DOI: https://doi.org/10.1038/s41423-024-01145-x
PMID: https://pubmed.ncbi.nlm.nih.gov/38443448
تاريخ النشر: 2024-03-05
تطوير العلاجات المناعية للسرطان المعتمدة على خلايا NK من خلال هندسة المستقبلات
الملخص
تجذب العلاجات المناعية المعتمدة على خلايا القاتل الطبيعي (NK) اهتمامًا متزايدًا في مجال علاج السرطان. أظهرت التجارب السريرية المبكرة نتائج واعدة، إلى جانب فعالية وسلامة المنتج المرضية. زادت التطورات الأخيرة بشكل كبير من الإمكانات العلاجية لخلايا NK من خلال منحها قدرات معززة على التعرف والتدمير الخلوي. تركز هذه المراجعة على هندسة مستقبلات السطح في علاج خلايا NK وتناقش تأثيرها، وتحدياتها، والاتجاهات المستقبلية.
تستند معظم الأساليب إلى الهندسة باستخدام مستقبلات المستضدات الهجينة للسماح لخلايا NK باستهداف مستضدات الأورام المحددة بشكل مستقل عن قيود مستضدات الكريات البيضاء البشرية. زادت هذه الطريقة من دقة وقوة التعرف والتخلص من خلايا السرطان بواسطة خلايا NK. بالإضافة إلى ذلك، فإن هندسة خلايا NK بمستقبلات خلايا T تعزز أيضًا التعرف على الإيبيتوبيات داخل الخلايا، مما يوسع نطاق الببتيدات المستهدفة. تم تحسين التعرف غير المباشر على ببتيدات الأورام بواسطة خلايا NK أيضًا من خلال تحسين تعبير مستقبلات الجزء الثابت من الغلوبولين المناعي وإشاراته. في الواقع، تتمتع خلايا NK المهندسة بقدرة محسنة على التعرف على وتدمير الخلايا المستهدفة المغلفة بأجسام مضادة محددة، مما يزيد من سميتها الخلوية المعتمدة على الأجسام المضادة. تم أيضًا استكشاف قدرة هندسة مستقبلات خلايا NK على تعزيز التوسع، والاستمرارية، والتسلل للخلايا المنقولة في بيئة الورم الدقيقة. تم أيضًا مناقشة استراتيجيات قائمة على المستقبلات لوظائف خلايا NK المستدامة داخل بيئة الورم، وتوفر هذه الاستراتيجيات آفاقًا لمواجهة تثبيط المناعة الناتج عن الورم.
المقدمة
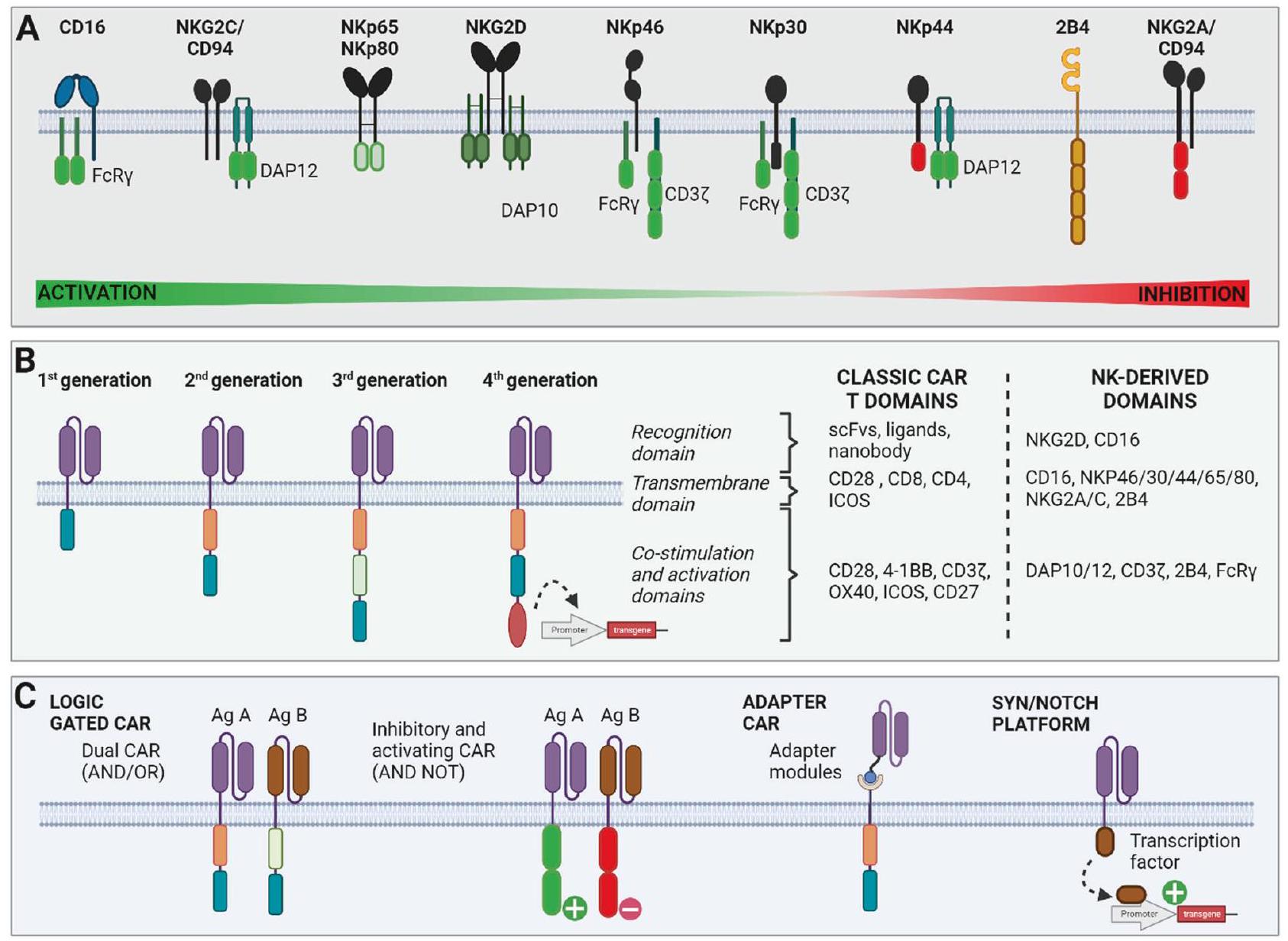
تم تحديد مجموعات فرعية جديدة من خلايا NK تتجاوز التصنيف المعتاد بناءً على CD16 و CD56 [3، 4]. من بينها، مجموعة تسمى خلايا NK التكيفية، التي تتميز بغياب تعبير مستقبل الغلوبولين المناعي Fc-epsilon (FCER1G) وزيادة تعبير مجموعة القاتل الطبيعي 2 C (NKG2C)، تظهر خصائص معززة، بما في ذلك زيادة التكاثر، وزيادة التوسع والقدرة السمية. على عكس خلايا T، لا تقتصر خلايا NK على التعرف على معقد التوافق النسيجي الكبير (MHC، المعروف أيضًا بمستضد الكريات البيضاء البشرية (HLA))، ويتطلب تنشيطها مستقبلات متعددة. في الواقع، يعتمد تنشيطها على ضبط دقيق للإشارات المثبطة والمنشطة. لمنع التنشيط غير المرغوب فيه للخلايا السليمة، تعبر خلايا NK عن مستقبلات الغلوبولين المناعي الشبيهة بالخلايا القاتلة (KIRs) والهيتيروديمر مجموعة القاتل الطبيعي 2 A (NKG2A)/CD94، الذي يتفاعل مع جزيئات HLA من الفئة I لتوفير إشارات مثبطة (الشكل 1). على العكس، تمتلك خلايا NK مجموعة من المستقبلات المنشطة، بما في ذلك مجموعة القاتل الطبيعي 2D (NKG2D)، وجزيء DNAX المساعد-1 (DNAM-1)،
الدروس المستفادة من التجارب السريرية الأولية مع خلايا NK
تحسين التعرف على خلايا الورم منح خلايا NK مستقبلات مستضدات هجينة
| هدف السيارة | مصدر NK | سرطانات مستهدفة | تاريخ | مرحلة | حالة | رقم | |
| الأورام الصلبة | بروتين سكري مشيمي جنيني 5T4 | لم يُكشف عنه | الأورام الصلبة المتقدمة | ٢٠٢٢ | أنا | التوظيف | NCT05194709 |
| CD70 | خلايا NK المشتقة من خلايا CB | سرطان الخلايا الكلوية المتقدم، الميزوثليوما وساركوما العظام | ٢٠٢٣ | أنا/اثنان | التوظيف | NCT05703854 | |
| CLDN6 | خلايا PB-NK للمرضى | الأورام الصلبة المتقدمة الإيجابية لـ CLDN6 | 2022 | أنا/اثنان | التوظيف | NCT05410717 | |
| DLL3 | لم يُكشف عنه | سرطان الرئة ذو الخلايا الصغيرة في المرحلة المتقدمة | ٢٠٢٢ | أنا | التوظيف | NCT05487651 | |
| HER2 | خط الخلايا NK | ورم دبقي متكرر إيجابي HER2 | 2017 | أنا | غير معروف | NCT03383978 | |
| ميسوثيلين | ذاتي | سرطان المبيض الظهاري المقاوم للعلاج | ٢٠٢١ | 0 | التوظيف | ChiCTR2100048100 | |
| خلايا NK المشتقة من الخلايا الجذعية المستحثة | سرطان المبيض الظهاري | 2018 | أنا | غير معروف | NCT03692637 | ||
| موك1 | لم يتم الكشف عنه | ورم صلب متكرر أو مقاوم إيجابي لمؤشر MUC1 | 2016 | 1/II | غير معروف. | NCT02839954 | |
| روابط NKG2D | لم يتم الكشف عنه | سرطان المبيض المتكرر المقاوم للبلاتين | ٢٠٢٣ | التوظيف | NCT05776355 | ||
| لم يتم الكشف عنه | سرطان القولون المستقيمي النقيلي المقاوم للعلاج | ٢٠٢٢ | أنا | التوظيف | NCT05213195 | ||
| لم يُكشف عنه | الأورام الصلبة النقيليّة | 2017 | أنا | مكتمل | NCT03415100 | ||
| لم يتم الكشف عنه | لم يُكشف عنه | سرطان الظهارة المبيضية | ٢٠٢٣ | أنا | لم يبدأ التوظيف بعد | NCT05856643 | |
| لم يتم الكشف عنه | سرطان الكبد الخلوي المتقدم | ٢٠٢٣ | أنا | لم يبدأ التوظيف بعد | NCT05845502 | ||
| لم يُكشف عنه | سرطان الثدي الثلاثي السلبي المتقدم | ٢٠٢٣ | أنا | لم يبدأ التوظيف بعد | NCT05686720 | ||
| PDL1 | خط الخلايا NK | سرطان المعدة أو الرأس والعنق المتكرر/النقائل | ٢٠٢١ | الثاني | التوظيف | NCT04847466 | |
| PSMA | خلايا NK المشتقة من الخلايا الجذعية المستحثة | سرطان البروستاتا المقاوم للعلاج بالحرمان من الأندروجين النقيلي | 2018 | أنا | غير معروف | NCT03692663 | |
| روبو1 | لم يتم الكشف عنه | سرطان البنكرياس | 2019 | أنا/اثنان | التوظيف | NCT03941457 | |
| لم يُكشف عنه | الأورام الصلبة | 2019 | أنا/اثنان | التوظيف | NCT03940820 | ||
| لم يتم الكشف عنه | ورم خبيث | 2019 | أنا/اثنان | التوظيف | NCT03931720 | ||
| تروب2 | خلايا NK المشتقة من خلايا CB | سرطان المبيض المقاوم للبلاتين، أدينوكارسينوما شبيهة بالميزونيفروس، وسرطان البنكرياس | ٢٠٢٣ | أنا/اثنان | لم يبدأ التوظيف بعد | NCT05922930 | |
| الأورام الخبيثة الدموية | CD33 | لم يتم الكشف عنه | سرطان الدم النخاعي الحاد المتكرر/المقاوم للعلاج | ٢٠٢١ | أنا | لم يتم التوظيف بعد | NCT05008575 |
| خط الخلايا NK | سرطانات الدم الحادة النخاعية المتكررة/المقاومة للعلاج | 2016 | أنا/اثنان | غير معروف | NCT02944162 | ||
| CD33/TIM3 | خلايا NK المشتقة من خلايا CB | سرطان الدم النخاعي الحاد | ٢٠٢١ | 0 | التوظيف | ChiCTR2100043081 | |
| CD33/CLL1 | لم يُكشف عنه | سرطان الدم النخاعي الحاد المتكرر/المقاوم للعلاج | ٢٠٢١ | 0 | التوظيف | ChiCTR2100047084 | |
| CD33/CCL1 | لم يتم الكشف عنه | سرطان الدم النخاعي الحاد | ٢٠٢٠ | أنا | التوظيف | NCT05215015 | |
| CD19 | لم يُكشف عنه | سرطان الغدد اللمفاوية الكبير B الخلوي المتكرر/المقاوم للعلاج | ٢٠٢٣ | أنا | لم يبدأ التوظيف بعد | NCT05673447 | |
| خلايا NK المشتقة من الخلايا الجذعية المستحثة | الأورام الخبيثة لبلايا B الإيجابية لـ CD19 | ٢٠٢٣ | أنا | التوظيف | NCT05336409 | ||
| ألوغيني | الأورام الدموية الخبيثة من نوع B-Cell | 2022 | أنا/اثنان | التوظيف | NCT05654038 | ||
| ألوغيني | الأورام الخبيثة لبائية الخلايا البالغة المتكررة/المقاومة للعلاج | ٢٠٢٢ | أنا | التوظيف | NCT05645601 |
| هدف السيارة | مصدر NK | سرطانات مستهدفة | تاريخ | مرحلة | حالة | رقم |
| لم يتم الكشف عنه | سرطان الدم اللمفاوي الحاد المتكرر/المقاوم للعلاج | 2022 | أنا | اكتمل التوظيف | NCT05563545 | |
| خلايا NK المشتقة من خلايا CB | سرطان الغدد اللمفاوية غير هودجكين من نوع B المقاوم للعلاج/المتكرر | 2022 | أنا | التوظيف | NCT05472558 | |
| لم يُكشف عنه | الأورام الخبيثة لبروتين B المتكررة/المقاومة للعلاج | ٢٠٢٢ | 。 | التوظيف | NCT05410041 | |
| ألوغيني | أورام الخلايا البائية | 2021 | أنا | التوظيف | NCT05020678 | |
| خلايا NK متطابقة النمط الوراثي (PB) | سرطان الغدد اللمفاوية غير هودجكين من نوع B الخبيث المقاوم/المتكرر | 2021 | أنا | التوظيف | NCT04887012 | |
| خلايا NK المشتقة من خلايا CB | الأورام الخبيثة اللمفاوية B | ٢٠٢١ | أنا | التوظيف | NCT04796675 | |
| لم يُكشف عنه | سرطان الدم اللمفاوي الحاد من نوع B الخلايا المتكررة/المقاومة للعلاج | ٢٠٢١ | أنا | التوظيف | NCT05379647 | |
| خلايا NK المشتقة من الخلايا الجذعية المستحثة | ليمفوما الخلايا البائية أو اللوكيميا اللمفاوية المزمنة. | ٢٠٢٠ | أنا | التوظيف | NCT04245722 | |
| لم يُكشف عنه | سرطان الغدد اللمفاوية غير هودجكين من نوع B-Cell المتكرر أو المقاوم للعلاج | ٢٠٢٠ | أنا | لم يتم التوظيف بعد | NCT04639739 | |
| لم يتم الكشف عنه | سرطان الغدد اللمفاوية من نوع B المتكرر والمقاوم للعلاج | 2019 | أنا | غير معروف | NCT03824951 | |
| خلايا NK المشتقة من الخلايا الجذعية المستحثة | سرطان الغدد اللمفاوية من نوع B المتكرر والمقاوم للعلاج | 2018 | أنا | غير معروف | NCT03690310 | |
| خلايا NK المشتقة من UC و CB | الأورام الخبيثة اللمفاوية B | 2017 | أنا/اثنان | مكتمل [38] | NCT03056339 | |
| خط الخلايا NK | لوكيميا ولينفومة إيجابية CD19 | 2016 | أنا/اثنان | غير معروف | NCT02892695 | |
| خلايا NK من الدم المحيطي (ألوغينية) | اللمفوما المتكررة والمقاومة للعلاج | 2013 | أنا | تم الانتهاء. | NCT01974479 | |
| خلايا NK من الدم المحيطي (ألوغينية) | سرطانات الدم اللمفاوية الحادة المتكررة و/أو المقاومة للعلاج | 2009 | أنا | تم الانتهاء. | NCT00995137 | |
| CD19/CD22 | خلايا NK المشتقة من الخلايا الجذعية المستحثة | سرطان الغدد اللمفاوية من نوع B المتكرر والمقاوم للعلاج | 2019 | أنا | لم يبدأ التوظيف بعد. | NCT03824964 |
| CD19/CD70 | خلايا NK المشتقة من خلايا CB | سرطان الغدد اللمفاوية غير هودجكين من نوع B الخبيث المقاوم/المتكرر | ٢٠٢٣ | أنا/اثنان | التوظيف | NCT05842707 |
| خلايا NK المشتقة من خلايا CB | سرطان الغدد اللمفاوية غير هودجكين من نوع B الخبيث المقاوم/المتكرر | 2022 | أنا | التوظيف | NCT05667155 | |
| CD70 | خلايا NK المشتقة من خلايا CB | الأورام الدموية المتكررة/المقاومة للعلاج | ٢٠٢١ | أنا/اثنان | التوظيف | NCT05092451 |
| CD7 | ألوغيني | الأورام الدموية CD7 | 2022 | أنا | لم يبدأ التوظيف بعد | NCT05377827 |
| خط الخلايا NK | لوكيميا وليمفوما متكررة أو مقاومة إيجابية CD7 | 2016 | أنا/اثنان | غير معروف | NCT02742727 | |
| لم يتم الكشف عنه | الأورام الخبيثة الدموية | ٢٠٢٣ | أنا | التوظيف | NCT05995028 | |
| CD123 | ألوغيني | سرطان الدم النخاعي الحاد المقاوم/المتكرر | 2022 | أنا | التوظيف | NCT05574608 |
| لم يُكشف عنه | سرطان الدم النخاعي الحاد المقاوم/المتكرر وورم الخلايا الشجيرية البلازمية | ٢٠٢٣ | أنا/اثنان | التوظيف | NCT06006403 | |
| CD5 ( + IL15) | خلايا NK المشتقة من خلايا CB | الأورام الدموية المتكررة/المقاومة للعلاج | ٢٠٢١ | أنا/اثنان | التوظيف | NCT05110742 |
| هدف السيارة | مصدر NK | سرطانات مستهدفة | تاريخ | مرحلة | حالة | رقم | |
| CD56 | لم يُكشف عنه | سرطان الغدد اللمفاوية من نوع NK/T-cell/لوكيميا خلايا NK المقاومة/الانتكاسية | ٢٠٢٣ | الثاني | التوظيف | NCT05941156 | |
| CD20 | خلايا NK المشتقة من الخلايا الجذعية المستحثة | سرطان الدم النخاعي الحاد المتكرر/المقاوم وليمفوما خلايا B | 2019 | أنا | نشط | NCT04023071 | |
| CD22 | خلايا NK المشتقة من الخلايا الجذعية المستحثة | سرطان الغدد اللمفاوية B-cell المتكرر والمقاوم للعلاج | 2018 | أنا | غير معروف | NCT03692767 | |
| CD33/CLL1 | لم يُكشف عنه | سرطان الدم النخاعي الحاد | ٢٠٢٣ | أنا | لم يبدأ التوظيف بعد | NCT05987696 | |
| CCL1 | خلايا NK المشتقة من الخلايا الجذعية المستحثة | سرطان الدم النخاعي الحاد | ٢٠٢٣ | أنا | التوظيف | NCT06027853 | |
| CD38/SLAMF7 | خلايا NK المشتقة من الخلايا الجذعية المستحثة | سرطان الدم النخاعي الحاد والورم النقوي المتعدد | ٢٠٢٠ | أنا | نشط | NCT04614636 | |
| بي سي إم إيه | ألوغيني | الورم النقوي المتعدد المتكرر/المقاوم للعلاج أو سرطان الدم النقوي | ٢٠٢٣ | أنا | التوظيف | NCT06045091 | |
| ألوغيني | ورم النخاع المتعدد المتكرر/المقاوم للعلاج | 2022 | أنا | التوظيف | NCT05652530 | ||
| خلايا NK المشتقة من الخلايا الجذعية المستحثة | الورم النقوي المتعدد | ٢٠٢١ | أنا | التوظيف | NCT05182073 | ||
| خلايا NK المشتقة من UC و CB | ورم النخاع العظمي المتكرر/المقاوم للعلاج | 2021 | أنا | التوظيف | NCT05008536 | ||
| خط الخلايا NK | ورم النخاع العظمي المتكرر/المقاوم للعلاج | 2019 | أنا/اثنان | التوظيف | NCT03940833 | ||
| خلايا NK المشتقة من الخلايا الجذعية المستحثة | لمفوما خلايا B المتكررة/المقاومة للعلاج | 2018 | أنا | غير معروف | NCT03559764 | ||
| روابط NKG2D | لم يُكشف عنه | سرطان الدم النخاعي الحاد المتكرر/المقاوم للعلاج | 2023 | تجنيد | NCT05734898 | ||
| خلايا NK المشتقة من الحبل السري | سرطان الدم النخاعي الحاد المتكرر/المقاوم للعلاج | 2022 | اكتمل التجنيد | NCT05247957 | |||
| خلايا NK المشتقة من الدم المحيطي | متلازمات خلل التنسج النقوي وسرطان الدم النخاعي الحاد | 2020 | I | تجنيد | NCT04623944 | ||
| غير مفصح عنه | غير مفصح عنه | الأورام الخبيثة للخلايا B | 2021 | I/II | تجنيد | NCT04747093 | |
| غير مفصح عنه | الأورام الخبيثة الدموية المتكررة/المقاومة للعلاج | 2021 | I | تجنيد | NCT04796688 | ||
| أخرى | روابط NKG2D – بروتين الغلاف SARS-CoV-2 | خلايا NK المشتقة من الحبل السري | كوفيد-19 | 2020 | I/II | تجنيد | NCT04324996 |
استجابات المناعة بواسطة خلايا NK المهندسة، يمكن أيضًا استخدام مستقبلات تنشيط خلايا NK بالاشتراك مع مستقبلات CAR. من الجدير بالذكر أن خلايا CAR-NK ثنائية الخصوصية التي تم هندستها مع CAR PD1-DAP10 وNKG2D أظهرت سمية خلوية قوية ضد خلايا سرطان المعدة سواء في المختبر أو في الجسم الحي [74]. في الواقع، استغل CAR المضاد-PD1-DAP10 إشارة NKG2D من خلال مجالات DAP10 وأظهر أنه يعزز تنشيط خلايا NK ضد خلايا الورم التي تعبر عن روابط PD1 وNKG2D [74].
| سيارة | تي سي آر | |
| مقيد بـ HLA | لا | نعم |
| كثافة المستضد المطلوبة | عالي | منخفض |
| استهداف داخل الخلايا | لا | نعم |
| تعدد الإرسال | نعم | نعم |
| جاهز للاستخدام | نعم | نعم |
| اختبار سريري | نعم | لا |
تزويد خلايا NK بمستقبلات T خارجية
[101]. علاوة على ذلك، بعد الحقن في نموذج فأر لورم النخاع المتعدد، لوحظ انخفاض في نمو الورم [101]. تقدم هذه النتيجة آفاقًا مثيرة للاهتمام للاختبار السريري في السنوات القادمة. ومع ذلك، قد تعيق علاجات NK-TCR العرض الضعيف لمستضدات الورم المرتبطة، والتي قد لا تصل إلى مستوى كافٍ لتحفيز تنشيط خلايا NK. من المثير للاهتمام أن تقارب TCR واهتمامه متشابهان في خلايا T الأولية وفي خطوط خلايا NK، مما يشير إلى أن خصائص TCR معينة يمكن أن تُنقل وتُستخدم لتحسين الاستجابات في خلايا NK [102]. يمكن أيضًا دمج CAR وTCR للحصول على تأثير تآزري. على سبيل المثال، تم تصميم خلايا NK مع TCR محدد لبروتين E7 من HPV16 جنبًا إلى جنب مع CAR محدد لـ TROP2 [103]. كان تركيب CAR يتكون من مجالين مساعدين (CD28 و4-1BB) ولكنه كان يفتقر إلى مجال تنشيط CD3؛ وبالتالي، كان تنشيط CAR ضروريًا ولكنه غير كافٍ لسمية خلايا NK. في الواقع، يعتمد تنشيط خلايا NK على التعرف على إبيتوبي E7، الذي يُعبر عنه حصريًا في خلايا الورم المصابة بـ HPV16، وتزداد السمية من خلال ارتباط CAR بالهدف TROP2 الذي يُعبر عنه في الغالب على خلايا الورم ولكن أيضًا في بعض الأنسجة السليمة. يؤدي الإشارات المجمعة إلى سمية مثالية ومحددة للورم مع الحفاظ على الأنسجة السليمة [103]. تؤسس هذه الاستراتيجية لبوابات منطقية متطورة وتطبيقات أوسع في المستقبل. على سبيل المثال، بدلاً من استخدام مستقبلين متميزين (واحد CAR وواحد TCR)، يمكن بناء مستقبلات هجينة. تم تطوير مثل هذه المستقبلات، سواء كانت مستقلة عن HLA (مستقبل HIT [104] وSTAR [105]) أو معتمدة على HLA (TCAR [106])، بالفعل في خلايا T وقد تُنقل إلى خلايا NK. من الجدير بالذكر أن TCR هجين يتكون من المجالات خارج الخلوية لسلسلة TCRa المدمجة مع مجال CD28 عبر الغشاء متبوعًا بمناطق الإشارة 2B4 وDAP10 وTCR.
تعزيز القدرة على قتل الخلايا السرطانية بواسطة خلايا NK
تم تطوير وتنفيذ (بروتيناز) بنجاح، مما أدى إلى تحسين القتل في النماذج قبل السريرية [112-114].
تحسين كيميائية الخلايا القاتلة الطبيعية
| المستقبل | الهدف | مصادر NK | تقنيات التعديل | المرض المستهدف | النتائج | المرجع |
| CXCR1 | IL-8 | PB | التحريض الكهربائي للـ mRNA | سرطان المبيض | زيادة الكيميائية في الجسم الحي زيادة السيطرة على الورم | [123] |
| CXCR2 | CXCL5 | PB | ناقل فيروسي عكسي | سرطان الخلايا الكلوية | زيادة الكيميائية في المختبر زيادة قتل الخلايا المستهدفة والالتصاق في المختبر | [120] |
| CXCR2 | CXCL1-3 و CXCL5-8 | NK92 | CRISPR-Cas9 | سرطان القولون البشري | زيادة الكيميائية في الجسم الحي نحو مواقع الأورام نشاط أقوى في قتل الخلايا والتكاثر تقليل الورم زيادة البقاء | [122] |
| CCR2B و CCR4 | CCL22 أو CCL2 | NK-92 و PB | ناقل فيروسي بطئ | لا شيء | زيادة الكيميائية في المختبر | [130] |
| CXCR4 | CXCL12 و SDF-1
|
YTS | ناقل فيروسي بطئ | الورم الدبقي | زيادة الكيميائية في المختبر وفي الجسم الحي تقليل/إزالة الورم زيادة البقاء | [68] |
| CXCR4 | SDF-1
|
PB | ناقل فيروسي بطئ | لا شيء | زيادة الكيميائية في المختبر | [126] |
| CXCR4
|
SDF-1
|
PB | نقل mRNA | لا شيء | زيادة الكيميائية في المختبر زيادة التوجه إلى نخاع العظام | [125] |
| CXCR4 و CCR7 | CXCL12 و CCL21 | NK92 | ناقل فيروسي بطئ | سرطانات القولون المستقيم | تقليل الورم زيادة البقاء | [127] |
| CCR5 | CCL5 | PB | ناقل فيروسي بطئ | سرطان القولون البشري | زيادة الكيميائية في المختبر وفي الجسم الحي | [132] |
| CCR7 | CCL19 و CCL21 | PB | تروغوسيتوز | لا شيء | زيادة الكيميائية في المختبر زيادة التوجه إلى العقد اللمفاوية | [129] |
| CCR7 | CCL19 و CCL21 | NK-92 | نقل DNA | سرطان الغدد اللمفاوية B | زيادة الكيميائية في المختبر وفي الجسم الحي زيادة السيطرة على الورم زيادة البقاء | [128] |
| CCR7 | CCL19 | PB | التحريض الكهربائي للـ mRNA | لا شيء | زيادة الكيميائية في المختبر | [109] |
تحسين بقاء خلايا NK وتكاثرها
في تجربة سريرية من المرحلة I/II تقييم خلايا CAR-NK المضادة لـ CD19 غير المتطابقة HLA التي تم نقلها بواسطة ناقل فيروسي عكسي يشفر IL-15 في الأورام اللمفاوية. من بين أول 11 مريضًا تم الإبلاغ عنهم، كان 8 (
مواجهة البيئة الدقيقة للورم المثبطة للمناعة
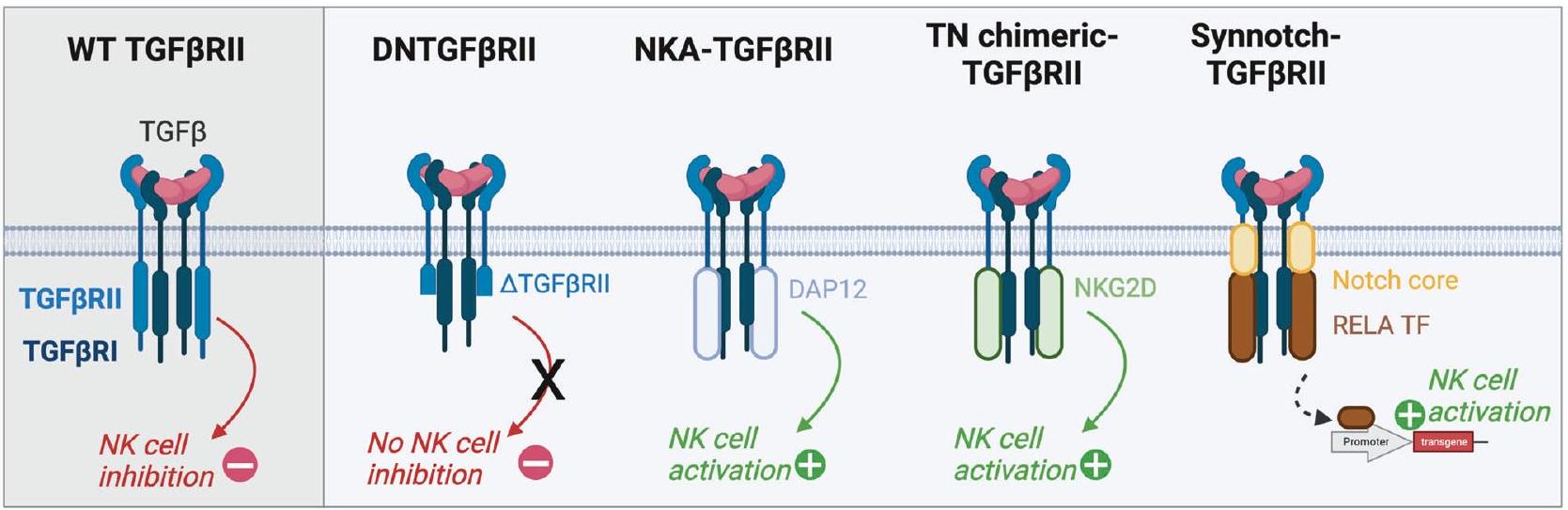
تقليل تعبير مستقبلات NK الذاتية
على سبيل المثال، في علاج الأورام الدموية، تم حذف جيني CD38 و CD7 في خلايا CAR-NK المهندسة. ومن المثير للاهتمام، أنه تم تطوير استراتيجية 2 في 1 من خلال إدخال مستقبل CAR في موضع الجين الذي يشفر هدفه، مما سمح بالتعبير عن CAR مع تجنب القتل الأخوي. كما ذُكر أعلاه في القسم 3.1، يمكن أيضًا تنفيذ مستقبلات CAR المثبطة لتجنب القتل الأخوي لخلايا CAR-NK الناتج عن التروغوسيتوز.
الاستنتاجات والآفاق
تم إظهار أن تعطيل SH2 المحتوي على (CISH) يعزز النشاط المضاد للورم لخلايا CAR-NK. بالإضافة إلى طرق الهندسة المتعددة، يمكن أن تساعد الذكاء الاصطناعي في تطوير علاجات مركبة مع العلاجات التقليدية أو العلاجات المناعية الأخرى من خلال التنبؤ باستجابات المرضى. ومن الجدير بالذكر أن المحفزات الخلوية قد حظيت باهتمام كبير في السنوات الأخيرة وتمت الموافقة عليها للاستخدام السريري. يمكنها ربط الروابط، مثل مستضدات الورم، وتنشيط المستقبلات على سطح خلايا NK. يمكن استخدام هذا النوع من الجزيئات لتعزيز تنشيط خلايا NK من خلال ربط المستقبلات المهندسة بخلايا الورم. من المهم أن تظل قضايا السلامة مرتبطة جوهريًا بالعلاجات الهندسية لخلايا NK، مثل التنشيط غير المستهدف. إن ضمان خصوصية علاجات خلايا NK أمر ضروري لتجنب إلحاق الضرر بالأنسجة السليمة، مما قد يؤدي إلى آثار جانبية خطيرة. لتحسين تنشيط خلايا NK واستهداف خلايا السرطان بشكل انتقائي، يجب تطوير دوائر منطقية مضبوطة بدقة. علاوة على ذلك، يجب تطوير نماذج حسابية للتنبؤ وضبط استجابات الخلايا ديناميكيًا. إن الاستخدام المتكرر للناقلات التكاملية لتعديل خلايا NK يأتي أيضًا مع خطر الطفرات الناتجة عن الإدخال، مما قد يؤدي إلى لمفوما خلايا NK. التوصية الحالية من إدارة الغذاء والدواء هي أن يكون عدد نسخ الناقل أقل من 5. علاوة على ذلك، يمكن أيضًا استخدام أدوات التحرير لتوليد انتقالات صبغية. مؤخرًا، تم تطوير عدة بروتوكولات تصنيع للتغلب على بعض القيود المذكورة أعلاه، وإذا تمت الموافقة عليها، يمكن أن تصبح ممارسات قياسية لتقليل فقد الكروموسومات والانتقالات في المنتجات المصنعة. كإجراء احترازي، لضمان تدمير الخلايا المهندسة في حالة حدوث أحداث سلبية، يجب تنفيذ مفتاح أمان كاسبيز قابل للتحفيز. يجب تحديد معايير مراقبة الجودة للتحقق من فعالية الهندسة، والنقاء، والنمط الظاهري، والسرطانية لخلايا NK. في الواقع، اعتمادًا على المتبرع، المصدر (PB أو UCB) وظروف الثقافة، يمكن أن تختلف مجموعات خلايا NK. بالنسبة لهذه النقاط الأخيرة، ستكون التقنيات الجديدة لتوسيع وتعديل وحفظ خلايا NK الأولية بالتبريد خارج الجسم مفيدة للغاية لتكبير الإنتاج للتطبيق السريري، حيث تتطلب الجرعات العالية والحقن المتعددة. من المتوقع أن تحسن التقدم المستقبلي المحقق في علاجات خلايا T، بما في ذلك البنى التحتية المطورة لإنتاج خلايا وناقلات فيروسية، بالإضافة إلى الأدوات المتاحة لهندسة الخلايا (من بينها استراتيجيات التحرير داخل الجسم)، علاج خلايا NK بشكل تآزري. ستوفر التجارب السريرية في المراحل المبكرة أيضًا معلومات قيمة وتعزز تحسين علاجات خلايا NK المستقبلية، التي من المحتمل أن تصل إلى الموافقة السريرية في السنوات القادمة. بشكل عام، لا تحمل هذه التقدمات الرائدة إمكانات هائلة لعلاج السرطان فحسب، بل توسع أيضًا نطاق العلاجات لأمراض أخرى، بما في ذلك العدوى.
REFERENCES
- Gill S, Olson JA, Negrin RS. Natural killer cells in allogeneic transplantation: effect on engraftment, graft- versus-tumor, and graft-versus-host responses. Biol Blood Marrow Transplant. 2009;15:765-76.
- Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47:820-33.
- Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue determinants of human NK cell development, function, and residence. Cell. 2020;180:749-763.e13.
- Buckle I, Johnson A, Rojas IL, Weinert V, Sester DP, Radford K, et al. High dimensional analysis reveals distinct NK cell subsets but conserved response to stimulation in umbilical cord blood and adult peripheral blood. Eur J Immunol. 2023;53:2250118.
- Wang W. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015;6. https://doi.org/10.3389/ fimmu.2015.00368.
- Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652-8.
- Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. 2016;7:86359-73.
- Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563-70.
- Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol. 2021;14:7.
- Bryceson YT, March ME, Ljunggren H-G, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107:159-66.
- Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex Vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother. 2015;38:24-36.
- Berrien-Elliott MM, Becker-Hapak M, Cashen AF, Jacobs M, Wong P, Foster M, et al. Systemic IL-15 promotes allogeneic cell rejection in patients treated with natural killer cell adoptive therapy. Blood. 2022;139:1177-83.
- Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123:3855-63.
- Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051-7.
- Heuser M, Tschan-Plessl A, Thol F, Schwarzer A, Kloos A, Kattre N, et al. Allogeneic, CD34 +, umbilical cordblood-derived NK cell adoptive immunotherapy for the treatment of acute myeloid leukemia patients with measurable residual disease. Blood. 2021;138:1745-1745.
- Choi I, Yoon SR, Park S-Y, Kim H, Jung S-J, Kang Y-L, et al. Donor-derived natural killer cell infusion after human leukocyte antigen-haploidentical hematopoietic cell transplantation in patients with refractory acute leukemia. Biol Blood Marrow Transplant. 2016;22:2065-76.
- Bachanova V, Maakaron JE, Cichocki F, McKenna DH, Cao Q, DeFor TE, et al. Gda201, a novel metabolically enhanced allogeneic natural killer (NK) cell product yields high remission rates in patients with relapsed/refractory non-Hodgkin lymphoma (NHL): 2-year survival and correlation with cytokine IL7. Blood. 2021;138:3854-3854.
- Nangia C, Soon-Shiong P, Rabizadeh S, Lee JH, Sender L, Jones F, et al. Complete responses in patients with second-line or greater metastatic triple negative breast cancer (TNBC) following first-in-human immunotherapy combining NK and T cell activation with off-the-shelf high-affinity CD16 NK cell line (haNK). Ann Oncol. 2019;30:v130.
- Fate Therapeutics. Fate therapeutics announces encouraging dose-escalation clinical data of FATE-NK100 and provides regulatory update on landmark IND appli- cation for FT500 [Press Release]. 2018. https://ir.fatetherapeutics.com/ press-releases?field_nir_news_date_value
. - Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19:185-99.
- Yoon DH, Koh Y, Park H, Hwang YK, Kim WS. A phase 1 study of the combination of MG4101, ex vivo -expanded allogeneic NK cells and rituximab for relapsed or refractory Non-Hodgkin Lymphoma. Blood. 2020;136:14-15.
- Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother. 2018;67:483-94.
- Liang S, Lin M, Niu L, Xu K, Wang X, Liang Y, et al. Cetuximab combined with natural killer cells therapy: an alternative to chemoradiotherapy for patients with advanced non-small cell lung cancer (NSCLC). Am J Cancer Res. 2018;8:879-91.
- Lin M, Luo H, Liang S, Chen J, Liu A, Niu L, et al. Pembrolizumab plus allogeneic NK cells in advanced non-small cell lung cancer patients. J Clin Invest. 2020;130:2560-9.
- Chung YY, Park SW, Im J-M, Yoo D-K, Cheon H-C, Kim J-E, et al. Abstract CT171: combinatorial allogeneic NK cell therapy with Pembrolizumab for cholangiocarcinoma; interim report of open label Phase1/2a study. Cancer Res. 2021;81:CT171-CT171.
- Adotevi O, Godet Y, Galaine J, Lakkis Z, Idirene I, Certoux JM, et al. In situ delivery of allogeneic natural killer cell (NK) combined with Cetuximab in liver metastases of gastrointestinal carcinoma: a phase I clinical trial. Oncolmmunology. 2018;7:e1424673.
- Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, et al. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin Cancer Res. 2017;23:6441-9.
- Talleur AC, Triplett BM, Federico S, Mamcarz E, Janssen W, Wu J, et al. Consolidation therapy for newly diagnosed pediatric patients with high-risk neuroblastoma using Busulfan/Melphalan, autologous hematopoietic cell transplantation, anti-GD2 antibody, granulocyte-macrophage
colony-stimulating factor, interleukin-2, and haploidentical natural killer cells. Biol Blood Marrow Transplant. 2017;23:1910-7. - Schmidt P, Raftery MJ, Pecher G. Engineering NK cells for CAR therapy-recent advances in gene transfer methodology. Front Immunol. 2021;11:611163.
- Gong Y, Klein Wolterink RGJ, Janssen I, Groot AJ, Bos GMJ, Germeraad WTV. Rosuvastatin enhances VSV-G lentiviral transduction of NK cells via upregulation of the low-density lipoprotein receptor. Mol Ther Methods Clin Dev. 2020;17:634-46.
- Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol. 2020;10:3123.
- Sengsayadeth S , Savani BN, Oluwole O, Dholaria B. Overview of approved CAR-T therapies, ongoing clinical trials, and its impact on clinical practice. eJHaem. 2022;3:6-10.
- Zhang
, Guo Y, Ji Y, Gao Y, Zhang M, Liu Y, et al. Cytokine release syndrome after modified CAR-NK therapy in an advanced non-small cell lung cancer patient: a case report. Cell Transpl. 2022;31:096368972210942. - Klingemann H. Are natural killer cells superior CAR drivers? Oncolmmunology. 2014;3:e28147.
- Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8:1083-9.
- Fate Therapeutics. Fate Therapeutics Showcases Positive Interim Phase 1 Data from FT596 Of-the-shelf, iPSC-derived CAR NK Cell Program for Relapsed/Refractory B-cell Lymphoma at 2021 ASH Annual Meeting [Press Release]. 2021. https://ir.fatetherapeutics.com/press-releases?field_nir_news_date_value% 5Bmin%5D=2021.
- Bachanova V, Cayci Z, Lewis D, Maakaron JE, Janakiram M, Bartz A, et al. Initial clinical activity of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood. 2020;136:8-8.
- Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545-53.
- Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19+B cell tumors: a phase
trial. Nat Med. 2024. https://doi.org/10.1038/s41591-023-02785-8. - Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8:297-310.
- Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, et al. cd 19- car engineered
cells are sufficient to overcome cell resistance in -cell malignancies. J Cell Mol Med. 2016;20:1287-94. - Oelsner S, Waldmann A, Billmeier A, Röder J, Lindner A, Ullrich E, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. 2019;145:1935-45.
- Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917-27.
- Mansour AG, Teng K-Y, Li Z, Zhu Z, Chen H, Tian L, et al. Off-the-shelf CAR-engineered natural killer cells targeting FLT3 enhance killing of acute myeloid leukemia. Blood Adv. 2023;7:6225-39.
- Chen Y, You F, Jiang L, Li J, Zhu X, Bao Y, et al. Gene-modified NK-92MI cells expressing a chimeric CD16-BB-
or CD64-BB- receptor exhibit enhanced cancer-killing ability in combination with therapeutic antibody. Oncotarget. 2017;8:37128-39. - Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, et al. Targeting CD20+ aggressive B-cell Non-Hodgkin lymphoma by anti-CD20 CAR mRNAmodified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3:333-44.
- Caruso S, De Angelis B, Del Bufalo F, Ciccone R, Donsante S, Volpe G, et al. Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J Hematol Oncol. 2022;15:163.
- Klaihmon P, Kang X, Issaragrisil S, Luanpitpong S. Generation and functional characterization of anti-CD19 chimeric antigen receptor-natural killer cells from human induced pluripotent stem cells. IJMS. 2023;24:10508.
- Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136:2308-18.
- Albinger N, Bexte T, Buchinger L, Wendel P, Al-Ajami A, Gessner A, et al. CRISPR/ Cas9 gene editing of immune checkpoint receptor NKG2A improves the efficacy of primary CD33-CAR-NK cells against AML. Blood. 2022;140:4558-9.
- Portillo AL, Hogg R, Poznanski SM, Rojas EA, Cashell NJ, Hammill JA, et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience. 2021;24:102619.
- Ao X, Yang Y, Li W, Tan Y, Guo W, Ao L, et al. Anti-aFR CAR-engineered NK-92 cells display potent cytotoxicity against aFR-positive ovarian cancer. J Immunother. 2019;42:284-96.
- Liu WN, So WY, Harden SL, Fong SY, Wong MXY, Tan WWS, et al. Successful targeting of PD-1/PD-L1 with chimeric antigen receptor-natural killer cells and nivolumab in a humanized mouse cancer model. Sci Adv. 2022;8:eadd1187.
- Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483.
- Zuo P, Li Y, He C, Wang T, Zheng X, Liu H, et al. Anti-tumor efficacy of anti-GD2 CAR NK-92 cells in diffuse intrinsic pontine gliomas. Front Immunol. 2023;14:1145706.
- Liu T, Dai X, Xu Y, Guan T, Hong L, Zaib T, et al. CD22 is a potential target of CARNK cell therapy for esophageal squamous cell carcinoma. J Transl Med. 2023;21:710.
- Chang Y-H, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777-86.
- Whalen KA, Rakhra K, Mehta NK, Steinle A, Michaelson JS, Baeuerle PA. Engaging natural killer cells for cancer therapy via NKG2D, CD16A and other receptors. mAbs. 2023;15:2208697.
- Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. 2019;27:1114-25.
- Cao B, Liu M, Huang J, Zhou J, Li J, Lian H, et al. Development of mesothelinspecific CAR NK-92 cells for the treatment of gastric cancer. Int J Biol Sci. 2021;17:3850-61.
- Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A novel chimeric PD1-NKG2D41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol Immunol. 2020;122:200-6.
- Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181-192.e5.
- Chambers AM, Lupo KB, Wang J, Cao J, Utturkar S, Lanman N, et al. Engineered natural killer cells impede the immunometabolic CD73-adenosine axis in solid tumors. eLife. 2022;11:e73699.
- Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. 2009;15:4857-66.
- Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptormodified natural killer cells against hepatocellular cancer cells in vitro. CMAR. 2020;12:3247-55.
- Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immuneinhibitory HLA-G. Oncolmmunology. 2017;6:e1250050.
- Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194:3201-12.
- Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvlll-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1a-secreting glioblastoma. J Immunother. 2015;38:197-210.
- Robbins Y, Greene S, Friedman J, Clavijo PE, Van Waes C, Fabian KP, et al. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. eLife. 2020;9:e54854.
- Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12:49.
- Zhuang
, Long EO. NK cells equipped with a chimeric antigen receptor that overcomes inhibition by HLA class I for adoptive transfer of CAR-NK cells. Front Immunol. 2022;13:840844. - Chang Y, Jin G, Luo W, Luo Q, Jung J, Hummel SN, et al. Engineered human pluripotent stem cell-derived natural killer cells with PD-L1 responsive immunological memory for enhanced immunotherapeutic efficacy. Bioact Mater. 2023;27:168-80.
- Goulding J, Yeh W-I, Hancock B, Blum R, Xu T, Yang B-H, et al. A chimeric antigen receptor uniquely recognizing MICA/B stress proteins provides an effective approach to target solid tumors. Med. 2023;4:457-477.e8.
- Li M, Zhi L, Yin M, Guo C, Zhang H, Lu C, et al. A novel bispecific chimeric PD1DAP10/NKG2D receptor augments NK92-cell therapy efficacy for human gastric cancer SGC-7901 cell. Biochem Biophys Res Commun. 2020;523:745-52.
- Raftery MJ, Franzén AS, Radecke C, Boulifa A, Schönrich G, Stintzing S, et al. Next generation CD44v6-specific CAR-NK cells effective against triple negative breast cancer. IJMS. 2023;24:9038.
- Aldea M, Andre F, Marabelle A, Dogan S, Barlesi F, Soria J-C. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discov. 2021;11:874-99.
- Song M-K, Park B-B, Uhm J-E. Resistance mechanisms to CAR T-cell therapy and overcoming strategy in B-cell hematologic malignancies. JJMS. 2019;20:5010.
- Zhou X, Rasche L, Kortüm KM, Mersi J, Einsele H. BCMA loss in the epoch of novel immunotherapy for multiple myeloma: from biology to clinical practice. Haematologica. 2022;108:958-68.
- Roex G, Campillo-Davo D, Flumens D, Shaw PAG, Krekelbergh L, De Reu H, et al. Two for one: targeting BCMA and CD19 in B-cell malignancies with off-the-shelf dual-CAR NK-92 cells. J Transl Med. 2022;20:124.
- Cronk RJ, Zurko J, Shah NN. Bispecific chimeric antigen receptor T cell therapy for B cell malignancies and multiple myeloma. Cancers. 2020;12:2523.
- Lim RM, Rong L, Zhen A, Xie J. A universal CAR-NK cell targeting various epitopes of HIV-1 gp160. ACS Chem Biol. 2020;15:2299-310.
- Garrison BS, Deng H, Yucel G, Frankel NW, Guzman-Ayala M, Gordley R, et al. FLT3 OR CD33 NOT EMCN logic gated CAR-NK cell therapy (SENTI-202) for precise targeting of AML. Blood. 2021;138:2799-2799.
- Li Y, Basar R, Wang G, Liu E, Moyes JS, Li L, et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat Med. 2022;28:2133-44.
- Hamieh M, Dobrin A, Cabriolu A, Van Der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568:112-6.
- Ramezani F, Panahi Meymandi AR, Akbari B, Tamtaji OR, Mirzaei H, Brown CE, et al. Outsmarting trogocytosis to boost CAR NK/T cell therapy. Mol Cancer. 2023;22:183.
- Luo H, Wu X, Sun R, Su J, Wang Y, Dong Y, et al. Target-dependent expression of IL12 by synNotch receptor-engineered NK92 cells increases the antitumor activities of CAR-T cells. Front Oncol. 2019;9:1448.
- Tseng H, Xiong W, Badeti S, Yang Y, Ma M, Liu T, et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat Commun. 2020;11:4810.
- Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173:1426-1438.e11.
- Li H-S, Wong NM, Tague E, Ngo JT, Khalil AS, Wong WW. High-performance multiplex drug-gated CAR circuits. Cancer Cell. 2022;40:1294-1305.e4.
- Li H-S, Israni DV, Gagnon KA, Gan KA, Raymond MH, Sander JD, et al. Multidimensional control of therapeutic human cell function with synthetic gene circuits. Science. 2022;378:1227-34.
- Zhang R, Liu Q, Zhou S, He H, Zhao M, Ma W. Engineering CAR-NK cells targeting CD33 with concomitant extracellular secretion of anti-CD16 antibody revealed superior antitumor effects toward myeloid leukemia. Cancer Lett. 2023;558:216103.
- Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination therapy with epCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J Immunol Res. 2018;2018:1-11.
- Wang F, Wu L, Yin L, Shi H, Gu Y, Xing N. Combined treatment with anti-PSMA CAR NK-92 cell and anti-PD-L1 monoclonal antibody enhances the antitumour efficacy against castration-resistant prostate cancer. Clin Transl Med. 2022;12. https://doi.org/10.1002/ctm2.901.
- Strecker MI, Wlotzka K, Strassheimer F, Roller B, Ludmirski G, König S, et al. AAVmediated gene transfer of a checkpoint inhibitor in combination with HER2targeted CAR-NK cells as experimental therapy for glioblastoma. Oncolmmunology. 2022;11:2127508.
- Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7:27764-77.
- Dong H, Ham JD, Hu G, Xie G, Vergara J, Liang Y, et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc Natl Acad Sci USA. 2022;119:e2122379119.
- Baulu E, Gardet C, Chuvin N, Depil S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci Adv. 2023;9:eadf3700.
- Rasul KH, Hussain A, Reilly H, Karvouni M, Dahlberg CIM, Al-Attar MS, et al. Assessment of T cell receptor complex expression kinetics in natural killer cells. CIMB. 2022;44:3859-71.
- Mensali N, Dillard P, Hebeisen M, Lorenz S, Theodossiou T, Myhre MR, et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine. 2019;40:106-17.
- Parlar A, Sayitoglu EC, Ozkazanc D, Georgoudaki A, Pamukcu C, Aras M, et al. Engineering antigen-specific NK cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur J Immunol. 2019;49:1278-90.
- Morton LT, Wachsmann TLA, Meeuwsen MH, Wouters AK, Remst DFG, van Loenen MM, et al. T cell receptor engineering of primary NK cells to therapeutically target tumors and tumor immune evasion. J Immunother Cancer. 2022;10:e003715.
- Kobayashi E, Ozawa T, Hamana H, Muraguchi A, Kishi H. Gene modified NK cell line as a powerful tool for evaluation of cloned TCRs for TCR-T cell therapy. Cell Immunol. 2023;383:104656.
- Poorebrahim M, Quiros-Fernandez I, Marmé F, Burdach SEG, Cid-Arregui A. A costimulatory chimeric antigen receptor targeting TROP2 enhances the cytotoxicity of NK cells expressing a T cell receptor reactive to human papillomavirus type 16 E7. Cancer Lett. 2023;566:216242.
- Mansilla-Soto J, Eyquem J, Haubner S, Hamieh M, Feucht J, Paillon N, et al. HLAindependent T cell receptors for targeting tumors with low antigen density. Nat Med. 2022;28:345-52.
- Liu Y, Liu G, Wang J, Zheng Z, Jia L, Rui W, et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci Transl Med. 2021;13:eabb5191.
- Birtel M, Voss R-H, Reinhard K, Rengstl B, Ouchan Y, Michel K, et al. A TCR-like CAR promotes sensitive antigen recognition and controlled T-cell expansion upon mRNA vaccination. Cancer Res Commun. 2022;2:827-41.
- Li S, Zhang C, Shen L, Teng X, Xiao Y, Yu B, et al. TCR extracellular domain genetically linked to CD28, 2B4/41BB and DAP10/CD3了, -engineered NK cells mediates antitumor effects. Cancer Immunol Immunother. 2023;72:769-74.
- Dixon KJ, Wu J, Walcheck B. Engineering anti-tumor monoclonal antibodies and Fc receptors to enhance ADCC by human NK cells. Cancers. 2021;13:312.
- Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M et al. Efficient mRNAbased genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front Immunol. 2016;7. https://doi.org/10.3389/fimmu.2016.00105.
- Clara JA, Levy ER, Reger R, Barisic S, Chen L, Cherkasova E, et al. High-affinity CD16 integration into a CRISPR/Cas9-edited CD38 locus augments CD38directed antitumor activity of primary human natural killer cells. J Immunother Cancer. 2022;10:e003804.
- Giles AJ, Hao S, Padget M, Song H, Zhang W, Lynes J, et al. Efficient ADCC killing of meningioma by avelumab and a high-affinity natural killer cell line, haNK. JCl Insight. 2019;4:e130688.
- Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. 2020;135:399-410.
- Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, et al. Identification of an ADAM17 Cleavage Region in Human CD16 (FcyRIII) and the Engineering of a Non-Cleavable Version of the Receptor in NK Cells. PLoS One. 2015;10:e0121788.
- Cichocki F, Bjordahl R, Goodridge JP, Mahmood S, Gaidarova S, Abujarour R, et al. Quadruple gene-engineered natural killer cells enable multi-antigen targeting for durable antitumor activity against multiple myeloma. Nat Commun. 2022;13:7341.
- Eitler J, Wotschel N, Miller N, Boissel L, Klingemann HG, Wels W, et al. Inability of granule polarization by NK cells defines tumor resistance and can be overcome by CAR or ADCC mediated targeting. J Immunother Cancer. 2021;9:e001334.
- Meng F, Zhang S, Xie J, Zhou Y, Wu Q, Lu B, et al. Leveraging CD16 fusion receptors to remodel the immune response for enhancing anti-tumor immunotherapy in iPSC-derived NK cells. J Hematol Oncol. 2023;16:62.
- Trick F, Benenson Y ADCC-and BCR-inspired receptors for antigen-specific NK cell activaiton. 2022. https://www.esgctcongress.com/poster-list.
- Tong L, Jiménez-Cortegana C, Tay AHM, Wickström S, Galluzzi L, Lundqvist A. NK cells and solid tumors: therapeutic potential and persisting obstacles. Mol Cancer. 2022;21:206.
- Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother. 2015;64:225-35.
- Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother cancer. 2017;5:73.
- Yang Y, Gordon N, Kleinerman ES, Huang G. Promoting NK cell trafficking to improve therapeutic effect of NK cell therapy on osteosarcoma. J Immunother Cancer. 2015;3:P24.
- Gao L, Yang L, Zhang S, Ge Z, Su M, Shi Y, et al. Engineering NK-92 cell by upregulating CXCR2 and IL-2 Via CRISPR-Cas9 improves its antitumor effects as cellular immunotherapy for human colon cancer. J Interferon Cytokine Res. 2021;41:450-60.
- Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75-85.
- Levy ER, Clara JA, Reger RN, Allan DSJ, Childs RW. RNA-seq analysis reveals CCR5 as a key target for CRISPR gene editing to regulate in vivo NK cell trafficking. Cancers. 2021;13:872.
- Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, et al. Enhanced bone marrow homing of natural killer cells following mRNA transfection with gain-of-function variant CXCR4R334X. Front Immunol. 2019;10:1262.
- Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, et al. Highly efficient generation of transgenically augmented CAR NK cells overexpressing CXCR4. Front Immunol. 2020;11:2028.
- Yang L, Huang C, Wang C, Zhang S, Li Z, Zhu Y, et al. Overexpressed CXCR4 and CCR7 on the surface of NK92 cell have improved migration and anti-tumor activity in human colon tumor model. Anti Cancer Drugs. 2020;31:333-44.
- Schomer NT, Jiang ZK, Lloyd MI, Klingemann H, Boissel L. CCR7 expression in CD19 chimeric antigen receptor-engineered natural killer cells improves migration toward CCL19-expressing lymphoma cells and increases tumor control in mice with human lymphoma. Cytotherapy. 2022;24:827-34.
- Somanchi SS, Somanchi A, Cooper UN, Lee DA. Engineering lymph node homing of ex vivo-expanded human natural killer cells via trogocytosis of the chemokine receptor CCR7. Blood. 2012;119:5164-72.
- Feigl FF, Stahringer A, Peindl M, Dandekar G, Koehl U, Fricke S, et al. Efficient redirection of NK cells by genetic modification with chemokine receptors CCR4 and CCR2B. IJMS. 2023;24:3129.
- Evgin L, Kottke T, Tonne J, Thompson J, Huff AL, Van Vloten J, et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci Transl Med. 2022;14:eabn2231.
- Li F, Sheng Y, Hou W, Sampath P, Byrd D, Thorne S, et al. CCL5-armed oncolytic virus augments CCR5-engineered NK cell infiltration and antitumor efficiency. J Immunother Cancer. 2020;8:e000131.
- Tao X, Zhang R, Du R, Yu T, Yang H, Li J, et al. EP3 enhances adhesion and cytotoxicity of NK cells toward hepatic stellate cells in a murine liver fibrosis model. J Exp Med. 2022;219:e20212414.
- Choi YH, Lim EJ, Kim SW, Moon YW, Park KS, An H-J. IL-27 enhances IL-15/IL-18mediated activation of human natural killer cells. J Immunother Cancer. 2019;7:168.
- Liu M, Meng Y, Zhang L, Han Z, Feng X. High-efficient generation of natural killer cells from peripheral blood with preferable cell vitality and enhanced cytotoxicity by combination of IL-2, IL-15 and IL-18. Biochem Biophys Res Commun. 2021;534:149-56.
- Lu T, Ma R, Dong W, Teng K-Y, Kollath DS, Li Z, et al. Off-the-shelf CAR natural killer cells secreting IL-15 target spike in treating COVID-19. Nat Commun. 2022;13:2576.
- Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61:1451-61.
- Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520-31.
- Bachier C, Borthakur G, Hosing C, Blum W, Rotta M, Ojeras P, et al. A phase 1 study of NKX101, an allogeneic CAR natural killer (NK) cell therapy, in subjects with relapsed/refractory (R/R) acute myeloid leukemia (AML) or higher-risk myelodysplastic syndrome (MDS). Blood. 2020;136:42-43.
- Silvestre RN, Eitler J, De Azevedo JTC, Tirapelle MC, Fantacini DMC, De Souza LEB, et al. Engineering NK-CAR. 19 cells with the IL-15/IL-15Ra complex improved proliferation and anti-tumor effect in vivo. Front Immunol. 2023;14:1226518.
- Bjordahl R, Gaidarova S, Goodridge JP, Mahmood S, Bonello G, Robinson M, et al. FT576: a novel multiplexed engineered off-the-shelf natural killer cell immunotherapy for the dual-targeting of CD38 and Bcma for the treatment of multiple myeloma. Blood. 2019;134:3214-3214.
- Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SMH, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. 2014;124:1081-8.
- Zhang D, Zhao Y, Kang Y, Hu J, Li R, Song J, et al. Enhanced NK cell adoptive antitumor effects against breast cancer in vitro via blockade of the transforming growth factor-
signaling pathway. Onco Targets Ther. 2015;2015:1553-9. - Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-
receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy. 2017;19:408-18. - Chaudhry K, Geiger A, Dowlati E, Lang H, Sohai DK, Hwang El, et al. Cotransducing B7H3 CAR-NK cells with the DNR preserves their cytolytic function against GBM in the presence of exogenous TGF-
. Mol Ther Methods Clin Dev. 2022;27:415-30. - Yang B, Liu H, Shi W, Wang Z, Sun S, Zhang G, et al. Blocking transforming growth factor-
signaling pathway augments antitumor effect of adoptive NK92 cell therapy. Int Immunopharmacol. 2013;17:198-204. - Burga RA, Yvon E, Chorvinsky E, Fernandes R, Cruz CRY, Bollard CM. Engineering the TGF
receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clin Cancer Res. 2019;25:4400-12. - Wang Z, Guo L, Song Y, Zhang Y, Lin D, Hu B, et al. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-
R II and NKG2D. Cancer Immunol Immunother. 2017;66:537-48. - Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther. 2014;22:1211-20.
- Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25:249-58.
- Lee MY, Robbins Y, Sievers C, Friedman J, Abdul Sater H, Clavijo PE, et al. Chimeric antigen receptor engineered NK cellular immunotherapy overcomes the selection of T-cell escape variant cancer cells. J Immunother Cancer. 2021;9:e002128.
- Mensali N, Dillard P, Fayzullin A, Köksal H, Gaudernack G, Kvalheim G, et al. Builtin” PD-1 blocker to rescue NK-92 activity from PD-L1-mediated tumor escape mechanisms. FASEB J. 2021;35:e21750.
- Susek KH, Schwietzer YA, Karvouni M, Gilljam M, Keszei M, Hussain A, et al. Generation of NK cells with chimeric-switch receptors to overcome PD1mediated inhibition in cancer immunotherapy. Cancer Immunol Immunother. 2023;72:1153-67.
- Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2020;107:437-45.
- Naeimi Kararoudi M, Nagai Y, Elmas E, de Souza Fernandes Pereira M, Ali SA, Imus PH, et al. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood. 2020;136:2416-27.
- Jiang J, Chen J, Liao C, Duan Y, Wang Y, Shang K, et al. Inserting EF1a-driven CD7-specific CAR at CD7 locus reduces fratricide and enhances tumor rejection. Leukemia. 2023;37:1660-70.
- Liao C, Wang Y, Huang Y, Duan Y, Liang Y, Chen J, et al. CD38-specific CAR integrated into CD38 locus driven by different promoters causes distinct antitumor activities of T and NK cells. Adv Sci. 2023;10:2207394.
- Hoerster K, Uhrberg M, Wiek C, Horn PA, Hanenberg H, Heinrichs S. HLA class I knockout converts allogeneic primary NK cells into suitable effectors for “off-the-shelf” immunotherapy. Front Immunol. 2021;11:586168.
- Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Investig. 2019;129:2094-106.
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8. https://doi.org/10.1126/ scitranslmed.aaf2341.
- He B, Mai Q, Pang Y, Deng S, He Y, Xue R, et al. Cytokines induced memory-like NK cells engineered to express CD19 CAR exhibit enhanced responses against
cell malignancies. Front Immunol. 2023;14:1130442. - Cho H, Kim KH, Lee H, Kim CG, Chung H, Choi YS, et al. Adaptive natural killer cells facilitate effector functions of daratumumab in multiple myeloma. Clin Cancer Res. 2021;27:2947-58.
- Woan KV, Kim H, Bjordahl R, Davis ZB, Gaidarova S, Goulding J, et al. Harnessing features of adaptive NK cells to generate iPSC-derived NK cells for enhanced immunotherapy. Cell Stem Cell. 2021;28:2062-2075.e5.
- Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CARNK cells. Blood. 2021;137:624-36.
- Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27:224-237.e6.
- Pekar L, Klausz K, Busch M, Valldorf B, Kolmar H, Wesch D, et al. Affinity maturation of B7-H6 translates into enhanced NK cell-mediated tumor cell lysis and improved proinflammatory cytokine release of bispecific immunoligands via NKp30 engagement. J Immunol. 2021;206:225-36.
- Klausz K, Pekar L, Boje AS, Gehlert CL, Krohn S, Gupta T, et al. Multifunctional NK cell-engaging antibodies targeting EGFR and NKp30 elicit efficient tumor cell killing and proinflammatory cytokine release. J Immunol. 2022;209:1724-35.
- Raynaud A, Desrumeaux K, Vidard L, Termine E, Baty D, Chames P, et al. AntiNKG2D single domain-based antibodies for the modulation of anti-tumor immune response. Oncolmmunology. 2021;10:1854529.
- Nikkhoi SK, Li G, Eleya S, Yang G, Vandavasi VG, Hatefi A. Bispecific killer cell engager with high affinity and specificity toward CD16a on NK cells for cancer immunotherapy. Front Immunol. 2023;13:1039969.
- Lipinski B, Arras P, Pekar L, Klewinghaus D, Boje AS, Krah S et al. NKp46 -specific single domain antibodies enable facile engineering of various potent NK cell engager formats. Protein Sci. 2023;32. https://doi.org/10.1002/pro.4593.
- Colomar-Carando N, Gauthier L, Merli P, Loiacono F, Canevali P, Falco M, et al. Exploiting natural killer cell engagers to control pediatric B-cell precursor acute lymphoblastic leukemia. Cancer Immunol Res. 2022;10:291-302.
- von Strandmann EP, Hansen HP, Reiners KS, Schnell R, Borchmann P, Merkert S, et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood. 2006;107:1955-62.
- Zhao Y, Stepto H, Schneider CK. Development of the first World Health Organization lentiviral vector standard: toward the production control and standardization of lentivirus-based gene therapy products. Hum Gene Ther Methods. 2017;28:205-14.
- Glaser V, Flugel C, Kath J, Du W, Drosdek V, Franke C, et al. Combining different CRISPR nucleases for simultaneous knock-in and base editing prevents translocations in multiplex-edited CAR T cells. Genome Biol. 2023;24:89.
- Tsuchida CA, Brandes N, Bueno R, Trinidad M, Mazumder T, Yu B et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell Biol. 2023 https://doi.org/10.1101/2023.03.22.533709.
- Wellhausen N, Agarwal S, Rommel PC, Gill SI, June CH. Better living through chemistry: CRISPR/Cas engineered T cells for cancer immunotherapy. Curr Opin Immunol. 2022;74:76-84.
- Wang X, Jasinski DL, Medina JL, Spencer DM, Foster AE, Bayle JH. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020;4:1950-64.
- Allan DSJ, Wu C, Mortlock RD, Chakraborty M, Rezvani K, Davidson-Moncada JK, et al. Expanded NK cells used for adoptive cell therapy maintain diverse clonality and contain long-lived memory-like NK cell populations. Mol Ther Oncolytics. 2023;28:74-87.
مساهمات المؤلفين
تمويل
المصالح المتنافسة
معلومات إضافية
© المؤلف(ون) 2024
- ¹مركز أبحاث السرطان في ليون، UMR INSERM U1052 CNRS 5286، مركز ليون بيران، ليون، فرنسا.
إيرفيميون، ليون، فرنسا. مركز ليون بيرار، ليون، فرنسا. جامعة كلود برنار ليون 1، ليون، فرنسا. البريد الإلكتروني:audrey.page@lyon.unicancer.fr; ستيفان.ديبيل@ليون.يونكانسر.فر
DOI: https://doi.org/10.1038/s41423-024-01145-x
PMID: https://pubmed.ncbi.nlm.nih.gov/38443448
Publication Date: 2024-03-05
Development of NK cell-based cancer immunotherapies through receptor engineering
Abstract
Natural killer (NK) cell-based immunotherapies are attracting increasing interest in the field of cancer treatment. Early clinical trials have shown promising outcomes, alongside satisfactory product efficacy and safety. Recent developments have greatly increased the therapeutic potential of NK cells by endowing them with enhanced recognition and cytotoxic capacities. This review focuses on surface receptor engineering in NK cell therapy and discusses its impact, challenges, and future directions.
Most approaches are based on engineering with chimeric antigen receptors to allow NK cells to target specific tumor antigens independent of human leukocyte antigen restriction. This approach has increased the precision and potency of NK-mediated recognition and elimination of cancer cells. In addition, engineering NK cells with T-cell receptors also mediates the recognition of intracellular epitopes, which broadens the range of target peptides. Indirect tumor peptide recognition by NK cells has also been improved by optimizing immunoglobulin constant fragment receptor expression and signaling. Indeed, engineered NK cells have an improved ability to recognize and destroy target cells coated with specific antibodies, thereby increasing their antibodydependent cellular cytotoxicity. The ability of NK cell receptor engineering to promote the expansion, persistence, and infiltration of transferred cells in the tumor microenvironment has also been explored. Receptor-based strategies for sustained NK cell functionality within the tumor environment have also been discussed, and these strategies providing perspectives to counteract tumor-induced immunosuppression.
INTRODUCTION
novel NK cell subpopulations beyond the usual classification have been identified based on CD16 and CD56 [3, 4]. Among them, a subset called adaptive NK cells, which are characterized by a lack of Fc-epsilon receptor Ig (FCER1G) expression and an overexpression of natural killer group 2 C (NKG2C), exhibit enhanced properties, including high proliferation, increased expansion and cytotoxic potential. Unlike T cells, NK cells are not restricted to major histocompatibility complex (MHC, also called human leukocyte antigen (HLA)) recognition, and their activation requires multiple receptors. Indeed, their activation relies on finely tuning inhibitory and activating signals. To prevent the undesirable activation of healthy cells, NK cells express killer cell immunoglobulin-like receptors (KIRs) and the natural killer group 2 A (NKG2A)/CD94 heterodimer, which interacts with HLA class I molecules to provide inhibitory signals (Fig. 1). Conversely, NK cells possess an array of activating receptors, including natural killer group 2D (NKG2D), DNAX accessory molecule-1 (DNAM-1),
LESSONS FROM INITIAL CLINICAL TRIALS WITH NK CELLS
IMPROVING TUMOR CELL RECOGNITION Endowing NK cells with chimeric antigen receptors
| CAR Target | NK source | Target cancers | Date | Phase | Status | Number | |
| Solid tumors | 5T4 Oncofetal Trophoblast Glycoprotein | Not disclosed | Advanced Solid Tumors | 2022 | I | Recruiting | NCT05194709 |
| CD70 | CB-derived NK cells | Advanced Renal Cell Carcinoma, Mesothelioma and Osteosarcoma | 2023 | I/II | Recruiting | NCT05703854 | |
| CLDN6 | Patient-PB-NK cells | CLDN6-positive Advanced Solid Tumors | 2022 | I/II | Recruiting | NCT05410717 | |
| DLL3 | Not disclosed | Extensive Stage Small Cell Lung Cancer | 2022 | I | Recruiting | NCT05487651 | |
| HER2 | NK cell line | Recurrent HER2-positive Glioblastoma | 2017 | I | Unknown | NCT03383978 | |
| Mesothelin | Autologous | Refractory Epithelial Ovarian Carcinoma | 2021 | 0 | Recruiting | ChiCTR2100048100 | |
| iPSC-derived NK cells | Epithelial Ovarian Cancer | 2018 | I | Unknown | NCT03692637 | ||
| Muc1 | Not disclosed | MUC1-Positive Relapsed or Refractory Solid Tumor | 2016 | 1/II | Unknown. | NCT02839954 | |
| NKG2D ligands | Not disclosed | Platinum-Resistant Recurrent Ovarian Cancer | 2023 | Recruiting | NCT05776355 | ||
| Not disclosed | Refractory Metastatic Colorectal Cancer | 2022 | I | Recruiting | NCT05213195 | ||
| Not disclosed | Metastatic Solid Tumors | 2017 | I | Completed | NCT03415100 | ||
| Not disclosed | Not disclosed | Ovarian epithelial carcinoma | 2023 | I | Not yet recruiting | NCT05856643 | |
| Not disclosed | Advanced Hepatocellular Carcinoma | 2023 | I | Not yet recruiting | NCT05845502 | ||
| Not disclosed | Advanced Triple Negative Breast Cancer | 2023 | I | Not yet recruiting | NCT05686720 | ||
| PDL1 | NK cell line | Recurrent/Metastatic Gastric or Head and Neck Cancer | 2021 | II | Recruiting | NCT04847466 | |
| PSMA | iPSC-derived NK cells | Metastatic Castration-Resistant Prostate Cancer | 2018 | I | Unknown | NCT03692663 | |
| ROBO1 | Not disclosed | Pancreatic Cancer | 2019 | I/II | Recruiting | NCT03941457 | |
| Not disclosed | Solid Tumors | 2019 | I/II | Recruiting | NCT03940820 | ||
| Not disclosed | Malignant Tumor | 2019 | I/II | Recruiting | NCT03931720 | ||
| TROP2 | CB-derived NK cells | Platinum Resistant Ovarian Cancer, Mesonephric-like Adenocarcinoma, and Pancreatic Cancer | 2023 | I/II | Not yet recruiting | NCT05922930 | |
| Hematologic malignancies | CD33 | Not disclosed | Relapsed/Refractory Acute Myeloid Leukemia | 2021 | I | Not yet ecruiting | NCT05008575 |
| NK cell line | Relapsed/Refractory Acute Myeloid Leukemias | 2016 | I/II | Unknown | NCT02944162 | ||
| CD33/TIM3 | CB-derived NK cells | Acute Myeloid Leukemia | 2021 | 0 | Recruiting | ChiCTR2100043081 | |
| CD33/CLL1 | Not disclosed | Relapsed/Refractory Acute Myeloid Leukemia | 2021 | 0 | Recruiting | ChiCTR2100047084 | |
| CD33/CCL1 | Not disclosed | Acute Myeloid Leukemia | 2020 | I | Recruiting | NCT05215015 | |
| CD19 | Not disclosed | Relapsed/Refractory Diffuse Large B-Cell Lymphoma | 2023 | I | Not yet recruiting | NCT05673447 | |
| iPSCs-derived NK cells | CD19-positive B-Cell Malignancies | 2023 | I | Recruiting | NCT05336409 | ||
| Allogenic | B-Cell Hematologic Malignancies | 2022 | I/II | Recruiting | NCT05654038 | ||
| Allogenic | Adult Relapsed/Refractory B-cell Malignancies | 2022 | I | Recruiting | NCT05645601 |
| CAR Target | NK source | Target cancers | Date | Phase | Status | Number |
| Not disclosed | Relapsed/Refractory Acute Lymphoblastic Leukemia | 2022 | I | Recruitment completed | NCT05563545 | |
| CB-derived NK cells | Refractory/Relapsed B-cell Non-Hodgkin Lymphoma L | 2022 | I | Recruiting | NCT05472558 | |
| Not disclosed | Relapsed/Refractory B-cell Malignancies | 2022 | । | Recruiting | NCT05410041 | |
| Allogenic | B-cell Malignancies | 2021 | I | Recruiting | NCT05020678 | |
| HLA haploidentical NK cells (PB) | Refractory/Relapsed B-cell Non-Hodgkin Lymphoma | 2021 | I | Recruiting | NCT04887012 | |
| CB-derived NK cells | B Lymphoid Malignancies | 2021 | I | Recruiting | NCT04796675 | |
| Not disclosed | Relapsed/refractory B-cell Acute Lymphoblastic Leukemia | 2021 | I | Recruiting | NCT05379647 | |
| iPSC-derived NK cells | B-cell Lymphoma or Chronic Lymphocytic Leukemia. | 2020 | I | Recruiting | NCT04245722 | |
| Not disclosed | Relapsed or Refractory B-Cell NonHodgkin Lymphoma | 2020 | I | Not yet ecruiting | NCT04639739 | |
| Not disclosed | Relapsed and Refractory B-Cell Lymphoma | 2019 | I | Unknown | NCT03824951 | |
| iPSC-derived NK cells | Relapsed and Refractory B-Cell Lymphoma | 2018 | I | Unknown | NCT03690310 | |
| UC and CB-derived NK cells | B Lymphoid Malignancies | 2017 | I/II | Completed [38] | NCT03056339 | |
| NK cell line | CD19 Positive Leukemia and Lymphoma | 2016 | I/II | Unknown | NCT02892695 | |
| PB NK cells (allogenic) | Relapsed and Refractory Lymphoma | 2013 | I | Completed. | NCT01974479 | |
| PB NK cells (allogenic) | Relapsed and/or Refractory Acute Lymphoid Leukemias | 2009 | I | Completed. | NCT00995137 | |
| CD19/CD22 | iPSC-derived NK cells | Relapsed and Refractory B-Cell Lymphoma | 2019 | I | Not yet recruiting. | NCT03824964 |
| CD19/CD70 | CB-derived NK cells | Refractory/Relapsed B-cell Non-Hodgkin Lymphoma | 2023 | I/II | Recruiting | NCT05842707 |
| CB-derived NK cells | Refractory/Relapsed B-cell Non-Hodgkin Lymphoma | 2022 | I | Recruiting | NCT05667155 | |
| CD70 | CB-derived NK cells | Relapse/Refractory Hematological Malignances | 2021 | I/II | Recruiting | NCT05092451 |
| CD7 | Allogenic | CD7 Hematologic Malignancies | 2022 | I | Not yet recruiting | NCT05377827 |
| NK cell line | CD7 Positive Relapsed or Refractory Leukemia and Lymphoma | 2016 | I/II | Unknown | NCT02742727 | |
| Not disclosed | Hematological Malignancies | 2023 | I | Recruiting | NCT05995028 | |
| CD123 | Allogenic | Refractory/Relapsed Acute Myeloid Leukemia | 2022 | I | Recruiting | NCT05574608 |
| Not disclosed | Refractory/Relapsed Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm | 2023 | I/II | Recruiting | NCT06006403 | |
| CD5 ( + IL15) | CB-derived NK cells | Relapse/Refractory Hematological Malignances | 2021 | I/II | Recruiting | NCT05110742 |
| CAR Target | NK source | Target cancers | Date | Phase | Status | Number | |
| CD56 | Not disclosed | Relapsed/Refractory NK/T-cell lymphoma/NK cell leukemia | 2023 | II | Recruiting | NCT05941156 | |
| CD20 | iPSC-derived NK cells | Relapsed/Refractory Acute Myelogenous Leukemia and B-Cell Lymphoma | 2019 | I | Active | NCT04023071 | |
| CD22 | iPSC-derived NK cells | Relapsed and Refractory B-Cell Lymphoma | 2018 | I | Unknown | NCT03692767 | |
| CD33/CLL1 | Not disclosed | Acute Myeloid Leukemia | 2023 | I | Not yet recruiting | NCT05987696 | |
| CCL1 | iPSC-derived NK cells | Acute Myeloid Leukemia | 2023 | I | Recruiting | NCT06027853 | |
| CD38/SLAMF7 | iPSC-derived NK cells | Acute myeloid leukemia and multiple myeloma | 2020 | I | Active | NCT04614636 | |
| BCMA | Allogenic | Relapsed/Refractory Multiple Myeloma or Plasma Cell Leukemia | 2023 | I | Recruiting | NCT06045091 | |
| Allogenic | Relapsed/Refractory Multiple Myeloma | 2022 | I | Recruiting | NCT05652530 | ||
| iPSCs-derived NK cells | Multiple myeloma | 2021 | I | Recruiting | NCT05182073 | ||
| UC and CB-derived NK cells | Relapse/Refractory Multiple Myeloma | 2021 | I | Recruiting | NCT05008536 | ||
| NK cell line | Relapse/Refractory Multiple Myeloma | 2019 | I/II | Recruiting | NCT03940833 | ||
| iPSC-derived NK cells | Relapsed/Refractory B-Cell Lymphoma | 2018 | I | Unknown | NCT03559764 | ||
| NKG2D ligands | Not disclosed | Relapsed/Refractory Acute Myeloid Leukemia | 2023 | Recruiting | NCT05734898 | ||
| CB-derived NK cells | Relapsed/Refractory Acute Myeloid Leukemia | 2022 | Recruitment completed | NCT05247957 | |||
| PB NK cells | Myelodysplastic Syndromes and Acute Myeloid Leukemia | 2020 | I | Recruiting | NCT04623944 | ||
| Not disclosed | Not disclosed | B-Cell Malignancies | 2021 | I/II | Recruiting | NCT04747093 | |
| Not disclosed | Relapsed/Refractory Hematological Malignancies | 2021 | I | Recruiting | NCT04796688 | ||
| Other | NKG2D ligands – SARS-CoV-2 envelope glycoprotein | CB-derived NK cells | COVID-19 | 2020 | I/II | Recruiting | NCT04324996 |
immune responses by engineered NK cells, NK cell activating receptors can also be used in combination with CAR receptors. Notably, bispecific CAR-NK cells engineered with a PD1-DAP10 CAR and NKG2D exhibited strong cytotoxicity against gastric cancer cells both in vitro and in vivo [74]. Indeed, anti-PD1-DAP10 CAR harnessed NKG2D signaling through the DAP10 domains and was shown to potentiate NK cell activation against PD1 and NKG2D ligand-expressing tumor cells [74].
| CAR | TCR | |
| HLA restricted | No | Yes |
| Antigen density required | High | Low |
| Intracellular targeting | No | Yes |
| Multiplexing | Yes | Yes |
| Off the shelf | Yes | Yes |
| Clinical test | Yes | No |
Endowing NK cells with ectopic TCRs
[101]. Moreover, after injection in a multiple myeloma mouse model, a reduction in tumor growth was observed [101]. This finding offers interesting perspectives for clinical testing in upcoming years. However, NK-TCR therapies may be hampered by the poor presentation of tumor-associated antigens, which may not reach a sufficient level to mediate NK cell activation. Interestingly, TCR affinity and avidity are similar in primary T cells and in NK cell lines, suggesting that the characteristics of a given TCR could be transposed and used to optimize responses in NK cells [102]. CAR and TCR can also be combined to obtain a synergistic effect. For instance, NK cells have been engineered with a TCR specific to the E7 protein of HPV16 jointly with a CAR specific to TROP2 [103]. The CAR construct comprised two costimulatory domains (CD28 and 4-1BB) but lacked the CD3了 activating domain; thus, CAR activation was necessary but not sufficient for NK cell-mediated cytotoxicity. Indeed, NK cell activation is dependent on E7 epitope recognition, which is exclusively expressed in HPV16-infected tumor cells, and cytotoxicity is enhanced by the binding of CAR to the TROP2 target expressed mostly on tumor cells but also in some healthy tissues. Combined signaling leads to optimal and tumor-specific cytotoxicity while sparing healthy tissues [103]. This strategy lays the foundation for sophisticated logic gates and broader applications in the future. For instance, instead of using two distinct receptors (one CAR and one TCR), hybrid receptors may be constructed. Such receptors, either HLA-independent (HIT receptor [104] and STAR [105]) or HLA-dependent (TCAR [106]), have already been developed in T cells and may be transposed to NK cells. Notably, a chimeric TCR composed of the extracellular domains of the TCRa chain fused to the CD28 transmembrane domain followed by the 2B4 and DAP10 signaling domains and the TCR
ENHANCING THE ADCC POTENTIAL OF NK CELLS
proteinase) has been developed and successfully implemented, which had improved killing in preclinical models [112-114].
IMPROVING NK CELL CHEMOTROPISM
| Receptor | Target | NK sources | Modification techniques | Target disease | Outcomes | Ref |
| CXCR1 | IL-8 | PB | mRNA electroporation | Ovarian cancer | Greater chemiotaxis in vivo Increased tumor control | [123] |
| CXCR2 | CXCL5 | PB | Retroviral vector | Renal cell carcinoma | Greater chemiotaxis in vitro Increase target cell killing and adhesion in vitro | [120] |
| CXCR2 | CXCL1-3 and CXCL5-8 | NK92 | CRISPR-Cas9 | Human Colon Cancer | Greater chemiotaxis in vivo into tumor sites Stronger cell-killing and proliferation activity Tumor reduction Increased survival | [122] |
| CCR2B and CCR4 | CCL22 or CCL2 | NK-92 and PB | Lentiviral vector | None | Greater chemiotaxis in vitro | [130] |
| CXCR4 | CXCL12 and SDF-1
|
YTS | Lentiviral vector | Glioblastoma | Greater chemiotaxis in vitro and in vivo Tumor reduction/clearance Increased survival | [68] |
| CXCR4 | SDF-1
|
PB | Lentiviral vector | None | Greater chemiotaxis in vitro | [126] |
| CXCR4
|
SDF-1
|
PB | mRNA transfection | None | Greater chemotaxis in vitro Increased the bone marrow homing | [125] |
| CXCR4 and CCR7 | CXCL12 and CCL21 | NK92 | Lentiviral vector | Colorectal cancers | Tumor reduction Increased survival | [127] |
| CCR5 | CCL5 | PB | Lentiviral vector | Human Colon Cancer | Greater chemiotaxis in vitro and in vivo | [132] |
| CCR7 | CCL19 and CCL21 | PB | Trogocytosis | None | Greater chemotaxis in vitro Increased the lymph node homing | [129] |
| CCR7 | CCL19 and CCL21 | NK-92 | DNA transfection | B-cell lymphoma | Greater chemiotaxis in vitro and in vivo Increased tumor control Increased survival | [128] |
| CCR7 | CCL19 | PB | mRNA electroporation | None | Greater chemiotaxis in vitro | [109] |
IMPROVING NK CELL PERSISTENCE AND PROLIFERATION
results were obtained in a phase I/II clinical trial evaluating HLAmismatched anti-CD19 CAR-NK cells transduced with a retroviral vector encoding IL-15 in lymphoid tumors. Among the first 11 reported patients, 8 (
COUNTERACTING THE IMMUNOSUPPRESSIVE TUMOR MICROENVIRONMENT
DECREASING THE EXPRESSION OF ENDOGENOUS NK RECEPTORS
instance, in the treatment of hematologic malignancies, the CD38 and CD7 genes were deleted in engineered CAR-NK cells [40,114,154,155]. Interestingly, a 2 -in-1 strategy was developed by inserting the CAR receptor in the locus of the gene encoding its target, which allowed for the expression of the CAR while avoiding fratricide [156, 157]. As mentioned above in Section 3.1, inhibitory CAR receptors can also be implemented to avoid trogocytosismediated CAR-NK cell fratricides [83, 85].
CONCLUSIONS AND PERSPECTIVES
inducible SH2-containing (CISH) disruption was shown to potentiate the antitumor activity of CAR-NK cells [164, 165]. In addition to multiple engineering methods, AI can help develop combination treatments with conventional treatments or other immunotherapies by predicting patient responses. Notably, cell engagers have gained much interest in recent years and have been approved for clinical use. They can bridge ligands, such as tumor antigens, and activate receptors on the NK cell surface [166-172]. This kind of molecule could be used to promote NK cell activation by linking engineered receptors to tumor cells. Importantly, safety issues remain intrinsically related to engineered NK cell therapies, such as off-target activation. Ensuring the specificity of NK cell therapies is essential to avoid damaging healthy tissue, which can lead to serious side effects. To refine NK cell activation and selectively target cancer cells, tightly tuned logic-gated circuits should be developed. Moreover, computational models should be developed to predict and dynamically adjust cell responses. The frequent use of integrative vectors to modify NK cells also comes with the risk of insertional mutagenesis, which may lead to NK cell lymphomas. The current FDA recommendation is to have a vector copy number less than 5 [173]. Moreover, editing tools can also be used to generate chromosomal translocations. Recently, several manufacturing protocols have been developed to overcome some of the abovementioned limitations and, if approved, could become standard practices to minimize chromosome loss and translocations in manufactured products [174-176]. As a safeguard, to ensure engineered cell destruction in the event of adverse events, an inducible caspase safety switch should be implemented [38, 177]. Quality control standards must be defined to check the engineering efficacy, purity, phenotype and tumorigenicity of NK cells. Indeed, depending on the donor, the source (PB or UCB) and the culture conditions, NK cell subsets can vary [178]. For these latter points, new techniques to expand, modify and cryopreserve primary NK cells ex vivo would be extremely useful for scaling up production for clinical application, as high doses and multiple injections are required [33]. The future progress achieved in T-cell therapies, including the infrastructures developed for the production of cell and viral vectors, as well as the tools available to engineer cells (among which in vivo editing strategies), are expected to synergistically improve NK cell therapy. Early-stage clinical trials will also provide valuable information and promote optimization for future NK cell therapies, which are likely to reach clinical approval in the coming years. Overall, these pioneering advancements not only hold tremendous potential for cancer treatment but also extend the spectrum of treatments for other diseases, including infections.
REFERENCES
- Gill S, Olson JA, Negrin RS. Natural killer cells in allogeneic transplantation: effect on engraftment, graft- versus-tumor, and graft-versus-host responses. Biol Blood Marrow Transplant. 2009;15:765-76.
- Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47:820-33.
- Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue determinants of human NK cell development, function, and residence. Cell. 2020;180:749-763.e13.
- Buckle I, Johnson A, Rojas IL, Weinert V, Sester DP, Radford K, et al. High dimensional analysis reveals distinct NK cell subsets but conserved response to stimulation in umbilical cord blood and adult peripheral blood. Eur J Immunol. 2023;53:2250118.
- Wang W. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015;6. https://doi.org/10.3389/ fimmu.2015.00368.
- Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652-8.
- Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. 2016;7:86359-73.
- Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563-70.
- Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol. 2021;14:7.
- Bryceson YT, March ME, Ljunggren H-G, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107:159-66.
- Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex Vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother. 2015;38:24-36.
- Berrien-Elliott MM, Becker-Hapak M, Cashen AF, Jacobs M, Wong P, Foster M, et al. Systemic IL-15 promotes allogeneic cell rejection in patients treated with natural killer cell adoptive therapy. Blood. 2022;139:1177-83.
- Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123:3855-63.
- Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051-7.
- Heuser M, Tschan-Plessl A, Thol F, Schwarzer A, Kloos A, Kattre N, et al. Allogeneic, CD34 +, umbilical cordblood-derived NK cell adoptive immunotherapy for the treatment of acute myeloid leukemia patients with measurable residual disease. Blood. 2021;138:1745-1745.
- Choi I, Yoon SR, Park S-Y, Kim H, Jung S-J, Kang Y-L, et al. Donor-derived natural killer cell infusion after human leukocyte antigen-haploidentical hematopoietic cell transplantation in patients with refractory acute leukemia. Biol Blood Marrow Transplant. 2016;22:2065-76.
- Bachanova V, Maakaron JE, Cichocki F, McKenna DH, Cao Q, DeFor TE, et al. Gda201, a novel metabolically enhanced allogeneic natural killer (NK) cell product yields high remission rates in patients with relapsed/refractory non-Hodgkin lymphoma (NHL): 2-year survival and correlation with cytokine IL7. Blood. 2021;138:3854-3854.
- Nangia C, Soon-Shiong P, Rabizadeh S, Lee JH, Sender L, Jones F, et al. Complete responses in patients with second-line or greater metastatic triple negative breast cancer (TNBC) following first-in-human immunotherapy combining NK and T cell activation with off-the-shelf high-affinity CD16 NK cell line (haNK). Ann Oncol. 2019;30:v130.
- Fate Therapeutics. Fate therapeutics announces encouraging dose-escalation clinical data of FATE-NK100 and provides regulatory update on landmark IND appli- cation for FT500 [Press Release]. 2018. https://ir.fatetherapeutics.com/ press-releases?field_nir_news_date_value
. - Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19:185-99.
- Yoon DH, Koh Y, Park H, Hwang YK, Kim WS. A phase 1 study of the combination of MG4101, ex vivo -expanded allogeneic NK cells and rituximab for relapsed or refractory Non-Hodgkin Lymphoma. Blood. 2020;136:14-15.
- Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother. 2018;67:483-94.
- Liang S, Lin M, Niu L, Xu K, Wang X, Liang Y, et al. Cetuximab combined with natural killer cells therapy: an alternative to chemoradiotherapy for patients with advanced non-small cell lung cancer (NSCLC). Am J Cancer Res. 2018;8:879-91.
- Lin M, Luo H, Liang S, Chen J, Liu A, Niu L, et al. Pembrolizumab plus allogeneic NK cells in advanced non-small cell lung cancer patients. J Clin Invest. 2020;130:2560-9.
- Chung YY, Park SW, Im J-M, Yoo D-K, Cheon H-C, Kim J-E, et al. Abstract CT171: combinatorial allogeneic NK cell therapy with Pembrolizumab for cholangiocarcinoma; interim report of open label Phase1/2a study. Cancer Res. 2021;81:CT171-CT171.
- Adotevi O, Godet Y, Galaine J, Lakkis Z, Idirene I, Certoux JM, et al. In situ delivery of allogeneic natural killer cell (NK) combined with Cetuximab in liver metastases of gastrointestinal carcinoma: a phase I clinical trial. Oncolmmunology. 2018;7:e1424673.
- Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P, et al. A pilot trial of humanized anti-GD2 monoclonal antibody (hu14.18K322A) with chemotherapy and natural killer cells in children with recurrent/refractory neuroblastoma. Clin Cancer Res. 2017;23:6441-9.
- Talleur AC, Triplett BM, Federico S, Mamcarz E, Janssen W, Wu J, et al. Consolidation therapy for newly diagnosed pediatric patients with high-risk neuroblastoma using Busulfan/Melphalan, autologous hematopoietic cell transplantation, anti-GD2 antibody, granulocyte-macrophage
colony-stimulating factor, interleukin-2, and haploidentical natural killer cells. Biol Blood Marrow Transplant. 2017;23:1910-7. - Schmidt P, Raftery MJ, Pecher G. Engineering NK cells for CAR therapy-recent advances in gene transfer methodology. Front Immunol. 2021;11:611163.
- Gong Y, Klein Wolterink RGJ, Janssen I, Groot AJ, Bos GMJ, Germeraad WTV. Rosuvastatin enhances VSV-G lentiviral transduction of NK cells via upregulation of the low-density lipoprotein receptor. Mol Ther Methods Clin Dev. 2020;17:634-46.
- Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol. 2020;10:3123.
- Sengsayadeth S , Savani BN, Oluwole O, Dholaria B. Overview of approved CAR-T therapies, ongoing clinical trials, and its impact on clinical practice. eJHaem. 2022;3:6-10.
- Zhang
, Guo Y, Ji Y, Gao Y, Zhang M, Liu Y, et al. Cytokine release syndrome after modified CAR-NK therapy in an advanced non-small cell lung cancer patient: a case report. Cell Transpl. 2022;31:096368972210942. - Klingemann H. Are natural killer cells superior CAR drivers? Oncolmmunology. 2014;3:e28147.
- Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8:1083-9.
- Fate Therapeutics. Fate Therapeutics Showcases Positive Interim Phase 1 Data from FT596 Of-the-shelf, iPSC-derived CAR NK Cell Program for Relapsed/Refractory B-cell Lymphoma at 2021 ASH Annual Meeting [Press Release]. 2021. https://ir.fatetherapeutics.com/press-releases?field_nir_news_date_value% 5Bmin%5D=2021.
- Bachanova V, Cayci Z, Lewis D, Maakaron JE, Janakiram M, Bartz A, et al. Initial clinical activity of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood. 2020;136:8-8.
- Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545-53.
- Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19+B cell tumors: a phase
trial. Nat Med. 2024. https://doi.org/10.1038/s41591-023-02785-8. - Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8:297-310.
- Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, et al. cd 19- car engineered
cells are sufficient to overcome cell resistance in -cell malignancies. J Cell Mol Med. 2016;20:1287-94. - Oelsner S, Waldmann A, Billmeier A, Röder J, Lindner A, Ullrich E, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. 2019;145:1935-45.
- Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917-27.
- Mansour AG, Teng K-Y, Li Z, Zhu Z, Chen H, Tian L, et al. Off-the-shelf CAR-engineered natural killer cells targeting FLT3 enhance killing of acute myeloid leukemia. Blood Adv. 2023;7:6225-39.
- Chen Y, You F, Jiang L, Li J, Zhu X, Bao Y, et al. Gene-modified NK-92MI cells expressing a chimeric CD16-BB-
or CD64-BB- receptor exhibit enhanced cancer-killing ability in combination with therapeutic antibody. Oncotarget. 2017;8:37128-39. - Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, et al. Targeting CD20+ aggressive B-cell Non-Hodgkin lymphoma by anti-CD20 CAR mRNAmodified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3:333-44.
- Caruso S, De Angelis B, Del Bufalo F, Ciccone R, Donsante S, Volpe G, et al. Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J Hematol Oncol. 2022;15:163.
- Klaihmon P, Kang X, Issaragrisil S, Luanpitpong S. Generation and functional characterization of anti-CD19 chimeric antigen receptor-natural killer cells from human induced pluripotent stem cells. IJMS. 2023;24:10508.
- Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136:2308-18.
- Albinger N, Bexte T, Buchinger L, Wendel P, Al-Ajami A, Gessner A, et al. CRISPR/ Cas9 gene editing of immune checkpoint receptor NKG2A improves the efficacy of primary CD33-CAR-NK cells against AML. Blood. 2022;140:4558-9.
- Portillo AL, Hogg R, Poznanski SM, Rojas EA, Cashell NJ, Hammill JA, et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience. 2021;24:102619.
- Ao X, Yang Y, Li W, Tan Y, Guo W, Ao L, et al. Anti-aFR CAR-engineered NK-92 cells display potent cytotoxicity against aFR-positive ovarian cancer. J Immunother. 2019;42:284-96.
- Liu WN, So WY, Harden SL, Fong SY, Wong MXY, Tan WWS, et al. Successful targeting of PD-1/PD-L1 with chimeric antigen receptor-natural killer cells and nivolumab in a humanized mouse cancer model. Sci Adv. 2022;8:eadd1187.
- Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483.
- Zuo P, Li Y, He C, Wang T, Zheng X, Liu H, et al. Anti-tumor efficacy of anti-GD2 CAR NK-92 cells in diffuse intrinsic pontine gliomas. Front Immunol. 2023;14:1145706.
- Liu T, Dai X, Xu Y, Guan T, Hong L, Zaib T, et al. CD22 is a potential target of CARNK cell therapy for esophageal squamous cell carcinoma. J Transl Med. 2023;21:710.
- Chang Y-H, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777-86.
- Whalen KA, Rakhra K, Mehta NK, Steinle A, Michaelson JS, Baeuerle PA. Engaging natural killer cells for cancer therapy via NKG2D, CD16A and other receptors. mAbs. 2023;15:2208697.
- Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. 2019;27:1114-25.
- Cao B, Liu M, Huang J, Zhou J, Li J, Lian H, et al. Development of mesothelinspecific CAR NK-92 cells for the treatment of gastric cancer. Int J Biol Sci. 2021;17:3850-61.
- Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A novel chimeric PD1-NKG2D41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol Immunol. 2020;122:200-6.
- Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181-192.e5.
- Chambers AM, Lupo KB, Wang J, Cao J, Utturkar S, Lanman N, et al. Engineered natural killer cells impede the immunometabolic CD73-adenosine axis in solid tumors. eLife. 2022;11:e73699.
- Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. 2009;15:4857-66.
- Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptormodified natural killer cells against hepatocellular cancer cells in vitro. CMAR. 2020;12:3247-55.
- Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immuneinhibitory HLA-G. Oncolmmunology. 2017;6:e1250050.
- Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194:3201-12.
- Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvlll-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1a-secreting glioblastoma. J Immunother. 2015;38:197-210.
- Robbins Y, Greene S, Friedman J, Clavijo PE, Van Waes C, Fabian KP, et al. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. eLife. 2020;9:e54854.
- Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12:49.
- Zhuang
, Long EO. NK cells equipped with a chimeric antigen receptor that overcomes inhibition by HLA class I for adoptive transfer of CAR-NK cells. Front Immunol. 2022;13:840844. - Chang Y, Jin G, Luo W, Luo Q, Jung J, Hummel SN, et al. Engineered human pluripotent stem cell-derived natural killer cells with PD-L1 responsive immunological memory for enhanced immunotherapeutic efficacy. Bioact Mater. 2023;27:168-80.
- Goulding J, Yeh W-I, Hancock B, Blum R, Xu T, Yang B-H, et al. A chimeric antigen receptor uniquely recognizing MICA/B stress proteins provides an effective approach to target solid tumors. Med. 2023;4:457-477.e8.
- Li M, Zhi L, Yin M, Guo C, Zhang H, Lu C, et al. A novel bispecific chimeric PD1DAP10/NKG2D receptor augments NK92-cell therapy efficacy for human gastric cancer SGC-7901 cell. Biochem Biophys Res Commun. 2020;523:745-52.
- Raftery MJ, Franzén AS, Radecke C, Boulifa A, Schönrich G, Stintzing S, et al. Next generation CD44v6-specific CAR-NK cells effective against triple negative breast cancer. IJMS. 2023;24:9038.
- Aldea M, Andre F, Marabelle A, Dogan S, Barlesi F, Soria J-C. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discov. 2021;11:874-99.
- Song M-K, Park B-B, Uhm J-E. Resistance mechanisms to CAR T-cell therapy and overcoming strategy in B-cell hematologic malignancies. JJMS. 2019;20:5010.
- Zhou X, Rasche L, Kortüm KM, Mersi J, Einsele H. BCMA loss in the epoch of novel immunotherapy for multiple myeloma: from biology to clinical practice. Haematologica. 2022;108:958-68.
- Roex G, Campillo-Davo D, Flumens D, Shaw PAG, Krekelbergh L, De Reu H, et al. Two for one: targeting BCMA and CD19 in B-cell malignancies with off-the-shelf dual-CAR NK-92 cells. J Transl Med. 2022;20:124.
- Cronk RJ, Zurko J, Shah NN. Bispecific chimeric antigen receptor T cell therapy for B cell malignancies and multiple myeloma. Cancers. 2020;12:2523.
- Lim RM, Rong L, Zhen A, Xie J. A universal CAR-NK cell targeting various epitopes of HIV-1 gp160. ACS Chem Biol. 2020;15:2299-310.
- Garrison BS, Deng H, Yucel G, Frankel NW, Guzman-Ayala M, Gordley R, et al. FLT3 OR CD33 NOT EMCN logic gated CAR-NK cell therapy (SENTI-202) for precise targeting of AML. Blood. 2021;138:2799-2799.
- Li Y, Basar R, Wang G, Liu E, Moyes JS, Li L, et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat Med. 2022;28:2133-44.
- Hamieh M, Dobrin A, Cabriolu A, Van Der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568:112-6.
- Ramezani F, Panahi Meymandi AR, Akbari B, Tamtaji OR, Mirzaei H, Brown CE, et al. Outsmarting trogocytosis to boost CAR NK/T cell therapy. Mol Cancer. 2023;22:183.
- Luo H, Wu X, Sun R, Su J, Wang Y, Dong Y, et al. Target-dependent expression of IL12 by synNotch receptor-engineered NK92 cells increases the antitumor activities of CAR-T cells. Front Oncol. 2019;9:1448.
- Tseng H, Xiong W, Badeti S, Yang Y, Ma M, Liu T, et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat Commun. 2020;11:4810.
- Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173:1426-1438.e11.
- Li H-S, Wong NM, Tague E, Ngo JT, Khalil AS, Wong WW. High-performance multiplex drug-gated CAR circuits. Cancer Cell. 2022;40:1294-1305.e4.
- Li H-S, Israni DV, Gagnon KA, Gan KA, Raymond MH, Sander JD, et al. Multidimensional control of therapeutic human cell function with synthetic gene circuits. Science. 2022;378:1227-34.
- Zhang R, Liu Q, Zhou S, He H, Zhao M, Ma W. Engineering CAR-NK cells targeting CD33 with concomitant extracellular secretion of anti-CD16 antibody revealed superior antitumor effects toward myeloid leukemia. Cancer Lett. 2023;558:216103.
- Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination therapy with epCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J Immunol Res. 2018;2018:1-11.
- Wang F, Wu L, Yin L, Shi H, Gu Y, Xing N. Combined treatment with anti-PSMA CAR NK-92 cell and anti-PD-L1 monoclonal antibody enhances the antitumour efficacy against castration-resistant prostate cancer. Clin Transl Med. 2022;12. https://doi.org/10.1002/ctm2.901.
- Strecker MI, Wlotzka K, Strassheimer F, Roller B, Ludmirski G, König S, et al. AAVmediated gene transfer of a checkpoint inhibitor in combination with HER2targeted CAR-NK cells as experimental therapy for glioblastoma. Oncolmmunology. 2022;11:2127508.
- Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7:27764-77.
- Dong H, Ham JD, Hu G, Xie G, Vergara J, Liang Y, et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc Natl Acad Sci USA. 2022;119:e2122379119.
- Baulu E, Gardet C, Chuvin N, Depil S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci Adv. 2023;9:eadf3700.
- Rasul KH, Hussain A, Reilly H, Karvouni M, Dahlberg CIM, Al-Attar MS, et al. Assessment of T cell receptor complex expression kinetics in natural killer cells. CIMB. 2022;44:3859-71.
- Mensali N, Dillard P, Hebeisen M, Lorenz S, Theodossiou T, Myhre MR, et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine. 2019;40:106-17.
- Parlar A, Sayitoglu EC, Ozkazanc D, Georgoudaki A, Pamukcu C, Aras M, et al. Engineering antigen-specific NK cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur J Immunol. 2019;49:1278-90.
- Morton LT, Wachsmann TLA, Meeuwsen MH, Wouters AK, Remst DFG, van Loenen MM, et al. T cell receptor engineering of primary NK cells to therapeutically target tumors and tumor immune evasion. J Immunother Cancer. 2022;10:e003715.
- Kobayashi E, Ozawa T, Hamana H, Muraguchi A, Kishi H. Gene modified NK cell line as a powerful tool for evaluation of cloned TCRs for TCR-T cell therapy. Cell Immunol. 2023;383:104656.
- Poorebrahim M, Quiros-Fernandez I, Marmé F, Burdach SEG, Cid-Arregui A. A costimulatory chimeric antigen receptor targeting TROP2 enhances the cytotoxicity of NK cells expressing a T cell receptor reactive to human papillomavirus type 16 E7. Cancer Lett. 2023;566:216242.
- Mansilla-Soto J, Eyquem J, Haubner S, Hamieh M, Feucht J, Paillon N, et al. HLAindependent T cell receptors for targeting tumors with low antigen density. Nat Med. 2022;28:345-52.
- Liu Y, Liu G, Wang J, Zheng Z, Jia L, Rui W, et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci Transl Med. 2021;13:eabb5191.
- Birtel M, Voss R-H, Reinhard K, Rengstl B, Ouchan Y, Michel K, et al. A TCR-like CAR promotes sensitive antigen recognition and controlled T-cell expansion upon mRNA vaccination. Cancer Res Commun. 2022;2:827-41.
- Li S, Zhang C, Shen L, Teng X, Xiao Y, Yu B, et al. TCR extracellular domain genetically linked to CD28, 2B4/41BB and DAP10/CD3了, -engineered NK cells mediates antitumor effects. Cancer Immunol Immunother. 2023;72:769-74.
- Dixon KJ, Wu J, Walcheck B. Engineering anti-tumor monoclonal antibodies and Fc receptors to enhance ADCC by human NK cells. Cancers. 2021;13:312.
- Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M et al. Efficient mRNAbased genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front Immunol. 2016;7. https://doi.org/10.3389/fimmu.2016.00105.
- Clara JA, Levy ER, Reger R, Barisic S, Chen L, Cherkasova E, et al. High-affinity CD16 integration into a CRISPR/Cas9-edited CD38 locus augments CD38directed antitumor activity of primary human natural killer cells. J Immunother Cancer. 2022;10:e003804.
- Giles AJ, Hao S, Padget M, Song H, Zhang W, Lynes J, et al. Efficient ADCC killing of meningioma by avelumab and a high-affinity natural killer cell line, haNK. JCl Insight. 2019;4:e130688.
- Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. 2020;135:399-410.
- Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, et al. Identification of an ADAM17 Cleavage Region in Human CD16 (FcyRIII) and the Engineering of a Non-Cleavable Version of the Receptor in NK Cells. PLoS One. 2015;10:e0121788.
- Cichocki F, Bjordahl R, Goodridge JP, Mahmood S, Gaidarova S, Abujarour R, et al. Quadruple gene-engineered natural killer cells enable multi-antigen targeting for durable antitumor activity against multiple myeloma. Nat Commun. 2022;13:7341.
- Eitler J, Wotschel N, Miller N, Boissel L, Klingemann HG, Wels W, et al. Inability of granule polarization by NK cells defines tumor resistance and can be overcome by CAR or ADCC mediated targeting. J Immunother Cancer. 2021;9:e001334.
- Meng F, Zhang S, Xie J, Zhou Y, Wu Q, Lu B, et al. Leveraging CD16 fusion receptors to remodel the immune response for enhancing anti-tumor immunotherapy in iPSC-derived NK cells. J Hematol Oncol. 2023;16:62.
- Trick F, Benenson Y ADCC-and BCR-inspired receptors for antigen-specific NK cell activaiton. 2022. https://www.esgctcongress.com/poster-list.
- Tong L, Jiménez-Cortegana C, Tay AHM, Wickström S, Galluzzi L, Lundqvist A. NK cells and solid tumors: therapeutic potential and persisting obstacles. Mol Cancer. 2022;21:206.
- Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother. 2015;64:225-35.
- Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother cancer. 2017;5:73.
- Yang Y, Gordon N, Kleinerman ES, Huang G. Promoting NK cell trafficking to improve therapeutic effect of NK cell therapy on osteosarcoma. J Immunother Cancer. 2015;3:P24.
- Gao L, Yang L, Zhang S, Ge Z, Su M, Shi Y, et al. Engineering NK-92 cell by upregulating CXCR2 and IL-2 Via CRISPR-Cas9 improves its antitumor effects as cellular immunotherapy for human colon cancer. J Interferon Cytokine Res. 2021;41:450-60.
- Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75-85.
- Levy ER, Clara JA, Reger RN, Allan DSJ, Childs RW. RNA-seq analysis reveals CCR5 as a key target for CRISPR gene editing to regulate in vivo NK cell trafficking. Cancers. 2021;13:872.
- Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, et al. Enhanced bone marrow homing of natural killer cells following mRNA transfection with gain-of-function variant CXCR4R334X. Front Immunol. 2019;10:1262.
- Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, et al. Highly efficient generation of transgenically augmented CAR NK cells overexpressing CXCR4. Front Immunol. 2020;11:2028.
- Yang L, Huang C, Wang C, Zhang S, Li Z, Zhu Y, et al. Overexpressed CXCR4 and CCR7 on the surface of NK92 cell have improved migration and anti-tumor activity in human colon tumor model. Anti Cancer Drugs. 2020;31:333-44.
- Schomer NT, Jiang ZK, Lloyd MI, Klingemann H, Boissel L. CCR7 expression in CD19 chimeric antigen receptor-engineered natural killer cells improves migration toward CCL19-expressing lymphoma cells and increases tumor control in mice with human lymphoma. Cytotherapy. 2022;24:827-34.
- Somanchi SS, Somanchi A, Cooper UN, Lee DA. Engineering lymph node homing of ex vivo-expanded human natural killer cells via trogocytosis of the chemokine receptor CCR7. Blood. 2012;119:5164-72.
- Feigl FF, Stahringer A, Peindl M, Dandekar G, Koehl U, Fricke S, et al. Efficient redirection of NK cells by genetic modification with chemokine receptors CCR4 and CCR2B. IJMS. 2023;24:3129.
- Evgin L, Kottke T, Tonne J, Thompson J, Huff AL, Van Vloten J, et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci Transl Med. 2022;14:eabn2231.
- Li F, Sheng Y, Hou W, Sampath P, Byrd D, Thorne S, et al. CCL5-armed oncolytic virus augments CCR5-engineered NK cell infiltration and antitumor efficiency. J Immunother Cancer. 2020;8:e000131.
- Tao X, Zhang R, Du R, Yu T, Yang H, Li J, et al. EP3 enhances adhesion and cytotoxicity of NK cells toward hepatic stellate cells in a murine liver fibrosis model. J Exp Med. 2022;219:e20212414.
- Choi YH, Lim EJ, Kim SW, Moon YW, Park KS, An H-J. IL-27 enhances IL-15/IL-18mediated activation of human natural killer cells. J Immunother Cancer. 2019;7:168.
- Liu M, Meng Y, Zhang L, Han Z, Feng X. High-efficient generation of natural killer cells from peripheral blood with preferable cell vitality and enhanced cytotoxicity by combination of IL-2, IL-15 and IL-18. Biochem Biophys Res Commun. 2021;534:149-56.
- Lu T, Ma R, Dong W, Teng K-Y, Kollath DS, Li Z, et al. Off-the-shelf CAR natural killer cells secreting IL-15 target spike in treating COVID-19. Nat Commun. 2022;13:2576.
- Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61:1451-61.
- Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520-31.
- Bachier C, Borthakur G, Hosing C, Blum W, Rotta M, Ojeras P, et al. A phase 1 study of NKX101, an allogeneic CAR natural killer (NK) cell therapy, in subjects with relapsed/refractory (R/R) acute myeloid leukemia (AML) or higher-risk myelodysplastic syndrome (MDS). Blood. 2020;136:42-43.
- Silvestre RN, Eitler J, De Azevedo JTC, Tirapelle MC, Fantacini DMC, De Souza LEB, et al. Engineering NK-CAR. 19 cells with the IL-15/IL-15Ra complex improved proliferation and anti-tumor effect in vivo. Front Immunol. 2023;14:1226518.
- Bjordahl R, Gaidarova S, Goodridge JP, Mahmood S, Bonello G, Robinson M, et al. FT576: a novel multiplexed engineered off-the-shelf natural killer cell immunotherapy for the dual-targeting of CD38 and Bcma for the treatment of multiple myeloma. Blood. 2019;134:3214-3214.
- Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SMH, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. 2014;124:1081-8.
- Zhang D, Zhao Y, Kang Y, Hu J, Li R, Song J, et al. Enhanced NK cell adoptive antitumor effects against breast cancer in vitro via blockade of the transforming growth factor-
signaling pathway. Onco Targets Ther. 2015;2015:1553-9. - Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-
receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy. 2017;19:408-18. - Chaudhry K, Geiger A, Dowlati E, Lang H, Sohai DK, Hwang El, et al. Cotransducing B7H3 CAR-NK cells with the DNR preserves their cytolytic function against GBM in the presence of exogenous TGF-
. Mol Ther Methods Clin Dev. 2022;27:415-30. - Yang B, Liu H, Shi W, Wang Z, Sun S, Zhang G, et al. Blocking transforming growth factor-
signaling pathway augments antitumor effect of adoptive NK92 cell therapy. Int Immunopharmacol. 2013;17:198-204. - Burga RA, Yvon E, Chorvinsky E, Fernandes R, Cruz CRY, Bollard CM. Engineering the TGF
receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clin Cancer Res. 2019;25:4400-12. - Wang Z, Guo L, Song Y, Zhang Y, Lin D, Hu B, et al. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-
R II and NKG2D. Cancer Immunol Immunother. 2017;66:537-48. - Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther. 2014;22:1211-20.
- Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25:249-58.
- Lee MY, Robbins Y, Sievers C, Friedman J, Abdul Sater H, Clavijo PE, et al. Chimeric antigen receptor engineered NK cellular immunotherapy overcomes the selection of T-cell escape variant cancer cells. J Immunother Cancer. 2021;9:e002128.
- Mensali N, Dillard P, Fayzullin A, Köksal H, Gaudernack G, Kvalheim G, et al. Builtin” PD-1 blocker to rescue NK-92 activity from PD-L1-mediated tumor escape mechanisms. FASEB J. 2021;35:e21750.
- Susek KH, Schwietzer YA, Karvouni M, Gilljam M, Keszei M, Hussain A, et al. Generation of NK cells with chimeric-switch receptors to overcome PD1mediated inhibition in cancer immunotherapy. Cancer Immunol Immunother. 2023;72:1153-67.
- Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2020;107:437-45.
- Naeimi Kararoudi M, Nagai Y, Elmas E, de Souza Fernandes Pereira M, Ali SA, Imus PH, et al. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood. 2020;136:2416-27.
- Jiang J, Chen J, Liao C, Duan Y, Wang Y, Shang K, et al. Inserting EF1a-driven CD7-specific CAR at CD7 locus reduces fratricide and enhances tumor rejection. Leukemia. 2023;37:1660-70.
- Liao C, Wang Y, Huang Y, Duan Y, Liang Y, Chen J, et al. CD38-specific CAR integrated into CD38 locus driven by different promoters causes distinct antitumor activities of T and NK cells. Adv Sci. 2023;10:2207394.
- Hoerster K, Uhrberg M, Wiek C, Horn PA, Hanenberg H, Heinrichs S. HLA class I knockout converts allogeneic primary NK cells into suitable effectors for “off-the-shelf” immunotherapy. Front Immunol. 2021;11:586168.
- Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Investig. 2019;129:2094-106.
- Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8. https://doi.org/10.1126/ scitranslmed.aaf2341.
- He B, Mai Q, Pang Y, Deng S, He Y, Xue R, et al. Cytokines induced memory-like NK cells engineered to express CD19 CAR exhibit enhanced responses against
cell malignancies. Front Immunol. 2023;14:1130442. - Cho H, Kim KH, Lee H, Kim CG, Chung H, Choi YS, et al. Adaptive natural killer cells facilitate effector functions of daratumumab in multiple myeloma. Clin Cancer Res. 2021;27:2947-58.
- Woan KV, Kim H, Bjordahl R, Davis ZB, Gaidarova S, Goulding J, et al. Harnessing features of adaptive NK cells to generate iPSC-derived NK cells for enhanced immunotherapy. Cell Stem Cell. 2021;28:2062-2075.e5.
- Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CARNK cells. Blood. 2021;137:624-36.
- Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27:224-237.e6.
- Pekar L, Klausz K, Busch M, Valldorf B, Kolmar H, Wesch D, et al. Affinity maturation of B7-H6 translates into enhanced NK cell-mediated tumor cell lysis and improved proinflammatory cytokine release of bispecific immunoligands via NKp30 engagement. J Immunol. 2021;206:225-36.
- Klausz K, Pekar L, Boje AS, Gehlert CL, Krohn S, Gupta T, et al. Multifunctional NK cell-engaging antibodies targeting EGFR and NKp30 elicit efficient tumor cell killing and proinflammatory cytokine release. J Immunol. 2022;209:1724-35.
- Raynaud A, Desrumeaux K, Vidard L, Termine E, Baty D, Chames P, et al. AntiNKG2D single domain-based antibodies for the modulation of anti-tumor immune response. Oncolmmunology. 2021;10:1854529.
- Nikkhoi SK, Li G, Eleya S, Yang G, Vandavasi VG, Hatefi A. Bispecific killer cell engager with high affinity and specificity toward CD16a on NK cells for cancer immunotherapy. Front Immunol. 2023;13:1039969.
- Lipinski B, Arras P, Pekar L, Klewinghaus D, Boje AS, Krah S et al. NKp46 -specific single domain antibodies enable facile engineering of various potent NK cell engager formats. Protein Sci. 2023;32. https://doi.org/10.1002/pro.4593.
- Colomar-Carando N, Gauthier L, Merli P, Loiacono F, Canevali P, Falco M, et al. Exploiting natural killer cell engagers to control pediatric B-cell precursor acute lymphoblastic leukemia. Cancer Immunol Res. 2022;10:291-302.
- von Strandmann EP, Hansen HP, Reiners KS, Schnell R, Borchmann P, Merkert S, et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood. 2006;107:1955-62.
- Zhao Y, Stepto H, Schneider CK. Development of the first World Health Organization lentiviral vector standard: toward the production control and standardization of lentivirus-based gene therapy products. Hum Gene Ther Methods. 2017;28:205-14.
- Glaser V, Flugel C, Kath J, Du W, Drosdek V, Franke C, et al. Combining different CRISPR nucleases for simultaneous knock-in and base editing prevents translocations in multiplex-edited CAR T cells. Genome Biol. 2023;24:89.
- Tsuchida CA, Brandes N, Bueno R, Trinidad M, Mazumder T, Yu B et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell Biol. 2023 https://doi.org/10.1101/2023.03.22.533709.
- Wellhausen N, Agarwal S, Rommel PC, Gill SI, June CH. Better living through chemistry: CRISPR/Cas engineered T cells for cancer immunotherapy. Curr Opin Immunol. 2022;74:76-84.
- Wang X, Jasinski DL, Medina JL, Spencer DM, Foster AE, Bayle JH. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020;4:1950-64.
- Allan DSJ, Wu C, Mortlock RD, Chakraborty M, Rezvani K, Davidson-Moncada JK, et al. Expanded NK cells used for adoptive cell therapy maintain diverse clonality and contain long-lived memory-like NK cell populations. Mol Ther Oncolytics. 2023;28:74-87.
AUTHOR CONTRIBUTIONS
FUNDING
COMPETING INTERESTS
ADDITIONAL INFORMATION
© The Author(s) 2024
- ¹Centre de Recherche en Cancérologie de Lyon, UMR INSERM U1052 CNRS 5286, Centre Léon Bérard, Lyon, France.
ErVimmune, Lyon, France. Centre Léon Bérard, Lyon, France. Université Claude Bernard Lyon 1, Lyon, France. email: audrey.page@lyon.unicancer.fr; stephane.depil@lyon.unicancer.fr