تسهيل تفاعل تطور الهيدروجين القلوي على واجهة الهترو Ru/RuO2 من خلال تشويب ذرات البلاتين الفردية Facilitating alkaline hydrogen evolution reaction on the hetero-interfaced Ru/RuO2 through Pt single atoms doping
استكشاف بديل نشط وفعال من حيث التكلفة للمواد المحفزة الكهربائية المدعومة بالكربون من جزيئات البلاتين النانوية لتفاعل تطور الهيدروجين القلوي (HER) لا يزال بعيد المنال حتى الآن. هنا، نبلغ عن محفز يعتمد على ذرات البلاتين الفردية (SAs) المدعومة في الواجهة غير المتجانسة.الدعم (المشار إليه بـ )، الذي يتميز بجهد منخفض لتجاوز HER، واستقرار ممتاز ونشاط محسّن بشكل ملحوظ من حيث التكلفة مقارنةً بالمنتجات التجارية و في 1 م KOH. تكشف التوصيفات الفيزيائية الكيميائية المتقدمة أن تفكك الماء البطيء يتم تسريعه بواسطةبينما تسهل Pt SAs والروثينيوم المعدني العملية اللاحقةتركيبة. تتوافق الحسابات النظرية مع النتائج التجريبية. علاوة على ذلك،يتطلب فقط 1.90 فولت للوصول إلىويقدم نشاطًا عالي السعر في إلكتروليزر الماء بغشاء تبادل الأنيونات، متفوقًا على المعيار.. تقدم هذه الدراسة إرشادات قابلة للتطبيق لتطوير المحفزات القائمة على المعادن النبيلة ذات الأداء العالي والتكلفة المنخفضة نحو التطبيق العملي.الإنتاج.
في الوقت الحاضر، أصبح البحث عن ناقل طاقة واعد ليحل محل الوقود الأحفوري التقليدي أكثر أهمية من أي وقت مضى للتعامل مع قضايا تدهور البيئة واستنزاف الموارد.الهيدروجينتعتبر ( ) بفضل مزايا انبعاثات غير الملوثات وكثافة الطاقة العالية البديل الأنسب. في الوقت نفسه، يوفر التحليل الكهربائي للماء المدعوم بالطاقة المتجددة وسيلة خضراء ومستدامة لكفاءةإنتاج. بالمقارنة مع التحليل الكهربائي للماء الذي يتم في وسط حمضي، فإن الماء القلوي تكنولوجيا التحليل الكهربائي أصبحت حالياً أكثر شعبية وقابلية للتطبيق في الصناعة العملية، وذلك بفضل مزاياها في وجود منشآت قوية وتكلفة منخفضة لبناء المحلل الكهربائي.ومع ذلك، فإن تفاعل تطور الهيدروجين الكاثودي (HER) في الإلكتروليت القلوي يتضمن عملية تفكك الماء السابقة (خطوة فولمر) وعملية تجمع الهيدروجين اللاحقة (خطوة هيروفسكي أو تافل)، حيث ستحد الديناميات البطيئة لخطوة فولمر بشكل كبير من أداء تقسيم الماء الكلي.. لذلك، من الأهمية بمكان تسريع هذا
حاجز تقييدي من خلال اكتشاف محفز كهربائي فعال تجاه تفاعل الهيدروجين القلوي.
على الرغم من أن المواد النانوية من البلاتين (Pt) ذات طاقة الربط الهيدروجيني المثلى معروفة على نطاق واسع بأنها أكثر المحفزات نشاطًا تجاه تفاعل تطور الهيدروجين (HER)، إلا أن أنشطتها التحفيزية في الوسط القلوي أقل بحوالي مرتبتين من حيث الحجم مقارنة بتلك في الوسط الحمضي. يمكن أن يُعزى ذلك بشكل رئيسي إلى نقص قدرة ذرات البلاتين على تفكك الماء، مما يؤدي إلى نقص في إمداد البروتونات.جيل. على العكس من ذلك، تم اعتبار أكاسيد/هيدروكسيدات المعادن المرشحين المثاليين لتفاعل تطور الأكسجين (OER) بسبب قدراتها الجيدة على كسر الرابطة القوية H-OH، بينما تكون غير فعالة في دفع عملية توليد الهيدروجين فيبشكل عام، يجب أن يوفر المحفز الكهربائي المصمم بشكل جيد لتفعيل تفاعل الهيدروجين في الإلكتروليت القلوي منصة تحتوي على مواقع نشطة مختلفة لخطوات تفكك الماء وامتصاص الهيدروجين. مستلهمين من هذه الإرشادات، تم تكريس العديد من الجهود مؤخرًا لبناء هندسة مركبة من خلال دمج البلاتين مع أكاسيد/هيدروكسيدات المعادن لتسريع تفاعل الهيدروجين القلوي بشكل متزامن. على سبيل المثال، أفاد سوبارامان وزملاؤه أن الحاجز الحركي لخطوة فولمر على مواد النانو من البلاتين يمكن أن يتم تعزيزها بشكل ملحوظ بواسطة هيدروكسيد النيكل المزخرف.العناقيد، مما يؤدي إلى تقليل الفائض الكهربائي لتفاعل تقليل الهيدروجين في محلول هيدروكسيد البوتاسيومنجح وانغ وزملاؤه أيضًا في تحضير جزيئات نانوية كثيفة من البلاتين مثبتة تحتوي على فراغات أكسجين غنية.الهجائن كعامل تحفيز كهربائي قاعدي لتفاعل الهيدروجين مع أداء معزز. بالإضافة إلى ذلك، أظهر هي وآخرون أن تفكك الماء وتوليد الهيدروجين يتسارعان على المواد النانوية من البلاتين المدمجة مع هيدروكسيدات الحديد والنيكل المزدوجة الطبقات. LDH) على الرغم من التقدم الذي تم تحقيقه، فإن الندرة المنخفضة والتكلفة العالية للبلاتين تحد بشدة من تطبيقاته على نطاق واسع في الصناعة العملية.في هذا الصدد، من الضروري أخذ النشاط العالي لتفاعل الهيدروجين وتقليل كمية استخدام البلاتين في الاعتبار في الوقت نفسه عند تحضير المحفزات الكهربائية الهجينة المعتمدة على البلاتين. مؤخرًا، تعتبر المحفزات ذات الذرات الفردية الموزعة بشكل كبير (SACs) من أكثر المرشحين وعدًا لمجموعة من التفاعلات الكهروكيميائية بسبب مزاياها المميزة مثل كفاءة استخدام الذرات القصوى، والمواقع النشطة المحددة جيدًا، وتكلفة التحضير المنخفضة بشكل كبير.لذلك، فإن الجمع بين ذرات البلاتين الفردية ودعامات أكسيد/هيدروكسيد المعادن يظهر آفاقاً مشرقة لتحقيق التوازن بين التكلفة المنخفضة والأداء العالي في التفاعلات الحفزية. في الوقت نفسه، باعتبارها واحدة من أرخص المعادن النبيلة، فإن الروثينيوم (Ru) الفعال من حيث التكلفة ومشتقاته تمتلك أقوى قدرة على تحفيز خطوة تفكك الماء، والتي يمكن استخدامها كدعامات لذرات البلاتين الفردية لإطلاق العنان بالكامل لإمكانات تحفيز تفاعل الهيدروجين القلوي، بينما لا توجد تقارير ذات صلة حتى الآن..
في هذا العمل، نبلغ عن تخليق وتوصيف وتحفيز كهربائي لمحفز نشط وفعال من حيث التكلفة لتفاعل تقليل الهيدروجين (HER) للاستخدام في الكاثود لمحللات الماء القلوية. يتضمن تصميم محفز HER لدينا ذرات معدن بلاتين فردية مدعومة في الواجهة غير المتجانسة.يدعم (المشار إليه بـ ). بشكل أكثر تحديدًا، يتطلب هذا المحفز ذو محتوى البلاتين المنخفض جهدًا زائدًا منخفضًا جدًا يبلغ 18 مللي فولت و63 مللي فولت عند كثافات التيار و ، على التوالي، متفوقة على معظم المحفزات الكهروكيميائية المعتمدة على المعادن النبيلة والمحررة من المعادن النبيلة التي تم الإبلاغ عنها مؤخرًا. تعتمد النشاط التحفيزي القائم على تكلفة المعادن النبيلة لـيصل إلى عامل يزيد عن 16 مقارنة بتلك الخاصة بالتجارة و ج عند الجهد الزائد 63 مللي فولت. من الجدير بالذكر،يوفر استقرارًا طويل الأمد لأكثر من 100 ساعة مع فقدان نشاط ضئيل. الآلية المقابلة التي تم التحقيق فيها من خلال تجارب مختلفة وخصائص تشغيلية تؤكد أن جميع ذرات البلاتين المعزولة، والروثينيوم وفي الدعم تلعب أدوارًا حيوية في تنشيط تفاعل الهيدروجين القلوي. بشكل ملموس، يتم تسريع خطوة تفكك الماء البطيئة بواسطة، الخطوة التالية في دمج الهيدروجين يتم تعزيزها بشكل أساسي على ذرات البلاتين الفردية وأيضًا تساهم جزئيًا من الروديوم، مما يؤدي بشكل متزامن إلى نشاط ملحوظ.نظرية الكثافة الوظيفية (DFT) تحدد الحسابات بشكل أكبر الوظائف المحددة للمواقع النشطة في، التي تؤكد الاكتشافات التجريبية. علاوة على ذلك، فإن دمج و NiFe LDH في إلكتروليزر الماء بغشاء تبادل الأنيون (AEMWE) يتطلب فقط 1.90 فولت للوصول إلى كثافة تيار كبيرة منويظهر نشاط سعر مرتفع قدره 247.1 دولارعند 2.1 فولت، يتفوق بشكل كبير على النظير القائم على Pt/C. هذه النتيجة البحثية تحفز اهتمامنا لمتابعة التوازن بين الجدوى الاقتصادية والنشاط العالي عند استكشاف المحفزات الكهربية القائمة على المعادن النبيلة نحو التطبيقات العملية.الإنتاج.
النتائج
الخصائص الهيكلية لـ
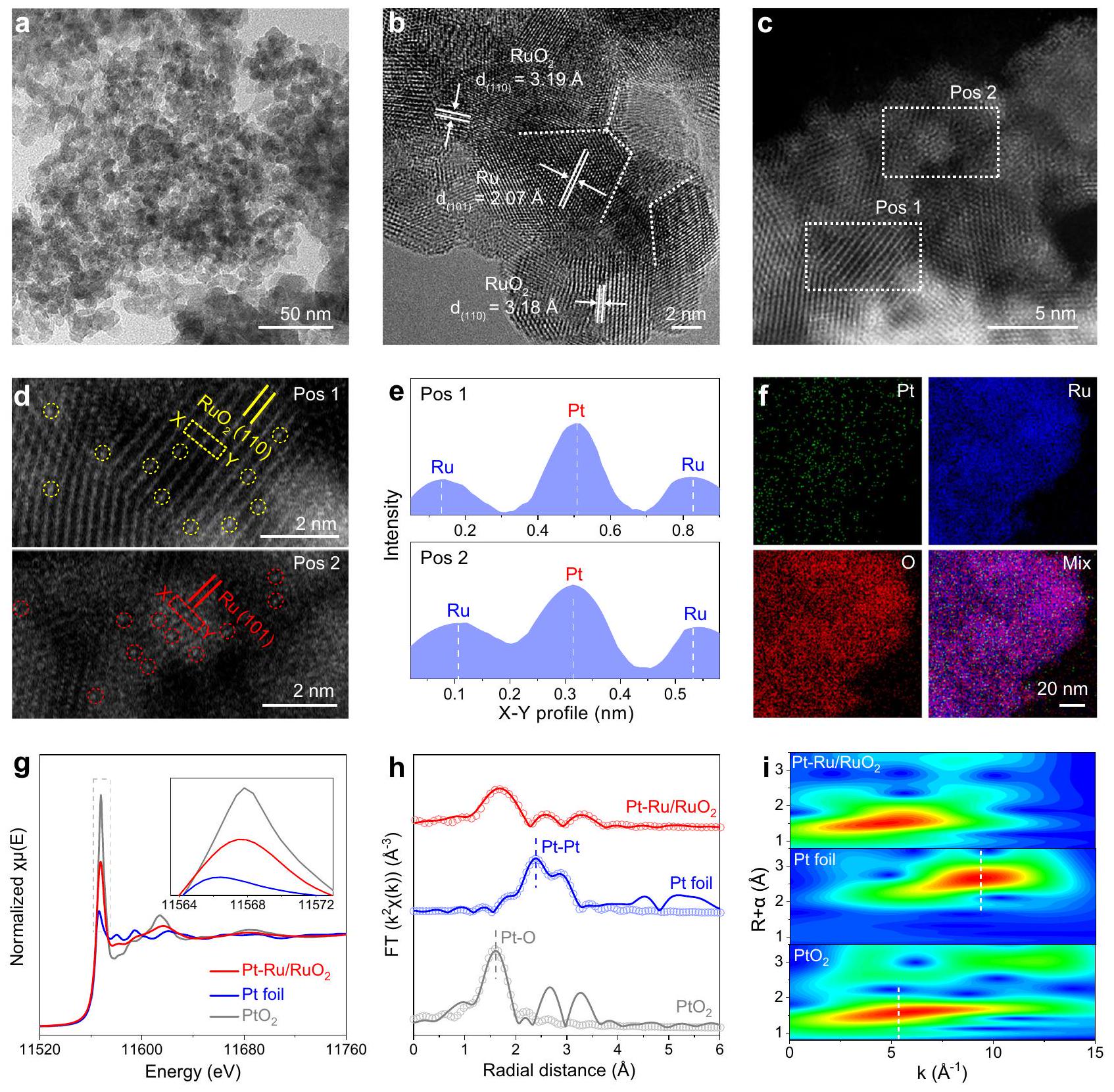
لتركيب، الـالمُغَنى بحدود الحبوب تم الحصول عليه أولاً عن طريق غمرالسابقة في المنصهر (الأشكال التكميلية 1، 2). ثم تم امتصاص أنواع البلاتين على المركب المُصنَّعتبع ذلك التكلس في الأرجون، حيث تم دمج ذرات البلاتين الفردية في هذه العملية وتم تقليله قليلاً إلى متصل (انظر الطرق للتفاصيل). كما هو موضح في الشكل 1a، الهيكل النانوي المسطح والكثيف لـتمت ملاحظته في البداية باستخدام المجهر الإلكتروني الناقل (TEM). تشير صورة مجهر TEM عالي الدقة (HR-TEM) النموذجية إلى أنيتكون من مناطق بلورية موزعة بشكل مستقل تنتمي إلى الروثينيوم و، مع وجود حدود حبيبية واضحة بينها. لتكون محددة، رو وتظهر المناطق البلورية تباعد الشبكة لـ و الذي يمكن أن يُنسب إلى و الوجوه، على التوالي (الشكل 1ب)بشكل عام، تشير وجود حدود الحبوب إلى الاتصال الوثيق بين مرحلتين، مما يكون ملائمًا لتحفيز تفاعل فعال مثل نقل الإلكترونات في التفاعلات الكهروكيميائية.. بالإضافة إلى ذلك، Ru/ تم إعدادها للمقارنة. كما هو موضح في الشكل التكميلي 3، يظهر مورفولوجيا وبنية متداخلة مشابهة لـ لكشف الهياكل البلورية التفصيلية لـ و تم استخدام حيود الأشعة السينية (XRD) والنتائج المقابلة تشير إلى وجود طورين ينسبان إلى Ru (رقم JCPDS 06-0663) و (رقم JCPDS 43-1027) تتواجد في هذه الأنماط، وهو ما يتماشى مع نتائج HR-TEM. علاوة على ذلك، لا يمكن العثور على أي قمة حيود مرتبطة بالبلاتين في هذه الأنماط. تم إجراء تحسينات ريتفيلد على القمم الأساسية لتلك الأنماط XRD لحساب المحتوى النسبي للروثينيوم ونتيجة لذلك، تم حساب نسبة المولارية للروثينيوم لتكونفي“، وأسلوب أكثر قليلاً من Ru ( يظهر على بسبب إدخال أنواع البلاتين (الأشكال التكميلية 4، 5 والجداول التكميلية 1، 2). بعد ذلك، تحدد المجهر الإلكتروني الماسح – التحليل الطيفي للطاقة المشتتة (SEM-EDS) وقياس الطيف الضوئي بالتحليل الطيفي للبلازما المقترنة (ICP-OES) أن النسبة الذرية للبلاتين هيفي (الشكل التوضيحي 6).
من أجل الكشف عن توزيع الذرات لأنواع البلاتين، تم استخدام تقنية المجهر الإلكتروني الماسح بتصحيح الشذوذ في مجال الظلام العالي الزاوية (AC HAADF-STEM) علىكما هو موضح في الشكل 1c و 1d، يمكن تخصيص منطقتين بلوريتين متميزتين مع ترتيبات ذرية واضحة تُسمى Pos 1 و Pos 2 إلى و الوجوه، على التوالي. ومن المثير للاهتمام، بسبب التباين العالي للزنك (Z-contrast) للبلاتين (Pt) مقارنة بالعناصر الأخرى، يتم تصور النقاط الساطعة المعزولة التي تتوافق مع ذرات البلاتين بشكل مباشر وتكون موزعة بشكل متجانس على كل من الروثينيوم (Ru) والمناطق، مما يدل على أن أنواع البلاتين توجد بشكل رئيسي كذرات مفردة علىيدعم. يتم تأكيد ذلك بشكل أكبر من خلال ملفات كثافة الذرات على طول المستطيلات المتقطعة المجمعة في الشكل 1d، حيث يتم ملاحظة ذرات البلاتين المعزولة ذات الكثافات الأعلى (الشكل 1e). بالإضافة إلى ذلك، تشير الخرائط العنصرية المقابلة إلى أن ذرات البلاتين موزعة بشكل متجانس على كامل (الشكل 1f والشكل التوضيحي 7). بعد ذلك، تم تحليل الهياكل الإلكترونية السطحية والتركيبات الكيميائية بواسطة مطيافية الأشعة السينية للأشعة السينية (XPS). يثبت طيف XPS الاستقصائي في البداية وجود عناصر البلاتين فيمن خلال عرض Pt واضحإشارة (الشكل التوضيحي 8أ). بالإضافة إلى ذلك، كل من روطيف XPS على و يمكن تفكيكه إلى
الشكل 1 | التوصيفات الهيكلية لـ Pt-Ru/RuO2. أ صورة TEM، ب صورة HR-TEM و (ج) صور AC HAADF-STEM لـ. د المناطق المتضخمة من Pos 1 و Pos 2 في (ج)، مع ذرات البلاتين المفردة المحددة في دوائر، على التوالي. هـ ملفات كثافة الذرات على طول المستطيلات المنقطة في (د). و الخرائط العنصرية المقابلة لـ . ج البلاتين المعاير-طيف XANES عند الحافةرقائق البلاتين و. هـ ب ت -طيف EXAFS الحافة والمنحنيات الملائمة المقابلة، و (i) الموجات المحولة-طيف EXAFS الموزون بـرقائق. ازدواجيات لـ و مما يوحي بتعايش الروديوم المعدني و. بعد إدخال Pt ، نسبة تم زيادته من 0.16 إلى 0.28، وهو ما يتوافق بشكل جيد مع نتائج XRD (الشكل التوضيحي 8b).الشكل التوضيحي الإضافي 8c يُظهرطيف على هذه العينات، حيث القمم عند طاقة الربط لـو 532.5 إلكترون فولت تتوافق مع الأكسجين الشبكي، الممتص وامتصت على التوالي. في هذه الأثناء، Ptتشير طيف XPS إلى أن Pt/C التجاري يحتوي بشكل أساسي على القمة المنسوبة إلى البلاتين المعدني. ومن الجدير بالذكر أن كل من البلاتين المعدني والبلاتين المؤكسد موجودان علىيتم تشكيل سبيكة ذات ذرة واحدة عندما يتم توزيع ذرات البلاتين بشكل متجانس في منطقة الروثينيوم، حيث تتميز البلاتين بخصائص معدنية وبالتالي لها حالة تكافؤ تساوي صفر.. في الوقت نفسه، فإن البلاتين المؤكسد ينشأ بشكل أساسي من ذرات البلاتين الفردية المنسقة بواسطة ذرات الأكسجين على (الشكل التوضيحي الإضافي 8d).
بعد ذلك، تم تحديد الحالات الكيميائية التفصيلية والهياكل الذرية المحلية باستخدام مطيافية امتصاص الأشعة السينية المعتمدة على إشعاع السنكروترون (XAS). حالة الأكسدة للبلاتين في Pt-Ru/تم اكتشافه لأول مرة بواسطة هيكل امتصاص الأشعة السينية بالقرب من الحافة (XANES) جنبًا إلى جنب مع رقائق البلاتين القياسية وكمرجع. يُعترف بأن موضع الخط الأبيض في -edge XANES حساس للغاية لحالة التكافؤ لـعنصركما هو موضح في الشكل 1 ج، فإن موضع طاقة الخط الأبيض لـ يقع بين رقائق البلاتين و مما يعني أن حالة الأكسدة لأنواع البلاتين تتراوح بين 0 و +4 في. علاوة على ذلك، فإن هيكل الامتصاص بالأشعة السينية الممتد (EXAFS) هو أداة قوية لاكتشاف بيئة التنسيق الذري. يمكن ملاحظته بوضوح أن يعرض قمة سائدة عندفي فضاء R الخاص بـ Pt-طيف EXAFS عند حافة، المرتبط برابطة Pt-O. بالإضافة إلى ذلك، لا يوجد قمة ملحوظة تتعلق بـيمكن تمييز الرابطة في ، مما يدل على غياب جزيئات أو تجمعات Pt (الشكل 1 ح). وقد تم تعزيز ذلك بشكل أكبر من خلال فضاء k لتحويل الموجات (WT)-EXAFS، حيث يظهر أقصى تشتت عند و قريب منتشتت في. أيضًا، لا يتوافق التشتت مع يتم العثور على الرابطة عند المقارنة مع رقائق البلاتين ( و )، مما يثبت عدم وجود بلورات متجمعة من البلاتين في (الشكل 1 i. علاوة على ذلك، تم إجراء ملاءمة EXAFS قائمة على النموذج علىورقة البلاتين. تؤكد النتائج أن القمة الرئيسية تقع عند هو رابطة، قمم أخرى عند و مشتقة من و الجزئيات، مما يشير إلى أن ذرات البلاتين المعزولة في يتم تنسيقها بواسطة ذرات الأكسجين المحيطة بـ المناطق المحيطة بذرات الروثينيوم من مناطق الروثينيوم، على التوالي (الشكل التكميلي 9 والجدول التكميلي 3)لذلك، من خلال دمج التحليل المفصل لتقنية AC HAADF-STEM وEXAFS، نستنتج أن أنواع البلاتين موجودة كذرات مفردة وموزعة بشكل متجانس على الواجهة.يدعم. في حالة أخرى، حالات التكافؤ للروثينيوم علىتم تقييمهم أيضًا بواسطة رو-طيف XANES الحافة مع ورقة روديوم، و للمقارنة. كما هو موضح في الشكل التكميلي 10a، فإن حافة الامتصاص علىيعرض تحولًا في الطاقة أقل مقارنة بـ، مما يشير إلى الحالة الأقل من حيث القيمة للأول، والتي تعود إلى النسبة الأعلى من الروديوم المعدني في وفقًا لنتائج XRD وXPS. من خلال إنشاء المنحنيات القياسية بين حالات أكسدة الروثينيوم وطاقة حواف الامتصاص البيضاء، تم تحديد متوسط حالات الأكسدة للروثينيوم على و تم تحديدهما على أنهما +2.8 و +3.3، على التوالي (الشكل التكميلي 10b). بالإضافة إلى ذلك، كل من Ru-طيف EXAFS عند حافة و تظهر القمم المتطابقة كماباستثناء القمم الدقيقة المنسوبة إلى رابطة رو-رو الموجودة في، مما يدل على وجود روديوم مخفض ويتماشى مع التوصيفات المذكورة أعلاه (الشكل التكميلي 10c).
الأداء الكهروكيميائي لـ
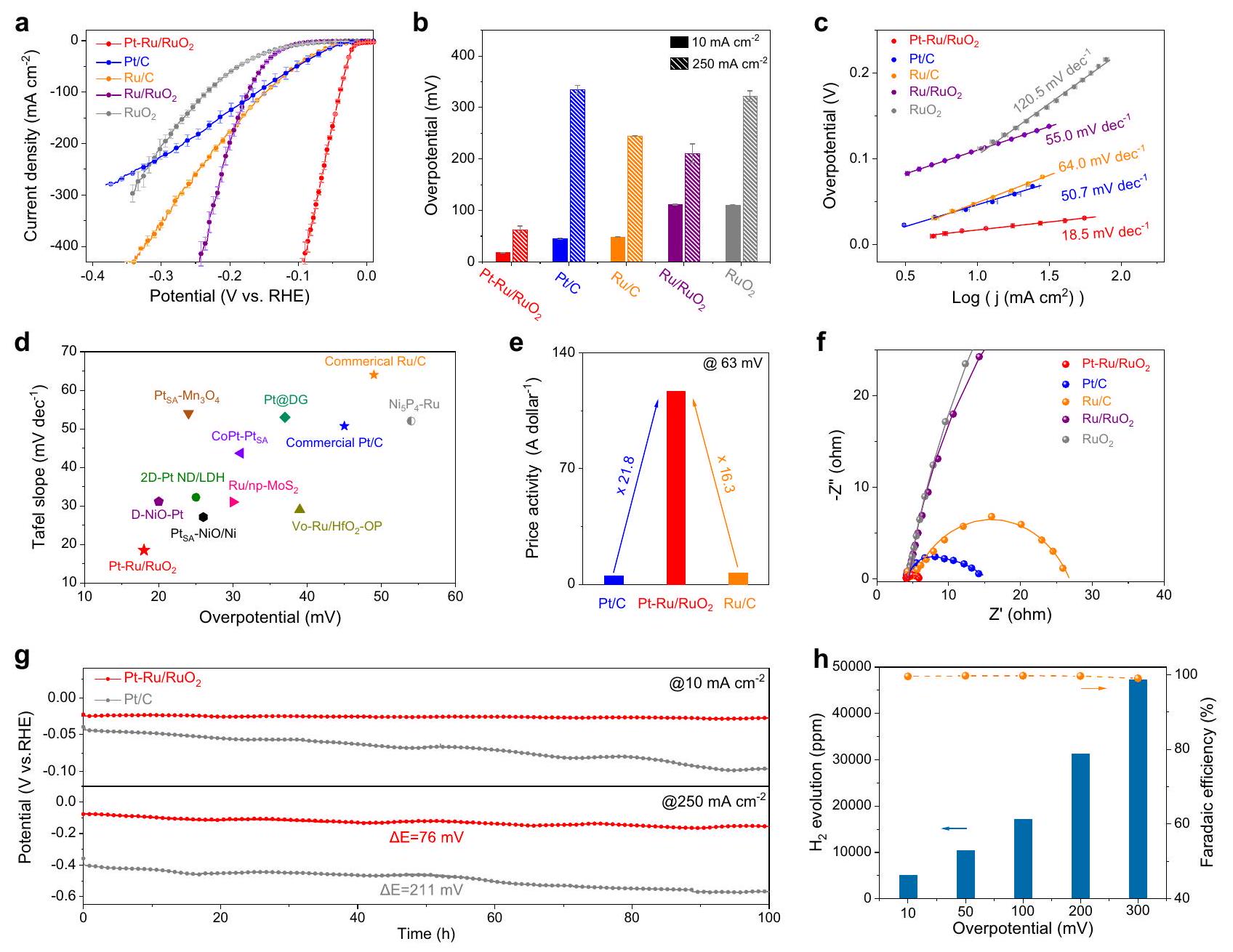
تم تقييم الأداء الكهروكيميائي لتلك المحفزات الكهربائية المعدة تجاه تفاعل تقليل الهيدروجين (HER) في محلول إلكتروليتي 1 م KOH باستخدام نظام ثلاثي الأقطاب. كما تم إجراء اختبارات مماثلة على Pt/C التجارية و Ru/C وللمقارنة الصريحة (الشكل التكميلي 11). تم معايرة جميع الجهود المقاسة في هذه الدراسة بعناية على مقياس القطب الهيدروجيني القابل للعكس (RHE) (الشكل التكميلي 12). تم الكشف عن القيم الفعلية للمقاومة لجميع المحفزات المدروسة في محلول 1 م KOH (الشكل التكميلي 13a) وتظهر منحنيات الفولتمترية الخطية غير المصححة (LSV) في الشكل التكميلي 13b. كما هو موضح في الشكل 2a، فإن منحنيات LSV مع التصحيحات تظهر اتجاه النشاط لـ عند الجهود المنخفضة. من الجدير بالذكر أن و تظهر زيادة متسارعة في النشاط مقارنةً بالعوامل المساعدة الأخرى مع زيادة الجهد، بسبب معدلات تفاعل الهيدروجين السريعة. لتكون أكثر تحديدًا،يوفر جهدًا زائدًا قدره 18 مللي فولت عند كثافة التيار، وهو ما يقل بشكل كبير عن تلك الخاصة بـ، و بشكل مثير للإعجاب، عند كثافة تيار أعلى منلا يزال يمتلك أقل جهد زائد (63 مللي فولت) بين جميع المحفزات الكهربائية المدروسة، مما يدل على نشاطه الملحوظ في تفاعل تطور الهيدروجين القلوي (الشكل 2ب). بالإضافة إلى ذلك، فإن المركب المُصنّع يظهر أداءً مشابهًا تقريبًا لتفاعل تطور الهيدروجين كما هو الحال في (الشكل التكميلي 14). علاوة على ذلك، يوضح الشكل 2ج ميل تافل بناءً على منحنيات LSV والقيم المقابلة هي و و و و لـ و و و على التوالي. أقل ميل تافل لـ يدل على أكثر تسهيل فعال لديناميكية تطور الهيدروجين . بالإضافة إلى ذلك، فإن قيمة ميل تافل المنخفضة هذه على تشير أيضًا إلى أنها تتبع آلية فولمر-تافل في تفاعل تطور الهيدروجين القلوي بعد دمج ذرات البلاتين الفردية ذات التغطية العالية للهيدروجين وقدرة الربط . لتسليط الضوء بشكل أكبر على مزايا الجهد الزائد وميل تافل، تم إدراج ملخص للأداءات على المحفزات الكهربائية القلوي المعتمدة على البلاتين والروثينيوم التي تم الإبلاغ عنها مؤخرًا في الشكل 2د والجدول التكميلي 4، حيث أن Pt-Ru/ يتفوق بشكل كبير على الآخرين. بالإضافة إلى ذلك، تم استخدام قياسات السعة الثنائية الطبقة () المشتقة من سلسلة من الفولتمترية الدورية (CV) بمعدلات مسح مختلفة للتحقيق في الأنشطة الجوهرية لهذه العينات. كما هو موضح في الشكل التكميلي 15، فإن القيمة الأكبر لـ لـ مقارنةً بـ تشير إلى أن ذرات البلاتين الفردية المدخلة تؤدي إلى مساحة سطح كيميائية كهربائية أكبر (ECSA) ومواقع نشطة أكثر تعرضًا . وبناءً عليه، من خلال تطبيع تيار تفاعل تطور الهيدروجين إلى ECSA عند الجهد الزائد لـ يمتلك أعلى نشاط محدد ()، وهو تقريبًا 5 مرات أعلى من ذلك الخاص بـ Pt/C القياسي () (الشكل التكميلي 16). بالإضافة إلى ذلك، بسبب قدرة الامتصاص الفعالة للهيدروجين من البلاتين والروثينيوم، تم استخدام طريقة ترسيب الجهد تحت الهيدروجين () لقياس ECSA لـ Pt/C وPt-Ru/RuO2 وRu/RuO2. كما هو موضح في الشكل التكميلي 17، بعد دمج ذرات البلاتين الفردية، يظهر قيمة ECSA مشابهة لـ Pt/C، والتي هي أكبر بكثير من و. وبالتالي، يظهر أيضًا أعلى نشاط محدد بين المحفزات المدروسة. علاوة على ذلك، فإن قيم ECSA والأنشطة المحددة المحسوبة بواسطة طريقة إزالة CO تتوافق بشكل كبير مع تلك المشتقة من طريقة (الشكل التكميلي 18). تم أيضًا التحقيق في الأنشطة المعادلة وفقًا لطريقة BET لاستبعاد التأثيرات الشكلية على تقييم الأداءات، ويمكن ملاحظتها في الشكل التكميلي 19 أن لا يزال يتفوق على . النشاط الكتلي هو عامل أساسي آخر لتقييم نظام المحفز الكهربائي نظرًا للعلاقة المباشرة مع التكلفة. الشكل التكميلي 20 رسم النشاطات الكتلية (المعادلة إلى كتلة تحميل البلاتين والروثينيوم) لهذه المواد المدروسة عند الجهد الزائد 63 مللي فولت. ونتيجة لذلك، يقدم نشاطًا كتليًا قدره ، وهو تقريبًا 14 و17 مرة أعلى من تلك الخاصة بـ و ()، مما يظهر مزاياه الكبيرة من حيث التكلفة. من المدهش أن النشاط السعري المحسوب (المعادلة إلى سعر البلاتين والروثينيوم) لـ (116.7 A دولار ) يمكن أن يصل أيضًا إلى 21.8 و16.3 مرة أكبر من تلك الخاصة بـ Pt/C التجارية () و (7.17 A دولار )، على التوالي، مما يعرض كفاءته الاقتصادية وإمكاناته المشرقة نحو التطبيقات الصناعية (الشكل 2هـ). علاوة على ذلك، تم إعداد المحفزات الأخرى بكميات مختلفة من مادة البلاتين المضافة عن طريق تقليل وزيادة مدخلات سلف البلاتين، وتم تحديد نسب ذرات البلاتين الخاصة بهم لتكون و بواسطة ICP، على التوالي. تظهر نتائج LSV في الشكل التكميلي 21 أنه بعد إضافة ، تم تعزيز نشاط تفاعل تطور الهيدروجين لـ بشكل واضح مقارنةً بـ بسبب دمج مواقع البلاتين الفردية النشطة للغاية. ومع ذلك، فإن نشاط تفاعل تطور الهيدروجين لـ ينخفض مع زيادة كمية مادة البلاتين المضافة من إلى ، وهو ما قد يُعزى إلى تكوين تجمعات البلاتين المتجمعة، مما يشير إلى أن الكمية المثلى من مادة البلاتين المضافة في نحو تفاعل تطور الهيدروجين القلوي هي . بالإضافة إلى ذلك، تم الحصول على منحنيات نايكويست من خلال إنشاء الدائرة المعادلة في قياسات الطيف الكهروكيميائي (EIS) تليها عمليات التناسب (الشكل التكميلي 22 والجدول التكميلي 5). كما هو موضح في الشكل 2و، مقارنةً بـ Pt/C ()، يمتلك Ru/C و
أقل مقاومة لنقل الشحنة () قدرها ، مما يعني معدل نقل الشحنة المعجل وبالتالي ديناميكيات أسرع لتفاعل تطور الهيدروجين. بعيدًا عن النشاط، فإن المتانة أيضًا حاسمة لتقييم محفز تفاعل تطور الهيدروجين، خاصةً عند استهداف تطبيقات إنتاج الهيدروجين الصناعية. تم إجراء اختبارات الاستقرار على وPt/C التجارية بواسطة قياسات الجهد الزمني عند كثافات تيار مختلفة. كما هو موضح في الشكل 2ز، يُرى أن نشاط Pt-Ru/ عند
يبقى تقريبًا ثابتًا دون تدهور واضح خلال التشغيل المستمر لمدة 100 ساعة، بينما يظهر الجهد الزائد لـ زيادة سريعة قدرها 55 مللي فولت بعد اختبار مستمر لمدة 100 ساعة. من خلال تحديد تركيزات الأيونات المذابة في المحاليل بعد الشكل 2 | أداءات تفاعل تطور الهيدروجين القلوي لـ . أ منحنيات الاستقطاب مع تصحيحات iR و(ب) الجهود الزائدة المقابلة لـ و و و عند كثافات التيار لـ و في 1 م
منحنيات تافل المشتقة من منحنيات الاستقطاب في (أ). د مقارنة الجهود الزائدة ومنحنيات تافل في المحفزات الكهربائية المعتمدة على البلاتين والروثينيوم التي تم الإبلاغ عنها مؤخرًا. هـ مقارنة الأنشطة السعرية على و عند الجهد الزائد 63 مللي فولت. أسعار البلاتين والروثينيوم مأخوذة من المسح الجيولوجي الأمريكي (التاريخ: متوسط سعر المعادن من مجموعة البلاتين في يناير-مارس 2022). و منحنيات نايكويست EIS و منحنيات التناسب لهذه العينات المسجلة عند -50 مللي فولت. ز قياسات الجهد الزمني لـ و عند كثافات التيار لـ و على التوالي، تم استخدام ورق الكربون كدعم للمحفز. كمية الناتجة وكفاءة فاراداي المقابلة على مدى مجموعة من الجهود الزائدة على . ملاحظة: تمثل أشرطة الخطأ الانحراف المعياري لثلاث قياسات مستقلة.
خلال تفاعل طويل الأمد، يُعزى سبب عدم النشاط لـ إلى تسرب أيونات البلاتين الشديد. بالمقابل، تحدث ذوبانات طفيفة لكل من أيونات البلاتين والروثينيوم على ، مما يؤدي إلى تعزيز استقرار تفاعل تطور الهيدروجين (الشكل التكميلي 23). تؤكد التوصيفات بعد التفاعل لـ TEM وHR-TEM أن الشكل العام والبنية المتداخلة لـ محفوظة جيدًا باستثناء تجمعات طفيفة. بالإضافة إلى ذلك، تشير الخرائط العنصرية إلى أن ذرات البلاتين لا تزال موزعة بشكل متجانس على والدراسات البلورية لا تظهر تغييرات واضحة في الهيكل البلوري من نمط XRD بعد اختبار الاستقرار عند (الشكل التكميلي 24). في الوقت نفسه، تشير طيف XPS لـ Pt وRu إلى انخفاض حالات التكافؤ للبلاتين والروثينيوم على ، وهو أمر شائع لمحفزات تفاعل تطور الهيدروجين بعد التشغيل طويل الأمد تحت إمكانيات الاختزال (الشكل التكميلي 25). علاوة على ذلك، تم إجراء اختبارات الاستقرار أيضًا عند كثافة تيار أعلى من . بشكل ملحوظ، مقارنةً بالزيادة الكبيرة في الجهد الزائد على
، يظهر فقط زيادة قدرها 76 مللي فولت في الجهد الزائد على بعد قياسات 100 ساعة. تظهر القياسات أعلاه الاستقرار الممتاز لتفاعل تطور الهيدروجين لـ تحت ظروف قلوية قوية وجهد عالي. تبرز تجارب المسح CV المتكررة أن منحنيات الاستقطاب لـ تبقى تقريبًا متطابقة قبل وبعد 5000 CV، بينما تظهر العينات الأخرى خسائر نشاط واضحة (الشكل التكميلي 26). بالإضافة إلى ذلك، لتجنب تداخل التفاعلات الجانبية وبالتالي التقدير المفرط للنشاط الفعلي لتفاعل تطور الهيدروجين، يتم مراقبة الكميات الناتجة من على عبر نطاق واسع من الجهود الزائدة بواسطة كروماتوغرافيا الغاز ويتم حساب كفاءات فاراداي المقابلة (FE) (الشكل التكميلي 27). كما هو موضح في الشكل 2ح، مع زيادة الجهود المطبقة، تظهر كمية تطور اتجاهًا سريعًا نحو الأعلى، بينما تبقى الكفاءات المرتبطة تقريبًا . وهذا يدل على أن الغالبية العظمى من الطاقة الكهروكيميائية تُستخدم لدفع تفاعل تطور الهيدروجين ولا تحدث تقريبًا أي تفاعلات جانبية على . بشكل عام، يتميز بمزايا النشاط العالي، والفعالية من حيث التكلفة، والاستقرار الملحوظ تجاه تفاعل تطور الهيدروجين القلوي، مما يظهر آفاقه الواعدة ليتم نشره في التطبيقات العملية لإنتاج الهيدروجين.
أفكار ميكانيكية حول تفاعل تطور الهيدروجين القلوي
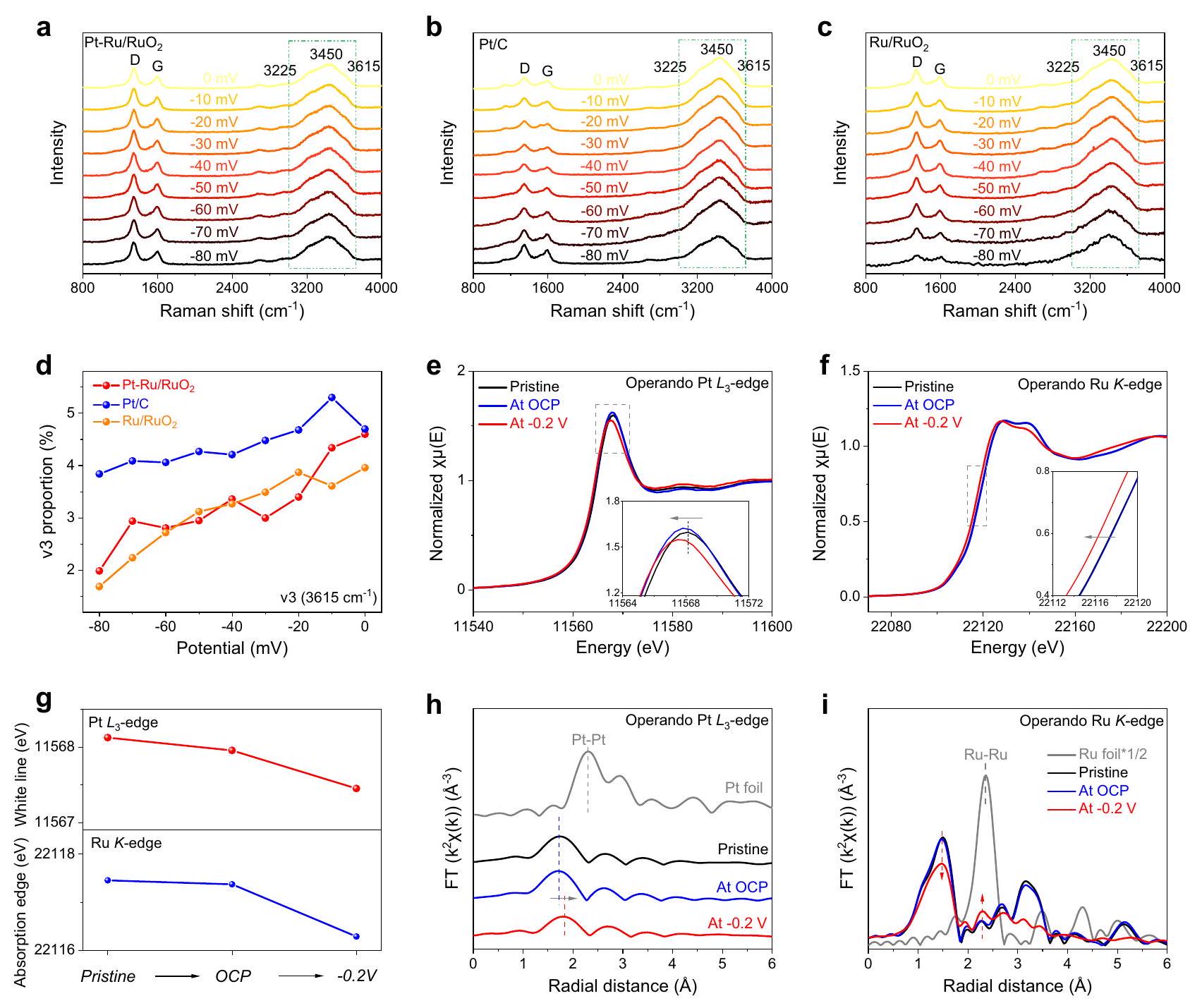
نظرًا للتصميم الجيد تم إثبات أنه محفز عالي الأداء لتفاعل الهيدروجين القلوي، وتم استكشاف مساهمات مواقع النشاط المختلفة. تم إجراء تجارب التسمم في البداية خارج عبر ثيوسيانات البوتاسيوم (KSCN) كمثبط لمنع الاتصال بين المواقع النشطة والوسطاء التفاعليينكما هو موضح في الشكل التوضيحي 28، الانخفاض الملحوظ في النشاط علىبعد الإضافة لـ، مما يوحي بالدور الأساسي لذرة البلاتين الفردية في تنشيط تفاعل تقليل الهيدروجين. بالإضافة إلى ذلك، مقارنةً بـفقدان النشاط الواضح علىتشير إلى أن الروثينيوم يساهم أيضًا في تفاعل الهيدروجين القلوي (الشكل التكميلي 29). للحصول على فهم أعمق لآلية تحسين ذرات البلاتين الفردية، تم إجراء قياسات EIS أثناء تفاعل الهيدروجين. تم تحفيز مخططات نيكويست بواسطة نموذج دائرة مكافئة مزدوجة متوازية، حيث كانت السعة الزائفة (ومقاومة امتصاص الهيدروجين ) يتم اعتمادها لتتبع الهيدروجين الممتص ( سلوك على سطح المحفز. من الجدير بالذكر أن ميل تافل ( مقابل الإمكانيات يعرض قيمة منخفضة بشكل ملحوظ من بالمقارنة مع و ، مما يشير إلى أن تسريع حركية الامتزاز يتم بواسطة ذرات البلاتين الفردية المدخلة. بالإضافة إلى ذلك، فإن دمجويحدد الإمكاناتشحنة الامتزاز، في إشارة إلى تركيز الامتصاص. يُلاحظ أنيمتلك مستوى أعلى بكثيرمن ذلك، مما يدعم أن ذرات البلاتين الفردية تساهم في التحسينالتغطية على السطح. علاوة على ذلك، فإن ميل تافل النسبي الأصغر وارتفاعمنمنكما يشير إلى أن الروبيديوم المخفض يمكن أن يحسن في الوقت نفسه الامتزاز (الشكل التوضيحي 30 والجدول التكميلي 6). هذه النتائج مدعومة أيضًا بـ تظهر النتائج في الشكل التوضيحي 17، بينما يمكن أن تزيد ذرات البلاتين الفردية ورماد الروثينيوم من مساحة امتصاص الهيدروجين، مما يشير إلى امتصاص المزيد من ذرات الهيدروجين ويؤدي إلى كفاءة أعلى.عملية الدمج. من الجدير بالذكر أنه على الرغم من أن البلاتين والروثينيوم هما مواقع نشطة لربط الهيدروجين، إلا أن الأول أكثر أهمية وضرورة نظرًا لتركيزه النسبي الأكبر من الممتص. وزيادة ملحوظة في نشاط HER الناتج عن ذرات البلاتين الفردية. من ناحية أخرى، من المعروف جيدًا أنه كلما كان جهد إزالة CO أقل، كانت قدرة تفكك الماء أفضل. كما هو موضح في الشكل التكميلي 18، فإن ذروة جهد إزالة CO على و أقل بوضوح من ذلك على، مما يؤكد أن القدرة الممتازة على تفكك الماء لا تعود إلى البلاتين المدعوم. الشكل التكميلي 31 يوضح أيضًا تأثيرات النظائر الحركية (KIE) لـ و في و الإلكتروليتات. القيمة الأصغر لـ KIE منيشير إلى سهولة انكسار رابطة HO-H، وهو ما يفيد عملية تفكك الماءمن أجل فهم شامل لقدرة تفكك الماء المعززة علىتم إجراء مطيافية رامان أثناء التشغيل عند إمكانيات مختلفة (الشكل التكميلي 32). كما هو موضح في الشكل 3a-c، يوجد ذروة عريضة واحدة عند انزياح رامان حوالييمكن ملاحظته في الطيف، والذي يمكن أن يُنسب إلى الماء عند واجهة المحفز-الكهارل. الإشارتان الكربونيتان لـوأشرطة G عند حوالي 1400 ومشتقة من لوحة الكربون الزجاجي (الشكل التكميلي 33a). بالإضافة إلى ذلك، من أجل التأكد من أن الليزر رامان كان مركّزًا على سطح المحفز الكهربائي، تم إجراء اختبارات رامان التشغيل تحت إمكانيات مختلفة في محلول 1 م KOH على لوحة الكربون الزجاجي بدون محفز كهربائي. يمكن رؤية أنه لا توجد تقريبًا أي إشارات للمياه الواجهة.يمكن العثور عليه في الشكل التكميلي 33b، والذي يعود إلى خاصية الزجاج الكربوني غير التفاعلية تجاه التفاعل الحفاز. علاوة على ذلك، يمكن تقسيم قمة الماء السطحي هذه إلى ثلاث قمم تقع تقريبًا عند و (v3)، على التوالي (الأشكال التكميلية 34-36). بشكل عام، يتم تخصيص v1 و v2 لجزيئات الماء المنسقة بتنسيق رباعي السطوح وثلاثي السطوح التي تشارك في تفاعل تقليل الهيدروجين، بينما ينتمي إلى المتدليرابطة جزيء الماء السطحي التي لا تتفاعل معمن المهم ملاحظة أننسب الذروة على كلا الجانبين و تظهر اتجاهًا هبوطيًا أسرع بشكل ملحوظ من ذلك لـمع زيادة إمكانيات HER، مما يدل على القدرة الأكثر كفاءة على و لإزالة رابطة الماء. بالإضافة إلى ذلك، فإن معدل الانخفاض في نسبة قمة v 3 على تقريبًا نفس الشيء كما هو على، مما يشير إلى أن ذرات البلاتين الفردية ليس لها أي مساهمة في خطوة تفكك الماء (الشكل 3d والجدول التكميلي 7)كما هو موضح في الأشكال التكميلية 37-39، تم إجراء اختبارات رامان في الظروف التشغيلية على الروثينيوم وبعد ضبط هذه الأطياف بعناية، يمكن ملاحظة بوضوح أنيعرض اتجاهًا هبوطيًا أسرع لـنسبة الذروة أكبر من تلك الموجودة على الروثينيوم في عملية تقليل الهيدروجين، مما يؤكد قدرته الأكبر على كسر رابطة الماء. بشكل ملحوظ،يعرض اتجاهًا هبوطيًا مشابهًا لـخلال المزيد من إمكانيات الكاثود الخاصة بها، مما يثبت أن هو الموقع النشط الرئيسي لتفعيل الماء وخطوة التفكك في بدلاً من الروبل. علاوة على ذلك، تم حساب أن معدل الانخفاض لـنسبة الذروة علىأبطأ قليلاً من ذلك على، والذي قد يُعزى إلى التفاعل الإلكتروني بين الروثينيوم وفي (الشكل التكميلي 40). استنادًا إلى التحقيقات السابقة، يمكن الاستنتاج تجريبيًا أن تكون مسؤولة بشكل أساسي عن خطوة تفكك الماء بينما تسهل كل من ذرات البلاتين الفردية والروثينيوم خطوة دمج الهيدروجين التالية، مما يؤدي بشكل جماعي إلى النشاط الملحوظ لتفاعل الهيدروجين القلوي.، وهذا يتم التحقق منه بشكل أكبر من خلال حسابات DFT.
علاوة على ذلك، تم قبول تقنيات XAS أثناء التشغيل لمراقبة التغيرات الديناميكية في الهياكل الإلكترونية والبيئات الذرية المحلية علىفي عملية HER (الشكل التكميلي 41). خلال القياسات، تم تقليل الجهد العامل من جهد الدائرة المفتوحة (OCP) إلى -0.2 فولت مقابل RHE. كما هو موضح في الشكل 3e، بمجرد فرض جهد -0.2 فولت، تمثل Ptموضع قمة الخط الأبيض الحاد يظهر انزياحًا سلبيًا واضحًا، مما يدل على أن ذرات البلاتين الفردية تم تقليلها إلى حالات أكسدة أقل أثناء. بالإضافة إلى ذلك، تم التحقق من التغيير الفوري في حالة الكيمياوية للروثينيوم بواسطة تقنية XANES عند حافة RuK. توضح الشكل 3f أن حافة الامتصاص تتحول سلبًا إلى موضع طاقة أقل تحت جهد العمل لتفاعل تقليل الهيدروجين -0.2 فولت، مما يشير إلى انخفاض حالة الفالنس للروثينيوم.خلال HER (الشكل 3g). ونتيجة لذلك، توضح حالات الفالنس المتناقصة باستمرار للبلاتين والروثينيوم خلال التفاعلات الحفزية التي تم اكتشافها بواسطة XANES في حالة التشغيل أن كل من ذرات البلاتين الفردية والدعم فيمفعلين ويشاركون معًا في تنشيط القلوي. من أجل تأكيد أن كل من البلاتين والروثينيوم هما مواقع نشطة خلال عملية تقليل الهيدروجين، فإن الجهد الكهربائي المنخفض الآخر لعملية تقليل الهيدروجين هو -0.018 فولت (حيث تكون كثافة التيار ) و -0.040 فولت تم تطبيقها على لإجراء العملية-حافة و Ru K-edge XANES. وبالمثل، فإن موضع قمة الخط الأبيض لكل من Pt وحافة الامتصاص لـ Ru ينتقل باستمرار إلى طاقة أقل مع زيادة الجهد السلبية، مما يشير إلى انخفاض حالاتهم التكافؤية في عملية HER (الشكل التكميلية 42). وبالتالي، فإن نتائج XANES التشغيلية المعتمدة على الجهد تؤكد مرة أخرى أن كلا من عناصر Pt و Ru فيهي مواقع نشطة تجاه تفاعل الهيدروجين القلوي. علاوة على ذلك، كما هو موضح في الشكل 3h، لا يمكن ملاحظة أي رابطة واضحة بين البلاتين والبلاتين في الظروف التشغيلية.-edge EXAFS الطيف أثناء HER، مما يشير إلى أن ذرات البلاتين علىتظل متباعدة ومعزولة طوال التفاعل. ومن الجدير بالذكر أنه يُلاحظ أن العنصر الرئيسيالرابطة تنتقل إلى وضع أكثر إيجابية عند -0.2 فولت، وهو ما قد يكون بسبب استرخاء الهيكل المحلي للبلاتين الناتج عن الامتصاص.في قدرتها على العمل. هذه أيضًا دليل قوي يتماشى مع النتائج التجريبية لتأكيد أن ذرات البلاتين الفردية تتحمل بشكل أساسي مسؤولية تعزيز الـامتصاص الذرات وتعزيز خطوة تافل في تفاعل الهيدروجين القلوي. روديوم في حالة التشغيلتظهر طيفيات EXAFS عند حافة الطاقة أن شدةيقل الرابطة بينما تزداد شدة رابطة الروثينيوم-روثينيوم مع زيادة الجهود التفاعلية المطبقة، مما يظهر تقليل الـيدعم خلال HER (الشكل 3i).
التحقيقات النظرية لـ
لتوضيح آلية النشاط الممتاز علىنحو تفاعل الهيدروجين القلوي، تم إجراء دراسة نظرية استنادًا إلى حسابات نظرية الكثافة (DFT). النماذج الذرية لـ
الشكل 3 | تحقيقات آلية القلوياتعلىطيف رامان أثناء التشغيل لـ (أ)، (ب) و (ج) تحت الجهود المطبقة في محلول 1 م من هيدروكسيد البوتاسيوم.نسبتصل إلى ذروتها فيعلى و . e The Pt -edge XANES و (f) الروثينيوم-edge XANES لحالةمواقع ذروة الخط الأبيض للبلاتين-edge XANES ومواقع حواف الامتصاص لـ Ru-edge XANES تحت الجهود المطبقة.النقطة-edge EXAFS و (i) الروثينيوم-edge EXAFS تحت الحالة النقية، OCP وظروف تشغيل HER عند -0.2 فولت.مقاسة في 1 م كوه تحت حالة نقية، جهد مفتوح وعمليات تقليل الهيدروجين
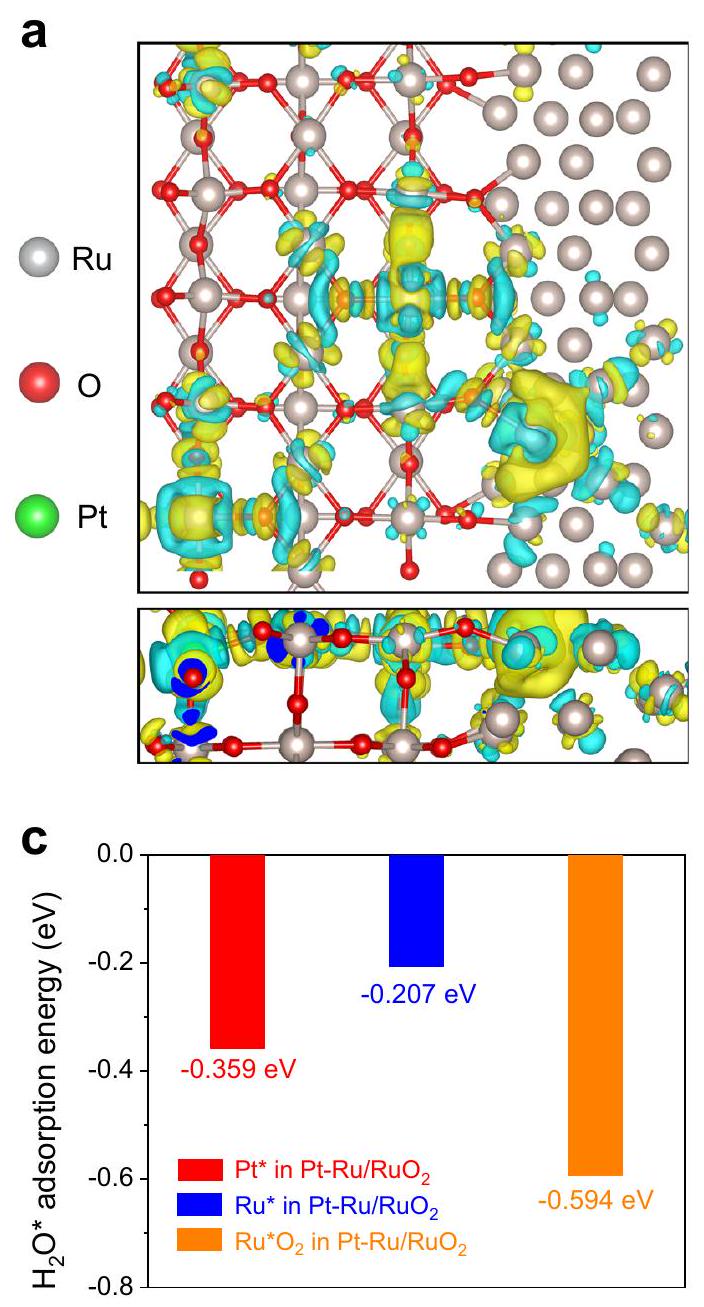
و تم بناؤها وتم تحسينها بشكل طاقي (الشكل التكميلي 43). كما هو موضح في الشكل 4a، فإن فرق كثافة الشحنةيظهر إعادة توزيع شحنات ملحوظة على طول الروابط الكيميائية بسبب الاختلاف في الكهربية، حيث تميل الإلكترونات إلى الانتقال من ذرات الروثينيوم المحيطة إلى ذرات البلاتين المركزية.. هذه النتيجة تؤكد وجود تفاعل إلكتروني فعال بين ذرات البلاتين المعزولة و. كما وُجد انتقال واضح للإلكترون من الروثينيوم إلىعلىالهياكل غير المتجانسة، مما يظهر أن الروثينيوم يمكن أن ينظم التركيب الإلكتروني لـويؤدي ذلك إلى تعزيز القدرة التحفيزية (الشكل التكميلي 44). بالإضافة إلى ذلك، يمكن تأكيد الهيكل الإلكتروني المحسن الناتج عن ذرات البلاتين الفردية من خلال كثافة الحالات المتوقعة (PDOS)، التي تظهر احتلالًا أعلى بالقرب من مستوى فيرمي ( ) على مقارنة مع ، مما يشير إلى تسريع نقل الإلكترونات وزيادة الموصلية الناتجة عن البلاتينالمساهمة المدارية. علاوة على ذلك، فإن -تُحسب مراكز النطاقات من PDOS. على وجه التحديد، المتوسط-قيم مركز النطاق لـ و هي -1.285 و -1.341 إلكترون فولت، على التوالي. وفقًا لـ نظرية مركز النطاق، يتم إضعاف ارتباط الوسط التفاعلي، وتكون حالة مضادة للارتباط أكثر مشغول بالتحول إلى السرعة المنخفضة-مركز نطاق التردد. وبالتالي، فإن المواقع النشطة علىتتعرض بسرعة، وتزداد عملية HER (الشكل 4 ب). بعد ذلك، آليات مواقع النشاط المختلفة في تمت دراستها من خلال استخدام نظرية دالة الكثافة لاستكشاف خصائصها الطاقية. نظرًا لأن خطوة فولمر هي الشرط المسبق والحاجز الرئيسي لتحفيز تفاعل الهيدروجين في الوسط القلوي، فمن الضروري قياس طاقة الامتزاز لـعلى كل موقع نشط، وهو عامل رئيسي لتقييم القدرة على تسريع عملية تفكك الماءكما هو موضح في الشكل 4c، يتضح بجلاء أن موقع الروثينيوممع طاقة امتصاص أعلى (-0.594 إلكترون فولت) هو أكثر ملاءمة من الناحية الديناميكية الحرارية لـالامتزاز مقارنة بمواقع البلاتين (-0.359 eV) ومواقع الروثينيوم (-0.207 eV)، مما يشير إلى أنمن الأكثر احتمالاً أن يتم امتصاصه في. الـطاقة الانفصال الحرة في الشكل التكميلية 45 تشير بشكل أكبر إلى أن ذرات البلاتين الفردية يصعب كسرها.الجزيء بسبب أكبر حاجز طاقة. بالإضافة إلى ذلك، على الرغم من أن الروثينيوم ولديها حواجز طاقة مشابهة، مع الأخذ في الاعتبار القدرة الأقوى على الامتصاصعلى، فإن تفكك الماء يكون أكثر كفاءة وأسهل في الاكتمال بواسطةبدلاً من رو. من ناحية أخرى،طاقة الامتزاز الحرة على جميع المواقع فيتم حسابها أيضًا لفحص قدراتهم على
الشكل 4 | حسابات DFT لـ. عرض الجزء العلوي والجانبي لاختلاف كثافة الشحنة على نموذج الهيكل غير المتجانس، تمثل المناطق الزرقاء والصفراء منطقة تراكم الإلكترونات ومنطقة نقص الإلكترونات،
تفعيل ما يليخطوة الدمج. كما هو موضح في الشكل 4d، مقارنةً بالمواقع النشطة الأخرى، توفر مواقع البلاتين القيمة المثلى الأكثر بواقع 0.115 إلكترون فولت، مما يدل على أن ذرات البلاتين المفردة المدمجة مفيدة لتعزيزخطوة الدمج. كما يُلاحظ أن طاقة الامتزاز الحرة على الروديوم أقرب إلى الصفر من، مما يشير إلى أن Ru يساهم أيضًا في الامتزاز و الجيل. لذلك، فإن النتائج التي كشفت عنها حسابات DFT تتماشى بشكل كبير مع النتائج التجريبية، والتي تظهر بشكل مشترك أن ذرات البلاتين الفردية، والروثينيوم وتعزيز أداء تفاعل الهيدروجين القلوي بشكل تعاوني.
أداء AEMWE
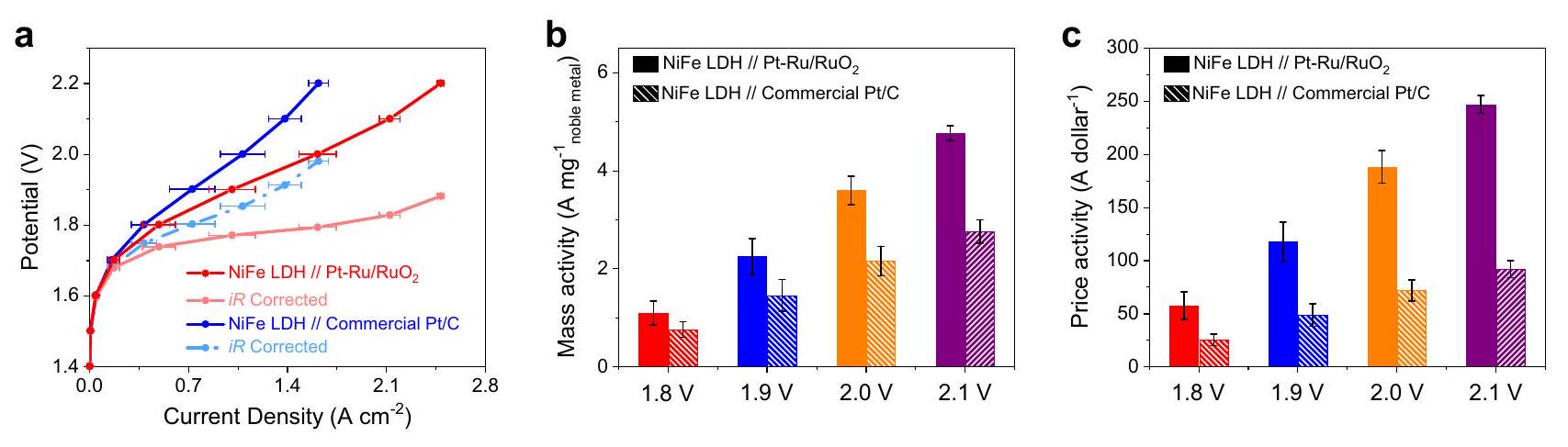
مدفوعين بالنشاط الممتاز لتفاعل تقليل الهيدروجين (HER)، قمنا بمزيد من بناء تجميع الإلكترود الغشائي (MEA) في التحليل الكهربائي للماء القلوي (AEMWE) من خلال استخدامكقطب سالب لتقييم أدائه في عملية تحليل الماء العملي. تحميل الكتلة لـتم التحكم فيه كـ“، في حين أن معيار Pt/C ( ) مع تم اختباره أيضًا في AEMWE للمقارنة. في الاختبارات الأولية، تم استخدام النيكل مباشرة كأنود (الشكل التوضيحي التكميلي 46). كما هو موضح في الشكل 5a،تظهر كثافات تيار أعلى من Pt/C التجارية عند نفس إمكانيات الخلية. يمكن تعزيز الأداء التحفيزي بشكل أكبر من خلال اعتماد هيدروكسيدات النيكل والحديد المزدوجة الطبقات (LDH) غير المكلفة في جانب الأنود. ونتيجة لذلك، تظهر منحنيات الاستقطاب أن NiFe LDH + يتطلب فقط 1.90 فولت و 1.77 فولت للوصول إلى كثافة التيارقبل وبعد تصحيح iR، على التوالي، وهو ما هو أقل بكثير من تلك الموجودة على NiFe LDH + Pt/C التجاري (1.98 فولت و 1.84 فولت). بالإضافة إلى ذلك، كما هو موضح في الشكل 5ب، فإن الأنشطة الكتلية لـوصل إلى 3.60 وعند 2.0 فولت و 2.1 فولت، على التوالي، وهي أكثر من مرتبتين من حيث الحجم أعلى من تلك الخاصة بالمنتجات التجارية (لا تصحيح iR). بناءً على الأداء الملحوظ، نعتبر أن تكلفة الروثينيوم أقل بكثير من البلاتين، وبالتاليتتمتع بكفاءة اقتصادية متفوقة مقارنةً بـ Pt/C التجاري. ومن المثير للإعجاب، بعد تعديل كثافة التيار وفقًا لسعر المعدن الثمين، فإن أنشطة الأسعار لـهي 188.2 و 247.1 دولارعند 2.0 فولت و 2.1 فولت، على التوالي، تصل إلى ما يقرب من 3 مرات أعلى من تلك الخاصة بـ Pt/C التجارية (72.0 و 92.0 دولار أمريكي) ) (الشكل 5ج). مزايا النشاط التحفيزي وتوفير التكاليف على توضيح جدواها وإمكان تطبيقها كعامل مساعد عملي لإنتاج الهيدروجين في الوسط القلوي.
نقاش
في الختام، محفز كهربائي نشط ومستقر للغاية من Pt-Ru/تم تصنيع HER القلوي بشكل مفصل في هذا العمل. تؤكد التجارب الشاملة والتوصيفات العملية، بالإضافة إلى حسابات DFT، أن جميع ذرات البلاتين المعزولة، والروثينيوم وتلعب أدوارًا حيوية خلال التفاعلات الحفزية. على وجه التحديد،
الشكل 5 | أداء AEMWE لـ .أ منحنيات الاستقطاب AEMWE مع وبدون تصحيح iR على NiFe LDH // Pt-Ru/RuO2 و NiFe LDH // Pt/C التجارية. المقارنات بين (ب) أنشطة الكتلة و (ج) أنشطة السعر بين
NiFe LDH // Pt-Ru/RuOو NiFe LDH // Pt/C التجاري عند إمكانيات خلوية مختلفة (دون تصحيح iR). ملاحظة: تمثل أشرطة الخطأ الانحراف المعياري لثلاث قياسات مستقلة. خطوة تفكك الماء البطيئة تتسارع بشكل أساسي بواسطةبينما يتم تسهيل خطوة دمج الهيدروجين التالية بواسطة ذرات البلاتين الفردية والروديوم، مما يساهم بشكل جماعي في تحسين تفاعل تقليل الهيدروجين في 1 م من هيدروكسيد البوتاسيوم. ونتيجة لذلك،تظهر جهدًا زائدًا ملحوظًا قدره 18 مللي فولت و63 مللي فولت عند كثافات التيار من و ، على التوالي، متفوقة على معظم المحفزات التي تم الإبلاغ عنها مؤخرًا. خاصة، النشاط القائم على التكلفة لـيمكن أن تصل إلى أكثر من 16 مرة أعلى من تلك الخاصة بالتجارة و ، مما يظهر كفاءتها الاقتصادية الجيدة. بالإضافة إلى ذلك، فإن استقرار HER لـيمكن أن يتم تمديده لأكثر من 100 ساعة مع فقدان نشاط ضئيل. علاوة على ذلك، فإن دمجو NiFe LDH في AEMWE يظهر نشاطًا سعريًا متفوقًا قدره 247.1 دولارعند 2.1 فولت، تصل إلى ما يقرب من 3 مرات أعلى من النظير القائم على Pt/C. تقدم هذه النتيجة استراتيجية محتملة لهندسة المركبات لتغيير التوازن بين النشاط العالي والكفاءة الاقتصادية عند تصنيع المحفزات القائمة على المعادن النبيلة نحو التطبيق العملي.الإنتاج.
طرق
المواد
كلوريد الروديوم الثلاثيكلوريد البلاتين الرباعي (صلب، 99.5%)صلب،أكسيد الروثينيومصلب،هيدروكسيد البوتاسيوم (KOH، صلب، )، وتجاري محفز ) تم شراؤها من إينوكيما. نترات الصوديوم ( صلب، ) وإيزوبروبانول ( سائلتم شراء ( ) من شركة سينوفارم للمواد الكيميائية المحدودة (شنغهاي، الصين). تم الحصول على محفز Pt/C التجاري (20 وزن%) من شركة جونسون ماثي (JM).
تحضير المحفز
في تخليق نموذجي،تم تسخينه في البداية عند لمدة 30 دقيقة في فرن تحت جو هوائي، ثم تم صب المسحوق في المنصهروتم الاحتفاظ بـلمدة 10 دقائق. بعد ذلك، تم غسل الصلب المبرد بالماء 3 مرات لإزالةوجفف overnight للحصول على المركب المُصنّع. ثم تم تصنيع 50 ملغتم تفريقه في 18 مل من الماء وتم إضافة 2 مل من الماء بالتنقيط إلى المحلول أعلاه تحت التحريك المستمر. بعد تكوين خليط متجانس، تم تسخينه إلىواستمر في التحريك لمدة 16 ساعة. أخيرًا، تم امتصاص أنواع البلاتينتم غسلها بالماء ثلاث مرات وجمعها بواسطة الطرد المركزي، تلا ذلك التكلس في الأرجون عندلمدة ساعتين بمعدل تسخين قدرهللحصول على. الـتم تحضيره بطريقة مشابهة لـباستثناء أن الـلم يتم إضافة السلف.
توصيف
تم تحديد صور TEM و HRTEM بواسطة جهاز JEOL 2100F عند جهد عمل قدره 200 كيلوفولت. صور AC HAADF-STEM وتم جمع الخرائط العنصرية المقابلة على مجهر ثيرمو ساينتيفيك ثيميس Z الذي يعمل بجهد 200 كف. تم جمع طيف SEM-EDS على جهاز زيس سوبرا 55 عند جهد تسريع قدره 5 كف. كانت الكتلة النسبية للبلاتين فيتم تحديد تركيزات الأيونات المذابة بعد اختبارات الاستقرار بواسطة ICP-OES على جهاز Agilent ICP-OES 730.
تم جمع بيانات حيود الأشعة السينية (XRD) على جهاز حيود مسحوق من نوع Bruker D8 Advance (يعمل عندمجهز بـ مصدر ( ) ومزود بنافذة من البريليوم في درجة حرارة الغرفة. تم إجراء تحسينات ريتفيلد لبيانات حيود الأشعة السينية باستخدام برنامج فول-بروف، وشملت المعلمات المحسنة معلمات الخلفية، أخطاء انزلاق الخط (انزلاق الصفري)، معاملات كاجليوتي (U و V و W)، عامل المقياس، معلمات الشبكة، موضع الذرات، معدل شغل الذرات ومعلمات إزاحة الذرات غير المتجانسة.
تم إجراء تحليل XPS على جهاز Thermo Scientific Escalab 250Xi مع مصدر أشعة Al-Kالمصدر. XAS للروثينيوم-حافة و Pt-edge تم إجراؤها في خط الشعاع 44A من مركز أبحاث الإشعاع السنكروتروني الوطني (NSRRC) في تايوان. تم إجراء تجارب XAS التشغيلية لـ Ruحافة وتم إجراء تجارب -edge في خط الشعاع 44A من NSRRC باستخدام أدوات XAS مصممة خصيصًا. تم جمع جميع تجارب XAS تحت درجة حرارة الغرفة وتحليلها باستخدام البرنامج القياسي Demeter.
تم جمع طيف رامان أثناء التشغيل على جهاز مطياف رامان HORIBA XploRA PLUS مع خلية كيميائية كهربائية، حيث تم إسقاط المحفزات بدون فحم أسود مباشرة على لوحة الكربون الزجاجي كقطب عمل، وتم استخدام قطب الكالوميل وقضيب الكربون كقطب مرجعي وقطب مضاد، على التوالي. كان يعمل من 800 إلىو0 إلى -80 مللي فولت مقابل RHE في محلول 1 م KOH لتقييم قدرة المحفزات على تفكك الماء.
القياس الكهروكيميائي
تم إجراء تقييم أداء HER على جهاز العمل الكهربائي CHI 760E مع تكوين قطب دوار (RDE) (أدوات البحث باين). تم استخدام قطب كربوني زجاجي بمساحة سطحية منتم اختيار إلكترود الكالوميل وقضيب الجرافيت كإلكترود العمل والمرجع والإلكترود المضاد، على التوالي. قبل الاختبار، تم أخذ عينة بوزن 5 ملغ ( أو ) و 5 ملغ من الكربون الأسود (Vulcan XC-72R) تم خلطها في 1.9 مل من محلول الإيزوبروبانول وتمت الموجات فوق الصوتية لمدة ساعة، تلاها إضافة 0.1 مل من محلول نافيون ( ، ألدرش) في الخليط أعلاه والتسنيق لمدة 5 دقائق لإنتاج حبر محفز متجانس. ثم،تم إسقاط حبر المحفز على القطب العامل وتجفيفه تحت ضوء الأشعة تحت الحمراء. تم التحكم في تحميلات الكتلة للمعادن لهذه العينات المحضرة عند. للمقارنة، فإن تحميل الكتلة من البلاتين والروثينيوم لبلاتين/كربون التجاري (20 وزن % ) والتجاري تم التحكم أيضًا في على القطب العامل أيضًا. تم معايرة جميع إمكانيات HER بالنسبة لمقياس RHE (الشكل التكميلي 12). تم إجراء المعايرة باستخدام سلك من البلاتين كقطب عامل، وقطب كولوميل كقطب مرجعي وقضيب من الجرافيت كقطب مضاد في-محلول إلكتروليتي مشبع (1 م كوه). تم إجراء مسح CV بمعدل مسح قدره 1 مللي فولت في الثانية وكان الجهد المتوسط الذي عبرت عنده التيارات الصفر هو قيمة المعايرة. تم إجراء LSV مع تصحيح iR بمعدل مسح قدرهتحت درجة حرارة الغرفة لتقييم نشاط تفاعل الهيدروجين. تم تعويض تصحيح iR تلقائيًا بواسطة المحطات الكهروكيميائية وتم التحكم في قيمة تصحيح iR كـلجميع منحنيات الاستقطاب. تم تحديد منحدرات تافل من خلال رسم الجهد الزائد مقابل لوغاريتم كثافة التيار.تم استكشاف منحدرات تافل على و ضمن نطاقات الجهد الزائد لـ، “ و على التوالي. تم تقييم الاستقرار باستخدام ورق الكربون المحمّل بالمواد الحفازة ) مع تحميلات الكتلة لإجراء الكرونوپوتنشيومترية عند كثافات تيار HER الثابتة لـ و تم إجراء اختبارات الاستقرار الدوري من خلال تكرار 5000 دورة CV على العينات المدروسة. تم قياس EIS على محطة Bio-Logic SP-200 في نطاق التردد من 0.01 إلى 100 كيلوهرتز عند -50 مللي فولت مقابل RHE؛ وتم قياس EIS في حالة التشغيل على جميع العينات في نطاق التردد من 0.01 إلى 100 كيلوهرتز عند إمكانيات مطبقة مختلفة. تم إنشاء الدوائر المعادلة المقابلة وتم الحصول على قيم المقاومة الملائمة من خلال تحليل برنامج EC-lab.
حساب المساحة السطحية الكهروكيميائية
تم حساب ECSAs من خلال تقييم سعة الطبقة الثنائية الكهربائية ) على السطح الحفاز للمحفزات المدروسة استنادًا إلى المعادلة (1):
أينتم قياسه من منحنيات الجهد المتناوب المعتمدة على معدل المسح في المنطقة غير الفارادائية من 0.5 إلى 0.6 فولت مقابل RHE في 1 م كOH بمعدلات مسح و يمثل المساحة السطحية الحقيقية للقطب المعدني الأملس، والتي كانت عمومًا تساوي المساحة الهندسية لقطب الكربون الزجاجي. ). السعة النوعية ( ) لسطح مستوٍ كان يُعتبر عمومًا في نطاق . في هذا العمل، قيمة لـتم قبولها. كما تم قياس ECSAs بواسطةطريقة، حيث تم فحص جميع المحفزات بواسطة CV فيمحلول KOH بتركيز 1 م مشبع بمعدل مسحتم إجراء اختبارات إزالة أول أكسيد الكربون عن طريق نفخ غاز أول أكسيد الكربون مباشرة على القطب المغلف بالكاتاليسور لمدة 20 دقيقة في محلول 1 م كOH، تلاها وضع القطب في إلكتروليت كOH جديد ومسح الجهد الكهربائي لإزالة أول أكسيد الكربون الممتص. تم التحكم في معدل المسح كـ.
حساب كفاءة فاراداي لتفاعل الهيدروجين
تم إسقاطه على ورق الكربون لإجراء تفاعل الهيدروجين في خلية محكمة الإغلاق. الجهود المطبقة على العينة لتوليدكانوا و -0.3 فولت مقابل RHE، على التوالي. كميات الناتج من تم تسجيلها بواسطة جهاز Agilent 7890 B المزود بـتم استخدام عمود غربال جزيئي وكاشف موصلية حرارية، وتم استخدام الأرجون كغاز حامل.تم تحديد المبلغ من خلال رسم البيانات الناتجةمنطقة الذروة في منحنيات التوافق القياسية المعتمدة. إن كفاءة استرداد الهيدروجين لـكان محسوب بناءً على المعادلة (2):
أين هو عدد الإلكترونات المنقولة لتوليد هو الشحنة الكلية، هو مقدار الناتج (بالمولات) و هو الثابت فاراداي.
التفاصيل الحاسوبية
تم إجراء جميع تحسينات الهيكل وحسابات طاقة الامتزاز استنادًا إلى نظرية الكثافة الوظيفية (DFT) كما تم تنفيذها في حزمة المحاكاة الأولية في فيينا (VASP).تم تنفيذ طريقة الموجة المعززة بواسطة البروجيكتور (PAW) لحساب التفاعلات بين النوى الأيونية والإلكترونات التكافؤية.تم استخدام نهج بيردو-بورك-إرنزرهوف (PBE) من التقريب العام المعمم المدفوع بالدوران (GGA) لوصف طاقة التبادل-الارتباط.تم إجراء جميع حسابات DFT لـنموذج الهيكل غير المتجانس. الهياكل الذرية لـ ( )- رو و ( 110 )- تم توليدها من قاعدة بيانات مشروع المواد (MP). بالإضافة إلى ذلك، 3 ذرات من الروديوم لـتم استبدال نموذج الهيكل غير المتجانس عشوائيًا بذرات البلاتين ذات أقل طاقة لتقريب المحاكاةالمحتوى الذري وبناءنموذج الهيكل غير المتجانس. تم استخدام طاقة الحركة الموجية القطعية البالغة 520 إلكترون فولت في الحسابات مع-حجم شبكة النقاط. تم استخدام طريقة التدرج المترافق في الاسترخاء الإلكتروني، وتم تعيين جميع معلمات تقارب DFT بما يتماشى مع مشروع المواد (MP).طاقة الامتزاز ) يُعرَّف على أنه
أينهي طاقة اللوح، هو طاقة الامتصاص الحر، و هو إجمالي طاقة نظام الامتصاص-اللوح في حالة التوازن.
الطاقة الحرة ( ) من الممتص و يُعرَف بأنه
أين هو طاقة الامتزاز للوحة، هو فرق الطاقة الصفرية بين الحالة الممتصة ومرحلة الغاز،هو مساهمة الإنتروبيا في التفاعل عند.
تحضير MEA لقياسات الخلايا الفردية AEMWE
(أيميونالغشاء، AEMIONTM أيونومر) تم إعداد مجموعة غشاء الإلكترود (MEA) بواسطة تقنية الطلاء بالرش اليدوي للأغشية المطلية بالكاتاليس (CCM) كما وصفها كلينغنهوف وآخرون.. لتغطية الغشاء بالرذاذ (AEMION ) تم تثبيته على لوحة تسخين تجارية مزودة بتحكم في درجة الحرارة (الكربون وخلايا الوقود) وضاغط (ويلش). تم ضبط درجة حرارة الطاولة على . علاوة على ذلك، قناع ( ) تم استخدامه لضمان تغطية الأنود والكاثود. كانت الحبر المستخدم للأنود والكاثود في كلا الحالتين تتكون من 50 ملغ من مسحوق المحفز، ماء فائق النقاء-PrOH و460 ملغ محلول أيونومر (AEMIONمحلول في الإيثانول و. في حالة الأنود، كمية إجمالية من تم رش الحبر على الغشاء، مستهدفًا تحميل NiFe-LDH بـفي الكاثود، حبر المحفز بكمية التحميل منتم رشها على الغشاء. بعد الرش، تم تجفيف CCM المُعد مسبقًا لمدة عشر دقائق عندتبع ذلك التبريد إلى درجة حرارة الغرفة والوزن. لتحقيق تحميل مرضٍ، تم إجراء خطوة الرش والوزن عدة مرات بكميات قليلة من حبر المحفز. كان الأنود دائمًا يتم رشه. تمت الطلاء أولاً. لضبط تحميل مناسب وتوفير مقارنات مناسبة بين المحفز المرجعي التجاري Pt/C والمحفيز المصنع، تم إجراء القياسات بدون NiFe-LDH كمحفز أنودي لتجنب نظام أكثر تعقيدًا. بعد الضبط المناسب لتحميل محفز الكاثود، تم إجراء القياسات مع محفز NiFe-LDH إضافي. تم إجراء القياسات باستخدام محطة اختبار Greenlight معإعداد خلية واحدة AEMWE. يتكون الإعداد من MEA المعدة، بالإضافة إلى أن جانب الأكسجين والهيدروجين مزودان بحشوات PTFE، وطبقات نقل مسامية (PTL، شعر النيكل عند الأنود وقماش الكربون عند الكاثود)، وألواح نهائية مع منافذ إلكتروليت، وألواح ثنائية الجهد (حقول تدفق متوازية، لوحة كربونية لجانب الهيدروجين ولوحة تيتانيوم لجانب الأكسجين). تم معالجة CCMs مسبقًا في 2 M KOH لمدة ساعتين قبل القياس. لتحقيق أفضل أداء، تم تبادل الأنيونات للCCMs المطلية بالرش في 2 M KOH ثلاث مرات، كل منها لمدة 20 دقيقة. لإزالة الفائض المتبقي من OH-، تم غسل الغشاء المتبادل للأنيونات بالماء المقطر ثلاث مرات لمدة 20 دقيقة. قبل تقييم النشاط من خلال تطبيق منحنيات الاستقطاب، تم تنشيط CCM عن طريق دورة جهد (50 مللي فولت بين 1.1 فولت و1.7 فولت جهد الخلية) وخطوة تثبيت جهد عند 1.83 فولت لمدة ساعة واحدة. تم إجراء قياسات الخلية الواحدة عندفي محلول 0.1 م من هيدروكسيد البوتاسيوم.
توفر البيانات
جميع البيانات ذات الصلة متاحة من المؤلفين المعنيين عند الطلب. يتم توفير بيانات المصدر مع هذه الورقة.
References
Suen, N.-T. et al. Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives. Chem. Soc. Rev. 46, 337-365 (2017).
Lei, C. et al. Efficient alkaline hydrogen evolution on atomically dispersed species anchored porous carbon with embedded Ni nanoparticles by accelerating water dissociation kinetics. Energy Environ. Sci. 12, 149-156 (2019).
Zeng, K. & Zhang, D. Recent progress in alkaline water electrolysis for hydrogen production and applications. Prog. Energy Combust. Sci. 36, 307-326 (2010).
Abbasi, T. & Abbasi, S. A. ‘Renewable’ hydrogen: prospects and challenges. Renew. Sust. Energy Rev. 15, 3034-3040 (2011).
Zheng, Y., Jiao, Y., Vasileff, A. & Qiao, S.-Z. The hydrogen evolution reaction in alkaline solution: from theory, single crystal models, to practical electrocatalysts. Angew. Chem. Int. Ed. 57, 7568-7579 (2018).
Cheng, F. et al. Accelerated water activation and stabilization metalorganic framework via constructing triangular active-regions for ampere-level current density hydrogen production. Nat. Commun. 13, 6486 (2022).
Xie, W.-F. & Shao, M.-F. Alkaline water electrolysis for efficient hydrogen production. J. Electrochem. 28, 22014008 (2022).
Durst, J. et al. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 7, 2255-2260 (2014).
Zheng, J., Sheng, W., Zhuang, Z., Xu, B. & Yan, Y. Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy. Sci. Adv. 2, e1501602 (2016).
Li, A. et al. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 5, 109-118 (2022).
Zhang, L. et al. Facet dependent oxygen evolution activity of spinel cobalt oxides. J. Electrochem. 28, 2108481 (2022).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212-222 (2021).
Subbaraman, R. et al. Enhancing hydrogen evolution activity in water splitting by tailoring -Ni( OH-Pt interfaces. Science 334, 1256-1260 (2011).
Wang, K. et al. Dense platinum / nickel oxide heterointerfaces with abundant oxygen vacancies enable ampere-level current density ultrastable hydrogen evolution in alkaline. Adv. Funct. Mater. 33, 2211273 (2023).
He, X. et al. Electronic modulation with Pt incorporated NiFe layered double hydroxide for ultrastable overall water splitting at 1000 mA . Appl. Catal. B: Environ. 331, 122683 (2023).
Zhu, Y. et al. Unusual synergistic effect in layered RuddlesdenPopper oxide enables ultrafast hydrogen evolution. Nat. Commun. 10, 149 (2019).
He, Y. et al. Amorphizing noble metal chalcogenide catalysts at the single-layer limit towards hydrogen production. Nat. Catal. 5, 212-221 (2022).
Jiang, K. et al. Single platinum atoms embedded in nanoporous cobalt selenide as electrocatalyst for accelerating hydrogen evolution reaction. Nat. Commun. 10, 1743 (2019).
Cheng, N. et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 7, 13638 (2016).
Jiang, K. et al. Rational strain engineering of single-atom ruthenium on nanoporous for highly efficient hydrogen evolution. Nat. Commun. 12, 1687 (2021).
Zhang, J. et al. Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction. Nat. Catal. 1, 985-992 (2018).
Zhang, Z. et al. Single-atom catalyst for high-performance methanol oxidation. Nat. Commun. 12, 5235 (2021).
Vukmirovic, M. B., Teeluck, K. M. & Adzic, R. R. Single platinum atoms electrocatalysts: oxygen reduction and hydrogen oxidation reactions. Croat. Chem. Acta 90, 225-230 (2017).
Zhu, S. et al. The role of ruthenium in improving the kinetics of hydrogen oxidation and evolution reactions of platinum. Nat. Catal. 4, 711-718 (2021).
Li, G. et al. The synergistic effect of Hf-O-Ru bonds and oxygen vacancies in for enhanced hydrogen evolution. Nat. Commun. 13, 1270 (2022).
Li, W. et al. Carbon-quantum-dots-loaded ruthenium nanoparticles as an efficient electrocatalyst for hydrogen production in alkaline media. Adv. Mater. 30, 1800676 (2018).
Liu, Y. et al. A general route to prepare low-ruthenium-content bimetallic electrocatalysts for pH -universal hydrogen evolution reaction by using carbon quantum dots. Angew. Chem. Int. Ed. 59, 1718-1726 (2020).
Yan, S. et al. Partially oxidized ruthenium aerogel as highly active bifunctional electrocatalyst for overall water splitting in both alkaline and acidic media. Appl. Catal. B: Environ. 307, 121199 (2022).
Wang, Z. et al. PtSe Pt heterointerface with reduced coordination for boosted hydrogen evolution reaction. Angew. Chem. Int. Ed. 60, 23388-23393 (2021).
Xu, Q. et al. Atomic heterointerface engineering overcomes the activity limitation of electrocatalysts and promises highly-efficient alkaline water splitting. Energy Environ. Sci. 14, 5228 (2021).
Schilling, A. C. et al. Accelerated reduction by single Pt atoms at the metal-oxide interface. ACS Catal. 10, 4215-4226 (2020).
Poerwoprajitno, A. R. et al. A single-Pt-atom-on-Ru-nanoparticle electrocatalyst for CO-resilient methanol oxidation. Nat. Catal. 5, 231-237 (2022).
Lai, W.-H. et al. Activating inert surface Pt single atoms via subsurface doping for oxygen reduction reaction. Nano Lett. 21, 7970-7978 (2021).
Huang, K. et al. Ru/Se-RuO2 composites via controlled selenization strategy for enhanced acidic oxygen evolution. Adv. Funct. Mater. 33, 2211102 (2023).
Zhu, Y. et al. Iridium single atoms incorporated in efficiently catalyze the oxygen evolution in acidic conditions. Nat. Commun. 13, 7754 (2022).
Liu, W. et al. Highly-efficient RuNi single-atom alloy catalysts toward chemoselective hydrogenation of nitroarenes. Nat. Commun. 13, 3188 (2022).
Luo, S. et al. A tensile-strained Pt-Rh single-atom alloy remarkably boosts ethanol oxidation. Adv. Mater. 33, 2008508 (2021).
Wang, H. et al. PdBi single-atom alloy aerogels for efficient ethanol oxidation. Adv. Funct. Mater. 31, 2103465 (2021).
Dai, J. et al. Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis. Nat. Commun. 13, 1189 (2022).
Tan, W. et al. Fine-tuned local coordination environment of Pt single atoms on ceria controls catalytic reactivity. Nat. Commun. 13, 7070 (2022).
Zeng, L. et al. Anti-dissolution Pt single sites with coordination for efficient alkaline water splitting electrolyzer. Nat. Commun. 13, 3822 (2022).
Chen, Y. et al. Plasma-assisted highly dispersed Pt single atoms on Ru nanoclusters electrocatalyst for pH-universal hydrogen evolution. Chem. Eng. J. 448, 137611 (2022).
Zhang, J. et al. OH spectator at IrMo intermetallic narrowing activity gap between alkaline and acidic hydrogen evolution reaction. Nat. Commun. 13, 5497 (2022).
Wan, C. et al. Amorphous nickel hydroxide shell tailors local chemical environment on platinum surface for alkaline hydrogen evolution reaction. Nat. Mater. 22, 1022-1029 (2023).
Zhu, Y. et al. Single-atom In-doped subnanometer Pt nanowires for simultaneous hydrogen generation and biomass upgrading. Adv. Funct. Mater. 30, 2004310 (2020).
Qi, K. et al. Single-atom cobalt array bound to distorted with ensemble effect for hydrogen evolution catalysis. Nat. Commun. 10, 5231 (2019).
Lai, W. et al. Boosting the interfacial hydrogen migration for efficient alkaline hydrogen evolution on Pt-based nanowires. J. Mater. Chem. A 10, 16834 (2022).
Li, J. et al. Ethylene-glycol ligand environment facilitates highly efficient hydrogen evolution of through proton concentration and hydrogen spillover. Energy Environ. Sci. 12, 2298 (2019).
Fu, H. Q. et al. Hydrogen spillover-bridged Volmer/Tafel processes enabling ampere-level current density alkaline hydrogen evolution reaction under low overpotential. J. Am. Chem. Soc. 144, 6028-6039 (2022).
Dai, Q. et al. Accelerated water dissociation kinetics by electronenriched cobalt sites for efficient alkaline hydrogen evolution. Adv. Funct. Mater. 32, 2109556 (2022).
Wang, K. et al. Kinetically accelerating elementary steps via bridged Ru-H state for the hydrogen-evolution in anion-exchange membrane electrolyzer. Adv. Funct. Mater. 33, 2212321 (2023).
Wu, K. et al. Atomically dispersed Ni-Ru-P interface sites for highefficiency pH-universal electrocatalysis of hydrogen evolution. Nano Energy 80, 105467 (2021).
Pi, Y. et al. Selective surface reconstruction of a defective iridiumbased catalyst for high-efficiency water splitting. Adv. Funct. Mater. 30, 2004375 (2020).
Lin, X. et al. 5f covalency synergistically boosting oxygen evolution of . Catal. J. Am. Chem. Soc. 144, 416-423 (2022).
Li, J. et al. Unveiling the nature of Pt single-atom catalyst during electrocatalytic hydrogen evolution and oxygen reduction reactions. Small 17, 2007245 (2021).
Li, Y. et al. Interstitial boron-triggered electron-deficient Os aerogels for enhanced pH-universal hydrogen evolution. Nat. Commun. 13, 1143 (2022).
Zhang, J. et al. Novel (Pt-Ox)-(Co-Oy) nonbonding active structures on defective carbon from oxygen-rich coal tar pitch for efficient HER and ORR. Adv. Mater. 34, 2206960 (2022).
Zhou, K. L. et al. Platinum single-atom catalyst coupled with transition metal/metal oxide heterostructure for accelerating alkaline hydrogen evolution reaction. Nat. Commun. 12, 3783 (2021).
Zhao, Y. et al. Modulating Pt-O-Pt atomic clusters with isolated cobalt atoms for enhanced hydrogen evolution catalysis. Nat. Commun. 13, 2430 (2022).
Liu, Y. et al. Unraveling the function of metal-amorphous support interactions in single-atom electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 61, e202114160 (2022).
Ma, T. et al. Crystalline lattice-confined atomic Pt in metal carbides to match electronic structures and hydrogen evolution behaviors of platinum. Adv. Mater. 34, 2206368 (2022).
Park, S. et al. Delaminated MBene sheets beyond usual 2D transition metal materials for securing Pt single atoms to boost hydrogen evolution. Energy Environ. Sci. 16, 4093-4104 (2023).
Ma, W. et al. Ru-W pair sites enabling the ensemble catalysis for efficient hydrogen evolution. Adv. Sci. 10, 2303110 (2023).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Perdew, J. P., Ernzerhof, M. & Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982-9985 (1996).
Jain, A. et al. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 50, 2295-2310 (2011).
Klingenhof, M. et al. Modular design of highly active unitized reversible fuel cell electrocatalysts. ACS Energy Lett. 6, 177-183 (2021).
شكر وتقدير
لقد حصل العمل الذي أدى إلى هذه النتائج على تمويل من المؤسسة الوطنية للعلوم الطبيعية في الصين (22179098) وصندوق البحث الأساسي للجامعات المركزية لج.م. وقد حصل المشروع الذي أدى إلى هذا الطلب على تمويل من برنامج الاتحاد الأوروبي HORIZON.3.1 – برنامج المجلس الأوروبي للابتكار (EIC) من خلال اتفاقية المنحة 101071111 – ANEMEL. يقر ب.س. و م.ك. بالدعم المالي من الوزارة الفيدرالية للتعليم والبحث والتطوير (Bundesministerium für Bildung und Forschung, BMBF) بموجب أرقام المنح O3SF0613D و 03HY13OB في مشاريع البحث التعاونية “AEMready” و “AEMDirekt”. يقر ز.هـ. بالدعم من مركز ماكس بلانك-POSTECH-هسينتشو للمواد المعقدة. يقر م.ي. بالدعم من المؤسسة الوطنية للعلوم الطبيعية في الصين (52302302).
مساهمات المؤلفين
قام ج.م. بتصور وإشراف البحث. ص.ز. وج.م. صمما التجارب. قام ص.ز.، م.ك.، ت.ك.، ج.و.، ي.ب.، س.ل.، ج.هـ.، ج.ل.، ز.هـ.، ب.س. وج.م. بتنفيذ التجارب وتحليل البيانات. قام ص.ز.، ج.غ.، ل.س. وم.ي. بإجراء الحسابات النظرية وتحليل النتائج النظرية. شارك ص.ز.، هـ.ج.، ل.س.، و.هـ.، ج.ب.، ز.هـ.، ب.س. وج.م. في جوانب مختلفة من التجارب والمناقشات. كتب ص.ز.، ز.هـ.، ب.س. وج.م. الورقة. ناقش جميع المؤلفين النتائج وعلقوا على المخطوطة.
تمويل
تم تمويل الوصول المفتوح وتنظيمه بواسطة مشروع DEAL.
يجب توجيه المراسلات والطلبات للحصول على المواد إلى مينغهاو يانغ، وزي وي هو، وبيتر ستراسر أو جي وي ما.
معلومات مراجعة الأقران تشكر مجلة Nature Communications يونغ وين تان والمراجع الآخر المجهول على مساهمتهما في مراجعة هذا العمل. يتوفر ملف مراجعة الأقران.
مختبر شنغهاي الرئيسي للبحث والتطوير وتطبيق المواد المعدنية الوظيفية، معهد الطاقة الجديدة للمركبات، كلية علوم المواد والهندسة، جامعة تونغجي، 201804 شنغهاي، الصين.الجامعة التقنية في برلين، قسم الكيمياء، 10623 برلين، ألمانيا.مدرسة الكيمياء والهندسة الكيميائية، جامعة يانغتشو، 225002 جيانغسو، الصين.المختبر الوطني الرئيسي للكيمياء الفيزيائية للأسطح الصلبة، كلية الكيمياء والهندسة الكيميائية، جامعة شيامن، 361005 شيامن، الصين.معهد باوستيل المركزي للبحوث، شركة باوشان للحديد والصلب المحدودة، 201999 شنغهاي، الصين.المختبر الوطني الرئيسي لتطوير وتطبيق تكنولوجيا الفولاذات السيارات، باوستيل، 201900 شنغهاي، الصين.مركز باو وو للألمنيوم الفني، معهد باوستيل المركزي للبحوث، شركة باوشان للحديد والصلب المحدودة، 201999 شنغهاي، الصين.مركز شنغهاي للأبحاث الهندسية للمعادن للنقل الخفيف، 201999 شنغهاي، الصين.مركز أبحاث الإشعاع السنكروتروني الوطني، هسينتشو 30076، تايوان.معهد ماكس بلانك لفيزياء الكيمياء للمواد الصلبة، شارع نوتنزر 40، 01187 دريسدن، ألمانيا. البريد الإلكتروني:menghaoyoung@tongji.edu.cn; zhiwei.hu@cpfs.mpg.de; pstrasser@tu-berlin.de; jiwei.ma@tongji.edu.cn
Exploring an active and cost-effective electrocatalyst alternative to carbonsupported platinum nanoparticles for alkaline hydrogen evolution reaction (HER) have remained elusive to date. Here, we report a catalyst based on platinum single atoms (SAs) doped into the hetero-interfaced support (referred to as ), which features a low HER overpotential, an excellent stability and a distinctly enhanced cost-based activity compared to commercial and in 1 M KOH . Advanced physico-chemical characterizations disclose that the sluggish water dissociation is accelerated by while Pt SAs and the metallic Ru facilitate the subsequent combination. Theoretical calculations correlate with the experimental findings. Furthermore, only requires 1.90 V to reach and delivers a high price activity in the anion exchange membrane water electrolyzer, outperforming the benchmark . This research offers a feasible guidance for developing the noble metal-based catalysts with high performance and low cost toward practical production.
At present, seeking a promising energy carrier to replace traditional fossil fuels is more critical than ever to deal with the issues of environmental deterioration and resource depletion . Hydrogen ( ) with the merits of non-pollutant emission and high energy density is regarded as the most suitable alternative. Meanwhile, the electrochemical water splitting powered by renewable energy provides a green and sustainable way for efficient production . Compared with water electrolysis conducted in the acidic medium, alkaline water
electrolysis technology is currently more popular and feasible in practical industry, due to its advantages of robust facility and cheap electrolyzer construction . However, the cathodic hydrogen evolution reaction (HER) in alkaline electrolyte involves the prior water dissociation process (Volmer step) and the later hydrogen combination process (Heyrovsky or Tafel step), in which the sluggish kinetics of the Volmer step will severely limit the overall water splitting performance . Therefore, it is of great significance to accelerate this
restrictive barrier by discovering an efficient electrocatalyst towards alkaline HER.
Although the platinum (Pt) nanomaterials with optimal hydrogen binding energy are widely acknowledged as the most active catalysts toward HER, their catalytic activities in the alkaline medium are approximately two orders of magnitude lower than those in the acidic medium. This can be mainly attributed to the shortage of water dissociation ability on Pt atoms and thus leads to the insufficient supply of protons for generation . Conversely, metal oxides/hydroxides have been regarded as the ideal candidates for oxygen evolution reaction (OER) because of their good capabilities for cleaving the strong H-OH bond, whereas they are inefficient for propelling the hydrogen generation process in . Generally, one well-designed electrocatalyst for activating HER in the alkaline electrolyte should provide a platform with different active sites for water dissociation and hydrogen adsorption steps to take place, respectively. Enlightened by this guideline, many efforts have recently been devoted to construct the composite engineering by integrating Pt with metal oxide/hydroxides to synergistically accelerate the alkaline HER. For instance, Subbaraman et al. reported that the kinetic barrier of the Volmer step on Pt nanomaterials can be availably promoted by the decorated nickel hydroxide clusters, yielding reduced HER overpotentials in KOH solution . Wang and coworkers also successfully prepared the dense Pt nanoparticles immobilized oxygen vacancy-rich heterojunctions as an alkaline HER electrocatalyst with an enhanced performance . In addition, He et al. demonstrated that the water dissociation and hydrogen generation are speeded up on Pt nanomaterials incorporated Ni -iron layered double hydroxides ( LDH) . Despite the progress that has been achieved, the low abundance and high cost of Pt severely restrict its large-scale applications in the practical industry . In this regard, it is necessary to take the high HER activity and minimized Pt usage amount into consideration simultaneously when preparing the Pt-based hybrid electrocatalysts. Recently, the highly dispersed single-atom catalysts (SACs) are considered as the most promising candidates for a range of electrochemical reactions because of their distinct merits such as maximized atom-utilization efficiency, well-defined active sites and significantly reduced preparation cost . Therefore, the combination of Pt single atoms and metal oxide/hydroxide supports shows brightening prospect for balancing the tip between low cost and high performance in catalytic reactions. Meanwhile, as one of the cheapest noble metals, the cost-effective ruthenium (Ru) and its derivatives possess the strongest ability to prompt the water dissociation step, which can be employed as the supports for Pt single atoms to fully unlock the potential for catalyzing the alkaline HER, while it has no related report to date .
In this work, we report the synthesis, characterization and electrocatalysis of an active and cost-efficient HER electrocatalyst for the use at cathode of alkaline water electrolyzers. Our HER catalyst design involves individual Pt single metal atoms doped into the heterointerfaced supports (referred to as ). More specifically, this catalyst with low-Pt content requires very low overpotentials of merely 18 mV and 63 mV at the current densities of and , respectively, outperforming most recently reported noble metal and noble metal-free electrocatalysts. The noble metal cost-based catalytic activity of reaches a factor of more than 16 compared to those of commercial and C at the overpotential of 63 mV . Notably, delivers a longterm stability over 100 h with negligible activity loss. Corresponding mechanism investigated by various experiments and operando characterizations validate that all isolated Pt atoms, Ru and in the support play vital roles in activating the alkaline HER. Concretely, the sluggish water dissociation step is accelerated by the , the following hydrogen combination step is primarily promoted on Pt single atoms and also partially contributed by Ru, synergistically resulting in a remarkable activity of . Density functional theory (DFT)
calculations further determine the specific functions of active sites in , which corroborate the experimental discoveries. Moreover, the integration of and NiFe LDH in the anion exchange membrane water electrolyzer (AEMWE) only requires 1.90 V to reach the large current density of and exhibits a high price activity of 247.1 A dollar at 2.1 V , much superior to the Pt/C-based counterpart. This research finding stimulates our interest to pursue the balance between cost-effectiveness and high activity when exploring noble metal-based electrocatalysts toward practical production.
Results
Structural characterizations of
To synthesize the , the enriched with grain boundaries was obtained first by immersing the precursor in the molten (Supplementary Figs. 1, 2). Then Pt species were absorbed on the synthesized followed by calcination in Ar , in which process the Pt single atoms were incorporated and was slightly reduced to interfaced (see Methods for details). As shown in Fig. 1a, the flat-spread and dense nanostructure of was initially characterized by transmission electron microscopy (TEM). A typical highresolution TEM (HR-TEM) image indicates that the consists of independently distributed crystal regions belong to Ru and , with evident grain boundaries among them. To be specific, Ru and crystal regions reveal the lattice spacings of and , which can be ascribed to the and facets, respectively (Fig. 1b) . Generally, the existence of grain boundary suggests the close contact between two phases, favorable for inducing efficient interaction such as electron transfer in electrochemical reactions . In addition, Ru/ was prepared for comparison. As depicted in Supplementary Fig. 3, shows a similar morphology and interfaced structure as the . To reveal the detailed crystal structures of and , X-ray diffraction (XRD) was employed and corresponding results suggest that two phases attributed to Ru (JCPDS No. 06-0663) and (JCPDS No. 43-1027) are coexisted in these patterns, which is in keep with the finding of HR-TEM. Moreover, no diffraction peak related to Pt can be found in these patterns. Rietveld refinements were further performed on the primary peaks of those XRD patterns to calculate the relative content of Ru and . As a result, the Ru molar ratio is calculated to be in , and a slightly more Ru ( ) appears on because of the introduction of Pt species (Supplementary Figs. 4, 5 and Supplementary Tables 1, 2) . Afterwards, scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS) and inductively coupled plasma-optical emission spectrometry (ICP-OES) determine that the atomic ratio of Pt is in (Supplementary Fig. 6).
In order to disclose the atomic distribution of Pt species, aberration-corrected high-angle annular dark field-scanning TEM (AC HAADF-STEM) was employed on . As revealed in Fig. 1c, d, two distinct crystal regions with clear atomic arrangements named as Pos 1 and Pos 2 can be assigned to the and facets, respectively. Interestingly, owing to the higher Z-contrast of Pt against other elements, the isolated bright spots correspond to Pt atoms are directly visualized and homogenously dispersed on both Ru and regions, demonstrating that Pt species mainly exist as the single atoms on the supports . This is further confirmed by the atomic intensity profiles along the dashed rectangles collected in Fig. 1d, from which the isolated Pt atoms with higher intensities are observed (Fig. 1e). Additionally, corresponding elemental mappings indicate that Pt atoms are homogeneously distributed over the entire (Fig. 1f and Supplementary Fig. 7). Afterwards, the surface electronic structures and chemical compositions were analyzed by X-ray photoelectron spectroscopy (XPS). The survey XPS spectrum initially proves the existence of Pt elements in through displaying an obvious Pt signal (Supplementary Fig. 8a). In addition, both Ru XPS spectra on and can be deconvoluted into
Fig. 1 | Structural characterizations of Pt-Ru/RuO2. a TEM, b HR-TEM and (c) AC HAADF-STEM images of . d The enlarged areas of Pos 1 and Pos 2 in (c), with the Pt single atoms marked in circles, respectively. e Atomic intensity profiles along the dashed rectangles in (d). f Corresponding elemental mappings of . g The normalized Pt -edge XANES spectra of , Pt foil and . h Pt -edge EXAFS spectra and corresponding fitting curves, and (i) wavelettransformed -weighted EXAFS spectra of foil and .
doublets of and , implying the coexistence of metallic Ru and . After the introduction of Pt , the ratio of is increased from 0.16 to 0.28 , which is in good accordance with the XRD results (Supplementary Fig. 8b) . Supplementary Fig. 8c shows the spectra on these samples, where the peaks at the binding energy of and 532.5 eV correspond to lattice O , absorbed and absorbed , respectively . Meanwhile, Pt XPS spectrum indicates that the commercial Pt/C primarily contains the peak ascribed to metallic Pt. It is worth noting that both metallic Pt and oxidized Pt are found on . The single-atom alloy is formed when Pt single atoms are homogeneously dispersed in the region of Ru , in which Pt features the metallic characteristics and thus has a valence state of zero . Meanwhile, the oxidized Pt is primarily originated from the Pt single atoms coordinated by O atoms on the (Supplementary Fig. 8d).
Subsequently, the detailed chemical states and the local atomic structures were determined by synchrotron-radiation-based X-ray absorption spectroscopy (XAS). The oxidation state of Pt in Pt-Ru/ was first detected by X-ray absorption near-edge structure (XANES) along with standard Pt foil and as the references. It is acknowledged that the white line position at the -edge XANES is highly sensitive to the valence state of element . As illustrated in Fig. 1 g , the white-line energy position of is located between Pt foil and , implying the oxidized state of Pt species is between 0 and +4 in . Moreover, the extended X-ray absorption fine structure (EXAFS) is a powerful tool for discovering the atomic coordination environment. It can be clearly observed that exhibits a dominant peak at in the R-space of the Pt -edge EXAFS spectrum, corresponding to Pt-O bond. Additionally, no discernible peak related to bond can be distinguished in , evidencing the absence of Pt nanoparticles or clusters (Fig. 1 h ). This was further consolidated by the k -space of wavelet transform (WT)-EXAFS, in which shows a maximum scattering at and , close to the scattering in . Also, no scattering corresponds to bond is found in comparison with Pt foil ( and ), proving the lack of agglomerated Pt in (Fig. 1 i. Furthermore, a model-based EXAFS fit was performed on and Pt foil. Results confirm that the main peak located at is bond, other peaks at and are derived from and moieties, which suggests that the isolated Pt atoms in are coordinated by surrounded O atoms from regions and surrounded Ru atoms from Ru regions, respectively (Supplementary Fig. 9 and Supplementary Table 3) . Therefore, combining the detailed AC HAADF-STEM and EXAFS analysis, we conclude that the Pt species are existed as single atoms and homogenously dispersed on the interfaced supports. In another case, the Ru valence states on were also evaluated by Ru -edge XANES spectra together with Ru foil, and for comparison. As shown in Supplementary Fig. 10a, the absorption edge on displays a lower energy shift compared with , indicating the lower valence state of the former, which is due to the higher fraction of metallic Ru in according to XRD and XPS results. Through establishing the standard curves between the Ru valence states and the energy of white-line absorption edges, the average oxidation states of Ru on and are determined as +2.8 and +3.3 , respectively (Supplementary Fig. 10b). Besides, both the Ru -edge EXAFS spectra of and show the identical peaks as except the subtle peaks attributed to Ru-Ru bond present at , which signifies the existence of reduced Ru and consistent with abovementioned characterizations (Supplementary Fig. 10c).
Electrochemical performance of
The electrochemical performances of those prepared electrocatalysts toward HER were evaluated in 1 M KOH electrolyte involving a threeelectrode system. Similar tests were also carried out on the commercial Pt/C, Ru/C and for explicit comparison (Supplementary Fig. 11). All measured potentials in this study were carefully calibrated to the reversible hydrogen electrode (RHE) scale (Supplementary Fig. 12). The actual resistance values for all studied catalysts were detected in 1 M KOH solution (Supplementary Fig. 13a) and the non-iR corrected linear sweep voltammetry (LSV) curves are shown in Supplementary Fig. 13b. As displayed in Fig. 2a, the LSV curves with corrections show the activity trend of at low potentials. It is noteworthy that and exhibit an accelerated rise in activity relative to other catalysts as the potential increases, on account of the speedy HER rates. To be specific, delivers an overpotential of 18 mV at the current density of , which is significantly lower than those of , and . Impressively, at a higher current density of still possesses the smallest overpotential ( 63 mV ) among all studied electrocatalysts, indicating its remarkable alkaline HER activity (Fig. 2b). In addition, the synthesized shows the almost same HER performance as the commercial (Supplementary Fig. 14). Moreover, Fig. 2c illustrates the Tafel slopes based on the LSV curves and the corresponding values are , , and for , and , respectively. The lowest tafel slope of indicates the most effective facilitation of the hydrogen evolution kinetic . In addition, such a low Tafel slope value on also indicates that it follows the Volmer-Tafel mechanism in alkaline HER after the incorporation of Pt single atoms with high hydrogen coverage and binding ability . In order to further highlight the advantages of overpotential and Tafel slope, a synopsis of performances on recently
reported Pt-based and Ru-based HER electrocatalysts in alkaline medium is listed in Fig. 2d and Supplementary Table 4, in which Pt-Ru/ is much superior to others. Additionally, the double-layer capacitance ( ) measurements derived from a series of cyclic voltammetry (CV) with various scanning rates were employed to investigate the intrinsic activities of these samples. As shown in Supplementary Fig. 15, the larger value of as compared to indicates the introduced Pt single atoms lead to a larger electrochemical surface area (ECSA) and more exposed active sites . Accordingly, by normalizing the HER current to ECSA at the overpotential of possesses the highest specific activity ( ), almost 5 times higher than that of benchmark Pt/C ( ) (Supplementary Fig. 16). In addition, due to the efficient hydrogen adsorption ability of Pt and Ru, the hydrogen underpotential deposition ( ) method was employed to measure the ECSA of Pt/C, Pt-Ru/RuO2 and Ru/RuO2. As demonstrated in Supplementary Fig. 17, after the incorporation of Pt single atoms, exhibits a similar ECSA value with Pt/C, which is much larger than the and . Consequently, also shows the highest specific activity among studied catalysts. Furthermore, the ECSA values and specific activities calculated by the CO stripping method are in highly accord with those derived from the method (Supplementary Fig. 18). BET-normalized activities were also investigated to exclude the morphological influences on evaluating performances, it can be observed in Supplementary Fig. 19 that still outperforms . Mass activity is another essential factor for the evaluation of an electrocatalyst system owing to the direct relationship with the cost. Supplementary Fig. 20 plotted the mass activities (normalized to the loading mass of Pt and Ru) of these studied materials at the overpotential of 63 mV . As a result, delivers a mass activity of , which is almost 14 and 17 times higher than those of the commercial and ( ), showing its greater merit of cost-effectiveness. Surprisingly, the calculated price activity (normalized to the price of Pt and Ru ) of (116.7 A dollar ) can also reach 21.8 and 16.3 folds greater than those of the commercial Pt/C ( ) and ( 7.17 A dollar ), respectively, further exhibiting its economic efficiency and brighten potential toward industrial applications (Fig. 2e). Moreover, the two other catalysts with various amounts of Pt dopant were prepared by halving and doubling the inputs of Pt precursor, their Pt atomic ratios are determined to be and by ICP, respectively. LSV results in Supplementary Fig. 21 show that after doping , the HER activity of is obviously enhanced compared to the due to the incorporation of highly active Pt single sites. However, the HER activity of decreases with further increasing the amount of Pt dopant from to , which may be attributed to the formation of aggregated Pt clusters, suggesting that the optimal amount of Pt dopant in toward alkaline HER is . In addition, the Nyquist plots are obtained by establishing the equivalent circuit in electrochemical impedance spectroscopy (EIS) measurements followed by fitting processes (Supplementary Fig. 22 and Supplementary Table 5). As depicted in Fig. 2f, compared to Pt/C ( ), Ru/C , and possesses the smallest charge transfer resistance ( ) of , implying the expedited charge transfer rate and thus faster HER kinetics.
Apart from the activity, durability is also critical to assess an HER electrocatalyst, especially when targeted for industrial hydrogen production applications. The stability tests on and commercial Pt/C were carried out by chronopotentiometry at various current densities. As presented in Fig. 2g, it is seen that the activity of Pt-Ru/ at remains almost steady without an obvious decay during the constant operation of 100 h , whereas the overpotential of exhibits a rapid 55 mV increase after the continuous 100 h test. By determining the dissolved ion concentrations in the electrolytes after a
Fig. 2 | Alkaline HER performances of . a Polarization curves with iR corrections and (b) corresponding overpotentials of , and at the current densities of and in 1 M Tafel plots derived from the polarization curves in (a). d Comparison of overpotentials and Tafel plots in recently reported Pt, Ru-based HER electrocatalysts. e Comparison of the price activities on and at the overpotential of 63 mV . The prices of Pt and Ru are sourced from U.S. Geological
Survey (Date: Average price of platinum-group metals in January-March 2022). f EIS Nyquist plots and fitting curves of these samples recorded at -50 mV . g Chronopotentiometric measurements of and at current densities of and , respectively, carbon paper was used as the catalyst support. Generated amount and corresponding Faradaic efficiency over a range of overpotentials on . Note: error bars represent the standard deviation of three independent measurements.
long-term reaction, the deactivated reason for is attributed to the severe Pt ion leaching. In contrast, slight dissolutions of both Pt and Ru ions are occurred on , resulting in the enhanced HER stability (Supplementary Fig. 23). Post-reaction characterizations of TEM and HR-TEM confirm that the overall morphology and the interfaced structure of are well preserved except for slight agglomerations. In addition, elemental mappings indicate that Pt atoms are still homogeneously dispersed on the supports and no obvious crystal structure changes are found from the XRD pattern after the stability test at (Supplementary Fig. 24). Meanwhile the Pt and Ru XPS spectra suggest the decreased valence states of Pt and Ru on , which is common for HER catalysts after long-term operation under the reduction potentials (Supplementary Fig. 25). Moreover, the stabilities tests were also conducted at a higher current density of . Significantly, compared with the dramatic increase of overpotential on , only a 76 mV increase of overpotential is shown on after 100 h measurements.
The above measurements demonstrate the excellent HER stability of under the conditions of strong alkaline and high voltage. The repeated CV scanning experiments further highlight that the
polarization curves of remain almost identical before and after 5000 CVs, whereas other samples display obvious activity losses (Supplementary Fig. 26). Besides, in order to avoid the interference of the side reactions and thus the overestimation of the actual HER activity, the generated amounts on over a wide range of overpotentials are monitored by gas chromatography and corresponding Faradaic efficiencies (FE) are calculated (Supplementary Fig. 27. As illustrated in Fig. 2h, with the increasing of applied potentials, the evolution amount shows a rapid upward tendency, meanwhile the associated FEs keep nearly . This signifies that the majority of electrochemical energy is employed to drive HER and almost no side reactions take place on . Overall, features the advantages of high activity, cost-effectiveness and remarkable stability toward alkaline HER, demonstrating its promising prospect to be deployed in the practical hydrogen production applications.
Mechanistic insights into alkaline HER
Considering the well-designed has been demonstrated as a high-performance alkaline HER catalyst, the contributions of various active sites were explored. Poisoning experiments were initially carried
out via potassium thiocyanate (KSCN) as the deactivator to block the contact between active sites and reaction intermediates . As shown in Supplementary Fig. 28, the significant decline of activity on after the addition of , implying the essential role of Pt single atom toward activating HER. Besides, compared with the , the obvious activity loss on indicates that Ru also contributes to alkaline HER (Supplementary Fig. 29). To further get a deep insight into the improved mechanism of Pt single atoms, the operando EIS measurements were performed during HER. The Nyquist plots were stimulated by a double-parallel equivalent circuit model, in which the pseudo-capacitance ( ) and hydrogen absorption resistance ( ) are adopted for tracking the absorbed hydrogen ( ) behavior on the catalyst surface . Notably, the Tafel slope ( vs. potentials) of exhibits a markedly decreased value of in comparison to and , suggesting that adsorption kinetics is facilitated by the introduced Pt single atoms. In addition, the integration of and potential determines the adsorption charge , referring to the absorption concentration. It is seen that possesses a much higher than that of , supporting that the Pt single atoms contribute to the improved coverage on the surface. Moreover, the relative smaller Tafel slope and higher of than also indicates the reduced Ru can simultaneously improve the adsorption (Supplementary Fig. 30 and Supplementary Table 6). These findings are also evidenced by results in Supplementary Fig. 17, while the introduced Pt single atoms and Ru can increase the hydrogen adsorption area, indicating more H atoms are absorbed and leading to efficient combination process. It is worth mentioning that although Pt and Ru are hydrogen-binding active sites, the former are more essential and indispensable considering the relatively larger concentration of adsorbed and remarkable enhancement of HER activity induced by Pt single atoms. On the other hand, it is well known that the lower the CO stripping potential, the better the water dissociation ability. As seen in Supplementary Fig. 18, the CO stripping peak potentials on and are obviously lower than that on , confirming the excellent water dissociation ability is not attributed to the doped Pt. Supplementary Fig. 31 further depicts the kinetic isotope effects (KIE) of and in and electrolytes. The smaller KIE value of implies the easier HO-H bond cleavage, which is beneficial for the water dissociation process . In order to comprehensively understand the enhanced water dissociation ability on , operando Raman spectroscopy was conducted at various potentials (Supplementary Fig. 32). As depicted in Fig. 3a-c, one broad peak at the Raman shift of around can be observed in the spectra, which can be ascribed to the water at the catalyst-electrolyte interface. The two carbon signals of and G bands at around 1400 and are derived from the glassy carbon plate (Supplementary Fig. 33a). In addition, in order to confirm that the Raman laser was focused on the surface of electrocatalyst, the operando Raman tests under various potentials in 1 M KOH solution were conducted on the glassy carbon plate without electrocatalyst. It can be clearly seen that almost no interfacial water signals ( ) can be found in Supplementary Fig. 33b, which is due to the inert property of the glassy carbon plate towards the catalytic reaction. Furthermore, this interfacial water peak can be further fitted into three peaks nearly located at and (v3), respectively (Supplementary Figs. 34-36). Generally, v1 and v2 are assigned to the tetrahedral and trihedral coordinated water molecules that participate in the HER, while belongs to the dangling bond of the interfacial water molecule that is inactive toward . It is significant to observe that the peak proportions on both and display a dramatically faster downward trend than that of with the increased HER potentials, indicating the more efficient capability on and for cleaving water bond. In addition, the decline rate of v 3 peak proportion on is almost the same as that on , which suggests that Pt single atoms have no contribution on water dissociation step (Fig. 3d and Supplementary Table 7) . As shown in Supplementary Figs. 37-39, the operando Raman tests were further conducted on Ru and . After carefully fitting these spectra, it can be obviously observed that exhibits a faster downward trend of peak proportion than that on Ru in HER process, confirming its greater capability for cleaving the water bond. Significantly, exhibits a similar downward trend as the during more HER cathodic potentials, which validates that is the main active site for water activation and dissociation step in instead of Ru. Furthermore, it is calculated that the decline rate of peak proportion on is slightly slower than that on , which may be attributed to the electronic interaction between Ru and in (Supplementary Fig. 40). Based on above investigations, it can be experimentally concluded that is primarily responsible for the water dissociation step while both Pt single atoms and Ru facilitate the followed hydrogen combination step, collectively leading to the remarkable alkaline HER activity of , and this is further validated by DFT calculations.
Furthermore, the operando XAS techniques were accepted for monitoring the dynamic changes of electronic structures and local atomic environments on in the HER process (Supplementary Fig. 41). During the measurements, the working potential was reduced from the open circuit potential (OCP) to -0.2 V vs. RHE. As shown in Fig. 3e, once the voltage of -0.2 V was imposed, corresponding Pt -edge white line peak position exhibits an obvious negative shift, manifesting that the Pt single atoms are reduced to lower oxidation states during . In addition, the real-time change of Ru chemical state was also checked by the operando RuK-edge XANES. Figure 3f illustrates that the absorption edge negatively shifts to lower energy position under the HER working potential of -0.2 V , suggesting the decreased Ru valence state of during HER (Fig. 3g). As a consequence, the continuously decreased valence states of Pt and Ru during catalytic reactions discovered by operando XANES results elucidate that both Pt single atoms and the support in are functionalized and jointly participate in activating the alkaline . In order to further confirm that both Pt and Ru are active sites during HER, the other low HER operating potentials of -0.018 V (at which the current density is ) and -0.040 V were applied on to conduct the operando -edge and Ru K-edge XANES. Similarly, both white line peak position of Pt and adsorption edge of Ru continuously shift to lower energy with the negatively increased potentials, indicating their decreased valence states in the HER process (Supplementary Fig. 42). Thus, the potentialdependent operando XANES results further verify that both Pt and Ru elements in are active sites toward the alkaline HER . Moreover, as shown in Fig. 3h, no obvious Pt-Pt bond can be observed in the operando -edge EXAFS spectra during HER, indicating that the Pt atoms on remain isolated dispersion throughout the reaction. Notably, it is seen that the main bond shifts to a more positive position at -0.2 V , which may due to the local structure relaxation of Pt caused by the absorbed at HER working potential . This is also a strong evidence consistent with experimental results to confirm that Pt single atoms principally take charge of promoting the atom adsorption and enhancing the Tafel step in alkaline HER. Operando Ru -edge EXAFS spectra further exhibit that the intensity of bond decreases while the intensity of Ru-Ru bond increases with the applied reaction potentials, manifesting the reduction of the supports during HER (Fig. 3i) .
Theoretical investigations of
To further elucidate the mechanism of the excellent activity on toward alkaline HER, a theoretical study was performed based on the density functional theory (DFT) calculations. The atomic models of
Fig. 3 | Mechanism investigations of alkaline on . The operando Raman spectra of (a) , (b) and (c) under applied potentials in 1 M KOH solution. The proportions of peaks at on and . e The Pt -edge XANES and (f) the Ru -edge XANES of condition of The white line peak positions of Pt -edge XANES and the absorption edge positions of Ru -edge XANES under applied potentials. The Pt -edge EXAFS and (i) the Ru -edge EXAFS under pristine state, OCP and HER operating condition of -0.2 V . measured in 1 M KOH under pristine state, OCP and HER operating
and were constructed and energetically optimized (Supplementary Fig. 43). As depicted in Fig. 4a, the charge density difference of shows a significant charge redistribution along the chemical bonds due to the different electronegativity, where the electron tends to move from the surrounded Ru to central Pt atoms . This result confirms an efficient electronic interaction between the isolated Pt atoms and the . It is also found an obvious electron transfer from Ru to on the heterostructure, manifesting that Ru can regulate the electronic structure of and results in an enhanced catalytic ability (Supplementary Fig. 44). In addition, the optimized electronic structure induced by the Pt single atoms can be confirmed by the projected density of states (PDOS), which show higher occupation near the Fermi level ( ) on compared with the , suggesting the accelerated electron transfer and promoted conductivity caused by Pt orbital contribution . Moreover, the corresponding -band centers are calculated from the PDOS. Specifically, the average -band center values for and are -1.285 and -1.341 eV , respectively. According to the -band center theory, the bonding of the reaction intermediate is weakened, and more antibonding state is
occupied with the downshifted -band center position. Thus, the active sites on are quickly exposed, and the HER process is boosted (Fig. 4 b) . Subsequently, the mechanisms of different active sites in were investigated by employing DFT to explore their energetic properties. Since the Volmer step is the precondition and main barrier for catalyzing the HER in alkaline media, it is vital to measure the adsorption energy of on each active site, which is a key factor to evaluate the ability of accelerating water dissociation process . As presented in Fig. 4c, it is clearly seen that the Ru site of with a higher adsorption energy ( -0.594 eV ) is thermodynamically more favorable for adsorption compared with the Pt sites ( -0.359 eV ) and Ru sites ( -0.207 eV ), indicating that is more possible to be absorbed on the . The dissociation free energy in Supplementary Fig. 45 further indicates that Pt single atoms are difficultly to break the molecule due to the largest energy barrier. In addition, although Ru and have similar energy barriers, considering the stronger ability of adsorbing on , the water dissociation is more efficient and easier to be completed by rather than Ru . On the other hand, the adsorption free energy on all sites in were also calculated to examine their abilities for
Fig. 4 | DFT calculations of . a The top and side views of charge density difference on heterostructure model, blue and yellow areas represent the electron-accumulated region and the electron-depleted region,
activating the following combination step. As shown in Fig. 4d, compared to the other active sites, Pt sites deliver the most optimal value of 0.115 eV , demonstrating that the incorporated Pt single atoms are favorable for boosting the combination step . It is also noted that the adsorption-free energy on Ru is closer to zero than , which suggests that Ru also contributes to the adsorption and generation. Therefore, the finding disclosed by DFT calculations is highly consistent with the experimental results, which jointly demonstrate that the Pt single atoms, Ru and cooperatively enhance the alkaline HER performance of .
AEMWE performance of
Encouraged by the excellent HER activity, we further constructed the membrane electrode assembly (MEA) in AEMWE by employing as the cathode to evaluate its performance in the practical water splitting. The mass loading of was controlled as , meanwhile the benchmark Pt/C ( ) with was also tested in AEMWE for the comparison. In the preliminary tests, Ni felt was directly used as the anode (Supplementary Fig. 46). As shown in Fig. 5a, exhibits higher current densities than commercial Pt/C at the same cell potentials. The catalytic performances can be further enhanced by adopting the inexpensive NiFe layered double hydroxides (LDH) in the anode side. As a result, the polarization curves show that NiFe LDH + only requires 1.90 V and 1.77 V to reach the current density of before and after iR correction, respectively, which is much lower than those on NiFe LDH + commercial Pt/C ( 1.98 V and 1.84 V ). In addition, as depicted in Fig. 5b, the mass activities of reach 3.60 and at 2.0 V and 2.1 V , respectively, which are more than two-order of magnitude higher than those of commercial (no iR correction). On the basis of the remarkable performances, we further consider that the cost of Ru is well below Pt , thus possesses a superior economic efficiency compared with the commercial Pt/C. Impressively, after normalizing the current density to the price of the noble metal, the price activities of are 188.2 and 247.1 A dollar at 2.0 V and 2.1 V , respectively, reaching almost 3 times higher than those of commercial Pt/C ( 72.0 and 92.0 A dollar ) (Fig. 5c). The advantages of catalytic activity and cost-saving on illustrate its feasibility and potential to be applied as the practical catalyst for the hydrogen production in alkaline.
Discussion
In conclusion, a highly active and stable electrocatalyst of Pt-Ru/ towards alkaline HER is elaborately fabricated in this work. Comprehensive experiments and operando characterizations, as well as DFT calculations, verify that all the isolated Pt atoms, Ru and play vital roles during the catalytic reactions. Specifically, the
Fig. 5 | AEMWE performances of . a The AEMWE polarization curves with and without iR correction on NiFe LDH // Pt-Ru/RuO2 and NiFe LDH // commercial Pt/C. The comparisons of (b) mass activities and (c) price activities between
NiFe LDH // Pt-Ru/RuO and NiFe LDH // commercial Pt/C at various cell potentials (without iR correction). Note: error bars represent the standard deviation of three independent measurements.
sluggish water dissociation step is primarily accelerated by while the following hydrogen combination step is facilitated by Pt single atoms and Ru, collectively contributing to the improved HER in 1 M KOH . As a result, exhibits remarkable overpotentials of 18 mV and 63 mV at the current densities of and , respectively, superior to most of the recently reported catalysts. Especially, the cost-based activity of can reach more than 16 times higher than those of commercial and , showing its good cost-effectiveness. In addition, the HER stability of can be prolonged to over 100 h with a negligible activity loss. Furthermore, the integration of and NiFe LDH in the AEMWE exhibits a superior price activity of 247.1 A dollar at 2.1 V , reaching almost 3 times higher than the Pt/Cbased counterpart. This finding offers a prospective strategy of composite engineering to tip the balance between the high activity and economic efficiency when fabricating the noble metal-based catalysts toward practical production.
Methods
Materials
Ruthenium trichloride ( , solid, 99.5%), platinum tetrachloride ( , solid, ), ruthenium oxide ( , solid, ), potassium hydroxide ( KOH , solid, ), and commercial catalyst ( ) were purchased from Innochem. Sodium nitrate ( , solid, ) and isopropanol ( , liquid, ) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Commercial Pt/C catalyst (20 wt%) was obtained from Johnson Matthey (JM) corporation.
Catalyst preparation
In a typical synthesis, was initially heated at for 30 min in an oven under air atmosphere, then powder was poured into the molten and kept at for 10 min . Afterwards, the cooled solid was washed by water 3 times to remove the and dried overnight to obtain the synthesized . Then 50 mg synthesized was dispersed in 18 mL water and dissolved in 2 mL water was dropwise added into the above solution under continuously stirring. After the formation of a homogeneous mixture, it was heated to and kept stirring for 16 h . Finally, the Pt species absorbed was washed by water for 3 times and collected by centrifugation, followed by calcination in Ar at for 2 h with a heating rate of to obtain the . The was prepared by a similar method as , except that the precursor was not added.
Characterization
The TEM and HRTEM images were determined by a JEOL 2100F instrument at a working voltage of 200 kV . AC HAADF-STEM images
and corresponding elemental mappings were collected on a Thermo Scientific Themis Z microscope operated at 200 kV . SEM-EDS spectra were collected on a Zeiss Supra 55 at an acceleration voltage of 5 kV . The mass fraction of Pt in and the dissolved ion concentrations after stability tests were determined by ICP-OES on an Agilent ICP-OES 730.
The XRD data was collected on a Bruker D8 Advance powder diffractometer (operating at ) equipped with a source ( ) and fitted with a beryllium window at room temperature. Rietveld refinements for XRD data were conducted by using the Full-Prof program, and the refined parameters included the background parameters, line shift errors (zero shift), Caglioti coefficients (U, V and W), scale factor, lattice parameters, atomic position, atomic rate occupancy and isotropic atomic displacement parameters.
The XPS was carried out on a Thermo Scientific Escalab 250Xi with an Al-K source. XAS of Ru -edge and Pt -edge were conducted at the 44A beamline of the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. The operando XAS experiments of Ru edge and -edge were performed at the 44A beamline of the NSRRC with a custom-built operando XAS instruments. All XAS experiments were collected under the room temperature and analyzed by using the standard program Demeter.
The operando Raman spectra were collected on a HORIBA XploRA PLUS Raman spectrometer with an electrochemical cell, catalysts without carbon black were directly dropped on glassy carbon plate as the working electrode, calomel electrode and carbon rod were used as the reference electrode and counter electrode, respectively. It operated from 800 to and 0 to -80 mV vs. RHE in 1 M KOH solution to evaluate the water dissociation ability of catalysts.
Electrochemical Measurement
The HER performance evaluation was carried out on the CHI 760E electrochemical workstation with a rotating disk electrode (RDE) configuration (Pine Research Instrumentation). A glassy carbon electrode with the surface area of , calomel electrode and graphite rod were selected as the working, reference and counter electrode, respectively. Before testing, 5 mg sample ( or ) and 5 mg carbon black (Vulcan XC-72R) were mixed in 1.9 mL isopropanol solution and sonication for 1 h , followed by adding 0.1 mL Nafion solution ( , Aldrich) in the above mixture and sonication for 5 min to produce a homogeneous catalyst ink. Then, of the catalyst ink was dropped onto the working electrode and dried under infrared light. The mass loadings of metals for these prepared samples were controlled at . For comparison, the Pt and Ru mass loadings for commercial Pt/C (20 wt ) and commercial were also controlled at on the working electrode as well. All HER potentials were calibrated with respect to the RHE scale (Supplementary Fig. 12). The calibration was performed by using a Pt wire as the working electrode, the calomel electrode as the reference electrode and a graphite rod as the counter electrode in a -saturated electrolyte ( 1 M KOH ). CV scanning was carried out with a scan rate of 1 mV s -1 and the average potential at which the currents crossed zero was the calibration value. LSV with iR correction was conducted at a scan rate of under room temperature to evaluate the HER activity. The iR correction was auto-compensated by the electrochemical stations and the iR correction value was controlled as for all polarization curves. Tafel slopes were determined by plotting the overpotential vs. the logarithm of current density ( ). The Tafel slopes were probed on and within the overpotential ranges of , and , respectively. Stability was evaluated by using catalysts loaded carbon paper ( ) with mass loadings of to carry out chronopotentiometry at the constant HER current densities of and , respectively. Cyclic stability tests were carried out by repeating 5000 CV cycles on the studied samples. EIS was measured on the Bio-Logic SP-200 workstation in the frequency range from 0.01 to 100 kHz at -50 mV vs. RHE; the operando EIS was measured on all samples in the frequency range from 0.01 to 100 kHz at various applied potentials. Corresponding equivalent circuits were established and fitting resistance values were obtained through the EC-lab Software Analysis.
Calculation of the electrochemically surface area
ECSAs were calculated by evaluating the electrochemical double-layer capacitance ( ) on catalytic surface of studied catalysts based on Eq. (1):
where was measured from the scan-rate-dependent CVs in the non-Faradaic region from 0.5 to 0.6 V vs. RHE in 1 M KOH with scan rates of and represents the real surface area of the smooth metal electrode, which was generally equal to the geometric area of the glassy carbon electrode ( ). The specific capacitance ( ) for a flat surface was generally considered to be in the range of . In this work, a value of was accepted. ECSAs were also measured by method, in which all catalysts were scanned by CV in saturated 1 M KOH solution with a scan rate of . CO stripping tests were conducted by bubbling the CO gas directly on the catalyst-coated electrode for 20 minutes in 1 M KOH solution, followed by placing the electrode in the fresh KOH electrolyte and scanning CV to remove the absorbed CO. The scanning rate was controlled as .
Calculation of HER Faradaic efficiency
was dropped on carbon paper to conduct HER in a sealed cell. The applied potentials on the sample to generate were and -0.3 V vs. RHE, respectively. The amounts of generated were recorded by Agilent 7890 B equipped with a molecular sieve column thermal conductivity detector and Ar was used as the carrier gas. amount was determined by plotting the generated peak area into the established standard fitting curves. The HER FE of was
calculated based on Eq. (2):
where is the number of electrons transferred for generating is the total charge, is the amount of generated (in moles) and is the Faradaic constant.
Computational details
All the structural optimizations and adsorption energy calculations were carried out based on the density-functional theory (DFT) as implemented in Vienna Ab initio Simulation Package (VASP) . The projector-augmented wave (PAW) method was implemented to calculate the interactions between the ionic cores and valence electrons . Perdew-Burke-Ernzerhof (PBE) approach of spinpolarized generalized gradient approximation (GGA) was used to describe the exchange-correlation energy . All DFT calculations were performed for the heterostructure model. The atomistic structures of ( )- Ru and ( 110 )- were generated from Materials Project (MP) database . Besides, 3 Ru atoms for heterostructure model were randomly replaced by Pt atoms with the lowest-energy configuration to approximately simulating atomic content and construct the heterostructure model. The cutoff plane-wave kinetic energy of 520 eV was used in calculations with the -point grid size. The conjugate-gradient method was used in electronic relaxation, and all DFT convergence parameters were set consistent with the Materials Project (MP) . The adsorption energy ( ) is defined as
where is the energy of slab, is the energy of free adsorbate, and is the total energy of adsorbate-slab system in the equilibrium state.
The free energy ( ) of adsorbed and is defined as
where is the adsorption energy of slab, is the difference of zero-point energy between adsorbed state and the gas phase, is the entropy contribution to the reaction at .
MEA preparation for AEMWE single-cell measurements
(AEMION Membrane, AEMIONTM Ionomer) The membrane electrode assembly (MEA) was prepared by manual spray coating technique of catalyst-coated membranes (CCM) as described by Klingenhof et al. . For spray coating the membrane (AEMION ) was fixed on a commercial heating vacuum plate equipped with temperature control (Carbon and Fuel Cell) and a compressor (Welch). The table temperature was set to . Furthermore, a mask ( ) was used to ensure vis-a-vis coating of the anode and cathode. The applied anode and cathode ink consisted in both cases of 50 mg catalyst powder, ultrapure water, -PrOH and 460 mg ionomer solution (AEMION solution in EtOH and . In the case of the anode, an overall amount of ink were sprayed on the membrane, aiming at a NiFe-LDH loading of . At the cathode catalyst ink with the loading amount of were spray coated on the membrane. After spray coating, the as-prepared CCM was dried for ten minutes at followed by cooling to RT and weighting. To achieve a satisfying loading spraying and weighing step were carried out several times with low amounts of the catalyst ink. The anode was always spray
coated first. To adjust a suitable loading and provide suitable comparisons between commercial Pt/C reference catalyst and the manufactured catalyst measurements without NiFe-LDH as anode catalyst were conducted avoiding a more complex system. After proper adjustment of the cathode catalyst loading, measurements with additional NiFe-LDH catalyst were conducted. The measurements were carried out using a Greenlight test station with a AEMWE single-cell setup. The setup consists of the prepared MEA, furthermore oxygen and hydrogen side are equipped with PTFE gaskets, porous transport layers (PTL, Nickel Felt at the Anode and Carbon Cloth at the cathode), endplates with electrolyte ports, bipotential plates (parallel flow fields, carbon plate for the hydrogen side and titanium plate for the oxygen side). The CCMs were pretreated in 2 M KOH over 2 h before the measurement. To achieve the best performance, the spray-coated CCMs were anion-exchanged in 2 M KOH three times, each 20 min . To remove the remaining surplus OH- the anion-exchanged membrane was washed with DI water three times for 20 min . Before evaluating the activity by applying polarization curves, the CCM was activated by potential cycling ( 50 mV between 1.1 V and 1.7 V cell voltage) and a potential holding step at 1.83 V over 1 h . The single-cell measurements were conducted at in 0.1 M KOH solution.
Data availability
All relevant data are available from the corresponding authors on request. Source data are provided with this paper.
References
Suen, N.-T. et al. Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives. Chem. Soc. Rev. 46, 337-365 (2017).
Lei, C. et al. Efficient alkaline hydrogen evolution on atomically dispersed species anchored porous carbon with embedded Ni nanoparticles by accelerating water dissociation kinetics. Energy Environ. Sci. 12, 149-156 (2019).
Zeng, K. & Zhang, D. Recent progress in alkaline water electrolysis for hydrogen production and applications. Prog. Energy Combust. Sci. 36, 307-326 (2010).
Abbasi, T. & Abbasi, S. A. ‘Renewable’ hydrogen: prospects and challenges. Renew. Sust. Energy Rev. 15, 3034-3040 (2011).
Zheng, Y., Jiao, Y., Vasileff, A. & Qiao, S.-Z. The hydrogen evolution reaction in alkaline solution: from theory, single crystal models, to practical electrocatalysts. Angew. Chem. Int. Ed. 57, 7568-7579 (2018).
Cheng, F. et al. Accelerated water activation and stabilization metalorganic framework via constructing triangular active-regions for ampere-level current density hydrogen production. Nat. Commun. 13, 6486 (2022).
Xie, W.-F. & Shao, M.-F. Alkaline water electrolysis for efficient hydrogen production. J. Electrochem. 28, 22014008 (2022).
Durst, J. et al. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 7, 2255-2260 (2014).
Zheng, J., Sheng, W., Zhuang, Z., Xu, B. & Yan, Y. Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy. Sci. Adv. 2, e1501602 (2016).
Li, A. et al. Enhancing the stability of cobalt spinel oxide towards sustainable oxygen evolution in acid. Nat. Catal. 5, 109-118 (2022).
Zhang, L. et al. Facet dependent oxygen evolution activity of spinel cobalt oxides. J. Electrochem. 28, 2108481 (2022).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212-222 (2021).
Subbaraman, R. et al. Enhancing hydrogen evolution activity in water splitting by tailoring -Ni( OH-Pt interfaces. Science 334, 1256-1260 (2011).
Wang, K. et al. Dense platinum / nickel oxide heterointerfaces with abundant oxygen vacancies enable ampere-level current density ultrastable hydrogen evolution in alkaline. Adv. Funct. Mater. 33, 2211273 (2023).
He, X. et al. Electronic modulation with Pt incorporated NiFe layered double hydroxide for ultrastable overall water splitting at 1000 mA . Appl. Catal. B: Environ. 331, 122683 (2023).
Zhu, Y. et al. Unusual synergistic effect in layered RuddlesdenPopper oxide enables ultrafast hydrogen evolution. Nat. Commun. 10, 149 (2019).
He, Y. et al. Amorphizing noble metal chalcogenide catalysts at the single-layer limit towards hydrogen production. Nat. Catal. 5, 212-221 (2022).
Jiang, K. et al. Single platinum atoms embedded in nanoporous cobalt selenide as electrocatalyst for accelerating hydrogen evolution reaction. Nat. Commun. 10, 1743 (2019).
Cheng, N. et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 7, 13638 (2016).
Jiang, K. et al. Rational strain engineering of single-atom ruthenium on nanoporous for highly efficient hydrogen evolution. Nat. Commun. 12, 1687 (2021).
Zhang, J. et al. Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction. Nat. Catal. 1, 985-992 (2018).
Zhang, Z. et al. Single-atom catalyst for high-performance methanol oxidation. Nat. Commun. 12, 5235 (2021).
Vukmirovic, M. B., Teeluck, K. M. & Adzic, R. R. Single platinum atoms electrocatalysts: oxygen reduction and hydrogen oxidation reactions. Croat. Chem. Acta 90, 225-230 (2017).
Zhu, S. et al. The role of ruthenium in improving the kinetics of hydrogen oxidation and evolution reactions of platinum. Nat. Catal. 4, 711-718 (2021).
Li, G. et al. The synergistic effect of Hf-O-Ru bonds and oxygen vacancies in for enhanced hydrogen evolution. Nat. Commun. 13, 1270 (2022).
Li, W. et al. Carbon-quantum-dots-loaded ruthenium nanoparticles as an efficient electrocatalyst for hydrogen production in alkaline media. Adv. Mater. 30, 1800676 (2018).
Liu, Y. et al. A general route to prepare low-ruthenium-content bimetallic electrocatalysts for pH -universal hydrogen evolution reaction by using carbon quantum dots. Angew. Chem. Int. Ed. 59, 1718-1726 (2020).
Yan, S. et al. Partially oxidized ruthenium aerogel as highly active bifunctional electrocatalyst for overall water splitting in both alkaline and acidic media. Appl. Catal. B: Environ. 307, 121199 (2022).
Wang, Z. et al. PtSe Pt heterointerface with reduced coordination for boosted hydrogen evolution reaction. Angew. Chem. Int. Ed. 60, 23388-23393 (2021).
Xu, Q. et al. Atomic heterointerface engineering overcomes the activity limitation of electrocatalysts and promises highly-efficient alkaline water splitting. Energy Environ. Sci. 14, 5228 (2021).
Schilling, A. C. et al. Accelerated reduction by single Pt atoms at the metal-oxide interface. ACS Catal. 10, 4215-4226 (2020).
Poerwoprajitno, A. R. et al. A single-Pt-atom-on-Ru-nanoparticle electrocatalyst for CO-resilient methanol oxidation. Nat. Catal. 5, 231-237 (2022).
Lai, W.-H. et al. Activating inert surface Pt single atoms via subsurface doping for oxygen reduction reaction. Nano Lett. 21, 7970-7978 (2021).
Huang, K. et al. Ru/Se-RuO2 composites via controlled selenization strategy for enhanced acidic oxygen evolution. Adv. Funct. Mater. 33, 2211102 (2023).
Zhu, Y. et al. Iridium single atoms incorporated in efficiently catalyze the oxygen evolution in acidic conditions. Nat. Commun. 13, 7754 (2022).
Liu, W. et al. Highly-efficient RuNi single-atom alloy catalysts toward chemoselective hydrogenation of nitroarenes. Nat. Commun. 13, 3188 (2022).
Luo, S. et al. A tensile-strained Pt-Rh single-atom alloy remarkably boosts ethanol oxidation. Adv. Mater. 33, 2008508 (2021).
Wang, H. et al. PdBi single-atom alloy aerogels for efficient ethanol oxidation. Adv. Funct. Mater. 31, 2103465 (2021).
Dai, J. et al. Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis. Nat. Commun. 13, 1189 (2022).
Tan, W. et al. Fine-tuned local coordination environment of Pt single atoms on ceria controls catalytic reactivity. Nat. Commun. 13, 7070 (2022).
Zeng, L. et al. Anti-dissolution Pt single sites with coordination for efficient alkaline water splitting electrolyzer. Nat. Commun. 13, 3822 (2022).
Chen, Y. et al. Plasma-assisted highly dispersed Pt single atoms on Ru nanoclusters electrocatalyst for pH-universal hydrogen evolution. Chem. Eng. J. 448, 137611 (2022).
Zhang, J. et al. OH spectator at IrMo intermetallic narrowing activity gap between alkaline and acidic hydrogen evolution reaction. Nat. Commun. 13, 5497 (2022).
Wan, C. et al. Amorphous nickel hydroxide shell tailors local chemical environment on platinum surface for alkaline hydrogen evolution reaction. Nat. Mater. 22, 1022-1029 (2023).
Zhu, Y. et al. Single-atom In-doped subnanometer Pt nanowires for simultaneous hydrogen generation and biomass upgrading. Adv. Funct. Mater. 30, 2004310 (2020).
Qi, K. et al. Single-atom cobalt array bound to distorted with ensemble effect for hydrogen evolution catalysis. Nat. Commun. 10, 5231 (2019).
Lai, W. et al. Boosting the interfacial hydrogen migration for efficient alkaline hydrogen evolution on Pt-based nanowires. J. Mater. Chem. A 10, 16834 (2022).
Li, J. et al. Ethylene-glycol ligand environment facilitates highly efficient hydrogen evolution of through proton concentration and hydrogen spillover. Energy Environ. Sci. 12, 2298 (2019).
Fu, H. Q. et al. Hydrogen spillover-bridged Volmer/Tafel processes enabling ampere-level current density alkaline hydrogen evolution reaction under low overpotential. J. Am. Chem. Soc. 144, 6028-6039 (2022).
Dai, Q. et al. Accelerated water dissociation kinetics by electronenriched cobalt sites for efficient alkaline hydrogen evolution. Adv. Funct. Mater. 32, 2109556 (2022).
Wang, K. et al. Kinetically accelerating elementary steps via bridged Ru-H state for the hydrogen-evolution in anion-exchange membrane electrolyzer. Adv. Funct. Mater. 33, 2212321 (2023).
Wu, K. et al. Atomically dispersed Ni-Ru-P interface sites for highefficiency pH-universal electrocatalysis of hydrogen evolution. Nano Energy 80, 105467 (2021).
Pi, Y. et al. Selective surface reconstruction of a defective iridiumbased catalyst for high-efficiency water splitting. Adv. Funct. Mater. 30, 2004375 (2020).
Lin, X. et al. 5f covalency synergistically boosting oxygen evolution of . Catal. J. Am. Chem. Soc. 144, 416-423 (2022).
Li, J. et al. Unveiling the nature of Pt single-atom catalyst during electrocatalytic hydrogen evolution and oxygen reduction reactions. Small 17, 2007245 (2021).
Li, Y. et al. Interstitial boron-triggered electron-deficient Os aerogels for enhanced pH-universal hydrogen evolution. Nat. Commun. 13, 1143 (2022).
Zhang, J. et al. Novel (Pt-Ox)-(Co-Oy) nonbonding active structures on defective carbon from oxygen-rich coal tar pitch for efficient HER and ORR. Adv. Mater. 34, 2206960 (2022).
Zhou, K. L. et al. Platinum single-atom catalyst coupled with transition metal/metal oxide heterostructure for accelerating alkaline hydrogen evolution reaction. Nat. Commun. 12, 3783 (2021).
Zhao, Y. et al. Modulating Pt-O-Pt atomic clusters with isolated cobalt atoms for enhanced hydrogen evolution catalysis. Nat. Commun. 13, 2430 (2022).
Liu, Y. et al. Unraveling the function of metal-amorphous support interactions in single-atom electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 61, e202114160 (2022).
Ma, T. et al. Crystalline lattice-confined atomic Pt in metal carbides to match electronic structures and hydrogen evolution behaviors of platinum. Adv. Mater. 34, 2206368 (2022).
Park, S. et al. Delaminated MBene sheets beyond usual 2D transition metal materials for securing Pt single atoms to boost hydrogen evolution. Energy Environ. Sci. 16, 4093-4104 (2023).
Ma, W. et al. Ru-W pair sites enabling the ensemble catalysis for efficient hydrogen evolution. Adv. Sci. 10, 2303110 (2023).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Perdew, J. P., Ernzerhof, M. & Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982-9985 (1996).
Jain, A. et al. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 50, 2295-2310 (2011).
Klingenhof, M. et al. Modular design of highly active unitized reversible fuel cell electrocatalysts. ACS Energy Lett. 6, 177-183 (2021).
Acknowledgements
The work leading to these results has received funding from the National Natural Science Foundation of China (22179098) and the Fundamental Research Funds for the Central Universities to J.M. The project leading to this application has received funding by European Union’s HORIZON.3.1 – The European Innovation Council (EIC) Programme through the grant agreement 101071111 – ANEMEL. P.S. and M.K. acknowledge the financial support by the federal ministry for education, research and development (Bundesministerium für Bildung und Forschung, BMBF) under Grant Numbers O3SF0613D and 03HY13OB in the collaborative research projects “AEMready” and “AEMDirekt”. Z.H. acknowledges the support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials. M.Y. acknowledges the support from the National Natural Science Foundation of China (52302302).
Author contributions
J.M. conceived and supervised the research. Y.Z. and J.M. designed the experiments. Y.Z., M.K., T.K., G.W., Y.P., S.L., J.H., J.L., Z.H., P.S. and J.M. performed the experiments and data analysis. Y.Z., C.G., L.S. and M.Y. performed the theoretical computations and analyzed the theoretical results. Y.Z., H.J., L.X., W.H., C.P., Z.H., P.S. and J.M. participated in various aspects of the experiments and discussions. Y.Z., Z.H., P.S. and J.M. wrote the paper. All authors discussed the results and commented on the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Correspondence and requests for materials should be addressed to Menghao Yang, Zhiwei Hu, Peter Strasser or Jiwei Ma.
Peer review information Nature Communications thanks Yongwen Tan and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Shanghai Key Laboratory for R&D and Application of Metallic Functional Materials, Institute of New Energy for Vehicles, School of Materials Science and Engineering, Tongji University, 201804 Shanghai, China. Technische Universität Berlin, Department of Chemistry, 10623 Berlin, Germany. School of Chemistry and Chemical Engineering, Yangzhou University, 225002 Jiangsu, China. State Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, 361005 Xiamen, China. Baosteel Central Research Institute, Baoshan Iron & Steel Co., Ltd., 201999 Shanghai, China. State Key Laboratory of Development and Application Technology of Automotive Steels, Baosteel, 201900 Shanghai, China. Baowu Aluminum Technical Center, Baosteel Central Research Institute, Baoshan Iron & Steel Co., Ltd., 201999 Shanghai, China. Shanghai Engineering Research Center of Metals for Lightweight Transportation, 201999 Shanghai, China. National Synchrotron Radiation Research Center, Hsinchu 30076, Taiwan. Max Planck Institute for Chemical Physics of Solids, Nöthnitzer Strasse 40, 01187 Dresden, Germany.
-e-mail: menghaoyoung@tongji.edu.cn; zhiwei.hu@cpfs.mpg.de; pstrasser@tu-berlin.de; jiwei.ma@tongji.edu.cn