تعتبر خلايا القاتل الطبيعي (NK) خلايا لمفاوية فطرية (ILCs) تساهم في الاستجابات المناعية تجاه الميكروبات والأورام. تاريخياً، كانت تصنيفاتها تعتمد على مجموعة محدودة من علامات البروتين السطحية. هنا، استخدمنا تسلسل RNA على مستوى الخلية الواحدة (scRNA-seq) وفهرسة الخلايا للترانسكريبتومات والإيبيتوبيات عن طريق التسلسل (CITE-seq) لتفكيك تباين خلايا NK. حددنا ثلاث مجموعات بارزة من خلايا NK في دم الإنسان السليم: NK1 وNK2 وNK3، والتي تم تمييزها إلى ستة مجموعات فرعية متميزة. توضح نتائجنا الخصائص الجزيئية، وعوامل النسخ الرئيسية، والوظائف البيولوجية، والسمات الأيضية، واستجابات السيتوكينات لكل مجموعة فرعية. تشير هذه البيانات أيضًا إلى أصلين تطوريين منفصلين لخلايا NK، مما يؤدي إلى مسارات نسخ متباينة. علاوة على ذلك، قمنا بتحليل توزيع مجموعات خلايا NK في الرئة، واللوزتين، واللمفاويات داخل الظهارة المعزولة من الأفراد الأصحاء وفي 22 نوعًا من الأورام. تهدف هذه المصطلحات الموحدة إلى تعزيز الوضوح والاتساق في الأبحاث المستقبلية، مما يحسن من المقارنات عبر الدراسات.
تعتبر خلايا NK من اللمفاويات في جهاز المناعة الفطري التي تنتمي إلى عائلة ILC.تم التعرف على خلايا NK في البداية لقدرتها على التعرف على الخلايا المصابة بالفيروسات والخلايا الورمية والقضاء عليها بشكل مستقل عن التحسس المسبق، ولكن تم الاعتراف منذ ذلك الحين بأدوارها المتعددة الأوجه. تشمل هذه الأدوار ليس فقط الاستجابات المناعية المباشرة، ولكن أيضًا الوظائف التنظيمية التي تؤثر على الجهاز المناعي التكيفي.
تعتبر تغايرية خلايا NK مركزية لوظائفها المتنوعة. على مر الزمن، حدد الباحثون مجموعات فرعية متميزة من خلايا NK، كل منها يتميز بإمكانات وظيفية ومسارات تطويرية فريدة. اعتمدت طرق التصنيف التقليدية هذه بشكل أساسي على تعبير علامات السطح. في هذا السياق، تُقسم خلايا NK البشرية عادةً إلى فئتين رئيسيتين بناءً على كثافة CD56، نظير جزيئي لجزيء التصاق الخلايا العصبية (NCAM)، على سطح الخلية: CD56 و CD56 خلايا NK. تمييزات إضافية في CD56يتم تحديد السكان على أساس تعبير جزء الكربوهيدرات CD57على سطح الخلية وغياب CD94-NKG2A وCD62L؛ الخلايا التي تتميز بهذه الخصائص تشكل مجموعة فرعية أكثر نضجًابالإضافة إلى ذلك، تظهر خلايا NK التكيفية، التي تشكل مجموعة فرعية مميزة من خلايا NK وتظهر خصائص مشابهة لتلك الخاصة بخلايا المناعة التكيفية، في سياقات مناعية معينة، مثل مواجهات فيروس السيتوميجالي البشري (HCMV).لقد أدى ظهور تقنيات الخلايا المفردة المتقدمة، وهي scRNA-seq وCITE-seq، إلى حدوث تحول جذري في فهمنا لخلايا NK. تكشف هذه التقنيات أن مشهد خلايا NK أكثر تعقيدًا ودقة مما كان مفهومة سابقًا، ويتميز بفروق دقيقة. ومع ذلك، على الرغم من هذه التقدمات، لا يوجد وصف موحد ومعياري لخلايا NK.
تظل تباين الخلايا غامضًا. تختلف التعريفات الحالية بين المختبرات وقد تؤدي إلى تناقضات في الأدبيات العلمية. يخلق هذا النقص في المصطلحات الموحدة تحديات كبيرة، خاصة في ترجمة الأبحاث عبر أنظمة النماذج أو مجموعات الأشخاص.
تؤكد الأهمية المتزايدة لخلايا NK في الأساليب العلاجية، خاصة في العلاج المناعي القائم على خلايا NK ضد السرطان، على ضرورة فهم شامل لتنوعها. قد يؤدي سوء التفسير أو إهمال مجموعات خلايا NK المحددة إلى عواقب كبيرة، مما قد يؤثر على فعالية أو سلامة العلاجات. في هذه الدراسة، قمنا بدمج بيانات scRNA-seq وCITE-seq منتم استخدام خلايا NK (718 متبرعًا) لإنشاء تصنيف أساسي لخلايا NK في الدم والرئة واللوزتين واللمفاويات داخل الظهارة لدى الأفراد الأصحاء، وفي 22 نوعًا من الأورام. تم استخراج هذه البيانات من 7 مجموعات بيانات عامة متميزة. يتم إدراج رمز الوصول لكل من مجموعات البيانات المستخدمة في الجدول التكميلي 3 و’توفر البيانات’. يهدف هذا التصنيف إلى أن يكون نقطة مرجعية للدراسات المستقبلية، مما يسهل اتباع نهج أكثر معيارية لفهم واستخدام خلايا NK في كل من الأبحاث والإعدادات السريرية.
النتائج
تتكون خلايا NK الدائرة في الإنسان من ثلاث مجموعات رئيسية
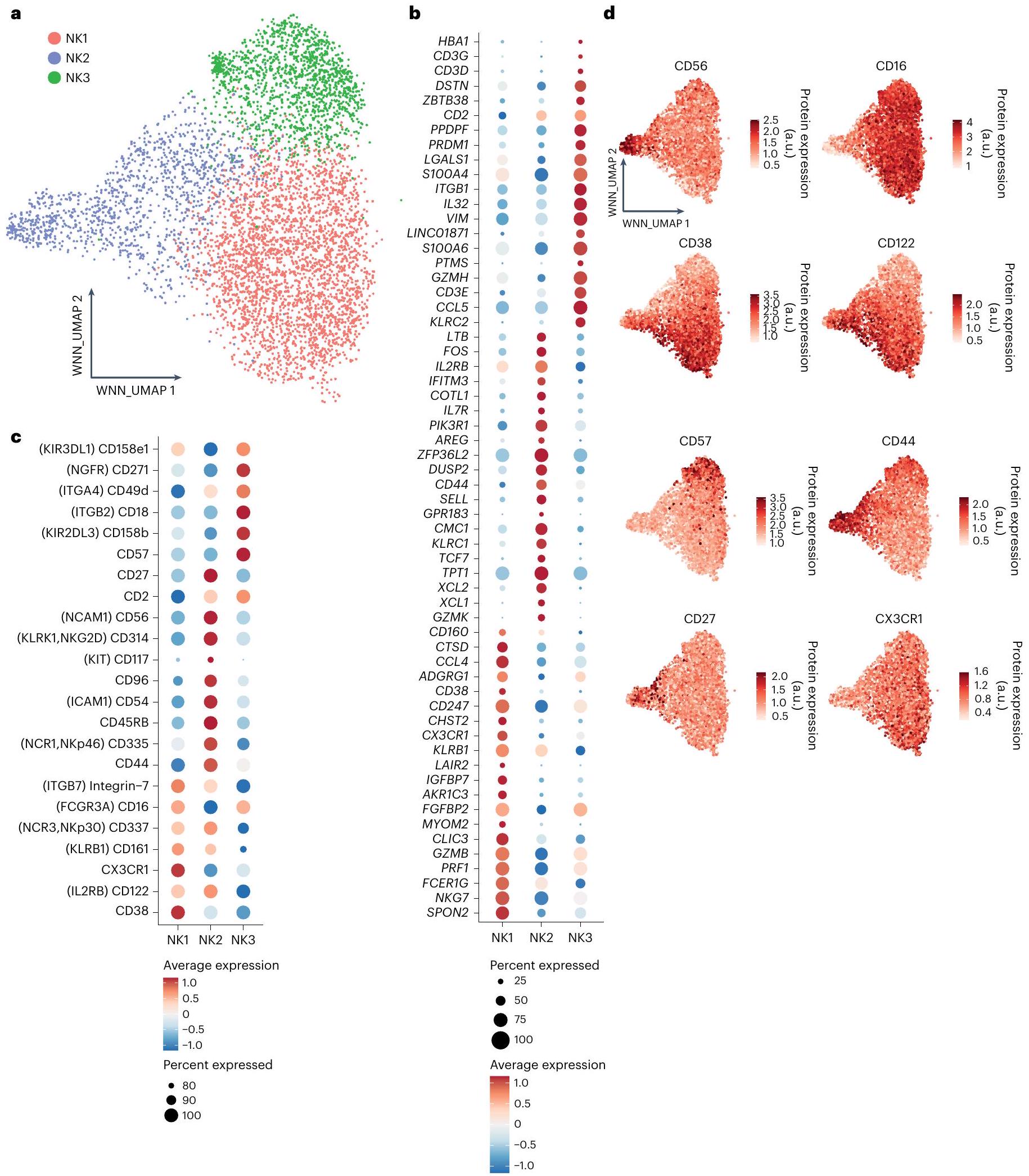
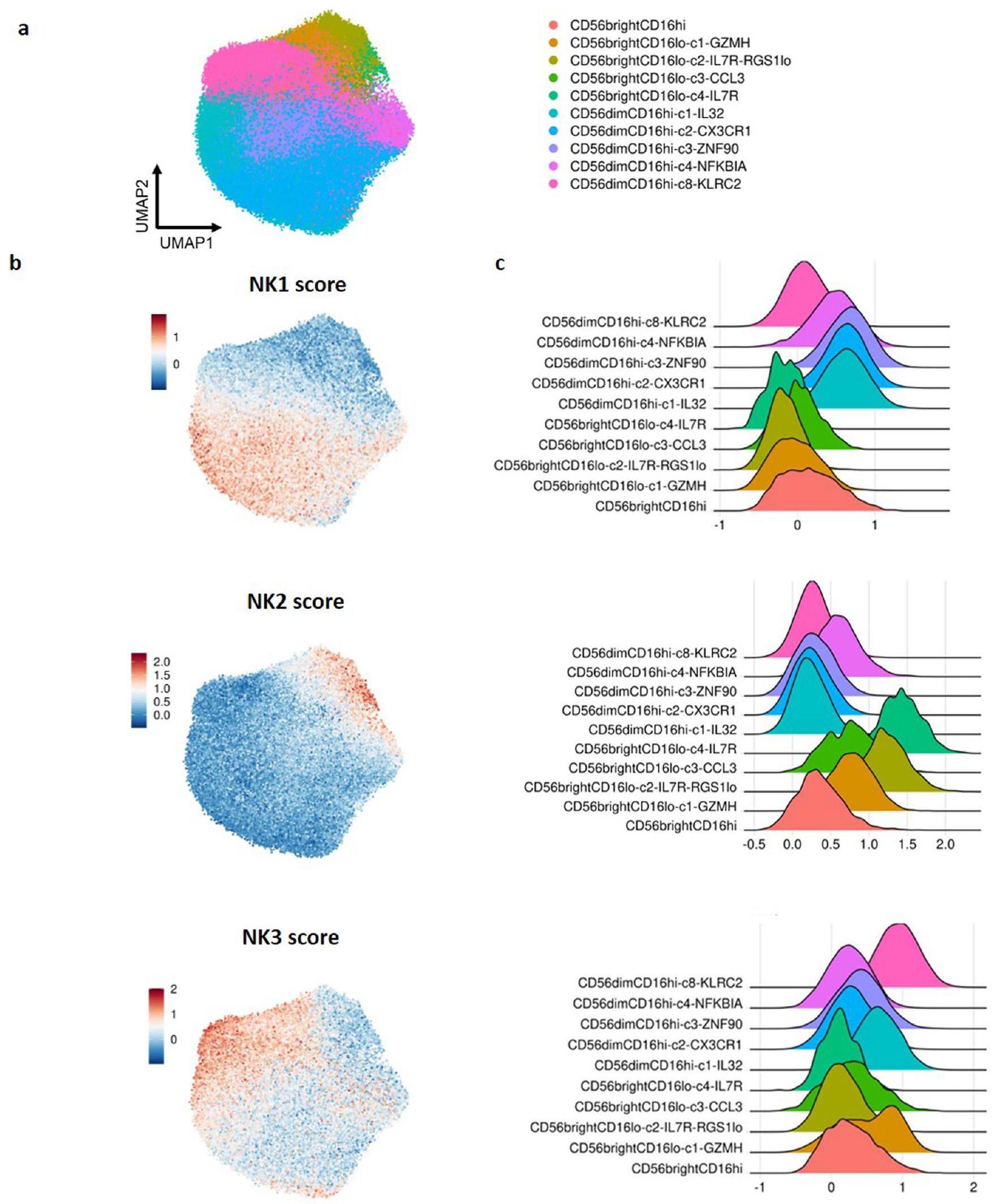
لتصنيف خلايا NK البشرية بشكل منهجي وشامل، استخدمنا مجموعة بيانات CITE-seq عالية الأبعاد، التي تشمل 228 علامة مشتقة من الأجسام المضادة (ADTs) والملفات التعبيرية لـ 5,708 خلايا NK من ثمانية متبرعين أصحاء.لدمج بيانات تعبير RNA والبروتين بشكل فعال، استخدمنا طريقة الجيران الأقرب الموزونة (WNN).في البداية، قمنا بعزل خلايا NK غير المتكاثرة في الخط الأساسي ثم أعيد تصنيفها لتوضيح التباين الأساسي بين خلايا NK في الدم. كشفت تحليلاتنا عن ثلاثة مجموعات رئيسية من خلايا NK: NK1 و NK2 و NK3 (الشكل 1أ). بعد ذلك، قمنا بتحليل توقيعاتها النسخية (الشكل 1ب) والبروتينية (الأشكال 1ج، 1د).
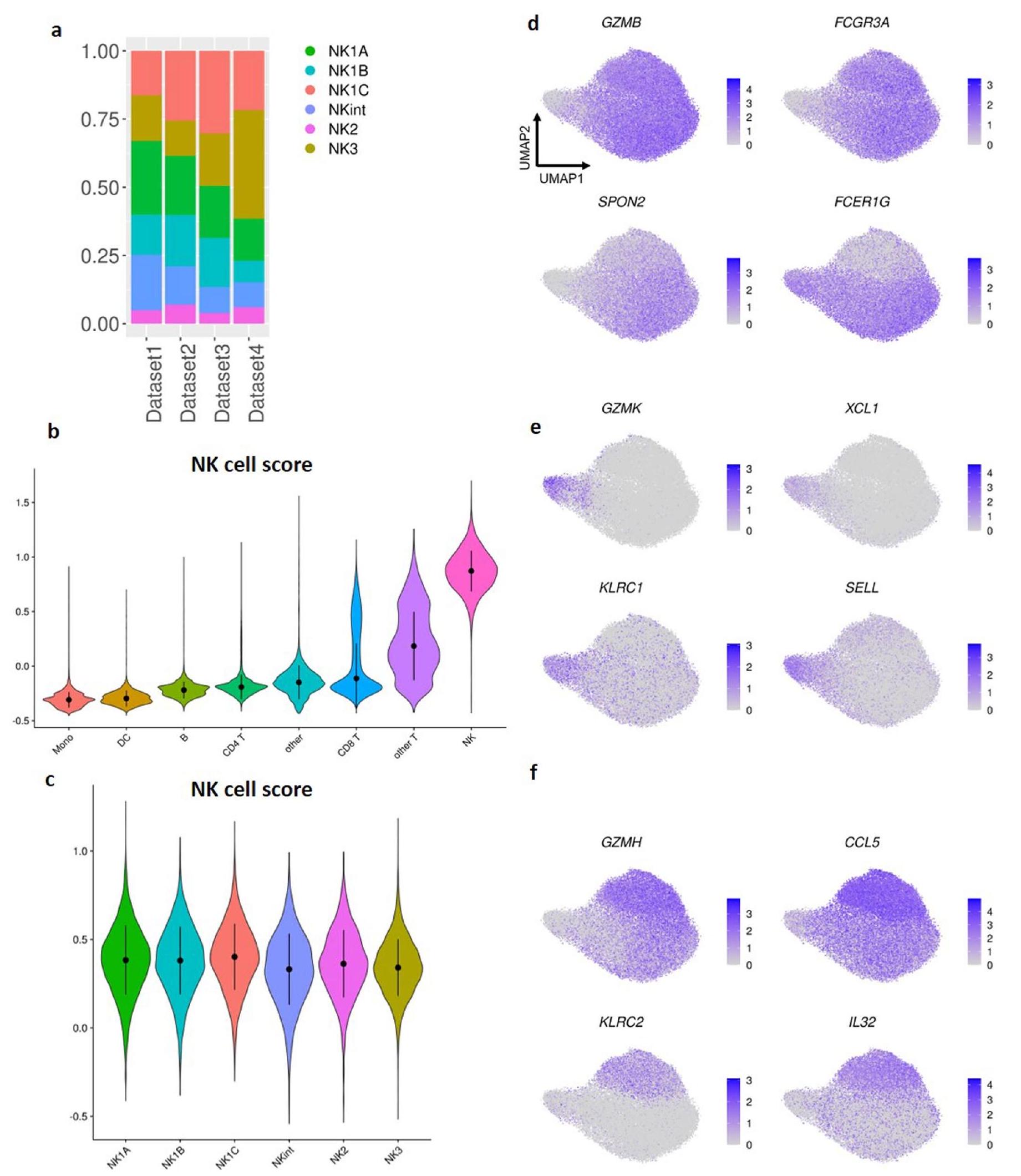
تميز تجمع NK1 بتعبير عالٍ عن البروتين CD16 و CX3CR1 و CD161،-الإنتجرين و CD38 (الشكل 1c، d). أبرز ملفه النسخي الجينات المرتبطة بهذه البروتينات، إلى جانب مستويات مرتفعة من الجينات التي تشفر الجزيئات السامة للخلايا (GZMB و PRF1) وعلامات نضج خلايا NK، مثل CD160 و CD247 و ADGRG1 و NKG7 و FCER1G و LAIR2 و SPON2 و CLIC3 و CHST2 (الشكل 1b). تعبر الخلايا في مجموعة NK1 عن مستويات أقل من CD56 مقارنة بالخلايا في مجموعة NK2 ومستويات أقل من CD57 مقارنة بالخلايا في مجموعة NK3.
تم تعريف مجموعة NK2 من خلال التعبير العالي عن CD56 و CD27 و CD44 و CD54 و CD45RB و CD314 (NKG2D) و CD335 (NKp46) وقلة أو عدم وجود تعبير عن CD16 و CD57 على مستوى البروتين (الشكل 1c، d). على مستوى النسخ الجيني، أظهرت خلايا NK2 تعبيرًا بارزًا عن جينات الريبوسوم (عائلات جينات RPL و RPS، الجدول التكميلي 1) وجينات ترميز البروتينات المشاركة في تخليق البروتين والسلامة الهيكلية (EEF1A1، TPT1)، مما يدل على زيادة تخليق البروتين والقدرة التكاثرية. كما أعربت هذه المجموعة عن مجموعة متنوعة من الجينات التي ترمز لمستقبلات السيتوكين (IL2RB، IL7R) ومستقبلات الغشاء (KLRC1 التي ترمز لـ NKG2A) وعوامل النسخ (TCF7) وعوامل قابلة للذوبان تعدل الاستجابات المناعية (XCL1، XCL2، AREG) وجزيئات متورطة في هجرة الخلايا وتوجيه الأنسجة (CD44، GPR183، SELL)، بالإضافة إلى غرانزيم K (GZMK) (الشكل 1b). كان تعبير علامات خلايا NK الكلاسيكية CD57 و CD16 منخفضًا أو غائبًا في خلايا NK2 مقارنة بخلايا NK1 و NK3، مما يشير إلى أن مجموعة NK2 تتكون من CD56.و CD56 في المراحل المبكرةخلايا NK.
بالنسبة لمجموعة NK3، شمل ملف تعبير البروتين CD16 وCD57 وCD271 (NGFR) وCD2 وCD18 وCD49d وم receptors الخلوية القاتلة المثبطة (KIRs) (CD158e وCD158b)، مع مستويات تعبير أقل من CD56 وNKp30 وNKp46 وCD161 وCD122 (الشكل 1c، d). من الناحية النسخية، تم تمييز خلايا NK3 من خلال التعبير التفضيلي للجينات التي تشفر عوامل النسخ (PRDM1 (الذي يشفر BLIMP1) وZBTB38)، والجزيئات السطحية والمستقبلات (CD2 وKLRC2 (الذي يشفر NKG2C))، ونقلات سلسلة CD3 (CD3D وCD3E وCD3G)، والسيتوكينات والكيموكينات المفرزة (IL32 وCCL5) و جرانزيم(الشكل 1ب). بشكل عام، تشبه توقيع البروتين والتعبير الجيني لمجموعة NK3 بشكل وثيق توقيع خلايا NK التكيفية، ويشير التعبير التفضيلي لهذه المجموعة عن CD57 وPRDM1 إلى أنها تشمل أيضًا خلايا NK ناضجة CD57CD56 خلايا NK التي لا يتم إنتاجها استجابةً لـ HCMV. ثم أكدنا قوة تصنيف خلايا NK في الدم البشري إلى مجموعات NK1 وNK2 وNK3 من خلال تطبيق التوقيعات الجينية المستمدة على خلايا NK في الدم من مجموعات بيانات أخرى متاحة(البيانات الموسعة الأشكال 1أ-ج و2أ-ج).
يمكن تقسيم ثلاث مجموعات خلايا NK الرئيسية إلى ستة مجموعات فرعية
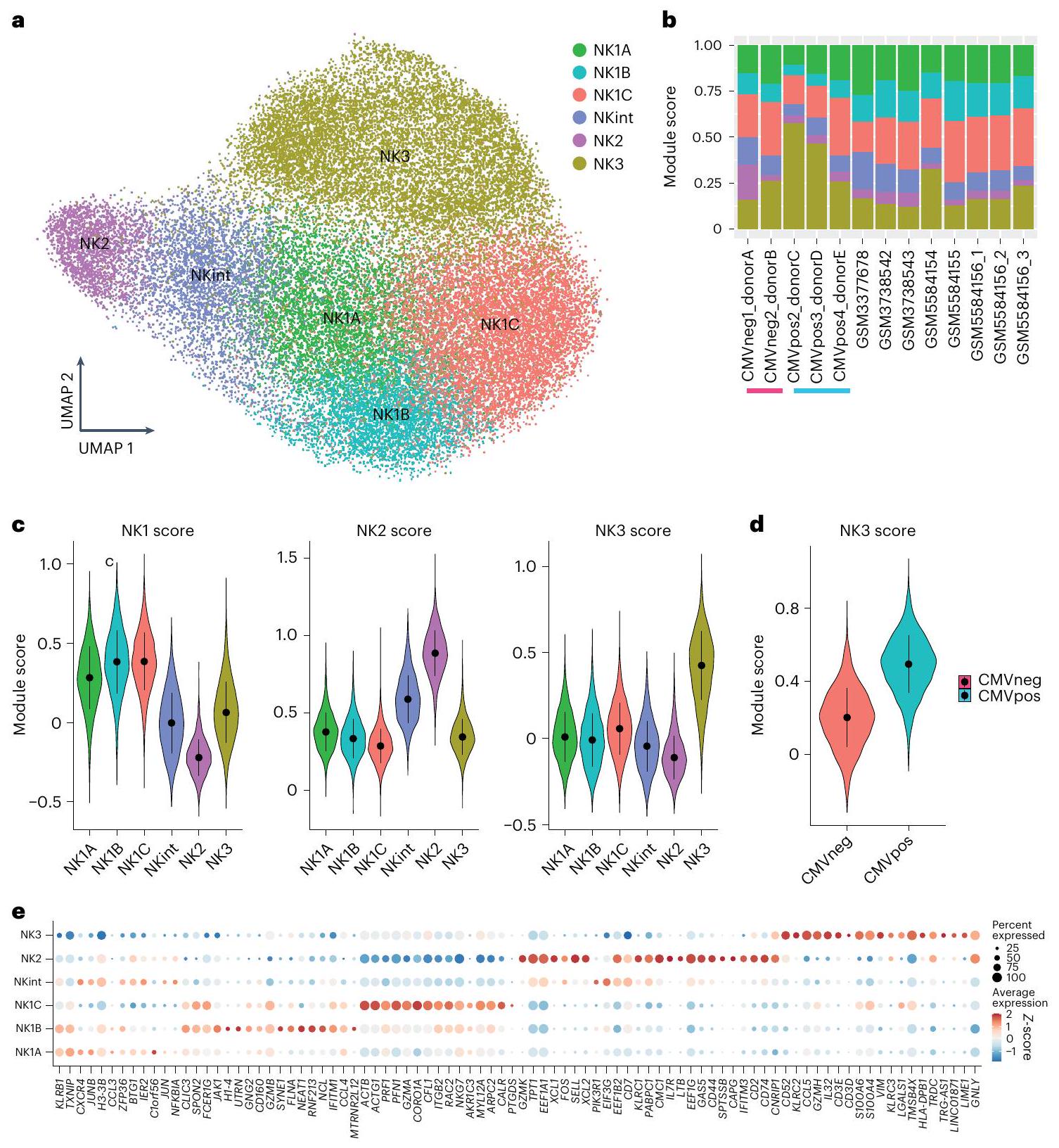
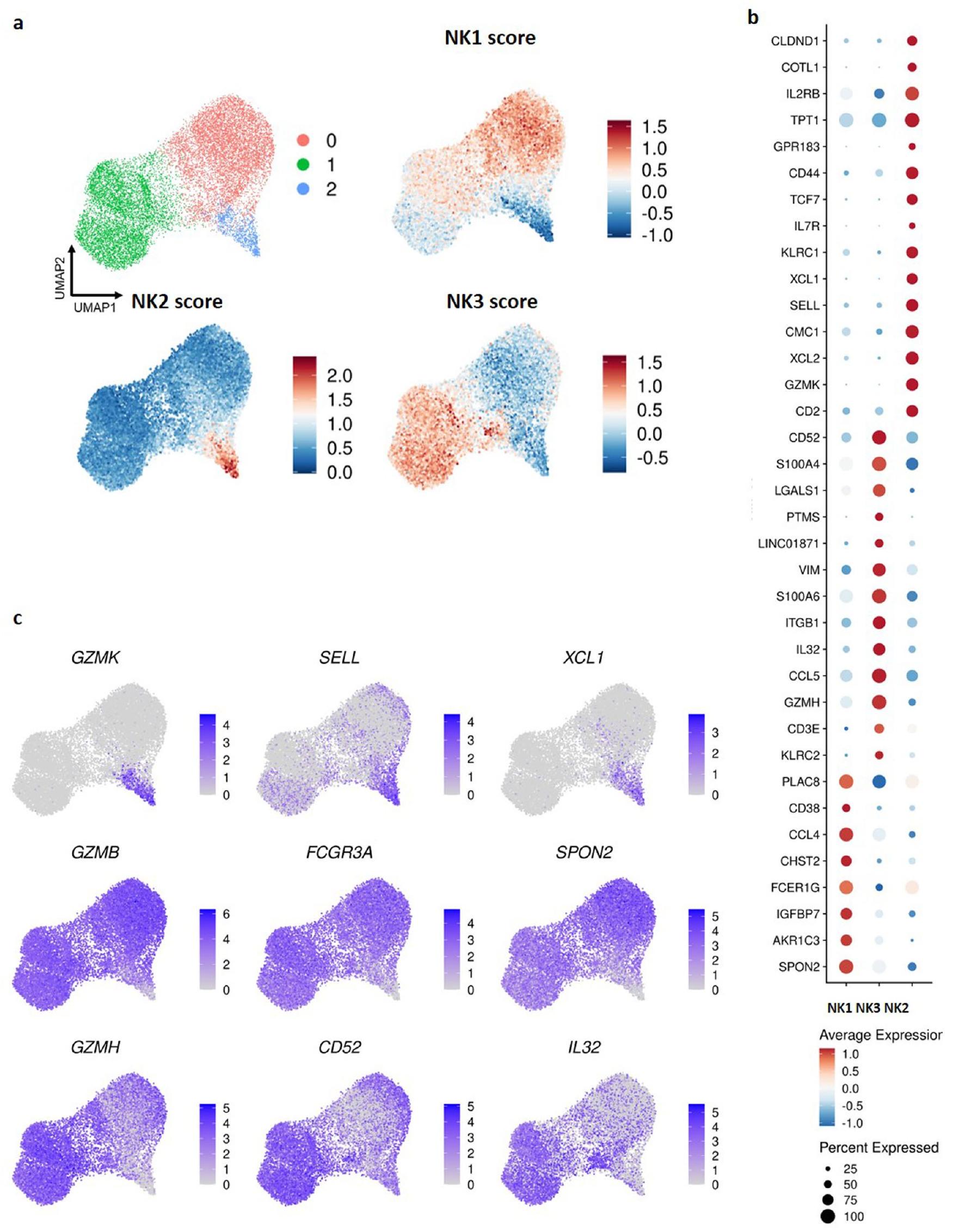
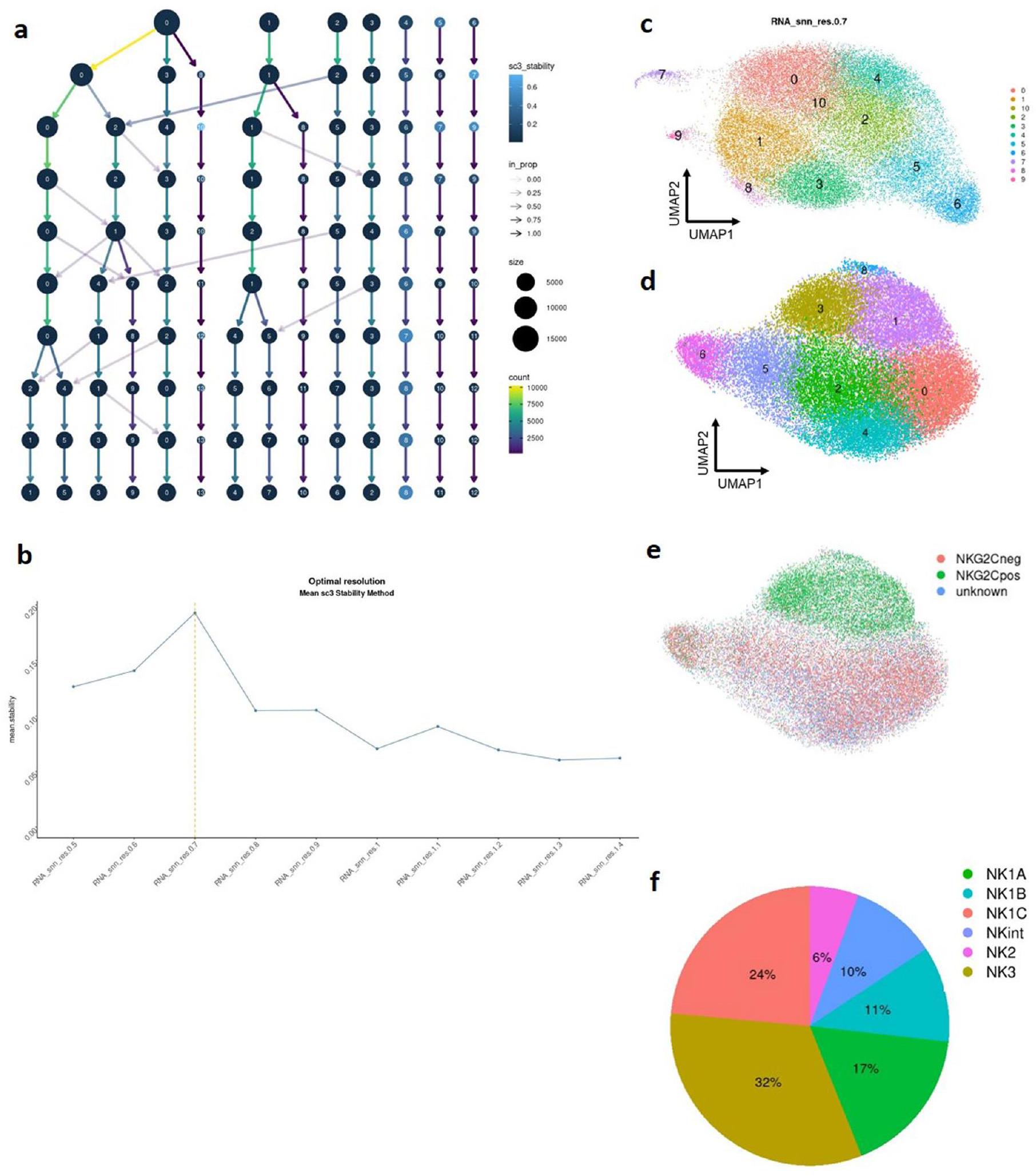
لتحديد تباين خلايا NK في الدم بشكل أكبر، قمنا بدمج بيانات scRNA-seq من خلايا NK المفروزة من 13 فردًا صحيًا عبر أربع مجموعات بيانات باستخدام نفس بروتوكول RNA-seq ( بروتوكول الكيمياء الجينومية v2)، وبالتالي شمل 36,270 خلية بعد تصفية الخلايا عالية الجودة. أدت هذه العملية إلى تحديد ثمانية مجموعات محددة جيدًا (البيانات الموسعة الشكل 3أ-د). شاركت ثلاث مجموعات (1 و3 و8) توقيع NK3 المميز بواسطة جينات مثل KLRC2 (التي تشفر NKG2C) وCD52 وIL32 وGZMH (الجدول التكميلي 1) وكانت غنية في خلايا تعبر عن NKG2C على سطحها (البيانات الموسعة الشكل 3هـ). من الجدير بالذكر أن NK3B (المجموعة 1) تميزت بتعبير أعضاء من عائلة جينات HLA-D وCD74 وCCL5 وCD7 وKLRC1، وأظهرت NK3A قدرات سامة معززة (من خلال تعبير GZMA وGZMB وPRF1) (الجدول التكميلي 1). أظهرت البيانات السابقة أن هناك تباينًا كبيرًا في التعبير الجيني والتعبير الجيني داخل خلايا NK التكيفية فيالأفراد. تم ملاحظة هذا التباين داخل نفس الشخص وعبر أشخاص مختلفين، مما يعكس التماثل في خلايا NK التكيفية بدلاً من البرامج الوظيفية المتميزة. قمنا بتوحيد هذه المجموعات الثلاث في مجموعة واحدة للتحليلات اللاحقة. أدى ذلك إلى تكوين نهائي لست مجموعات (الشكل 2أ والبيانات الموسعة الشكل 3و). يمكن استكشاف مجموعة البيانات المدمجة في دراستنا على:https://collections.cellatlas.io/meta-nk.
بعد التأكد من أن تصحيح الدفعة كان كافيًا وضمان أن تسميات مجموعتنا النهائية خالية من تأثيرات الدفعة على مستوى مجموعة البيانات (البيانات الموسعة الشكل 4أ) ومانح (الشكل 2ب)، قمنا بتسجيل جميع مجموعات CD45 من مجموعة البيانات 5 باستخدام توقيع 13 جين (CD160 وCD244 وCHST12 وCST7 وGNLY وIL18RAP وIL2RB وKLRC1 وKLRC3 وKLRD1 وKLRF1 وPRF1 وXCL2) الذي يتميز بخلايا NK البشرية، وبالتالي التحقق من قدرة هذا التوقيع على تمييز خلايا NK عن مجموعات فرعية أخرى (البيانات الموسعة الشكل 4ب). استخدمنا أيضًا توقيع 13 جين لتسجيل المجموعات الست من خلايا NK، وبالتالي التحقق من قوة هذا التوقيع عبر جميع مجموعات NK (البيانات الموسعة الشكل 4ج). ثم قمنا بتقييم كل مجموعة مقابل التوقيعات الجينية المعروفة لـ NK1 وNK2 وNK3 (الشكل 2ج). أظهرت ثلاث مجموعات (المجموعة 2 والمجموعة 4 والمجموعة 0) ارتباطًا قويًا مع توقيع NK1، مما دفع إلى إعادة تصنيفها كـ NK1A وNK1B وNK1C، على التوالي. كما هو متوقع، أظهرت المجموعة التي تحتوي على مجموعات فرعية من NK3 ارتباطًا واضحًا مع توقيع NK3. من الجدير بالذكر أنه تم ملاحظة درجة NK3 أعلى في خلايا NK3 المشتقة منالأفراد (الشكل 2د). في الوقت نفسه، أظهرت المجموعة 6 ارتباطًا قويًا مع توقيع NK2، مما يبرر تصنيفها كـ NK2. أخيرًا، أظهرت المجموعة 5 ارتباطًا متوسطًا مع كل من توقيع NK1 وNK2 وبالتالي أعيدت تسميتها كـ NK الوسيطة (NKint). يتماشى هذا إعادة التعيين مع أنماط التعبير الجيني التي حددتها التوقيعات المحددة مسبقًا، مما يسهل فهمًا أوضح للمنظر الوظيفي داخل مجموعة خلايا NK في الدم.
تحليل أهم 20 علامة تعريفية للمجموعات الست (الشكل 2هـ) قدم ملفًا تفصيليًا للتعبير الجيني لكل مجموعة. أظهرت خلايا NK1، كما هو ملاحظ في الشكل 1ب، توقيعًا أساسيًا يدل على الكيموكينات (CCL3 وCCL4) والبروتينات الحيوية للسُمية الخلوية وتنظيمها (PRF1 وGZMA وGZMB وNKG7) وديناميات الهيكل الخلوي (RAC2 وARPC2 وCFL1) والالتصاق الخلوي (ITGB2 وCALR). عبرت مجموعات NK1 الفرعية عن علامات محددة فريدة لكل مجموعة. تميزت NK1A بتعبير عالٍ عن CXCR4 وJUN وJUNB، التي تشفر AP-1
الشكل 1 | يكشف تحليل CITE-seq عن ثلاث مجموعات بارزة من خلايا NK في الدم المحيطي لدى الأفراد الأصحاء. بناءً على مجموعة البيانات 5. أ، تصور WNN وUMAP (WNN_UMAP) لخلايا NK المفروزة من دم الإنسان الصحي مع مجموعات تم تحديدها بواسطة التجميع الهرمي غير المراقب (استنادًا إلى scRNA-seq وتعبير 228 بروتين سطحي). ب، رسم نقطي لأهم 20 جينًا مميزًا تم التعبير عنها لثلاث مجموعات رئيسية من خلايا NK في الدم البشري. تم تحليل تعبير الجين باستخدام اختبار ويلكوكسون ذو الجانبين مع تعديل بونفيروني. تم
إزالة الجينات الريبوسومية والميتوكندريا من أجل الوضوح. اللون يشير إلى-مستويات تعبير الجين المقاسة. ج، رسم نقطي لأهم البروتينات المميزة التي تم التعبير عنها لثلاث مجموعات رئيسية من خلايا NK في الدم البشري. تم تحليل تعبير البروتين باستخدام اختبار ويلكوكسون ذو الجانبين مع تعديل بونفيروني. يتم عرض أسماء البروتينات البديلة بين قوسين. اللون يشير إلى-مستويات تعبير البروتين المقاسة. د، تصور WNN_UMAP لتعبير السطح للبروتينات الرئيسية المميزة التي تم التعبير عنها على سطح خلايا NK1 وNK2 وNK3. وحدات عشوائية.
الشكل 2 | يمكن تقسيم ثلاث مجموعات خلايا NK الأكثر أهمية إلى ستة مجموعات فرعية. بناءً على مجموعات البيانات 1-4 أ. أ، تصور UMAP لخلايا NK المفروزة من دم الإنسان الصحي، مع مجموعات تم تحديدها بواسطة التجميع الهرمي غير المراقب. ب، رسم بياني عمودي يوضح نسبة الخلايا داخل كل مجموعة في جميع المتبرعين. تظهر الأعمدة الزرقاء والوردية تحت الأفراد الإيجابيين والسالبين لـ HCMV، على التوالي. ج، رسم بياني على شكل كمان لتسجيل المجموعات الست من خلايا NK بالنسبة لتوقيعات NK1 وNK2 وNK3 المعروفة (
عينات). في الرسوم البيانية على شكل كمان، النقطة هي القيمة المتوسطة. تمثل أشرطة الخطأ المتوسط +/- الانحراف المعياري. د، رسم بياني على شكل كمان لتسجيل مجموعات NK3 مع توقيع NK3 في الأفراد الإيجابيين والسالبين لـ HCMV ( عينات). هـ، رسم نقطي لأهم 20 جينًا مميزًا لكل مجموعة من خلايا NK. تم تحليل تعبير الجين باستخدام اختبار ويلكوكسون ذو الجانبين مع تعديل بونفيروني. تم إزالة الجينات الريبوسومية والميتوكندريا من أجل الوضوح. اللون يشير إلى-مستويات تعبير الجين المقاسة.
عوامل النسخ؛ تميزت NK1B بعلامة السطح CD160، وRNA غير المشفر الطويل NEAT1 وبروتين الغشاء المتداخل المستحث بواسطة الإنترفيرون 1/FITM1؛ أظهرت NK1C قدرة سامة معززة، مع مستويات أعلى من ترانسكريبتات الغرانزيم والبرفورين، ونمط تعبير مميز يتعلق بتمثيل البروستاجلاندين (PTGDS وAKR1C3) وأعلى نمط نشط للهيكل الخلوي (ACTB وACTG1 وCFL1 وRAC2 وARPC2).
تظهر مجموعات NK2 وNKint، التي تشترك توقيعاتها الأساسية في جينات تشفر الكيموكينات (XCL1 وXCL2) والغرانزيمات (GZMK) والبروتينات المعنية في تنظيم النسخ والإشارات (NFKBIA وFOS وBTG1 وGAS5) وتخليق البروتين (TPT1 وعائلة جينات EEF) والبروتينات السطحية (CD44 وCD74 وCD7 وKLRC1)، أيضًا علامات مميزة. عبرت NK2
عن و، بينما أظهرت NKint تعبيرًا قويًا عن CXCR4 وJUNB وZFP36 وIER2 وEIF3G.
عبرت NK3، جنبًا إلى جنب مع التوقيع المحدد مسبقًا (KLRC2 وCCL5 وGZMH وIL32 وCD3E وCD3D وS100A4 وLGALS1)، عن علامات إضافية (CD52 وTMSB4X) وشاركت بعض العلامات مع مجموعة NK2، مثل NKG2E (KLRC3) والغرانوليسين (GNLY). يبرز هذا المشهد الجيني المعقد الوظائف المتنوعة والآليات التنظيمية التي تلعب دورًا داخل مجموعات خلايا NK.
أظهر التحقيق الإضافي في توزيع هذه المجموعات بين 13 متبرعًا صحيًا هيمنة خلايا NK1، التي تشكل حوالي من خلايا NK الدائرة. مثلت خلايا NK2 وNK3 و، على التوالي (الجدول التكميلي
الجدول 2). أظهر تحليل أكثر تفصيلاً على مستوى الفئات الفرعية (الشكل 2ب والشكل الممتد 3ف) أن ما يقرب من نصف مجموعة NK1 تتكون من خلايا NK1C، مما يترجم إلى من جميع خلايا NK المتداولة. مثلت مجموعة NK2 جزءًا صغيرًا من إجمالي خلايا NK ( ) مقارنةً بـ NKint ( من إجمالي خلايا NK). أظهر تجمع NK3، الذي يتميز بالتعبير المميز عن علامات تشير إلى كل من خلايا NK الناضجة بشكل نهائي والخلايا التكيفية (مثل PRDM1 و B3GAT1 (الذي يشفر إنزيمًا رئيسيًا في تخليق CD57)) جنبًا إلى جنب مع الجينات المرتبطة بشكل فريد بخلايا NK التكيفية (CD3E و ZBTB38) (الشكل 1ج، د)، تباينًا كبيرًا في انتشاره عبر الأفراد (الشكل 2ب). تم إجراء نهج دراستنا لتحديد التجمعات دون النظر في حالة HCMV. وبالتالي، للتعرف على التأثير المحتمل لـ HCMV على تجمع NK3، أجرينا تحليلات منفصلة لتكرار ونقاط التنبؤ لخلايا NK3 في الأفراد الإيجابيين لـ وفي أولئك الذين ليس لديهم . من الجدير بالذكر أن خلايا تجمع NK3 لوحظت في كل من المتبرعين بـ و (الشكل 2ب والشكل الممتد 3ف). ومع ذلك، لوحظت درجة NK3 أعلى بشكل أساسي في خلايا NK3 المشتقة من الأفراد بـ (الشكل 2د). بشكل عام، توفر هذه الرؤى منظورًا كميًا حول توزيع وتباين مجموعات خلايا NK في مجرى الدم.
الميزات الجزيئية لمجموعات خلايا NK
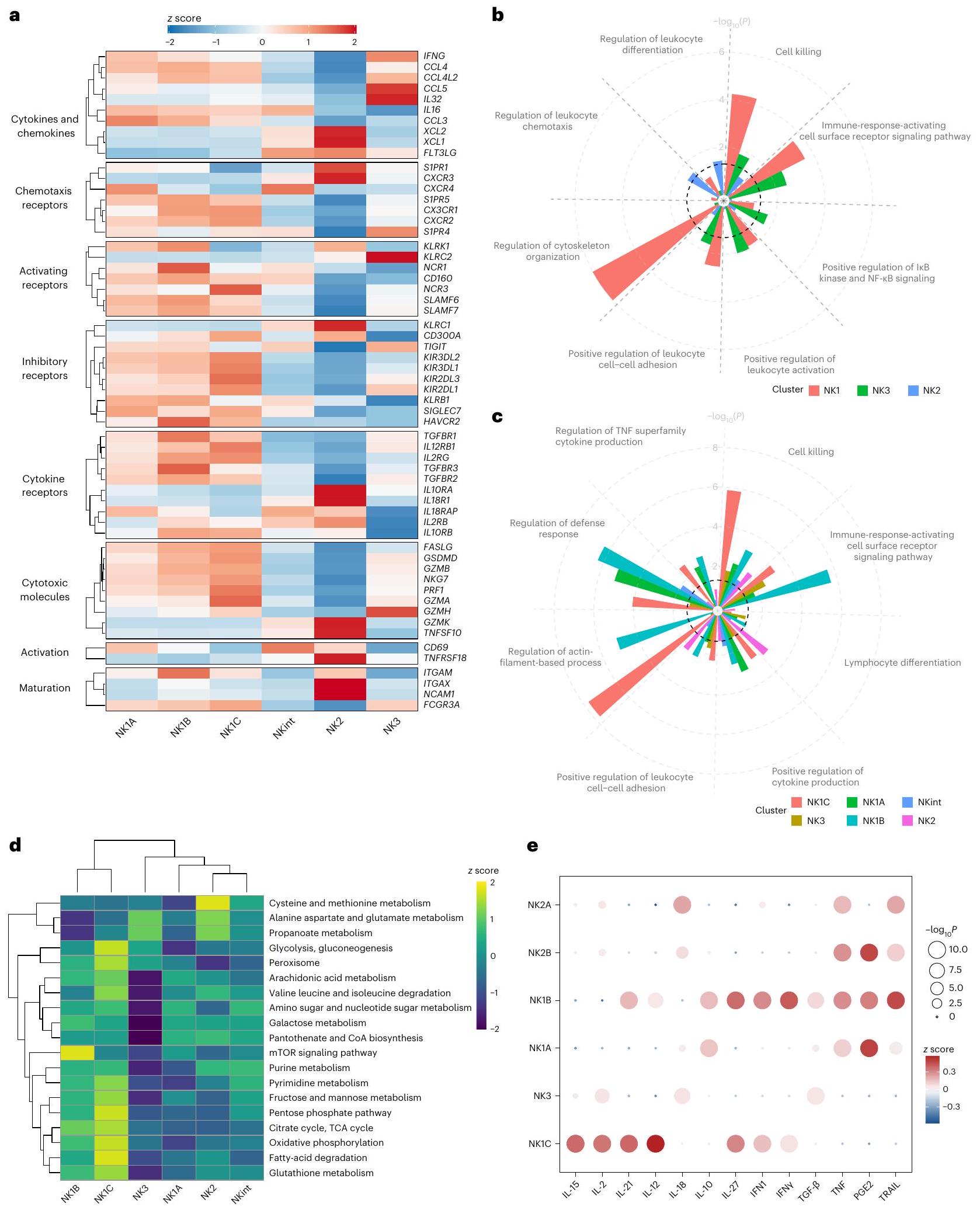
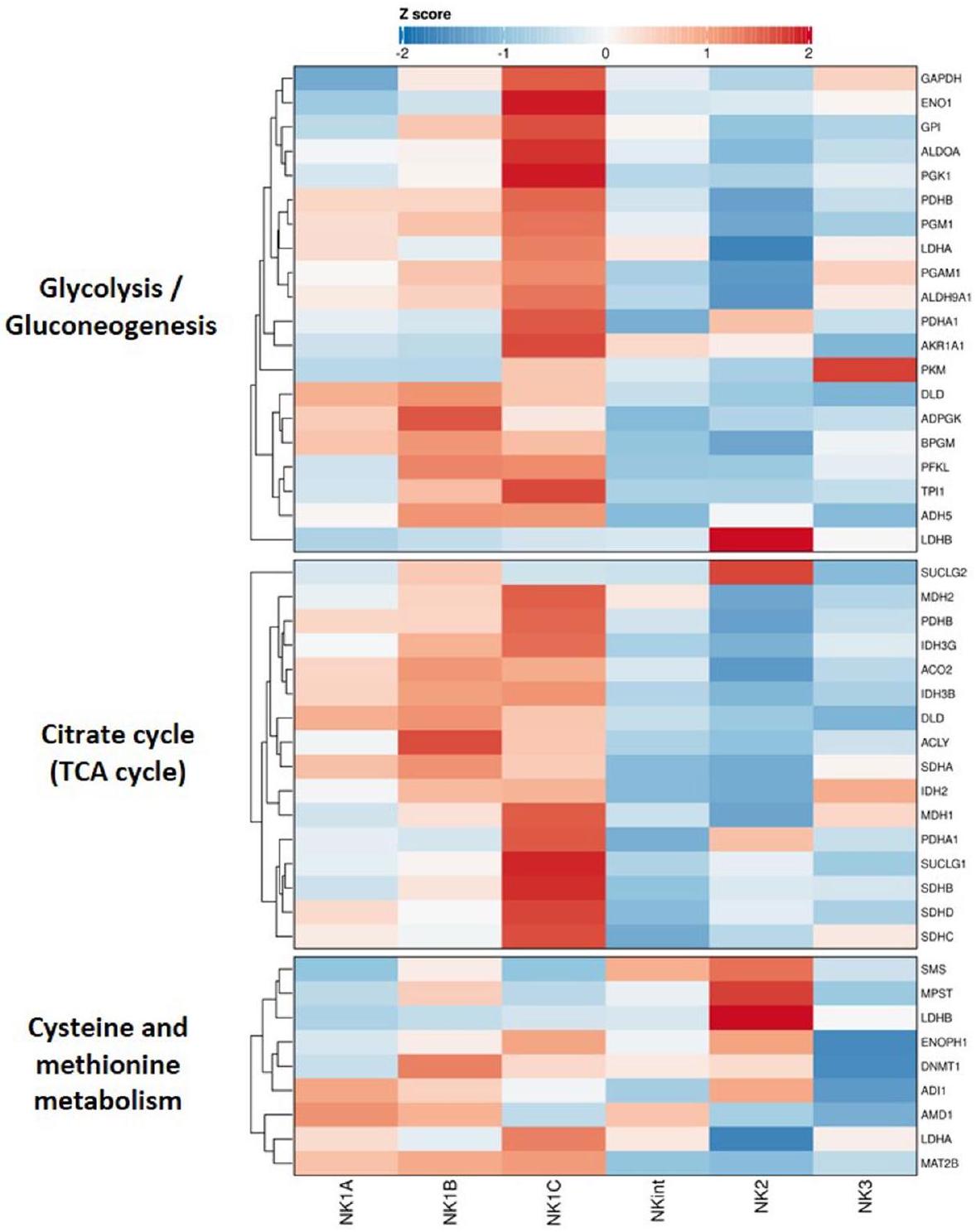
بعد التأكد من أن الفئات الفرعية الست تعبر عن علامات تعريف الفئات (الشكل الممتد 4د-ف)، قمنا بحساب درجات التعبير لمستويات التعبير عن علامات مختلفة ذات صلة في الفئات الفرعية لخلايا NK، مع الأخذ في الاعتبار فقط تلك الجينات المعبر عنها فوق عتبة محددة – الكشف في أكثر من 5% من خلايا NK المتداولة – للإدراج في خريطة الحرارة. أسفر هذا النهج عن خريطة حرارة كشفت، بالإضافة إلى العلامات المحددة مسبقًا، عن خصائص مميزة إضافية للفئات الفرعية (الشكل 3أ).
قمنا بفحص إنتاج السيتوكينات والكيموكينات ووجدنا أن الفئات الفرعية NK1 تتميز بترجمة قوية لـ CCL4 وCCL3 وCCL4L2 وIL16، بينما أظهرت خلايا NK2 وNKint ترجمة سائدة لـ FLT3LG جنبًا إلى جنب مع و . أخيرًا، تميزت خلايا NK3 بمستويات عالية من التعبير عن IFNG وIL32 وCCL5. كان التعبير التفاضلي واضحًا أيضًا في مستقبلات الكيمياء الحيوية وبروتينات الالتصاق بين الخلايا. على وجه الخصوص، تميزت الفئات الفرعية بأنماط مختلفة من مستقبلات السفينغوزين-1-فوسفات (S1PR1 لـ NK2، S1PR4 لـ NK3 وS1PR5 لـ NK1). علاوة على ذلك، كان لعائلة مستقبلات الكيموكين CXC دور في تمييز الفئات الفرعية (CXCR2 وCX3CR1 لـ NK1، CXCR3 لـ NK2 وCXCR4 لكل من NK1A وNKint). أظهرت مستقبلات التنشيط الكلاسيكية لخلايا NK أيضًا أنماط تعبير محددة للفئات الفرعية. كانت مستويات عالية من NKp46 (NCR1) وCD160 وNKp30 (NCR3) وجينات مستقبلات جزيئات تنشيط اللمفاويات سمة مميزة لمجموعة NK1. شاركت NK2 تعبيرًا بارزًا عن NKG2D (KLRK1) مع NK1A وNK1B. كما هو متوقع، تم التعبير عن NKG2C (KLRC2) بشكل رئيسي من قبل مجموعة NK3. تباينت ملفات التعبير عن مستقبلات المثبطة بين الفئات الفرعية. عبرت خلايا NK1 عن مستويات أعلى من KIRs المثبطة، جنبًا إلى جنب مع TIM3 (HAVCR2) وCD161 (KLRB1) وSIGLEC7، بينما كانت خلايا NK2 وNKint تتمتع بمستوى مرتفع من التعبير عن NKG2A (KLRC1)، وبدت خلايا NK3 أنها تمتلك مستويات أعلى من التعبير عن TIGIT. من حيث تعبير مستقبلات السيتوكينات، أظهرت مجموعات NK1 مستويات مرتفعة من التعبير عن TGFBR1 وTGFBR2 وTGFBR3، بالإضافة إلى IL12RB1 وIL1ORB وIL2RG. بالمقابل، تميزت خلايا NK2 بتفضيلها لـ IL2RB (متسقة مع تعبيرها القوي عن CD122 على مستوى البروتين، الشكل 1ج)، وIL1ORA، وتعبير مميز عن IL18R وبروتينها المساعد IL18RAP. تشير هذه النتائج إلى أن الفئات الفرعية لديها مستويات متفاوتة من الحساسية للسيتوكينات والكيموكينات.
كانت الملفات السمية الملحوظة متوافقة مع الملاحظات السابقة: أظهرت مجموعات NK1 طيفًا من الجزيئات السامة والبروتينات المرتبطة – GZMA وGZMB وPRF1 وNKG7 وGSDMD وFASLG – بينما أظهرت NK2 وNKint تعبيرًا قويًا عن وTRAIL (TNFSF1O). عبرت مجموعة NK3 عن مستويات متوسطة من الجزيئات السامة وتميزت بتعبير مرتفع عن GZMH.
كانت علامات التنشيط أيضًا بمثابة علامات تفاضلية، حيث كان CD69 الأكثر بروزًا في NK1B وNKint، بينما كان TNFRSF18 (الذي يشفر GITR) أكثر وضوحًا في NK2. علاوة على ذلك، كانت العلامات الكلاسيكية لنضوج NK متوافقة مع الأوصاف السابقة: كان CD56 (NCAM1) أكثر انتشارًا في NK2، وزاد تعبير CD16 (FCGR3A) تدريجيًا من NK2 إلى NK1C. بالإضافة إلى ذلك، كانت مستويات CD11B (ITGAM) أعلى في NK1B، بينما كان تعبير CD11C (ITGAX) أكثر وضوحًا في NK2.
كشف تحليل إثراء مصطلحات علم الجينات (GO) عن تخصصات وظيفية مميزة داخل الفئات الفرعية لخلايا NK. كانت خلايا NK1 معنية بشكل أساسي في عمليات مثل الالتصاق بين الخلايا، استجابة التنشيط، الإشارة، النشاط الهيكلي الخلوي والسمية الخلوية المعتمدة على الخلايا (الشكل 3ب). تؤكد هذه النتائج على أن خلايا NK1 لها وظائف فعالة سمية محورية. بالمقابل، كانت خلايا NK2 مرتبطة بتنظيم كيمياء الحركة المعززة وتمايز الكريات البيضاء، مما يشير إلى قدرتها على اختراق الأنسجة وعملية نضوج مستمرة. أظهرت خلايا NK3 زيادة في تنشيط الكريات البيضاء. ثم استكشفنا وظائف الفئات الفرعية الست الرئيسية لخلايا NK (الشكل 3ج). وُجد أن خلايا NK1B تستجيب بشكل كبير للتنشيط من خلال مستقبلات السطح، مما يشير إلى إمكاناتها كأهداف رئيسية في استراتيجيات العلاج المناعي. كانت كل من مجموعات NK1A وNK1B غنية بشكل ملحوظ في إنتاج عامل نخر الورم (TNF) والسيتوكينات. من الجدير بالذكر أن مجموعة NK1C تبدو الأكثر سمية، كما يتضح من نشاطها الهيكلي الخلوي الملحوظ وتوقيع قتل الخلايا. كانت اكتشافًا مثيرًا هو الزيادة الكبيرة في أنشطة دورة حمض ثلاثي الكربوكسيل (TCA) في مجموعة NK1C، مما دفع إلى مزيد من التحقيق باستخدام تحليل تباين مجموعة الجينات على مستوى الخلية الواحدة (scGSVA).
فصل تحليل التجميع بناءً على تحليل إثراء مسار الأيض الفئات الفرعية لخلايا NK إلى فئتين عريضتين، حيث كانت NK1B وNK1C تتجمعان بشكل أقرب معًا وبشكل منفصل عن الفئات الفرعية NK1A وNK2 وNK3 وNKint (الشكل 3د). يبدو أن مجموعة NK1C ‘هايبر ميتابوليك’، مع زيادة ملحوظة عبر الأيض الكربوني المركزي، بما في ذلك التحلل السكري ودورة TCA، والفوسفوريلاكس الأكسيدية الميتوكوندرية (OXPHOS)، مما قد يدعم النشاط السمي المعزز. وبالمثل، تظهر مجموعة NK1B أيضًا زيادة في دورة TCA وOXPHOS، وإن كان إلى حد أقل من NK1C، على عكس الفئات الفرعية الأخرى لخلايا NK. بالإضافة إلى ذلك، يتم تعريف خلايا NK1B بشكل أوضح من خلال زيادة في مسار إشارة mTOR. أخيرًا، يتم إثراء أيض السيستين والميثيونين في مجموعة NK2. وجدت تحليل التفكيك لهذا المسار (الشكل الممتد 5أ) أن التوقيع كان مدفوعًا جزئيًا بمستويات عالية من إنزيم اللاكتات ديهيدروجيناز B (LDHB) وسميرين سينثاز (SMS) و3-ميركابتوبيروفات سلفورترانسفيراز (MPST). يبدو أن LDHB (الذي يحول بشكل تفضيلي اللاكتات إلى البيروفات وNAD إلى NADH) هو الشكل المهيمن من اللاكتات ديهيدروجيناز في مجموعة NK2؛ بينما يتم التعبير عن LDHA (الذي يحول بشكل تفضيلي البيروفات إلى اللاكتات وNADH إلى NAD ) بشكل مرتفع عبر الفئات الفرعية الأخرى لخلايا NK. تشير هذه البيانات إلى أن ملفات الأيض المختلفة تكمن وراء الفئات الفرعية لخلايا NK وتستدعي مزيدًا من التحقيق.
لتوضيح الاستجابات المتفاوتة للفئات الفرعية الست لخلايا NK لتحفيز السيتوكينات، استخدمنا محلل إشارة السيتوكين (CytoSig)، الذي يتنبأ باستجابة الخلايا لإشارات السيتوكين. أظهرت هذه التحليل أن مجموعة NK2 تظهر رد فعل بارز تجاه الإنترلوكين-18 (IL-18)، وهو ما يتماشى مع التعبير القوي عن IL18R وبروتينه المرتبط IL18RAP في هذه الفئة (الشكل 3e). بدت خلايا NKint وNK1A وNK1B أكثر عرضة لـ IL-10 وPGE2، وهما إشارتان يمكن أن تخففا من الاستجابات المناعية، لا سيما في بيئة الورم.“. بالمقابل، أظهرت NK1B و NK3 استجابة أكبر لعامل النمو المحول بيتا (TGF- ). TGF- يشتهر بتأثيراته المثبطة للمناعة على خلايا NK، لا سيما داخل بيئة الورم الدقيقة، حيث يمكن أن تعيق وظائفها السامة للخلاياوفقًا للدراسات السابقة، أظهرت خلايا NK3 حساسية منخفضة تجاه IL-12 (المرجع 9). أخيرًا، أظهرت خلايا NK1C أقوى استجابة لمجموعة من السيتوكينات، وهي
الشكل 3 | علامات الاهتمام، الوظائف والتمثيل الغذائي التي تميز تجمعات خلايا NK. استنادًا إلى مجموعات البيانات 1-4a. أ، خريطة حرارية تظهر التعبير التفاضلي لعلامات الاهتمام بين مجموعات خلايا NK. مقياس الألوان يعتمد على-تعبير الجينات المقاس على مقياس.-تتراوح توزيع الدرجات من -2 (أزرق) إلى 2 (أحمر). ب، ج، مصطلحات GO المختارة التي تظهر الثراء في ثلاث مجموعات رئيسية (وست مجموعات فرعية رئيسية) من خلايا NK في دم الإنسان السليم. مصحح باستخدام طريقة بنجاميني-هوشبرغ.تم حساب القيم بواسطة اختبار هايبرجيوما. الخط المنقط الأسود يشير إلى عتبة الدلالة، والتي هي. خريطة حرارية تظهر الثراء التفاضلي لمسارات الأيض المختارة بين مجموعات خلايا NK. يعتمد مقياس الألوان علىدرجة درجة الإثراء المنظم لكل مسار أيضي. هـ، التقييم (درجات) الاستجابة لمختلف السيتوكينات والكيموكينات في كل مجموعة فرعية، تم قياسها بواسطة Cytosig. IFN، إنترفيرون؛ PGE2، بروستاجلاندين E2.تم حساب القيم من خلال المقارنةيقارن الدرجات في مجموعة فرعية واحدة من NK مع تلك في مجموعات فرعية أخرى باستخدام اختبار ستودنت ذو الجانبين-اختبارات، وتم تحديد القيم التي تتجاوز 10 عند 10 لتسهيل التصور.
IL-2 و IL-15 و IL-12، التي ترتبط تقليديًا بتنشيط وتكاثر خلايا NK.
مسارات النسخ الفرعية لمجموعات خلايا NK
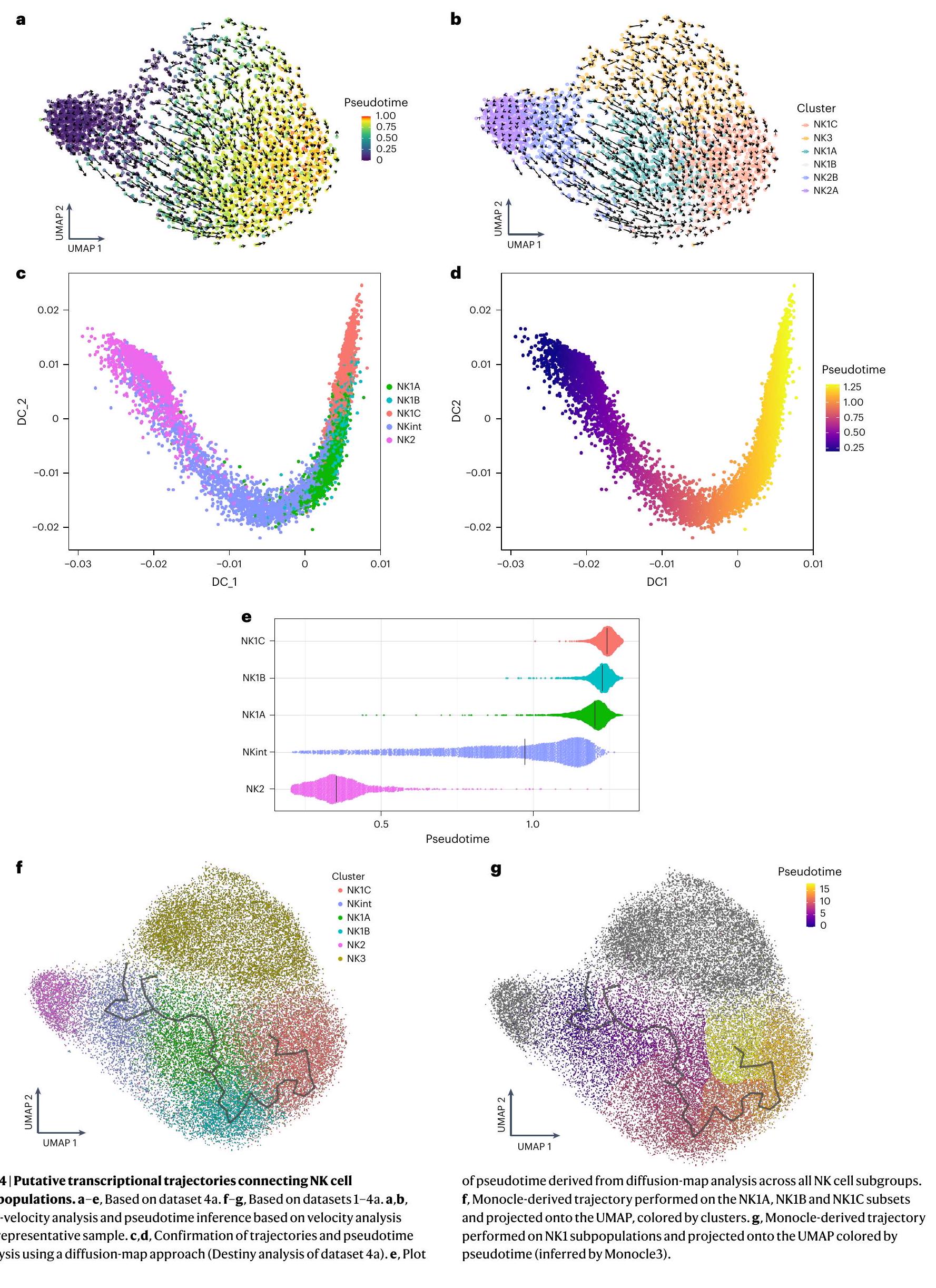
لقد كشفت الدراسة الشاملة لستة مجموعات فرعية من خلايا NK عن خصائصها المميزة من حيث العلامات، استجابة السيتوكينات والوظائف، بالإضافة إلى وجود استمرارية في المناظر النسخية لها، لا سيما بين مجموعتي NKint وNK1A. يبدو أن هذه الاستمرارية تربط بين الحالات النسخية لـ NKint وNK1C. للتحقيق في المسارات النسخية المحتملة التي تربط بين هذه المجموعات الفرعية، قمنا بإجراء تحليل متعدد الأبعاد.
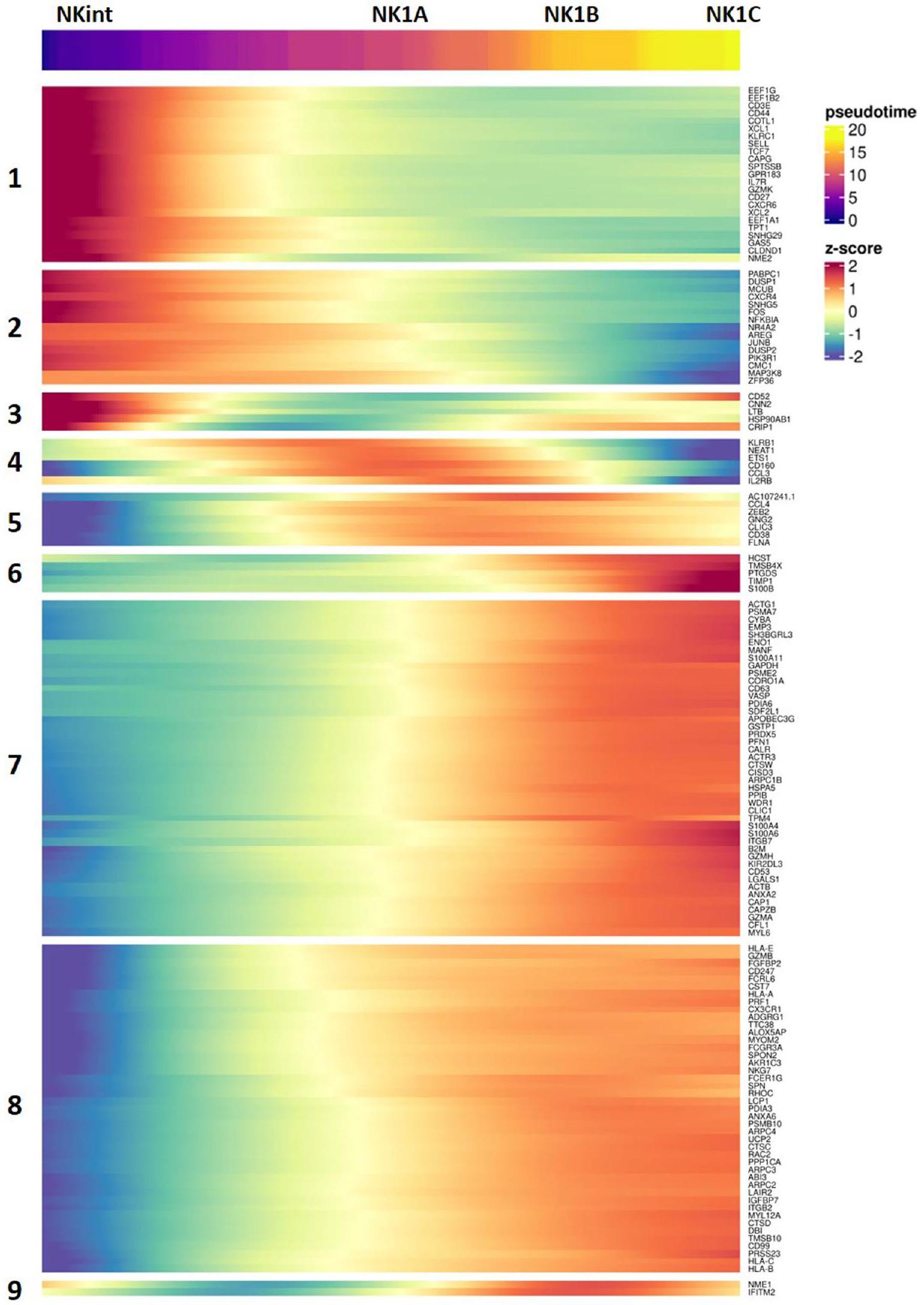
أولاً، تم استخدام سرعة RNA للتنبؤ بالحالات المستقبلية للخلايا الفردية. أشار هذا التحليل إلى أن الغالبية العظمى من خلايا NK2 من المحتمل أن تستمر كخلايا NK2، مكونة مسار NK2 محدد (الشكل 4a،b). ومع ذلك، أشار أيضًا إلى مسار تمايز محتمل من NKint إلى NK1C. كان هذا المسار يتميز بتقدم زائف واضح من NKint إلى NK1C، مع الانتقال عبر تجمعات وسيطة (NK1A و NK1B) (الشكل 4a،b). خلايا NK3، التي تظهر ديناميات نسخ شبيهة بالتكاثر بسبب تفاعلها معتم استبعادها من تحليل المسار التالي، لتجنب تأثير سلوكها النسخي الفريد على النتائج. تم استخدام تحليل المسار الإضافي باستخدام خرائط الانتشار (Destiny ) واستنتاج المسار (Monocle3 (المرجع 20)) أكد المسار الذي اقترحته تحليل سرعة RNA (الشكل 4c-g). استنتاج الزمن الزائف أوضح بوضوح مسارًا من NKint إلى NK1C (الشكل 4d,e,g). ومن الجدير بالذكر أن الزمن الزائف المستنتج من خلال تحليل خريطة الانتشار أظهر فجوة كبيرة بين NK2 و NKint (الشكل 4e)، مما يعزز مفهوم وجود مسارين متميزين: أحدهما حيث تبقى خلايا NK2 في الغالب NK2، والآخر يؤدي من NKint إلى NK1C. يتماشى هذا المسار الأخير مع التجميع غير المراقب القائم على الأيض الذي تم مناقشته سابقًا (الشكل 3d)، والذي جمع بين NK1B و NK1C بشكل وثيق بسبب نشاطهما القوي في أيض الكربون المركزي. بالمقابل، تجمعت NKint و NK1A معًا وأظهرت نشاطًا أيضيًا أقل.
استنادًا إلى تحليل مونوكل، ركزنا على أفضل 150 جينًا أظهرت تغييرات ملحوظة على طول مسار نضوج خلايا NK من NKint إلى NK1C، كما هو موضح بواسطةقيمة أقل من 0.05 ودرجة ارتباط / موران عالية. كشفت هذه الدراسة التفصيلية عن تسعة وحدات جينية، تم تنشيط كل منها بشكل متسلسل مع تقدم الخلايا عبر مراحل النضج (الشكل التمديدي 6a).
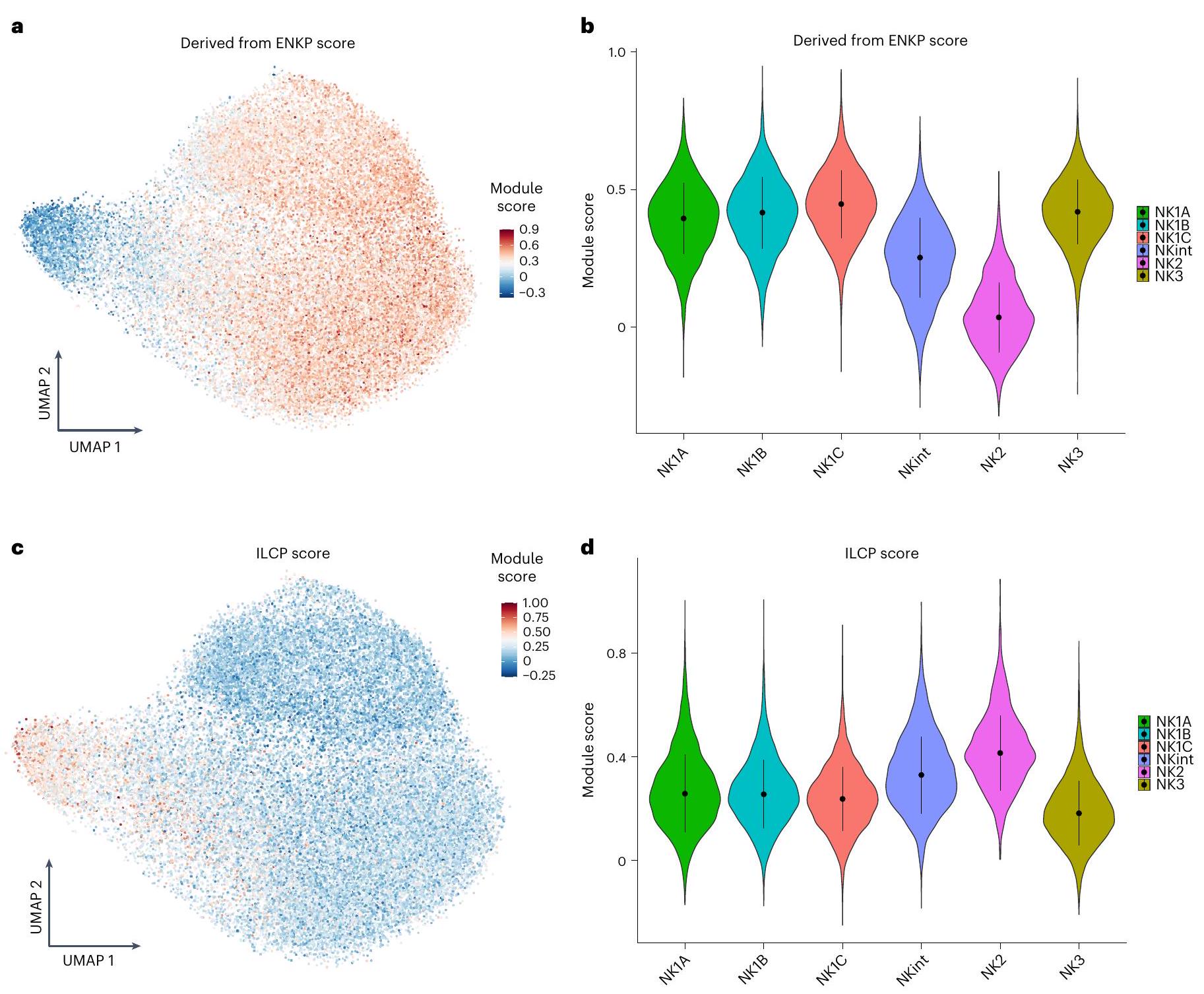
تحليل سرعة RNA يتنبأ أيضًا بمسار تطوري رئيسي آخر، مما يشير إلى أن جزءًا كبيرًا من خلايا NK2 من المحتمل أن تحافظ على حالة خلايا NK2. ويدعم هذا الاحتمال أدلة تشير إلى أن تجمعات خلايا NK في الفئران تنشأ من سلالتين متميزتين: سلف أولي، يُعرف باسم سلف خلايا NK المبكرة (ENKP)، وسلف بديل، يُسمى السلف الشائع للخلايا اللمفاوية الفطرية (ILCP)، الذي يمكنه أيضًا أن يعطي نشأة لأنواع أخرى من خلايا ILC.من خلال رسم درجات وحدات النسخ لخلايا الدم البشرية ENKPs على تمثيل تقريبي موحد وProjection (UMAP)، لاحظنا أن كل من مجموعات NK1 وNK3 أظهرت توقيعات نسخية تتماشى بشكل وثيق مع تلك الخاصة بخلايا NK التي تنشأ من ENKPs (الشكل 5a، b). بالإضافة إلى ذلك، تم تقييم درجات مجموعات NK1 وNK2 وNK3 بناءً على توقيعات ILCP البشرية المتاحة حديثًا في الدم.كشفت أن توقيعهم غني بـ NK2s (الشكل 5c، d). تدعم هذه الملاحظات مجتمعة وجود مسارين تطوريين متباينين: أحدهما لـ NK1 و NK3، ينشأ من ENKPs، والآخر لـ NK2، ينشأ من ILCPs.
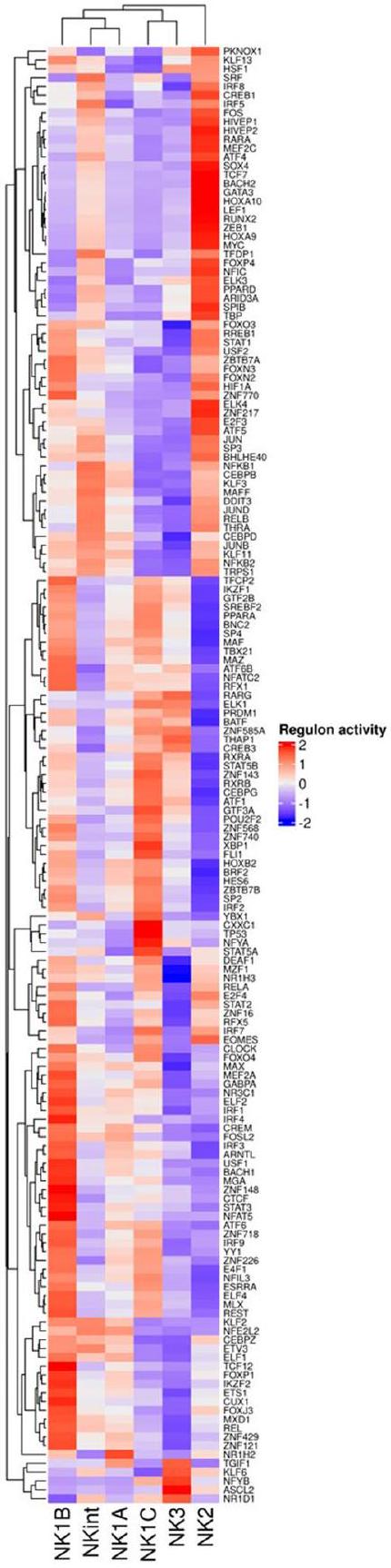
لتوضيح الجينات التنظيمية الرئيسية التي تحدد ستة مجموعات فرعية من خلايا NK، قمنا بإجراء تحليل لشبكة تنظيم الجينات باستخدام سير عمل استنتاج الشبكة التنظيمية على مستوى الخلية الواحدة والتجميع (SCENIC).كانت الخطوة الأولية تتضمن تصنيف الأنظمة التنظيمية المحددة في مجموعة البيانات الخاصة بنا. يتكون كل نظام تنظيمي من عامل نسخ أو عامل مساعد وجيناته المستهدفة المرتبطة به (الشكل التوضيحي الممتد 7a). بعد ذلك، قمنا بمقارنة قائمتنا من الأنظمة التنظيمية مع قاعدة بيانات أكثر موثوقية لعوامل النسخ المؤكدة.. كانت هذه المقارنة حاسمة لاستبعاد عوامل النسخ غير الموثوقة والبروتينات التي ترتبط بـ RNA و
الحمض النووي الريبي غير المحدد، والتركيز على تحليلنا فقط على عوامل النسخ الحقيقية. كشفت التجميع غير المراقب بناءً على نشاط التنظيم أولاً عن ميزتين بارزتين: أولاً، أن NK2 تفرعت بعيدًا عن المجموعات الفرعية الأخرى، مما يدعم نظرية أصل وجودي متميز لـ NK2. ثانيًا، قام التجميع أولاً بتجميع تحت المجموعات NKint وNK1A، مما يشير إلى أنهما قد تمثلان مراحل مبكرة من تمايز خلايا NK1، ثم جمع خلايا NK1B التي بدت أكثر تمايزًا، وأخيرًا شمل تحت المجموعات NK1C وNK3 التي تت correspond إلى حالات خلايا NK الأكثر تقدمًا.
تُبرز هذه النمط التفريقي المتسلسل الآليات التنظيمية المعقدة التي تحكم تطور خلايا NK وتمايزها. عوامل النسخ التي تلعب دورًا حيويًا في نضوج خلايا NKمثل T-bet (TBX21) و BLIMP1 (PRDM1)أظهر زيادة تدريجية عبر NK2 و NKint و NK1A و NK1B إلى NK1C. على العكس من ذلك، كانت MYC و TCF7 و RUNX2 و GATA3 معبرًا عنها بشكل رئيسي في مجموعات NK2، مما يتماشى مع نتائج الأبحاث السابقة.تميزت NK3 بالتعبير القوي عن و ، ووجود BLIMP1 (PRDM1) المستمر. لذلك، فإن نمط التعبير الملحوظ لمُنظِّمات النضج الرئيسية يدعم الفرضية القائلة بوجود سلالات متميزة من سلالات خلايا NK.
توزيع مجموعات خلايا NK في الأنسجة السليمة
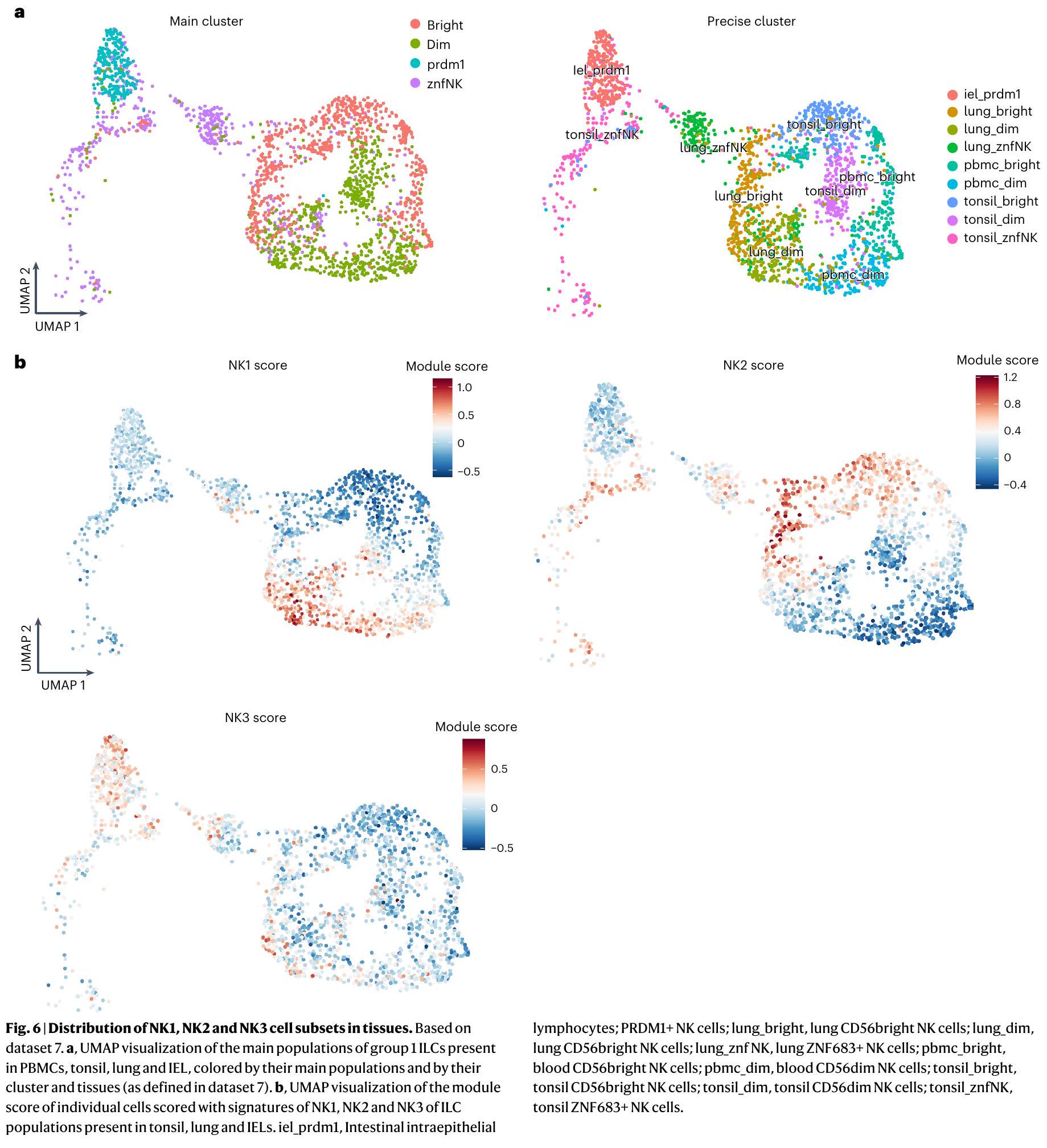
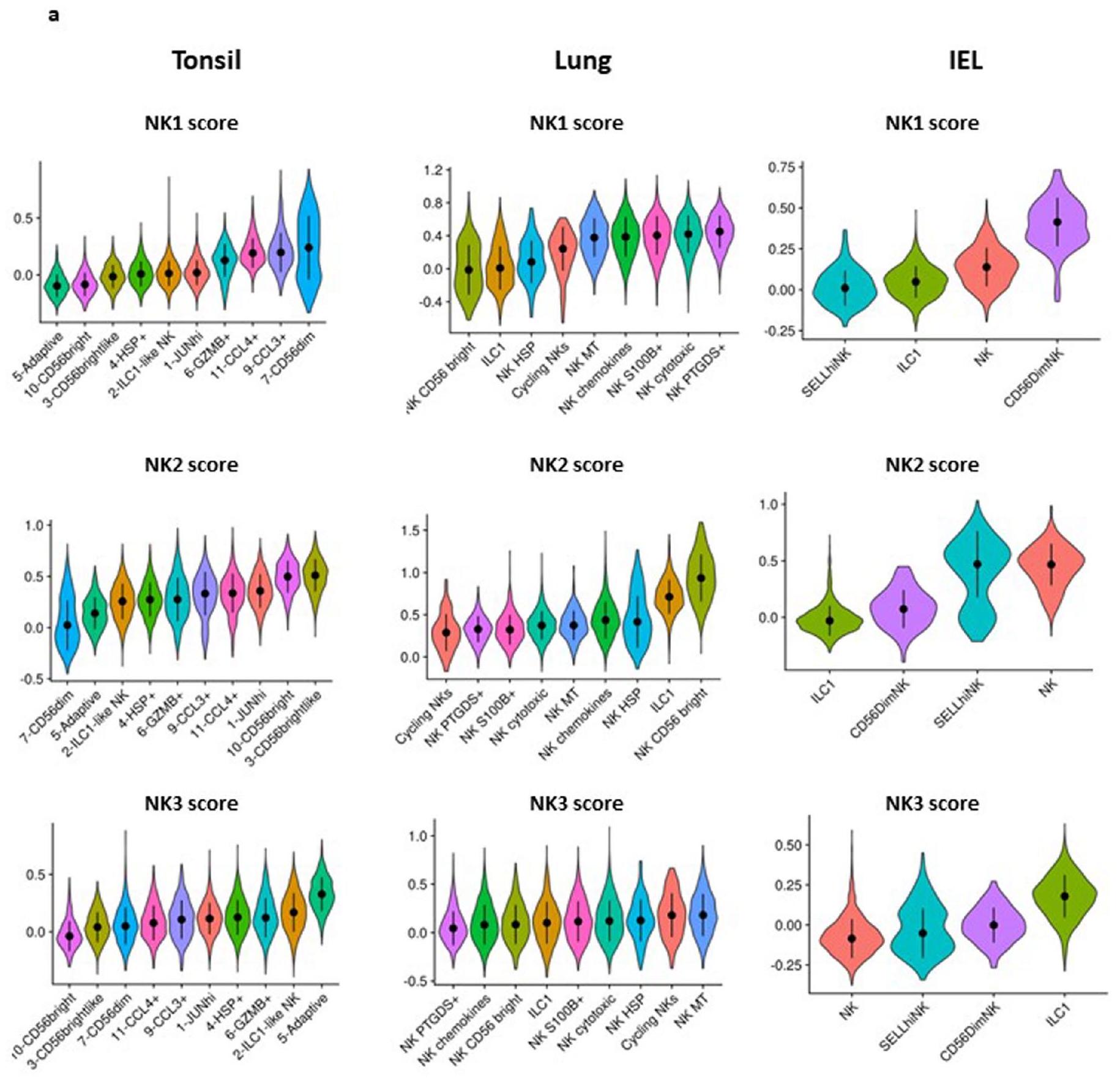
توجد خلايا NK في الأنسجة بالإضافة إلى الدم المحيطي. العلاقة بين خلايا NK المتداولة، خلايا NK المتسللة إلى الأنسجة وخلايا ILC المقيمة في الأنسجة هي مجال بحث ناشئ. تختلف خلايا ILC بشكل كبير اعتمادًا على بيئتها والإشارات المحلية، مثل السيتوكينات، التي تتعرض لها، مما يؤدي إلى ملفات تعريف ILC مميزة في أنسجة وأمراض مختلفة. تم نشر وصف مفصل لهذه التغيرات في ILC مؤخرًا. لذلك قمنا بتحليل بيانات scRNA-seq في الدراسة السابقة للتحقيق في توزيع مجموعات NK1 و NK2 و NK3 في اللوزتين والرئتين واللمفاويات داخل الظهارة (IELs) المعزولة من الأفراد الأصحاء (الشكل 6a). من المRemarkably، تطابقت توقيعات NK1 و NK2 مع CD56 و CD56 المجموعات، على التوالي، التي تم تحديدها في الرئة واللوزتين و IELs (الشكل 6b). بشكل أكثر تحديدًا، يمكن تصنيف الغالبية العظمى من المجموعات الفرعية المختلفة من خلايا CD56 و CD56 في هذه الأنسجة على أنها NK1 و NK2، على التوالي (بيانات موسعة الشكل 8a). لم يكن بالإمكان تخصيص بعض المجموعات الفرعية المتميزة من خلايا NK في اللوزتين (المعلمة JUNhi، NK شبيهة بـ ILC1، HSP) والرئتين (المعلمة NKs دورية، NK HSP، ILC1) إلى مجموعات NK1 أو NK2 أو NK3. نظرًا لأننا قمنا بإزالة مجموعتين فرعيتين، خلايا NK دورية وخلايا NK التي أظهرت خصائص الإجهاد، للتحليل الذي أدى إلى تحديد مجموعات NK1 و NK2 و NK3، توقعنا أن بعض المجموعات، وهي اللوزتين] JUNhi، اللوزتين، الرئة NK HSP و NKs دورية في الرئة، قد لا يمكن توضيحها. من الجدير بالذكر أن مجموعات اللوزتين الشبيهة بـ ILC1 ومجموعات الرئة ILC1 لم تتطابق أيضًا مع أي من ملفات تعريف NK1 و NK2 و NK3، مما يؤكد أن التوقيعات النسخية اللاحقة تشبه بشكل تفضيلي تلك الخاصة بخلايا NK. ومع ذلك، فإن الزيادة الجزئية في الرئة ILC1 مع توقيعات NK2 تعزز الفكرة بأن هناك أنطولوجيا مشتركة بين هاتين المجموعتين (بيانات موسعة الشكل 8a). أخيرًا، تظهر بياناتنا أوجه التشابه بين مجموعات IEL ILC1 و NK3 (الشكل 6a، b وبيانات موسعة الشكل 8a)، كما يتضح بشكل خاص من التعبير القوي عن PRDM1 في كل من مجموعات IEL ILC1 و NK3. تشير هذه النتائج إلى أن أوجه التشابه بين IEL ILC1s وخلايا NK وانحراف IEL ILC1s عن خلايا ILC1 المقيمة الأخرى يجب إعادة تحليلها.
توزيع مجموعات خلايا NK في السرطان
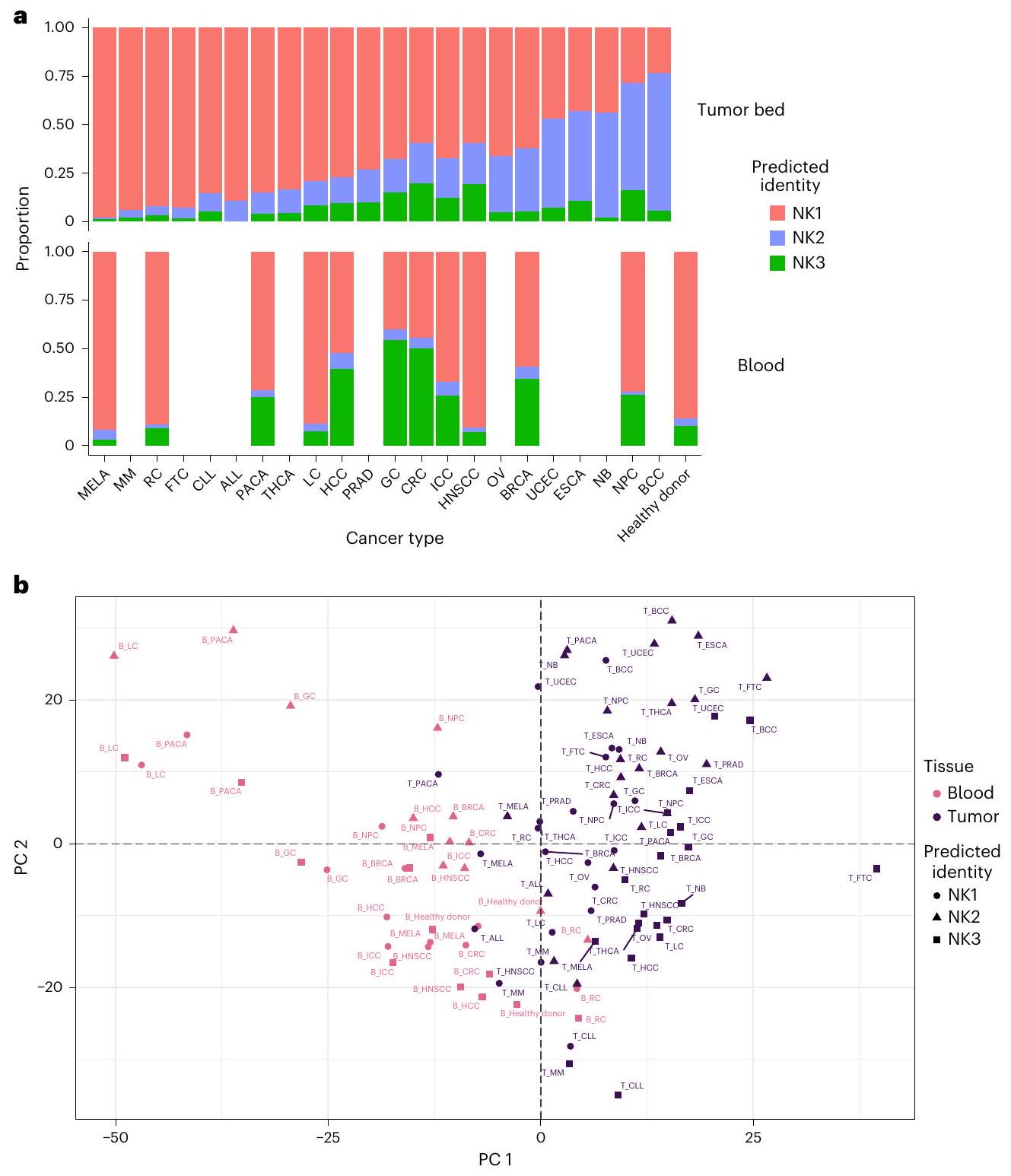
كانت نقطة مهمة في تحليلنا هي توفير معيار للمقارنات المستقبلية مع الحالات المرضية. لذلك، قمنا بتحليل أنماط توزيع مجموعات خلايا NK1 و NK2 و NK3 في 22 نوعًا من السرطان (الشكل 7). لهذا الغرض، استخدمنا نهج نقل العلامات الكلاسيكي (انظر الطرق). بعد التحقق من دقة الطريقة المستخدمة لتوضيح المجموعات الفرعية (بيانات موسعة الشكل 9a-e)، قمنا بالتحقيق في النسب والقرب النسخي لهذه المجموعات عبر الأنسجة وأنواع السرطان. يختلف توزيع مجموعات خلايا NK في هذه الأورام الـ 22 حسب نوع الورم (الشكل 7a، اللوحة العلوية). هذا التوزيع
الشكل 5 | الأنطوجين المحتمل للسكان الرئيسيين من خلايا NK. بناءً على مجموعات البيانات 1-4a. أ، تصور UMAP لدرجة الوحدة للخلايا الفردية التي تم تقييمها بتوقيعات مستمدة من السلف الرئيسي لخلايا NK (ENKP) المحددة في الفئران. ب، مخططات كمان لدرجات الوحدة للخلايا الفردية التي تم تقييمها بتوقيعات ENKP. تُظهر البيانات كوسيط انحراف معياري عينات. ب، د، في مخططات الكمان النقطة
هي القيمة الوسيطة. تمثل أشرطة الخطأ الوسيط– الانحراف المعياري. ج، تصور UMAP لدرجة الوحدة للخلايا الفردية التي تم تقييمها بتوقيعات ILCP من دم الإنسان. د، مخططات كمان لدرجات الوحدة للخلايا الفردية التي تم تقييمها بتوقيعات ILCPs من دم الإنسان. تُظهر البيانات كوسيط انحراف معياري ( عينات).
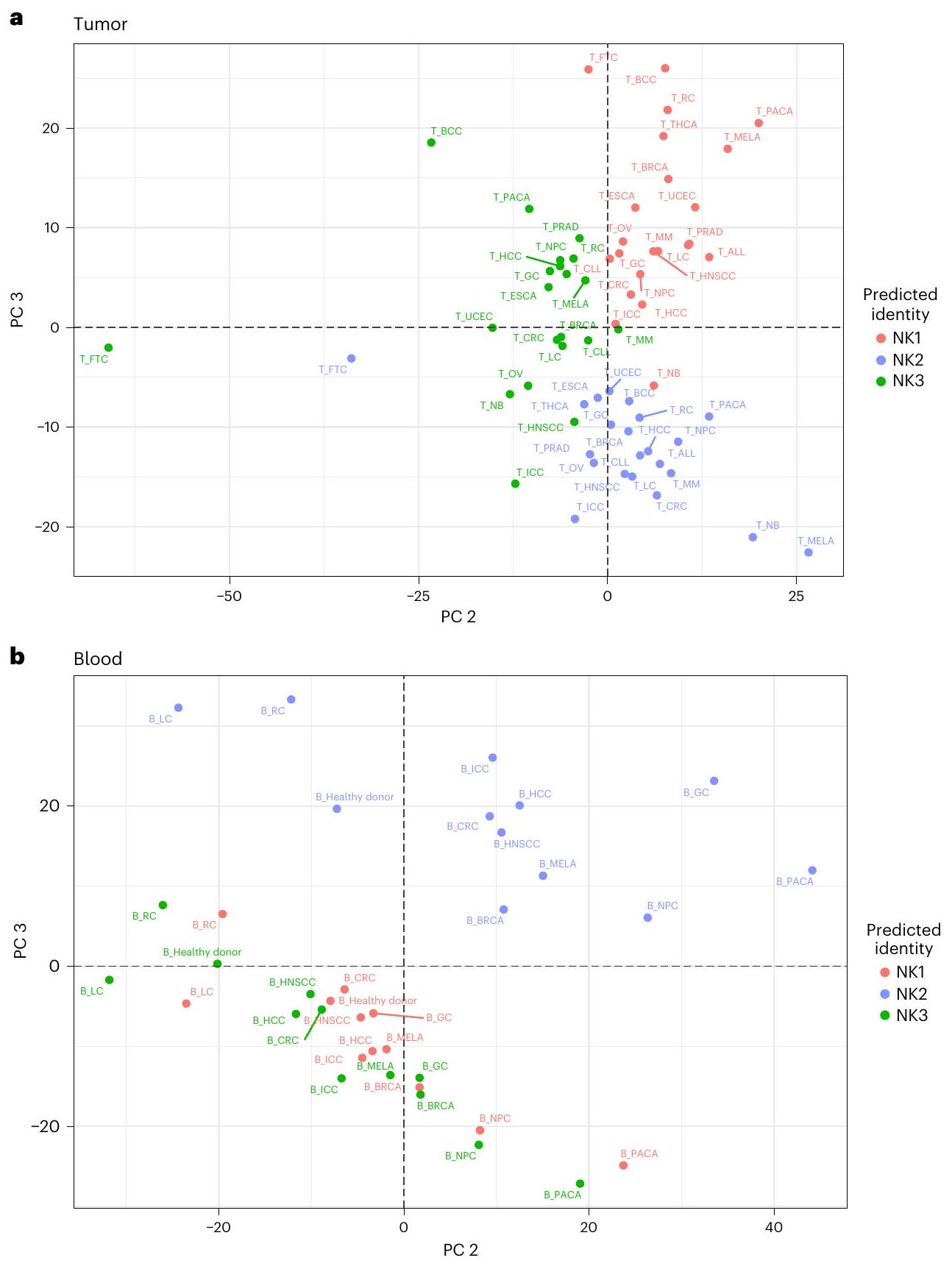
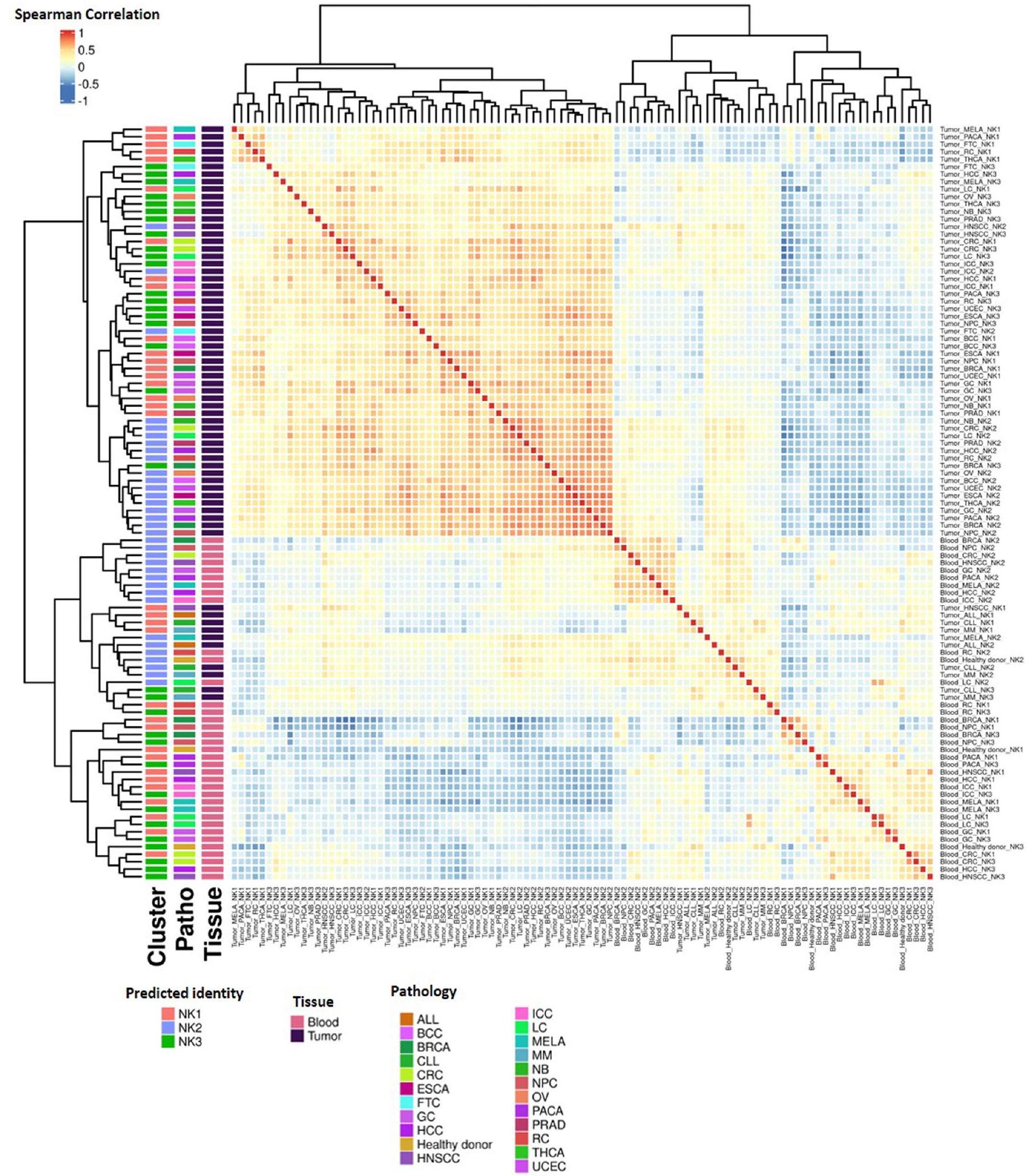
لا يتوافق مع ما تم العثور عليه في الدم (الشكل 7a، اللوحة السفلية). تم تأكيد هذا الاختلاف بين خلايا NK المتداولة وخلايا NK المرتبطة بالورم من خلال تحليل المكونات الرئيسية (PCA) (الشكل 7b، PC1)، كما تم تأكيد دقة توضيح NK1 و NK2 و NK3 في سرير الورم (الشكل 8a، b، PC2 و PC3). تم تأكيد الانحراف بين NK2 والمجموعات الفرعية الأخرى أيضًا في الدم من الأشخاص المصابين بالسرطان، لكن تأثير الورم على التمييز بين مجموعات NK1 و NK2 و NK3 أقوى في سرير الورم مقارنة بالدم (الشكل 8a، b). أكدت علاقة سبيرمان المحسوبة عبر مجموعات NK، نوع السرطان والأنسجة وتجمعها الهرمي غير المراقب أن خلايا NK تنفصل أولاً حسب نوع الأنسجة ثم حسب المجموعة التي تنتمي إليها (بيانات موسعة الشكل 10a). يشير التجميع الأفضل لـ NK1s و NK2s و NK3s في سرير الورم مقارنة بالدم أيضًا إلى ظاهرة متفاقمة في ظروف الورم.
نقاش
على الرغم من أن scRNA-seq و CITE-seq قد تقدمت بشكل كبير في استكشاف تنوع خلايا NK البشرية، إلا أن تعريفات أنواع خلاياها و
أنواعها الفرعية قد اختلفت عبر المنشورات لعدة أسباب، بما في ذلك بروتوكولات تجريبية مختلفة، وطرق اكتساب البيانات وأدوات التحليل. أدى ذلك إلى تعقيد في الأدبيات وحتى خلاف حول ما إذا كانت بعض المجموعات الخلوية الحقيقية أو عيوب ناتجة عن منهجية معالجة معينة. على سبيل المثال، ما يبدو أنه نوع فرعي من خلايا NK قد يكون نتيجة استجابة للإجهاد تم تحفيزها أثناء عزل الخلايا أو بواسطة ظروف الثقافة. وبالتالي، من المهم إنشاء إطار عمل متفق عليه لمجموعة أساسية من أنواع خلايا NK من خلال تجميع مجموعات البيانات التي تم الحصول عليها من مختبرات متعددة وتحليلها بشكل شامل.
أدى دمج بيانات خلايا NK من CITE-seq و scRNA-seq في تحليلنا الشامل، بما في ذلك بيانات من أكثر من 225,000 خلية NK، إلى تمييز ثلاث مجموعات رئيسية من خلايا NK في الدم المحيطي، والتي أطلقنا عليها هنا NK1 و NK2 و NK3. هذه المجموعات غنية جدًا في خلايا NK التقليدية CD56 و CD56 التقليدية و خلايا NK التكيفية المدفوعة بـ HCMV، على التوالي. يتماشى ملف التعبير الجيني لمجموعة NK1 الموصوفة هنا مع تلك الخاصة بخلايا hNK_Bl1 الموصوفة سابقًا، مع تعبير قوي عن FGFBP2 و GZMB و SPON2 و. يتداخل ملف التعبير الجيني لمجموعة NK2
مع تلك الخاصة بـ hNK_BI2، المحددة بمستويات عالية من COTL1 و CD44 و XCL1 و LTB و. من الجدير بالذكر أن نظائر NK1 و NK2 قد تم تحديدها أيضًا في دم الفئران: mNK_Bl1 و mNK_BI2، على التوالي. أظهرت خلايا NK3 نمطًا من التعبير الجيني يتداخل مع تلك الخاصة بخلايا NK التكيفية المدفوعة بـ HCMV الموصوفة سابقًا، المحددة بمستويات عالية من KLRC2 و CD3E و ZBTB38 (المراجع 12، 30). ومع ذلك، على الرغم من أن هذه الجينات التكيفية هي المحركات الرئيسية لتوقيع مجموعة NK3، يمكن أيضًا العثور على خلايا المخصصة لمجموعة NK3 في دراستنا بترددات أقل في الأفراد. لذلك، فإن مجموعة NK3 المحددة هنا
ليست محدودة بخلايا NK التكيفية المدفوعة بـ HCMV. بالإضافة إلى ذلك، بالنظر إلى أن التوقيع النسخي الذي يقتصر على خلايا NK التكيفية محدود بالنسبة لمستوى إعادة تشكيل الإيبيجينوم الذي تخضع له هذه الخلايا، قد تكون الخطوة التالية في حل هوية خلايا NK التكيفية على المستوى الإيبيجينومي من خلال طرق تسلسل ATAC أحادي الخلية. يبرز هذا فوائد دمج أساليب أحادية الخلية متعددة النماذج عند تعريف مجموعات خلوية متميزة. بدلاً من ذلك، يمكن أيضًا تمييز خلايا NK التكيفية عن خلايا NK التقليدية CD56 بناءً على نقص تعبير PLZF.
الشكل 7 | توزيع مجموعات خلايا NK1 و NK2 و NK3 في دم الأشخاص المصابين بالسرطان وفي سرير الورم. بناءً على مجموعات البيانات و 6. أ، رسم بياني عمودي يوضح نسبة المجموعات الثلاث الرئيسية من خلايا NK في الدم وفي سرير الورم في 22 نوعًا من السرطان ( عينات). ب، PCA على خلايا NK المتسللة إلى الورم والدم، مجمعة حسب مجموعة NK، ظروف السرطان والأنسجة. يعتمد PCA على متوسط مستويات التعبير لـ 2,000 جين الأكثر اختلافًا عبر الأنسجة والظروف. يتم تلوين المجموعات بناءً على نوع الأنسجة التي تنتمي إليها. PC1 و PC2 يفسران و من التباين، على التوالي ( عينات). MELA، ميلانوما؛ MM، ورم متعدد النخاع؛ RC، سرطان الكلى؛ FTC، سرطان قناة فالوب؛ CLL، اللوكيميا اللمفاوية المزمنة؛ ALL، اللوكيميا اللمفاوية الحادة؛ PACA، سرطان البنكرياس؛ THCA، سرطان الغدة الدرقية؛ LC، سرطان الرئة؛ HCC، سرطان الكبد الخلوي؛ PRAD، سرطان البروستاتا؛ GC، سرطان المعدة؛ CRC، سرطان القولون والمستقيم؛ ICC، سرطان القناة الصفراوية داخل الكبد؛ HNSCC، سرطان الخلايا الحرشفية في الرأس والعنق؛ OV، سرطان المبيض؛ BRCA، سرطان الثدي؛ UCEC، سرطان بطانة الرحم؛ ESCA، سرطان المريء؛ NB، الورم العصبي الدبقي؛ NPC، سرطان البلعوم الأنفي؛ BCC، سرطان الخلايا القاعدية.
يمكن تقسيم مجموعة NK1 بشكل موثوق إلى ثلاث مجموعات فرعية، تُسمى NK1A وNK1B وNK1C؛ كما تم وصف مجموعة NKint ذات النمط الظاهري الوسيط بين NK1 وNK2. تماشيًا مع دراستنا الشاملة، حددت مجموعات بيانات scRNA-seq المنشورة مؤخرًا أيضًا عدة مجموعات فرعية. أظهرت المجموعة الفرعية NK1A أعلى مستويات التعبير لـ CXCR4 وJUN وJUNB، مما يعكس الوصف المنشور سابقًا لـ CD56 النشط. (مرجع 31) و CD56 الوسيط (المرجع 32) المجموعات. كان CD160 و IFITM1 الأكثر وفرة في مجموعة NK1B، وهي مجموعة تم التنبؤ بأن لديها أعلى استجابة للكيموكينات والسيتوكينات. أظهرت مجموعة NK1C أعلى مستويات التعبير عن PRF1 و PFN1 و ACTB و NKG7، بما يتماشى مع أوصاف CD56 الناضجة والنهائية. (مرجع 31)، CD56 المتأخر (مرجع 32)، العنقود 2 (مرجع 33) و CD56 CD57خلايا NK.
أفادت الدراسات السابقة بوجود مجموعة فرعية متوسطة تربط CD56 (NK2) و CD56 خلايا NK. تشترك هذه المجموعة المتوسطة في توقيع أساسي يشمل تعبير CD44 وXCL1 وGZMK، لكنها تتميز بارتفاع تعبير CXCR4، بما يتماشى مع وصفنا لـ NKint. يشير التعبير العالي لـ KLRC1 (NKG2A) ولكن التعبير المنخفض لـ CD56 إلى أن مجموعة NKint لديها تشابهات قوية مع NKG2A المبكرة.تعداد الخلايا. التعبير الوسيط لـ CD56، الذي يقع بين تعبير NK2 و NK1 ومستويات أقل من البيرفورين والجرانزيم B، بالإضافة إلى تعبير CD27، الذي تم اكتشافه في بداية المسار النسخي المفترض الذي يربط NKint بـ NK1C، يشير أيضًا إلى CD27 الذي تم تعريفه سابقًا.CD56سي دي 94سكان كوريا الشمالية.
الشكل 8 | أنماط التعبير الجيني المميزة لمجموعات خلايا NK1 و NK2 و NK3 في دم الأشخاص المصابين بالسرطان وفي موضع الورم. استنادًا إلى مجموعات البيانات 1-4a و 6.a، تم إجراء تحليل المكونات الرئيسية (PCA) لخلايا NK في الدم، مصنفة حسب مجموعة خلايا NK وظروف السرطان. يعتمد تحليل المكونات الرئيسية على متوسط مستويات التعبير لـ 2000 جين الأكثر اختلافًا في التعبير عبر الأنسجة والظروف. تم تلوين المجموعات بناءً على مجموعات خلايا NK. أوضحت PC2 و PC3 8.1% و 7% من التباين، على التوالي (عينات). ب، تحليل المكونات الرئيسية لخلايا NK المتسللة إلى الورم، مصنفة حسب مجموعة خلايا NK وظروف السرطان. يعتمد تحليل المكونات الرئيسية على متوسط مستويات التعبير لـ 2000 جين الأكثر اختلافًا في التعبير عبر الأنسجة والظروف. تم تلوين المجموعات بناءً على فئات خلايا NK. تفسر PC2 و PC3 و من التباين، على التوالي ( عينات).
كشفت دراساتنا الشاملة عن مسارين تنمويين متميزين لخلايا NK. يشير المسار الأول إلى طريق يمكن لخلايا NK2 من خلاله الحفاظ على هويتها؛ بينما يتضمن المسار الثاني عملية نضوج تدريجية تتطور فيها خلايا NKint إلى مراحل NK1A وNK1B وNK1C. بالنسبة لخلايا NK2، التي يبدو أنها مشتقة من ILCPs، من الجدير بالذكر أن الأبحاث السابقة قد حددت مجموعة نقيّة من المتقدّرات البشرية لخلايا NK، تُسمى NKO (المرجع 37)، وأن توقيع NKO البشري يتطابق مع توقيع ILCPs، مما يشير إلى أن NKOs قد تت correspond إلى ILCPs النخاعية. كما هو الحال مع خلايا NK1، يرتبط نضوجها بتحول ملحوظ في المشهد النسخي، يتميز بشكل خاص بزيادة في التعبير. الجينات المرتبطة بالسُمية الخلوية مثل و . في الوقت نفسه، لاحظنا تصعيدًا في أنشطة التمثيل الغذائي للكربون المركزي على طول مسار النضج هذا. يتميز هذا التصعيد بزيادة في عملية التحلل السكري، ونشاط دورة TCA، وOXPHOS. تؤكد تحليل درجات الوحدة أيضًا هذه المسارات التنموية المتميزة، كاشفة عن ارتباط قوي بين مجموعة NK2 وILCPs في الدم، كما يتضح من علامات التوقيع المشتركة بينها بما في ذلك ، CD44، LTB، IL7R و GPR183 (الأشكال 2e و 5c، d). وبالمثل، تظهر تجمعات NK1 و NK3 ارتباطًا واضحًا بـ ENKPs، مما يتماشى مع الملاحظة أن Ly49Hخلايا NK في الفئران، التي تستجيب لفيروس CMV في الفئران وتعتبر مماثلة لخلايا NK التكيفية البشرية المدرجة في تحت مجموعة NK3، تنشأ في الغالب من ENKPsعلى مستوى عوامل النسخ، تتمتع NK1 و NK3 بخصائص فريدة تشبه بعض سمات ENKP، مثل انخفاض التعبير عن GATA3 و EOMES و TCF7 مع زيادة التعبير عن KLF2. تؤكد هذه التحليل المتعدد الأبعاد على المسارات والآليات المعقدة التي تحكم تمايز ووظائف خلايا NK.
تحقيقنا يسلط الضوء أيضًا على عدة أبعاد جزيئية لبيولوجيا خلايا NK، مما يستدعي مزيدًا من البحث. إحدى النتائج الرئيسية هي الملفات المميزة للجرانزيمات والبرفورين عبر ثلاث مجموعات رئيسية من خلايا NK. تظهر خلايا NK1 تعبيرًا قويًا عن GZMA وGZMB وPRF1، التي تم دراستها بشكل موسع.. على العكس، تعبر خلايا NK2 بشكل رئيسي عنالمعروف بدوره في الموت الخلوي المستقل عن الكاسبازاتوفي التحكم في المناعة الذاتيةتتميز مجموعة NK3 بالتعبير عن، ترميز إنزيم غرانزيم H، الذي يبدأ أيضًا موت الخلايا المستقل عن الكاسبازاتوهو فعال في تحفيز الموت الخلوي السريع في خلايا الورم. هذا يبرز الإمكانات المضادة للأورام الكبيرة لخلايا NK3. لكن لا يزال هناك المزيد لفهمه حول بيولوجيا الجرانزيمات، كما يتضح من العرض الأخير لدور الجرانزيم A في تحفيز إنتاج الغازدرمين-. كانت نتيجة ملحوظة أخرى هي ملف السيتوكينات لمجموعة NK2 التي قامت بشكل أساسي بترجمة FLT3LG جنبًا إلى جنب مع و التي تشفر البروتينات التي تجذب الخلايا الشجرية وتعزز وظيفتها في تقديم المستضداتإن ملف الإنتغرين لمجموعة NK1 والتغير في تعبيرها على طول مسار نضوج NK1 يشير إلى أن هذه الإنتغرينات قد تكون لها دور أساسي في تعزيز تفاعلات الاتصال مع خلايا أخرى، وتنظيم السمية الخلوية لـ NK1 أو تسهيل دخول خلايا NK1 إلى الأنسجة (على سبيل المثال، يتشكل ثنائي ITGB7 مع ITGA4 للالتصاق بـ MAdCAM-1 للدخول إلى الأمعاء).يمكن أن يكون لفهم أفضل لآلية التعبير وتنظيم هذه الإنتغرينات تطبيقات سريرية كبيرة، مثل تعزيز المناعة المضادة للأورام في سرطان القولون..
تشير نتائجنا أيضًا إلى وجود اختلافات ملحوظة في استجابات السيتوكين بين مجموعات خلايا NK. في سياق علاج خلايا NK بالتبني، يعزز تكييف IL-21 التكاثر، السمية الخلوية وإنتاج الإنترفيرون-و TNF في خلايا NKإن الاستجابة الأقوى لمجموعات NK1B و NK1C لـ IL-21 تجعلها واعدة بشكل خاص للعلاجات المعتمدة على خلايا NK. بالإضافة إلى ذلك، لقد أظهر IL-15 أنه يعزز من استقلاب خلايا NK وطول عمرها.، متماشياً مع خصائص مجموعة NK1C الفرعية، التي تظهر نشاطاً استقلابياً قوياً واستجابة ملحوظة لـ IL-15. يمكن استغلال هذه الاستجابات السيتوكينية بشكل أكبر من خلال استخدام محركات خلايا NK المسلحة بالسيتوكينات.، مما يعزز فهمنا للاستجابات المحددة للمجموعات الفرعية لتحسين وتنويع هذه الأساليب العلاجية الجديدة.
تظهر بياناتنا أيضًا أن ملفات الجينات لمجموعات NK1 و NK2 و NK3 تمتد إلى ما هو أبعد من الدم المحيطي للأفراد الأصحاء، مما سمح لنا بوصف تباين خلايا NK في الأنسجة. في الواقع، تمكنا من تحديد مجموعات خلايا NK1 و NK2 و NK3 في الرئة واللوزتين و IELs. كما تم توضيح أهمية ملفات NK1 و NK2 و NK3 من خلال التمييز بين مجموعات خلايا NK المتسللة إلى الأنسجة و ILC1s.
من الجدير بالذكر أننا تمكنا أيضًا من تحليل توزيع مجموعات خلايا NK في 22 نوعًا من السرطان. أظهر ذلك أن توزيع مجموعات خلايا NK يختلف اعتمادًا على نوع الورم ولا يظهر ارتباطًا صارمًا مع التوزيع في الدم. الدلالة الفورية لهذه الملاحظة هي القيمة النسبية لمراقبة خلايا NK في الدم المحيطي لتقييم مناعة خلايا NK لدى الأشخاص المصابين بالسرطان. ومن المثير للاهتمام أن نسبة خلايا NK2 قد زادت في معظم الأورام التي تم اختبارها، لا سيما في سرطان المبيض وسرطان الثدي وسرطان بطانة الرحم وسرطان المريء والورم العصبي الدبقي وسرطان البلعوم الأنفي وسرطان الخلايا القاعدية. على الرغم من أن خلل خلايا NK في سرير الورم مثبت جيدًا.لم يتم تحديد أي ملف تعريف محدد يتوافق مع خلايا NK غير الوظيفية. إن العجز المبلغ عنه في القدرة السيتوليتية لخلايا NK في سرير الورم يتماشى مع التحول نحو ملف NK2، حيث يكون تعبير الجزيئات المشاركة في الآلية السيتوليتية منخفضًا. من المهم أيضًا أن نأخذ في الاعتبار الملف الأيضي لـ NK2 واستجابته للسيتوكينات مقارنة بـ NK1 و NK3. على وجه الخصوص، تم تطوير عدة عوامل علاجية لتحفيز خلايا NK باستخدام السيتوكينات أو السيتوكينات المتحورة.مثل محرضات خلايا NK المجهزة بمتحولات IL-2ومن الضروري أن نأخذ في الاعتبار حساسية السيتوكين لخلايا NK المرتبطة بالورم.
تقدم خلية NK هنا كمرجع للدراسات المستقبلية حول خلايا NK في الدم في الصحة والمرض، ولكنها أيضًا أداة لفهم تنوع خلايا NK في الأنسجة بالنسبة لخلايا NK الدائرة، ونشوء خلايا NK في الأنسجة، والعلاقة بين خلايا NK وILC1s في الأنسجة في الصحة والمرض.
المحتوى عبر الإنترنت
أي طرق، مراجع إضافية، ملخصات تقارير Nature Portfolio، بيانات المصدر، بيانات موسعة، معلومات تكميلية، شكر وتقدير، معلومات مراجعة الأقران؛ تفاصيل مساهمات المؤلفين والمصالح المتنافسة؛ وبيانات توفر البيانات والرموز متاحة علىhttps://doi.org/10.1038/s41590-024-01883-0.
References
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054-1066 (2018).
Lanier, L. L., Testi, R., Bindl, J. & Phillips, J. H. Identity of Leu-19 (CD56) leukocyte differentiation antigen and neural cell adhesion molecule. J. Exp. Med. 169, 2233-2238 (1989).
Phillips, J. H. & Lanier, L. L. A model for the differentiation of human natural killer cells. Studies on the in vitro activation of Leugranular lymphocytes with a natural killer-sensitive tumor cell, K562. J. Exp. Med. 161, 1464-1482 (1985).
Beziat, V., Descours, B., Parizot, C., Debre, P. & Vieillard, V. NK cell terminal differentiation: correlated stepwise decrease of NKG2A and acquisition of KIRs. PLoS ONE 5, e11966 (2010).
Bjorkstrom, N. K. et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56 NK-cell differentiation uncoupled from NK-cell education. Blood 116, 3853-3864 (2010).
Juelke, K. et al. CD62L expression identifies a unique subset of polyfunctional CD56 NK cells. Blood 116, 1299-1307 (2010).
Yu, J. et al. CD94 surface density identifies a functional intermediary between the CD56 and CD56 human NK-cell subsets. Blood 115, 274-281 (2010).
Lee, J. et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirusinfected individuals. Immunity 42, 431-442 (2015).
Schlums, H. et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 42, 443-456 (2015).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587 (2021).
Tang, F. et al. A pan-cancer single-cell panorama of human natural killer cells. Cell 186, 4235-4251 (2023).
Ruckert, T., Lareau, C. A., Mashreghi, M. F., Ludwig, L. S. & Romagnani, C. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol. 23, 1551-1563 (2022).
Crinier, A. et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity 49, 971-986 e975 (2018).
Jiang, P. et al. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nat. Methods 18, 1181-1191 (2021).
Konjevic, G. M., Vuletic, A. M., Mirjacic Martinovic, K. M., Larsen, A. K. & Jurisic, V. B. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine 117, 30-40 (2019).
Patterson, C., Hazime, K. S., Zelenay, S. & Davis, D. M. Prostaglandin impacts multiple stages of the natural killer cell antitumor immune response. Eur. J. Immunol. 54, e2350635 (2023).
Castriconi, R. et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc. Natl Acad. Sci. USA 100, 4120-4125 (2003).
Viel, S. et al. TGF- inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 9, ra19 (2016).
Angerer, P. et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241-1243 (2016).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381-386 (2014).
Ding, Y. Different developmental pathways generate functionally distinct populations of natural killer cells. Nat. Immunol. (in the press).
Jaeger, N. Diversity of group 1 innate lymphoid cells in human tissues. Nature Immunol. (in the press).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083-1086 (2017).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650-665 (2018).
Bi, J. & Wang, X. Molecular regulation of NK cell maturation. Front. Immunol. 11, 1945 (2020).
Gordon, S. M. et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36, 55-67 (2012).
Kallies, A. et al. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood 117, 1869-1879 (2011).
Collins, P. L. et al. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell 176, 348-360 (2019).
Torcellan, T. et al. Circulating NK cells establish tissue residency upon acute infection of skin and mediate accelerated effector responses to secondary infection. Immunity 57, 124-140 (2024).
Holmes, T. D. et al. The transcription factor Bcl11b promotes both canonical and adaptive NK cell differentiation. Sci. Immunol. 6, eabc9801 (2021).
Yang, C. et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 10, 3931 (2019).
Netskar, H. et al. Pan-cancer profiling of tumor-infiltrating natural killer cells through transcriptional reference mapping. Nat. Immunol. (in the press).
Smith, S. L. et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv. 4, 13881406 (2020).
Scheiter, M. et al. Proteome analysis of distinct developmental stages of human natural killer (NK) cells. Mol. Cell Proteom. 12, 1099-1114 (2013).
Melsen, J. E. et al. Single-cell transcriptomics in bone marrow delineates CD56 GranzymeK subset as intermediate stage in NK cell differentiation. Front. Immunol. 13, 1044398 (2022).
Vossen, M. T. et al. CD27 defines phenotypically and functionally different human NK cell subsets. J. Immunol. 180, 3739-3745 (2008).
Crinier, A. et al. Single-cell profiling reveals the trajectories of natural killer cell differentiation in bone marrow and a stress signature induced by acute myeloid leukemia. Cell Mol. Immunol. 18, 1290-1304 (2021).
Lieberman, J. & Fan, Z. Nuclear war: the granzyme A-bomb. Curr. Opin. Immunol. 15, 553-559 (2003).
Trapani, J. A. & Sutton, V. R. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 15, 533-543 (2003).
Guo, Y., Chen, J., Shi, L. & Fan, Z. Valosin-containing protein cleavage by granzyme K accelerates an endoplasmic reticulum stress leading to caspase-independent cytotoxicity of target tumor cells. J. Immunol. 185, 5348-5359 (2010).
Zhao, T. et al. Granzyme K cleaves the nucleosome assembly protein SET to induce single-stranded DNA nicks of target cells. Cell Death Differ. 14, 489-499 (2007).
Jiang, W., Chai, N. R., Maric, D. & Bielekova, B. Unexpected role for granzyme K in CD56 NK cell-mediated immunoregulation of multiple sclerosis. J. Immunol. 187, 781-790 (2011).
Fellows, E., Gil-Parrado, S., Jenne, D. E. & Kurschus, F. C. Natural killer cell-derived human granzyme H induces an alternative, caspaseindependent cell-death program. Blood 110, 544-552 (2007).
Hou, Q. et al. Granzyme H induces apoptosis of target tumor cells characterized by DNA fragmentation and Bid-dependent mitochondrial damage. Mol. Immunol. 45, 1044-1055 (2008).
Zhou, Z. et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368, eaaz7548 (2020).
Barry, K. C. et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 24, 1178-1191 (2018).
Bottcher, J. P. et al. NK cells stimulate recruitment of cDC 1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022-1037 (2018).
Hegewisch-Solloa, E. et al. Differential integrin adhesome expression defines human NK cell residency and developmental stage. J. Immunol. 207, 950-965 (2021).
Zhang, Y. et al. Integrin inhibits colorectal cancer pathogenesis via maintaining antitumor immunity. Cancer Immunol. Res. 9, 967-980 (2021).
Oyer, J. L. et al. Natural killer cells stimulated with PM21 particles expand and biodistribute in vivo: clinical implications for cancer treatment. Cytotherapy 18, 653-663 (2016).
Li, L. et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci. Adv. 9, eadd6997 (2023).
Demaria, O. et al. Antitumor immunity induced by antibody-based natural killer cell engager therapeutics armed with not-alpha IL-2 variant. Cell Rep. Med 3, 100783 (2022).
Dean, I. et al. Rapid functional impairment of natural killer cells following tumor entry limits anti-tumor immunity. Nat. Commun. 15, 683 (2024).
Vivier, E. et al. Natural killer cell therapies. Nature 626, 727-736 (2024).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
(c) The Author(s) 2024 Aix Marseille Université, CNRS, INSERM, Centre d’Immunologie de Marseille-Luminy, Marseille, France. Leiden University Medical Center, Willem-Alexander Children’s Hospital, Laboratory for Pediatric Immunology, Leiden, the Netherlands. Leiden University Medical Center, Department of Immunology, Leiden, the Netherlands. Wellcome Sanger Institute, Wellcome Genome Campus, Cambridge, UK. Biosciences Institute, Newcastle University, Newcastle upon Tyne, UK. T Cell Biology and Development Unit, Laboratory of Genome Integrity, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA. Center for Infectious Medicine, Department of Medicine Huddinge, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden. Department of Medicine Huddinge, Karolinska Institutet, Stockholm, Sweden. Division of Clinical Immunology and Transfusion Medicine, Karolinska University Hospital, Stockholm, Sweden. Sweden Broegelmann Research Laboratory, Department of Clinical Science, University of Bergen, Bergen, Norway. Department of Experimental Medicine (DIMES), University of Genoa, Genoa, Italy. Laboratory of Clinical and Experimental Immunology, IRCCS Istituto Giannina Gaslini, Genova, Italy. Department of Medicine, University of Minnesota, Minneapolis, MN, USA. Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, USA. Department of Life Sciences, Imperial College London, Sir Alexander Fleming Building, South Kensington, London, UK. Laboratory of Innate Immunity, Institute of Microbiology, Infectious Diseases and Immunology (I-MIDI), Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany. Mucosal and Developmental Immunology, Deutsches Rheuma-Forschungszentrum (DRFZ), an Institute of the Leibniz Association, Berlin, Germany. Department of Dermatology and NIHR Biomedical Research Centre, Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, UK. Department of Immunology & Immunotherapy, The Marc and Jennifer Lipschultz Precision Immunology Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA. Department of Oncological Sciences, The Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA. Department of Microbiology and Immunology and the Parker Institute for Cancer Immunotherapy, University of California, San Francisco, San Francisco, CA, USA. Precision Immunotherapy Alliance, The University of Oslo, Oslo, Norway. The Institute for Cancer Research, Oslo University Hospital, Oslo, Norway. Tumor Immunology Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy. School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin, Ireland. Institute of Medical Immunology, Charité Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt Universität zu Berlin, Berlin, Germany. Innate Immunity, Deutsches Rheuma-Forschungszentrum Berlin (DRFZ), ein Leibniz Institut, Berlin, Germany. Berlin University Alliance, Berlin, Germany. MRC Mitochondrial Biology Unit, University of Cambridge, Cambridge, UK. IRCCS Ospedale Policlinico San Martino, Genova, Italy. Innate Pharma Research Laboratories, Innate Pharma, Marseille, France. APHM, Hôpital de la Timone, Marseille-Immunopôle, Marseille, France. Paris-Saclay Cancer Cluster, Le Kremlin-Bicêtre, France. e-mail: vivier@ciml.univ-mrs.fr
طرق
استرجاع بيانات تسلسل RNA أحادي الخلية والمعالجة المسبقة
للمجموعات البيانية 1-4، تم استرجاع بيانات تسلسل RNA أحادي الخلية من الدراسات المشار إليها في الجدول التكميلي 3. تم محاذاة بيانات التسلسل أحادي الخلية مع الجينوم البشري المرجعي GRCh38 وتم قياسها باستخدام Cell Ranger (الإصدار 6.1.2، 10x Genomics). تم استخدام البيانات المفلترة الأولية التي تم إنشاؤها من Cell Ranger للتصفية والتحليلات اللاحقة. أولاً، تم فحص كل عينة بشكل فردي لإزالة الخلايا ذات الجودة المنخفضة والتلوث الخلوي. تم الاحتفاظ بالجينات التي تم اكتشافها في أكثر من ثلاث خلايا، وتم إزالة الخلايا التي تعبر عن أقل من 200 ميزة مميزة. ثم، لكل عينة، تم تطبيع البيانات وتعديلها وتم تجميع الخلايا وفقًا لبروتوكول Seurat القياسي. تم تحديد التلوث المتبقي باستخدام حزمة SingleR (الإصدار 1.4.1). تم استرجاع البيانات الوصفية التفصيلية (بما في ذلك معرف المريض وحالة CMV) من الدراسات الأصلية. بالنسبة لمجموعة البيانات 4، نظرًا لأن البيانات الأصلية كانت غنية بنسبةبين NKG2Cو NKG2Cتم تقليل عينات خلايا NK لتتناسب مع النسبة البيولوجية الأولية لكل عينة (متبرع). بالنسبة لمجموعات البيانات 5 و 6 و 7، تم استخدام كائنات Seurat المعالجة مسبقًا.
تصحيح تأثير الدفعة والتجميع غير المراقب
ثم تم دمج العينات. لتقليل تأثير الدفعة أثناء عملية التجميع، تم الاحتفاظ بـ 11,965 جينًا موجودًا في كل من العينات لخطوة التجميع في التحليل. لحساب الفرق في عمق التسلسل بين العينات، تم تطبيع بيانات العد باستخدام دالة Multibatchnorm مع المعامل ‘batch= sample’ من batchelor (v1.10.0). تم تحديد أفضل 5,000 جين متغير عالي (HVGs) في كل عينة باستخدام دالة FindVariableFeatures في Seurat (v4.0.0). ثم، لاختيار أفضل 2,000 ميزة للاحتفاظ بها من أجل التكامل، تم استخدام دالة SelectIntegrationFeatures من Seurat مع إعداد المعامل ‘nfeatures = 2000’. ثم تم توسيع وتوسيع تعبير الجين باستخدام دالة ScaleData من مكتبة Seurat. بعد ذلك، تم إجراء تحليل المكونات الرئيسية (PCA) على مصفوفة HVG لتقليل الضوضاء وكشف المحاور الرئيسية للتباين باستخدام دالة RunPCA، وتم الاحتفاظ بأفضل 30 مكونًا للتحليل. تم تصحيح تأثيرات الدفعة باستخدام خوارزمية تصحيح harmony (v0.1.0) عبر العينات.تم حساب تقليل الأبعاد باستخدام UMAP ورسم الجيران الأقرب المشترك على تمثيلات PCA المصححة بواسطة Harmony. تم اختيار معلمة الدقة لوظيفة FindClusters في Seurat لتعظيم متوسط استقرار sc3 للتجميع لمدى دقة يتراوح منإلىتم تحديد مجموعة الخلايا المتكاثرة باستخدام وظيفة CellCycleScoring من Seurat. ثم تمت إزالة الخلايا في هذه المجموعات، وتم حساب تصور UMAP جديد لتحسين تصور المجموعات والخلايا المتبقية. الكائن النهائي المستخدم في تحليل مجموعات البياناتمتاح على: https://collections.cellatlas.io/meta-nk.
تم تحديد جينات العلامة المحددة للتجمع باستخدام دالة FindAllMarkers في Seurat مع المعامل ‘method= wilcox, only.pos’صحيح،عتبة logfc.
تسجيل النقاط بالتوقيعات
لتسجيل الخلايا بالنسبة لتوقيعات محددة، تم إدخال أفضل 20 علامة محددة للتجمع (محسوبة كما هو موضح أعلاه) في دالة AddModuleScore. باختصار، تم حساب مستوى التعبير المتوسط لكل جين في برامج التعبير المحددة لكل خلية، ثم تم طرح التعبير المجمع لمجموعات الجينات الضابطة. تم تصنيف جميع الجينات التي تم تحليلها بناءً على مستوى التعبير المتوسط، وتم اختيار الجينات الضابطة عشوائيًا من كل فئة.
تحليل سرعة RNA
لتقليل تأثيرات الدفعات وأخذ الفروق في جودة العينات بعين الاعتبار، تم إجراء تحليل سرعة RNAتم إجراء ذلك بشكل منفصل على العينات المختلفة. أولاً، تم إعادة حساب المعرفات الجزيئية الفريدة المدمجة وغير المدمجة باستخدام حزمة بايثون. فيلوسيتو(v0.2.2). بعد ذلك، تم تقدير سرعة RNA باستخدام دالة scvelo المدمجة في حزمة R velociraptor (v3.18). تم تقييد حسابات السرعة للجينات التي تم استخدامها سابقًا لدمج البيانات. لتسهيل التصور، تم إسقاط أوقات السرعة الزائفة على إحداثيات UMAP.
تحليل خريطة الانتشار
خوارزميات خريطة الانتشار المنفذة في حزمة R المسماة destiny (v3.4.0) تم استخدامها لاستنتاج الوقت الزائف. قمنا بإزالة خلايا NK3 من التحليل بسبب ديناميكيتها شبه الكلونية المحددة. لإزالة تأثير دفعة مجموعة البيانات، تم إجراء التحليل على أكبر مجموعة بيانات (المجموعة 4) فقط. لمنع تأثير الدفعة الفردية (على مستوى العينة)، تم تنفيذ دالة RunFastMNN في حزمة R batchelor. (v1.10.0) تم استخدامه. ثم تم استخدام مصفوفة التعبير المصححة كمدخلات لتوليد خرائط الانتشار باستخدام دالة DiffusionMap مع تعيين المعلمات إلى ‘censor_valنطاق الرقابةقام خوارزمية Destiny تلقائيًا بتحديد ثلاث خلايا ‘جذر’. اخترنا الخلية الجذرية الأولى كالجذر الرئيسي لأنها تقع في بداية الخط المتجه المستنتج من سرعة RNA، ثم قمنا بحساب زمن الانتشار الزائف لجميع الخلايا باستخدام دالة DPT.
تحليل مسار النسخ
لتأكيد المسارات النسخية المحددة ولتفهم أفضل للتغيرات على طول المسار من NKint إلى NK1C، قمنا بإجراء تحليل الزمن الزائف باستخدام Monocle3 (المرجع 20) v1.3.1 على كل عينة من مجموعات البيانات 1-4a معًا. تم إزالة خلايا NK3 وNK2 من التحليل، للتركيز فقط على عملية نضوج NK1. تم تشغيل دالة learn_graph مع المعامل ‘ncenter = 150’ لمنع التفرع المفرط للمسار. تم اختيار نقطة البداية للمسار كنقطة النهاية للفرع في مجموعة NKint، كما تم تحديده من خلال تحليلات سرعة RNA وخريطة الانتشار. ثم تم حساب الزمن الزائف باستخدام دالة order_cells. بعد ذلك، قمنا بإجراء اختبار Moran’s / لاكتشاف الجينات المهمة التي تظهر ارتباطًا على طول الرسم البياني الرئيسي، واختيار أفضل 150 جينًا مع قيمة وأعلى درجة ارتباط Moran’s / ورسم تعبيرها ( الدرجة) على طول الزمن الزائف باستخدام دالة Heatmap من مكتبة ComplexHeatmap (v2.6.2).
تحليل SCENIC
تم تحليل التنظيمات النشطة في المجموعات الفرعية المختلفة بواسطة SCENIC (v0.12.1). تم استخدام البيانات التي تم تحليلها لتحديد المجموعات الفرعية الرئيسية الستة من NK كمدخل لتنفيذ خوارزمية SCENIC بلغة بايثون (pyscenic) . باختصار، تم استنتاج علاقات التعبير المشترك بين عوامل النسخ وأهدافها المحتملة باستخدام دالة grn مع خوارزمية إعادة بناء الشبكة التنظيمية ‘grnboost2’ المختارة. تشكل عوامل النسخ وجيناتها المستهدفة معًا تنظيمًا. ثم، تم استخدام ctx لتنقيح التنظيمات باستخدام الأهداف التي لا تحتوي على إثراء لموتيف مطابق لعامل النسخ، مما يفصل بشكل فعال الأهداف المباشرة عن غير المباشرة بناءً على وجود بصمة تنظيمية سيس. بعد ذلك، تم استخدام الأمر aucell لحساب نشاط التنظيم لكل خلية. ثم، تم التحقق من قائمة التنظيمات مع قاعدة بيانات موثوقة من عوامل النسخ المؤكدة لإزالة عوامل النسخ غير الموثوقة والبروتينات التي ترتبط بـ RNA وDNA بشكل غير محدد، ولتحديد التحليل لعوامل النسخ الحقيقية. ثم تم توسيع نشاط التنظيم وتوسيع نطاقه قبل التصور باستخدام دالة Heatmap من مكتبة ComplexHeatmap.
تقييم توقيعات ENKP
لتقييم الخلايا بتوقيعات خلايا NK المشتقة من ENKP، قمنا باستخراج قوائم الجينات الأكثر تمثيلًا التي تم التعبير عنها بشكل مختلف في خلايا NK المشتقة من ENKP وتحويلها إلى نظيرها البشري.
نظرًا لأنها لا تمتلك نظيرًا بشريًا، وبسبب تقاربها التطوري، تم استبدال الجينات في عائلة Klra بنظيراتها البشرية KIR . ثم تم تقييم الخلايا باستخدام دالة AddModuleScore على 20 جينًا الأكثر أهمية. لتقييم ILCP، تم استرجاع توقيعات الجينات مباشرة من المنشور الأصلي .
تحليل إثراء GO
قمنا بإجراء تحليل إثراء GO باستخدام حزمة clusterProfiler (v3.18.1). تم اختيار ثمانية أوصاف مثيرة للاهتمام من بين أعلى 20 توصيفًا تمييزًا لـ GO لكل مجموعة. تم حساب درجات الإثراء ( القيم) للتوصيفات الثمانية المختارة بواسطة اختبار إحصائي هايبرجومتري مع عتبة دلالة 0.05. تم رسم البيانات كـ القيم بعد تصحيح بنجاميني-هوشبرغ. تم تعيين عتبة الدلالة عند .
استجابة السيتوكين
لمقارنة استجابة السيتوكين عبر مجموعات خلايا NK، قمنا بتطبيع عدد الجينات الخام إلى -عدد مقاييس لكل مليون، تلاها مركزية متوسطة لتمكين المقارنة المباشرة عبر الخلايا. ثم تم تحليل البيانات في CytoSig (v0.0.3)، مع المعامل -s 2 لتضمين مجموعة أكثر شمولاً من التوقيعات. القيم تم اشتقاقها من مقارنة الدرجات بين مجموعة خلايا NK واحدة والأخرى، باستخدام اختبارات Student’s -tests.
تحليل CITE-seq
لتحليل بيانات CITE-seq (مجموعة البيانات 5)، تم استخدام البيانات التي تم معالجتها مسبقًا كما هو موضح في المرجع 10. باختصار، بعد إزالة الخلايا التي تحتوي على عدد غير عادي من الميزات (الجينات و/أو ADTs)، تم استخدام HTODemux لاكتشاف وإزالة الثنائيات . ثم، تم إجراء تصحيح الدفعة باستخدام SCTransform تلاه سير عمل PCA العكسي . تم استخدام نفس العملية لـ ADTs، ولكن تم إجراء التطبيع بواسطة تحويل CLR داخل كل خلية. ثم، تم تشغيل PCA على كل من RNA وطرق البروتين، وتم استخدام أعلى 40 أو أعلى 50 بُعدًا، على التوالي، لبناء -رسوم الجار الأقرب. تم استخدام هذه الرسوم كمدخل لإجراء WNN . بناءً على تعليقات المؤلف، تم استخراج خلايا NK وتمت إزالة خلايا NK المتكاثرة. تم أيضًا إزالة الخلايا التي نشأت بعد التطعيم (في الأيام 3 و7) من التحليل، لذا تم الاحتفاظ فقط بالخلايا غير المعالجة. تم إعادة تجميع 5,708 خلية NK المتبقية بدقة عالية جدًا (). تم تحديد الجينات وADTs المعبر عنها بشكل مختلف باستخدام اختبار ويلكوكسون ذو الجانبين مع تعديل بونفيروني المحسوب باستخدام دالة FindAllMarkers، كما هو موضح أعلاه. لتحسين تصور تعبير ADT على UMAP، تم استخدام دالة FeaturePlot من Seurat مع المعاملات ‘min.cutoff = ‘q01’، max.cutoff = ‘q99″ لمنع التأثير القوي للنقاط الشاذة على مقياس اللون.
تحسين التجميع
لتحديد أكثر دقة مناسبة للتجميع بطريقة غير متحيزة، تم استخدام حزمة clustree لت quantifying مقياس استقرار SC3. يتم استخدام هذا المقياس لتقييم استقرار التجميع على مستويات مختلفة من التفاصيل . تقيس هذه الطريقة مدى ثبات تجميعات الخلايا عبر دقة التجميع المختلفة وت quantifies استقرار كل مجموعة عند مستويات محددة من الدقة (الشكل 3a من البيانات الموسعة). من خلال تحديد الدقة التي تعظم استقرار SC3، حددنا أكثر تكوين تجميع موثوق (الشكل 3b من البيانات الموسعة)، واستقرينا في النهاية على قيمة دقة 0.7، والتي تتوافق مع 11 مجموعة (الشكل 3c من البيانات الموسعة).
تحليل المسار الأيضي
لمقارنة الأيض عبر المجموعات الفرعية، تم استخدام scGSVA (https://github. com/guokai8/scGSVA)، وهو التنفيذ أحادي الخلية لـ GSVA . لهذه الدراسة، تم الاحتفاظ فقط بالمسارات الأيضية الرئيسية لـ
التي تم اكتشاف عدة جينات فيها بشكل كافٍ (على سبيل المثال تم اكتشافها في أكثر من 10% من الخلايا في على الأقل من مجموعات NK).
تحسين توقع الهوية من خلال نقل التسمية
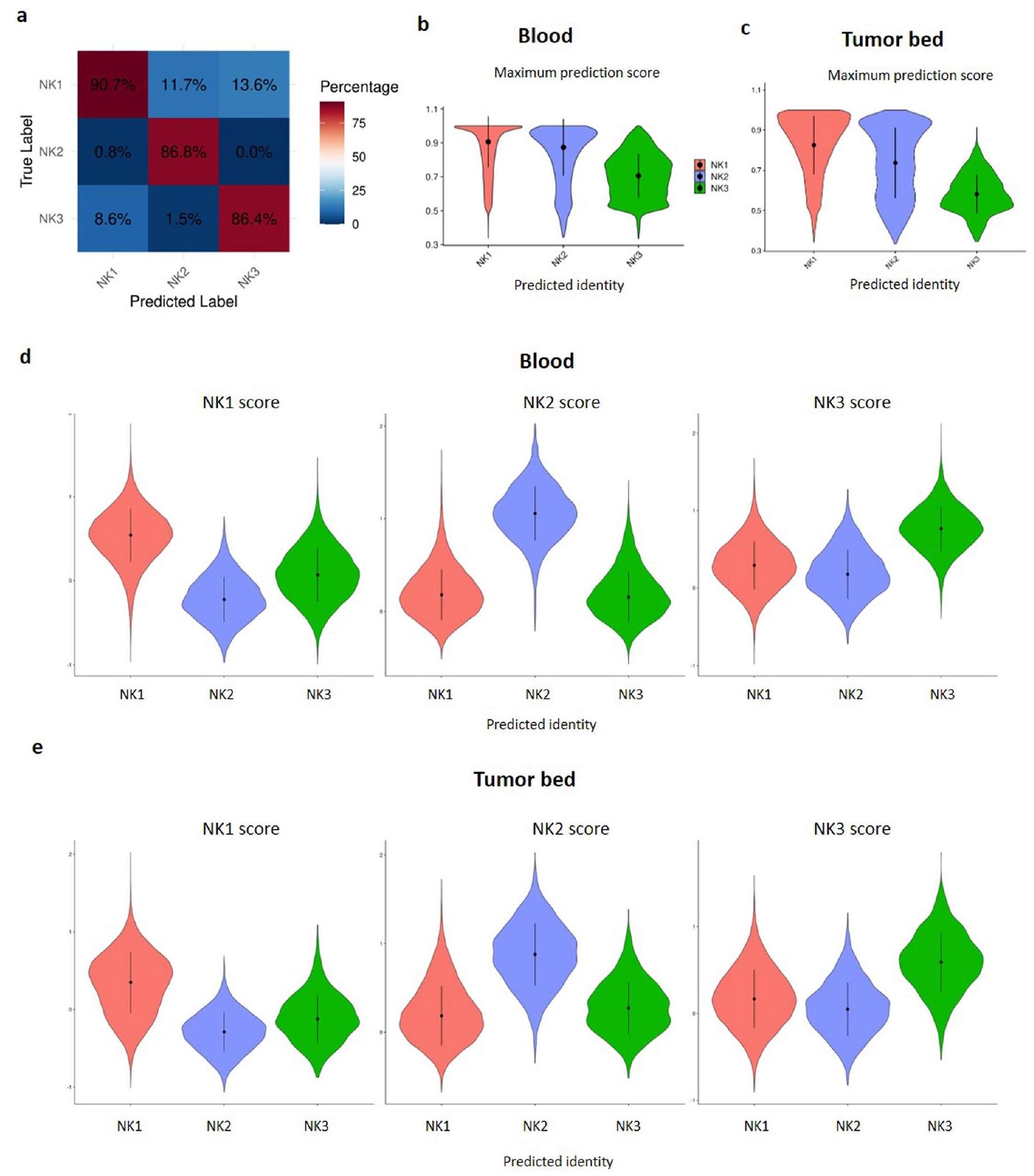
للحصول على تصنيفات خلايا NK1 وNK2 وNK3 في مجموعة البيانات 7، استخدمنا بروتوكول Seurat المعتمد لنقل التسمية. في البداية، تم بناء مرجع باستخدام مجموعات البيانات ، مما مكن من نقل التسمية إلى مجموعة البيانات 7. من الجدير بالذكر أن خلايا NKint تم تصنيفها كـ NK1، مما يعكس موقعها الأولي في مسار نضوج NK1. من خلال عملية الدمج ونقل التسمية (انظر https://satijalab.org)، قمنا بفحص موثوقية الطريقة ودقة التسمية من خلال تطبيقها على مجموعات البيانات . ثم قمنا بتقييم دقة التسمية على مجموعة فرعية من كل مجموعة، والتي تم استبعادها من تدريب المرجع (الشكل 9a من البيانات الموسعة). أظهرت هذه التقييمات دقة توقع لا تقل عن عبر المجموعات، مع كون تحديد NK1 دقيقًا بشكل خاص ( دقة). تم تقييم سلامة نقل التسمية إلى مجموعة البيانات 7 من خلال فحص أعلى درجة توقع للخلايا الفردية داخل كل من بيئات الدم والورم (الشكل 9b، ج من البيانات الموسعة). أكد ذلك أن مجموعة NK1 كانت الأكثر توقعًا بثقة. بالإضافة إلى ذلك، قمنا بتقييم توافق الخلايا مع توقيعات NK1 وNK2 وNK3، مجمعين إياها حسب هوياتها المتوقعة لتأكيد إثراء كل مجموعة متوقعة بتوقيعها المقابل (الشكل 9d، هـ من البيانات الموسعة).
تحليل PCA وتحليل التغاير
لتحليل PCA على مجموعة البيانات 7، تم اعتماد نهج منظم من ثلاث خطوات. في البداية، تم تصنيف الخلايا حسب نوع الأنسجة (ورم أو دم) وتصنيف السرطان. بعد ذلك، تم تحديد أعلى 2000 ميزة متغيرة ضمن كل فئة باستخدام وظيفة FindVariableFeatures. بعد ذلك، حددت وظيفة FindIntegrationFeatures أكثر 2000 جين متغير عبر الفئات. بعد التقييس، قمنا بحساب متوسط التعبير لهذه الميزات الـ 2000 المختارة لمجموعات الخلايا، المصنفة حسب الهوية المتوقعة، نوع السرطان والأنسجة، باستخدام وظيفة AverageExpression من Seurat. ثم تم إعادة تقييس البيانات لتحليل PCA، الذي تم إجراؤه باستخدام مكتبة ade4. تم تكرار هذه الإجراءات بشكل مستقل لخلايا NK في الورم والدم وشملت اختبار كروسكال-واليس لتحديد المكونات الرئيسية (PC2 و PC3) التي تميز بشكل أفضل بين ثلاث مجموعات رئيسية من خلايا NK في كل من سياقات الدم والورم. بالنسبة لتحليل التغاير، تم استخدام نفس الخطوات التحضيرية، تلتها حساب معامل الارتباط سبيرمان بين كل مجموعة باستخدام وظيفة cor. تم استخدام حزمة Pheatmap لتصور هذه الارتباطات.
ملخص التقرير
معلومات إضافية حول تصميم البحث متاحة في ملخص تقارير مجموعة نيتشر المرتبط بهذه المقالة.
توفر البيانات
تم إيداع جميع بيانات تسلسل RNA أحادي الخلية وبيانات CITE-seq المستخدمة في هذه الدراسة في أرشيف التعبير الجيني. تم إدراج رمز الوصول لكل مجموعة بيانات مستخدمة في الجدول التكميلي 3. تتوافق مجموعات البيانات 1-7 مع أرقام الوصول التالية، على التوالي: GSE119562، GSE130430، GSE184329، GSE197037، GSE164378، GSE212890 و GSE240441. تم محاذاة بيانات تسلسل الخلايا الفردية مع الجينوم البشري المرجعي GRCh38. لجعل بياناتنا أكثر وصولاً إلى المجتمع البحثي الأوسع، قمنا بإنشاء بوابة تفاعلية (https://collections.cellatlas.io/meta-nkصُمم لتحليل البيانات أحادية الخلية لدينا بسهولة وتصورها.
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289-1296 (2019).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494-498 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408-1414 (2020).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421-427 (2018).
Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247-2276 (2020).
Barten, R., Torkar, M., Haude, A., Trowsdale, J. & Wilson, M. J. Divergent and convergent evolution of NK-cell receptors. Trends Immunol. 22, 52-57 (2001).
Stoeckius, M. et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888-1902 (2019).
Kiselev, V. Y. et al. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483-486 (2017).
Zappia, L. & Oshlack, A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 7, giyO83 (2018).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
شكر وتقدير
مختبر E.V. في CIML وAssistance-Publique des Hôpitaux de Marseille مدعوم بتمويل من المجلس الأوروبي للبحث (ERC) في إطار برنامج الأبحاث والابتكار Horizon 2020 التابع للاتحاد الأوروبي (TILC، اتفاقية منحة رقم 694502 وMInfla-TILC، اتفاقية منحة رقم 875102)، والوكالة الوطنية للبحث بما في ذلك مشروع PIONEER (ANR-17-RHUS-0007)، وMSDAvenir، وInnate Pharma، والمنح المؤسسية الممنوحة لـ CIML (INSERM، CNRS وجامعة إكس-مارسيليا). مختبر D.M.D. ممول من المجلس الطبي للبحوث (MR/WO31698/1) وWellcome (110091/Z/15/Z). L.M. ممول من Associazione Italiana contro il Cancro (AIRC)، مشروع 5 xmille رقم 21147. S.S. ممول من Ministero dell’Istruzione, dell’Università e della Ricerca (PRIN 2017WC8499_004) وFondazione AIRC (AIRCرقم المشروع 21147). يتم دعم D.G.R. من خلال تمويل من مجلس البحوث الطبية (MRC) (MC_UU_00028) ومؤسسة ويلكوم – الأكاديمية of Medical Sciences (WT-AMS) (SBF009ไ1119). مختبر C.R. مدعوم من منحة ERC المتقدمة ‘MEM-CLONK’ (101055157) ومنح Deutsche Forschungsgemeinschaft (DFG) SFB TRR241 BO2 و RO 3565/7-1. تم دعم K.J.M. من قبل مجلس البحث النرويجي، مركز التميز: تحالف العلاج المناعي الدقيق (332727) والمعهد الوطني الأمريكي للسرطان (PO1 CA111412، P009500901). تم دعم العمل من قبل المجلس الأوروبي للبحث (ERC AdG ILCAdapt، 101055309 إلى A.D.) ومن قبل DFG (SFB 1444/427826188 و TRR 241/375876048 إلى A.D.، SPP1937/Di764 /9-2 إلى A.D.). نحن ممتنون لمرافق فرز الخلايا في بنجامين فرانكلين (BFFC) على الدعم في فرز الخلايا. يتم دعم B.F.F.C. من قبل منح أدوات DFG INST 335/597-1 FUGG و INST 335/777-1 FUGG. يتم دعم Y.B. من خلال تمويل من مجلس البحث السويدي ومؤسسة السرطان السويدية. نشكر أعضاء مجموعة CB2M (علم الأحياء الحاسوبي، الإحصاء الحيوي والنمذجة) في مركز مارسيليا-لوميني للمناعة (CIML) على مساعدتهم ودعمهم في التحليل الحيوي والإحصائي للبيانات.
مساهمات المؤلفين
قام كل من L.R. و J.E.M. و B.E. و D.S. و C.V. بإجراء التحليل البيوانفورماتيكي. قام E.V. بتصور المشروع بمساعدة جميع المؤلفين المشاركين الآخرين. شارك كل من L.R. و J.E.M. و B.E. و D.B.-L. و A.B. و N.K.B. و Y.T.B. و R.C. و F.C. و M.C. و D.M.D. و A.D. و Y.D. و M.H. و A.H. و L.L.L. و K.-J.M. و J.S.M. و L.M. و E.N.-M. و L.A.J.O. و C.R. و D.G.R. و S.S. و D.S. و C.V. و E.V. في كتابة المخطوطة. جميع المؤلفين مدرجون بالترتيب الأبجدي، باستثناء L.R. و J.E.M. و B.E.
المصالح المتنافسة
إي. في. و سي. في. هما موظفان في إنات فارما. ك.-ج. م. هو مستشار في فيت ثيرابيوتيكس وفايسليكس ويتلقى دعمًا بحثيًا من فيت ثيرابيوتيكس وأونكوببتيدز لدراسات غير مرتبطة بهذا العمل. يعلن المؤلفون الآخرون عدم وجود مصالح متنافسة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى إريك فيفييه.
معلومات مراجعة الأقران تشكر مجلة نيتشر إيمونولوجي المراجعين المجهولين على مساهمتهم في مراجعة هذا العمل. جيمي دي. ك. ويلسون، بالتعاون مع فريق نيتشر إيمونولوجي.
الشكل البياني الموسع 1 | تصنيف خلايا NK إلى 3 عائلات رئيسية قوي في أطلس خلايا NK في الدم الأخرى. استنادًا إلى مجموعة البيانات 6. أ، تقريب وتوقع متعدد الأبعاد الموحد (UMAP) لخلايا NK في الدم من أطلس خلايا NK في بانكانسر لتانغ وآخرين. المجموعات الفرعية التي تشكل أقل منخلايا NK الدائرة
تم استبعادها مما أسفر عن إجمالي 84,343 خلية NK بشرية من الدم للتحليل. ب، UMAP لخلية NK من الدم من مجموعة البيانات 5 تم تقييمها بتوقيعات NK1 و NK2 و NK3. ج، تصور RidgePlot لتقييم المجموعات المحددة بواسطة تانغ وآخرون.عينات).
الشكل البياني الموسع 2 | تصنيفالخلايا إلى 3 عائلات رئيسية قوية في عينات أخرى من خلايا NK في الدم. بناءً على مجموعة البيانات 4b. أ، UMAP بناءً على عينتين مستقلتين من خلايا NK المفروزة من دم إنسان سليم مع تجمعات تم تحديدها بواسطة التجميع الهرمي غير المراقب وتقييمها باستخدام NK1،
توقيعات NK2 و NK3. ب، ج، رسم نقطي وتصوير UMAP لبعض من أكثر العلامات تمييزًا المعبر عنها على المستوى النسخي من قبل الثلاثة مجموعات الرئيسية من خلايا NK في دم الإنسان.
الشكل البياني الموسع 3 | يمكن تقسيم خلايا NK في دم الإنسان إلى ستة مجموعات فرعية. استنادًا إلى مجموعة البياناتورسم تخطيطي لـ Clustree لاستقرار المجموعات في sc 3 عند دقة تجميع مختلفة (منإلى ). متوسط استقرار sc3 كدالة لمدى دقة تجميع الكتل. ج، تصور UMAP لرسم خلايا NK المفروزة من دم إنسان سليم مع الكتل المحددة بواسطة التجميع الهرمي غير المراقب عند دقة
0.7 (الدقة المثلى وفقًا لاستقرار sc3). د، تصور UMAP للفرعيات من خلايا NK من دم الأفراد الأصحاء مع تحديد المجموعات بواسطة التجميع الهرمي غير المراقب بعد إزالة الخلايا المتكاثرة والسكان الذين يمثلون أقل منمن إجمالي خلايا NK. هـ، تصور UMAP للتعبير عن بروتين NKG2C.رسم بياني دائري يوضح نسبة كل مجموعة فرعية في مجموعة خلايا NK في الدم.
الشكل البياني الموسع 4 | يمكن تقسيم خلايا NK في دم الإنسان إلى ستة المجموعات الفرعية. أ، ج ود-و: استنادًا إلى مجموعة البيانات 1، 2، 3 و4 ب: استنادًا إلى مجموعة البيانات 5. أ، رسم بياني عمودي يوضح نسبة الخلايا داخل كل مجموعة في مجموعات البيانات.عينات) ب، مخطط الكمان لتسجيل جميع CD45الخلايا من مجموعة البيانات 5 مع 13 جينًا مميزًا لخلايا NK البشرية كما حددها كرينير
أ
الشكل البياني الموسع 5 | علامات الاهتمام، الوظائف والتمثيل الغذائي التي تميز تجمعات خلايا NK. استنادًا إلى مجموعة البيانات 1 و2 و3 و4 أ. أ، خريطة حرارية تظهر التعبير التفاضلي للجينات المكونة لثلاث مسارات تمثيل غذائي ذات اهتمام بين مجموعات خلايا NK. مقياس الألوان يعتمد على تعبير الجينات المقاس بمقياس z-score. يتراوح توزيع z-score من -2 (أزرق) إلى 2 (أحمر). ن
الشكل التوضيحي الممتد 6 | تحليل المسار الذي يؤدي من NKint إلى NK1C. استنادًا إلى مجموعة البيانات 1 و2 و3 و4a. أ، خريطة حرارية ديناميكية لتطور أعلى 150 علامة تتطور بشكل أكبر على طول الزمن الزائف للمسار الذي يؤدي من مجموعة NKint إلى مجموعة NK1C.
أ
الشكل البياني الموسع 7| جينات المنظمين الرئيسيين المميزة لكل مجموعة فرعية من خلايا NK في الدم والنشوء المحتمل للسكان الرئيسيين من خلايا NK. استنادًا إلى مجموعة البيانات 1 و2 و3 و4a. أ، خريطة حرارية تظهر التعبير التفاضلي لعوامل النسخ الحقيقية المكتشفة في مجموعات خلايا NK الفرعية. مقياس الألوان يعتمد على درجة z لنشاط التنظيم. تتراوح توزيع درجات z من -2 (أزرق) إلى 2 (أحمر).
الشكل البياني الموسع | توزيع مجموعات خلايا NK1 و NK2 و NK3 في الأنسجة. استنادًا إلى مجموعة البيانات 7. أ، تصور ViolinPlot لدرجة الوحدة للخلايا الفردية التي تم تقييمها بتوقيعات NK1 و NK2 و NK3 من مجموعات ILC. موجود في اللوزتين والرئة و IELs ومجموعة في عناقيد (كما هو محدد في مجموعة البيانات 7). تمثل أشرطة الخطأ الوسيط – الانحراف المعياري. (الرئة: عينات، اللوزة:عينات، IEL:عينات).
الشكل البياني الموسع 9 | توزيع مجموعات خلايا NK1 و NK2 و NK3 في دم مرضى السرطان وفي سرير الورم. أ: استنادًا إلى مجموعة البيانات 1، 2، 3، 4أ. ب-هـ: استنادًا إلى مجموعة البيانات 6.أ، خريطة حرارية توضح دقة نقل التسمية لتوضيح المجموعات المختبرة علىمن الخلايا التي تم الاحتفاظ بها لتدريب المرجع. ب، تصور ViolinPlot لأقصى درجة توقع لكل خلية في عينات NK من الدم في مجموعة البيانات 6. يتم تجميع الخلايا حسب هويتها المتوقعة. تمثل أشرطة الخطأ الوسيط. – الانحراف المعياري. ج، تصور ViolinPlot لأقصى درجة توقع لكل خلية في عينات NK المتسللة إلى الورم من مجموعة البيانات 6. الخلايا يتم تجميعها حسب هويتها المتوقعة. تمثل أشرطة الخطأ الوسيط +/- الانحراف المعياري. د، تصور ViolinPlot لدرجة الوحدة لخلايا NK الفردية في مجموعة البيانات 6 التي تم تقييمها بتوقيعات NK1 و NK2 و NK3. يتم تجميع الخلايا حسب هويتها المتوقعة. تمثل أشرطة الخطأ الوسيط +/- الانحراف المعياري. هـ، تصور ViolinPlot لدرجة الوحدة لخلايا NK المتسللة إلى الورم الفردية في مجموعة البيانات 6 التي تم تقييمها بتوقيعات NK1 و NK2 و NK3. يتم تجميع الخلايا حسب هويتها المتوقعة. تمثل أشرطة الخطأ الوسيط.-الانحراف المعياري. ن
الشكل 10 من البيانات الموسعة | أنماط التعبير الجيني المميزة لمجموعات خلايا NK1 و NK2 و NK3 في دم مرضى السرطان وفي موضع الورم. استنادًا إلى مجموعة البيانات 6.a، خريطة حرارية توضح ارتباط سبيرمان بين NK1،
تعداد خلايا NK2 و NK3 في الأفراد الأصحاء وعبر 22 نوعًا مختلفًا من السرطان في كل من الدم والورم. تمثل أشرطة الخطأ الوسيط +/- الانحراف المعياري.عينات).
محفظة الطبيعة
المؤلف (المؤلفون) المراسلون:إريك فيفييه
آخر تحديث بواسطة المؤلف(ين):16/05/24
ملخص التقرير
تسعى Nature Portfolio إلى تحسين إمكانية تكرار العمل الذي ننشره. يوفر هذا النموذج هيكلًا للاتساق والشفافية في التقرير. لمزيد من المعلومات حول سياسات Nature Portfolio، يرجى الاطلاع على سياسات التحرير وقائمة مراجعة سياسة التحرير.
يرجى عدم ملء أي حقل بـ “غير قابل للتطبيق” أويرجى الرجوع إلى نص المساعدة لمعرفة النص الذي يجب استخدامه إذا كان العنصر غير ذي صلة بدراستك. للتقديم النهائي: يرجى التحقق بعناية من إجاباتك للتأكد من دقتها؛ لن تتمكن من إجراء تغييرات لاحقًا.
الإحصائيات
لجميع التحليلات الإحصائية، تأكد من أن العناصر التالية موجودة في أسطورة الشكل، أسطورة الجدول، النص الرئيسي، أو قسم الطرق.
تم التأكيد
X حجم العينة بالضبط ( ) لكل مجموعة/شرط تجريبي، معطاة كرقم منفصل ووحدة قياس بيان حول ما إذا كانت القياسات قد أُخذت من عينات متميزة أو ما إذا كانت نفس العينة قد تم قياسها عدة مرات X الاختبار الإحصائي المستخدم وما إذا كان أحادي الجانب أو ثنائي الجانب. يجب وصف الاختبارات الشائعة فقط بالاسم؛ وصف التقنيات الأكثر تعقيدًا في قسم الطرق. وصف لجميع المتغيرات المشتركة التي تم اختبارها وصف لأي افتراضات أو تصحيحات، مثل اختبارات الطبيعية والتعديل للمقارنات المتعددة Xوصف كامل للمعلمات الإحصائية بما في ذلك الاتجاه المركزي (مثل المتوسطات) أو تقديرات أساسية أخرى (مثل معامل الانحدار) والتباين (مثل الانحراف المعياري) أو تقديرات مرتبطة بعدم اليقين (مثل فترات الثقة) لاختبار الفرضية الصفرية، إحصائية الاختبار (على سبيل المثال، ) مع فترات الثقة، أحجام التأثير، درجات الحرية وقيمة ملحوظة أعطِالقيم كقيم دقيقة كلما كان ذلك مناسبًا. لتحليل بايزي، معلومات حول اختيار القيم الأولية وإعدادات سلسلة ماركوف مونت كارلو xللتصاميم الهرمية والمعقدة، تحديد المستوى المناسب للاختبارات والتقارير الكاملة عن النتائج X تقديرات أحجام التأثير (مثل حجم تأثير كوهين)بيرسون )، مما يشير إلى كيفية حسابها
تحتوي مجموعتنا على الويب حول الإحصائيات لعلماء الأحياء على مقالات تتناول العديد من النقاط المذكورة أعلاه.
البرمجيات والشيفرة
معلومات السياسة حول توفر كود الكمبيوتر جمع البيانات تحليل البيانات cellranger v6.1.2؛ SingleR v1.4.1؛ batchelor v1.10.0؛ Seurat v4.0.0؛ destiny v3.4.0؛ Monocle3 v1.3.1؛ ade4 v 1.7.16؛ clusterprofiler v 3.18.1؛ Cytosig v0.0.3؛ بالنسبة للمخطوطات التي تستخدم خوارزميات أو برامج مخصصة تكون مركزية في البحث ولكن لم يتم وصفها بعد في الأدبيات المنشورة، يجب أن تكون البرامج متاحة للمحررين والمراجعين. نحن نشجع بشدة على إيداع الشيفرة في مستودع مجتمعي (مثل GitHub). راجع إرشادات Nature Portfolio لتقديم الشيفرة والبرامج لمزيد من المعلومات.
بيانات
معلومات السياسة حول توفر البيانات يجب أن تتضمن جميع المخطوطات بيانًا حول توفر البيانات. يجب أن يوفر هذا البيان المعلومات التالية، حيثما ينطبق:
رموز الانضمام، معرفات فريدة، أو روابط ويب لمجموعات البيانات المتاحة للجمهور
وصف لأي قيود على توفر البيانات
بالنسبة لمجموعات البيانات السريرية أو بيانات الطرف الثالث، يرجى التأكد من أن البيان يتماشى مع سياستنا.
تم إيداع جميع بيانات تسلسل RNA أحادي الخلية وبيانات CITE-seq المستخدمة في هذه الدراسة في أرشيف التعبير الجيني. تم إدراج رمز الوصول لكل مجموعة بيانات مستخدمة في الجدول التكميلي 3. تتوافق مجموعات البيانات من 1 إلى 7 مع أرقام الوصول التالية: GSE119562، GSE130430، GSE184329، GSE197037، GSE164378، GSE212890، GSE240441. تم محاذاة بيانات تسلسل الخلايا المفردة مع الجينوم البشري المرجعي GRCh38. يمكن أيضًا الاطلاع على البيانات المعالجة مسبقًا وتنزيلها من الرابط التالي:https://collections.cellatlas.io/meta-nk
معلومات السياسة حول الدراسات التي تشمل مشاركين بشريين أو بيانات بشرية. انظر أيضًا معلومات السياسة حول الجنس، الهوية/التقديم الجنسي، والتوجه الجنسي والعرق، والعرقية والعنصرية.
التقارير عن الجنس والنوع الاجتماعي
غير متوفر
التقارير عن العرق أو الإثنية أو غيرها من المجموعات الاجتماعية ذات الصلة
غير متوفر
خصائص السكان
غير متوفر
التوظيف
غير متوفر
رقابة الأخلاقيات
غير متوفر
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
التقارير المتخصصة في المجال
يرجى اختيار الخيار أدناه الذي يناسب بحثك بشكل أفضل. إذا لم تكن متأكدًا، اقرأ الأقسام المناسبة قبل اتخاذ قرارك. علوم الحياة Xالعلوم السلوكية والاجتماعيةالعلوم البيئية والتطورية والإيكولوجية
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبياً.
حجم العينة
غير متوفر
استثناءات البيانات
غير متوفر
استنساخ
غير متوفر
التوزيع العشوائي
غير متوفر
مُعَمي
غير متوفر
تصميم دراسة العلوم السلوكية والاجتماعية غير متاح
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبياً.
وصف الدراسة
عينة البحث
استراتيجية أخذ العينات
جمع البيانات
توقيت
استثناءات البيانات
عدم المشاركة
العشوائية
تصميم دراسة العلوم البيئية والتطورية والبيئية غير متاح
يجب على جميع الدراسات الإفصاح عن هذه النقاط حتى عندما يكون الإفصاح سلبياً.
وصف الدراسة
عينة البحث
استراتيجية أخذ العينات
جمع البيانات
التوقيت والمقياس المكاني
استبعاد البيانات
إعادة الإنتاج
العشوائية
عمى
هل شمل البحث العمل الميداني؟
نعم
لا
العمل الميداني، الجمع والنقل غير متاح
ظروف الميدان
الموقع
الوصول والاستيراد/التصدير
اضطراب
التقارير عن مواد وأنظمة وطرق محددة
نحتاج إلى معلومات من المؤلفين حول بعض أنواع المواد والأنظمة التجريبية والأساليب المستخدمة في العديد من الدراسات. هنا، يرجى الإشارة إلى ما إذا كانت كل مادة أو نظام أو طريقة مدرجة ذات صلة بدراستك. إذا لم تكن متأكدًا مما إذا كان عنصر القائمة ينطبق على بحثك، يرجى قراءة القسم المناسب قبل اختيار رد.
الأجسام المضادة
الأجسام المضادة المستخدمة غير متوفر التحقق غير متوفر
خطوط خلايا حقيقية النواة
معلومات السياسة حول خطوط الخلايا والجنس والنوع في البحث
مصدر(s) خط الخلايا
غير متوفر
المصادقة
غير متوفر
تلوث الميكوبلازما
غير متوفر
الخطوط التي يتم التعرف عليها بشكل خاطئ بشكل شائع (انظر سجل ICLAC)
غير متوفر
علم الحفريات وعلم الآثار غير متاح
أصل العينة
إيداع العينة
طرق التأريخ
قم بتحديد هذا المربع لتأكيد أن التواريخ الخام والمعايرة متاحة في الورقة أو في المعلومات التكميلية.
رقابة الأخلاقيات
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
الحيوانات وغيرها من الكائنات البحثية غير متاحة
معلومات السياسة حول الدراسات التي تشمل الحيوانات؛ توجيهات ARRIVE الموصى بها للإبلاغ عن أبحاث الحيوانات، والجنس والنوع في البحث
الحيوانات المخبرية
الحيوانات البرية
التقارير عن الجنس
عينات تم جمعها من الميدان
رقابة الأخلاقيات
يرجى ملاحظة أنه يجب أيضًا تقديم معلومات كاملة حول الموافقة على بروتوكول الدراسة في المخطوطة.
البيانات السريرية
معلومات السياسة حول الدراسات السريرية يجب أن تتوافق جميع المخطوطات مع إرشادات ICMJE لنشر الأبحاث السريرية ويجب أن تتضمن جميع التقديمات قائمة مراجعة CONSORT مكتملة. تسجيل التجارب السريريةغير متوفر
بروتوكول الدراسةغير متوفر
جمع البياناتغير متوفر
النتائجغير متوفر
البحث ذو الاستخدام المزدوج الذي يثير القلق
معلومات السياسة حول أبحاث الاستخدام المزدوج المثيرة للقلق
المخاطر
هل يمكن أن يشكل الاستخدام العرضي أو المتعمد أو المتهور للمواد أو التقنيات الناتجة عن العمل، أو تطبيق المعلومات المقدمة في المخطوطة، تهديدًا لـ:
لا
تجارب مثيرة للقلق
هل يتضمن العمل أيًا من هذه التجارب المثيرة للقلق:
إكسأظهر كيف تجعل اللقاح غير فعال X توفير المقاومة للمضادات الحيوية المفيدة علاجياً أو للعوامل المضادة للفيروسات X تعزيز شدة الفوعة لمرض معدٍ أو جعل غير المعدي فائق الفوعة Xزيادة قابلية انتقال مسببات الأمراض X تغيير نطاق المضيف لمرض معدٍ إكستمكين التهرب من أساليب التشخيص/الكشف X تمكين تسليح عامل بيولوجي أو سم Xأي تركيبة أخرى محتملة الضار من التجارب والعوامل
النباتات
غير متوفر
مخزونات البذور
أنماط جينية نباتية جديدة
المصادقة
تسلسل شريحة الكروماتين
غير متوفر
إيداع البيانات
أكد أن كل من البيانات الخام والبيانات النهائية المعالجة قد تم إيداعها في قاعدة بيانات عامة مثل GEO. أكد أنك قد قمت بإيداع أو توفير الوصول إلى ملفات الرسم البياني (مثل ملفات BED) للقيم المستدعاة.
روابط الوصول إلى البيانات
قد تبقى خاصة قبل النشر. الملفات في تقديم قاعدة البيانات جلسة متصفح الجينوم (على سبيل المثال UCSC)
توضح تسميات المحاور العلامة والفلوركروم المستخدم (مثل CD4-FITC). المقاييس على المحاور مرئية بوضوح. قم بتضمين الأرقام على المحاور فقط للرسم البياني في الأسفل الأيسر من المجموعة (المجموعة هي تحليل للعلامات المتطابقة). جميع الرسوم البيانية هي رسوم بيانية متساوية الارتفاع مع نقاط شاذة أو رسوم بيانية بالألوان الزائفة. تم توفير قيمة عددية لعدد الخلايا أو النسبة المئوية (مع الإحصائيات).
المنهجية
تحضير العينة آلة برمجيات وفرة تجمع الخلايا استراتيجية البوابة
غير متوفر
قم بتحديد هذا المربع لتأكيد أنه تم تقديم شكل يوضح استراتيجية البوابة في المعلومات التكميلية.
Natural killer (NK) cells are innate lymphoid cells (ILCs) contributing to immune responses to microbes and tumors. Historically, their classification hinged on a limited array of surface protein markers. Here, we used single-cell RNA sequencing (scRNA-seq) and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) to dissect the heterogeneity of NK cells. We identified three prominent NK cell subsets in healthy human blood: NK1, NK2 and NK3, further differentiated into six distinct subgroups. Our findings delineate the molecular characteristics, key transcription factors, biological functions, metabolic traits and cytokine responses of each subgroup. These data also suggest two separate ontogenetic origins for NK cells, leading to divergent transcriptional trajectories. Furthermore, we analyzed the distribution of NK cell subsets in the lung, tonsils and intraepithelial lymphocytes isolated from healthy individuals and in 22 tumor types. This standardized terminology aims at fostering clarity and consistency in future research, thereby improving cross-study comparisons.
NK cells are lymphocytes of the innate immune system that belong to the ILC family . NK cells were initially recognized for their capability to identify and eliminate virus-infected and tumor cells independently of prior sensitization, but their multifaceted roles have since been acknowledged. These include not only direct immune responses, but also regulatory functions that influence the adaptive immune system.
The heterogeneity of NK cells is central to their varied functions. Over time, researchers have identified distinct NK cell subgroups, each characterized by unique functional potentials and developmental pathways. These traditional classification methods mainly relied on surface marker expression. Along this line, human NK cells are typically divided into two main categories on the basis of the density of CD56, the isoform of the neural cell adhesion molecule (NCAM) , on the
cell surface: CD56 and CD56 NK cells. Further distinctions in the CD56 population are made on the basis of expression of the CD57 carbohydrate moiety on the cell surface and the absence of CD94-NKG2A and CD62L; cells with these features comprise a more mature subset . Additionally, adaptive NK cells, which make up a distinct NK cell subset demonstrating characteristics akin to those of adaptive immune cells, emerge in certain immune contexts, such as human cytomegalovirus (HCMV) encounters . The advent of advanced single-cell technologies, namely scRNA-seq and CITE-seq, has precipitated a paradigm shift in our understanding of NK cells. These technologies reveal that the NK cell landscape is more intricate and nuanced than previously understood and is marked by subtle distinctions. However, despite these advancements, a unified and standardized description of NK
cell heterogeneity remains elusive. Current definitions vary between laboratories and could lead to discrepancies in scientific literature. This lack of standardized terminology creates major challenges, particularly in translating research across model systems or cohorts of people.
The increasing relevance of NK cells in therapeutic approaches, especially in NK-cell-based immunotherapy against cancer, underscores the necessity of a comprehensive understanding of their heterogeneity. Misinterpretation or neglect of specific NK cell subsets could have substantial implications, potentially affecting the effectiveness or safety of therapies. In this study, we integrated scRNA-seq and CITE-seq data from NK cells ( 718 donors) to establish a baseline classification of NK cells in the blood, lung, tonsil and intraepithelial lymphocytes of healthy individuals, and in 22 tumor types. These data were extracted from 7 distinct publicly available datasets. The accession code of each of the datasets used is listed in Supplementary Table 3 and ‘Data Availability’. This classification is intended to serve as a reference point for future studies, thereby facilitating a more standardized approach to understanding and using NK cells in both research and clinical settings.
Results
Human circulating NK cells comprise three main populations
To systematically and comprehensively categorize human blood NK cells, we used a high-dimensional CITE-seq dataset, encompassing 228 antibody-derived tags (ADTs) and the transcriptional profiles of 5,708 NK cells from eight healthy donors . To effectively integrate both RNAand protein-expression data, we used the weighted nearest neighbors (WNN) method . Initially, we isolated non-proliferating NK cells at the baseline and then reclustered them to elucidate the foundational heterogeneity among blood NK cells. Our analysis revealed three primary NK cell subsets: NK1, NK2 and NK3 (Fig. 1a). We subsequently analyzed their transcriptional (Fig. 1b) and proteomic signatures (Fig. 1c,d).
The NK1 cluster was marked by high protein expression of CD16, CX3CR1, CD161, -integrin and CD38 (Fig. 1c,d). Its transcriptional profile highlighted genes corresponding to these proteins, along with elevated levels of genes encoding cytotoxic molecules (GZMB and PRF1) and markers of NK cell maturity, such as CD160, CD247,ADGRG1,NKG7, FCER1G,LAIR2,SPON2,CLIC3 and CHST2 (Fig.1b).Cells in the NK1 cluster express lower levels of CD56 compared to cells in the NK2 cluster and lower levels of CD57 compared to cells in the NK3 cluster.
The NK2 cluster was defined by high expression of CD56, CD27, CD44, CD54, CD45RB, CD314 (NKG2D) and CD335 (NKp46) and little or no expression of CD16 and CD57 at the protein level (Fig. 1c,d). At the transcriptome level, NK2 cells showed pronounced expression of ribosomal genes (RPL and RPS gene families, Supplementary Table 1) and genes encoding proteins involved in protein synthesis and structural integrity (EEF1A1, TPT1), indicative of heightened protein synthesis and proliferative capacity. This subset also expressed various genes encoding cytokine receptors (IL2RB,IL7R), membrane receptors (KLRC1 encoding NKG2A), transcription factors (TCF7), soluble factors that modulate immune responses (XCL1, XCL2, AREG) and molecules implicated in cell migration and tissue homing (CD44, GPR183, SELL), along with granzyme K (GZMK) (Fig. 1b). Expression of the classic NK cell markers CD57 and CD16 was reduced or absent on NK2 cells compared with NK1 and NK3 cells, indicating that the NK2 population comprised CD56 and early-stage CD56 NK cells.
For the NK3 cluster, the protein-expression profile included CD16, CD57, CD271 (NGFR), CD2, CD18, CD49d and inhibitory killer cell immunoglobulin-like receptors (KIRs) (CD158e, CD158b), with lower expression levels of CD56, NKp30, NKp46, CD161 and CD122 (Fig.1c,d). Transcriptionally, NK3 cells were characterized by the preferential expression of genes encoding transcription factors (PRDM1 (encoding BLIMP1) and ZBTB38), surface molecules and receptors (CD2 and KLRC2 (encoding NKG2C)), CD3 chain transcripts (CD3D, CD3E, CD3G), secreted cytokines and chemokines (IL32, CCL5) and
granzyme (Fig. 1b). Altogether, the combined protein and transcriptional signature of the NK3 cluster closely resembles that of adaptive NK cells, and this cluster’s preferential expression of CD57 and PRDM1 suggests that it also includes mature CD57 CD56 NK cells that are not produced in response to HCMV. We then confirmed the robustness of the classification of human blood NK cells into the NK1, NK2 and NK3 clusters by applying the derived transcriptional signatures to blood NK cells from other available datasets (Extended Data Figs. 1a-c and 2a-c).
The three primary NK cell populations can be split into six subsets
To further delineate the heterogeneity of blood NK cells, we integrated scRNA-seq data from sorted NK cells from 13 healthy individuals across four datasets using the same RNA-seq protocol ( genomics v2 chemistry protocol), therefore including 36,270 cells after high-quality cell filtering. This procedure resulted in the identification of eight well-defined clusters (Extended Data Fig. 3a-d). Three clusters (1,3 and 8) shared an NK3 signature marked by genes such as KLRC2 (encoding NKG2C), CD52, IL32 and GZMH (Supplementary Table 1) and were enriched in cells expressing NKG2C on their surface (Extended Data Fig. 3e). Notably, NK3B (cluster 1) was distinguished by expression of members of the HLA-D gene family, CD74, CCL5, CD7 and KLRC1, and NK3A exhibited enhanced cytotoxic capabilities (through expression of GZMA, GZMB and PRF1) (Supplementary Table 1). Previous data have shown that there is dramatic epigenetic and transcriptional heterogeneity within adaptive NK cells in individuals. This heterogeneity was observed within the same person and across different people, reflecting the clonality of adaptive NK cells rather than functionally distinct programs . We consolidated these three clusters into a single cluster for subsequent analyzes. This led to a final configuration of six clusters (Fig. 2a and Extended Data Fig. 3f). The integrated dataset in our study can be explored at: https://collections.cellatlas.io/meta-nk.
Upon confirming that batch correction was adequate and ensuring that our final cluster designations were free from batch effects both at the dataset (Extended Data Fig. 4a) and donor (Fig. 2b) levels, we scored all CD45 populations from dataset 5 with the previously defined 13-gene signature (CD160, CD244, CHST12, CST7, GNLY, IL18RAP, IL2RB, KLRC1,KLRC3,KLRD1,KLRF1,PRF1,XCL2) that is characteristic of human NK cells, therefore validating the ability of this signature to discriminate NK cells from other subsets (Extended Data Fig. 4b). We also used the 13-gene signature to score the six subsets of NK cells, thus verifying the robustness of this signature across all NK populations (Extended Data Fig. 4c). Then, we evaluated each cluster against established transcriptional signatures for NK1, NK2 and NK3 (Fig. 2c). Three clusters (cluster 2, cluster 4 and cluster 0) exhibited a strong correlation with the NK1 signature, prompting their reclassification as NK1A, NK1B and NK1C, respectively. As expected, the cluster containing the NK3 subpopulations displayed a clear association with the NK3 signature. Notably, a higher NK3 score was observed in NK3 cells derived from individuals (Fig. 2d). Simultaneously, cluster 6 showed a strong correlation with NK2 signature, justifying its classification as NK2. Finally, cluster 5 displayed an intermediate association with both NK1 and NK2 signatures and was thus renamed intermediate NK (NKint). This reassignment is consistent with the gene-expression patterns delineated by pre-defined signatures, facilitating a clearer understanding of the functional landscape within the blood NK cell repertoire.
Analysis of the top 20 defining markers for the six clusters (Fig. 2e) provided a detailed transcriptional profile for each cluster. NK1 cells, as noted in Figure 1b, showed a core signature indicative of chemokines (CCL3,CCL4) and proteins critical for cytotoxicity and its regulation (PRF1, GZMA, GZMB, NKG7), cytoskeletal dynamics (RAC2, ARPC2,CFL1) and cellular adhesion (ITGB2, CALR). NK1 subpopulations expressed unique subset-specific markers. NK1A was characterized by high expression of CXCR4 and the JUN and JUNB, which encode AP-1
Fig. 1 | CITE-seq analysis reveals three prominent subsets of peripheral blood NK cells in healthy individuals. Based on dataset 5. a, WNN and UMAP (WNN_UMAP) visualization of NK cells sorted from healthy human blood with clusters identified by unsupervised hierarchical clustering (based on scRNA-seq and expression of 228 surface proteins). b, Dot plot of the 20 most distinguishing genes expressed for the three major subsets of human blood NK cells. Gene expression was analyzed using the using the two-sided Wilcoxon rank-sum test with Bonferroni adjustment. Ribosomal genes and mitochondrial genes were
removed for clarity. The color indicates the -score scaled gene expression levels. c, Dot plot of the most distinguishing proteins expressed for the three major subsets of human blood NK cells. Protein expression was analyzed using the twosided Wilcoxon rank-sum test with Bonferroni adjustment. Alternative protein names are shown in parentheses. The color indicates the -score scaled protein expression levels. d, WNN_UMAP visualization of the surface expression of the major discriminating proteins expressed at the surface of NK1, NK2 and NK3 cells. a.u., arbitrary units.
Fig. 2 | The three most important NK cell populations can be subdivided into six subgroups. Based on datasets 1-4a.a, UMAP visualization of NK cells sorted from healthy human blood, with clusters identified by unsupervised hierarchical clustering. b, Bar graph showing the proportion of cells within each cluster in all donors. Blue and pink bars are shown under HCMV-positive and HCMV-negative individuals, respectively. c, Violin plot of the scoring of the six NK clusters with respect to established NK1, NK2 and NK3 signatures (
samples). In the violin plots, the point is the median value. The error bars present the median +/-standard deviation. d, Violin plot of the scoring of the NK3 clusters with the NK3 signature in HCMV-positive and HCMV-negative individuals ( samples).e, Dot plot of the 20 most distinguishing genes for each subset of NK cells. Gene expression was analyzed using the two-sided Wilcoxon rank-sum test with Bonferroni adjustment. Ribosomal genes and mitochondrial genes were removed for clarity. The color indicates the -score scaled gene expression levels.
transcription factors; NK1B was distinguished by the surface marker CD160, the long non-coding RNA NEAT1 and the interferon-induced transmembrane protein 1/FITM1; NK1C exhibited enhanced cytotoxic potential, with higher levels of granzyme and perforin transcripts, a distinct expression profile related to prostaglandin metabolism (PTGDS,AKR1C3) and the most active cytoskeletal profile (ACTB,ACTG1, CFL1, RAC2, ARPC2).
NK2 and NKint populations, whose core signatures shared genes encoding chemokines (XCL1, XCL2), granzymes (GZMK), proteins involved in transcription and signaling regulation (NFKBIA,FOS,BTG1, GAS5) and protein synthesis (TPT1, EEF gene family) and surface proteins (CD44, CD74, CD7, KLRC1), also displayed distinct markers. NK2
expressed and , whereas NKint exhibited strong expression of CXCR4,JUNB,ZFP36,IER2 and EIF3G.
NK3, along with the previously defined signature (KLRC2, CCL5, GZMH,IL32, CD3E, CD3D,S100A4,LGALS1), expressed additional markers (CD52, TMSB4X) and shared certain ones with the NK2 population, such as NKG2E (KLRC3) and granulysin (GNLY). This intricate transcriptional landscape underscores the diverse functionalities and regulatory mechanisms at play within the NK cell subsets.
Further investigation into the distribution of these populations among the 13 healthy donors revealed a predominance of NK1 cells, constituting approximately of circulating NK cells. NK2 and NK3 cells represented and , respectively (Supplementary
Table2). A moregranular analysis at the subpopulation level (Fig. 2b and Extended Data Fig.3f) showed that nearly half of the NK1 population was made up of NK1C cells, translating to of all circulating NK cells. The NK2 population represented a minor fraction of total NK cells ( ) as compared to NKint ( of total NK cells). The NK3 cluster, characterized by distinctive expression of markers indicative of both adaptive and terminally mature NK cells (such as PRDM1 and B3GAT1 (encoding an enzyme key for the biosynthesis of CD57)) along with genes uniquely associated with adaptive NK cells (CD3E and ZBTB38) (Fig.1c,d), exhibited considerable variability in its prevalence across individuals (Fig. 2b). Our study’s approach to cluster identification was conducted without consideration of HCMV status. Consequently, to discern the potential impact of HCMV on the NK3 cluster, we conducted separate analyzes of the frequency and predictive scores of NK3 cells in individuals positive for and in those without . Notably, cells in the NK3 cluster were observed in both and donors (Fig. 2b and Extended Data Fig. 3f). However, a higher NK3 score was predominantly observed in NK 3 cells derived from individuals (Fig. 2d). Altogether, these insights provide a quantitative perspective on the distribution and variability of NK cell subsets in the bloodstream.
Molecular features of NK cell subsets
After confirming that the six subpopulations expressed the populations’ defining markers (Extended Data Fig. 4d-f), we computed the scores for the expression levels of various pertinent markers in the NK cell subpopulations, considering only those genes expressed above a defined threshold-detection in more than 5% of circulating NK cellsfor inclusion in the heatmap. This approach yielded a heatmap that, beyond previously identified markers, unveiled additional distinctive characteristics for the subpopulations (Fig. 3a).
We examined cytokine and chemokine production and found that NK1 subpopulations were characterized by robust transcription of CCL4,CCL3,CCL4L2 and IL16, whereas NK2 and NKint cells exhibited predominant transcription of FLT3LG along with and . Finally, NK3 cells were marked by high transcription levels of IFNG,IL32 and CCL5. Differential expression was also apparent in chemotaxis receptors and cell-cell adhesion proteins. In particular, the subsets were distinguished by different patterns of sphingosine-1-phosphate receptors (S1PR1 for NK2, S1PR4 for NK3 and S1PR5 for NK1). Furthermore, the CXC chemokine receptor family had a role in distinguishing the subpopulations (CXCR2 and CX3CR1 for NK1, CXCR3 for NK2 and CXCR4 for both NK1A and NKint). Classic activating receptors of NK cells also exhibited subset-specific expression patterns. High levels of NKp46 (NCR1), CD160, NKp30 (NCR3) and signaling lymphocyte activation molecule receptor genes were characteristic of the NK1 population. NK2 shared a pronounced expression of NKG2D (KLRK1) with NK1A and NK1B. As expected, NKG2C (KLRC2) was predominantly expressed by the NK3 population. Inhibitory-receptor expression profiles diverged between subsets. NK1 cells expressed higher levels of inhibitory KIRs, along with TIM3 (HAVCR2), CD161 (KLRB1) and SIGLEC7, whereas NK2 and NKint cells had an elevated expression level of NKG2A (KLRC1), and NK3 cells appeared have higher expression levels of TIGIT. In terms of cytokine-receptor expression, NK1 populations exhibited heightened expression levels of TGFBR1, TGFBR2 and TGFBR3, as well as IL12RB1, IL1ORB and IL2RG. By contrast, NK2 cells were characterized by a preference for IL2RB (consistent with their strong expression of CD122 at the protein level, Fig. 1c),IL1ORA, and a distinct expression of IL18R and its accessory protein IL18RAP. These findings suggest that the subsets have varying levels of sensitivity to cytokines and chemokines.
The observed cytotoxic profiles were in line with prior observations: NK1 populations exhibited a spectrum of cytotoxic molecules and associated proteins-GZMA, GZMB, PRF1, NKG7, GSDMD and FASLG-whereas NK2 and NKint displayed strong expression of and TRAIL (TNFSF1O). The NK3 subset expressed intermediate levels of cytotoxic molecules and was distinguished by high GZMH expression.
Activation markers also served as differential markers, with CD69 being most prominent in NK1B and NKint, whereas TNFRSF18 (encoding GITR) was more pronounced in NK2. Moreover, classic markers of NK maturation aligned with earlier descriptions: CD56 (NCAM1) was more prevalent in NK2, and CD16 (FCGR3A) expression increased progressively from NK2 to NK1C. In addition, CD11B (ITGAM) levels were higher in NK1B, whereas CD11C (ITGAX) expression was more pronounced in NK2.
Gene Ontology (GO) term enrichment analysis revealed distinct functional specializations within NK subpopulations. NK1 cells were primarily involved in processes such as cell-cell adhesion, activation response, signaling, cytoskeletal activity and cell-mediated cytotoxicity (Fig. 3b). These findings underscore NK1 cells’ have pivotal cytotoxic effector functions. By contrast, NK2 cells were linked to enhanced chemotaxis regulation and leukocyte differentiation, suggestive of their ability to infiltrate tissues and an ongoing maturation process. NK3 cells displayed an upregulation in leukocyte activation. We then explored the functions of the six main NK cell subpopulations (Fig. 3c). NK1B cells were found to be highly responsive to activation through surface receptors, indicating their potential as primary targets in immunotherapeutic strategies. Both NK1A and NK1B populations were significantly enriched for production of tumor necrosis factor (TNF) and cytokines. Notably, the NK1C subset seems to be the most cytotoxic, as indicated by its pronounced cytoskeletal activity and cell-killing signature. An intriguing discovery was the considerable enrichment of tricarboxylic acid (TCA) cycle activities in the NK1C subset, prompting further investigation using single-cell gene set variation analysis (scGSVA).
Clustering analysis based on metabolic-pathway-enrichment analysis separated the NK cell subpopulations into two broad categories, with NK1B and NK1C clustering more closely together and separately from the NK1A, NK2, NK3 and NKint subsets (Fig. 3d). The NK1C subset appears to be ‘hypermetabolic,’ with notable enrichment across the central carbon metabolism, including glycolysis and the TCA cycle, and mitochondrial oxidative phosphorylation (OXPHOS), which could support enhanced cytotoxic activity. Similarly, the NK1B subset also exhibits enrichment in the TCA cycle and OXPHOS, albeit to a lesser extent than does NK1C, in contrast to the other NK cell subpopulations. In addition, NK1B cells are more clearly defined by an enrichment in the mTOR signaling pathway. Finally, cysteine and methionine metabolism are enriched in the NK2 subset. Decomposition analysis of this pathway (Extended Data Fig. 5a) found that the signature was in part driven by high expression levels of lactate dehydrogenase B (LDHB), spermine synthase (SMS) and 3-mercaptopyruvate sulfurtransferase (MPST). LDHB (which preferentially converts lactate to pyruvate and NAD to NADH) seems to be the predominant isoform of lactate dehydrogenase in the NK2 subset; LDHA (which preferentially converts pyruvate to lactate and NADH to NAD ) is highly expressed across the other NK subpopulations. These data suggest that different metabolic profiles underlie NK cell subpopulations and warrant further investigation.
To elucidate the varying responses of the six NK cell subsets to cytokine stimulation, we used the cytokine signaling analyzer (CytoSig) , which predicts the responsiveness of cells to cytokine signals. This analysis indicated that the NK2 population exhibits a pronounced reaction to interleukin-18 (IL-18), consistent with the strong expression of IL18R and its associated protein IL18RAP in this subset (Fig. 3e). NKint, NK1A and NK1B cells seemed to be more susceptible to IL-10 and PGE2, which are signals that can dampen immune responses, in particular in the tumor microenvironment . By contrast, NK1B and NK3 showed a greater response to transforming growth factor beta (TGF- ). TGF- is notorious for its immunosuppressive effects on NK cells , particularly within the tumor microenvironment, where it can hinder their cytotoxic functions . Consistent with previous studies, NK3 showed reduced sensitivity to IL-12 (ref. 9). Finally, NK1C cells demonstrated the most robust response to a suite of cytokines, namely
Fig. 3 | Markers of interest, functions and metabolism characterizing NK cell populations. Based on datasets 1-4a. a, Heatmap showing the differential expression of markers of interest among NK cell subsets. The color scale is based on -score-scaled gene expression. The -score distribution ranges from -2 (blue) to 2 (red). b,c, Selected GO terms showing enrichment in the three major populations (and six major subsets) of healthy human blood NK cells. Benjamini-Hochberg-corrected values were calculated by a hypergeometric test. The black dotted line indicates the significance threshold, which is .
d, Heatmap showing the differential enrichment for selected metabolic pathways among NK cell subsets. The color scale is based on the score of the normalized enrichment score for each metabolic pathway. e, Assessment ( scores) of the response to different cytokines and chemokines in each subgroup, quantified by Cytosig. IFN, interferon; PGE2, prostaglandin E2. values were computed by comparing scores in one NK subset with those in other subsets using two-sided Student’s -tests, and values exceeding 10 were capped at 10 to facilitate visualization.
IL-2, IL-15 and IL-12, that is traditionally associated with the activation and proliferation of NK cells .
Transcriptional trajectories of NK cell subpopulations
The comprehensive examination of six NK cell subsets has revealed not only their distinctive characteristics in terms of markers, cytokine response and functionalities, but also a continuum in their transcriptional landscapes, particularly between the NKint and NK1A subsets. This continuum seems to bridge the transcriptional states of NKint with NK1C. To investigate the potential transcriptional pathways connecting these subsets, we performed a multifaceted analysis.
First, RNA velocity was used to predict the future states of individual cells. This analysis indicated that the majority of NK2 cells would likely persist as NK2 cells, forming a specific NK2 trajectory (Fig. 4a,b). However, it also pointed towards a potential differentiation pathway from NKint into NK1C. This path was characterized by a clear pseudotime progression from NKint to NK1C, transitioning through intermediary populations (NK1A and NK1B) (Fig. 4a,b). NK3 cells, which exhibit clonal-like transcriptional dynamics owing to their interaction with , were excluded from the following trajectory analysis, to avoid having their unique transcriptional behavior skew the findings. Further trajectory analysis using diffusion maps (Destiny ) and trajectory inference (Monocle3 (ref. 20)) corroborated the pathway suggested by the RNA-velocity analysis (Fig. 4c-g). Pseudotime inference clearly outlined a trajectory from NKint to NK1C (Fig. 4d,e,g). Notably, the pseudotime inferred through diffusion-map analysis highlighted a considerablegap between NK2 and NKint (Fig. 4e), reinforcing the concept of two distinct trajectories: one in which NK2 cells predominantly remain NK2, and another leading from NKint to NK1C. This latter trajectory aligns with the metabolism-based unsupervised clustering previously discussed (Fig.3d), which grouped NK1B and NK1C closely together owing to their strong central carbon metabolism activity. By contrast, NKint and NK1A clustered together and exhibited lower metabolic activity.
Building on the Monocle analysis, we homed in on the top 150 genes that exhibited significant changes along the NK cell maturation trajectory from NKint to NK1C, as indicated by a value below 0.05 and a high Moran’s / correlation score. This detailed examination revealed nine gene modules, each of which was sequentially activated as the cells progressed through maturation stages (Extended Data Fig. 6a).
The RNA-velocity analysis also predicts another major developmental pathway, indicating that a considerable portion of NK2 cells is likely to maintain the NK2-cell state. Consistent with this possibility, evidence has been presented suggesting that mouse NK cell populations arise from two distinct lineages: a primary progenitor, known as the early NK cell progenitor (ENKP), and an alternative one, called the innate lymphoid common progenitor (ILCP), which is also capable of giving rise to other types of ILCs . By mapping the transcriptional module scores of human blood ENKPs onto a uniform manifold approximation and projection (UMAP) representation, we observed that both NK1 and NK3 populations displayed transcriptional signatures that closely align with those of NK cells originating from ENKPs (Fig. 5a,b). In addition, the scoring of NK1, NK2 and NK3 subsets on the basis of recently available blood human ILCP signatures revealed that their signature is enriched in NK2s (Fig. 5c,d). Altogether, these observations support the existence of two divergent ontogenic pathways: one for NK1 and NK3, originating from ENKPs, and another for NK2, originating from ILCPs.
To elucidate the master regulatory genes that define the six NK cell subpopulations, we conducted a gene regulatory network analysis using the single-cell regulatory network inference and clustering (SCENIC) workflow . The initial step involved cataloging the regulons identified in our dataset. Each regulon consists of a transcription factor or cofactor and its associated target genes (Extended Data Fig. 7a). Next, we compared our list of regulons with a more robust database of verified transcription factors . This comparison was crucial for excluding unreliable transcription factors and proteins that bind to RNA and
DNA non-specifically, and to focus our analysis solely on bona fide transcription factors. Unsupervised clustering based on regulon activity first revealed two striking features: first, that NK2 branched away from the other subsets, supporting the theory of a distinct ontological origin for NK2. Second, clustering first grouped the NKint and NK1A subpopulations, suggesting that they might represent early stages of NK1 cell differentiation, then grouped the NK1B cells that appeared more differentiated, and finally included the NK1C and NK3 subsets that correspond to more advanced NK cell states.
This sequential differentiation pattern highlights the complex regulatory mechanisms that govern NK cell development and differentiation. Transcription factors that are pivotal in NK cell maturation , such as T-bet (TBX21) and BLIMP1 (PRDM1) , showed a progressive increase across NK2, NKint, NK1A, NK1B to NK1C continuum. Conversely, MYC, TCF7, RUNX2 and GATA3 were predominantly expressed in NK2 subsets, aligning with previous research findings . NK3 was distinguished by robust expression of and , and the continued presence of BLIMP1 (PRDM1). Therefore, the observed expression pattern of key master regulators of maturation substantiates the hypothesis that there are distinct lineages of NK cell progenitors.
Distribution of NK cell subsets in healthy tissue
NK cells are found in tissues in addition to the peripheral blood . The link between circulating NK cells, tissue-infiltrating NK cells and tissue-resident ILCs is an emerging area of research. ILCs vary greatly depending on their environment and the local signals, such as cytokines, that they are exposed to, resulting in distinct ILC profiles in different tissues and diseases . A detailed description of these ILC variations was recently published . We therefore analyzed the scRNA-seq data in the earlier study to investigate the distribution of NK1, NK2 and NK3 subsets in tonsils, lungs and intraepithelial lymphocytes (IELs) isolated from healthy individuals (Fig. 6a). Remarkably, the NK1 and NK2 signatures coincided with the CD56 and CD56 subsets, respectively, identified in lung, tonsil and IELs (Fig. 6b). More specifically, the vast majority of the different subgroups of CD56 and CD56 cells in these tissues could be characterized as NK1 and NK2, respectively (Extended Data Fig. 8a). A few discrete subsets of NK cells in the tonsils (labeled JUNhi, ILC1-like NK, HSP ) and lungs (labeled cyclic NKs, NK HSP, ILC1) could not be assigned to the NK1, NK2 or NK3 subsets. Because we had removed two subsets, cyclic NK cells and NK cells that exhibited characteristics of stress, for the analysis that led to the identification of the NK1, NK2 and NK3 subsets, we expected that some subsets, namely tonsil]JUNhi, tonsil , lung NK HSP and cyclic NKs in the lung, could not be annotated. Notably, the tonsil ILC1-like and lung ILC1 subsets also did not match any of the NK1, NK2 and NK3 profiles, confirming that the later transcriptomic signatures preferentially resemble those of NK cells. However, the partial enrichment of lung ILC1s with NK2 signatures reinforces the idea that there is a shared ontology between these two populations (Extended Data Fig. 8a). Finally, our data show the similarities between the IEL ILC1 and NK3 subsets (Fig. 6a,b and Extended Data Fig. 8a), as illustrated in particular by the strong expression of PRDM1 in both the IEL ILC1 and NK3 subsets. These results indicate that the similarities between IEL ILC1s and NK cells and the divergence of IEL ILC1s from other tissue-resident ILC1s should be reanalyzed.
Distribution of NK cell subsets in cancer
An important point of our analysis was to provide a benchmark for future comparisons with diseased conditions. Therefore, we analyzed the distribution patterns of NK1, NK2 and NK3 cell subsets in 22 cancer types (Fig. 7). To that end, we used a classical label-transfer approach (see Methods). After verifying the accuracy of the method used to annotate the subgroups (Extended Data Fig. 9a-e), we investigated the proportions and transcriptional proximity of these subsets across tissues and cancer types. The distribution of NK cell subsets in these 22 tumors varies by tumor type (Fig. 7a, top panel). This distribution
Fig. 5|Putative ontogeny of the main NK populations. Based on datasets 1-4a. a, UMAP visualization of the module score of individual cells scored with signatures derived from the main NK cell progenitor (ENKP) identified in mice. b, Violin plots of the module scores of individual cells scored with ENKP signatures. Data are shown as median s.d. samples . b,d, In the violin plots the point
is the median value. The error bars present the median -standard deviation.c, UMAP visualization of the module score of individual cells scored with human blood ILCP signatures. d, Violin plots of the module scores of individual cells scored with signatures of human blood ILCPs Data are shown as median s.d. ( samples).
does not correlate with that found in the blood (Fig. 7a, bottom panel). This difference between circulating and tumor-associated NK cells was confirmed by principal component analysis (PCA) (Fig. 7b, PC1), as was the accuracy of NK1, NK2 and NK3 annotation at the tumor bed (Fig. 8a,b, PC2 and PC3). The divergence between NK2 and the other subsets was also confirmed in blood from people with cancer, but the influence of the tumor on the distinction between the NK1, NK2 and NK3 subsets is stronger at the tumor bed than in the blood (Fig. 8a,b). The Spearman correlation calculated across NK groups, type of cancer and tissues and their unsupervised hierarchical clustering confirmed that NK cells first segregate by tissue type and then by the subset to which they belong (Extended Data Fig. 10a). The better grouping of NK1s, NK2s and NK3s in the tumor bed than in the blood also suggests an exacerbated phenotype in tumor conditions.
Discussion
AlthoughscRNA-seq and CITE-seq have considerably advanced exploration of the diversity of human NK cells, definitions of their cell types and
subtypes have varied across publications for several reasons, including differing experimental protocols, data-acquisition methods and analysis tools. This has led to complexity in the literature and even disagreement as to whether certain cell subsets are real or artifacts arising from a particular processing methodology. For example, what seems to be an NK cell subtype could be the result of a stress response that was triggered during cell isolation or by culture conditions. Thus, it is important to establish a consensus framework for a basic set of NK cell types by pooling datasets obtained from multiple laboratories and analyzing them holistically.
The integration of CITE-seq and scRNA-seq NK cell data in our meta-analysis, including data from a total of more than 225,000 NK cells, led us to discern three major NK cell populations in peripheral blood, herein called NK1, NK2 and NK3. These populations are highly enriched in canonical CD56 , canonical CD56 and HCMV-driven adaptive NK cells, respectively. The gene-expression profile of the NK1 population described here aligns with that of the previously described hNK_Bl1 cells, with strong expression of FGFBP2, GZMB, SPON2 and . The gene-expression profile of the NK2 population overlaps
with that of hNK_BI2, defined by high levels of COTL1, CD44, XCL1, LTB and . Notably, the equivalents of NK1 and NK2 have also been characterized in mouse blood: mNK_Bl1 and mNK_BI2, respectively . NK3 cells exhibited a pattern of gene expression overlapping with that of previously described HCMV-driven adaptive NK cells, defined by high levels of KLRC2, CD3E and ZBTB38(refs.12,30). However, although these adaptive genes are the main drivers of the NK3 cluster signature, cells assigned to the NK3 cluster in our study can be also found at lower frequencies in individuals. Therefore, the NK3 cluster defined here
is not limited to HCMV-driven adaptive NK cells. Additionally, considering that the transcriptional signature that is exclusive to adaptive NK cells is limited relative to the level of epigenetic remodeling that these cells undergo, the next step in resolving adaptive NK cell identity might be at the epigenetic level through single-cell ATAC sequencing methods . This highlights the benefits of combining multimodal single-cell approaches when defining distinct cell subsets. Alternatively, adaptive NK cells can also be distinguished from canonical CD56 NK cells on the basis of deficient PLZF expression .
Fig. 7 | Distribution of NK1, NK2 and NK3 cell subsets in the blood of people with cancer and at the tumor bed. Based on datasets and 6. a, Bar graph showing the proportion of the three main NK populations in the blood and at the tumor bed in 22 cancer types ( samples). b, PCA on tumor-infiltrating and blood NK cells, grouped by NK population, cancer conditions and tissue. The PCA is based on the mean expression levels of the 2,000 genes most differentially expressed across tissue and conditions. Groups are colored on the basis of their tissue of origin. PC1 and PC2 explained and of the variance, respectively ( samples). MELA, melanoma; MM, multiple myeloma; RC,
renal carcinoma; FTC, fallopian tube carcinoma; CLL, chronic lymphocytic leukemia; ALL, acute lymphocytic leukemia; PACA, pancreatic carcinoma; THCA,: thyroid carcinoma; LC, lung cancer; HCC, hepatocellular carcinoma; PRAD, prostate cancer; GC, gastric cancer; CRC, colorectal cancer; ICC, intrahepatic cholangiocarcinoma; HNSCC, head and neck squamous cell carcinoma; OV, ovarian cancer; BRCA, breast cancer; UCEC, uterine corpus endometrial carcinoma; ESCA, esophageal cancer; NB, neuroblastoma; NPC, nasopharyngeal carcinoma; BCC, basal cell carcinoma.
The NK1 population could be reliably divided into three subsets, called NK1A, NK1B and NK1C; an NKint population with an intermediate phenotype between NK1 and NK2 was also characterized. In line with our meta-study, recently published scRNA-seq datasets also delineated multiple subclusters. The NK1A subset exhibited the highest expression levels of CXCR4,JUN and JUNB, mirroring the description of the previously published active CD56 (ref.31) and intermediate CD56 (ref. 32) clusters. CD160 and IFITM1 were most abundant in the NK1B subset, a population that was predicted to have the highest response to chemokines and cytokines. The NK1C subset displayed the highest expression levels of PRF1, PFN1, ACTB and NKG7, concordant with the descriptions of mature and terminal CD56 (ref. 31), late CD56 (ref. 32), cluster 2 (ref. 33) and CD56 CD57 NK cells .
Previous studies have reported an intermediate subset linking CD56 (NK2) and CD56 NK cells . This intermediate subset shares a core signature including expression of CD44, XCL1 and GZMK, but is distinguished by elevated expression of CXCR4, in line with our description of NKint. The high expression of KLRC1 (NKG2A) but low expression of CD56 indicates that the NKint population has strong similarities with the early NKG2A cell population . The intermediate expression of CD56, which lies between that of NK2 and NK1 and the lower levels of perforin and granzyme B, as well as the expression of CD27, which was detected at the beginning of the putative transcription pathway connecting NKint to NK1C, also point to a previously defined CD27 CD56 CD94 NK population .
Fig. 8|Distinct transcriptional phenotypes of NK1, NK2 and NK3 cell subsets in the blood of people with cancer and at the tumor bed. Based on datasets 1-4a and 6.a, PCA of blood NK cells, grouped by NK population and cancer conditions. The PCA is based on the mean expression levels of the 2,000 genes most differentially expressed across tissue and conditions. Groups are colored on the basis of NK cell subsets. PC2 and PC3 explained 8.1% and 7% of the
variance, respectively ( samples). b, PCA of tumor-infiltrating NK cells, grouped by NK population and cancer conditions. The PCA is based on the mean expression levels of the 2,000 genes most differentially expressed across tissues and conditions. Groups are colored on the basis of NK cell subsets. PC2 and PC3 explained and of the variance, respectively ( samples).
Our comprehensive trajectory studies revealed two distinct developmental pathways for NK cells. The first trajectory indicates a path through which NK2 cells can maintain their identity; the second involves a progressive maturation process in which NKint cells evolve into the NK1A, NK1B and NK1C stages. For NK2 cells, which seem to derive from ILCPs, it is noteworthy previous research has identified a medullary population of human NK progenitors, termed NKO (ref.37), and that the human NKO signature matches that of ILCPs, suggesting that NKOs might correspond to medullary ILCPs. As with NK1 cells, their maturation is associated with a notable shift in the transcriptional landscape, characterized in particular by an increase in expression
of cytotoxicity-related genes such as and . Concurrently, we observed an escalation in central carbon metabolism activities along this maturation trajectory. This escalation is marked by enhanced glycolysis, TCA cycle activity and OXPHOS. The module score analysis further corroborates these distinct developmental paths, revealing a strong association between the NK2 population and blood ILCPs, as evidenced by their shared signature markers including , CD44, LTB, IL7R and GPR183 (Figs. 2e and 5c,d). Similarly, NK1 and NK3 populations show a pronounced connection to ENKPs, aligning with the observation that Ly49H NK cells in mice, which respond to mouse CMV and are analogous to the human adaptive NK cells included in
the NK3 subset, predominantly originate from ENKPs . At the level of transcription factors, NK1 and NK3 have unique characteristics akin to certain ENKP traits, such as reduced expression of GATA3, EOMES and TCF7 alongside an increased expression of KLF2. This multifaceted analysis underscores the intricate pathways and mechanisms governing NK cell differentiation and functionality.
Our investigation also sheds light on several molecular dimensions of NK cell biology, warranting additional research. A key finding is the distinct profiles of granzymes and perforin across the three primary NK populations. NK1 cells exhibit robust expression of GZMA, GZMB and PRF1, which have been extensively studied . Conversely, NK2 cells predominantly express , known for its role in caspase-independent apoptosis and in controlling autoimmunity . The NK3 subset is characterized by expression of , encoding granzyme H , which also initiates caspase-independent cell death and is effective in inducing rapid apoptosis in tumor cells . This underlines the considerable antitumor potential of NK3 cells. But more remains to be understood about the biology of granzymes, as illustrated by the recent demonstration of the role of granzyme A in triggering production of gasdermin- . Another noteworthy finding was the cytokine profile of the NK2 subset that predominantly transcribed FLT3LG along with and , which encode proteins that attract dendritic cells and promote their antigen-presentation function . The integrin profile of the NK1 subset and the change in their expression along the NK1-maturation trajectory suggest that these integrins could be instrumental in enhancing contact interactions with other cells, regulating NK1 cytotoxicity or facilitating NK1 cells’ entry into tissues (for example, ITGB7 dimerizes with ITGA4 to adhere to MAdCAM-1 for intestinal entry) . A better understanding of the mechanism of expression and regulation of these integrins could have major clinical applications, such as enhancing antitumor immunity in colorectal cancer .
Our results also indicate that there are notable differences in cytokine responses among NK cell subsets. In the context of adoptive NK cell therapy, IL-21 conditioning enhances proliferation, cytotoxicity and production of interferon- and TNF in NK cells . The stronger response of NK1B and NK1C subsets to IL-21 makes them particularly promising for NK cell-based therapies. Additionally, IL-15 has been shown to boost NK cell metabolism and longevity , aligning with the characteristics of the NK1C subset, which exhibits strong metabolic activity and a pronounced response to IL-15. These cytokine responses can be further exploited through the use of cytokine-armed NK cell engagers , enhancing our understanding of subset-specific responses to improve and diversify these new therapeutic approaches.
Our data also show that the gene profiles of the NK1, NK2 and NK3 subsets extend beyond the peripheral blood of healthy individuals, and allowed us to describe the heterogeneity of NK cells in tissues. Indeed, we were able to identify NK1, NK2 and NK3 cell subsets in the lung, tonsils and IELs. The relevance of NK1, NK2 and NK3 profiles was also illustrated by distinguishing between subsets of tissue-infiltrating NK cells and ILC1s.
Notably, we were also able to analyze the distribution of NK cell subsets in 22 cancer types. This showed that the distribution of NK cell subsets varies depending on the tumor type and does not show a strict correlation with the distribution in the blood. The immediate implication of this observation is the relative value of monitoring NK cells in peripheral blood to assess NK cell immunity in people with cancer. Interestingly, the proportion of NK2 cells was increased in most tumors tested, particularly in ovarian cancer, breast cancer, endometrial carcinoma of the uterus, esophageal cancer, neuroblastoma, nasopharyngeal carcinoma and basal cell carcinoma. Although NK cell dysfunction at the tumor bed is well established , no specific profile corresponding to dysfunctional NK cells has been characterized. The reported impairment of the cytolytic capacity of NK cells at the tumor bed is consistent with a shift towards the NK2 profile, in which the expression of molecules involved in the cytolytic machinery is low. It is also important to consider
the metabolic profile of NK2 and its response to cytokines compared with NK1 and NK3. In particular, several therapeutic agents have been developed to stimulate NK cells using cytokines or mutant cytokines , such as NK cell engagers armed with IL-2 variants , and it is crucial to consider the cytokine sensitivity of tumor-associated NK cells.
The NK cell at las presented here not only serves as a reference for future studies on NK cells in blood in health and disease, but is also a tool for understanding NK cell diversity in tissues in relation to circulating NK cells, the ontogeny of NK cells in tissues and the relationship between NK cells and ILC1s in tissues in health and disease.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41590-024-01883-0.
References
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054-1066 (2018).
Lanier, L. L., Testi, R., Bindl, J. & Phillips, J. H. Identity of Leu-19 (CD56) leukocyte differentiation antigen and neural cell adhesion molecule. J. Exp. Med. 169, 2233-2238 (1989).
Phillips, J. H. & Lanier, L. L. A model for the differentiation of human natural killer cells. Studies on the in vitro activation of Leugranular lymphocytes with a natural killer-sensitive tumor cell, K562. J. Exp. Med. 161, 1464-1482 (1985).
Beziat, V., Descours, B., Parizot, C., Debre, P. & Vieillard, V. NK cell terminal differentiation: correlated stepwise decrease of NKG2A and acquisition of KIRs. PLoS ONE 5, e11966 (2010).
Bjorkstrom, N. K. et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56 NK-cell differentiation uncoupled from NK-cell education. Blood 116, 3853-3864 (2010).
Juelke, K. et al. CD62L expression identifies a unique subset of polyfunctional CD56 NK cells. Blood 116, 1299-1307 (2010).
Yu, J. et al. CD94 surface density identifies a functional intermediary between the CD56 and CD56 human NK-cell subsets. Blood 115, 274-281 (2010).
Lee, J. et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirusinfected individuals. Immunity 42, 431-442 (2015).
Schlums, H. et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 42, 443-456 (2015).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587 (2021).
Tang, F. et al. A pan-cancer single-cell panorama of human natural killer cells. Cell 186, 4235-4251 (2023).
Ruckert, T., Lareau, C. A., Mashreghi, M. F., Ludwig, L. S. & Romagnani, C. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol. 23, 1551-1563 (2022).
Crinier, A. et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity 49, 971-986 e975 (2018).
Jiang, P. et al. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nat. Methods 18, 1181-1191 (2021).
Konjevic, G. M., Vuletic, A. M., Mirjacic Martinovic, K. M., Larsen, A. K. & Jurisic, V. B. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine 117, 30-40 (2019).
Patterson, C., Hazime, K. S., Zelenay, S. & Davis, D. M. Prostaglandin impacts multiple stages of the natural killer cell antitumor immune response. Eur. J. Immunol. 54, e2350635 (2023).
Castriconi, R. et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc. Natl Acad. Sci. USA 100, 4120-4125 (2003).
Viel, S. et al. TGF- inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 9, ra19 (2016).
Angerer, P. et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241-1243 (2016).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381-386 (2014).
Ding, Y. Different developmental pathways generate functionally distinct populations of natural killer cells. Nat. Immunol. (in the press).
Jaeger, N. Diversity of group 1 innate lymphoid cells in human tissues. Nature Immunol. (in the press).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083-1086 (2017).
Lambert, S. A. et al. The human transcription factors. Cell 172, 650-665 (2018).
Bi, J. & Wang, X. Molecular regulation of NK cell maturation. Front. Immunol. 11, 1945 (2020).
Gordon, S. M. et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36, 55-67 (2012).
Kallies, A. et al. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood 117, 1869-1879 (2011).
Collins, P. L. et al. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell 176, 348-360 (2019).
Torcellan, T. et al. Circulating NK cells establish tissue residency upon acute infection of skin and mediate accelerated effector responses to secondary infection. Immunity 57, 124-140 (2024).
Holmes, T. D. et al. The transcription factor Bcl11b promotes both canonical and adaptive NK cell differentiation. Sci. Immunol. 6, eabc9801 (2021).
Yang, C. et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 10, 3931 (2019).
Netskar, H. et al. Pan-cancer profiling of tumor-infiltrating natural killer cells through transcriptional reference mapping. Nat. Immunol. (in the press).
Smith, S. L. et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv. 4, 13881406 (2020).
Scheiter, M. et al. Proteome analysis of distinct developmental stages of human natural killer (NK) cells. Mol. Cell Proteom. 12, 1099-1114 (2013).
Melsen, J. E. et al. Single-cell transcriptomics in bone marrow delineates CD56 GranzymeK subset as intermediate stage in NK cell differentiation. Front. Immunol. 13, 1044398 (2022).
Vossen, M. T. et al. CD27 defines phenotypically and functionally different human NK cell subsets. J. Immunol. 180, 3739-3745 (2008).
Crinier, A. et al. Single-cell profiling reveals the trajectories of natural killer cell differentiation in bone marrow and a stress signature induced by acute myeloid leukemia. Cell Mol. Immunol. 18, 1290-1304 (2021).
Lieberman, J. & Fan, Z. Nuclear war: the granzyme A-bomb. Curr. Opin. Immunol. 15, 553-559 (2003).
Trapani, J. A. & Sutton, V. R. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 15, 533-543 (2003).
Guo, Y., Chen, J., Shi, L. & Fan, Z. Valosin-containing protein cleavage by granzyme K accelerates an endoplasmic reticulum stress leading to caspase-independent cytotoxicity of target tumor cells. J. Immunol. 185, 5348-5359 (2010).
Zhao, T. et al. Granzyme K cleaves the nucleosome assembly protein SET to induce single-stranded DNA nicks of target cells. Cell Death Differ. 14, 489-499 (2007).
Jiang, W., Chai, N. R., Maric, D. & Bielekova, B. Unexpected role for granzyme K in CD56 NK cell-mediated immunoregulation of multiple sclerosis. J. Immunol. 187, 781-790 (2011).
Fellows, E., Gil-Parrado, S., Jenne, D. E. & Kurschus, F. C. Natural killer cell-derived human granzyme H induces an alternative, caspaseindependent cell-death program. Blood 110, 544-552 (2007).
Hou, Q. et al. Granzyme H induces apoptosis of target tumor cells characterized by DNA fragmentation and Bid-dependent mitochondrial damage. Mol. Immunol. 45, 1044-1055 (2008).
Zhou, Z. et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368, eaaz7548 (2020).
Barry, K. C. et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 24, 1178-1191 (2018).
Bottcher, J. P. et al. NK cells stimulate recruitment of cDC 1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022-1037 (2018).
Hegewisch-Solloa, E. et al. Differential integrin adhesome expression defines human NK cell residency and developmental stage. J. Immunol. 207, 950-965 (2021).
Zhang, Y. et al. Integrin inhibits colorectal cancer pathogenesis via maintaining antitumor immunity. Cancer Immunol. Res. 9, 967-980 (2021).
Oyer, J. L. et al. Natural killer cells stimulated with PM21 particles expand and biodistribute in vivo: clinical implications for cancer treatment. Cytotherapy 18, 653-663 (2016).
Li, L. et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci. Adv. 9, eadd6997 (2023).
Demaria, O. et al. Antitumor immunity induced by antibody-based natural killer cell engager therapeutics armed with not-alpha IL-2 variant. Cell Rep. Med 3, 100783 (2022).
Dean, I. et al. Rapid functional impairment of natural killer cells following tumor entry limits anti-tumor immunity. Nat. Commun. 15, 683 (2024).
Vivier, E. et al. Natural killer cell therapies. Nature 626, 727-736 (2024).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
(c) The Author(s) 2024 Aix Marseille Université, CNRS, INSERM, Centre d’Immunologie de Marseille-Luminy, Marseille, France. Leiden University Medical Center, Willem-Alexander Children’s Hospital, Laboratory for Pediatric Immunology, Leiden, the Netherlands. Leiden University Medical Center, Department of Immunology, Leiden, the Netherlands. Wellcome Sanger Institute, Wellcome Genome Campus, Cambridge, UK. Biosciences Institute, Newcastle University, Newcastle upon Tyne, UK. T Cell Biology and Development Unit, Laboratory of Genome Integrity, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA. Center for Infectious Medicine, Department of Medicine Huddinge, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden. Department of Medicine Huddinge, Karolinska Institutet, Stockholm, Sweden. Division of Clinical Immunology and Transfusion Medicine, Karolinska University Hospital, Stockholm, Sweden. Sweden Broegelmann Research Laboratory, Department of Clinical Science, University of Bergen, Bergen, Norway. Department of Experimental Medicine (DIMES), University of Genoa, Genoa, Italy. Laboratory of Clinical and Experimental Immunology, IRCCS Istituto Giannina Gaslini, Genova, Italy. Department of Medicine, University of Minnesota, Minneapolis, MN, USA. Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, MO, USA. Department of Life Sciences, Imperial College London, Sir Alexander Fleming Building, South Kensington, London, UK. Laboratory of Innate Immunity, Institute of Microbiology, Infectious Diseases and Immunology (I-MIDI), Campus Benjamin Franklin, Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany. Mucosal and Developmental Immunology, Deutsches Rheuma-Forschungszentrum (DRFZ), an Institute of the Leibniz Association, Berlin, Germany. Department of Dermatology and NIHR Biomedical Research Centre, Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, UK. Department of Immunology & Immunotherapy, The Marc and Jennifer Lipschultz Precision Immunology Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA. Department of Oncological Sciences, The Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA. Department of Microbiology and Immunology and the Parker Institute for Cancer Immunotherapy, University of California, San Francisco, San Francisco, CA, USA. Precision Immunotherapy Alliance, The University of Oslo, Oslo, Norway. The Institute for Cancer Research, Oslo University Hospital, Oslo, Norway. Tumor Immunology Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy. School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin, Ireland. Institute of Medical Immunology, Charité Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt Universität zu Berlin, Berlin, Germany. Innate Immunity, Deutsches Rheuma-Forschungszentrum Berlin (DRFZ), ein Leibniz Institut, Berlin, Germany. Berlin University Alliance, Berlin, Germany. MRC Mitochondrial Biology Unit, University of Cambridge, Cambridge, UK. IRCCS Ospedale Policlinico San Martino, Genova, Italy. Innate Pharma Research Laboratories, Innate Pharma, Marseille, France. APHM, Hôpital de la Timone, Marseille-Immunopôle, Marseille, France. Paris-Saclay Cancer Cluster, Le Kremlin-Bicêtre, France. e-mail: vivier@ciml.univ-mrs.fr
Methods
scRNA-seq data retrieval and preprocessing
For datasets 1-4, scRNA-seq data were retrieved from the studies referenced in Supplementary Table 3. Single-cell sequencing data were aligned with the GRCh38 human reference genome and quantified using Cell Ranger (v6.1.2, 10x Genomics). The preliminary filtered data generated from Cell Ranger were used for downstream filtering and analyzes. First, each sample was examined individually to remove low-quality cells and cell contaminations. Genes detected in more than three cells were retained, and cells expressing fewer than 200 distinct features were removed. Then, for each sample, data were normalized and scaled and cells were clustered following the standard Seurat protocol. The remaining contaminations were identified using the SingleR package (v1.4.1). The detailed metadata (including patient identifier and CMV status) were retrieved from the original studies. For dataset 4 , because the original data were enriched at a ratio of between NKG2C and NKG2C NK cells, the samples were downsampled to match the initial biological ratio of each sample (donor). For datasets 5, 6 and 7, the preprocessed Seurat objects were used.
Batch-effect correction and unsupervised clustering
The samples were then merged. To reduce the batch effect during the clustering process, the 11,965 genes present in each of the samples were kept for the clustering step of the analysis. To account for the difference in sequencing depth between samples, count data were normalized using the Multibatchnorm function with the parameter ‘batch= sample’ of batchelor (v1.10.0). The top 5,000 highly variable genes (HVGs) were identified in each sample using the FindVariableFeatures function in Seurat (v4.0.0). Then, to choose the 2,000 best features to keep for integration, the SelectIntegrationFeatures function of Seurat was used with the parameter setting ‘nfeatures = 2000’. Gene expression was then scaled and centered using the ScaleData function of the Seurat library. Next, PCA was performed on the HVG matrix to reduce noise and reveal the main axes of variation using the RunPCA function, and the top 30 components were retained for analysis. The batch effects were corrected using harmony (v0.1.0) correction algorithm across samples . UMAP dimensional reduction and the shared nearest neighbor graph were calculated on harmony-corrected PCA embeddings. The resolution parameter of the FindClusters function of Seurat was chosen to maximize the mean sc3 stability of the clustering for a granularity ranging from to . The cluster of proliferating cells was identified using the CellCycleScoring function of Seurat. Cells in these clusters were then removed, and a new UMAP visualization was calculated to better visualize the remaining clusters and cells. The final object used for the analysis of datasets is available at: https:// collections.cellatlas.io/meta-nk.
The cluster-specific marker genes were identified using the FindAllMarkers function of Seurat with the parameter ‘method= wilcox, only.pos TRUE, , logfc.threshold .
Scoring with signatures
To score cells with respect to specific signatures, the top 20 cluster-specific markers (calculated as defined above) were entered into the AddModuleScore function. In brief, the mean expression level for each gene in the defined expression programs was calculated for each cell, and the aggregated expression of control gene sets was then subtracted. All analyzed genes were binned on the basis of the mean expression level, and control genes were randomly selected from each bin.
RNA-velocity analysis
To limit batch effects and to take into account the differences in the quality of the samples, the RNA-velocity analysis was carried out separately on the different samples. First, the spliced and unspliced unique molecular identifiers were recounted using the Python package
velocyto (v0.2.2). Subsequently, RNA velocity was estimated using the scvelo function implemented in the R package velociraptor (v3.18). Velocity calculations were restricted to genes previously used for data integration. To facilitate visualization, velocity pseudotimes were projected onto the UMAP coordinates.
Diffusion-mapanalysis
Diffusion-map algorithms implemented in the R package destiny (v3.4.0) were used to infer pseudotime. We removed NK3 cells from the analysis owing to their specific quasi-clonal dynamic. To eliminate the dataset batch effect, the analysis was performed on the biggest dataset (dataset 4) alone. To prevent individual batch effect (at the sample level), the RunFastMNN function implemented in the R package batchelor (v1.10.0) was used. The corrected expression matrix was then used as input to generate diffusion maps using the DiffusionMap function with the parameters set to ‘censor_val , censor_range . The Destiny algorithm automatically identified three ‘root’ cells. We selected the first root cell as the main root because it is located at the start of the directed streamline inferred by RNA velocity, and we then calculated the diffusion pseudotime for all the cells using the DPT function.
Transcriptional trajectory analysis
To confirm the identified transcriptional trajectories and to better understand the changes along the trajectory from NKint to NK1C, we performed pseudotime analysis using Monocle3 (ref. 20) v1.3.1 on every sample from datasets 1-4a together. NK3 and NK2 cells were removed from the analysis, to focus only on the NK1-maturation process. The learn_graph function was run with the parameter ‘ncenter = 150’ to prevent over-branching of the trajectory. The starting point of the trajectory was chosen as the endpoint of the branch in the NKint population, as identified by the RNA-velocity and diffusion-map analyses. The pseudotime was then calculated using the order_cells function. Then, we performed Moran’s / test to detect significant genes showing correlation along the principal graph, selected the top 150 genes with a value and the highest Moran’s / correlation score and plotted their expression ( score) along the pseudotime using the Heatmap function of the ComplexHeatmap library (v2.6.2).
SCENIC analysis
Activated regulons in the different subsets were analyzed by SCENIC (v0.12.1). The data analyzed for the identification of the main six NK subpopulations was used as input for the python implementation of the SCENIC algorithm (pyscenic) . In brief, the gene-gene co-expression relationships between transcription factors and their potential targets were inferred using the grn function with the gene regulatory network reconstruction algorithm ‘grnboost2’ selected. A transcription factor and its target genes together make up a regulon. Then, ctx was used to refine the regulons by using targets that do not have an enrichment for a corresponding motif of the transcription factor, effectively separating direct from indirect targets on the basis of the presence of a cis-regulatory footprint. Next, the command aucell was used to calculate the regulon activity for each cell. Then, the list of regulons was cross-checked with a robust database of verified transcription factors to remove unreliable transcription factors and proteins that bind to RNA and DNA non-specifically, and to limit the analysis to bona fide transcription factors. The regulon activity was then scaled and centered before visualization using the Heatmap function of the ComplexHeatmap library.
ENKP signatures scoring
To score the cells with ENKP-derived NK cell signatures, we extracted the lists of the most representative genes differentially expressed in ENKP-derived NK cells and converted them to their human equivalent.
Because they do not have a human equivalent, and owing to their evolutionary convergence, the genes in the Klra family were replaced with the equivalent human KIR genes . Cells were then scored using the AddModuleScore function on the 20 most significant genes. For ILCP scoring, gene signatures were directly retrieved from the original publication .
GO enrichment analysis
We performed GO enrichment analysis with the clusterProfiler package (v3.18.1). Eight descriptions of interest were chosen among the top 20 most discriminating GO annotations for each cluster. Enrichment scores ( values) for the eight selected GO annotations were calculated by a hypergeometric statistical test with a significance threshold of 0.05 . The data were plotted as the values after Benjamini-Hochberg correction. The significance threshold was set at .
Cytokine responsiveness
To compare cytokine responsiveness across NK cell subsets, we normalized the raw gene counts to -scaled counts per million, followed by mean centralization to enable direct comparison across cells. The data were then analyzed in CytoSig (v0.0.3), with the parameter -s 2 to include a more comprehensive set of signatures. values were derived from the comparison of scores between one NK cell subset and the others, using Student’s -tests.
CITE-seq analysis
For the analysis of CITE-seq data (dataset 5), data that had been preprocessed as described in ref. 10 were used. In brief, after removing the cells with an outlier number of features (genes and or ADTs), HTODemux was used to detect and remove doublets . Then, the batch correction was performed using SCTransform followed by the reciprocal PCA workflow . The same process was used for ADTs, but normalization was performed by CLR transformation within each cell. Then, the PCA was run on both RNA and protein modalities, and the top 40 or top 50 dimensions, respectively, were used to construct -nearest neighbor graphs. This graph was then used as input for the WNN procedure . On the basis of the author’s annotations, NK cells were extracted and proliferating NK cells were removed. Cells that arose after vaccination (on days 3 and 7) were also removed from the analysis, so only untreated cells were kept. The remaining 5,708 NK cells were reclustered with very high granularity ( ). Differentially expressed genes and ADTs were identified using the two-sided Wilcoxon rank-sum test with Bonferroni adjustment calculated using the FindAllMarkers function, as described above. For better visualization of ADT expression on the UMAP, the FeaturePlot function of Seurat was used with the parameters ‘min.cutoff = ‘q01’, max.cutoff = ‘q99″ to prevent outliers from affecting the color scale too strongly.
Optimization of clustering
To determine the most appropriate granularity of clustering in an unbiased way, the clustree package was used to quantify the SC3 stability metric. This metric is used to evaluate clustering stability at various levels of detail . This approach measures how consistently cell groupings hold up across different clustering resolutions and quantifies the stability of each cluster at chosen levels of granularity (Extended Data Fig. 3a). By pinpointing the granularity that maximized SC3 stability, we determined the most reliable clustering configuration (Extended Data Fig. 3b), ultimately settling on a granularity value of 0.7 , which corresponded to 11 clusters (Extended Data Fig. 3c).
Metabolic-pathway analysis
To compare metabolism across subsets, the scGSVA (https://github. com/guokai8/scGSVA), which is the single-cell implementation of GSVA , was used. For this study, only the major metabolic pathways for
which multiple genes were sufficiently detected (for example detected in more than 10% of the cells in at least of the NK populations) were retained.
Enhanced identity prediction through label transfer
To obtain classifications of NK1, NK2 and NK3 cells in dataset 7, we used Seurat’s established protocol for label transfer. Initially, a reference was constructed using datasets , enabling the annotation transfer to dataset 7. Of note, NKint cells were categorized as NK1, reflecting their initial position in the NK1-maturation trajectory. Through the integration and label transfer process (see https://satijalab.org), we examined the method’s reliability and annotation precision by applying it to datasets . We then assessed the labeling accuracy on a subset of each population, which was excluded from reference training (Extended Data Fig. 9a). This evaluation demonstrated a minimum prediction accuracy of across populations, with NK1 identification being particularly accurate ( accuracy). The integrity of label transfer to dataset 7 was further assessed by examining the highest prediction score for individual cells within both blood and tumor environments (Extended Data Fig. 9b,c). This confirmed that the NK1 population was the most confidently predicted. Additionally, we assessed the cells’ congruence with NK1, NK2 and NK3 signatures, grouping them by their predicted identities to confirm the enrichment of each predicted population with its corresponding signature (Extended Data Fig. 9d,e).
PCA and covariance analysis
For PCA analysis on dataset 7, a structured three-step approach was adopted. Initially, cells were categorized by tissue type (tumor or blood) and cancer classification. Following normalization, the top 2,000 variable features within each category were identified using the FindVariableFeatures function. Subsequently, the FindIntegrationFeatures function pinpointed the 2,000 most variable genes across categories. Post-scaling, we computed the mean expression of these 2,000 selected features for cell groups, classified by predicted identity, cancer type and tissue, using Seurat’s AverageExpression function. The data were then scaled again for PCA analysis, which was conducted with the ade4 library. This procedure was replicated for tumor and blood NK cells independently and included a Kruskal-Wallis test to determine the principal components (PC2 and PC3) that best differentiated the three primary NK cell populations in both blood and tumor contexts. For covariance analysis, the same preparatory steps were used, followed by calculation of the Spearman correlation among each group using the cor function. The Pheatmap package was used for the visualization of these correlations.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the scRNA-seq and CITE-seq data used in this study have been deposited in the Gene Expression Omnibus. The accession code for each of the datasets used is listed in Supplementary Table 3. Datasets 1-7 correspond to the following accession numbers, respectively: GSE119562, GSE130430, GSE184329, GSE197037, GSE164378, GSE212890 and GSE240441. Single-cell sequencing data were aligned with the GRCh38 human reference genome. To make our data more accessible to the broader research community, we have created an interactive portal (https://collections.cellatlas.io/meta-nk) designed for easy analysis and visualization of our single-cell data.
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289-1296 (2019).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494-498 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408-1414 (2020).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421-427 (2018).
Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247-2276 (2020).
Barten, R., Torkar, M., Haude, A., Trowsdale, J. & Wilson, M. J. Divergent and convergent evolution of NK-cell receptors. Trends Immunol. 22, 52-57 (2001).
Stoeckius, M. et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888-1902 (2019).
Kiselev, V. Y. et al. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483-486 (2017).
Zappia, L. & Oshlack, A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 7, giyO83 (2018).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
Acknowledgements
E.V.’s laboratory at CIML and Assistance-Publique des Hôpitaux de Marseille is supported by funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (TILC, grant agreement no. 694502 and MInfla-TILC, grant agreement no. 875102), the Agence Nationale de la Recherche including the PIONEER Project (ANR-17-RHUS-0007), MSDAvenir, Innate Pharma and institutional grants awarded to the CIML (INSERM, CNRS and Aix-Marseille University). D.M.D.’s laboratory is funded by the Medical Research Council (MR/WO31698/1) and Wellcome (110091/Z/15/Z). L.M. is funded by Associazione Italiana contro il Cancro (AIRC), 5 xmille project no. 21147. S.S. is funded by Ministero dell’Istruzione, dell’Università e della Ricerca (PRIN 2017WC8499_004) and Fondazione AIRC (AIRC project no. 21147). D.G.R. is supported by funding from the Medical Research Council (MRC) (MC_UU_00028) and Wellcome Trust-Academy
of Medical Sciences (WT-AMS) (SBF009ไ1119). C.R.’s laboratory is supported by the ERC Advanced Grant ‘MEM-CLONK’ (101055157) and the Deutsche Forschungsgemeinschaft (DFG) grants SFB TRR241 BO2 and RO 3565/7-1. K.J.M. was supported by the Research Council of Norway, Center of Excellence: Precision Immunotherapy Alliance (332727) and the US National Cancer Institute (PO1 CA111412, P009500901). The work was supported by the European Research Council (ERC AdG ILCAdapt, 101055309 to A.D.) and by the DFG (SFB 1444/427826188 and TRR 241/375876048 to A.D., SPP1937/Di764 /9-2 to A.D.). We are grateful to the Benjamin Franklin Flow Cytometry Facility (BFFC) for support in cell sorting. B.F.F.C. is supported by DFG Instrument Grants INST 335/597-1 FUGG und INST 335/777-1 FUGG. Y.B. is supported by funding from the Swedish Research Council and Swedish Cancer Foundation. We thank the members of the CB2M (Computational Biology, Biostatistics and Modeling) group at the Marseille-Luminy Immunology Centre (CIML) for their help and support in the bioinformatics and statistical data analysis.
Author contributions
L.R., J.E.M., B.E., D.S. and C.V. performed the bioinformatic analysis. E.V. conceived the project with the help of all other co-authors. L.R., J.E.M., B.E., D.B.-L., A.B., N.K.B., Y.T.B., R.C., F.C., M.C., D.M.D., A.D., Y.D., M.H., A.H., L.L.L., K.-J.M., J.S.M., L.M., E.N.-M., L.A.J.O., C.R., D.G.R., S.S., D.S., C.V. and E.V. participated in the writing of the manuscript. All authors are listed alphabetically, with the exception of L.R., J.E.M. and B.E.
Competing interests
E.V. and C.V. are employees of Innate Pharma. K.-J.M. is a consultant at Fate Therapeutics and Vycellix and receives research support from Fate Therapeutics, Oncopeptides for studies unrelated to this work. The other authors declare no competing interests.
Correspondence and requests for materials should be addressed to Eric Vivier.
Peer review information Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. Jamie D. K. Wilson, in collaboration with the Nature Immunology team.
Extended Data Fig. 1 | The classification of NK cells into 3 main families is robust in other blood NK cell atlas. Based on Dataset 6. a, Uniform Manifold Approximation and Projection (UMAP) of blood NK cells from Tang et al. pancancer NK cells atlas. Subsets constituting less than of circulating NK cells
were excluded which resulted in a total of 84,343 human blood NK cells for analysis. b, UMAP of blood NK cells from Dataset 5 scored with NK1, NK2, and NK3 signatures. c, RidgePlot visualization of the scoring of the clusters defined by Tang et al. ( samples).
Extended Data Fig. 2 | The classification of cells into 3 main families is robust in other blood NK cell samples. Based on Dataset 4b. a, UMAP based on 2 independent samples of NK cells sorted from healthy human blood with clusters identified by unsupervised hierarchical clustering and their scoring with NK1,
NK2, and NK3 signatures. b, c, Dot plot and UMAP visualization of some of the most discriminatory markers expressed at the transcriptional level by the three major subsets of human blood NK cells.
Extended Data Fig. 3 | The NK cells in human blood can be divided into six subgroups. Based on Dataset and 4 a a, Clustree plot of sc 3 stability of clusters at different clustering resolution (from to ). , Mean sc3 stability as a function of the granularity of clustering resolution. c, UMAP visualization of the plot of NK cells sorted from healthy human blood with clusters identified by unsupervised hierarchical clustering at a granularity of
0.7 (optimal resolution according to sc3 stability). d, UMAP visualization of the subpopulations of NK cells from the blood of healthy individuals with clusters identified by unsupervised hierarchical clustering after removing proliferating cells and populations representing less than of total NK cells. e, UMAP visualization of NKG2C protein expression. , Pie chart showing the proportion of each subgroup in the NK cell population in blood.
Extended Data Fig. 4 | The NK cells in human blood can be divided into six
subgroups. a, c and d-f: Based on Based on Dataset 1,2,3 and 4a b: Based on Dataset 5.a, Bar graph showing the proportion of cells within each cluster in the datasets. ( samples) b, Violin plot of the scoring of all CD45 cells from Dataset 5 with the 13 genes characteristic of human NK cells as defined by Crinier
a
Extended Data Fig. 5 | Markers of interest, functions and metabolism characterizing NK cell populations. Based on Dataset 1,2,3 and 4 a. a, Heatmap showing the differential expression of the genes composing three metabolic pathways of interest among NK cell subsets. The color scale is based on z-score-scaled gene expression. The z-score distribution ranges from – 2 (blue) to 2 (red).
a
Extended Data Fig. 6 | Dissection of the trajectory leading from NKint to NK1C. Based on Dataset 1,2,3 and 4a. a, Dynamic heatmap of the evolution of the top 150 markers that evolve most along the pseudotime of the trajectory leading from the NKint subset to the NK1C subset.
a
Extended Data Fig. 7| Master regulators genes characteristic for each subset of NK cells in the blood and putative ontogeny of the main NK populations. Based on Dataset 1,2,3 and 4a. a, Heatmap showing the differential expression of true transcription factors detected in NK cell subsets. The color scale is based on the z-score of the regulon activity. The z-score distribution ranges from – 2 (blue) to 2 (red).
Extended Data Fig. | Distribution of NK1, NK2 and NK3 cell subsets in tissues. Based on Dataset 7. a, ViolinPlot visualization of the module score of individual cells scored with signatures of NK1, NK2 and NK3 of ILC populations
present in tonsil, lung and IELs and grouped by clusters (as defined in Dataset 7). The error bars present the median – standard deviation. (Lung: samples, Tonsil: samples, IEL: samples).
Extended Data Fig. 9 | Distribution of NK1, NK2 and NK3 cell subsets in the blood of cancer patients and at the tumor bed. a: Based on dataset 1,2,3,4a. b-e: Based on dataset 6.a, Heatmap depicting accuracy of the label transfer for subset annotation tested on of the cells heldout to train the reference. b, ViolinPlot visualization of the maximum prediction score per cell in the blood NK samples of Dataset 6 . Cells are grouped by their predicted identity. The error bars present the median – standard deviation. c, ViolinPlot visualization of the maximum prediction score per cell in the tumor-infiltrated NK samples of Dataset 6. Cells
are grouped by their predicted identity. The error bars present the median +/standard deviation. d, ViolinPlot visualization of the module score of individual blood NK cells of Dataset 6 scored with signatures of NK1, NK2 and NK3. Cells are grouped by their predicted identity. The error bars present the median +/standard deviation. e, ViolinPlot visualization of the module score of individual tumor-infiltrated NK cells of Dataset 6 scored with signatures of NK1, NK2 and NK3. Cells are grouped by their predicted identity. The error bars present the median -standard deviation.
a
Extended Data Fig. 10 | Distinct transcriptionnal phenotypes of NK1, NK2 and NK3 cell subsets in the blood of cancer patients and at the tumor bed.
Based on dataset 6.a, Heatmap showing the Spearman correlation between NK1,
NK2 and NK3 populations in healthy individuals and across 22 different cancer types in both blood and tumor. The error bars present the median +/-standard deviation. ( samples).
natureportfolio
Corresponding author(s): Eric Vivier
Last updated by author(s): 16/05/24
Reporting Summary
Nature Portfolio wishes to improve the reproducibility of the work that we publish. This form provides structure for consistency and transparency in reporting. For further information on Nature Portfolio policies, see our Editorial Policies and the Editorial Policy Checklist.
Please do not complete any field with “not applicable” or . Refer to the help text for what text to use if an item is not relevant to your study. For final submission: please carefully check your responses for accuracy; you will not be able to make changes later.
Statistics
For all statistical analyses, confirm that the following items are present in the figure legend, table legend, main text, or Methods section.
Confirmed
X The exact sample size ( ) for each experimental group/condition, given as a discrete number and unit of measurement A statement on whether measurements were taken from distinct samples or whether the same sample was measured repeatedly X The statistical test(s) used AND whether they are one- or two-sided Only common tests should be described solely by name; describe more complex techniques in the Methods section. A description of all covariates tested X A description of any assumptions or corrections, such as tests of normality and adjustment for multiple comparisons
X A full description of the statistical parameters including central tendency (e.g. means) or other basic estimates (e.g. regression coefficient) AND variation (e.g. standard deviation) or associated estimates of uncertainty (e.g. confidence intervals) For null hypothesis testing, the test statistic (e.g. ) with confidence intervals, effect sizes, degrees of freedom and value noted Give values as exact values whenever suitable. For Bayesian analysis, information on the choice of priors and Markov chain Monte Carlo settings
x For hierarchical and complex designs, identification of the appropriate level for tests and full reporting of outcomes
X Estimates of effect sizes (e.g. Cohen’s , Pearson’s ), indicating how they were calculated
Our web collection on statistics for biologists contains articles on many of the points above.
Software and code
Policy information about availability of computer code
Data collection
Data analysis cellranger v6.1.2; SingleR v1.4.1; batchelor v1.10.0; Seurat v4.0.0; destiny v3.4.0; Monocle3 v1.3.1; ade4 v 1.7.16; clusterprofiler v 3.18.1; Cytosig v0.0.3; For manuscripts utilizing custom algorithms or software that are central to the research but not yet described in published literature, software must be made available to editors and reviewers. We strongly encourage code deposition in a community repository (e.g. GitHub). See the Nature Portfolio guidelines for submitting code & software for further information.
Data
Policy information about availability of data
All manuscripts must include a data availability statement. This statement should provide the following information, where applicable:
Accession codes, unique identifiers, or web links for publicly available datasets
A description of any restrictions on data availability
For clinical datasets or third party data, please ensure that the statement adheres to our policy
All the scRNA-seq and CITE-seq data used in this study have been deposited in the Gene Expression Omnibus. The accession code of each of the datasets used are listed in the Supplementary Table 3. Datasets 1 to 7 are respectively correspond to the following accession numbers: GSE119562, GSE130430, GSE184329, GSE197037, GSE164378, GSE212890, GSE240441. Single-cell sequencing data were aligned with the GRCh38 human reference genome. The preprocessed data can also be consulted and downloaded at the following link: https://collections.cellatlas.io/meta-nk
Policy information about studies with human participants or human data. See also policy information about sex, gender (identity/presentation), and sexual orientation and race, ethnicity and racism.
Reporting on sex and gender
n/a
Reporting on race, ethnicity, or other socially relevant groupings
n/a
Population characteristics
n/a
Recruitment
n/a
Ethics oversight
n/a
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Field-specific reporting
Please select the one below that is the best fit for your research. If you are not sure, read the appropriate sections before making your selection.
X Life sciences Behavioural & social sciences Ecological, evolutionary & environmental sciences
All studies must disclose on these points even when the disclosure is negative.
Sample size
n/a
Data exclusions
n/a
Replication
n/a
Randomization
n/a
Blinding
n/a
Behavioural & social sciences study design n/a
All studies must disclose on these points even when the disclosure is negative.
Study description
Research sample
Sampling strategy
Data collection
Timing
Data exclusions
Non-participation
Randomization
Ecological, evolutionary & environmental sciences study design n/a
All studies must disclose on these points even when the disclosure is negative.
Study description
Research sample
Sampling strategy
Data collection
Timing and spatial scale
Data exclusions
Reproducibility
Randomization
Blinding
Did the study involve field work?
Yes
No
Field work, collection and transport n/a
Field conditions
Location
Access & import/export
Disturbance
Reporting for specific materials, systems and methods
We require information from authors about some types of materials, experimental systems and methods used in many studies. Here, indicate whether each material, system or method listed is relevant to your study. If you are not sure if a list item applies to your research, read the appropriate section before selecting a response.
Antibodies
Antibodies used
n/a
Validation
n/a
Eukaryotic cell lines
Policy information about cell lines and Sex and Gender in Research
Cell line source(s)
n/a
Authentication
n/a
Mycoplasma contamination
n/a
Commonly misidentified lines (See ICLAC register)
n/a
Palaeontology and Archaeology n/a
Specimen provenance
Specimen deposition
Dating methods
Tick this box to confirm that the raw and calibrated dates are available in the paper or in Supplementary Information.
Ethics oversight
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Animals and other research organisms n/a
Policy information about studies involving animals; ARRIVE guidelines recommended for reporting animal research, and Sex and Gender in Research
Laboratory animals
Wild animals
Reporting on sex
Field-collected samples
Ethics oversight
Note that full information on the approval of the study protocol must also be provided in the manuscript.
Clinical data
Policy information about clinical studies
All manuscripts should comply with the ICMJE guidelines for publication of clinical research and a completed CONSORT checklist must be included with all submissions.
Clinical trial registration n/a
Study protocol n/a
Data collection n/a
Outcomes n/a
Dual use research of concern
Policy information about dual use research of concern
Hazards
Could the accidental, deliberate or reckless misuse of agents or technologies generated in the work, or the application of information presented in the manuscript, pose a threat to:
No
Experiments of concern
Does the work involve any of these experiments of concern:
X Demonstrate how to render a vaccine ineffective
X Confer resistance to therapeutically useful antibiotics or antiviral agents
X Enhance the virulence of a pathogen or render a nonpathogen virulent
X Increase transmissibility of a pathogen
X Alter the host range of a pathogen
X Enable evasion of diagnostic/detection modalities
X Enable the weaponization of a biological agent or toxin
X Any other potentially harmful combination of experiments and agents
Plants
n/a
Seed stocks
Novel plant genotypes
Authentication
ChIP-seq
n/a
Data deposition
Confirm that both raw and final processed data have been deposited in a public database such as GEO. Confirm that you have deposited or provided access to graph files (e.g. BED files) for the called peaks.
Data access links
May remain private before publication.
Files in database submission
Genome browser session
(e.g. UCSC)
Methodology
n/a
Replicates
Sequencing depth
Antibodies
Peak calling parameters
Data quality
Software
Plots
Confirm that:
The axis labels state the marker and fluorochrome used (e.g. CD4-FITC). The axis scales are clearly visible. Include numbers along axes only for bottom left plot of group (a ‘group’ is an analysis of identical markers). All plots are contour plots with outliers or pseudocolor plots. A numerical value for number of cells or percentage (with statistics) is provided.
Methodology
Sample preparation
Instrument
Software
Cell population abundance
Gating strategy
n/a
Tick this box to confirm that a figure exemplifying the gating strategy is provided in the Supplementary Information.
Magnetic resonance imaging n/a
Experimental design
Design type
Design specifications
Statistic type for inference
(See Eklund et al. 2016)