تشكيل IrOx غير المشبع في SrIrO3 عن طريق إضافة الكوبالت لتفاعل تطور الأكسجين الحمضي The formation of unsaturated IrOx in SrIrO3 by cobalt-doping for acidic oxygen evolution reaction
تحليل الماء الكهروكيميائي هو طريق واعد لإنتاج الهيدروجين المستدام. ومع ذلك، فإن الجهد الزائد العالي لتفاعل تطور الأكسجين الأنودي يشكل تحديًا كبيرًا.لقد أظهرت المحفزات من نوع البيروفيسكايت المعتمدة على – إمكانات كبيرة لتفاعل تطور الأكسجين الحمضي، لكن أصول نشاطها العالي لا تزال غير واضحة. هنا، نقوم بتطوير محفز مخلوط بالكوبيكنظام لتعزيز نشاط تفاعل تطور الأكسجين وتوضيح أصل النشاط التحفيزي. تكشف التجارب في الموقع أن الكوبالت ينشط أكسجين الشبكة السطحية، مما يعرض بسرعةالمواقع النشطة، بينما يعمل تشبع الكوبالت في الكتلة على تحسين طاقة ارتباط الممتزاتالمشوب المزدوجتظهر نشاطًا كهربائيًا تحفيزيًا عاليًا في تفاعل تطور الأكسجين، متجاوزة بشكل ملحوظ المنتجات التجاريةالعوامل المساعدة في كل من المحلل الكهربائي التقليدي ومحلل الماء بغشاء تبادل البروتون.
تحليل الماء لإنتاج الهيدروجين يوفر ميزة إنتاج وقود نظيف ومستدام دون انبعاثات كربونية.حتى الآن، يُعتبر التحليل الكهربائي للماء باستخدام غشاء تبادل البروتون (PEM) واحدًا من أكثر الطرق رسوخًا في مجال إنتاج الهيدروجين الأخضر.. ومع ذلك، فإن الجهد الزائد العالي المرتبط عادةً بتفاعل تطور الأكسجين الأنودي (OER) يشكل تحديًا كبيرًا لتحسين كفاءة إنتاج الهيدروجين من التحليل الكهربائي للماءعلاوة على ذلك، فإن تقدم المحفزات الخاصة بتفاعل الأكسدة الكهربائية للأكسجين المصممة للوسط الحمضي يمثل تحديًا أكبر مقارنة بتلك المخصصة للوسط القلوي. نظرًا لأن المحفزات الكهربائية النشطة للغاية لتفاعل الأكسدة الكهربائية للأكسجين تتكون أساسًا من أكاسيد أو هيدروكسيدات المعادن، فإن معظمها يظهر استقرارًا ضعيفًا تحت الظروف الحمضية.لذلك، فإن تطوير محفزات OER الفعالة التي تعمل في وسط حمضي أمر حاسم.
تشمل المحفزات الحمضية الفعالة الحالية لأكسدة الماء Ru وIr وأكاسيد المعادن القائمة على Mn.. على وجه الخصوص،معهيكل البيروفسكايت (حيث تمثل A عادةً معدنًا قلويًا أرضيًا وB تمثل معدنًا انتقاليًا) يظهر نشاطًا عاليًا في التحفيز الحمضي لتفاعل الأكسدة. أداءفي السنوات الأخيرة، تم إجراء أبحاث واسعة لفهم آلية التحفيز OER لـقام جارامييو وآخرون بتحضير السطح (001)فيلم عبر استراتيجية ترسيب بالليزر، وتمت ملاحظة تحسين تدريجي في أدائه التحفيزي في عملية التحفيز.من خلال دمج الحسابات النظرية والتجارب، اكتشفوا أن النشاط التحفيزي العالي لـيمكن أن يُعزى إلى المكشوف المواقع التي تتبع ذوبان Sr. في دراسات شاملة إضافية، أكد الباحثون التعديلات الهيكلية لـ تحت عملية OER الحمضية باستخدام مطيافية الكتلة للأيونات الثانوية (SIMS) ، ومجهر القوة الذرية في الموقع (AFM) ، ومطيافية امتصاص الأشعة السينية (XAS) ، على التوالي . اقترحوا وجود علاقة بين عملية ذوبان Sr و ما تم تشكيلهأنشطة السطح. علاوة على ذلك، تحقيق يتضمنيشير أيضًا إلى أن نشاط OER ينشأ من الحالة غير المتبلورةهيكل يتكون من ذوبان. تبدو هذه الدراسات أنها تحدد المكون النشط لـالبيروفسكايتات القائمة على – كالأم amorphous
الهيكل. ومع ذلك، فإن معظم الدراسات تتجاهل تأثير ذوبان موقع B على استقرار الأكسجين السطحي وبنية تنسيق Ir-O، والأسباب الدقيقة التي تساهم في الأداء العالي لتحفيز OER المخصب بالكوبالت.تظل المحفزات غير واضحة. تنبع هذه المشكلة بشكل أساسي من التحدي الذي يواجهه الباحثون في توضيح جانبين رئيسيين من عملية التحفيز OER: (1) الدور الرئيسي لذوبان الكوبالت في العمليات التحفيزية و (2) تأثير الكوبالت السطحي والكتلي علىالمواقع.
لمعالجة التحديين الرئيسيين المذكورين أعلاه، قمنا بتصميم نظام SrIrO3 المضاف إليه الكوبالت في موقع B لفهم آلية الذوبان في مواقع المحفز وأصولالنشاط التحفيزي، كما هو موضح في الشكل 1a. كشفت تجارب مطيافية الكتلة بالتحليل الطيفي المتصل بالحث (ICP-MS) عن ذوبان أيوني متزامن لكل من الكوبالت (Co) والسترونتيوم (Sr)، الناتج عن تآكل الحمض قبل عملية OER. عند جهد OER (1.60 فولت مقابل RHE)، وُجد أن ظاهرة الذوبان كانت ضئيلة. جنبًا إلى جنب مع الحسابات النظرية، تم إجراء سلسلة من التجارب في الموقع بما في ذلك رسم خرائط رامان في الموقع، وXAS في الموقع، ومطيافية الكتلة الكهروكيميائية التفاضلية (DEMS). هذه أظهرت النتائج دور الكوبالت في طريقتين حرجتين: (1) يقلل الكوبالت السطحي من استقرار جسر الأكسجين Co -O-Ir في، مما أدى إلى التعرض السريع للتنسيق المنخفض الهيكل؛ (2) يقوم مصفوفة الكوبالت الضخمة بتحسين طاقة ارتباط OOH وبالتالي تقليل الجهد الزائد. وهكذا، فإن الكوبالت المخلوط الذي تم تصنيعهأظهرت نشاطًا عاليًا في OER، متجاوزة بشكل ملحوظ المنتجات التجاريةالعوامل المساعدة في إلكتروليز الماء باستخدام غشاء تبادل البروتون. ستعزز الرؤى المستخلصة من هذا البحث بشكل كبير فهم الأداء التحفيزي العالي لتفاعل أكسدة الماء.محفزات البيروفسكايت القائمة على، مما يوفر رؤى رئيسية لتصميم وتحضير محفزات OER الحمضية عالية الأداء.
النتائج
تصنيع وتوصيفمُشَبَّع بـعيناتتم تحضير العينات المخدرة بكميات مختلفة من الكوبالت باستخدام طريقة السول-جل، وتم غسلها بالكامل بحمض قبل الاستخدام. وتم الإشارة إلى العينات كما يلي:، ، و .
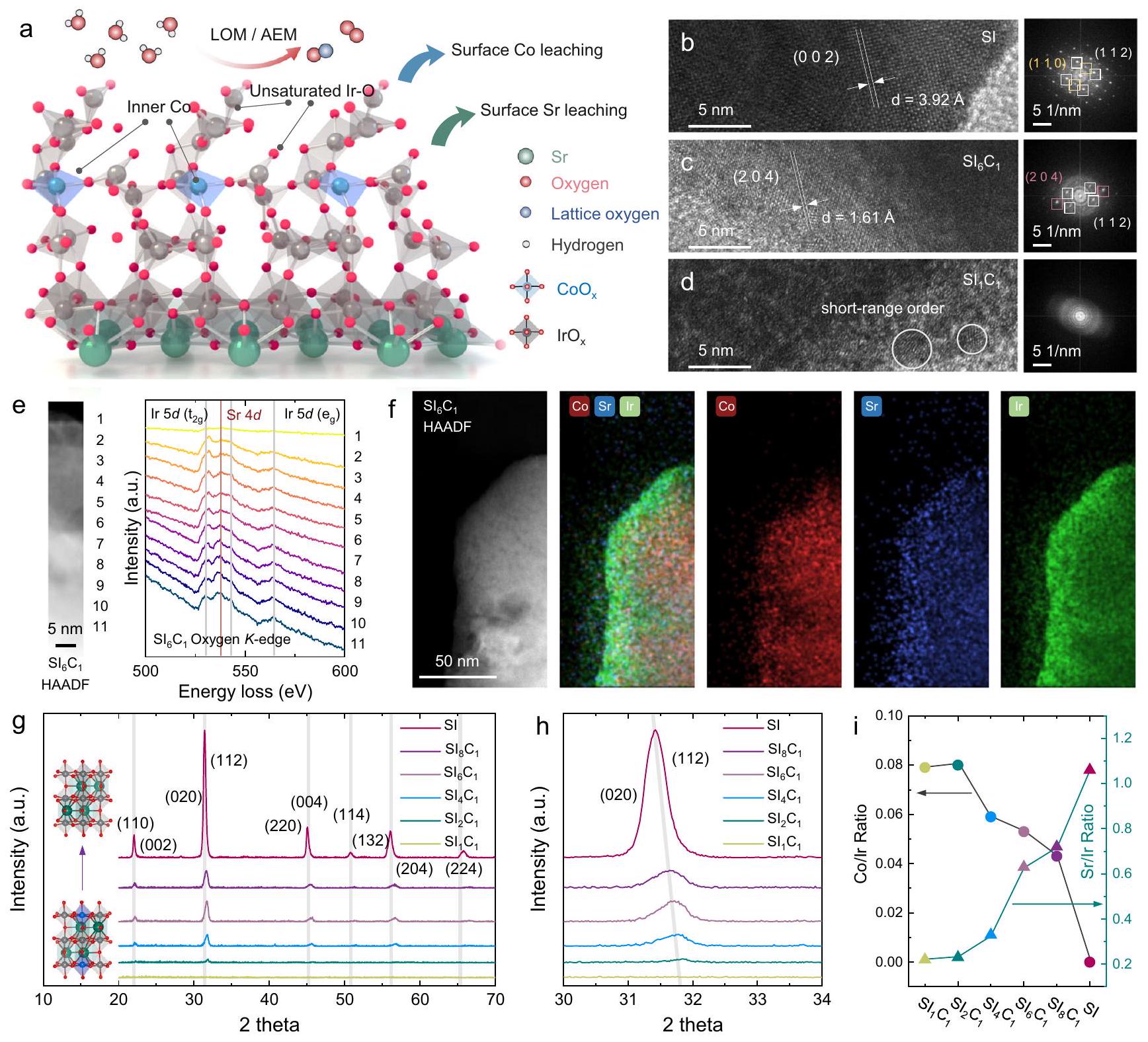
الشكل 1 | توصيف العينات. أ مخطط آلية التحفيز OER للمواد المزدوجة.محفزصور HRTEM والرسوم البيانية FFT المقابلة لـ SI و ; e -طيف EELS الحافة من; صورة HAADF لـ و
صور خرائط EDS المقابلة؛طيف حيود الأشعة السينية (XRD)، “، وعينات SI. مخطط ICP-MS لـ و نسب، ، وعينات SI.
تم تحليل الهياكل البلورية لهذه العينات من خلال مجهر الإلكترون الناقل عالي الدقة (HRTEM) وحيود الأشعة السينية (XRD) في الشكل.“، على التوالي. لاختبار HRTEM، تم أخذ ثلاث عينات (، و ) تم اختيارها. على وجه التحديد، أظهرت SI هيكلًا بلوريًا متعامدًا. أظهر تحويل فورييه السريع (FFT) أن الطائرات البلورية الرئيسية المعرضة تشمل (112)، (110)، و(002). على الرغم من انخفاض بلورية السطح، لا تزال هناك مستويات بلورية مكشوفة بوضوح مثل (112) و(204) و(020). في هذه الأثناء، عرضت بلورات سطحية ضعيفة مع وجود بعض المناطق التي تظهر ترتيب بلوري قصير المدى فقط، وعدم وجود بقع حيود واضحة في نمط تحويل فورييه. للتحقيق في سبب انخفاض البلورية الملحوظة فيعينة، قمنا بإجراء تحليلات طيف امتصاص طاقة الإلكترون (EELS)، والنتائج المعروضة في الشكل 1e والشكل التكميلي 3. يمكن أن تُنسب القمم عند 532 و542 و564 إلكترون فولت إلى Ir 5 d، بينما القمة عند 536 إلكترون فولت تعود إلىيكشف التحليل المقارن أن قمة Sr 4d لعينة SI عادة ما تكون أعلى من تلك الخاصة بـعينة، مما يشير إلى أن وجود Sr يساهم في الحفاظ على الهيكل البلوري. علاوة على ذلك، حددنا تباينًا كبيرًا بين تركيزات Sr على السطح وفي الكتلة داخل SI وعينات، حيث يكون هذا التأثير ملحوظًا بشكل خاص فيعينة. وهذا يشير إلى أن ذوبان الكوبالت يؤثر أيضًا على نسبة السترونتيوم الموجودة على السطح. تم إجراء مزيد من التحقيقات من خلال رسم خرائط الطيف الكهروضوئي للطاقة المشتتة (EDS) (الشكل 1f). تؤكد النتائج أن ذوبانوتشكيل لاحق لـ.
أظهر تحليل حيود الأشعة السينية (XRD) أن قمم الحيود لـ SI كانت متوافقة مع البيروفسكايتات ذات الشكل الزائف المكعب (Pnma) النموذجية. كما أظهرت SI المدعمة بالكوبالت أيضًا قمم حيود مشابهة، مما يؤكد نجاح تخليق المحفز القائم على البيروفسكايت.كما هو موضح في الشكل 1h، أدى تشويب الكوبالت إلى تقليل كبير في بلورية SI وأسفر عن انتقال قمة الحيود إلى الزاوية العالية، مما يدل على انكماش الشبكة في SI المشوب بالكوبالت. على وجه الخصوص، و عرضت تقريبًا عدم وجود قمم حيود، ربما بسبب الذوبان الكبير لسطح الكوبالت والسترونتيوم نتيجة عملية الغسل الحمضي. تشير هذه النتائج إلى أن زيادة نسبة الكوبالت قد تعطل بنية البيروفسكايت الزائفة المكعبة بعد الغسل الحمضي، وهو استنتاج يتماشى مع نتائج HRTEM. تم تحليل التركيب العام للعينات باستخدام ICP-MS، كما هو موضح في الشكل 1i. ومن الجدير بالذكر أن هناك تفاوتًا كبيرًا بين النسبة الأولية للكوبالت والإيريديوم والنسبة النهائية لتركيب العينات. العينات ذات البلورية الضعيفة، تحديدًا و ، أظهر أعلىنسبة، معأعلى قليلاً من. قد يُعزى ذلك إلى الصعوبة في الحفاظ على هيكل البيروفسكايت في، مما أدى إلى كمية كبيرة من ذوبان الكوبالت خلال عملية غسل الحمض، وبالتالي، تقليل نسبة. على الرغم من امتلاكها لأعلىالنسب، كانت هذان العيّنتان لا تزالان أقل بكثير من النسبة الأولية، مما يؤكد مرة أخرى عدم استقرار الكوبالت السطحي في الظروف الحمضية.نسب، ، و أظهر اتجاهًا متناقصًا منإلىكما هو موضح في الشكل 1i مما يشير إلى أن الحفاظ على الشبكة يساعد في تثبيت ذرات الكوبالت في الطور الكتلي. من بين العينات المختلفة المعتمدة على SI، و عرضت الأدنىنسبة، ونسبة زادت بشكل ملحوظ مع انخفاض نسبة الدوبامين من الكوبالت.كانت نسبة العينة SI أعلى قليلاً من 1، مما يشير إلى نسبة أعلى قليلاً من Sr مقارنة بـ Ir في الهيكل الكلي، كما اقترحت نتائج EELS الموضحة في الشكل التكميلي 3. وتم تأكيد هذه الاستنتاجات بشكل أكبر من خلال طرق توصيف إضافية مثل المجهر الإلكتروني الماسح (SEM) وطيف الأشعة السينية للألكترونات (XPS) وطيف رامان، كما هو موضح في الشكل التكميلي 5a و 5b.
أداء تحفيزي عالي في تفاعل الأكسدة الكهربية للأكسجين لمركب Co-doped SrIrO
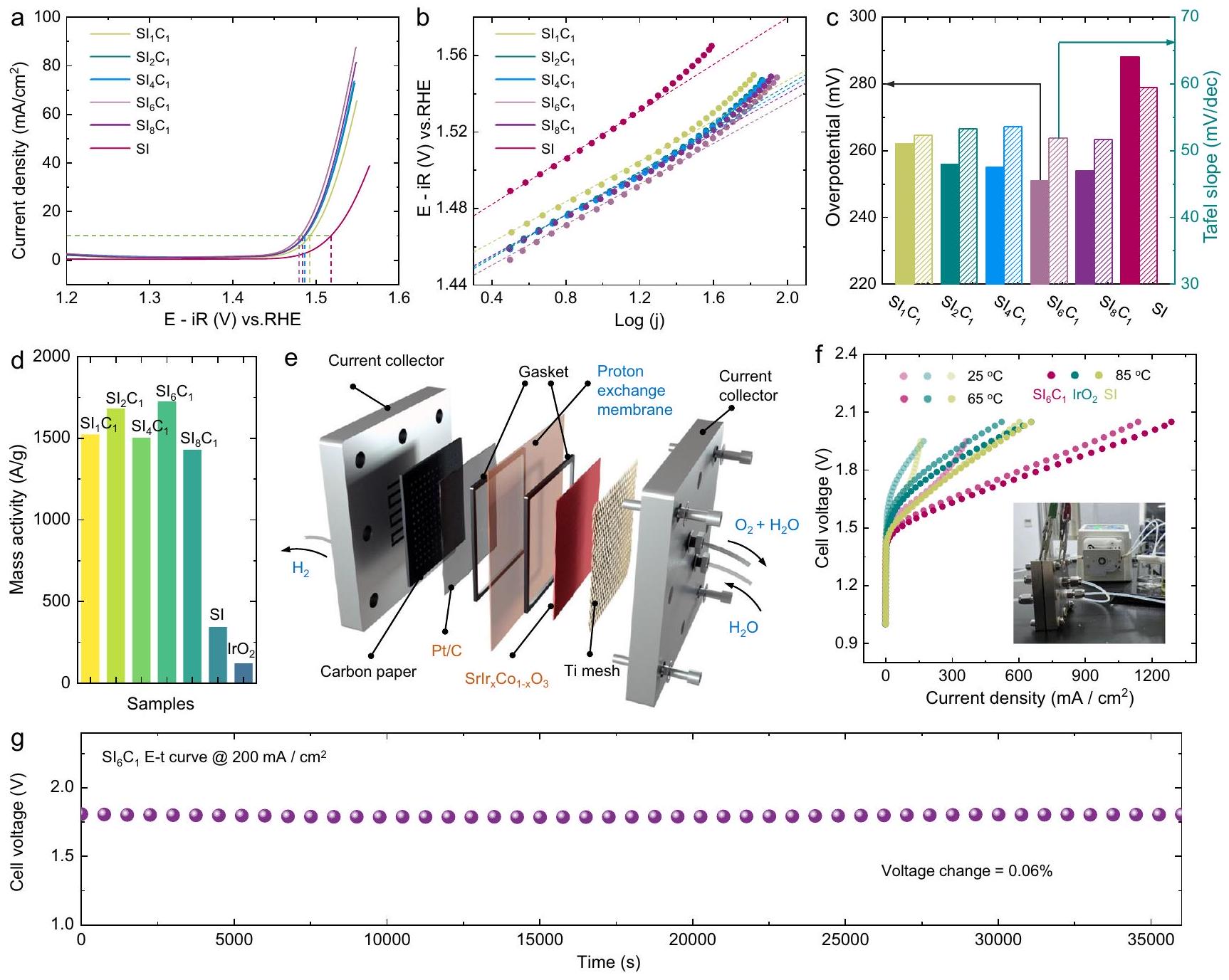
أداءات التحفيز لإنتاج الأكسجين“، وتمت دراسة عينات SI في وسط حمضي. كانت منحنيات الفولتامترية المسحية الخطية (LSV) لـتم تسجيل عينات SI بعد 10 دورات من الفولتمترية الدورية (CV)، كما هو موضح في الشكل 2a. تشير النتائج إلى أنيتطلب فقط 245 مللي فولت للوصول إلى كثافة تيار، وهو ما يزيد بحوالي 5 مرات عن أداء السيليكون عند نفس الجهد. نظرًا لأن عملية التحفيز لتفاعل تطور الأكسجين تحدث بشكل أساسي على السطح، فإن أداء و العينات عمومًا متفوقة وتظهر اتجاهًا مشابهًا. هذا يتماشى مع نتائج التوصيف السابقة، مما يشير إلى أنه على الرغم من اختلاف نسب التخصيب بالكوبيك، فإن هياكل سطح المحفزات قابلة للمقارنة. كما هو موضح في الشكل 2ب، ج، و تظهر العينات انحدارات تافل ضمن النطاقديسمبر، أقل بكثير من ذلك في SI ). وهذا يشير إلى أن المشاركة المشتركة يمكن أن تعزز حركية تفاعل OER. كما هو موضح في الشكل 2d، مقارنة النشاط الكتلي لـ و تظهر العينات أن العينات المضافة بالكوبيوم جميعها أظهرت نشاطًا عاليًا، متجاوزة بشكل كبير تلك الخاصة بـ SI و.
لدراسة التطبيقات المحتملة لمحفزات سلسلة SI في التحليل الكهربائي للماء، قمنا بمزيد من الفحص لأداء التحفيز الكهربائي لثلاث عينات (، و ) لتحليل الماء بواسطة غشاء تبادل البروتون. يتم تقديم المخطط التخطيطي لمحلل الماء بغشاء تبادل البروتون في الشكل 2e. تكشف نتائج أداء تحليل الماء بغشاء تبادل البروتون، الموضحة في الشكل 2f، أن النشاط التحفيزي لـ أعلى بكثير من تلك الخاصة بـ SI و. يمكن أن تحقق كثافة تيار تتجاوز عند جهد خلية 2.0 فولت“، وهو أعلى بكثير من تلك الخاصة بمحفز SI ( ) و محفز ) في نفس الظروف. كما اختبرنا الاستقرار على المدى الطويل لـ العامل المساعد لتحليل الماء باستخدام كهرباء غشاء البوليمر كما هو موضح في الشكل 2g والشكل التكميلي 6. تشير النتائج إلى أنالعامل المساعد يظهر استقرارًا عاليًا، مع معدل تدهور في الأداء لـ، والذي يمكن مقارنته بذاك المحفز SI والتجاريالمحفز. تشير هذه الاستقرار العالي الاستثنائي إلى أنيظهر المحفز إمكانية التطبيق العملي في التحليل الكهربائي للماء باستخدام أغشية تبادل البروتون.
أدوار التشارك في التفاعل والذوبان في SrIrOدُرِسَت من خلال الحسابات النظرية
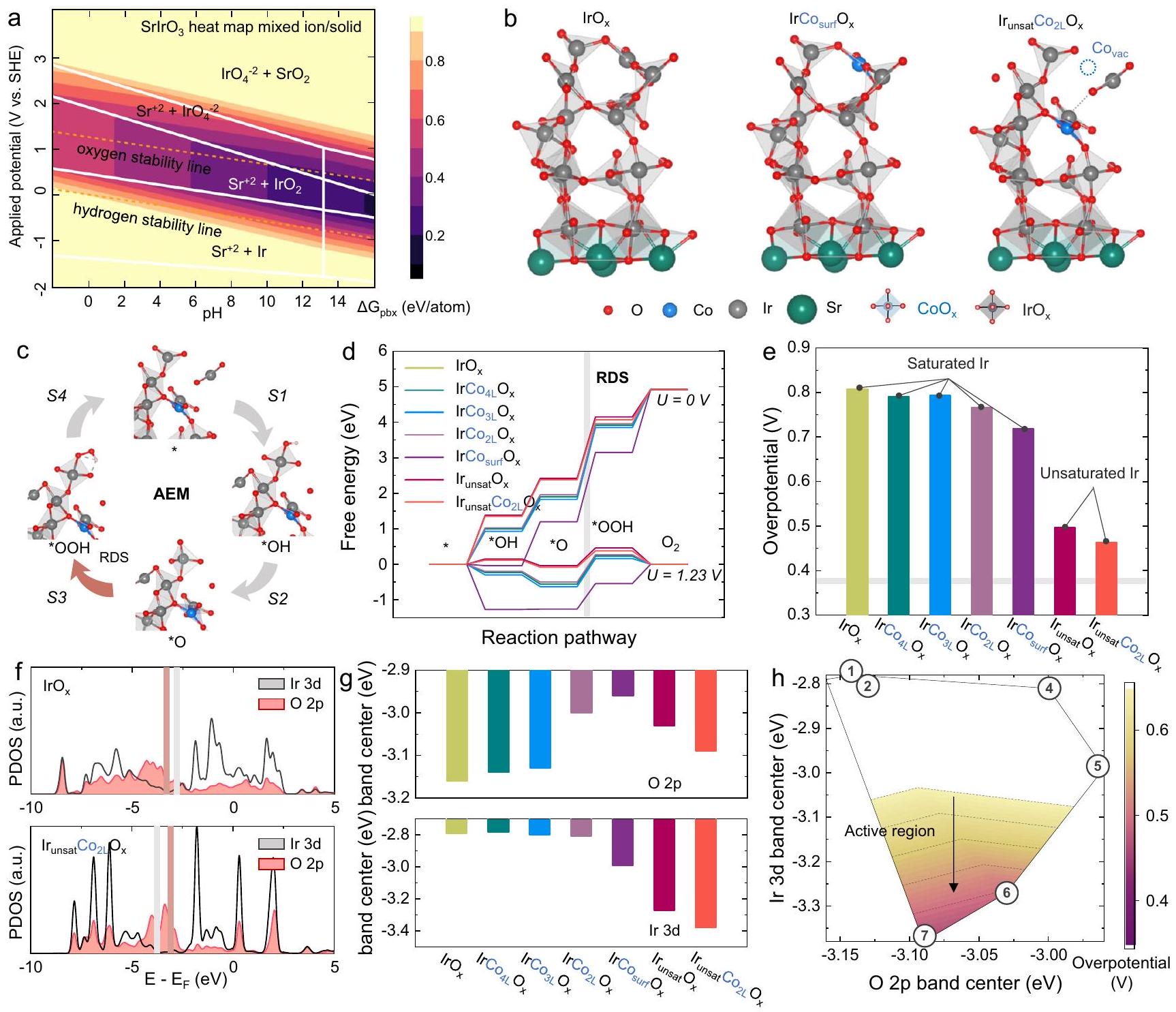
أثر التداخل مع الكوبالت علىتمت دراسته من خلال دراسة نظرية. حيث أثبتت العديد من الدراسات المبكرة أنتتعرض المحفزات المعتمدة على Sr لذوبان كبير أثناء. تم فحص ذلك من خلال تحليل مخططات بوربايكس لـ و ، مع التركيز بشكل خاص على استقرارها في الوسط الحمضي (الشكل 3 أ والشكل التوضيحي 7). تكشف النتائج عن تشابه ملحوظ في خصائص الاستقرار لـ و تحت الظروف الحمضية، يظهر السترونتيوم ميلاً ديناميكياً حرارياً نحو الذوبان، مما يكشف عن العديد منالمواقع على السطح. بالإضافة إلى ذلك، كوبر في الـكما يظهر عدم استقرار في الوسط الحمضي، مما قد يؤدي أيضًا إلى ذوبان كبير، متسق مع نتائج رسم الخرائط EDS و ICP-MS في الشكل 1.
كما هو موضح في مخطط بورباي، فإن هيكل المحفز يتأثر بشكل كبير بالجهد المطبق. ومع ذلك، لا تظهر المحفزان اختلافات حرارية ملحوظة بالقرب من منحنى استقرار الأكسجين، على الرغم من أنهما يظهران اختلافات بارزة في الاتجاهات الحرارية عبر قيم pH مختلفة. وهذا يشير إلى أن تأثيرقد يكون الذوبان عند إمكانيات OER المختلفة أقل بكثير من تأثير pH المحلول، لا سيما في الوسط الحمضي القوي. تشير الأبحاث السابقة إلى أن عدد تنسيق Ir-O للسطحعلىأثناء تحفيز OER هوبينما تم الكشف عن وجود محتوى Sr بكميات ضئيلة في العديد من الدراسات، لا يزال تحديد الحالة الدقيقة لـ Sr على مقياس دون النانومتر يمثل تحديًا.من خلال دمج النتائج من مخططات بوربايكس والتوصيفات المذكورة أعلاه، تم بناء سبعة نماذج للتحقيق في المواقع الحفازة النظرية لأكسدة الماء وأداء التحفيز.ومشوب بـ (الشكل 3ب والشكل التوضيحي 8).
تم إجراء الحسابات النظرية على النماذج، كما هو موضح في الشكل 3c. تكشف مخططات الطاقة الحرة المحسوبة (الشكل 3d) أن الخطوة المحددة للسرعة لجميع النماذج هي تكوين *OOH. إن إضافة الكوبالت، سواء على السطح أو في الطور الكتلي، والتي ست
الشكل 2 | القياسات الكهروكيميائية للعينات. أ منحنيات استقطاب OER لـ، وعينات SI بتحميل كتلة منفي و (ب) المنحدرات تافل المقابلة. قيم المقاومة لـ ، و كانت 3.9، 3.7، 3.6، 4.7، 3.2، 4.6 و، على التوالي. ج مقارنة بين الفولتية الزائدة وانحدارات تافل لـ، ، وعينات SI. د مقارنة نشاط الكتلة لـ OER من ، و عينات. الرسم التخطيطي لجهاز التحليل الكهربائي للماء باستخدام غشاء البوليمر.أداء التحليل الكهربائي للماء باستخدام غشاء تبادل البروتونصورة لجهاز التحليل الكهربائي للماء باستخدام غشاء تبادل البروتون (PEM). استقرار التحليل الكهربائي للماء باستخدام غشاء PEMعينة. لا تغير هيكل التنسيق لـالأوكتاهيدرون، يمكن أن يعزز نشاط التحفيز لتفاعل الأكسدة (OER) إلى حد معين. ومع ذلك، فإن مثل هذه التعزيزات في النشاط محدودة، خاصة بالنسبة للكوبالت المدعوم بكميات كبيرة، حيث يتم تقليل جهد الأكسدة (OER) بمقدار فقط. على النقيض، السطح غير المشبع، الذي يتشكل بعد ذوبان الكوبالت، يظهر تحسنًا كبيرًا في النشاط التحفيزي لتطور الأكسجين، مع انخفاض الجهد الزائد بـ (الشكل 3e). تشير هذه النتائج إلى أن الكوبالت قد لا يشارك مباشرة في التحفيز، بل يعزز إعادة بناء السطح من خلال ذوبان المواقع، مما يؤدي إلى تعرض سريع لمزيد من التنسيق المنخفض.المواقع النشطة.
للمزيد من التحقيق في أصل النشاط، تم إجراء تحليلات كثافة الحالات (DOS) على هذه النماذج (الشكل 3f). تشير البيانات إلى أن تشويب الكوبالت يؤثر بشكل كبير على الأكسجين الجسر Co-O-Ir، مما يؤدي إلى تحريكمركز النطاق أقرب إلى مستوى فيرمي. وفقًا للدراسات ذات الصلة، فإن مثل هذا التحول فييؤثر مركز النطاق بشكل كبير على استقرار سطح المحفز وقد يكون مرتبطًا بذوبان الأكسجين. هذا يشير إلى أن التأثير الرئيسي لتطعيم الكوبالت هو تغيير استقرار السطح لـ، وتعزيز إعادة بناء السطح وتشكيل المواقع النشطة. بالإضافة إلى ذلك، فإن التخصيب والانحلال للكوبيوم يؤثران بشكل كبير على الإيريديوم.فرقة مركز (الشكل 3g). يؤثر إزاحة المدارات 3d المعدنية عادةً بشكل مباشر على طاقة الربط مع المادة الماصة. في هذه الدراسة، كانت الإزاحة السلبية للـ Ir أدى مركز الشريط إلى انخفاض في *OOH الطاقة الحرة، مما عزز بشكل كبير نشاط التحفيز لتفاعل الأكسدة (الشكل 3g). من خلال إقامة العلاقة بين Ir و تم صياغة مخطط بركان جديد لتفاعل الأكسدة (OER) بناءً على مراكز النطاق والجهد الزائد النظري (الشكل 3h). كشفت البيانات أن المعتدلمركز النطاق و Ir السفليتلعب مراكز النطاق أدوارًا حاسمة في النشاط التحفيزي لـمحفزات قائمة على …
توصيفات في الموقع للتغيرات الهيكلية السطحية للمحفزات
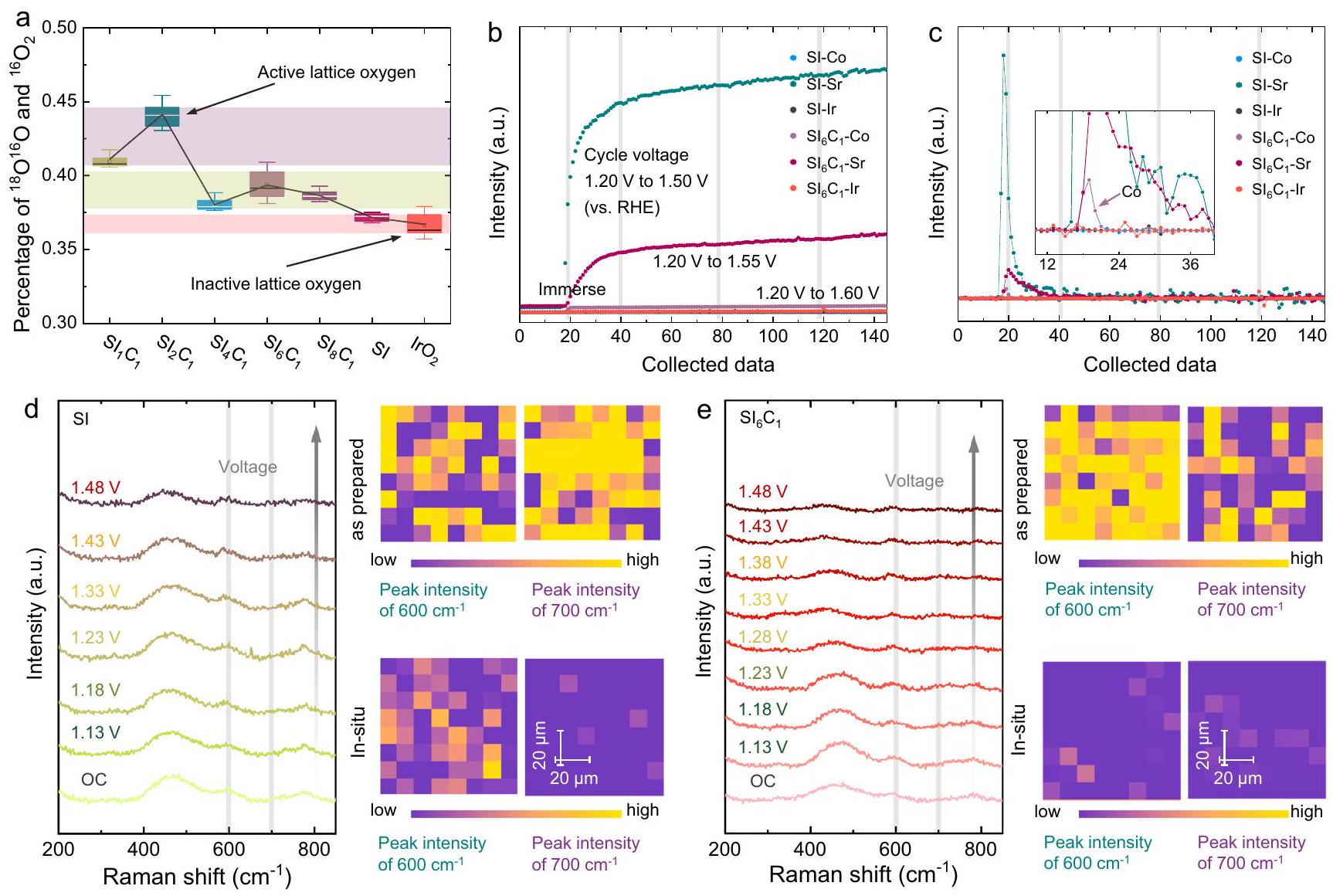
لتوضيح الآلية التحفيزية، تم إجراء اختبارات DEMS أولاً، كما هو موضح في الشكل 4a. أسطح، ، و تم وضع علامات على العينات بـكما هو موضح في الأشكال التكميلية 10-12. لكل عينة،تم جمع الإشارات في دورات الفولتمترية الدورية المختلفة، ونسبةإلىتم استخدامه للقضاء على الوفرة الطبيعية لـفي الهواء. كما هو موضح في الشكل التكميلي 13، فإن اتجاه تفاعل تطور الأكسجين الشبكي (LOER) لمعظم المحفزات عبر دورات الفولتمترية الدورية المختلفة مشابه، ويبدو أن نسبة تشريب الكوبالت تسهل
الشكل 3 | الحسابات النظرية لـمحفزات البيروفسكايت القائمة على. مخطط بوربايكس لـ. ب نماذج حسابية محتملة لـ (تطعيم السطح Co) و (الطبقة الثانية من التداخل مع IrO غير المشبع ) لحسابات DFT. ج مخطط آلية تطور الممتزات. د طاقة التفاعل الحرة لتفاعل الأكسدة. مخططات لنماذج مختلفة. ج الجهد الزائد لنماذج حسابية مختلفة. ف مخططات كثافة الحالات لـ و ، و مركز نطاق الطاقة ومخطط البراكين لنماذج حسابية مختلفة. إطلاق الأكسجين الشبكي. أظهرت النتائج أن إضافة الكوبالت تقلل من استقرار الأكسجين السطحي فيمتسق معنتائج حساب المركز. ومن الجدير بالذكر أن LOER لـ و هو الأكثر بروزًا، مع يتجاوز قليلاً. لقد تم تفسير هذه الظاهرة سابقًا من خلال خصائص XRD و ICP-MS، على أنها يواجه صعوبة في الحفاظ على شبكة البيروفسكايت، تنخفض نسبة تشويب الكوبالت. على النقيض، SI وعرض أصغر عملية LOER، والتي ترتبط باستقرار أكسجين شبكة سطح المحفز. أشارت النتائج إلى أن إضافة الكوبالت والذوبان يمكن أن ينشّط أكسجين شبكة المحفز، مما يسرع من تكوين.
بالإضافة إلى LOER، تم إجراء تجارب ICP-MS في الموقع للكشف عن ظاهرة ذوبان الأيونات أثناء OER، كما هو موضح في الشكل 4b، c. لقد لاحظنا أن كلا من و undergo SI ذوبًا كبيرًا عند غمره في إلكتروليت حمضي. إن ربط هذه الملاحظة مع نتائج XRD وRaman السابقة يشير إلى أن عملية الذوبان يتم تحفيزها بواسطة الذوبان السريع لـ الأكاسيد ذات الصلة أو المركبات غير المتجانسة. علاوة على ذلك، تشير الاتجاهات الثابتة اللاحقة للذوبان إلى أن ذوبان الأيونات لـ لا يحدث العامل المساعد بالضرورة أثناء العملية التحفيزية. ومن الجدير بالذكر أن ظاهرة ذوبان Sr في SI أكثر أهمية من تلك الخاصة بـ، والذي يُعزى إلى التركيز الأعلى من السطح والكتلة من Sr في SI. علاوة على ذلك،يرافق ذلك أيضًا كمية صغيرة من ذوبان الكوبالت، مما يؤكد أكثر أن ذوبان الكوبالت يعزز تكوين غير المشبع..
لتأكيد المعلومات الهيكلية للعامل الحفاز، تم إجراء دراسة رسم خرائط رامان في الموقع، كما هو موضح في الشكل 4d، e. القمة حوليمكن أن يُعزى إلىاهتزاز الشد -أوكسي (الذي يتضمن الأكسجين الجسر غير البروتوني، )، والقمم المميزة حول و يمكن أن يُعزى إلى النمط المعتاد و قمم الاهتزاز. بالإضافة إلى ذلك، فإن القمم المميزة حول 300 ويمكن أن يُعزى إلى أكسيد أو مركب مرتبط بالسترونتيومتظهر الشكل أنه قبل وبعد عملية OER، يظهر SI بوضوحذروة اهتزاز الشد -أوكسي، مؤكداً وجود البيروفكيت الهيكل. ومع ذلك، بالنسبة لـ ، انخفاض كبير في الـ Ir-يتم ملاحظة ذروة اهتزاز الشد -أوكسي. قد يُعزى ذلك إلى
الشكل 4 | التوصيفات في الموقع للعينات. أنسبة العينات المختبرة بواسطة DEMS.رسم تخطيطي لـ ICP-MS في الموقعو (ج) مخطط التحويل التفاضلي الخاص به، في المجموعة: مخطط موسع. د، هـ قياسات رامان في الموقع ورسم خرائط رامان لعينات SI وعينات.
تدمير الـهيكل بواسطة Codissolution لتشكيل التنسيق المنخفض الهيكل، مما يؤدي إلى تقليل كبير في شدة الذروة. علاوة على ذلك، فإن القمم المميزة لأكاسيد/ مركبات مرتبطة بالسترونشيوم بالقرب من 300 وتقريبًا تختفي من جهد الدائرة المفتوحة (OC). مع نتائج ICP-MS في الموقع، يمكن تأكيد ذلك بشكل أكبر بأنه ناتج عن ذوبان أكاسيد/مركبات مرتبطة بالسترونتيوم.
أدلة رئيسية على النشاط العالي للتنسيق المنخفضفي كودوبيدلـ OER
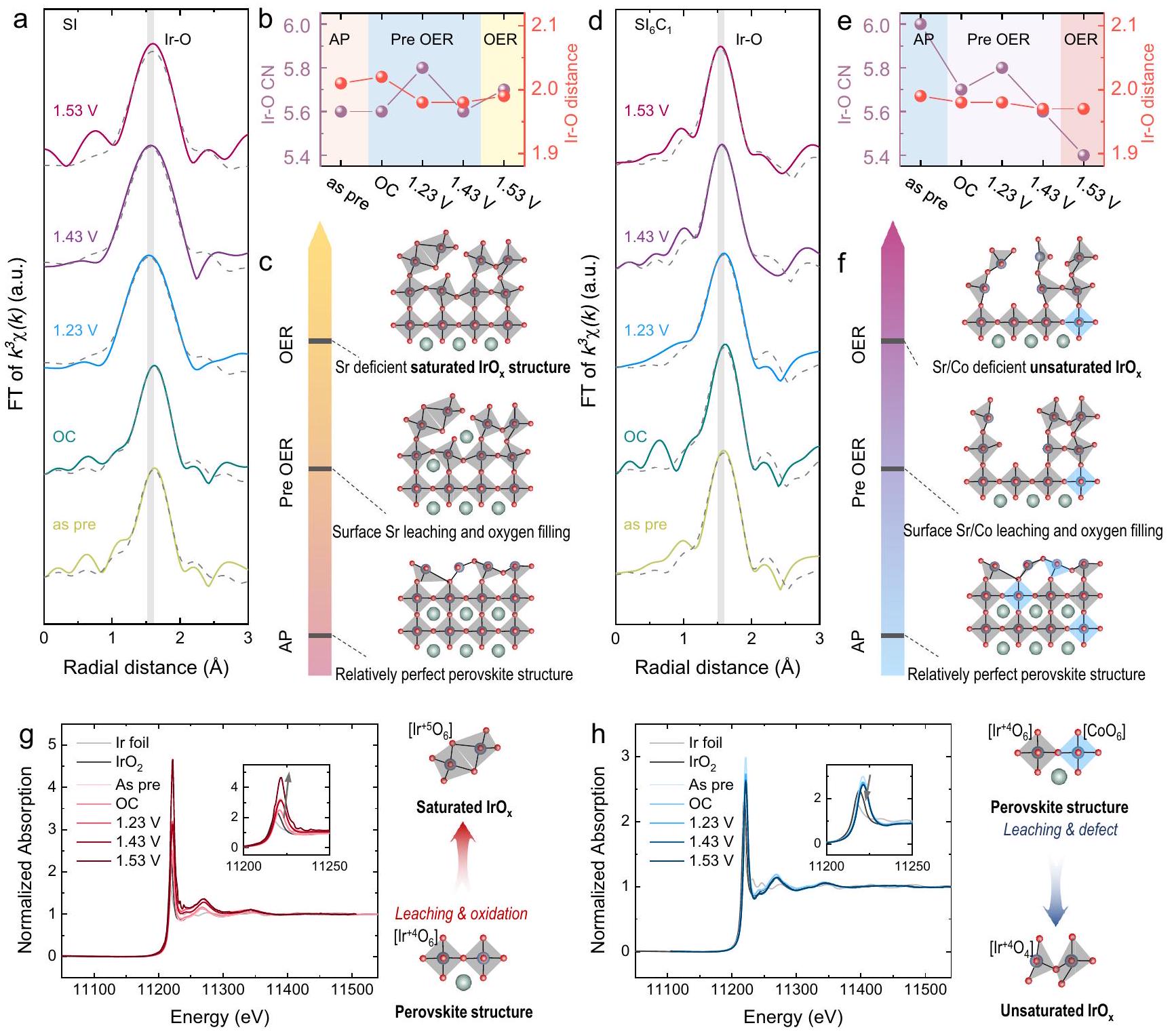
لتوضيح آلية التكوين لـعلى السطح، تم تسجيل طيف EXAFS شامل في الموقع عند Ir- حافة لمراقبة تطور التنسيق المحلي لـ Ir، كما هو موضح في الشكل 5a-e. في البداية، قمنا بفحص تغييرات التنسيق لعينة SI، كما هو موضح في الشكل 5a و b. تمتلك عينة SI في حالتها الأصلية عدد تنسيق Ir-O يبلغ 5.6، مما يشير إلى وجود هيكل إكسايدري Ir-O سليم بشكل كبير داخل المحفز. خلال مرحلة ما قبل OER (مقابل RHE)، أظهرت أرقام تنسيق Ir-O لعينة SI نمطًا من الزيادة الأولية تليها انخفاض.
أظهرت التقارير السابقة زيادة ملحوظة في عدد تنسيق Ir-O لـعينات الفيلم الرقيق أثناء عملية تطوير الأكسجين، والتي تم نسبها إلى عملية إعادة تعبئة الأكسجين. في هذه الدراسة، أدى ذوبان Sr إلى تغييرات في الهيكل السطحي لـ Sl. ومع ذلك، لم يكن هناك فرق كبير في LOER في عينة SI. لذلك، خلال عملية أكسدة OER، كانت سرعة تجاوز التعبئة معدل LOER، مما أدى إلى الاتجاه الملحوظ في أعداد التنسيق. خلال مرحلة OER (1.63 فولت مقابل RHE)، أظهر عدد تنسيق Ir-O لعينة SI زيادة طفيفة، مما يشير إلى أنه عند الجهود العالية، ظل الأكسجين الشبكي لعينة SI غير نشط، مما أدى إلى الزيادة الأعلى.معدل التعبئة مقارنة بمعدل LOER. بالإضافة إلى ذلك، عند 1.23 فولت مقابل RHE، خفض كبير في الـتم ملاحظة طول الرابطة في عينة SI، مما يشير بشكل محتمل إلى تحول في الهيكل المشترك لزوايا الأوكتاهيدرال خلالعملية التعبئةباختصار، كما هو موضح في الشكل 5c، يخضع آلية الموقع السطحي لعينة SI لعملية ديناميكية من هيكل بيروفسكايت مكتمل نسبيًا إلى ذوبان الموقع وملء المواد الممتصة، مما ينتقل في النهاية إلى حالة مشبعة.هيكل.
قمنا بعد ذلك بتحليل التغيرات في التنسيق فيعينة، كما هو موضح في الشكل 5d، e. عدد تنسيق Ir-O الأولي لـكان 6.0، مما يشير إلى هيكل أوكتاهيدرالي Ir-O أكثر كمالاً مقارنةً بـ SI. ومع ذلك، قبل مرحلة OER (OC، مقابل RHE)، عدد تنسيق Ir-O لـ أظهر العينة اتجاهًا ملحوظًا في الانخفاض. وهذا يتماشى مع تشكيل التنسيق المنخفض.كما لوحظ في نتائج ICP-MS في الموقع. علاوة على ذلك، خلال مرحلة OER، كان عدد تنسيق Ir-O لـانخفضت بشكل أكبر إلى 5.4، وهو أقل بكثير من SI، مما يؤكد أن الأكسجين النشط في الشبكةسهلت بشكل أكبر تشكيل مواد ذات تنسيق منخفض عالية النشاط.
لتحقيق صحة توليد غير المشبعتم تحليل طيف XANES في الموقع في الهيكل حافة لدراسة حالة الأكسدة للـ Ir، كما هو موضح في الشكل 5 ج، ح. الـ Ir-أظهر حافة الامتصاص لـ SI زيادة في شدة الخط الأبيض خلال عملية OER، مما يشير إلى زيادة في حالة الأكسدة لـ Ir. بشكل مختلف، فإن Ir-حد الامتصاص لـأظهر انخفاضًا طفيفًا في شدة الخط الأبيض، مما يدل على انخفاض في حالة الأكسدة للبلاتين. بالاقتران مع معلومات حالة التكافؤ لعينات المرجع Ir و IrO.يمكن استنتاج آلية تغيير حالة التكافؤ لإيريديوم خلال عملية تطوير الأكسجين. خلال عملية تطوير الأكسجين لـ SI، يحدث ذوبان السترونشيوم وملءأدى إلى تشبع تنسيق Ir-O، مما يظهر حالة تكافؤ أعلى. على العكس، بالنسبة لـ، أدى حل الشركة إلى
الشكل 5 | توصيف XAS في الموقع للعينات. أ مخطط تركيب R-space لعينة SI. التغيرات في تنسيق Ir -O و طول الرابطة لعينة السيليكون.رسم بياني لتغيير الهيكل لعينة SI؛رسم بياني لتناسب R-spaceعينة. مخطط تنسيق Ir-O وتغير طول رابطة Ir-Oعينة.
رسم بياني لتغيير الهيكلعينة.إر-رسم بياني لحافة الامتصاص لعينة SI، في المجموعة: رسم بياني مكبر.إر-رسم بياني لحافة الامتصاصعينة، في المجموعة: رسم توضيحي مكبر. تكوين Ir-O غير المشبع، مما يؤدي إلى انخفاض في متوسط حالة الفالنس لإيريديوم. هذه الملاحظة تدعم بشكل أكبر أن الكوبالت يمكن أن يعزز توليد غير المشبع.الهياكل، متوافقة مع النتائج من التوصيفات السابقة.
نقاش
آلية التحفيز OER على SrIrO المضاف إليه الكوبالتمحفز
لقد أوضحت دراستنا عملية إعادة بناء السطح لمحفزات سلسلة SIC من خلال الحسابات النظرية وسلسلة شاملة من التوصيفات في الموقع. نحن الآن نتابع لمناقشة وتلخيص آليات OER المحتملة المرتبطة بمحفزات سلسلة SIC. هناك آليتان معروفتان على نطاق واسع لـ OER، وهما آلية تطور الامتصاص (AEM) وآلية الأكسجين الشبكي (LOM)، كما هو موضح في الشكل 6a.لقد أظهرت حساباتنا النظرية أن السطحمركز الفرقةالنموذج أقرب إلى مستوى فيرمي مقارنة بنموذج الإيريديوم، مما يشير إلى أن الأكسجين الجسري في Co-O-Ir يميل حرارياً إلى أن يتم أكسدته.كشفت مطيافية رامان في الموقع أنذروة اهتزاز الشد -أوكسي (تشير إلى الأكسجين الجسري) فيتم تقليل المحفز النظامي بشكل ملحوظ خلال عمليات OC و OER عند مقارنته بنظام Ir. علاوة على ذلك، وجدت اختبارات DEMS أن محتوى أعلى من Co يعزز LOM.
على الرغم من هذه النتائج، تشير نتائج نظام إدارة البيانات لدينا إلى أن النموذج المعتمد على الطاقة يبقى الآلية السائدة، بينما يساهم النموذج اللوجستي بشكل كبير في تشكيل غير المشبع.. وبالتالي، نقترح آلية تحفيزية مميزة، وهي آلية تطور الممتزات المعززة بالأكسجين الشبكي (LOPAEM)، كما هو موضح في الشكل 6c. على عكس الآليات التحفيزية التقليدية لتفاعل الأكسدة (OER)، تتضمن LOPAEM العمل التآزري لكلا الآليتين. بشكل محدد، عندما تصبح سرعة أكسدة الأكسجين الشبكي في بعض المحفزات سريعة بشكل مفرط، سيؤدي ذلك إلى تكوين عدد كبير من فراغات الأكسجين السطحية (أي، مواقع المعادن غير المشبعة)، مما يغير المواقع التحفيزية الأصلية للمحفز. قد تظهر هذه المواقع المعدنية غير المشبعة أداءً أكثر كفاءة في AEM، وبالتالي تحقيق LOPAEM. تشير نتائجنا إلى أن LOM في محفزات نظام Co/Ir هو خطوة أساسية لتفعيل المحفز. تشكيل Ir-
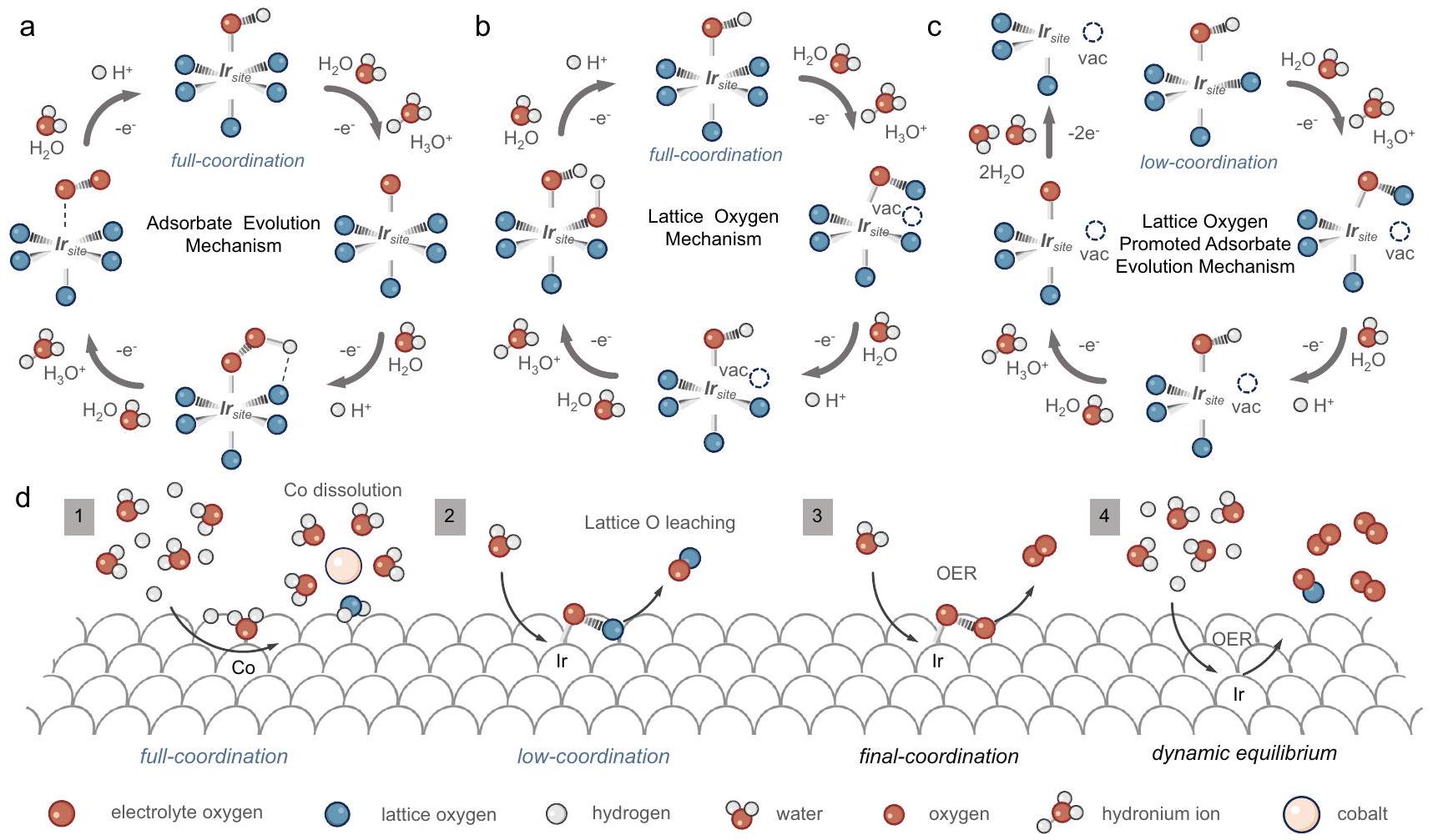
الشكل 6 | آلية OER المقترحة المحتملة. أ آلية تطور الممتزات. ب آلية الأكسجين الشبكي. ج آلية تطور الممتزات المعززة بالأكسجين الشبكي. د الآلية الحفزية لـ Co-doped SrIrOمحفز
يقلل بشكل كبير من جهد التأكسد الكهربائي للكاتاليس، مما يجعله أكثر ملاءمة من الناحية الديناميكية الحرارية. وهذا يتماشى مع حساباتنا النظرية ونتائج DEMS. علاوة على ذلك، فإن عملية LOM غير قادرة على إنتاج هيكل تنسيق Ir-O أقل، حيث إن ذوبان الأكسجين الشبكي يحول…مركز بعيد عن مستوى فيرمي، مما يمنع المزيد من الأكسدة (كما هو موضح في قسم الحسابات النظرية).
في ضوء النتائج المذكورة أعلاه، قمنا بتلخيص الآلية لـالمحفز النظامي كما هو موضح في الشكل 6d. في البداية، يذوب الكوبالت على سطح المحفز تحت ظروف حمضية، مما يزيل جزءًا من الأكسجين الشبكي الرابط لتشكيل هيكل بتنسيق أقل. بعد ذلك، يذوب الأكسجين الشبكي خلال عملية OER، مكونًا حالة غير مشبعةهيكل يظهر نشاط AEM عالي. أخيرًا، تحت التوازن الديناميكي لملء الممتزات وLOM، يقوم المحفز بإجراء AEM لتسهيل OER بكفاءة.
باختصار، قمنا بتصميم محفز عالي النشاط من خلال إضافة الكوبالت والذوبان الديناميكي لـأيونات ثنائية المعدن لدراسة الآلية التحفيزية لـبيروفسكايت قائم على. تظهر الحسابات النظرية والتوصيفات في الموقع (DEMS، رسم خرائط رامان في الموقع، XAS في الموقع و ICP-MS في الموقع) أن الذوبان الديناميكي للكوبالت أمر حاسم لتشكيل مواد غير مشبعة عالية النشاط.تظهر المحفزات التي تم تصنيعها حديثًا حركيات تفاعل OER أعلى منوتجاريالعامل المساعد في كل من المحلل الكهربائي ومحلل الماء PEM، يكشف عن آلية تعزيز النشاط التحفيزي من خلال ضبط المواقع التحفيزية. هذه الدراسة ذات أهمية كبيرة لفهم الأداء التحفيزي العالي لعملية الأكسدة الكهربائية للأكسجين (OER) للعوامل المساعدة المعتمدة على الإيريديوم، وستوفر أساسًا مهمًا لتصميم وتحضير عوامل مساعدة عالية الأداء لعملية الأكسدة الكهربائية للأكسجين في البيئات الحمضية.
طرق
المواد الكيميائية
المواد الكيميائية المستخدمة في هذه الدراسة تم استلامها جميعًا من الشركة المصنعة. هكساهيدروكلوريد البوتاسيوم (IV) [ماكلين] ، نترات الكوبالت (III) سداسي الماء [، AR، سيغما-ألدريتش، نترات السترانشيوم (II)تم استخدام حمض الستريك أحادي الهيدرات (AR، LookChem.) كمواد أولية.
تحضير المحفز SI. تم تحضير المحلول A عن طريق إذابة وحمض الستريك في 5.0 مل من الماء المقطر. تم تحضير المحلول B عن طريق إذابةفي 4.0 مل من الإيثيلين غليكول. ثم تم إضافة المحلول A قطرة قطرة مع التحريك إلى المحلول B. تم تجفيف المزيج الناتج عندلمدة 12 ساعة للحصول على منتج صلب بني كمواد سابقة. بعد ذلك، تم تسخين المادة السابقة في الهواء عندلـلـ لمدة 3 ساعات ، و لمدة 6 ساعات بمعدل تسخين قدره. بعد ذلك، الفائضتمت إزالة الشوائب عن طريق التفاعل مع محلول HCl بتركيز 1.0 م لمدة 12 ساعة للحصول علىمحفز
إعدادالمحفز. تم تحضير المحلول A عن طريق إذابة وحمض الستريك في 5.0 مل من الماء المقطر. تم تحضير المحلول B عن طريق إذابة و في 4.0 مل من الإيثيلين غليكول. كانت الخطوات التالية مماثلة لتحضير المحفز SI الموصوف أعلاه.
إعدادالمحفز. تم تحضير المحلول A عن طريق إذابة وحمض الستريك في 5.0 مل من الماء المقطر. تم تحضير المحلول B عن طريق إذابة و في 4.0 مل من الإيثيلين غليكول. كانت الخطوات التالية مماثلة لتحضير المحفز SI الموصوف أعلاه.
إعدادالمحفز. تم تحضير المحلول A عن طريق إذابة وحمض الستريك في 5.0 مل من الماء المقطر. تم تحضير المحلول B عن طريق إذابة و في 4.0 مل من الإيثيلين غليكول. كانت الخطوات التالية مماثلة لتحضير المحفز SI الموصوف أعلاه.
إعدادالمحفز. تم تحضير المحلول A عن طريق إذابة وحمض الستريك في 5.0 مل من الماء المقطر. تم تحضير المحلول B عن طريق إذابة و في 4.0 مل من الإيثيلين غليكول. كانت الخطوات التالية مماثلة لتحضير المحفز SI الموصوف أعلاه.
إعدادالمحفز. تم تحضير المحلول A عن طريق إذابة وحمض الستريك في 5.0 مل من الماء المقطر. تم تحضير المحلول B عن طريق إذابة و في 4.0 مل من الإيثيلين غليكول. كانت الخطوات التالية متطابقة مع تحضير المحفز SI الموصوف أعلاه.
توصيف المواد. تم إجراء توصيف هيكل البلورة على المستوى الذري باستخدام مجهر إلكتروني ناقل مصحح للانحراف (JEM-ARM200P، اليابان) يعمل عند 300 كيلوفولت. تم استخدام تحليل الأشعة السينية المشتتة للطاقة (EDX) لقياس المحتوى النسبي للعناصر. تم الحصول على أنماط حيود الأشعة السينية (XRD) لـتم تسجيلها على جهاز تحليل الأشعة السينية (Smart lab) باستخدامإشعاع ) بحجم خطوة وزمن الخطوة 0.2 ثانية فينطاق. تم إجراء مطيافية الإلكترونات الضوئية بالأشعة السينية (XPS) باستخدام مطياف الإلكترونات الضوئية بالأشعة السينية Thermo Scientific K-Alpha، وتم معايرة جميع طيفيات XPS باستخدام الكربونخط عند 284.8 إلكترون فولت. الشكل السطحي لـتمت ملاحظته باستخدام مجهر إلكتروني مسح (TESCAN MIRA LMS). بالنظر إلى المقاومة العالية للأحماض لـتم تحضير مضاد الماء الملكي عن طريق خلط حمض الهيدروكلوريك وحمض النيتريك فينسبة للتجربة. في هذا المحلول، 1.0 ملغ منتم إذابة المسحوق في 10 مل من الماء الملكي وتركه لمدة 1-3 أسابيع بعد معالجة بالموجات فوق الصوتية بشكل شامل. أخيرًا، تم تحديد نسب كل عنصر فيتم تحديدها بواسطة تحليل ICP-MS (iCAP RQ).
توصيفات في الموقع وتأكيد LOER. طيف امتصاص الأشعة السينية لـ Ir-edge تم الحصول عليها في شعاع BL17B و BL20U1 من منشأة الإشعاع السنكروتروني في شنغهاي (SSRF). تم جمع الطيف إما في وضع النقل أو وضع الفلورية باستخدام كاشف لايتل. تم جمع العينات المرجعية في وضع النقل. تم طحن العينات وتطبيقها بشكل موحد على شريط لاصق خاص. تم إجراء توصيفات XANES في الموقع في وضع الفلورية في نفس الشعاع. تم رش العينات على ورق الكربون بتحميل قدره كقطب عمل. تم إجراء القياسات تحت نفس الظروف التي أجريت فيها قياسات OER في خلية مصممة ذاتيًا. تم إجراء خصائص الطيف Raman في الموقع باستخدام مجهر رامان كونفوكالي inVia من شركة Renishaw. تم ضبط طاقة الليزر عند 532 نانومتر مع 1% من الطاقة عند شبكة منتم معايرة قمة السيليكون قبل الاختبار. خلال عملية OER، تم إجراء قياسات مطيافية الكتلة الكهروكيميائية التفاضلية (DEMS) باستخدام جهاز QAS 100 من شركة شنغهاي لينغلو للأدوات لتحديد المنتجات التفاعلية المتطايرة من محفزات سلسلة SI ومحفز موسوم بـمشبعوكانت أسلاك Pt تعمل كإلكترود مرجعي (RE) وإلكترود مضاد (CE)، على التوالي. تم تحضير الإلكترود العامل (WE) عن طريق ترسيب الذهب (Au) علىغشاء PTFE المسامي السميك، يليه ترسيبحبر المحفز ( ) على سطح الذهب. تم تحقيق وسم النظائر الحفازة من خلال تدوير القطب في لمدة ثمانية دورات باستخدام الفولتمترية الدورية (CV) بمعدل مسحفي نطاق مقابل . بعد ذلك، الـتم شطف القطب المسمى بـلإزالة المتبقي. أخيرًا، تم اختبار القطب كهربائيًا ضد فيحل عند إمكانيات مختلفة بمعدل مسح. تم تطبيع إشارة DEMS بواسطة كثافة التيار (في الوقت نفسه، تم إجراء قياسات فورية لمنتجات الغاز ذات الأوزان الجزيئية المختلفة الناتجة خلال عملية OER باستخدام مطياف الكتلة. تم إجراء تجارب ICP-MS في الموقع باستخدام جهاز Thermo Scientific iCAP RQ. إعداد التجربة تكونت من خلية قياسية ذات ثلاثة أقطاب مع قطب عمل من الكربون الزجاجي بقطر 3 مم، متوافقة مع الاختبارات الكهروكيميائية. كان القطب المرجعي المستخدم هو قطب الكالوميل المشبع (Hg/ )، وكانت القطب المضاد عبارة عن قطب من رقائق البلاتين. لضمان الكشف الدقيق عن توزيع الأيونات، تم استخدام محرك خلط لمنع تسرب الأخطاء والانحلال، مع أخذ عينات البيانات كل 15 ثانية.
الخصائص الكهروكيميائية. لتحضير حبر المحفز، تم خلط 0.5 ملغ من المحفز مع 1.0 مل منمحلول نافيون ومعتدل. بعد ذلك، تم إيداع حجم الحبر المحضر على قطب كربون زجاجي (GCE) بقطر 5 مم وتم تجفيفه باستخدام مصباح الأشعة تحت الحمراء. قبل الاستخدام، تم تلميع GCE بـمسحوق الألومينا وتم شطفه ثلاث مرات بمزيج من الماء عالي النقاء والإيثانول. تم إجراء القياسات الكهروكيميائية في نظام ثلاثي الأقطاب باستخدام محطة عمل كهروكيميائية (CHI 760E). كانت القطب المرجعي المستخدم هوإلكترود في 0.5 مالمحلول الكهربائي، بينما كان قضيب الكربون يعمل كالكاثود. كانت الإلكترود العاملة هي GCE مع المحفز. تم إجراء LSV و CV فيحل بمعدل مسحتم استخدام خلية تحليل الماء بالكهرباء PEM محلية الصنع مع غشاء تبادل البروتون لتقييم أداء محفز سلسلة SI. خطوة تحضير أحبار المحفز هي نفسها الطريقة المذكورة أعلاه. كانت إجمالي تحميل المحفز على القطب 1.0 ملغ وتم إيداع جميع أحبار المحفز على ورق الكربون. ). درجة حرارة الخلية ( 25,65 , و تم الحفاظ على ذلك بواسطة لوحة تسخين كهربائية وتم قياسه بواسطة مسبار حرارة في الإلكتروليت.
طرق حسابية. تم إجراء حسابات نظرية الكثافة المعتمدة على دوران المغناطيس (DFT) باستخدام موجات مستوية وبدائل فوق ناعمة (USPP) مع تصحيح دالة تبادل بيردو-بورك-إرنزرهوف (PBE) كما هو مطبق في Quantum ESPRESSO.تم استخدام حد طاقة قدره 25 ري للتوسع في الموجات المستوية لدالة الموجة الإلكترونية. تم استرخاء الهياكل الذرية للنماذج بالكامل حتى تم تحقيق التوافق الذاتي مع معايير التقاربللطاقة وبور للإحداثيات الذرية. لمنع التفاعل بين الطبقات، يتم استخدام لوح فراغي منتم استخدامه لعزل السطح. من أجل تحسين هندسة الكتلة،تم استخدام مجموعة نقاط كيبونكهورست-باك، بينماتم استخدام المجموعة لحسابات الهيكل الإلكتروني. التصحيح لكل مادة ممتصة وسطح، مع قيم نموذجية منلـ، * أو على التوالي. لمحاكاة غير المشبعة المعقدة الهيكل، نبدأ ببناء وتحسين هيكل اللوح الأصلي . ثم، تم إزالة ذرات السطح Sr من لوح لمزيد من التحسين. وفقًا لتقرير سابق، الـعدد التنسيق علىالسطح حوالي 4.5، تم تحقيقه عن طريق إزالة 4 ذرات Sr وذرات الأكسجين الثلاثة المجاورة لها. يمكن تطبيق نفس الإجراء على النظام المدعوم بكوبالت، حيث تؤدي إزالة ذرة كوبالت أيضًا إلى إزالة 2 من ذرات الأكسجين المجاورة.
تحليل XAS. تم معالجة بيانات هيكل الامتصاص الدقيق للأشعة السينية الممتدة (EXAFS) المكتسبة وفقًا للإجراءات القياسية باستخدام وحدة ATHENA من حزمة برامج Demeter.تم الحصول على طيف EXAFS عن طريق طرح الخلفية بعد الحافة من الامتصاص الكلي ثم تم تطبيعه بالنسبة لخطوة قفزة الحافة.تم تحويل البيانات إلى الفضاء الحقيقي (R) باستخدام نافذة هانينغ ) لفصل المساهمات من قذائف التنسيق المختلفة. لتحديد المعلمات الهيكلية الكمية حول الذرات المركزية، تم إجراء ملاءمة معلمات منحنى المربعات الصغرى باستخدام وحدة ARTEMIS من حزمة برامج Demeter.
توفر البيانات
البيانات التي تم توليدها في هذه الدراسة متاحة في المعلومات التكميلية ويمكن الحصول عليها من المؤلفين عند الطلب. يتم توفير بيانات المصدر مع هذه الورقة.
References
Hong, W. T. et al. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8, 1404-1427 (2015).
Hwang, J. et al. Perovskites in catalysis and electrocatalysis. Science 358, 751-756 (2017).
Chen, Z. et al. Advances in oxygen evolution electrocatalysts for proton exchange membrane water electrolyzers. Adv. Energy Mater. 12, 2103670 (2022).
Xue, Y. et al. Sulfate-functionalized as highly efficient oxygen evolution reaction electrocatalyst in acid. Adv. Funct. Mater. 31, 2101405 (2021).
Zhao, Z. L. et al. Boosting the oxygen evolution reaction using defect-rich ultra-thin ruthenium oxide nanosheets in acidic media. Energy Environ. Sci. 13, 5143-5151 (2020).
Shih, A. J. et al. Water electrolysis. Nat. Rev. Methods Primers 2, 84 (2022).
Huynh, M., Bediako, D. K. & Nocera, D. G. A functionally stable manganese oxide oxygen evolution catalyst in acid. J. Am. Chem. Soc 136, 6002-6010 (2014).
Gunasooriya, G. K. K. & Nørskov, J. K. Analysis of acid-stable and active oxides for the oxygen evolution reaction. ACS Energy Lett. 5, 3778-3787 (2020).
Cao, L. et al. Dynamic oxygen adsorption on single-atomic ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
Danilovic, N. et al. Using surface segregation to design stable Ru-Ir oxides for the oxygen evolution reaction in acidic environments. Angew. Chem. Int. Ed. 53, 14016-14021 (2014).
Hubert, M. A. et al. Acidic oxygen evolution reaction activitystability relationships in Ru-based pyrochlores. ACS Catal. 10, 12182-12196 (2020).
Cui, X. et al. Robust interface Ru centers for high-performance acidic oxygen evolution. Adv. Mater. 32, 1908126 (2020).
Shan, J. et al. Charge-redistribution-enhanced nanocrystalline Ru@lrO electrocatalysts for oxygen evolution in acidic media. Chem 5, 445-459 (2019).
Lin, C. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021).
Frydendal, R., Paoli, E. A., Chorkendorff, I., Rossmeisl, J. & Stephens, I. E. Toward an active and stable catalyst for oxygen evolution in acidic media: Ti-stabilized . Adv. Energy Mater. 5, 1500991 (2015).
Morgan Chan, Z . et al. Electrochemical trapping of metastable ions for activation of oxygen evolution catalysts. Proc. Natl. Acad. Sci. 115, E5261-E5268 (2018).
Kuo, D.-Y. et al. Influence of strain on the surface-oxygen interaction and the oxygen evolution reaction of . J. Phys. Chem. C 122, 4359-4364 (2018).
Lee, K., Osada, M., Hwang, H. Y. & Hikita, Y. Oxygen evolution reaction activity in catalysts: correlations between structural parameters and the catalytic activity. J. Phys. Chem. Lett 10, 1516-1522 (2019).
Shin, S. et al. Single-phase perovskite nanofibers as a highly efficient electrocatalyst for a pH -universal oxygen evolution reaction. ACS Appl. Energy Mater 5, 6146-6154 (2022).
Yang, L. et al. Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing octahedral dimers. Nat. Commun. 9, 5236 (2018).
Seitz, L. C. et al. A highly active and stable IrO/ catalyst for the oxygen evolution reaction. Science 353, 1011-1014 (2016).
Ben-Naim, M. et al. Understanding degradation mechanisms in oxygen evolution electrocatalysts: chemical and structural microscopy at the nanoscale. Adv. Funct. Mater. 31, 2101542 (2021).
Akbashev, A. R. et al. Probing the stability of during active water electrolysis via operando atomic force microscopy. Energy Environ. Sci. 16, 513-522 (2023).
Wan, G. et al. Amorphization mechanism of electrocatalyst: how oxygen redox initiates ionic diffusion and structural reorganization. Sci. Adv. 7, eabc7323 (2021).
Chen, Y. et al. Exceptionally active iridium evolved from a pseudocubic perovskite for oxygen evolution in acid. Nat. Commun. 10, 572 (2019).
Tang, R. et al. Oxygen evolution reaction electrocatalysis on grown using molecular beam epitaxy. J. Mater. Chem. A 4, 6831-6836 (2016).
Chen, H. et al. Optimization of active sites via crystal phase, composition, and morphology for efficient low-iridium oxygen evolution catalysts. Angew. Chem. Int. Ed. 59, 19654-19658 (2020).
Liang, X. et al. Electrocatalytic water oxidation activity-stability maps for perovskite oxides containing 3d, 4d and 5d transition metals. Angew. Chem. Int. Ed. 62, e202311606 (2023).
Zhang, R. et al. A dissolution/precipitation equilibrium on the surface of iridium-based perovskites controls their activity as oxygen evolution reaction catalysts in acidic media. Angew. Chem. Int. Ed 131, 4619-4623 (2019).
Song, C. W., Suh, H., Bak, J., Bae, H. B. & Chung, S.-Y. Dissolutioninduced surface roughening and oxygen evolution electrocatalysis of alkaline-earth iridates in acid. Chem 5, 3243-3259 (2019).
Tomita, K., Miyata, T., Olovsson, W. & Mizoguchi, T. Strong excitonic interactions in the oxygen K-edge of perovskite oxides. Ultramicroscopy 178, 105-111 (2017).
Grimaud, A. et al. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 4, 2439 (2013).
Mefford, J. T. et al. Water electrolysis on perovskite electrocatalysts. Nat. Commun. 7, 11053 (2016).
Pavlovic, Z., Ranjan, C., Gao, Q., van Gastel, M. & Schlögl, R. Probing the structure of a water-oxidizing anodic iridium oxide catalyst using Raman spectroscopy. ACS Catal. 6, 8098-8105 (2016).
Korotcov, A. V., Huang, Y. S., Tiong, K. K. & Tsai, D. S. Raman scattering characterization of well-aligned and nanocrystals. J. Raman Spec. 38, 737-749 (2007).
Ni, S., Yang, X. & Li, T. Hydrothermal synthesis and photoluminescence properties of . Mater. Lett. 65, 766-768 (2011).
Biedermann, N. et al. High-pressure phase behavior of : an experimental and computational Raman scattering study. Phys. Chem. Miner. 44, 335-343 (2017).
Yoo, J. S., Rong, X., Liu, Y. & Kolpak, A. M. Role of lattice oxygen participation in understanding trends in the oxygen evolution reaction on perovskites. ACS Catal. 8, 4628-4636 (2018).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457 (2017).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and opensource software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537-541 (2005).
شكر وتقدير
تم دعم هذا العمل من قبل NSFC (52373215) وبرنامج العلوم والتكنولوجيا في سيتشوان (2023NSFSC0086) وصناديق البحث الأساسية للجامعات المركزية (YJ2021156). نشكر أيضًا محطة BL17B1 وBL20U1 في منشأة الإشعاع السنكروتروني في شنغهاي (SSRF) على المساعدة في التوصيفات.
مساهمات المؤلفين
تم تصور وتصميم التجارب بواسطة ج.-ر.ل. وج.-و.ز.، مع مدخلات من جميع المؤلفين. قام س.و.، ج.ز.، د.و.، ج.س. و ف.ي.ف. بإجراء تجارب الكيمياء الكهربائية والتوصيف. ساهم ك.ي. في دراسات XAS في الموقع واختبار PEM. قدم ه.ز. المساعدة في توصيف EELS و HAADF.
المصالح المتنافسة
يعلن المؤلفون عدم وجود مصالح متنافسة.
معلومات إضافية
معلومات إضافية النسخة الإلكترونية تحتوي على المواد التكميلية متاحة على https://doi.org/10.1038/s41467-024-46801-y. يجب توجيه المراسلات والطلبات للحصول على المواد إلى جاو-رين لي.
معلومات مراجعة الأقران تشكر Nature Communications تاو تشينغ و المراجعين المجهولين الآخرين على مساهمتهم في مراجعة الأقران لهذا العمل. يتوفر ملف مراجعة الأقران.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح. هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسب 4.0 الدولية، التي تسمح بالاستخدام والمشاركة والتكيف والتوزيع وإعادة الإنتاج بأي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح ما إذا تم إجراء تغييرات. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي للمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمواد. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي للمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، ستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http://creativecommons.org/licenses/by/4.0/.
(ج) المؤلفون 2024
كلية علوم المواد والهندسة، جامعة سيتشوان، تشنغدو 610065، الصين. مدرسة الكيمياء، جامعة صن يات صن، قوانغتشو 510275، الصين. قسم الهندسة الميكانيكية، جامعة مدينة هونغ كونغ، 83 طريق تات تشي، كولون، منطقة هونغ كونغ الإدارية الخاصة 999077، الصين. مختبر المواد الرئيسية لتحويل الطاقة، معهد شنغهاي للسيراميك، الأكاديمية الصينية للعلوم (SICCAS)، 585 طريق هيسو، شنغهاي 200050، الصين. مركز المجهر الإلكتروني، مدرسة العلوم الفيزيائية والتكنولوجيا، جامعة لانتشو، لانتشو 730099، الصين. الجامعة الوطنية للبحوث النووية MEPhI (معهد موسكو للهندسة الفيزيائية)، كاشيرسكو الشارع 31، موسكو 115409، روسيا. ساهم هؤلاء المؤلفون بالتساوي: جيا-وي تشاو، كايهانغ يوي، هونغ زانغ. البريد الإلكتروني: ligaoren@scu.edu.cn
Electrocatalytic water splitting is a promising route for sustainable hydrogen production. However, the high overpotential of the anodic oxygen evolution reaction poses significant challenge. -based perovskite-type catalysts have shown great potential for acidic oxygen evolution reaction, but the origins of their high activity are still unclear. Herein, we develop a Co-doped system to enhance oxygen evolution reaction activity and elucidate the origin of catalytic activity. In situ experiments reveal Co activates surface lattice oxygen, rapidly exposing active sites, while bulk Co doping optimizes the adsorbate binding energy of . The Co -doped demonstrates high oxygen evolution reaction electrocatalytic activity, markedly surpassing the commercial catalysts in both conventional electrolyzer and proton exchange membrane water electrolyzer.
Water splitting for hydrogen production offers the advantage of producing clean and sustainable fuel without carbon emissions . To date, proton exchange membrane (PEM) water electrolysis is one of the most established ways in the field of green hydrogen production . However, the high overpotential typically associated with the anodic oxygen evolution reaction (OER) poses a significant challenge to enhancing hydrogen production efficiency from water electrolysis . Furthermore, the advancement of OER catalysts designed for acidic medium poses a greater challenge compared to those intended for alkaline medium. Since highly OER-active electrocatalysts are mainly comprised of metal oxides or hydroxides, most of which exhibit poor stability under acidic conditions . Therefore, the development of efficient OER catalysts functioning in acidic medium is critical.
Current, effective acidic OER catalysts include Ru, Ir, and Mnbased metal oxides . In particular, with an perovskite structure (where A typically represents an alkaline earth metal and B represents a transition metal) shows high acidic OER catalytic
performance . In recent years, extensive researchers have been conducted to understand the OER catalytic mechanism of . Jaramillo et al. prepared the (001) plane film via a laser epitaxy strategy, and observed an incremental improvement in its OER catalytic performance throughout the catalytic process . Through combining theoretical calculations and experiments, they discovered that the high catalytic activity of could be attributed to the exposed sites following Sr dissolution. In further comprehensive studies, researchers confirmed the structural modifications of under acidic OER process using secondary ion mass spectrometry (SIMS), in situ atomic force microscopy (AFM), and X-ray absorption spectroscopy (XAS), respectively . They proposed a correlation between the Sr dissolution process and the formed surface activities. Moreover, an investigation involving further indicates that the OER activity also originates from the amorphous structure formed by the dissolution of . These studies appear to identify the active component of -based perovskites as the amorphous
structure. Nevertheless, most studies overlook the impact of B-site dissolution on surface oxygen stability and Ir-O coordination structure, the precise reasons contributing to the high OER catalytic performance of Co -doped catalysts remain unclear. This issue primarily stems from the challenge faced by researchers in elucidating two key aspects of the OER catalytic process: (1) The key role of Co dissolution in catalytic processes and (2) the influence of surface and bulk Co on sites.
To address the two key challenges mentioned above, we designed a B-site Co-doped SrIrO3 system to discern the dissolution mechanism at catalyst sites and the origins of catalytic activity, as shown in Fig. 1a. In situ inductively coupled plasma mass spectrometry (ICP-MS) experiments revealed simultaneous ion dissolution of Co and Sr , caused by acid corrosion prior to OER process. At the OER potential ( 1.60 V vs. RHE), the dissolution phenomenon was found to be negligible. Along with theoretical calculations, a series of in situ experiments including in situ Raman mapping, in situ XAS, and differential electrochemical mass spectrometry (DEMS) were conducted. These
results highlighted the role of Co in two critical ways: (1) Surface Co reduces the stability of the Co -O-Ir bridge oxygen in , leading to the rapid exposure of the low-coordination structure; (2) Bulk lattice Co optimizes the OOH binding energy of , consequently reducing the overpotential. Thus, the synthesized Co -doped demonstrated high OER activity, markedly surpassing commercial catalysts in PEM water electrolyzer. The insights obtained from this research would significantly enhance the understanding of high OER catalytic performance of -based perovskite catalysts, providing key insights for designing and preparing high-performance acidic OER catalysts.
Results
Fabrication and characterizations of -doped samples doped with different amounts of Co were synthesized by using the sol-gel method, and were fully acid washed before use. The samples were denoted as follows: , , and .
Fig. 1 | Characterizations of samples. a OER catalytic mechanism diagram of Codoped catalyst; HRTEM images and corresponding FFT diagrams of SI , and ; e -edge EELS spectra of ; f HAADF image of and
corresponding EDS mapping images; XRD spectra of , , and SI samples. i ICP-MS diagram of and ratios of , , and SI samples.
The crystal structures of these samples were analyzed through highresolution transmission electron microscopy (HRTEM) and X-ray diffraction (XRD) in Fig. , respectively. For HRTEM testing, three samples ( , and ) were selected. Specifically, SI exhibited an orthorhombic crystal structure. Fast Fourier transform (FFT) showed that the primary exposed crystal planes included (112), (110), and (002). Despite its low surface crystallinity, still clearly exposed crystal planes such as (112), (204), and (020). Meanwhile, displayed poor surface crystallinity with only a few areas showing short-range ordered crystallinity and no clear diffraction spots in the FFT pattern. To investigate the reason for the diminished crystallinity observed in sample, we conducted electron energy loss spectroscopy (EELS) analyses, the results of which are presented in Fig. 1e and Supplementary Fig. 3. The peaks at 532,542 , and 564 eV can be ascribed to the Ir 5 d , while the peak at 536 eV is attributable to the . Comparative analysis reveals that the Sr 4d peak of the SI sample is generally higher than that of the sample, suggesting that the presence of Sr contributes to maintaining the crystalline structure. Moreover, we identified a significant discrepancy between the surface and bulk Sr concentrations within the SI and samples, with this effect being particularly pronounced in the sample. This indicates that Co dissolution also impacts the proportion of Sr presenting on the surface. Further investigations were undertaken through energy-dispersive Xray spectroscopy (EDS) mapping (Fig. 1f). The results confirm that the dissolution of and the subsequent formation of .
XRD revealed that the diffraction peaks of SI were consistent with typical pseudocubic (Pnma) perovskites. Also, Co-doped SI also demonstrated similar diffraction peaks, confirming the successful synthesis of the perovskite-based catalyst . As shown in Fig. 1h, Co doping significantly reduced the crystallinity of SI and resulted in the diffraction peak shifting to the high angle, indicating lattice contraction in the Co -doped SI . In particular, and displayed almost no diffraction peaks, possibly due to the substantial dissolution of surface Co and Sr by acid washing process. These findings suggest that excessive Co doping may disrupt the pseudocubic perovskite structure after acid washing, a conclusion in line with the HRTEM results. The overall composition of the samples was analyzed by using ICP-MS, as shown in Fig. 1i. Notably, there was a significant discrepancy between the Co and Ir initial ratio and the final composition ratio of the samples. Samples with poor crystallinity, specifically and , exhibited the highest ratio, with slightly higher than . This could possibly be attributed to the difficulty in maintaining the perovskite structure in , which led to a large amount of Co dissolution during the acid washing process and, consequently, a reduced ratio. Despite possessing the highest ratios, these two samples were still significantly lower than the initial ratio, further confirming the instability of surface Co in acidic conditions. The ratios of , , and showed a decreasing trend from to as shown in. Figure 1i suggesting that lattice maintenance assists in stabilizing Co atoms in the bulk phase. Among various SI-based samples, the and displayed the lowest ratio, and the ratio significantly increased with the decrease of Co doping. The ratio of the SI sample was slightly higher than 1, indicating a slightly higher Sr proportion compared to Ir in bulk structure, as suggested by the EELS results shown in Supplementary Fig. 3. These conclusions were further confirmed by additional characterization methods such as scanning electron microscope (SEM), X-ray photoelectron spectroscopy (XPS), and Raman spectroscopy, as shown in Supplementary Fig. 5a, b.
High OER catalytic performance of Co-doped SrIrO
The OER catalytic performances of the , and SI samples were investigated in acidic medium. The linear sweep voltammetry (LSV) curves for , and SI samples were recorded after 10 cyclic voltammetry (CV) cycles, as shown in Fig. 2a. The results indicate that necessitates an
overpotential of only 245 mV to reach a current density of , which is approximately 5 times higher than that of SI at the same potential. Given that the OER catalytic process primarily occurs on the surface, the performance of and samples is generally superior and exhibits a similar trend. This aligns with previous characterization results, suggesting that despite varying Co doping ratios, the catalysts’ surface structures are comparable. As presented in Fig. 2b, c, the and samples show Tafel slopes within the range dec , significantly lower than that of SI ( ). This suggests that Co participation can enhance the reaction kinetics of OER. As shown in Fig. 2d, a comparison of the mass activity of the and samples reveals that the Co-doped samples all demonstrated high activity, significantly surpassing those of SI and .
To study the potential applications of SI series catalysts in water electrolysis, we further examined the electrocatalytic performance of three samples ( , and ) for water electrolysis by PEM. The schematic diagram of the PEM water electrolyzer is presented in Fig. 2e. The performance results of the PEM water electrolysis, shown in Fig. 2f, reveal that the catalytic activity of is significantly higher than those of SI and . It can achieve a current density exceeding at 2.0 V cell voltage at , which is much higher than those of the SI catalyst ( ) and catalyst ( ) under the same conditions. We further tested the long-term stability of catalyst for PEM water electrolysis as shown in Fig. 2g and Supplementary Fig. 6. The results indicate that the catalyst exhibits high stability, with a performance decay rate of , which is comparable to that of the SI catalyst and commercial catalyst. This exceptional high stability indicates that the catalyst exhibits the potential for practical application in PEM water electrolysis.
The roles of Co doping and dissolution in SrIrO studied by theoretical calculations
The impact of Co doping on was investigated through a theoretical study. As numerous early studies had substantiated that based catalysts undergo significant Sr dissolution during . This was examined by analyzing the Pourbaix diagrams of and , particularly focusing on their stability in acidic medium (Fig. 3a and Supplementary Fig. 7). The findings reveal a marked similarity in the stability properties of and . Under acidic conditions, Sr displays a thermodynamic inclination towards dissolution, thereby exposing a multitude of sites on the surface. Additionally, Co in the also exhibits instability in acidic medium, which could also result in substantial dissolution, consistent with the EDS mapping and ICP-MS results in Fig. 1.
As indicated by the Pourbaix diagram, the structure of catalyst is significantly influenced by the applied voltage. However, the two catalysts do not exhibit notable thermodynamic differences near the oxygen stability curve, although they display pronounced differences in thermodynamic tendencies across varying pH values. This suggests that the effect of dissolution at different OER potentials may be significantly less than the effect of electrolyte pH , particularly in strongly acidic medium. Previous research suggests that the Ir-O coordination number of surface on during OER catalysis is . While the presence Sr content at the trace amount has been detected in numerous studies, determining the exact state of Sr at the subnanometer scale remains a challenge . By combining the results from the Pourbaix diagrams and above characterizations, seven models were constructed to investigate the theoretical OER catalytic sites and catalytic performance of and Co -doped (Fig. 3b and Supplementary Fig. 8).
The theoretical calculations were conducted on the models, as shown in Fig. 3c. The computed free energy diagrams (Fig. 3d) reveal that the rate-determining step for all models is the formation of *OOH. The Co doping, whether at the surface and bulk phase, which would
Fig. 2 | Electrochemical measurements of samples. a OER polarization curves of , and SI samples with a mass loading of in and (b) corresponding Tafel slopes. The resistance values for , and were 3.9, 3.7, 3.6, 4.7, 3.2, 4.6 and , respectively. c Comparison of overvoltages and Tafel slopes of , , and SI samples. d Comparison of OER mass activity of , and samples. e Schematic diagram of PEM water electrolysis device. PEM water electrolysis performance of , and SI samples, in set: PEM water electrolysis device photograph. g PEM water electrolysis stability of sample.
not change the coordination structure of octahedron, can only enhance OER catalytic activity to a certain extent. However, such activity enhancements are restricted, especially for bulk-doped Co, where the OER overpotential is reduced by only . In contrast, the surface unsaturated , which forms following Co dissolution, exhibits a significant improvement in the catalytic activity for oxygen evolution, with the overpotential decreasing by (Fig. 3e). These findings suggest that Co may not directly participate in the catalysis, but rather promote the surface reconstruction through site dissolution, leading to rapid exposure of more low-coordination active sites.
To further investigate the activity origin, density of states (DOS) analyses were performed on these models (Fig. 3f). The data suggest that Co doping significantly affects the Co-O-Ir bridge oxygen, shifting the band center closer to the Fermi level. According to the related studies, such a shift in the band center substantially affect the surface stability of the catalyst and may be associated with oxygen dissolution . This indicates that the primary effect of Co doping is to alter the surface stability of , and promote the surface reconstruction and the formation of active sites. Additionally, the doping and dissolution of Co significantly influence the Ir band
center (Fig. 3g). The displacement of the metal 3d orbitals typically directly impacts the binding energy to the adsorbate. In this study, the negative shift of the Ir band center led to a decrease in *OOH free energy, significantly enhancing the OER catalytic activity (Fig. 3g). By establishing the relationship between the Ir and band centers and the theoretical overpotential, a new OER volcano plot was formulated (Fig. 3h). The data revealed that the moderate band center and the lower Ir band center play crucial roles in the catalytic activity of -based catalysts.
In situ characterizations of surface structural changes of catalysts
To elucidate the catalytic mechanism, DEMS tests were first conducted, as shown in Fig. 4a. The surfaces of , , and samples were labeled with , as shown in Supplementary Figs. 10-12. For each sample, signals at different cyclic voltammetry cycles were collected, and the ratio of to was utilized to eliminate the natural abundance of in the air. As shown in Supplementary Fig. 13, the lattice oxygen evolution reaction (LOER) trend of most catalysts across different cyclic voltammetry cycles is similar, and the Co doping ratio appears to facilitate the
Fig. 3 | Theoretical calculations of -based perovskite catalysts. a Pourbaix diagram of . b Possible computational models of (surface Co doping) and (second layer Co doping with unsaturated IrO ) for DFT calculations. c Adsorbate evolution mechanism (AEM) diagram. d OER free energy
diagrams of different models. e Overpotential of different computational models. f Density of states diagrams for and , and energy band center and volcano plot for different computational models.
release of lattice oxygen. The results indicated that Co doping reduces the stability of the surface oxygen in , consistent with center calculation results. Notably, the LOER of and is the most prominent, with slightly surpassing . This phenomenon has been previously explained by XRD and ICP-MS characterizations, as has difficulty in maintaining the perovskite lattice, the proportion of Co doping decreases. In contrast, SI and exhibit the smallest LOER process, which is linked to the stability of the catalyst surface lattice oxygen. The results indicated that Co doping and dissolution can activate the catalyst lattice oxygen, thereby accelerating the formation of .
In addition to LOER, in situ ICP-MS experiments were performed to detect the phenomenon of ion dissolution during OER, as shown in Fig. 4b, c. We observed that both and SI undergo considerable dissolution when immersed in an acidic electrolyte. Coupling this observation with prior XRD and Raman results, it suggests that the dissolution process is triggered by the rapid dissolution of related oxides or compound heterophases. Furthermore, the subsequent steady dissolution trend indicates that ion dissolution of the
catalyst does not necessarily occur during the catalytic process. It is noteworthy that the Sr dissolution phenomenon of SI is more significant than that of , which is attributed to the higher concentration of surface and bulk Sr in SI . Moreover, is also accompanied by a small amount of Co dissolution, thus further confirming that Co dissolution promotes the formation of unsaturated .
To confirm the structural information of the catalyst, an in situ Raman mapping study was conducted, as shown in Fig. 4d, e. The peak around can be attributed to the -oxo stretching vibration of (involving the unprotonated bridge oxygen, ), and the characteristic peaks around and can be attributed to the typical and vibrational peaks of . In addition, the characteristic peaks around 300 and can be attributed to Srrelated oxide or compound . The figure shows that before and after the OER process, SI exhibits an obvious -oxo stretching vibration peak at , confirming the existence of the perovskite structure. However, for , a significant decrease in the Ir- -oxo stretching vibration peak is observed. This may be attributed to the
Fig. 4 | In situ characterizations of samples. a The percentage of the samples test by DEMS. In situ ICP-MS diagram of and (c) its differential transformation diagram, in set: enlarged diagram. d, e In situ Raman and Raman mapping of SI samples and samples.
destruction of the structure by Codissolution to form the lowcoordination structure, leading to a significant reduction in peak intensity. Moreover, the characteristic peaks of Sr-related oxides/ compounds near 300 and nearly disappear from the open circuit voltage (OC). Together with the results of in situ ICP-MS, it can be further confirmed to be caused by the dissolution of Sr-related oxides/compounds.
Key evidences of highly active low-coordination in Codoped for OER
To elucidate the formation mechanism of on the surface, comprehensive in situ EXAFS spectra was recorded at the Ir- edge to monitor the evolution of local coordination of Ir, as shown in Fig. 5a-e. Initially, we examined the coordination changes of the SI sample, as demonstrated in Fig. 5a, b. The pristine state SI sample owns a Ir-O coordination number of 5.6, indicating the existence of substantial intact Ir-O octahedral structure within the catalyst. During the pre-OER stage ( vs. RHE), the Ir-O coordination numbers of SI sample exhibited a pattern of initial increase followed by a decrease.
Previous reports showed a significant augmentation in the Ir-O coordination number of thin film samples during OER, which was ascribed to the oxygen refilling process . In this study, the dissolution of Sr led to alterations in the surface structure of Sl . However, there was no significant LOER in SI sample. Therefore, during the OER oxidation process, the rate of filling exceeded the rate of LOER, resulting in the observed trend in coordination numbers. During the OER stage ( 1.63 V vs. RHE), the Ir-O coordination number of the SI sample exhibited a minor increase, suggesting that at high potentials, the lattice oxygen of SI remained inactive, leading to the higher filling rate than the LOER rate . Additionally, at 1.23 V vs. RHE, a
significant reduction in the bond length of the SI sample was observed, potentially indicating a transformation of the octahedral corner-sharing structure during the filling process . In sum, as shown in Fig. 5c, the surface site mechanism of the SI sample undergoes a dynamic process from a relatively complete perovskite structure to site dissolution and adsorbate filling, ultimately transitioning into a saturated structure.
We next analyzed the coordination changes in the sample, as illustrated in Fig. 5d, e. The initial Ir-O coordination number of was 6.0, suggesting a more perfect Ir-O octahedral structure compared with SI. However, prior to the OER stage (OC, vs. RHE), the Ir-O coordination number of the sample exhibited a notable decreasing trend. This is consistent with the formation of lowcoordination as observed in in situ ICP-MS results. Furthermore, during the OER stage, the Ir-O coordination number of further decreased to 5.4 , significantly lower than SI , confirming that the active lattice oxygen in further facilitated the formation of highly active low-coordination .
To validate the generation of the unsaturated structure, in situ XANES spectra were analyzed at the edge to study the oxidation state of Ir, as shown in Fig. 5 g , h. The Ir- absorption edge of SI displayed an increasing white line intensity during the OER process, indicating an increase in the oxidation state of Ir. Differently, the Ir- absorption edge of exhibited a slight decrease in white line intensity, signifying a reduction in the oxidation state of Ir. Combined with the valence state information of reference samples Ir and IrO , the mechanism of Ir valence state change during OER can be inferred. During the OER process of SI, the dissolution of Sr and the filling of led to the saturation of the Ir -O coordination, displaying a higher valence state. Conversely, for , the dissolution of Co resulted in
Fig. 5 | In situ XAS characterization of samples. a R-space fitting diagram for SI sample. Changes in Ir -O coordination and bond length of SI sample. Structure change diagram of SI sample; R-space fitting diagram of sample. e Ir-O coordination and Ir-O bond length variation diagram of sample.
f Structure change diagram of sample. Ir- Absorption edge diagram of SI sample, in set: enlarged diagram. Ir- absorption edge diagram of sample, in set: enlarged diagram.
the formation of unsaturated Ir-O, leading to a decrease in the average Ir valence state. This observation further substantiates that Co can enhance the generation of unsaturated structures, aligning with the results from prior characterizations.
Discussion
OER catalytic mechanism on Co-doped SrIrO catalyst
Our study has elucidated the surface reconstruction process of SIC series catalysts by theoretical calculations and a comprehensive series of in situ characterizations. We now proceed to discuss and summarize the potential OER mechanisms inherent to SIC series catalysts. There are two widely recognized mechanisms for OER, specifically the adsorbate evolution mechanism (AEM) and the lattice oxygen mechanism (LOM), as shown in Fig. 6a, . Our theoretical calculations have demonstrated that the surface band center of the model is closer to the Fermi level compared to the Ir model, which suggests that the bridging oxygen of Co-O-Ir is thermodynamically predisposed to be oxidized . In situ Raman spectroscopy revealed
that the -oxo stretching vibration peak (indicative of bridging oxygen) in the system catalyst was notably reduced during the OC and OER processes when compared to the Ir system. Further, DEMS tests have found that a higher content of Co promotes the LOM.
Despite these findings, our DEMS results suggest that the AEM remains the dominant mechanism, while the LOM significantly contributes to the formation of unsaturated . Consequently, we propose a distinctive catalytic mechanism, the lattice oxygen promoted adsorbate evolution mechanism (LOPAEM), as illustrated in Fig. 6c. Unlike the conventional catalytic mechanisms of OER, LOPAEM involves the synergistic action of both mechanisms. Specifically, when the oxidation rate of lattice oxygen in certain catalysts becomes excessively fast, it will lead to the formation of a large number of surface oxygen vacancies (i.e., unsaturated metal sites), thereby altering the original catalytic sites of the catalyst. These unsaturated metal sites may exhibit more efficient AEM performance, thus achieving LOPAEM. Our findings suggest that the LOM in Co/Ir system catalysts is an integral step for catalyst activation. The formation of Ir-
Fig. 6 | Proposed possible OER mechanism. a Adsorbate evolution mechanism. b Lattice oxygen mechanism. c Lattice oxygen promoted adsorbate evolution mechanism. d Catalytic mechanism of Co-doped SrIrO catalyst.
substantially reduces the catalyst’s AEM overpotential, thereby making it thermodynamically more favorable. This is in agreement with our theoretical calculations and DEMS results. Moreover, the LOM process is unable to produce a lower Ir-O coordination structure as the dissolution of lattice oxygen shifts the center away from the Fermi level, precluding further oxidation (as detailed in the theoretical calculation section).
In light of the above findings, we have summarized the mechanism for the system catalyst as represented in Fig. 6d. Initially, Co on the catalyst’s surface dissolves under acidic conditions, removing a portion of the bridging lattice oxygen to form an structure with lower coordination. Subsequently, lattice oxygen dissolves during the OER process, forming an unsaturated structure that exhibits high AEM activity. Lastly, under the dynamic balance of adsorbate filling and LOM, the catalyst conducts AEM to efficiently facilitate the OER.
In summary, we designed a highly active catalyst through Co doping and dynamic dissolution of bimetallic ions to study the catalytic mechanism of -based perovskite. Theoretical calculations and in situ characterizations (DEMS, in situ Raman mapping, in situ XAS and in situ ICP-MS) show that dynamic dissolution of Co is crucial for forming highly active unsaturated . The as-synthesized catalysts exhibit higher OER reaction kinetics than and commercial catalyst in both electrolyzer and PEM water electrolyzer, revealing the mechanism of catalytic activity enhancement by tuning catalytic sites. This work is of great significance for understanding the high OER catalytic performance of Ir-based catalysts, and will provide an important basis for the design and preparation of high-performance acidic OER catalysts.
Methods
Chemicals
The chemical reagents utilized in this study were all received from the manufacturer. Potassium hexachloroiridate (IV) [ , Macklin], cobalt(III) nitrate hexahydrate [ , AR, Sigma-Aldrich],
strontium(II) nitrate [ , AR, Guangdong chemical reagent)], citric acid monohydrate (AR, LookChem.) were utilized as precursors.
Preparation of SI Catalyst. Solution A was prepared by dissolving and citric acid in 5.0 mL of deionized water. Solution B was prepared by dissolving in 4.0 mL of ethylene glycol. Solution A was then added dropwise with stirring to solution B. The resulting mixture was dried at for 12 h to obtain a brown solid product as a precursor. Subsequently, the precursor was calcined in air at for for for 3 h , and for 6 h with a heating rate of . Afterward, the excess impurities were removed by reacting with a 1.0 M HCl solution for 12 h to obtain catalyst.
Preparation of catalyst. Solution A was prepared by dissolving and citric acid in 5.0 mL of deionized water. Solution B was prepared by dissolving and in 4.0 mL of ethylene glycol. The subsequent steps were identical to the preparation of the SI catalyst described above.
Preparation of catalyst. Solution A was prepared by dissolving and citric acid in 5.0 mL of deionized water. Solution B was prepared by dissolving and in 4.0 mL of ethylene glycol. The subsequent steps were identical to the preparation of the SI catalyst described above.
Preparation of catalyst. Solution A was prepared by dissolving and citric acid in 5.0 mL of deionized water. Solution B was prepared by dissolving and in 4.0 mL of ethylene glycol. The subsequent steps were identical to the preparation of the SI catalyst described above.
Preparation of catalyst. Solution A was prepared by dissolving and citric acid in 5.0 mL of deionized water. Solution B was prepared by dissolving and in 4.0 mL of ethylene glycol. The subsequent steps were identical to the preparation of the SI catalyst described above.
Preparation of catalyst. Solution A was prepared by dissolving and citric acid in 5.0 mL of deionized water. Solution B was prepared by dissolving and in 4.0 mL of ethylene glycol. The subsequent steps were identical to the preparation of the SI catalyst described above.
Materials characterizations. Characterization of the atomic-level crystal structure was performed using an aberration-corrected scanning transmission electron microscope (JEM-ARM200P, JAPAN) operated at 300 kV . Energy-dispersive X-ray (EDX) analysis was used to measure the relative elemental content. X-ray diffraction (XRD) patterns of were recorded on an X-ray diffractometer (Smart lab) using radiation ( ) with a step size of and a step time of 0.2 s in the range. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific K-Alpha X-ray photoelectron spectrometer, and all XPS spectra were calibrated using the C line at 284.8 eV . The surface morphology of was characterized using a scanning electron microscope (TESCAN MIRA LMS). Considering the high acid resistance of , anti aqua regia was prepared by mixing hydrochloric acid and nitric acid in a ratio for the experiment. In this solution, 1.0 mg of powder was dissolved in 10 mL of aqua regia and left to stand for 1-3 week after thorough ultrasonic treatment. Finally, the proportions of each element in were determined by ICP-MS (iCAP RQ) analysis.
In situ characterizations and LOER confirmation. X-ray absorption spectra of Ir -edge were obtained at the BL17B and BL20U1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF). The spectra were collected either in transmission mode or fluorescence mode using a Lytle detector. The corresponding reference samples were collected in transmission mode. The samples were ground and uniformly applied to special adhesive tape. In situ XANES characterizations were performed in fluorescence mode at the same beamline. The samples were sprayed onto carbon paper at a loading of as the working electrode. The measurements were conducted under the same conditions as the OER measurements in a self-designed cell. The in situ Raman spectroscopy characterizations were carried out using an inVia confocal Raman microscope from Renishaw. The laser power was set at 532 nm with 1% power at a grating of , the silicon peak was calibrated before testing. During the OER process, differential electrochemical mass spectrometry (DEMS) measurements were conducted using the QAS 100 apparatus from Shanghai Linglu Instruments to determine the volatile reaction products of the SI series catalysts and catalyst labeled with . Saturated and Pt wires served as the reference electrode (RE) and counter electrode (CE), respectively. The working electrode (WE) was prepared by sputtering Au onto a -thick porous PTFE membrane, followed by depositing of catalyst ink ( ) onto the Au surface. The catalyst isotopic labeling was achieved by cycling the electrode in for eight cycles using cyclic voltammetry (CV) with a scan rate of in the range of vs. . Subsequently, the -labeled electrode was rinsed with to remove residual . Finally, the electrode was electrochemically tested against in a solution at different potentials with a scan rate of . The DEMS signal was normalized by current density ( ). Simultaneously, real-time measurements of gas products with different molecular weights generated during the OER process were conducted using mass spectrometry. In situ ICP-MS experiments were performed using a Thermo Scientific iCAP RQ instrument. The experimental setup
consisted of a standard three-electrode cell with a 3 mm glassy carbon working electrode, consistent with the electrochemical testing. The reference electrode used was a saturated calomel electrode (Hg/ ), and the counter electrode was a platinum foil electrode. To ensure accurate detection of ion distribution, a stirrer was employed to prevent leaching and dissolution errors, with data sampling occurring every 15 s .
Electrochemical characterizations. To prepare the catalyst ink, a 0.5 mg amount of catalyst was mixed with 1.0 ml of a Nafion solution and neutralized. Subsequently, a volume of the prepared ink was deposited onto a glassy carbon electrode (GCE) with a diameter of 5 mm and dried using an infrared lamp. Prior to use, the GCE was polished with alumina powder and rinsed three times with a mixture of high purity water and ethanol. Electrochemical measurements were conducted in a three-electrode system using an electrochemical workstation ( CHI 760E). The reference electrode used was an electrode in a 0.5 M electrolyte, while a carbon rod served as the counter electrode. The working electrode was the GCE with the catalyst. LSV and CV was performed in solution at a scan rate of . A home-made PEM water electrolysis cell with a proton exchange membrane was used to evaluate the performance of SI series catalyst. The preparation step of catalyst inks is the same as above method. The total catalyst loading on the electrode was 1.0 mg and all the catalyst inks were deposited on carbon paper ( ). The cell temperature ( 25,65 , and ) was maintained by an electric heating plate and measured by a temperature probe in electrolyte.
Computational methods. Spin-polarized density functional theory (DFT) calculations were performed in the plane wave and ultrasoft pseudopotential (USPP) with Perdew-Burke-Ernzerhof (PBE) exchange functional correction as implemented in Quantum ESPRESSO . An energy cutoff of 25 Ry was employed for the plane wave expansion of the electronic wavefunction. The atomic structures of the models were fully relaxed until self-consistency was achieved with a convergence criteria of for the energy and Bohr for the atomic coordinates. To prevent interaction between layers, a vacuum slab of was used to isolate the surface. For bulk geometry optimization, a Monkhorst-Pack kpoint set was used, while a set was used for electronic structure calculations. The correction for every adsorbate and surface, with typical values of for , * O and respectively. To simulate the complex unsaturated structure, we start by constructing and optimizing the slab structure of the original . Then, the surface Sr atoms were removed from the slab for further optimization. According to a previous report , the coordination number on the surface is approximately 4.5 , achieved by removing 4 Sr atoms and their corresponding 3 neighboring O atoms. The same procedure can be applied to the Co-doped system, where removing a Co atom also removes 2 neighboring O atoms.
XAS analysis. The acquired extended X-ray absorption fine structure (EXAFS) data were processed following standard procedures using the ATHENA module of the Demeter software package . The EXAFS spectra were obtained by subtracting the post-edge background from the overall absorption and then normalized with respect to the edgejump step. The data were Fourier transformed to real (R) space using a Hanning window ( ) to separate the contributions from different coordination shells. To determine the quantitative structural parameters around the central atoms, least-squares curve parameter fitting was performed using the ARTEMIS module of the Demeter software package.
Data availability
The data generated in this study are provided in the Supplementary Information and are available from the authors upon request. Source data are provided with this paper.
References
Hong, W. T. et al. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8, 1404-1427 (2015).
Hwang, J. et al. Perovskites in catalysis and electrocatalysis. Science 358, 751-756 (2017).
Chen, Z. et al. Advances in oxygen evolution electrocatalysts for proton exchange membrane water electrolyzers. Adv. Energy Mater. 12, 2103670 (2022).
Xue, Y. et al. Sulfate-functionalized as highly efficient oxygen evolution reaction electrocatalyst in acid. Adv. Funct. Mater. 31, 2101405 (2021).
Zhao, Z. L. et al. Boosting the oxygen evolution reaction using defect-rich ultra-thin ruthenium oxide nanosheets in acidic media. Energy Environ. Sci. 13, 5143-5151 (2020).
Shih, A. J. et al. Water electrolysis. Nat. Rev. Methods Primers 2, 84 (2022).
Huynh, M., Bediako, D. K. & Nocera, D. G. A functionally stable manganese oxide oxygen evolution catalyst in acid. J. Am. Chem. Soc 136, 6002-6010 (2014).
Gunasooriya, G. K. K. & Nørskov, J. K. Analysis of acid-stable and active oxides for the oxygen evolution reaction. ACS Energy Lett. 5, 3778-3787 (2020).
Cao, L. et al. Dynamic oxygen adsorption on single-atomic ruthenium catalyst with high performance for acidic oxygen evolution reaction. Nat. Commun. 10, 4849 (2019).
Danilovic, N. et al. Using surface segregation to design stable Ru-Ir oxides for the oxygen evolution reaction in acidic environments. Angew. Chem. Int. Ed. 53, 14016-14021 (2014).
Hubert, M. A. et al. Acidic oxygen evolution reaction activitystability relationships in Ru-based pyrochlores. ACS Catal. 10, 12182-12196 (2020).
Cui, X. et al. Robust interface Ru centers for high-performance acidic oxygen evolution. Adv. Mater. 32, 1908126 (2020).
Shan, J. et al. Charge-redistribution-enhanced nanocrystalline Ru@lrO electrocatalysts for oxygen evolution in acidic media. Chem 5, 445-459 (2019).
Lin, C. et al. In-situ reconstructed Ru atom array on with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012-1023 (2021).
Frydendal, R., Paoli, E. A., Chorkendorff, I., Rossmeisl, J. & Stephens, I. E. Toward an active and stable catalyst for oxygen evolution in acidic media: Ti-stabilized . Adv. Energy Mater. 5, 1500991 (2015).
Morgan Chan, Z . et al. Electrochemical trapping of metastable ions for activation of oxygen evolution catalysts. Proc. Natl. Acad. Sci. 115, E5261-E5268 (2018).
Kuo, D.-Y. et al. Influence of strain on the surface-oxygen interaction and the oxygen evolution reaction of . J. Phys. Chem. C 122, 4359-4364 (2018).
Lee, K., Osada, M., Hwang, H. Y. & Hikita, Y. Oxygen evolution reaction activity in catalysts: correlations between structural parameters and the catalytic activity. J. Phys. Chem. Lett 10, 1516-1522 (2019).
Shin, S. et al. Single-phase perovskite nanofibers as a highly efficient electrocatalyst for a pH -universal oxygen evolution reaction. ACS Appl. Energy Mater 5, 6146-6154 (2022).
Yang, L. et al. Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing octahedral dimers. Nat. Commun. 9, 5236 (2018).
Seitz, L. C. et al. A highly active and stable IrO/ catalyst for the oxygen evolution reaction. Science 353, 1011-1014 (2016).
Ben-Naim, M. et al. Understanding degradation mechanisms in oxygen evolution electrocatalysts: chemical and structural microscopy at the nanoscale. Adv. Funct. Mater. 31, 2101542 (2021).
Akbashev, A. R. et al. Probing the stability of during active water electrolysis via operando atomic force microscopy. Energy Environ. Sci. 16, 513-522 (2023).
Wan, G. et al. Amorphization mechanism of electrocatalyst: how oxygen redox initiates ionic diffusion and structural reorganization. Sci. Adv. 7, eabc7323 (2021).
Chen, Y. et al. Exceptionally active iridium evolved from a pseudocubic perovskite for oxygen evolution in acid. Nat. Commun. 10, 572 (2019).
Tang, R. et al. Oxygen evolution reaction electrocatalysis on grown using molecular beam epitaxy. J. Mater. Chem. A 4, 6831-6836 (2016).
Chen, H. et al. Optimization of active sites via crystal phase, composition, and morphology for efficient low-iridium oxygen evolution catalysts. Angew. Chem. Int. Ed. 59, 19654-19658 (2020).
Liang, X. et al. Electrocatalytic water oxidation activity-stability maps for perovskite oxides containing 3d, 4d and 5d transition metals. Angew. Chem. Int. Ed. 62, e202311606 (2023).
Zhang, R. et al. A dissolution/precipitation equilibrium on the surface of iridium-based perovskites controls their activity as oxygen evolution reaction catalysts in acidic media. Angew. Chem. Int. Ed 131, 4619-4623 (2019).
Song, C. W., Suh, H., Bak, J., Bae, H. B. & Chung, S.-Y. Dissolutioninduced surface roughening and oxygen evolution electrocatalysis of alkaline-earth iridates in acid. Chem 5, 3243-3259 (2019).
Tomita, K., Miyata, T., Olovsson, W. & Mizoguchi, T. Strong excitonic interactions in the oxygen K-edge of perovskite oxides. Ultramicroscopy 178, 105-111 (2017).
Grimaud, A. et al. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 4, 2439 (2013).
Mefford, J. T. et al. Water electrolysis on perovskite electrocatalysts. Nat. Commun. 7, 11053 (2016).
Pavlovic, Z., Ranjan, C., Gao, Q., van Gastel, M. & Schlögl, R. Probing the structure of a water-oxidizing anodic iridium oxide catalyst using Raman spectroscopy. ACS Catal. 6, 8098-8105 (2016).
Korotcov, A. V., Huang, Y. S., Tiong, K. K. & Tsai, D. S. Raman scattering characterization of well-aligned and nanocrystals. J. Raman Spec. 38, 737-749 (2007).
Ni, S., Yang, X. & Li, T. Hydrothermal synthesis and photoluminescence properties of . Mater. Lett. 65, 766-768 (2011).
Biedermann, N. et al. High-pressure phase behavior of : an experimental and computational Raman scattering study. Phys. Chem. Miner. 44, 335-343 (2017).
Yoo, J. S., Rong, X., Liu, Y. & Kolpak, A. M. Role of lattice oxygen participation in understanding trends in the oxygen evolution reaction on perovskites. ACS Catal. 8, 4628-4636 (2018).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457 (2017).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and opensource software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537-541 (2005).
Acknowledgements
This work was supported by NSFC (52373215), Sichuan Science and Technology Program (2023NSFSC0086), and Fundamental Research Funds for the Central Universities (YJ2021156). We also thank BL17B1 and BL20U1 station at Shanghai Synchrotron Radiation Facility (SSRF) for the help in characterizations.
Author contributions
Experiments were conceived and designed by G.-R.L. and J.-W.Z., with inputs from all authors. S.W., J.Z., D.W., J.C. and V.Y.F. performed the electrochemistry and characterization experiments. K.Y. contributed to the in situ XAS studies and PEM test. H.Z. provided help in EELS and HAADF characterizations.
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains
supplementary material available at https://doi.org/10.1038/s41467-024-46801-y.
Correspondence and requests for materials should be addressed to Gao-Ren Li.
Peer review information Nature Communications thanks Tao Cheng and
the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Reprints and permissions information is available at
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/ licenses/by/4.0/.
(c) The Author(s) 2024
College of Materials Science and Engineering, Sichuan University, Chengdu 610065, China. School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China. Department of Mechanical Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong SAR 999077, China. CAS Key Laboratory of Materials for Energy Conversion, Shanghai Institute of Ceramics, Chinese Academy of Sciences (SICCAS), 585 Heshuo Road, Shanghai 200050, China. Electron Microscopy Centre, School of Physical Science and Technology, Lanzhou University, Lanzhou 730099, China. National Research Nuclear University MEPhI (Moscow Engineering Physics Institute), Kashirskoe sh. 31, Moscow 115409, Russia. These authors contributed equally: Jia-Wei Zhao, Kaihang Yue, Hong Zhang. e-mail: ligaoren@scu.edu.cn