تعديل تساهمية روابط Ru-O من خلال إعادة البناء الديناميكي لتحقيق كفاءة عالية في تطور الأكسجين الحمضي Modulating the covalency of Ru-O bonds by dynamic reconstruction for efficient acidic oxygen evolution
تطوير محفزات أكسيد الروديوم القادرة على كبح مشاركة الأكسجين الشبكي في عملية التفاعل الحفزي أمر حاسم للحفاظ على استقرار تفاعل تطور الأكسجين (OER) في الظروف الحمضية. هنا، نقوم ببناء مربوط بجزيئات النانومحفز كهربائي نانويتحقيق تحسين ديناميكي لـخلال عملية التفاعل وتحسين الاستقرار التحفيزي. الاستفادة من الخصائص الفريدة لإزالة الليثيوم الكهروكيميائية لـالدعم، يتم تنظيم التساهمية لرابطة Ru-O بشكل فعال خلال عملية OER. الرابطة التساهمية الضعيفة بين Ru-O تعيق مشاركة الأكسجين الشبكي في التفاعل الحفاز وتضمن التشغيل المستمر لمواقع Ru النشطة. علاوة على ذلك، فإن رابطة Ru-O الممتدة في التصميم المحسنالمحفز يقلل من حاجز طاقة التكوين لل intermediates *OOH، مما يسرع من تقدم تفاعل OER. ونتيجة لذلك،المحفز يتطلب فقط جهدًا زائدًا قدرهفيفيويعمل بشكل مستقر لمدة 2000 ساعة عندفي التحليل الكهربائي للماء باستخدام غشاء تبادل البروتون. يفتح هذا العمل آفاقًا جديدة لتصميم محفزات قائمة على الروثينيوم بكفاءة.
يعتبر التحليل الكهربائي للماء باستخدام غشاء تبادل البروتون (PEMWE) بكثافة تيار عالية وفقدان مقاومة منخفض تقنية واعدة لإنتاج الهيدروجين في المستقبل.حالياً، تُستخدم محفزات أكسيد المعادن النبيلة الروثينيوم (Ru) والإيريديوم (Ir) بشكل واسع في الأنودات لخلية التحليل الكهربائي للماء باستخدام غشاء البوليمر (PEMWE).. مقارنة بـيقدم نشاطًا عاليًا وفعالية من حيث التكلفة، مما يظهر إمكانيات كبيرة للتطبيق في تفاعلات تطور الأكسجين الحمضي (OER). ومع ذلك، تميل المحفزات إلى اتباع آلية الأكسجين الشبكي (LOM) وتوليد العديد من عيوب الأكسجين خلال تفاعل OER، مما يؤدي إلى انهيار الهيكل البلوري.. بالإضافة إلى ذلك، يمكن أن تتأكسد مواقع المعدن Ru بشكل مفرط إلى مواد قابلة للذوبانأنواع، التي تفصل من الشبكة البلورية تحت جهد أكسدة مرتفع، مما يؤدي إلى ضعف الاستقرار التحفيزي. من منظور بنية البلورات، عدم الاستقرار لـمرتبط ارتباطًا وثيقًا بتوزيع الشحنة لـرابطةلذلك، فإن تنظيم الحالة الإلكترونية لرابطة Ru-O لقمع مشاركة الأكسجين الشبكي في عملية OER هو استراتيجية فعالة لتعزيز النشاط والاستقرار.محفزات.
بشكل عام، مواقع الروثينيوم الغنية بالإلكترونات فيتفعيل الأكسجين الشبكي وتوليد العيوب، بينما تميل الحالات الناقصة من الإلكترونات إلى الأكسدة إلى حالات تكافؤ مرتفعة بشكل مفرط وتذوباستراتيجيات الدعم التقليدية المانحة للإلكترونات تعدل توزيع الإلكترونات لـ
رابطة رو-أو، تمنع ذوبان مركز المعدن وتتبعه آلية امتصاص أكسجين مستقرة نسبيًا (AEM)في الوقت الحالي، لا يمكن للدعائم المانحة للإلكترونات ذات القدرة التنظيمية المحدودة تلبية الطلب على التلاعب بروابط Ru-O المتغيرة ديناميكيًا تحت ظروف OER المعقدة ذات الجهد العالي.. لذلك، فإن تصميم الدعامات القادرة على تنظيم الهيكل الإلكتروني بشكل ديناميكي استجابةً للتغيرات في عملية التفاعل الحفزي، فإنها جذابة للغاية. لمواجهة هذا التحدي، تعمل أكاسيد المعادن الانتقالية مع إدخال الكاتيونات على تحسين البيئة الميكروية المحلية لروابط المعادن والأكسجين بشكل ديناميكي خلال التفاعل الحفزي من خلال استخراج وإدخال الكاتيونات. .من الجدير بالذكر أن أكسيد الكوبالت الليثيوم ) مع هيكل طبقي فريد يظهر ترتيب كاتيون منظم وثبات حراري متفوق الحافة الضيقة المشتركةهيكل ثماني السطوح فييقلل من حاجز طاقة الهجرة لـويضمن الانتشار ثنائي الأبعاد لـفي الطائرة، مما يسهل إعادة البناء الديناميكي أثناء عملية التحفيز. ومع ذلك، فإن الهيكل الضخم متعدد الطبقات لـتظهر قدرة بطيئة على نقل الإلكترونات ومساحة سطح محدودة، مما لا يمكنها من تلبية متطلبات دعم المحفز بكفاءة.. في هذا الصدد، ثنائي الأبعادتقلل الأوراق النانوية بشكل ملحوظ من مسار نقل الإلكترونات بين البلورات وتعزز الموصلية. بالإضافة إلى ذلك، فإن هيكل النانو شيت ثنائي الأبعاد يشكل قناة سريعة لنقل الإلكترونات عمودية على مستوى التعرض، مما يؤدي إلى تفاعل قوي بين المعدن والدعم.. لذلك، استخدام ثنائي الأبعادالأغشية النانوية كدعم لـمن المتوقع أن يحقق تنظيمًا ديناميكيًا للهيكل الإلكتروني خلال التفاعلات الحفزية ويظهر نشاطًا حفزيًا عاليًا.
هنا، نقترح استراتيجية فعالة لتحسين توزيع الشحنة لـمن خلال عملية التطور الديناميكية الفريدة لـ دعم ( ” ). الـدعم النانوورقة مع القدرة على تبرع الإلكترونات يحفز نقل الإلكترونات من مواقع الكوبالت إلى الروثينيوم، مما يوفر تعويضًا إلكترونيًا لاستقرار حالة التكافؤ لـالأهم من ذلك، الانتشار ثنائي الأبعاد واستخراج أيونات الليثيوم داخل الطبقة البينية لـتحت جهد الأكسدة الأكسجينية، تتسبب النانوورقة في إعادة البناء الديناميكية وتطور الـواجهة المحفز. عملية التحسين الذاتي الديناميكية الفريدة تضعف بشكل معتدل من تساهمية رابطة Ru-O، مما يثبط مشاركة الأكسجين الشبكي ويحقق توازنًا جيدًا بين النشاط التحفيزي والاستقرار. تسهل مواقع Ru المحسّنة تشكيل وسيط *OOH، مما يقلل بشكل كبير من حاجز الطاقة التحفيزي للخطوة المحددة لمعدل التفاعل. وبالتالي،المحفز الكهربائي يوفر كثافة تيار قدرهاعند جهد زائد قدرهويحافظ على الاستقرار لأكثر من 2300 ساعة في. بالإضافة إلى ذلك،يمكن أن تعمل الأنود بشكل مستمر لمدة 2000 ساعة عندفي جهاز التحليل الكهربائي PEM. يعزز هذا العمل تطبيق المحفزات القائمة على الروثينيوم في PEMWE.
النتائج
آلية التفاعل والتحليل الهيكلي
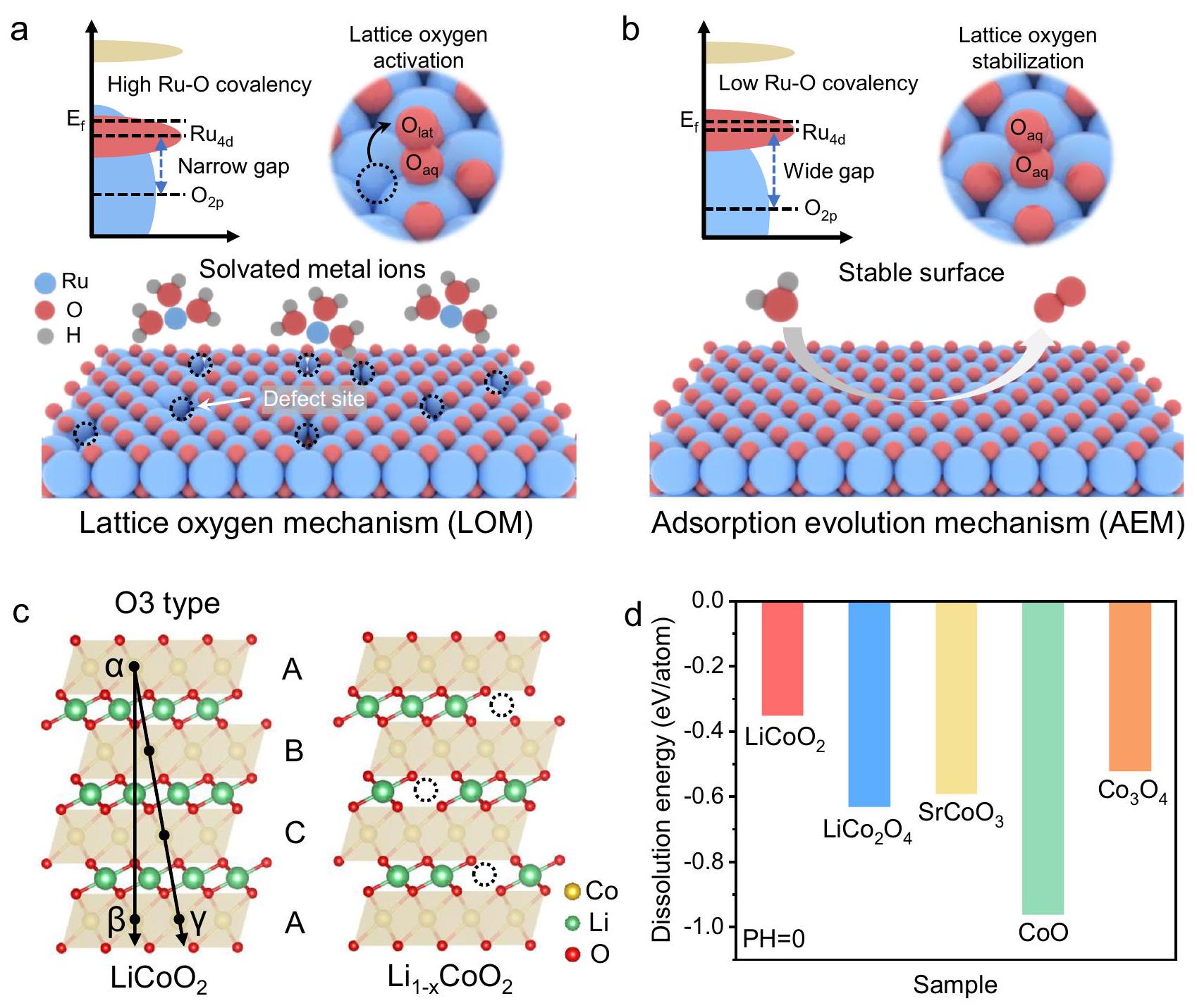
نظرًا للاختلافات في التساهمية لروابط Ru-O، تتبع أكاسيد الروثينيوم إما آلية الأكسجين الشبكي (LOM) أو آلية الأكسجين الممتص (AEM) خلال عملية OER الحمضية (الشكل 1a، b).في مسار LOM، يشارك الأكسجين الشبكي المنشط ذو التساهمية الأقوى في عملية OER، مما يؤدي إلى توليد عيوب الأكسجين.يمكن أن يؤدي العدد المفرط من فراغات الأكسجين إلى انفصال عن الشبكة البلورية في مواقع معدن الروثينيوم، مما يقلل بشكل كبير من الاستقرار التحفيزي. ومع ذلك، فإن أكاسيد الروثينيوم ذات الروابط التساهمية الضعيفة Ru-O تتبع آلية AEM، مما يحقق عملية OER حمضية مستقرة.. لذلك، فإن التنظيم الديناميكي لروابط Ru-O خلال التفاعل الحفاز أمر حاسم لتعزيز استقرار المحفز. نوع O3المادة التي تحتوي على إدخال عنصر الليثيوم تظهر موصلية متفوقة واستقرار هيكلي. والأهم من ذلك، استخراج أيونات الليثيوم بين الطبقات عند جهد تفاعل الأكسدة. يحفز نقل الإلكترون، مما يمكّن من التنظيم في الوقت الحقيقي لمدى التساهمية لروابط المعدن-الأكسجين (الشكل 1c). بالإضافة إلى ذلك، نستخدم حسابات نظرية الكثافة الوظيفية (DFT) لدراسة استقرار أكاسيد الكوبالت المختلفة في البيئات الحمضية.يقدم حاجز انحلال غير مواتٍ حراريًا، مما يشير إلى أنه لا يزال بإمكانه الحفاظ على الاستقرار في بيئة حمضية (الشكل 1d). مستلهمًا من هذه النتائج، يتم تنظيم تفاعل تساهمي لرابطة Ru-O بشكل ديناميكي فيباستخدام الطبقاتمن المتوقع أن يحقق الدعم استقرارًا تحفيزيًا عاليًا.
توصيف المواد
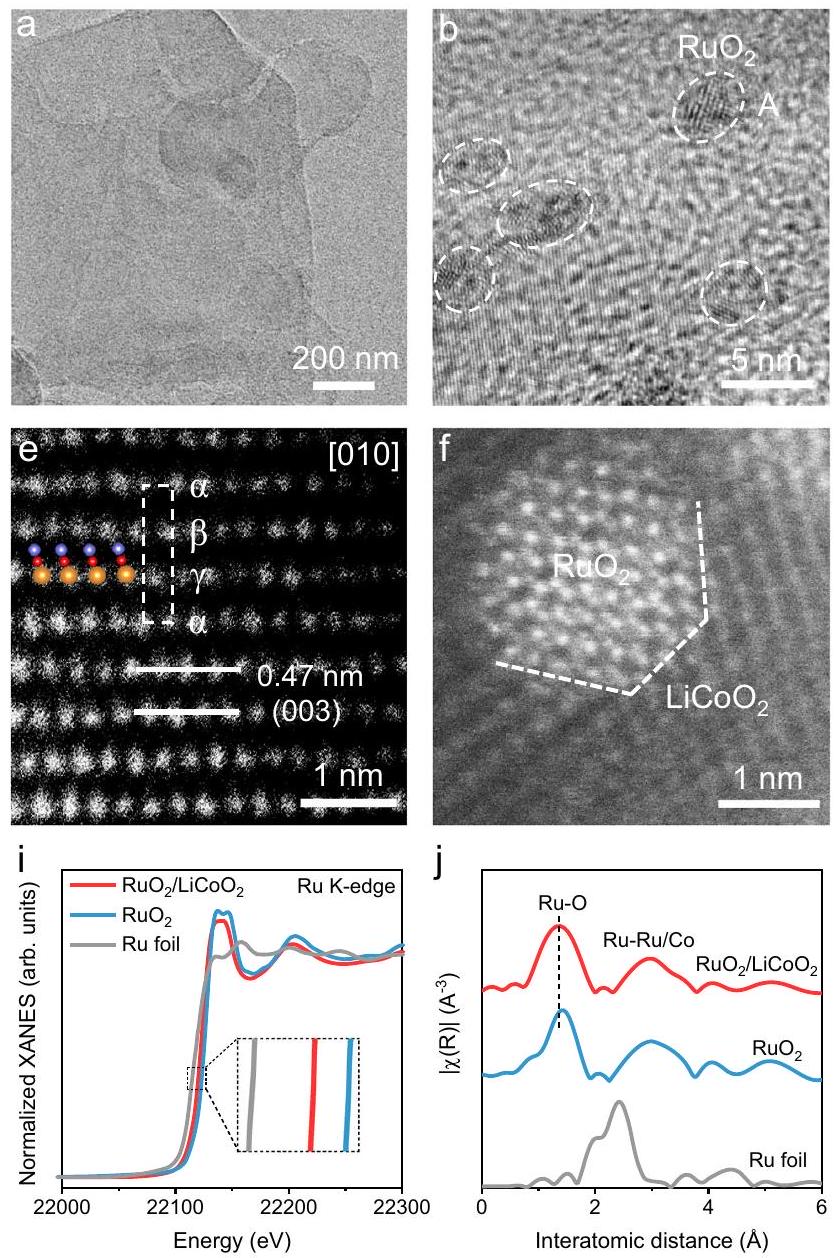
تُستخدم استراتيجية الكلسنة السهلة الامتصاص للتحضيرأغشية نانوية محملةجزيئات نانوية ). باختصار، تُستخرج الأوراق النانوية من خلال تقشير مساعد بالموجات فوق الصوتية للكتل المُصنّعة في الطور الصلب. (الأشكال التكميلية 1، 2). بعد ذلك، يتم امتصاص أيونات الروثينيوم على سطح من خلال الربط الكهروستاتيكي ثم تم حرقه لتشكيلالمحفز الكهربائي. وفقًا لصور المجهر الإلكتروني الماسح (SEM) والمجهر الإلكتروني الناقل (TEM)،يعرض هيكلًا نانويًا فائق الرقة (الشكل 2أ والأشكال التكميلية 3-5). من المثير للاهتمام أنتكون الجسيمات النانوية محصورة بإحكام فيالشبكة في صورة المجهر الإلكتروني عالي الدقة (HRTEM)، والتي تختلف عن الامتصاص الفيزيائي البسيط (الشكل 2ب). يوجد اتصال شبكي واضح بينالجزيئات النانوية والدعم، حيث تت correspond خطوط الشبكة مع المسافات بين الطبقات 0.31 و 0.20 نانومتر إلى (110) و (104) طائرة، على التوالي (الشكل 2ج، ح) . الـالدعم يوفر احتجاز الشبكة لـجزيئات النانو لتشكيل تفاعل فريد بين المعدن والدعم. تظهر المجهرية القوية الذرية (AFM) أن سمك الـالطبقة النانوية بسماكة 5.1 نانومتر في الشكل 2د. الهيكل النانوي فائق الرقة لـيزيد من تعرض المزيد من مواقع النشاط Ru. يتم استخدام مجهر المسح الإلكتروني بتقنية الحقل الداكن الزاوي العالي المصحح بالانحراف بدقة ذرية (HAADF-STEM) لتحليل التركيب الذري لـمركب. يتم ترتيب ذرات الكوبالت فيوضع التكديس في صورة HAADF-STEM على محور المنطقة [010]، مما يشير إلى تشكيل نوع O3ومتوافق مع نتائج حيود الأشعة السينية (الشكل 2e والأشكال التكميلية 6، 7). المسافة التي تبلغ حوالي 0.47 نانومتر تتوافق مع المسافة النموذجية بين الطبقات . بالإضافة إلى ذلك، يتم تضمين مصفوفة الذرات اللامعة من الروثينيوم في الشبكة الخاصة بـويظهر تطابقًا ممتازًا في الشبكة عند الواجهة (الشكل 2f). تؤكد توزيع عناصر الروثينيوم والكوبالت والأكسجين في طيف التحليل الطيفي للطاقة المشتتة (EDS) المزيد من تجانس الهيكل غير المتجانس (الشكل 2g). تُظهر التحليلات السابقة أن الهيكل المنظم هيكليًادعم النانو شيت مرتبط ارتباطًا وثيقًا بـالجزيئات النانوية، التي توفر إمكانية تحسين الهيكل الإلكتروني.
لفهم هيكل التنسيق والحالات الإلكترونية بشكل أفضلتم إجراء مطيافية امتصاص الأشعة السينية (XAS). كما هو موضح في الشكل 2i، حالة الأكسدة لـأقل من ذلك لـفي طيف امتصاص الأشعة السينية بالقرب من حافة Ru K (XANES)، مما يشير إلى تشكيل قناة لنقل الإلكترونات بين و . هذا يتماشى أيضًا مع نتائج المشتق الأول لطيف XANES عند حافة كا Ru وطيف التحليل الطيفي للأشعة السينية (XPS) عالي الدقة (الأشكال التكميلية 8، 9). دعم النانوورقة مع تعويض الشحنة ينظم بفعالية حالة الأكسدة لمواقع الروثينيوم، مما يمنع الأكسدة المفرطة خلال عملية التحفيز. وفقًا لطيف هيكل الامتصاص الدقيق للأشعة السينية الممتد عند حافة K للروثينيوم (FT-EXAFS)، فإن طول رابطة الروثينيوم-الأكسجين يتقلص قليلاً بعد الاتصال بين و (من 1.93 إلى )، والذي يُنسب إلى تشكيل التفاعلات السطحية (الشكل 2j والأشكال التكميلية 10، 11 والجداول التكميلية 1، 2) . حافة كايل Co XANES من يتم نقله إلى طاقات أعلى مقارنة بـفي الشكل 2k، وهو متسق مع طيف نطاق التكافؤ XPS و
الشكل 1 | مخطط تخطيطي لآلية تفاعل الأكسدة والاختزال وتحليل هيكل أكسيد الكوبالت الليثيوم. أ مخطط LOM لـ في OER الحمضي. ب مخطط AEM لـ RuO2 في OER الحمضي. ج نماذج هيكلية لـ و طاقة تذويب الحواجز لأكاسيد الكوبالت.
نتائج الرنين المغناطيسي الإلكتروني (EPR). هذا يؤكد قدرة التبرع بالإلكترونات لـ الناقل (الأشكال التكميلية 12، 13). علاوة على ذلك، فإن القمم الرئيسية لـ عند 1.4 ويتوافق مع و أصداء التنسيق في طيف FT-EXAFS عند حافة كوبالت K، على التوالي (الشكل 21)من الجدير بالذكر أنالسندات فيتمتد أكثر من، وهو ما يتماشى مع نتائج التحويل الموجي الطيفية (الأشكال التكميلية 14-16). تم التحقق من انتقال الإلكترون من الكوبالت إلى الروثينيوم من خلال جمع طيف فقدان طاقة الإلكترون (EELS) لـ و فيواجهة (الشكل التوضيحي 17). لذلك، فإن التفاعل بين الواجهات و يعوض إلكترونياً مواقع الروثينيوم ويحسن استقرار المحفز الكهربائي.
أداء التحفيز الكهربائي لـ OER
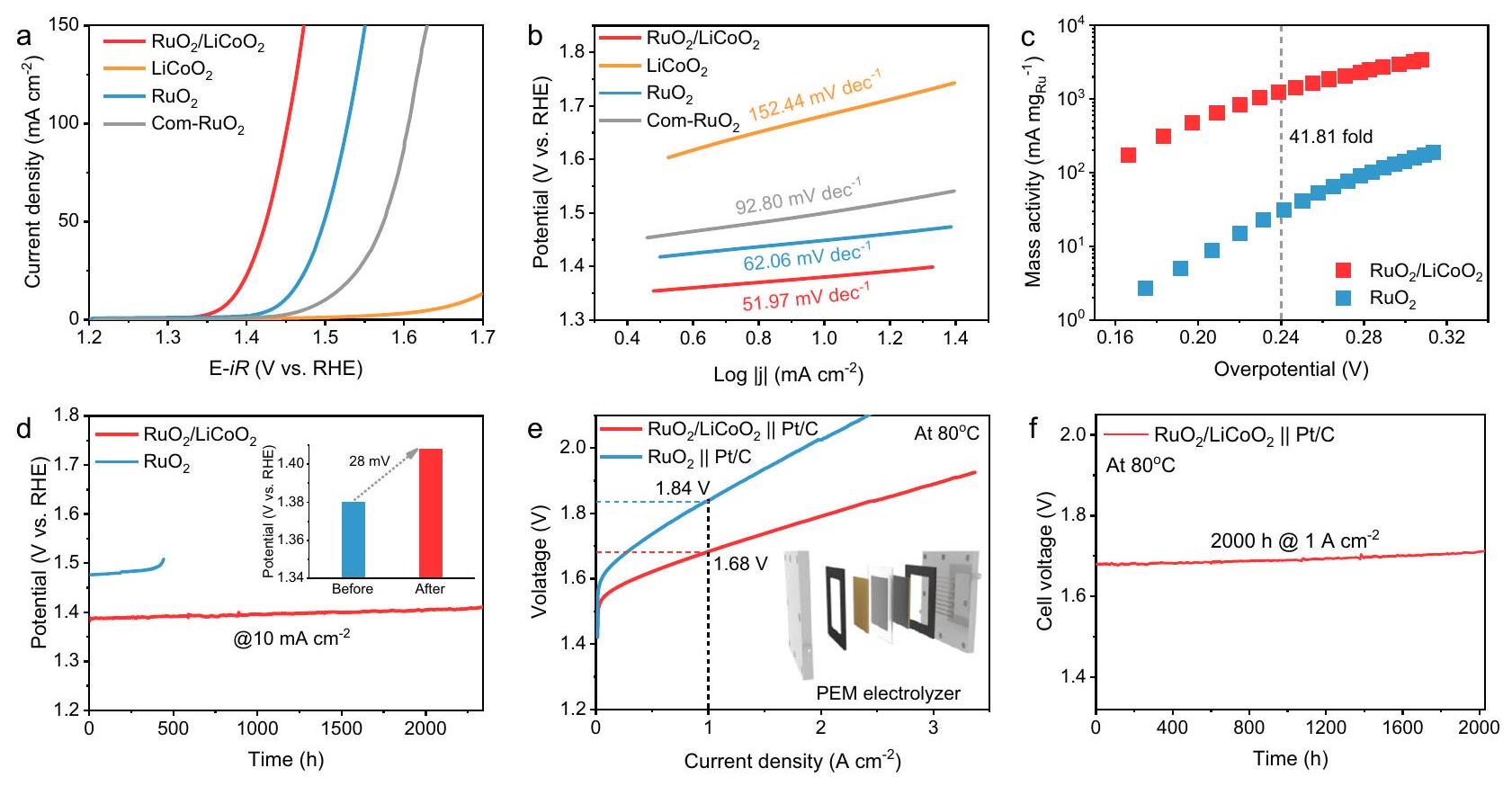
مستوحاة من المزايا الهيكلية لـتم تقييم أداء OER عبر نظام ثلاثي الأقطاب في وجود المحفز. في الشكل 3أ، المحفز الكهربائي يظهر جهدًا زائدًا قدرهفي، وهو أصغر بكثير من التجاريالمشار إليه بـ Com-RuO2 و (الأشكال التكميلية 18-24). بالمقارنة، فإن النشاط العالي الحموضة لتفاعل الأكسدة الكهربائي للأكسجين يتجاوز أيضًا معظم المحفزات الكهروكيميائية المعتمدة على المعادن النبيلة التي تم الإبلاغ عنها سابقًا (المكملات
الأشكال 25 والجدول التكميلي 3). ميل تافل لـيظهر ديناميات تفاعل محسّنة مقارنة بـ و (الشكل 3ب) في إمكانيات الطاقة المتجددةيعرض كفاءة تحويل متوسطة سريعة. في الوقت نفسه، زاوية الطور لـتنخفض بسرعة عند إمكانيات مختلفة في مخطط بود، مما يؤكد بشكل أكبر الانتشار السريع للشحنات على سطح المحفز (الأشكال التكميلية 26-28 والجدول التكميلية 4). النشاط الكتلي لـالمحفز هو 41.81 مرة منعند جهد زائد قدره 240 مللي فولت في الشكل 3ج. بالإضافة إلى ذلك، فإن تردد التحويل (TOF) والتيار الناتج عن عملية الأكسدة الكهربائية (OER) تم تطبيعهما حسب المساحة السطحية النشطة كهربائيًا (ECSA) لـالمحفزات الكهربائية أعلى بكثير من (الأشكال التكميلية 29-32 والجدول التكميلية 5). يتم اختبار متانة المحفز باستخدام الكرونو بوتنشيومترية لتقييم الإمكانية للتطبيقات العملية. يمكن أن يعمل المحفز الكهربائي بشكل مستقر لمدة 2300 ساعة عند كثافة تياربينماتقريبًا تفقد نشاطها بعد 400 ساعة (الشكل 3د والشكل التكميلي 33). تظهر النتائج أعلاه أنالدعم فيالمحفز الكهربائي يتجنب الذوبان فيفي تفاعل الأكسدة الكهربائي الحمضي لتحقيق نشاط تحفيزي حمضي مستدام.
لتقييم الإمكانيات التطبيقية الصناعية لـتم تجميع جهاز تحليل كهربائي من نوع PEM معو
الشكل 2 | توصيف هيكل. صورة TEM، (ب، ج) صور HRTEM، و (د) صور AFM لـ (إدراج: ملف الارتفاع لـصور HAADFSTEM لـ. تخطيط العناصر باستخدام EDSالنموذج الذري لـ و . قمت بتطبيع طيف XANES لحافة الروثينيوم K
و (ج) طيف FT-EXAFS لحافة Ru K لـ، ورقة Ru، على التوالي. k طيف XANES عند حافة Co K المنظم و (I) طيف FT-EXAFS عند حافة Co K لـ، وورق الألمنيوم، على التوالي.
Pt/C كعوامل حفازة للأنود والكاثود، على التوالي. على وجه التحديد،يتطلب فقط جهد خلية قدره 1.68 فولت للوصول إلى كثافة تيار قدرهاوهو ما هو أقل بكثير منPt/C (1.84 فولت) (الشكل 3e). تُظهر تحليل الفائض الجهد أن تحسين نقل الكتلة يخفف من آثار الاستقطاب التركيز في بيئة التفاعل المحلية، مما يعزز بشكل غير مباشر الحركيات الحفزية (الشكل التكميلية 34). علاوة على ذلك، فإن النشاط الكتلي لـتقريبًا 21.33 مرة أكبر من ذلك لـعند جهد خلية قدره 1.7 فولت (الشكل التوضيحي 35). النشاط الكتلي وتكلفةأقل بكثير من تلك الخاصة بالتجارةPt/C (الشكل التوضيحي 36). من اللافت للنظر أن جهاز التحليل الكهربائي PEM الذي يستخدميمكن أن تعمل بشكل مستقر لمدة 2000 ساعة عند كثافة تيارمع تدهور ضئيل في الشكل 3f، مما يشير إلى إمكانيات الـمحفز للتطبيقات العملية. المتانة على المدى الطويل لـفي PEMWE يتجاوز أيضًا أداء معظم المحفزات الكهربية عالية الأداء التي تم الإبلاغ عنها مؤخرًا (الجدول التكميلي 6). هذه النتائج توضح أنيظهر المحفز الكهربائي أداءً متميزًا كخيار لإنتاج الهيدروجين في إلكتروليزر PEM، مما يوفر دعمًا قويًا لإنتاج الطاقة المستدامة.
التحول الهيكلي بعد الاستقرار
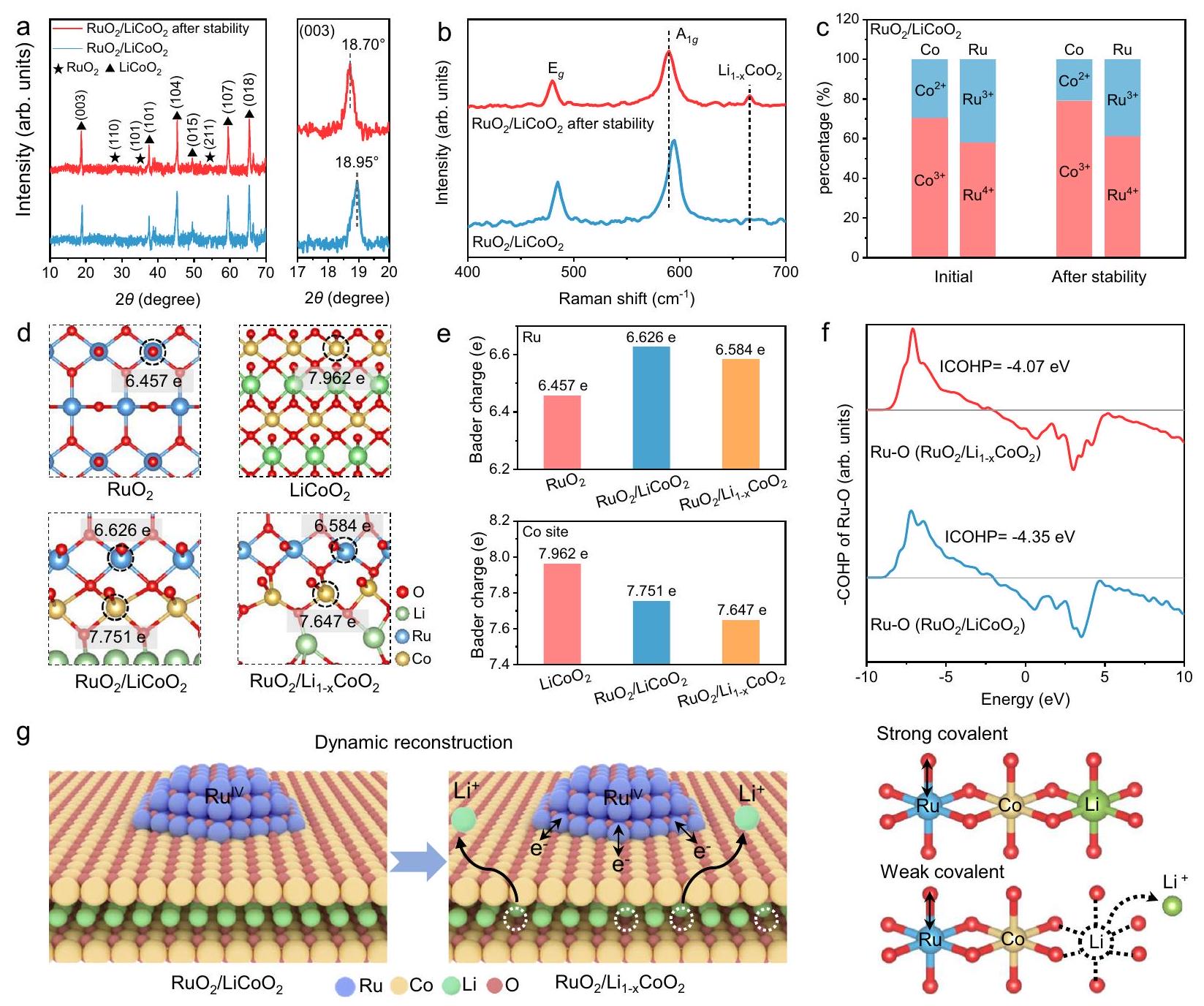
الهيكل الفيزيائي لـتمت دراسة المحفز الكهربائي بعد اختبار استقرار OER لاستكشاف العوامل المؤثرة على النشاط التحفيزي المحسن. تظهر صور SEM و TEM أنتظل الجسيمات النانوية مدعومة بإحكام على سطحالأغشية النانوية فيالمحفز الكهربائي بعد اختبار الاستقرار (الأشكال التكميلية 37، 38). بالإضافة إلى ذلك، يمكن ملاحظة طبقة غير متبلورة معاد بناؤها بوضوح على سطحأوراق نانوية في. تُنسب هذه الطبقة إلى هجرة واستخراج أيونات الليثيوم بين الطبقات أثناء تطبيق الجهد، مما يؤدي إلى تكوين سطح طبقة غير متبلورة. مقارنةً بالأصلذروة مستوى البلورة (003) منبعد أن تنتقل استقرار OER إلى زاوية أقل، وهو ما يمكن أن يُعزى إلى تشوه الشبكة الناتج عن استخراج أيونات الليثيوم (الشكل 4 أ والشكل التكميلي 39). بالإضافة إلى ذلك، طيف رامان لـيكتشف قمتين مميزتين لـ و أوضاع عند 484 و ، مما يتوافق معانحناء والتمدد، على التوالي (الشكل 4ب والشكل التكميلي 40). بسبب زيادة القطبية لـالرابطة الناتجة عن استخراج الليثيوم، و قمم مميزة لـتظهر انزياحًا أزرق بعد استقرار OER (الشكل التكميلي 41). على وجه الخصوص، القمة الجديدة عندبعد الاستقرار تُعزى إلى روابط Co-O في الهيكل السبينللطبقة إعادة بناء السطح، مما يعزز قطبيةالروابط. أيونات الليثيوم بين الطبقات تنفصل عنمن خلال مسار انتشار ثنائي الأبعاد تحت جهد OER، مما يؤدي إلى إعادة بناء هيكلية ديناميكية وتغيير بيئة التنسيق السطحية للمحفز.
الشكل 3 | الأداء الكهروكيميائي لـ. منحنيات استقطاب OER و (ب) مخططات تافل لـ, وفيالمحلول الكهربائي، على التوالي. يتم تصحيح الجهد بواسطةتعويض iR تلقائي (R هو).النشاط الكتلي لذرات Ru فيوكنتيجة للجهد الزائد. د منحنيات الجهد الزمني لـ وعند، على التوالي (الإدراج: تغييرات الجهد لـقبل وبعد الاستقرار). هـ منحنيات الاستقطاب لـ PEMWE معوالمحفز في الماء النقي عندبدون تصحيح iR.منحنى الجهد الزمني لـ PEMEW باستخدامالمحفز عند.
تتم تحليل الحالات الإلكترونية لـالمحفزات قبل وبعد استقرار OER لفهم تأثير عملية إعادة البناء. نسبةبعد استقرار OER تزداد مقارنة بـالأصلية، مما يعني تقليل كثافة الإلكترون لمواقع Co (الشكل 4c والشكل التكميلي 42). يؤدي الاستخراج الديناميكي لأيونات الليثيوم إلى نقل الإلكترون، مما يؤدي إلى أكسدة جزئية لمواقع Co وزيادة التساهمية فيالرابطة. بالإضافة إلى ذلك، تزداد نسبةفيبشكل طفيف منإلىبعد قياس الاستقرار. بالمقارنة، فإن نسبةفيبعد قياس الاستقرار هي، وهو أعلى بكثير من نسبة(الأشكال التكملية 43، 44). على الرغم من الأكسدة الطفيفة لـالدعم، إلا أنه يمكنه الحفاظ على حالة الأكسدة لـمن خلال التبرع المستمر بالإلكترونات، مما يمنع إذابة مواقع Ru (الأشكال التكملية 45، 46). تم بناء نموذج هيكلي لـبعد إزالة جزئية لليثيوملكشف التغيرات في الهيكل الإلكتروني من خلال حسابات DFT (الشكل التكميلي 47). تشير شحنة بادر إلى أن Ru فييحصل على حوالي 0.169 e من الإلكترونات منالدعم (الشكل 4d). بسبب استخراج أيونات الليثيوم، فإن كثافة الشحنة عند مواقع Ru فيأقل من تلك الموجودة في(6.626 e)، بينما تبقى أعلى من تلك الموجودة في(الشكل 4e). تظهر تحليل كثافة السكان للمدار البلوري (COHP) أن تساهمية رابطة Ru-O فيأقل من تلك الموجودة في(الشكل 4f. تضمن عملية إزالة الليثيوم الفريدة تنظيمًا ديناميكيًا لتساهمية رابطة Ru-O خلال العملية التحفيزية لتقليل مشاركة الأكسجين الشبكي (الشكل 4g).
التوصيف في الموقع للتغيرات الهيكلية للمحفز
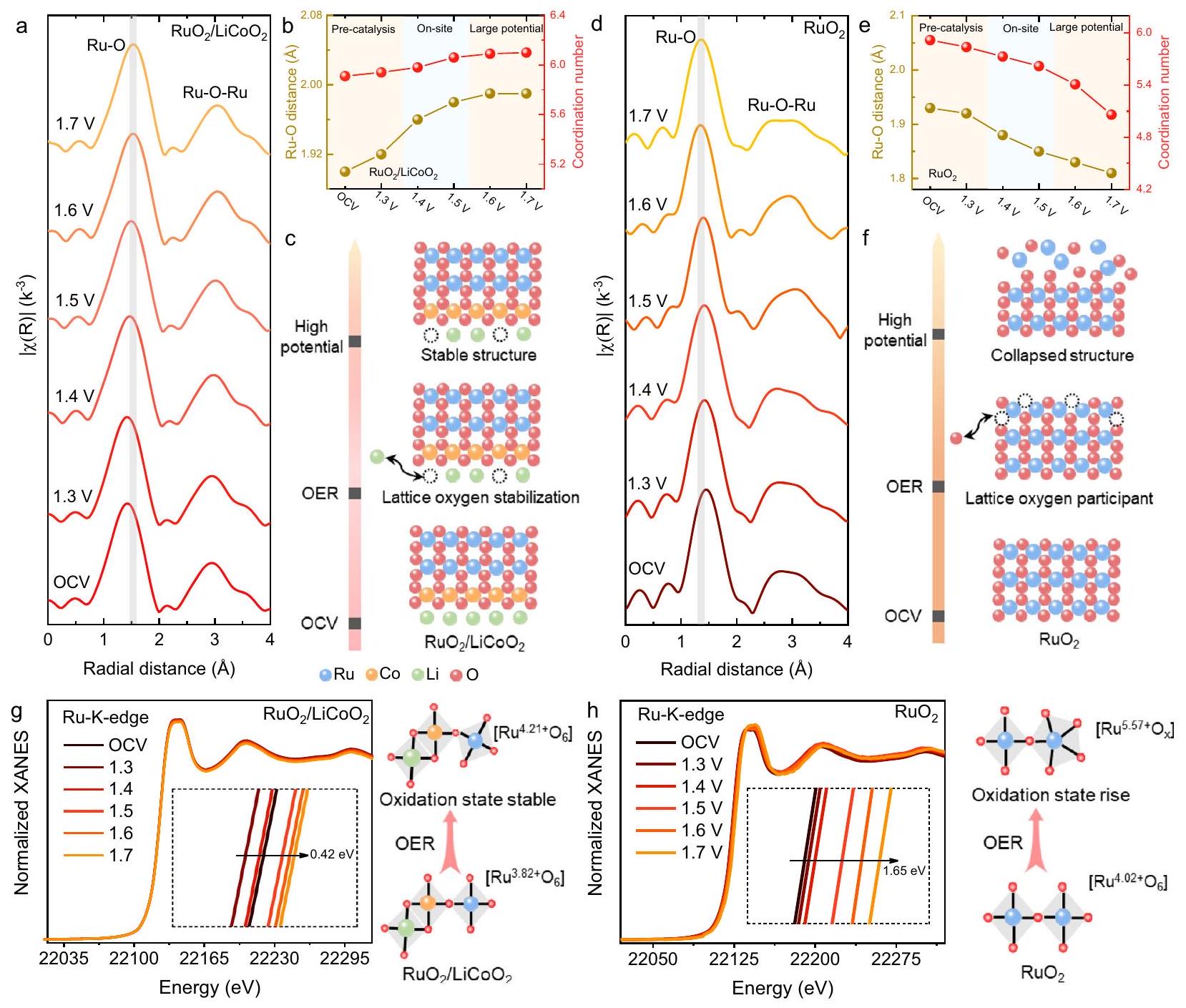
تم تنفيذ XAS في الموقع لاستكشاف آلية التطور الديناميكي للمحفزات الكهروكيميائية في عملية OER (الشكل التكميلي 48). في طيف FT-EXAFS عند حافة Ru K، يتم تمديد رابطة Ru-O لـ
بشكل مناسب مع زيادة الجهد من
جهد الدائرة المفتوحة (OCV) إلى 1.7 فولت (من 1.90 إلى Å. على النقيض من ذلك، تميل رابطةإلى أن تكون أكثر تساهمية خلال عملية إزالة الليثيوم، مما يؤدي إلى انزياح سلبي لقشرة تنسيق Co-O فيوتشكيل طبقة إعادة بناء سطحية (من 1.92 إلىÅ. تؤكد تحويلة الموجة لحافة Ru K وحافة Co K EXAFS أيضًا هذه النتيجة بشكل بديهي (الأشكال التكملية 52-54). وبالتالي، ينظم الاستخراج الديناميكي لأيونات الليثيوم تساهمية رابطة Ru-O من خلال التفاعلات السطحية، مما يمكن أن يقيد مشاركة الأكسجين الشبكي ويحافظ على الاستقرار الهيكلي لـ(الشكل 5c). من الجدير بالذكر أن رابطة Ru-O فيتخضع لتقصير كبير منÅÅ. علاوة على ذلك، تشير التغيرات الطفيفة في عدد تنسيق Ru-O فيإلى استقرار الهيكل التنسيقي لـ. بالمقارنة، يمكن ملاحظة عدد تنسيق مخفض لرابطةفيوهو ما يُعزى إلى وجود أكسجين معيب (الشكل 5f). بسبب التساهمية القوية لرابطة Ru-O، يتبعمسار LOM وينتج عددًا كبيرًا من العيوب الأكسجينية خلال عملية OER، مما يؤدي إلى انهيار هيكل سطح المحفز.لتوضيح التغيرات في الهيكل الإلكتروني للمحفز الكهروكيميائي عند الجهود العالية، يتم الكشف عن حالات الأكسدة لـ
ومن خلال أطياف XANES في الموقع. يرتفع متوسط حالة التكافؤ لعينات Ru فيبشكل طفيف من +3.82 إلى +4.21 مع انتقال الجهد من OCV إلى 1.7 فولت مقابل RHE في الشكل 5g (الشكل التكميلي 58). يؤكد هذا بشكل أكبر أن التفاعل الإلكتروني القوي بينوالواجهة يمنع الأكسدة المفرطة لـ. بالإضافة إلى ذلك، يمكن أن تحافظ الأنواع Co في المحفزعلى حالة أكسدة +3.16 عند جهد مرتفعالشكل 4 | إعادة بناء المحفز
بعد الاستقرار. أ أنماط XRD و (ب) أطياف رامان لـوبعد الاستقرار، على التوالي.محتوىوفيقبل وبعد استقرار OER. د تحليل شحنة بادر لـو، على التوالي. هـ مقارنة كميات الشحنة لـ ووو، على التوالي.من Ru-O فيو، على التوالي. ز مخطط توضيحي للتطور الديناميكي لـ.
جهد 1.7 فولت (الأشكال التكملية 59، 60). نتيجة لذلك، يمكن أن ينظم استخراج أيونات الليثيوم توزيع الإلكترونات لـالدعم بشكل معتدل دون تدمير الهيكل الرئيسي. من المRemarkably، حالة التكافؤ لـ Ru تزداد بسرعة من +4.02 إلى +5.57 فيمع تغيير الجهد من OCV إلى 1.7 فولت مقابل RHE، وهو ما يُعزى إلى الأكسدة المفرطة الناتجة عن تلف الهيكل(الشكل 5h، الأشكال التكملية 61، 62). بناءً على النتائج السابقة، يمكن أن تعدل عملية إعادة البناء الديناميكية لـبيئة الشحنة لرابطةوت stabilize الأكسجين الشبكي، مما يؤدي إلى تحسين أداء OER.
أصل الأداء التحفيزي العالي
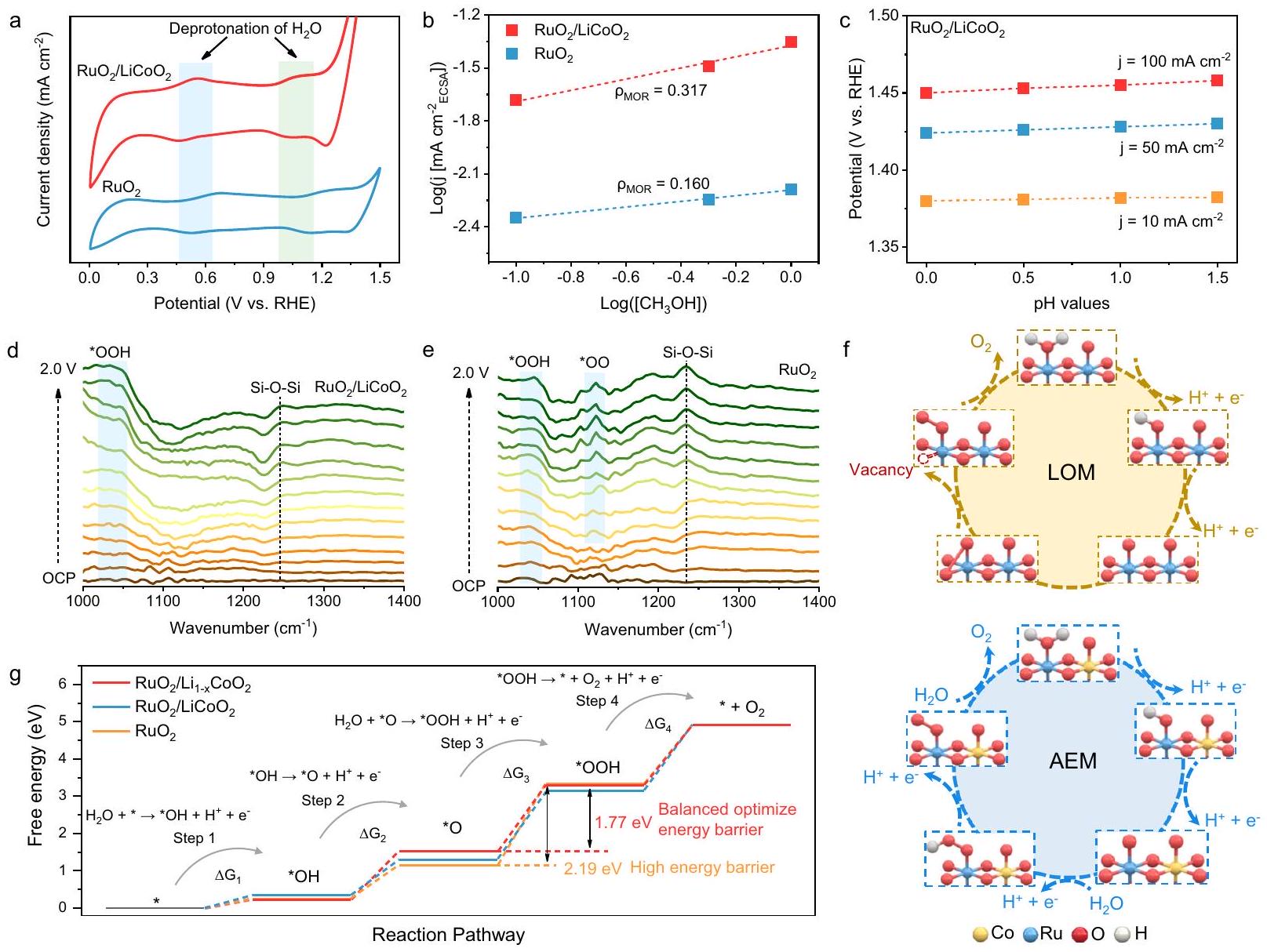
لتحقيق ظواهر نقل الوسائط التفاعلية على سطح المحفز الكهروكيميائي، يتم إجراء سلسلة من القياسات الكهروكيميائية. يمكن أن تظهر منحنيات الفولتمترية الدورية لـوزوجين من قمم الأكسدة والاختزال حول 0.5 فولت و 1.0 فولت على التوالي، والتي تتوافق مع تشكيل وسائط تفاعلية *OH معإزالة البروتون (الشكل 6a). من الواضح أن قمة الأكسدة والاختزال لـتنخفض أكثر من تلك الموجودة في، مما يشير إلى إزالة بروتون مواتية لـوتعزيز عملية التفاعل. بعد ذلك، يتم استخدام الميثانول كجزيء استكشافي للكشف عن تغطية وسائط *OH على سطح المحفز (الشكل التكميلي 63).
تفاعل أكسدة الميثانول (MOR) أكثر نشاطًا على-سطوح المحفزات المهيمنة بسبب آلية الهجوم النووي على المواقع الكهربية.تظهر تغطية سطحية عالية لـمقارنة بـفي الشكل 6b، مما يعني أنتحتوي على المزيد من مواقع OER النشطة الفعالة. علاوة على ذلك، فإن التغير في النشاط التحفيزي للمحفزعند قيم pH مختلفة ضئيل، وهو سمة من سمات آلية AEM النموذجية (الشكل 6c والشكل التكميلي 64). على النقيض من ذلك، تظهرسلوكًا يعتمد على pH لنشاط OER، مما يدل على حدوث آلية LOM غير مستقرة. تشير اختبار القطب الدوار والقرص وتحليل حاجز الطاقة للامتصاص/الإزالة للوسائط المحتوية على الليثيوم إلى أنه من الصعب على أيونات الليثيوم المستخرجة المشاركة في عملية OER لـ(الأشكال التكملية 65-68). علاوة على ذلك، تحافظعلى نفس الجهد في المحاليل الكهربائية ذات تركيزات أيونات الليثيوم المختلفة بعد 50 ساعة من اختبار الاستقرار، مما يؤكد أكثر أن الكمية الصغيرة من الليثيوم المستخرج لها تأثير ضئيل على أداء المحفز (الشكل التكميلي 69).
يتم مراقبة آلية تفاعل OER الحمضي لـوبواسطة مطيافية الامتصاص بالأشعة تحت الحمراء المعززة السطحية (ATR-SEIRAS) (الشكل التكميلي 70). يمكن ملاحظة نطاق امتصاص قوي في حوالي ، والذي يتم التعرف عليه على أنه
الشكل 5 | توصيف XAS في الموقع لـ يؤكد العملية الديناميكية. أ طيف EXAFS لحد Ru K في الموقع لـ مع الجهود المطبقة من OCV إلى 1.7 فولت. ب التغيرات في طول الرابطة وشدة غلاف Ru-O لـ / عند جهود مختلفة. ج مخطط تخطيطي لتطور الهيكل لـ عند جهود مختلفة. د طيف EXAFS لحد Ru K في الموقع لـ مع
الجهود المطبقة من OCV إلى 1.7 فولت. هـ التغيرات في طول الرابطة وشدة غلاف Ru-O لـ عند جهود مختلفة. مخطط تخطيطي لتطور الهيكل لـ عند جهود مختلفة. طيف XANES لحد Ru K في الموقع لـ . إدراج: تغييرات حالة الأكسدة لـ Ru في . ح طيف XANES لحد Ru K في الموقع لـ . إدراج: تغييرات حالة الأكسدة لـ Ru في .
تمدد في وسيط *OOH في الشكل 6د . نظرًا لأن *OOH هو وسيط نموذجي لآلية AEM، فإن عملية OER لـ تهيمن عليها بشكل رئيسي AEM. بالمقابل، فإن القمم المميزة عند 1033 و في تتوافق مع و وسطاء، مما يشير إلى مسار مشترك بين LOM و AEM (الشكل 6هـ) . وبالتالي، فإن تأثير إعادة البناء الديناميكي يحقق التحول الكامل لـ التحفيز الكهربائي من آلية LOM إلى AEM، مما يمنع إذابة المحفز ويعزز استقرار OER (الشكل 6و). تم حساب حواجز الطاقة الحرة لجزيئات تفاعل OER على المحفز الكهربائي لاستكشاف اختلافات النشاط (الأشكال التكميلية 71-73). وفقًا للشكل 6ز، فإن الخطوة المحددة لمعدل OER (RDS) لـ ، ، ، و هي حاجز طاقة تشكيل الوسيط *OOH. يتطلب المحفز الكهربائي حاجز طاقة قدره 1.77 إلكترون فولت لتجاوز RDS، وهو أقل بكثير من ذلك لـ . حاجز التكوين للوسيط *OO لـ أعلى بكثير من ذلك لـ *OOH، مما يعزز آلية AEM (الأشكال التكميلية 74-81). بالإضافة إلى ذلك، تم تقليل التداخل بين مدارات Ru d و O p، مما يؤكد ضعف الترابط في رابطة Ru-O. هذا يعيق
الأكسجين الشبكي من المشاركة في عملية تفاعل OER، مما يعزز الاستقرار الهيكلي (الأشكال التكميلية 82، 83). نتيجة لذلك، فإن تعديل دعم يحسن طاقة الربط لـ
مع الوسائط الرئيسية، مما يؤدي إلى عملية حرارية مواتية وزيادة النشاط الجوهري.
نقاشباختصار، لقد نجحنا في التحكم في بيئة الربط لـ خلال التفاعل التحفيزي من خلال استخدام إعادة البناء الديناميكي لـ الدعم لتحقيق توازن بين النشاط والاستقرار في OER الحمضي. ينظم التفريغ الكهربائي الديناميكي توزيع الإلكترونات وهيكل التنسيق عند الواجهة، مما يعزز التحسين الذاتي لـ المحفز خلال عملية OER. يؤدي ضعف الترابط في رابطة Ru-O إلى انتقال كامل لـ المحفز الكهربائي من مسار تفاعل LOM إلى AEM، مما يحسن الاستقرار التحفيزي. نتيجة لذلك، يصل المحفز الكهربائي إلى كثافة تيار قدرها عند جهد زائد منخفض قدره . على وجه الخصوص، يعمل جهاز التحليل الكهربائي PEM باستخدام بشكل مستقر لمدة 2000 ساعة عند كثافة تيار عالية قدرها
. توفر هذه الدراسةالشكل 6 | آلية التفاعل على . أ منحنيات الفولتمترية الدورية لـ و المحفزات في . ب الرسم البياني للدالة بين كثافة تيار MOR وتركيز الميثانول على و المحفزات. ج اعتماد pH على جهد OER عند كثافات تيار مختلفة لـ و . طيف ATR-SEIRAS في الموقع لـ و هـ عند جهود مطبقة مختلفة. ف المخططات التخطيطية لآلية OER على و ، على التوالي. مخططات الطاقة الحرة لجزيئات OER لـ ، و ، و
، على التوالي.
طريقة لحل التوازن بين النشاط والاستقرار لمحفزات الأكسيد المعتمد على الروثينيوم في OER الحمضي.
طرققسم التجارب إعداد المواد
إعداد رقائق نانوية. تم خلط الكمية المتوازنة من (صلب، كاشف ACS، ) و فائض (صلب، كاشف ACS، ) عن طريق الطحن الكروي لمدة 6 ساعات عند . بعد ذلك، تم تسخين المكونات لمدة 4 ساعات عند في الهواء بمعدل تسخين قدره للحصول على مسحوق الكتلي. تم تفريق المساحيق الكتلية لـ في الماء المقطر وتمت معالجتها بالموجات فوق الصوتية لمدة 4 ساعات في حمام ماء مثلج، مع الحفاظ على درجة الحرارة عند . تم جمع السائل العلوي عن طريق الطرد المركزي عند . أخيرًا، تم الحصول على مساحيق رقائق
عن طريق التجفيف بالتجميد للسائل العلوي عند .إعداد . تم إذابة 50 ملغ من و 10 ملغ من في 20 مل من الماء المنزوع الأيونات وتم تحريكها مغناطيسيًا في درجة حرارة الغرفة لمدة 12 ساعة. ثم، تم جمع مسحوق المكون عن طريق الطرد المركزي عند . أخيرًا، تم تسخين المسحوق الناتج في الهواء عند لمدة 3 ساعات بمعدل تسخين قدره للحصول على . للمقارنة، تم تغيير كمية إلى 5 ملغ و 15 ملغ للحصول على محفز كهربائي
بتحميلات مختلفة لـ
.التوصيف. تم توصيف شكل المحفزات بواسطة مجهر إلكتروني مس扫描 (SEMJEOLJSM-6700F) ومجهر إلكتروني ناقل (TEM FEI Tecnai G2 F20). تم التقاط صور مجهر إلكتروني مسح ضوئي عالي الزاوية مصحح التشوه (AC HAADF-STEM) في JEM-ARM200F المجهز بنظام JED-2300T SDD. تم استخدام بيانات حيود الأشعة السينية (XRD) التي تم الحصول عليها من معدات Bruker D8 Advance لتحليل الهيكل البلوري. تم تحليل التركيب العنصري بواسطة ICP (ICP-MS، مطياف الكتلة المتصل بالبلازما). تم إجراء تحليل مطياف الأشعة السينية (XPS) على نظام Escalab 250 Xi باستخدام أشعة X من Al Kα. تم قياس مطياف امتصاص الأشعة السينية (XAS) لحد Co K و Ru K في خط الشعاع TLS 17C1 و TPS 44A1 في مركز أبحاث الإشعاع المتناظر الوطني (NSRRC) في تايوان.القياسات الكهروكيميائية. تم اختبار الأداء الكهروكيميائي على محطة عمل كهروكيميائية باستخدام نظام ثلاثي الأقطاب (Autolab PGSTAT302، Metrohm). تم استخدام قضيب الجرافيت وقطب الزئبق المشبع كقطب مضاد (CE) وقطب مرجعي (RE)، على التوالي. تم خلط المحفز، الكربون الأسود، وفلوريد البولي فينيليدين (PVDF) بنسبة وزن قدرها ، وتم استخدام N -ميثيل-2-بيروليدون (NMP) كمذيب. تم طلاء المعجون اللزج بالتساوي على ورق الكربون وتجفيفه تحت فراغ. تحميل المحفز هو ومساحة التحميل هي . تم تحويل الجهد المقاس إلى قطب هيدروجين قابل للعكس وفقًا للمعادلة
.بعد ذلك، تم قياس CV بمعدل مسح قدره . تم إجراء الفولتمترية المسحية الخطية (LSV) في محلول مشبع بـ (pH هو ) بمعدل مسح قدره . تم إعداد الإلكتروليت واستخدامه على الفور وتم تخزينه في زجاجة زجاجية في درجة حرارة الغرفة. تم تصحيح الجهد وفقًا لصيغة . تم إجراء طيف الامتصاص الكهروكيميائي (EIS) في نطاق تردد قدره مع سعة 5 مللي فولت. تم الحصول على سعة الطبقة المزدوجة عن طريق جمع منحنيات CV بمعدلات مسح من 10 إلى . تم إجراء اختبارات OCP مع مشبعة بهيدروجين عالي النقاء لتأكيد جهد القطب المرجعي. تم إجراء تجارب المعايرة في درجة حرارة الغرفة () لتقليل تأثيرات درجة الحرارة.
اختبار القطب الدوار ذو القرص (RRDE) لمحفز . تم تفريق المحفز في محلول مختلط من كحول إيزوبروبيل و نافيون ()، تلاه معالجة بالموجات فوق الصوتية لمدة ساعة للحصول على حبر محفز موزع بشكل موحد. تم إسقاط من حبر المحفز على قطب قرصي (مساحة ) وتجفيفه تحت فراغ في درجة حرارة الغرفة للحصول على قطب عمل. تم استخدام قضيب كربوني و كقطب مضاد وقطب مرجعي، على التوالي. تم إجراء الفولتمترية المسحية الخطية في محلول مع تركيزات مختلفة من أيونات الليثيوم بمعدل مسح قدره وسرعة دوران قدرها 1600 دورة في الدقيقة.
يمكن تقييم المساحة السطحية الكهروكيميائية (ECSA) لمحفز كهربائي بواسطة سعة الطبقة المزدوجة الكهروكيميائية () وفقًا للمعادلة التالية:
حيث تم تحديد من خلال أخذ نصف ميل الفرق في التيار () الذي تم رسمه كدالة لمعدل المسح في تجربة CV. هو السعة السطحية العامة المحددة ().
تم حساب تردد التحويل (TOF) للمحفز الكهربائي بواسطة المعادلة التالية:
تم الحصول على دوران الفورمات لكل وحدة مساحة هندسية من كثافة التيار الهندسية لخصائص الاستقطاب LSV وفقًا للمعادلة:
تجارب ATR-SEIRAS الكهروكيميائية في الموقع. تم إجراء قياسات ATR-SEIRAS باستخدام مطياف FT-IR من نوع Nicolet iS50، حيث تم الحصول على كل طيف من خلال تجميع 32 تداخلاً، مما حقق دقة طيفية منتضمنت عملية تحضير القطب العامل خطوتين رئيسيتين. أولاً، تم ترسيب فيلم رقيق جداً من الذهب كيميائياً على بلورة السيليكون لتعزيز حساسية إشارة الأشعة تحت الحمراء وموصلية الإلكترونات. ثم، تم إعداد معلق محفز بتحميل قدرهتم تطبيقه على سطح الذهب. تم تحضير هذا المعلق عن طريق توزيع 7 ملغ من المحفز و3 ملغ من الكربون الأسود في 1 مل من الإيثانول، تلاه إضافةمن نافيون بعد 30 دقيقة من الصوتنة. تم وضع الأقطاب الكهربائية العاملة المجمعة في خلية كيميائية كهربائية ثلاثية الأقطاب، حيثتم استخدامه كإلكترود مرجعي، وقضيب جرافيت كإلكترود مضاد، وغاز الأرجون المشبعكإلكتروليت لتفاعل OERتم إجراء جميع القياسات باستخدام تقنية الفولتمترية ذات المسح الخطي (LSV) للتحقيق في وسائط تفاعل الأكسدة (OER) عند مختلف الجهود المطبقة.
قياسات XAFS في الموقع. تم استخدام مطيافية امتصاص الأشعة السينية في الموقع (XAS)، بما في ذلك كل من XANES وEXAFS عند حواف Ru وCo، كانت تمت الدراسة في وضع العائد الكلي للفلوريسcence تحت ظروف بيئية في BL-12B2 من SPring-8، NSRRC. تم إجراء القياسات باستخدام حاوية مصممة خصيصًا من مادة التفلون مزودة بنافذة مختومة بشريط كيبون، مما يسمح لأشعة X بالمرور من خلال الشريط والكهارل. هذا الإعداد ضمن أن إشارات XAS تم التقاطها بفعالية في وضع العائد الكلي للفلوريسcence في المركز الوطني لأبحاث الإشعاع السنكروتروني (NSRRC)، SPring-8. تم إجراء التجارب تحت تكوين ثلاثي الأقطاب، بما يتماشى مع ظروف التوصيف الكهروكيميائي.لعمليات معالجة البيانات، تم تحقيق التطبيع الطيفي من خلال إزالة خط الأساس قبل الحافة وضبط منطقة ما بعد الحافة.-تمت معالجة تذبذبات EXAFS الموزونة بواسطة تحويل فورييه لتسهيل تحليل EXAFS، حيث تم تقديم جميع طيف EXAFS بدون تصحيح الطور. تم إجراء ملاءمة بيانات تحويل فورييه (FT) باستخدام Artemis (الإصدار 0.9.25)، مع استخدام عامل الوزن مع نطاق k من ونطاق R من عدد التنسيق، طول الرابطة، عامل ديباي-والر، وانزياح الطاقة (، و تم تحديدها من خلال التناسب دون أي معلمات ثابتة، في حين أن عامل تقليل السعة (تم تعيينه إلى 0.85.
قياسات PEMWE.تم استخدامه كعوامل حفازة للأنود في إلكتروليزر PEM. تم استخدام Pt/C التجاري كعامل حفاز للكاثود. تم تصنيع مجموعة الأقطاب الكهربائية الغشائية عبر تقنية الغشاء المغطى بالحفاز، مع تغطية منطقة هندسية قدرهاتم dispersing مسحوق المحفز في الإيزوبروبانول والماء المقطر وحل Nafion لتحضير الحبر. تم الحصول على الحبر الموزع بشكل موحد من خلال الاستحلاب الشريطي وتفكيك الخلايا بالموجات فوق الصوتية. تم الحصول على و تم رش أحبار المحفز على كلا الجانبين من غشاء تبادل البروتون، على التوالي. كانت تحميلاتالأنود وكاثود Pt/C هما و على التوالي. تم استخدام نافيون 115 كغشاء تبادل بروتون (PEM) وتم معالجته بـ و فيلمدة ساعة واحدة متتالية. كان حجم غشاء تبادل البروتونوكانت سماكة الغشاءتم ضغط الغشاء المرشوش، وطبقة انتشار غاز الأنود (ألياف التيتانيوم)، وطبقة انتشار غاز الكاثود (ورق الكربون) بالحرارة عندوضغط 10 ميغاباسكال للحصول على تجميع إلكترود الغشاء. بعد ذلك، تم تجميع واختبار إلكتروليزر الماء PEM عنداستخدام الماء النقي كإلكتروليت. تم الحصول على منحنى الاستقطاب لـ PEMWE عند معدل مسح، وتم إجراء اختبار كرونو بوتنشيومتري فيلتقييم الاستقرار.
حسابات DFT. تم إجراء جميع حسابات نظرية الكثافة (DFT) باستخدام حزمة المحاكاة الأولية فيينا (VASP)تم تطبيق نموذج الجهد الزائف لموجة العرض (PAW) بالاشتراك مع دالة التبادل والتفاعل التقريبية العامة (PBE GGA).للوصف المناسب للإلكترونات d المحلية لـ Co، تم استخدام طريقة DFT + U، مع تضمين تصحيح هوبارد-U بقيمة Ueff(Co)، تم تحديدها من خلال نظرية الاستجابة الخطية. تم إجراء الحسابات باستخدام مجموعة أساس الموجة المسطحة مع حد طاقة يبلغ 500 إلكترون فولت، وشبكة نقاط كيه من نوع مونكهورست-باك منتم استخدامه لأخذ عينات من منطقة بريلوان. تم اعتبار استقطاب الدوران، وتم إجراء استرخاء هيكلي كامل حتى تم الوصول إلى معيار تقارب الطاقة.لكل ذرة، مع الحفاظ على القوة النهائية المؤثرة على كل ذرة تحت. بالإضافة إلى ذلك، تم إنشاء مخططات بورباي باستخدام بيئة المحاكاة الذرية (ASE)، حيث تم اشتقاق طاقات التكوين المدخلة من حسابات DFT لنماذج الكتلة والسطح.
يمكن حساب طاقة الامتزاز للوسطاء التفاعليين باستخدام المعادلة التالية:
أين الإعلانات، أهي طاقة الربط، هو تغيير طاقة النقطة الصفرية، هو تغيير الإنتروبيا. في هذا العمل، قيم الـ و تم الحصول عليها من خلال حساب تردد الاهتزاز.
يمكن حساب الطاقة الحرة لجيبس لخطوات التفاعل من خلال المعادلات الأربع التالية:
في هذا العمل،تم حسابها عند.
توفر البيانات
البيانات المصدرية التي تستند إليها الأشكال مقدمة كملف بيانات مصدرية. يتم تقديم البيانات المصدرية مع هذه الورقة.
References
Li, A. et al. Atomically dispersed hexavalent iridium oxide from reduction for oxygen evolution catalysis. Science 384, 666-670 (2024).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
Wang, L. et al. Optimizing edge active sites via intrinsic in-plane iridium deficiency in layered iridium oxides for oxygen evolution electrocatalysis. Adv. Mater. 36, 2312608 (2024).
Qin, Y. et al. electronic structure and lattice strain dual engineering for enhanced acidic oxygen evolution reaction performance. Nat. Commun. 13, 3784 (2022).
Zhu, J. et al. Regulative electronic states around ruthenium/ruthenium disulphide heterointerfaces for efficient water splitting in acidic media. Angew. Chem. Int. Ed. 60, 12328-12334 (2021).
Song, H. et al. lattice matching strategy enables robust water oxidation electrocatalysis in acidic media via two distinct oxygen evolution mechanisms. ACS Catal. 14, 3298-3307 (2024).
Jin, H. et al. Dynamic rhenium dopant boosts ruthenium oxide for durable oxygen evolution. Nat. Commun. 14, 354 (2023).
Deng, L. et al. Valence oscillation of Ru active sites for efficient and robust acidic water oxidation. Adv. Mater. 35, 2305939 (2023).
Lee, K. et al. Modulating the valence electronic structure using earth-abundant aluminum for high-performance acidic oxygen evolution reaction. Chem. 9, 3600-3612 (2023).
Chen, D. et al. Heteroanion induced structural asymmetricity centered on Ru sites switches the rate-determining step of acid water oxidation. Energy Environ. Sci. 17, 1885-1893 (2024).
. et al. Accelerating adjacent dual sites construction by copper switch for efficient alkaline hydrogen evolution. Adv. Energy Mater. 13, 2302668 (2023).
Li, L. et al. Lanthanide-regulating Ru-O covalency optimizes acidic oxygen evolution electrocatalysis. Nat. Commun. 15, 4974 (2024).
Xu, Y. et al. Strain-modulated Ru-O covalency in Ru-Sn oxide enabling efficient and stable water oxidation in acidic solution. Angew. Chem. Int Ed. Engl. 63, e202316029 (2024).
Liang, X. et al. Electrocatalytic water oxidation activity-stability maps for perovskite oxides containing 3d, 4d and 5d transition metals. Angew. Chem. Int. Ed. 62, e202311606 (2023).
Qin, Y. et al. Orthorhombic : a superior electrocatalyst for acidic oxygen evolution reaction. Nano Energy 115, 108727 (2023).
Xiao, K., Wang, Y., Wu, P., Hou, L. & Liu, Z.-Q. Activating lattice oxygen in spinel through filling oxygen vacancies with fluorine for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 62, e202301408 (2023).
Lee, G. R. et al. Efficient and sustainable water electrolysis achieved by excess electron reservoir enabling charge replenishment to catalysts. Nat. Commun. 14, 5402 (2023).
Long, X . et al. nano-heterostructures stabilized by the sacrificing oxidation strategy of substrate for boosting acidic oxygen evolution reaction. Appl. Catal. B Environ. 343, 123559 (2024).
Du, K. et al. Interface engineering breaks both stability and activity limits of for sustainable water oxidation. Nat. Commun. 13, 5448 (2022).
Jia, H., Yao, N., Yu, C., Cong, H. & Luo, W. Unveiling the electrolyte cations dependent kinetics on CoOOH -catalyzed oxygen evolution reaction. Angew. Chem. Int. Ed. 62, e202313886 (2023).
Sun, Y. et al. Navigating surface reconstruction of spinel oxides for electrochemical water oxidation. Nat. Commun. 14, 2467 (2023).
Zheng, X. et al. Ru-Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212-222 (2021).
Hu, E. et al. Oxygen-redox reactions in cathode without bonding during charge-discharge. Joule 5, 720-736 (2021).
Zheng, X. et al. Enriched d-band holes enabling fast oxygen evolution kinetics on atomic-layered defect-rich lithium cobalt oxide nanosheets. Adv. Funct. Mater. 32, 2200663 (2022).
Zheng, X. et al. Electronic structure engineering of toward enhanced oxygen electrocatalysis. Adv. Energy Mater. 9, 1803482 (2019).
Zheng, X. et al. Multifunctional active-center-transferable platinum/ lithium cobalt oxide heterostructured electrocatalysts towards superior water splitting. Angew. Chem. Int. Ed. 59, 14533-14540 (2020).
Yan, G. et al. Ultrathin two-dimensional medium-entropy oxide as a highly efficient and stable electrocatalyst for oxygen evolution reaction. Nano Res 17, 2555-2562 (2024).
Zhu, W. et al. Stable and oxidative charged Ru enhance the acidic oxygen evolution reaction activity in two-dimensional rutheniumiridium oxide. Nat. Commun. 14, 5365 (2023).
Ping, X. et al. Locking the lattice oxygen in RuO(2) to stabilize highly active Ru sites in acidic water oxidation. Nat. Commun. 15, 2501 (2024).
Yao, N. et al. Atomically dispersed Ru oxide catalyst with lattice oxygen participation for efficient acidic water oxidation. Chem. 9, 1882-1896 (2023).
Deng, L. et al. Activity-stability balance: the role of electron supply effect of support in acidic oxygen evolution. Small 19, e2302238 (2023).
Zhang, X.-L. et al. Efficient acidic hydrogen evolution in proton exchange membrane electrolyzers over a sulfur-doped marcasitetype electrocatalyst. Sci. Adv. 9, eadh2885 (2023).
Rao, P. et al. Single atomic cobalt electrocatalyst for efficient oxygen reduction reaction. eScience 2, 399-404 (2022).
Dan, M. et al. Dual-axial engineering on atomically dispersed catalysts for ultrastable oxygen reduction in acidic and alkaline solutions. Proc. Natl Acad. Sci. USA 121, e2318174121 (2024).
Jian, L., Wang, G., Liu, X. & Ma, H. Unveiling an S-scheme heterojunction for robust water purification. eScience 4, 100206 (2024).
Wang, Z. et al. Optimizing the oxygen-catalytic performance of spinel by regulating the bond competition at octahedral sites. Adv. Funct. Mater. 33, 2214275 (2023).
Sun, K. et al. Manipulating the spin state of Co sites in metal-organic frameworks for boosting CO2 photoreduction. J. Am. Chem. Soc. 146, 3241-3249 (2024).
Yang, F. et al. Sub-3 nm Pt@Ru toward outstanding hydrogen oxidation reaction performance in alkaline media. J. Am. Chem. Soc. 145, 27500-27511 (2023).
Tian, X. et al. Synergy of dendrites-impeded atomic clusters dissociation and side reactions suppressed inert interface protection for ultrastable Zn anode. Adv. Mater. 36, 2400237 (2024).
Sun, Z. et al. Lattice strain and mott-schottky effect of the chargeasymmetry single-atom alloy catalyst for semi-hydrogenation of alkynes with high efficiency. ACS Nano 18, 13286-13297 (2024).
Yu, Z.-Y. et al. General synthesis of tube-like nanostructured perovskite oxides with tunable transition metal-oxygen covalency for efficient water electrooxidation in neutral media. J. Am. Chem. Soc. 144, 13163-13173 (2022).
Kuang, J., Deng, B., Jiang, Z., Wang, Y. & Jiang, Z.-J. Sr-stabilized solid solution nano-electrocatalysts with superior activity and excellent durability for oxygen evolution reaction in acid Media. Adv. Mater. 36, 2306934 (2024).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Wei, Y. et al. Triggered lattice-oxygen oxidation with active-site generation and self-termination of surface reconstruction during water oxidation. Proc. Natl Acad. Sci. USA 120, e2312224120 (2023).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659-23669 (2023).
Rong, C. et al. Defect-balanced active and stable for proton exchange membrane water electrolysis at ampere-level current density. Energy Environ. Sci. 17, 4196-4204 (2024).
Delmo, E. P. et al. In situ infrared spectroscopic evidence of enhanced electrochemical reduction and coupling on oxide-derived copper. J. Am. Chem. Soc. 146, 1935-1945 (2024).
Lin, Y. et al. In situ identification and time-resolved observation of the interfacial state and reactive intermediates on a cobalt oxide nanocatalyst for the oxygen evolution reaction. ACS Catal. 12, 5345-5355 (2022).
Zhang, T. et al. Spatial configuration of Fe-Co dual-sites boosting catalytic intermediates coupling toward oxygen evolution reaction. Proc. Natl Acad. Sci. USA 121, e2317247121 (2024).
Hao, Y. et al. Designing neighboring-site activation of single atom via tunnel ions for boosting acidic oxygen evolution. Nat. Commun. 15, 8015 (2024).
Hung, S.-F. et al. Unraveling geometrical site confinement in highly efficient iron-doped electrocatalysts toward oxygen evolution reaction. Adv. Energy Mater. 8, 1701686 (2018).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Hjorth Larsen, A. et al. The atomic simulation environment-a Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017).
شكر وتقدير
تم دعم هذا العمل من قبل المؤسسة الوطنية للعلوم الطبيعية في الصين (52371226، L.L.L و 52371226، S.J.P)، وصندوق الابتكار العلمي والتكنولوجي الخاص لذروة الكربون والحياد الكربوني في مقاطعة جيانغسو (BK20220039، S.J.P). كما تم دعم هذا العمل من قبل برنامج العلماء الشباب عبر الأجيال 2030 التابع للمجلس الوطني للعلوم والتكنولوجيا في تايوان بموجب المنحة NSTC 112-2628-E-007-014-MY4 للدكتور هان يي-تشين. يشكر المؤلفون مركز أبحاث الإشعاع السنكروتروني الوطني، هسينتشو، تايوان، على توفير خط الأشعة السنكروترونية XAS TLS 17C1 و TPS 44A1.
مساهمات المؤلفين
صمم الدراسة L.W. و S.Z. و L.L. و F.H. و S.P. أجرى التجارب L.W. و S.P. شارك في توصيف العينات L.W. و S.F.H. و J.J.M. و C.Z. و Y.Z. و T.Y.C. و H.Y.C. حلل البيانات L.W. و Y.W. و S.B. و S.L. و Y.W. و S.P. كتب L.W. الورقة. تصور S.P. الفكرة وراجع المخطوطة.
ملاحظة الناشر: تظل شركة سبرينغر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسبية-غير التجارية-بدون اشتقاقات 4.0 الدولية، التي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع، وإعادة إنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(ج) المؤلفون 2025
¹كلية علوم المواد والتكنولوجيا، جامعة نانجينغ للطيران والفضاء، نانجينغ 210016، الصين. ²قسم الكيمياء التطبيقية، جامعة يانغ مينغ تشياو تونغ الوطنية، هسينتشو، تايوان.المختبر الرئيسي للغرويات الاصطناعية والبيولوجية، وزارة التعليم، كلية الهندسة الكيميائية ومواد الهندسة، جامعة جيانغنان، ووكسي، جيانغسو 214122، الصين.المركز الوطني لأبحاث إشعاع السنكروترون، هسينتشو، تايوان.قسم علوم المواد والهندسة، جامعة تسينغ هوا الوطنية، هسينتشو، تايوان.مختبر كونفوشيوس لتخزين الطاقة، كلية الطاقة والبيئة ومركز تخزين الطاقة Z، جامعة جنوب شرق، نانجينغ 211189، الصين. البريد الإلكتروني:lilinlin@nuaa.edu.cn; fenghu@nuaa.edu.cn; pengshengjie@seu.edu.cn
Modulating the covalency of Ru-O bonds by dynamic reconstruction for efficient acidic oxygen evolution
Received: 25 August 2024
Accepted: 25 March 2025
Published online: 13 April 2025
Check for updates
Luqi Wang , Sung-Fu Hung , , Sheng Zhao , Yue Wang , Suwan Bi , Shaoxiong Li (D¹, Jian-Jie Ma², Chenchen Zhang³, Ying Zhang , Linlin Li , Tsung-Yi Chen , Han-Yi Chen , Feng Hu ®, Yuping Wu & Shengjie Peng
Developing ruthenium-based oxide catalysts capable of suppressing lattice oxygen participation in the catalytic reaction process is crucial for maintaining stable oxygen evolution reaction (OER) under acidic conditions. Herein, we delicately construct a nanoparticle-anchored nanosheet electrocatalyst ( ), achieving dynamic optimization of during the reaction process and improving catalytic stability. Benefiting from the unique electrochemical delithiation characteristics of the support, the covalency of the Ru-O bond is effectively regulated during the OER process. The weakened Ru-O covalent bond inhibits the participation of lattice oxygen in the catalytic reaction and ensures the continuous operation of the Ru active sites. Moreover, the extended Ru-O bond in the optimized catalyst reduces the formation energy barrier of the *OOH intermediates, accelerating the progress of the OER. As a result, the catalyst requires only an overpotential of at in and operates stably for 2000 h at in a proton exchange membrane water electrolysis. This work opens new avenues for designing efficient ruthenium-based catalysts.
Proton exchange membrane water electrolysis (PEMWE) with high current density and low resistance loss is regarded as a promising hydrogen production technology in the future . Currently, noble metal ruthenium (Ru) and iridium (Ir) oxide catalysts are extensively used in the anodes of PEMWE . Compared to offers high activity and cost-effectiveness, presenting significant potential for application in acidic oxygen evolution reactions (OER) . However, catalysts tend to follow the lattice oxygen mechanism (LOM) and generate numerous oxygen defects during the OER reaction, leading to crystal structure collapse . Additionally, the metal Ru sites can be over-oxidized into soluble species, which separate
from the crystal lattice under high oxidation potential, resulting in poor catalytic stability . From a crystal structure perspective, the instability of is closely related to the charge distribution of the bond . Therefore, regulating the electronic state of the Ru-O bond to suppress the involvement of lattice oxygen in the OER process is an effective strategy for enhancing the activity and stability of catalysts.
Generally, electron-rich Ru sites in activate lattice oxygen and generate defects, while electron-deficient states tend to oxidize to excessively high valence states and dissolve . Traditional electrondonating support strategies modulate the electron distribution of the
Ru-O bond, preventing the dissolution of the metal center and following a relatively stable adsorption oxygen mechanism (AEM) . At present, electron-donating supports with limited regulatory capacity cannot meet the demand for manipulating dynamically changing Ru-O bonds under complex high-potential OER conditions . Therefore, designing supports capable of dynamically regulating the electronic structure of in response to changes in the catalytic reaction process is highly attractive. To address this challenge, transition metal oxides with cation intercalation dynamically optimize the local microenvironment of metal-oxygen bonds during the catalytic reaction through the extraction and insertion of cations . Notably, lithium cobalt oxide ( ) with a unique layered structure exhibits an ordered cation arrangement and superior thermodynamic stability . The tight edge-shared octahedral structure in reduces the migration energy barrier of and ensures twodimensional diffusion of in the plane, facilitating dynamic reconstruction during the catalytic process . However, the multi-layered bulk structure of exhibits slow electron transport capability and limited surface area, which cannot meet the requirements for efficient catalyst support . In this regard, two-dimensional nanosheets markedly shorten the intercrystalline electron transmission path and enhance conductivity . Additionally, the twodimensional nanosheet structure forms a fast electron transmission channel perpendicular to the exposure plane, resulting in strong metal-support interaction . Therefore, using two-dimensional nanosheets as a support for is expected to achieve dynamic regulation of the electronic structure during catalytic reactions and exhibit high catalytic activity.
Herein, we propose an effective strategy to improve the charge distribution of through the unique dynamic evolution process of the support ( ). The nanosheet support with electron-donating ability induces electron transfer from Co to Ru sites, providing electron compensation to stabilize the valence state of . More importantly, the two-dimensional diffusion and extraction of Li ions within the interlayer of the nanosheet under OER potential cause the dynamic reconstruction and evolution of the catalyst interface. The unique dynamic self-optimization process moderately weakens the covalency of the Ru-O bond, suppressing the participation of lattice oxygen and achieving a good balance between catalytic activity and stability. The optimized Ru sites facilitate the formation of the *OOH intermediate, significantly lowering the catalytic energy barrier of the rate-determining step. Consequently, the electrocatalyst provides a current density of at an overpotential of and maintains stability for over 2300 h in . In addition, as anode can operate continuously for 2000 h at in a PEM electrolyzer. This work advances the application of ruthenium-based catalysts in PEMWE.
Results
Reaction mechanism and structural analysis
Due to the differences in the covalency of Ru-O bonds, Ru oxides follow either the lattice oxygen mechanism (LOM) or the adsorption oxygen mechanism (AEM) during the acidic OER process (Fig. 1a, b) . In the LOM pathway, activated lattice oxygen with stronger covalency participates in the OER process, resulting in the generation of oxygen defects . An excessive number of oxygen vacancies can induce detachment from the crystal lattice at the Ru metal sites, which greatly reduces the catalytic stability. However, Ru oxides with weak covalent Ru-O bonds follow the AEM mechanism, achieving a stable acidic OER process . Therefore, dynamically regulating the covalency of Ru-O bonds during the catalytic reaction is crucial for enhancing catalyst stability. The O3-type material with Li element intercalation exhibits superior conductivity and structural stability. More importantly, the extraction of interlayer lithium ions at the OER potential
triggers electron transfer, enabling real-time regulation of the covalency of metal-oxygen bonds (Fig. 1c). Additionally, we employ density functional theory (DFT) calculations to study the stability of different Co-based oxides in acidic environments. presents a thermodynamically unfavorable dissolution barrier, indicating that it can still maintain stability in an acidic environment (Fig. 1d). Inspired by these results, dynamically regulating the Ru-O bond covalency in using the layered support is expected to achieve high catalytic stability.
Materials characterization
A facile adsorption calcination strategy is employed to prepare nanosheets with loaded nanoparticles ( ). Briefly, nanosheets are obtained through ultrasonic-assisted exfoliation of solid-phase synthesized bulk (Supplementary Figs.1,2). Subsequently, ruthenium ions are adsorbed on the surface through electrostatic binding and then calcined to form the electrocatalyst. According to the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images, exhibits an ultrathin nanosheet structure (Fig. 2a and Supplementary Figs. 3-5). Interestingly, the nanoparticles are tightly confined in the lattice in the high-resolution TEM (HRTEM) image, which is different from simple physical adsorption (Fig. 2b). A clear lattice contact exists between nanoparticles and the support, in which lattice stripes with interplanar spacings of 0.31 and 0.20 nm correspond to the (110) and (104) planes, respectively (Fig. 2c, h) . The support provides lattice confinement for the nanoparticles to form a unique metalsupport interaction. Atomic force microscopy (AFM) shows that the thickness of the nanosheet is 5.1 nm in Fig. 2d. The ultrathin nanosheet structure of maximizes the exposure of more Ru active sites. Atomic-resolution aberration-corrected highangle annular dark-field scanning TEM (HAADF-STEM) is used to analyze the atomic structure of the composite. The Co atoms are arranged in an stacking mode in the HAADF-STEM image along the [010] zone axis, indicating the formation of O3-type and consistent with the XRD results (Fig. 2e and Supplementary Figs. 6, 7) . The spacing of about 0.47 nm corresponds to the typical interlayer spacing of . In addition, the bright Ru atomic array is embedded in the lattice of and exhibits superb lattice matching at the interface (Fig. 2f). The distribution of Ru, Co, and O elements in the energy dispersive spectroscopy (EDS) spectrum further confirms the uniformity of the heterostructure (Fig. 2g). The above analysis shows that the structurally ordered nanosheet support is closely connected to the nanoparticles, which provides the possibility for the optimization of the electronic structure.
To further understand the coordination structure and electronic states of , X-ray absorption spectroscopy (XAS) was performed. As shown in Fig. 2i, the oxidation state of is lower than that of in the Ru K-edge X-ray absorption near-edge structure (XANES) spectra, implying the formation of an electron transfer channel between and . This is also consistent with the results of the first derivative of the Ru K-edge XANES spectra and high-resolution X-ray photoelectron spectroscopy (XPS) (Supplementary Figs. 8, 9). The nanosheet support with charge compensation effectively regulates the oxidation state of Ru sites, preventing excessive oxidation during the catalytic process. According to the Fourier transformed Ru K-edge extended X-ray absorption fine structure (FT-EXAFS) spectra, the Ru-O bond length is slightly shortened after contact between and (from 1.93 to ), which is attributed to the formation of interfacial interactions (Fig. 2j and Supplementary Figs. 10, 11 and Supplementary Tables 1, 2) . The Co K-edge XANES of is transferred to higher energies compared to in Fig. 2k, which is consistent with the XPS valence band spectrum and
Fig. 1 | Schematic diagram of OER mechanism and structural analysis of lithium cobalt oxide. a LOM schematic of in acidic OER. b AEM schematic of RuO2 in acidic OER. c Structural models of and . d Dissolution energy barriers of cobalt-based oxides.
electron paramagnetic resonance (EPR) results. This verifies the electron-donating ability of the carrier (Supplementary Figs. 12, 13). Moreover, the main peaks of at 1.4 and correspond to and coordination shells in the Co K-edge FT-EXAFS spectra, respectively (Fig. 21) . It is worth noting that the bonds in are stretched more than , which is consistent with the wavelet transform spectral results (Supplementary Figs. 14-16). The electron transfer from Co to Ru are verified by collecting electron energy loss spectra (EELS) of and at the interface (Supplementary Fig. 17). Therefore, the interfacial interaction between and electronically compensates the Ru sites and improves the stability of the electrocatalyst.
OER electrocatalytic performance
Inspired by the structural advantages of the catalyst, the OER performance was evaluated via a three-electrode system in . In Fig. 3a, the electrocatalyst exhibits an overpotential of at , which is much smaller than commercial denoted Com-RuO2 and (Supplementary Figs. 18-24). In comparison, the highly acidic OER activity of also surpasses most previously reported noble metal-based electrocatalysts (Supplementary
Figs. 25 and Supplementary Table 3). The Tafel slope of shows improved reaction kinetics compared to and (Fig. 3b) . At the OER potential, exhibits fast intermediate conversion efficiency. Meanwhile, the phase angle of rapidly decreases at different potentials in the Bode plot, further confirming the rapid charge diffusion on the catalyst surface (Supplementary Figs. 26-28 and Supplementary Table 4). The mass activity of the catalyst is 41.81 times that of at an overpotential of 240 mV in Fig. 3c. In addition, the turnover frequency (TOF) and OER current normalized by the electrochemically active surface area (ECSA) of electrocatalysts are significantly higher than (Supplementary Figs. 29-32 and Supplementary Table 5). The catalyst durability is tested using chronopotentiometry to evaluate the potential for practical applications. The electrocatalyst can operate stably for 2300 h at a current density of , while almost loses its activity after 400 h (Fig. 3d and Supplementary Fig. 33). The above results show that the support in the electrocatalyst avoids the dissolution of in acidic OER to achieve sustained acidic OER catalytic activity.
To evaluate the industrial application potential of , a PEM electrolyzer was assembled with and
Fig. 2 | Characterizing the structure of . a TEM, (b, c) HRTEM, and (d) AFM images of (inset: height profile of ). e, f HAADFSTEM images of . The EDS elemental mapping of . h The atomic model of and . i Normalized Ru K-edge XANES spectra
and (j) Ru K-edge FT-EXAFS spectra of , and Ru foil, respectively. k Normalized Co K-edge XANES spectra and (I) Co K-edge FT-EXAFS spectra of , and Co foil, respectively.
Pt/C as anode and cathode catalysts, respectively. Specifically, only requires a cell voltage of 1.68 V to reach a current density of , which is significantly lower than Pt/C ( 1.84 V ) (Fig. 3e). Overpotential analysis shows that improved mass transport alleviates concentration polarization effects in the local reaction environment, indirectly enhancing catalytic kinetics (Supplementary Fig. 34). Furthermore, the mass activity of is approximately 21.33 times greater than that of at a cell voltage of 1.7 V (Supplementary Fig. 35). The mass activity and cost of are also much lower than those of commercial Pt/C (Supplementary Fig. 36). Strikingly, the PEM electrolyzer using can operate stably for 2000 h at a current density of with negligible decay in Fig. 3f, indicating the potential of the catalyst for practical applications. The long-term durability of in PEMWE also exceeds that of most recently reported various high-performance electrocatalysts (Supplementary Table 6). These findings demonstrate that the electrocatalyst exhibits outstanding performance as a choice for hydrogen production in the PEM electrolyzer, providing strong support for sustainable energy production.
Structural transformation after stability
The physical structure of the electrocatalyst after OER stability testing was investigated to explore the influencing factors for
the improved catalytic activity. SEM and TEM images show that nanoparticles remain tightly supported on the surface of nanosheets in the electrocatalyst after stability testing (Supplementary Figs. 37, 38). Besides, a reconstructed amorphous layer can be clearly observed on the surface of nanosheets in . This layer is attributed to the migration and extraction of interlayer Li-ions during voltage application, resulting in the formation of a surface amorphous layer. Compared with the original , the (003) crystal plane peak of after OER stability shifts to a lower angle, which may be attributed to the lattice distortion caused by the extraction of Li ions (Fig. 4a and Supplementary Fig. 39). Additionally, the Raman spectrum of detects two characteristic peaks of and modes at 484 and , corresponding to bending and stretching, respectively (Fig. 4b and Supplementary Fig. 40). Due to the enhanced polarity of the bond induced by Li extraction, the and characteristic peaks of exhibit a blue shift after OER stability (Supplementary Fig. 41) . In particular, the new peak at after stabilization is attributed to the Co -O bonds of the spinel structure of the surface reconstruction layer, which enhances the polarity of the bonds. The interlayer Li ions detach from through a two-dimensional diffusion path under the OER potential, performing dynamic structural reconstruction and altering the interface coordination environment of the catalyst.
Fig. 3 | Electrocatalytic performance of . a OER polarization curves and (b) Tafel plots of , and in electrolyte, respectively. The voltage is corrected by an automatic of iR compensation ( R is ). Mass activity of Ru atoms in and as a function of overpotential. d Chronopotentiometric curves of and at , respectively (Inset: Potential changes of before and after stabilization). e The polarization curves of PEMWE with and catalyst in pure water at without iRcorrection. Chronopotentiometric curve of PEMEW using catalyst at .
The electronic states of catalysts before and after OER stability are further analyzed to understand the influence of the reconstruction process. The ratio of after OER stabilization increases compared with the pristine , implying the reduction of electron density for Co sites (Fig. 4c and Supplementary Fig. 42) . The dynamic extraction of Li ions induces electron transfer, leading to partial oxidation of Co sites and increased covalency in the bond. Additionally, the proportion of in slightly increases from to after the stability measurement . As a comparison, the proportion of in after the stability measurement is , which is much higher than that of (Supplementary Figs. 43, 44). Despite the slight oxidation of the support, it can maintain the oxidation state of by continuously donating electrons, thereby preventing the dissolution of Ru sites (Supplementary Figs. 45, 46). A structural model of after partial delithiation is constructed to reveal the changes in electronic structure by DFT calculations (Supplementary Fig. 47). The Bader charge indicates that Ru in obtains about 0.169 e of electrons from the support (Fig. 4d) . Due to the extraction of Li ions, the charge density at the Ru sites in is lower than that of ( 6.626 e ), while remaining higher than that of (Fig. 4e). Crystal orbital Hamiltonian population (COHP) analysis shows that the Ru-O bond covalency of is lower than that of (Fig. 4 f. The unique delithiation process ensures dynamic regulation of the covalency of the Ru-O bond during the catalytic process to suppress the participation of lattice oxygen (Fig. 4g).
In situ characterization of catalyst structural changes
In-situ XAS was implemented to explore the dynamic evolution mechanism of electrocatalysts in the OER process (Supplementary Fig. 48). In the fitted Ru K-edge FT-EXAFS spectra, the Ru-O bond of is appropriately extended as the potential increases from
open circuit voltage (OCV) to 1.7 V (from 1.90 to ), indicating that the covalency of Ru-O is weakened (Fig. 5a, b and Supplementary Fig. 49 and Supplementary Table 7) . In contrast, the bond tends to be more covalent during the delithiation process, resulting in a negative shift of the Co-O coordination shell in and the formation of a surface reconstruction layer (from 1.92 to ) (Supplementary Figs. 50, 51 and Supplementary Table 8) . The wavelet transform of Ru K-edge and Co K-edge EXAFS spectra also intuitively confirm this result (Supplementary Figs. 52-54). Thus, the dynamic extraction of lithium ions regulates the covalency of the Ru-O bond through interfacial interactions, which can restrict the participation of lattice oxygen and maintain the structural stability of (Fig. 5c). Notably, the Ru-O bond in undergoes a significant shortening from to as the voltage transitions from OCV to 1.7 V (Fig. 5d, e and Supplementary Figs. 55-57 and Supplementary Table 9). The excessive enhancement of the covalency of the Ru-O bond provides conditions for triggering lattice oxygen to participate in the OER reaction . Moreover, the slight change in the Ru-O coordination number in indicates the stability of the coordination structure of . As a comparison, a reduced coordination number can be observed for the bonds in , which is attributed to the presence of defective oxygen (Fig. 5f). Due to the strong covalency of the Ru-O bond, follows the LOM path and generates a large number of oxygen defects during the OER process, leading to the collapse of the catalyst surface structure.
To elucidate the electronic structure changes of the electrocatalyst at high potentials, the oxidation states of and are detected by in situ XANES spectra. The average valence state of Ru species in rises gently from +3.82 to +4.21 as the potential transitions from OCV to 1.7 V vs. RHE in Fig. 5g (Supplementary Fig. 58). This further confirms that the strong electronic interaction between the and interface inhibits the excessive oxidation of . In addition, the Co species in the catalyst can still maintain an oxidation state of +3.16 at a high
Fig. 4 | Reconstruction of catalyst after stabilization. a XRD patterns and (b) Raman spectra of and after stability, respectively. The content of , and in before and after OER stability. d Bader charge analysis of , and , respectively. e Comparison of charge amounts of , , and , respectively. of Ru-O in and , respectively. g Schematic diagram of the dynamic evolution of .
voltage of 1.7 V (Supplementary Figs.59,60). As a result, the extraction of lithium ions can moderately regulate the electron distribution of support without destroying the main structure. Remarkably, the valence state of Ru rapidly increases from +4.02 to +5.57 in switching the voltage from OCV to 1.7 V vs. RHE, which is attributed to the excessive oxidation originating from the damage of the structure (Fig. 5h, Supplementary Figs. 61, 62). Based on the above results, the dynamic reconstruction process of can effectively modify the charge environment of the bond and stabilize the lattice oxygen, resulting in improved OER performance.
Origin of high catalytic performance
To investigate the transfer phenomena of reaction intermediates on the electrocatalyst surface, a series of electrochemical measurements are performed. The cyclic voltammetry curves of and can observe two pairs of redox peaks around 0.5 V and 1.0 V respectively, which correspond to the formation of *OH reaction intermediates with deprotonation (Fig. 6a). Obviously, the redox peak of moves downwards than that of , indicating favorable deprotonation and promoting the reaction process . Subsequently, methanol is used as a probe molecule to detect the coverage of *OH intermediates on the catalyst surface (Supplementary Fig. 63). The
methanol oxidation reaction (MOR) is more active on -dominated catalyst surfaces due to the mechanism of nucleophilic attack on electrophilic sites . exhibits high surface coverage compared to in Fig. 6b, implying that contains more effective OER active sites. Furthermore, the change in catalytic activity of the catalyst at different pH values is negligible, which is a characteristic of the typical AEM mechanism (Fig. 6c and Supplementary Fig. 64) . In contrast, exhibits a pH -dependent behavior of OER activity, demonstrating the occurrence of an unstable LOM mechanism. The rotating ring-disk electrode test and the adsorption/desorption energy barrier analysis of Li-containing intermediates indicate that it is difficult for the extracted Li ions to participate in the OER process of (Supplementary Figs. 65-68). Furthermore, maintains the same potential in electrolytes with different Li-ion concentrations after 50 h of stability testing, further confirming that the small amount of extracted Li has a negligible effect on the performance of the catalyst (Supplementary Fig. 69).
The mechanism of the acidic OER reaction of and is further monitored by Operando attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) (Supplementary Fig. 70) . A strong absorption band can be observed in at about , which is identified as
Fig. 5 | In-suit XAS characterization of confirming the dynamic process. a In situ Ru K-edge EXAFS spectra of with applied potentials from OCV to 1.7 V .b Changes in bond length and intensity of the Ru-O shell of / at different potentials. c Schematic diagram of the structural evolution of at different potentials. d In situ Ru K-edge EXAFS spectra of with
applied potentials from OCV to 1.7 V . e Changes in bond length and intensity of the Ru-O shell of at different potentials. Schematic diagram of the structural evolution of at different potentials. In situ Ru K-edge XANES spectra of . Inset: Oxidation state changes of Ru in . h In situ Ru K-edge XANES spectra of . Inset: Oxidation state changes of Ru in .
stretching in the *OOH intermediate in Fig. 6d . Since *OOH is a typical intermediate of the AEM mechanism, the OER process of is mainly dominated by AEM. In contrast, the characteristic peaks at 1033 and in correspond to and intermediates, respectively, indicating a combined path of LOM and AEM (Fig. 6e) . Thus, the dynamic reconstruction effect realizes the complete transformation of electrocatalysis from LOM to AEM mechanism, thereby preventing the dissolution of the catalyst and enhancing OER stability (Fig. 6f). The Gibbs free energy barriers of OER reaction intermediates on the electrocatalyst are calculated to explore the activity differences (Supplementary Figs. 71-73). According to Fig. 6g, the OER rate-determining step (RDS) of , , and is the formation energy barrier of the intermediate * OOH . The electrocatalyst only requires an energy barrier of 1.77 eV to overcome RDS, which is much lower than that of . The formation barrier of the * OO intermediate of is much higher than that of * OOH , which is more conducive to the AEM mechanism (Supplementary Figs. 74-81). In addition, the overlap between Ru d and O p orbitals is reduced, further confirming the weakened covalency of the Ru-O bond. This hinders
lattice oxygen from participating in the OER reaction process, thereby enhancing structural stability (Supplementary Figs. 82, 83). As a result, the modification of support optimizes the binding energy of with key intermediates, resulting in a favorable thermodynamic process and enhanced intrinsic activity.
Discussion
In summary, we have successfully manipulated the bonding environment of during the catalytic reaction by utilizing the dynamic reconstruction of the support to achieve a balance between activity and stability in acidic OER. Dynamic electrochemical delithiation regulates the electron distribution and coordination structure at the interface, promoting the self-optimization of the catalyst during the OER process. The weakened covalency of the Ru-O bond triggers a complete transition of the electrocatalyst from the LOM to the AEM reaction pathway, improving the catalytic stability. As a result, the electrocatalyst reaches a current density of at a low overpotential of . In particular, the PEM electrolyzer using operates stably for 2000 h at a high current density of . This work provides a
Fig. 6 | Reaction mechanism on . a Cyclic voltammetry curves of and catalysts in . b The function plot between MOR current density and methanol concentration on and catalysts. c The pH dependence of the OER potential at various current densities for and . Operando ATR-SEIRAS spectra of and e at various applied potentials. f Schematic diagrams of the OER mechanism on and , respectively. OER Gibbs free energy diagrams of , and , respectively.
method to solve the balance between the activity and stability of ruthenium-based oxide electrocatalysts in acidic OER.
Methods
Experimental section Materials preparation
Preparation of Nanosheets. The stoichiometric amount of (solid, ACS reagent, ) and excess (solid, ACS reagent, ) was mixed by ball milling for 6 h at . Subsequently, the precursors were calcined for 4 h at in air with a heating rate of to obtain bulk powder. The bulk powders were dispersed in distilled water and sonicated for 4 h in an icewater bath, maintaining the temperature at . The supernatant was collected by centrifugation at . Finally, nanosheet powders were obtained by freeze-drying the supernatant at .
Preparation of . The 50 mg of and 10 mg of were dissolved in 20 mL of deionized water and magnetically stirred at room temperature for 12 h . Then, the precursor powder was collected by centrifugation at . Finally, the obtained powder was calcined in air at for 3 h with a heating rate of to obtain . For comparison, the amount of was changed to 5 mg and 15 mg to obtain a electrocatalyst with different loadings.
Characterization. The morphology of the catalysts was characterized by a scanning electron microscope (SEMJEOLJSM-6700F) and transmission electron microscope (TEM FEI Tecnai G2 F20). Aberration-corrected high-angle annular dark-field scanning transmission electron microscope (AC HAADF-STEM) images were taken at JEM-ARM200F equipped with a JED-2300T SDD. X-ray diffraction (XRD) data obtained from Bruker D8 Advance equipment was used to analyze the crystal structure. The elemental compositions were analyzed by ICP (ICP-MS, Inductively coupled plasma-mass spectrometry). X-ray photoelectron spectroscopy (XPS) analysis was performed on an Escalab 250 Xi system using Al Kα X-rays. The Co K-edge and Ru K-edge X-ray absorption spectroscopy (XAS) was measured at the beamline of TLS 17C1 and TPS 44A1 at the National Synchrotron Radiation Research Center (NSRRC) in Taiwan.
Electrochemical measurements. The electrochemical performance was tested on an electrochemical workstation using a three-electrode system (Autolab PGSTAT302, Metrohm). Graphite rod and saturated Hg/ electrode were used as counter electrode (CE) and reference electrode (RE), respectively. The catalyst, carbon black, and polyvinylidene fluoride (PVDF) were mixed in a weight ratio of , and N -methyl-2-pyrrolidone (NMP) was used as the solvent. The viscous slurry was evenly coated on carbon paper and dried under a vacuum. The catalyst loading is and the loading area is . The measured potential was converted into a reversible hydrogen electrode according to the equation .
Subsequently, CV was measured at a scan rate of . The linear sweep voltammetry (LSV) was performed in -saturated solution ( pH is ) at a scan rate of . The electrolyte was prepared and used immediately and stored in a glass bottle at room temperature. The potential was corrected according to the formula. The electrochemical impedance spectroscopy (EIS) was performed in the frequency range of with an amplitude of 5 mV . The double-layer capacitance was obtained by collecting CV curves with scan rates of 10 to . OCP tests were performed with saturated with high-purity hydrogen to confirm the reference electrode potential. Calibration experiments were performed at room temperature ( ) to reduce temperature effects.
Rotating ring disk electrode (RRDE) test of catalyst. The catalyst was dispersed in a mixed solution of isopropyl alcohol and Nafion ( ), followed by ultrasonication for 1 h to obtain a uniformly dispersed catalyst ink. of catalyst ink was dropped onto a disk electrode (area ) and dried under vacuum at room temperature to obtain a working electrode. A carbon rod and were used as the counter electrode and reference electrode, respectively. The linear sweep voltammetry was carried out in solution with different Li-ion concentrations at a scan rate of and a rotation speed of 1600 rpm .
The electrochemical surface area (ECSA) of an electrocatalyst can be evaluated by electrochemical double capacitance ( ) according to the following equation:
where was determined by taking half the slope of the current differences ( that were plotted as a function of the scan rate in a CV experiment. is the general surface specific capacitance ( ).
The turnover frequency (TOF) of the electrocatalyst was calculated by the following equation:
The formate turnover per geometric area was obtained from the geometric current density for the LSV polarization curves according to the equation:
Electrochemical in situ ATR-SEIRAS experiments. ATR-SEIRAS measurements were conducted using a Nicolet iS50 FT-IR spectrometer, with each spectrum obtained by accumulating 32 interferograms, achieving a spectral resolution of . The working electrode preparation involved two key steps. First, an ultra-thin Au film was chemically deposited onto a silicon crystal to enhance infrared signal sensitivity and electron conductivity. Then, a catalyst slurry with a loading of was applied onto the Au surface. This slurry was prepared by dispersing 7 mg of catalyst and 3 mg of carbon black in 1 mL ethanol, followed by the addition of of Nafion after 30 min of sonication. The assembled working electrodes were placed in a three-electrode electrochemical cell, where served as the reference electrode, a graphite rod as the counter electrode, and Ar-saturated as the electrolyte for the OER reaction . All measurements were performed using linear sweep voltammetry (LSV) to investigate OER reaction intermediates at various applied potentials.
In situ XAFS measurements. In-situ X-ray absorption spectroscopy (XAS), including both XANES and EXAFS at the Ru and Co K-edges, was
conducted in total fluorescence yield mode under ambient conditions at BL-12B2 of SPring-8, NSRRC. The measurements were carried out using a custom-designed Teflon container equipped with a Kepton tape-sealed window, allowing X-rays to pass through both the tape and electrolyte. This setup ensured that XAS signals were effectively captured in total fluorescence yield mode at the National Synchrotron Radiation Research Center (NSRRC), SPring-8. The experiments were performed under a three-electrode configuration, consistent with the electrochemical characterization conditions . For data processing, spectral normalization was achieved by removing the pre-edge baseline and adjusting the post-edge region. The -weighted EXAFS oscillations underwent Fourier transformation to facilitate EXAFS analysis, with all EXAFS spectra presented without phase correction. The Fourier-transformed (FT) data fitting was conducted using Artemis (version 0.9.25), employing a weighting factor with a k -range of and an R-range of . The coordination number, bond length, Debye-Waller factor, and energy shift ( , and ) were determined through fitting without any fixed parameters, while the amplitude reduction factor ( ) was set to 0.85 .
PEMWE measurements. was used as anode catalysts in PEM electrolyzers. Commercial Pt/C was used as a cathode catalyst. The membrane electrode assembly was fabricated via the catalystcoated membrane technique, covering a geometric area of . The catalyst powder was dispersed in isopropanol, deionized water, and Nafion solution to prepare the ink. The uniformly dispersed ink was obtained by slice emulsification and ultrasonic cell disruption. The well-dispersed and catalyst inks were sprayed on both sides of the PEM, respectively. The loadings of anode and Pt/C cathode are and , respectively. Nafion 115 was used as a proton exchange membrane (PEM) and was treated with and at for 1 h in sequence. The size of the proton exchange membrane was and the membrane thickness was . The sprayed membrane, anode gas diffusion layer (Ti felt), and cathode gas diffusion layer (carbon paper) were hotpressed at and 10 MPa pressure to obtain a membrane electrode assembly. Subsequently, a PEM water electrolyzer was assembled and tested at using pure water as the electrolyte. The polarization curve of PEMWE was obtained at a scan rate of , and a chronopotentiometric test was performed at to evaluate the stability.
DFT calculations. All density functional theory (DFT) calculations were conducted using the Vienna Ab initio Simulation Package (VASP) . The projector augmented wave (PAW) pseudopotential was applied in combination with the PBE generalized gradient approximation (GGA) exchange-correlation functional . To appropriately describe the localized d-electrons of Co, the DFT + U method was employed, incorporating a Hubbard-U correction of Ueff(Co) , determined via linear response theory. The calculations were performed with a plane wave basis set energy cutoff of 500 eV , and a Monkhorst-Pack k-point grid of was used for Brillouin zone sampling. Spin polarization was considered, and full structural relaxation was conducted until the energy convergence criterion reached per atom, with the final force acting on each atom kept below . Additionally, Pourbaix diagrams were generated using the Atomic Simulation Environment (ASE), where input formation energies were derived from DFT calculations of bulk and surface models .
The adsorption energy of reaction intermediates can be computed using the following Equation:
Where ads , an is the binding energy, is the zero-point energy change, is the entropy change. In this work,
the values of and were obtained by vibration frequency calculation.
The Gibbs free energy of the reaction steps can be calculated by the following four Equations:
In this work, were calculated at .
Data availability
The source data underlying Figures are provided as a Source Data file. Source data are provided with this paper.
References
Li, A. et al. Atomically dispersed hexavalent iridium oxide from reduction for oxygen evolution catalysis. Science 384, 666-670 (2024).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100-108 (2023).
Wang, L. et al. Optimizing edge active sites via intrinsic in-plane iridium deficiency in layered iridium oxides for oxygen evolution electrocatalysis. Adv. Mater. 36, 2312608 (2024).
Qin, Y. et al. electronic structure and lattice strain dual engineering for enhanced acidic oxygen evolution reaction performance. Nat. Commun. 13, 3784 (2022).
Zhu, J. et al. Regulative electronic states around ruthenium/ruthenium disulphide heterointerfaces for efficient water splitting in acidic media. Angew. Chem. Int. Ed. 60, 12328-12334 (2021).
Song, H. et al. lattice matching strategy enables robust water oxidation electrocatalysis in acidic media via two distinct oxygen evolution mechanisms. ACS Catal. 14, 3298-3307 (2024).
Jin, H. et al. Dynamic rhenium dopant boosts ruthenium oxide for durable oxygen evolution. Nat. Commun. 14, 354 (2023).
Deng, L. et al. Valence oscillation of Ru active sites for efficient and robust acidic water oxidation. Adv. Mater. 35, 2305939 (2023).
Lee, K. et al. Modulating the valence electronic structure using earth-abundant aluminum for high-performance acidic oxygen evolution reaction. Chem. 9, 3600-3612 (2023).
Chen, D. et al. Heteroanion induced structural asymmetricity centered on Ru sites switches the rate-determining step of acid water oxidation. Energy Environ. Sci. 17, 1885-1893 (2024).
. et al. Accelerating adjacent dual sites construction by copper switch for efficient alkaline hydrogen evolution. Adv. Energy Mater. 13, 2302668 (2023).
Li, L. et al. Lanthanide-regulating Ru-O covalency optimizes acidic oxygen evolution electrocatalysis. Nat. Commun. 15, 4974 (2024).
Xu, Y. et al. Strain-modulated Ru-O covalency in Ru-Sn oxide enabling efficient and stable water oxidation in acidic solution. Angew. Chem. Int Ed. Engl. 63, e202316029 (2024).
Liang, X. et al. Electrocatalytic water oxidation activity-stability maps for perovskite oxides containing 3d, 4d and 5d transition metals. Angew. Chem. Int. Ed. 62, e202311606 (2023).
Qin, Y. et al. Orthorhombic : a superior electrocatalyst for acidic oxygen evolution reaction. Nano Energy 115, 108727 (2023).
Xiao, K., Wang, Y., Wu, P., Hou, L. & Liu, Z.-Q. Activating lattice oxygen in spinel through filling oxygen vacancies with fluorine for electrocatalytic oxygen evolution. Angew. Chem. Int. Ed. 62, e202301408 (2023).
Lee, G. R. et al. Efficient and sustainable water electrolysis achieved by excess electron reservoir enabling charge replenishment to catalysts. Nat. Commun. 14, 5402 (2023).
Long, X . et al. nano-heterostructures stabilized by the sacrificing oxidation strategy of substrate for boosting acidic oxygen evolution reaction. Appl. Catal. B Environ. 343, 123559 (2024).
Du, K. et al. Interface engineering breaks both stability and activity limits of for sustainable water oxidation. Nat. Commun. 13, 5448 (2022).
Jia, H., Yao, N., Yu, C., Cong, H. & Luo, W. Unveiling the electrolyte cations dependent kinetics on CoOOH -catalyzed oxygen evolution reaction. Angew. Chem. Int. Ed. 62, e202313886 (2023).
Sun, Y. et al. Navigating surface reconstruction of spinel oxides for electrochemical water oxidation. Nat. Commun. 14, 2467 (2023).
Zheng, X. et al. Ru-Co pair sites catalyst boosts the energetics for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202205946 (2022).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212-222 (2021).
Hu, E. et al. Oxygen-redox reactions in cathode without bonding during charge-discharge. Joule 5, 720-736 (2021).
Zheng, X. et al. Enriched d-band holes enabling fast oxygen evolution kinetics on atomic-layered defect-rich lithium cobalt oxide nanosheets. Adv. Funct. Mater. 32, 2200663 (2022).
Zheng, X. et al. Electronic structure engineering of toward enhanced oxygen electrocatalysis. Adv. Energy Mater. 9, 1803482 (2019).
Zheng, X. et al. Multifunctional active-center-transferable platinum/ lithium cobalt oxide heterostructured electrocatalysts towards superior water splitting. Angew. Chem. Int. Ed. 59, 14533-14540 (2020).
Yan, G. et al. Ultrathin two-dimensional medium-entropy oxide as a highly efficient and stable electrocatalyst for oxygen evolution reaction. Nano Res 17, 2555-2562 (2024).
Zhu, W. et al. Stable and oxidative charged Ru enhance the acidic oxygen evolution reaction activity in two-dimensional rutheniumiridium oxide. Nat. Commun. 14, 5365 (2023).
Ping, X. et al. Locking the lattice oxygen in RuO(2) to stabilize highly active Ru sites in acidic water oxidation. Nat. Commun. 15, 2501 (2024).
Yao, N. et al. Atomically dispersed Ru oxide catalyst with lattice oxygen participation for efficient acidic water oxidation. Chem. 9, 1882-1896 (2023).
Deng, L. et al. Activity-stability balance: the role of electron supply effect of support in acidic oxygen evolution. Small 19, e2302238 (2023).
Zhang, X.-L. et al. Efficient acidic hydrogen evolution in proton exchange membrane electrolyzers over a sulfur-doped marcasitetype electrocatalyst. Sci. Adv. 9, eadh2885 (2023).
Rao, P. et al. Single atomic cobalt electrocatalyst for efficient oxygen reduction reaction. eScience 2, 399-404 (2022).
Dan, M. et al. Dual-axial engineering on atomically dispersed catalysts for ultrastable oxygen reduction in acidic and alkaline solutions. Proc. Natl Acad. Sci. USA 121, e2318174121 (2024).
Jian, L., Wang, G., Liu, X. & Ma, H. Unveiling an S-scheme heterojunction for robust water purification. eScience 4, 100206 (2024).
Wang, Z. et al. Optimizing the oxygen-catalytic performance of spinel by regulating the bond competition at octahedral sites. Adv. Funct. Mater. 33, 2214275 (2023).
Sun, K. et al. Manipulating the spin state of Co sites in metal-organic frameworks for boosting CO2 photoreduction. J. Am. Chem. Soc. 146, 3241-3249 (2024).
Yang, F. et al. Sub-3 nm Pt@Ru toward outstanding hydrogen oxidation reaction performance in alkaline media. J. Am. Chem. Soc. 145, 27500-27511 (2023).
Tian, X. et al. Synergy of dendrites-impeded atomic clusters dissociation and side reactions suppressed inert interface protection for ultrastable Zn anode. Adv. Mater. 36, 2400237 (2024).
Sun, Z. et al. Lattice strain and mott-schottky effect of the chargeasymmetry single-atom alloy catalyst for semi-hydrogenation of alkynes with high efficiency. ACS Nano 18, 13286-13297 (2024).
Yu, Z.-Y. et al. General synthesis of tube-like nanostructured perovskite oxides with tunable transition metal-oxygen covalency for efficient water electrooxidation in neutral media. J. Am. Chem. Soc. 144, 13163-13173 (2022).
Kuang, J., Deng, B., Jiang, Z., Wang, Y. & Jiang, Z.-J. Sr-stabilized solid solution nano-electrocatalysts with superior activity and excellent durability for oxygen evolution reaction in acid Media. Adv. Mater. 36, 2306934 (2024).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Wei, Y. et al. Triggered lattice-oxygen oxidation with active-site generation and self-termination of surface reconstruction during water oxidation. Proc. Natl Acad. Sci. USA 120, e2312224120 (2023).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659-23669 (2023).
Rong, C. et al. Defect-balanced active and stable for proton exchange membrane water electrolysis at ampere-level current density. Energy Environ. Sci. 17, 4196-4204 (2024).
Delmo, E. P. et al. In situ infrared spectroscopic evidence of enhanced electrochemical reduction and coupling on oxide-derived copper. J. Am. Chem. Soc. 146, 1935-1945 (2024).
Lin, Y. et al. In situ identification and time-resolved observation of the interfacial state and reactive intermediates on a cobalt oxide nanocatalyst for the oxygen evolution reaction. ACS Catal. 12, 5345-5355 (2022).
Zhang, T. et al. Spatial configuration of Fe-Co dual-sites boosting catalytic intermediates coupling toward oxygen evolution reaction. Proc. Natl Acad. Sci. USA 121, e2317247121 (2024).
Hao, Y. et al. Designing neighboring-site activation of single atom via tunnel ions for boosting acidic oxygen evolution. Nat. Commun. 15, 8015 (2024).
Hung, S.-F. et al. Unraveling geometrical site confinement in highly efficient iron-doped electrocatalysts toward oxygen evolution reaction. Adv. Energy Mater. 8, 1701686 (2018).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169-11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953-17979 (1994).
Hjorth Larsen, A. et al. The atomic simulation environment-a Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52371226, L.L.L and 52371226, S.J.P), and the Scientific and Technological Innovation Special Fund for Carbon Peak and Carbon Neutrality of Jiangsu Province (BK20220039, S.J.P). This work was also supported by the 2030 Cross-Generation Young Scholars Program of the National Science and Technology Council in Taiwan under the grant NSTC 112-2628-E-007-014-MY4 to Dr. Han Yi-Chen. The authors thank the National Synchrotron Radiation Research Center, Hsinchu, Taiwan, for providing synchrotron XAS beamline TLS 17C1 and TPS 44A1.
Author contributions
L.W., S.Z., L.L., F.H. and S.P. designed the study. L.W. and S.P. conducted the experiments. L.W., S.F.H., J.J.M., C.Z., Y.Z., T.Y.C. and H.Y.C. participated in the characterization of the samples. L.W., Y.W., S.B., S.L., Y.W. and S.P. analyzed data. L.W. wrote the paper. S. P. conceived the idea and revised the manuscript.
Correspondence and requests for materials should be addressed to Linlin Li, Feng Hu or Shengjie Peng.
Peer review information Nature Communications thanks Sung Jong Yoo, Ronghui Qi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2025
¹College of Materials Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing 210016, China. ²Department of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu, Taiwan. Key Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi, Jiangsu 214122, China. National Synchrotron Radiation Research Center, Hsinchu, Taiwan. Department of Materials Science and Engineering, National Tsing Hua University, Hsinchu, Taiwan. Confucius Energy Storage Lab, School of Energy and Environment & Z Energy Storage Center, Southeast University, Nanjing 211189, China. e-mail: lilinlin@nuaa.edu.cn; fenghu@nuaa.edu.cn; pengshengjie@seu.edu.cn