ترقية ثاني أكسيد الكربون إلى عطور مستدامة من خلال التحفيز المتسلسل بواسطة البيروفكسايت Upgrading CO2 to sustainable aromatics via perovskite-mediated tandem catalysis
التحول الاتجاهي لثاني أكسيد الكربون ) مع الهيدروجين المتجدد إلى منتجات ثقيلة الكربون محددة ( ) ذات القيمة العالية تقدم مسارًا مستدامًا لصناعة المواد الكيميائية ذات الصفر الصافي. ومع ذلك، لا يزال من التحدي تحقيق النشاط العالي والانتقائية في الوقت نفسه بسبب عدم التوازن الهدرجة ومعدلات الاقتران على مواقع نشطة مكملة في محفز ثنائي الوظيفة، مما يتسبب في تفاعل ثانوي غير متوقع. هنا نبلغ عنتحويل مزدوج موجه بواسطة البيروفكسايتنحو العطور الثقيلة ذات التركيز العاليتحويل (اختيارية استثنائية للعطور بين الهيدروكربونات (> 85%)، وعدم وجود تدهور واضح لمدة 1000 ساعة. يتم تحقيق ذلك من خلال فك تشابك الـمجال الهدرجة من مجال اقتران C-C في النظام المتسلسل لمحفزات قائمة على الحديد. على عكس أكاسيد الحديد النشطة الأخرى التي تظهر توزيعًا واسعًا لمنتجات الهيدروكربونات بسبب تكوين الكربيد،بشكل مصمم يتمتع بمقاومة متفوقة للتكربن، مما يمنع حدوث غير متحكم فيهالترابط على الأكسيد وعزل تكوين العطرية في الزيوليت. تكشف الأدلة الطيفية في الموقع والحسابات النظرية عن كيمياء سطح غنية بالأكسجين.التي تهرب بسهولة من سطح الأكسيد لمزيد من الدقةالترابط داخل الزيوليت، وبالتالي توجيه-مسار تفاعل العطرية لتمكين إنتاج عائد مرتفع من العطور.
ثاني أكسيد الكربون ( ) هو الغاز الدفيء الأكثر شهرة الذي يؤدي إلى الاحتباس الحراري ومجموعة من القضايا المناخية والبيئية للتخفيف من هذا التغير المناخي الناتج عن الأنشطة البشرية، هناك حاجة ملحة لتطوير حلول لـانبعاث. استخدام النفاياتكمادة خام وتحويلها إلى مواد كيميائية ذات قيمة مضافة باستخدام الطاقة المتجددة، لذلك فإن كلا الجانبين من إعادة التدوير يعدان واعدين للغاية.وتخفيف اعتماد المجتمع على الوقود الأحفوري الموارد. من بين مختلف استراتيجيات الاستخدام، التحفيز الحراريتحويل يستفيد من القدرة التخفيضية للهيدروجين المتجددتمتلك قدرة فريدة على إنتاج جزيئات معقدة مثل البارافينات طويلة السلسلة والأروماتيات الثقيلة.من منظور تقني اقتصادي، تعتبر العطرية منتجات مستهدفة مثالية كإضافات في وقود الطائرات الاصطناعي وجزيئات أساسية في العديد من تطبيقات الكيمياء الدقيقة.. وبالتالي، الاتجاه المدعوم بالطاقة المتجددة
تحويلتقدم المواد العطرية بديلاً مستدامًا وصديقًا للبيئة لعمليات الكيمياء التقليدية المعتمدة على الموارد الأحفورية، وبالتالي يمكن أن تسهم بشكل كبير في تقليل الكربون في قطاعات النقل الجوي وتصنيع المواد الكيميائية.
تم بذل العديد من الجهود لتطوير محفزات مركبة من أكاسيد المعادن/الزيوليت عالية الفعالية مع الميثانول المنتج على الأكاسيد، مثل و وهكذا، منالهدرجة، ومن ثم تحويلها إلى مركبات عطرية على مكون الزيوليت مثلت stems من المجالات النشطة المنفصلة مكانيًا وتعمل بالتوازي؛ بينما و تُفعّل على الأكسيد كمنطقة الهدرجة، مما ينتج بشكل رئيسي الهيدروجين.نوعتقوم منطقة الزيوليت بحبس هذه الأنواع في مسامها الحمضية المحصورة وتحفز التحولات المعقدة اللاحقة نحو المركبات العطرية.يمكن تنظيم الانتقائية بسهولة من خلال ضبط هيكل وحموضة الزيوليتات. ومع ذلك، فإن النموذجية التحويل في مثل هذه العمليات الحفزية غير مرضٍ (حوالي )، والتي تقتصر بشكل رئيسي على تفاعلات الهدرجة البطيئة المستخدمة عادة أكاسيد قائمة على السبينل في العمليات المتتالية. علاوة على ذلك، هجرة أن الأنواع من أكسيد المعدن إلى الزيوليت غالبًا ما تؤدي إلى تقصير عمر المحفز. هذه التحديات تبرز الحاجة الملحة لأساليب مبتكرة لا تعزز فقطكفاءة التحويل ولكن أيضًا معالجة استقرار وطول عمر النظام الحفاز.
على العكس، تم الإبلاغ على نطاق واسع عن المحفزات القائمة على الحديد بفعاليتها المتفوقة لـالهدرجة إلى العطرية؛ ولكن بشكل عام، تعاني أيضًا من انتقائية غير مرغوب فيها عند دمجها مع H-ZSM-5. وقد أطلق على هذه العملية أيضًا اسم مسار فيشر-تروبش المعزز. لتكون أكثر تحديدًا، الأوليفينات الخفيفة ( ) تم إنتاجها على نطاق أكاسيد الحديد مع انتقائية في نطاق . مثل هذه الوسائط تنتشر في الزيوليت لفتح تفاعل اقتران C-C وتفاعل التدوير لإنتاج المركبات العطرية. ومع ذلك، فإن هذا المسار المركب يشمل شبكة تفاعلات معقدة: الهدرجة وغير المنضبطةيحدث الاقتران على أكسيد الحديد، بينما المزيديحدث الاقتران داخل الزيوليت. وغالبًا ما تؤدي هذه التعقيدات إلى عدم التوافق.معدلات الاقتران بين الأكسيد والزيوليت والتحويل غير المقصود للأوليفينات داخل قنوات الزيوليت. ونتيجة لذلك، نادراً ما حققت أكاسيد الحديد انتقائية مرضية للأروماتيات بين الهيدروكربونات.في هذا التحويل المتسلسل المحفز المركب لـ. بينما استراتيجيات التحكم الدقيقة للغاية مثل إدخال كمية مناسبة من المعادن القلوية، كما هو موضح من قبل صن وآخرون.لقد أظهرت أنها تحسن الانتقائية، لكنها تقدم تحديًا جديدًا: تميل المعادن القلوية إلى الهجرة إلى الزيوليت، مما يقلل من الأداء التحفيزي مع مرور الوقت.. لتجاوز هذه القضايا، فصل الـمجال الهدرجة منقد يكون مجال الاقتران في النظام المتسلسل استراتيجية محتملة أخرى للمحفزات القائمة على الحديد. بعبارة أخرى، من خلال الحفاظ على نشاط الهدرجة القوي مع تخفيف الـقدرة الاقتران على جانب أكسيد الحديد، قد تقدم إمكانيات كبيرة للتحويل الثنائي الاتجاه بكفاءة عاليةإلى العطور.
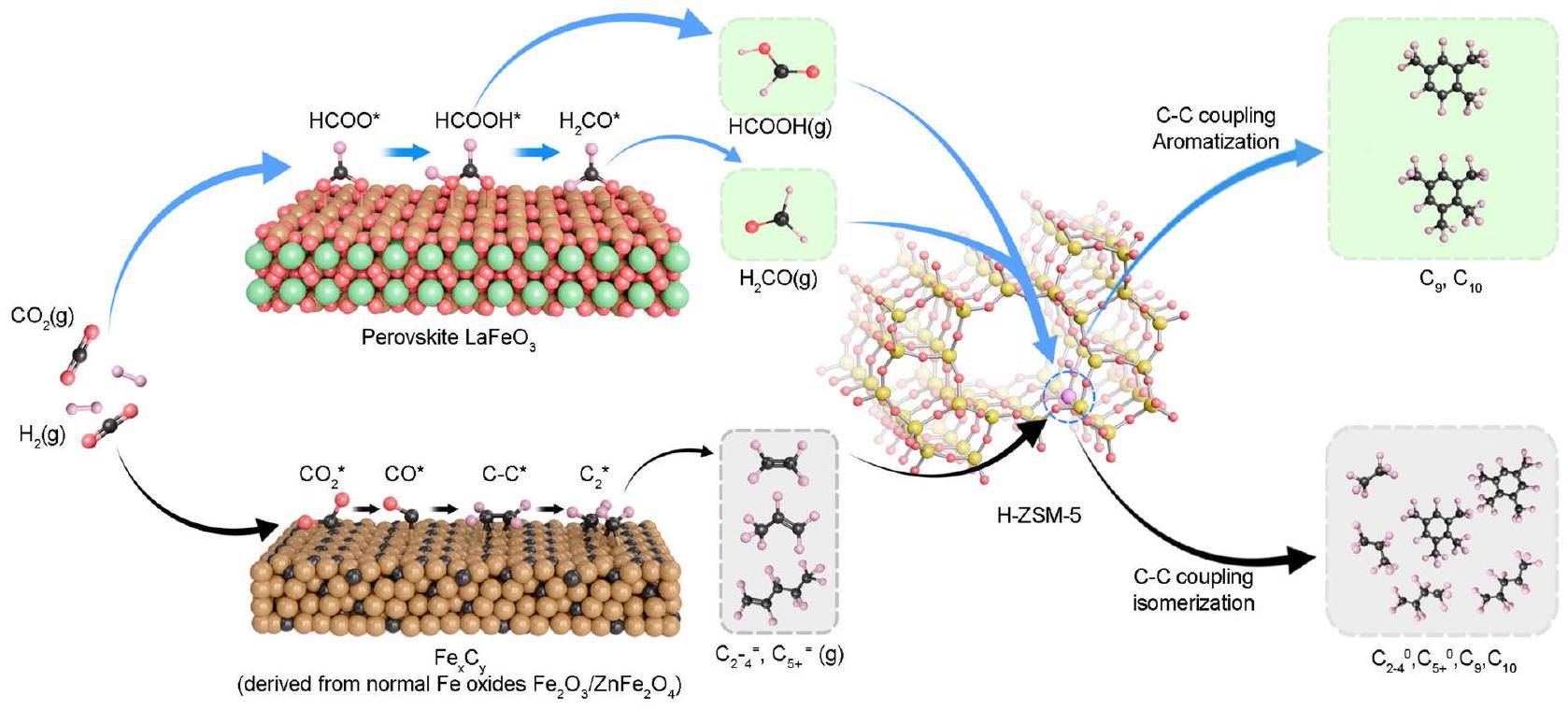
استنادًا إلى الاعترافات المذكورة أعلاه، نقدم نظامًا مركبًا جديدًا من أكسيد الحديد/الزيوليت يتميز بـالبيروفسكايت، الذي يتميز بشكل واضح بتصميمه المتقنإطار ثماني السطوح وهياكل نطاق الحديد-3d، مما يوفر استقرارًا فائقًا ومقاومة استثنائية للكربنة. وهذا يتناقض بشكل حاد مع المحفزات التقليدية القائمة على الحديد، التي تظهر عمومًا ارتفاعًا فيالانتقائية. نظامنا يقمع بشكل فريد تكوين كربيد الحديد، مما يضمن أنالهدرجة علىيحقق بشكل أساسي حاسمالمنتجات ( )، بينما يحدث الاقتران بشكل أساسي داخل زيوسيت H-ZSM-5، باستخدام الأكسجينات المزالة كمواد أولية. هذه التآزر بينالبيروفيسكايت وH-ZSM-5 يؤديان إلى تحكم لا مثيل له في انتقائية الهيدروكربونات، مع تجاوز إجمالي العطرياتوانتقاء أول أكسيد الكربون-إنجاز لم يتم تحقيقه بعد من قبل غيره من المحفزات القائمة على الحديد. علاوة على ذلك، فإنه يدفع معدل التحويل إلى أكثر من، متجاوزًا بشكل كبير التقليديتمت ملاحظته مع أكاسيد غير الحديد. ومن الجدير بالذكر أنيظهر محفز H-ZSM-5 أيضًا متانة استثنائية، حيث يحافظ على الأداء لأكثر من 1000 ساعة مع معدل تدهور لعائدات العطرية أقل بكثير من تلك الموجودة في أنظمة المركبات الأخرى المبلغ عنها. إن التنفيذ الناجح لـبالتزامن التحويل لا يبرز فقط الإمكانيات الواسعة للبيروفسكايت القائم على المعادن النشطة في الصناعة إعادة التدوير ولكنها أيضًا تبشر بعصر جديد من التحفيز الثنائي المدعوم بالبيروفسكايت. توفر هذه الخطوة الرائدة مسارًا مستدامًا وقابلًا للتوسع للاستخدام الواسع.الاستخدام والتركيب الكيميائي، مما يمهد الطريق لصناعة كيميائية خالية من الكربون.
النتائج والمناقشة
تصميم وبنيةبيروفسكايت
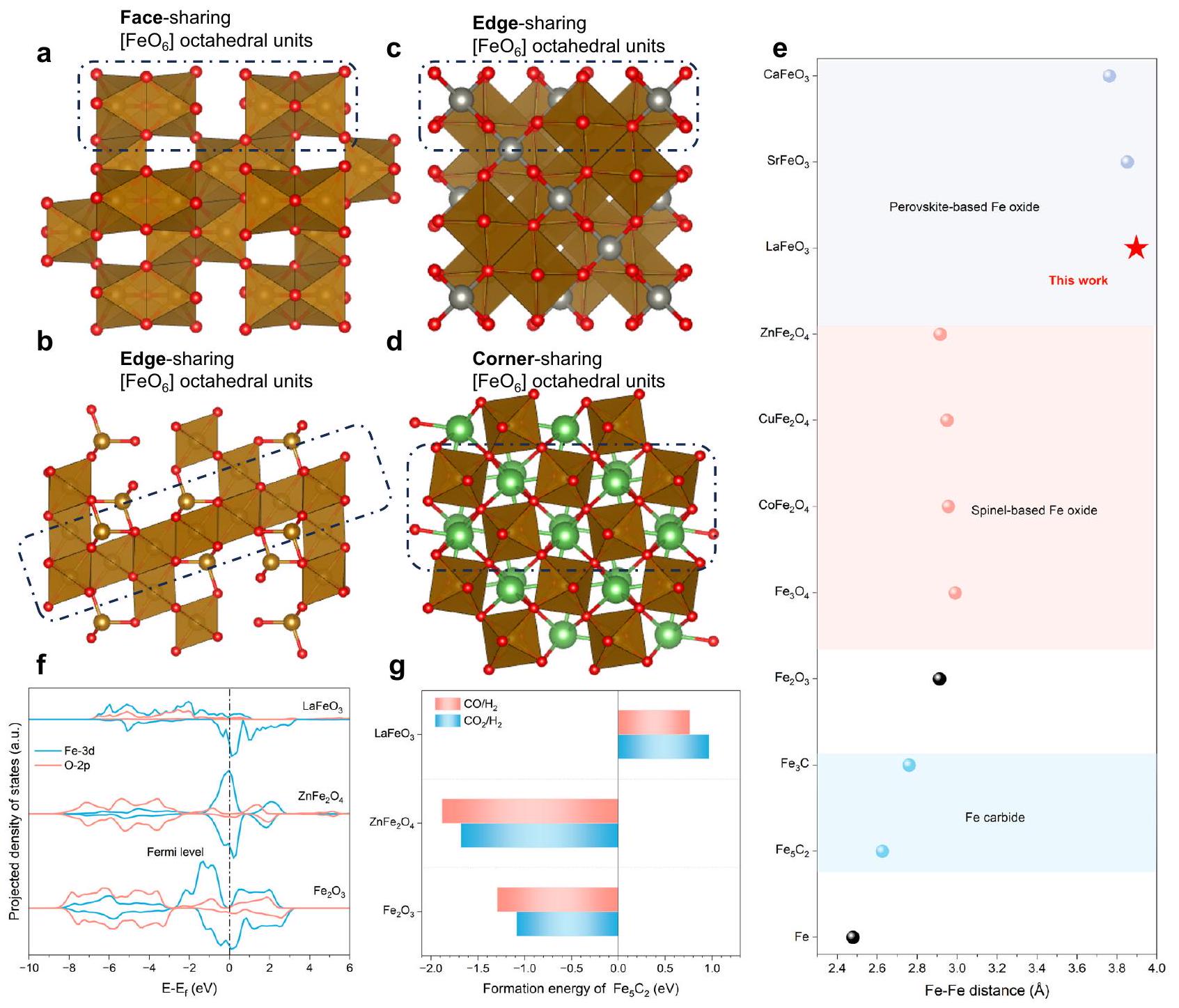
أكاسيد البيروفسكايت في صيغة عامة منلديك إطار من المشاركة في الزواياأوكتاهيدرا تحتوي على كاتيونات A كبيرة مملوءة في الفراغات“. في مثل هذه البنية، يفضل الحديد (Fe) شغل موقع B وبالتالي يمكّن من بناء مشاركة الزاوية [إطار العمل في البيروفسكيتات القائمة على الحديد، والذي يختلف تمامًا عن الأطر المشتركة في الوجوه أو الحواف في أكاسيد الحديد العامة (على سبيل المثال، ) و -نوع السبينل (الشكل 1أ-د). تؤدي هذه الاختلافات الطوبولوجية إلى تمييز واضحالمسافات، التي من المتوقع أن تؤثر على هجرة الحديد إذا تم كربنة الأكسيد (الشكل 1e). من بين مختلف أكاسيد الحديد، لديه الأكثر انضغاطًا [ ] الاتصال وبالتالي الأقصر مسافةسبينلات من نوع -type أو سبينل عكسيتحتوي بشكل أساسي على حواف متشاركة ] الأوكتاهدرا، تعرض الوسيطة المسافات في نطاق؛ عادةً ما تمتلك البيروفسكايت القائمة على الحديد فترة أطول بكثيرمسافاتخلال الانتقال الطوري من أكاسيد الحديد إلى كربيدات الحديد،يجب أن تقل المسافة إلى حوالي. لذلك، فإن المسافة المطلوبة لهجرة الحديد خلال الانتقال الطوري تكون ممتدة بشكل ملحوظ بالنسبة للبيروفسكيتات القائمة على الحديد. في الوقت نفسه، يعمل الكاتيون في موقع A كأعمدة تثبيت في البيروفسكيت [نحن نتوقع أن الجوانب الهيكلية لكل من البيروفيسكايت حسب التصميم يمكن أن تؤدي إلى تقليل هجرة الحديد خلال عملية الكربنة المحتملة.
كإثبات للمفهوم،تم اختيار La لأنه معدن نادر الأرض بأكبر نصف قطر أيوني، مما يؤدي إلى طول ملحوظ مسافة، ولدى اللانثانوم مدارات 4f فارغة فريدة يمكن أن تنظم استقرار إطار من خلال تأثير المجال البلوري. لتوضيح هذا التأثير، تم إجراء حسابات نظرية الكثافة الوظيفية (DFT) لتحليل الكثافات الحركية للدالة.، و (الشكل 1f). يتبين أن دمج اللانثانوم في الزوايا المشتركة [ ] المصفوفة تقلل بشكل كبير من عدد حالات Fe-3d بالقرب من مستوى فيرمي عند مقارنتها بـ يظهر اتجاه معكوس في حالات Fe -3d للسبينيل. بينما تشكيل أولاً يتضمن التفاعل بين حالات الحديد بالقرب من مستوى فيرمي وحالات الكربون، يمكن أن يؤدي الانخفاض في حالات Fe-3d بالقرب من مستوى فيرمي إلى الحاجة إلى طاقات أكبر بكثير لتشكيلوبذلك تم قمع الكربنة لـطاقات التكوين المحسوبة لـتأكيد أكبر على الاستقرار الأعلى لـمن و تحت أجواء اختزالية نموذجية تتكون من أو (الشكل 1g). تحت كلا الغلافين الجويين، طاقات التكوين لـ (لكل مول) من و سلبية، تظهر ميلاً قوياً لتشكيل كربيدات الحديد. في تناقض حاد، فإن طاقات التشكيل لـمنأكثر إيجابية بمقدار 2.06 و 2.64 إلكترون فولت من و ، على التوالي. توفر النتائج النظرية رؤى حرارية حول مقاومة الكربنة المتفوقة لـالذي ينشأ من انخفاض نشاط الحديد من خلال التعديل الإلكتروني للان.
لتحقيق التصميم أعلاه، جزيئات نقية من المرحلةمحفز بحجمتم تخليقه بواسطة طريقة المستحلب الدقيق وتم حل هيكله البيروفسكيتي المنظم (PDF-#37-1493) بواسطة مجهر الإلكترون الناقل الماسح بتباين الطور المتكامل ومجهر الحقل المظلم الحلقي عالي الزاوية (HAADF-
الشكل 1 | تصميمالبيروفسكايت كأكسيد قائم على الحديد مقاوم للتCarbonization. نموذج الهيكل الذري لـ (أ)، (ب) ، (ج) ، و (د) ، موضحًا أوضاع الاتصال المختلفة لـالأوكتاهدرا. الكرات باللون الأحمر والبني والفضي والأخضر تمثل، وذرات لا، على التوالي. e مسافاتالكربيدات، ومختلف أكاسيد الحديد.الكثافة المتوقعة للحالات (PDOSs) لـ، و تم حساب طاقات التكوين لكل مولمن خلال تقليل و عن الأجواء التفاعلية النموذجية.
STEM و iDPC-STEM) و حيود الأشعة السينية (XRD) (الشكل التوضيحي 1). تم أيضًا حل تشوه بنية البيروفسكايت المكعبة المثالية فيالذي يشير إليه الميل من خلال ملاحظة iDPCSTEM (الشكل التكميلي 2). [ ] الميل يكون من أجل استيعاب الفجوة الكبيرة لأيونات اللانثانيد. هيكل الهيماتيت (PDF-#33-0664) وبنية السبينل (PDF-#221012)تم الحصول على المحفزات كعينات تحكم (الأشكال التكميلية 3 و 4). جميع العينات تظهر كجسيمات نانوية بحجم يتراوح من.
أداء التحفيز لـتحويل على مختلفمحفز
تم تقييم المحفزات الثلاثة المذكورة أعلاه أولاً تحتظروف الهدرجة ). كما هو موضح في الشكل 2أ، كلاهما و تنتج بشكل أساسي الهيدروكربونات في و نطاقات (الانتقائية الإجمالية ). يتماشى توزيع الهيدروكربونات هذا مع نموذج ASF، مما يتوافق مع احتمال نمو السلسلة ( ) يتجاوز 0.7 (الشكل التوضيحي 5). على النقيض، يولد الميثان ( ) كمنتج هيدروكربوني أساسي ، مع تقليل كبيرمن 0.09. منذ أن تم إنفاق وتعاني الأكاسيد من انتقال ملحوظ في الطور إلى كربيدات الحديد، التي ثبت أنها عوامل حفازة ممتازة لـ الاقتران في تخليق فيشر-تروبش (FTS) (الأشكال التكميلية 6 و 7)، بينما تم إنفاق لا (الشكل التوضيحي 8)، تم قمعه بشكل ملحوظالاقتران على البيروفسكايت يُنسب ذلك إلى مقاومته العالية للتكربون. بشكل عام، تكشف النتائج أعلاه عن الإمكانيات الكبيرة لـ كمنطقة تحفيزية معزولة لا تمثل سوىالهدرجة في التحفيز المتزامن.
عند التكامل مع H-ZSM-5 (الشكل التوضيحي التكميلي 9)، وهو زيلوت ألومينوسيليكات محترم قادر على التحفيزالاقتران وإيزومرة الهيدروكربونات ولكن غير قادر على التحفيزتمت ملاحظة تغييرات ملحوظة في ملفات المنتجات فقط من خلال الهدرجة (الشكل 2أ). كلاهما و تظهر زيادات فينسبة التحويل ( و ) وانتقائية العطور ( و )، والتي هي، مع ذلك، أقل عمقًا من (الشكل 2أ). و البرافينات هي منتجات ثانوية رئيسية من الهيدروكربونات في النظامين المركبين (الشكل التكميلي 10). المنافسة بين الاثنينتساهم مسارات الاقتران على كربيدات الحديد المستمدة في الموقع وH-ZSM-5، على التوالي، في هذا التوزيع الواسع للمنتجات متعددة الكربون. بينما بالنسبة لـإجمالي العطرية
الشكل 2 | الأداء التحفيزي لـالتحويل. الانتقائية في الهيدروكربونات،تحويل، انتقائية CO لمحفزات الأكسيد النقية (اللوحة اليسرى: ، و )، زيوسيت H-ZSM-5 (67) فقط، وأنظمة التحفيز المركبة المتسلسلة (اللوحة اليمنى: (67)، (67)، و نسبة الكتلة للأكسيد إلى H-ZSM-5 هي 2:1.تقييم تحفيزي لـ (67) بنسب كتلة مختلفة. ج الأداء التحفيزي على مع نسبة Si/Al مختلفة. أداء التحفيز
مقارنة بين (نجمة حمراء) وذكرت المحفزات غير الحديدية (دائرة وردية) والحديدية (دائرة زرقاء) فيما يتعلق باختيار العطرية في الهيدروكربونات والتحويل. الخط المنقط الأسود هو دليل للعين. البيانات المعروضة فيتم الحصول عليه تحتبسرعة فضائية قدرهافيتقييم الاستقرار التحفيزي لـمحفز مع ظروف تفاعل أكثر صرامة بسرعة فضاء تبلغفي، و . تصل الانتقائية في جميع الهيدروكربونات إلىبينما يتكون كل من ثلاثي ميثيل البنزين وأربعة ميثيل البنزين، وهما مضافان مرغوب فيهما ذوا قيمة حرارية عالية في وقود الطائرات، من تقريباًمن إجمالي العطريات (الشكل 2 أ والشكل التكميلي 10). بالإضافة إلى ذلك،نسبة التحويل تظهر زيادة حادة منإلى. هذا يعني أن H-ZSM-5 يمكنه استخدام بعض الوسائط بكفاءة على طول المسار البطيء مسارات الهدرجة على الحالة النقية. يشير إلى أن الأروماة لمثل هذه الوسائط في المسام الحمضية لـ H-ZSM5 قد تكون أسرع من الهيدروجين العميق نحو على أسطح الأكسيدأثرتمت دراسة نسبة الكتلة لـ H-ZSM-5 بشكل أعمق (الشكل 2ب). تم اعتماد نسبة كتلة معتدلة منيؤدي إلى مثاليتحويل وانتقائية العطور. إن زيادة أو تقليل نسبة الكتلة يؤدي كلاهما إلى تدهور غير مرغوب فيه في أداء التحفيز، والذي يُعزى إلى عدم توازن الهدرجة ومعدلات الاقتران على أكسيد المكمل ومجالات الزيوليت.
لقد تم الإبلاغ عن أن خاصية الحمضية للزيوليت لها تأثير قوي على الأخيرة.تفاعل الاقتران وتثبيط CO -المنتج الثانوي. لذلك، قمنا أيضًا بإجراءمع نسبة Si/Al مختلفة (الشكل التكميلي 11). كما هو موضح في الشكل 2c، مع زيادة الخصائص الحمضية في الزيوليت، تم تثبيط المنتج الثانوي CO منإلى و تم زيادة التحويل من 50.8 إلىيمكن استنتاج أن المواد الوسيطة المنتجة على مدىقد يكون له ردتان لاحقتان. واحدة هي الانتشار في الزيوليت لمزيد منالترابط لإنتاج العطرية و آخر يتحلل إلى CO كمنتج ثانوي عندما تكون مواقع H-ZSM-5 الحمضية غير كافية.
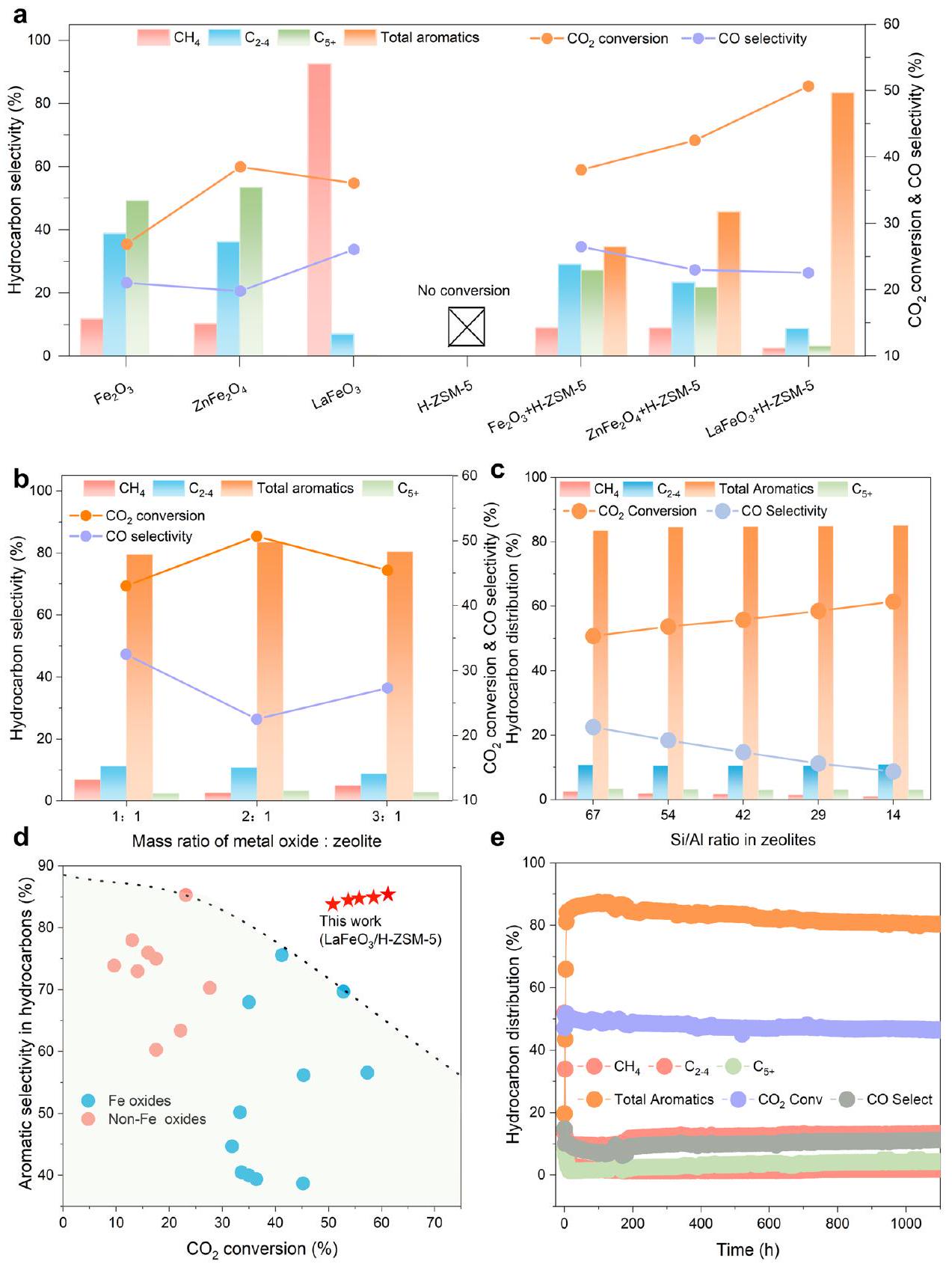
لإظهار هذه التفوق بشكل أكبر، نقارن الأداء التحفيزي لـمع غيرها من المحفزات المركبة المتطورة المبلغ عنها لـالتحويل إلى العطرية تحت ظروف مشابهة (الجدول التكميلي 1). كما هو موضح في الشكل 2d،ZSM-5 يظهر ليس فقط على الأقل عدة أضعاف أعلىمعدل التحويل أعلى من المحفزات غير أكسيد الحديد ولكنه أيضًا أعلى انتقائية للأروماتيات في الهيدروكربونات. يُعزى هذان الجانبان إلى مواقع نشطة للغاية قائمة على الحديد لـالهدرجة وهيكل البيروفكيت المقاوم للتCarburization الفريد من نوعه عن طريق التصميم، على التوالي. ونتيجة لذلك،و H-ZSM-5 يمكّنان معًا من تقييد صارم للتسلسلالهدرجة وتكوين العطرية في مجالات مفصولة مكانيًا، مما يؤدي إلى أداء تحفيزي متفوق لتحويل التتابع الاتجاهي لـإلى العطور. تم أيضًا إخضاعه لاختبار متانة التفاعل المستمر في ظروف تفاعل أكثر صرامة. وقد تم الكشف عن أن انتقائية العطرية ومعدل التحويل الحفاظ على و ، على التوالي، لأكثر من 1000 ساعة على التوالي (الشكل 2e). عند التركيز على معدل التحويل الانتقائي للمواد العطرية الكلية، والذي يعمل كمؤشر متكامل للأداء التحفيزي،يعرض معدل تدهور منخفض للغاية لمعدل الإنتاج الأولي (STY)في الساعة)، مما يتوافق مع تدهور أداء أبطأ بمعدل 3.4-17.6 مرة مقارنةً بالمواد المحفزة المركبة المبلغ عنها لـ-تحويل إلى العطرية (الشكل التوضيحي 12). بينما بالمقارنة مع المحفزات التقليدية القائمة على الحديد أو الزنك التي تعاني بسهولة من ترسب الكربون و/أو التلبيد وبالتالي تتعطل بسرعةهيكل البيروفسكايتيظهر إمكانيات كبيرة للتطبيقات الصناعية بسبب الاستقرار القياسي العالي، بالإضافة إلى معدل إنتاج جيد من العطرية بانتقائية عالية. علاوة على ذلك، تم إجراء المزيد من اختبارات الاستقرار بما في ذلك H-ZSM-5 التقليدي مع H-ZSM-5 بحجم نانو كما هو موضح في الشكل التكميلي 13.
الاستقرار الهيكلي لـتحت جو تفاعلي
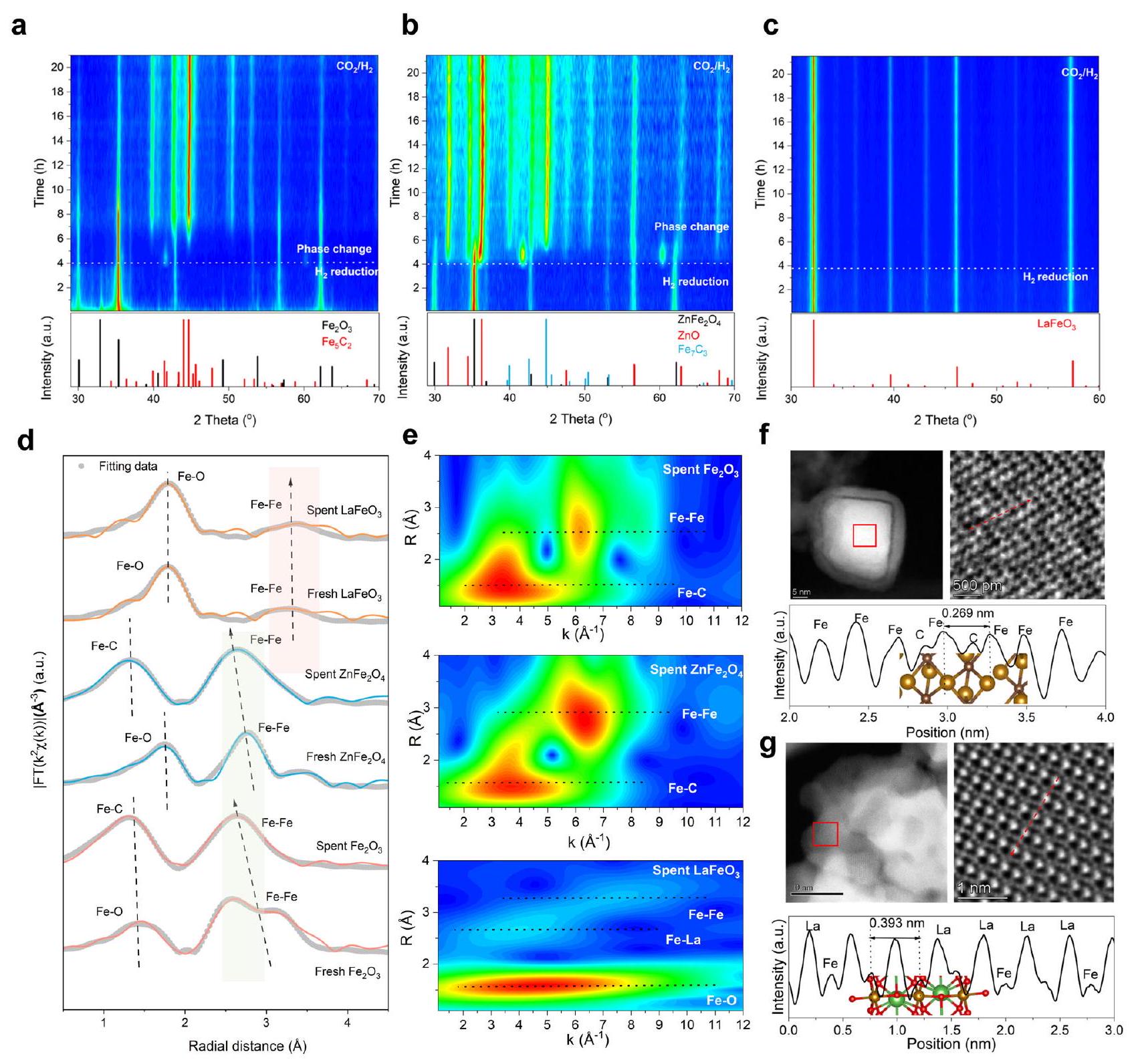
الانتقائية العطرية العالية والاستقرار التحفيزي المتفوق لـالبيروفسكايت يتماشى جيدًا مع التوقعات التييمكن أن يكون البيروفيسكايت محفزًا مقاومًا للتكربن بشكل مختلف تمامًا عن أكاسيد الحديد العادية والسبينيل. للحصول على رؤى تجريبية إضافية حول تطور المحفز تحت جو تفاعلي، تم إجراء تشخيصات طيفية وميكروسكوبية تجمع بين التقنيات في الموقع وخارج الموقع. تكشف حيود الأشعة السينية في الموقع (XRD) أولاً أن و يمكن أن تحافظ على هياكلها الضخمة الأصلية عند البدايةتقليل ولكن التحول تدريجياً إلىومزيج من ZnO وعلى التوالي، بمجرد أن يصبح الغاز الكربوني ( ) يتم إدخاله إلى غرفة المراقبة في الموقع لتشبه الأجواء التفاعلية (الشكل 3 أ، ب والشكل التكميلي 14). بشكل مميز، هيكل البيروفسكايت لـ يتم الاحتفاظ به جيدًا دون توليد أي مراحل بلورية جديدة تحت أي من أو الجو (الشكل 3c والشكل التكميلي 15). بالإضافة إلى تطور الهيكل الكتلي، تم دراسة تطور السطح بواسطة مطيافية الأشعة السينية للأشعة السطحية (XPS) في الموقع. و تظهر أنماطًا مشابهة لجزء منيتم تقليله أولاً إلى حالة ذات قيمة أقلبواسطةثمإذا تم إعادة أكسدته جزئيًا إلىفي مختلط (الشكل التوضيحي الإضافي 16أ، ب). ظهور يتوافق جيدًا مع التكافؤ الاسمي للحديد في كربيداته. بينما بالنسبة لـأكسيد الحديد البيروفيسكايت، حيث أن الأجواء الاختزالية فقط تؤدي إلى مزيد من عدم التشبعلكن لا (الشكل التكميلي 16c). ويصاحب ذلك زيادة في كميات فراغات الأكسجين (الشكل التوضيحي الإضافي 17). قد تكون مواقع لـتفعيلوخطوات الهدرجة اللاحقةالذي سنتناول دوره في التحفيز في قسم لاحق.
تم استخدام تقنيات الأشعة السينية في السنكروترون خارج الموقع للتحقيق في الحالة الكيميائية والهياكل المحلية الدقيقة لـ محفزات أكسيد الحديد المستهلكة. هياكل امتصاص الأشعة السينية عند حافة الحديد K المنضبطة تكشف عن حالة الأكسدة المنخفضة بشكل ملحوظ للحديد في و بعد تفاعل لمدة 24 ساعة، بما يتماشى مع ملاحظة XPS في الموقع (الشكل التكميلي 18). في FT-EXAFS، القمة الثانية التي تُعزى إلىتنقل التنسيق إلى مواقع على مسافات أقصر لـ و بعد تفاعل لمدة 24 ساعة (الشكل 3د)تم استخراج معلومات هيكلية أكثر تفصيلاً تحت بيانات FTEXAFS الخام بعد التوافق (الجدول التكميلي 2).أرقام التنسيق في النفايات و كلاهما يزيد إلى أكثر من 7، مما يشير إلى تشكيل كربيدات الحديد مع المزيد من الحديد المتجمع. على عكس و ، يعرض حالة أكسدة تغيرت بشكل طفيفالمسافة (في الأصل أطول من و )، و عدد التنسيق بعد نفس المعالجة. يوفر WT-EXAFS رؤى إضافية حول تطور المحفز (الشكل التكميلي 19 والشكل 3e). تشتت الحديد-حديد في المحفز المستهلكلديه شدة أقل من العادي المستهلكوالسبينيل، مما يشير إلى ضعف التفاعل وبالتالي قمع هجرة الحديد وتجمعه.
أخيرًا، تم إجراء مجهر إلكتروني عالي الدقة للكشف عن التطور الشكلي لمحفزات أكسيد الحديد المستهلكة. بالنسبة لـكمية كبيرة منتم الكشف عنها مع الحديد المتجمع في الفجوات الكربونية، مما يظهر قصرمسافة 0.269 نانومتر (الشكل 3f)؛ بينمايظهر أنه يحتفظ بالشكل الجزيئي مع تغير ضئيل في حجم الجسيمات، بالإضافة إلى الهيكل الدقيق للبيروفسكايت مع ترتيب بديل للانثانوم والحديد (الشكل 3g). توضح ملفات شدة صور iDPC-STEM على طول الخط الوهمي الحفاظ على الطولالمسافة (0.393 نانومتر) ووجود (الشكل التوضيحي التكميلي 20). جميع نتائج الحي والحي الخارجي من التحليل بالأشعة السينية، الطيفية، والميكروسكوبية تؤكد على الاستقرار الهيكلي المتفوق ومقاومة الكربنة العالية للبيروفسكايت.في جو تفاعلي، مما يثبت عقلانية تصميمنا الهيكلي وتوقعنا النظري بأن طويلاًيمكن أن يؤدي تعديل المسافة وهيكل الإلكترونيات من خلال استخدام أعمدة اللانثانوم إلى تقليل هجرة الحديد بشكل فعال.
فهم آلي لـتحويل على. ( إلىإلى العطور)
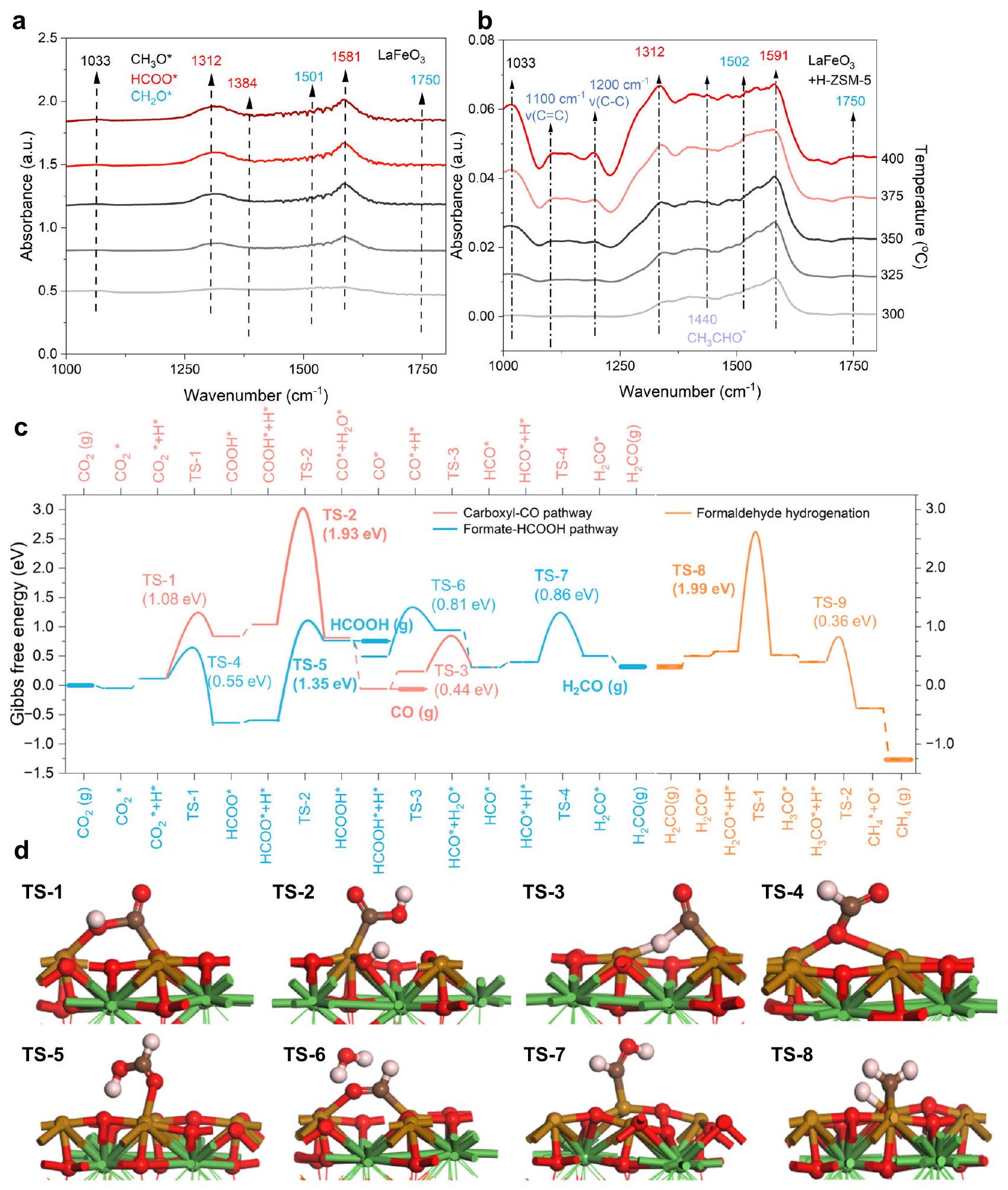
بالإضافة إلى الاستقرار الهيكلي، القدرة الفريدة لـلتحفيز انتقائيالتحويل إلىالأنواع، فضلاً عن التآزر بينو H-ZSM-5 في تمكين الاتجاه العالي الكفاءةتم الكشف عن تحويل – إلى عطر. تم استخدام مطيافية الأشعة تحت الحمراء التحويلية فورييه بالانعكاس المنتشر في الموقع (DRIFTS) لأول مرة لاستكشاف الأنواع الرئيسية التي قد تهيمن على التفاعلات على أسطح الأكاسيد مع تغير درجة الحرارة. علىفقطأنواع مؤكسدة مثل الفورماتفي و “; النجمة تشير إلى الحالة الممتصة على السطح)، الفورمالديهايد (في ) وميثوكسي ( في ) لوحظت ( الشكل 4أ) لاحظ أن شدة القمم المميزة المذكورة أعلاه تزداد إلى الحد الأقصى حتى ترتفع درجة الحرارة إلى ما فوق (الشكل التوضيحي 21). بالإضافة إلى ذلك، لم تُلاحظ أي إشارات تتعلق بأنواع متعددة الكربون فقط عند دمجه مع H-ZSM-5، تزداد شدة نطاقات الانعكاس المرتبطة بـتقل الأنواع المؤكسجة بمقدار مرتبة (الشكل 4ب)، مصحوبة بظهور بارز لـفرقة فيعلى التوالي، مما يعني أن H-ZSM-5 لديه القدرة على تحويلأنواع مؤكسدة إلى منتجات متعددة الكربون (الأشكال التكميلية 22 و23). بالإضافة إلى ذلك، أيضًايمكن العثور على الأنواع فيلقد تم اقتراح آلية الألدول أو برينس سابقًا لتفسير هذه القدرة.. لكلاهما و ZSM-5، تظل شدة الأطياف المميزة غير متغيرة بعد فترة تحريض قصيرة، مما يشير إلى حالة مستقرة نسبياً.الأسطح.
مثل كيمياء السطح التي تهيمن عليها الأكسجيناتمختلف تمامًا عن ذلك على (مشتق من )، مجموعة من أوضاع التمدد ( ) و أوضاع التمدد/الانحناء
الشكل 3 | التطور الهيكلي لمختلف محفزات الحديد تحت أجواء تفاعلية. أنماط XRD في الموقع لـ (أ)، (ب) و (ج) تحت أجواء اختزالية مختلفة. شروط الاختبار: و (الأربع ساعات الأولى) أو (الـ 20 ساعة التالية). د هياكل الامتصاص الضوئي الدقيقة الممتدة بواسطة تحويل فورييه عند حافة الحديد K (FT-EXAFSs). هـ تحويل الموجات (WT)-EXAFSs عند حافة الحديد K للنفايات، و المحفزات. زاوية عالية مجهر الإلكترون الناقل بتقنية المسح في مجال الظلام الحلقي (STEM)، صور STEM بتقنية التباين الطوري التفاضلي المتكامل (iDPC)، وملفات شدة صور iDPC-STEM على طول خط وهمي من النفايات (f) و (ز) تظهر الإضافات الهيكل الذري مع كرات باللون الأحمر والأسود والبني والأخضر تمثلوذرات اللانثانوم، على التوالي. ( ) تشير إلى وجود هيدروكربونات متنوعة (الشكل التكميلي 23). هذه الميزات نموذجية لمحفزات كربيد المعادن النشطة في تفاعل تحويل الغاز إلى سوائل (FTS) التي تتضمن البلمرة. وبالتالي، تُنسب هذه الأنماط الملحوظة إلى تكوين كربيدات الحديد. ومن الجدير بالذكر أن شدةتزداد الحزم باستمرار مع ارتفاع درجة حرارة التفاعل، مما يشير إلى احتباس وتراكم الأنواع الكربونية التي تؤدي في النهاية إلى تمرير السطح. وهذا يتماشى مع التوقف السريع للتفاعل العادي.والعوامل الحفازة المشتقة منها.
في ضوء كيمياء السطح الغنية بالأكسجين الذي يؤدي بشكل رئيسي إلى تمت دراسة المنتجات وطاقة التفاعل باستخدام حسابات DFT. (220) سطح مع أو بدون سطح تم اعتبارها لمحاكاة (الشكل التوضيحي 24)، وكانت طاقات جيبس الحرة للتكوين للأنواع/الحالات السطحية ذات الصلة، بما في ذلك حالات الانتقال (TSs)، بعد تحسين الهندسة هي تم حسابها (الأشكال التكميلية 25-28). بشكل عام، فإن قوى الامتزاز للأنواع السطحية على السطح بدونضعيفة بشكل ملحوظ، مما يجعلسطح خالٍ من التفاعلالهدرجة (الجدول التكميلي 3). لذلك، نركز بعد ذلك على السطح معلتحقيق في الديناميكا الحرارية والحركية لمختلف مسارات التفاعل.
كما هو موضح في الشكل 4c،الهدرجة تتضمن أولاً الهيدروجينالامتزاز على موقع الأكسجين والامتزاز على بالقرب من موقع الحديد. بينما قناتين لـ الهدرجة مفتوحة إما أو كربوكسيل ( )، تتفرع مسارات التفاعل إلى فرعين رئيسيين، حيث يسير أحدهما عبر والآخر من خلال حمض الفورميك ( )، حتى تشكيل الفورمال ( ” ). بمجرد التشكيل، تندمج الفرعان مرة أخرى، يتبع ذلك هدرجة متسلسلة لتكوين، و في النهاية (الشكل 4c). كلا من مسارات الكربوكسيل والفورمات هي بشكل أساسي
الشكل 4 | التحقيق الميكانيكي فيتحويل علىطيف DRIFTS في الموقع المعتمد على درجة الحرارة على (أ) و (ب) -ZSM-5. جداول الطاقة الحرة لـالهدرجة على سطح (220) سطح مع ، موضحًا مسارات الكربوكسيل (بالأحمر) والفورمات (بالأزرق) نحو الفورمالديهايد ومسار هدرجة الفورمالديهايد (بالبرتقالي) لإنتاجلإظهار تأثير إزالة الجزيئات، فإن حالات الجزيئات في الطور الغازي هي
مُميز بخطوط أفقية عريضة. يمكن توضيح المنافسة بين إزالة الجزيئات والهيدروجين الإضافي في الحالات التي يكون فيها HCOOH الغازي ويُفترض أن تكون المواد المتفاعلة للهيدروجين اللاحق. هيكل حالات الانتقال d TS فوقخلالإلىالكرات باللون الأسود والأحمر والبني والأخضر تمثلوذرات اللانثانوم، على التوالي. مقيد بالخطوة الثانية من الهيدروجين، التي يجب أن تتجاوز حاجز تنشيط قدره 1.93 إلكترون فولت (TS-2) و1.35 إلكترون فولت (TS-5) لإنتاج و على التوالي. لذلك، فإن مسار الفورمات أكثر وصولاً من الناحية الحركية من مسار الكربوكسيل على يحتوي على (220)، وهو ما يتماشى مع الإشارات القوية لـ HCOO* (الموجودة حصريًا في مسار الفورمات) في قياسات DRIFTS في الموقع. مسار ثانوي يبدأ من الفورمات، وهو ما يسمى تتم الهدرجة عبر أكسيد الفورمالديهايد (تمت أيضًا دراسة ( ) ولكن هناك حاجز عالٍ يبلغ 2.02 إلكترون فولت (الشكل التكميلي 29).
ديناميكا حرارية وت kinetics التفاعل تبرر العديد من الملاحظات التجريبية. أولاً، يؤدي المسار السائد للفورمات إلى وفرة من الأنواع المؤكسدة كما تم استكشافه بواسطة DRIFTS في الموقع. ثانياً، فإن الامتصاص الضعيف لـ و بالإضافة إلى الحاجز العالي لمزيد من الهدرجة لـ، TS-8)، يحدد الفجوة في تحويل بين النقي و . على النقيض، فإن الإزالة السهلة لهذه الأنواع تقلل من تغطيتها السطحية، أو بمعنى آخر، تزيد من عقوبات الطاقة الحرة للهيدروجين اللاحق إذا افترضنا البدء بحمض الفورميك أوالغاز، وكلاهما تم اكتشافه تجريبيًا على النقيخلال تفاعل الهدرجة (الشكل التوضيحي 30). بينما بالنسبة لـتحويل HCOOH المنفصل و/أوفي المسام الحمضية لـ H-ZSM-5، يكون من الناحية الديناميكية الحرارية مفضلاً جداً نحو المركبات العطرية (الجدول التكميلي 4) وقد تم إثبات أن حواجز الت aromatization منخفضة جداً.. لذلك، فإن التحسين في يتم تحقيق التحويل بعد تضمين H-ZSM-5 في النظام بالكامل. ثالثًا، بسبب الحاجز الأدنى لـالتحلل (عكسي؛ TS-3) أكثر من ذلك منالهدرجة (الأمام، TS-7)، تشكيل CO من خلال مسار غير مباشر (يصبح أكثر جدوى من المسار المباشر للكربوكسيل. قد يضيء هذا الفهم النظري الجهود المستقبلية في تنظيم الامتصاص والتحويل من الشكل السطحي للفورمال لتحقيق انتقائية منخفضة لثاني أكسيد الكربون.
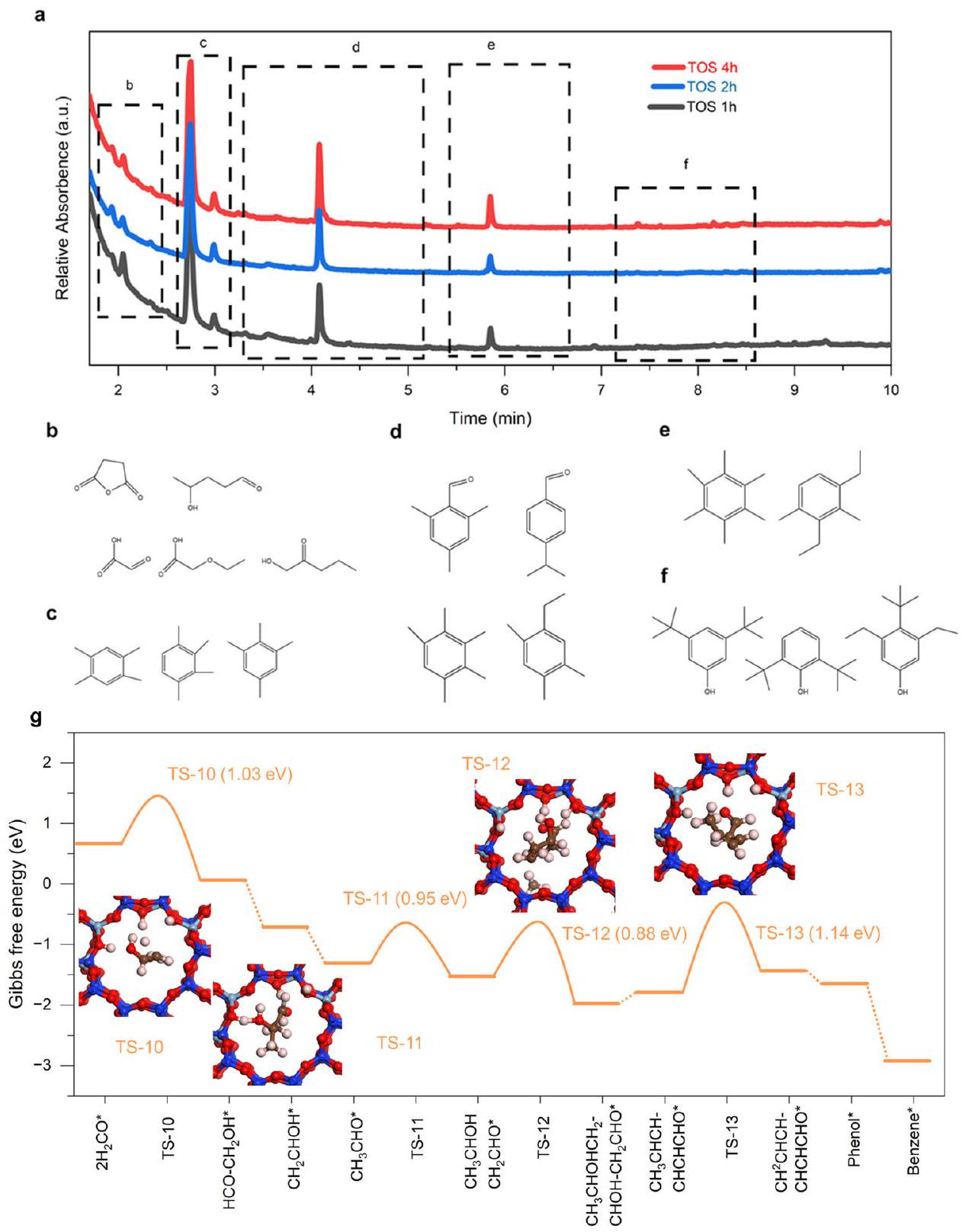
لتأكيدك intermediates محورية في هذه الحفز المتسلسل، استكشفنا المزيد من تفاعل الألدول-العطري داخل H-ZSM-5تم تنفيذ إجراء تجريبي تم تطويره بواسطة الطريقة السابقة للكشف عن الأنواع داخل الزيوليت خلال أوقات مختلفة من التشغيل (TOS). يمكن العثور على بعض مكثفات الألدول، والألدهيدات العطرية، والأروماتيات من نوع الفينولات جنبًا إلى جنب مع متعدد الميثيل بنزين (الشكل 5a-f). يُظهر انخفاض مكثفات الألدول من 1 ساعة إلى 4 ساعات أن تفاعل الألدول يلعب دورًا مهمًا في فترة التحفيز الأولية. بالإضافة إلى ذلك، تظهر الأدلة أن الألدهيدات العطرية، والفينولات تظهر أيضًا كميات كبيرة في TOS لمدة 1 ساعة و2 ساعة في التحفيز، مما يشير إلى إمكانية تحويل مكثفات الألدول إلى الألدهيدات العطرية وبعض الأكسيدات الفينولية، مما يظهر مسار تفاعل الألدول-العطر كما توقعنا. بالإضافة إلى ذلك، تم النظر في مسار التفاعل التفصيلي أيضًا باستخدام حسابات DFT داخل H-ZSM-5. كما هو موضح في الشكل 5g، h، افترضنا أنانتشرت الأنواع بسرعة في H-ZSM-5، مما أثار سلسلة من التحولات إلى مكثفات الألدول. على وجه التحديد، اثنان منتقوم الجزيئات في البداية بامتصاص بالقرب من موقع Al-OH، مما يبدأ الأولالاقتران. تؤدي هذه العملية إلى تشكيلالنوع (الذي تم ملاحظته أيضًا في الشكل 4ب)، والذي يدخل بعد ذلك في تفاعل الألدول، مما يسهل المزيد من الاقتران حتى تتشكل الأكسيدات طويلة السلسلة ذات الست كربونات. بعد ذلك، تخضع هذه لعملية التدوير والتعطير، وهي عملية تتأثر بالاحتجاز والخصائص الحمضية لـ H-ZSM-5، مما يؤدي في النهاية إلى تكوين البنزين (الشكل التكميلي 31). من منظور الديناميكا الحرارية، فإن ملف الطاقة داخل H-ZSM-5 أقل بكثير من ذلك لـ الأنواع علىالسطح. تشير هذه الملاحظة إلى أن الزيوليت يغير بشكل فعال مسار التفاعل منإلى ما هو أكثر رغبةتسلسل العطور. تتماشى هذه الرؤى الحاسوبية بشكل متماسك مع نتائجنا التجريبية، مما يوفر مبررًا قويًا لمسار التحول الملحوظ ويبرز الدور الحاسم لـك intermediates في هذه العملية المعقدة للتفاعل المتسلسل.
بشكل عام، تؤكد النتائج التجريبية والنظرية تصميمنا العقلاني لـالبيروفسكايت كمجال أكسيدي عالي النشاط ومقاوم للكربنة للتحويل الاتجاهي المزدوج المحفز بالمركباتإلى العطور (الشكل 6). يُعزى الاختيار العالي للعطور إلى تشكيل مفصول عن عمد لـالأكسجينات مثل HCOOH وعلىو تشكيل العطرية في الزيوليت الحمضي، على التوالي. وبالتالي، يتم تحقيق السيطرة الصارمة على انتقائية الهيدروكربونات نحو العطرية الثقيلة ذات القيمة المضافة، مما يمنح هذامسار الاستخدام عملية اقتصادية ممتازة. بينما بالنسبة لأكاسيد الحديد العادية أو السبينل مثل أو إن التحول الطوري إلى كربيدات الحديد أمر لا مفر منه، وقد تم اقتراح آلية FTS تسلسلية، مما يؤدي إلى توزيع واسع للمنتجات يتوافق مع توزيع ASF. ستؤدي الانتقائية غير المرضية إلى زيادة كبيرة في تكلفة فصل المنتجات.
في هذه الدراسة، نقدم تحقيقات تجريبية ونظرية تركيبية تبرر تصميمالبيروفسكايت لتسهيل التحويل الاتجاهي المتسلسلمع كفاءة تحويل عالية (اختيار استثنائي للروائح ) ، وثبات التحفيز لمدة 1000 ساعة. إن اقتران البيروفسكايت النشط والمقاوم للكربنة مع الزيوليت يؤدي إلى فصل صارم لـ الهدرجة والاقتران، يقدم نهجًا مختلفًا جذريًا عن الطرق الصناعية التقليدية لتخليق العطور ويعرض مبدأ تصميم جديد لمجال الأكسيد في التحفيز المتزامن. في سياق أوسع، نظرًا للتنوع الكبير والكيميائيات المثمرة للبيروفسكايت والزيوليت، فإن استراتيجيتنا تمنح الاستدامةاستغلال العديد من الفرص من خلال تحسين الاتجاه بشكل أكبر-تحويل إلى العطرية أو هندسة عمليات متتالية جديدة لإنتاج منتجات ذات قيمة مضافة أخرى. نظرًا لأن الكهرباء المتجددة والهيدروجين الأخضر أصبحا بتكلفة منخفضة بشكل متزايد، فإن التحويل الاتجاهي المدعوم بالبيروفسكايت لـجاهز لإعادة تدوير غازات الدفيئة المدفوعة بالطاقة المتجددة وتخفيض الكربون في صناعة الكيمياء.
طرق
تركيبأكسيد
تمت تخليقه عبر طريقة الترسيب المشترك. عادةً، 16.16 جرام منتمت إضافة 45 مل من الماء المقطر (DI) مع التحريك حتى تكوين محلول شفاف. في المحلول أعلاه، 40 مل منثم أضيف المحلول المائي بالتنقيط تحت التحريك في درجة حرارة الغرفة. بعد أن تم تركه لمدة 6 ساعات، تم ترشيح السائل المعلق، وغسله أكثر من 50 مرة بماء منزوع الأيونات، وتجفيفه طوال الليل فيتم حرق العينات الناتجة في فرن مفلطحة عندلمدة ساعتين.
تركيبأكسيد
تم تخليق المحفز بواسطة طريقة الترسيب المشترك. في المجموع، و تم خلطها جيدًا وتسخينها مسبقًا إلى. بعد ذلك، تمت الإضافة بالتنقيط إلى الخليط وتم التحريك المستمر بسرعة 720 دورة في الدقيقة، وسيتم تركه ليتقدم لمدة 5 ساعات عند درجة حموضة حوالي 10. تم الحصول على كعكة الفلتر من عملية الترشيح بالشفط للمحلول المتقدم، وتم غسلها أكثر من 50 مرة لاستبعاد السطح النشط. ثم تم تجفيف الراسب طوال الليل في الفرن عندبعد التبريد إلى درجة حرارة الغرفة، تم إرسال المحفز إلى فرن الموفل بعد الطحن الكافي. تم رفع درجة الحرارة من درجة حرارة الغرفة إلىفي 120 دقيقة.
تركيبأكسيد
تم تصنيع العينات باستخدام تقنية المستحلبات الدقيقة. في البداية، تم تحضير ثلاثة مستحلبات دقيقة متميزة، كل منها يحتوي على مراحل مائية مختلفة: الأول احتوى على كلوريد اللانثانوم (III) هيدراته السبع. )، كان الثاني يحتوي على كلوريد الحديد (III) رباعي الماء ( )، واشتمل الثالث على . تم تثبيت كل ميكروإيمولسيون بتركيز ثابت قدره 0.12 م من ديوكتيل سلفوسكسينات الصوديوم (أيروسول-OT) وتمت إضافته مع الإيزوكتان كمرحلة زيتية. بعد ذلك، تم دمج الميكروإيمولسيون المحتوية على اللانثانوم مع تلك الغنية بالحديد لتشكيل إيمولسيون وسيطة، تم تسميتها بالميكروإيمولسيون I. بعد ساعة من الخلط الميكانيكي، تم تقديم المستحلب الدقيق المحتوي على –
الشكل 5 | مسار تفاعل الألدول العطري اللاحق داخل الزيوليتات منكوسيط رئيسي. أ. الأنواع الكربونية المحددة المحتفظ بها في LaFeO3/H-ZSM-5(14) المستهلكة خلال أوقات تفاعل مختلفة. ب-و. الأنواع التي تم تحديدها بواسطة الكروماتوغرافيا الغازية-مطياف الكتلة.كمفتاح
الوسطاء في مسار التفاعل لتكوين العطريات (الألدول-العطرية) داخل الزيوليت. بعض الهياكل الانتقالية النموذجية خلالإلى العطور فوقمحفز ZSM-5. الكرات باللون الأبيض والبني والأحمر والأزرق الفاتح والأزرق تمثل ذرات الهيدروجين والكربون والأكسجين والألمنيوم والسيليكون، على التوالي. إلى المستحلب الدقيق I. خضت هذه الخلطة بشكل مكثف في ظروف محيطية لمدة ساعتين، مما ساعد على تشكيلالسابقة. تم إضافة الميثانول لاحقًا لتحفيز فصل الطور ولتكون بمثابة عامل غسيل. بعد أكثر من 50 دورة غسيل، تم طرد المحلول لفصلالمقدمة، التي تم تجفيفها طوال الليل فيالكريستالي النهائيتم الحصول على المنتج بعد التكلس عندلمدة ساعتين.
تركيب زينون-حجم نانو-H-ZSM-5
تم تحضير H-ZSM-5 بحجم نانو مع نسب مختلفة من Si/Al باستخدام الطريقة الهيدروحرارية. تم استخدام كميات مناسبة من رباعي إيثيل أورثوسيليكات (TEOS ( 11.2 جرام )) وهيدروكسيد رباعي بروبيل الأمونيوم (TPAOHنترات الألمنيومهيدروكسيد الصوديوم ( )، بروبان-2-أول (IPA ( 0.1 جرام ))، ويوريا تم إذابتها في الماء المقطرتحت التحريك القوي لمدة لا تقل عن 6 ساعات، في درجة حرارة الغرفة. تم نقل المحلول إلى
مفاعل أوتوكلاف من الفولاذ المقاوم للصدأ مبطن بتفلون تم وضعه لاحقًا في فرن مبرمج حراريًا. لعملية التبلور، تم رفع درجة حرارة الفرن من درجة حرارة الغرفة إلىمعدل تسخين قدرهوتم عقده في لمدة 48 ساعة. بمجرد اكتمال التبلور، تم تبريد درجة حرارة مفاعل الأوتوكلاف بسرعة باستخدام حمام ماء بارد. ثم تم جمع البلورات الناتجة من خلال عملية الترشيح (غسلت عدة مرات بماء منزوع الأيونات لإزالة السائل الأم) وتم تجفيفها طوال الليل في الفرن عند لإزالة عامل التشكيل TPAOH، تم تسخين البلورات في الهواء عندلمدة 5 ساعات. باستخدام طريقة تبادل الكاتيون (تم تكرار الخطوة ثلاث مرات على الأقل)، تم تحويل الشكل الصوديومي من ZSM-5 (أي 10 جرام) إلى NH4-ZSM-5 بعد أن تم تفريقه في محلول 1 م من NH4NO3 (أي 100 مل) تحت تحريك قوي لمدة 6 ساعات. أخيرًا،-تم تحويل ZSM-5 إلى H-ZSM-5 من خلال خطوة تسخين بسيطة تمت عندلمدة 5 ساعات في بيئة الهواء. وتم الإشارة إلى المسحوق الناتج باسم H-ZSM-5.
الشكل 6 | توضيح تخطيطي لـمسارات التحويل على الثنائي الوظيفةالمحفز المركب والعامل المساعد، الذي فيهمشتق من التحول في الموقع لأكسيد الحديد التقليدي محفز (على سبيل المثال، و ) تحت الجو التفاعلي. الكرات باللون الوردي، الأسود، الأحمر، الأرجواني، الأصفر، البني، والأخضر تمثل وذرات اللانثانوم، على التوالي.
تقييم أداء التحفيز
تم إجراء تفاعلات الهدرجة في مفاعل سرير ثابت تحت ضغط 30 بار من الغاز المختلط. الغاز المختلط معنسبةتركيب الغاز،، و ما لم يُذكر خلاف ذلك. بشكل عام، فإن المحفز بحجم حبة يتراوح بين 355-850 ميكرومتر (تم تحميل المواد المصفاة المخففة مع رمل الكوارتز المسحوق في مفاعل ثابت السرير بقطر داخلي يبلغ 10 مم. تم إجراء التفاعل عند و ما لم يُذكر خلاف ذلك. تم الاحتفاظ بجميع نواتج التفاعل في الطور الغازي وتم تحليلها عبر الإنترنت بواسطة جهازين كروماتوغرافيين غازيين (Agilent 7890A) مزودين بعمود capillary HP-PLOT/Q متصل بكاشف تأين اللهب (FID) وعمود TDX-1 (المصنوع من DICP) متصل بكاشف الموصلية الحرارية (TCD).تم استخدامه كجسر مرجعي بين FID و TCD. تم استخدام Ar كمعيار داخلي. الـالتحويل وانتقائية أول أكسيد الكربون، الهيدروكربوناتواختيار DME بين المنتجات الكربونية بدون CO (بما في ذلكوتم حساب (DME) باستخدام المعادلات التالية:
مولات منعند المدخل؛مولات منفي المنفذ؛
: مولات من CO عند المخرج؛
أينيمثل المخرج مولات من منتج الهيدروكربون الفردي عند المخرج. كان توازن الكربون بين 95.0 و.
قياس طيف الأشعة تحت الحمراء باستخدام تقنية الانعكاس المنتشر في الموقع (DRIFTS)
تم استكشاف تغيير أنواع التفاعل باستخدام طريقة DRIFTS في الموقع. تم تقليل شريحة المحفز في الهيدروجين.في الحجم،متوازن) عند و لمدة ساعة واحدة، ثم تم تمرير النيتروجين بنفس معدل التدفق ودرجة الحرارة لمدة 20 دقيقة. تم تسجيل طيف DRIFTS باستخدام مطياف نيكوليت 6700 من خلال جمع
64 مسح فيالقرار. بعد ذلك، تم تفاعل المحفز مع (متوازن) عندوضغط الهواء. تم تسجيل طيف DRIFTS في الموقع كل 5 دقائق حتى استمرت التفاعل لمدة 60 دقيقة.
توصيف المحفزات
تم إجراء أشكال العينات بواسطة المجهر الإلكتروني الماسح بتأثير الحقل (FESEM، SU-8010) والمجهر الإلكتروني الناقل (TEM، HT7700 120 kV).
تم إجراء تجارب مجهر الإلكترون الناقل الماسح (STEM) على مقياس ذري باستخدام مجهر إلكتروني ناقل مصحح بالسيزيوم (FEI Titan Cubed Themis G2 300) يعمل عند 300 كيلو فولت بزاوية تقارب نصفية تبلغ 23.6 مللي راديان وكان تيار الشعاع أقل من 30 بيكو أمبير. كان المجهر مزودًا بمصحح انكسار كروي DCOR+ لنبضة الإلكترون، والذي تم محاذاته قبل التجارب باستخدام عينة ذهبية قياسية. كانت مدة مسح البروبي…لكل صورة خريطة EDS. كان وقت الإقامةلكل بكسل مع حجم خريطةبكسلات؛ استغرق عملية رسم خرائط EDS الكاملة حوالي 0.5 ساعة للوصول إلى نسبة إشارة إلى ضوضاء عالية بشكل مناسب. تم قياس معاملات التشويه التالية كالتالي: A1 =; A5 و R5 .
تم تشغيل تقنية التباين الطوري المتكامل بدقة ذرية (iDPCSTEM) أيضًا عند 300 كيلوفولت بزاوية تقارب نصفية قدرها 15 مللي راديان. تم ضبط تيار الشعاع بين 1 بيكو أمبير و 0.5 بيكو أمبير. تم إجراء تجارب iDPCSTEM باستخدام مجهر إلكتروني ناقل مسح مصحح Cs (FEI Titan Cubed Themis G2 300) يعمل عند 300 كيلوفولت. كان المجهر مزودًا بمصحح انحراف كروي DCOR+ لشعاع الإلكترون الذي تم محاذاته قبل التجارب باستخدام عينة ذهب قياسية. تم قياس معاملات الانحراف التالية كالتالي: A1 = 1.41 نانومتر؛ A2; ، و . كانت زاوية التقارب نصفية 15 مللي راديان، وكان تيار الشعاع أقل من 0.5 بيكو أمبير (كانت القياسات محدودة بدقة كوب فاراداي)، وكانت زاوية الجمع بين 4-22 مللي راديان، ووقت الإقامة لمسح المجس كان.
تم الحصول على أنماط حيود الأشعة السينية لمختلف أكاسيد المعادن والزيوليتات باستخدام جهاز حيود الأشعة السينية Bruker D8 Advance باستخدام إشعاع CuKa. ) عند 40 مللي أمبير و 40 كيلو فولت. تم اختبار طيف الكترون الأشعة السينية (XPS) للعينات على جهاز Escalab 250 Xi XPS بمصدر أشعة سينية Al Ka (Thermo Fisher Scientific). تم إجراء مساحة السطح وفقًا لطريقة برونور-إيميت-تيلر للعينات باستخدام جهاز تحليل المساحة السطحية المحددة والمسامية المتوسطة (TriStar II).
تم تحليل قياسات هياكل الامتصاص الدقيق للأشعة السينية (XAFS) في محطة BL14W1 في منشأة الإشعاع السنكروتروني في شنغهاي (SSRF،في الحد الأقصى، بلورات مزدوجة من Si (111). تم معايرة الطاقة وفقًا لحافة الامتصاص لرقائق الحديد النقي. قبل تجارب XAFS، تم نقل العينات المحضرة إلى صندوق قفازات لتشكيلها دون التعرض للهواء. تم معالجة جميع بيانات XANES وEXAFS باستخدام برنامج أثينا.
تم تحديد تركيبة الألدول-أروماتيك المحتفظ بها داخل المحفز المستهلك باستخدام تقنية الذوبان بالفلوريد الهيدروجيني بواسطة جهاز الكروماتوغرافيا الغازية-مطياف الكتلة. تم استخدام كميات مناسبة من حمض الفلوريد الهيدروجيني.تمت إضافته ببطء وحذر إلى 1 جرام من المحفز المركب المختلط جسديًا (وبعد ذلك بضع قطرات من كلوريد الميثيلين ( ) أُضيفت إلى المحلول أعلاه وتم الطرد المركزي حتى أصبح طبقة من زيت الهيدروكربون المنفصل مرئية. ثم تم استخراج هذا الزيت بعناية من المحلول الأم وتحليله باستخدام جهاز الكروماتوغرافيا الغازية – مطياف الكتلة. تم تكرار هذه الخطوات على عينات تم الحصول عليها في أوقات مختلفة أثناء التشغيل.
طرق نظرية
تم إجراء حسابات نظرية الكثافة الوظيفية (DFT) باستخدام الوظيفة المعدلة من بيرديو-بورك-إرنزرهوف (RPBE) ضمن تقريب التدرج العام (GGA) المطبق في حزمة المحاكاة الأولية في فيينا.تم تطبيق طريقة الموجة المعززة بواسطة البروجيكتور لوصف تفاعلات الإلكترون-الأيون.، وتم استخدام طريقة D3 Grimme لتصحيح تفاعل فان der Waals. استخدمنا طاقة قطع الموجة المسطحة تبلغ 400 إلكترون فولت وسمك Gaussian بمقدار 0.1 إلكترون فولت. تم تطبيق شروط الحدود الدورية، وأكثر منتم استخدام فراغ من الفضاء لتجنب تفاعل الصور المجاورة.
تم تقييم طاقات الامتزاز باستخدام ثمانية طبقاتالخلايا الفائقة مع تقييد الطبقات الأربعة السفلية، وتم استخدام شبكات نقاط كيه من نوع مونكهورست-باك مع عتبة تقارب قدرهاللتكرار في مجال ذاتي متسق (SCF). تم تحسين جميع الهياكل حتى كانت مكونات القوة أقل من تم حساب الترددات الاهتزازية للجزيئات الحرة والمواد الممتصة باستخدام وحدات الصوت في كود VASP 5.4.4. تم تطبيق تصحيح حراري قياسي لتحديد تصحيحات الطاقة الحرة، بما في ذلك تصحيح تأثير الطاقة عند النقطة الصفرية، والضغط، والطاقة الداخلية، والإنتروبيا. تم الإشارة إلى جميع طاقات تشكيل المواد الممتصة/الحالات السطحية، بالإضافة إلى المنتجات في الطور الغازي إلى، و . بسبب أخطاء DFT في تقدير الجزيئات في الطور الغازي التي تحتوي على أو العمود الفقري، مثل و على مستوى PBE/RPBE، تم تطبيق تصحيح الرابطة المزدوجة بمقدار +0.33 eV على الطاقات الخام لـ و طريقة الحصول على مثل هذا التصحيح تعتمد على الدراسات السابقةقمنا بحساب حالة الانتقال باستخدام طريقة الحزام المرن المدفوع بصورة متصاعدة (CI-NEB). تم أيضًا حساب الترددات الاهتزازية لحالة الانتقال ويجب ملاحظة أنه يوجد تردد تخيلي واحد فقط.
استخدمنا طاقات الامتزاز الحرة،بدلاً من الطاقات الإلكترونية لبناء مخطط الطاقة الحرة لجيبس. هنا افترضنا أن المنتجات الغازية في المسار تم حسابها عند ضغوط جزئية كما هو موضح في الشكل 2a. تم حساب الإنثالبي، والإنتروبيا، والطاقة الحرة لجيبس لكل نوع من الأنواع من خلال تحليل تردد الاهتزاز استنادًا إلى تقريب الوضع الطبيعي التوافقي باستخدام طريقة الفرق المحدود في VASP 5.4.4. الطاقة الحرة لجيبس لنوع معين هي
أين
أين
أين
أين هو عزم القصور الذاتي، هو عدد التماثل الدوراني، و هو كتلة الجزيء. بالنسبة للجزيئات الممتصة وحالات الانتقال على السطح، تم تحويل المساهمات الدورانية والترجمية إلى أوضاع اهتزازية. كما قمنا بتقريب أن مصطلح PV للأنواع السطحية غير مهم لأنه صغير جداً بالنسبة للمصطلحات الطاقية، وبالتالي، اعتبرنا في هذه الحالة.
استقرار و فيما يتعلق بـ تم تحديدها بالمعادلات التالية:
تم اعتماد تغيير الطاقة الحرة لجيبس للتفاعلات المذكورة أعلاه لتحديد احتمال و لتشكيل.
توفر البيانات
جميع البيانات متاحة في المخطوطة أو المعلومات التكميلية. البيانات الإضافية متاحة من المؤلفين المراسلين عند الطلب.
References
Hepburn, C. et al. The technological and economic prospects for utilization and removal. Nature 575, 87-97 (2019).
Aresta, M., Dibenedetto, A. & Angelini, A. Catalysis for the valorization of exhaust carbon: from to chemicals, materials, and fuels. Technological use of . Chem. Rev. 114, 1709-1742 (2014).
Appel, A. M. et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of fixation. Chem. Rev. 113, 6621-6658 (2013).
Wang, W., Wang, S., Ma, X. & Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 40, 3703-3727 (2011).
Li, Z. et al. Highly selective conversion of carbon dioxide to aromatics over tandem catalysts. Joule 3, 570-583 (2019).
Li, Z. et al. Ambient-pressure hydrogenation of CO 2 into long-chain olefins. Nat. Commun. 13, 2396 (2022).
Wang, L. et al. Cobalt-nickel catalysts for selective hydrogenation of carbon dioxide into ethanol. ACS Catal. 9, 11335-11340 (2019).
Hu, J. et al. Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol. Nat. Catal. 4, 242-250 (2021).
Tian, G., Zhang, C. & Wei, F. COx conversion to aromatics: a minireview of nanoscale performance. Nanoscale Horiz. 7, 1478-1487 (2022).
Ji, Y. et al. Oxygenate-based routes regulate syngas conversion over oxide-zeolite bifunctional catalysts. Nat. Catal. 5, 594-604 (2022).
Wang, Y. et al. Direct conversion of CO2 to aromatics with high yield via a modified Fischer-Tropsch synthesis pathway. Appl. Catal. B Environ. 269, 118792 (2020).
Arslan, M. T. et al. Highly selective conversion of CO 2 or CO into precursors for kerosene-based aviation fuel via an aldol-aromatic mechanism. ACS Catal. 12, 2023-2033 (2022).
Wang, Y. et al. Rationally designing bifunctional catalysts as an efficient strategy to boost CO2 hydrogenation producing valueadded aromatics. ACS Catal. 9, 895-901 (2018).
Wei, J. et al. Precisely regulating Brønsted acid sites to promote the synthesis of light aromatics via CO2 hydrogenation. Appl. Catal. B Environ. 283, 119648 (2021).
Wang, T. et al. ZnZrOx integrated with chain-like nanocrystal HZSM5 as efficient catalysts for aromatics synthesis from CO2 hydrogenation. Appl. Catal. B Environ. 286, 119929 (2021).
Ramirez, A. et al. Effect of zeolite topology and reactor configuration on the direct conversion of CO2 to light olefins and aromatics. ACS Catal. 9, 6320-6334 (2019).
Tian, H. et al. Tandem catalysts composed of different morphology HZSM-5 and metal oxides for CO2 hydrogenation to aromatics. Fuel 314, 123119 (2022).
Zhang, J. et al. Hydrogenation of CO2 into aromatics over a ZnCrO x-zeolite composite catalyst. Chem. Commun. 55, 973-976 (2019).
Li, Y. et al. Direct conversion of carbon dioxide into liquid fuels and chemicals by coupling green hydrogen at high temperature. Appl. Catal. B Environ. 324, 122299 (2023).
Zhu, J. et al. Deconvolution of the particle size effect on CO2 hydrogenation over iron-based catalysts. ACS Catal. 10, 7424-7433 (2020).
Zhu, J. et al. Dynamic structural evolution of iron catalysts involving competitive oxidation and carburization during CO2 hydrogenation. Sci. Adv. 8, eabm3629 (2022).
Zhao, B. et al. Direct transformation of syngas to aromatics over Na-Zn-Fe5C2 and hierarchical HZSM-5 tandem catalysts. Chem 3, 323-333 (2017).
. et al. Selective conversion of syngas to aromatics over Fe3O4@MnO2 and hollow HZSM-5 bifunctional catalysts. ACS Catal. 9, 5147-5156 (2019).
Tian, G. et al. Accelerating syngas-to-aromatic conversion via spontaneously monodispersed Fe in ZnCr 2 O 4 spinel. Nat. Commun. 13, 5567 (2022).
Xu, Y. et al. A hydrophobic FeMn@ Si catalyst increases olefins from syngas by suppressing C1 by-products. Science 371, 610-613 (2021).
Song, G., Li, M., Yan, P., Nawaz, M. A. & Liu, D. High conversion to aromatics via CO2-FT over a CO-reduced Cu-Fe2O3 catalyst integrated with HZSM-5. ACS Catal. 10, 11268-11279 (2020).
Wei, J. et al. Directly converting CO2 into a gasoline fuel. Nat. Commun. 8, 15174 (2017).
Cui, X. et al. Selective production of aromatics directly from carbon dioxide hydrogenation. ACS Catal. 9, 3866-3876 (2019).
Wang, C. et al. Fischer-Tropsch synthesis to olefins boosted by MFI zeolite nanosheets. Nat. Nanotechnol. 17, 714-720 (2022).
Cheng, K. et al. Impact of the spatial organization of bifunctional metal-zeolite catalysts on the hydroisomerization of light alkanes. Angew. Chem. 132, 3620-3628 (2020).
Wang, Y. et al. Visualizing element migration over bifunctional metal-zeolite catalysts and its impact on catalysis. Angew. Chem. 133, 17876-17884 (2021).
Rong, Y. et al. Challenges for commercializing perovskite solar cells. Science 361, eaat8235 (2018).
Peña, M. A. & Fierro, J. Chemical structures and performance of perovskite oxides. Chem. Rev. 101, 1981-2018 (2001).
Liao, X. et al. Highly efficient reduction of O2-containing CO2 via chemical looping based on perovskite nanocomposites. Nano Energy 78, 105320 (2020).
Zhu, X., Li, K., Neal, L. & Li, F. Perovskites as geo-inspired oxygen storage materials for chemical looping and three-way catalysis: a perspective. ACS Catal. 8, 8213-8236 (2018).
Wang, J. et al. Elucidating surface and bulk phase transformation in Fischer-Tropsch synthesis catalysts and their influences on catalytic performance. ACS Catal. 9, 7976-7983 (2019).
Wang, S. et al. Highly selective hydrogenation of CO 2 to propane over GaZrO x/H-SSZ-13 composite. Nat. Catal. 5, 1-13 (2022).
Gao, P. et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 9, 1019-1024 (2017).
Cheng, Y. et al. High-yield production of aromatics over CuFeO2/ hierarchical HZSM-5 via CO2 Fischer-Tropsch synthesis. Green Chem. 25, 3570-3584 (2023).
Meng, C. et al. Oxygen-deficient metal oxides supported nanointermetallic InNi 3 CO .5 toward efficient CO 2 hydrogenation to methanol. Sci. Adv. 7, eabi6012 (2021).
Yang, H. et al. Selective synthesis of olefins via CO2 hydrogenation over transition-metal-doped iron-based catalysts. Appl. Catal. B Environ. 321, 122050 (2023).
Guo, L. et al. Heteroatom doped iron-based catalysts prepared by urea self-combustion method for efficient CO2 hydrogenation. Fuel 276, 118102 (2020).
Zhang, W., Fu, Q., Luo, Q., Sheng, L. & Yang, J. Understanding single-atom catalysis in view of theory. JACS Au 1, 2130-2145 (2021).
Shang, X. et al. Xylene synthesis through tandem CO2 hydrogenation and toluene methylation over a composite ZnZrO zeolite catalyst. Angew. Chem. 62, e202309377 (2023).
Li, Y. et al. Interfacial Fe5C2-Cu catalysts toward low-pressure syngas conversion to long-chain alcohols. Nat. Commun. 11, 61 (2020).
Gao, W. et al. Photo-driven syngas conversion to lower olefins over oxygen-decorated Fe5C2 catalyst. Chem 4, 2917-2928 (2018).
Li, S., Meitzner, G. D. & Iglesia, E. Structure and site evolution of iron oxide catalyst precursors during the Fischer-Tropsch synthesis. J. Phys. Chem. B 105, 5743-5750 (2001).
Goldstein, J. & Moren, A. Diffusion modeling of the carburization process. Metall. Trans. A 9, 1515-1525 (1978).
Simonetti, S., Moro, L., Brizuela, G. & Juan, A. A computational study of the carburization phenomena in a Fe-Ni alloy. J. Phys. D Appl. Phys. 41, 125006 (2008).
Ribeiro, M. C. et al. Fischer-Tropsch synthesis: influence of Mn on the carburization rates and activities of Fe-based catalysts by TPR-
EXAFS/XANES and catalyst testing. J. Phys. Chem. C 115, 4783-4792 (2011).
51. Zabilskiy, M. et al. The unique interplay between copper and zinc during catalytic carbon dioxide hydrogenation to methanol. Nat. Commun. 11, 2409 (2020).
52. Studt, F. et al. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nat. Chem. 6, 320-324 (2014).
53. Zhou, H. et al. Engineering the Cu/Mo2C T x (MXene) interface to drive CO2 hydrogenation to methanol. Nat. Catal. 4, 860-871 (2021).
54. Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
55. Chen, Y.-H. et al. First-principle study of H 2 adsorption on LaFeO 3 (110) surface. J. Nanomater. 2014, 16-16 (2014).
56. Wang, H. et al. Bifunctional catalysts with versatile zeolites enable unprecedented para-xylene productivity for syngas conversion under mild conditions. Chem. Catal. 2, 779-796 (2022).
57. Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
58. Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
59. Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
60. Christensen, R., Hansen, H. A. & Vegge, T. Identifying systematic DFT errors in catalytic reactions. Catal. Sci. Technol. 5, 4946-4949 (2015).
61. Granda-Marulanda, L. P. et al. A semiempirical method to detect and correct DFT-based gas-phase errors and its application in electrocatalysis. ACS Catal. 10, 6900-6907 (2020).
شكر وتقدير
تم دعم هذا العمل من قبل المؤسسة الوطنية للعلوم الطبيعية في الصين (رقم المنحة 22278238، C.Z. رقم المنحة 22109020، H.J.P. الأرقام 22275110، 22322203، X.C.)، وصندوق البحث المشترك بين تسينغhua وتويوتا (X.C. و F.W.)، وبرنامج البحث والتطوير الرئيسي في منغوليا الداخلية وأوردوس (20211140095، C.Z.)، وصناديق الابتكار من CNPC (2020990028، C.Z.)، والبرنامج الوطني الرئيسي للبحث والتطوير في الصين (رقم 22238004، F.W. و X.C.).
مساهمات المؤلفين
صمم الدراسات جي. تي.، زي. إل.، إتش. جي. بي.، سي. زي.، إكس. سي. و إف. دبليو. قام جي. تي. وإكس. إف. بتخليق المحفزات. أجرى جي. تي. وإكس. إف. اختبارات التحفيز. جي. تي.، زي. إل. قام كل من X.F. وK.S. وH.M. وN.W. وM.Z. وX.L. وL.L. وZ.L. وH.X. وX.C. وB.Y. بإجراء التوصيفات. ساهم G.T. وX.L. في حسابات DFT. كتب G.T. وH.J.P. الورقة. وافق جميع المؤلفين على النسخة النهائية من المخطوطة.
المصالح المتنافسة
يعلن المؤلفون عدم وجود مصالح متنافسة.
معلومات إضافية
معلومات إضافية النسخة الإلكترونية تحتوي على المواد التكميلية متاحة على https://doi.org/10.1038/s41467-024-47270-z. يجب توجيه المراسلات والطلبات للحصول على المواد إلى تشينكسي زانغ، شياو تشين، هونغ-جيه بينغ أو في وي.
معلومات مراجعة الأقران تشكر مجلة Nature Communications المراجعين المجهولين على مساهمتهم في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
معلومات إعادة الطباعة والتصاريح متاحة على http://www.nature.com/reprints ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
مختبر بكين الرئيسي لهندسة وتكنولوجيا التفاعلات الكيميائية الخضراء، قسم الهندسة الكيميائية، جامعة تسينغhua، 100084 بكين، الصين.مختبر أوردوس، أوردوس، منغوليا الداخلية 017010، الصين.معهد الحياد الكربوني، جامعة تسينغhua، 100084 بكين، الصين.معهد العلوم الأساسية والحدودية، جامعة علوم وتكنولوجيا الإلكترونيات في الصين، تشنغدو 611731 سيتشوان، الصين.المختبر الرئيسي لتكنولوجيا خلايا الوقود في مقاطعة قوانغدونغ، كلية الكيمياء والهندسة الكيميائية، جامعة جنوب الصين للتكنولوجيا، قوانغتشو 510640، الصين.كلية الكيمياء، جامعة تاييوان للتكنولوجيا، تاييوان 030024، الصين.كلية البيئة والحياة، جامعة بكين للتكنولوجيا، 100124 بكين، الصين.ساهم هؤلاء المؤلفون بالتساوي: قوه تيان، تشنغوين لي.البريد الإلكتروني:cxzhang@tsinghua.edu.cn;chenx123@tsinghua.edu.cn;hjpeng@uestc.edu.cn;wf-dce@tsinghua.edu.cn
The directional transformation of carbon dioxide ( ) with renewable hydrogen into specific carbon-heavy products ( ) of high value presents a sustainable route for net-zero chemical manufacture. However, it is still challenging to simultaneously achieve high activity and selectivity due to the unbalanced hydrogenation and coupling rates on complementary active sites in a bifunctional catalyst, thus causing unexpected secondary reaction. Here we report perovskite-mediated directional tandem conversion of towards heavy aromatics with high conversion ( ), exceptional aromatics selectivity among hydrocarbons (> 85%), and no obvious deactivation for 1000 hours. This is enabled by disentangling the hydrogenation domain from the C-C coupling domain in the tandem system for Iron-based catalyst. Unlike other active Fe oxides showing wide hydrocarbon product distribution due to carbide formation, by design is endowed with superior resistance to carburization, therefore inhibiting uncontrolled coupling on oxide and isolating aromatics formation in the zeolite. In-situ spectroscopic evidence and theoretical calculations reveal an oxygenate-rich surface chemistry of , that easily escape from the oxide surface for further precise coupling inside zeolites, thus steering -Aromatics reaction pathway to enable a high yield of aromatics.
Carbon dioxide ( ) is the most well-known greenhouse gas leading to global warming and a range of climate and environmental issues . To mitigate such an anthropogenic climate change, there exists an urgent need to develop solutions to emission. Utilizing waste as a feedstock and transforming it to value-added chemicals with renewable energy are therefore very promising, with both aspects of recycling and alleviating society’s dependence on fossil
resources . Among various utilization strategies, thermocatalytic conversion that leverages the reducing power of renewable hydrogen ( ) possesses a unique capability of producing complex molecules such as long-chain paraffins and heavy aromatics . From a technoeconomic perspective, aromatics are ideal target products as additives in synthetic aviation fuels and platform molecules in many fine chemical applications . Thus, renewable-powered directional
conversion of to aromatics presents a sustainable and eco-friendly alternative to conventional fossil-resource-based chemical processes and thus could considerably contribute to decarbonizing the air transportation and chemical manufacture sectors.
Many efforts have been made to develop highly effective metal oxide/zeolite composite catalysts with methanol produced on the oxides, such as and and so on, from hydrogenation, and subsequently transform them into aromatics over the zeolite component such as . The high aromatics selectivity stems from the spatially separated active domains and process in tandem; while and are activated on the oxide as the hydrogenation domain, generating mainly hydrogenated species , the zeolite domain traps these species in its confined acidic pores and catalyzes the subsequent complicated transformations toward aromatics . The selectivity can be easily regulated by tuning the structure and acidicity of zeolites. Nevertheless, the typical conversion in such catalytic processes is unsatisfactory (around ), which is mainly limited by the sluggish hydrogenation reactions on commonly used spinel-based oxides in the tandem processes . Moreover, the migration of species from metal oxide to zeolite often results in a shortened lifespan for the catalyst. These challenges underscore a critical need for innovative approaches that not only enhance conversion efficiency but also address the stability and longevity of the catalytic system.
On the contrary, Fe-based catalysts have been widely reported with superior activity for hydrogenation to aromatics ; but in general, also suffer from undesirable selectivity when combined with H-ZSM-5. This process was also called enhanced Fischer-Tropsch route. To be more specific, light olefins ( ) were produced over iron-based oxides domain with a selectivity at the range of . Such intermediates diffuse into zeolite to open C-C coupling and cyclization reaction to produce aromatics . However, this composite route encompasses a complex reaction network: hydrogenation and uncontrolled coupling occur on the iron oxide, while further coupling takes place inside the zeolite. This complexity often results in mismatched coupling rates between oxide and zeolite and unintended isomerization of olefins within the zeolite channels. As a result, Fe oxides have seldom achieved satisfactory selectivity for aromatics among hydrocarbons ( in this composite catalyzed tandem conversion of . While extremely precise control strategies like the introduction of an appropriate amount of alkali metal, as illustrated by Sun et al. , have shown to improve selectivity, they introduce a new challenge: alkali metals tend to migrate to the zeolite, diminishing catalytic performance over time . To circumvent these issues, disentangling the hydrogenation domain from the coupling domain in the tandem system may be another potential strategy for Iron-based catalyst. In other words, by maintaining robust hydrogenation activity while tempering the coupling propensity on the iron oxide side, may offer great potential for highly efficient directional tandem conversion of to aromatics .
Building on the acknowledgments above, we present a novel composite iron-oxide/zeolite system featuring perovskite, distinctively characterized by a well-crafted octahedral framework and Fe-3d band structures, offering superior stability and exceptional resistance to carburization. This stands in stark contrast to traditional iron-based catalysts, which generally exhibit high selectivity. Our system uniquely suppresses Fe carbide formation, ensuring that hydrogenation on yields primarily crucial products ( ), while coupling predominantly takes place within the H-ZSM-5 zeolite, utilizing desorbed oxygenates as precursors. This synergy between perovskite and H-ZSM-5 leads to unparalleled control over hydrocarbon selectivity, with total aromatics exceeding and CO selectivity -a milestone yet to be achieved by other Fe-based catalysts. Furthermore, it propels the conversion rate to over , significantly surpassing the conventional observed with non-Fe oxides. Notably, the H-ZSM-5 catalyst also demonstrates extraordinary durability, maintaining performance for over 1000 h with the decay rate of aromatics yield markedly lower than that of other reported composite systems. The successful implementation of in tandem conversion not only underscores the vast potential of active-metal-based perovskites in industrial recycling but also heralds a new era of perovskite-mediated tandem catalysis. This breakthrough provides a sustainable and scalable pathway for extensive utilization and chemical synthesis, paving the way for a decarbonized chemical industry.
Results and discussion
The design and structure of perovskite
Perovskite oxides in a general formula of have a framework of corner-sharing [ ] octahedra with large A cations filled in the interspace . In such a structure, Fe prefers to occupy the B site and thus enables the construction of a corner-sharing [ ] framework in Fe-based perovskites, which is quite different from the face- or edgesharing frameworks in general Fe oxides (e.g., ) and -type spinels (Fig. 1a-d). Such a topological difference results in distinct distances, which are expected to affect the migration of Fe if the oxide was to be carburized (Fig. 1e). Among various Fe oxides, has the most compact [ ] connection and thus the shortest distance of -type spinels or inverse-spinel mainly contain edge-sharing [ ] octahedra, displaying intermediate distances in the range of ; Fe-based perovskites normally possess much longer distances of . During the phase transition from Fe oxides to Fe carbides, the distance has to decrease to around . Therefore, the required migration distance of Fe during the phase transition is notably elongated for Fe-based perovskites. Meanwhile, the A-site cation serves as pillars pinning in the perovskite [ ] framework. We anticipate that both structural aspects of perovskite by design could lead to suppressed Fe migration during possible carburization.
As a proof of the concept, is selected because La is a rare earth metal with the largest ion radius , which gives rise to a remarkably long distance , and La has unique empty 4 f orbitals that could further regulate the stability of framework through a crystal field effect. To elucidate such effect, density functional theory (DFT) calculations were performed to analyze the PDOSs of , and (Fig. 1f). It is unveiled that the incorporation of La into the corner-sharing [ ] matrix substantially reduces the number of Fe-3d states near the Fermi level when compared to . An opposite trend in Fe -3d states is shown for spinel . While the formation of firstly involves the interaction between Fe states near the Fermi level and carbon states , the reduction in Fe-3d states near the Fermi level can lead to significantly more energies required to form and thus suppressed carburization of . Calculated formation energies of further confirm the higher stability of than and under typical reductive atmospheres consisting of or (Fig. 1g). Under both atmospheres, formation energies of (per mole) from and are negative, showing a strong tendency to form Fe carbides. In a sharp contrast, formation energies of from are more positive by 2.06 and 2.64 eV than and , respectively. The theoretical results provide thermodynamic insights into the superior carburization resistance of , which originates from the reduced activity of Fe through electronic modulation of La.
To realize the above design, phase-pure particulate catalyst with a size of was synthesized by a microemulsion method and its ordered perovskite structure (PDF-#37-1493) was resolved by high-angle annular dark field and integrated differential phase contrast scanning transmission electron microscopy (HAADF-
Fig. 1 | The design of perovskite as a carburization-resistant Fe based oxide. Atomic structural model of (a) , (b) , (c) , and (d) , showing different connection modes of octahedra. Spheres in red, brown, silver, and green represent , and La atoms, respectively. e
distances of carbides, and various Fe oxides. Projected density of states (PDOSs) of , and . g Calculated formation energies of per mole through the reduction of and by typical reactive atmospheres.
STEM & iDPC-STEM) and X-ray diffraction (XRD) (Supplementary Fig. 1). The distortion of ideal cubic perovskite structure is also resolved in , which is indicated by tilting through iDPCSTEM observation (Supplementary Fig. 2). The [ ] tilting is in order to accommodate the interspace large-size La cations. Hematitestructured (PDF-#33-0664) and spinel-structured (PDF-#221012) catalysts were obtained as control samples (Supplementary Figs. 3 and 4). All samples are shown as nanoparticles with a size ranging from .
Catalytic performance of conversion on different catalyst
The above three catalysts were firstly assessed under hydrogenation conditions ( ). As depicted in Fig. 2a, both and predominantly yield hydrocarbons in the and ranges (total selectivity ). Such hydrocarbon distribution aligns with ASF model, corresponding to a chain growth probability ( ) exceeding 0.7 (Supplementary Fig. 5). In contrast, yields methane ( ) as the primary hydrocarbon product , with a substantially reduced of 0.09 . Since the spent
and oxides suffer from notable phase transition to Fe carbides, which have been proved as excellent catalysts for coupling in Fischer-Tropsch synthesis (FTS) (Supplementary Figs. 6 and 7), while the spent does not (Supplementary Fig. 8), the notably suppressed coupling on perovskite is ascribed to its high resistance to carburization. Overall, the above results reveal the great potential of as an isolated catalytic domain that only accounts for hydrogenation in tandem catalysis.
Upon integration with H-ZSM-5 (Supplementary Fig. 9), an esteemed aluminosilicate zeolite able to catalyze coupling and hydrocarbon isomerization but unable to catalyze hydrogenation alone, marked shifts in product profiles were observed (Fig. 2a). Both and display increases in conversion ratio ( and ) and aromatics selectivity ( and ), which are, however, less profound than (Fig. 2a). and paraffins are major hydrocarbon by-products in the two composite systems (Supplementary Fig. 10). The competition between the two coupling pathways on in situ derived Fe carbides and H-ZSM-5, respectively, accounts for such broad distribution of multi-carbon products. While for , the total aromatics
Fig. 2 | Catalytic performance of conversion. a The selectivity in hydrocarbons, conversion, CO selectivity of pristine oxide catalysts (left panel: , and ), solely H-ZSM-5 (67) zeolite, and composite tandem catalytic systems (right panel: (67), (67), and ) The mass ratio of oxide to H-ZSM-5 is 2:1. Catalytic evaluation of (67) with different mass ratios. c Catalytic performance over with different Si/Al ratio. d Catalytic performance
comparison between (red star) and reported non-Fe (pink cycle) and Fe (blue cycle) oxide catalysts, regarding aromatics selectivity in hydrocarbons and conversion. The black dashed line is a guide for the eye. The data shown in was obtained under with a space velocity of at . e Catalytic stability evaluation of catalyst with a more rigorous reaction condition with a space velocity of at , and .
selectivity in all hydrocarbons reaches while tri-methylbenzene and tetra-methylbenzene, both of which are desirable high-calorificvalue additives in aviation fuels, comprise almost of total aromatics (Fig. 2a and Supplementary Fig. 10). In addition, the conversion ratio shows a sharp increase from to . This implies that H-ZSM-5 can efficiently utilize certain intermediates along the sluggish hydrogenation pathways on pristine . It suggests that aromatization of such intermediates in the acidic pores of H-ZSM5 might be faster than deep hydrogenation toward on oxide surfaces . The effect of : H-ZSM-5 mass ratio was further investigated (Fig. 2b). Adopting a moderate mass ratio of yields the
optimal conversion and aromatics selectivity. Increasing or decreasing the mass ratio both induces unwanted catalytic performance deterioration, which is ascribed to unbalanced hydrogenation and coupling rates on the complementary oxide and zeolite domains.
It has been reported that the zeolite acidic property has a strong influence with the latter coupling reaction and inhibition of CO -by product . Therefore, we also conducted with different Si/Al ratio (Supplementary Fig. 11). As shown in Fig. 2c, with the increasing acidic properties in zeolite, the CO-by product has been inhibited from to and conversion has been increased
from 50.8 to . It can be deduced that intermediates produced over may have two subsequent reactions. One is diffuse into zeolite for further coupling to produce aromatics and another is decomposed into CO-by product when the H-ZSM-5 acidic sites are deficient.
To further demonstrate such superiority, we compare the catalytic performance of with other reported state-of-theart composite catalysts for conversion to aromatics under similar conditions (Supplementary Table 1). As shown in Fig. 2d, ZSM-5 exhibits not only at least a-fold higher conversion rate than non-Fe oxide catalysts but also the highest aromatics selectivity in hydrocarbon. The two aspects are attributed to highly active Fe-based sites for hydrogenation and the unique carburization-resistant perovskite structure by design, respectively. As a consequence, and H-ZSM-5 jointly enable strict confinement of cascade hydrogenation and aromatics formation at spatially separated domains, leading to superior catalytic performance for directional tandem conversion of to aromatics. was also subjected to a continuous reaction durability test at more rigorous reaction condition. It is revealed that the aromatics selectivity and conversion rate maintain as and , respectively, for over 1000 h on stream (Fig. 2e). When focusing on STY of total aromatics, which serves as an integrated index of the catalytic performance, exhibits an extremely low decay rate of initial STY ( per hour), which corresponds to 3.4-17.6 times slower performance deterioration than reported composite catalysts for -to-aromatics conversion (Supplementary Fig. 12). While compared to conventional Fe- or Zn-based catalysts that easily suffer from carbon deposition and/or sintering and thus rapidly deactivate , the perovskite-structured demonstrates a great potential for industrial applications due to the record high stability, as well as a decent STY of aromatics at high selectivity. Besides, more stability tests including conventional H-ZSM-5 with nanosized H-ZSM-5 were also carried out shown in Supplementary Fig. 13.
Structural stability of under reactive atmosphere
The high aromatic selectivity and superior catalytic stability of perovskite align well with the anticipation that perovskite could be an especially different carburization-resistant catalyst from normal and spinel Fe oxides. To further obtain experimental insights into the catalyst evolution under reactive atmosphere, combinatorial in situ and ex situ spectroscopic and microscopic characterizations were performed. In situ X-ray diffraction (XRD) first reveals that and could maintain their original bulk structures upon initial reduction but gradually transform to and a mixture of ZnO and , respectively, once carbonaceous gas ( ) being introduced into the in situ observation chamber to resemble the reactive atmosphere (Fig. 3a, b and Supplementary Fig. 14). Distinctively, the perovskite structure of is well retained with no new crystal phases generated under either or atmospheres (Fig. 3c and Supplementary Fig. 15). In addition to the bulk structure evolution, the surface evolution was investigated by in situ X-ray photoelectron spectroscopy (XPS). and show similar patterns that a portion of is firstly reduced to lower-valence by and then is, if partially, re-oxidized to in mixed (Supplementary Fig. 16a, b). The emergence of corresponds well with the nominal valence of Fe in its carbides. While for perovskite Fe oxide, either reductive atmosphere only results in more unsaturated but no (Supplementary Fig. 16c). This is accompanied with increased amounts of oxygen vacancy (Supplementary Fig. 17). might serve as the sites for activation of and subsequent hydrogenation steps , of which the role in catalysis we will discussed in a later section.
Ex situ synchrotron X-ray characterizations were further employed to investigate the chemical status and fine local structures of
spent Fe oxide catalysts. Normalized Fe K-edge X-ray absorption nearedge structures unravel the significantly reduced oxidation state of Fe in and after 24-h reaction, in line with the in situ XPS observation (Supplementary Fig. 18). In FT-EXAFS, the second-shell peak that is assigned to coordination shifts to positions at shorter distances for and after 24-h reaction (Fig. 3d) . More detailed structural information beneath the raw FTEXAFS data was further extracted after fitting (Supplementary Table 2)., coordination numbers in spent and both increase to above 7, indicating the formation of Fe carbides with more agglomerated Fe . Unlike and , exhibits marginally changed oxidation state of distance (originally longer than and ), and coordination number after the same treatment. WT-EXAFS provides additional insights into the catalyst evolution (Supplementary Fig. 19 and Fig. 3e). The Fe-Fe scattering in spent has lower intensity than spent normal and spinel , indicating a weaker interaction and thus suppressed Fe migration and aggregation .
Lastly, high-resolution electron microscopy was carried out to reveal the morphological evolution of spent Fe oxide catalysts. For the spent , a large quantity of were detected with carboninterstitial aggregated Fe , showing a short distance of 0.269 nm (Fig. 3f); while is shown to retain the particulate morphology with negligible particle size variation, as well as the fine perovskite structure with an alternate La and Fe arrangement (Fig. 3g). Intensity profiles of the iDPC-STEM images along imaginary line depict the preservation of long distance ( 0.393 nm ) and the existence of (Supplementary Fig. 20). All above in situ and ex situ diffraction, spectroscopic, and microscopic results validate the superior structural stability and high carburization resistance of perovskite in reactive atmosphere, thereby substantiating the rationality of our structural design and theoretical prediction that a long distance and electronic structure modulation through La pillaring can effectively suppress Fe migration.
Mechanistic understandings of conversion on . ( to to aromatics)
In addition to structural stability, the unique ability of to selectively catalyzing conversion to species, as well as the synergy between and H-ZSM-5 in enabling high-efficiency directional -to-aromatic conversion, is disclosed. In situ diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS) was firstly employed to probe key species potentially dominating the reactions on oxide surfaces with varying temperature. On , only oxygenated species such as formate ( at and ; the asterisk indicates the surface-adsorbed state), formaldehyde ( at ), and methoxy ( at ) were observed (Fig. 4a) . Note that the intensity of the above characteristic peaks increases to maximum until temperature rises to above (Supplementary Fig. 21). Besides, no signals related to multicarbon species are observed for solely . When combined with H-ZSM-5, the intensities of reflection bands associated with above oxygenated species decrease by an order of magnitude (Fig. 4b), accompanied by the emergence of a prominent band at , respectively, implying that H-ZSM-5 has an ability to convert oxygenated species to multicarbon products (Supplementary Figs. 22 and 23). Besides, also species can be found at . The Aldol or Prins mechanism has been previously suggested to account for this ability . For both and ZSM-5, the intensities of characteristic bands maintain unchanged after a short induction period, indicating a relatively steady state of surfaces.
Such an oxygenate-dominated surface chemistry of is quite distinct from that on (derived from ), a variety of stretching modes ( ) and stretching/bending modes
Fig. 3 | Structural evolution of different Fe catalysts under reactive atmospheres. In situ XRD patterns of (a) , (b) and (c) under different reductive atmosphere. Test conditions: and (the first 4 h ) or (the subsequent 20 h ). d Fe K-edge Fourier-transform extended X-ray absorption fine structures (FT-EXAFSs). e Fe K-edge wavelet transform (WT)-EXAFSs of spent , and catalysts. High-angle
annular dark-field scanning transmission electron microscopy (STEM), integrated differential phase contrast (iDPC) STEM images, and intensity profiles of the iDPCSTEM images along imaginary line of spent (f) and (g) . The insets show the atomic structure with spheres in red, black, brown, and green representing , and La atoms, respectively.
( ) indicate the presence of various hydrocarbons (Supplementary Fig. 23). These features are typical for active metal carbide catalysts in FTS that involve polymerization. Thus, these observed patterns are attributed to the formation of Fe carbides. Notably, the intensities of bands continuously rise with increasing reaction temperature, indicating the retention and accumulation of carbonaceous species that eventually passivate the surface. This is consistent with the rapid deactivation of normal and its derived catalysts.
In light of the oxygenate-rich surface chemistry of that mainly leads to products, reaction energetics were investigated using DFT calculations. The (220) surface with or without surface was considered for simulations (Supplementary Fig. 24), and formation Gibbs free energies of relevant surface species/states, including transition states (TSs), after geometry optimization were
computed (Supplementary Figs. 25-28). In general, adsorption strengths of surface species on the surface without are notably weak, making the -free surface inert for hydrogenation (Supplementary Table 3). Therefore, we then focus on the surface with to investigate the thermodynamics and kinetics of various reaction pathways.
As depicted in Fig. 4c, the hydrogenation firstly involves hydrogen adsorption on the O site and adsorption on the near the Fe site. While two channels for hydrogenation open to either or carboxyl ( ), reaction pathways bifurcate to two major branches, with one proceeding through and the other through formic acid ( ), until the formation of formyl ( ). Once forming , the two branches merge again, thereafter followed by sequential hydrogenation to form , and at the end (Fig. 4c). Both the carboxyl and formate pathways are primarily
Fig. 4 | Mechanistic investigation of conversion on . Temperaturedependent in situ DRIFTS spectra over (a) and (b) -ZSM-5. c Free energy diagrams of hydrogenation on the surface of the (220) surface with , showing carboxyl (red) and formate (blue) pathways toward formaldehyde and the formaldehyde hydrogenation pathway (orange) to produce . To indicate the effect of molecular desorption, the states of gas-phase molecules are
highlighted as bold horizontal lines. The competition between molecule desorption and further hydrogenation could be elucidated in the cases where gaseous HCOOH and are assumed to be reactants for subsequent hydrogenation. d TS, transition states structure over during to . Spheres in black, red, brown, and green represent , and La atoms, respectively.
limited by the second hydrogenation step, which has to overcome an activation barrier of 1.93 eV (TS-2) and 1.35 eV (TS-5) to yield and , respectively. Therefore, the formate pathway is more kinetically accessible than the carboxyl pathway on
containing (220), which is consistent with the strong signals of HCOO* (exclusively presented in the formate pathway) in the in situ DRIFTS measurements. A minor pathway initiating from formate, namely hydrogenation proceeds via formaldehyde oxide
( ), was also investigated but there presents a high barrier of 2.02 eV (Supplementary Fig. 29).
The reaction thermodynamics and kinetics rationalize several experimental observations. First, the dominant formate pathway results in a plenty of oxygenated species as probed by in situ DRIFTS. Second, the weak adsorption of and , as well as the high barrier for further hydrogenation of , TS-8), dictates the disparity in conversion between pure and . On pure , the easy desorption of these species reduces their surface coverages, or equivalently, increase the free energy penalties of subsequent hydrogenation if we assume to start with HCOOH or gas, both of which were experimentally detected on pure during hydrogenation reaction (Supplementary Fig. 30). While for , the conversion of desorbed HCOOH and/or in the acidic pore of H-ZSM-5 is thermodynamically very favorable toward aromatics (Supplementary Table 4) and the barriers of aromatization have been demonstrated fairly low . Therefore, the enhancement in conversion is realized after including H-ZSM-5 in the whole system. Third, due to the lower barrier of decomposition (backward; TS-3) than that of hydrogenation (forward, TS-7), the formation of CO through an indirect pathway ( ) becomes more viable than the direct carboxyl pathway. Such a theoretical insight may enlighten future efforts in regulating the adsorption and conversion of surface formyl to realize low CO selectivity.
To substantiate as pivotal intermediates in this tandem catalysis, we further explored the aldol-aromatic reaction within H-ZSM-5 ( . One experimental procedure developed by the previous method has been carried out to detect the species inside zeolite during different time on stream (TOS). It can be found that some of aldol condensates, aromatic aldehydes and phenols-type aromatics along with multi-methylbenzene (Fig. 5a-f). The decrease of aldol condensates from 1 h to 4 h demonstrates that aldol reaction plays a significant role in the initial induction period. In addition, evidence shows that aromatic aldehydes, phenols also exhibit considerable amounts in the TOS of 1 h and 2 h in induction, indicating the feasibility for the transformation of aldol condensates into aromatic aldehydes and some phenolic oxygenates, which shows that a reaction pathway of aldol-aromatic as we speculated. Besides, the detailed reaction route was also considered by using DFT calculation inside H -ZSM-5. As delineated in Fig. 5g, h, we assumed that species rapidly diffused into H-ZSM-5, instigating a series of transformations into aldol condensates. Specifically, two molecules initially adsorb near the Al-OH site, initiating the first coupling. This process results in the formation of species (also observed in Fig. 4b), which then enters the aldol reaction, facilitating further coupling until long-chain six-carbon oxygenates are formed. Subsequently, these undergo cyclization and aromatization, a process influenced by the confinement and acidic properties of H-ZSM-5, ultimately leading to the formation of benzene (Supplementary Fig. 31). From a thermodynamic perspective, the energy profile within H-ZSM-5 is significantly lower than that of the species on the surface. This observation indicates that the zeolite effectively alters the reaction pathway from to a more desirable Aromatics sequence. Such computational insights coherently align with our experimental results, offering a robust rationale for the observed transformation pathway and underscoring the crucial role of as intermediates in this sophisticated tandem catalysis process.
Overall, the experimental and theoretical results validate our rational design of perovskite as a highly active and carburization-resistant oxide domain for composite-catalyzed directional tandem conversion of to aromatics (Fig. 6). The high aromatics selectivity is attributed to deliberately decoupled formation of oxygenates such as HCOOH and on and
formation of aromatics in the acidic zeolite, respectively. Consequently, strict control of hydrocarbon selectivity toward value-added heavy aromatics is realized, endowing this utilization route excellent process economy. While for normal or spinel Fe oxides such as or , phase transformation to Fe carbides is inevitable and a sequential FTS mechanism has been proposed, giving rise to broad product distribution complying the ASF distribution. The unsatisfactory selectivity will substantially increase the cost of product separation.
In this study, we present combinatorial experimental and theoretical investigations that rationalize the design of perovskite to mediate directional tandem conversion of with high conversion efficiency ( ), exceptional aromatics selectivity ( ), and catalytic stability for 1000 h . The coupling of active and carburizationresistant perovskite with zeolite renders strictly decoupling of hydrogenation and coupling, offering a radically different approach from conventional industrial routes for aromatics synthesis and showcasing a new design principle of the oxide domain in tandem catalysis. In a broader context, given the large variety and fruitful chemistries of perovskites and zeolites, our strategy endows sustainable utilization many opportunities by further improving the directional -to-aromatics conversion or engineering novel tandem processes to yield other value-added products. Since renewable electricity and green hydrogen are both at increasingly low cost, perovskite-mediated directional tandem conversion of is ready for renewable-driven greenhouse gas upcycling and chemical industry decarbonizing.
Methods
Synthesis of oxide
was synthesized via co-precipitation method. Typically, 16.16 g of was added to 45 ml of deionized water (DI) under stirring until the formation of a clear solution. In the above solution, 40 ml of aqueous solution was then added dropwise under stirring at room temperature. After being aged for 6 h , the suspension liquid was filtrated, washed more than 50 times with DI water, and dried overnight at . The resulting samples were calcinated in muffle furnace at for 2 h .
Synthesis of oxide
catalyst was synthesized by co-precipitation method. In total, and were thoroughly mixed and preheated to . Thereafter, was added dropwise to the mixture and continuous stirred at the speed of 720 rpm , and it would be aging for 5 h at the pH around 10 . The filter cake was obtained from suction filtration operation of aged solution, and washed more than 50 times to exclude the surfactant. Then the precipitate was dried out overnight in the oven at . After cooling to the room temperature, the catalyst was sent to muffle furnace after sufficient grinding. The temperature was raised from room temperature to in 120 min .
Synthesis of oxide
samples were synthesized using the microemulsion technique. Initially, three distinct microemulsions were formulated, each with varying aqueous phases: the first contained Lanthanum (III) chloride heptahydrate ( ), the second had iron (III) chloride tetrahydrate ( ), and the third comprised . Each microemulsion was stabilized with a consistent 0.12 M concentration of dioctyl sulfosuccinate sodium (aerosol-OT) and supplemented with isooctane as the oil phase. Subsequently, the Lanthanum-containing microemulsion was combined with the iron-enriched one to form an intermediate emulsion, designated as microemulsion I. After an hour of mechanical mixing, the -containing microemulsion was introduced
Fig. 5 | The subsequent Aldol-aromatic reaction pathway inside zeolites from as a key intermediate. a Identified carbonaceous species retained in the spent LaFeO3/H-ZSM-5(14) during different reaction time. b-f The species determined by gas chromatography-mass spectrometry. as the key
intermediates in the reaction pathway to form aromatics (aldol-aromatic) inside zeolite. Some typical transition structure during to aromatics over ZSM-5 catalyst. Spheres in white, brown, red, light blue, and blue represent H, C, O, Al and Si , atoms, respectively.
into microemulsion I. This mixture underwent vigorous stirring at ambient conditions for 2 h , facilitating the formation of a precursor. Methanol was later added to induce phase separation and serve as a washing agent. After more than 50 times washing cycles, the solution was centrifuged to isolate the precursor, which was dried overnight at . The final crystalline product was obtained post-calcination at for 2 h .
Synthesis of nano-sized-H-ZSM-5
Nano-sized H-ZSM-5 with different Si/Al ratio was prepared by using the hydrothermal method. Appropriate amounts of Tetraethyl orthosilicate (TEOS ( 11.2 g )), Tetra propylammonium hydroxide (TPAOH , Aluminum nitrate , Sodium hydroxide ( ), Propan-2-ol (IPA ( 0.1 g )), and urea were dissolved in DI water under vigorous stirring for at least 6 h , at room temperature. The solution was transferred into a
Teflon-lined stainless steel autoclave reactor that was further placed in a temperature-programmed oven. For crystallization, the temperature of the oven was raised from room temperature to with a heating rate of and held at for 48 h . Once the crystallization is complete, the temperature of the autoclave reactor was quenched rapidly using a cold water bath. The obtained crystals were then collected through the filtration process (washed several times with DI water to remove the mother liquid) and dried overnight in the oven at . To remove the templating agent TPAOH, the crystals were calcined in air at for 5 h . Using a cation exchange method (step repeated at least three times), the Na-form of ZSM-5 (i.e., 10 g ) was converted to NH4-ZSM-5 after being dispersed in a 1 M NH4NO3 solution (i.e., 100 ml ) under vigorous stirring for 6 h . Finally, -ZSM-5 was transformed into H-ZSM-5 through a simple calcination step performed at for 5 h in the environment of air. The resulting powder was denoted as H-ZSM-5.
Fig. 6 | Schematic illustration of the conversion pathways on the bifunctional composite catalyst and the catalyst, in which is derived from in situ transformation of conventional Fe oxide
catalyst (e.g., and ) under the reactive atmosphere. Spheres in pink, black, red, magenta, yellow, brown, and green represent , and La atoms, respectively.
Catalytic performance evaluation
hydrogenation reactions were carried out in a fixed-bed reactor under 30 bar of mixed gas. The mixed gas with a ratio of (gas composition, , and ) unless otherwise stated. Generally, the catalyst with grain size of 355-850 um ( meshes) diluted with powdered quartz was loaded into a fixed-bed reactor with an inner diameter of 10 mm . The reaction was carried out at and unless otherwise specified. All the reaction products were kept in the gas phase and analyzed online by two gas chromate-graphs (Agilent 7890A) equipped with a HP-PLOT/Q capillary column connected to a flame ionization detector (FID) and a TDX-1 column (produced from DICP) connected to a thermal conductivity detector (TCD). was taken as a reference bridge between FID and TCD. Ar was used as an inner standard. The conversion and the CO selectivity, hydrocarbons and DME selectivity among the carbon products without CO (including and DME) were calculated with the followed equations:
: moles of at the inlet; : moles of at the outlet;
: moles of CO at the outlet;
where outlet represents moles of individual hydrocarbon product at the outlet. The carbon balance was between 95.0 and .
In situ diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS) measurement
The change of reaction species was explored with an in situ DRIFTS method. The catalyst wafer was reduced in hydrogen ( in volume, balanced) at and for 1 h , and then swept in nitrogen at the same flow rate and temperature for 20 min . The DRIFTS spectra were recorded with a Nicolet 6700 spectrometer by collecting
64 scans at resolution. After that, the catalyst was reacted with ( balanced) at , and atmospheric pressure. The in situ DRIFTS spectra were recorded every 5 min until the reaction lasted for 60 min .
Catalyst characterizations
The morphologies of samples were performed by field-emission scanning electron microscopy (FESEM, SU-8010), transmission electron microscopy (TEM, HT7700 120 kV ).
The atomic scale scan transmission electron microscopy (STEM) high angle annular dark field (HAADF) experiments were performed using a Cs-corrected scanning transmission electron microscope (FEI Titan Cubed Themis G2 300) operated at 300 kV with a convergence semi-angle of 23.6 mrad and the beam current was lower than 30 pA . The microscope was equipped with a DCOR+ spherical aberration corrector for the electron probe which was aligned before the experiments using a standard gold sample. The dwell time of probe scanning was for all the EDS mapping image. The dwell time was per pixel with a map size of pixels; a complete process of EDS mapping took roughly 0.5 h to reach an appropriately high signal-to-noise ratio. The following aberration coefficients were measured as: A1 = ; , A5 and R5 .
Atomic-resolution integrated differential phase contrast (iDPCSTEM) was also operated at 300 kV with a convergence semi-angle was 15 mrad . The beam current was set between 1 pA and 0.5 pA . The iDPCSTEM experiments were performed using a Cs-corrected scanning transmission electron microscope (FEI Titan Cubed Themis G2 300) operated at 300 kV . The microscope was equipped with a DCOR+ spherical aberration corrector for the electron probe which was aligned before the experiments using a standard gold sample. The following aberration coefficients were measured as: A1 = 1.41 nm ; A2 ; , and . The convergence semi-angle was 15 mrad , the beam current was lower than 0.5 pA (the measurement was limited by the precision of the Faraday cup), the collection angle was 4-22 mrad , and the dwell time of probe scanning was .
The X-ray diffraction patterns of various metal oxides and zeolites were obtained by Bruker D8 Advance X-ray diffractometer using CuKa radiation ( ) at 40 mA and 40 kV . X-ray photoelectron spectroscopy (XPS) of samples was tested on Escalab 250 Xi XPS with an Al Ka X-ray resource (Thermo Fisher Scientific). The Brunauer-Emmett-Teller surface area of samples were performed by specific surface area and mesoporous analyzer (TriStar II).
The X-ray absorption fine structures (XAFS) measurements were analyzed at the BL14W1 station in Shanghai Synchrotron Radiation Facility (SSRF, in maximum, Si (111) double-crystals). The energy was calibrated accordingly to the absorption edge of pure Fe foil. Before XAFS experiments, the as-prepared samples were transferred to a glove box for tableting without exposure to air. All XANES and EXAFS data were processed using Athena program.
Aldol-Aromatics composition retained inside the spent catalyst was determined by GC-MS using the hydrofluoric dissolution technique. Appropriate amounts of hydrofluoric acid ( ) was slowly and carefully added to 1 g of physically mixed composite catalyst ( and H-ZSM-5). Then a few drops of methylene chloride ( ) were added to the above solution and centrifuged until a layer of separated hydrocarbon oil was visible. This oil was then carefully extracted from the mother solution and analyzed on a GC-MS. These steps were repeated on samples obtained at different times on stream.
Theoretical methods
Density functional theory (DFT) calculations were performed by using the revised Perdew-Burke-Ernzerhof (RPBE) functional within the generalized gradient approximation (GGA) implemented in Vienna Ab Initio Simulation Package . The projector-augmented wave method was applied to describe the electron-ion interactions , and the D3 Grimme’s method was employed to correct van der Waals interaction. We used a plane-wave cutoff energy of 400 eV and the Gaussian smearing with a width of 0.1 eV . Periodic boundary conditions were applied, and more than of vacuum space was used to avoid the interaction of the adjacent images.
The adsorption energies were evaluated using eight-layer supercells with the bottom four layers constrained, and Monkhorst-Pack k-point grids were used with a convergence threshold of for the iteration in self-consistent field (SCF) . All structures were optimized until force components were less than . The vibrational frequencies of free molecules and adsorbates were calculated by using the phonon modules in the VASP 5.4.4 code. A standard thermodynamic correction was applied to determine the free energy corrections, including the correction of the effect from zero-point energy, pressure, inner energy, and entropy. All the formation energies of surface adsorbates/states, as well as gas-phase products were referenced to , and . Due to the DFT errors in estimating gas-phase molecules containing or backbone, such as and , at a PBE/RPBE level, a double bond correction of +0.33 eV was applied to raw energies of and . The way of obtaining such a correction is based on previous studies . We calculated the transition state using the climbing image nudged elastic band (CI-NEB) method. The vibrational frequencies of the transition state are also calculated and it should be noted that there is only one imaginary frequency.
We used adsorption free energies, , instead of electronic energies to construct Gibbs free energy diagram. Here we made the assumption that the gaseous products in the pathway were calculated at partial pressures as Fig. 2a depicted. The enthalpy, entropy and Gibbs free energy of each species were calculated by vibrational frequency analysis based on harmonic normal mode approximation using the finite difference method in VASP 5.4.4. The Gibbs free energy for a given species is
Where
where
where
where is the moment of inertia, is the rotational symmetry number, and is the mass of the molecule. For adsorbed molecules and transition states on the surface, the rotational and translational contributions were converted into vibration modes. We also approximated that the PV term of the surface species is negligible because it is very small with regard to the energetic terms, and thus, we considered in this case.
The stability of and with respect to were determined with the following equations:
The Gibbs free energy change of the above reactions were adopted to quantify the likelihood of and to form .
Data availability
All data are available in the manuscript or the Supplementary Information. Additional data are available from the corresponding authors upon request.
References
Hepburn, C. et al. The technological and economic prospects for utilization and removal. Nature 575, 87-97 (2019).
Aresta, M., Dibenedetto, A. & Angelini, A. Catalysis for the valorization of exhaust carbon: from to chemicals, materials, and fuels. Technological use of . Chem. Rev. 114, 1709-1742 (2014).
Appel, A. M. et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of fixation. Chem. Rev. 113, 6621-6658 (2013).
Wang, W., Wang, S., Ma, X. & Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 40, 3703-3727 (2011).
Li, Z. et al. Highly selective conversion of carbon dioxide to aromatics over tandem catalysts. Joule 3, 570-583 (2019).
Li, Z. et al. Ambient-pressure hydrogenation of CO 2 into long-chain olefins. Nat. Commun. 13, 2396 (2022).
Wang, L. et al. Cobalt-nickel catalysts for selective hydrogenation of carbon dioxide into ethanol. ACS Catal. 9, 11335-11340 (2019).
Hu, J. et al. Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol. Nat. Catal. 4, 242-250 (2021).
Tian, G., Zhang, C. & Wei, F. COx conversion to aromatics: a minireview of nanoscale performance. Nanoscale Horiz. 7, 1478-1487 (2022).
Ji, Y. et al. Oxygenate-based routes regulate syngas conversion over oxide-zeolite bifunctional catalysts. Nat. Catal. 5, 594-604 (2022).
Wang, Y. et al. Direct conversion of CO2 to aromatics with high yield via a modified Fischer-Tropsch synthesis pathway. Appl. Catal. B Environ. 269, 118792 (2020).
Arslan, M. T. et al. Highly selective conversion of CO 2 or CO into precursors for kerosene-based aviation fuel via an aldol-aromatic mechanism. ACS Catal. 12, 2023-2033 (2022).
Wang, Y. et al. Rationally designing bifunctional catalysts as an efficient strategy to boost CO2 hydrogenation producing valueadded aromatics. ACS Catal. 9, 895-901 (2018).
Wei, J. et al. Precisely regulating Brønsted acid sites to promote the synthesis of light aromatics via CO2 hydrogenation. Appl. Catal. B Environ. 283, 119648 (2021).
Wang, T. et al. ZnZrOx integrated with chain-like nanocrystal HZSM5 as efficient catalysts for aromatics synthesis from CO2 hydrogenation. Appl. Catal. B Environ. 286, 119929 (2021).
Ramirez, A. et al. Effect of zeolite topology and reactor configuration on the direct conversion of CO2 to light olefins and aromatics. ACS Catal. 9, 6320-6334 (2019).
Tian, H. et al. Tandem catalysts composed of different morphology HZSM-5 and metal oxides for CO2 hydrogenation to aromatics. Fuel 314, 123119 (2022).
Zhang, J. et al. Hydrogenation of CO2 into aromatics over a ZnCrO x-zeolite composite catalyst. Chem. Commun. 55, 973-976 (2019).
Li, Y. et al. Direct conversion of carbon dioxide into liquid fuels and chemicals by coupling green hydrogen at high temperature. Appl. Catal. B Environ. 324, 122299 (2023).
Zhu, J. et al. Deconvolution of the particle size effect on CO2 hydrogenation over iron-based catalysts. ACS Catal. 10, 7424-7433 (2020).
Zhu, J. et al. Dynamic structural evolution of iron catalysts involving competitive oxidation and carburization during CO2 hydrogenation. Sci. Adv. 8, eabm3629 (2022).
Zhao, B. et al. Direct transformation of syngas to aromatics over Na-Zn-Fe5C2 and hierarchical HZSM-5 tandem catalysts. Chem 3, 323-333 (2017).
. et al. Selective conversion of syngas to aromatics over Fe3O4@MnO2 and hollow HZSM-5 bifunctional catalysts. ACS Catal. 9, 5147-5156 (2019).
Tian, G. et al. Accelerating syngas-to-aromatic conversion via spontaneously monodispersed Fe in ZnCr 2 O 4 spinel. Nat. Commun. 13, 5567 (2022).
Xu, Y. et al. A hydrophobic FeMn@ Si catalyst increases olefins from syngas by suppressing C1 by-products. Science 371, 610-613 (2021).
Song, G., Li, M., Yan, P., Nawaz, M. A. & Liu, D. High conversion to aromatics via CO2-FT over a CO-reduced Cu-Fe2O3 catalyst integrated with HZSM-5. ACS Catal. 10, 11268-11279 (2020).
Wei, J. et al. Directly converting CO2 into a gasoline fuel. Nat. Commun. 8, 15174 (2017).
Cui, X. et al. Selective production of aromatics directly from carbon dioxide hydrogenation. ACS Catal. 9, 3866-3876 (2019).
Wang, C. et al. Fischer-Tropsch synthesis to olefins boosted by MFI zeolite nanosheets. Nat. Nanotechnol. 17, 714-720 (2022).
Cheng, K. et al. Impact of the spatial organization of bifunctional metal-zeolite catalysts on the hydroisomerization of light alkanes. Angew. Chem. 132, 3620-3628 (2020).
Wang, Y. et al. Visualizing element migration over bifunctional metal-zeolite catalysts and its impact on catalysis. Angew. Chem. 133, 17876-17884 (2021).
Rong, Y. et al. Challenges for commercializing perovskite solar cells. Science 361, eaat8235 (2018).
Peña, M. A. & Fierro, J. Chemical structures and performance of perovskite oxides. Chem. Rev. 101, 1981-2018 (2001).
Liao, X. et al. Highly efficient reduction of O2-containing CO2 via chemical looping based on perovskite nanocomposites. Nano Energy 78, 105320 (2020).
Zhu, X., Li, K., Neal, L. & Li, F. Perovskites as geo-inspired oxygen storage materials for chemical looping and three-way catalysis: a perspective. ACS Catal. 8, 8213-8236 (2018).
Wang, J. et al. Elucidating surface and bulk phase transformation in Fischer-Tropsch synthesis catalysts and their influences on catalytic performance. ACS Catal. 9, 7976-7983 (2019).
Wang, S. et al. Highly selective hydrogenation of CO 2 to propane over GaZrO x/H-SSZ-13 composite. Nat. Catal. 5, 1-13 (2022).
Gao, P. et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 9, 1019-1024 (2017).
Cheng, Y. et al. High-yield production of aromatics over CuFeO2/ hierarchical HZSM-5 via CO2 Fischer-Tropsch synthesis. Green Chem. 25, 3570-3584 (2023).
Meng, C. et al. Oxygen-deficient metal oxides supported nanointermetallic InNi 3 CO .5 toward efficient CO 2 hydrogenation to methanol. Sci. Adv. 7, eabi6012 (2021).
Yang, H. et al. Selective synthesis of olefins via CO2 hydrogenation over transition-metal-doped iron-based catalysts. Appl. Catal. B Environ. 321, 122050 (2023).
Guo, L. et al. Heteroatom doped iron-based catalysts prepared by urea self-combustion method for efficient CO2 hydrogenation. Fuel 276, 118102 (2020).
Zhang, W., Fu, Q., Luo, Q., Sheng, L. & Yang, J. Understanding single-atom catalysis in view of theory. JACS Au 1, 2130-2145 (2021).
Shang, X. et al. Xylene synthesis through tandem CO2 hydrogenation and toluene methylation over a composite ZnZrO zeolite catalyst. Angew. Chem. 62, e202309377 (2023).
Li, Y. et al. Interfacial Fe5C2-Cu catalysts toward low-pressure syngas conversion to long-chain alcohols. Nat. Commun. 11, 61 (2020).
Gao, W. et al. Photo-driven syngas conversion to lower olefins over oxygen-decorated Fe5C2 catalyst. Chem 4, 2917-2928 (2018).
Li, S., Meitzner, G. D. & Iglesia, E. Structure and site evolution of iron oxide catalyst precursors during the Fischer-Tropsch synthesis. J. Phys. Chem. B 105, 5743-5750 (2001).
Goldstein, J. & Moren, A. Diffusion modeling of the carburization process. Metall. Trans. A 9, 1515-1525 (1978).
Simonetti, S., Moro, L., Brizuela, G. & Juan, A. A computational study of the carburization phenomena in a Fe-Ni alloy. J. Phys. D Appl. Phys. 41, 125006 (2008).
Ribeiro, M. C. et al. Fischer-Tropsch synthesis: influence of Mn on the carburization rates and activities of Fe-based catalysts by TPR-
EXAFS/XANES and catalyst testing. J. Phys. Chem. C 115, 4783-4792 (2011).
51. Zabilskiy, M. et al. The unique interplay between copper and zinc during catalytic carbon dioxide hydrogenation to methanol. Nat. Commun. 11, 2409 (2020).
52. Studt, F. et al. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nat. Chem. 6, 320-324 (2014).
53. Zhou, H. et al. Engineering the Cu/Mo2C T x (MXene) interface to drive CO2 hydrogenation to methanol. Nat. Catal. 4, 860-871 (2021).
54. Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
55. Chen, Y.-H. et al. First-principle study of H 2 adsorption on LaFeO 3 (110) surface. J. Nanomater. 2014, 16-16 (2014).
56. Wang, H. et al. Bifunctional catalysts with versatile zeolites enable unprecedented para-xylene productivity for syngas conversion under mild conditions. Chem. Catal. 2, 779-796 (2022).
57. Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
58. Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
59. Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
60. Christensen, R., Hansen, H. A. & Vegge, T. Identifying systematic DFT errors in catalytic reactions. Catal. Sci. Technol. 5, 4946-4949 (2015).
61. Granda-Marulanda, L. P. et al. A semiempirical method to detect and correct DFT-based gas-phase errors and its application in electrocatalysis. ACS Catal. 10, 6900-6907 (2020).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 22278238, C.Z. Grant No. 22109020, H.J.P. Nos. 22275110, 22322203, X.C.), the Tsinghua-Toyota Joint Research Fund (X.C. and F.W.). the Key Research and Development Program of Inner Mongolia and Ordos (20211140095, C.Z.), and CNPC Innovation Funds (2020990028, C.Z.), the National Key Research and Development Program of China (No. 22238004, F.W. and X.C.).
Author contributions
G.T., Z.L., H.J.P., C.Z., X.C. and F.W. designed the studies. G.T. and X.F. synthesized catalysts. G.T. and X.F. performed catalytic tests. G.T., Z.L.,
X.F., K.S., H.M., N.W., M.Z., X.L., L.L., Z.L., H.X., X.C. and B.Y. conducted characterizations. G.T. and X.L. contributed to DFT calculations. G.T. and H.J.P. wrote the paper. All the authors approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains
supplementary material available at https://doi.org/10.1038/s41467-024-47270-z.
Correspondence and requests for materials should be addressed to Chenxi Zhang, Xiao Chen, Hong-Jie Peng or Fei Wei.
Peer review information Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Reprints and permissions information is available at http://www.nature.com/reprints
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Beijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, 100084 Beijing, China. Ordos Laboratory, Ordos, Inner Mongolia 017010, China. Institute for Carbon Neutrality, Tsinghua University, 100084 Beijing, China. Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 611731 Sichuan, China. Key Laboratory of Fuel Cell Technology of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. College of Chemistry, Taiyuan University of Technology, Taiyuan 030024, China. Faculty of Environment and Life, Beijing University of Technology, 100124 Beijing, China. These authors contributed equally: Guo Tian, Zhengwen Li. e-mail: cxzhang@tsinghua.edu.cn; chenx123@tsinghua.edu.cn; hjpeng@uestc.edu.cn; wf-dce@tsinghua.edu.cn