DOI: https://doi.org/10.1038/s41467-024-47348-8
PMID: https://pubmed.ncbi.nlm.nih.gov/38684659
تاريخ النشر: 2024-04-29
تحويل الأراضي إلى الزراعة يؤدي إلى تجانس تصنيفي لمجتمعات الميكروبات التربة على مستوى العالم
تم القبول: 28 مارس 2024
نُشر على الإنترنت: 29 أبريل 2024
الملخص
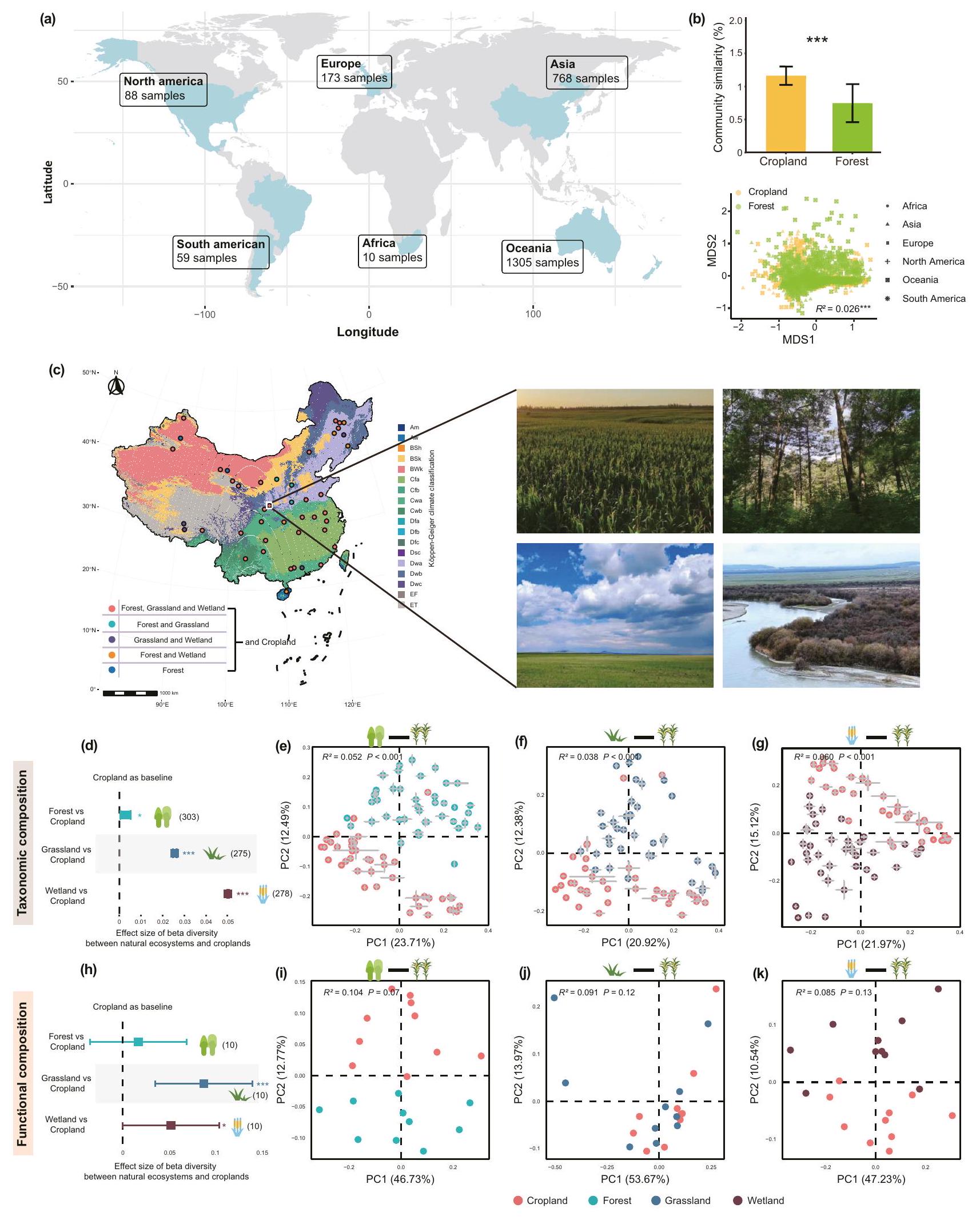
تساهم الزراعة في تراجع تنوع الأنواع المحلية وفي تجانس الكائنات الحية فوق وتحت الأرض. هنا، نقوم بإجراء مسح قاري باستخدام 1185 عينة من التربة ونقارن المجتمعات الميكروبية من النظم البيئية الطبيعية (الغابات، المراعي، والأراضي الرطبة) مع الأراضي الزراعية المحولة. نجمع نتائج مسحنا القاري مع تحليل ميتا عالمي للبيانات المتاحة للتسلسل التي تغطي أكثر من 2400 عينة عبر ست قارات. تظهر نتائجنا المجمعة أن تحويل الأراضي إلى أراضٍ زراعية يؤدي إلى تجانس تصنيفي ووظيفي لبكتيريا التربة، مدفوعًا بشكل رئيسي بزيادة النطاقات الجغرافية للأنواع في الأراضي الزراعية. نجد أن
مقاييس
النتائج
الزراعة تسبب تجانس حيوي في الملفات التصنيفية والوظيفية
أقل في الأراضي الزراعية مقارنة بالتربة الطبيعية (الشكل 1d، h والأشكال التكميلية 1b، 2) مما يوضح أن تربة الأراضي الزراعية أكثر تشابهًا من تربة النظم البيئية الطبيعية المزدوجة. على سبيل المثال،
آثار الزراعة على الأنماط البكتيرية المحددة والوظائف
s.e.m. من أحجام التأثير المقدرة. يتم عرض حجم العينة بعدد أزواج البيانات لكل مجموعة. تعتمد الأهمية الإحصائية على
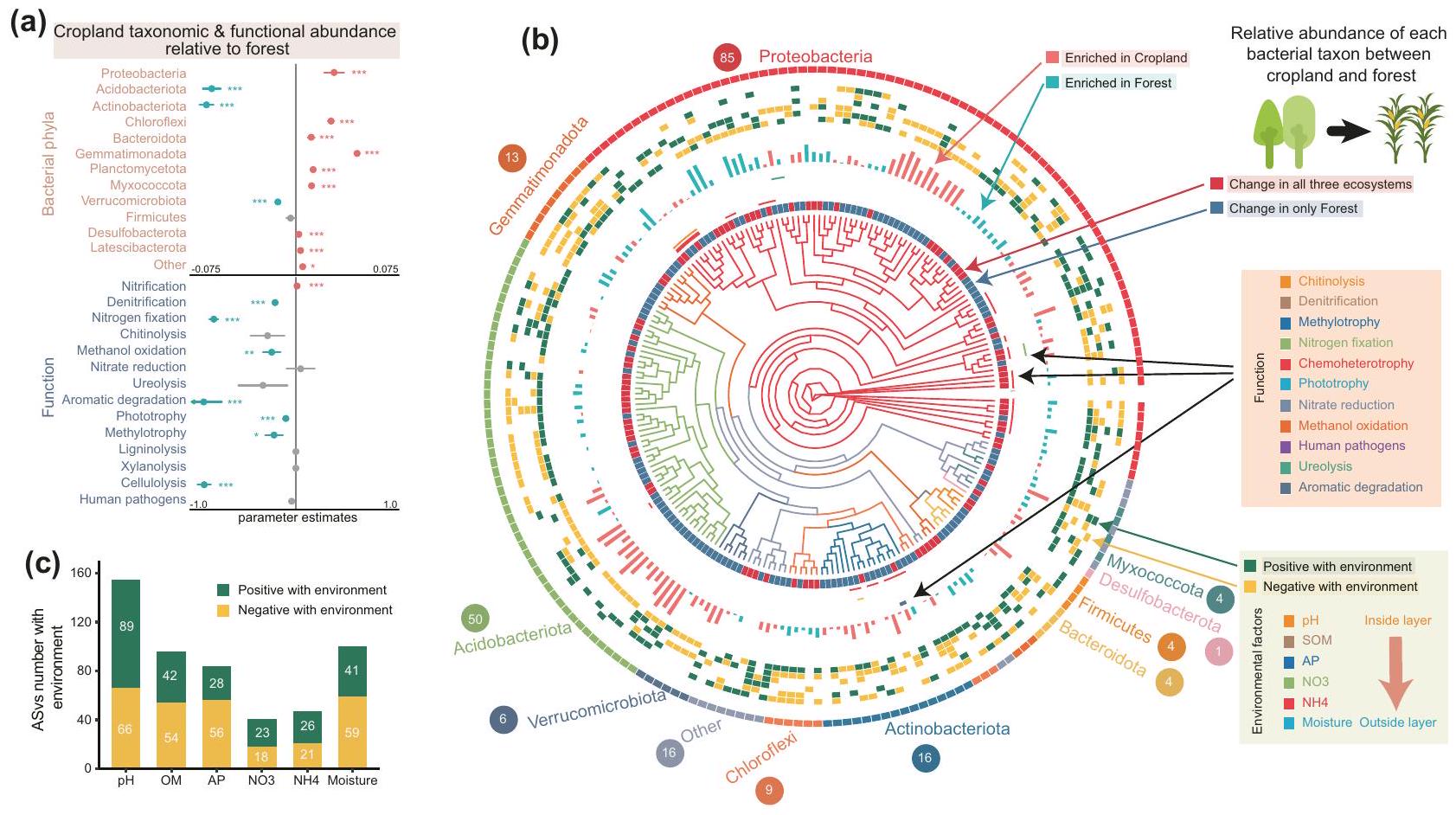
الحلقة الثانية تمثل الأنماط الفيلوجينية التي زادت أو انخفضت بشكل كبير تحت آثار الزراعة. تمثل الكتل الملونة في الحلقة الثالثة أداء وظيفة معينة بواسطة على الأقل نمط فيلوجيني واحد. تمثل الأشرطة في الحلقة الرابعة أحجام التأثير الإيجابية والسلبية لآثار الزراعة على الوفرة النسبية للأنماط الفيلوجينية. تمثل الكتل الملونة في الحلقة الخامسة ارتباط سبيرمان بين الوفرة النسبية للأنماط الفيلوجينية وخصائص التربة الفيزيائية والكيميائية (من الحلقات الداخلية إلى الخارجية:
تم التأثير عليها من قبل التحويل الزراعي (
الاتجاه مقارنةً مع الأنظمة البيئية الطبيعية الثلاثة الأخرى عند التجميع عبر الفئات الوظيفية من المستوى 3 من خلال تعليقات COG (الشكل التوضيحي 8). زادت الفئات الوظيفية “الترجمة، هيكل الريبوسوم وتكوين الريبوسوم” و”الهيكل الخلوي” بينما انخفضت “آليات الدفاع” في الأراضي الزراعية. ومع ذلك، أظهرت بعض الجينات الخاصة بتفكيك الكربون تأثيرات غير متسقة عند التحويل الزراعي (بعض الجينات كانت غنية أو محذوفة)، بينما لم يتم الكشف عن اختلافات كبيرة في عملية التمثيل الغذائي للكربون بشكل عام تحت استخدام الأراضي الزراعية (الشكل 3 أ، ب). من المرجح أن يكون ذلك بسبب الازدواجية العالية للوظائف المنتشرة على نطاق واسع، وبالتالي
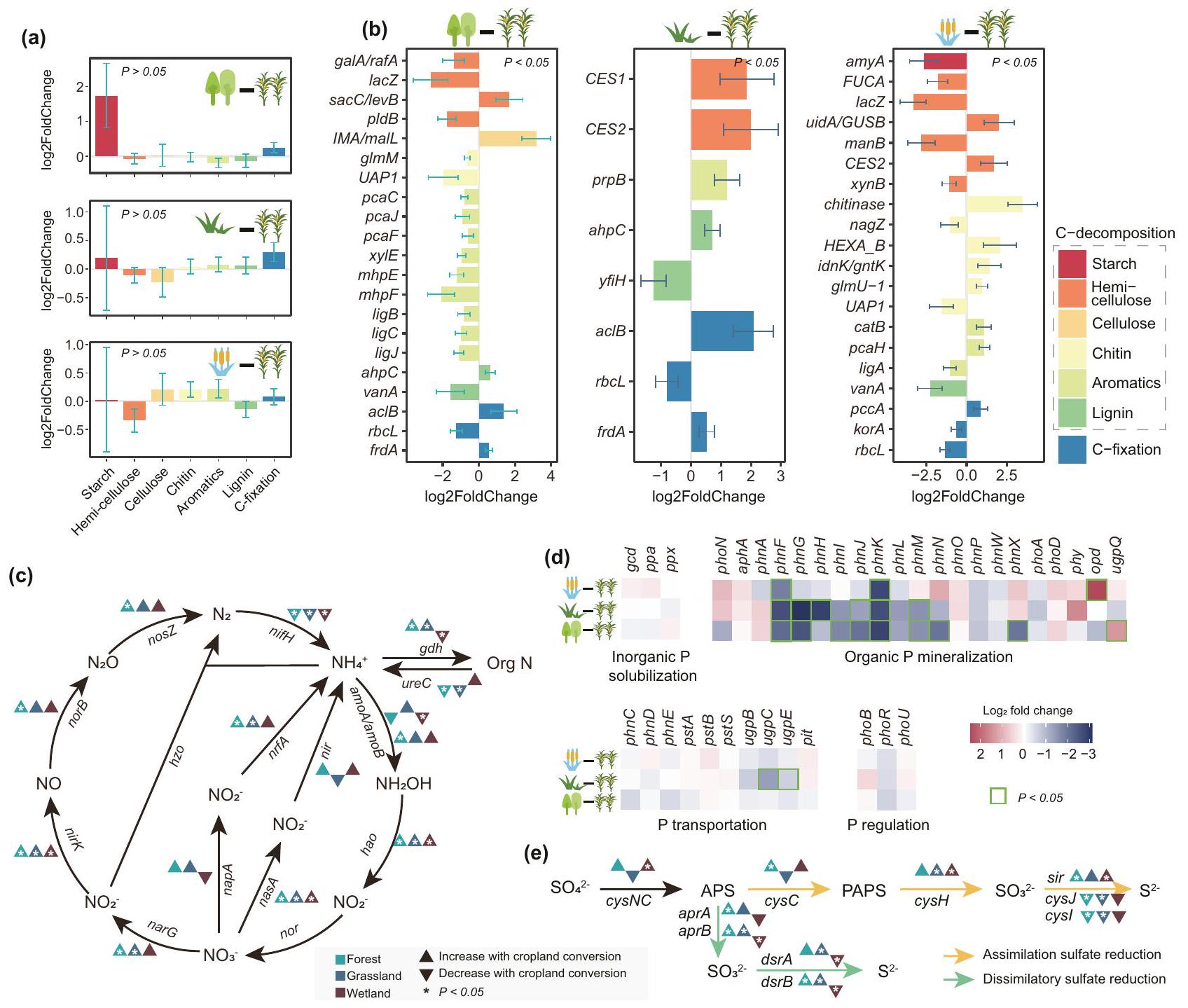
نجوم. د دورة الفوسفور من تسلسل الميتاجينوم. تمثل خرائط الحرارة
الحماية من التغيرات التصنيفية الناتجة عن استخدام الأراضي الزراعية. في الواقع، فإن الوظائف العامة مثل التنفس، والتحلل الكربوني الشامل، والتمثيل الحيوي غالبًا ما تبدو أكثر استقرارًا تجاه التغيرات في التركيب التصنيفي الميكروبي مقارنةً بالوظائف الأيضية الضيقة مثل تحلل الركائز المحددة.
جينات اختزال الكبريتات غير المتجانسة (apr و
الآليات الكامنة وراء تغير المجتمعات البكتيرية
يسمح بانتشار الأنواع الساكنة ويعجل بزيادة تنوع الأنواع
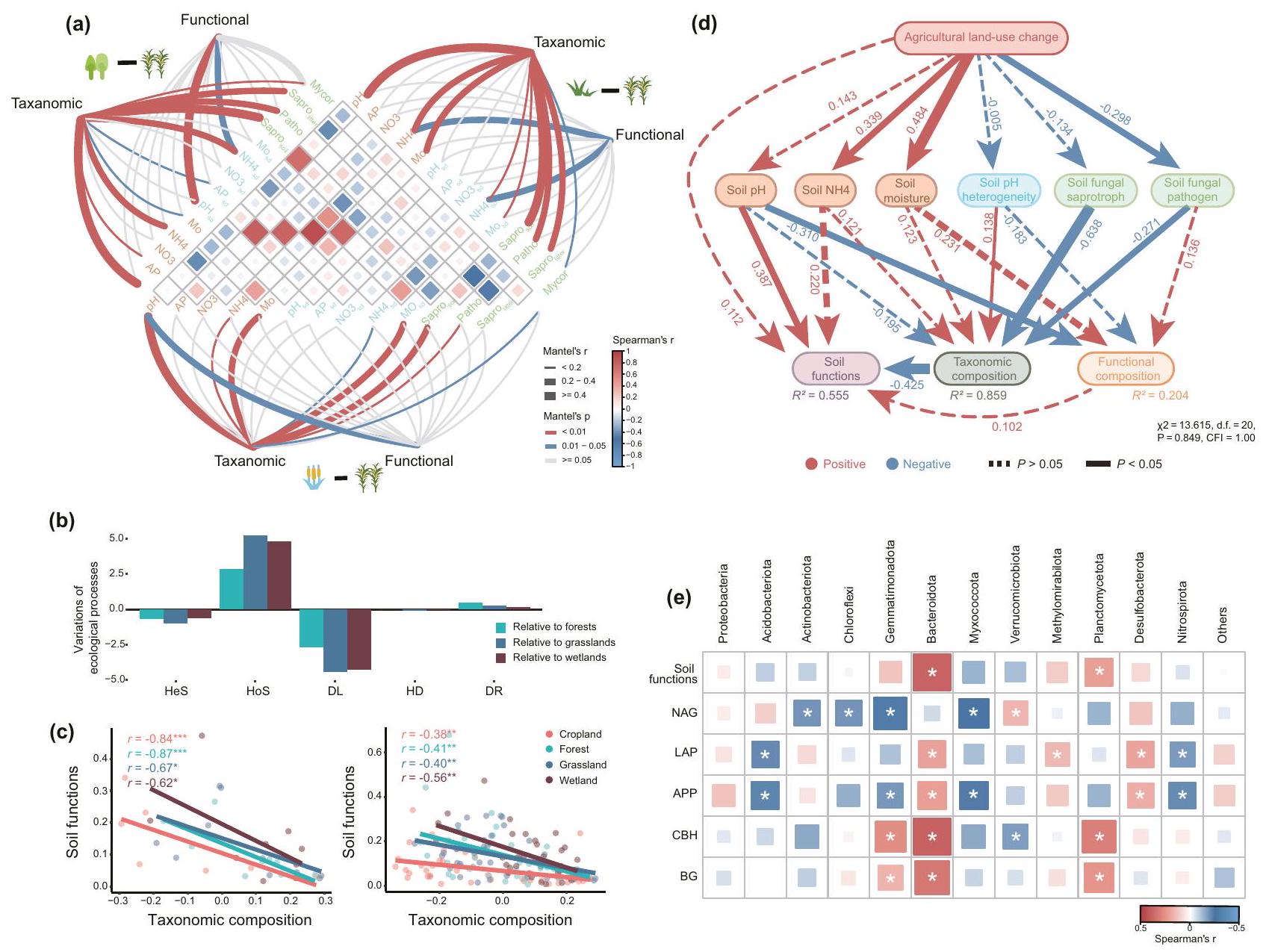
تشير الخطوط إلى أهمية (
مرتبط بتكوين التصنيف البكتيري (الشكل 4a، d). لعب تصفية الرقم الهيدروجيني للتربة الدور الأقوى في تشكيل التركيب التصنيفي والوظيفي (الشكل 4a، d). علاوة على ذلك، تشير العلاقة بين المجتمعات الفطرية والبكتيرية إلى دور مهم للتفاعلات الحيوية في التوسط في تغييرات التركيب الميكروبي الناتجة عن الزراعة. على الرغم من أن هذه المتغيرات يمكن أن تفسر
روابط بين المجتمعات البكتيرية ووظائف التربة
نقاش
الناجم عن التحويل الزراعي
التضاريس
طرق
استطلاع قاري وأخذ عينات
حيث تم تجميد مجموعة واحدة عند
تحليل ميتا على نطاق عالمي
متغيرات البيئة التربة
أنشطة إنزيمات التربة
تسلسل الميتاجينوم بالأسلوب الشامل
لتسلسل لتحليل التغيرات في الإمكانات الوظيفية لمجتمع الميكروبات (
تقدير العمليات البيئية
التحليلات الإحصائية
ملخص التقرير
توفر البيانات
توفر الشيفرة
References
- Benton, T. G., Bieg, C., Harwatt, H., Pudasaini, R. & Wellesley, L. in Three Levers for Food System Transformation in Support of Nature 02-03 (Chatham House, 2021).
- Tsiafouli, M. A. et al. Intensive agriculture reduces soil biodiversity across Europe. Glob. Change Biol. 21, 973-985 (2015).
- Wang, Y. et al. Forest conversion to plantations: a meta-analysis of consequences for soil and microbial properties and functions. Glob. Change Biol. 27, 5643-5656 (2021).
- Gossner, M. M. et al. Land-use intensification causes multitrophic homogenization of grassland communities. Nature 540, 266-269 (2016).
- Roswell, M., Dushoff, J. & Winfree, R. A conceptual guide to measuring species diversity. Oikos 130, 321-338 (2021).
- Hooper, D. U. et al. A global synthesis reveals biodiversity loss as a major driver of ecosystem change. Nature 486, 105-108 (2012).
- Johnson, Christopher et al. Biodiversity losses and conservation responses in the Anthropocene. Science 356, 270-275 (2017).
- Lees, A. C., Attwood, S., Barlow, J. & Phalan, B. Biodiversity scientists must fight the creeping rise of extinction denial. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-020-01285-z (2020).
- Solar, R. Rd. C. et al. How pervasive is biotic homogenization in human-modified tropical forest landscapes? Ecol. Lett. 18, 1108-1118 (2015).
- Daru, B. H. et al. Widespread homogenization of plant communities in the Anthropocene. Nat. Commun. 12, 6983 (2021).
- Finderup Nielsen, T., Sand-Jensen, K., Dornelas, M. & Bruun, H. H. More is less: net gain in species richness, but biotic homogenization over 140 years. Ecol. Lett. 22, 1650-1657 (2019).
- Olden, J. D. & Rooney, T. P. On defining and quantifying biotic homogenization. Glob. Ecol. Biogeogr. 15, 113-120 (2006).
- Karp, D. S. et al. Intensive agriculture erodes beta-diversity at large scales. Ecol. Lett. 15, 963-970 (2012).
- McKinney, M. L. & Lockwood, J. L. Biotic homogenization: a few winners replacing many losers in the next mass extinction. Trends Ecol. Evol. 14, 450-453 (1999).
- Gámez-Virués, S. et al. Landscape simplification filters species traits and drives biotic homogenization. Nat. Commun. 6, 8568 (2015).
- Hautier, Y. et al. Local loss and spatial homogenization of plant diversity reduce ecosystem multifunctionality. Nat. Ecol. Evol. 2, 50-56 (2018).
- McKinney, M. L. Urbanization as a major cause of biotic homogenization. Biol. Conserv. 127, 247-260 (2006).
- Newbold, T. et al. Climate and land-use change homogenise terrestrial biodiversity, with consequences for ecosystem functioning and human well-being. Emerg. Top. Life Sci. 3, 207-219 (2019).
- Nowakowski, A. J., Frishkoff, L. O., Thompson, M. E., Smith, T. M. & Todd, B. D. Phylogenetic homogenization of amphibian assemblages in human-altered habitats across the globe. Proc. Natl Acad. Sci. 115, E3454-E3462 (2018).
- Felipe-Lucia, M. R. et al. Land-use intensity alters networks between biodiversity, ecosystem functions, and services. Proc. Natl Acad. Sci. 117, 28140-28149 (2020).
- Rodrigues, J. L. M. et al. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc. Natl Acad. Sci. 110, 988-993 (2013).
- Cornell, C. R. et al. Land use conversion increases network complexity and stability of soil microbial communities in a temperate grassland. ISME J. https://doi.org/10.1038/s41396-023-01521-x (2023).
- Hartmann, M. & Six, J. Soil structure and microbiome functions in agroecosystems. Nat. Rev. Earth Environ. https://doi.org/10.1038/ s43017-022-00366-w (2022).
- Guan, N. et al. Microbial response to environmental stresses: from fundamental mechanisms to practical applications. Appl. Microbiol. Biotechnol. 101, 3991-4008 (2017).
- Banerjee, S. et al. Biotic homogenization, lower soil fungal diversity and fewer rare taxa in arable soils across Europe. Nat. Commun. 15, 327 (2024).
- Ling, N., Wang, T. & Kuzyakov, Y. Rhizosphere bacteriome structure and functions. Nat. Commun. 13, 836 (2022).
- Li, X. et al. Acidification suppresses the natural capacity of soil microbiome to fight pathogenic Fusarium infections. Nat. Commun. 14, 5090 (2023).
- Li, X. et al. Agriculture erases climate constraints on soil nematode communities across large spatial scales. Glob. Change Biol. 12 https://doi.org/10.1111/gcb. 14821 (2019).
- Liu, J. et al. Conversion of steppe to cropland increases spatial heterogeneity of soil functional genes. ISME J. https://doi.org/10. 1038/s41396-023-01496-9 (2023).
- Chen, Y. et al. Conversion of natural grassland to cropland alters microbial community assembly across northern China. Environ. Microbiol. https://doi.org/10.1111/1462-2920.16127 (2022).
- Wattenburger, C. J. & Buckley, D. H. Land use alters bacterial growth dynamics in soil. Environ. Microbiol. https://doi.org/10.1111/ 1462-2920.16514 (2023).
- Wang, H. et al. Large-scale homogenization of soil bacterial communities in response to agricultural practices in paddy fields, China. Soil Biol. Biochem. 164, 108490 (2022).
- Delgado-Baquerizo, M. et al. Soil microbial communities drive the resistance of ecosystem multifunctionality to global change in drylands across the globe. Ecol. Lett. 20, 1295-1305 (2017).
- Wagg, C., Bender, S. F., Widmer, F. & van der Heijden, M. G. A. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc. Natl. Acad. Sci. USA. 111, 5266-5270 (2014).
- Louca, S., Parfrey, L. W. & Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272-1277 (2016).
- Galand, P. E., Pereira, O., Hochart, C., Auguet, J. C. & Debroas, D. A strong link between marine microbial community composition and function challenges the idea of functional redundancy. ISME J. 12, 2470-2478 (2018).
- Biggs, C. R. et al. Does functional redundancy affect ecological stability and resilience? A review and meta-analysis. Ecosphere 11, e03184 (2020).
- Tian, L. et al. Deciphering functional redundancy in the human microbiome. Nat. Commun. 11, 6217 (2020).
- Wang, J., Chadwick, D. R., Cheng, Y. & Yan, X. Global analysis of agricultural soil denitrification in response to fertilizer nitrogen. Sci. Total Environ. 616-617, 908-917 (2018).
- Zhang, Y. et al. Land-use type affects N2O production pathways in subtropical acidic soils. Environ. Pollut. 237, 237-243 (2018).
- Zhang, J. et al. Agricultural land use affects nitrate production and conservation in humid subtropical soils in China. Soil Biol. Biochem. 62, 107-114 (2013).
- Kearns, P. J. & Shade, A. Trait-based patterns of microbial dynamics in dormancy potential and heterotrophic strategy: case studies of resource-based and post-press succession. ISME J. 12, 2575-2581 (2018).
- Kana, B. D. & Mizrahi, V. Resuscitation-promoting factors as lytic enzymes for bacterial growth and signaling. FEMS Immunol. Med. Microbiol. 58, 39-50 (2010).
- Wall, D. H., Nielsen, U. N. & Six, J. Soil biodiversity and human health. Nature 528, 69-76 (2015).
- Foley, Jonathan et al. Global Consequences of Land Use. Science 309, 570-574 (2005).
- Maxwell, S. L., Fuller, R. A., Brooks, T. M. & Watson, J. E. J. N. N. Biodiversity: The ravages of guns, nets and bulldozers. Nature 536, 143 (2016).
- Labouyrie, M. et al. Patterns in soil microbial diversity across Europe. Nat. Commun. 14, 3311 (2023).
- Sax, D. F. & Gaines, S. D. Species diversity: from global decreases to local increases. Trends Ecol. Evol. 18, 561-566 (2003).
- de Carvalho, T. S. et al. Land use intensification in the humid tropics increased both alpha and beta diversity of soil bacteria. Ecology 97, 2760-2771 (2016).
- Clavel, J., Julliard, R. & Devictor, V. Worldwide decline of specialist species: toward a global functional homogenization? Front. Ecol. Environ. 9, 222-228 (2011).
- Díaz, S. et al. Incorporating plant functional diversity effects in ecosystem service assessments. Proc. Natl Acad. Sci. 104, 20684 (2007).
- Bongers, F. J. et al. Functional diversity effects on productivity increase with age in a forest biodiversity experiment. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-021-01564-3 (2021).
- van der Plas, F. et al. Biotic homogenization can decrease landscape-scale forest multifunctionality. Proc. Natl Acad. Sci. USA 113, 3557-3562 (2016).
- Le Provost, G. et al. The supply of multiple ecosystem services requires biodiversity across spatial scales. Nat. Ecol. Evol. https:// doi.org/10.1038/s41559-022-01918-5 (2022).
- Peng, Z. et al. The neglected roles of adjacent natural ecosystems in maintaining bacterial diversity in agroecosystems. Glob. Change Biol. n/a, e16996 (2023).
- Xu, W.-B. et al. Regional occupancy increases for widespread species but decreases for narrowly distributed species in metacommunity time series. Nat. Commun. 14, 1463 (2023).
- Newbold, T. et al. Widespread winners and narrow-ranged losers: Land use homogenizes biodiversity in local assemblages worldwide. PLOS Biol. 16, e2006841 (2018).
- Bell, T. H. & Bell, T. Many roads to bacterial generalism. FEMS Microbiol. Ecol. 97, fiaa240 (2021).
- Mueller, R. C., Rodrigues, J. L. M., Nüsslein, K. & Bohannan, B. J. M. Land use change in the Amazon rain forest favours generalist fungi. Funct. Ecol. 30, 1845-1853 (2016).
- Ladouceur, E. et al. Linking changes in species composition and biomass in a globally distributed grassland experiment. Ecol. Lett. https://doi.org/10.1111/ele. 14126 (2022).
- Fahrig, L. et al. Functional landscape heterogeneity and animal biodiversity in agricultural landscapes. Ecol. Lett. 14, 101-112 (2011).
- Montoya-Sánchez, V. et al. Landscape heterogeneity and soil biota are central to multi-taxa diversity for oil palm landscape restoration. Commun. Earth Environ. 4, 209 (2023).
- Louca, S. et al. Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2, 936-943 (2018).
- Louca, S. et al. High taxonomic variability despite stable functional structure across microbial communities. Nat. Ecol. Evol. 1 https:// doi.org/10.1038/s41559-016-0015 (2017).
- Rillig, M. C. et al. The role of multiple global change factors in driving soil functions and microbial biodiversity. Science 366, 886-890 (2019).
- Jurburg, S. D. & Salles, J. F. Functional Redundancy and Ecosystem Function-The Soil Microbiota as a Case Study (IntechOpen, 2015).
- Wilhelm, R. C., Singh, R., Eltis, L. D. & Mohn, W. W. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J. 13, 413-429 (2019).
- Kong, Y. et al. DNA stable-isotope probing delineates carbon flows from rice residues into soil microbial communities depending on
fertilization. Appl. Environ. Microbiol. AEM.02151-02119, https://doi. org/10.1128/AEM.02151-19 (2020). - Peng, J., Zhou, X., Rensing, C., Liesack, W. & Zhu, Y.-G. Soil microbial ecology through the lens of metatranscriptomics. Soil Ecol. Lett. 6, 230217 (2023).
- Hungate, Bruce et al. Quantitative microbial ecology through stable isotope probing. Appl. Environ. Microbiol. 81, 7570-7581 (2015).
- Rousk, J. et al. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4, 1340-1351 (2010).
- Boer, W. D., Folman, L. B., Summerbell, R. C. & Boddy, L. Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol. Rev. 29, 795-811 (2005).
- Haq, I. U., Zhang, M., Yang, P. & van Elsas, J. D. in Advances in Applied Microbiolog 89 (eds. Sariaslani, S. & Gadd, G. M.) 185-215 (Academic Press, 2014).
- Jiao, S. et al. Linking soil fungi to bacterial community assembly in arid ecosystems. iMeta 1, e2 (2022).
- Wu, L. et al. Reduction of microbial diversity in grassland soil is driven by long-term climate warming. Nat. Microbiol. 7, 1054-1062 (2022).
- Meng, Q. et al. Understanding production potentials and yield gaps in intensive maize production in China. Field Crops Res. 143, 91-97 (2013).
- Lauber Christian, L., Hamady, M., Knight, R. & Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111-5120 (2009).
- Brockett, B. F. T., Prescott, C. E. & Grayston, S. J. Soil moisture is the major factor influencing microbial community structure and enzyme activities across seven biogeoclimatic zones in western Canada. Soil Biol. Biochem. 44, 9-20 (2012).
- Delgado-Baquerizo, M. et al. It is elemental: soil nutrient stoichiometry drives bacterial diversity. Environ. Microbiol. 19, 1176-1188 (2017).
- Bell, C. W. et al. High-throughput fluorometric measurement of potential soil extracellular enzyme activities. J. Vis. Exp. 81, e50961 (2013).
- Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581-583 (2016).
- Modin, O. et al. Hill-based dissimilarity indices and null models for analysis of microbial community assembly. Microbiome 8, 1-16 (2020).
- Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590-D596 (2013).
- Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643-D648 (2014).
- Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59-60 (2015).
- Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359 (2012).
- Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10, e65088 (2021).
- Stegen, J. C. et al. Quantifying community assembly processes and identifying features that impose them. ISME J. 7, 2069-2079 (2013).
- Jiao, S., Yang, Y., Xu, Y., Zhang, J. & Lu, Y. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. https://doi. org/10.1038/s41396-019-0522-9 (2019).
- Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685-688 (2020).
- Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixedeffects models using lme4. J. Stat. Softw. 67, 48 (2015).
- Põlme, S. et al. FungalTraits: a user-friendly traits database of fungi and fungus-like stramenopiles. Fungal Diversity https://doi.org/10. 1007/s13225-020-00466-2 (2021).
- Rosseel, Y. lavaan: an R package for structural equation modeling. J. Stat. Softw. 48, 36 (2012).
- Pong. Pong2021/Agricultural-impacts-on-soil-microbiome- Function: Land Conve Rsion to Agriculture Induces Taxonomic Homogenization of Soil Microbia L Communities Globally (Zenodo, 2024).
شكر وتقدير
مساهمات المؤلفين
المصالح المتنافسة
معلومات إضافية
http://www.nature.com/reprints
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
© المؤلف(ون) 2024
المختبر الوطني الرئيسي لمقاومة إجهاد المحاصيل والإنتاج عالي الكفاءة، المختبر الرئيسي لم microbiology الزراعية والبيئية في شنشي، كلية علوم الحياة، جامعة شمال غرب A&F، 712100 يانغلينغ، شنشي، جمهورية الصين الشعبية. كلية الموارد الطبيعية والبيئة، جامعة شمال غرب الزراعة والغابات، 712100 يانغلينغ، شنشي، جمهورية الصين الشعبية. مجموعة تفاعلات النبات والتربة، أغروسكيب، زيورخ، سويسرا. قسم علم النبات والميكروبات، جامعة زيورخ، زيورخ، سويسرا. البريد الإلكتروني: weigehong@nwsuaf.edu.cn; shuojiao@nwsuaf.edu.cn
DOI: https://doi.org/10.1038/s41467-024-47348-8
PMID: https://pubmed.ncbi.nlm.nih.gov/38684659
Publication Date: 2024-04-29
Land conversion to agriculture induces taxonomic homogenization of soil microbial communities globally
Accepted: 28 March 2024
Published online: 29 April 2024
Abstract
Agriculture contributes to a decline in local species diversity and to above- and below-ground biotic homogenization. Here, we conduct a continental survey using 1185 soil samples and compare microbial communities from natural ecosystems (forest, grassland, and wetland) with converted agricultural land. We combine our continental survey results with a global meta-analysis of available sequencing data that cover more than 2400 samples across six continents. Our combined results demonstrate that land conversion to agricultural land results in taxonomic and functional homogenization of soil bacteria, mainly driven by the increase in the geographic ranges of taxa in croplands. We find that
scales
Results
Agriculture causes biotic homogenization of taxonomic and functional profiles
lower in cropland than in natural soil (Fig. 1d, h and Supplementary Figs. 1b, 2) demonstrating that cropland soils are more similar than paired natural ecosystem soils. For example, the
Agricultural effects on specific bacterial phylotypes and functions
s.e.m. of the estimated effect sizes. Sample size is showed by number of data pairs for each group. Statistical significance is based on
the second ring represent phylotypes with significant increase or decrease under agricultural impacts. Colored blocks of the third ring represent the performance of a particular function by at least one phylotypes. The bars of the fourth ring represent the positive and negative effect sizes of agricultural impacts on relative abundances of phylotypes. Colored blocks of the fifth ring represent Spearman’s correlation between the relative abundance of phylotypes and soil physicochemical properties (from inner to outer rings:
were affected by agricultural conversion (
direction compared with other three natural ecosystems when aggregating over level 3 functional categories through COG annotations (Supplementary Fig. 8). The functional categories “translation, ribosomal structure and biogenesis”, and “cytoskeleton” increased while “defense mechanisms” diminished in croplands. However, specific carbon-degrading genes exhibited inconsistent effects upon agricultural conversion (some genes were enriched or deleted), while significant differences in the overall carbon metabolism were not detected under agricultural land-use (Fig. 3a, b). This is most likely due to the high redundancy of broadly distributed functions, thereby
asterisks. d Phosphorus cycling from metagenomic shotgun sequence. Heatmaps represent the
buffering against taxonomic changes induced by agricultural land-use. Indeed, broad functions such as respiration, overall carbon catabolism and anabolism often seem more stable to shifts in microbial taxonomic composition than narrow metabolic functions such as the degradation of specific substrate
dissimilatory sulfate reduction genes (apr and
Mechanisms underlying changed bacterial communities
allow for the proliferation of dormant taxa and accelerate increases in species richness
lines indicate significant (
correlated with bacterial taxonomic composition (Fig. 4a, d). Soil pH filtering played the strongest role in shaping taxonomic and functional composition (Fig. 4a, d). Moreover, the association of fungal and bacterial communities suggest an important role for biotic interactions in mediating agricultural-induced microbial composition changes. Although these variables could explain
Links between bacterial communities and soil functions
Discussion
caused by agricultural conversion
topography
Methods
Continental survey and sampling
where one set was frozen at
Global-scale meta-analysis
Soil environmental variables
Soil enzyme activities
Shotgun metagenome sequencing
sequencing to analyze changes in microbial community functional potential (
Estimation of ecological processes
Statistical analyses
Reporting summary
Data availability
Code availability
References
- Benton, T. G., Bieg, C., Harwatt, H., Pudasaini, R. & Wellesley, L. in Three Levers for Food System Transformation in Support of Nature 02-03 (Chatham House, 2021).
- Tsiafouli, M. A. et al. Intensive agriculture reduces soil biodiversity across Europe. Glob. Change Biol. 21, 973-985 (2015).
- Wang, Y. et al. Forest conversion to plantations: a meta-analysis of consequences for soil and microbial properties and functions. Glob. Change Biol. 27, 5643-5656 (2021).
- Gossner, M. M. et al. Land-use intensification causes multitrophic homogenization of grassland communities. Nature 540, 266-269 (2016).
- Roswell, M., Dushoff, J. & Winfree, R. A conceptual guide to measuring species diversity. Oikos 130, 321-338 (2021).
- Hooper, D. U. et al. A global synthesis reveals biodiversity loss as a major driver of ecosystem change. Nature 486, 105-108 (2012).
- Johnson, Christopher et al. Biodiversity losses and conservation responses in the Anthropocene. Science 356, 270-275 (2017).
- Lees, A. C., Attwood, S., Barlow, J. & Phalan, B. Biodiversity scientists must fight the creeping rise of extinction denial. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-020-01285-z (2020).
- Solar, R. Rd. C. et al. How pervasive is biotic homogenization in human-modified tropical forest landscapes? Ecol. Lett. 18, 1108-1118 (2015).
- Daru, B. H. et al. Widespread homogenization of plant communities in the Anthropocene. Nat. Commun. 12, 6983 (2021).
- Finderup Nielsen, T., Sand-Jensen, K., Dornelas, M. & Bruun, H. H. More is less: net gain in species richness, but biotic homogenization over 140 years. Ecol. Lett. 22, 1650-1657 (2019).
- Olden, J. D. & Rooney, T. P. On defining and quantifying biotic homogenization. Glob. Ecol. Biogeogr. 15, 113-120 (2006).
- Karp, D. S. et al. Intensive agriculture erodes beta-diversity at large scales. Ecol. Lett. 15, 963-970 (2012).
- McKinney, M. L. & Lockwood, J. L. Biotic homogenization: a few winners replacing many losers in the next mass extinction. Trends Ecol. Evol. 14, 450-453 (1999).
- Gámez-Virués, S. et al. Landscape simplification filters species traits and drives biotic homogenization. Nat. Commun. 6, 8568 (2015).
- Hautier, Y. et al. Local loss and spatial homogenization of plant diversity reduce ecosystem multifunctionality. Nat. Ecol. Evol. 2, 50-56 (2018).
- McKinney, M. L. Urbanization as a major cause of biotic homogenization. Biol. Conserv. 127, 247-260 (2006).
- Newbold, T. et al. Climate and land-use change homogenise terrestrial biodiversity, with consequences for ecosystem functioning and human well-being. Emerg. Top. Life Sci. 3, 207-219 (2019).
- Nowakowski, A. J., Frishkoff, L. O., Thompson, M. E., Smith, T. M. & Todd, B. D. Phylogenetic homogenization of amphibian assemblages in human-altered habitats across the globe. Proc. Natl Acad. Sci. 115, E3454-E3462 (2018).
- Felipe-Lucia, M. R. et al. Land-use intensity alters networks between biodiversity, ecosystem functions, and services. Proc. Natl Acad. Sci. 117, 28140-28149 (2020).
- Rodrigues, J. L. M. et al. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc. Natl Acad. Sci. 110, 988-993 (2013).
- Cornell, C. R. et al. Land use conversion increases network complexity and stability of soil microbial communities in a temperate grassland. ISME J. https://doi.org/10.1038/s41396-023-01521-x (2023).
- Hartmann, M. & Six, J. Soil structure and microbiome functions in agroecosystems. Nat. Rev. Earth Environ. https://doi.org/10.1038/ s43017-022-00366-w (2022).
- Guan, N. et al. Microbial response to environmental stresses: from fundamental mechanisms to practical applications. Appl. Microbiol. Biotechnol. 101, 3991-4008 (2017).
- Banerjee, S. et al. Biotic homogenization, lower soil fungal diversity and fewer rare taxa in arable soils across Europe. Nat. Commun. 15, 327 (2024).
- Ling, N., Wang, T. & Kuzyakov, Y. Rhizosphere bacteriome structure and functions. Nat. Commun. 13, 836 (2022).
- Li, X. et al. Acidification suppresses the natural capacity of soil microbiome to fight pathogenic Fusarium infections. Nat. Commun. 14, 5090 (2023).
- Li, X. et al. Agriculture erases climate constraints on soil nematode communities across large spatial scales. Glob. Change Biol. 12 https://doi.org/10.1111/gcb. 14821 (2019).
- Liu, J. et al. Conversion of steppe to cropland increases spatial heterogeneity of soil functional genes. ISME J. https://doi.org/10. 1038/s41396-023-01496-9 (2023).
- Chen, Y. et al. Conversion of natural grassland to cropland alters microbial community assembly across northern China. Environ. Microbiol. https://doi.org/10.1111/1462-2920.16127 (2022).
- Wattenburger, C. J. & Buckley, D. H. Land use alters bacterial growth dynamics in soil. Environ. Microbiol. https://doi.org/10.1111/ 1462-2920.16514 (2023).
- Wang, H. et al. Large-scale homogenization of soil bacterial communities in response to agricultural practices in paddy fields, China. Soil Biol. Biochem. 164, 108490 (2022).
- Delgado-Baquerizo, M. et al. Soil microbial communities drive the resistance of ecosystem multifunctionality to global change in drylands across the globe. Ecol. Lett. 20, 1295-1305 (2017).
- Wagg, C., Bender, S. F., Widmer, F. & van der Heijden, M. G. A. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc. Natl. Acad. Sci. USA. 111, 5266-5270 (2014).
- Louca, S., Parfrey, L. W. & Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272-1277 (2016).
- Galand, P. E., Pereira, O., Hochart, C., Auguet, J. C. & Debroas, D. A strong link between marine microbial community composition and function challenges the idea of functional redundancy. ISME J. 12, 2470-2478 (2018).
- Biggs, C. R. et al. Does functional redundancy affect ecological stability and resilience? A review and meta-analysis. Ecosphere 11, e03184 (2020).
- Tian, L. et al. Deciphering functional redundancy in the human microbiome. Nat. Commun. 11, 6217 (2020).
- Wang, J., Chadwick, D. R., Cheng, Y. & Yan, X. Global analysis of agricultural soil denitrification in response to fertilizer nitrogen. Sci. Total Environ. 616-617, 908-917 (2018).
- Zhang, Y. et al. Land-use type affects N2O production pathways in subtropical acidic soils. Environ. Pollut. 237, 237-243 (2018).
- Zhang, J. et al. Agricultural land use affects nitrate production and conservation in humid subtropical soils in China. Soil Biol. Biochem. 62, 107-114 (2013).
- Kearns, P. J. & Shade, A. Trait-based patterns of microbial dynamics in dormancy potential and heterotrophic strategy: case studies of resource-based and post-press succession. ISME J. 12, 2575-2581 (2018).
- Kana, B. D. & Mizrahi, V. Resuscitation-promoting factors as lytic enzymes for bacterial growth and signaling. FEMS Immunol. Med. Microbiol. 58, 39-50 (2010).
- Wall, D. H., Nielsen, U. N. & Six, J. Soil biodiversity and human health. Nature 528, 69-76 (2015).
- Foley, Jonathan et al. Global Consequences of Land Use. Science 309, 570-574 (2005).
- Maxwell, S. L., Fuller, R. A., Brooks, T. M. & Watson, J. E. J. N. N. Biodiversity: The ravages of guns, nets and bulldozers. Nature 536, 143 (2016).
- Labouyrie, M. et al. Patterns in soil microbial diversity across Europe. Nat. Commun. 14, 3311 (2023).
- Sax, D. F. & Gaines, S. D. Species diversity: from global decreases to local increases. Trends Ecol. Evol. 18, 561-566 (2003).
- de Carvalho, T. S. et al. Land use intensification in the humid tropics increased both alpha and beta diversity of soil bacteria. Ecology 97, 2760-2771 (2016).
- Clavel, J., Julliard, R. & Devictor, V. Worldwide decline of specialist species: toward a global functional homogenization? Front. Ecol. Environ. 9, 222-228 (2011).
- Díaz, S. et al. Incorporating plant functional diversity effects in ecosystem service assessments. Proc. Natl Acad. Sci. 104, 20684 (2007).
- Bongers, F. J. et al. Functional diversity effects on productivity increase with age in a forest biodiversity experiment. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-021-01564-3 (2021).
- van der Plas, F. et al. Biotic homogenization can decrease landscape-scale forest multifunctionality. Proc. Natl Acad. Sci. USA 113, 3557-3562 (2016).
- Le Provost, G. et al. The supply of multiple ecosystem services requires biodiversity across spatial scales. Nat. Ecol. Evol. https:// doi.org/10.1038/s41559-022-01918-5 (2022).
- Peng, Z. et al. The neglected roles of adjacent natural ecosystems in maintaining bacterial diversity in agroecosystems. Glob. Change Biol. n/a, e16996 (2023).
- Xu, W.-B. et al. Regional occupancy increases for widespread species but decreases for narrowly distributed species in metacommunity time series. Nat. Commun. 14, 1463 (2023).
- Newbold, T. et al. Widespread winners and narrow-ranged losers: Land use homogenizes biodiversity in local assemblages worldwide. PLOS Biol. 16, e2006841 (2018).
- Bell, T. H. & Bell, T. Many roads to bacterial generalism. FEMS Microbiol. Ecol. 97, fiaa240 (2021).
- Mueller, R. C., Rodrigues, J. L. M., Nüsslein, K. & Bohannan, B. J. M. Land use change in the Amazon rain forest favours generalist fungi. Funct. Ecol. 30, 1845-1853 (2016).
- Ladouceur, E. et al. Linking changes in species composition and biomass in a globally distributed grassland experiment. Ecol. Lett. https://doi.org/10.1111/ele. 14126 (2022).
- Fahrig, L. et al. Functional landscape heterogeneity and animal biodiversity in agricultural landscapes. Ecol. Lett. 14, 101-112 (2011).
- Montoya-Sánchez, V. et al. Landscape heterogeneity and soil biota are central to multi-taxa diversity for oil palm landscape restoration. Commun. Earth Environ. 4, 209 (2023).
- Louca, S. et al. Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2, 936-943 (2018).
- Louca, S. et al. High taxonomic variability despite stable functional structure across microbial communities. Nat. Ecol. Evol. 1 https:// doi.org/10.1038/s41559-016-0015 (2017).
- Rillig, M. C. et al. The role of multiple global change factors in driving soil functions and microbial biodiversity. Science 366, 886-890 (2019).
- Jurburg, S. D. & Salles, J. F. Functional Redundancy and Ecosystem Function-The Soil Microbiota as a Case Study (IntechOpen, 2015).
- Wilhelm, R. C., Singh, R., Eltis, L. D. & Mohn, W. W. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J. 13, 413-429 (2019).
- Kong, Y. et al. DNA stable-isotope probing delineates carbon flows from rice residues into soil microbial communities depending on
fertilization. Appl. Environ. Microbiol. AEM.02151-02119, https://doi. org/10.1128/AEM.02151-19 (2020). - Peng, J., Zhou, X., Rensing, C., Liesack, W. & Zhu, Y.-G. Soil microbial ecology through the lens of metatranscriptomics. Soil Ecol. Lett. 6, 230217 (2023).
- Hungate, Bruce et al. Quantitative microbial ecology through stable isotope probing. Appl. Environ. Microbiol. 81, 7570-7581 (2015).
- Rousk, J. et al. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4, 1340-1351 (2010).
- Boer, W. D., Folman, L. B., Summerbell, R. C. & Boddy, L. Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol. Rev. 29, 795-811 (2005).
- Haq, I. U., Zhang, M., Yang, P. & van Elsas, J. D. in Advances in Applied Microbiolog 89 (eds. Sariaslani, S. & Gadd, G. M.) 185-215 (Academic Press, 2014).
- Jiao, S. et al. Linking soil fungi to bacterial community assembly in arid ecosystems. iMeta 1, e2 (2022).
- Wu, L. et al. Reduction of microbial diversity in grassland soil is driven by long-term climate warming. Nat. Microbiol. 7, 1054-1062 (2022).
- Meng, Q. et al. Understanding production potentials and yield gaps in intensive maize production in China. Field Crops Res. 143, 91-97 (2013).
- Lauber Christian, L., Hamady, M., Knight, R. & Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111-5120 (2009).
- Brockett, B. F. T., Prescott, C. E. & Grayston, S. J. Soil moisture is the major factor influencing microbial community structure and enzyme activities across seven biogeoclimatic zones in western Canada. Soil Biol. Biochem. 44, 9-20 (2012).
- Delgado-Baquerizo, M. et al. It is elemental: soil nutrient stoichiometry drives bacterial diversity. Environ. Microbiol. 19, 1176-1188 (2017).
- Bell, C. W. et al. High-throughput fluorometric measurement of potential soil extracellular enzyme activities. J. Vis. Exp. 81, e50961 (2013).
- Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581-583 (2016).
- Modin, O. et al. Hill-based dissimilarity indices and null models for analysis of microbial community assembly. Microbiome 8, 1-16 (2020).
- Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590-D596 (2013).
- Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643-D648 (2014).
- Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59-60 (2015).
- Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359 (2012).
- Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10, e65088 (2021).
- Stegen, J. C. et al. Quantifying community assembly processes and identifying features that impose them. ISME J. 7, 2069-2079 (2013).
- Jiao, S., Yang, Y., Xu, Y., Zhang, J. & Lu, Y. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. https://doi. org/10.1038/s41396-019-0522-9 (2019).
- Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685-688 (2020).
- Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixedeffects models using lme4. J. Stat. Softw. 67, 48 (2015).
- Põlme, S. et al. FungalTraits: a user-friendly traits database of fungi and fungus-like stramenopiles. Fungal Diversity https://doi.org/10. 1007/s13225-020-00466-2 (2021).
- Rosseel, Y. lavaan: an R package for structural equation modeling. J. Stat. Softw. 48, 36 (2012).
- Pong. Pong2021/Agricultural-impacts-on-soil-microbiome- Function: Land Conve Rsion to Agriculture Induces Taxonomic Homogenization of Soil Microbia L Communities Globally (Zenodo, 2024).
Acknowledgements
Author contributions
Competing interests
Additional information
http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
© The Author(s) 2024
State Key Laboratory for Crop Stress Resistance and High-Efficiency Production, Shaanxi Key Laboratory of Agricultural and Environmental Microbiology, College of Life Sciences, Northwest A&F University, 712100 Yangling, Shaanxi, P. R. China. College of Natural Resources and Environment, Northwest A&F University, 712100 Yangling, Shaanxi, P. R. China. Plant-Soil Interactions Group, Agroscope, Zurich, Switzerland. Department of Plant and Microbial Biology, University of Zurich, Zurich, Switzerland. e-mail: weigehong@nwsuaf.edu.cn; shuojiao@nwsuaf.edu.cn