
- Hyland, E. E., Kelly, P. Q., McKillop, A. M., Dherange, B. D. & Levin, M. D. Unified access to pyrimidines and quinazolines enabled by N-N cleaving carbon atom insertion. J. Am. Chem. Soc. 144, 19258-19264 (2022).
- Reisenbauer, J. C., Green, O., Franchino, A., Finkelstein, P. & Morandi, B. Late-stage diversification of indole skeletons through nitrogen atom insertion. Science 377, 1104-1109 (2022).
- Bartholomew, G. L., Carpaneto, F. & Sarpong, R. Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion. J. Am. Chem. Soc. 144, 22309-22315 (2022).
- Woo, J. et al. Scaffold hopping by net photochemical carbon deletion of azaarenes. Science 376, 527-532 (2022).
- Fout, A. R., Bailey, B. C., Tomaszewski, J. & Mindiola, D. J. Cyclic denitrogenation of N -heterocycles applying a homogeneous titanium reagent. J. Am. Chem. Soc. 129, 12640-12641 (2007).
- Patel, S. C. & Burns, N. Z. Conversion of aryl azides to aminopyridines. J. Am. Chem. Soc. 144, 17797-17802 (2022).
- Cabrera-Pardo, J. R., Chai, D. I., Liu, S., Mrksich, M. & Kozmin, S. A. Label-assisted mass spectrometry for the acceleration of reaction discovery and optimization. Nat. Chem. 5, 423-427 (2013).
- Cabrera-Pardo, J. R., Chai, D. I. & Kozmin, S. A. Silver-promoted benzannulations of siloxyalkynes with pyridinium and isoquinolinium salts. Adv. Synth. Catal. 355, 2495-2498 (2013).
- Morofuji, T., Kinoshita, H. & Kano, N. Connecting a carbonyl and a
-conjugated group through a -phenylene linker by ( ) benzene ring formation. Chem. Commun. 55, 8575-8578 (2019). - Morofuji, T., Inagawa, K. & Kano, N. Sequential ring-opening and ring-closing reactions for converting para-substituted pyridines into meta-substituted anilines. Org. Lett. 23, 6126-6130 (2021).
- Morofuji, T., Nagai, S., Watanabe, A., Inagawa, K. & Kano, N. Streptocyanine as an activation mode of amine catalysis for the conversion of pyridine rings to benzene rings. Chem. Sci. 14, 485-490 (2023).
- Zhu, C., Kuniyil, R. & Ackermann, L. Manganese(I)-catalyzed C-H activation/Diels-Alder/retro-Diels-Alder domino alkyne annulation featuring transformable pyridines. Angew. Chem. Int. Ed. 58, 5338-5342 (2019).
- Narayan, A. R. H. & Sarpong, R. Indolizinones as synthetic scaffolds: fundamental reactivity and the relay of stereochemical information. Org. Biomol. Chem. 10, 70-78 (2012).
- Zhang, F.-G., Chen, Z., Tang, X. & Ma, J.-A. Triazines: syntheses and inverse electron-demand Diels-Alder reactions. Chem. Rev. 121, 14555-14593 (2021).
- Suzuki, S., Segawa, Y., Itami, K. & Yamaguchi, J. Synthesis and characterization of hexaarylbenzenes with five or six different substituents enabled by programmed synthesis. Nat. Chem. 7, 227-233 (2015).
- Song, C. et al. Visible-light-induced [4+2] annulation of thiophenes and alkynes to construct benzene rings. Angew. Chem. Int. Ed. 58, 12206-12210 (2019).
- Kotha, S. & Banerjee, S. Recent developments in the retro-DielsAlder reaction. RSC Adv. 3, 7642-7666 (2013).
- Png, Z. M., Zeng, H., Ye, Q. & Xu, J. Inverse-electron-demand Diels-Alder reactions: principles and applications. Chem. Asian J. 12, 2142-2159 (2017).
- Duret, G., Fouler, V. L., Bisseret, P., Bizet, V. & Blanchard, N. DielsAlder and formal Diels-Alder cycloaddition reactions of ynamines and ynamides. Eur. J. Org. Chem. 2017, 6816-6830 (2017).
- Pradhan, S., Mohammadi, F. & Bouffard, J. Skeletal transformation of unactivated arenes enabled by a low-temperature dearomative (3 + 2) cycloaddition. J. Am. Chem. Soc. 145, 12214-12223 (2023).
- Shi, J., Li, L. & Li, Y. o-Silylaryl triflates: a journey of Kobayashi aryne precursors. Chem. Rev. 121, 3892-4044 (2021).
- Medina, J. M., Mackey, J. L., Garg, N. K. & Houk, K. N. The role of aryne distortions, steric effects, and charges in regioselectivities of aryne reactions. J. Am. Chem. Soc. 136, 15798-15805 (2014).
- Neochoritis, C. G., Zarganes-Tzitzikas, T. & Stephanidou-Stephanatou, J. Dimethyl acetylenedicarboxylate: a versatile tool in organic synthesis. Synthesis 46, 537-585 (2014).
- El-Sheref, E. M. & Brown, A. B. Utility of acetylenedicarboxylate in organic synthesis. J. Heterocycl. Chem. 54, 825-843 (2017).
- Nilova, A., Campeau, L.-C., Sherer, E. C. & Stuart, D. R. Analysis of benzenoid substitution patterns in small molecule active pharmaceutical ingredients. J. Med. Chem. 63, 13389-13396 (2020).
- Heber, D., Rösner, P. & Tochtermann, W. Cyclooctyne and 4-cyclooctyn-1-ol-versatile building blocks in organic synthesis. Eur. J. Org. Chem. 2005, 4231-4247 (2005).
- Jacek, K. et al. Fragment-based drug discovery of potent protein kinase C iota inhibitors. J. Med. Chem. 61, 4386-4396 (2018).
- Meunier, B. Hybrid molecules with a dual mode of action: dream or reality? Acc. Chem. Res. 41, 69-77 (2008).
- Decker, M. (ed.) Design of Hybrid Molecules for Drug Development (Elsevier, 2017).
- Roberts, R. A., Metze, B. E., Nilova, A. & Stuart, D. R. Synthesis of arynes via formal dehydrogenation of arenes. J. Am. Chem. Soc. 145, 3306-3311 (2023).
© The Author(s) 2024
طرق
الإجراء العام لتعديل البيريدين باستخدام الأرينات كديينوفيلات (GP1)
الإجراء العام لتحرير البيريدين باستخدام الألكينات المنشطة كدائنوفيلات (GP2)
الإجراء العام لتحرير البيريدين من خلال عملية من خطوتين (GP3)
توفر البيانات
شكر وتقدير
جمعية الأبحاث (DFG) لدعمها هذا العمل.
مساهمات المؤلفين
تمويل
المصالح المتنافسة
معلومات إضافية
معهد الكيمياء العضوية، جامعة مونستر، مونستر، ألمانيا. كلية الكيمياء والعلوم الجزيئية، جامعة ووهان، ووهان، جمهورية الصين الشعبية. ساهم هؤلاء المؤلفون بالتساوي: تشيانغ تشينغ، ديبكانتا بهاتاشاريا. البريد الإلكتروني: studer@uni-muenster.de
DOI: https://doi.org/10.1038/s41557-023-01428-2
PMID: https://pubmed.ncbi.nlm.nih.gov/38238464
Publication Date: 2024-01-18
Skeletal editing of pyridines through atom-pair swap from CNtoCC
Accepted: 15 December 2023
Published online: 18 January 2024
(D) Check for updates
Abstract
Skeletal editing is a straightforward synthetic strategy for precise substitution or rearrangement of atoms in core ring structures of complex molecules; it enables quick diversification of compounds that is not possible by applying peripheral editing strategies. Previously reported skeletal editing of common arenes mainly relies on carbene- or nitrene-type insertion reactions or rearrangements. Although powerful, efficient and applicable to late-stage heteroarene core structure modification, these strategies cannot be used for skeletal editing of pyridines. Here we report the direct skeletal editing of pyridines through atom-pair swap from CN to CC to generate benzenes and naphthalenes in a modular fashion. Specifically, we use sequential dearomatization, cycloaddition and rearomatizing retrocycloaddition reactions in a one-pot sequence to transform the parent pyridines into benzenes and naphthalenes bearing diversified substituents at specific sites, as defined by the cycloaddition reaction components. Applications to late-stage skeletal diversification of pyridine cores in several drugs are demonstrated.
developed strategy through sequential dearomatization, cycloaddition and rearomatizing retrocyclization enabling an atom-pair swap from CN to CC in pyridines. The method is a one-pot modular approach for pyridine editing with a broad substrate scope that is applicable to late-stage modification.
less well explored, probably because the substrate scope is limited for most reported methods, rendering it difficult to apply these reactions to late-stage diversification of more complex systems (Fig. 1b)
pyridine core into a
Results and discussion
of benzenoid pharmaceutical compounds
condition
of triflic acid and trifluoroacetic anhydride gave thianthrenium salt 55. This readily isolable salt was reacted with 4-phenylpyridine-derived oxazino pyridine in acetonitrile using
from 4-phenylpyridine and 55 also provided the product 56, though in a lower yield (32%).
Conclusion
Online content
References
- Baumann, M. & Baxendale, I. R. An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem. 9, 2265-2319 (2013).
- Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257-10274 (2014).
- Kallitsis, J. K., Geormezi, M. & Neophytides, S. G. Polymer electrolyte membranes for high-temperature fuel cells based on aromatic polyethers bearing pyridine units. Polym. Int. 58, 1226-1233 (2009).
- Zhou, F.-Y. & Jiao, L. Recent developments in transition-metal-free functionalization and derivatization reactions of pyridines. Synlett 32, 159-178 (2021).
- Murakami, K., Yamada, S., Kaneda, T. & Itami, K. C-H functionalization of azines. Chem. Rev. 117, 9302-9332 (2017).
- Dolewski, R. D., Hilton, M. C. & McNally, A. 4-Selective pyridine functionalization reactions via heterocyclic phosphonium salts. Synlett 29, 8-14 (2018).
- Proctor, R. S. J. & Phipps, R. J. Recent advances in Minisci-type reactions. Angew. Chem. Int. Ed. 58, 13666-13699 (2019).
- Cao, H., Cheng, Q. & Studer, A. Radical and ionic meta-C-H functionalization of pyridines, quinolines, and isoquinolines. Science 378, 779-785 (2022).
- Boyle, B. T., Levy, J. N., Lescure, L. D., Paton, R. S. & McNally, A. Halogenation of the 3-position of pyridines through Zincke imine intermediates. Science 378, 773-779 (2022).
- Liu, Z. et al. Borane-catalyzed C3-alkylation of pyridines with imines, aldehydes, or ketones as electrophiles. J. Am. Chem. Soc. 144, 4810-4818 (2022).
- Zhou, X.-Y., Zhang, M., Liu, Z., He, J.-H. & Wang, X.-C. C3-selective trifluoromethylthiolation and difluoromethylthiolation of pyridines and pyridine drugs via dihydropyridine intermediates. J. Am. Chem. Soc. 144, 14463-14470 (2022).
- Zhang, M. et al. C3-cyanation of pyridines: constraints on electrophiles and determinants of regioselectivity. Angew. Chem. Int. Ed. 62, e202216894 (2022).
- Fan, Z. et al. Molecular editing of aza-arene
bonds by distance, geometry and chirality. Nature 610, 87-93 (2022). - Jurczyk, J. et al. Single-atom logic for heterocycle editing. Nat. Synth. 1, 352-364 (2022).
- Joynson, B. W. & Ball, L. T. Skeletal editing: interconversion of arenes and heteroarenes. Helv. Chim. Acta 106, e2022001 (2023).
- Hu, Y., Stumpfe, D. & Bajorath, J. Recent advances in scaffold hopping. J. Med. Chem. 60, 1238-1246 (2017).
- Buchner, E. & Curtius, T. H. Ueber die einwirkung von diazoessigäther auf aromatische kohlenwasserstoffe. Ber. Dtsch. Chem. Ges. 18, 2377-2379 (1885).
- Ciamician, G. L. & Dennstedt, M. Ueber die einwirkung des chloroforms auf die kaliumverbindung pyrrols. Ber. Dtsch. Chem. Ges. 14, 1153-1163 (1881).
- Dherange, B. D., Kelly, P. Q., Liles, J. P., Sigman, M. S. & Levin, M. D. Carbon atom insertion into pyrroles and indoles promoted by chlorodiazirines. J. Am. Chem. Soc. 143, 11337-11344 (2021).
- Hyland, E. E., Kelly, P. Q., McKillop, A. M., Dherange, B. D. & Levin, M. D. Unified access to pyrimidines and quinazolines enabled by N-N cleaving carbon atom insertion. J. Am. Chem. Soc. 144, 19258-19264 (2022).
- Reisenbauer, J. C., Green, O., Franchino, A., Finkelstein, P. & Morandi, B. Late-stage diversification of indole skeletons through nitrogen atom insertion. Science 377, 1104-1109 (2022).
- Bartholomew, G. L., Carpaneto, F. & Sarpong, R. Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion. J. Am. Chem. Soc. 144, 22309-22315 (2022).
- Woo, J. et al. Scaffold hopping by net photochemical carbon deletion of azaarenes. Science 376, 527-532 (2022).
- Fout, A. R., Bailey, B. C., Tomaszewski, J. & Mindiola, D. J. Cyclic denitrogenation of N -heterocycles applying a homogeneous titanium reagent. J. Am. Chem. Soc. 129, 12640-12641 (2007).
- Patel, S. C. & Burns, N. Z. Conversion of aryl azides to aminopyridines. J. Am. Chem. Soc. 144, 17797-17802 (2022).
- Cabrera-Pardo, J. R., Chai, D. I., Liu, S., Mrksich, M. & Kozmin, S. A. Label-assisted mass spectrometry for the acceleration of reaction discovery and optimization. Nat. Chem. 5, 423-427 (2013).
- Cabrera-Pardo, J. R., Chai, D. I. & Kozmin, S. A. Silver-promoted benzannulations of siloxyalkynes with pyridinium and isoquinolinium salts. Adv. Synth. Catal. 355, 2495-2498 (2013).
- Morofuji, T., Kinoshita, H. & Kano, N. Connecting a carbonyl and a
-conjugated group through a -phenylene linker by ( ) benzene ring formation. Chem. Commun. 55, 8575-8578 (2019). - Morofuji, T., Inagawa, K. & Kano, N. Sequential ring-opening and ring-closing reactions for converting para-substituted pyridines into meta-substituted anilines. Org. Lett. 23, 6126-6130 (2021).
- Morofuji, T., Nagai, S., Watanabe, A., Inagawa, K. & Kano, N. Streptocyanine as an activation mode of amine catalysis for the conversion of pyridine rings to benzene rings. Chem. Sci. 14, 485-490 (2023).
- Zhu, C., Kuniyil, R. & Ackermann, L. Manganese(I)-catalyzed C-H activation/Diels-Alder/retro-Diels-Alder domino alkyne annulation featuring transformable pyridines. Angew. Chem. Int. Ed. 58, 5338-5342 (2019).
- Narayan, A. R. H. & Sarpong, R. Indolizinones as synthetic scaffolds: fundamental reactivity and the relay of stereochemical information. Org. Biomol. Chem. 10, 70-78 (2012).
- Zhang, F.-G., Chen, Z., Tang, X. & Ma, J.-A. Triazines: syntheses and inverse electron-demand Diels-Alder reactions. Chem. Rev. 121, 14555-14593 (2021).
- Suzuki, S., Segawa, Y., Itami, K. & Yamaguchi, J. Synthesis and characterization of hexaarylbenzenes with five or six different substituents enabled by programmed synthesis. Nat. Chem. 7, 227-233 (2015).
- Song, C. et al. Visible-light-induced [4+2] annulation of thiophenes and alkynes to construct benzene rings. Angew. Chem. Int. Ed. 58, 12206-12210 (2019).
- Kotha, S. & Banerjee, S. Recent developments in the retro-DielsAlder reaction. RSC Adv. 3, 7642-7666 (2013).
- Png, Z. M., Zeng, H., Ye, Q. & Xu, J. Inverse-electron-demand Diels-Alder reactions: principles and applications. Chem. Asian J. 12, 2142-2159 (2017).
- Duret, G., Fouler, V. L., Bisseret, P., Bizet, V. & Blanchard, N. DielsAlder and formal Diels-Alder cycloaddition reactions of ynamines and ynamides. Eur. J. Org. Chem. 2017, 6816-6830 (2017).
- Pradhan, S., Mohammadi, F. & Bouffard, J. Skeletal transformation of unactivated arenes enabled by a low-temperature dearomative (3 + 2) cycloaddition. J. Am. Chem. Soc. 145, 12214-12223 (2023).
- Shi, J., Li, L. & Li, Y. o-Silylaryl triflates: a journey of Kobayashi aryne precursors. Chem. Rev. 121, 3892-4044 (2021).
- Medina, J. M., Mackey, J. L., Garg, N. K. & Houk, K. N. The role of aryne distortions, steric effects, and charges in regioselectivities of aryne reactions. J. Am. Chem. Soc. 136, 15798-15805 (2014).
- Neochoritis, C. G., Zarganes-Tzitzikas, T. & Stephanidou-Stephanatou, J. Dimethyl acetylenedicarboxylate: a versatile tool in organic synthesis. Synthesis 46, 537-585 (2014).
- El-Sheref, E. M. & Brown, A. B. Utility of acetylenedicarboxylate in organic synthesis. J. Heterocycl. Chem. 54, 825-843 (2017).
- Nilova, A., Campeau, L.-C., Sherer, E. C. & Stuart, D. R. Analysis of benzenoid substitution patterns in small molecule active pharmaceutical ingredients. J. Med. Chem. 63, 13389-13396 (2020).
- Heber, D., Rösner, P. & Tochtermann, W. Cyclooctyne and 4-cyclooctyn-1-ol-versatile building blocks in organic synthesis. Eur. J. Org. Chem. 2005, 4231-4247 (2005).
- Jacek, K. et al. Fragment-based drug discovery of potent protein kinase C iota inhibitors. J. Med. Chem. 61, 4386-4396 (2018).
- Meunier, B. Hybrid molecules with a dual mode of action: dream or reality? Acc. Chem. Res. 41, 69-77 (2008).
- Decker, M. (ed.) Design of Hybrid Molecules for Drug Development (Elsevier, 2017).
- Roberts, R. A., Metze, B. E., Nilova, A. & Stuart, D. R. Synthesis of arynes via formal dehydrogenation of arenes. J. Am. Chem. Soc. 145, 3306-3311 (2023).
© The Author(s) 2024
Methods
General procedure for pyridine editing with arynes as dienophiles (GP1)
General procedure for pyridine editing with activated alkynes as dienophiles (GP2)
General procedure for pyridine editing through two-pot process (GP3)
Data availability
Acknowledgements
Forschungsgemeinschaft (DFG) for supporting this work.
Author contributions
Funding
Competing interests
Additional information
Organisch-Chemisches Institut, Universität Münster, Münster, Germany. College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, P. R. China. These authors contributed equally: Qiang Cheng, Debkanta Bhattacharya. e-mail: studer@uni-muenster.de