تحفيز الحديد الضوئي عبر نقل الشحنة من ligand إلى المعدن المفتوح بواسطة حمض برونستيد Iron photocatalysis via Brønsted acid-unlocked ligand-to-metal charge transfer
إصلاح منصات التحفيز الضوئي المستدامة القائمة على المعادن ثلاثية الأبعاد باستخدام الضوء المرئي لتفعيل المواد الكيميائية غير النشطة أمر مرغوب فيه للغاية. هنا، نوضح استخدام حمض برونستيد لفتح تحفيز ضوئي قوي وعملي لنقل الشحنة من الرابطة الحديدية إلى المعدن (LMCT) لتفعيل مجموعة متنوعة من هالوألكيل كربوكسيليت غير النشطة. أو Cl ) لإنتاج الجذور الحرة. تتيح هذه العملية الفلورو-بوليهالوألكيليشن للألكينات غير المنشطة من خلال دمج Selectfluor المتاح بسهولة كمصدر للفلور. يمكن الحصول على فلوريدات الألكيل القيمة بما في ذلك جزيئات الأدوية المحتملة بسهولة من خلال هذه البروتوكولات. تشير الدراسات الآلية إلى أن الأنواع الحقيقية لجمع الضوء قد تنشأ من التجميع في الموقع لـومذيب الأسيتونيتريل، حيث أن الحمض برونستيد يزيد فعلاً من كفاءة LMCT بين مركز الحديد وعبر تفاعلات الروابط الهيدروجينية. نتوقع أن تكون هذه المنصة من الحديد التي تم فتحها بواسطة حمض برونستيد خيارًا مستدامًا مثيرًا لتنفيذ تنشيط المركبات الخاملة.
تطوير استراتيجيات تحفيزية مستدامة لتوليد وتحويل الجذور من المواد الكيميائية الرخيصة وخاصة الخاملة هو ما يسعى إليه الكيميائيون المتخصصون في الجذور باستمرار.مع نهضة الكيمياء الضوئيةلقد تم استخدام نقل الشحنة من ligand إلى المعدن (LMCT) الناتج عن الضوء في المجمعات المعدنية النشطة ضوئيًا كأداة قوية لتوليد أنواع الجذور المفتوحة. حتى الآن، كانت استكشافات الليغاندات للمجمعات المعدنية النشطة ضوئيًا تركز بشكل رئيسي علىالكحوليات، والأحماض الكربوكسيلية الأليفاتية سهلة التفعيل. توسيع نطاق الليجند لـ LMCT ليشمل مركبات أكثر خمولًا هو أحد أهم المهام لتطوير كيمياء LMCT المحفزة بالضوء. مؤخرًا، تم استخدام استراتيجيات LMCT للنحاس المحفز بالأشعة فوق البنفسجية أو الضوء الأرجواني من أجل الوظائف اللامائية لحمض البنزويك المستقر (مقابل قطب الزئبق المشبع (SCE) تم الإبلاغ عنه من قبل ريتير وماكميلانمن الجدير بالذكر أن هناك العديد من المواد الكيميائية الرخيصة بكميات كبيرة التي تتمتع بإمكانات أكسدة عالية للغاية مثل ثلاثي فلورو الأسيتيت. مقابل SCE) ، مما يجعل التفعيل والتحويل عبر الضوء المرئي المحفز
LMCT صعب للغايةلذلك، فإن تعزيز تفاعل LMCT للمواد الكيميائية الضخمة غير النشطة للغاية كعوامل ربط لإنتاج الجذور الحرة أمر مرغوب فيه للغاية (الشكل 1أ).
الحديد كأحد أرخص وأكثر المعادن وفرةلقد أظهر أنشطة ضوئية مثيرة للإعجاب في الكيمياء الضوئيةالذي تعتبر قدرته التحفيزية على إنتاج الأنواع الجذرية من نوع LMCT البديل المثالي لقدرة نقل الإلكترون الفردي (SET) المعتمدة على معقدات البوليبيريديل من المعادن النبيلة المثارة مثل الروثينيوم والإيريديوم. مع الطلب الملح على الكيمياء المستدامة، نتوقع تطوير منصة تحفيزية قوية من الحديد تعتمد على LMCT يمكن تطبيقها في تنشيط المواد الكيميائية الساكنة وبناء هياكل جزيئية ذات قيمة مضافة. بالنظر إلى الوضع المتميز للفلور و/أو المجموعات المحتوية على الفلور في الأدوية الجديدة، والكيماويات الزراعية، والمواد الوظيفية، والتركيب العضوي.نأمل في الاستفادة من الوجود الشاملأو Clكـمصدر جذريوالألكين كحلقة ربط صناعية لأداء الفلورو-بوليهالوألكيلاشن المحفز بالحديد تحت تأثير الضوء المرئي باستخدام مادة فلورة متاحة بسهولة وتجارية. سيوفر ذلك تكلفة منخفضة و
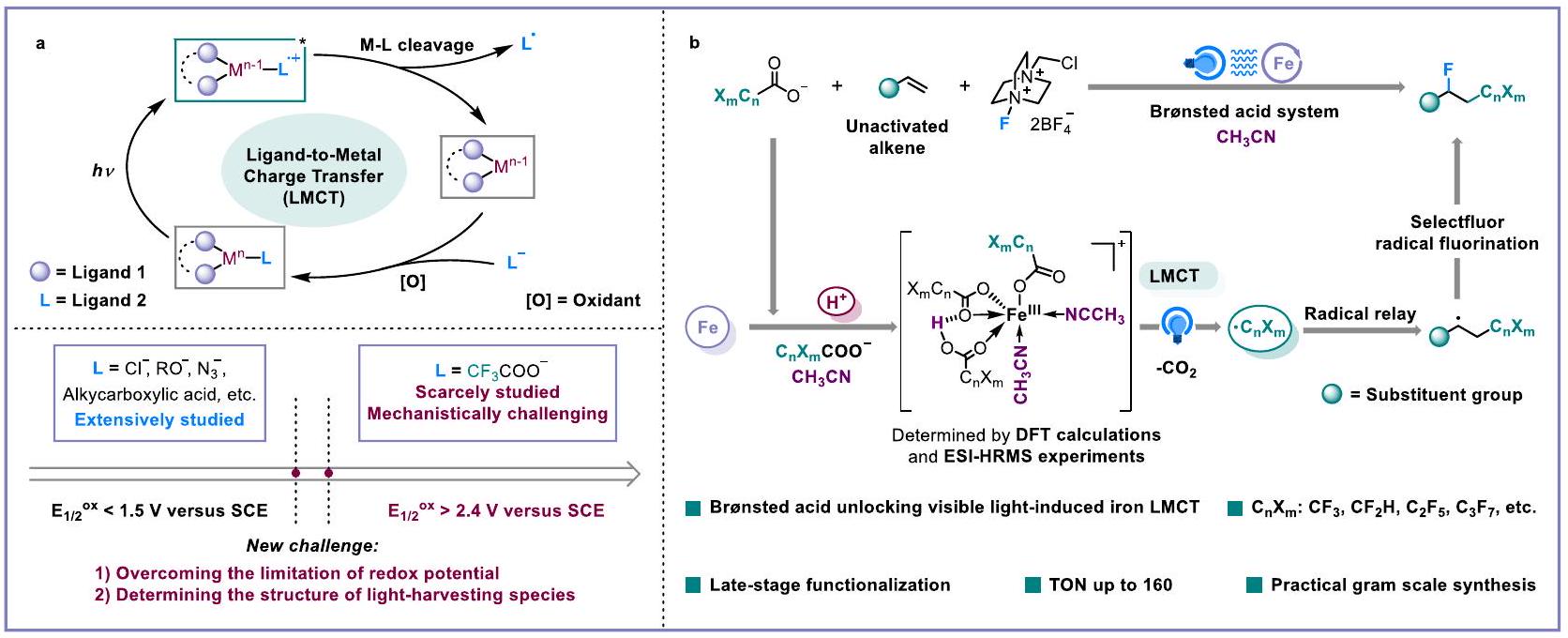
الشكل 1 | LMCT المعتمد على المعادن المستحثة بالضوء لإنتاج وتحويل الجذور. أ أحدث ما توصلت إليه تقنية LMCT المستحثة بالضوء. ب هذا العمل: حمض برونستيد يفتح LMCT الحديدي لتفاعل الفلورو-بوليهالوألكيلاشن للألكينات. DFT نظرية الكثافة الوظيفية، ESI-HRMS مطيافية الكتلة عالية الدقة بالاستنشاق الكهربائي.
طريقة عالمية لتخليق الهالوألكانات القيمة. ومع ذلك، فإن التأثيرات الاستقرائية السلبية للهالوجينات تضعف بشكل كبير القدرة على التنسيق وكثافة الإلكترونات لمجموعات الكربوكسيل، مما يجعل من الصعب جدًا تجميع الحديد بشكل فعال وأنواع جمع الضوء المعتمدة على LMCT لتوليدالجذور الحرة تحت إشعاع الضوء المرئي.
لمعالجة هذه القضية المعقدة، من خلال دراسات آلية مفصلة، نكشف هنا عن استخدام حمض برونستيد لفتح منصة تحفيزية عملية لتفاعل LMCT للفلوروبولي هالوألكلة للألكينات غير المنشطة مع وفرة.كماالمصدر و Selectfluor الكهربية كعامل الفلورة (الشكل 1ب). يمكن تحويل مجموعة متنوعة من الألكينات غير المنشطة بنجاح إلى فلوريدات ألكيل مع انتقائية مكانية ممتازة وعوائد متوسطة إلى جيدة. ومن الجدير بالذكر أن ميزات الفلورة المتأخرة للبوليهالوألكيل المعقدة الشبيهة بالأدوية، والتخليق على نطاق جرام، وكمية التحميل المنخفضة من محفز الحديد (عدد الدوران، TON: 160)، تظهر آفاق تطبيق محتملة في اكتشاف الأدوية والكيمياء التركيبية.
النتائج
تطوير التفاعل
لتطوير بروتوكول عام لتفعيل LMCT الحديد المفتوح بواسطة حمض برونستيدلتشكيلالمتطرفون، اقترحنا أن موازنة تركيز حمض برونستيد والليغاندهو أمر حيوي. لذلك، نتوقع اعتماد استراتيجية التوليد في الموقع لإطلاق الحمض برونستيد و(الشكل 2أ، الأشكال التكميلية 13 و ). من خلال تم تحديد NMR، ووجد أن الجمع بين أنهدريد ثلاثي فلورو الأسيتيك (TFAA) والنيوكليوفيل المحتوي على OH وهو الإيزوبروبانول يمكن أن يوفر الحمض القوي برونستيد حمض ثلاثي فلورو الأسيتيك (TFA). عندما تم استبدال الإيزوبروبانول بأوكسي دي بنزين، كانت الأسترات المحتملة لثلاثي فلورو الأسيتيك ( و ) وتم الكشف عن المنتج الثانوي 5 بواسطة تأين الرذاذ الكهربائي – مطياف الكتلة عالي الدقة (ESI-HRMS). كنا نعتقد أن تم توليده من الاستبدال النووي المهاجم بين و . ستؤدي هذه العملية إلى تجديد الأوكسي دي بنزين، وقد يؤدي 4 في النهاية إلى إنتاج 5 مع إطلاق TFA. ثم قررنا اختيار TFAA الرخيصة، والإيزوبروبانول، والأوكسي دي بنزين، ولبناء نظام تفاعل مناسب لحمض برونستيد وثلاثي فلوروأسيتيت. بعد جهودنا المستمرة، حددنا ظروفًا مناسبة وقابلة للإدارة تم استخدامها مباشرة من المنتجات التجارية.كعامل مساعد وSelectfluor كعامل فلورة جذرية لتحقيق فلورة ثلاثي الفلورو ميثيل بنجاح للالكينات غير المنشطة باستخدام نظام تفاعل حمض برونستيد وثلاثي فلور الأسيتيت وضوء LED الأزرق إشعاع (الشكل 2ب-1، الجداول التكميلية 4 و 5). عندما تمت إزالة الأوكسي دي بنزين من الظروف القياسية، فقطتم الكشف عن عائد المنتج المرغوب فيه من الفلوروتريفلووروميثيليشن، مصحوبًا بالاستهلاك الكامل للألكين 6. بعد الكشف بواسطة ESI-HRMS، تم اقتراح إمكانية الأمينوتريفلووروميثيليشن والفلوروأمينات الألكينات. و ) اعتُبرت التحولات الرئيسية (الشكل 2ب-ii، الأشكال التكميلية 16 و21 و22). وبالمثل، في غياب الأوكسيديبنزين، فإن استخدام N-فلوروبنزينسولفونيميد كعامل فلورة كهربائي أضعف من Selectfluor يفضل أيضًا تكوين الروابط بسبب تأثير تطابق القطبية بينالألكينات الوسيطة غير النشطة والراديكالية (الأشكال التكميلية 17 و 23). استنادًا إلى التقارير السابقةتتضمن جيل 9 انتشار الجذور بشكل جذري كاتيون جذري مع الألكين، وبالتالي قد يكون الدور الثاني للأوكسي دي بنزين هو العمل كعازل للأكسدة والاختزال لإخماد النشاط الكهربائي في الوقت المناسب.الكاتيون الجذري من سيلكتفلور (الشكل 2ب-iii). من المهم، بالمقارنة مع نظام TFA المباشر وظروف TFAA-أوكسي ديفينزين (بدون إيزوبروبانول)، أن معدل التفاعل الأولي الأسرع وفترة الحضانة الأقصر في ظروفنا القياسية تثبت ضرورة ومزايا حمض برونستيد المتوازن والليغاند لدينا.النظام (الشكل 2ج، الشكل التكميلي 15).
الدراسات الميكانيكية
تم إجراء دراسات آلية مفصلة لتحديد الأنواع الحقيقية لجمع الضوء. خلال التحقيق في محفز ملح الحديد، وجدنا أن و تصرفت بقدرة تحفيزية مرضية لفلورة الألكينات ثلاثية الفلورو ميثيل (الجدول التكميلي 2، المدخلات 1 و 2). لذلك، اقترحنا أن هذه الأملاح الحديدية يمكن أن تتحول إلى الحديد والأنواع المستندة إلى جمع الضوء في المنشأةحمضوثلاثي فلوروأسيتيت () النظام. تم إثبات هذا الاقتراح من خلال دراسات UV-Vis مفصلة (الشكل التوضيحي 26). كشفت تجارب ستوكيومترية إضافية أنه تحت إشعاع LED الأزرق، يمكن فتحه وإطلاقهالجذور الحرة فقط في وجود حمض برونستيد قوي (الشكل 2د، المدخلات 1-4). الأحماض الضعيفة أو الخارجيةلم يعزز تفعيللإجراء الفلوروتريفلووروميثيليشن للألكينات (المدخلات 5-7). لقد تجنب إضافة حمض برونستيد بنجاح الحاجة إلى ضوء الأشعة فوق البنفسجية لتحويل (المدخلان 8 و 9). أشارت نتائج دراسات UV-Vis أيضًا إلى أن التحلل الضوئي لـسيحدث فقط عندماتعرضت لظروف حمضية. في غياب حمض برونستيد، إضافيلا يزال غير قادر على المساعدة في LMCT الناتج عن الضوء المرئي لـ (الشكل 2e). بالإضافة إلى ذلك، استخدام
الشكل 2 | تصميم وتحديد نقل الشحنة من ligand الحديد غير المقيد بحمض برونستيد. أ توليد حمض برونستيد في الموقع و. ب أكسيديبنزين كعازل ريدوكس. i الظروف القياسية: ألكين ( ) ، سيلكتفلورأوكسي دي بنزين (0.1 مليمول)، إيزوبروبانول (0.4 مليمول)، و، مصابيح LED زرقاء، وقت تفاعل 12 ساعة؛ (ii) بدون أكسيد ثنائي الفينيل تحت ظروف قياسية؛ (iii) مع أوكسي دي بنزين كعازل ريدوكس. ج. ميزة النظام المتوازن. (ط) ظروف قياسية؛ (2) بدون إيزوبروبانول تحت ظروف قياسية؛ (3) مع TFA بدلاً من TFAA وإيزوبروبانول في ظروف قياسية. د. تجارب ستوكيومترية للأنواع الوسيطة Fe (III). TfOH حمض ثلاثي فلور الميثان سلفونيك، N.D. لم يتم الكشف عنه. هـ. تجارب UV-Vis.تجارب تشغيل/إيقاف الضوء.دراسات حركية لـ. و TFA لتوليدفي الموقع وخلق جو حمضي أعطى أيضًا نتائج مشابهة في الأشعة فوق البنفسجية والمرئية (الشكل التوضيحي 27). كما وجدنا أن كمية الحمض برونستيد تحدد عائد الفلوروتريفلووروميثيليشن – ثلاثة مكافئات بالنسبة إلىيجب أن يكون التحميل مثاليًا لـالإنتاج الجذري في تظهر هذه النتائج بالتأكيد أن حمض برونستيد قوي قادر بالفعل على فتح LMCT الصعبة.لـجيل راديكاليمُدمَج معتحت ظروفنا القياسية هو نوع محتمل من LMCT. متطلبات الاستمرارية
الشكل 3 | تحديد الأنواع القائمة على الحديد لجمع الضوء. أ مقارنة بين عدة مذيبات مختلفة.تحديدكأنيونات. ج حسابات نظرية الكثافة الوظيفية (DFT). د آلية محتملة.
الشكل 4 | نطاق الألكينات غير المفعلة.ظروف التفاعل العامة: الألكينسيلكتفلورأوكسي دي بنزين (0.1 مليمول)، إيزوبروبانول (0.4 مليمول) وLEDات زرقاء،وقت رد الفعل.
الشكل 5 | تجارب التعديل الوظيفي في المراحل المتأخرة وتكبير الحجم. أ تعديل المشتقات الصيدلانية.ظروف التفاعل العامة: ألكين (0.2 ملليمول)،سيلكتفلورأوكسي دي بنزين (0.1 مليمول)، إيزوبروبانول (0.4 مليمول) وLEDات زرقاء، وقت تفاعل 12 ساعة.LEDات زرقاء، وقت تفاعل 24 ساعة.تجارب على نطاق جرام وTON. الشرط A: الألكين ( ) ، ( سيلكتفلور (7.5 مليمول) أوكسي دي بنزين (2.5 مليمول)، إيزوبروبانول (10.0 مليمول) و ، مصابيح LED زرقاء، وقت تفاعل 72 ساعة. الحالة ب: ألكين (62، سيلكتفلور ( 0.006 مليمول ) أكسيد ثنائي البنزين ( 1.0 مليمول ) إيزوبروبانول ( 4.0 مليمول ) وLEDات زرقاء،زمن الاستجابة. عدد دوران TON. تم تأكيد الإشعاع من خلال تجارب تشغيل وإيقاف الضوء على مر الزمن (الشكل 2f). من المهم أن منحنيات الحركة لـو TFAA تظهر أن معدل التفاعل الأولي سيتحسن عند زيادة تحميل المحفز الحديدي والمصدر (الشكل 2g، الأشكال التكميلية 31 و32).
بالإضافة إلى ذلك، خلال تحقيق المذيبات، لاحظنا أنه فقط عندما تمتلك المذيبات القدرة على التنسيق مثل، و يمكن الحصول على المنتج المرغوب فيه من الفلوروتريفلوروميثيليشن. على النقيض من ذلك، فشلت المذيبات مثل ثنائي كلورو الميثان (DCM) وثنائي كلورو الإيثان (DCE) في توليدالجذور (الشكل 3أ). ألهمنا ذلك للتفكير فيما إذاكان متورطًا في تجميع الحديد والأنواع المعتمدة على جمع الضوء (الشكل 3 أ، ب، الجدول التكميلي 3، الشكل التكميلي 33). لتحديد الهيكل الحقيقي للحديد بسرعة والأنواع المعتمدة على جمع الضوء، قمنا بإجراء حسابات نظرية الكثافة الوظيفية (DFT) (الأشكال التكميلية 34-37). كما هو موضح في الشكل 3c، فإن الـ 12 التي يمكن اعتبارها مزيجًا منمعوجزيئان منمن خلال تأثير الروابط الهيدروجينية والتنسيق، يتصرف فعلاً كـ LMCT الفعال بين الحديد والمركبات أحادية الأسنان.تحت إشعاع الضوء الأزرق. وجودتم تحديده بشكل أكبر من خلال تجارب ESI-HRMS (الشكل التوضيحي 24). بدون تأثير الرابطة الهيدروجينية لحمض برونستيد، فإن إطلاقأصبح الجذري 13 من الإثارة 12 غير مواتٍ حراريًا (الأشكال التكميلية 38-42). علاوة على ذلك، في وجود الروابط الهيدروجينية داخل الجزيء، انخفضت طاقة مدار LUMO للحديد بشكل كبير بمقدار 0.86 إلكترون فولت، مما يفيد عملية LMCT المرغوبة (الشكل التكميلية 44). بالنظر إلى أن احتمالالموجود في نظام التفاعل وبناءً على الهيكلاستبدال أحد الروابط الهيدروجينيةمجموعات معلا يمكن استبعاد أن تعمل كبديل غير ضروري لتجمعات من أنواع جمع الضوء المعتمدة على الحديد (انظر الأشكال التكميلية 46-49 لمناقشات مفصلة).
تم توضيح دورة الآلية التفصيلية لهذا البروتوكول في الشكل 3d (الأشكال التكميلية 52 و53). في وجود حمض برونستيد،وأسيتونيتريل،سيحدث تفاعل الحديد المحرر من الحمض المقترح للأنواع التي تجمع الضوء 12 تحت إشعاع الضوء الأزرق، مما يوفرالجذر 13 والمستوى المتوسط 14. بعد إصدارالغاز من الوسيط الجذري 13 (الشكل التكميلي 11)، المطلوبتم إنتاج الجذير، الذي تم احتجازه لاحقًا بواسطة الألكين لتشكيل المنتج الجذري 15. وفرت عملية الفلورة الجذرية بين 15 و Selectfluor المنتج المطلوب.
الشكل 6 | التطبيقات الاصطناعية.ظروف التفاعل العامة: ألكين (0.2 ملليمول)،سيلكتفلورأوكسي دي بنزين (0.1 مليمول)، إيزوبروبانول (0.4 مليمول) و، مصابيح LED زرقاء، 12 – وقت رد الفعل.LEDات زرقاء،وقت رد الفعل.بدلاً منLEDات زرقاء،وقت رد الفعل. منتج الفلوروتريفلووروميثيليشن 16. المنتج الناتجالكاتيون الجذري 10 تطلب تنظيمًا بواسطة العازل الأحمروكسي ديفينزين لتجنب إنتاج روابط C -N ذات الصلة (الأشكال التكميلية 16 و21 و22). الكاتيونات الجذرية و يجب أن تكون مسؤولة عن إعادة تدوير الحديد (III) من خلال خطوات SET، وهو ما تم تبريره أيضًا من خلال حسابات DFT (الأشكال التكميلية 50 و51).
نطاق الركائز
بعد إنشاء نظام حمض برونستيد الذي يفتح الحديد LMCT، تم إنتاج مجموعة متنوعة من الفلورايد الألكيلية القيمة عبر طريقتنا (الشكل 4). تم تحمل العديد من الأوليفينات الطرفية الأليفاطية التي تحتوي على مجموعات بنزوات متنوعة بشكل جيد لتقديم منتجات الفلورايد ثلاثي الفلورو ميثيل مع عوائد مرضية وانتقائية كيميائية واحدة (7 و 18-36)، على الرغم من أن البنزوئات قد تكون عرضة للتفاعل في وجود حمض برونستيد قوي. بالإضافة إلى ذلك، كان الألكين المعدل بالسلفونات أيضًا كفؤًا (37). ومن الجدير بالذكر أن قوة الأكسدة العالية لـ Selectfluor تحت نظامنا لم تعرقل تحويل تلك الألكينات التي تربط الأنيسولات الغنية بالإلكترونات لبناء المنتجات المرغوبة (38 و 39). كما قمنا بتجميع الترايازول والفثاليميد عالية القيمة مع الألكينات، مما وفر عوائد معتدلة من الفلورايد ثلاثي الفلورو ميثيل (40-42). كانت مشتقات الأليلبنزين و1-هكساديكين أيضًا ممكنة (43 و 44). بالإضافة إلى ذلك،تمت عملية تفاعل الألكين ثنائي الاستبدال بنجاح لتغني تنوع الفلورايدات الألكيلية (45).
التعديل الوظيفي في المراحل المتأخرة
لإظهار التوافق الاصطناعي الممتاز لهذا البروتوكول، تم إخضاع مجموعة واسعة من الأوليفينات المعقدة المشتقة من الجزيئات الصيدلانية لظروف تحفيز الضوء LMCT للحديد غير المقيد بحمض برونستيد (الشكل 5أ). تم تركيب أدوية تخفيف الألم الضرورية مثل الكيتوبروفين، الإيبوبروفين، والفينبوفين مع الفلورايد الألكيلي بعوائد متوسطة إلى جيدة (46-48). بالإضافة إلى ذلك، يمكن أيضًا تحمل مشتقات المبيد الجرثومي إيثيل بارابين ووكيل مضاد الالتهاب إيزوكسيباك بسلاسة ( و ). ومن الجدير بالذكر أن الفلورايد ثلاثي الفلوروميثيل لمشتق الثاليدوميد كان قادرًا على تحقيق عائد بنسبة 75% (51)، حيث أن الثاليدوميد هو علاج فعال لمرض الحمامي العقدية الجلدي. أدت إضافة مجموعة الألكين إلى DL-مينثول (دواء تقليدي صيني لإزالة السموم) إلى إنتاج ركيزة مرضية للفلورايد ثلاثي الفلوروميثيل (52). كما قمنا بتمديد هذا البروتوكول لتحويل الوسائط الطبية الرئيسية حمض ترانس-4-(ميثوكسي كربونيل) سيكلوهكسان كربوكسيلي ودي أستون فركتوز لزيادة الفرصة لمزيد من التعديلات ( و54). تم تحويل الأوليفينات ذات هياكل الإسترون والسكرين إلى منتجاتها المقابلة بعوائد متوسطة ( و56). ومن الجدير بالذكر أن بنزبرومارون وبروبينسيد، وهما دواءان لعلاج النقرس، يحتويان على مشتقات أوليفين كانت ركيزات مناسبة للفلورايد ثلاثي الفلوروميثيل في المراحل المتأخرة (57 و58). بالإضافة إلى ذلك، يمكن أن توفر المسكنات الخافضة للحرارة مثل حمض الأسيتيل ساليسيليك والفوربروفين عوائد جيدة (59-61). نجاح
تجارب النطاق الكبير وTON (TON: 160) أظهر الإمكانية التطبيقية لهذا التركيب المثير (الشكل 5ب).
بالإضافة إلى الفلورايد ثلاثي الفلوروميثيل للأوليفينات، يمكن أن توسع منصتنا من الحديد غير المقيد بحمض برونستيد LMCT أيضًا التركيب المتباين وتحل التحولات الجذرية الصعبة بما في ذلك الفلورايد ثنائي الفلوروميثيل (63، 68، 72، 75، و80)، الفلورايد كلوريد ثنائي الفلوروميثيل (64، 69، 73، 76، و81)، الفلورايد ثلاثي الكلوروميثيل (67، 71، 78، و82)، الفلورايد ثنائي الكلوروميثيل (70، 74، 77، و84)، الفلورايد بانتافلورو إيثيليشن (65)، الفلورايد كلوريد رباعي الفلورو إيثيليشن (79 و83)، وفلورايد هيبتافلورو بروبيل (66) لجزيئات الأدوية (الشكل 6).
نقاش
في الختام، تم الكشف عن بروتوكول عملي للحديد غير المقيد بحمض برونستيد LMCT لتنشيط مجموعة متنوعة من الكربوكسيليت المحتوية على الهالوجين غير النشطة إلى الجذور من خلال دراسات آلية مفصلة، والتي تم تطبيقها على الفلورايد بولي هالوألكيليشن للأوليفينات غير النشطة. تأثير الرابطة الهيدروجينية لحمض برونستيد وتنسيق المذيب مهمان للغاية لضمان التجميع الفعال للحديد و-المبني على الأنواع التي تجمع الضوء. الدراسات الإضافية حول تنشيط المركبات غير النشطة الأكثر تحديًا عبر LMCT للمعادن 3d غير المقيدة بحمض برونستيد جارية حاليًا في مختبرنا.
طرق
الإجراء العام للفلورايد بولي هالوألكيليشن للأوليفينات
تم شحن دورق شلنكي سعة 25 مل مزود بعصا مغناطيسية بـ وSelectfluor ( ). تم إخلاء الدورق وإعادة ملئه بـ ثلاث مرات. ثم تم شحن الوعاء بـ جاف جدًا، أوليفين (0.2 مليمول)، أو Cl (1.0 أو 1.4 مليمول)، أكسيد ثنائي البنزين (17.0 ملغ، 0.1 مليمول) وإيزوبروبانول ( ). تم تحريك خليط التفاعل تحت جو نيتروجين وتم تسليط الضوء بواسطة مصابيح LED زرقاء لمدة 12 أو 24 أو 36 ساعة. بعد انتهاء التفاعل، تم الكشف عن بواسطة TCD-GC. ثم تم إخماد نظام التفاعل بواسطة ثلاثي إيثيل الأمين، وتم تخفيفه بـ EtOAc. بعد التركيز تحت الفراغ، تم تنقية الرواسب الناتجة بواسطة كروماتوغرافيا عمود السيليكا للحصول على المنتجات.
توفر البيانات
يعلن المؤلفون أن البيانات المتعلقة بتوصيف المواد والمنتجات، والأساليب العامة، ودراسات التحسين، والإجراءات التجريبية، والدراسات الآلية، وبيانات HRMS وطيف NMR، والدراسات الحاسوبية متاحة ضمن المقالة ومعلوماتها التكميلية بالإضافة إلى البيانات التكميلية. جميع البيانات متاحة من المؤلف المراسل عند الطلب. يتم توفير بيانات المصدر مع هذه الورقة.
References
Hartwig, J. F. & Larsen, M. A. Undirected, homogeneous C-H bond functionalization: challenges and opportunities. ACS Cent. Sci. 2, 281-292 (2016).
Jiang, H. & Studer, A. Intermolecular radical carboamination of alkenes. Chem. Soc. Rev. 49, 1790-1811 (2020).
Yoon, T. P., Ischay, M. A. & Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2, 527-532 (2010).
Narayanam, J. M. & Stephenson, C. R. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 40, 102-113 (2011).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322-5363 (2013).
Liu, P., Liu, W. & Li, C.-J. Catalyst-free and redox-neutral innate trifluoromethylation and alkylation of aromatics enabled by light. J. Am. Chem. Soc. 139, 14315-14321 (2017).
Marzo, L., Pagire, S. K., Reiser, O. & Konig, B. Visible-light photocatalysis: does it make a difference in organic synthesis? Angew. Chem. Int. Ed. 57, 10034-10072 (2018).
Fu, M.-C., Shang, R. & Fu, Y. Photocatalytic decarboxylative alkylations mediated bytriphenylphosphine and sodium iodide. Science 363, 1429-1434 (2019).
Holmberg-Douglas, N. & Nicewicz, D. A. Photoredox-catalyzed C-H functionalization reactions. Chem. Rev. 122, 1925-2016 (2022).
Bellotti, P., Huang, H.-M., Faber, T. & Glorius, F. Photocatalytic latestage C-H functionalization. Chem. Rev. 123, 4237-4352 (2023).
Golden, D. L. et al. Benzylic C-H esterification with limiting C-H substrate enabled by photochemical redox buffering of the Cu catalyst. J. Am. Chem. Soc. 145, 9434-9440 (2023).
Abderrazak, Y., Bhattacharyya, A. & Reiser, O. Visible-light-induced homolysis of Earth-abundant metal-substrate complexes: a complementary activation strategy in photoredox catalysis. Angew. Chem. Int. Ed. 60, 21100-21115 (2021).
Chang, L., An, Q., Duan, L., Feng, K. & Zuo, Z. Alkoxy radicals see the light: newparadigms of photochemical synthesis. Chem. Rev. 122, 2429-2486 (2022).
Juliá, F. Ligand-to-metal charge transfer (LMCT) photochemistry at 3d-metal complexes: an emerging tool for sustainable organic synthesis. ChemCatChem 14, e202200916 (2022).
Li, Q. Y. et al. Decarboxylative cross-nucleophile coupling via ligand-to-metal charge transfer photoexcitation of carboxylates. Nat. Chem. 14, 94-99 (2022).
Tsurugi, H. & Mashima, K. Renaissance of homogeneous cerium catalysts with unique couple: redox-mediated organic transformations involving homolysis of -ligand covalent bonds. J. Am. Chem. Soc. 143, 7879-7890 (2021).
Zhao, R. & Shi, L. A renaissance of ligand-to-metal charge transfer by cerium photocatalysis. Org. Chem. Front. 5, 3018-3021 (2018).
Zhou, W.-J. et al. Light runs across iron catalysts in organic transformations. Chem. Eur. J. 26, 15052-15064 (2020).
Nicewicz, D., Roth, H. & Romero, N. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett 27, 714-723 (2015).
Denkler, L. M., et al. A general iron-catalyzed decarboxylative oxygenation of aliphatic carboxylic acids. Angew. Chem. Int. Ed. e202403292 (2024). During the revisions of our manuscript, Bunescu, A. et al. published this related work.
Su, W., Xu, P. & Ritter, T. Decarboxylative hydroxylation of benzoic acids. Angew. Chem. Int. Ed. 60, 24012-24017 (2021).
Xu, P., Lopez-Rojas, P. & Ritter, T. Radical decarboxylative carbometalation of benzoic acids: a solution to aromatic decarboxylative fluorination. J. Am. Chem. Soc. 143, 5349-5354 (2021).
Chen, T. Q. et al. A unified approach to decarboxylative halogenation of (hetero)aryl carboxylic acids. J. Am. Chem. Soc. 144, 8296-8305 (2022).
Dow, N. W. et al. Decarboxylative borylation and cross-coupling of (hetero)aryl acids enabled by copper charge transfer catalysis. J. Am. Chem. Soc. 144, 6163-6172 (2022).
Xu, P., Su, W. & Ritter, T. Decarboxylative sulfoximination of benzoic acids enabled by photoinduced ligand-to-copper charge transfer. Chem. Sci. 13, 13611-13616 (2022).
Depecker, C., Marzouk, H., Trevin, S. & Devynck, J. Trifluoromethylation of aromatic compounds via Kolbe electrolysis in pure organic solvent. Study on laboratory and pilot scale. New. J. Chem. 23, 739-742 (1999).
Bian, K.-J. et al. Photocatalytic hydrofluoroalkylation of alkenes with carboxylic acids. Nat. Chem. 15, 1683-1692 (2023). During the
revisions of our manuscript, West, J. G. et al. published this related work.
Qi, X.-K. et al. Photoinduced hydrodifluoromethylation and hydromethylation of alkenes enabled by ligand-to-iron charge transfer mediated decarboxylation. ACS Catal. 14, 1300-1310 (2024). During the revisions of our manuscript, Xia, W. et al. published this related work.
Zhang, W. et al. Leaving group assisted strategy for photoinduced fluoroalkylations using N-hydroxybenzimidoyl chloride esters. Angew. Chem. Int. Ed. 58, 624-627 (2019).
Lv, D. et al. Iron-catalyzed radical asymmetric aminoazidation and diazidation of styrenes. Angew. Chem. Int. Ed. 60, 12455-12460 (2021).
Zhang, Z. et al. Controllable C-H alkylation of polyethers via iron photocatalysis. J. Am. Chem. Soc. 145, 7612-7620 (2023).
Feng, G., Wang, X. & Jin, J. Decarboxylative C-C and C-N bond formation by ligand-accelerated iron photocatalysis. Eur. J. Org. Chem. 2019, 6728-6732 (2019).
Li, Z., Wang, X., Xia, S. & Jin, J. Ligand-accelerated iron photocatalysis enabling decarboxylative alkylation of heteroarenes. Org. Lett. 21, 4259-4265 (2019).
Xia, S., Hu, K., Lei, C. & Jin, J. Intramolecular aromatic C-H acyloxylation enabled by iron photocatalysis. Org. Lett. 22, 1385-1389 (2020).
Jin, Y. et al. Photo-induced direct alkynylation of methane and other light alkanes by iron catalysis. Green. Chem. 23, 9406-9411 (2021).
Kang, Y.-C., Treacy, S. M. & Rovis, T. Iron-catalyzed photoinduced LMCT: a abstraction enables skeletal rearrangements and C(sp )-H alkylation. ACS Catal. 11, 7442-7449 (2021).
Bian, K.-J., Kao, S.-C., Nemoto, D. Jr., Chen, X.-W. & West, J. G. Photochemical diazidation of alkenes enabled by ligand-to-metal charge transfer and radical ligand transfer. Nat. Commun. 13, 7881 (2022).
Lu, Y.-C. & West, J. G. Chemoselective decarboxylative protonation enabled by cooperative Earth-abundant element catalysis. Angew. Chem. Int. Ed. 135, e202213055 (2022).
Tu, J.-L. et al. Iron-catalyzed ring-opening of cyclic carboxylic acids enabled by photoinduced ligand-to-metal charge transfer. Green. Chem. 24, 5553-5558 (2022).
Zhang, M., Zhang, J., Li, Q. & Shi, Y. Iron-mediated ligand-to-metal charge transfer enables 1,2-diazidation of alkenes. Nat. Commun. 13, 7880 (2022).
Zhang, Q. et al. Iron-catalyzed photoredox functionalization of methane and heavier gaseous alkanes: scope, kinetics, and computational studies. Org. Lett. 24, 1901-1906 (2022).
Kao, S.-C. et al. Photochemical iron-catalyzed decarboxylative azidation via the merger of ligand-to-metal charge transfer and radical ligand transfer catalysis. Chem. Catal. 3, 100603 (2023).
Zhang, W.-M., Feng, K.-W., Hu, R.-G., Guo, Y.-J. & Li, Y. Visible-lightinduced iron redox-catalyzed selective transformation of biomass into formic acid. Chem 9, 430-442 (2023).
Xiong, N., Li, Y. & Zeng, R. Merging photoinduced iron-catalyzed decarboxylation with copper catalysis for and couplings. ACS Catal. 13, 1678-1685 (2023).
Lutovsky, G. A., Gockel, S. N., Bundesmann, M. W., Bagley, S. W. & Yoon, T. P. Iron-mediated modular decarboxylative crossnucleophile coupling. Chem 9, 1610-1621 (2023).
Muller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881-1886 (2007).
Honer, M., Schubiger, P. A. & Ametamey, S. M. Molecular imaging with PET. Chem. Rev. 108, 1501-1516 (2008).
O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 37, 308-319 (2008).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320-330 (2008).
Wang, J. et al. Fluorine in pharmaceutical industry: fluorinecontaining drugs introduced to the market in the last decade (20012011). Chem. Rev. 114, 2432-2506 (2008).
Caron, S . Where does the fluorine come from? A review on the challenges associated with the synthesis of organofluorine compounds. Org. Process Res. Dev. 24, 470-480 (2020).
Josephson, B. et al. Light-driven post-translational installation of reactive protein side chains. Nature 585, 530-537 (2020).
Xu, W., Jiang, H., Leng, J., Ong, H. W. & Wu, J. Visible-light-induced selective defluoroborylation of polyfluoroarenes, gem-difluoroalkenes, and trifluoromethylalkenes. Angew. Chem. Int. Ed. 59, 4009-4016 (2020).
Intermaggio, N. E., Millet, A., Davis, D. L. & MacMillan, D. W. Deoxytrifluoromethylation of alcohols. J. Am. Chem. Soc. 144, 11961-11968 (2022).
Qing, F.-L. et al. A fruitful decade of organofluorine chemistry: new reagents and reactions. CCS Chem. 4, 2518-2549 (2022).
Xu, P., Fan, W., Chen, P. & Liu, G. Enantioselective radical trifluoromethylation of benzylic C-H bonds via cooperative photoredox and copper catalysis. J. Am. Chem. Soc. 144, 13468-13474 (2022).
Beatty, J. W., Douglas, J. J., Cole, K. P. & Stephenson, C. R. J. A scalable and operationally simple radical trifluoromethylation. Nat. Commun. 6, 7919 (2015).
Beatty, J. W. et al. Photochemical perfluoroalkylation with pyridine -oxides: mechanistic insights and performance on a kilogram scale. Chem 1, 456-472 (2016).
Yin, D., Su, D. & Jin, J. Photoredox catalytic trifluoromethylation and perfluoroalkylation of arenes using trifluoroacetic and related carboxylic acids. Cell Rep. Phys. Sci. 1, 100141 (2020).
Zhang, K., Rombach, D., Notel, N. Y., Jeschke, G. & Katayev, D. Radical trifluoroacetylation of alkenes triggered by a visible-lightpromoted bond fragmentation of trifluoroacetic anhydride. Angew. Chem. Int. Ed. 60, 22487-22495 (2021).
Giri, R. et al. Photoredox activation of anhydrides for the solventcontrolled switchable synthesis of gem-difluoro compounds. Angew. Chem. Int. Ed. 61, e202209143 (2022).
Wu, W., You, Y. & Weng, Z. Recent advances in the synthesis of fluoroalkylated compounds using fluoroalkyl anhydrides. Chin. Chem. Lett. 33, 4517-4530 (2022).
Zhang, M. et al. Photocatalytic fluoroalkylations of (hetero)arenes enabled by the acid-triggered reactivity umpolung of acetoxime esters. Chem. Catal. 2, 1793-1806 (2022).
Fernández-García, S., Chantzakou, V. O. & Juliá-Hernández, F. Direct decarboxylation of trifluoroacetates enabled by iron photocatalysis. Angew. Chem. Int. Ed. 63, e202311984 (2024). During the revisions of our manuscript, Juliá-Hernández, F. et al. published this related work.
Yu, W., Xu, X.-H. & Qing, F.-L. Silver-mediated oxidative fluorotrifluoromethylation of unactivated alkenes. Adv. Synth. Catal. 357, 2039-2044 (2015).
Liu, Z. et al. Radical carbofluorination of unactivated alkenes with fluoride ion. J. Am. Chem. Soc. 140, 6169-6175 (2018).
Pitts, C. R., Ling, B., Snyder, J. A., Bragg, A. E. & Lectka, T. Aminofluorination of cyclopropanes: a multifold approach through a common, catalytically generated intermediate. J. Am. Chem. Soc. 138, 6598-6609 (2016).
Riener, M. & Nicewicz, D. A. Synthesis of cyclobutane lignans via an organic single electron oxidant-electron relay system. Chem. Sci. 4, 2625-2629 (2013).
الشكر
تم دعم هذا المشروع من قبل المؤسسة الوطنية للعلوم الطبيعية في الصين، أرقام المنح 22101265 (L.N.)، 21903071 (S.L.) و21822303 (Y.L.);
مؤسسة العلوم ما بعد الدكتوراه في الصين، أرقام المنح 2022M712866 (L.N.)، 2023M733212 (S.J.); صندوق مشترك لبرنامج البحث والتطوير للتقنيات الرئيسية في مقاطعة خنان، رقم المنحة 222301420006 (Y.L.); مشاريع الترويج للبحث والتطوير الرئيسي في مقاطعة خنان، رقم المنحة 222102310042 (L.N.); وزارة العلوم والتكنولوجيا لجمهورية الصين الشعبية (Y.L.).
مساهمات المؤلفين
X.J. وL.N. تصوروا العمل. صمم X.J. وY.H. التجارب وحللا البيانات. قام X.J. وY.H. وK.J. وJ.H. وJ.Z. وS.J. وJ.S. بتنفيذ التجارب الاصطناعية. ساهم Y.L. وS.L. في حسابات DFT. وصف X.J. وL.N. المخطوطة الأصلية وقام جميع المؤلفين بمراجعتها.
كلية الكيمياء، ومختبر بينغيوان، جامعة تشنغتشو، تشنغتشو، خنان، جمهورية الصين الشعبية.المختبر الوطني الرئيسي للأدوية المضادة للفيروسات، مختبر بينغيوان، جامعة خنان العادية، شينشيانغ، خنان، جمهورية الصين الشعبية.مدرسة الكيمياء والهندسة الكيميائية، مختبر تشونغتشينغ الرئيسي لنظرية الكيمياء والآلية، جامعة تشونغتشينغ، تشونغتشينغ، جمهورية الصين الشعبية.البريد الإلكتروني: lanyu@cqu.edu.cn; lishijunzong@zzu.edu.cn; nlb@zzu.edu.cn
Iron photocatalysis via Brønsted acidunlocked ligand-to-metal charge transfer
Received: 27 September 2023
Accepted: 15 July 2024
Published online: 20 July 2024
(T) Check for updates
Xiaoyu Jiang , Yu Lan , Yudong Hao , Kui Jiang , Jing He , Jiali Zhu , Shiqi Jia , Jinshuai Song , Shi-Jun Li & Linbin Niu
Reforming sustainable 3d-metal-based visible light catalytic platforms for inert bulk chemical activation is highly desirable. Herein, we demonstrate the use of a Brønsted acid to unlock robust and practical iron ligand-to-metal charge transfer (LMCT) photocatalysis for the activation of multifarious inert haloalkylcarboxylates ( or Cl ) to produce radicals. This process enables the fluoro-polyhaloalkylation of non-activated alkenes by combining easily available Selectfluor as a fluorine source. Valuable alkyl fluorides including potential drug molecules can be easily obtained through this protocol. Mechanistic studies indicate that the real light-harvesting species may derive from the in situ-assembly of , and acetonitrile solvent, in which the Brønsted acid indeed increases the efficiency of LMCT between the iron center and via hydrogen-bond interactions. We anticipate that this Brønsted acid-unlocked iron LMCT platform would be an intriguing sustainable option to execute the activation of inert compounds.
Developing sustainable catalytic strategies for radical generation and transformation from cheap and particularly inert bulk chemicals is what radical chemists are continually pursuing . With the renaissance of photochemistry , photo-induced ligand-to-metal charge transfer (LMCT) of photochemically active metal complex has been served as a robust tool for the generation of open-shell radical species. Up to now, exploration of the ligands for photochemically active metal complexes have mainly focused on , alcohols, and easily-activated aliphatic carboxylic acids . Expanding the ligand scope of LMCT to more inert compounds is one of the most important tasks for the development of photo-induced LMCT chemistry. Recently, UV or purple light-induced copper LMCT strategies for the decarboxylative functionalization of stable benzoic acid ( versus saturated calomel electrode (SCE)) were reported by Ritter and MacMillan . Notably, there are still numerous cheap bulk chemicals with extremely high oxidative potentials such as trifluoroacetate ( versus SCE) , making the activation and transformation via visible light-induced
LMCT quite challenging . Therefore, enhancing the LMCT reactivity of extremely inert bulk chemicals as ligands for radical production is highly desirable (Fig. 1a).
Iron as one of the cheapest and most abundant metals , has displayed impressive photochemical activities in photochemistry , whose LMCT catalytic ability for radical species production is the ideal alternative to the single electron transfer (SET) capacity of the excited noble-metal ruthenium and iridium polypyridyl complexes. With the urgent demand for sustainable chemistry, we anticipate the development of a robust iron LMCT catalytic platform that can be applied for inert bulk chemical activation and value-added molecular skeleton construction. Considering the privileged status of fluorine and/or fluorine-containing groups in new pharmaceuticals, agrochemicals, functional materials, and organic synthesis , we hope to utilize the ubiquitous or Cl as a radical source and alkene as a synthetic linker to perform visible light-induced iron-catalyzed fluoro-polyhaloalkylation with an easily available and commercial fluorination reagent. This would provide a low-cost and
Fig. 1 | Photo-induced metal-based LMCT for radical production and transformation. a The state-of-the-art of photo-induced LMCT. b This work: Brønsted acid unlocking iron LMCT for fluoro-polyhaloalkylation of alkenes. DFT density functional theory, ESI-HRMS electrospray ionization high-resolution mass spectrometry.
universal method for the synthesis of valuable haloalkanes . However, the negative inductive effects of halogens seriously weaken the coordination ability and electron density of carboxyl groups, making it very difficult to effectively assemble iron and -based lightharvesting species via LMCT to generate radicals under visible light irradiation.
To address this intricate issue, through detailed mechanistic studies, we herein disclose the use of a Brønsted acid to unlock a practical iron LMCT catalytic platform for fluoro-polyhaloalkylation of nonactivated alkenes with abundant as the source and electrophilic Selectfluor as the fluorination reagent (Fig. 1b). A variety of non-activated alkenes can be successfully converted into alkyl fluorides with excellent regioselectivity and moderate to good yields. Notably, the features of late-stage fluoro-polyhaloalkylation of complex drug-like molecules, gram-scale synthesis, and low loading amount of iron catalyst (Turnover Number, TON: 160), show potential application prospects in pharmaceutical discovery and synthetic chemistry.
Results
Reaction development
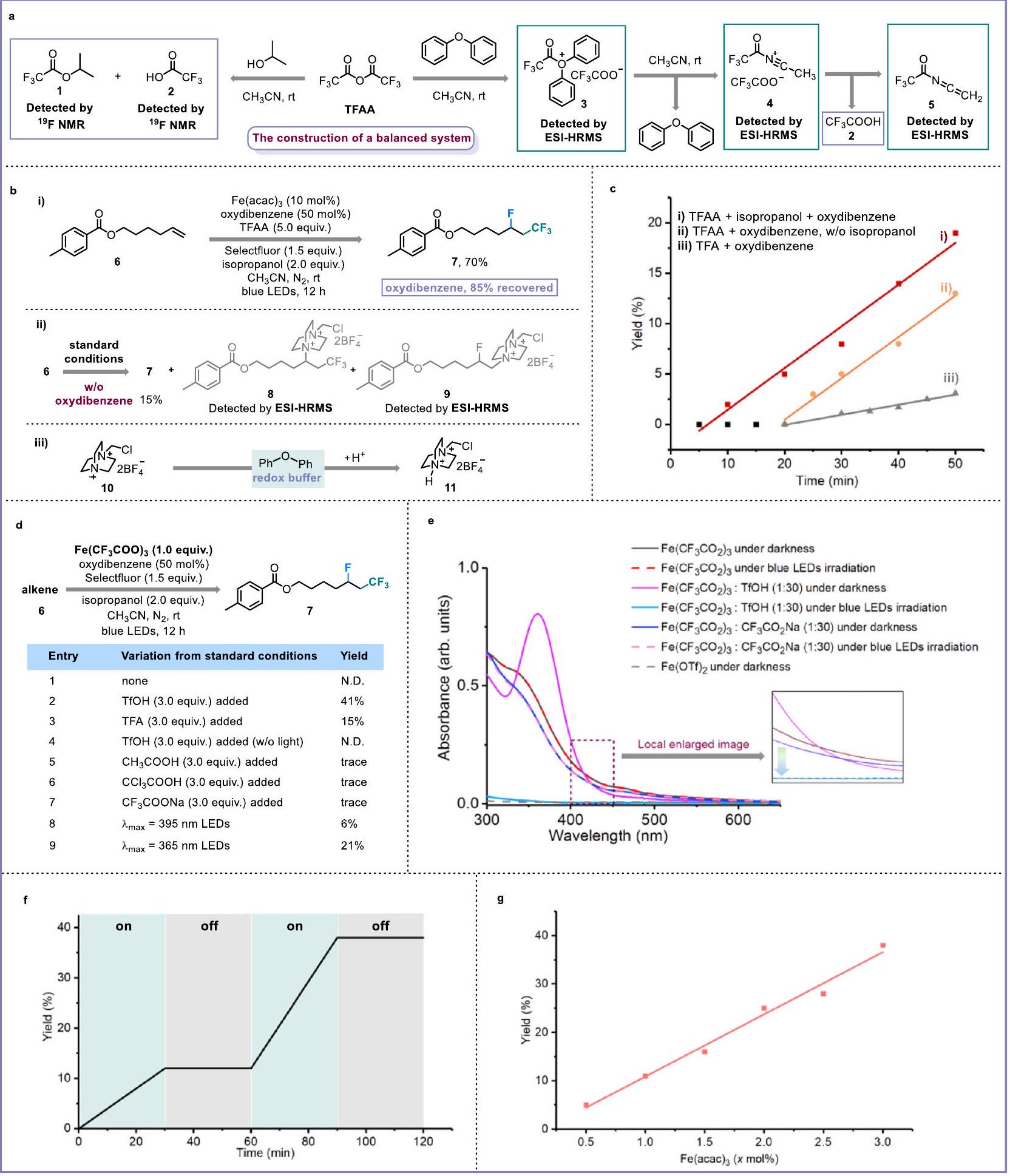
To develop a general Brønsted acid-unlocked iron LMCT protocol for the activation of to form radicals, we proposed that balancing the concentration of Br ønsted acid and ligand is vital. Therefore, we expect to adopt the in situ generation strategy for the release of the Brønsted acid and (Fig. 2a, Supplementary Figs. 13 and ). Through NMR identification, it was found that the combination of trifluoroacetic anhydride (TFAA) and the OH nucleophile-containing isopropanol could afford the strong Brønsted acid trifluoroacetic acid (TFA). When isopropanol was replaced by oxydibenzene, the possible trifluoroacetates ( and ) and by-product 5 were detected by electrospray ionization-high resolution mass spectros (ESI-HRMS). We thought that was generated from the nucleophilic substitution between and . This process would accomplish the regeneration of oxydibenzene, and 4 might eventually deliver 5 with the release of TFA. Then, we decided to choose cheap TFAA, isopropanol, oxydibenzene, and to construct a suitable Brønsted acid and trifluoroacetate reaction system. Following our continuous effort, we identified suitable and manageable conditions that directly employed commercial as the catalyst and Selectfluor as the radical fluorination reagent to successfully achieve the fluorotrifluoromethylation of non-activated alkenes using a Brønsted acid and trifluoroacetate reaction system and blue LED
irradiation (Fig. 2b-i, Supplementary Tables 4 and 5). When the oxydibenzene was removed from the standard conditions, only yield of desired fluorotrifluoromethylation product was detected, accompanied by the complete consumption of alkene 6. After ESI-HRMS detection, the possible aminotrifluoromethylation and fluoroamination of alkenes ( and ) were considered as the major transformations (Fig. 2b-ii, Supplementary Figs. 16, 21, and 22). Similarly, in the absence of oxydibenzene, utilizing N-Fluorobenzenesulfonimide as a weaker electrophilic fluorination reagent than Selectfluor also prefers the bond formation owing to the polarity matching effect between the radical intermediate and non-activated alkenes (Supplementary Figs. 17 and 23). Based on previous reports , the generation of 9 involves the radical propagation of radical cation with an alkene, and thus the second role of oxydibenzene might serve as the redox buffer to timely quench the electrophilic radical cation from the Selectfluor (Fig. 2b-iii). Significantly, compared with the direct TFA system and TFAA-oxydibenzene conditions (without isopropanol), the faster initial reaction rate and shorter induction period of our standard conditions evidence the necessity and advantages of our balanced Brønsted acid and ligand system (Fig. 2c, Supplementary Fig. 15).
Mechanistic studies
Detailed mechanistic studies were performed to identify the real lightharvesting species. During the investigation of the iron salt catalyst, we found that and behaved satisfying catalytic ability for alkene fluorotrifluoromethylation (Supplementary Table 2, Entries 1 and 2). Therefore, we proposed that these iron salts could be transformed into iron and -based light-harvesting species in the constructed acid and trifluoroacetate ( ) system. This proposal was evidenced by detailed UV-Vis studies (Supplementary Fig. 26). Further stoichiometric experiments revealed that under the blue LED irradiation, can be unlocked and release radicals only in the presence of a strong Brønsted acid (Fig. 2d, Entries 1-4). Weak Brønsted acids or exogenous did not promote the activation of for the fluorotrifluoromethylation of alkene (Entries 5-7). The addition of a Brønsted acid successfully avoided the need for UV light for the transformation of (Entries 8 and 9). The results of UV-Vis studies also indicated that the photodecomposition of would occur only when was subjected to acidic conditions. In the absence of a Brønsted acid, extra could still not assist with the visible light-induced LMCT of (Fig. 2e). Besides, using
Fig. 2 | Design and identification of Brønsted acid-unlocked iron ligand-tometal charge transfer. a In situ generating Brønsted acid and . b Oxydibenzene as redox buffer. i Standard conditions: alkene ( ), , Selectfluor , oxydibenzene ( 0.1 mmol ), isopropanol ( 0.4 mmol ), and , blue LEDs, 12 h reaction time; (ii) without oxydibenzene under standard conditions; (iii) with
oxydibenzene as redox buffer. c The advantage of balanced system. (i) standard conditions; (ii) without isopropanol under standard conditions; (iii) with TFA instead of TFAA and isopropanol in standard conditions. d Stoichiometric experiments of Fe (III)-intermediate species. TfOH trifluoromethanesulfonic acid, N.D. not detected. e UV-Vis experiments. Light on/off experiments. Kinetic studies of .
and TFA to generate in situ and to create an acidic atmosphere also gave similar UV-Vis results (Supplementary Fig. 27). We also found that the amount of Brønsted acid determined the yield of fluorotrifluoromethylation-three equivalents relative to the loading should be optimal for radical production in
the stoichiometric experiments (Supplementary Fig. 30). These results certainly reveal that a strong Brønsted acid is indeed capable of unlocking the challenging LMCT of for radical generation and combined with under our standard conditions is a potential LMCT species. The requirement of continuous
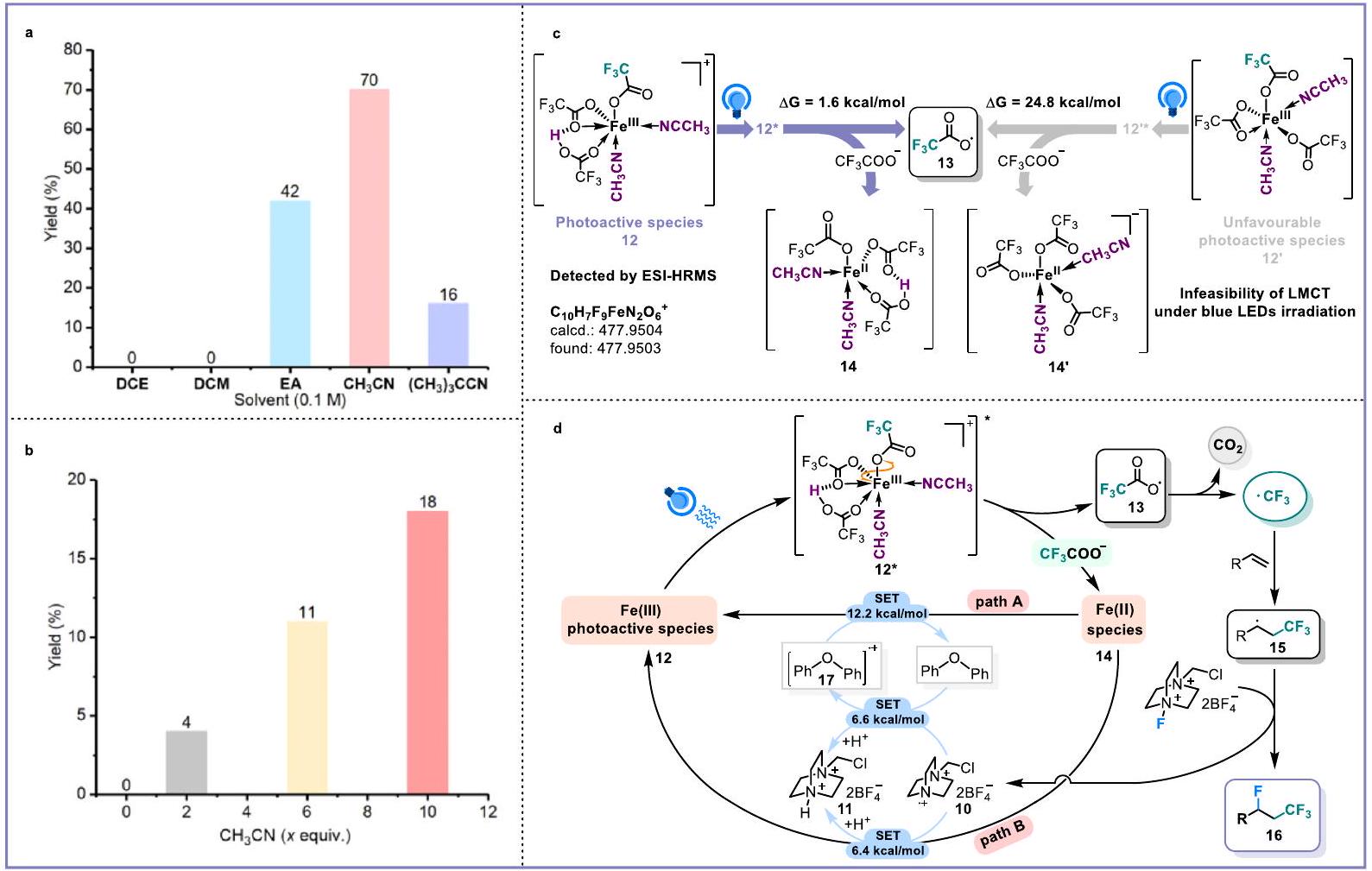
Fig. 3 | Identification of iron-based light-harvesting species. a Comparison of several different solvents. Identification of as ligands. c Density Functional Theory (DFT) calculations. d Possible mechanism.
Fig. 4 | Scope of unactivated alkenes. General reaction conditions: alkene , Selectfluor , oxydibenzene ( 0.1 mmol ), isopropanol ( 0.4 mmol ) and , blue LEDs, reaction time.
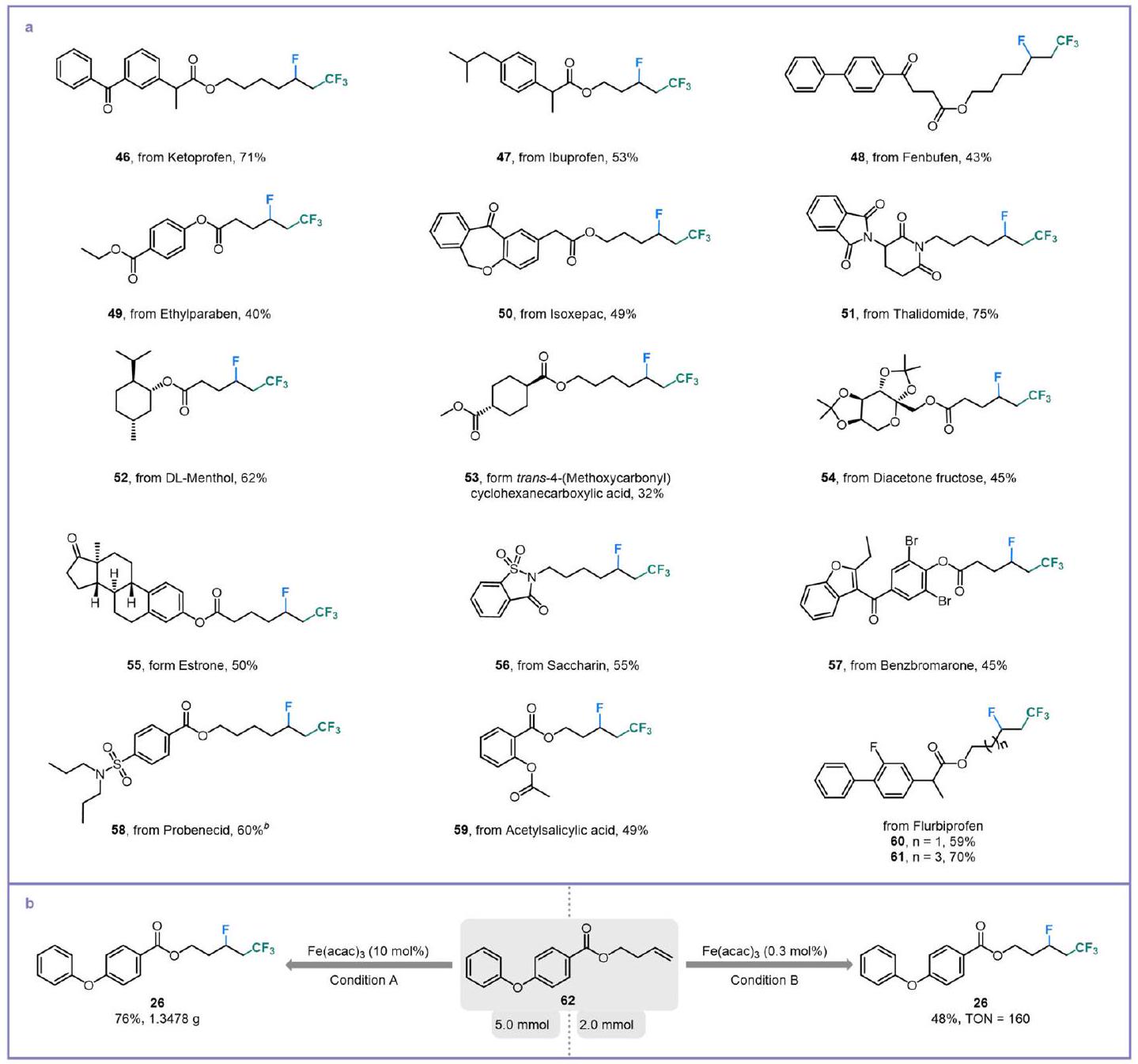
Fig. 5 | Late-stage functionalization and scale-up experiments. a Modification of pharmaceutical derivatives. General reaction conditions: alkene ( 0.2 mmol ), , Selectfluor , oxydibenzene ( 0.1 mmol ), isopropanol ( 0.4 mmol ) and , blue LEDs, 12 h reaction time. Blue LEDs, 24 -h reaction time. Gram-scale and TON experiments. Condition A: alkene ( ), ( , Selectfluor ( 7.5 mmol ), , oxydibenzene ( 2.5 mmol ), isopropanol ( 10.0 mmol ) and , blue LEDs, 72-h reaction time. Condition B: alkene ( 62 , , Selectfluor
( 0.006 mmol ), oxydibenzene ( 1.0 mmol ), isopropanol ( 4.0 mmol ) and , blue LEDs, reaction time. TON turnover number.
irradiation was confirmed by the light on/off experiments over time (Fig. 2f). Importantly, the kinetic curves of and TFAA show that the initial reaction rate would improve when increasing the loading of the iron catalyst and source (Fig. 2g, Supplementary Figs. 31 and 32).
Additionally, during the solvent investigation, we observed that only when the solvents possess coordination ability like , and , can desired fluorotrifluoromethylation product be obtained. In contrast, dichloromethane (DCM) and 1,2-dichloroethane (DCE) as solvents failed in generating radicals (Fig. 3a). It inspired us to consider whether was involved in the assembly of iron and -based light-harvesting species (Fig. 3a, b, Supplementary Table 3, Supplementary Fig. 33). To quickly determine the real structure of iron and -based light-harvesting species, we performed density functional theory (DFT) calculations (Supplementary Figs. 34-37). As shown in Fig. 3c, the 12 that could be regarded as the combination of with and two molecules of through the hydrogen bond and coordination effect, respectively, indeed behaves the efficient LMCT between iron and monodentate-coordinated under blue light irradiation. The presence of was further identified by ESI-HRMS experiments
(Supplementary Fig. 24). Without the hydrogen bond effect of the Brønsted acid, the release of radical 13 from excited 12′ became thermodynamically unfavorable (Supplementary Figs. 38-42). Moreover, in the presence of intramolecular hydrogen bonding, the LUMO orbital energy of iron significantly decreased by 0.86 eV , which is beneficial to the desired LMCT process (Supplementary Fig. 44). Considering that the possibility of existing in the reaction system and based on the structure of , the replacement of one of hydrogen bond-binding groups with to serve as an alternative but unnecessary assembly of iron-based light-harvesting species could not be excluded (see Supplementary Figs. 46-49 for detailed discussions).
The detailed mechanism cycle of this protocol was illustrated in Fig. 3d (Supplementary Figs. 52 and 53). In the presence of Brønsted acid, and acetonitrile, nsted acid-unlocked iron LMCT of proposed light-harvesting species 12 would occur under blue light irradiation, delivering radical 13 and intermediate 14. After the release of gas from radical intermediate 13 (Supplementary Fig. 11), the desired radical was produced, which was further trapped by the alkene to form radical adduct 15. Radical fluorination between 15 and Selectfluor provided the desired
Fig. 6 | Synthetic applications. General reaction conditions: alkene ( 0.2 mmol ), , Selectfluor , oxydibenzene ( 0.1 mmol ), isopropanol ( 0.4 mmol ) and , blue LEDs, 12 –
h reaction time. Blue LEDs, reaction time. instead of , blue LEDs, reaction time.
fluorotrifluoromethylation product 16. The generated radical cation 10 required the regulation by the redox buffer oxydibenzene to avoid the yielding of relevant C -N bonds (Supplementary Figs. 16, 21, and 22). The radical cations and should be responsible for recycling iron(III) through the SET steps, which was also rationalized by the DFT calculations (Supplementary Figs. 50 and 51).
Scope of substrates
Having established the system of Brønsted acid-unlocked iron LMCT, various valuable alkyl fluorides were produced via our method (Fig. 4). Numerous aliphatic terminal olefins containing diverse benzoate groups were well tolerated to deliver the fluorotrifluoromethylation products with satisfying yields and single chemoselectivity ( 7 and 18-36), even though the benzoate might be susceptible to reaction in the presence of a strong Brønsted acid. Besides, a sulfonate-modified alkene was also competent (37). Notably, the strong oxidizability of Selectfluor under our system did not hinder the transformation of those alkenes connecting electron-rich anisoles for the construction of desired products ( 38 and 39). We also assembled high-value triazole and phthalimide to alkenes, which provided moderate fluorotrifluoromethylation yields (40-42). The allylbenzene derivative and 1 -hexadecene were also feasible ( 43 and 44). Additionally, -disubstituted olefin was successfully functionalized to enrich the diversity of alkyl fluorides (45).
Late-stage functionalization
To further showcase the excellent synthetic compatibility of this protocol, a wide variety of complex olefins derived from pharmaceutical molecules were subjected to this Brønsted acid-unlocked iron LMCT photocatalysis condition (Fig. 5a). The indispensable pain-relief drugs such as ketoprofen, ibuprofen, and fenbufen were installed with alkyl fluorides in moderate to good yields (46-48). Additionally, derivatives of the germicide ethyl-paraben and anti-inflammatory agent isoxepac could also be tolerated smoothly ( and ). It is worth noting that the fluorotrifluoromethylation of thalidomide’s derivate was capable of affording 75% yield (51), in which thalidomide is an effective cure for the erythema nodosum leprosum. The introduction of an alkenyl group into DL-menthol (a traditional Chinese medicine for detoxification) produced a satisfying substrate for fluorotrifluoromethylation (52). We also extended this protocol to the transformation of the key medicine intermediates trans-4-(methoxycarbonyl)cyclohexanecarboxylic acid and diacetone fructose to increase the opportunity for further modifications ( and 54). Alkenes with estrone and saccharin scaffolds were both converted into their corresponding products with moderate yields ( and 56). Notably, benzbromarone and probenecid, drugs for the treatment of gout, contain olefin derivatives that were suitable substrates for late-stage fluorotrifluoromethylation ( 57 and 58). Additionally, antipyretic analgesics such as acetylsalicylic acid and flurbiprofen could provide good yields (59-61). The success
of the gram-scale and TON experiments (TON: 160) showed the application potential of this intriguing synthesis (Fig. 5b).
In addition to fluorotrifluoromethylation of alkenes, our platform of a Brønsted acid-unlocked iron LMCT could also broaden the divergent synthesis and solve the challenging radical transformations including fluoro-difluoromethylation (63, 68, 72, 75, and 80), fluoro-chlorodifluoromethylation (64, 69, 73, 76, and 81), fluorotrichloromethylation (67,71,78, and 82), fluoro-dichloromethylation (70, 74, 77, and 84), fluoro-pentafluoroethylation (65), fluoro-chlorotetrafluoroethylation (79 and 83), and fluoroheptafluoropropylation (66) of drug molecules (Fig. 6).
Discussion
In conclusion, a practical Brønsted acid-unlocked iron LMCT protocol for the activation of various inert halogen-containing carboxylates to radicals was disclosed through detailed mechanistic studies, which was applied to fascinating fluoro-polyhaloalkylation of nonactivated alkenes. Hydrogen bond effect of the Brønsted acid and the coordination of the solvent are highly important to ensure the effective assembly of iron and -based light-harvesting species. Further studies on more challenging inert compound activation via Brønsted acid-unlocked 3d-metal LMCT are currently in progress in our laboratory.
Methods
General procedure for fluoro-polyhaloalkylation of alkenes
A 25 mL Schlenk flask equipped with a magnetic bar was charged with and Selectfluor ( ). The flask was evacuated and refilled with for three times. The vessel was then charged with extra dry , alkene ( 0.2 mmol ), or Cl ( 1.0 or 1.4 mmol ), oxydibenzene ( 17.0 mg , 0.1 mmol ) and isopropanol ( ). The reaction mixture was stirred under nitrogen atmosphere and irradiated by blue LEDs for 12 or 24 or 36 h . After completion of the reaction, was detected by TCD-GC. Then the reaction system was quenched by triethylamine, diluted with EtOAc. After concentrated under vacuum, the resulting residue was purified by silica gel flash column chromatography to give the products.
Data availability
The authors declare that the data relating to the characterization of materials and products, general methods, optimization studies, experimental procedures, mechanistic studies, HRMS data and NMR spectra, computational studies are available within the article and its Supplementary Information as well as supplementary data. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Hartwig, J. F. & Larsen, M. A. Undirected, homogeneous C-H bond functionalization: challenges and opportunities. ACS Cent. Sci. 2, 281-292 (2016).
Jiang, H. & Studer, A. Intermolecular radical carboamination of alkenes. Chem. Soc. Rev. 49, 1790-1811 (2020).
Yoon, T. P., Ischay, M. A. & Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2, 527-532 (2010).
Narayanam, J. M. & Stephenson, C. R. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 40, 102-113 (2011).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322-5363 (2013).
Liu, P., Liu, W. & Li, C.-J. Catalyst-free and redox-neutral innate trifluoromethylation and alkylation of aromatics enabled by light. J. Am. Chem. Soc. 139, 14315-14321 (2017).
Marzo, L., Pagire, S. K., Reiser, O. & Konig, B. Visible-light photocatalysis: does it make a difference in organic synthesis? Angew. Chem. Int. Ed. 57, 10034-10072 (2018).
Fu, M.-C., Shang, R. & Fu, Y. Photocatalytic decarboxylative alkylations mediated bytriphenylphosphine and sodium iodide. Science 363, 1429-1434 (2019).
Holmberg-Douglas, N. & Nicewicz, D. A. Photoredox-catalyzed C-H functionalization reactions. Chem. Rev. 122, 1925-2016 (2022).
Bellotti, P., Huang, H.-M., Faber, T. & Glorius, F. Photocatalytic latestage C-H functionalization. Chem. Rev. 123, 4237-4352 (2023).
Golden, D. L. et al. Benzylic C-H esterification with limiting C-H substrate enabled by photochemical redox buffering of the Cu catalyst. J. Am. Chem. Soc. 145, 9434-9440 (2023).
Abderrazak, Y., Bhattacharyya, A. & Reiser, O. Visible-light-induced homolysis of Earth-abundant metal-substrate complexes: a complementary activation strategy in photoredox catalysis. Angew. Chem. Int. Ed. 60, 21100-21115 (2021).
Chang, L., An, Q., Duan, L., Feng, K. & Zuo, Z. Alkoxy radicals see the light: newparadigms of photochemical synthesis. Chem. Rev. 122, 2429-2486 (2022).
Juliá, F. Ligand-to-metal charge transfer (LMCT) photochemistry at 3d-metal complexes: an emerging tool for sustainable organic synthesis. ChemCatChem 14, e202200916 (2022).
Li, Q. Y. et al. Decarboxylative cross-nucleophile coupling via ligand-to-metal charge transfer photoexcitation of carboxylates. Nat. Chem. 14, 94-99 (2022).
Tsurugi, H. & Mashima, K. Renaissance of homogeneous cerium catalysts with unique couple: redox-mediated organic transformations involving homolysis of -ligand covalent bonds. J. Am. Chem. Soc. 143, 7879-7890 (2021).
Zhao, R. & Shi, L. A renaissance of ligand-to-metal charge transfer by cerium photocatalysis. Org. Chem. Front. 5, 3018-3021 (2018).
Zhou, W.-J. et al. Light runs across iron catalysts in organic transformations. Chem. Eur. J. 26, 15052-15064 (2020).
Nicewicz, D., Roth, H. & Romero, N. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett 27, 714-723 (2015).
Denkler, L. M., et al. A general iron-catalyzed decarboxylative oxygenation of aliphatic carboxylic acids. Angew. Chem. Int. Ed. e202403292 (2024). During the revisions of our manuscript, Bunescu, A. et al. published this related work.
Su, W., Xu, P. & Ritter, T. Decarboxylative hydroxylation of benzoic acids. Angew. Chem. Int. Ed. 60, 24012-24017 (2021).
Xu, P., Lopez-Rojas, P. & Ritter, T. Radical decarboxylative carbometalation of benzoic acids: a solution to aromatic decarboxylative fluorination. J. Am. Chem. Soc. 143, 5349-5354 (2021).
Chen, T. Q. et al. A unified approach to decarboxylative halogenation of (hetero)aryl carboxylic acids. J. Am. Chem. Soc. 144, 8296-8305 (2022).
Dow, N. W. et al. Decarboxylative borylation and cross-coupling of (hetero)aryl acids enabled by copper charge transfer catalysis. J. Am. Chem. Soc. 144, 6163-6172 (2022).
Xu, P., Su, W. & Ritter, T. Decarboxylative sulfoximination of benzoic acids enabled by photoinduced ligand-to-copper charge transfer. Chem. Sci. 13, 13611-13616 (2022).
Depecker, C., Marzouk, H., Trevin, S. & Devynck, J. Trifluoromethylation of aromatic compounds via Kolbe electrolysis in pure organic solvent. Study on laboratory and pilot scale. New. J. Chem. 23, 739-742 (1999).
Bian, K.-J. et al. Photocatalytic hydrofluoroalkylation of alkenes with carboxylic acids. Nat. Chem. 15, 1683-1692 (2023). During the
revisions of our manuscript, West, J. G. et al. published this related work.
Qi, X.-K. et al. Photoinduced hydrodifluoromethylation and hydromethylation of alkenes enabled by ligand-to-iron charge transfer mediated decarboxylation. ACS Catal. 14, 1300-1310 (2024). During the revisions of our manuscript, Xia, W. et al. published this related work.
Zhang, W. et al. Leaving group assisted strategy for photoinduced fluoroalkylations using N-hydroxybenzimidoyl chloride esters. Angew. Chem. Int. Ed. 58, 624-627 (2019).
Lv, D. et al. Iron-catalyzed radical asymmetric aminoazidation and diazidation of styrenes. Angew. Chem. Int. Ed. 60, 12455-12460 (2021).
Zhang, Z. et al. Controllable C-H alkylation of polyethers via iron photocatalysis. J. Am. Chem. Soc. 145, 7612-7620 (2023).
Feng, G., Wang, X. & Jin, J. Decarboxylative C-C and C-N bond formation by ligand-accelerated iron photocatalysis. Eur. J. Org. Chem. 2019, 6728-6732 (2019).
Li, Z., Wang, X., Xia, S. & Jin, J. Ligand-accelerated iron photocatalysis enabling decarboxylative alkylation of heteroarenes. Org. Lett. 21, 4259-4265 (2019).
Xia, S., Hu, K., Lei, C. & Jin, J. Intramolecular aromatic C-H acyloxylation enabled by iron photocatalysis. Org. Lett. 22, 1385-1389 (2020).
Jin, Y. et al. Photo-induced direct alkynylation of methane and other light alkanes by iron catalysis. Green. Chem. 23, 9406-9411 (2021).
Kang, Y.-C., Treacy, S. M. & Rovis, T. Iron-catalyzed photoinduced LMCT: a abstraction enables skeletal rearrangements and C(sp )-H alkylation. ACS Catal. 11, 7442-7449 (2021).
Bian, K.-J., Kao, S.-C., Nemoto, D. Jr., Chen, X.-W. & West, J. G. Photochemical diazidation of alkenes enabled by ligand-to-metal charge transfer and radical ligand transfer. Nat. Commun. 13, 7881 (2022).
Lu, Y.-C. & West, J. G. Chemoselective decarboxylative protonation enabled by cooperative Earth-abundant element catalysis. Angew. Chem. Int. Ed. 135, e202213055 (2022).
Tu, J.-L. et al. Iron-catalyzed ring-opening of cyclic carboxylic acids enabled by photoinduced ligand-to-metal charge transfer. Green. Chem. 24, 5553-5558 (2022).
Zhang, M., Zhang, J., Li, Q. & Shi, Y. Iron-mediated ligand-to-metal charge transfer enables 1,2-diazidation of alkenes. Nat. Commun. 13, 7880 (2022).
Zhang, Q. et al. Iron-catalyzed photoredox functionalization of methane and heavier gaseous alkanes: scope, kinetics, and computational studies. Org. Lett. 24, 1901-1906 (2022).
Kao, S.-C. et al. Photochemical iron-catalyzed decarboxylative azidation via the merger of ligand-to-metal charge transfer and radical ligand transfer catalysis. Chem. Catal. 3, 100603 (2023).
Zhang, W.-M., Feng, K.-W., Hu, R.-G., Guo, Y.-J. & Li, Y. Visible-lightinduced iron redox-catalyzed selective transformation of biomass into formic acid. Chem 9, 430-442 (2023).
Xiong, N., Li, Y. & Zeng, R. Merging photoinduced iron-catalyzed decarboxylation with copper catalysis for and couplings. ACS Catal. 13, 1678-1685 (2023).
Lutovsky, G. A., Gockel, S. N., Bundesmann, M. W., Bagley, S. W. & Yoon, T. P. Iron-mediated modular decarboxylative crossnucleophile coupling. Chem 9, 1610-1621 (2023).
Muller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881-1886 (2007).
Honer, M., Schubiger, P. A. & Ametamey, S. M. Molecular imaging with PET. Chem. Rev. 108, 1501-1516 (2008).
O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 37, 308-319 (2008).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320-330 (2008).
Wang, J. et al. Fluorine in pharmaceutical industry: fluorinecontaining drugs introduced to the market in the last decade (20012011). Chem. Rev. 114, 2432-2506 (2008).
Caron, S . Where does the fluorine come from? A review on the challenges associated with the synthesis of organofluorine compounds. Org. Process Res. Dev. 24, 470-480 (2020).
Josephson, B. et al. Light-driven post-translational installation of reactive protein side chains. Nature 585, 530-537 (2020).
Xu, W., Jiang, H., Leng, J., Ong, H. W. & Wu, J. Visible-light-induced selective defluoroborylation of polyfluoroarenes, gem-difluoroalkenes, and trifluoromethylalkenes. Angew. Chem. Int. Ed. 59, 4009-4016 (2020).
Intermaggio, N. E., Millet, A., Davis, D. L. & MacMillan, D. W. Deoxytrifluoromethylation of alcohols. J. Am. Chem. Soc. 144, 11961-11968 (2022).
Qing, F.-L. et al. A fruitful decade of organofluorine chemistry: new reagents and reactions. CCS Chem. 4, 2518-2549 (2022).
Xu, P., Fan, W., Chen, P. & Liu, G. Enantioselective radical trifluoromethylation of benzylic C-H bonds via cooperative photoredox and copper catalysis. J. Am. Chem. Soc. 144, 13468-13474 (2022).
Beatty, J. W., Douglas, J. J., Cole, K. P. & Stephenson, C. R. J. A scalable and operationally simple radical trifluoromethylation. Nat. Commun. 6, 7919 (2015).
Beatty, J. W. et al. Photochemical perfluoroalkylation with pyridine -oxides: mechanistic insights and performance on a kilogram scale. Chem 1, 456-472 (2016).
Yin, D., Su, D. & Jin, J. Photoredox catalytic trifluoromethylation and perfluoroalkylation of arenes using trifluoroacetic and related carboxylic acids. Cell Rep. Phys. Sci. 1, 100141 (2020).
Zhang, K., Rombach, D., Notel, N. Y., Jeschke, G. & Katayev, D. Radical trifluoroacetylation of alkenes triggered by a visible-lightpromoted bond fragmentation of trifluoroacetic anhydride. Angew. Chem. Int. Ed. 60, 22487-22495 (2021).
Giri, R. et al. Photoredox activation of anhydrides for the solventcontrolled switchable synthesis of gem-difluoro compounds. Angew. Chem. Int. Ed. 61, e202209143 (2022).
Wu, W., You, Y. & Weng, Z. Recent advances in the synthesis of fluoroalkylated compounds using fluoroalkyl anhydrides. Chin. Chem. Lett. 33, 4517-4530 (2022).
Zhang, M. et al. Photocatalytic fluoroalkylations of (hetero)arenes enabled by the acid-triggered reactivity umpolung of acetoxime esters. Chem. Catal. 2, 1793-1806 (2022).
Fernández-García, S., Chantzakou, V. O. & Juliá-Hernández, F. Direct decarboxylation of trifluoroacetates enabled by iron photocatalysis. Angew. Chem. Int. Ed. 63, e202311984 (2024). During the revisions of our manuscript, Juliá-Hernández, F. et al. published this related work.
Yu, W., Xu, X.-H. & Qing, F.-L. Silver-mediated oxidative fluorotrifluoromethylation of unactivated alkenes. Adv. Synth. Catal. 357, 2039-2044 (2015).
Liu, Z. et al. Radical carbofluorination of unactivated alkenes with fluoride ion. J. Am. Chem. Soc. 140, 6169-6175 (2018).
Pitts, C. R., Ling, B., Snyder, J. A., Bragg, A. E. & Lectka, T. Aminofluorination of cyclopropanes: a multifold approach through a common, catalytically generated intermediate. J. Am. Chem. Soc. 138, 6598-6609 (2016).
Riener, M. & Nicewicz, D. A. Synthesis of cyclobutane lignans via an organic single electron oxidant-electron relay system. Chem. Sci. 4, 2625-2629 (2013).
Acknowledgements
This project was supported by the National Natural Science Foundation of China, Grant Nos. 22101265 (L.N.), 21903071 (S.L.) and 21822303 (Y.L.);
China Postdoctoral Science Foundation, Grant Nos. 2022M712866 (L.N.), 2023M733212 (S.J.); Joint Fund of Key Technologies Research & Development Program of Henan Province, Grant No. 222301420006 (Y.L.); Promotion Projects for Key Research & Development in Henan Province, Grant No. 222102310042 (L.N.); the Ministry of Science and Technology of the People’s Republic of China (Y.L.).
Author contributions
X.J. and L.N. conceived the work. X.J. and Y.H. designed the experiments and analyzed the data. X.J., Y.H., K.J., J.H., J.Z., S.J., and J.S. performed the synthetic experiments. Y.L. and S.L. contributed to the DFT calculations. X.J. and L.N. described original manuscript and all authors revised.
Correspondence and requests for materials should be addressed to Yu Lan, Shi-Jun Li or Linbin Niu.
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
College of Chemistry, and Pingyuan Laboratory, Zhengzhou University, Zhengzhou, Henan, PR China. State Key Laboratory of Antiviral Drugs, Pingyuan Laboratory, Henan Normal University, Xinxiang, Henan, PR China. School of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Chemical Theory and Mechanism, Chongqing University, Chongqing, PR China. e-mail: lanyu@cqu.edu.cn; lishijunzong@zzu.edu.cn; nlb@zzu.edu.cn