المجلة: Scientific Reports، المجلد: 14، العدد: 1
DOI: https://doi.org/10.1038/s41598-024-51290-6
PMID: https://pubmed.ncbi.nlm.nih.gov/38225280
تاريخ النشر: 2024-01-15
DOI: https://doi.org/10.1038/s41598-024-51290-6
PMID: https://pubmed.ncbi.nlm.nih.gov/38225280
تاريخ النشر: 2024-01-15
تخليق، توصيفات هيكلية، تقييم بيولوجي في المختبر، والتحقيقات الحاسوبية لمشتقات البيرازول كعوامل محتملة مضادة للسكري ومضادة للأكسدة
في هذه الدراسة، تم تخليق مشتقّين من البيرازول؛ 2-(5-ميثيل-1H-بيرازول-3-كربونيل)-N-فينيل هيدرازين-1-كربوكسايد (Pyz-1) و4-أمينو-5-(5-ميثيل-1H-بيرازول-3-يل)-4H-1,2,4-تريازول-3-ثيول (Pyz-2) وتم توصيفهما بواسطة
يتميز داء السكري من النوع 2 (T2DM) بارتفاع مستويات الجلوكوز في الدم، مما يمكن أن يؤدي إلى مضاعفات كبيرة مثل أمراض القلب والأوعية الدموية، والاعتلال العصبي، واعتلال الشبكية، وأمراض الكلى.
مستويات سكر الدم. وبالتالي،
إن البيرازول يشكل فئة كبيرة من الحلقات غير المتجانسة التي يمكن استكشافها لتطوير مواد دوائية جديدة. وقد أظهرت دراسة شاملة لهذه الفئة أن البيرازول يمكن أن يكون موجودًا في العديد من الأدوية المعروفة من فئات مختلفة ذات أنشطة علاجية متنوعة.
تجريبي
المواد الكيميائية والأدوات
تم شراء المواد الكيميائية (الميثانول، الإيثانول، إيزوسيانات الفينيل، هيدروكسيد البوتاسيوم، هيدرازين هيدرات، ثنائي كبريتيد الكربون وحمض الهيدروكلوريك) من شركات فلوكا، سيغما وألدريش. تم قياس نقاط الانصهار باستخدام جهاز قياس نقطة الانصهار الرقمي Buchi B-545. تم استخدام TLC مع هلام السيليكا 60 F254 للتحقق من التفاعلات. تم تسجيل طيف الأشعة تحت الحمراء باستخدام مطياف Perkin-Elmer VERTEX 70 FT-IR الذي يغطي المجال.
كيمياء
إجراء تخليق 2-(5-mيثيل-1H-بيرازول-3-كربونيل)-N-فينيل هيدرازين-1-كربوكسياميد (Pyz-1) تم تخليق 5-mيثيل-1H-بيرازول-3-كربوهيدرازيد (1) وفقًا للطريقة المبلغ عنها سابقًا.
إجراء تخليق 4-أمينو-5-(5-mيثيل-1H-بيرازول-3-يل)-4H-1,2,4-تريازول-3-ثيول (Pyz-2)
إلى محلول مبرد بالثلج من 5-ميثيل-1H-بيرازول-3-كاربوهيدرازيد (1) (
إلى محلول مبرد بالثلج من 5-ميثيل-1H-بيرازول-3-كاربوهيدرازيد (1) (
التفاصيل الحاسوبية
تم إجراء سلسلتين من الحسابات في محاولة لاستنتاج وتفسير البيانات التجريبية. أولاً، تم إجراء حسابات DFT لجمع المعلومات المتعلقة بتفاعل المركبات المُصنّعة. ثم، تم استخدام محاكاة ربط الجزيئات لتحديد أوضاع الربط و
الطاقة بين المركبات (
الطاقة بين المركبات (
الربط الجزيئي
لتحقيق في أوضاع الربط المحتملة للكيانات الكيميائية المختارة، تم استخدام محاكاة الربط باستخدام AutodockVina v1.5.6
علم الأحياء
ال
أنشطة مضادات الأكسدة
تم تحديد أنشطة مضادات الأكسدة للمركب المذكور في المختبر بواسطة DPPH و ABTS و FRAP ونشاط بيروكسيد الهيدروجين.
اختبار تثبيط أكسيداز الزانثين (XO)
نشاط تثبيط أكسيداز الزانثين
التحليل الإحصائي
تم إجراء التحليل الإحصائي باستخدام برنامج GraphPad Prism8. تم استخدام تحليل التباين الأحادي (ANOVA) لتحديد الفرق المعنوي. تم تقديم البيانات الكمية كمتوسط.
النتائج والمناقشة الكيمياء
أولاً، بروتوكول التخليق لـ
تم تأكيد الهياكل الجزيئية لـ Pyz-1 و Pyz-2 باستخدام
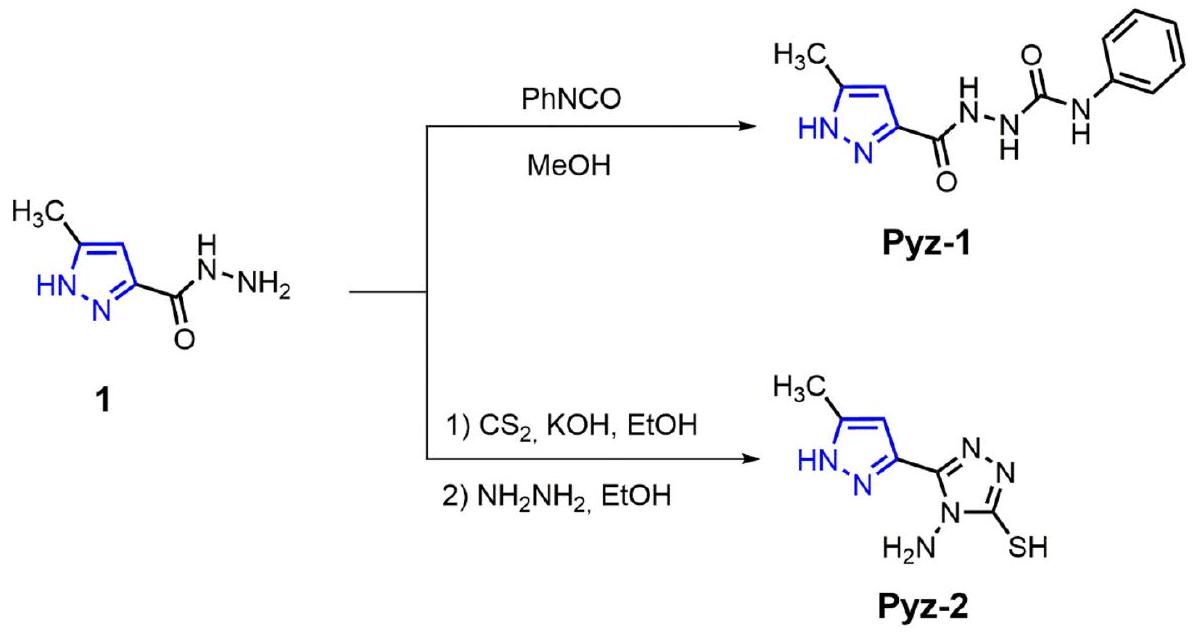
المخطط 1. المسار الاصطناعي لتحضير المركبات Pyz-1 وPyz-2.
ال
تظهر طيف الكتلة (ESI) قمة مرتبطة بالأيونات الجزيئية عند
النتائج الحاسوبية
الخصائص الإلكترونية
المعلمات الإلكترونية والديناميكية الحرارية هي وسيلة فعالة لشرح استقرار وتفاعل الجزيئات. الكميات الفيزيائية لـ Pyz-1 و Pyz-2 المحسوبة على مستوى نظرية B3LYP/6-311G++ (d,p) موضحة في الجدول 1، والهياكل المحسنة لأكثر المتوافقات استقرارًا لـ
المعلمات الإلكترونية والديناميكية الحرارية هي وسيلة فعالة لشرح استقرار وتفاعل الجزيئات. الكميات الفيزيائية لـ Pyz-1 و Pyz-2 المحسوبة على مستوى نظرية B3LYP/6-311G++ (d,p) موضحة في الجدول 1، والهياكل المحسنة لأكثر المتوافقات استقرارًا لـ
تم حساب الطاقات الكلية للجزيئات المذكورة في العنوان لـ
تحليل FMO
في الكيمياء الحاسوبية، تعتبر طاقات وتوزيعات المدارات الجزيئية المتفاعلة (FMO) أوصافًا حيوية للتفاعلية. المدار الجزيئي الأعلى شغلاً (HOMO) والمدار الجزيئي الأدنى غير الشاغل (LUMO) لديهما
| بيز-1 | بيز-2 | |||
| غاز | ماء | غاز | ماء | |
| طاقة (أو) | – ٨٨٩.٥٦٠٢٣٣ | – ٨٨٩٫٥٨٠٧٣٣ | – 960.268510 | – ٩٦٠٫٢٨٧٩٦٠ |
| القطبية (au) | 193.402 | ٢٥٤.٧٤٩ | ١٣٩.٣٢٩ | ١٨٤٫٥٨٨ |
| لحظة ثنائي القطب (D) | 2.660 | ٢.٩٩١ | ٦.٥٨٥ | 8.747 |
|
|
-889.292364 | -889.313353 | – ٩٦٠.١٠١٢٤٥ | – ٩٦٠.١٢١٦٧٦ |
|
|
-889.360095 | – ٨٨٩.٣٨١٦٦١ | -960.155981 | -960.173090 |
|
|
167.498 | 167.191 | ١٠٤.٣٦٨ | ١٠٣.٧٥٢ |
| السيرة الذاتية (
|
65.152 | 65.480 | ٤٥٫٢٣٩ | ٤٣.١٥٥ |
| س (كال/molK) | ١٤٢.٥٥٢ | ١٤٣.٧٦٦ | ١١٥.٢٠٣ | ١٠٨.٢١٠ |
الجدول 1. طاقات الحالة الأساسية والمعلمات الديناميكية الحرارية لـ Pyz-1 و Pyz-2.
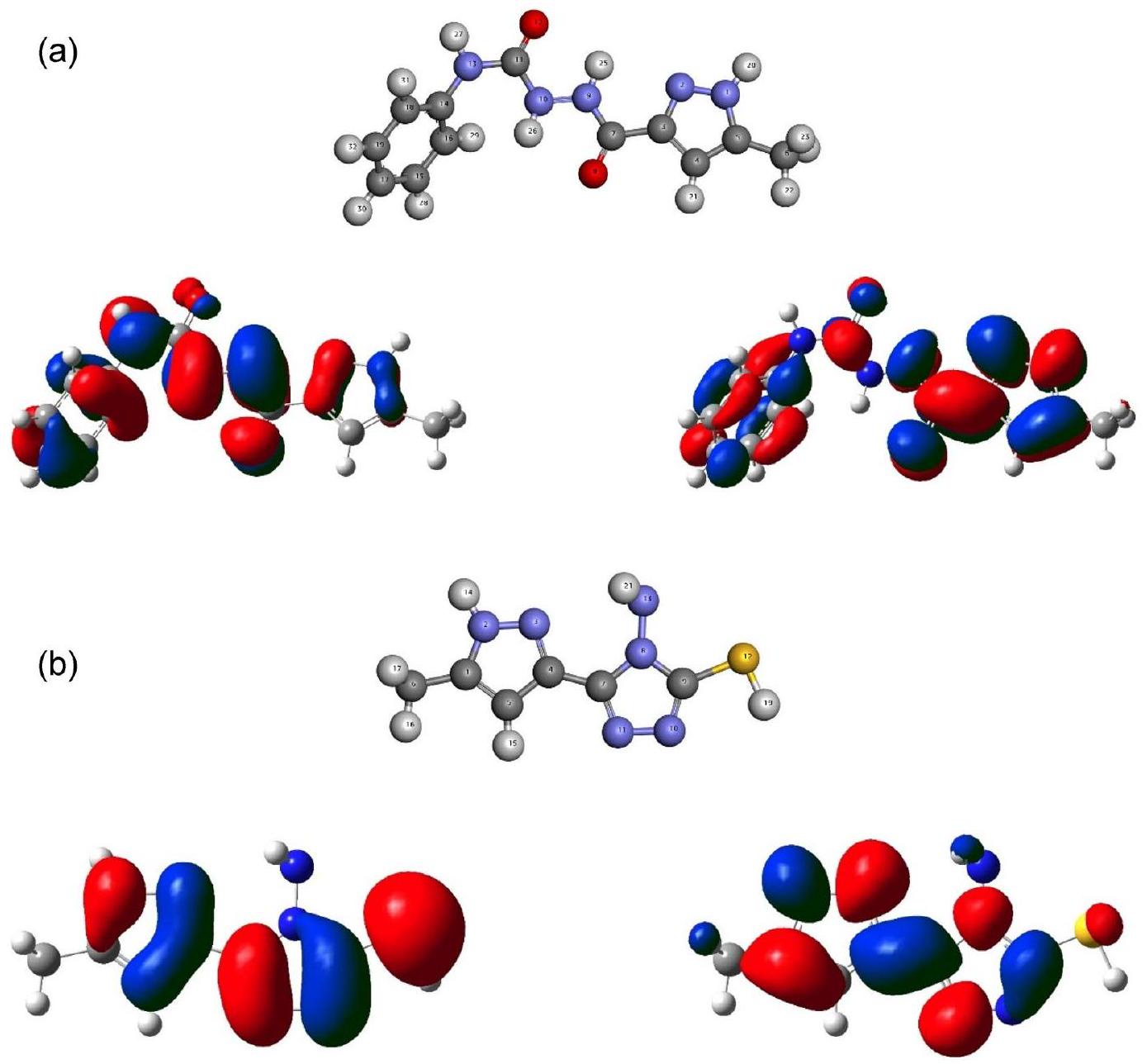
الشكل 1. الهياكل الجزيئية المحسّنة وتوزيعات كثافة FMO لـ (أ) Pyz-1 و (ب) Pyz-2.
أدوار هامة في الخصائص الكهربائية والإلكترونية والبصرية، والتفاعل الكيميائي والمواقع النشطة لمركب معين
تحدد الفجوة الطاقية بين مدارات HOMO و LUMO الاستقرار الحركي للمركبات المذكورة. وبالتالي، فإن الجزيئات ذات الفجوة الطاقية الصغيرة تكون قابلة للاستقطاب بشكل كبير وغالبًا ما ترتبط بارتفاع التفاعل الكيميائي بالإضافة إلى انخفاض الاستقرار الحركي.
| Pyz-1 | Pyz-2 | |||
| غاز | ماء | غاز | ماء | |
|
|
-6.150 | -6.404 | -6.102 | -6.394 |
|
|
-1.032 | -1.269 | -0.937 | -1.043 |
|
|
5.118 | 5.136 | 5.166 | 5.352 |
الجدول 2.
وصف التفاعل
نظرًا لمجموعة واسعة من التطبيقات في مجالات مثل البيولوجيا والكيمياء وتصميم الأدوية، فقد اكتسب تطوير أوصاف التفاعل الكيميائي زخمًا كبيرًا.
الصلابة العالمية
خريطة الجهد الكهربائي الجزيئي (MEP)
الجهد الكهربائي الجزيئي هو تمثيل ثلاثي الأبعاد يسمح بتصور توزيع الشحنات، وتوقع التفاعل تجاه الهجمات الإلكترونيات والنيوكليوفيلية والقطبية النسبية لجزيء معين.
تم ملاحظة قطبية عالية لجزيئات
| معامل | Pyz-1 | Pyz-2 |
| I | 6.150 | 6.102 |
| A | 1.032 | 0.937 |
|
|
-3.591 | -3.520 |
|
|
5.118 | 5.165 |
|
|
1.260 | 1.199 |
|
|
0.104 | 0.085 |
|
|
4.091 | 4.178 |
|
|
0.702 | 0.681 |
|
|
-1.260 | -1.199 |
|
|
-1795 | -1761 |
الجدول 3. معلمات التفاعل الكيميائي الكمي لـ
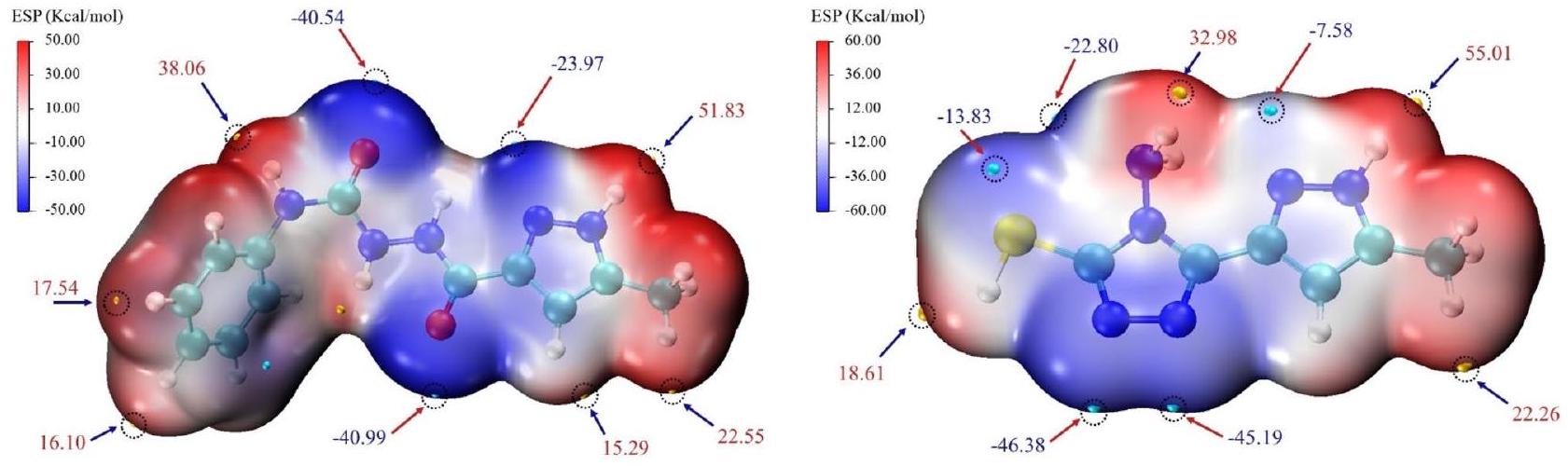
الشكل 2. سطح الجهد الكهربائي الجزيئي المحسوب (MEP) لـ Pyz-1 و Pyz-2.
و Pyz-2. تكشف هذه الملاحظات عن ميل قوي لجذب الذرات المشحونة سلبًا كهربائيًا والتصرف كمجموعة مانحة للروابط الهيدروجينية. علاوة على ذلك، بالنسبة لـ
تحليل NBO
يمثل تحليل المدار الطبيعي (NBO) أداة حسابية لفهم وتفسير أكثر سهولة وملاءمة للحلول الحسابية لمعادلة شرودنجر في مفاهيم الترابط الكيميائي.
لتقييم تفاعلات المانح-المستقبل، تم إجراء مصفوفة Fock من الدرجة الثانية. تم تقدير طاقة الاستقرار
حيث
بالنسبة لكلا المركبين، تم حساب أهم تحليلات التفاعل بين المدار NBO من نوع لويس المملوء (الترابط) والمدار NBO غير لويس غير المملوء (المضاد للترابط) وتم تقديم تفاعلات المانح-المستقبل التي لها طاقة استقرار فوق
الأنشطة البيولوجية
النشاط المضاد للسكري
تمت دراسة المركبات Pyz-1 و Pyz-2 في المختبر لفعالياتها المضادة للسكري ضد
| المركبات |
|
|
|
|
|
|
|
|
|
|
| بيز-1 |
|
|
| بيز-2 |
|
|
| أكاربوز |
|
|
الجدول 4.
نشاط مضادات الأكسدة
تم تحديد خصائص مضادات الأكسدة للمركبات المستهدفة باستخدام DPPH و ABTS،
لتحليل DPPH، نشاط مضادات الأكسدة لـ
أظهرت نتائج التأثير المثبط على زانثين أوكسيداز للمركبات المذكورة في العنوان أن Pyz-1 أظهر نشاطًا كبيرًا في XO مع
دراسات الارتباط
التفاعل الرابط لـ
كما هو ملاحظ من الجدول 6، فإن جميع قيم طاقات الربط سالبة، مما يثبت الأنظمة ويفضل التفاعلات بين البروتينات والليغاندات.
ضد
من بين المركبات البديلة المقترحة حديثًا لعلاج السكري،
من بين المركبات البديلة المقترحة حديثًا لعلاج السكري،
الشكل 3 قدم الأحماض الأمينية المتفاعلة
| مركب | IC50 (
|
||||
| DPPH | ABTS | فراپ |
|
إكس أو | |
| بيز-1 |
|
|
|
|
|
| بيز-2 |
|
|
|
|
|
| حمض الأسكوربيك |
|
|
|
|
– |
| ألوبورينول | – | – | – | – |
|
الجدول 5. أنشطة مضادات الأكسدة لـ
| الليغاندات |
|
|
||
| الألفة (كيلو كالوري/مول) | Rmsdl.b | الألفة (كيلو كالوري/مول) | Rmsdl.b | |
|
|
-5.3 | 0.000 | -5.3 | 0.000 |
| بيز-1 | -5.7 | 0.000 | -6.7 | 0.000 |
| بيز-2 | -4.3 | 0.000 | -5.6 | 0.000 |
| ACA | -5.9 | 0.000 | -6.2 | 0.000 |
الجدول 6. نتائج الربط ل affinity والـ RMSD لقيم الأوضاع المختلفة في 3A4A و 2GJP.
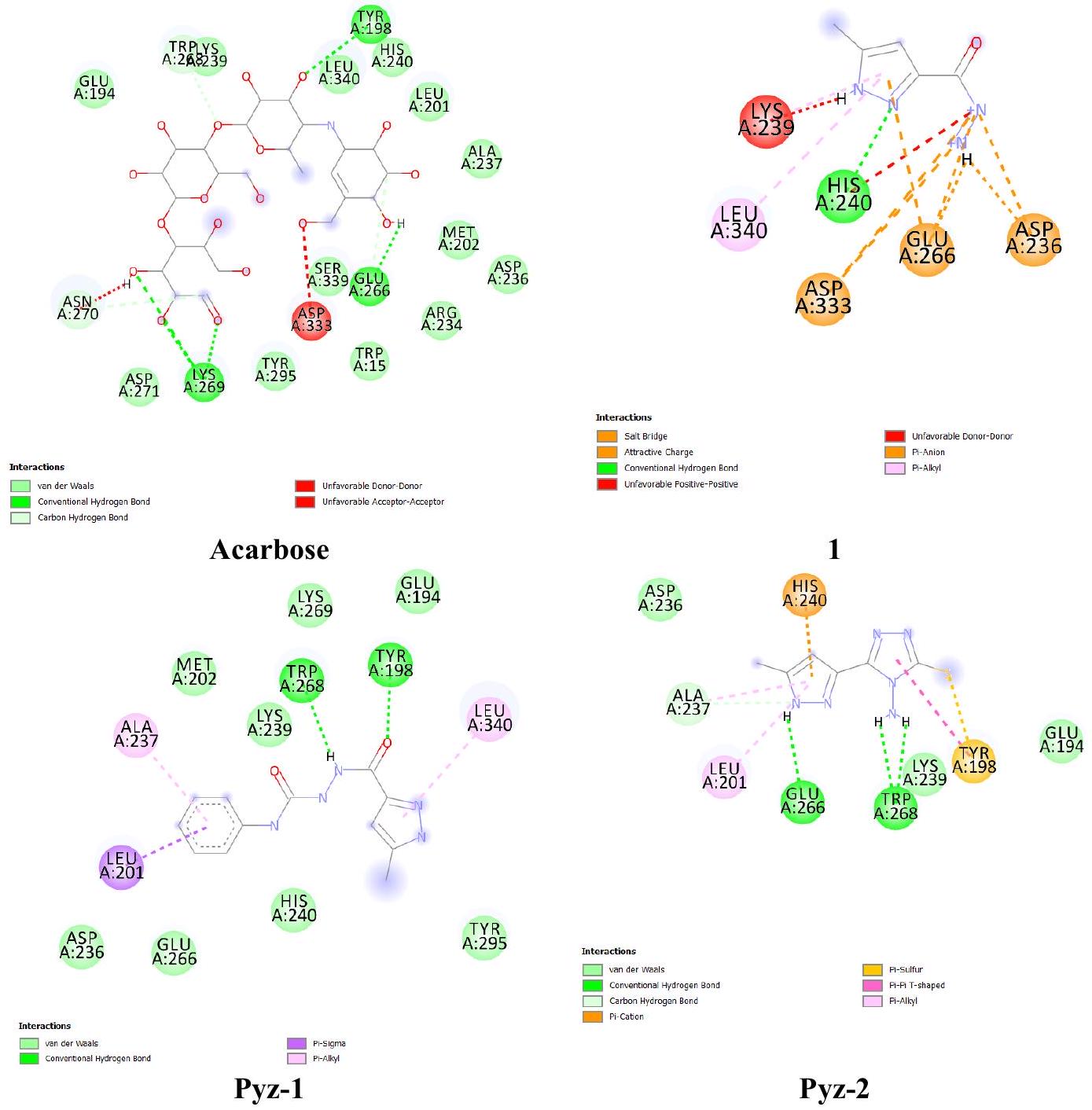
الشكل 3. الارتباط بين المركبات المرسومة (1، Pyz-1، Pyz-2 و Acarbose) و
تُلاحظ أيضًا مع الحمض الأميني Asp333. فيما يتعلق بالمركب
ضد
من الجدول 6، وُجد أن التحكم الإيجابي ACA كان الأكثر فعالية
من الجدول 6، وُجد أن التحكم الإيجابي ACA كان الأكثر فعالية
تحليل ADME-T
في هذه الدراسة، دراسة حسابية لـ
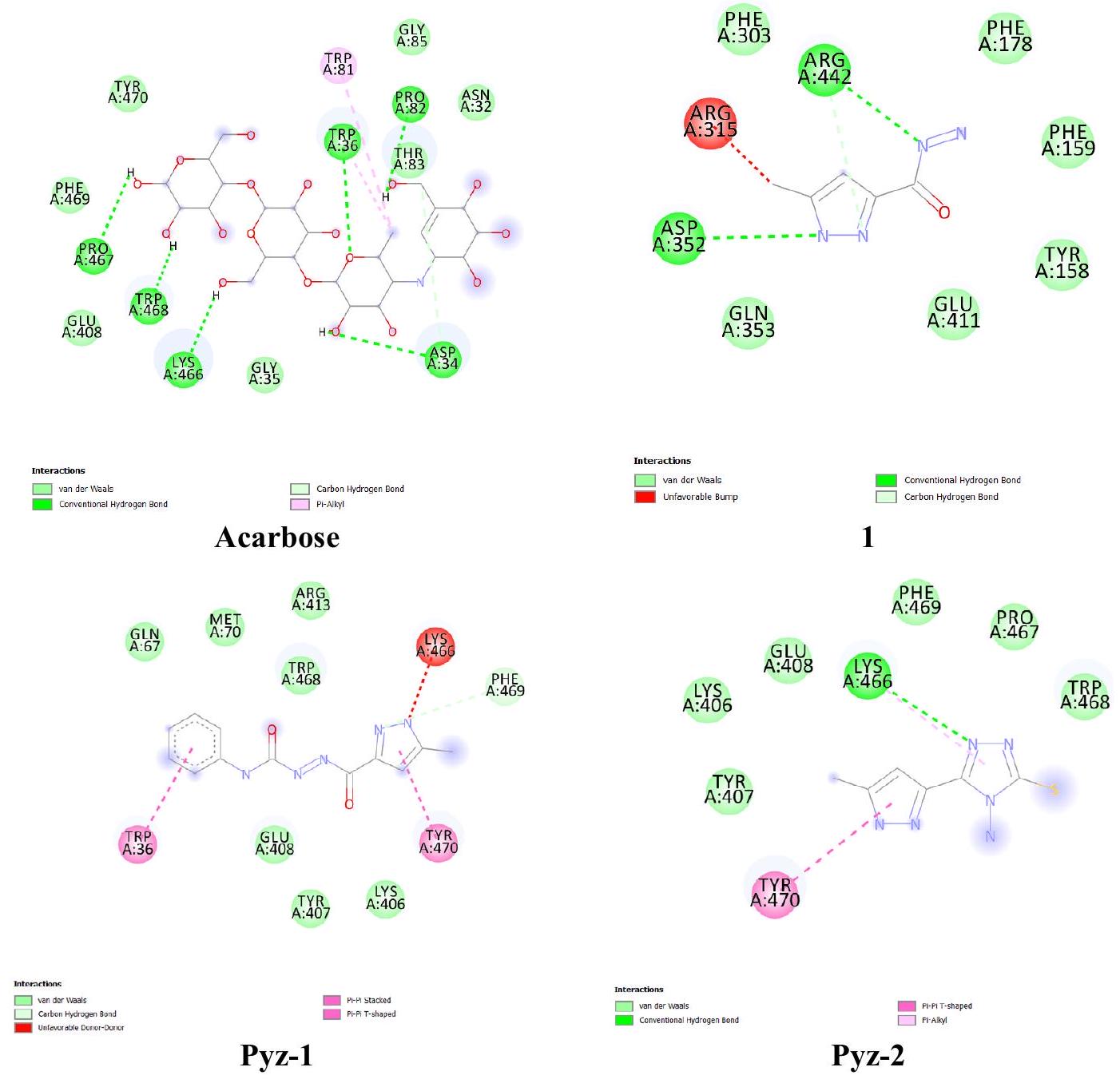
الشكل 4. الارتباط بين المركبات المرسومة (1، Pyz-1، Pyz-2 و Acarbose) و
من المرجح أن يكون امتصاص المركب أفضل إذا حقق الجزيء ثلاثة على الأقل من القواعد الأربعة التالية: (i) مجموعات مانحة للهيدروجين
أيضًا، ملفات ADMET لـ
عند النظر إلى قيم المعلمات المحسوبة، نجد أن هذه القيم ضمن المعايير الموصى بها. وبالتالي، يمكننا أن نستنتج أن جزيئات العنوان تتمتع بخصائص قابلة للاستخدام كدواء وأن تطبيقها كدواء نظري آمن.
الخاتمة
في الختام، تم تخليق مشتقات جديدة من البيرازول Pyz-1 وPyz-2، وتمت دراستها وتقييمها من حيث أنشطتها المضادة للسكري ومضادات الأكسدة. تم تقييم Pyz-1 وPyz-2 في المختبر من حيث أنشطتها المضادة للسكري ومضادات الأكسدة. بالنسبة لنتيجة النشاط المضاد للسكري،
| معامل | بيز-1 | بيز-2 |
| الوزن الجزيئي (غرام/مول) | 259.26 | 196.23 |
| الدهون المحبة (LogP) | 1.044 | 0.64 |
| مانحو الروابط الهيدروجينية | ٤ | ٣ |
| مستقبلات الروابط الهيدروجينية | ٣ | 2 |
| مساحة السطح القطبي (Psa) | ٩٨.٩١ | ٨٨.٣١ |
| التوزيع عند الرقم الهيدروجيني 7.4 (لوغ د) | 1.447 | 0.245 |
| ربط البروتينات البلازمية (Ppb) | ٢٩.٣٢٪ | 30.68% |
| سمية الفم الحادة في الجرذان | 0.026 | 0.008 |
| نفاذية كاكو-2 | -5.303 | 0.023 |
| مثبط Cyp1a2 | 0.028 | 0.083 |
| ركيزة Cyp1a2 | 0.334 | 0.054 |
| مثبط Cyp2c19 | 0.034 | 0.059 |
| ركيزة Cyp2c19 | 0.052 | 0.038 |
| مثبط Cyp2c9 | 0.019 | 0.91 |
| ركيزة Cyp2c9 | 0.908 | 0.001 |
| مثبط Cyp2d6 | 0.003 | 0.147 |
| ركيزة Cyp2d6 | 0.282 | 0.012 |
| مثبط Cyp3a4 | 0.011 | 0.101 |
| ركيزة Cyp3a4 | 0.045 | 0.023 |
الجدول 7. ملف ADMET لـ Pyz-1 و Pyz-2.
أظهر Pyz-2 قدرة مثبطة ملحوظة مع
توفر البيانات
جميع البيانات التي تم توليدها أو تحليلها خلال هذه الدراسة مدرجة في هذه المقالة المنشورة وملفات المعلومات التكميلية الخاصة بها.
تاريخ الاستلام: 9 أكتوبر 2023؛ تاريخ القبول: 3 يناير 2024
نُشر على الإنترنت: 15 يناير 2024
نُشر على الإنترنت: 15 يناير 2024
References
- Chatterjee, S., Khunti, K. & Davies, M. J. Type 2 diabetes. The Lancet 389, 2239-2251 (2017).
- Ademiluyi, A. O. & Oboh, G. Soybean phenolic-rich extracts inhibit key-enzymes linked to type 2 diabetes (
-amylase and a-glucosidase) and hypertension (angiotensin I converting enzyme) in vitro. Exp. Toxicol. Pathol. 65, 305-309 (2013). - Gao, Z. et al. Automatic interpretation and clinical evaluation for fundus fluorescein angiography images of diabetic retinopathy patients by deep learning. British J. Ophthalmol. 107, 1852-1858 (2023).
- Yang, Y. Y., Shi, L. X., Li, J. H., Yao, L. Y. & Xiang, D. X. Piperazine ferulate ameliorates the development of diabetic nephropathy by regulating endothelial nitric oxide synthase. Mol. Med. Rep. 19, 2245-2253 (2019).
- Chen, J. et al. Bone marrow stromal cell-derived exosomal circular RNA improves diabetic foot ulcer wound healing by activating the nuclear factor erythroid 2-related factor 2 pathway and inhibiting ferroptosis. Diabetes Med. 1, e15031 (2023).
- Pai, Y. W. et al. Glycaemic control for painful diabetic peripheral neuropathy is more than fasting plasma glucose and glycated haemoglobin. Diabetes Metab. 47, 101158 (2021).
- Chen, Y., Tan, S., Liu, M. & Li, J. LncRNA TINCR is downregulated in diabetic cardiomyopathy and relates to cardiomyocyte apoptosis. Scand. Cardiovasc. J. 52, 335-339 (2018).
- Wang, H., Yang, T., Wu, J., Chen, D. & Wang, W. Unveiling the Mystery of SUMO-activating enzyme subunit 1: A groundbreaking biomarker in the early detection and advancement of hepatocellular carcinoma. Transplant. Proc. 1, 945-951 (2023).
- Asmat, U., Abad, K. & Ismail, K. Diabetes mellitus and oxidative stress-A concise review. Saudi Pharm. J. 24, 547-553 (2016).
- Araki, E. & Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 1, 90-96 (2010).
- Matough, F. A., Budin, S. B., Hamid, Z. A., Alwahaibi, N. & Mohamed, J. The role of oxidative stress and antioxidants in diabetic complications. Sultan Qaboos Univ. Med. J. 12, 5 (2012).
- Karrouchi, K. et al. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 23, 134 (2018).
- Bennani, F. E. et al. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 97, 103470 (2020).
- Karrouchi, K. et al. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N’-(4-(dimethylamino) benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1219, 128541 (2020).
- Chaudhary, M. et al. Chloro and bromo-pyrazole curcumin Knoevenagel condensates augmented anticancer activity against human cervical cancer cells: design, synthesis, in silico docking and in vitro cytotoxicity analysis. J. Biomol. Struct. Dyn. 38, 200-218 (2020).
- Tighadouini, S. et al. Synthesis, crystal structure, DFT studies and biological activity of (Z)-3-(3-bromophenyl)-1-(1, 5-dimethyl-1 H-pyrazol-3-yl)-3-hydroxyprop-2-en-1-one. Chem. Central J. 12, 1-11 (2018).
- Karrouchi, K. et al. Synthesis, a-glucosidase inhibition, anticancer, DFT and molecular docking investigations of pyrazole hydrazone derivatives. Polycycl. Aromat. Compd. 1, 1-20 (2022).
- Karrouchi, K. et al. Synthesis, characterization, free-radical scavenging capacity and antioxidant activity of novel series of hydrazone, 1, 3, 4-oxadiazole and 1, 2, 4-triazole derived from 3, 5-dimethyl-1H-pyrazole. Lett. Drug Des. Discov. 16, 712-720 (2019).
- Abu-Melha, S. et al. Clean grinding technique: A facile synthesis and in silico antiviral activity of hydrazones, pyrazoles, and pyrazines bearing thiazole moiety against SARS-CoV-2 main protease (Mpro). Molecules 25, 4565 (2020).
- Pogaku, V., Krishna, V. S., Sriram, D., Rangan, K. & Basavoju, S. Ultrasonication-ionic liquid synergy for the synthesis of new potent anti-tuberculosis 1, 2, 4-triazol-1-yl-pyrazole based spirooxindolopyrrolizidines. Bioorg. Med. Chem. Lett. 29, 1682-1687 (2019).
- Karrouchi, K. et al. Synthesis, crystal structure, DFT,
-glucosidase and -amylase inhibition and molecular docking studies of (E)-N’-(4-chlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1245, 131067 (2021). - Mortada, S. et al. Synthesis, spectroscopic and DFT studies of 5-methyl-1H-pyrazole-3-carbohydrazide N-glycoside as potential anti-diabetic and antioxidant agent. J. Mol. Struct. 1267, 133652 (2022).
- Karrouchi, K. et al. Synthesis, X-ray, spectroscopy, molecular docking and DFT calculations of (E)-N’-(2, 4-dichlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1228, 129714 (2021).
- Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785 (1988).
- Becke, A. D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 98, 1372-1377 (1993).
- Krishnan, R. B. J. S., Binkley, J. S., Seeger, R. & Pople, J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650-654 (1980).
- McLean, A. D. & Chandler, G. S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 72, 5639-5648 (1980).
- Kainat, S. et al. Theoretical modeling of B12N12 nanocage for the effective removal of paracetamol from drinking water. Computation 11, 183 (2023).
- Shahab, M. et al. Structure based virtual screening and molecular simulation study of FDA-approved drugs to inhibit human HDAC6 and VISTA as dual cancer immunotherapy. Sci. Rep. 13, 14466 (2023).
- Ali, Q. et al. Theoretical insight of ciprofloxacin removal from water using boron nitride (B12N12) nanocage. Surf. Interfaces. 31, 101982 (2022).
- Cances, E., Mennucci, B. & Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 107, 3032-3041 (1997).
- Cossi, M., Barone, V., Cammi, R. & Tomasi, J. Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 255, 327-335 (1996).
- Barone, V., Cossi, M. & Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 19, 404-417 (1998).
- Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580-592 (2012).
- Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33-38 (1996).
- Reed, A. E., Curtiss, L. A. & Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899-926 (1988).
- Reed, A. E., Weinstock, R. B. & Weinhold, F. Natural population analysis. J. Chem. Phys. 83, 735-746 (1985).
- F. Weinhold, J. E. Carpenter, The natural bond orbital Lewis structure concept for molecules, radicals, and radical ions. In The Structure of Small Molecules and Ions (pp. 227-236) (Springer, Boston, MA, 1988).
- Parthasarathi, R., Subramanian, V., Roy, D. R. & Chattaraj, P. K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 12, 5533-5543 (2004).
- Chattaraj, P. K. & Giri, S. Electrophilicity index within a conceptual DFT framework. Phys. Chem. 105, 13-39 (2009).
- Islam, N., Kaya, S. (Eds.). Conceptual Density Functional Theory and Its Application in the Chemical Domain (CRC Press, 2018).
- Frisch, M. E., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R. et al. **Gaussian, Inc., Wallingford CT, 2016.
- Dennington, R., Keith, T., & Millam, J. GaussView, version 5 (2009).
- Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785-2791 (2009).
- Systemes, D. BIOVIA Discovery Studio Visualizer 2016. San Diego, CA: Dassault Systemes, 2016.
- Schrodinger, L. L. C. The PyMOL molecular graphics system. Version, 1(5) (2010).
- Ahmed, A. et al. Novel adamantyl clubbed iminothiazolidinones as promising elastase inhibitors: design, synthesis, molecular docking, ADMET and DFT studies. RSC Adv. 12, 11974-11991 (2022).
- Karrouchi, K. et al. Experimental and computational interaction studies of (E)-N’-benzylidene-5-methyl-1H-pyrazole-3-carbohydrazide with
-glucosidase and -amylase enzymes: A detailed structural, spectroscopic, and biophysical study. Polycycl. Aromat. Compd. 43, 1812-1832 (2023). - Pillai, R. R. et al. Synthesis, spectroscopic characterization, reactive properties by DFT calculations, molecular dynamics simulations and biological evaluation of Schiff bases tethered 1, 2, 4-triazole and pyrazole rings. J. Mol. Struct. 1177, 47-54 (2019).
- Kostić, D. A. et al. Xanthine oxidase: Isolation, assays of activity, and inhibition. J. Chem. 2015, 1-9 (2015).
- Arivazhagan, M., Manivel, S., Jeyavijayan, S. & Meenakshi, R. Vibrational spectroscopic (FTIR and FT-Raman), first-order hyperpolarizablity, HOMO, LUMO, NBO, Mulliken charge analyses of 2-ethylimidazole based on Hartree-Fock and DFT calculations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1, 493-501 (2015).
- Karelson, M., Lobanov, V. S. & Katritzky, A. R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 96, 1027-1044 (1996).
- Streitwieser, A. & Salzberg, H. W. Molecular orbital theory for organic chemists. J. Electrochem. Soc. 109, 116 (1962).
- Chattaraj, P. K., Nath, S., Maiti, B. Reactivity Descriptors 295-322. (Marcel Dekker, New York, 2003).
- El-Hadki, H. et al. Theoretical Study of Reaction Between Nitrilimine and 1,4 oxazine 2 Carboxylate by MP2 and DFT Methods. Oriental J. Chem. 34, 2992 (2018).
- Parr, R. G., Szentpály, L. V. & Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1, 1922-1924 (1999).
- Luque, F. J., López, J. M. & Orozco, M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 1, 343-345 (2000).
- El Hadki, A. et al. Removal of oxytetracycline by graphene oxide and boron-doped reduced graphene oxide: A combined density function theory, molecular dynamics simulation and experimental study. FlatChem. 27, 100238 (2021).
- Weinhold, F., Landis, C. R. & Glendening, E. D. What is NBO analysis and how is it useful?. Int. Rev. Phys. Chem. 1, 399-440 (2016).
- Weinhold, F. & Landis, C. R. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 1, 91-104 (2001).
- Agwupuye, J. A. et al. Electronic structure investigation of the stability, reactivity, NBO analysis, thermodynamics, and the nature of the interactions in methyl-substituted imidazolium-based ionic liquids. J. Mol. Liq. 337, 116458 (2021).
- Yu, Z. et al. Insights from molecular dynamics simulations and steered molecular dynamics simulations to exploit new trends of the interaction between HIF-1a and p300. J. Biomol. Struct. Dyn. 38, 1-12 (2020).
- Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 64, 4-17 (2012).
- Xiong, G. et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucl. Acids Res. 49, W5-W14 (2021).
- van Breemen, R. B. & Li, Y. Caco-2 cell permeability assays to measure drug absorption. Expert. Opin. Drug Metab. Toxicol. 1, 175-185 (2005).
- Prabakaran, G., Manivarman, S. & Bharanidharan, M. Catalytic synthesis, ADMET, QSAR and molecular modeling studies of novel chalcone derivatives as highly potent antioxidant agents. Mater. Today: Proc. 48, 400-408 (2022).
- Fettach, S. et al. Biological, toxicological and molecular docking evaluations of isoxazoline-thiazolidine-2, 4-dione analogues as new class of anti-hyperglycemic agents. J. Biomol. Struct. Dyn. 41, 1072-1084 (2021).
- Fettach, S. et al. Synthesis,
-glucosidase and -amylase inhibitory activities, acute toxicity and molecular docking studies of thiazolidine-2, 4-diones derivatives. J. Biomol. Struct. Dyn. 1, 1-12 (2021).
شكر وتقدير
يود المؤلفون أن يعبروا عن امتنانهم لجامعة الملك سعود، الرياض، المملكة العربية السعودية، لدعمها العمل من خلال مشروع دعم الباحثين رقم (RSPD2024R740).
مساهمات المؤلفين
تصور، س.م؛ المنهجية، س.م وك.ك؛ البرمجيات، إ.هـ.هـ؛ الموارد، أ.ع؛ الحصول على التمويل، م.أ.ب؛ التحليل الرسمي، ح.م وي.أ؛ إعداد المسودة الأصلية، ك.ك، س.م، م.أ.ب وإ.هـ.هـ؛ التصور، س.ر وأ.م؛ الكتابة – المراجعة والتحرير، ك.ك؛ الإشراف، م.أ وم.إ.أ.ف. جميع المؤلفين قرأوا ووافقوا على النسخة المنشورة من المخطوطة.
المصالح المتنافسة
يعلن المؤلفون عدم وجود مصالح متنافسة.
معلومات إضافية
معلومات إضافية النسخة الإلكترونية تحتوي على مواد إضافية متاحة علىhttps://doi.org/10.1038/s41598-024-51290-6.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى K.K.
معلومات إعادة الطباعة والتصاريح متاحة علىwww.nature.com/reprints.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
معلومات إعادة الطباعة والتصاريح متاحة علىwww.nature.com/reprints.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا ما تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارةhttp://creativecommons.org/licenses/by/4.0/.
© المؤلف(ون) 2024
© المؤلف(ون) 2024
مختبر علم الأدوية والسموم، فريق بحث التحليل البيوفارماوي والسموم، كلية الطب والصيدلة، جامعة محمد الخامس، الرباط، المغرب. مختبر الكيمياء التحليلية والبروماطولوجيا، فريق صياغة ومراقبة جودة المنتجات الصحية، كلية الطب والصيدلة، جامعة محمد الخامس في الرباط، الرباط، المغرب. CERNE2D: مختبر الطيفية، النمذجة الجزيئية، المواد، النانومواد، المياه والبيئة (LS3MN2E)، كلية العلوم، جامعة محمد الخامس، الرباط، المغرب. معهد المواد المكثفة والنانوساينس، الكيمياء الجزيئية، المواد والتحفيز (IMCN/MOST)، جامعة لوفان الكاثوليكية، 1348 لوفان-لا-نوف، بلجيكا. قسم الكيمياء الصيدلانية، كلية الصيدلة، جامعة الملك سعود، 11451 الرياض، المملكة العربية السعودية. المختبر المركزي لعلم الدم، مستشفى ابن سينا، كلية الطب والصيدلة، جامعة محمد الخامس، الرباط، المغرب. مختبر الكيمياء التطبيقية والبيئة (LCAE)، كلية العلوم، جامعة محمد الأول، 60000 وجدة، المغرب. مختبر الكيمياء الطبية، كلية الطب والصيدلة، جامعة محمد الخامس، الرباط، المغرب. ساهم هؤلاء المؤلفون بالتساوي: أزلراب مصرار ومي ال عباس فوزي. البريد الإلكتروني:خالد.كروشي@um5s.net.ma
Journal: Scientific Reports, Volume: 14, Issue: 1
DOI: https://doi.org/10.1038/s41598-024-51290-6
PMID: https://pubmed.ncbi.nlm.nih.gov/38225280
Publication Date: 2024-01-15
DOI: https://doi.org/10.1038/s41598-024-51290-6
PMID: https://pubmed.ncbi.nlm.nih.gov/38225280
Publication Date: 2024-01-15
Synthesis, structural characterizations, in vitro biological evaluation and computational investigations of pyrazole derivatives as potential antidiabetic and antioxidant agents
In this study, a two pyrazole derivatives; 2-(5-methyl-1H-pyrazole-3-carbonyl)-N-phenylhydrazine-1-carboxamide (Pyz-1) and 4-amino-5-(5-methyl-1H-pyrazol-3-yl)-4H-1,2,4-triazole-3-thiol (Pyz-2) were synthesized and characterized by
Type 2 diabetes mellitus (T2DM) is characterized by high blood glucose levels, which can lead to major complications such as cardiovascular disease, neuropathy, retinopathy and kidney disease
blood glucose levels. Thus,
Indeed, pyrazole constitute a large class of heterocycles which can be explored for the development of new drug substances. An extensive study of this class has demonstrated that pyrazole can be present in various known drugs of different classes with different therapeutic activities
Experimental
Reagents and instruments
Chemical reagents (Methanol, Ethanol, Phenyl isocyante, Potassium hydroxide, hydrazine hydrate, Carbon disulfide and Hydrochloric acid) were purchased from Fluka, Sigma and Aldrich chemicals. Melting points were measured using a Buchi B-545 digital capillary melting point apparatus. TLC with silica gel 60 F254 were used to check the reactions. The IR spectra were recorded by using Perkin-Elmer VERTEX 70 FT-IR spectrometer covering field
Chemistry
Procedure for the synthesis of 2-(5-methyl-1H-pyrazole-3-carbonyl)-N-phenylhydrazine-1-carboxamide (Pyz-1) 5-methyl-1H-pyrazole-3-carbohydrazide (1) was synthesized according the previously reported method
Procedure for the synthesis of 4-amino-5-(5-methyl-1H-pyrazol-3-yl)-4H-1,2,4-triazole-3-thiol (Pyz-2)
To an ice cooled solution of 5-methyl-1H-pyrazole-3-carbohydrazide (1) (
To an ice cooled solution of 5-methyl-1H-pyrazole-3-carbohydrazide (1) (
Computational details
Two series of calculations were conducted in an attempt to deduce and interpret the experimental data. First, The DFT computations were carried out in order to gather information regarding the reactivity of the synthesized compounds. Then, a molecular docking simulation was used to determine the binding modes and
energies between the compounds (
energies between the compounds (
Molecular docking
To investigate possible binding modes of selected chemical entities, a docking simulation employing the AutodockVina v1.5.6
Biology
The
Antioxidant activities
The antioxidant activities of the title compound were determined in vitro by DPPH, ABTS, FRAP and hydrogen peroxide activity (
Xanthine oxidase inhibition assay (XO)
The xanthine oxidase inhibitory activity of
Statistical analysis
Statistical analyze was performed using GraphPad Prism8 program. One-way ANOVA was used to determine the significant difference. Quantitative data were presented as mean
Results and discussion Chemistry
First, the synthesis protocol of
The molecular structures of Pyz-1 and Pyz-2 were confirmed by using
Scheme 1. Synthetic route for preparation of compounds Pyz-1 and Pyz-2.
The
The mass spectra (ESI) show a peak related to the molecular ions at
Computational results
Electronic properties
Electronic and thermodynamic parameters are an effective way to explain the stability and reactivity of molecules. The physical quantities of Pyz-1 and Pyz-2 calculated at B3LYP/6-311G++ (d,p) level of theory are given in Table 1, the optimized structures of the most stable conformer of
Electronic and thermodynamic parameters are an effective way to explain the stability and reactivity of molecules. The physical quantities of Pyz-1 and Pyz-2 calculated at B3LYP/6-311G++ (d,p) level of theory are given in Table 1, the optimized structures of the most stable conformer of
The total energies of title molecules have been calculated for
FMO’s analysis
Within computational chemistry, the energies and allocations of FMO’s are imperative reactivity descriptors. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) have
| Pyz-1 | Pyz-2 | |||
| Gas | Water | Gas | Water | |
| Energy (au) | – 889.560233 | – 889.580733 | – 960.268510 | – 960.287960 |
| Polarizability (au) | 193.402 | 254.749 | 139.329 | 184.588 |
| Dipole Moment (D) | 2.660 | 2.991 | 6.585 | 8.747 |
|
|
-889.292364 | -889.313353 | – 960.101245 | – 960.121676 |
|
|
-889.360095 | – 889.381661 | -960.155981 | -960.173090 |
|
|
167.498 | 167.191 | 104.368 | 103.752 |
| Cv (
|
65.152 | 65.480 | 45.239 | 43.155 |
| S (cal/molK) | 142.552 | 143.766 | 115.203 | 108.210 |
Table 1. Ground state energies and thermodynamical parameters of Pyz-1 and Pyz-2.
Figure 1. Optimized molecular structures and FMO’s density distributions of (a) Pyz-1 and (b) Pyz-2.
significant roles in the electrical, electronic, optical properties, chemical reactivity and active sites of a given compound
The energy difference between the HOMO and LUMO orbitals determines the kinetic stability of the title compounds. Thus, molecules with a small energy gap are highly polarizable and are usually associated with high chemical reactivity as well as low kinetic stability
| Pyz-1 | Pyz-2 | |||
| Gas | Water | Gas | Water | |
|
|
-6.150 | -6.404 | -6.102 | -6.394 |
|
|
-1.032 | -1.269 | -0.937 | -1.043 |
|
|
5.118 | 5.136 | 5.166 | 5.352 |
Table 2.
Reactivity descriptors
Due to their wide range of applications in fields such as biology, chemistry and drug design, the development of chemical reactivity descriptors has gained considerable momentum
Global hardness
Molecular electrostatic potential (MEP) map
Molecular electrostatic potential is a three-dimensional representation allowing the visualization of charge distributions, the prediction of reactivity towards electrophilic and nucleophilic attacks and the relative polarity of a given molecule
A high polarity is noted for
| Parameter | Pyz-1 | Pyz-2 |
| I | 6.150 | 6.102 |
| A | 1.032 | 0.937 |
|
|
-3.591 | -3.520 |
|
|
5.118 | 5.165 |
|
|
1.260 | 1.199 |
|
|
0.104 | 0.085 |
|
|
4.091 | 4.178 |
|
|
0.702 | 0.681 |
|
|
-1.260 | -1.199 |
|
|
-1795 | -1761 |
Table 3. Quantum chemical reactivity parameters of
Figure 2. Computed molecular electrostatic potential surface (MEP) of Pyz-1 and Pyz-2.
and Pyz-2. These observations reveal a strong tendency to attract negatively charged atoms electrostatically and to act as a hydrogen bond donor group. Furthermore, For
NBO analysis
Natural bond orbital (NBO) analysis represents a computational tool for a more understandable and user-friendly interpretation of the computational solutions of the Schrödinger equation in chemical bonding concepts
To assess the donor-acceptor interactions, the second-order Fock matrix was performed. The stabilization energy
where
For both compounds, the most important interaction analyses between the occupied Lewis-type NBO orbital (bonding) and the unoccupied non-Lewis-type NBO orbital (anti-bonding) have been calculated and donor-acceptor interactions having a stabilization energy above
Biological activities
Antidiabetic activity
Compounds Pyz-1 and Pyz-2 were studied in vitro for their antidiabetic activities against
| Compounds |
|
|
|
|
|
|
|
|
|
|
| Pyz-1 |
|
|
| Pyz-2 |
|
|
| Acarbose |
|
|
Table 4.
Antioxidant Activity
The antioxidant properties of the target compounds were determined by using DPPH, ABTS,
For DPPH assay, the antioxidant activity of
The results inhibitory effect on xanthine oxidase of title compounds, showed that Pyz-1 exhibited a considerable XO activity with
Docking studies
The binding interaction of the
As noted from Table 6, all values of binding energies are negative which stabilize the systems and favor interactions between the proteins and the ligands.
Against
Among the newly suggested alternative compounds for treating diabetes,
Among the newly suggested alternative compounds for treating diabetes,
Figure 3 presented the interacted amino acid residues of
| Compound | IC50 (
|
||||
| DPPH | ABTS | FRAP |
|
XO | |
| Pyz-1 |
|
|
|
|
|
| Pyz-2 |
|
|
|
|
|
| Ascorbic acid |
|
|
|
|
– |
| Allopurinol | – | – | – | – |
|
Table 5. Antioxidant activities of
| Ligands |
|
|
||
| Affinity (Kcal/mol) | Rmsdl.b | Affinity (Kcal/mol) | Rmsdl.b | |
|
|
-5.3 | 0.000 | -5.3 | 0.000 |
| Pyz-1 | -5.7 | 0.000 | -6.7 | 0.000 |
| Pyz-2 | -4.3 | 0.000 | -5.6 | 0.000 |
| ACA | -5.9 | 0.000 | -6.2 | 0.000 |
Table 6. Docking results of the binding affinity and RMSD values of different poses in 3A4A and 2GJP.
Figure 3. Binding between the docked compounds (1, Pyz-1, Pyz-2 and Acarbose) and
are also observed with the amino acid Asp333. Regarding compound
Against
From the Table 6, the positive control ACA was found to be most effective
From the Table 6, the positive control ACA was found to be most effective
ADME-T analysis
In this study, a computational study of
Figure 4. Binding between the docked compounds (1, Pyz-1, Pyz-2 and Acarbose) and
absorption of a compound is more likely to be better if the molecule achieve at least three out of four of the following rules: (i) HB donor groups
Also, ADMET profiles of
Looking at the calculated parameter values, we find that these values are within the recommended standard. Thus, we can conclude that the molecules of the title had characteristics of drugability and their application as a theoretical drug is safe.
Conclusion
In conclusion, two novel pyrazole derivatives Pyz-1 and Pyz-2 were synthesized, characterized and evaluated for their antidiabetic and antioxidant activities. Pyz-1 and Pyz-2 were evaluated in vitro for their anti-diabetic, antioxidant activities. For anti-diabetic activity result,
| Parameter | Pyz-1 | Pyz-2 |
| Molecular Weight (g/mol) | 259.26 | 196.23 |
| Lipophilicity (LogP) | 1.044 | 0.64 |
| H-bond donors | 4 | 3 |
| H-bond acceptors | 3 | 2 |
| Polar Surface Area (Psa) | 98.91 | 88.31 |
| Distribution At Ph 7.4 (Logd) | 1.447 | 0.245 |
| Plasma Protein Binding (Ppb) | 29.32% | 30.68% |
| Rat Acute Oral Toxicity | 0.026 | 0.008 |
| Caco-2 Permeability | -5.303 | 0.023 |
| Cyp1a2 Inhibitor | 0.028 | 0.083 |
| Cyp1a2 Substrate | 0.334 | 0.054 |
| Cyp2c19 Inhibitor | 0.034 | 0.059 |
| Cyp2c19 Substrate | 0.052 | 0.038 |
| Cyp2c9 Inhibitor | 0.019 | 0.91 |
| Cyp2c9 Substrate | 0.908 | 0.001 |
| Cyp2d6 Inhibitor | 0.003 | 0.147 |
| Cyp2d6 Substrate | 0.282 | 0.012 |
| Cyp3a4 Inhibitor | 0.011 | 0.101 |
| Cyp3a4 Substrat | 0.045 | 0.023 |
Table 7. ADMET profile of Pyz-1 and Pyz-2.
Pyz-2 exhibited remarkable inhibitory ability with
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Received: 9 October 2023; Accepted: 3 January 2024
Published online: 15 January 2024
Published online: 15 January 2024
References
- Chatterjee, S., Khunti, K. & Davies, M. J. Type 2 diabetes. The Lancet 389, 2239-2251 (2017).
- Ademiluyi, A. O. & Oboh, G. Soybean phenolic-rich extracts inhibit key-enzymes linked to type 2 diabetes (
-amylase and a-glucosidase) and hypertension (angiotensin I converting enzyme) in vitro. Exp. Toxicol. Pathol. 65, 305-309 (2013). - Gao, Z. et al. Automatic interpretation and clinical evaluation for fundus fluorescein angiography images of diabetic retinopathy patients by deep learning. British J. Ophthalmol. 107, 1852-1858 (2023).
- Yang, Y. Y., Shi, L. X., Li, J. H., Yao, L. Y. & Xiang, D. X. Piperazine ferulate ameliorates the development of diabetic nephropathy by regulating endothelial nitric oxide synthase. Mol. Med. Rep. 19, 2245-2253 (2019).
- Chen, J. et al. Bone marrow stromal cell-derived exosomal circular RNA improves diabetic foot ulcer wound healing by activating the nuclear factor erythroid 2-related factor 2 pathway and inhibiting ferroptosis. Diabetes Med. 1, e15031 (2023).
- Pai, Y. W. et al. Glycaemic control for painful diabetic peripheral neuropathy is more than fasting plasma glucose and glycated haemoglobin. Diabetes Metab. 47, 101158 (2021).
- Chen, Y., Tan, S., Liu, M. & Li, J. LncRNA TINCR is downregulated in diabetic cardiomyopathy and relates to cardiomyocyte apoptosis. Scand. Cardiovasc. J. 52, 335-339 (2018).
- Wang, H., Yang, T., Wu, J., Chen, D. & Wang, W. Unveiling the Mystery of SUMO-activating enzyme subunit 1: A groundbreaking biomarker in the early detection and advancement of hepatocellular carcinoma. Transplant. Proc. 1, 945-951 (2023).
- Asmat, U., Abad, K. & Ismail, K. Diabetes mellitus and oxidative stress-A concise review. Saudi Pharm. J. 24, 547-553 (2016).
- Araki, E. & Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 1, 90-96 (2010).
- Matough, F. A., Budin, S. B., Hamid, Z. A., Alwahaibi, N. & Mohamed, J. The role of oxidative stress and antioxidants in diabetic complications. Sultan Qaboos Univ. Med. J. 12, 5 (2012).
- Karrouchi, K. et al. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 23, 134 (2018).
- Bennani, F. E. et al. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 97, 103470 (2020).
- Karrouchi, K. et al. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N’-(4-(dimethylamino) benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1219, 128541 (2020).
- Chaudhary, M. et al. Chloro and bromo-pyrazole curcumin Knoevenagel condensates augmented anticancer activity against human cervical cancer cells: design, synthesis, in silico docking and in vitro cytotoxicity analysis. J. Biomol. Struct. Dyn. 38, 200-218 (2020).
- Tighadouini, S. et al. Synthesis, crystal structure, DFT studies and biological activity of (Z)-3-(3-bromophenyl)-1-(1, 5-dimethyl-1 H-pyrazol-3-yl)-3-hydroxyprop-2-en-1-one. Chem. Central J. 12, 1-11 (2018).
- Karrouchi, K. et al. Synthesis, a-glucosidase inhibition, anticancer, DFT and molecular docking investigations of pyrazole hydrazone derivatives. Polycycl. Aromat. Compd. 1, 1-20 (2022).
- Karrouchi, K. et al. Synthesis, characterization, free-radical scavenging capacity and antioxidant activity of novel series of hydrazone, 1, 3, 4-oxadiazole and 1, 2, 4-triazole derived from 3, 5-dimethyl-1H-pyrazole. Lett. Drug Des. Discov. 16, 712-720 (2019).
- Abu-Melha, S. et al. Clean grinding technique: A facile synthesis and in silico antiviral activity of hydrazones, pyrazoles, and pyrazines bearing thiazole moiety against SARS-CoV-2 main protease (Mpro). Molecules 25, 4565 (2020).
- Pogaku, V., Krishna, V. S., Sriram, D., Rangan, K. & Basavoju, S. Ultrasonication-ionic liquid synergy for the synthesis of new potent anti-tuberculosis 1, 2, 4-triazol-1-yl-pyrazole based spirooxindolopyrrolizidines. Bioorg. Med. Chem. Lett. 29, 1682-1687 (2019).
- Karrouchi, K. et al. Synthesis, crystal structure, DFT,
-glucosidase and -amylase inhibition and molecular docking studies of (E)-N’-(4-chlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1245, 131067 (2021). - Mortada, S. et al. Synthesis, spectroscopic and DFT studies of 5-methyl-1H-pyrazole-3-carbohydrazide N-glycoside as potential anti-diabetic and antioxidant agent. J. Mol. Struct. 1267, 133652 (2022).
- Karrouchi, K. et al. Synthesis, X-ray, spectroscopy, molecular docking and DFT calculations of (E)-N’-(2, 4-dichlorobenzylidene)-5-phenyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1228, 129714 (2021).
- Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785 (1988).
- Becke, A. D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 98, 1372-1377 (1993).
- Krishnan, R. B. J. S., Binkley, J. S., Seeger, R. & Pople, J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650-654 (1980).
- McLean, A. D. & Chandler, G. S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 72, 5639-5648 (1980).
- Kainat, S. et al. Theoretical modeling of B12N12 nanocage for the effective removal of paracetamol from drinking water. Computation 11, 183 (2023).
- Shahab, M. et al. Structure based virtual screening and molecular simulation study of FDA-approved drugs to inhibit human HDAC6 and VISTA as dual cancer immunotherapy. Sci. Rep. 13, 14466 (2023).
- Ali, Q. et al. Theoretical insight of ciprofloxacin removal from water using boron nitride (B12N12) nanocage. Surf. Interfaces. 31, 101982 (2022).
- Cances, E., Mennucci, B. & Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 107, 3032-3041 (1997).
- Cossi, M., Barone, V., Cammi, R. & Tomasi, J. Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem. Phys. Lett. 255, 327-335 (1996).
- Barone, V., Cossi, M. & Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 19, 404-417 (1998).
- Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580-592 (2012).
- Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33-38 (1996).
- Reed, A. E., Curtiss, L. A. & Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88, 899-926 (1988).
- Reed, A. E., Weinstock, R. B. & Weinhold, F. Natural population analysis. J. Chem. Phys. 83, 735-746 (1985).
- F. Weinhold, J. E. Carpenter, The natural bond orbital Lewis structure concept for molecules, radicals, and radical ions. In The Structure of Small Molecules and Ions (pp. 227-236) (Springer, Boston, MA, 1988).
- Parthasarathi, R., Subramanian, V., Roy, D. R. & Chattaraj, P. K. Electrophilicity index as a possible descriptor of biological activity. Bioorg. Med. Chem. 12, 5533-5543 (2004).
- Chattaraj, P. K. & Giri, S. Electrophilicity index within a conceptual DFT framework. Phys. Chem. 105, 13-39 (2009).
- Islam, N., Kaya, S. (Eds.). Conceptual Density Functional Theory and Its Application in the Chemical Domain (CRC Press, 2018).
- Frisch, M. E., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R. et al. **Gaussian, Inc., Wallingford CT, 2016.
- Dennington, R., Keith, T., & Millam, J. GaussView, version 5 (2009).
- Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785-2791 (2009).
- Systemes, D. BIOVIA Discovery Studio Visualizer 2016. San Diego, CA: Dassault Systemes, 2016.
- Schrodinger, L. L. C. The PyMOL molecular graphics system. Version, 1(5) (2010).
- Ahmed, A. et al. Novel adamantyl clubbed iminothiazolidinones as promising elastase inhibitors: design, synthesis, molecular docking, ADMET and DFT studies. RSC Adv. 12, 11974-11991 (2022).
- Karrouchi, K. et al. Experimental and computational interaction studies of (E)-N’-benzylidene-5-methyl-1H-pyrazole-3-carbohydrazide with
-glucosidase and -amylase enzymes: A detailed structural, spectroscopic, and biophysical study. Polycycl. Aromat. Compd. 43, 1812-1832 (2023). - Pillai, R. R. et al. Synthesis, spectroscopic characterization, reactive properties by DFT calculations, molecular dynamics simulations and biological evaluation of Schiff bases tethered 1, 2, 4-triazole and pyrazole rings. J. Mol. Struct. 1177, 47-54 (2019).
- Kostić, D. A. et al. Xanthine oxidase: Isolation, assays of activity, and inhibition. J. Chem. 2015, 1-9 (2015).
- Arivazhagan, M., Manivel, S., Jeyavijayan, S. & Meenakshi, R. Vibrational spectroscopic (FTIR and FT-Raman), first-order hyperpolarizablity, HOMO, LUMO, NBO, Mulliken charge analyses of 2-ethylimidazole based on Hartree-Fock and DFT calculations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1, 493-501 (2015).
- Karelson, M., Lobanov, V. S. & Katritzky, A. R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 96, 1027-1044 (1996).
- Streitwieser, A. & Salzberg, H. W. Molecular orbital theory for organic chemists. J. Electrochem. Soc. 109, 116 (1962).
- Chattaraj, P. K., Nath, S., Maiti, B. Reactivity Descriptors 295-322. (Marcel Dekker, New York, 2003).
- El-Hadki, H. et al. Theoretical Study of Reaction Between Nitrilimine and 1,4 oxazine 2 Carboxylate by MP2 and DFT Methods. Oriental J. Chem. 34, 2992 (2018).
- Parr, R. G., Szentpály, L. V. & Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1, 1922-1924 (1999).
- Luque, F. J., López, J. M. & Orozco, M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 1, 343-345 (2000).
- El Hadki, A. et al. Removal of oxytetracycline by graphene oxide and boron-doped reduced graphene oxide: A combined density function theory, molecular dynamics simulation and experimental study. FlatChem. 27, 100238 (2021).
- Weinhold, F., Landis, C. R. & Glendening, E. D. What is NBO analysis and how is it useful?. Int. Rev. Phys. Chem. 1, 399-440 (2016).
- Weinhold, F. & Landis, C. R. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 1, 91-104 (2001).
- Agwupuye, J. A. et al. Electronic structure investigation of the stability, reactivity, NBO analysis, thermodynamics, and the nature of the interactions in methyl-substituted imidazolium-based ionic liquids. J. Mol. Liq. 337, 116458 (2021).
- Yu, Z. et al. Insights from molecular dynamics simulations and steered molecular dynamics simulations to exploit new trends of the interaction between HIF-1a and p300. J. Biomol. Struct. Dyn. 38, 1-12 (2020).
- Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 64, 4-17 (2012).
- Xiong, G. et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucl. Acids Res. 49, W5-W14 (2021).
- van Breemen, R. B. & Li, Y. Caco-2 cell permeability assays to measure drug absorption. Expert. Opin. Drug Metab. Toxicol. 1, 175-185 (2005).
- Prabakaran, G., Manivarman, S. & Bharanidharan, M. Catalytic synthesis, ADMET, QSAR and molecular modeling studies of novel chalcone derivatives as highly potent antioxidant agents. Mater. Today: Proc. 48, 400-408 (2022).
- Fettach, S. et al. Biological, toxicological and molecular docking evaluations of isoxazoline-thiazolidine-2, 4-dione analogues as new class of anti-hyperglycemic agents. J. Biomol. Struct. Dyn. 41, 1072-1084 (2021).
- Fettach, S. et al. Synthesis,
-glucosidase and -amylase inhibitory activities, acute toxicity and molecular docking studies of thiazolidine-2, 4-diones derivatives. J. Biomol. Struct. Dyn. 1, 1-12 (2021).
Acknowledgements
The authors are grateful to King Saud University, Riyadh, Saudi Arabia for funding the work through the Researchers Supporting Project number (RSPD2024R740).
Author contributions
Conceptualization, S.M.; methodology, S.M. and K.K.; software, E.H.H.; resources, A.O.; funding acquisition, M.A.B.; formal analysis, H.M. and Y.A.; writing-original draft preparation, K.K., S.M., M.A.B and E.H.H.; Visualization, S.R. and A.M.; writing-review and editing, K.K.; supervision, M.A. and M.E.A.F. All authors have read and agreed to the published version of the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Supplementary Information The online version contains supplementary material available at https://doi.org/ 10.1038/s41598-024-51290-6.
Correspondence and requests for materials should be addressed to K.K.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
© The Author(s) 2024
© The Author(s) 2024
Laboratory of Pharmacology and Toxicology, Biopharmaceutical and Toxicological Analysis Research Team, Faculty of Medicine and Pharmacy, Mohammed V University, Rabat, Morocco. Laboratory of Analytical Chemistry and Bromatology, Team of Formulation and Quality Control of Health Products, Faculty of Medicine and Pharmacy, Mohammed V University in Rabat, Rabat, Morocco. CERNE2D: Laboratory of Spectroscopy, Molecular Modelling, Materials, Nanomaterials, Water and Enviroment (LS3MN2E), Faculty of Sciences, Mohammed V University, Rabat, Morocco. Institute of Condensed Matter and Nanosciences, Molecular Chemistry, Materials and Catalysis (IMCN/MOST), Université Catholique de Louvain, 1348 Louvain-la-Neuve, Belgium. Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, 11451 Riyadh, Saudi Arabia. Central Laboratory of Hematology, Ibn Sina Hospital, Faculty of Medicine and Pharmacy, Mohammed V University, Rabat, Morocco. Laboratoire de Chimie Appliquée et Environnement (LCAE), Faculté des Sciences, Université Mohammed I, 60000 Oujda, Morocco. Laboratory of Medicinal Chemistry, Faculty of Medicine and Pharmacy, Mohammed V University, Rabat, Morocco. These authors contributed equally: Azlarab Masrar and My El Abbes Faouzi. email: khalid.karrouchi@um5s.net.ma