تحسين تزاوج d-p في المحفز المساعد Ni-Bx لزيادة إنتاج الهيدروجين الضوئي Fine-tuning d-p hybridization in Ni-Bx cocatalyst for enhanced photocatalytic H2 production
تحسين تزاوج d-p في المحفز المساعد Ni-Bx لزيادة إنتاج الهيدروجين الضوئي
تم الاستلام: سبتمبر 2024
تم القبول: 14 يناير 2025
نُشر على الإنترنت: 22 يناير 2025
الهيدروجينإنتاج الهيدروجين من المحفزات الضوئية باستخدام الطاقة الشمسية هو تحدٍ حاسم لتحقيق اقتصاد خالٍ من الكربون ومستدام.. ومع ذلك، فإن المحفزات الضوئية العارية عادة ما تظهر أداءً ضعيفًا-كفاءة الإنتاج بسبب إعادة التركيب الكبيرة للحاملات الناتجة عن الضوء وبطء التفاعل بين الواجهاتديناميكا الاختزاللمعالجة هذه القيود، يتم استخدام المحفزات المساعدة بشكل شائع لتسريع فصل الإلكترونات والثقوب وتوفير مواقع نشطة لتعزيز الواجهة.-عملية التطورمن الناحية المثالية، أداء متفوق-يمكن لموقع الإنتاج النشط أن يشكل بشكل فعال موقعًا نشطًا مناسبًا-الرابطة، التي تسرع في الوقت نفسه من التقاط البروتونات وحريةتطلق الجزيئات، مما يعزز بشكل عام-عملية الإنتاجلسوء الحظ، تعاني معظم المواقع النشطة من المحفزات المبلغ عنها من إما قوة مفرطة أو ضعف مفرط في مواقع النشاط. الروابط بسبب تكويناتها الإلكترونية غير المواتية، مما يحد بشدة من أداء التطور. لذلك، الإلكترونية المعقولة
التلعب حركيات تطور الهيدروجين دورًا حيويًا في تنظيم عملية تطور الهيدروجين الضوئي. ومع ذلك، فإن تحقيق تنظيم دقيق لتوازن امتصاص الهيدروجين وإطلاق الهيدروجينلا يزال يمثل تحديًا كبيرًا. هنا، نقترح ضبطًا دقيقًااستراتيجية التهجين لتحسين الدقةالحركيات في Ni-محفز ضوئي CdS المعدل (Ni-تظهر مطيافية امتصاص الأشعة السينية الدقيقة والحسابات النظرية أن زيادة كمية ذرات البورون في المساعد المساعد Ni-Bx تعزز تدريجياً التفاعل المداري بين و ، مما أدى إلى -توسيع النطاق وقابلية التحكم-مركز النطاق على مواقع النيكل النشطة. ما سبق هو تسلسلتحسين النطاق يسمح بتعديل دقيق لـ الديناميات في النيكل – ، مما يظهر في النهاية قدرة ملحوظة-نشاط التطور (AQE ). تؤكد مطيافية الامتصاص العابر بالفيمتوثانية الديناميات السريعة لنقل الإلكترونات في المحفز الضوئي. يوفر هذا العمل رؤى حول التصميم الأمثل للمستقبل-محفزات التطور. تحسين المواقع النشطة لتحقيق عملية امتصاص/إطلاق متوازنة للهيدروجين هو عامل رئيسي في تحسين التحفيز الضوئيأداء الإنتاج.
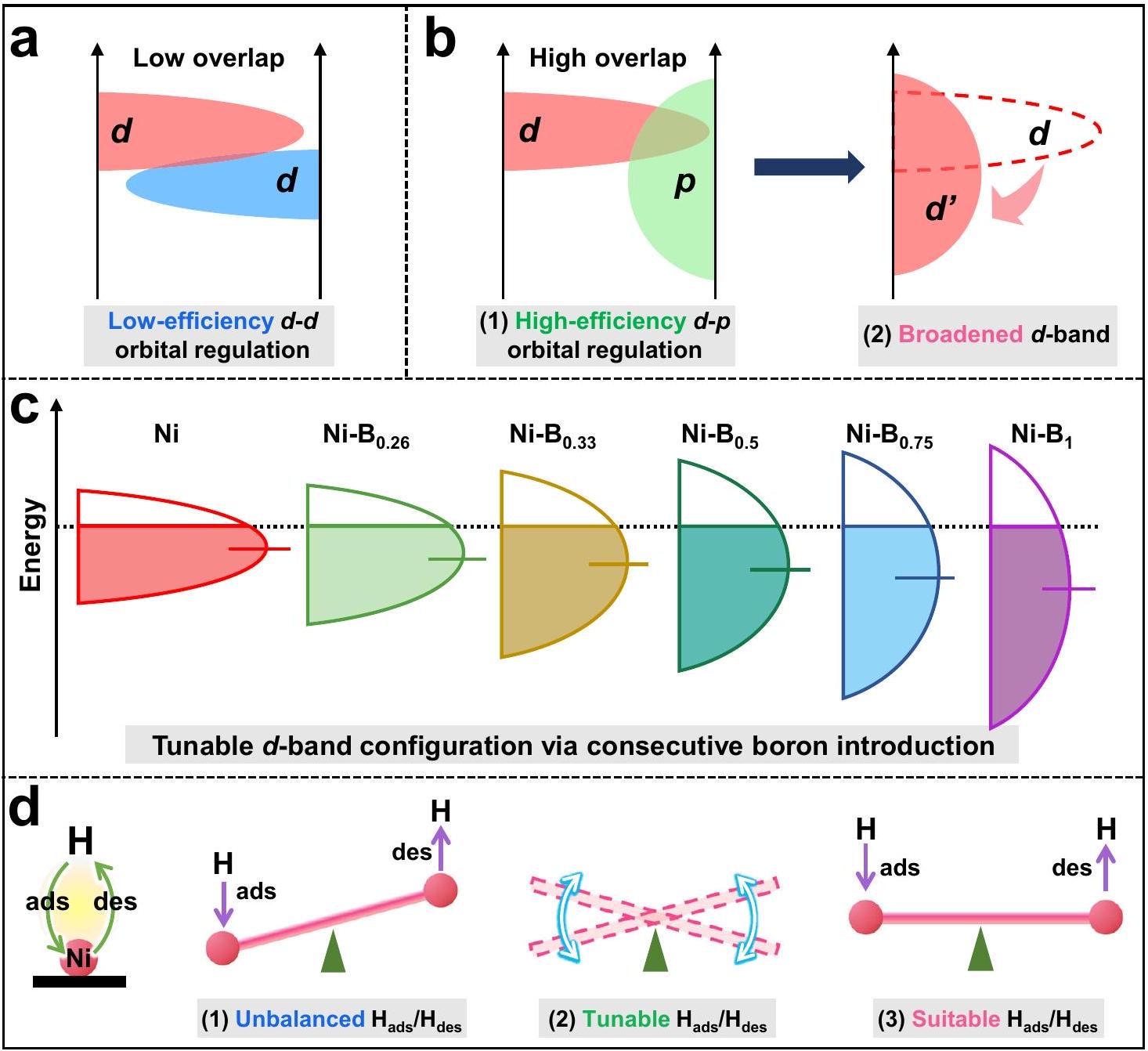
وفقًا لـ-نظرية النطاق، يتم تحديد ديناميات امتصاص/إزالة الهيدروجين عند المواقع النشطة بشكل أساسي بواسطة-تكوين الإلكترونيات في نطاق المعادن الانتقالية، الذي يحكم الموقع النشط-قوة الرابطة وبدورها تتحكم في-عملية التطور. ومع ذلك، بالنسبة لمعظم الفلزات الانتقالية، فإن-تتميز الإلكترونات في النطاق بخصائص موضعية نموذجية، حيث أن الغالبية من-الإلكترونات المحصورة ضمن نطاق طاقة ضيق، مما يسبب خمول الإلكترونات وبالتالي يعيق الأداء التحفيزي. حتى الآن، تم القيام بالعديد من الجهود لتعديل -تكوين إلكتروني للفرقة لتحسين أداء التحفيز لموقع الفلز الانتقالي النشطعلى سبيل المثال، يمكن أن يؤدي السبك مع ذرات المعادن الانتقالية غير المتجانسة إلى تعديل-تكوين النطاق للموقع النشط عبر
الهجينة المدارية. ومع ذلك، مثل هذهالهجينة المدارية عادة ما تنتج كفاءة تعديل منخفضة بسبب التشابه فيالتكوينات المدارية والخصائص الإلكترونية بين عناصر المعادن الانتقالية (الشكل 1أ). لمزيد من التعديل على-تكوين إلكتروني ذو نطاق، الـتم تطوير استراتيجية تنظيم المدارات، التي يمكن أن تعدل بشكل أكثر فعالية -تكوين النطاق لمواقع المعادن الانتقالية من خلال تعزيز التفاعلات المدارية (الشكل 1ب(1)). في هذه الحالة، تم تقديم-المداري يزيد من المنطقة المتداخلة ويعمل على توسيع عرض النطاق الموزع لـ-نطاق، مما يؤدي إلى تشكيل -نطاق طاقة النطاق (الشكل 1ب(2)). ونتيجة لذلك، فإن القوة الشديدةلقد تم الإبلاغ على نطاق واسع عن تأثير التهجين المداري في تحسين الأداء التحفيزي لمواقع المعادن الانتقالية النشطة. على سبيل المثال، يمكن تحسين النشاط التحفيزي لذرات Nb بشكل فعال من خلالالهجينة المدارية داخلالجزيء النشط. وبالمثل، يمكن أن يؤدي إدخال ذرات الفوسفور في الأوزميوم إلى تعزيز نشاط تفكيك الماء لذرات الأوزميوم بشكل كبير عبر الهجين في محفز فوسفيد الأوزميوم. ومع ذلك، فإن ما ذُكر أعلاهتستهدف استراتيجيات التنظيم عادةً الذرات الفردية المحدودة بعدد التنسيق أو أنظمة المركبات المعدنية الثابتة التركيب.، الذي يظهر تعديلًا معزولًا لـ-تكوين النطاق، مما يحد من المرونة في التعديل-ديناميات التطور. لذلك، من الممكن أن يكون هناك استراتيجية التعديل يمكن أن تحسن بدقةتكوين المدارات -band لذرات المعادن الانتقالية، مما يحقق التحفيز الأمثل-ديناميات التطور.
مقارنةً بالتقليدي-كتل الذرات (، إلخ)، يتمتع ذرة البورون (B) بميزة نقص الإلكترونات النموذجية، مما يمكنها من تشكيل روابط فلزية مع ذرات المعادن الانتقالية وإنتاج سبائك بوريد المعادن القابلة للتعديل في التركيب.. في هذه الدراسة، تم إجراء تم ابتكار استراتيجية التهجين لتعديلتكوين نطاق موقع النيكل النشط في التركيبة القابلة للتعديل (الشكل 1ج)، بهدف تحسين التوازن الديناميكي في مواقع النشاط Ni لتعزيز-أداء التطورالمساعد المساعد (الشكل 1د). هنا،تم تصنيع مساعد حفاز بنسب قابلة للتعديل من البورون والنيكل بمهارة على سطح كبريتيد الكادميوم (CdS) لإنتاجالمحفزات الضوئية من خلال مسار ذاتي التحفيز يتم تفعيله بواسطة الضوء بشكل معتدل. تكشف الدراسات المنهجية أن زيادة محتوى البورون فييمكن أن يزيد المساعد المساعد بشكل متتابع-نطاق ترددات النيكل، مما يسمح بتقليل التردد بشكل قابل للتحكم-مركز النطاق لموقع النيكل النشط لإضعاف التحفيزالسندات، مما يعززإزالة الامتصاص لتحسين التحفيز-نشاط التطور. نتيجة لذلك، الأمثلالعامل الضوئي أظهر نشاطًا ضوئيًا استثنائيًا-أداء التطوربالإضافة إلى فقاعات الهيدروجين المرئية بكثرة، والتي هي أعلى بمقدار 5.5 مرة
الشكل 1 | استراتيجية التعديل الدقيقالهجين فيمساعد حفاز مناسبالحركيات. رسم تخطيطي يوضح الكفاءة المنخفضةتنظيم مداري. ب مخطط توضيحي يوضح التوسع-نطاق عبر كفاءة عاليةتنظيم مداري. ج رسم تخطيطي لسلسلة النيكل -توسيع النطاق إلى الانخفاض-مركز النطاق في التركيب القابل للتعديلرسم تخطيطي يوضح الديناميكية القابلة للتعديل لامتصاص الهيدروجين للوصول إلى المستوى المناسبتوازن.
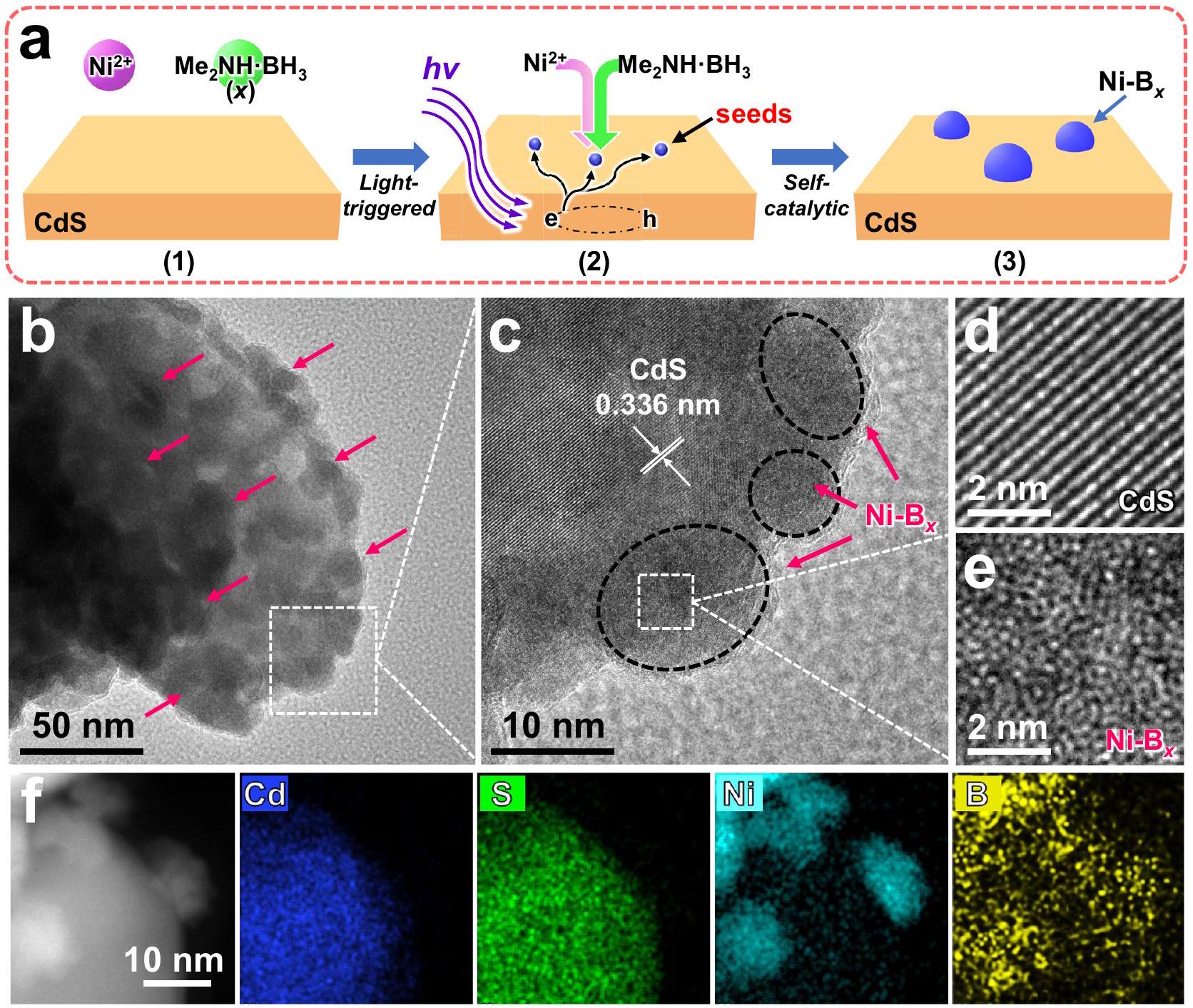
الشكل 2 | الاستراتيجية الاصطناعية وخصائص الشكل. أ مخطط يوضح التكوين الذاتي التحفيزي الناتج عن الضوء لـالمحفزات المساعدة على CdS لتصنيعالمحفز الضوئي. ب TEM، ج-هـ HRTEM، و f HAADF-STEM، وصور رسم خرائط EDS لـ.
من ذلكمحفز ضوئي )، ونشاط التحفيز الضوئي أيضًا تجاوز نشاط معيار Pt/CdS وغيرها من المحفزات الضوئية المتطورة (الجدول التكميلي 1). هذا العمل حدد تسلسلًااستراتيجية التعديل لتحسين الديناميكية بدقةتوازن المواقع النشطة لتطور الهيدروجين، مما يوفر رؤى لتصميم المحفزات المحتملة.
قابل للتعديل في التركيبتُودع الجسيمات النانوية بدقة على سطح CdS باستخدام طريقة تحفيزية ذاتية مستحثة بالضوء سهلة (الشكل 2أ). أولاً، يتم توزيع جزيئات CdS العارية بشكل متجانس في محلول مختلط من خلات النيكل.وثنائي ميثيل أمين البورانللحصول على تعليق برتقالي مستقر (الشكل 2أ(1)). عند التعرض للضوء المرئي لمدة -2 دقيقة، يمكن تحفيز أشباه الموصلات CdS بسهولة لإنتاج إلكترونات مثارة بالضوء، والتي يمكن أن تهاجر بنجاح إلى سطح CdS لتحفيز التفاعل بين و لإنشاءالبذور (الشكل 2أ(2)). بعد إيقاف الضوء، فإنيمكن للبذور أن تحفز بشكل عفوي التفاعل الحفزي التالي لـ و ، مما يؤدي في النهاية إلى التكوين في الموقع لـجزيئات نانوية على سطح CdS لصنع Ni-محفز ضوئي (الشكل 2a(3)). آلية التكوين الذاتي التحفيزي الناتجة عن الإلكترونات لـ Ni-تظهر الجسيمات النانوية بشكل أكبر من خلال التجارب الداعمة (الشكل التكميلي 1)، حيث تتبعيتم إضافته كمزود للإلكترونات إلى المحلول المختلط من و لإنتاج الـالبذور. هذه البذور بعد ذلك تحفز التكوين العفوي للون الأسودجزيئات نانويةعلاوة على ذلك، استنادًا إلى التغيرات اللونية الملحوظة (الشكل التوضيحي 2)، يتحول معلق CdS البرتقالي تدريجيًا إلى لون داكن عميق خلال عملية التحفيز الذاتي، مما يشير إلى التكوين الفعال للون الأسود.الجزيئات النانوية على سطح CdS. بالإضافة إلى ذلك، وفقًا لتغيرات اللون (الشكل التكميلي 3)،يمكن أيضًا تصنيع المساعد المساعد بشكل فعال على محفزات ضوئية مضيفة أخرى (التي تُعتبر نموذجية و أشباه الموصلات) من خلال الطريقة الاصطناعية الحالية. لذلك، يمكن الاستنتاج بشكل منطقي أنتم تخليق المحفز الضوئي بنجاح من خلال مسار التخليق الذاتي المحفز بالضوء المذكور أعلاه.
يتم استخدام مجهر الإلكترون الناقل (TEM) لفحص البنية المجهرية مباشرةً لـالمحفز الضوئي، والصور الناتجة معروضة في الشكل 2ب-و. تظهر الصور بوضوح أن هناك العديد من الظلال الداكنةجزيئات نانوية ) موزعة بشكل موحد على سطح CdS (الشكل 2ب)، مما يظهر ترتيبًا ذريًا غير منظم (الشكل 2ج-هـ)، وهو ما يتوافق مع نتائج تحويل فورييه السريع ونتائج حيود الأشعة السينية (XRD) لنيكل النقي – (الأشكال التكميلية 4 و 5). بالإضافة إلى ذلك، تم البناء الناجح لـيمكن أن يتضح ذلك أكثر من خلال رسم خرائط EDS (الشكل 2f)، حيث تتداخل إشارات النيكل والبورون وتوزع بشكل متساوٍ على سطح CdS، مما يوفر دليلاً قوياً على تحقيق الـالمحفز الضوئي. علاوة على ذلك، استنادًا إلى نتائج التحليل الطيفي للانبعاث الضوئي الناتج عن البلازما المقترنة بالحث (ICP-OES) (الجدول التكميلي 2)، يتضح أن التركيبة قابلة للتعديلالمحفزات المساعدة (مع الذرةيمكن تصنيع النسبة التي تتراوح من 0.15 إلى 0.89 بنجاح عن طريق ضبط كميةمصدر البورون. علاوة على ذلك، وفقًا لنتائج طيف الأشعة فوق البنفسجية والمرئية (الشكل التوضيحي 6)، فإن امتصاص الضوء من 520 إلى 800 نانومتر منأكبر بكثير من CdS وعينات CdS، مع زيادة شدة الامتصاص مع ارتفاع محتوى B، مما يؤكد تحميل التركيبة القابلة للتعديل بنجاحمساعد حفاز. بالإضافة إلى ذلك، تظهر نتائج حيود الأشعة السينية (XRD) ورامان (RAMAN) والأشعة تحت الحمراء (IR) والتصوير المجهري الإلكتروني الماسح بالانبعاث الميداني (FESEM) (الأشكال التكميلية 7-10) أن يعرض هيكل بلوري داخلي مشابه وشكل ظاهر لـ CdS و Ni/CdS، مما يشير إلى أن الاستراتيجية الاصطناعية المعتمدة على الضوء لها تأثير ضئيل على المضيف.. لذلك، تظهر النتائج أعلاه مجتمعة فعالية تصنيع التركيبة القابلة للتعديلالمساعد المساعد على CdS من خلال مسار ذاتي التحفيز تحت تأثير الضوء المعتدل لتخليقمحفز ضوئي
أداء فوتوكاتاليتيكي فعال وواجهته-آلية التطور
أنشطة التركيب القابل للتعديلتمت دراسة المحفزات المساعدة من خلال اختبارات تطور الهيدروجين الضوئي، كما هو موضح في الشكل 3. يظهر CdS النقي أداءً ضعيفًا-نشاط التطوربسبب إعادة التركيب الكثيف لزوج الإلكترون والثقب ومواقع التفاعل السطحية الضعيفة (الشكل 3أ(1)). حتى بعد تعديل المحفز المساعد Ni، فإن
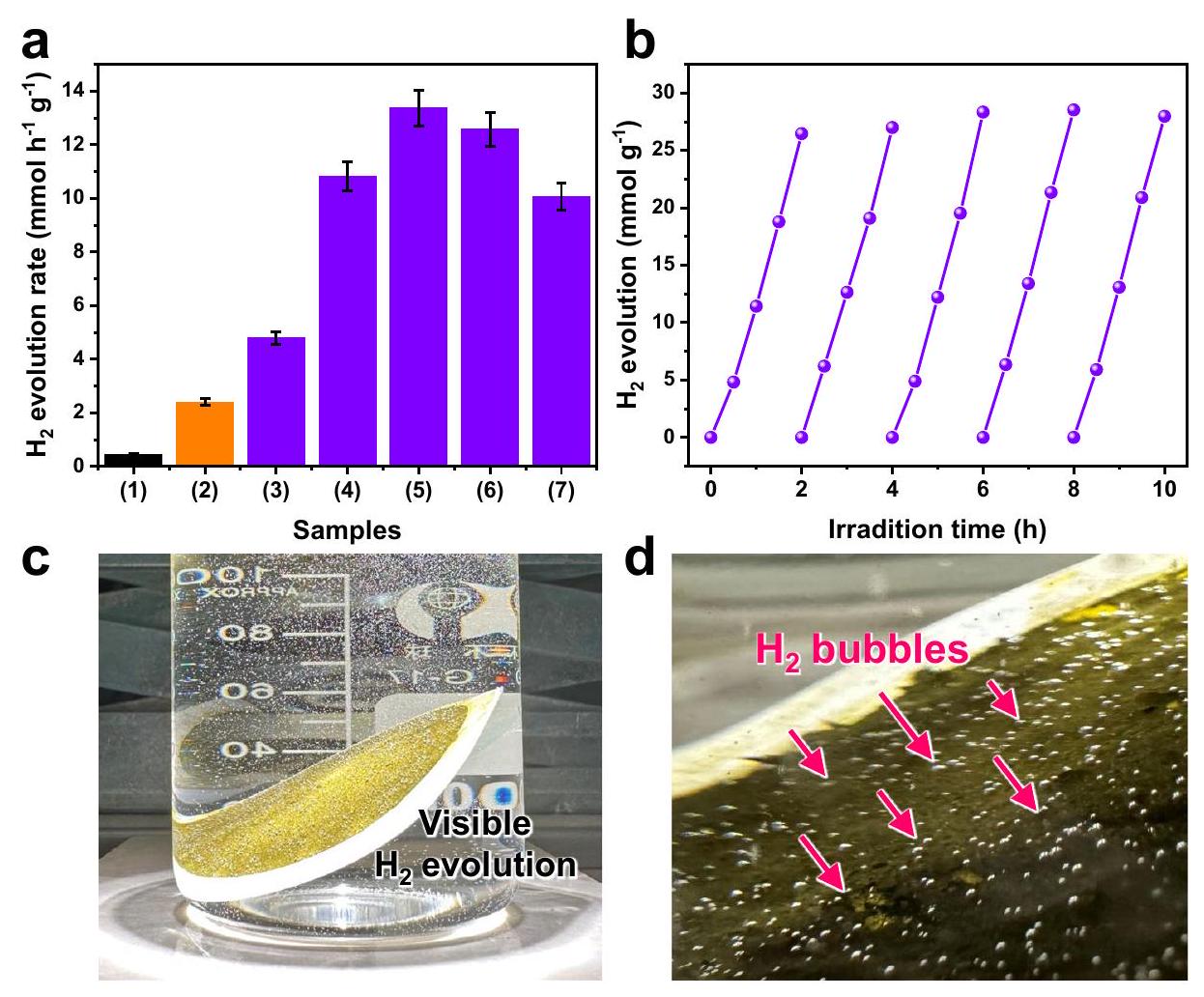
محفز Ni/CdS يظهر فقط تحسن طفيف في الأداء، الشكل )، مما يوحي بأن مواقع النيكل النشطة النقية لا تزال توفر حدًا محدودًا نشاط تطوري. مع المزيد من دمج ذرات البورون في المساعد المساعد نيكل، فإن الناتجتظهر المحفزات الضوئية ارتفاعًا ملحوظًا-معدلات الجيل مقارنة بـ CdS و، وتمتلك اتجاه مخطط البركان (الشكل 3أ(3-7)). على وجه التحديد، فإن الأمثلعينة ( ) يحقق تحفيز ضوئي ملحوظ -معدل الإنتاج لـ، والتي هي 29.1 و 5.6 مرة أعلى من CdS و Ni/CdS، على التوالي. علاوة على ذلك، استنادًا إلى أدائها في الدورات، فإنيقدم متانة مستدامة مع عاليةنشاط التطور (الشكل 3ب) ويمكن الحفاظ عليه بشكل جيد بعد اختبار أداء التحفيز الضوئي (الشكل التكميلي 11). من المثير للإعجاب، تحت إشعاع ضوء الشمس المحاكى، يمكن رؤية مستمريمكن ملاحظة الفقاعات على سطحعينة، تأكيد الكفاءة العالية-نشاط التطورالمساعد المساعد (الشكل 3c، d والفيلم التكميلي). بالإضافة إلى ذلك،نشاط التطور الحالييتجاوز CdS ذلك الخاص بالمعيار Pt/CdS (الشكل التكميلي 12) وغيرها من المحفزات الضوئية المتطورة القائمة على CdS (الجدول التكميلي 1). لذلك، فإن الدمج المتتالي للبورون في النيكل لإنتاجيمكن أن يحسن المساعد المساعد بشكل كبير من نشاط توليد الهيدروجين الضوئي المحفز لـ CdS.
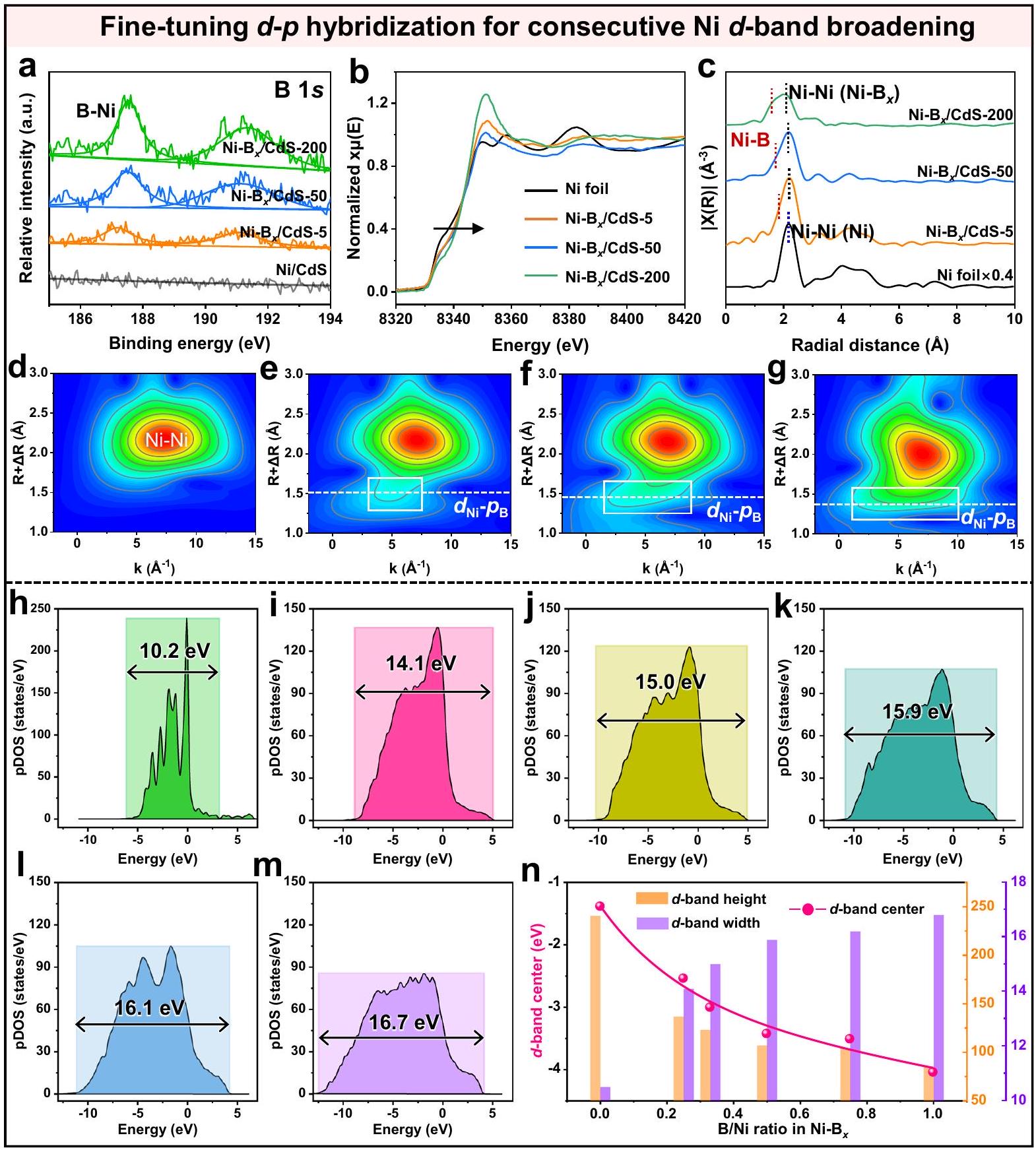
وفقًا لنتائج الأداء المذكورة أعلاه، فإنيمكن تحسين نشاط تطور الهيدروجين لمساعد النيكل بشكل كبير من خلال إدخال البورون، والذي يُعزى أساسًا إلى حقيقة أن زيادة دمج البورون تعززتفاعل لتعديل-تكوين النطاق لنيكل، كما يتضح من خلال التحقيقات النظرية والتجريبية التالية. استنادًا إلى نتائج مطيافية الإلكترون الضوئي بالأشعة السينية (XPS) لـ B 1s (الشكل 4a) و Ni 2p (الشكل التكميلي 13)، يمكن ملاحظة أنه مع زيادة كمية التخصيب بالبورون فيالمساعد المساعد، يظهر ذروة النيكل النموذجية تحولًا متزامنًا نحو قيمة أكثر سلبية، مما يعني تعزيزًا متتابعًا
الشكل 3 | التحفيز الضوئي-أنشطة الإنتاج. محفز ضوئي-نشاط تطور الهيدروجين لعينات مختلفة: (1) CdS، (2) Ni/CdS، (3) Ni-Bx/CdS-5، (4) Ni-Bx/CdS-20، (5) ، (6)، (7)تم حساب أشرطة الخطأ (المتوسط ± الانحراف المعياري) استنادًا إلى ثلاثة تجارب فوتوكاتاليتيكية مستقلة.
تجارب. ب تشغيلات الدراجات الضوئية المحفزة-نشاط التطور لـ .c التحفيز الضوئي المرئي-اختبار التطور. صور للعرض فقاعات على .
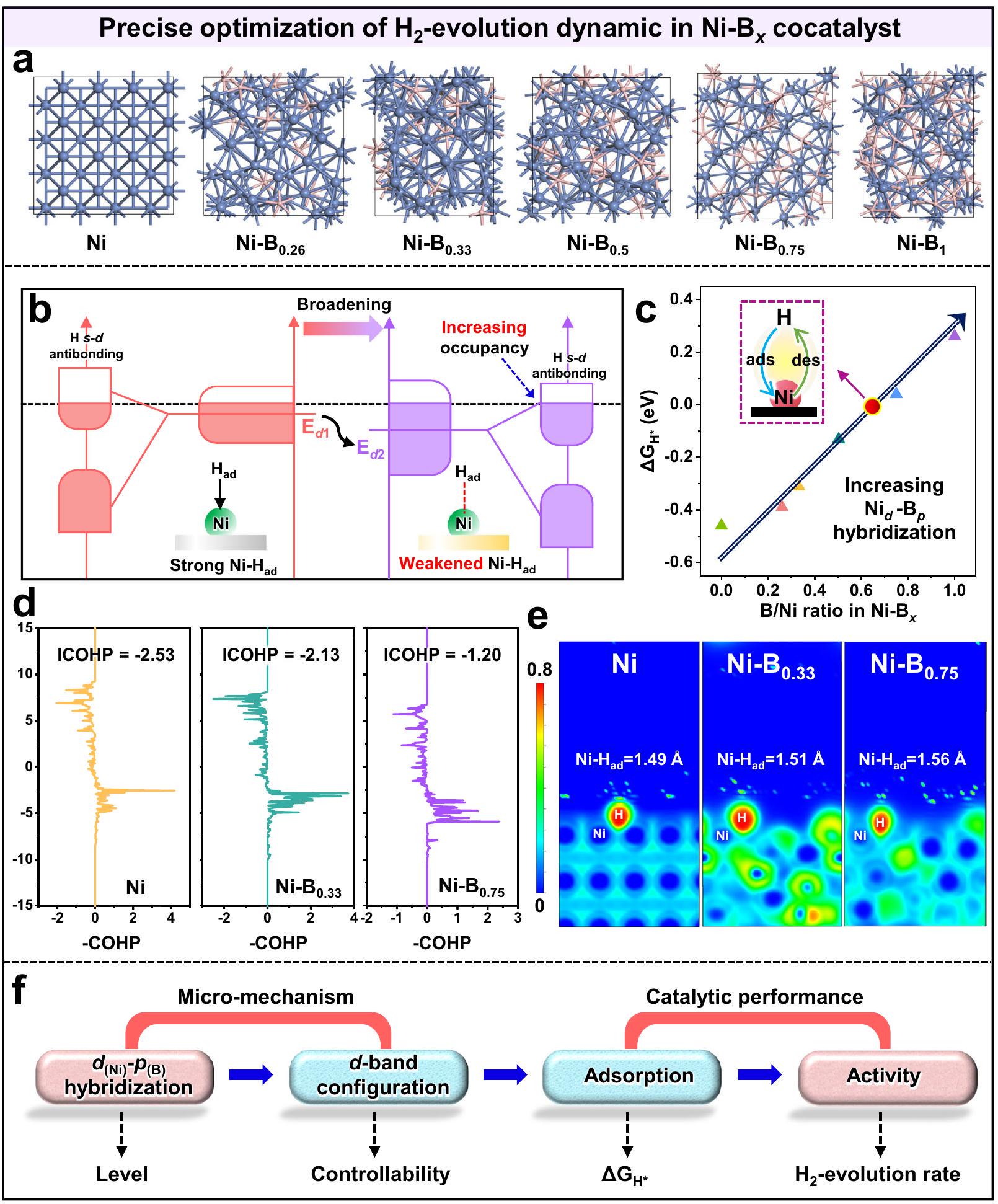
الشكل 4 | الضبط الدقيقالهجين فيللتوالي-توسيع النطاق. أ طيف XPS عالي الدقة لذرة B 1s. ب طيف XANES المُعَدل. ج طيف FTEXAFS في فضاء R. تحويلات الموجة لبيانات EXAFS لحد Ni K من د رقائق Ni. Ni-Bx/CdS-5، fNi-Bx/CdS-50، و g Ni-Bx/CdS-200. الكثافة المتوقعة لحالة Ni 3d المداري لـ، و . تم حسابه-ارتفاعات النطاقات،-عرض النطاقات، و-قيم مركز النطاقات المختلفة. التفاعل الإلكتروني بين النيكل والبورون فيالمساعد المساعد. بالإضافة إلى ذلك، يمكن أيضًا التحقق من النتائج المذكورة أعلاه من خلال تحليل شحنات بادير في الشكل التكميلية 14. مع زيادة كمية التسمم بالبورون في (الأشكال التكميلية 15 و 16)، تزداد القيمة المتوسطة لشحنة بادر لذرات النيكل تدريجياً، بينما تنخفض القيمة لذرات البورون بشكل متناسب، مما يؤكد مرة أخرى تعزيز التفاعل الإلكتروني. الضبط الدقيقالهجينة المدارية فيتُظهر الأدلة الإضافية على المساعد المساعد من خلال تحليل الحالات الإلكترونية وبيئات التنسيق باستخدام مطيافية امتصاص الأشعة السينية. كما هو موضح في الشكل 4ب، تكشف طيف امتصاص الأشعة السينية القريب من الحافة (XANES) المنظم عن حافة امتصاص النيكل K لعينة مختلفة. يمكن ملاحظة بسهولة أنه مع زيادة كمية التخصيب بالبورون في المساعد المساعد Ni-Bx، فإن النيكل يظهر حافة الامتصاص K تحولًا إيجابيًا تدريجيًا، مما يدل على التفاعل الإلكتروني المعزز بشكل متتابع بين ذرات النيكل والبورون فيالمساعد المساعد. علاوة على ذلك،الهجينة المدارية فيتمت دراسة المساعد المساعد من خلال تحليل غلاف التنسيق للنيكل وفقًا لنتائج هيكل الامتصاص الدقيق للأشعة السينية (EXAFS) التالية. استنادًا إلى بيانات FT-EXAFS (الشكل 4c)، يظهر رقائق النيكل ذروة مهيمنة فردية عندفي فضاء R، مما يشير إلى الجوهرالتنسيق في النيكل المعدني. بعد إدخال البورون في النيكل لتشكيلالمساعد المساعد، الذروة عند (يمثل المسافة بين طبقة القشرة الأولى B وذرات النيكل) يمكن ملاحظته بسهولة، مما يتوافق مع وجود التنسيق فيمساعد حفازمن المهم أنه مع زيادة كمية التخصيب بالبورون،تظهر قمم التنسيق تدريجياً انزياحاً سلبياً من -1.8 إلى، مما يعني تعزيز التفاعل المداري فيالمساعد المساعد. علاوة على ذلك، يتم إجراء تحويل الموجات الصغيرة-EXAFS، وتظهر النتائج في الشكل 4d-f. يظهر عينة النيكل إشارة معزولة تتوافق مع. مع الإدخال المتتالي لـ B في Ni، إشارة التنسيق يظهر بوضوح ويزداد تدريجياً. بالإضافة إلى ذلك، مع زيادة كميات ذرات البورون فيإشارةيظهر تحولًا سلبيًا في فضاء R، مما يثبت التفاعل المتزايد تدريجيًا بين النيكل والبورون فيالمساعد المساعد. لذلك، يمكن أن يؤدي الإدخال المتتالي لـ B في Ni إلى تعزيز فعّال لـالهجين المداري فيوبذلك تحقيق التنظيم المتتالي لموقع النشاط Ni.
الضبط الدقيقالهجينة المدارية فييمكن أن يوسع بشكل متتالي-عرض نطاق موقع النيكل النشط، كما يتضح أكثر من خلال نتائج كثافة الحالة (DOS) التالية (الشكل 4h-m، الشكل التوضيحي 17). وفقًا لنتائج كثافة الحالة المتوقعة، من الواضح أن-فرقة النيكل النقي تظهر طابعًا محليًا عاليًا، حيث أن معظم الـ-الإلكترونات محصورة في نطاق طاقة ضيق من -7.2 إلى 3.1 إلكترون فولت (الشكل 4h، الشكل التكميلية 18(1)). بعد التتابعالتفاعل المداري في، و 1.00، كل الـ-فرقة من مختلفتتسع بشكل ملحوظ (الشكل 4i-m)، أي كلما زادت كمية ذرات البورون المدخلة، زادت الاتساع.-تم الحصول على النطاق. خاصةً عندما يتم رفع نسبة الذرات B/Ni إلى 1.00، يتم توزيع النيكليمكن توسيع نطاق -band بشكل كبير ليشمل نطاق طاقة واسع من -12.7 إلى 3.9 إلكترون فولت (الشكل 4 م).درجة توسيع النطاقتُقاس العينات باستخدام-عرض النطاق الكامل عند نصف الحد الأقصى (FWHM)، كما هو موضح في الشكل التوضيحي 19. يمكن ملاحظة أن قيمة FWHM للنيكل-الحزام يزداد تدريجياً مع زيادة محتوى البورون، مما يثبت مرة أخرى التتابع-توسيع النطاق في. المتتالية أعلاهتوسيع النطاق فييمكن تعيينه إلى المعززالتهجين المداري، كما يتضح من النتائج الإحصائية لطول رابطة Ni-B (الشكل التكميلي 20) وفقًا للحسابات النظرية. في هذه الحالة،طول الرابطةالهيكل يتراوح من 1.8 إلى. مع زيادة كمية B في نسبة الـ الروابط ذات الطول القصير (1.8-2.0 Å) تزداد تدريجياً (الشكل التكميلي 21)، مما يشير إلى تعزيز التفاعل فيعبر إدخال B المتتالي، الذي يتماشى مع بيانات دالة التوزيع الشعاعي (الشكل التوضيحي 22) لروابط Ni-B فينماذج. وبالتالي، المتتالية-توسيع النطاق يؤدي حتمًا إلى توليد إلكترونات غير متوازنة فوق مستوى فيرمي، مما يؤدي إلى انخفاض المستوى-مركز النطاق (الشكل التوضيحي 18(2، 3)). يمكن إثبات النتائج المذكورة أعلاه من خلال ما يلي-حسابات مركز النطاق في الشكل 4 ن. كما أننسبة الني ترتفع في، الـ-عرض النطاق يزيد من 10.2 إلى 16.6 إلكترون فولت، بينما-تنخفض ارتفاع الشريط من 240.6 إلى 85.4 إلكترون فولت، وبالتالي الحصول على الانزلاق التدريجي-مركز النطاق من -1.4 إلى -4.0 إلكترون فولت. في الوقت نفسه، مع الانخفاض المتتالي لـ-مركز الشريط، عدد شغل الإلكترونات لـيظهر المداري زيادة تدريجية أيضًا (الشكل التوضيحي 23). وبالتالي، تثبت النتائج أعلاه أنيمكن أن تعزز المقدمة تدريجياًالتفاعل المداري لتوسيع-نطاق النيكل، مما يؤدي إلى تعديل النشاط التحفيزي بشكل فعال-مركز النطاقمساعد حفاز
التتابعي-تعديل مركز النطاق في النيكل-يمكن أن يقوم المساعد المساعد بتحسين ديناميات امتصاص/إطلاق الهيدروجين بدقة على موقع النيكل النشط لتعزيز الواجهة-تفاعل التطور (الشكل 5). هنا، طاقة الامتزاز الحرة للهيدروجين ( ) وتحليل كثافات هاملتونية المدارات البلورية (COHP) يتم إجراؤها للكشف عن -ديناميات التطور وفقًا لنماذج السطح لـ (الشكل 5أ). نظريًا، بالنسبة للنيكل النقي مع مستوى عالٍ من -مركز الشريط، الحالات المضادة للرابطة التي تتشكل بعدالتفاعل يظهر شغفًا منخفضًا للإلكترونات (الشكل 5ب (يسار))، مما يؤدي إلى امتصاص قوي للغاية للهيدروجين على النيكل، مما يثبطإزالة الامتصاص. وهذا يؤكد أكثر من خلال الحسابات السلبيةقيمة -0.46 إلكترون فولت لموقع النيكل في النيكل نموذج (الشكل 5ج). عند دمج B في Ni لتشكيل، الـ– يتم خفض مركز النطاق للنيكل بشكل قابل للتحكم، مما يزيد من شغل المدارات الإلكترونية المضادة للرابطة، مما يؤدي إلى عدم استقرار الـيربط ويعزز-عملية إزالة الامتصاص (الشكل 5ب (يمين)). وبالتالي، استنادًا إلىالنتائج في الشكل 5ج،تزداد قيمة مواقع النيكل النشطة تدريجياً وتصل إلى قيمة مثالية تبلغ 0.04 إلكترون فولت لـ، تحسين توازن امتصاص/إطلاق الهيدروجين على موقع النيكل النشط وبالتالي تعزيز-نشاط التطور. تدعم هذه النتائج المزيد من تحليلات COHP (ICOHP)، كما هو موضح في الشكل 5d. يمكن ملاحظة أنه مع إدخال البورون في النيكل، فإن القيمة المدمجة لـ ICOHP لـالرابطة ترتفع تدريجياً من -2.53 إلى -1.20، مما يشير إلى ضعف امتصاص الهيدروجين على موقع النيكل لتعزيز-عملية إزالة الامتصاص (الشكل 5d والشكل التوضيحي 24). علاوة على ذلك، فإن الشكل الممتديمكن أن يثبت طول الرابطة وزيادة وظيفة توطين الإلكترون (ELF) على النيكل أن قوة امتصاص الهيدروجين على موقع النيكل يمكن أن تضعف بشكل فعال من خلال الإدخال المتتابع للبورون في النيكل، مما يسهل ذلك.إزالة الامتصاص (الشكل 5e). لذلك، يمكن الاستنتاج من النتائج السابقة أن زيادة إدخال البورون في النيكل يمكن أن تعزز بشكل فعال الهجين المداري لتعديل التحفيز-تكوين النطاق لموقع النيكل النشط، والذي يعد الآلية الجوهرية لتحسين ديناميكية امتصاص/إطلاق الهيدروجين على النيكل لتعزيزنشاط التطور (الشكل 5f).
نقل الإلكترونات الضوئية الاتجاهية والتحقيق في ديناميكياتها
عملية نقل الفوتونات الإلكترونية الاتجاهيةتمت دراسة تأثير الإشعاع الضوئي باستخدام مجهر القوة الكهروستاتيكية كيلفن في الموقع (KPFM) وتقنية التحليل الطيفي للأشعة السينية في الموقع (XPS) (الأشكال التكميلية 25 و26). يمكن ملاحظة بوضوح من اختبارات فرق الجهد التلامسي (CPD) (الشكل التكميلية 25) أن CdS العاري يظهر جهد سطح قدره. بعد التعديل على على سطح CdS، لوحظ انخفاض في فرق الجهد الكهربائي (0.14 فولت)، مما يشير إلى انتقال الإلكترونات الحرة من CdS إلى، والذي يمكن أن يُعزى إلى ارتفاع وظيفة العمل لـ مقارنة بـ CdS (الشكل التوضيحي 27). عندما يتم تعريضه للضوء المرئي، مما يؤدي إلى زيادة فرق الجهد السطحييلاحظ، مما يتوافق مع الفوتوإلكترون المثار بالضوء من CdS، الذي يحفز بدوره هجرة الفوتوإلكترون إلىالمساعد المساعد، كما يتضح أكثر من خلال تحليل XPS المعرض للإشعاع في الموقع (الشكل التوضيحي 26). عندما يتم تشغيل الضوء، فإن طاقة الربط لـتتحول القمم بشكل إيجابي إلى قيمة عالية، بينما تلك منتتحول القمم سلبًا، مما يؤكد بقوة انتقال الفوتوالكترونات من المضيف CdS إلىالمساعد المساعد في.
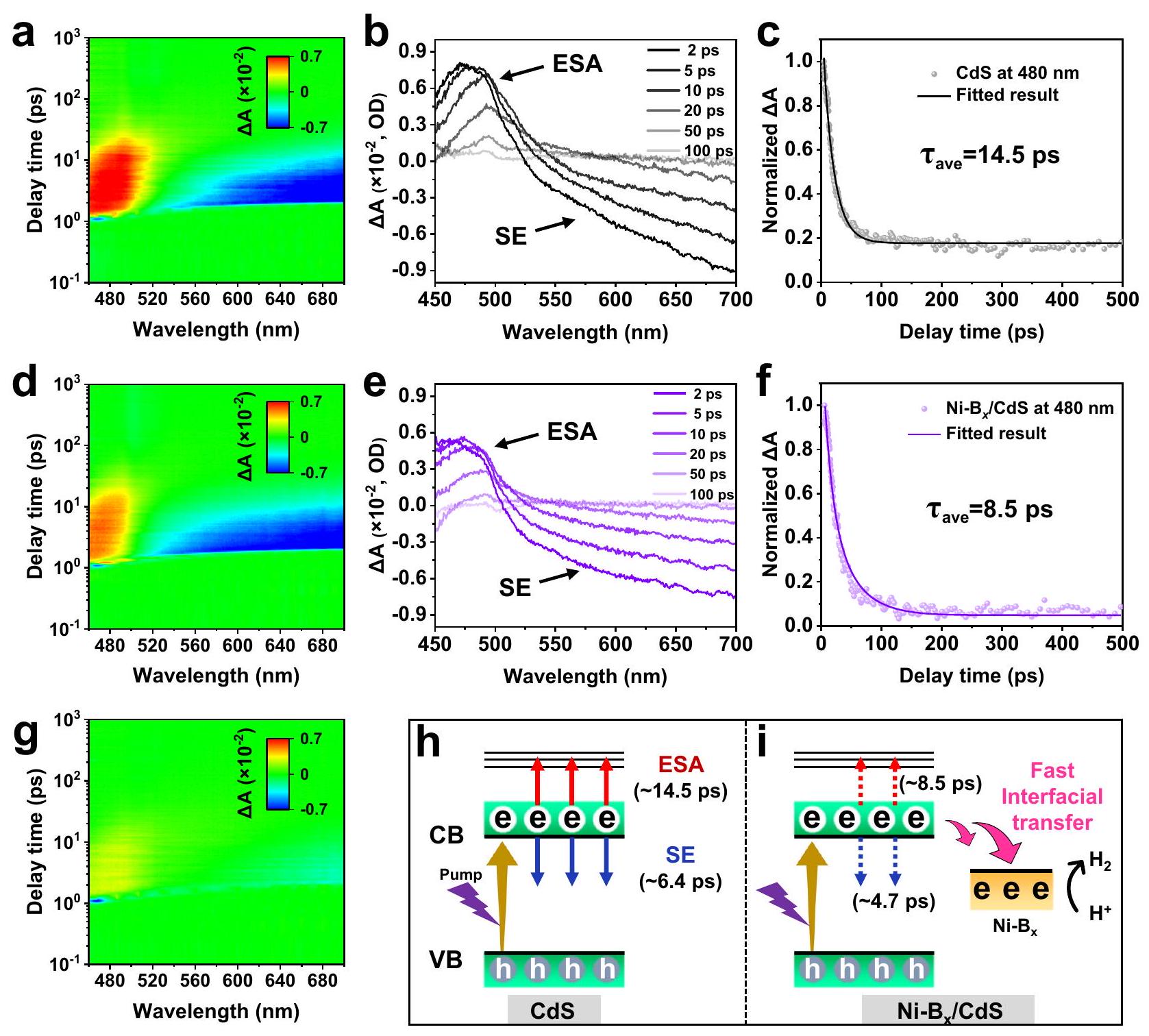
يتم استخدام مطيافية الامتصاص العابر في الفيمتوثانية (fs-TAS) أيضًا للتحقيق في ديناميات نقل الإلكترونات الضوئية لمجموعة متنوعة من العينات (الشكل 6 والشكل التكميلي 28). بالنسبة لـ CdS (الشكل 6a و 6b)، يظهر طيف الامتصاص العابر إشارة إيجابية (حوالي 480 نانومتر، والتي تنتمي بشكل رئيسي إلى امتصاص الحالة المثارة (ESA)، وتمثل انتقال الإلكترون المثار من مستوى طاقة منخفض إلى مستوى طاقة مرتفع في نطاق التوصيل (CB). علاوة على ذلك، الإشارة السلبية ( ) في نطاق يمكن أن يُعزى ذلك إلى الانبعاث المحفز (SE)، الذي يتوافق مع الانبعاث الإلكتروني من مستوى التوصيل إلى مستويات الطاقة المنخفضة.. بالإضافة إلى ذلك، يظهر إشارة تبييض سلبية في حالة الأرض عند حوالي 470 نانومتر خلال الـ 700 فيمتوثانية الأولى (الشكل التوضيحي 29)، ثم يتأخر بسرعة أو يتداخل مع إشارة ESA في نطاق بعد 900 فيمتوثانية. بعد تعديلالمساعد المساعد على سطح CdS (الشكل 6d، e)، إشارات ESA و SE لـالعينات أضعف بوضوح من تلك الخاصة بمحفز CdS النقي، مما يشير إلى أن الإلكترونات المثارة بواسطة نبضة المضخة على سطح CdS يمكن أن تهاجر بفعالية إلى Ni-B.المساعد المساعد، مما يؤدي إلى قمع عملية ESA و SE.
الشكل 5 | محسّن بدقةديناميات فيمساعد حفاز. هياكل سطحية محسّنة للنيكل ولحساب DFT. ب الرسم البياني الذي يوضح توسيع-حزام لزيادة شغل الحالة المضادة للرابطة لإضعافالسندات. ج تم حسابهاقيم الهياكل المتعددة. بيانات d COHP لـ روابط. نتيجة وظيفة الإلكترون المحلية (ELF) بعد امتصاص الهيدروجين على النيكل. آلية للتحكم-تكوين النطاق من خلالالهجين لتنظيمالحركيات نحو تعزيز-نشاط التطور. نقل الفوتوإلكترون من CdS إلىتم تأكيد وجود المساعد المساعد بشكل أكبر من خلال استخدام ثنائي كرومات البوتاسيوم كجامع للإلكترونات (الشكل 6g والشكل التكميلي 30). وقد وُجد أنه، مع إضافةيظهر CdS إشارات ESA و SE محدودة جدًا، مما يعني أن الإلكترونات المثارة في نطاق التوصيل لـ CdS يمكن استهلاكها بكفاءة بواسطةالناقل. علاوة على ذلك، لقياس كفاءة نقل الفوتوإلكترون في CdS و Ni-التأخير الحركي تم ملاءمة منحنيات العينات لعمليات ESA و SE (الشكل 6c، f، h، i). يمكن ملاحظة أن CdS العاري يظهر عمرًا متوسطًا طويلًا (14.5 بيكوثانية لعملية EAS و6.4 بيكوثانية لعملية SE)، مما يشير إلى أن الإلكترونات المثارة في نطاق التوصيل لـ CdS يصعب استهلاكها أو نقلها. مع تعديلالمساعد المساعد، بسبب النقل السريع للإلكترونات من CdS CB إلىمتوسط أعمار الإلكترونات المثارة في كل من ESA (8.5 بيكوثانية) و SE (4.7 بيكوثانية)
الشكل 6 | آلية نقل الإلكترونات الناتجة عن الضوء والديناميات. مخططات الألوان الزائفة، إشارات الامتصاص العابرة في الفيمتوثانية خلال 100 بيكوثانية، ومنحنيات الانحلال المقابلة لـ CdS (أ-ج) و Ni-Bx/CdS (د-و) (ESA و SE تمثل امتصاص الحالة المثارة والانبعاث المحفز، على التوالي).بيانات fs-TAS لـ CdS مع الإضافة لـرسم توضيحي يوضح تحقيق نقل الشحنات الواجهة المعجل في. تقلصت العمليات بشكل كبير، مما يشير بقوة إلى النقل السريع للفوتونات الإلكترونية في. علاوة على ذلك، تتماشى هذه النتائج مع النتائج المستخلصة من اختبارات الفوتوإلكتروكيميائية وطيف الفوتولومينسنس في الحالة العابرة (TRPL) (الشكل التكميلي 31). لذلك، تثبت النتائج المذكورة أعلاه بقوة أن الإلكترونات المثارة ضوئيًا من CdS يتم التقاطها بكفاءة بواسطة المساعد المساعد Ni-Bx، مما يؤدي إلى تفعيل عملية نقل الفوتوإلكترونات الاتجاهية على الواجهة.
باختصار، ضبط دقيقاستراتيجية التهجين مقترحة لتعديل-تكوينات النطاق لذرات النيكل، مما يؤدي إلى تحسين ديناميات امتصاص/إطلاق الهيدروجين بدقة في المواقع النشطة للنيكل في النيكل-المساعد المساعد. تكشف النتائج التجريبية والنظرية أن الزيادة المتتاليةمقدمة فييعزز تدريجياًالتهجين المداري، الذي يمكن أن يوسع بشكل فعال-توزيع النطاق لنيكل للتلاعب بـمركز النطاقالمساعد المساعد. يمكن أن تعمل هذه النتائج على تحسين ديناميكية امتصاص/إطلاق الهيدروجين لموقع النيكل النشط بدقة لتعزيز التحفيز الضوئي.-نشاط التطور لـ. كما هو متوقع، الـالمحفز الضوئي يظهر مستوى عالٍ-أداء التطور لـ مع إمكانية العرض المستمرإطلاق الفقاعات، والذي هو 5.6 مرات أكثر من Ni/CdS. تكشف هذه الدراسة عن آلية متعمقة قابلة للتعديل.-تكوينات النطاق عبر التعديلالتفاعل الإلكتروني، مما يوفر رؤى حول تصميم المحفز ومبادئ التطبيق.
طرق
تحضيرمحفز ضوئي
قابل للتعديل في التركيبتم تحضير المحفز الضوئي باستخدام استراتيجية تحفيز ذاتي مستحث بالضوء. أولاً، تم إضافة 100 ملغ من CdS التجاري إلى 80 مل من محلول الميثانول-الماء. ) تحت التحريك المغناطيسي المستمر. بعد ذلك، حل ( ) يتم إضافته إلى التعليق أعلاه بواسطة حقنة، حيث يتم التحكم في نسبة الكتلة بين Ni و CdS لتكون . ثم، تحت التحريك المغناطيسي، كميات مختلفة ( ) من تم إضافة (المصدر B) إلى النظام أعلاه، على التوالي. بعد ذلك، تم إخلاء النظام أعلاه بـ لمدة 15 دقيقة ثم تم ختمه بسدادة مطاطية. يتم تحفيز التفاعل من خلال إشعاع لمدة دقيقتين باستخدام أربعة مصابيح LED. ) تحت تحريك. بعد إيقاف الضوء، يتم تحريك النظام أعلاه مغناطيسيًا لمدة ساعتين في ظروف مظلمة لتمكين التكوين الذاتي التحفيزي لـجزيئات النانو على CdS. أخيرًا، يتم تصفية المنتجات، وغسلها، وتجفيفها طوال الليل فيالمحفزات الضوئية التي تم الحصول عليها تُشار إليها بـ، حيث تمثل X الإضافة كميات من و 200 ملغ، على التوالي. في هذا الصدد،العينة تظهر أفضل أداء، لذا تم تسميتها بـالمحفز الضوئي. للمقارنة، يتم تحضير محفز Ni/CdS الضوئي من خلال طريقة الترسيب الضوئي المباشر التقليدية لمدة ساعتين دون إضافةمصدر البورون. يمكن الحصول على جميع المحفزات الضوئية المذكورة أعلاه بعائد يزيد عن. جميع المواد الكيميائية من درجة التحليل (AR) وتم استخدامها دون مزيد من التنقية. CdS ( ) ومعقد بوران ثنائي ميثيل الأمين تم شراؤها من شركة علاء الدين للتكنولوجيا الكيميائية المحدودة (شنغهاي، الصين). أسيتات النيكل رباعي الماء (تم شراءه من شركة ماكلين للتكنولوجيا الحيوية المحدودة (شنغهاي، الصين).
توصيف
تم الحصول على طيف الانعكاس المنتشر للأشعة فوق البنفسجية والمرئية (UV-vis DRS) للعينات على جهاز الطيف الضوئي للأشعة فوق البنفسجية والمرئية (UV-2600، شيمادزو، اليابان). تم تحليل الطور البلوري لمختلف العينات بواسطة جهاز حيود الأشعة السينية (XRD-6100، شيمادزو، اليابان). تم دراسة أشكال جميع العينات المحضرة باستخدام المجهر الإلكتروني الماسح (FESEM) (JMS7500، جيول، اليابان). تم دراسة البنية الدقيقة لـ Ni-Bx/CdS بواسطة المجهر الإلكتروني الناقل (TEM) (Titan G2، FEI، الولايات المتحدة الأمريكية). تم تنفيذ تحليل محتوى العناصر باستخدام ICP-OES. تم جمع أطياف تحويل فورييه للأشعة تحت الحمراء على جهاز طيفي (Nicolet iS50، ثيرمو ساينتيفيك، الولايات المتحدة الأمريكية). تم قياس XPS للعينات على جهاز الطيف الإلكتروني (ESCALAB 250Xi، ثيرمو فيشر، الولايات المتحدة الأمريكية) بمصدر Al Kα (1486.6 eV)، وتم الإشارة إلى جميع طاقات الربط إلى خط عند 248.4 إلكترون فولت. تم الحصول على طيف هيكل الامتصاص الدقيق للأشعة السينية (XAFS) لحافة النيكل في وضع النقل على الطاولة XAFS-500. تم إجراء قياس كفاءة القوة الكهربية الضوئية (KPFM) المعرضة للضوء على جهاز SPM-9700 (شيمادزو، اليابان). تم الحصول على أطياف الفلورية الزمنية (TRPL) باستخدام مطياف عمر الفلورية (FLS1000، إدنبرة، المملكة المتحدة).
تحفيز ضوئي-اختبار الإنتاج
التحفيز الضوئيتم تقييم نشاط التحلل الضوئي في دورق بايركس ثلاثي العنق سعة 100 مل، تم الحفاظ عليه عند درجة حرارة الغرفة والضغط الجوي. لكل تجربة، تم إضافة 0.05 جرام من المحفز الضوئي إلى 80 مل من محلول مائي يحتوي علىحمض اللبنيك كعامل إزالة الثقوب. ثم تم تطهير القارورة بغاز النيتروجين عالي النقاء.لإزالة الهواء (15 دقيقة). تم تحريك النظام مغناطيسيًا وتعرض لأربعة مصابيح LED بقوة 3 واط.لإجراء التحفيز الضوئيالإنتاج. تحت 0.5 ساعة من الإضاءة، تم تحليل الغاز المنبعث (0.4 مل) باستخدام جهاز كروماتوغرافيا الغاز شيمادزو GC-2014C (اليابان،كغاز ناقل). بعد اختبارات دورية لمدة ساعتين، التحفيز الضوئيتم الحصول على بيانات نشاط تطور الهيدروجين لمختلف المحفزات الضوئية.
توفر البيانات
البيانات التجريبية التي تدعم نتائج هذه الدراسة متاحة من المؤلف المراسل عند الطلب المعقول. يتم توفير بيانات المصدر مع هذه الورقة.
References
Wang, J. et al. Enabling enhanced photocatalytic hydrogen evolution in water by doping perovskite with Pt . ACS Energy Lett. 9, 653-661 (2024).
Wang, X. et al. Sulfone-containing covalent organic frameworks for photocatalytic hydrogen evolution from water. Nat. Chem. 10, 1180-1189 (2018).
Bie, C. et al. A bifunctional catalyst enhances photocatalytic evolution and pyruvic acid synthesis. Angew. Chem. Int. Ed. 61, e202212045 (2022).
Jin, N. et al. Type-I CdS/ZnS core/shell quantum dot-gold heterostructural nanocrystals for enhanced photocatalytic hydrogen generation. J. Am. Chem. Soc. 145, 21886-21896 (2023).
Zhou, Q. et al. Photocatalytic sacrificial evolution dominated by micropore-confined exciton transfer in hydrogen-bonded organic frameworks. Nat. Catal. 6, 574-584 (2023).
Cao, S. et al. Ultrasmall CoP nanoparticles as efficient cocatalysts for photocatalytic formic acid dehydrogenation. Joule 2, 549-557 (2018).
Gao, D. et al. Optimizing atomic hydrogen desorption of sulfur-rich cocatalyst for boosting photocatalytic evolution. Adv. Mater. 34, 108475 (2022).
Pérez, J. et al. Strategies to break linear scaling relationships. Nat. Catal. 2, 971-976 (2019).
He, L. et al. Molybdenum carbide-oxide heterostructures: in situ surface reconfiguration toward efficient electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 59, 3544-3548 (2019).
Shah, A. H. et al. The role of alkali metal cations and platinumsurface hydroxyl in the alkaline hydrogen evolution reaction. Nat. Catal. 5, 923-933 (2022).
Huang, Z. et al. Edge sites dominate the hydrogen evolution reaction on platinum nanocatalysts. Nat. Catal. 7, 678-688 (2024).
Xie, Y. et al. Evidence for an interface of hybrid cocatalysts favoring photocatalytic hydrogen evolution kinetics. ACS Appl. Mater. Interfaces 15, 59309-59318 (2023).
Wang, M. et al. Self-optimized H-adsorption affinity of CuRu alloy cocatalysts towards efficient photocatalytic evolution. J. Mater. Sci. Technol. 174, 168-175 (2024).
Su, H. et al. A 2D bimetallic Ni-Co hydroxide monolayer cocatalyst for boosting photocatalytic evolution. Chem. Commun. 58, 6180-6183 (2022).
Gao, D. et al. Reversing free-electron transfer of cocatalyst for optimizing antibonding-orbital occupancy enables high photocatalytic evolution. Angew. Chem. Int. Ed. 62, e2O23O4559 (2023).
Zhao, T. et al. Heterostructured V-doped electrocatalysts for hydrogen evolution in anion exchange membrane water electrolyzers. Small 18, 2204758 (2022).
Liu, J. et al. Optimizing hydrogen adsorption by d-d Orbital modulation for efficient hydrogen evolution catalysis. Adv. Energy Mater. 12, 2103301 (2022).
Yan, Y. et al. Orienting electron fillings in d orbitals of cobalt single atoms for effective zinc-air battery at a subzero temperature. Adv. Funct. Mater. 34, 2316100 (2024).
Wu, X. et al. Tuning the d-band center of via octahedral and tetrahedral codoping for oxygen evolution reaction. ACS Catal. 14, 5888-5897 (2024).
Tian, J. et al. Sabatier relations in electrocatalysts based on highentropy alloys with wide-distributed d-band centers for batteries. Angew. Chem. Int. Ed. 62, e202310894 (2023).
Gao, D. et al. Tailoring antibonding-orbital occupancy state of selenium in Se-enriched cocatalyst for exceptional evolution of photocatalyst. Adv. Funct. Mater. 33, 2209994 (2023).
Liu, H. et al. Tailoring d-band center of high-valent metal-oxo species for pollutant removal via complete polymerization. Nat. Commun. 15, 2327 (2024).
Wang, C. et al. Octahedral nanocrystals of Ru-doped PtFeNiCuW/ CNTs high-entropy alloy: high performance toward pH-universal hydrogen evolution reaction. Adv. Mater. 36, 2400433 (2024).
Zhang, X. et al. Enhancing photocatalytic production with Au co-catalysts through electronic structure modification. Nat. Commun. 15, 3212 (2024).
Li, A. et al. Opening the bandgap of metallic half-heuslers via the introduction of d-d orbital interactions. Adv. Sci. 10, 2302086 (2023).
Wei, M. et al. High-entropy alloy nanocrystal assembled by nanosheets with d -d electron interaction for hydrogen evolution reaction. Energy Environ. Sci. 16, 4009 (2023).
Joshi, U. et al. Ruthenium-tungsten composite catalyst for the efficient and contamination-resistant electrochemical evolution of hydrogen. ACS Appl. Mater. Interfaces 10, 6354-6360 (2018).
Han, Z. et al. Engineering d-p orbital hybridization in single-atom metal-embedded three-dimensional electrodes for Li-S batteries. Adv. Mater. 33, 2105947 (2021).
Li, F. et al. Rhodium and carbon sites with strong orbital interaction for efficient bifunctional catalysis. ACS Nano 17, 24282-24289 (2023).
Zhu, W. et al. Weakened d-p orbital hybridization in in situ reconstructed heterointerfaces for accelerated ammonia electrosynthesis from nitrates. Energy Environ. Sci. 16, 2483 (2023).
Feng, Y. et al. Alleviating the competitive adsorption of hydrogen and hydroxyl intermediates on Ru by d-p orbital hybridization for hydrogen electrooxidation. Chem. Sci. 15, 2123 (2024).
Zhang, Y. et al. d-p hybridization-induced “trapping-coupling-conversion” enables high-efficiency Nb single-atom catalysis for Li-S batteries. J. Am. Chem. Soc. 145, 1728-1739 (2023).
Li, Q. et al. Strong d-p orbital hybridization of Os-P via ultrafast microwave plasma assistance for anion exchange membrane electrolysis. Adv. Funct. Mater. 34, 2408517 (2024).
Zhao, J. et al. Tailoring d-p orbital hybridization to decipher the essential effects of heteroatom substitution on redox kinetics. Angew. Chem. Int. Ed. 63, e202404968 (2024).
Kang, Y. et al. Porous nanoarchitectures of nonprecious metal borides: from controlled synthesis to heterogeneous catalyst applications. ACS Catal. 12, 14773-14793 (2022).
Qi, Y. et al. Insights into the activity of nickel boride/nickel heterostructures for efficient methanol electrooxidation. Nat. Commun. 13, 4602 (2022).
Lee, E. et al. Nonprecious metal borides: emerging electrocatalysts for hydrogen production. Acc. Chem. Res. 55, 56-64 (2022).
Long, H. et al. Amorphization-induced reverse electron transfer in NiB cocatalyst for boosting photocatalytic production. Appl. Catal. B Environ. 340, 123270 (2024).
Park, H. et al. Graphene- and phosphorene-like boron layers with contrasting activities in highly active for hydrogen evolution. J. Am. Chem. Soc. 139, 12915-12918 (2017).
Kang, Y. et al. Mesoporous metal-metalloid amorphous alloys: the first synthesis of open 3D mesoporous Ni-B amorphous alloy spheres via a dual chemical reduction method. Small 16, 1906707 (2020).
Kang, Y. et al. Amorphous alloy architectures in pore walls: mesoporous amorphous NiCoB alloy spheres with controlled compositions via a chemical reduction. ACS Nano 14, 17224-17232 (2020).
Xiang, X. et al. Ultrafast electron transfer from CdS quantum dots to atomically-dispersed Pt for enhanced evolution and valueadded chemical synthesis. Appl. Catal. B Environ. 340, 123196 (2024).
Zhang, J. et al. Electron transfer kinetics in CdS/Pt heterojunction photocatalyst during water splitting. Chin. J. Catal. 42, 530-2538 (2022).
Wu, F. et al. Enhanced spin-polarized electric field modulating p-band center on Ni-doped CdS for boosting photocatalytic hydrogen evolution. Small 20, 2309439 (2024).
Xiang, X. et al. Cadmium chalcogenide (CdS, CdSe, CdTe) quantum dots for solar-to-fuel conversion. Adv. Photonics Res. 3, 2200065 (2022).
Ge, F. et al. Elucidating facet-dependent photocatalytic activities of metastable CdS and Au@CdS core-shell nanocrystals. ACS Appl. Mater. Interfaces 16, 32847-32856 (2024).
Boonta, W. et al. Rhenium(I) complex-containing amphiphilic metallopolymer stabilizing CdS quantum dots for synergistically boosting photoreduction of . ACS Catal. 18, 12391-12402 (2023).
Yang, Y. et al. Enhanced photocatalytic -production activity of CdS nanoflower using single atom Pt and graphene quantum dot as dual cocatalysts. Chin. J. Struct. Chem. 41, 2206006-2206014 (2022).
Wang, F. et al. Modulating electronic structure of atomically dispersed nickel sites through boron and nitrogen dual coordination boosts oxygen reduction. Adv. Funct. Mater. 33, 2213863 (2023).
Wang, N. et al. Temperature-induced low-coordinate Ni single-atom catalyst for boosted electroreduction activity. Small 19, 2301469 (2023).
Wu, J. et al. Composition engineering of amorphous nickel boride nanoarchitectures enabling highly efficient electrosynthesis of hydrogen peroxide. Adv. Mater. 34, 2202995 (2022).
Zhang, Z. et al. Amplified internal electric field of S-scheme heterojunction for efficient photoreduction. J. Energy Chem. 92, 521-533 (2024).
Biglarbeigi, P. et al. Unraveling spatiotemporal transient dynamics at the nanoscale via wavelet transform-based kelvin probe force microscopy. ACS Nano 17, 21506-21517 (2023).
Zhang, F. et al. In situ metal-oxygen-hydrogen modified B- -X S-scheme heterojunction effectively enhanced charge separation for photo-assisted uranium reduction. Adv. Sci. 11, 2305439 (2024).
Cao, S. et al. Insights into photocatalytic mechanism of production integrated with organic transformation over S-scheme heterojunction. Adv. Sci. 11, 2305439 (2024).
Cheng, C. et al. In-situ formatting donor-acceptor polymer with giant dipole moment and ultrafast exciton separation. Nat. Commun. 15, 1313 (2024).
Deng, X . et al. Ultrafast electron transfer at the S-scheme interface for photoreduction. Nat. Commun. 15, 4807 (2024).
Ostovar, B. et al. The role of the plasmon in interfacial charge transfer. Sci. Adv. 10, eadp3353 (2024).
Qiu, J. et al. COF/ -scheme photocatalyst with enhanced light absorption and -production activity and fs-TA investigation. Adv. Mater. 36, 2400288 (2024).
Dana, J. et al. Unusually strong biexciton repulsion detected in quantum confined nanocrystals with two and three pulse femtosecond spectroscopy. ACS Nano 15, 9039-9047 (2021).
شكر وتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2022YFB3803600 (J.Y.))، ومؤسسة العلوم الطبيعية الوطنية في الصين (U23A2O1O2 (Z.Z.)، U22A2O147 (H.Y.) و22075220 (H.Y.)) ومؤسسة العلوم الطبيعية في مقاطعة هوبى في الصين (2022CFA001 (H.Y.)). نشكر كلية علوم المواد والكيمياء، جامعة علوم الأرض في الصين (CUG)، ووهان على مرافق TEM وتحليل البيانات من الدكتور مينغشينغ قونغ.
مساهمات المؤلفين
قام H.L. و H.Y. بتصميم وتخطيط التجارب. قام H.L. و X.Z. بتنفيذ التخليق، والتوصيفات، وحسابات النظرية للمواد. قام H.L. بإجراء اختبار التحفيز الضوئي. ساهم H.L. و X.Z. و Z.Z. و J.Z. و J.Y. و H.Y. في تحليل البيانات. أشرف J.Y. و H.Y. على المشروع. كتب H.L. و H.Y. المخطوطة. قام J.Y. و H.Y. بمراجعة وتدقيق المخطوطة. ناقش جميع المؤلفين النتائج وعلقوا على المخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى جياغو يوي أو هوغن يوي.
تُشكر مجلة Nature Communications Xinlong Tian و Tae Kyu Kim و Zhenjiang Li و Shizhang Qiao و Ying Zhou على مساهمتهم في مراجعة الأقران لهذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر تظل Springer Nature محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي للاستخدام غير التجاري بدون اشتقاقات 4.0، والتي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع، وإعادة إنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي للمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي للمقالة واستخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، ستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(ج) المؤلفون 2025
مختبر الوقود الشمسي، كلية علوم المواد والكيمياء، جامعة علوم الأرض الصينية، ووهان، جمهورية الصين.المختبر الرئيسي لاستخدام موارد الطاقة الجديدة والعناصر الأرضية النادرة التابعة للجنة الشؤون القومية، كلية الفيزياء وهندسة المواد، جامعة داليان مينزو، داليان، جمهورية الصين. البريد الإلكتروني: yujiaguo93@cug.edu.cn; yuhuogen@cug.edu.cn
Fine-tuning hybridization in cocatalyst for enhanced photocatalytic production
Received: September 2024
Accepted: 14 January 2025
Published online: 22 January 2025
Hydrogen ( ) production over photocatalysts utilizing solar energy is a critical challenge for achieving a carbon-free, sustainable economy . However, bare photocatalysts typically exhibit poor -production efficiency due to the significant recombination of photogenerated carriers and sluggish interfacial -reduction kinetics . To address these limitations, cocatalysts are commonly employed to accelerate electron-hole separation and provide active sites to boost the interfacial -evolution process . Ideally, an outperforming -production active site can effectively form an appropriate active site- bond, which simultaneously accelerates the proton capture and free molecules release, thereby promoting the overall -production process . Unfortunately, most active sites of reported cocatalysts suffer from either too-strong or too-weak active site- bonds owing to their unfavorable electron configurations, severely limiting their evolution performance . Therefore, reasonable electronic
The -evolution kinetics play a pivotal role in governing the photocatalytic hydrogen-evolution process. However, achieving precise regulation of the H -adsorption and H -desorption equilibrium ( ) still remains a great challenge. Herein, we propose a fine-tuning hybridization strategy to precisely optimize the kinetics in a Ni- modified CdS photocatalyst (Ni- ). X-ray absorption fine-structure spectroscopy and theoretical calculations reveal that increasing B-atom amount in the Ni-Bx cocatalyst gradually strengthens the orbital interaction between and , resulting in a consecutive -band broadening and controllable -band center on Ni active sites. The above consecutive -band optimization allows for precise modulation of the dynamics in the Ni- , ultimately demonstrating a remarkable -evolution activity of (AQE ). The femtosecond transient absorption spectroscopy further confirms the rapid electron-transfer dynamics in the photocatalyst. This work provides insights into the optimal design of prospective -evolution catalysts.
optimization of the active sites to achieve a balanced H-adsorption/ desorption process is a key factor in improving the photocatalytic production performance .
According to the -band theory, the H-adsorption/desorption dynamics at the active sites are primarily determined by the -band electronic configuration of transition metal atoms, which governs the active site- bond strength and, in turn, controls the -evolution process . However, for most transition metals, the -band electrons possess a typical localized feature, with the majority of -electrons confined within a narrow energy range, which causes electron inertness and thus inhibits the catalytic performance . To date, numerous endeavors have been undertaken to adjust the -band electronic configuration to optimize the catalytic performance of the transition metal active site . For instance, alloying with hetero-transition metal atoms can modulate the -band configuration of the active site via
orbital hybridization . However, such orbital hybridization normally yields low modulation efficiency due to the similarities in orbital configurations and electronic properties among transition metal elements (Fig. 1a). To further adjust the -band electronic configuration, the orbital regulation strategy has been developed , which can more effectively adjust the -band configuration of transition metal sites through enhanced orbital interactions (Fig. 1b(1)). In this case, the introduced -orbital increases the overlapped region and effectively broadens the distributed bandwidth of the -band, causing the formation of a wider electronic -band energy range (Fig.1b(2)). As a result, the strong orbital hybridization effect has been widely reported to improve the catalytic performance of the transition metal active sites. For example, the catalytic activity of Nb atoms can be effectively improved via the orbital hybridization within the active moiety . Similarly, the introduction of P atoms into Os can significantly boost the water-splitting activity of the Os atoms via hybridization in the osmium phosphide catalyst . However, the aforementioned regulation strategies typically focus on coordination-number-limited single atoms or composition-fixed metal compound systems , which exhibits isolated modulation of the -band configuration, thereby limiting the flexibility in adjusting -evolution dynamics. Therefore, it is conceivable that a consecutive modulation strategy could precisely optimize the -band orbital configuration of transition metal atoms, thereby achieving the optimal catalytic -evolution dynamics.
Compared with traditional -block atoms ( , etc.), the boron (B) atom possesses a typical electron-deficient feature, enabling it to form metalloid bonds with transition metal atoms and produce composition-tunable metal boride alloys . In this study, a consecutive hybridization strategy is pioneered to modulate the band configuration of the Ni active site in composition-adjustable (Fig. 1c), aiming to precisely optimize the dynamic balance at Ni active sites to promote the -evolution performance of cocatalyst (Fig. 1d). Herein, the cocatalyst with tunable B/Ni ratios is skillfully fabricated on the cadmium sulfide (CdS) surface to produce photocatalysts via a mild photo-triggered selfcatalytic route. Systematic studies reveal that increasing B content in cocatalyst can consecutively increase the -band bandwidth of Ni , thereby controllably downshifting the -band center of Ni active site to weaken the catalytic bonds, accordingly boosting the desorption to improve the catalytic -evolution activity. As a result, the optimal photocatalyst displayed an extraordinary photocatalytic -evolution performance of along with abundant viewable hydrogen bubbles, which is 5.5 times higher
Fig. 1 | The strategy of fine-tuning hybridization in cocatalyst for the suitable kinetics. a Schematic diagram illustrating the low-efficiency orbital regulation. b Schematic diagram illustrating the broadened -band via high-efficiency orbital regulation. c Schematic diagram for the consecutive Ni -band broadening to downshift -band center in composition-adjustable . d Schematic illustrating the tunable hydrogen-adsorption dynamic to reach the suitable balance.
Fig. 2 | Synthetic strategy and morphology characterization. a Schematic diagram illustrating the light-induced self-catalytic formation of cocatalysts on CdS to fabricate photocatalyst. b TEM, c-e HRTEM, f HAADF-STEM, and EDS mapping images of .
than that of photocatalyst ( ), and its -evolution activity also surpassed that of the benchmark Pt/CdS and other state-of-the-art photocatalysts (Supplementary Table 1). This work determined a consecutive modulation strategy to precisely optimize the dynamic balance of active sites for hydrogen evolution, offering insights for the design of prospective catalysts.
Results and discussion
Synthetic strategy and structural characterizations of CdS photocatalyst
The composition-adjustable nanoparticles are meticulously deposited on the CdS surface using a facile light-induced self-catalytic approach (Fig. 2a). Firstly, bare CdS particles are homogeneously dispersed into a mixed solution of nickel acetate and dimethylamine borane to obtain a stable orange suspension (Fig. 2a(1)). Upon exposure to visible light for -2 min , the CdS semiconductor can be easily excited to produce light-excited electrons, which can successfully migrate to the CdS surface to trigger the reaction between and to generate the seeds (Fig. 2a(2)). After the light is turned off, the above seeds can spontaneously induce the following catalytic reaction of and , ultimately leading to the in situ formation of nanoparticles on CdS surface to fabricate the Ni- photocatalyst
(Fig. 2a(3)). The above electron-induced self-catalytic formation mechanism of Ni- nanoparticles is further demonstrated via the supporting experiments (Supplementary Fig. 1), in which trace is added as an electron provider into the mixed solution of and to produce the seeds. These seeds then catalyze the spontaneous formation of black nanoparticles . Furthermore, based on the color changes observed (Supplementary Fig. 2), the orange CdS suspension gradually turns deep dark during the selfcatalytic process, suggesting the efficacious formation of black nanoparticles on CdS surface. In addition, according to the color changes (Supplementary Fig. 3), the cocatalyst can also be effectively fabricated on other host photocatalysts (the typical and semiconductors) via the present synthetic method. Therefore, it can be rationally concluded that the photocatalyst is successfully synthesized through the above photoinduced self-catalytic synthesis route.
Transmission electron microscopy (TEM) is employed to directly examine the microstructure of the photocatalyst, and the resulting images are displayed in Fig. 2b-f. The images clearly show that numerous dark nanoparticles ( ) are uniformly distributed on the CdS surface (Fig. 2b), exhibiting a disordered atomic arrangement (Fig. 2c-e), which is consistent with the results of fast Fourier transform and X-ray diffraction (XRD) results of pure Ni-
(Supplementary Figs. 4 and 5). Additionally, the successful construction of can further be demonstrated by the EDS mapping (Fig. 2f), where the Ni and B signals overlap and are evenly distributed on the CdS surface, providing compelling evidence for the achievement of the photocatalyst. Furthermore, based on the inductively coupled plasma optical emission spectroscopy (ICP-OES) results (Supplementary Table 2), it is shown that the compositiontunable cocatalysts (with the atomic ratio range from 0.15 to 0.89 ) can be successfully fabricated by adjusting the amount of boron source. Moreover, according to the UV-vis spectra results (Supplementary Fig. 6), the light absorption from 520 to 800 nm of is significantly greater than that of CdS and CdS samples, with the absorption intensity increasing as the B content rises, reaffirming the successful loading of the composition-adjustable cocatalyst . In addition, XRD, RAMAN, IR, and field emission scanning electron microscopy (FESEM) results (Supplementary Figs. 7-10) demonstrate that the displays a similar internal crystalline structure and apparent morphology to CdS and Ni/CdS, indicating that the photoinduced synthetic strategy has minimal impact on the host . Therefore, the above results collectively demonstrate the effective fabrication of the composition-adjustable cocatalyst on CdS through a mild photoinduced self-catalytic route for synthesizing the photocatalyst.
Efficient photocatalytic performance and its interfacial -evolution mechanism
The activities of the composition-tunable cocatalysts are investigated via the photocatalytic hydrogen evolution tests, as displayed in Fig. 3. The pure CdS exhibits a poor -evolution activity of owing to the heavy recombination of electron-hole pair and inferior interfacial active sites (Fig. 3a(1)) . Even after the modification of Ni cocatalyst, the
Ni/CdS photocatalyst only shows a slight performance improvement ( , Fig. ), implying the fact that the pristine Ni active sites still provide a limited -evolution activity. With the further integration of boron atoms into Ni cocatalyst, the resulting photocatalysts exhibit significantly elevated -generation rates compared to CdS and , and possess a volcano-plot trend (Fig. 3a(3-7)). Specifically, the optimum sample ( ) achieves a remarkable photocatalytic -production rate of , which is 29.1 and 5.6 times over the CdS and Ni/CdS, respectively. Furthermore, based on its cycling performance, the presents sustained durability with high evolution activity (Fig. 3b) and can be well maintained after the photocatalytic performance testing (Supplementary Fig. 11). Impressively, under simulated sunlight irradiation, continuous viewable bubbles can be observed on the surface of sample, reconfirming the highefficiency -evolution activity of cocatalyst (Fig. 3c, d and Supplementary Movie). In addition, the -evolution activity of the present CdS surpasses that of the benchmark Pt/CdS (Supplementary Fig. 12) and other state-of-the-art CdS-based photocatalysts (Supplementary Table 1). Therefore, the consecutive integration of boron into Ni to produce cocatalyst can significantly improve the photocatalytic hydrogengeneration activity of CdS.
According to the above performance results, the -evolution activity of the Ni cocatalyst can be greatly improved by introducing boron, which is primarily ascribed to the fact that increasing boron integration strengthens the interaction to modulate the -band configuration of Ni , as demonstrated via the following theoretical and experimental investigations. Based on the X-ray photoelectron spectroscopy (XPS) results of B 1s (Fig. 4a) and Ni 2p (Supplementary Fig. 13), it can be found that with the increase of B-doping amount in the cocatalyst, the typical Ni peak exhibits a simultaneous shift towards a more negative value, implying a consecutively enhanced
Fig. 3 | Photocatalytic -production activities. a Photocatalytic -evolution activity of various samples: (1) CdS, (2) Ni/CdS, (3) Ni-Bx/CdS-5, (4) Ni-Bx/CdS-20, (5) , (6) , (7) . The error bars (mean ± standard deviation) were calculated based on three independent photocatalytic
experiments. b Cycling runs of the photocatalytic -evolution activity of the . c Visible photocatalytic -evolution test of . d Photographs for the viewable bubbles on .
Fig. 4 | Fine-tuning hybridization in for consecutive -band broadening. a High-resolution B 1s XPS spectra. b Normalized XANES spectra. c FTEXAFS spectra in R space. Wavelet transforms of Ni K-edge EXAFS data of d Ni foil,
e Ni-Bx/CdS-5, fNi-Bx/CdS-50, and g Ni-Bx/CdS-200. Projected DOS of Ni 3d orbital for , and . n Calculated -band heights, -band widths, and -band center values of different .
electronic interaction between Ni and B in the cocatalyst. In addition, the above results can also be validated through the Bader charges analysis in Supplementary Fig. 14. As the B-doping amount increases in the (Supplementary Figs. 15 and 16), the average Bader charge value of Ni atoms increases gradually, while that of boron atoms decreases correspondingly, reconfirming the strengthened electronic interaction. The fine-tuning orbital hybridization in cocatalyst is further evidenced by the following electronic states and coordination environments analysis using X-ray absorption spectroscopy. As displayed in Fig. 4b, the normalized X-ray absorption near-edge structure (XANES) spectra reveal the Ni adsorption K -edge for various samples. It can be easily observed that with the increase of B-doping amount in Ni-Bx cocatalyst, the Ni
adsorption K-edge gradually exhibits a positive shift, demonstrating the consecutively enhanced electronic interaction of Ni and B atoms in cocatalyst. Moreover, the orbital hybridization in cocatalyst is investigated via the Ni coordination shell analysis according to the following X-ray absorption fine structure (EXAFS) results. Based on the FT-EXAFS data (Fig. 4c), the Ni foil exhibits the individual dominant peak at in R space, suggesting the inherent coordination in metallic Ni . After the introduction of B into Ni to form cocatalyst, the peak at (representing the distance between first shell layer B and Ni atoms) can be easily observed, corresponding to the presence of coordination in cocatalyst . Importantly, with the increase of the B-doping amount, the coordination peaks gradually exhibit a negative shift from -1.8
to , implying the strengthened orbital interaction in the cocatalyst. Moreover, wavelet transform-EXAFS is further conducted and the results are shown in Fig. 4d-f. The Ni sample exhibits an isolated signal corresponding to . With the consecutive introduction of B into Ni , the signal of coordination clearly appears and gradually enhances. In addition, with the increase of B-atom amounts in , the signal of shows a negative shift in R space, proving the gradually enhanced interaction between Ni and B in the cocatalyst. Therefore, the consecutive introduction of B into Ni can effectively enhance the orbital hybridization in , thereby achieving the consecutive regulation of the Ni active site.
The fine-tuning orbital hybridization in can consecutively broaden the -band width of Ni active site, as further demonstrated by the following density of states (DOS) results (Fig. 4h-m, Supplementary Fig. 17). According to the projected DOS results, it is obvious that the -band of the pristine Ni shows a highly localized character, where most of the -electrons are confined to a narrow energy range of -7.2 to 3.1 eV (Fig. 4h, Supplementary Fig. 18(1)). After the consecutive orbital interaction in , and 1.00, all the -band of different are significantly broadened (Fig. 4i-m), that is, the more B atoms are introduced, the broader -band is obtained. Especially, when the atomic B/Ni ratio is raised to 1.00 , the distributed Ni -band can be significantly broadened to a wide energy range of -12.7 to 3.9 eV (Fig. 4 m ). The -band broadening degree of samples is quantified using the -band full width at half maxima (FWHM), as shown in Supplementary Fig. 19. It can be observed that the FWHM value of the Ni -band increases progressively with higher B content, again proving the consecutive -band broadening in . The above consecutive band broadening in can be assigned to the enhanced orbital hybridization, as demonstrated by the statistical results of Ni-B bond length (Supplementary Fig. 20) according to the theoretical calculation. In this case, the bond length of the structure ranges from 1.8 to . With increasing B amount in , the percentage of bonds with short length (1.8-2.0 Å) gradually increases (Supplementary Fig. 21), indicating the strengthened interaction in via consecutive B introduction, which is in line with the radial distribution function data (Supplementary Fig. 22) of Ni-B bonds in the models. Consequently, the consecutive -band broadening inevitably causes the generation of unbalanced electrons above the fermi level, resulting in the downshift of the -band center (Supplementary Fig. 18(2, 3)). The abovementioned results can be proved by the following -band center calculations in Fig. 4 n . As the Ni ratio rises in , the -band width increases from 10.2 to 16.6 eV , while the -band height decreases from 240.6 to 85.4 eV , therefore obtaining the gradually downshifted -band center from -1.4 to -4.0 eV . Meanwhile, with the consecutive downshift of the -band center, the electron occupancy number of the orbital exhibits a gradual increase as well (Supplementary Fig. 23). Thus, the above results prove that the introduction can gradually strengthen the orbital interaction to broaden the -band of Ni , thereby effectively modulating the catalytic -band center of the cocatalyst.
The consecutive -band center modulation in Ni- cocatalyst can precisely optimize the H -adsorption/desorption dynamics on the Ni active site to promote the interfacial -evolution reaction (Fig. 5). Herein, the hydrogen-adsorption free energy ( ) and crystal orbital Hamilton populations (COHP) analyses are conducted to reveal the -evolution dynamics according to the surface models of (Fig. 5a). Theoretically, for the pristine Ni with a high -band center, the antibonding states formed after the interaction exhibit low electron occupancy (Fig. 5b(left)), resulting in excessively strong H adsorption on Ni , which suppresses desorption. This is further confirmed by the calculated negative value of -0.46 eV for the Ni site in the Ni
model (Fig. 5c). Upon incorporating B into Ni to form , the -band center of Ni is controllably downshifted, increasing the antibonding orbital electronic occupancy, which destabilizes the bonds and promotes the -desorption process (Fig. 5b(right)). Consequently, based on the results in Fig. 5c, the value for the active Ni sites gradually increases and reaches an optimal value of 0.04 eV for , optimizing the H -adsorption/desorption balance on the Ni active site and thereby enhancing the -evolution activity. These results are further supported by the COHP (ICOHP) analyses, as displayed in Fig. 5d. It can be observed that with the introduction of B into Ni, the integrated ICOHP value of the bond gradually rises from -2.53 to -1.20 , indicating a weakening of H adsorption on the Ni site to promote the -desorption process (Fig. 5d and Supplementary Fig. 24). Moreover, the elongated bond length and the increased electron localization function (ELF) on Ni can further prove that the hydrogen-adsorption strength on Ni site further demonstrate that the H -adsorption strength on Ni active site can be effectively weakened through the successive introduction of B into Ni , thus facilitating desorption (Fig. 5e). Therefore, it can be concluded from the above results that increasing B introduction into Ni can effectively strengthen the orbital hybridization to modulate the catalytic -band configuration of Ni active site, which is the intrinsic mechanism to optimize the H -adsorption/desorption dynamic on Ni to boost the -evolution activity (Fig. 5f).
Directional photoelectron transfer and its dynamics investigation
The directional photoelectron transfer process of under light irradiation is investigated using in situ Kelvin probe force microscopy (KPFM) and in situ XPS (Supplementary Figs. 25 and 26). It can be clearly observed from the contact potential difference (CPD) tests (Supplementary Fig. 25) that bare CdS displays a surface potential of . After the modification of cocatalyst on the CdS surface, a decreased CPD ( 0.14 V ) is observed, indicating the transfer of free electrons from CdS to , which can be attributed to the higher work function of compared to CdS (Supplementary Fig. 27). When the is irradiated with visible light, an increased surface potential difference is observed, corresponding to the light-excited photoelectron of CdS, which further induces photoelectron migration to the cocatalyst, as further evidenced by the in situ irradiated XPS characterization (Supplementary Fig. 26). When the light is turned on, the binding energy of peaks shifts positively to a high value, while that of peaks shift negatively, strongly verifying the photoelectrons transfer from host CdS to cocatalyst in the .
Femtosecond transient absorption spectroscopy (fs-TAS) is further employed to investigate the photoelectron transfer dynamics of various samples (Fig. 6 and Supplementary Fig. 28). For CdS (Fig. 6a, b), the transient absorption spectrum shows the positive signal ( ) at around 480 nm , which mainly belongs to the excited state absorption (ESA), representing the excited-electron transition from low to high energy level in the conduction band (CB) . Furthermore, the negative signal ( ) in the range of can be ascribed to the stimulated emission (SE), corresponding to the electronic emission from CB to the low energy levels . In addition, a negative ground-state bleach signal at around 470 nm appears within the initial 700 fs (Supplementary Fig. 29) , and then quickly delays or overlaps with the ESA signal in the range of after 900 fs . After the modification of cocatalyst on the CdS surface (Fig. 6d, e), the ESA and SE signals of the sample are clearly weaker than that of pure CdS photocatalyst, indicating that the pump pulse-excited electrons on CdS surface can effectively migrate to Ni-B cocatalyst, resulting in the suppression of the ESA and SE process. The
Fig. 5 | Precisely optimized dynamics in cocatalyst. a Optimized surface structures of Ni and for DFT calculation. b Diagram illustrating the broadening of -band to increase the antibonding occupancy to weaken bonds. c Calculated values of multiple structures. d COHP data of
bonds. e Electron local function (ELF) result after H adsorption on Ni. f A mechanism for controlling -band configuration through hybridization to regulate kinetics towards enhancing -evolution activity.
photoelectron transfer from CdS to cocatalyst is further confirmed by using potassium dichromate as the electron scavenger (Fig. 6g and Supplementary Fig. 30). It is found that, with the addition of , CdS exhibits very limited ESA and SE signals, implying the fact that the excited electrons on the CB of CdS can be efficiently consumed by scavenger. Moreover, to quantify the photoelectron transfer efficiency in CdS and Ni- , the kinetic delay
curves of samples for ESA and SE processes are fitted (Fig. 6c, f, h, i). It can be observed that the bare CdS shows a long average lifetime ( 14.5 ps for the EAS process and 6.4 ps for the SE process), indicating the fact that the excited electrons in the CB of CdS are difficult to be consumed or transferred. With the modification of cocatalyst, owing to the rapid electron transfer from CdS CB to , the average lifetimes of excited electrons in both ESA ( 8.5 ps) and SE ( 4.7 ps)
Fig. 6 | Photogenerated electron transfer mechanism and dynamics. Pseudocolor plots, femtosecond transient absorption signals within 100 ps , and corresponding decay curves of CdS (a-c) and Ni-Bx/CdS (d-f) (ESA and SE represent
excited state absorption and stimulated emission, respectively). fs-TAS data of CdS with the addition of Diagram illustrating the achievement of expedited interfacial charge transfer in the .
processes are significantly reduced, strongly suggesting the fast photoelectron transfer in . Furthermore, these results are consistent with the findings from photoelectrochemical tests and transient-state photoluminescence (TRPL) spectra (Supplementary Fig. 31). Therefore, the above results forcefully prove that the photoexcited electrons of CdS are efficiently captured by the Ni-Bx cocatalyst, thereby triggering the interfacial directional photoelectron transfer process.
In summary, a fine-tuning hybridization strategy is proposed to modulate the -band configurations of the Ni atoms, thereby precisely optimizing the H -adsorption/desorption dynamics of the Ni active sites in the Ni- cocatalyst. Experimental and theoretical results disclose that the consecutively increasing introduction in gradually enhances the orbital hybridization, which can effectively broaden the -band distribution of Ni to manipulate the band center of cocatalyst. Such results can precisely optimize the H -adsorption/desorption dynamic of the Ni active site to boost the photocatalytic -evolution activity of the . As expected, the photocatalyst shows a high -evolution performance of with continuously viewable bubble release, which is 5.6 times over the Ni/CdS. This study reveals an in-depth mechanism of tunable -band configurations via modulating electronic interaction, providing insights into the catalyst design and application principles.
Methods
Preparation of photocatalyst
The composition-tunable photocatalyst is prepared by a photoinduced self-catalytic strategy. First, 100 mg of commercial CdS was added into 80 mL methanol-water solution ( ) under constant magnetic stirring. Next, solution ( ) is added into the above suspension by an injector, where the mass ratio of Ni to CdS is controlled to be . Then, under magnetic stirring, various amount ( ) of (B source) is added to the above system, respectively. Subsequently, the above system was evacuated with for 15 min and then was sealed with a rubber septum. The reaction is triggered via 2 -min irradiation by using four LED illuminants ( ) under
stirring. After the light is turned off, the above system is magnetically stirred for 2 h under dark conditions to enable the self-catalytic formation of nanoparticles on CdS . Finally, the products are filtered, washed, and dried overnight at . The obtained photocatalysts are referred to , where X represents the added amounts of and 200 mg , respectively. In this regard, the sample shows the best performance, so it is named as photocatalyst. For comparison, the Ni/CdS photocatalyst is prepared by a 2 h of traditional direct photodeposition approach in the lack of adding boron source. All the above photocatalysts could be obtained with a yield of over . All the chemicals are of analytical grade (AR) and were used without further purification. CdS ( ) and borane dimethylamine complex were purchased from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Nickel acetate tetrahydrate ( ) was purchased from Macklin Biochemical Technology Co., Ltd. (Shanghai, China).
Characterization
The ultraviolet-visible diffuse reflectance spectra (UV-vis DRS) of the samples were obtained on the UV-vis spectrophotometer (UV-2600, Shimadzu, Japan). The crystal phase of various samples was analyzed by X-ray diffractometer (XRD-6100, Shimadzu, Japan). The morphologies of all the as-prepared samples were investigated via FESEM (JMS7500, JEOL, Japan). The microstructure of the Ni-Bx/CdS was studied by TEM (Titan G2, FEI, USA). The elemental content analysis was implemented using ICP-OES. The Fourier transform infrared spectra were collected on a spectrometer (Nicolet iS50, Thermo Scientific, USA). The XPS of samples was measured on the electron spectrometer (ESCALAB 250Xi, Thermo Fisher, USA) with Al Kα source ( 1486.6 eV ), and all the binding energies were referenced to the adventitious line at 248.4 eV . X-ray absorption fine structure (XAFS) spectra of the Ni K-edge are obtained in transmission mode on Table XAFS-500. Photo-irradiated KPFM was performed on SPM-9700 (Shimadzu, Japan). The time-resolved photoluminescence (TRPL) spectra were acquired using a fluorescence lifetime spectrophotometer (FLS1000, Edinburgh, UK).
Photocatalytic -production test
The photocatalytic -evolution activity was assessed in a 100 mL three-neck Pyrex flask, maintained at ambient temperature and atmospheric pressure. For each experiment, 0.05 g of the photocatalyst was added to 80 mL of an aqueous solution containing lactic acid as a hole scavenger. The flask was then purged with highpurity nitrogen gas ( ) to eliminate air ( 15 min ). The system was stirred magnetically and exposed to four 3 W LED lamps to conduct photocatalytic production. Under 0.5 h of illumination, the evolved gas ( 0.4 mL ) was analyzed using a Shimadzu GC-2014C gas chromatograph (Japan, as the carrier gas). After the 2 h periodic tests, the photocatalytic -evolution activity data of various photocatalysts were obtained.
Data availability
The experimental data that support the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Wang, J. et al. Enabling enhanced photocatalytic hydrogen evolution in water by doping perovskite with Pt . ACS Energy Lett. 9, 653-661 (2024).
Wang, X. et al. Sulfone-containing covalent organic frameworks for photocatalytic hydrogen evolution from water. Nat. Chem. 10, 1180-1189 (2018).
Bie, C. et al. A bifunctional catalyst enhances photocatalytic evolution and pyruvic acid synthesis. Angew. Chem. Int. Ed. 61, e202212045 (2022).
Jin, N. et al. Type-I CdS/ZnS core/shell quantum dot-gold heterostructural nanocrystals for enhanced photocatalytic hydrogen generation. J. Am. Chem. Soc. 145, 21886-21896 (2023).
Zhou, Q. et al. Photocatalytic sacrificial evolution dominated by micropore-confined exciton transfer in hydrogen-bonded organic frameworks. Nat. Catal. 6, 574-584 (2023).
Cao, S. et al. Ultrasmall CoP nanoparticles as efficient cocatalysts for photocatalytic formic acid dehydrogenation. Joule 2, 549-557 (2018).
Gao, D. et al. Optimizing atomic hydrogen desorption of sulfur-rich cocatalyst for boosting photocatalytic evolution. Adv. Mater. 34, 108475 (2022).
Pérez, J. et al. Strategies to break linear scaling relationships. Nat. Catal. 2, 971-976 (2019).
He, L. et al. Molybdenum carbide-oxide heterostructures: in situ surface reconfiguration toward efficient electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 59, 3544-3548 (2019).
Shah, A. H. et al. The role of alkali metal cations and platinumsurface hydroxyl in the alkaline hydrogen evolution reaction. Nat. Catal. 5, 923-933 (2022).
Huang, Z. et al. Edge sites dominate the hydrogen evolution reaction on platinum nanocatalysts. Nat. Catal. 7, 678-688 (2024).
Xie, Y. et al. Evidence for an interface of hybrid cocatalysts favoring photocatalytic hydrogen evolution kinetics. ACS Appl. Mater. Interfaces 15, 59309-59318 (2023).
Wang, M. et al. Self-optimized H-adsorption affinity of CuRu alloy cocatalysts towards efficient photocatalytic evolution. J. Mater. Sci. Technol. 174, 168-175 (2024).
Su, H. et al. A 2D bimetallic Ni-Co hydroxide monolayer cocatalyst for boosting photocatalytic evolution. Chem. Commun. 58, 6180-6183 (2022).
Gao, D. et al. Reversing free-electron transfer of cocatalyst for optimizing antibonding-orbital occupancy enables high photocatalytic evolution. Angew. Chem. Int. Ed. 62, e2O23O4559 (2023).
Zhao, T. et al. Heterostructured V-doped electrocatalysts for hydrogen evolution in anion exchange membrane water electrolyzers. Small 18, 2204758 (2022).
Liu, J. et al. Optimizing hydrogen adsorption by d-d Orbital modulation for efficient hydrogen evolution catalysis. Adv. Energy Mater. 12, 2103301 (2022).
Yan, Y. et al. Orienting electron fillings in d orbitals of cobalt single atoms for effective zinc-air battery at a subzero temperature. Adv. Funct. Mater. 34, 2316100 (2024).
Wu, X. et al. Tuning the d-band center of via octahedral and tetrahedral codoping for oxygen evolution reaction. ACS Catal. 14, 5888-5897 (2024).
Tian, J. et al. Sabatier relations in electrocatalysts based on highentropy alloys with wide-distributed d-band centers for batteries. Angew. Chem. Int. Ed. 62, e202310894 (2023).
Gao, D. et al. Tailoring antibonding-orbital occupancy state of selenium in Se-enriched cocatalyst for exceptional evolution of photocatalyst. Adv. Funct. Mater. 33, 2209994 (2023).
Liu, H. et al. Tailoring d-band center of high-valent metal-oxo species for pollutant removal via complete polymerization. Nat. Commun. 15, 2327 (2024).
Wang, C. et al. Octahedral nanocrystals of Ru-doped PtFeNiCuW/ CNTs high-entropy alloy: high performance toward pH-universal hydrogen evolution reaction. Adv. Mater. 36, 2400433 (2024).
Zhang, X. et al. Enhancing photocatalytic production with Au co-catalysts through electronic structure modification. Nat. Commun. 15, 3212 (2024).
Li, A. et al. Opening the bandgap of metallic half-heuslers via the introduction of d-d orbital interactions. Adv. Sci. 10, 2302086 (2023).
Wei, M. et al. High-entropy alloy nanocrystal assembled by nanosheets with d -d electron interaction for hydrogen evolution reaction. Energy Environ. Sci. 16, 4009 (2023).
Joshi, U. et al. Ruthenium-tungsten composite catalyst for the efficient and contamination-resistant electrochemical evolution of hydrogen. ACS Appl. Mater. Interfaces 10, 6354-6360 (2018).
Han, Z. et al. Engineering d-p orbital hybridization in single-atom metal-embedded three-dimensional electrodes for Li-S batteries. Adv. Mater. 33, 2105947 (2021).
Li, F. et al. Rhodium and carbon sites with strong orbital interaction for efficient bifunctional catalysis. ACS Nano 17, 24282-24289 (2023).
Zhu, W. et al. Weakened d-p orbital hybridization in in situ reconstructed heterointerfaces for accelerated ammonia electrosynthesis from nitrates. Energy Environ. Sci. 16, 2483 (2023).
Feng, Y. et al. Alleviating the competitive adsorption of hydrogen and hydroxyl intermediates on Ru by d-p orbital hybridization for hydrogen electrooxidation. Chem. Sci. 15, 2123 (2024).
Zhang, Y. et al. d-p hybridization-induced “trapping-coupling-conversion” enables high-efficiency Nb single-atom catalysis for Li-S batteries. J. Am. Chem. Soc. 145, 1728-1739 (2023).
Li, Q. et al. Strong d-p orbital hybridization of Os-P via ultrafast microwave plasma assistance for anion exchange membrane electrolysis. Adv. Funct. Mater. 34, 2408517 (2024).
Zhao, J. et al. Tailoring d-p orbital hybridization to decipher the essential effects of heteroatom substitution on redox kinetics. Angew. Chem. Int. Ed. 63, e202404968 (2024).
Kang, Y. et al. Porous nanoarchitectures of nonprecious metal borides: from controlled synthesis to heterogeneous catalyst applications. ACS Catal. 12, 14773-14793 (2022).
Qi, Y. et al. Insights into the activity of nickel boride/nickel heterostructures for efficient methanol electrooxidation. Nat. Commun. 13, 4602 (2022).
Lee, E. et al. Nonprecious metal borides: emerging electrocatalysts for hydrogen production. Acc. Chem. Res. 55, 56-64 (2022).
Long, H. et al. Amorphization-induced reverse electron transfer in NiB cocatalyst for boosting photocatalytic production. Appl. Catal. B Environ. 340, 123270 (2024).
Park, H. et al. Graphene- and phosphorene-like boron layers with contrasting activities in highly active for hydrogen evolution. J. Am. Chem. Soc. 139, 12915-12918 (2017).
Kang, Y. et al. Mesoporous metal-metalloid amorphous alloys: the first synthesis of open 3D mesoporous Ni-B amorphous alloy spheres via a dual chemical reduction method. Small 16, 1906707 (2020).
Kang, Y. et al. Amorphous alloy architectures in pore walls: mesoporous amorphous NiCoB alloy spheres with controlled compositions via a chemical reduction. ACS Nano 14, 17224-17232 (2020).
Xiang, X. et al. Ultrafast electron transfer from CdS quantum dots to atomically-dispersed Pt for enhanced evolution and valueadded chemical synthesis. Appl. Catal. B Environ. 340, 123196 (2024).
Zhang, J. et al. Electron transfer kinetics in CdS/Pt heterojunction photocatalyst during water splitting. Chin. J. Catal. 42, 530-2538 (2022).
Wu, F. et al. Enhanced spin-polarized electric field modulating p-band center on Ni-doped CdS for boosting photocatalytic hydrogen evolution. Small 20, 2309439 (2024).
Xiang, X. et al. Cadmium chalcogenide (CdS, CdSe, CdTe) quantum dots for solar-to-fuel conversion. Adv. Photonics Res. 3, 2200065 (2022).
Ge, F. et al. Elucidating facet-dependent photocatalytic activities of metastable CdS and Au@CdS core-shell nanocrystals. ACS Appl. Mater. Interfaces 16, 32847-32856 (2024).
Boonta, W. et al. Rhenium(I) complex-containing amphiphilic metallopolymer stabilizing CdS quantum dots for synergistically boosting photoreduction of . ACS Catal. 18, 12391-12402 (2023).
Yang, Y. et al. Enhanced photocatalytic -production activity of CdS nanoflower using single atom Pt and graphene quantum dot as dual cocatalysts. Chin. J. Struct. Chem. 41, 2206006-2206014 (2022).
Wang, F. et al. Modulating electronic structure of atomically dispersed nickel sites through boron and nitrogen dual coordination boosts oxygen reduction. Adv. Funct. Mater. 33, 2213863 (2023).
Wang, N. et al. Temperature-induced low-coordinate Ni single-atom catalyst for boosted electroreduction activity. Small 19, 2301469 (2023).
Wu, J. et al. Composition engineering of amorphous nickel boride nanoarchitectures enabling highly efficient electrosynthesis of hydrogen peroxide. Adv. Mater. 34, 2202995 (2022).
Zhang, Z. et al. Amplified internal electric field of S-scheme heterojunction for efficient photoreduction. J. Energy Chem. 92, 521-533 (2024).
Biglarbeigi, P. et al. Unraveling spatiotemporal transient dynamics at the nanoscale via wavelet transform-based kelvin probe force microscopy. ACS Nano 17, 21506-21517 (2023).
Zhang, F. et al. In situ metal-oxygen-hydrogen modified B- -X S-scheme heterojunction effectively enhanced charge separation for photo-assisted uranium reduction. Adv. Sci. 11, 2305439 (2024).
Cao, S. et al. Insights into photocatalytic mechanism of production integrated with organic transformation over S-scheme heterojunction. Adv. Sci. 11, 2305439 (2024).
Cheng, C. et al. In-situ formatting donor-acceptor polymer with giant dipole moment and ultrafast exciton separation. Nat. Commun. 15, 1313 (2024).
Deng, X . et al. Ultrafast electron transfer at the S-scheme interface for photoreduction. Nat. Commun. 15, 4807 (2024).
Ostovar, B. et al. The role of the plasmon in interfacial charge transfer. Sci. Adv. 10, eadp3353 (2024).
Qiu, J. et al. COF/ -scheme photocatalyst with enhanced light absorption and -production activity and fs-TA investigation. Adv. Mater. 36, 2400288 (2024).
Dana, J. et al. Unusually strong biexciton repulsion detected in quantum confined nanocrystals with two and three pulse femtosecond spectroscopy. ACS Nano 15, 9039-9047 (2021).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFB3803600 (J.Y.)), National Natural Science Foundation of China (U23A2O1O2 (Z.Z.), U22A2O147 (H.Y.) and 22075220 (H.Y.)) and the Natural Science Foundation of Hubei Province of China (2022CFA001 (H.Y.)). We thank the Faculty of Materials Science and Chemistry, China University of Geosciences (CUG), Wuhan for its TEM facilities and the data analysis of Dr. Mingxing Gong.
Author contributions
H.L. and H.Y. conceived and designed the experiments. H.L. and X.Z. carried out the synthesis, characterizations, and theory calculations of the materials. H.L. carried out the photocatalytic test. H.L., X.Z., Z.Z., J.Z., J.Y. and H.Y. contributed to data analysis. J.Y. and H.Y. supervised the project. H.L. and H.Y. wrote the manuscript. J.Y. and H.Y. revised and reviewed the manuscript. All authors discussed the results and commented on the manuscript.
Correspondence and requests for materials should be addressed to Jiaguo Yu or Huogen Yu.
Peer review information Nature Communications thanks Xinlong Tian, Tae Kyu Kim, Zhenjiang Li, Shizhang Qiao, and Ying Zhou reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2025
Laboratory of Solar Fuel, Faculty of Materials Science and Chemistry, China University of Geosciences, Wuhan, PR China. Key Laboratory of New Energy and Rare Earth Resource Utilization of State Ethnic Affairs Commission, School of Physics and Materials Engineering, Dalian Minzu University, Dalian, PR China. ae-mail: yujiaguo93@cug.edu.cn; yuhuogen@cug.edu.cn