تخصيص مركز نطاق d للأنواع المعدنية الأكسو ذات التكافؤ العالي لإزالة الملوثات من خلال البلمرة الكاملة Tailoring d-band center of high-valent metal-oxo species for pollutant removal via complete polymerization
تقدم إزالة الملوثات المدفوعة بالبوليمرة في عمليات الأكسدة المتقدمة (AOPs) طريقة مستدامة لتحقيق تقليل التلوث واستعادة الموارد في آن واحد، مما يدعم نهج تنقية المياه منخفض الكربون. ومع ذلك، فإن تنظيم مثل هذه العملية لا يزال يمثل تحديًا كبيرًا بسبب الفهم المجهري غير الكافي لآليات التفاعل المعتمدة على الهيكل الإلكتروني. هنا، تستكشف هذه الدراسة أصل بوليمرة الملوثات الحفازة باستخدام سلسلة من المعادن الانتقالية.محفزات ذرات مفردة من (كوبالت، والنحاس، والحديد) ويحدد -مركز النطاق لموقع النشاط كعامل رئيسي لنقل البوليمرات للملوثات. يتم إنتاج الأنواع المعدنية الأكسجينية عالية التكافؤ، الناتجة عن تنشيط بيروكسيمونوكبريت، والتي تُعتبر محفزًا لإزالة الملوثات عبر نقل البوليمرات. تعمل الجذور الفينوكسي، التي تم تحديدها من خلال أساليب التقاط الدوران والاختزال المبتكرة، كوسيط رئيسي في تفاعلات البوليمرة. والأهم من ذلك، يمكن ضبط قدرة الأكسدة للأنواع المعدنية الأكسجينية عالية التكافؤ بسهولة من خلال تنظيم قوة ارتباطها لبيروكسيمونوكبريت من خلال-تعديل مركز النطاق. يتم تحقيق نسبة نقل البوليمرization بنسبة 100% عن طريق خفض-مركز النطاق. يقدم هذا العمل نموذجًا لتعديل الهيكل الإلكتروني للأنواع المعدنية الأكسجينية عالية التكافؤ بشكل ديناميكي وتحسين إزالة الملوثات من مياه الصرف الصحي من خلال البلمرة.
أزمة “المياه والطاقة والصرف الصحي” واضحة بالنظر إلى أنيعيش مليون شخص بدون مياه نظيفة، و1.1 مليار فرد يفتقرون إلى الوصول إلى الكهرباء، و2.5 مليار لا يتمتعون بالصرف الصحي الكافي.في سعيها لتوفير مياه صالحة للشرب وآمنة صناعيًا، تم اعتماد عملية الأكسدة المتقدمة (AOP) بشكل متزايد كإضافة للعملية البيولوجية التقليدية للتخلص من الملوثات العضوية السائدة.. العمليات المؤكسدة المتقدمة (AOPs) التي لديها القدرة على إنتاج الأنواع التفاعلية (على سبيل المثال، و تسمى العمليات المؤكسدة المتقدمة المعدنية (M-AOPs) التي تهدف إلى تحويل مجموعة واسعة من الملوثات إلى معادن. على الرغم من مزاياها، فإن لممارسات M-AOPs بعض العيوب، مثل تكوين وسائط أكثر استقرارًا وسُمية. زيادة وقت العلاج والتكاليف، والأهم من ذلك، زيادة انبعاثات الكربون. عادةً، في عمليات M-AOPs، يتم تحويل الكربونات في الملوثات العضوية في النهاية إلى ، والذي يتم إطلاقه بعد ذلك في الغلاف الجوي. تؤدي هذه العملية إلى عدم القدرة على استعادة الطاقة الكيميائية الكبيرة الموجودة في مياه الصرف الصحي.لذلك، من أجل تحقيق معالجة مياه الصرف الصحي المستدامة وتعزيز استعادة الموارد في الوقت نفسه، هناك حاجة ملحة لتغيير النموذج التقليدي لعمليات الأكسدة المتقدمة (M-AOPs) لتحقيق تقليل التلوث واستعادة الموارد بطريقة منخفضة الكربون.
مؤخراً، حظي تنظيم آلية تحويل الملوثات من التمعدن (M-AOPs) إلى البلمرة (P-AOPs) في عمليات الأكسدة المتقدمة (AOPs) باهتمام متزايد.. تهدف P-AOPs إلى تحويل الملوثات العضوية من المحاليل المائية إلى منتجات قابلة لإعادة التدوير على الأسطح الصلبة من خلال تفاعلات نقل البوليمر (PT). وبالتالي، يمكنها تحقيق الحفاظ على الطاقة وتقليل الانبعاثات في تنقية المياه في الوقت نفسه. سابقًا، تم الإبلاغ عن عمليات PT بشكل رئيسي في أنظمة التحفيز القائمة على أكاسيد المعادن والمواد الكربونية باستخدام البيرسلفات أو كأكسيد مع نسبة PT متنوعة (المعادلة 1، الميزة هي الإزالة المتزامنة للكربون العضوي الكلي (TOC) والملوثات)ومع ذلك، فإن تحقيق إزالة ملوثات بكفاءة عالية من خلال التحفيز الضوئي لا يزال يمثل تحديًا كبيرًا بسبب الفهم المحدود جدًا للعلاقة بين الهيكل والوظيفة بين التركيب الإلكتروني للمحفز ونسبة التحفيز الضوئي.
لتوضيح الميزات التحفيزية المعتمدة على الهيكل في عمليات الأكسدة المتقدمة غير المتجانسة، تُعتبر المحفزات ذات الذرة الواحدة (SACs) المنصة المختارة على نطاق واسع بسبب هياكلها التحفيزية الواضحة والمرنة.حاليًا، تم إجراء أبحاث واسعة النطاق حول تقنيات المعالجة المتقدمة المعتمدة على SACs لإزالة الملوثات.. ومع ذلك، كانت هناك تناقضات ملحوظة تم تجاهلها على نطاق واسع في تقارير العمليات المعتمدة على SACs. أي أن استهلاك المؤكسد الفعلي كان أقل من الاستهلاك النظري للمؤكسد لإزالة الملوثات في و ، مما يدل على سمة نموذجية لتفاعل البلمرة. ومع ذلك، لم يكن مثل هذا الظاهرة موجودة في Fe-SACs (الجدول التكميلي 1). مثل هذا الانحراف الحاد يوحي بأن الأنواع المعدنية قد تغير ديناميكيًا مسارات إزالة التمعدن/البلمرة للملوثات. ومع ذلك، فإن الفهم الأساسي حول كيفية بدء عملية PT وما هي ميزة المحفز التي تحكم نسبة PT لا يزال غامضًا. وهذا يشكل عقبة كبيرة في تقديم مبدأ توجيهي عام لتصميم أنظمة تحفيزية تقريبًا الانتقائية لإزالة الملوثات عبر PT.
فيما يلي، نقدم تحقيقًا منهجيًا متعمقًا في عمليات الأكسدة المتقدمة (AOPs) المعتمدة على بيروكسيمونوكبريتات (PMS) عبر سلسلة من المعادن الانتقالية (TM:، و SACs القائمة على الحديد )-SACs وكشف الآلية الأساسية لتحقيق نسبة PT عن طريق تعديل-مركز النطاق لمركز التحفيز. في البداية، الخصائص الفيزيائية والكيميائية المماثلة وTM- المتطابقة-المواقع النشطة لأربعة TM-SA/PN-g-C3تم تأكيد المحفزات من خلال مجموعة من التوصيفات. بعد ذلك، تم إنشاء طريقة مبتكرة لتحديد الجذور الفينوكسي في عمليات الأكسدة المتقدمة. بعد ذلك، تم تسليط الضوء على الأدوار الحاسمة للأنواع المعدنية عالية التكافؤ، مثل، III ، و ، في بلمرة الملوثات تم توضيحها. أخيرًا، الـآلية تعتمد على مركز النطاق لتحقيقتم الكشف عن نسبة PT من خلال مزيج من الحسابات النظرية والنتائج التجريبية. بشكل عام، يكشف هذا العمل عن آلية لإزالة الملوثات في SACs-AOPs ويقترح إرشادات أساسية لتصميم أنظمة التحفيز المعتمدة على SACs معالانتقائية لإزالة الملوثات من خلال عملية PT القوية.
النتائج
تركيب وتوصيف TM-SA/PN-g-C3
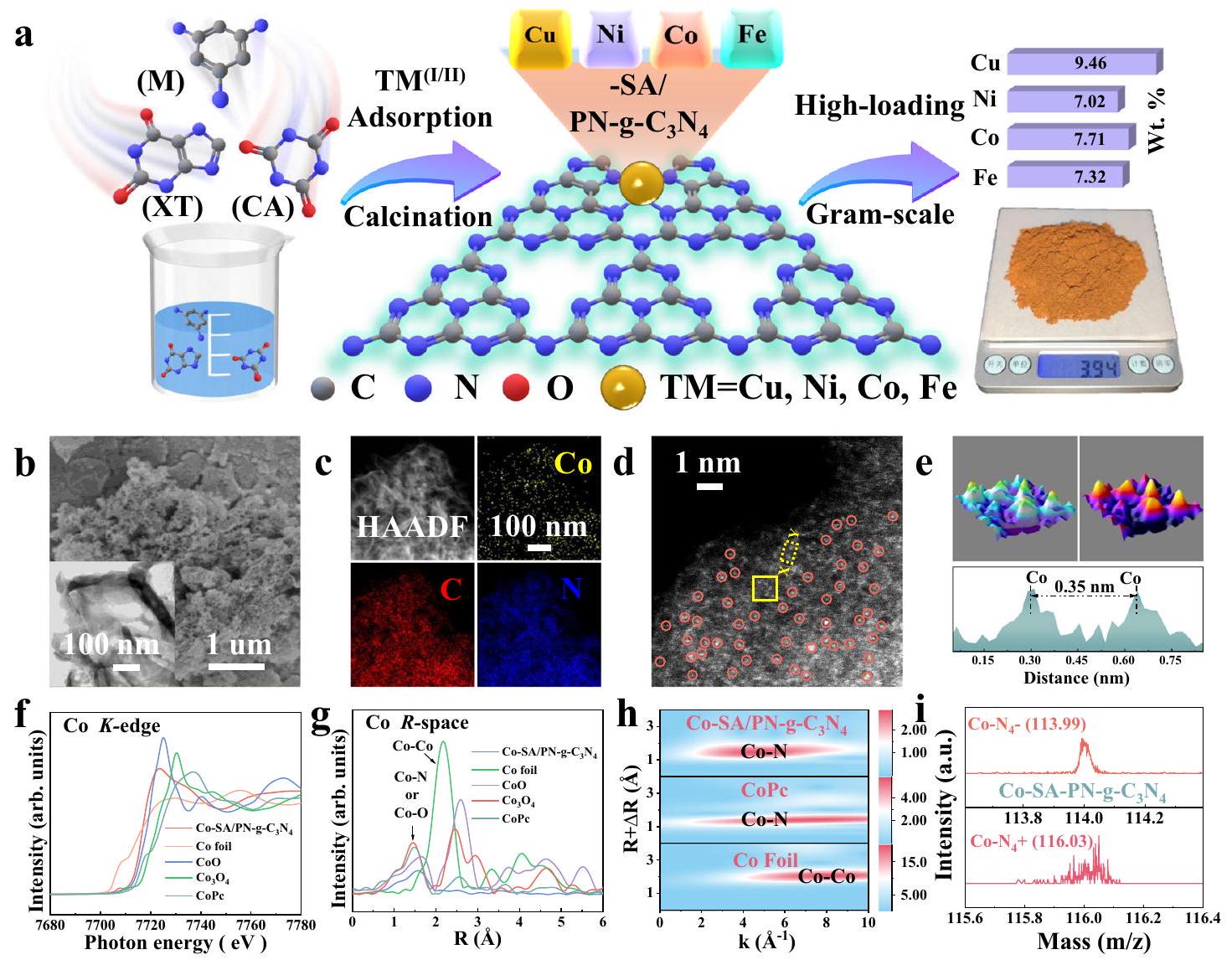
تم استخدام استراتيجية التحلل الحراري المدعوم بالروابط الهيدروجينية لتصنيع سلسلة من الكربون النيتري الغرافيتي الغني بالنيتروجين المدمج بمعادن انتقالية مفردة (TM-SA/PN-g-C3الخ catalysts. هذه الطريقة السهلة سمحت بالإنتاج على نطاق جرامي من TM-SACs مع تحميلات معدنية عالية (طرق إضافية وشكل 1a)تشير تحليلات طيف الانبعاث الذري باستخدام البلازما المقترنة بالحث (ICP-AES) إلى أن تحميل المعدن كان 9.46 و 7.02 و 7.71 ولـ“، و Fe، على التوالي (الشكل التوضيحي 1a). غياب القمم البلورية المميزة للمعادن في أنماط حيود الأشعة السينية (XRD) لمسحوق TM-SA/PN-g-C3 العيّنات (الشكل التوضيحي التكميلي 1ب) استبعدت وجود جزيئات بلورية كبيرة. وبالمثل، لم تكشف المجهر الإلكتروني الناقل عالي الدقة (HR-TEM) عن أي جزيئات نانوية مرئية (الشكل 1b والشكل التكميلي 2). تكشف صور المجهر الإلكتروني الماسح (SEM) أن TM-SA/PN-g- تتميز العينات بشكل مشابه من الرقائق المنحنية مع هيكل مسامي. الهيكل الرئيسي للمسامات المتوسطة دعم TM-SA/PN-g-C3.مع مساحات سطحية عالية كما هو موضح من خلال قياسات برونور-إيميت-تيلر (BET) (الشكل 1ب والأشكال التكميلية 3 و4). تؤكد صور خرائط التحليل الطيفي للطاقة المشتتة (EDS) التوزيعات المتجانسة لعناصر المعادن والكربون والنيتروجين عبر هذه الهياكل (الشكل 1ج والشكل التكميلية 5). ثم تم استخدام مجهر الإلكترون الناقل الماسح بتصحيح الشذوذ وزاوية عالية (AC HAADFSTEM) بدقة دون أنغستروم لتحديد تشتت أنواع المعادن.كما هو موضح في الشكل 1d والشكل التكميلي 6، تم توزيع ذرات المعادن الفردية عالية الكثافة، التي تمثل كنقاط ساطعة معزولة محددة بدوائر حمراء، بشكل موحد في TM-SA/ PN-g-عينات. يمكن تأكيد مثل هذه الملاحظات بشكل أكبر من خلال خرائط التوافق ثلاثي الأبعاد (3D) للخطوط المتساوية وتناسب دالة غاوسية متداخلة ذرية (الشكل 1e والشكل التكميلية 6). علاوة على ذلك، توزيع الشدة على طولفي يكشف عن المسافة الذرية لـ، وكانت SACs من الحديد تقريبًا0.35 و 0.35 نانومتر، على التوالي (الشكل 1d والشكل التوضيحي 6). بشكل عام، تؤكد هذه النتائج تحميل الذرات لـ، و Fe على TM-SA/PN-g-C3عينات بدون وجود هياكل بلورية مشتقة من المعادن.
الهياكل الإلكترونية والذرية لذرات المعادن في TM-SA/PN-g-C3تم استكشاف العينات بشكل أكبر باستخدام طيف امتصاص الأشعة السينية الدقيقة (XAFS). مع أخذ Co-SA/PN-g-C3 في الاعتباركمثال، تُظهر ملفات هيكل امتصاص الأشعة السينية بالقرب من الحافة (XANES) في الشكل 1f أن شدة امتصاص الطاقة للخط الأبيض لـكان أعلى من ذلك لرقائق الكوبالت وأقل من ذلك لأكسيد الكوبالت، مما يشير إلى أن حالة التكافؤ لذرة الكوبالت تقع بين و إن الحالة الأقل من حيث التكافؤ للكوبيالت مقارنةً بسابقه تشير إلى تفاعل ملحوظ بين المعدن والدعم ضمنعينة. بالإضافة إلى ذلك، طيف الامتصاص الدقيق للأشعة السينية الممتد لتحويل فورييه (FT-EXAFS) لـ-مساحة لـ Co-SA/PN-g-C3عرضت ذروة رئيسية عند حواليالانحراف عن مسار تشتت المعدنحواليفي غشاء Co. تؤكد هذه النتيجة التوزيع المعزول لذرات Co على PN-g-C3الدعم (الشكل 1g). يتماشى مع تحليل FT-EXAFS، حيث تُظهر مخططات تحويل الموجات (WT) بصريًا فقط الحد الأقصى للشدة المرتبط بـ أو تفاعل، بدون أيالتفاعل من أجل (الشكل 1هـ).
علاوة على ذلك، فإن التكوين التنسيقي الكمي لـ Co في Co-SA/PN-g-C3تم تحليلها بواسطة ملاءمة EXAFS. تشير المحاكاة إلى وجود قمة رئيسية ناتجة عن أو التنسيق في القشرة الأولى مع عدد تنسيق محسوب يقارب 4.0 (الأشكال التكميلية 9 و 10). تم تفصيل نتائج الملاءمة المثلى من بيانات EXAFS بإيجاز في الجدول التكميلية 2. ومع ذلك، بسبب الاحتمال لوجود ذرات الأكسجين في البيئة الدقيقة، ظل الهيكل التنسيقي الدقيق لذرة الكوبالت المفرد غير واضح. لذلك، تم استخدام قياس مطيافية الكتلة للأيونات الثانوية بتقنية زمن الرحلة (TOF-SIMS) باستخدامتم استخدام تقنية قذف الأيونات بالليزر للتحقق من هيكل التنسيق للكوبيليت فيعن طريق استكشاف الوزن الجزيئيمنأو Oوحدة (الشكل التكميلي 11a)²5. أيونات ثانوية سلبية وإيجابية نموذجية ( ) تم اكتشافها في Co-SA/PN-مؤكداً الذريالموقع في (الشكل 1i). مثل هذا TM- تم التحقق من الهيكل أيضًا للـ TM-SA الأخرى، وعينات Fe ) باستخدام طرق التحليل المماثلة (الأشكال التكميلية 7-11 والملاحظة التكميلية 1). علاوة على ذلك، فإن C و-edge XANES الطيف يؤكد أن إضافة المعادن كان لها تأثير ضئيل على التركيب الذري لـ PN-g-C3الركيزة (الشكل التوضيحي التكميلي 12).
الشكل 1 | تخليق وتوصيف TM (النحاس، النيكل، الكوبالت، والحديد)-SA/PN-gرسم تخطيطي يوضح إجراءات تحضير TM، و Fe )-SA/PN-g- عينات.صورة SEM (الإدراج: صورة HR-TEM)، (ج) صورة HAADFSTEM مع خريطة العناصر EDS المقابلة، و (د) صورة AC HAADFSTEM لـتُحدد الذرات الفردية بدوائر حمراء معزولة في (د). خريطة خطوط العزل ثلاثية الأبعاد وتناسب دالة غاوسية متداخلة مع الذرات
المستطيل الأصفر من (د) وملف الكثافة على طول في (د). شركة-حافة XANES و(g) المحولة فورييه-مساحة EXAFS لـ Co -SA/PN-g-ورق الألمنيوم، و CoPc. رسوم WT-EXAFS لـ“، وورق الألمنيوم. طيف الأيونات عالي الدقة TOF-SIMS لـوحدة هيكلية. يتم توفير بيانات المصدر كملف بيانات مصدر.
تظهر هذه التوصيفات أن TM-SA/PN-g-C3محفزاتNi و Co و Fe) أظهرت خصائص فيزيائية كيميائية مماثلة وبنية تنسيق (مثل TM- )، الذي وفر منصة مثالية لكشف الخصائص التحفيزية المعتمدة على الهيكل الإلكتروني في SACs-AOPs.
الأدوار المحورية للأنواع المعدنية الأكسيدية عالية التكافؤ في إزالة الملوثات
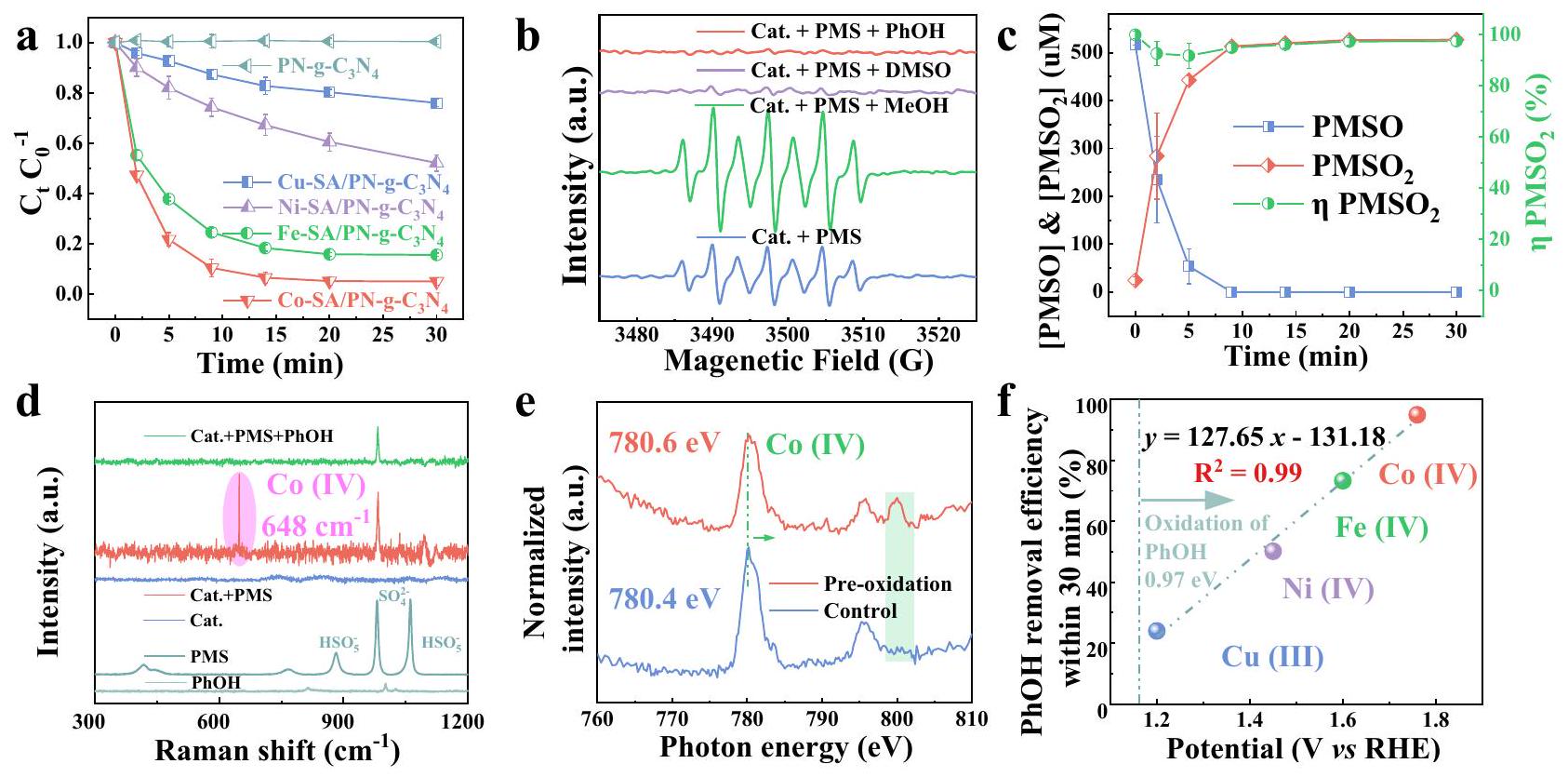
القدرات التحفيزية الشبيهة بفنتون لأربعة TM-SA/PN-g-C3تم فحص المحفزات لإزالة الفينول (PhOH) في وجود PMS. لم يُعزى الانخفاض في تركيز الفينول إلى الامتزاز بواسطة PN-g- الركيزة (الشكل التكميلي 13) أو التحلل الذي يحفزه أيونات المعادن المستخرجة (الشكل التكميلي 14)، مما يشير إلى الدور الرئيسي للنوع المعدني الفردي المدمج في إزالة الفينول. كما هو موضح في الشكل 2a، كانت تسلسل التفاعل الملحوظ للمواد الحفازة يتبع ترتيب ، وهو ما يتماشى مع ترتيب قيم تردد الدوران (TOF، النشاط المحدد بالكتلة، الشكل التوضيحي 15). إن فهم الأصل الجزيئي لمثل هذه التسلسلات التفاعلية أمر حاسم لتصنيع SAC بكفاءة عالية لتحفيز شبيه فنتون، لكنه يواجه تحديًا كبيرًا بسبب آلية تنشيط PMS المثيرة للجدل.. لذلك، تم إجراء تجارب لتوضيح آلية التفاعل باستخدام الرنين المغناطيسي الإلكتروني (EPR)، وإخماد الأنواع التفاعلية، ووضع العلامات النظيرية، وحبس ميثيل فينيل سلفوكسيد (PMSO)، وطيف رامان في الموقع، وطيف امتصاص الأشعة السينية الناعمة (XAS). كما هو موضح في الشكل 2b، إشارة EPR نموذجية مكونة من سبع خطوط
تم ملاحظة 5,5-dيميثيل-1-بيروليدون-N-أوكسيلي (DMPOX) في وجودو PMS، الذي يمكن أن يُعزى إلى أكسدة 5,5-dimethyl-1-pyrroline N-oxide (DMPO) بواسطة المعادن عالية التكافؤأو كمية مفرطة من أو الأنواع التفاعلية المسؤولة عن توليد DMPOX ساهمت أيضًا في إزالة PhOH بسبب الانخفاض الملحوظ في شدة إشارة DMPOX عند إدخال PhOH (الشكل 2b). تظهر نتائج تجارب إخماد الأنواع التفاعلية أن الميثانول (MeOH) والتيرت-بيوتانول (TBA) لم يثبطا بشكل ملحوظ كفاءة إزالة PhOH خلال 30 دقيقة (الشكل التكميلي 16)، مما يعني أن لا شيء منولاتم إنتاجه. هذه النتيجة تم التحقق منها أيضًا من خلال دراسات EPR، التي تظهر زيادة بدلاً من انخفاض في شدة إشارة DMPOX بعد إضافة الميثانول (الشكل 2ب). علاوة على ذلك، تشير قمم EPR الثلاثية المميزة إلى ظهور، وهو نوع تفاعلي مقترح بشكل شائع في PMS-AOP (الشكل التكميلي 17). وقد تم الإبلاغ على نطاق واسع عن أكسيد الديوتيريوم (يمكن أن تعزز كفاءة الأكسدة لـلأنه يطيل عمر بحوالي 18 مرة ( في مقابل في. ومع ذلك، فإن تبادل المذيب (إلىلم تعزز تحلل PhOH، مما يشير إلى المساهمة الضئيلة لـ (الشكل التوضيحي التكميلي 18). بالإضافة إلى ذلك، فإن مساهمة كما تم استبعاد الجذور المرتبطة بالسطح من خلال تجارب EPR (الأشكال التكميلية 19 و 20، والملاحظة التكميلية 2). وبالتالي، تظهر هذه النتائج أن الأنواع عالية التكافؤ من الكوبالت، أي، قد تكون مسؤولة بشكل أساسي عن إزالة PhOH وتوليد DMPOX في Co-SA/PN-g-C3نظام إدارة الممتلكات.
الشكل 2 | الدور المحوري لـلإزالة الملوثات. مقارنة بين كفاءة إزالة الملوثات بين TM (، و Fe )-SA/PN-g- تمثل أشرطة الخطأ الانحراف المعياري، الذي تم الحصول عليه من تكرار التجربة ثلاث مرات.طيف EPR للأنظمة المختلفة مع DMPO كعامل لاحتجاز الدوران. ج تجربة مجس PMSO في Co-SA/PN-g-Cالنظام. تمثل أشرطة الخطأ الانحراف المعياري، الذي تم الحصول عليه من تكرار التجربة مرتين. ظروف التفاعل في (أ-ج) [PMS] =قط.، ، أولي، و . دراسة رامان في الموقع (اللون الوردي يمثل إشارة ). يقوم تحليل Soft-XAS (الظل الأخضر يمثل إشارة ). توافق العلاقة بين جهد اختزال الأكسجين للمعادن عالية التكافؤ وكفاءة إزالة الفينول (PhOH) خلال 30 دقيقة. تم توفير بيانات المصدر كملف بيانات مصدر.
لتقديم دليل مقنع على الدور الحاسم لـتم إجراء تجربة التبريد باستخدام ثنائي ميثيل سلفوكسيد (DMSO). وقد تم التعرف على DMSO على نطاق واسع كمانع فعال للمعادن عالية التكافؤ من خلال تفاعل نقل الأكسجين.تظهر نتائج تجربة التبريد في الشكل التكميلية 16 أن حوالي 10 مللي مول من DMSO قد أوقف تمامًا تحلل الفينول. يجب ملاحظة أن DMSO لم يقض فقط علىولكنها تفاعلت أيضًا مباشرة مع PMS بمعدل ثابت للتفاعل قدره. ومع ذلك، فإن التنشيط الحفزي لـ PMS بواسطة Co-SA/ PN-g- لا يزال يُلاحظ في وجود DMSO، بسبب معدل التحلل الأعلى بشكل ملحوظ لـ PMS في نظام Co-SA/ PN-g-نظام DMSO مقارنةً بـ DMSO وحده (الشكل التكميلي 21)تدعم هذه النتائج الدور الحاسم لـفي إزالة PhOH في نظام Co-SA/PN-g-C3نظام PMS، الذي تم التحقق منه بشكل أكبر من خلال الانخفاض الكبير في شدة قمة EPR لـ DMPOX عند إدخال DMSO (الشكل 2b). علاوة على ذلك، تم استخدام PMSO، الذي يمكن أكسدته بواسطةعبر مسار نقل الأكسجين لإنتاج المنتج المحدد ميثيل فينيل سلفون ()، ككاشف كيميائي لتأكيد وجود. كما هو موضح في الشكل 2c، تم العثور على عائد، (، نسبة المول منالتي تم إنتاجها إلى PMSO المفقود، حواليعلى مدى زمن التفاعل، مما يثبتككائن أكسدة رئيسي تم توليده في نظام Co-SA/PN-g-C3نظام PMS. تم تأكيد وجودبشكل أكبر من خلال دراسات رامان في الموقع. يمكن أن يُعزى القمة الجديدة عندبعد إدخال PMS إلى اهتزاز الشد لـالهيكل، الذي اختفى تمامًا عند وجود PhOH (الشكل 2d)، مما يشير إلى الدور الحاسم لـفي إزالة PhOH. بالإضافة إلى ذلك، تم التحقق من حدوثمن خلال تحليلات XAS. كما هو موضح في الشكل 2e، تم تحويل قمة الامتصاص لـ Co-SA/PN-gعند 780.4 eV بشكل إيجابي بمقدار 0.2 eV بعد إضافة PMS، مما يدل على أكسدة الأنواع السطحية لـ Co. في الوقت نفسه، تؤكد قمة جديدة عند حوالي 800 eV توليد.
يمكن الحفاظ على الأنواع السطحية لـعلى Co-SA/PN-من خلال الأكسدة المسبقة مع PMS في غياب الملوثات. أظهر هذا المحفز المؤكسد مسبقًا قدرة أكسدة مباشرة مثيرة للإعجاب لإزالة PhOH ()، أعلى بكثير من قدرة الامتصاص لـالجديدة (الشكل التكميلي 29). تم استخدام مطيافية الإلكترون الضوئي بالأشعة السينية (XPS) لتتبع التغيير في حالة التكافؤ لـ Co طوال عملية التفاعل الحفاز التي تشمل تنشيط PMS وتحلل PhOH (الشكل التكميلي 30c). توفر الاتجاهات الملحوظة للزيادة الأولية تليها انخفاض لاحق دليلًا واضحًا على مشاركة الأنواع عالية التكافؤ من Co في تحلل PhOH. مشابهة للأنظمة الشبيهة بفنتون القائمة على المعادن عالية التكافؤ، يمكن أن تزيل الأنواع الكهربيةذات الميزات عالية الانتقائية الملوثات بفعالية (مثل PhOH و) مع مجموعات مانحة للإلكترون (-OH و) فقط، لكنها فشلت في إزالة حمض البنزويك مع مجموعة سحب الإلكترون (-COOH) (الأشكال التكملية 31-33). من المهم أن الدور السائد للمعادن عالية التكافؤ (مثل، و) لإزالة PhOH تم تأكيده أيضًا في المحفزات الأخرى TM-SA/PN-g-C3تحت نفس الظروف لـ Co-SA/ PN-g-. يمكن العثور على النتائج والتفاصيل الدقيقة في الأشكال التكملية 13-35 والملاحظة التكملية 2. أثرت الهياكل الإلكترونية الفريدة لهذه المعادن عالية التكافؤ بشكل كبير على إمكاناتها الأكسدة، كما كشفت اختبارات الفولتمترية الدورية (CV) (الشكل التكميلي 36)، التي أظهرت علاقة خطية بين كفاءة إزالة PhOH والجهد (، الشكل 2f)، مما يوضح الدور المحوري للمعادن عالية التكافؤ في إزالة الملوثات في أنظمة PMS الحفازة SACs.
آليات إزالة الملوثات المدفوعة بالبوليمرة
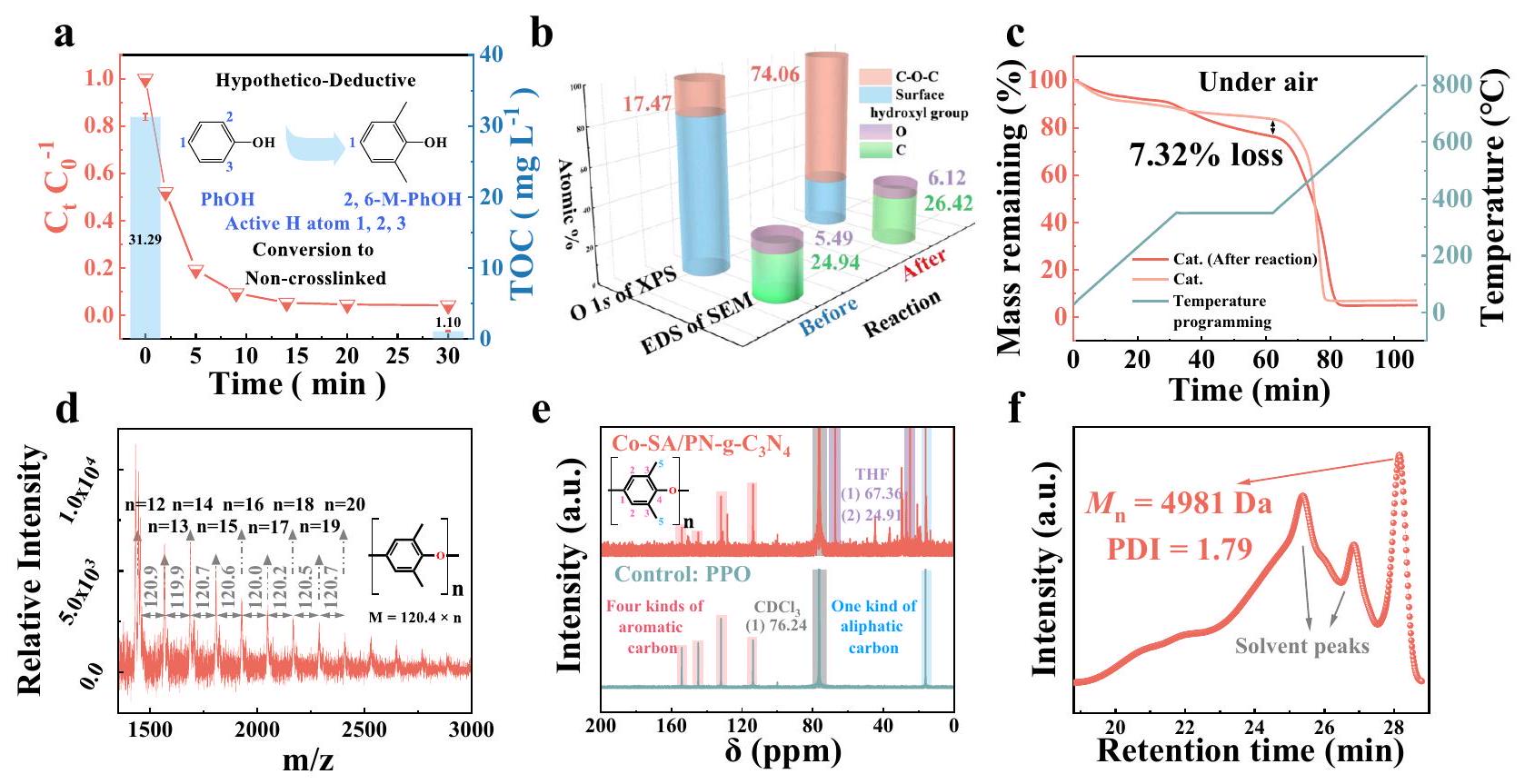
في نظام Co-SA/PN-g-PMS، تمت إزالة TOC المائي بشكل متزامن مع PhOH بكفاءة عالية (، الشكل 3a). أدى هذا الاختفاء غير العادي لـ TOC المائي إلى تراكم بوليمرات متقاطعة من PhOH على سطح المحفزات، والتي يمكن التعرف عليها مباشرة بواسطة الليزر المساعد على إزالة المادة/
الشكل 3 | خصائص منتجات البوليمرة لـ 2،6-M-PhOH على Co-
SA/PN-g-C3. أُزيل TOC وPhOH بشكل متزامن وفعال في محلول Co-SA/PN-g-C3PMS (المخطط هو رسم تخطيطي لطريقة استنتاج فرضية باستخدام). تمثل الرسوم البيانية العمودية قيم TOC. تمثل أشرطة الخطأ الانحراف المعياري، الذي تم الحصول عليه من خلال تكرار التجربة ثلاث مرات. ب SEM-EDS و O 1s من XPS، (ج) منحنيات TGA لـ Co-SA/PN-g-
قبل وبعد التفاعل. د MALDI-TOF-MS، (هـ) تحليل هيكلي قائم على NMR (هـ)، و(و) GPC لمنتجات الإيلاشن على سطح Co-SA/PN-g-C3. تمثل الظلال الوردية والزرقاء والرمادية والبنفسجية في (هـ) أربعة أنواع من الكربون العطري، ونوع واحد من الكربون الأليفاتي،، وTHF، على التوالي. ظروف التفاعل في (أ-ف) [PMS] =Cat.، البدايةو. تم توفير بيانات المصدر كملف بيانات المصدر.
اختبارات مطيافية الكتلة بتقنية زمن الرحلة الأيونية (MALDI-TOF-MS) (الشكل التكميلي 37). ومع ذلك، تم أكسدة PhOH مع ثلاثة مواقع هيدروجين نشطة في هيكله الجزيئي (أي، مواضع ortho وpara لمجموعة الهيدروكسيل) بسهولة لتشكيل بوليمر متقاطع على شكل شبكة، والذي كان غير قابل للذوبان في المذيبات العضوية مثل ثلاثي هاليد الميثان (THMs)، والأسيتونيتريل (ACN)، والتتراهيدروفوران (THF) (الشكل التكميلي 37a). لذلك، تم اختيار 2،6-ثنائي ميثيل فينول (2،6-Mمع موقع نشط واحد فقط، والذي يميل إلى تشكيل منتج بوليمرة غير متقاطع، للتحقيق فيتفاعلات البوليمرة التي تم تحفيزها (الشكل 3a)، والتي تم دراستها من خلال عدة تحليلات بما في ذلك SEM-EDS وXPS وتحليل الوزن الحراري (TGA). كما هو موضح في الشكل 3b والشكل التكميلي 40، بعد اختفاءفي المحلول، لوحظ زيادة في عناصر C وO علىمن خلال SEM-EDS. تم التحقق من هذه النتيجة من خلال نتائج XPS O، التي أظهرت انتقالًا مميزًا في الأنواع السطحية O من مجموعة الهيدروكسيل إلى(الشكل 3b والأشكال التكملية 43a و44a). تكشف نتائج XPS C 1s بعد التفاعل أيضًا عن زيادة كبيرة فيوالروابط (الأشكال التكملية 45 و46). بالمقابل، لم تُلاحظ تغييرات كبيرة في طيف SEM-EDS أو XPS لركيزة PN-gقبل وبعد التفاعل (الأشكال التكملية 42 و46)، مما يدل على الدور الحاسم لمركز المعدن في بوليمرة الملوثات. أيضًا، تشير التغييرات الطفيفة في طيف XPS N 1s إلى أن بيئة التنسيق للذرة الواحدة ظلت مستقرة في التفاعل (الأشكال التكملية 43b و44b). تكشف هذه النتائج عن تراكم كبير للمنتجات العضوية على سطح المحفز. يمكن أن تتعرض هذه المنتجات للتفكك الحراري في عملية برمجة درجة الحرارة معفقدان الوزن مقارنةً بـكما كشفت تحليلات TGA (الشكل 3c والأشكال التكملية 47 و48). لفهم خصائص المنتجات المتكونة على سطح Co-SA/PN-g-C3، تم فصل المواد المودعة باستخدام مذيبات THF وتم تحليلها بواسطة MALDI-TOF-MS، والكروماتوغرافيا بالاستبعاد الهلامي (GPC)، والرنين المغناطيسي النووي (NMR)، وتحليل تحويل فورييه
للمطيافية تحت الحمراء (FTIR). كانت المنتجات المصفاة، التي تتكون من وحدات متكررة معمن 120.4، تعرض هيكلًا مميزًا من بولي فينيلين أكسيد (PPO)، كما يتضح من MALDI-TOF-MS (الشكل 3d). بالإضافة إلى ذلك، تم إثبات هيكل PPO لمنتج البوليمرة أيضًا من خلال الكربونات العطرية/الأليفاتيكية المماثلة ومجموعات العطريةمقارنةً بعينة PPO القياسية، كما كشفت تحليلات NMR وFTIR، على التوالي (الشكل 3e والشكل التكميلي 51). تم تحديد متوسطلمنتجات السلسلة على أنه 4981 Da (الشكل 3f).
بشكل عام، تم تصنيف المواد العضوية المتراكمة على سطح Co-SA/PN-g-C3كـ PPO، والذي تم تأكيده أيضًا في إزالة البوليمرة لـبواسطة المحفزات الأخرى TM (وFe)-SA/PN-g-(الأشكال التكملية 37-52 والملاحظة التكملية 3). لن تؤثر البوليمرات على سطح المحفز على مواقع التفاعل السطحية، ويمكن جمعها بسهولة من خلال بروتوكول الإيلاشن-التجفيف مع نسبة استرداد منولـوCo -SACs، على التوالي (الأشكال التكملية 53 و54، والملاحظة التكملية 3). ومع ذلك، لا يزال من غير الواضح كيف تم تحفيز إزالة البوليمرات للملوثات بواسطة الأنواع المعدنية عالية التكافؤ.
تحديد الجذور الفينوكسي في تشكيل البوليمر
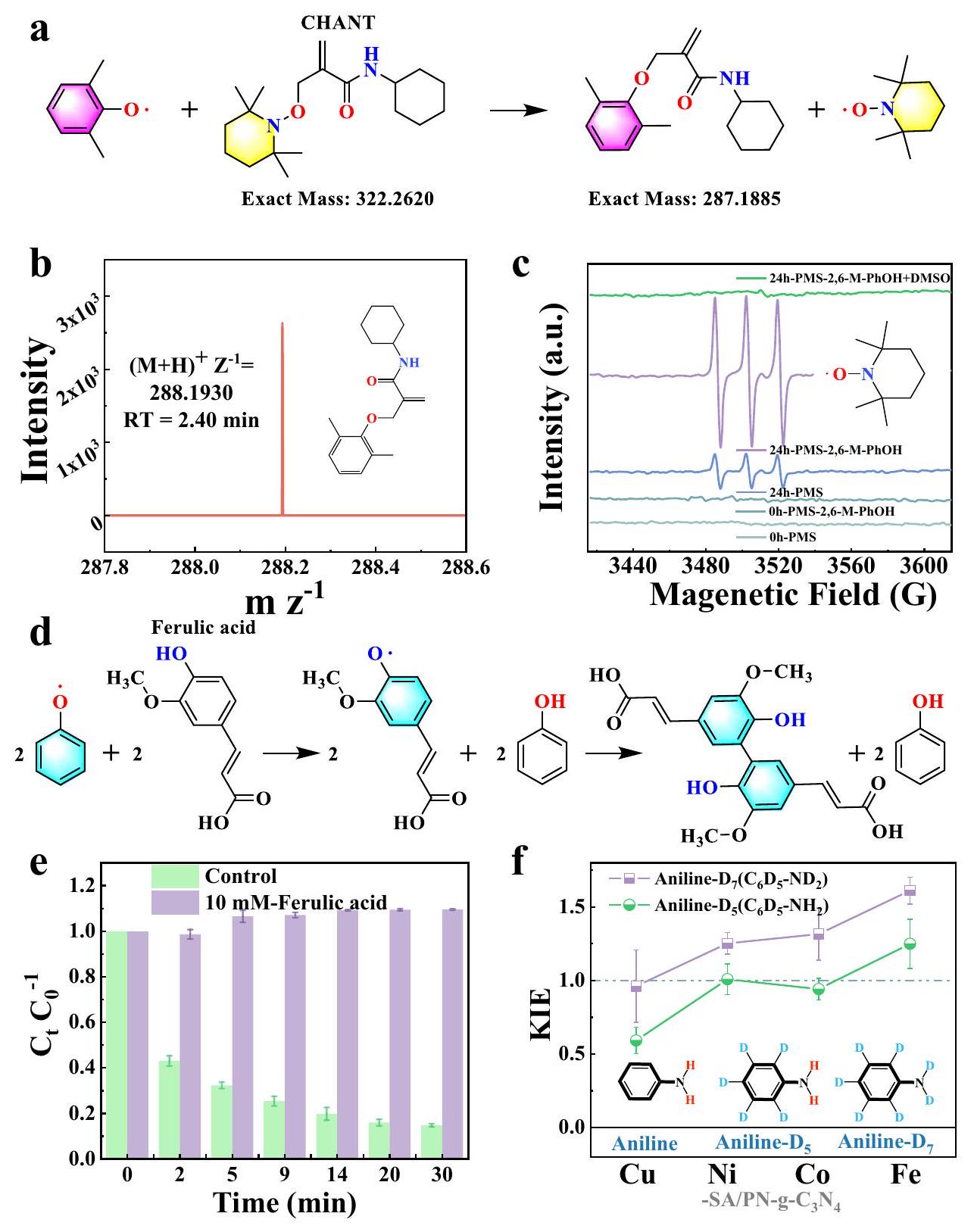
على الرغم من أن الجذور الفينوكسي قد اعتُبرت وسيطًا رئيسيًا في تشكيل البوليمر، إلا أن ظهور الجذور الفينوكسي وكيفية الكشف عن الجذر منخفض النشاط في أنظمة البوليمرة الشبيهة بفنتون لا يزال يمثل تحديًا. وبالتالي، تم تطبيق بروتوكولين مبتكرين، حيث تم استخدام مصائد قائمة على (2،2،6،6-تترا ميثيل بيبيريدين-1-يل) أوكسي (TEMPO) تحتوي على ألكيل (CHANT) كعامل مصيد وحمض الفيروليك (FA) كعامل إخماد، لتحديد الجذور الفينوكسي في أنظمة PMS-AOPs.
يتم استخدام الربط المتقاطع وحبس الدوران بشكل شائع لتثبيت الجذور الحرة العابرة في تحليل EPR (الشكل التكميلي 55 أ، ب). ومع ذلك، فإن هذه الطرق تعاني من نقص في المعلومات الهيكلية للجذور الحرة المحبوسة، وحساسية ضعيفة، وارتفاع حدوث إيجابيات زائفة بسبب التفاعلات الجانبيةلتجاوز هذه العيوب، قمنا بتطوير بروتوكول للكشف عن الجذور الحرة الفينوكسي باستخدام فخ الجذور الحرة الذي تم الإبلاغ عنه مؤخرًا، CHANT (الشكل التكميلي 56).. في مثل هذا البروتوكول، تم دمج الكروماتوغرافيا السائلة عالية الأداء مع مطيافية الكتلة المت tandem (UPLC-MS) مع تحليل EPR لتمكين الكشف الدقيق والموثوق (الشكل التكميلي 55c). كما هو موضح في الشكل 4a، أدى تفاعل جذر الفينوكسي مع CHANT إلى إنتاج منتج مستقر وغير جذري وأطلق الجذر المستمر TEMPO، الذي تم الكشف عنه بواسطة UPLC-MS معمن 288.1930 (الشكل 4ب) وتم استكشافه بواسطة EPR مع القمم الثلاثية النموذجية (الشكل 4ج)، على التوالي. عندما تم استخدام DMSO، وهو مادة مدمرة لـتمت إضافته، واختفى إشارة EPR للجذر المستمر TEMPO تمامًا (الشكل 4c). ثم، تم استخدام TEMPO، كعامل تقاطع تقليدي، أيضًا لالتقاط الجذور الفينوكسي. تظهر النتائج أن الجذور الفينوكسي يمكن أن تتفاعل مع TEMPO لتكوين مركب مستقر معمن 278.2075 (الشكل التوضيحي 59d). بالإضافة إلى ذلك، مستلهمًا من وظيفة FA المضادة للأكسدة في مجال الكيمياء النباتيةقمنا بتطوير FA كعامل لإزالة الجذور الحرة الفينوكسي لتحويل الجذور الحرة الفينوكسي مرة أخرى إلى سلف الفينول غير النشط من الناحية الحمراء (الشكل 4d). تظهر النتائج أن حوالي 10 مللي مول من FA قد منعت تمامًا إزالة PhOH في Co-SA/PN-g-C3.نظام PMS (الشكل 4e والشكل التكميلي 61). على الرغم من أن FA يمكن أن يتفاعل مع PMS مباشرة، إلا أن التنشيط الحفزي لـ PMS بواسطة Co-SA/PN-g-C3لا يزال يُلاحظ في وجود FA (الشكل التكميلي 61f)، مما يشير إلى الدور الحاسم لجذور الفينوكسي في تفاعل البوليمرة للملوثات. وبالتالي، تدعم هذه النتائج بقوةآلية تكوين الجذور الفينوكسي الناتجة عن التحفيز في عملية البلمرة لـ
الشكل 4 | تحديد الجذور الفينوكسي. أ نهج متعدد الاستخدامات لعزل الجذور الفينوكسي باستخدام CHANT. ب كروماتوغرافيا UPLC-MS و (ج) طيف EPR لاستكشاف ناتج التفاعل بين CHANT والجذور الفينوكسي. د توضيح تخطيطي لتقليل الجذور الفينوكسي بواسطة FA. هـ تأثير FA على تحلل PhOH. تمثل أشرطة الخطأ الانحراف المعياري، الذي تم الحصول عليه من تكرار التجربة مرتين.أثر النظير الحركي للأنيلين
الملوثات، تم تأكيد عمومية مثل هذا الآلية أيضًا بالنسبة للمعادن الانتقالية الأخرى، و Fe )-SA/PN-g- المحفزات (الأشكال التكميلية 55-61، الملاحظة التكميلية 4).
علاوة على ذلك، تم الإبلاغ عن أن المعادن ذات التكافؤ العالي يمكن أن تؤكسد الملوثات إلى جذور الفينوكسي عبر مسار استخراج الهيدروجين (HA) أو نقل الإلكترون المرتبط بالبروتون (PCET). القيمة الصغيرة لتأثير النظائر الحركية (KIE) استبعدت مسار HA، مما يشير إلى أن أكسدة الركيزة كانت مدفوعة بـ PCET لتوليد الجذور العضوية المقابلة في الأربعة TM-SA/PN-g-C3.أنظمة PMS (الشكل 4 ف والشكل التكميلي 62). بشكل عام، نكتشف الدور الوسيط للمعادن عالية التكافؤ التي تحفز الجذور الفينوكسيّة لإزالة الملوثات من خلال البلمرة. بالإضافة إلى ذلك، من المتوقع أن تكون الطريقة التي تم وضعها لتحديد الجذور الفينوكسيّة في هذا العمل ذات إمكانيات تطبيقية كبيرة لمجموعة واسعة من التفاعلات التي تشمل الجذور الحرة العضوية، بما في ذلك التحفيز الضوئي، والتحفيز الكهربائي، والتركيب العضوي..
تنظيم-مركز النطاق لإزالة الملوثات بواسطة البلمرة
أنواع المعادن عالية التكافؤ، تشمل III ، و قادرة على بدء إزالة الملوثات من خلال مسار PCET المماثل. ومع ذلك، فإن هذه الأنواع تظهر أيضًا سلوكًا يعتمد على الهيكل الإلكتروني، مما يؤدي إلى الأكسدة الزائدة وتحلل الملوثات إلى أحماض عضوية جزيئية صغيرة.
جدير بالذكر أن نسبة PT للـ Fe-SACs كانت أقل بكثير، حيث بلغت فقط، مقارنة بقيم ، و لـ“، و SACs المعتمدة على Co، على التوالي (الشكل 5 أ والشكل التكميلي 32). تشير هذه النتيجة إلى أن الملوثات يمكن أن تتبلمر بالكامل على سطح المعادن الانتقالية (، و Co )-SA/PN-g-C المحفزات. ومع ذلك، بالنسبة لـ Fe-SA/PN-g-C3قد يوجد نظام PMS، وهو آلية إضافية لإزالة الملوثات، ربما تتضمن مسار التحلل المعدني، في هذه العملية. وقد تم تأكيد هذه الفرضية بشكل أكبر من خلال الانخفاض الملحوظ فيللبوليمر الشبيه بالسلسلة على سطح Fe-SA/PN-g-C3المحفز، كما هو موضح من خلال تحليلات GPC (الشكل 5 أ والشكل التكميلي 52). تشير هذه النتيجة إلى المزيد من تحلل الملوثات إلى مواد جزيئية صغيرة بواسطةالأنواع. وبالتالي، فإن فهم كيفية تنظيم التركيب الإلكتروني للمركز الحفاز لآلية إزالة الملوثات أمر أساسي لتوفير إرشادات أساسية لتصميم أنظمة حفازة ذات وظائف مدهشة لإزالة البوليمرات الملوثة من مياه الصرف الصحي.
الميزة الأكسيدية المميزة لـتم الكشف عن ذلك من خلال مزيج من حسابات نظرية الكثافة الوظيفية (DFT) وتصميمات تجريبية. كما هو موضح في الشكل 5ب، يتضمن تكوين الجذور الفينوكسي من خلال آلية نقل الإلكترون المزدوج (PCET) ثلاث خطوات: امتصاص PMS على موقع TM-SA (الخطوة 1)، ثم نقل إلكترونين يؤدي إلى إنتاج معادن عالية التكافؤ (الخطوة 2)، وأكسدة الملوث لإنتاج الجذور الفينوكسي عبر مسار PCET (الخطوة 3). في النهاية، ستخضع الجذور الفينوكسي المتكونة لتفاعل بلمرة إضافي على سطح المحفز. وبالتالي، يمكن أن يظهر الهيكل الإلكتروني لمركز التحفيز تأثيرات كبيرة على آلية إزالة الملوثات من خلال التأثير على عمليات الامتصاص ونقل الإلكترون المذكورة أعلاه.
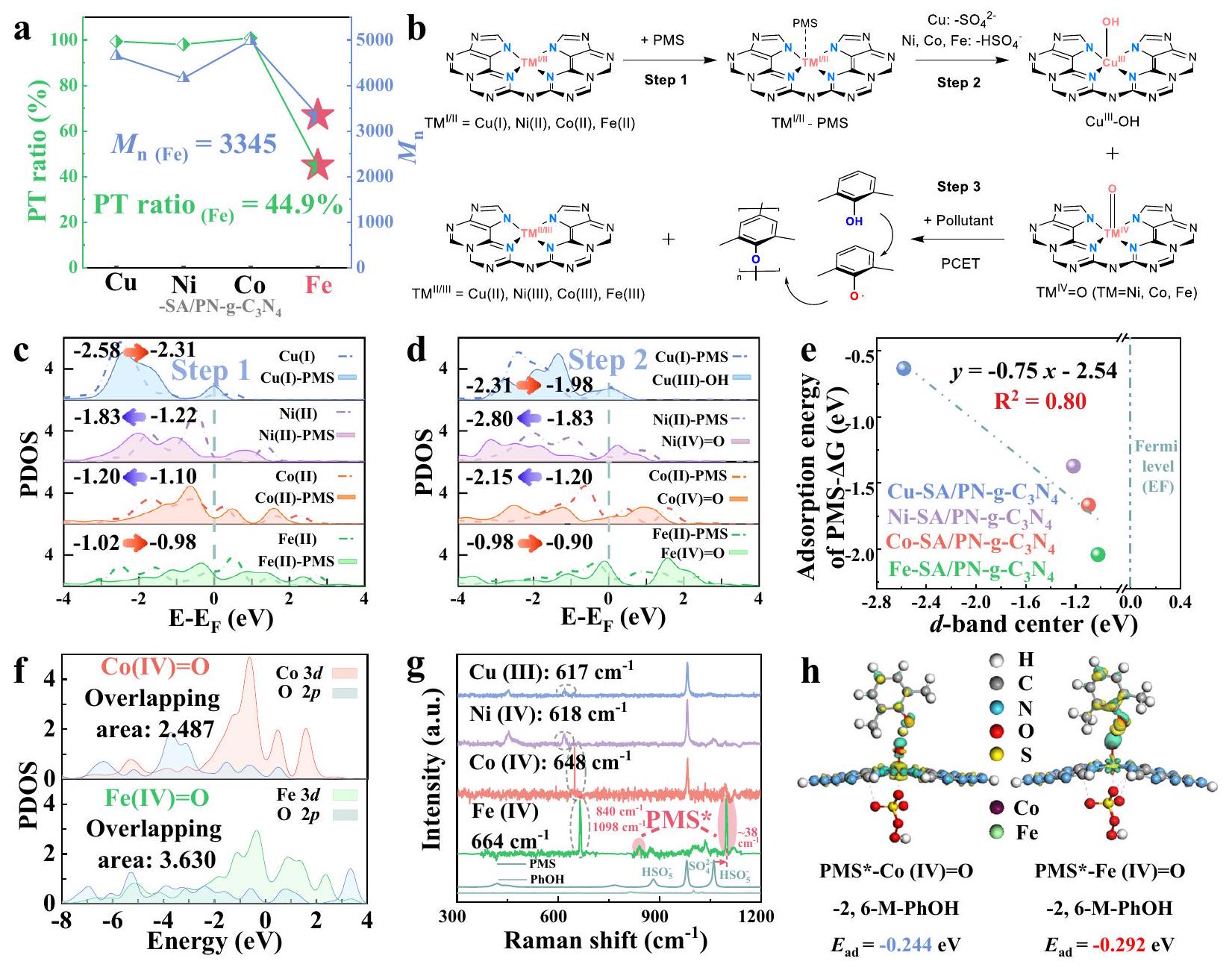
في عملية امتصاص PMS الأولية (الخطوة 1)، تكشف تحليلات كثافة الحالة المتوقعة (PDOS) أن موقع الحديد يمتلك أعلى-مركز النطاق مقارنةً بـ، و شركة. أيضًا، الـمركز نطاق الحديد يرتفع أكثر ويقترب من مستوى فيرمي ) بعد امتصاص PMS ( Fe (II)-PMS) لتكوين الأكسو المعدني عالي التكافؤ . بالمقابل، تظهر مواقع الكوبالت والنيكل اتجاهات تنازلية خلال العمليات المذكورة أعلاه (الشكل 5c، d). عادةً، قرب مركز النطاق إلىيشير عمومًا إلى زيادة ميل المحفز للتبرع بالشحنة للمواد الممتصةوبالتالي علاقة خطية دقيقة بين الـتمت ملاحظة مركز نطاق المعادن وطاقة الامتزاز لـ PMS (الشكل 5e و
الشكل التوضيحي التكميلي 65). بالإضافة إلى ذلك، الترتيب غير المتماثل لـالإلكترونات المدارية في قناة الدوران ) يؤكد بشكل أكبر على قوة الترابط الأقوى ونقل الإلكترون الأكبر بسبب استقطاب الدوران (الشكل التوضيحي 67). عند المقارنة مع نوعيظهر بشكل واضح تداخلًا متزايدًا لـ و المدارات (3.630 مقابل 2.487، الشكل 5f)، مما يؤدي إلى توسيع أكبر في انتشار الإلكترونات علىالتكوين وقدرة الربط الأقوى للحديد المحوري لـتم التحقق من الألفة العالية لموقع الحديد لـ PMS من خلال التوصيفات رامان في الموقع [ذروة جديدة عند، الذي يتوافق مع الأنواع البيروكسو المفعلة (PMS*)]، والتي لوحظت على Fe-SA/PN-gفقط (الشكل 5g). علاوة على ذلك، الـذروة الاهتزاز المميزة لـ PMS* شهدت انزياحًا نحو الأزرق بـ (من 1060 إلى )، مما يدل على تفاعل قوي بين PMS و Fe- موقع على المحفز. ظلت هذه القمم غائبة في TM-SA/PN-g-C3 الأخرىطيف المحفزات. عند إضافة الملوثات، يحدث الاختفاء المتزامن للإشارات التي تتوافق مع و أكدوا على دورهم التعاوني في إزالة الملوثات، من خلال العمل عبر كل من مسارات PT والتعدين (الشكل التوضيحي التكميلي 27).
لفهم تكوين PMS* على سطح المحفز، تم إجراء حسابات DFT. تكشف رؤانا الحاسوبية عن ميل ملحوظ لـ PMS نحو موقع Fe(IV) في اتجاه محوري، مما يؤدي إلى تكوين PMS*-معقد، تكوين مفضل أكثر من (الشكل التوضيحي 70). في المرحلة التالية، كانت الملوثات تميل إلى شغل موقع الأكسجين المتاح. ومن الجدير بالذكر أن طاقة الامتزاز المرتبطة بـ PMS*كان التعقيد ملحوظًا أكبر من ذلك المرتبط بـ PMS*-Co(IV)=O، كما يتضح من قيم الطاقة التي بلغت -0.292 eV للحديد و -0.244 eV للكوبالت (الشكل 5h). عادةً ما تتوافق مثل هذه الطاقة العالية للامتزاز مع زيادة احتمالية حدوث الأكسدة. وقد تم دعم صحة هذا الادعاء من خلال النقص الملحوظ في الإلكترونات في الملوثات عند الاقتراب من (الشكل 5h والملاحظة التكميلية 6).
بشكل عام، تظهر النتائج التجريبية والمحاكاة أعلاه أن المستوى الأعلى-مركز نطاق الحديد سيؤدي إلى تفاعل أقوى بين و مدارات لتشكيلفي الاتجاه المحوري، مما قد يؤدي بعد ذلك إلى بدء تمعدن الملوثات في Fe-SA/PN-g-C3نظام إدارة الممتلكات.
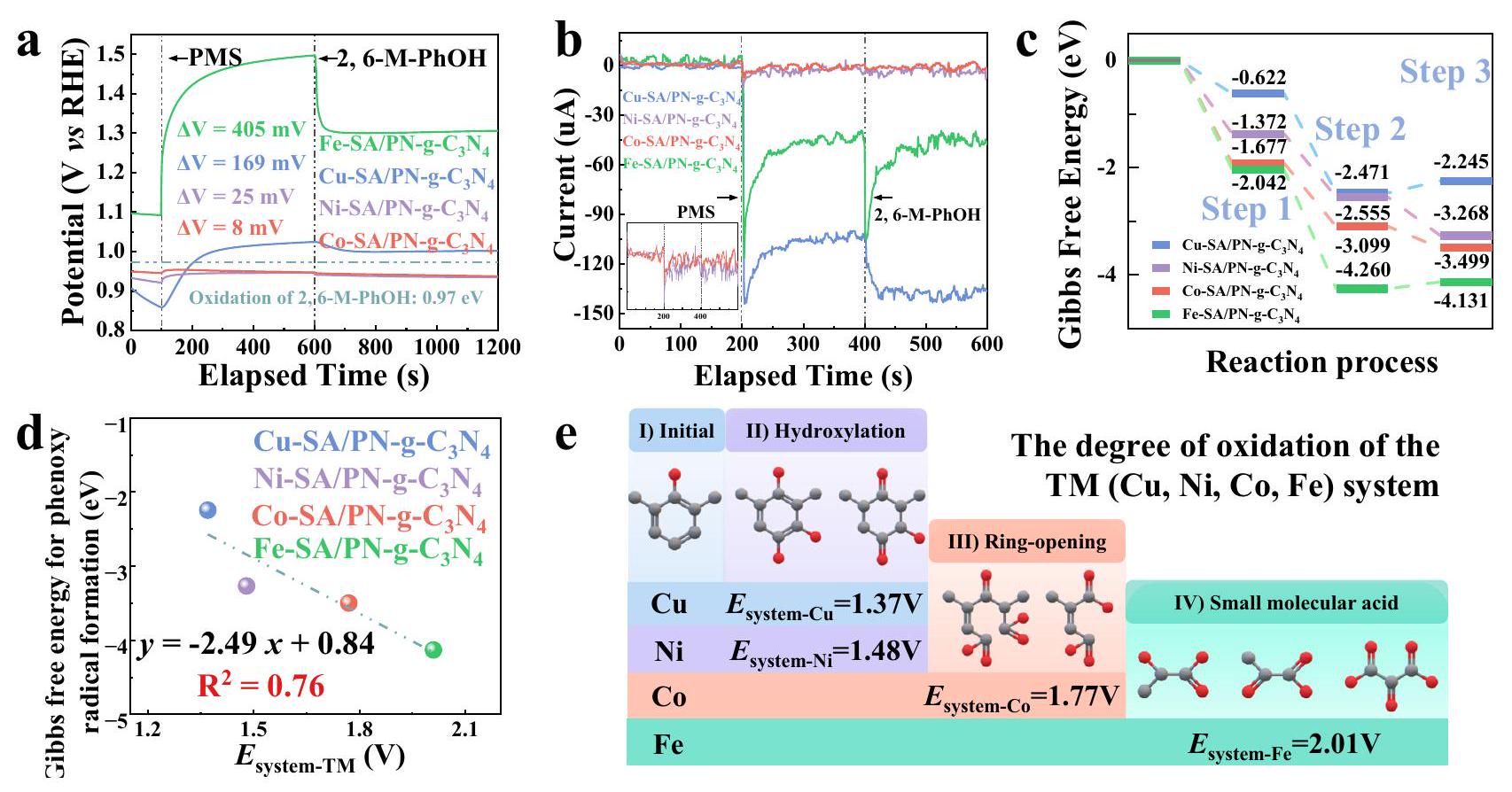
للتوليد الأكسيدي لجذور الفينوكسي (الخطوة 3)، لوحظ أن امتصاص سطح PMS كان له تأثير في إطالة رابطة O-O مع PMS، مما يعزز القدرة الأكسيدية لـ TM-SA/PN-g.أنظمة PMS عندما كانت المعادن عالية التكافؤ متورطة. أثرت الضغوط التأكسدية المختلفة بشكل كبير على السلوكيات اللاحقة لجذور الفينوكسي. لتأكيد هذا التغيير في القدرة التأكسدية بشكل تجريبي (بعد إنشاء نظام إدارة الطاقة (PMS*), تم إجراء قياسات جهد الدائرة المفتوحةكما هو موضح في الشكل 6a، تشير الزيادات الإيجابية في جهد الدائرة المفتوحة عند إدخال PMS إلى ارتفاع القدرة التأكسدية عبر جميع TM-SA/PN-g الأربعة.أنظمة PMS. تم ترتيب زيادات الجهد بترتيب تنازلي من حيث سعتها كالتالي:. يبدو أن الحديد (Fe) هو الأعلى-مركز النطاق وأعلى تقارب لـ PMS أظهروا الزيادة الأكثر أهمية في القدرة التأكسدية بعد تكوين PMS*الشكل 6 أ والشكل التكميلية 75). القدرة المرتفعة على الأكسدة لـ Fe-SA/PN-g-C3تم التحقق من نظام PMS بعد تكوين معقد FePMS* من خلال مجموعة متنوعة من التوصيفات الكهروكيميائية. أوضحت التحليلات الكرونوأمبيرومترية انخفاضًا سريعًا في اتجاهات التيار بعد حقن PMS، مما يشير إلى انتقال فوري للإلكترونات من PMS إلى المراكز المعدنية للمعادن الانتقالية (TM) (التي تتكون من، و الحديد ) في SA/PN-g- العوامل المساعدة، التي تعزز ولادة كيانات المعادن عالية التكافؤ (الشكل 6ب). من الجدير بالذكر أنه عندماتمت الإضافة لاحقًا، وانخفضت الاستجابة الحالية في البداية، لكنها زادت تدريجيًا لـ Fe-SA/PN-g-C3.بينما كانت تظهر فقط اتجاهًا هبوطيًا لـ (الشكل 6ب). كانت هذه الفروق الواضحة شهادة على القدرة التأكسدية القوية لمركب Fe-PMS*، الذي يمكنه استخراج
الشكل 5 | يوضح أدوار الـ-مركز النطاق في روابط المؤكسدات والملوثات. أ. نسب PT ولمنتجات الإيلاution في الأربعة TM-SA/PN-gأنظمة. ظروف التفاعل في ، [قط.] ، أولي، و . رسم تخطيطي لتكوين الجذور الفينوكسي. PDOS لـ TM (، والحديد ) في TM-SA/PN-g- أنظمة (ج) الخطوة 1 و(د) الخطوة 2. هـ العلاقة بين-مركز النطاق وطاقة الامتزاز لـ PMS.الـ PDOS لـ و TM (الحديد والكوبالت) يذكر في
TM(IV) الهيكل، والمنطقة المتداخلة المدمجة موضحة في الزاوية العليا اليسرى. ج تم تحديد أنواع سطح Fe-PMS* (المعلمة باللون الأحمر) بواسطة طيف رامان.فرق كثافة الشحنة والطاقة النظرية المرتبطة بالامتصاص ) لامتصاص PMS في الاتجاه المحوري، حيث تشير المناطق الخضراء الفاتحة والصفراء إلى مناطق تراكم الإلكترونات ونقصها، على التوالي (سطح متساويبورتم توفير بيانات المصدر كملف بيانات المصدر. إلكترونات منتجلى ذلك كزيادة في كثافة التيارلوصف القدرة التأكسدية لـ TM-SA/PN-g-C3 بشكل كمينظام إدارة الممتلكات، الذي يمثل مجموع جهد الاختزال للمعادن عالية التكافؤ (الشكل 2f) والزيادة في جهد الدائرة المفتوحة بعد إضافة PMS (الشكل 6أ)، تم تعريفه لذلك (المعادلة 2). الـتم تحديد القيم على أنها، و 1.37 فولت لـونظم التفاعل المعتمدة على Cu SAC، على التوالي.
كما هو موضح في مخطط تغير الطاقة الحرة لجيبس (الشكل 6c)، يمكن ملاحظة تقدم حراري تلقائي لتكوين جذر الفينوكسي لأربعة TM-SA/PN-g-C3.أنظمة إدارة المشاريع مع الميل في ترتيبالعلاقة الخطية بينوأظهرت الطاقة الحرة لجيبس لتكوين الجذور الفينوكسي أن الجذور الفينوكسي هي الوسيط الرئيسي في إزالة الملوثات الفينولية في TM-SA/PN-g.أنظمة PMS (الشكل 6d). توفر التوضيحات الإضافية من تحليلات UPLC-MS مسارًا تحويليًا لـبالتزامن مع زيادة القدرة التأكسدية المتأصلة في
TM-SA/PN-g-C3نظام PMS. تم التعرف على التحولات الجزيئية التي تشمل الهيدروكسلة، وتفكيك الحلقة، والت derivation اللاحق للأحماض العضوية الأساسية مثل حمض الجليكوليك، وحمض البيروفيك، وحمض الميسوكسيليك داخل الـ، و Fe-SA/PN-g-C3 الأنظمة، على التوالي (الشكل 6e والأشكال التكميلية 76-80). تدعم هذه النتائج أنه، بالإضافة إلى مسار البوليمرة الموثق جيدًا، فإن Fe-SA/PN-g-C3يمكن لنظام PMS ذو القدرة العالية على الأكسدة أن يحفز الأكسدة المفرطة للملوثات. وت culminates هذه العملية في توليد الأحماض العضوية التي تستمر في المحلول الكلي، مما يؤدي إلى أدنى نسبة PT.
لذلك، تخصيص-مركز النطاق للأنواع المعدنية الأكسو ذات القيمة العالية يمكن أن تحقق إزالة فعالة للملوثات من خلال البلمرة الكاملة، والتي تظهر متانة مثيرة للإعجاب ضد الظروف البيئية المتغيرة. أنشطة التحفيز لـ، و SA/PN-g-C3العوامل المساعدة، التي تتميز بمسار تحفيز البلمرة، تأثرت بشكل ضئيل بالأنونات المتواجدة (مثل،، ، و ) ومصفوفات المياه (مثل مياه الصنبور، ومياه البحيرات، والمياه المعالجة الثانوية من محطات معالجة مياه الصرف الصحي). تؤكد هذه النتيجة على قوة هذه المحفزات في بيئات مائية متنوعة. على النقيض من ذلك، فإن النشاط التحفيزي لـكان مثبطًا قليلاً بسبب مشاركة نسبة أعلى من تكوين أنواع المعادن-الأكسو الفلزية، وتكوين الجذور الفينوكسي في المعادن الانتقالية المختلفة، و (شركة) -SA/PN-g- أنظمة PMS. د العلاقة بين قدرة الأكسدة لنظام التفاعل وطاقة غيبس الحرة لتكوين الجذور الفينوكسي. هـ درجة الأكسدة في TM المختلفة.، و Co )-SA/PN-g-C3 أنظمة إدارة المشاريع. يتم توفير بيانات المصدر كملف بيانات مصدر. تحلل التمعدن ). بالإضافة إلى ذلك، كان لنطاق pH المائي من 4.0 إلى 9.0 تأثير ضئيل على كفاءة الإزالة أو نسبة PT لـ PhOH في الأربعة TM-SA/PN-g- تظهر هذه النتائج القدرة المثيرة للإعجاب على مقاومة التداخل لتفاعل البوليمرة في معالجة مياه الصرف الصحي الحقيقية، مما يؤكد تنوعه وفعاليته في إزالة الملوثات (الأشكال التكميلية 81-86 والملاحظة التكميلية 5).
نقاش
باختصار، يوفر عملنا رؤى أساسية حول تنظيم مسارات البلمرة الحفزية والتعدين لإزالة الملوثات من خلال تعديل-مركز النطاق للـ TM (TM: ، و (Fe)-SACs (الشكل التوضيحي 87). تثبت كل من حسابات DFT والتحقيقات التجريبية أن الأنواع المعدنية الأكسجينية ذات التكافؤ العالي (على سبيل المثال،، و ) مع أدنى يمكن أن تحقق مراكز النطاق تقريبًا نسبة إزالة الملوثات تصل إلى 100%، بينمامع مستوى أعلى-يمكن أن يعزز مركز النطاق إعادة ربط PMS، مما يؤدي إلى قدرة أكسدة أعلى لتحويل الملوثات إلى أحماض جزيئية صغيرة مع أدنى نسبة PT (بالإضافة إلى ذلك، يمكن أن تؤدي الأنواع المعدنية عالية التكافؤ إلى أكسدة الإلكترون الواحد للملوثات لتوليد الجذور الفينوكسي الوسيطة الرئيسية لتفاعلات البلمرة اللاحقة، والتي تم تحديدها من خلال مجموعة من طرق التقاط الدوران المبتكرة وطرق الإخماد. يوفر هذا العمل رؤى عميقة حول العلاقة بين الهيكل والوظيفة بين الهيكل الإلكتروني للمركز الحفاز ونسبة PT لإزالة الملوثات على المستوى الذري. ستكون النتائج مفيدة في تطوير تقنيات تنقية المياه المستدامة لتحقيق تقليل التلوث واستعادة الموارد بطريقة منخفضة الكربون.
طرق
المواد الكيميائية
نترات الحديد غير المائينترات النحاس ثلاثي الهيدراتنترات الكوبالت سداسي الماء نيترات النيكل سداسي الهيدرات، نترات الكروم سداسي الهيدراتنترات الزنك سداسي الماءميلامينثيوسلفات الصوديوم ( ) ، يوديد البوتاسيوم ( )، ميثانول ( )، الإيثانول ( )، و TBA ( ) تم شراؤها من شركة سينوفارم للمواد الكيميائية، الصين. PhOH ( ), 2,6-M-فوه ( حمض البنزويك ) ، زانثين ( حمض السيانوريك ) ، متلازمة ما قبل الحيض ( ) ، ذرةأنيلين )، DMSO ( )، ميثيل فينيل سلفوكسيد (PMSO، 97%)، ميثيل فينيل سلفون ( ), 2,2,6,6-تترا ميثيل-4-بيبيريدينول (TEMP، 98%) و DMPO (97%) تم الحصول عليها من شركة Aladdin Chemistry، الصين. تم شراء أوراق الكربون من شركة Toray، اليابان. تم الحصول على أنيلين-D5 (98% ذرة D) وأنيلين-D7 (98% ذرة D) من شركة Shanghai Civi Chemical Technology، الصين وشركة J&K Scientific، الصين، على التوالي. محلول نافيون ( ) و ACN (>99%) تم شراؤها من شركة سيغما-ألدريتش، الصين. ماء منزوع الأيونات (DI) ( ) تم استخدامه في جميع التجارب. تم استخدام جميع المواد الكيميائية كما هي دون مزيد من التنقية.
تحضير المحفز
لتحضير الـ TM- (TM: ، و Fe ) SA/PN-g- تم إذابة المحفزات، حمض السيانوريك (9.6 مللي مول) وزانثين (2.4 مللي مول) أولاً في 80 مل من الماء المقطر تحت تأثير الموجات فوق الصوتية. ثم تم خلط 80 مل من محلول الميلاتونين (1.2 مللي مول) بسرعة مع المحلول السابق لتكوين تعليق، والذي تم معالجته بالموجات فوق الصوتية لمدة 20 دقيقة وتم تحريكه مغناطيسياً لمدة 4 ساعات إضافية. بعد ذلك، تم إعداد محلول سلف المعدن عن طريق إذابة 9.6 مللي مول من ملح المعدن المقابل في 40 مل من الماء المقطر، والذي أضيف إلى التعليق السابق وتم تحريكه لمدة ساعة واحدة لتحقيق التجميع الذاتي لبوليمر المعدن-carbon. تم طرد التجميعات في جهاز الطرد المركزي وغسلها بالماء المقطر عدة مرات وتجفيفها في فراغ عندلمدة 10 ساعات. أخيرًا، تم طحن العينات المجففة في هاون وتم حرقها عندتحتالغلاف الجوي بمعدل تسخينللحصول على العلامة التجارية – (TM = ، و Co ) SA/PN-g- المحفزات. PN-g-ركيزة تم التحضير وفقًا للإجراءات المذكورة أعلاه في غياب المواد الأولية المعدنية.
توصيفات
تم وصف طرق التوصيف بما في ذلك XRD و BET و XPS و SEM و HR-TEM و HAADF-STEM و FTIR و EPR و soft-XAS و XAFS بالتفصيل في الطرق التكميلية.
تجارب تحلل الملوثات
تم إجراء تجارب تحلل الملوثات على الأقل في نسختين في أكواب سعة 50 ملتحت التحريك المغناطيسي. في اختبار نموذجي، كتلة معينة من TM-SA/PN-g-تمت إضافة إلى المحلول المائي الذي يحتوي على الكميات المطلوبة من الملوث وPMS. ما لم يُذكر خلاف ذلك، كانت النسبة المولية للأكسدة إلى الملوثات ثابتة عند 2:1، وتم ضبط الرقم الهيدروجيني الابتدائي عند 7.0 بواسطةأو NaOH. في عملية التفاعل، تم أخذ 1 مل من تعليق التفاعل وتم إيقافه بواسطةثم تم تصفية الخليط من خلالغشاء لمزيد من التحليل.
طرق تحليلية
الطرق التحليلية النوعية والكمية بما في ذلك قياسات UPLC، قياسات UPLC-MS، قياسات TOC، تجارب التوقف، تجارب KIE، تجارب ما قبل الأكسدة، قياسات MALDI-TOF، توصيف C-NMR، قياسات GPC، والاختبارات الكهروكيميائية متاحة في الطرق التكميلية.
حسابات الكيمياء الكمومية
تم إجراء جميع حسابات DFT على CASTEP، راجع الطرق التكميلية لمزيد من التفاصيل. يتم تقديم الإحداثيات الذرية للبنية المحسّنة مع هذه الورقة.
توفر البيانات
البيانات التي تدعم نتائج الدراسة مدرجة في النص الرئيسي وملفات المعلومات التكميلية. يمكن الحصول على البيانات الخام من المؤلف المراسل عند الطلب. يتم توفير بيانات المصدر مع هذه الورقة.
References
Naddaf, M. The world faces a water crisis- 4 powerful charts show how. Nature 615, 774-775 (2023).
The water crisis is worsening. Researchers must tackle it together. Nature. 613, 611-612 (2023).
Gleick, P. H. Moving to a sustainable future for water. Nat. Water 1, 486-487 (2023).
Lu, L. et al. Wastewater treatment for carbon capture and utilization. Nat. Sustain. 1, 750-758 (2018).
Dickin, S., Bayoumi, M., Giné, R., Andersson, K. & Jiménez, A. Sustainable sanitation and gaps in global climate policy and financing. npj Clean Water. 3, 24 (2020).
Gibb, B. C. Weird and wonderful water. Nat. Chem. 8, 733-734 (2016).
Hodges, B. C., Cates, E. L. & Kim, J. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 13, 642-650 (2018).
Zhang, S., Zheng, H. & Tratnyek, P. G. Advanced redox processes for sustainable water treatment. Nat. Water 1, 666-681 (2023).
Shang, Y., Xu, X., Gao, B., Wang, S. & Duan, X. Single-atom catalysis in advanced oxidation processes for environmental remediation. Chem. Soc. Rev. 5, 5281-5322 (2021).
Ren, W. et al. Origins of electron-transfer regime in persulfatebased nonradical oxidation processes. Environ. Sci. Technol. 56, 78-97 (2022).
Lee, J., von Gunten, U. & Kim, J. H. Persulfate-based advanced oxidation: critical assessment of opportunities and roadblocks. Environ. Sci. Technol. 54, 3064-3081 (2020).
Wu, X. & Kim, J. Outlook on single atom catalysts for persulfate-based advanced oxidation. ACS ES&T Eng. 2, 1776-1796 (2022).
Wang, C. et al. Selective oxidation of various phenolic contaminants by activated persulfate via the hydrogen abstraction pathway. ACS ES&T Eng. 1, 1275-1286 (2021).
Wang, C. et al. Catalytic reactivity of with different facets in the hydrogen abstraction of phenol by persulfate. Appl. Catal., B. 270, 118819 (2020).
Liu, L. et al. Nonradical activation of peroxydisulfate promoted by oxygen vacancy-laden NiO for catalytic phenol oxidative polymerization. Appl. Catal., B. 254, 166-173 (2019).
Liu, T. et al. Oxidative polymerization of bisphenol A (BPA) via H -abstraction by and persulfate: importance of the surface complexes. Chem. Eng. J. 435, 134816 (2022).
Zhang, Y. et al. Simultaneous nanocatalytic surface activation of pollutants and oxidants for highly efficient water decontamination. Nat. Commun. 13, 3005 (2022).
Huang, M. et al. Facilely tuning the intrinsic catalytic sites of the spinel oxide for peroxymonosulfate activation: from fundamental investigation to pilot-scale demonstration. Proc. Natl. Acad. Sci. USA. 119, e2092285177 (2022).
Wang, J. et al. Interlayer structure manipulation of iron oxychloride by potassium cation intercalation to steer activation pathway. J. Am. Chem. Soc. 144, 4294-4299 (2022).
Zhang, Y. et al. Distinguishing homogeneous advanced oxidation processes in bulk water from heterogeneous surface reactions in organic oxidation. Proc. Natl. Acad. Sci. USA. 120, e1992560176 (2023).
Qiao, B. et al. Single-atom catalysis of CO oxidation using . Nat. Chem. 3, 634-641 (2011).
Yu, X. et al. A green edge-hosted zinc single-site heterogeneous catalyst for superior fenton-like activity. Proc. Natl. Acad. Sci. USA. 120, e2073739176 (2023).
Cheng, C . et al. Generation of and its contribution to fenton-like reactions on a single-atom iron-N-C catalyst. Angew. Chem. Int. Ed. Engl. 62, e202218510 (2023).
Gao, Y. et al. Activity trends and mechanisms in peroxymonosulfate-assisted catalytic production of singlet oxygen over atomic metal-N-C catalysts. Angew. Chem. Int. Ed. Engl. 133, 22687-22695 (2021).
Wang, B. et al. A site distance effect induced by reactant molecule matchup in single-atom catalysts for fenton-like reactions. Angew. Chem. Int. Ed. Engl. 61, e202207268 (2022).
Mi, X. et al. Almost peroxymonosulfate conversion to singlet oxygen on single-atom sites. Angew. Chem. Int. Ed. Engl. 60, 4588-4593 (2021).
Pan, J. et al. Improving peroxymonosulfate activation by copper ion-saturated adsorbent-based single atom catalysts for the degradation of organic contaminants: electron-transfer mechanism and the key role of Cu single atoms. J. Mater. Chem. A. 9, 1164-11613 (2021).
Qian, K. et al. Single-atom Fe catalyst outperforms its homogeneous counterpart for activating peroxymonosulfate to achieve effective degradation of organic contaminants. Environ. Sci. Technol. 55, 7034-7043 (2021).
Chen, F. et al. Molecular engineering toward pyrrolic N -rich ( ) single-atom sites for enhanced heterogeneous fenton-like reaction. Adv. Funct. Mater. 31, 2007877 (2021).
Han, G. et al. Abrading bulk metal into single atoms. Nat. Nanotechnol. 17, 403-407 (2022).
Hai, X. et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 17, 174-181 (2022).
Xia, C. et al. General synthesis of single-atom catalysts with high metal loading using graphene quantum dots. Nat. Chem. 13, 887-894 (2021).
Kim, H. et al. Activation of hydrogen peroxide by a titanium oxidesupported iron catalyst: evidence for surface Fe(IV) and its selectivity. Environ. Sci. Technol. 54, 15424-15432 (2020).
Zhu, L. et al. Designing 3D-MoS sponge as excellent cocatalysts in advanced oxidation processes for pollutant control. Angew. Chem. Int. Ed. Engl. 59, 13968-13976 (2020).
Shao, P. et al. Revisiting the graphitized nanodiamond-mediated activation of peroxymonosulfate: singlet oxygenation versus electron transfer. Environ. Sci. Technol. 55, 16078-16087 (2021).
Ren, W. et al. The intrinsic nature of persulfate activation and N-doping in carbocatalysis. Environ. Sci. Technol. 54, 6438-6447 (2020).
Wang, L. et al. A polymer tethering strategy to achieve high metal loading on catalysts for Fenton reactions. Nat. Commun. 14, 7841 (2023).
Zong, Y. et al. Unraveling the overlooked involvement of high-valent cobalt-oxo species generated from the cobalt(II)-activated peroxymonosulfate process. Environ. Sci. Technol. 54, 16231-16239 (2020).
Sun, H., Xu, X., Song, Y., Zhou, W. & Shao, Z. Designing high-valence metal sites for electrochemical water splitting. Adv. Funct. Mater. 31, 2009779 (2021).
Li, F., Lu, Z., Li, T., Zhang, P. & Hu, C. Origin of the excellent activity and selectivity of a single-atom copper catalyst with unsaturated sites via peroxydisulfate activation: Cu (III) as a dominant oxidizing species. Environ. Sci. Technol. 56, 8765-8775 (2022).
Williams, P. J. H. et al. New approach to the detection of short-lived radical intermediates. J. Am. Chem. Soc. 144, 15969-15976 (2022).
Ranguelova, K. & Mason, R. P. The fidelity of spin trapping with DMPO in biological systems. Magn. Reson. Chem. 49, 152-158 (2011).
Pou, S., Hassett, D. J., Britigan, B. E., Cohen, M. S. & Rosen, G. M. Problems associated with spin trapping oxygen-centered free radicals in biological systems. Anal. Biochem. 177, 1-6 (1989).
Amić, A., Marković, Z., Dimitrić Marković, J. M., Milenković, D. & Stepanić, V. Antioxidative potential of ferulic acid phenoxyl radical. Phytochemistry. 170, 112218 (2020).
Bednar, T. N. & Nagib, D. A. Radical arenes. Nat. Chem. 15, 3-4 (2023).
Nguyen, J. D., D’Amato, E. M., Narayanam, J. M. R. & Stephenson, C. R. J. Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions. Nat. Chem. 4, 854-859 (2012).
Geng, J. et al. Radical polymerization inside living cells. Nat. Chem. 11, 578-586 (2019).
Chen, Z. et al. Tailoring the -dand centers enables nanosheets to be highly active for hydrogen evolution catalysis. Angew. Chem. Int. Ed. Engl. 57, 5076-5080 (2018).
Greiner, M. T. et al. Free-atom-like states in single-atom alloy catalysts. Nat. Chem. 10, 1008-1015 (2018).
Sun, Y. et al. Spin-related electron transfer and orbital interactions in oxygen electrocatalysis. Adv. Mater. 32, e2003297 (2020).
Zhou, Q. et al. Generating dual-active species by triple-atom sites through peroxymonosulfate activation for treating micropollutants in complex water. Proc. Natl. Acad. Sci. USA. 120, e1994882176 (2023).
Yang, M. et al. Unveiling the origins of selective oxidation in singleatom catalysis via intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635-11645 (2022).
Yu, X. et al. Enhanced nonradical catalytic oxidation by encapsulating cobalt into nitrogen doped graphene: highlight on interfacial interactions. J. Mater. Chem. A 9, 7198-7207 (2021).
Clark, S. J. et al. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 220, 567-570 (2005).
شكر وتقدير
تم دعم هذا العمل من قبل المؤسسة الوطنية للعلوم الطبيعية في الصين (22376072 إلى M.J.H.، 52192684 إلى H.Q.Y.، 51821006 إلى H.Q.Y.، 22106159 إلى M.J.H.، و52027815 إلى H.Q.Y.)، ومؤسسة الابتكار وريادة الأعمال للطلاب في جامعة العلوم والتكنولوجيا في الصين (CY2022G11 إلى H.Z.L. وCY2023GO20 إلى H.Z.L.). نشكر محطات الإشعاع الضوئي BL1W1B في منشأة الإشعاع المتزامن في بكين (BSRF)، وBL14W1 في منشأة الإشعاع المتزامن في شنغهاي (SSRF)، وBL12B-a، وBL11U، وBL10B في المختبر الوطني للإشعاع المتزامن (NSRL)، ومختبر Shiyanjia (www.shiyanjia.comو الدكتور مي صن من مجموعة HRTEM (مركز الأدوات للعلوم الفيزيائية في جامعة العلوم والتكنولوجيا في الصين) للمساعدة في التوصيفات. تم إجراء الحسابات العددية على نظام الحوسبة الفائقة في مركز الحوسبة الفائقة بجامعة العلوم والتكنولوجيا في الصين.
مساهمات المؤلفين
M.J.H. و J.J.C. و H.Q.Y. قاموا بتصميم الدراسة والإشراف على المشروع. H.Z.L. نفذ عملية التخليق، والتوصيف، واختبار الأداء التحفيزي. X.X.S. و J.J.C. أجريا الدراسات النظرية. B.B.W. و X.S.W. ساعدا في تخليق عامل التقاط CHANT. H.L.L. ساعد في تخليق المحفزات. M.J.H. و H.Z.L. و J.J.C. و H.Q.Y. قاموا بتحليل النتائج وشاركوا في كتابة الورقة. جميع المؤلفين ساهموا في مناقشة النتائج والمخطوطة.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى مينغجي هوانغ، جيه-جيه تشين أو هان-تشينغ يو.
معلومات مراجعة الأقران تشكر مجلة Nature Communications دونغيا لي والمراجعين الآخرين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا ما تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/رخص/بواسطة/4.0/.
مختبر المفاتيح لعلوم تحويل الملوثات الحضرية، قسم علوم البيئة والهندسة، جامعة العلوم والتكنولوجيا في الصين، هيفي، الصين.مدرسة علوم البيئة والهندسة، جامعة هواتشونغ للعلوم والتكنولوجيا، ووهان، الصين.قسم الكيمياء، جامعة العلوم والتكنولوجيا في الصين، هيفي، الصين.ساهم هؤلاء المؤلفون بالتساوي: هونغ-تشي ليو، شياو-شوان شو.
Tailoring -band center of high-valent metaloxo species for pollutant removal via complete polymerization
Received: 8 November 2023
Accepted: 7 March 2024
Published online: 14 March 2024
(A) Check for updates
Hong-Zhi Liu , Xiao-Xuan Shu , Mingjie Huang Bing-Bing Wu , Jie-Jie Chen , Xi-Sheng Wang , Hui-Lin Li & Han-Qing Yu ®
Abstract
Polymerization-driven removal of pollutants in advanced oxidation processes (AOPs) offers a sustainable way for the simultaneous achievement of contamination abatement and resource recovery, supporting a low-carbon water purification approach. However, regulating such a process remains a great challenge due to the insufficient microscopic understanding of electronic structure-dependent reaction mechanisms. Herein, this work probes the origin of catalytic pollutant polymerization using a series of transition metal ( , Co , and Fe ) single-atom catalysts and identifies the -band center of active site as the key driver for polymerization transfer of pollutants. The high-valent metal-oxo species, produced via peroxymonosulfate activation, are found to trigger the pollutant removal via polymerization transfer. Phenoxyl radicals, identified by the innovative spin-trapping and quenching approaches, act as the key intermediate in the polymerization reactions. More importantly, the oxidation capacity of high-valent metal-oxo species can be facilely tuned by regulating their binding strength for peroxymonosulfate through -band center modulation. A 100% polymerization transfer ratio is achieved by lowering the -band center. This work presents a paradigm to dynamically modulate the electronic structure of high-valent metal-oxo species and optimize pollutant removal from wastewater via polymerization.
The “Water-Energy-Sanitation” crisis is evident considering that million people live without clean water, 1.1 billion individuals lack access to electricity, and 2.5 billion do not have adequate sanitation . In pursuit of delivering potable and industrially safe water, the advanced oxidation process (AOP) has been increasingly adopted as supplement to the conventional biological process to eliminate the prevailing organic pollutants . The AOPs with a capacity to produce reactive species (e.g., and ) to mineralize a vast array of pollutants are termed M-AOPs. Despite their advantages, M-AOPs have some drawbacks, such as forming more stable and toxic intermediates,
the increase in treatment time and costs, and more importantly, the increase in carbon emissions . Typically, in M-AOPs, carbons in organic contaminants are eventually converted into , which is then released into the atmosphere. This process results in the inability to recover the substantial chemical energy contained within wastewater . Therefore, to achieve sustainable wastewater treatment and promote resource recovery simultaneously, the conventional M-AOPs are in urgent need of a paradigm shift to simultaneously realize contamination abatement and resource recovery in a lowcarbon manner.
Recently, regulating the conversion mechanism of pollutants from mineralization (M-AOPs) to polymerization (P-AOPs) in AOPs has drawn increasing attention . P-AOPs aim to convert organic pollutants from aqueous solutions into recyclable products to solid surfaces via polymerization transfer (PT) reactions. Thus, they can simultaneously achieve energy conservation and emission reduction for water purification. Previously, the PT processes were predominantly reported in the metal oxides and carbonaceous materials-based catalytic systems by using persulfates or as the oxidant with a varied PT ratio (Eq. 1, the feature is the synchronous removal of total organic carbon (TOC) and pollutants) . However, achieving a highly efficient pollutant removal via PT is still a great challenge due to the very limited understanding about the structure-function relationship between the electronic structure of the catalyst and the PT ratio.
To elucidate the structure-dependent catalytic features in heterogeneous AOPs, single-atom catalysts (SACs) are the widely selected platform due to their explicit and flexible catalytic structures . Currently, extensive research on SACs-based AOPs for pollutant removal has been conducted . However, a widely overlooked inconsistency existed in the reported SACs-based AOPs. That is, the actual oxidant consumption was lower than the theoretical oxidant consumption for pollutant removal in and , indicative of a typical polymerization reaction feature. However, such a phenomenon did not exist in Fe-SACs (Supplementary Table 1). Such a sharp divergence implies that the metal species might dynamically change the mineralization/polymerization removal pathways of pollutants. However, the fundamental understanding about how the PT process is initialized and what feature of the catalyst governs the PT ratio is still elusive. This poses a great obstacle in delivering a general guiding principle for designing catalytic systems with nearly selectivity for pollutant removal via PT.
Herein, we present a systematic in-depth investigation into peroxymonosulfate (PMS)-based heterogeneous catalytic AOPs over a series of transition metal (TM: , and Fe )-SACs and reveal the underlying mechanism for achieving a PT ratio by modulating the -band center of the catalytic center. Initially, the similar physicochemical properties and identical TM- active sites of the four TM-SA/PN-g-C3 catalysts were confirmed with a suite of characterizations. Subsequently, an innovative method was established to identify phenoxyl radicals in AOPs. Thereafter, the critical roles of high-valent metal-oxo species, e.g., III , and , in the polymerization of pollutants were elucidated. Last, the -band center-dependent mechanism for achieving PT ratio was revealed with a combination of theoretical calculations and experimental results. Overall, this work unravels a mechanism for pollutant removal in the SACs-AOPs and proposes a foundational guideline for designing SACs-based catalytic systems with selectivity for pollutant removal via the robust PT process.
Results
Synthesis and characterizations of TM-SA/PN-g-C3
A hydrogen-bonding-assisted pyrolysis strategy was employed to fabricate a series of single transition metal-embedded pyrrolic N-rich graphitic carbon nitride (TM-SA/PN-g-C3 , etc.) catalysts. This facile method allowed for the gram-scale production of TM-SACs with high metal loadings (Supplementary Methods and Fig. 1a) . Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis indicates that the metal loading was 9.46, 7.02, 7.71, and for , and Fe , respectively (Supplementary Fig. 1a). The absence of characteristic crystal peaks of metals in the powder X-ray diffraction (XRD) patterns of the TM-SA/PN-g-C3
samples (Supplementary Fig. 1b) excluded the presence of large crystalline particles . Likewise, high-resolution transmission electron microscopy (HR-TEM) did not detect any visible nanoparticles (Fig. 1b and Supplementary Fig. 2). Scanning electron microscopy (SEM) images reveal that the TM-SA/PN-g- samples possessed similar curved flake-like morphology with a porous structure. The main mesopore structure endorsed the TM-SA/PN-g-C3 with high surface areas as indicated by the Brunauer-Emmett-Teller (BET) measurements (Fig. 1b and Supplementary Figs. 3 and 4). Energy-dispersive spectroscopy (EDS) mapping images verify the uniform distributions of the metal, C , and N elements across these architectures (Fig. 1c and Supplementary Fig. 5). Then, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADFSTEM) with sub-angstrom resolution was utilized to identify the dispersion of metal species . As shown in Fig. 1d and Supplementary Fig. 6, high-density single metal atoms represented as isolated bright dots marked with red circles were uniformly dispersed in the TM-SA/ PN-g- samples. Such observations could be further confirmed by the three-dimensional (3D) isoline and atomic overlapping Gaussian function fitting mappings (Fig. 1e and Supplementary Fig. 6). Furthermore, the intensity distribution along in reveals the atomic spacing for , and Fe -SACs were approximately , 0.35 , and 0.35 nm , respectively (Fig. 1d and Supplementary Fig. 6). Overall, these results confirm the atomic loading of , and Fe on the TM-SA/PN-g-C3 samples without the existence of metalderived crystalline structures.
The electronic and atomic structures of metal atoms in the TM-SA/PN-g-C3 samples were further explored using X-ray absorption fine structure (XAFS) spectra. Taking Co-SA/PN-g-C3 as an example, the X-ray absorption near edge structure (XANES) profiles in Fig. 1f shows that the energy absorption intensity of the white line for was higher than that for Co foil and lower than that for CoO , indicating that the valence state of the Co atom lay between and . The lower valence state of Co than its precursor signifies a notable metal-support interaction within the sample. Besides, Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra of -space for the Co-SA/PN-g-C3 exhibited a principal peak at about , deviating from the metal scattering path of at about in Co foil. This result confirms the isolated distribution of Co atoms on the PN-g-C3 support (Fig. 1g). Consistent with the FT-EXAFS analysis, wavelet transform (WT) contour plots visually show only the intensity maximum related to or interaction, without any interaction for the (Fig. 1h).
Furthermore, the quantitative coordination configuration of Co in the Co-SA/PN-g-C3 was analyzed by EXAFS fitting. The simulation indicates a primary peak stemming from or first shell coordination with a calculated coordination number of approximately 4.0 (Supplementary Figs. 9 and 10). The optimal-fitting results from the EXAFS data are concisely detailed in Supplementary Table 2. Nevertheless, due to the potential presence of O atoms in the microenvironment, the precise coordination structure of the Co single atom remained unclear. Therefore, time-of-flight secondary ion mass spectrometry (TOF-SIMS) measurement using ion beam sputtering was utilized to validate the coordination structure of Co in the by probing the molecular weight of the or O unit (Supplementary Fig. 11a)²5. Typical secondary negative and positive ions ( ) were detected in the Co-SA/PN-, affirming the atomic site in the (Fig. 1i). Such TM- structure was also verified for the other TM-SA/ , and Fe ) samples with the similar analysis methods (Supplementary Figs. 7-11 and Supplementary Note 1). Moreover, the C and -edge XANES spectra confirm that metal doping had minimal impact on the atomic structure of the PN-g-C3 substrate (Supplementary Fig. 12).
Fig. 1 | Synthesis and characterization of the TM (Cu, Ni, Co, and Fe)-SA/PN-g. a Schematic illustration of the preparation procedures of the TM ( , and Fe )-SA/PN-g- samples. SEM image (inset: HR-TEM image), (c) HAADFSTEM image with the corresponding EDS elemental mapping, and (d) AC HAADFSTEM image of , the single atoms are marked with isolated red circles in (d). e3D isolines and atom-overlapping Gaussian-function fitting mapping
of the yellow rectangle from (d) and intensity profile along in (d). Co -edge XANES and (g) Fourier-transformed -space EXAFS of Co -SA/PN-g- , Co foil, , and CoPc. WT-EXAFS plots of , and Co foil. i TOF-SIMS high-resolution ion spectra for structural unit. Source data are provided as a Source Data file.
These characterizations show that the TM-SA/PN-g-C3 catalysts ( , Ni, Co , and Fe ) exhibited similar physicochemical properties and coordination structure (e.g., TM- ), which provided an ideal platform for revealing the electronic structure-dependent catalytic feature in SACs-AOPs.
Pivotal roles of high-valent metal-oxo species in pollutant removal
The Fenton-like catalytic capabilities of the four TM-SA/PN-g-C3 catalysts were examined for phenol ( PhOH ) removal in the presence of PMS. The reduction in PhOH concentration was not attributed to adsorption by the PN-g- substrate (Supplementary Fig. 13) or degradation catalyzed by the leached metal ions (Supplementary Fig. 14), suggesting the principal role of the embedded single metal species in PhOH removal. As shown in Fig. 2a, the observed reactivity sequence of the catalysts followed the order of , which is consistent with the order of turnover frequency values (TOF, mass-specific activity, Supplementary Fig. 15). Understanding the molecular origin of such a reactivity sequence is critical for highly efficient SAC fabrication for Fenton-like catalysis, but it faces a great challenge due to the contentious PMS activation mechanism . Therefore, experiments were conducted to elucidate the reaction mechanism with electron paramagnetic resonance (EPR), reactive species quenching, isotope labeling, methyl phenyl sulfoxide (PMSO) trapping, in situ Raman spectroscopy, and soft X-ray absorption spectra (XAS). As shown in Fig. 2b, a typical seven-line EPR signal of
5,5-dimethyl-1-pyrrolidone-N-oxyl (DMPOX) was observed in the coexistence of and PMS, which could be attributed to 5,5-dimethyl-1-pyrroline N-oxide (DMPO) oxidation by high-valent metals or excessive amount of or . The reactive species responsible for DMPOX generation also contributed to PhOH removal because of the significantly depressed signal intensity of DMPOX when PhOH was introduced (Fig. 2b). Reactive species quenching experimental results show that methanol ( MeOH ) and tert-butanol (TBA) negligibly inhibited the removal efficiency of PhOH within 30 min (Supplementary Fig. 16), implying that neither nor was produced. This result is also validated by the EPR studies, which show an increase rather than decrease in DMPOX signal intensity after MeOH addition (Fig. 2b). Furthermore, the characteristic triplet EPR peaks indicated the emergence of , a commonly proposed reactive species in PMS-AOP (Supplementary Fig. 17). It is widely reported that deuterium oxide ( ) can enhance the oxidation efficiency of because it prolongs the lifetime of by approximately 18 times ( in vs. in . However, the solvent exchange ( to ) did not enhance PhOH degradation, suggesting the negligible contribution of (Supplementary Fig. 18). In addition, the contribution of and surface-attached radicals were also excluded by the EPR experiments (Supplementary Figs. 19 and 20, and Supplementary Note 2). Thus, these results demonstrate that high-valent Co species, i.e., , might be primarily responsible for PhOH removal and DMPOX generation in the Co-SA/PN-g-C3 PMS system.
Fig. 2 | The pivotal role of for pollutant removal. a Comparison of pollutant removal efficiency between TM ( , and Fe )-SA/PN-g- . Error bars represent the standard deviation, obtained by repeating the experiment three times. EPR spectra of the different systems with DMPO as the spin-trapping agent. c PMSO probe experiment in Co-SA/PN-g-C system. Error bars represent the standard deviation, obtained by repeating the experiment two times. Reaction conditions in (a-c) [PMS] = Cat. , , initial , and . d In situ Raman study (the pink shade represents the signal of ). e Soft-XAS analyzes (the green shade represents the signal of ). Fitting of the relationship between the oxygen reduction potential of high-valent metals and the removal efficiency of PhOH within 30 min . Source data are provided as a Source Data file.
To provide compelling evidence for the crucial role of , the quenching experiment using dimethyl sulfoxide (DMSO) was conducted. DMSO has been widely recognized as an effective inhibitor of high-valent metals through the oxygen transfer reaction . The quenching experiment results in Supplementary Fig. 16 show that ca. 10 mM DMSO completely inhibited PhOH degradation. It should be noted that DMSO not only eliminated , but also directly reacted with PMS at a reaction rate constant of . However, the catalytic activation of PMS by Co-SA/ PN-g- was still observed in the presence of DMSO, due to a significantly higher decomposition rate of PMS in the mixed Co-SA/ PN-g- DMSO system compared to DMSO alone (Supplementary Fig. 21) . These results support the critical role of in PhOH removal in the Co-SA/PN-g-C3 PMS system, which was further verified by the substantial decrease in the EPR peak intensity of DMPOX when DMSO was introduced (Fig. 2b). Furthermore, PMSO, which can be oxidized by via the oxygen transfer pathway to yield the specific methyl phenyl sulfone ( ) product , was applied as a chemical probe to confirm the presence of . As shown in Fig. 2c, the yield of , ( , the molar ratio of produced to PMSO lost, was found to be approximately over the reaction time, validating as the primary oxidative species generated in the Co-SA/PN-g-C3 PMS system. The presence of was further confirmed by in situ Raman studies. The new peak at after the introduction of PMS could be attributed to the stretching vibration of the structure, which completely disappeared when PhOH was present (Fig. 2d), indicating the critical role of in PhOH removal. Additionally, the occurrence of was verified by XAS analyzes. As shown in Fig. 2e, the absorption peak of the original Co-SA/PN-g at 780.4 eV positively shifted by 0.2 eV after PMS addition, demonstrating the oxidation of the surface Co species. Meanwhile, a new peak at approximately 800 eV further confirms the generation of .
The surface species can be preserved on the Co-SA/PN- through pre-oxidation with PMS in the absence of pollutants. Such a pre-oxidized catalyst exhibited an impressive direct oxidative capacity for PhOH removal ( ), significantly higher than the adsorption capacity of the fresh (Supplementary Fig. 29). X-ray photoelectron spectroscopy (XPS) was used to track the change in the valence state of Co throughout the catalytic reaction process involving PMS activation and PhOH degradation (Supplementary Fig. 30c). The observed trend of initial increase followed by subsequent decrease provides clear evidence for the involvement of the high-valent Co species in PhOH degradation. Similar to other highvalent metal-based Fenton-like systems , electrophilic with high-selective features could effectively remove pollutants (e.g., PhOH and ) with electron-donating groups ( -OH and ) only, but failed to remove benzoic acid with an electronwithdrawing group (-COOH) (Supplementary Figs. 31-33). Importantly, the dominant role of high-valent metals (e.g., , and ) for PhOH removal was also confirmed in the other TM-SA/PN-g-C3 catalysts under the same conditions for the Co-SA/ PN-g- . Detailed results and discussions can be found in Supplementary Figs. 13-35 and Supplementary Note 2. The unique electronic structures of these high-valent metals significantly influenced their redox potentials, as revealed by the cyclic voltammetry (CV) tests (Supplementary Fig. 36), which showed a linear correlation between the removal efficiency of PhOH and potential ( , Fig. 2f), further demonstrating the pivotal role of high-valent metals for pollutant removal in the SACs-catalytic PMS systems.
In the Co-SA/PN-g- PMS system, the aqueous TOC was synchronously removed with PhOH at a high efficiency ( , Fig. 3a). Such unusual disappearance of aqueous TOC resulted in the accumulation of PhOH cross-linking polymers on the surface of catalysts, which could be directly identified by the matter-assisted laser desorption/
Fig. 3 | Characteristics of the polymerization products of 2,6-M-PhOH on Co-
SA/PN-g-C3 . a TOC and PhOH were removed synchronously and efficiently in Co-SA/PN-g-C3 PMS aqueous solution (Inset is schematic diagram of a hypothesis deductive method using ). The bar graphs correspond to the TOC values. Error bars represent the standard deviation, obtained by repeating the experiment three times. b SEM-EDS and O 1s of XPS, (c) TGA curves of Co-SA/PN-g-
before and after the reaction. d MALDI-TOF-MS, (e) NMR-based structural analysis (e), and (f) GPC of elution products on the surface of the Co-SA/PN-g-C3 . The pink, blue, gray, and purple shades in e represent four kinds of aromatic carbon, one kind of aliphatic carbon, , and, THF, respectively. Reaction conditions in (a-f) [PMS] = Cat. , initial , and . Source data are provided as a Source Data file.
ionization time-of-flight mass spectrometry (MALDI-TOF-MS) tests (Supplementary Fig. 37). However, PhOH with three active hydrogen sites in its molecular structure (i.e., the ortho- and para-positions of the hydroxyl group) was readily oxidized to form a network-like crosslinked polymer, which was insoluble in organic solvents such as trihalomethanes (THMs), acetonitrile (ACN), and tetrahydrofuran (THF) (Supplementary Fig. 37a). Therefore, 2,6-dimethylphenol (2,6-M with only one active site, which tends to form non-crosslinked polymerization product, was selected to investigate the initiated polymerization reactions (Fig. 3a), which were studied by multiple characterizations including SEM-EDS, XPS, and thermal gravimetric analysis (TGA). As shown in Fig. 3b and Supplementary Fig. 40, after the disappearance of in solution, an increase in C and O elements on the was observed by SEMEDS. This result was verified by the XPS O results, which showed a distinct transition in surface O species from the hydroxyl group to (Fig. 3b and Supplementary Figs. 43a and 44a). The postreaction XPS C 1s results also reveal a significant increase in and bonds (Supplementary Figs. 45 and 46). In contrast, no considerable changes were noted in SEM-EDS or XPS spectra of the PN-g substrate before and after the reaction (Supplementary Figs. 42 and 46), signifying the crucial role of the metal center in pollutant polymerization. Also, negligible changes in XPS N 1s spectra suggest that the coordination environment of the single atom remained stable in the reaction (Supplementary Figs. 43b and 44b). These findings reveal a substantial accumulation of organic products on the catalyst surface. Such products could be pyrolyzed in the temperature programming process with weight loss compared to the pristine as revealed by the TGA analyzes (Fig. 3c and Supplementary Figs. 47 and 48). To further understand the properties of the products formed on the Co-SA/PN-g-C3 surface, the deposited substances were separated using THF solvent and characterized by MALDI-TOF-MS, gel permeation chromatography (GPC), nuclear magnetic resonance (NMR), and Fourier transform
infrared spectroscopy (FTIR) analyzes. The eluted products, consisting of repeat units with an of 120.4 , exhibited a distinct polyphenylene oxide (PPO) structure, as evidenced by MALDI-TOF-MS (Fig. 3d). Additionally, the PPO structure of the polymerization product was also proven by the similar aromatic/aliphatic carbons and the aromatic groups compared with the standard PPO sample, as revealed by the NMR and FTIR analyzes, respectively (Fig. 3e and Supplementary Fig. 51). The average of the chain-like products was identified as 4981 Da (Fig. 3f).
Overall, the accumulated organics on the Co-SA/PN-g-C3 surface were characterized as PPO, which was also confirmed in the polymerization removal of by the other TM ( , and Fe )-SA/PN-g- catalysts (Supplementary Figs. 37-52 and Supplementary Note 3). The polymers on the catalyst surface would not affect the surface reactive sites, and they could be facilely collected by an elution-drying protocol with a recovery ratio of , and for and Co -SACs, respectively (Supplementary Figs. 53 and 54, and Supplementary Note 3). However, how the polymerization removal of pollutants was triggered by high-valent metal species is still elusive.
Identification of phenoxyl radicals in polymer formation
Although phenoxyl radicals have been considered a key intermediate in polymer formation13-15, whether phenoxyl radicals emerged and how to detect the low-activity radical in the Fenton-like polymerization systems remains challenging. Thus, two innovative protocols, in which (2,2,6,6-tetramethylpiperidin-1-yl) oxyl (TEMPO)-based traps containing alkyl (CHANT) were employed as the trapping agent and ferulic acid (FA) as the quenching agent, were applied to identify phenoxyl radicals in the PMS-AOPs.
Cross-coupling and spin-trapping are commonly used to stabilize transient radicals in EPR analysis (Supplementary Fig. 55a, b). However, these methods have deficiencies of limited structural information for trapped free radicals, poor sensitivity, and a high
incidence of false positives due to side reactions . To overcome these drawbacks, we developed a detection protocol for phenoxyl radicals using a recently reported free radical trap, CHANT (Supplementary Fig. 56. In such a protocol, ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS) was combined with EPR analysis to enable accurate and reliable detection (Supplementary Fig. 55c). As shown in Fig. 4a, the reaction of a phenoxyl radical with CHANT yielded a stable, non-radical product and released the TEMPO persistent radical, which were detected by UPLC-MS with of 288.1930 (Fig. 4b) and probed by EPR with the typical triplet peaks (Fig. 4c), respectively. When DMSO, a scavenger for , was added, the EPR signal for the TEMPO persistent radical completely disappeared (Fig. 4c). Then, TEMPO, as a traditional cross-coupling agent, was also used to capture
phenoxyl radicals. The results show that phenoxyl radicals could cross-couple with TEMPO to form a stable adduct with of 278.2075 (Supplementary Fig. 59d). In addition, inspired by the antioxidant function of FA in the field of phytochemistry , we developed FA as a phenoxyl radical scavenger to convert phenoxyl radicals back to the redox-inert phenol precursor (Fig. 4d). The results show that ca. 10 mM FA completely inhibited the removal of PhOH in the Co-SA/PN-g-C3 PMS system (Fig. 4e and Supplementary Fig. 61). Although FA can react with PMS directly, the catalytic activation of PMS by Co-SA/PN-g-C3 was still observed in the presence of FA (Supplementary Fig. 61f), suggesting the critical role of phenoxyl radicals in the polymerization reaction of pollutants. Thus, these results firmly support the -induced formation mechanism of phenoxyl radicals in the polymerization process of
Fig. 4 | Identification of phenoxyl radicals. a A versatile approach for sequestering phenoxyl radicals using CHANT. b UPLC-MS chromatogram and (c) EPR spectra for probing the reaction product between CHANT and phenoxyl radicals. d Schematic illustration of phenoxyl radicals quenching by FA. e The effect of FA on PhOH degradation. Error bars represent the standard deviation, obtained by repeating the experiment two times. kinetic isotope effect for aniline
pollutants, the universality of such a mechanism was also confirmed for the other TM ( , and Fe )-SA/PN-g- catalysts (Supplementary Figs. 55-61, Supplementary Note 4).
Moreover, it was reported that high-valent metals could oxidize pollutants into phenoxyl radicals via the hydrogen abstraction (HA) or proton-coupled electron transfer (PCET) pathway. The minor kinetic isotope effect (KIE) value excluded the HA pathway, suggesting that substrate oxidation was driven by PCET to generate the corresponding organic radicals in the four TM-SA/PN-g-C3 PMS systems (Fig. 4 f and Supplementary Fig. 62). Overall, we discover the mediating role of the high-valent metals triggered phenoxyl radicals for the polymerization removal of pollutants. In addition, the method established for phenoxyl radical identification in this work is anticipated to have great application potential for a wide range of reactions involving organic free radicals, including photocatalysis, electrocatalysis, and organic synthesis .
Regulating -band center for polymerization removal of pollutants
High-valent metal species, encompassing III , and , are capable of initiating the polymerization removal of pollutants through the identical PCET pathway. However, these species also exhibit an electronic-structure-dependent behavior, leading to over-oxidation and decomposition of pollutants into small molecular organic acids.
Notably, the PT ratio for the Fe-SACs was substantially lower, at merely , compared to the values of , and for the , and Co -based SACs, respectively (Fig. 5a and Supplementary Fig. 32). This result suggests that pollutants could be completely polymerized on the surface of the TMs ( , and Co )-SA/PN-g-C catalysts. However, for the Fe-SA/PN-g-C3 PMS system, an additional pollutant removal mechanism, possibly involving the mineralization degradation pathway, might exist in this process. This hypothesis was further corroborated by the markedly lower of the chain-like polymer on the surface of the Fe-SA/PN-g-C3 catalyst, as indicated by the GPC analyzes (Fig. 5a and Supplementary Fig. 52). This finding implies the further degradation of pollutants into small molecule substances by the species. Thus, understanding how the electronic structure of the catalytic center regulated the removal mechanism of the pollutants is essential to provide fundamental guidance for catalytic system design with impressive functions for pollutant polymerization removal from wastewater.
The distinct oxidative feature of was revealed through a combination of density functional theory (DFT) calculations and experimental designs. As shown in Fig. 5b, the formation of phenoxyl radicals through the PCET mechanism includes three steps: adsorption of PMS on the TM-SA site (Step 1), the subsequent two-electron transfer leading to the production of high-valent metals (Step 2), and the oxidation of pollutant to yield phenoxyl radicals via the PCET pathway (Step 3). Eventually, the formed phenoxyl radicals would undergo further polymerization reaction on the catalyst surface. Thus, the electronic structure of the catalytic center could exhibit significant impacts on the pollutant removal mechanism by affecting the abovementioned adsorption and electron transfer processes.
In the initial PMS adsorption process (Step 1), the projected density of states (PDOS) analyzes reveal that Fe site possesses the highest -band center compared to that of , and Co . Also, the band center of Fe further rises and approaches the Fermi-level ( ) after PMS adsorption ( Fe (II)-PMS) to form the high-valent metal-oxo . In contrast, Co and Ni sites exhibit decreasing trends throughout the aforementioned processes (Fig. 5c, d). Typically, a band center proximity to the generally indicates an enhanced propensity of the catalyst to donate charge to the adsorbate , and thus a precisely linear correlation between the -band center of metals and the adsorption energy of PMS was observed (Fig. 5e and
Supplementary Fig. 65). Besides, the asymmetric arrangement of orbital electrons in the spin channel ( ) further validates the stronger bond strength and larger electron transfer due to spin polarization (Supplementary Fig. 67) . When compared with the species, conspicuously exhibits an augmented overlap of the and orbitals ( 3.630 vs. 2.487, Fig. 5f), leading to a more extensive electron delocalization over the configuration and a stronger binding ability of the axial Fe for . The high affinity of Fe site for PMS was verified by the in situ Raman characterizations [a new peak at , corresponding to the activated peroxo-species (PMS*)], which was observed on the Fe-SA/PN-g only (Fig. 5g). Moreover, the characteristic vibration peak of PMS* underwent a blue-shift of (from 1060 to ), indicating a strong interaction between PMS and the Fe- site on the catalyst . Such peaks remained absent in the other TM-SA/PN-g-C3 catalyst spectra. Upon addition of pollutants, the simultaneous disappearance of signals corresponding to and emphasized their collaborative role in pollutant elimination, operating through both PT and mineralization pathways (Supplementary Fig. 27).
To understand the configuration of PMS* on the catalyst surface, DFT calculations were carried out. Our computational insights reveal a marked affinity of PMS towards the Fe(IV) site in an axial orientation, culminating in the formation of the PMS*- complex, a more favored configuration over the (Supplementary Fig. 70). At the subsequent stage, the pollutants tended to occupy the available O site. Notably, the adsorption energy linked with the PMS* complex was discernibly greater than that associated with PMS*-Co(IV)=O, as exemplified by the energy values of -0.292 eV for Fe and -0.244 eV for Co (Fig. 5h). Such an elevated adsorption energy typically resonated with an increased likelihood of undergoing oxidation. The validity of this assertion was underpinned by the observable substantial electron depletion in pollutants when in the proximity of (Fig. 5h and Supplementary Note 6).
Overall, the above simulation and experimental results demonstrate that the higher -band center of Fe would result in a stronger interaction between and orbitals to form in the axial direction, which might subsequently initiate the mineralization of pollutants in the Fe-SA/PN-g-C3 PMS system.
For the oxidative generation of phenoxyl radicals (Step 3), the surface adsorption of PMS was observed to elongate the O-O bond with PMS, thus enhancing the oxidative capacity of the TM-SA/PN-g PMS systems when high-valent metals were involved. The different oxidative stresses significantly affected the subsequent behaviors of the phenoxyl radicals. To empirically ascertain this oxidative potential alteration ( ) post the creation of PMS*, open-circuit voltage measurements were conducted . As shown in Fig. 6a, the positive shifts in open-circuit voltage upon introducing PMS indicate the elevation of the oxidative capacity across all four TM-SA/PN-g PMS systems. In a decreasing order of their amplitude, the voltage increments were arranged as: . Apparently, Fe with the highest -band center and highest affinity for PMS showed the most significant increase in the oxidative capacity after PMS* formation ( , Fig. 6a and Supplementary Fig. 75). The elevated oxidation capacity of the Fe-SA/PN-g-C3 PMS system after the FePMS* complex formation was verified by various electrochemical characterizations. The chronoamperometric analyzes elucidated a swift dip in current trends post PMS infusion, implying an immediate electron transference from PMS to the metallic centers of the TM (comprising , and Fe ) in the SA/PN-g- catalysts, fostering the birth of high-valent metal entities (Fig. 6b). It is worth noting that when was subsequently added, the current response initially dropped, but gradually increased for the Fe-SA/PN-g-C3 , while it merely displayed a downtrend for the (Fig. 6b). Such a vivid difference was a testament to the formidable oxidative caliber of the Fe-PMS* complex, which could robustly extract
Fig. 5 | Depicting the roles of the -band center in oxidant and pollutant bindings. a The PT ratios and of the elution products in the four TM-SA/PN-g systems. Reaction conditions in , [Cat.] , initial , and . b Schematic diagram of phenoxyl radicals formation. PDOS of TM ( , and Fe ) in TM-SA/PN-g- systems of (c) step 1 and (d) step 2. e The relationship between the -band center and the adsorption energy of PMS. The PDOS of and TM (Fe and Co ) states on
TM(IV) structure, and the integrated overlapping area is labeled in the top left. g The identification of surface Fe-PMS* species (marked in red shade) by the Raman spectra. Charge density difference and corresponding theoretical adsorption energy ( ) for PMS adsorption in the axial direction, in which light green and yellow areas indicate electron accumulation and depletion regions, respectively (isosurface bohr ). Source data are provided as a Source Data file.
electrons from , manifested as an enhanced current density uptick . To quantitatively describe the oxidation capacity of the TM-SA/PN-g-C3 PMS system, , which represents the sum of the redox potential of the high-valent metals (Fig. 2f) and the increment in open circuit voltage after PMS addition ( , Fig. 6a), was therefore defined (Eq. 2). The values were determined to be , and 1.37 V for the , and Cu SAC -based reaction systems, respectively.
As shown in the Gibbs free energy change plot (Fig. 6c), a spontaneous thermodynamic progression for phenoxyl radical formation could be observed for the four TM-SA/PN-g-C3 PMS systems with the tendency in the order of . The linear relationship between the and the Gibbs free energy for phenoxyl radicals formation demonstrated phenoxyl radicals as the key intermediate in removing phenolic pollutants in the TM-SA/PN-g /PMS systems (Fig. 6d). Further elucidation from UPLC-MS analyzes provides a transformative trajectory of in tandem with the augmentation of the oxidative capacity inherent to the
TM-SA/PN-g-C3 PMS system. Molecular transformations encompassing hydroxylation, ring cleavage, and subsequent derivation of elementary organic acids such as glycolic, pyruvic, and mesoxalic acid were discerned within the , and Fe-SA/PN-g-C3 systems, respectively (Fig. 6e and Supplementary Figs. 76-80). These findings substantiate that, beyond the well-documented polymerization pathway, the Fe-SA/PN-g-C3 PMS system with the paramount oxidation capacity can induce the over-oxidation of pollutants. This process culminates in the generation of organic acids that persist in the bulk solution, leading to the most diminished PT ratio.
Therefore, tailoring -band center of high-valent metal-oxo species can achieve effective pollutant removal via complete polymerization, which exhibits impressive robustness against varying environmental conditions. The catalytic activities of the , and SA/PN-g-C3 catalysts, which feature a polymerization catalytic pathway, were negligibly affected by the co-existed anions (e.g., , , and ) and water matrices (e.g., tap water, lake water, and secondary effluent from wastewater treatment plants). This result underscores the robustness of these catalysts in diverse aqueous environments. In contrast, the catalytic activity of was slightly inhibited due to the involvement of a higher proportion of
valent metal-oxo species formation, and phenoxyl radical formation in the different TM ( , and Co )-SA/PN-g- PMS systems. d The relationship between the oxidation capacity of the reaction system and the Gibbs free energy for phenoxyl radical formation. e The degree of oxidation in the different TM ( , and Co )-SA/PN-g-C3 PMS systems. Source data are provided as a Source Data file.
mineralization degradation ( ). In addition, an aqueous pH ranging from 4.0 to 9.0 had little effect on the removal efficiency or the PT ratio of PhOH in the four TM-SA/PN-g- systems. These results demonstrate the impressive anti-interference ability of the polymerization reaction in real wastewater treatment, confirming its versatility and effectiveness in pollutant removal (Supplementary Figs. 81-86 and Supplementary Note 5).
Discussion
In summary, our work provides fundamental insights into the regulation of the catalytic polymerization and mineralization pathways for pollutant removal through modulating the -band center of the TM (TM: , and Fe )-SACs (Supplementary Fig. 87). Both DFT calculations and experimental investigations prove that the high-valent metal-oxo species (e.g., , and ) with lower -band centers could achieve nearly 100% PT ratio for pollutant removal, whereas with a higher -band center could strengthen the re-binding of PMS, thus resulting in a higher oxidative capacity to over-oxidize pollutants into small molecular acids and with the lowest PT ratio ( ). In addition, the high-valent metal-oxo species could trigger the one-electron oxidation of pollutants to generate the key phenoxyl radicals intermediate for further polymerization reactions, which was identified by a combination of innovative spin-trapping and quenching methods. This work provides deep insights into the structure-function relationship between the electronic structure of the catalytic center and the PT ratio for pollutant removal at the atomic level. The results would be helpful for developing sustainable water purification technologies to simultaneously realize contamination abatement and resource recovery in a lowcarbon manner.
Methods
Chemicals
Iron nitrate nonahydrate , copper nitrate trihydrate , cobalt nitrate hexahydrate , nickel nitrate hexahydrate , ), chromium nitrate hexahydrate , zinc nitrate hexahydrate , melamine , sodium thiosulfate ( ), potassium iodide ( ), MeOH ( ), ethanol ( ), and TBA ( ) were purchased from Sinopharm Chemical Reagent Co., China. PhOH ( ), 2,6-M-PhOH ( ), benzoic acid ( ), xanthine ( ), cyanuric acid ( ), PMS ( ), atom , aniline ( ), DMSO ( ), methyl phenyl sulfoxide (PMSO, 97%), methyl phenyl sulfone ( ), 2,2,6,6-tetramethyl-4-piperidinol (TEMP, 98%), and DMPO (97%) were acquired from Aladdin Chemistry Co., China. Carbon papers were purchased from Toray Co., Japan. Aniline-D5 (98 atom% D) and Anline-D7 (98 atom% D) were obtained from Shanghai Civi Chemical Technology Co., China and J&K Scientific Co., China, respectively. Nafion solution ( ) and ACN (>99%) were bought from Sigma-Aldrich Co., China. Deionized (DI) water ( ) was used in all the experiments. All chemicals were used as received without further purification.
Catalyst preparation
For the synthesis of the TM-(TM: , and Fe ) SA/PN-g- catalysts, cyanuric acid ( 9.6 mM ) and xanthine ( 2.4 mM ) were first dissolved into 80 mL DI water under ultrasonication. Then, 80 mL of melamine solution ( 1.2 mM ) was quickly mixed with the above solution to form a suspension, which was ultrasonically by 20 min and magnetically stirred for a further 4 h . Subsequently, the metal precursor solution was prepared by dissolving 9.6 mM of the corresponding metal salt in 40 mL DI water, which was added to the above suspension and stirred for 1 h to achieve the self-assembly of the metal-carbon polymer. The assemblies were centrifuged and washed with DI water several times and dried in a vacuum at for 10 h . Finally, the dried samples were ground in a mortar and calcined at under atmosphere with a heating rate of to obtain the TM-(TM= , and Co ) SA/PN-g- catalysts. The PN-g- substrate
was prepared according to the above procedures in the absence of metal precursors.
Characterizations
The characterization methods including XRD, BET, XPS, SEM, HR-TEM, HAADF-STEM, FTIR, EPR, soft-XAS, and XAFS are described in detail in Supplementary Methods.
Pollutant degradation experiments
Pollutant degradation experiments were conducted at least in duplicate in 50 mL beakers at under magnetic stirring. In a typical test, a certain mass of TM-SA/PN-g- was added to the aqueous solution containing the desired amounts of pollutant and PMS. Unless otherwise stated, the molar ratio of oxidants to pollutants was fixed at 2:1, and the initial pH was adjusted to 7.0 by or NaOH . In the reaction process, 1 mL of the reaction suspension was taken out and quenched by , the mixture was then filtered through a membrane for further analysis.
Analytical methods
The qualitative and quantitative analytical methods including UPLC, UPLC-MS measurement, TOC measurements, quenching experiments, KIE Experiments, pre-oxidation experiments, MALDI-TOF measurement, C-NMR characterization, GPC measurement, and electrochemical tests are available in Supplementary Methods.
Quantum chemical calculations
All DFT calculations were performed on CASTEP , seeing Supplementary Methods for more details. The atomic coordinates of the optimized structure are provided with this paper.
Data availability
The data that supports the findings of the study are included in the main text and supplementary information files. Raw data can be obtained from the corresponding author upon request. Source data are provided with this paper.
References
Naddaf, M. The world faces a water crisis- 4 powerful charts show how. Nature 615, 774-775 (2023).
The water crisis is worsening. Researchers must tackle it together. Nature. 613, 611-612 (2023).
Gleick, P. H. Moving to a sustainable future for water. Nat. Water 1, 486-487 (2023).
Lu, L. et al. Wastewater treatment for carbon capture and utilization. Nat. Sustain. 1, 750-758 (2018).
Dickin, S., Bayoumi, M., Giné, R., Andersson, K. & Jiménez, A. Sustainable sanitation and gaps in global climate policy and financing. npj Clean Water. 3, 24 (2020).
Gibb, B. C. Weird and wonderful water. Nat. Chem. 8, 733-734 (2016).
Hodges, B. C., Cates, E. L. & Kim, J. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 13, 642-650 (2018).
Zhang, S., Zheng, H. & Tratnyek, P. G. Advanced redox processes for sustainable water treatment. Nat. Water 1, 666-681 (2023).
Shang, Y., Xu, X., Gao, B., Wang, S. & Duan, X. Single-atom catalysis in advanced oxidation processes for environmental remediation. Chem. Soc. Rev. 5, 5281-5322 (2021).
Ren, W. et al. Origins of electron-transfer regime in persulfatebased nonradical oxidation processes. Environ. Sci. Technol. 56, 78-97 (2022).
Lee, J., von Gunten, U. & Kim, J. H. Persulfate-based advanced oxidation: critical assessment of opportunities and roadblocks. Environ. Sci. Technol. 54, 3064-3081 (2020).
Wu, X. & Kim, J. Outlook on single atom catalysts for persulfate-based advanced oxidation. ACS ES&T Eng. 2, 1776-1796 (2022).
Wang, C. et al. Selective oxidation of various phenolic contaminants by activated persulfate via the hydrogen abstraction pathway. ACS ES&T Eng. 1, 1275-1286 (2021).
Wang, C. et al. Catalytic reactivity of with different facets in the hydrogen abstraction of phenol by persulfate. Appl. Catal., B. 270, 118819 (2020).
Liu, L. et al. Nonradical activation of peroxydisulfate promoted by oxygen vacancy-laden NiO for catalytic phenol oxidative polymerization. Appl. Catal., B. 254, 166-173 (2019).
Liu, T. et al. Oxidative polymerization of bisphenol A (BPA) via H -abstraction by and persulfate: importance of the surface complexes. Chem. Eng. J. 435, 134816 (2022).
Zhang, Y. et al. Simultaneous nanocatalytic surface activation of pollutants and oxidants for highly efficient water decontamination. Nat. Commun. 13, 3005 (2022).
Huang, M. et al. Facilely tuning the intrinsic catalytic sites of the spinel oxide for peroxymonosulfate activation: from fundamental investigation to pilot-scale demonstration. Proc. Natl. Acad. Sci. USA. 119, e2092285177 (2022).
Wang, J. et al. Interlayer structure manipulation of iron oxychloride by potassium cation intercalation to steer activation pathway. J. Am. Chem. Soc. 144, 4294-4299 (2022).
Zhang, Y. et al. Distinguishing homogeneous advanced oxidation processes in bulk water from heterogeneous surface reactions in organic oxidation. Proc. Natl. Acad. Sci. USA. 120, e1992560176 (2023).
Qiao, B. et al. Single-atom catalysis of CO oxidation using . Nat. Chem. 3, 634-641 (2011).
Yu, X. et al. A green edge-hosted zinc single-site heterogeneous catalyst for superior fenton-like activity. Proc. Natl. Acad. Sci. USA. 120, e2073739176 (2023).
Cheng, C . et al. Generation of and its contribution to fenton-like reactions on a single-atom iron-N-C catalyst. Angew. Chem. Int. Ed. Engl. 62, e202218510 (2023).
Gao, Y. et al. Activity trends and mechanisms in peroxymonosulfate-assisted catalytic production of singlet oxygen over atomic metal-N-C catalysts. Angew. Chem. Int. Ed. Engl. 133, 22687-22695 (2021).
Wang, B. et al. A site distance effect induced by reactant molecule matchup in single-atom catalysts for fenton-like reactions. Angew. Chem. Int. Ed. Engl. 61, e202207268 (2022).
Mi, X. et al. Almost peroxymonosulfate conversion to singlet oxygen on single-atom sites. Angew. Chem. Int. Ed. Engl. 60, 4588-4593 (2021).
Pan, J. et al. Improving peroxymonosulfate activation by copper ion-saturated adsorbent-based single atom catalysts for the degradation of organic contaminants: electron-transfer mechanism and the key role of Cu single atoms. J. Mater. Chem. A. 9, 1164-11613 (2021).
Qian, K. et al. Single-atom Fe catalyst outperforms its homogeneous counterpart for activating peroxymonosulfate to achieve effective degradation of organic contaminants. Environ. Sci. Technol. 55, 7034-7043 (2021).
Chen, F. et al. Molecular engineering toward pyrrolic N -rich ( ) single-atom sites for enhanced heterogeneous fenton-like reaction. Adv. Funct. Mater. 31, 2007877 (2021).
Han, G. et al. Abrading bulk metal into single atoms. Nat. Nanotechnol. 17, 403-407 (2022).
Hai, X. et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 17, 174-181 (2022).
Xia, C. et al. General synthesis of single-atom catalysts with high metal loading using graphene quantum dots. Nat. Chem. 13, 887-894 (2021).
Kim, H. et al. Activation of hydrogen peroxide by a titanium oxidesupported iron catalyst: evidence for surface Fe(IV) and its selectivity. Environ. Sci. Technol. 54, 15424-15432 (2020).
Zhu, L. et al. Designing 3D-MoS sponge as excellent cocatalysts in advanced oxidation processes for pollutant control. Angew. Chem. Int. Ed. Engl. 59, 13968-13976 (2020).
Shao, P. et al. Revisiting the graphitized nanodiamond-mediated activation of peroxymonosulfate: singlet oxygenation versus electron transfer. Environ. Sci. Technol. 55, 16078-16087 (2021).
Ren, W. et al. The intrinsic nature of persulfate activation and N-doping in carbocatalysis. Environ. Sci. Technol. 54, 6438-6447 (2020).
Wang, L. et al. A polymer tethering strategy to achieve high metal loading on catalysts for Fenton reactions. Nat. Commun. 14, 7841 (2023).
Zong, Y. et al. Unraveling the overlooked involvement of high-valent cobalt-oxo species generated from the cobalt(II)-activated peroxymonosulfate process. Environ. Sci. Technol. 54, 16231-16239 (2020).
Sun, H., Xu, X., Song, Y., Zhou, W. & Shao, Z. Designing high-valence metal sites for electrochemical water splitting. Adv. Funct. Mater. 31, 2009779 (2021).
Li, F., Lu, Z., Li, T., Zhang, P. & Hu, C. Origin of the excellent activity and selectivity of a single-atom copper catalyst with unsaturated sites via peroxydisulfate activation: Cu (III) as a dominant oxidizing species. Environ. Sci. Technol. 56, 8765-8775 (2022).
Williams, P. J. H. et al. New approach to the detection of short-lived radical intermediates. J. Am. Chem. Soc. 144, 15969-15976 (2022).
Ranguelova, K. & Mason, R. P. The fidelity of spin trapping with DMPO in biological systems. Magn. Reson. Chem. 49, 152-158 (2011).
Pou, S., Hassett, D. J., Britigan, B. E., Cohen, M. S. & Rosen, G. M. Problems associated with spin trapping oxygen-centered free radicals in biological systems. Anal. Biochem. 177, 1-6 (1989).
Amić, A., Marković, Z., Dimitrić Marković, J. M., Milenković, D. & Stepanić, V. Antioxidative potential of ferulic acid phenoxyl radical. Phytochemistry. 170, 112218 (2020).
Bednar, T. N. & Nagib, D. A. Radical arenes. Nat. Chem. 15, 3-4 (2023).
Nguyen, J. D., D’Amato, E. M., Narayanam, J. M. R. & Stephenson, C. R. J. Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions. Nat. Chem. 4, 854-859 (2012).
Geng, J. et al. Radical polymerization inside living cells. Nat. Chem. 11, 578-586 (2019).
Chen, Z. et al. Tailoring the -dand centers enables nanosheets to be highly active for hydrogen evolution catalysis. Angew. Chem. Int. Ed. Engl. 57, 5076-5080 (2018).
Greiner, M. T. et al. Free-atom-like states in single-atom alloy catalysts. Nat. Chem. 10, 1008-1015 (2018).
Sun, Y. et al. Spin-related electron transfer and orbital interactions in oxygen electrocatalysis. Adv. Mater. 32, e2003297 (2020).
Zhou, Q. et al. Generating dual-active species by triple-atom sites through peroxymonosulfate activation for treating micropollutants in complex water. Proc. Natl. Acad. Sci. USA. 120, e1994882176 (2023).
Yang, M. et al. Unveiling the origins of selective oxidation in singleatom catalysis via intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635-11645 (2022).
Yu, X. et al. Enhanced nonradical catalytic oxidation by encapsulating cobalt into nitrogen doped graphene: highlight on interfacial interactions. J. Mater. Chem. A 9, 7198-7207 (2021).
Clark, S. J. et al. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 220, 567-570 (2005).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22376072 to M.J.H., 52192684 to H.Q.Y., 51821006 to H.Q.Y., 22106159 to M.J.H., and 52027815 to H.Q.Y.), and the Students’ Innovation and Entrepreneurship Foundation of University of Science and Technology of China (CY2022G11 to H.Z.L. and CY2023GO20 to H.Z.L.). We thank the photoemission endstations BL1W1B in Beijing Synchrotron Radiation Facility (BSRF), BL14W1 in Shanghai Synchrotron Radiation Facility (SSRF), BL12B-a, BL11U, and BL10B in National Synchrotron Radiation Laboratory (NSRL), the Shiyanjia Lab (www.shiyanjia.com), and Dr. Mei Sun from the HRTEM group (Instrument center for Physical Science in University of Science and Technology of China) for help with characterizations. The numerical calculations were performed on the supercomputing system in the Supercomputing Center of University of Science and Technology of China.
Author contributions
M.J.H., J.J.C., and H.Q.Y. conceived and designed the study and supervised the project. H.Z.L. carried out the synthesis, characterization, and catalytic performance test. X.X.S. and J.J.C. conducted the theoretical studies. B.B.W. and X.S.W. helped to synthesize CHANT trapping agent. H.L.L. helped to synthesize the catalysts. M.J.H., H.Z.L., J.J.C., and H.Q.Y. analyzed the results and co-wrote the paper. All authors contributed to discussion of the results and the manuscript.
Correspondence and requests for materials should be addressed to Mingjie Huang, Jie-Jie Chen or Han-Qing Yu.
Peer review information Nature Communications thanks Dongya Li and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
CAS Key Laboratory of Urban Pollutant Conversion, Department of Environmental Science and Engineering, University of Science and Technology of China, Hefei, China. School of Environmental Science and Engineering, Huazhong University of Science and Technology, Wuhan, China. Department of Chemistry, University of Science and Technology of China, Hefei, China. These authors contributed equally: Hong-Zhi Liu, Xiao-Xuan Shu.