تعديل مجالات التنسيق في الموقع لمحفز الكوبالت ذي الذرة الواحدة من أجل تحسين تفاعل اختزال الأكسجين In situ modulating coordination fields of single-atom cobalt catalyst for enhanced oxygen reduction reaction
محفزات ذرات مفردة، خاصة تلك التي تحتوي على المعادن-المجموعات، تحمل وعدًا كبيرًا في تسهيل تفاعل اختزال الأكسجين. ومع ذلك، فإن التوزيع المتماثل للإلكترونات داخل المعدن-تؤدي النصفية إلى قوة امتصاص غير مرضية للوسائط، مما يحد من تحسين أدائها. هنا، نقدم محفزات ذرات مفردة من الكوبالت منظمة بتنسيق ذري تتضمن كسر التناظرجزء، الذي يعمل على كسر توزيع الإلكترونات المتناظر. تكشف التوصيفات في الموقع عن التطور الديناميكي لكسر التناظر في الكلور.جزء إلى تنسيق مخفضالهيكل، مما يؤدي إلى تحسين فعال لـملء الإلكترونات لمواقع الكوبالت نحو حالة مختزلة-شغل الإلكترونات في النطاق ) تحت ظروف التفاعل لعملية اختزال الأكسجين السريعة بأربعة إلكترونات. ونتيجة لذلك، توفر المحفزات أحادية الذرة من الكوبالت المنظمة بالتنسيق جهد نصف كبير يبلغ 0.93 فولت ونشاط كتلي قدره من المهم أن بطارية الزنك-هواء التي تستخدم المحفزات أحادية الذرة من الكوبالت المنظمة بالتنسيق كالكاثود تظهر أيضًا كثافة طاقة كبيرة وثباتًا ممتازًا.
تعتبر تفاعل اختزال الأكسجين الكهروكيميائي (ORR) مكونًا محوريًا في تقنيات الطاقة الناشئة مثل خلايا الوقود وبطاريات المعدن-الهواء، مما يقدم طريقًا واعدًا وصديقًا للبيئة لتحويل الطاقة المستدامة وتخزينها الحديثة.لسوء الحظ، فإن عملية ORR هي عملية متعددة الخطوات تتضمن اقتران متعدد الإلكترونات والبروتونات وتعاني من حركية بطيئة وآليات معقدة.على الرغم من أن المواد المعتمدة على البلاتين قد أظهرت تاريخياً كفاءة تحفيزية عالية في تسهيل تفاعل اختزال الأكسجين، إلا أن التكلفة العالية، والندرة، وعدم الاستقرار الجيد للبلاتين قد أعاقت تطبيقها التجاري الواسع.. وبالتالي، يجب أن تمتلك المواد التي تسعى لتكون محفزات كهربائية فعالة لتفاعل اختزال الأكسجين (ORR) تفاعلية كافية لقطع الـرابطة، بناء هندسي مُحسّن لتسهيل الكفاءةالانتشار ونقل الكتلة، ومواقع تفاعلية قوية لضمان المتانة على المدى الطويلبين هذه، المحفزات ذات الذرة الواحدة (SACs)، وخاصة تلك التي تحتوي على مواقع معدنية مفردة معزولة مثبتة في مصفوفة كربونية مضافة بالنيتروجين -C)، وقد حظيت باهتمام كبير في ORR بفضل خصائصها الإلكترونية القابلة للتعديل، والتكوينات الهندسية المميزة، وكفاءة الاستخدام الذري القصوى، وانخفاض. وبالتالي، فإن السعي لتطوير نموذج يحتذى بهتحفيزات كهربائية قائمة على ORR ذات انتقائية عالية، وتفاعل كافٍ، وقابلية تطبيق صناعي، تحظى باهتمام متزايد، لكنها لا تزال تمثل تحديًا كبيرًا. المواد، وخاصة تلك التي تتميز بتكوينات مستوية متناظرة تحتوي على أربعة مواقع معدنية منسقة بالنيتروجينالمجموعات)، تم التحقيق فيها بشكل مكثف وقد أظهرت أداءً تحفيزيًا مفضلًا. ومع ذلك، فإن نشاطهم في ORR وأربعة إلكترونات (تظل الانتقائية لا تفي بالمطلوب عند مقارنتها بـ
معيار Pt/C. يُعزى هذا النقص إلى القدرة غير المواتية على الامتصاص للوسائط المتعلقة بالأكسجين، الناتجة عن التوزيع المتماثل للإلكترونات داخل المستوىأجزاءمن الملحوظ أن النشاط والانتقائية لـ SACs مرتبطان ارتباطًا وثيقًا بإشغال الإلكترونات لـالمدارات، حيث تشارك فيالارتباط مع الأنواع المرتبطة بالأكسجين. للأسف، بالنسبة لـالمعادن الانتقالية، كلما-إلكترونات النطاق في الشكل المتماثليمكن أن تؤدي الأجزاء إلى تقليل تداخل المدارات معالإلكترونات، مما يتطلب طاقة أعلى لتكوين وتطور أنواع *OOHلمعالجة هذا التحدي، يُعتبر تعديل بيئة التنسيق لمراكز المعادن باستخدام عناصر ذات أشعة ذرية مختلفة والكهرسلبية استراتيجية واعدة لكسر التناظر المسطح لكثافة الإلكترون.علاوة على ذلك، تم التنبؤ بإعادة ترتيب السطح المعتمد على الجهد للكواشف الكهربية في الحسابات النظرية وتم ملاحظته في التجارب، مما يمكن أن يحسن في الموقع توزيع الإلكترونات في المواقع النشطة لتسريع امتصاص وتفكك الأنواع التفاعلية.بشكل خاطئ، المستقر المتماثلتعيق الجزء النصفى تحسين الذات للمراكز النشطة خلال التفاعل، مما يشكل عقبة أمام تحسين نشاطها في تفاعل اختزال الأكسجين. وبالتالي، فإن كسر توزيع الإلكترونات المتماثل لمراكز المعادن ذات الذرة الواحدة يعد مثيرًا لتفعيل إعادة بناء الهيكل الذاتي، وتحسين قوة الامتصاص للأنواع الأكسجينية في الموقع، وبالتالي تعزيز خصائص تفاعل اختزال الأكسجين بأربعة إلكترونات.
هنا، قمنا بنجاح بتصنيع مواقع الكوبالت المنظمة بالتنسيق (CR-Co) المحصورة في أوراق الكربون النانوية المضافة بالنيتروجين من خلال استراتيجية تحلل حراري بسيطة من خطوتين. على المستوى الذري، كل من الانتقال الرباعي الكهربائي المحلي (1s إلىالانتقال) وانتقال ثنائي القطب الكهربائي المحلي (1s إلى“الانتقال) يتم تحقيقه من خلال إدخال روابط الكلور لتشكيل “كسر التماثلالجزء في مصفوفة الكربون المضافة بالنيتروجين (CR-Co/ClNC)، مما يدل على كسر التوزيع المتناظر للإلكترونات. تكشف مطيافية الامتصاص الدقيق للأشعة السينية (XAFS) أنالجزيء يحرر ديناميكياً تنسيقين من النيتروجين لتشكيل مركب غير مشبعالهيكل في حالة التفاعل المبكرة. خاصة، يمكن أن يقوم هذا إعادة ترتيب الهيكل الذاتي بتحسين الوضع في الموقعملء الإلكترونات لمواقع الكوبالت نحو مستوى منخفض-شغل الإلكترون في النطاق ( ) خلال التفاعل. وهذا بدوره يعزز تحول الـ رابطة من الأنواع المحتوية على الأكسجين الممتصوتطور إضافي إلى ( ) الهيكل. لقد تم تأكيد هذا التحول من خلال نتائج مطيافية الأشعة تحت الحمراء باستخدام إشعاع السنكروترون في الموقع (SRIR)، مما يعزز بعد ذلك خصائص اختزال الأكسجين بأربعة إلكترونات (ORR). يُظهر المحفز CR-Co/CINC المصمم بشكل جيد تحسينًا ملحوظًاانتقائية ORR (98%) مع جهد نصف الموجة 0.93 فولت ونشاط كتلي مرتفع منعند 0.85 فولت. هذا يمثل زيادة تقريبية بمقدار 47 مرة مقارنةً بما هو موجود في السوق.كإثبات للمفهوم، توفر بطارية الزنك-هواء المجمعة مع محفز CR-Co/CINC كثافة طاقة فائقة مقارنة بـ Pt/C ( )، مما يشير إلى إمكانيات ممتازة للتطبيق العملي في بطاريات المعدن-الهواء.
النتائج والمناقشة
خصائص الشكل والبنية للمواد المحفزة الكهربائية
تم تحضير المحفز الكهربائي الكوبالت الموزع ذريًا، حيث تم حصر مواقع الكوبالت المنظمة بالتنسيق داخل رقائق الكربون المضافة بالنيتروجين، من خلال استراتيجية بلمرة سهلة من خطوتين. في البداية، تم خلط ZIF-8 مع مسحوق KCl وتعريضه للبلمرة تحت جو من غاز الأرجون المتدفق لإنتاج رقائق كربون مضافة بالنيتروجين (NC) تحتوي على مواقع نشطة متاحة. بعد ذلك، لتنظيم مجال تنسيق الكوبالت، تم إدخال تنسيق مواقع المعادن في ملح الكوبالت المحتوي على الكلور على سطح رقاقة NC من خلال البلمرة عند درجات حرارة مختلفة، حيثتم اختيارها للحصول على CR-Co/ CINC. في هذه العملية، تم احتجاز ذرات الكوبالت وتثبيتها في NC في نفس الوقت بسبب تأثير الربط القوي مع الإلكترونات الوحيدة الزوج من نوع النيتروجين. في الوقت نفسه،تم إعداد العينة كـ المرجع من خلال رفع درجة حرارة التحلل الحراري إلى لاستبعاد عنصر الكلور .
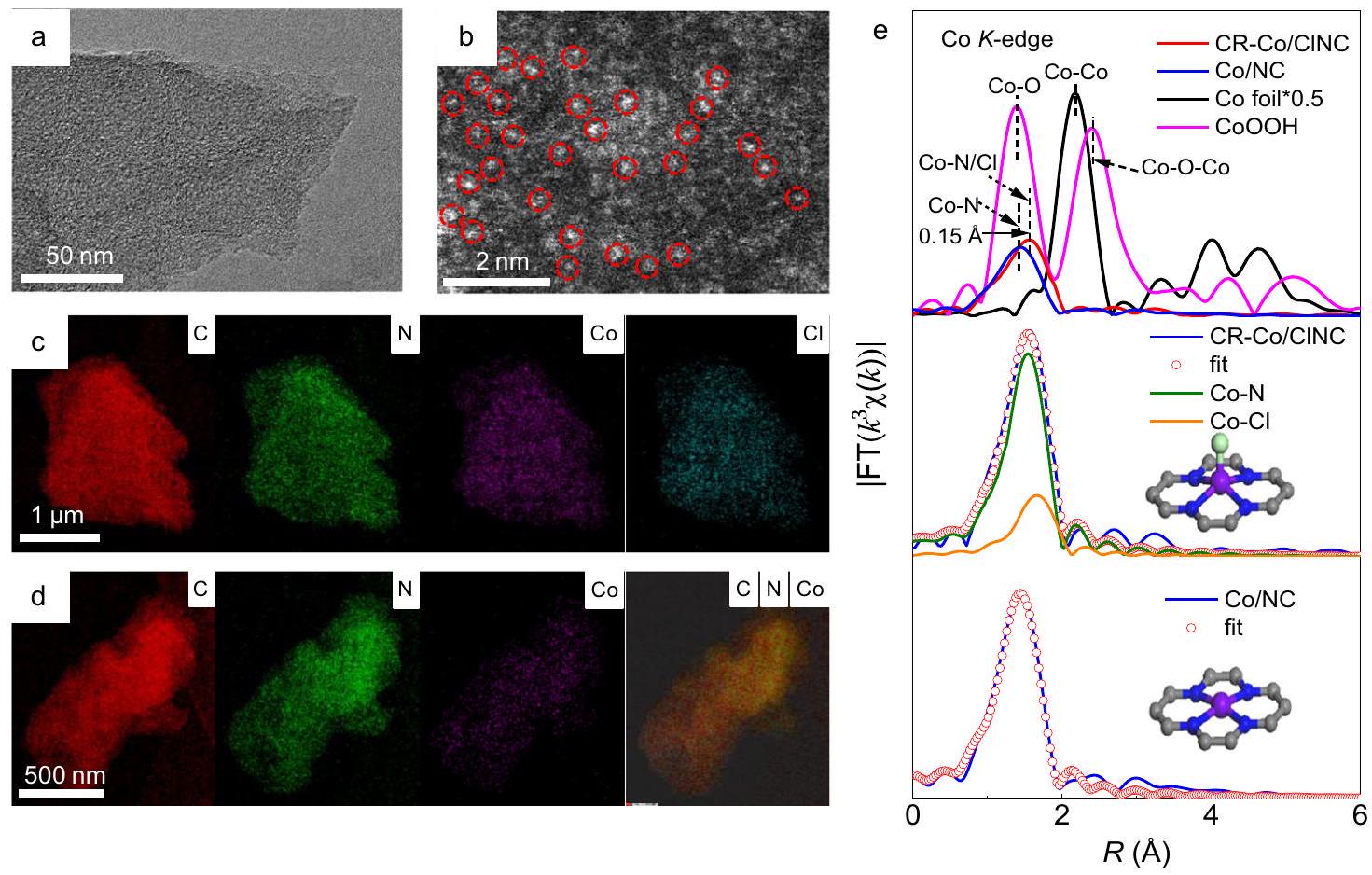
تظهر صور المجهر الإلكتروني الناقل (TEM) أن المحفزات CR-Co/CINC و Co/NC المكتسبة تمتلك هيكلًا شبيهًا بالورقة (الشكل 1a والأشكال التكميلية 1 و 2) دون وجود جزيئات نانوية واضحة. يؤكد المجهر الإلكتروني الناقل ذو الزاوية العالية (HAADF-STEM، الشكل 1b) لـ CR-Co/CINC وجود نقاط ساطعة كثيفة ومعزولة محددة بدوائر حمراء، مما يدل على وجود ذرات الكوبالت الموزعة ذريًا والمثبتة على ركيزة NC. يمكن إثبات ذلك بشكل أكبر من خلال نتائج حيود الأشعة السينية (XRD) في الشكل التكميلية 3، التي تظهر غياب قمم الحيود المرتبطة بأنواع الكوبالت. لتوضيح الهيكل الشكلي لمواقع الكوبالت بشكل أكبر، تكشف صور رسم الخرائط العنصرية عن التوزيع المتجانس لـ، و Cl في الكل معمارية CINC (الشكل 1c) وتوزيع متجانس للكربون والنيتروجين والكوبالت فيعينة (الشكل 1د). الـتظهر منحنيات الامتصاص الفيزيائي مساحة سطح محددة أكبر قليلاً لـ CR-Co/CINC وهندسة مسامية مشابهة بالمقارنة مع (الشكل التوضيحي 4). من المهم أن تحميلات الكوبالت لـ CR-Co/CINC و Co/NC كانت و على التوالي، كما تم تحديده من تحليل طيف الانبعاث الضوئي الناتج عن البلازما المقترنة بالحث (ICP-OES). تؤكد هذه النتائج التشتت المتجانس لذرات الكوبالت على مواقع السطح لمادة NC وإدخال الكلور في.
بالإضافة إلى ذلك، في الدقة العاليةطيف مطيافية الإلكترونات السينية (XPS) لـ CR-Co/CINC، القمة المميزة عند 197.7 eV تتوافق معالأنواع، بينما تصل إلى و تُنسب إلى رابطة. محتوى Cl في يتم تحديده ليكون 2.15 بواسطة XPS. تظهر طيف N 1s (الشكل التكميلي 6) أيضًا قمة عند 399.4 إلكترون فولت، والتي يمكن نسبها إلى رابطة. بشكل جماعي، مظهر و تشير الروابط في طيف XPS لـ CR-Co/ CINC إلى تنسيق ذرات الكوبالت مع كل من ذرات الكلور والنيتروجين. والأهم من ذلك، تم الحصول على طيف الامتصاص الدقيق للأشعة السينية الممتدة (EXAFS) لتحديد بيئة التنسيق للكوبالت بشكل أكبر (الشكل 1e). لاستبعاد الضوضاء عالية التردد في النطاق العاليالمنطقة وضمان وجود نقاط حرة مستقلة كافية أثناء ملاءمة EXAFS،نطاق منتم اختيارها لجميع عينات Co خلال تحويل فورييه السريع (الشكل التوضيحي 7). على عكس مرجع رقائق Co و CoOOH، فإن القمة البارزة الموجودة فييمكن تعيينه إلىرابطة، بسبب وجود عناصر النيتروجين والكلور، دونظهر ذروة التشتت في كلاهما و التحقق من انتشار ذري لكوبالت في دعم النانو كربون. بعد إدخال Cl في ، الشركةالقمة تظهر تحولًا إيجابيًا من مقارنة بـ عينة، مما يشير إلى تشكيلهيكل التنسيق بسبب نصف القطر الذري الأكبر للكلور. علاوة على ذلك، لتوضيح الهيكل المحلي للتنسيق لمواقع الكوبالت، تم ملاءمة المنحنيات لـوزن Co-طيف EXAFS عند حافة في الشكل 1e والشكل التكميلي 8، جنبًا إلى جنب مع البيانات المقابلة لـ و المُدرجة في الجدول التكميلي 1، تُظهر أن عدد التنسيق (CN) لـرابطة لـ هو 4.1 . لـ 3.9 و 1.0 لـرابطة ومحورتم الحصول على التنسيق الموضح في الشكل 1e، على التوالي. لتسليط الضوء على الهيكل المحلي لموقع معدن الكوبالت وتنسيق الكلور المحوري، يتم عرض نموذج الهيكل المحلي في الشكل 1e. تشير النتائج السابقة إلى أن بيئة التنسيق المحلية المقترحة لـ CR-Co/ClNC تتكون من تنسيق كسر التناظر Cl-Co.جزء، مما يشير إلى التنظيم الناجح لمجال التنسيق لمركز الكوبالت من خلال إدخال عنصر الكلور ذو الكهربية العالية.
للحصول على رؤى على مستوى الذرة حول الإلكترونيات المحلية لـ Co، تم إجراء مطيافية امتصاص الأشعة السينية الناعمة بالقرب من حافة الطيف (XANES) لـ N، كما هو موضح في الشكل 2a. القمة عند يُنسب إلى إثارة مدار هجين لـ، بينما الذروة فيينشأ منالانتقالاتعند إدخال مواقع الكوبالت في ركيزة NC، ظهرت قمم جديدة عند 399.4 إلكترون فولت.
الشكل 1 | خصائص الشكل والبنية. أ صور TEM و ب صور HAADF-STEM لمحفز CR-Co/CINC. صور خرائط TEM-EDS لـ CR-Co/CINC و طيف FT-EXAFS لـ Co/NC.-حافة لـالعامل المساعد و عينات مرجعية، والمنحنيات المناسبة لـ و NC. يظهر الرسم التوضيحي وتم الإشارة إلى ذرات الكربون بالأزرق السماوي، والبنفسجي، والأزرق الداكن، والرمادي.
الشكل 2 | الحالة الكيميائية والتركيب الذري المحلي. أ-طيف XANES عند الحافة و NC. ب N طيف XPS لـ CR-Co/ClNC و Co/NC. ج Coطيف XANES الحافة لعينة CR-Co/CINC وعينات المرجع. يظهر الشكل الداخلي منطقة ما قبل الحافة الملائمة.المتوسط الملائم الرسمي-عدد الإلكترونات في النطاق. تمثل أشرطة الخطأ الانحرافات المعيارية لثلاث حسابات مكررة. e Coطيف XPS لـ و . تمت ملاحظته لكل من CR-Co/CINC والذي يمكن أن يُعزى إلى إثارة. ل، الـيظهر الذروة تحولًا إيجابيًا طفيفًا من بالمقارنة مع ، مما يشير إلى تقليل نقل الإلكترون منبعد إدخال عناصر Cl. علاوة على ذلك، يمكن تحليل طيف XPS N 1s عالي الدقة لكلا المحفزين إلى شكل بيريدينيكأنواع النيتروجين البيرولي والنيتروجين الجرافيتي (الشكل 2 ب)النسب المقابلة لـ تم تلخيص الأنواع N المفككة في الشكل التكميلية 9. يكشف عن وجود أنواع N بنسب متشابهة، مما يستبعد تأثير نوع N في الركيزة على أداء ORR. في الوقت نفسه، فإن التحول العالي للطاقة في طاقة الربط ( ) بالنسبة لـ Co -N في CR-Co/CINC يثبت أن التفاعل المخفض بين Co و N يُعزى إلى ذرات Cl المدخلة، وهو ما يتوافق جيدًا مع نتائج XANES الحافة.
الشكل 3 | أداء اختزال الأكسجين الكهروكيميائي. أ منحنيات الاستقطاب لـ CR-Co/ClNC والمراجع تحت-مشبعمعدل المسح لـالتباين بين CR-Co/CINC والمراجع لـ و أشرطة الخطأ هي الانحرافات المعيارية لثلاث حسابات مكررة.منحدرات الطاولة والنشاط الكتلي (MA) وتردد الدوران (TOF) لـ CR-Co/CINC و
عينات مرجعية. أشرطة الخطأ هي الانحرافات المعيارية لثلاث حسابات مكررة. عدد نقل الإلكترون (أعلى) والعائد (الحد الأدنى) مقابل إمكانيات CR-Co/CINC و Pt/C. استجابات التيار الزمني الكرونوأمبريومترية الحالية لـ و .
للحصول على فهم عميق للتغيرات في البنية الإلكترونية المحلية لمواقع الكوبالت بعد إدخال الكلور على المستوى الذري، تم قياس طيف XANES للكوبالتتم اعتماد -edge (الشكل 2c). يظهر طيف XANES لـ CR-Co/ClNC أشكال قمم مختلفة وكثافات، مما يشير إلى بيئة تنسيق وهيكل إلكتروني محلي مختلف مقارنةً بـتشير الميزة الموسعة في الحافة السابقة (الذروة A) لـ CR-Co/ CINC (في الصورة المصغرة، الصورة المكبرة في الشكل 2c) إلى وجود كل من انتقال الرباعي الكهربائي المحليإلىالانتقال) وانتقال ثنائي القطب الكهربائي المحلي (1s إلىالانتقال) في المحفز، مما يؤكد بوضوح توزيع الإلكترونات غير المركزي لمواقع الكوبالت فيأقصى قيمة للمشتقة الأولى لـيتجاوز CINC ذلك الخاص بـ CoOOH، مما يكشف عن حالة أكسدة أعلى للكوبالت في CR-Co/CINC مقارنة بـ CoOOH (الشكل التكميلي 10). بالإضافة إلى ذلك، فإن التحول العالي للطاقة لحافة الامتصاص وزيادة شدة قمة الخط الأبيض في CR-Co/CINC يوضحان زيادة في حالة التكافؤ للكوبالت بعد إدخاليمكن الحصول على حالة التكافؤ من خلال التناسب الخطي لموقع حافة الامتصاص (الشكل التكميلي 11). يوضح الشكل 2d مواقع حواف الامتصاص للعوامل الحفازة كدالة لـ-عدد الإلكترونات في النطاق المستخرج منمعيار. من الجدير بالذكر،يعرض عددًا أقل بشكل ملحوظ من-إلكترونات النطاق (5.80) مقارنة بـ (5.96)، مما يشير إلى تزاوج ملحوظ لـالدول، مما يؤدي بشكل فعال إلى كسر توزيع الإلكترونات المتماثل لمجموعات Co-N. الكوبالتذروة في CR-Co/CINC تنتقل إلى طاقة ارتباط أعلى من تلك الخاصة بـ (الشكل 2e)، مما يشير إلى حالة تكافؤ أعلى للكوبيليوم بعد تنظيم الكلور، وهو ما يتماشى مع نتائج XANES السابقة. وبالتالي، تُظهر هذه النتائج أن الكسر في التناظر تعمل المجموعات في CR-Co/ClNC على كسر التوزيع الإلكتروني المتماثل لمواقع الكوبالت في نموذج نموذجي.نصف، مما يؤدي إلى تقليل العدد من-إلكترونات النطاق، وفي النهاية تحسين أداء المواقع النشطة.
أداء اختزال الأكسجين الكهروكيميائي
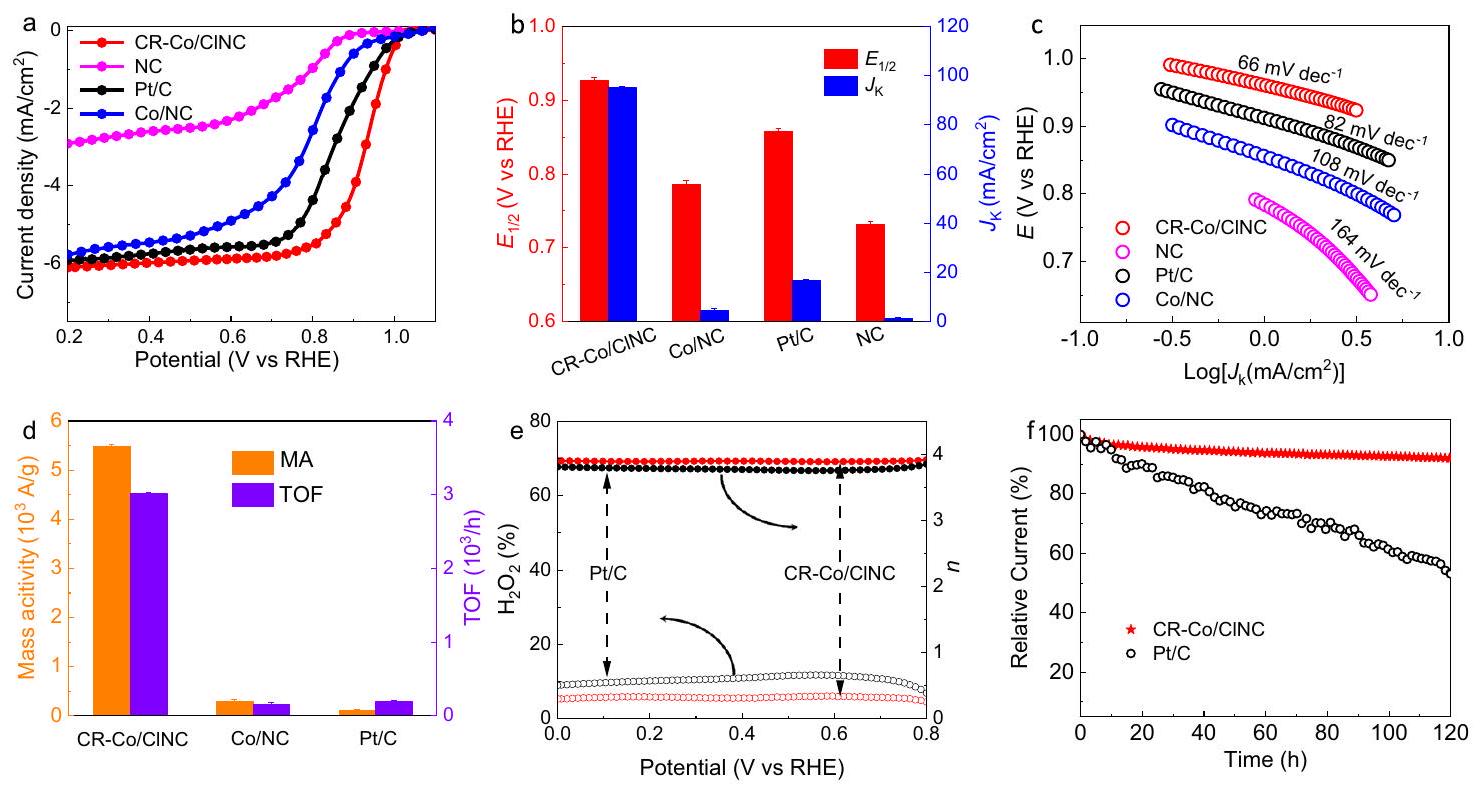
لتقييم أداء ORR، تم إخضاع العينات المحضرة تجارياً وPt/C للتقييم باستخدام إلكترود قرص دوار (RDE) في-محلول إلكتروليتي 0.1 م KOH مشبع. مقارنة منحنيات الفولتاموجرافيا بالمسح الخطي (LSV) لـ CR-Co/CINC، Co/NC، NC، وتُظهر المحفزات في الشكل 3أ. يظهر نشاطًا مثاليًا في معدل تقليل الأكسجين مع جهد نصف الموجة (0.93 فولت مقابل قطب هيدروجين قابل للعكس (RHE، الجهود المذكورة أدناه جميعها بالنسبة لـ RHE) (الشكل 3ب)، مما يتفوق بوضوح على تلك الخاصة بالمنتجات التجارية و NC. يتطلب جهد بدء أعلى قدره 1.008 فولت لـ CR-Co/CINC لتحقيق 5% من التيار المحدود بالانتشار (الشكل التكميلي 12)، متجاوزًا قليلاً جهد Pt/C (0.994 فولت). علاوة على ذلك، تصل كثافة التيار المحدود بنقل الكتلة لـ CR-Co/CINC إلى ، متجاوزًا ما تم ملاحظته بالنسبة لمحفز Pt/C التجاري ( ). يبدو أن كثافة التيار المحدود الأعلى هذه هي نتيجة لمعدلات إزالة سريعة للمنتج على قطب CR-Co/CINC. بالإضافة إلى ذلك، يتمتع CR-Co/CINC بكثافة تيار حركي متفوقة ( حتىعند 0.85 فولت،طوي ذلك من. على النقيض، يعرض نشاط ORR أدنى مع تقليلوأكثر سلبية ( 0.79 فولت )، مما يدل على النشاط الداخلي الأمثل لتفاعل اختزال الأكسجين (ORR) لـ CR-Co/CINC المنسوب إلى التعديل المناسب لحقل التنسيق لمواقع الكوبالت مع عناصر الكلور. في الوقت نفسه، فإن أداء ORR الذي حققته CR-Co/CINC يتفوق على معظم SACs التي تم الإبلاغ عنها مؤخرًا (الجدول التكميلي 2). تم تأكيد سرعة حركية ORR لـ CR-Co/CINC بشكل أكبر من خلال أقل ميل لتافل. مقارنة بـ “، و NC ( ) (الشكل 3ج). بالإضافة إلى ذلك، كما هو موضح في الشكل 3د، يوفر CR-Co/ClNC تردد دوران مرتفع (TOF) قدره ونشاط جماهيري كبير (MA) منعند 0.85 فولت، أعلى بنحو 15 و 47 مرة من تلك الخاصة بالمنتجات التجارية و لإزالة تأثير أخطاء الاختبار، بما في ذلك تحميل المحفز، وتجانس فيلم الإلكترود، ومستوى سطح الإلكترود الكربوني الزجاجي، واختراق الإلكتروليت على سطح الإلكترود، CR-Co/ CINC وتم اختبارها ثلاث مرات في العملية التكرارية للحصول على أشرطة الخطأ لـ ECSA (الأشكال التكميلية 13-15). علاوة على ذلك، كانت المساحة السطحية النشطة كهربائياً (ECSA) مشابهة لـ CR-Co/CINC (مع شريط خطأ من ) و مع شريط خطأ من ) تشير إلى أن مصدر النشاط المعزز لـ هو هيكل يتكون من تنسيق الكلور المحوري، الذي يحسن التركيب الإلكتروني لتحقيق زيادة في النشاط الجوهري لموقع النشاط، بدلاً من
الشكل 4 | التحليلات في الموقع لـ XAFS و SRIR. أ تحليل ملاءمة المنحنى لطيف EXAFS وطيف XANES لكوبالت-edge المسجل عند إمكانيات تطبيق مختلفة خلال عملية ORR لـ CR-Co/CINC. في الزاوية، منطقة الذروة البيضاء المكبرة. ج المتوسط الملائم الرسمي-عدد الإلكترونات في نطاق الكوبالت عند CR-Co/ClNC تحت ظروف ex situ، 1.00 فولت و 0.90 فولت بناءً على حافة الامتصاص لطيف XANES لحافة الكوبالت. تمثل أشرطة الخطأ الانحرافات المعيارية لثلاث حسابات مكررة. قياسات SRIR في الموقع تحت إمكانيات مختلفة لـ (د) CR-Co/CINC و (هـ). مخططات ORR لـ. زيادة في عدد المواقع النشطة (الأشكال التكميلية 16-18). تظهر هذه النتائج أن إدخال ذرات الكلور المحورية في النظام المكسور التناظرأجزاء ذات منخفضيمكن أن يعزز ملء الإلكترونات في النطاق بشكل كبير نشاط تفاعل اختزال الأكسجين.
بصرف النظر عن نشاط التحفيز في تفاعل اختزال الأكسجين، فإن الانتقائية التحفيزية هي مؤشر مهم لتقييم أداء اختزال الأكسجين الكهروكيميائي. كما هو موضح في الشكل 3e، فإن عدد نقل الإلكتروناتتم حساب CR-Co/CINC ليكونوفقًا لقياسات قطب القرص الدوار (RRDE)، مما يؤكد تفضيل محفز CR-Co/CINC لـالمسار. علاوة على ذلك،تم إثبات مسار ORR بشكل أكبر من خلال معادلة كوتيشي-ليفش (K-L) المطبقة في منطقة التحكم في الانتشار عند سرعات دوران مختلفة (الأشكال التكميلية 19 و20). من الواضح أن إنتاج البيروكسيد لـ CR-Co/CINC في نفس نطاق الجهد أقل من 3%، وهو أقل من تلك العينات الأخرى (الشكل التكميلية 21)، مما يشير إلى أعلى انتقائية لأربعة إلكترونات تجاه مسار ORR لـ CR-Co/CINC. بالإضافة إلى ذلك، تم قياس استقرار ودوام محفز CR-Co/CINC من خلال قياس التيار الزمني (CA) واختبار التحمل المعجل (ADT). يظهر CR-Co/CINC استقرارًا موثوقًا مع مقاومة أفضل للميثانول وانخفاض في تآكل كثافة التيار (<8%) بعد 120 ساعة من اختبار CA عند 0.7 فولت، ويعاني محفز CR-Co/CINC فقط من فقدان قدره 20 مللي فولت فيبعد ADT، مما يشير إلى متانته على المدى الطويل لـ ORR (الشكل 3f والأشكال التكميلية 22 و23). تم إجراء التحليلات الشكلية والإلكترونية، مثل HAADF-STEM وXAFS لـ CR-Co/CINC (الأشكال التكميلية 24 و25) بعد التحليل الكهربائي طويل الأمد، دون ملاحظة أنواع Co والجسيمات النانوية. تشير هذه النتائج إلى القوة الهيكلية الفائقة والثبات الممتاز لـ CR-Co/CINC.
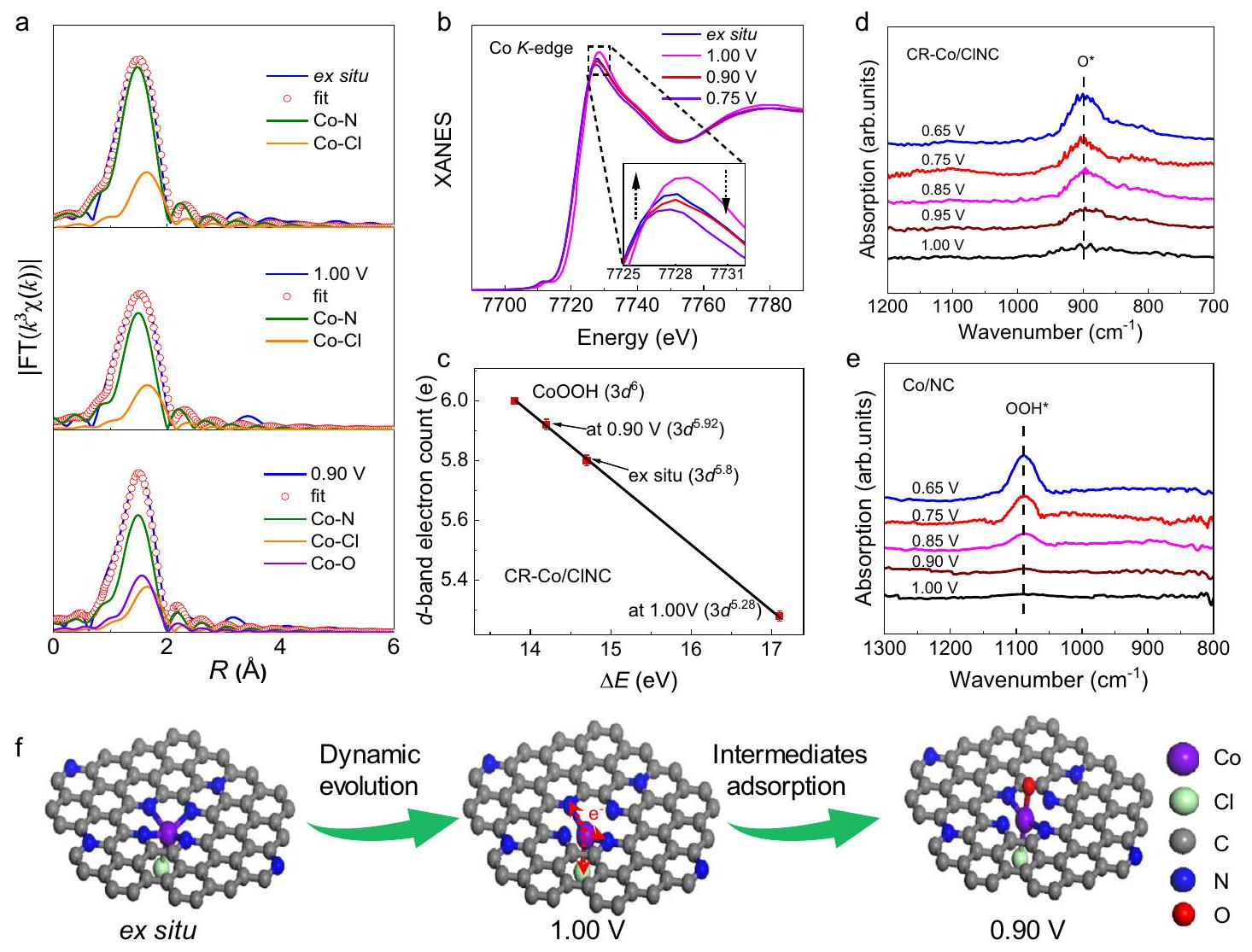
توصيفات في الموقع لتطور المواقع النشطة لتوضيح الآليات الأساسية المسؤولة عن الانتقائية الاستثنائية لأربعة إلكترونات لـ CR-Co/CINC خلال عملية اختزال الأكسجين (ORR)، تم إجراء قياسات XAFS في الموقع لـ Coتم إجراء -edge باستخدام خلية محلية الصنع في الموقع (الشكل التوضيحي 26a)أولاً، تم توضيح تطور بيئة التنسيق المحلية لمواقع الكوبالت تحت الفولتية المطبقة من خلال نتائج EXAFS في الموقع (الشكل التكميلي 26). من الواضح أن هناك قمة سائدة عندأظهرتخفيف في شدة الذروة وتحول إيجابي لـلـ CR-Co/ CINC حيث تغير الجهد من ex situ (مغمور في المحلول بدون جهد مطبق) إلى ظروف 1.00 فولت. تشير شدة الذروة المنخفضة في EXAFS بوضوح إلى الانخفاض المدفوع بالجهد في تنسيق مواقع الكوبالت الفردية وعدم وجود امتصاص من قبل الأنواع المحتوية على الأكسجين في حالة التفاعل المبكرة. تعرض الذروة زيادة ملحوظة في الشدة وانزياح طفيف سلبي في الموقع عند تطبيق جهود 0.90 و 0.75 فولت، مما يوحي بامتصاص الأنواع المحتوية على الأكسجين خلال عملية ORR. كميًا، تظهر نتائج تركيب EXAFS في الشكل 4a، الأشكال التكميلية 27-30، والجدول التكميلية 3 عدد تنسيق يساوي أربعة.رابطة واحدة ومحوريةرابطة تحت حالات خارج الموقع، تشبه نتيجة EXAFS المقاسة تحت ظروف الهواء. من المثير للاهتمام أنه عند تطبيق جهد قدره 1.00 فولت، فإن عدد التنسيق لـمن الواضح أن الروابط تقلصت إلى اثنتين، مما يشير إلى أن التطور الهيكلي المدفوع بالجهد لمواقع الكوبالت الفردية يحدث حقًا تحت ظروف اختزال الأكسجين عن طريق تحرير مراكز الكوبالت من الركيزة N-C لتشكيل Cl-Co-الموقع النشط. من المهم جداً أن تكون هذه التنسيق غير المشبعالبنية تفضل امتصاص جزيئات الأكسجين على السطح. مع استمرار انخفاض الجهد إلى 0.90 فولت،التنسيق مع إضافة أخرىيتم الاحتفاظ بالتنسيق من أجل الشركة المواقع، التي قد تكون ناتجة عن امتصاص الوسائط التفاعلية الرئيسية المحتوية على الأكسجين.
غالبًا ما تصاحب التطورات الديناميكية في هيكل التنسيق المحلي تحسين الهيكل الإلكتروني لموقع النشاط تحت ظروف العمل. يتم استخدام تحليل XANES لتوضيح التغيرات في الهيكل الإلكتروني. طيف XANES في الموقع لـ Coيتم توضيح حافة الامتصاص لـ CR-Co/CINC عند فولتages مختلفة في الشكل 4b. بالمقارنة مع الظروف الخارجية، تظهر حافة الامتصاص انزياحًا طفيفًا نحو الطاقة الإيجابية، ويظهر ذروة الخط الأبيض زيادة متواضعة في الشدة عند فولتية مطبقة قدرها 1.0 فولت. لت quantifying التغيير فيتعبئة الإلكترونات في النطاق، فإن تحولات حافة الامتصاص مرتبطة بـ-عدد الإلكترونات في النطاق لـ Co باستخدام كمعيار (الشكل 4c والشكل التكميلي 31). لوحظ عدد ملء إلكترونات Co 3d الأقل في CR-Co/CINC (5.28) عند 1.0 فولت. تم تطوير الديناميكية يشهد انخفاضًا سريعًا قدره 0.52 إلكترون في حالة التفاعل المبكرة (التي تتوافق مع ” ديناميكيتطور الإلكترون )، مما يوحي بأن هناك المزيد من -مدارات شاغرة للربط معمدارات لتنظيم امتصاص الأنواع المحتوية على الأكسجين. ومن الجدير بالذكر أن الكوبالتعدد ملء الإلكترونات في CR-Co/CINC ينخفض بشكل أسرع وأكثر عنفًا مع الجهد المطبق مقارنةً بـ (الشكل التوضيحي 32)، والذي يكون مفيدًا لتحسين حركيات الامتصاص للوسائط. تكشف هذه النتائج عن تعديل سريع في الموقع لـتحدث الإلكترونات في نطاق – عند حالة التفاعل المبكرة لـ CR-Co/CINC، كما أن التماثل مكسورالجزء يتطور بسرعة إلى الحالة المنسقة المخفضة.
لتحقيق خصائص الامتزاز للأنواع الوسيطة الرئيسية على مواقع الكوبالت مع الديناميكيةتطور الإلكترون، تم اعتماد تقنية SRIR السنكروترونية الحساسة للسطح في الموقع بواسطة خلية مخصصة. كما هو موضح في الشكل 4d، عندما يكون الجهد المطبق أقل من 1.00 فولت لـ CR-Co/CINC، تظهر نطاق امتصاص جديد عند يظهر سلوكًا يعتمد على الجهد. يمكن أن يُعزى ذلك إلى تراكم الوسائط الحاسمة *O على الجزئيات تحت ظروف ORR لأن اهتزاز الشد لعينات الأكسجين (*O) يكون عادة في نطاقللمزيد من التحقق من تأثير رابطة الكلور المنسقة، تم تسجيل إشارات SRIR في الموقع لـتم الحصول عليها أيضًا تحت نفس الإمكانيات النموذجية للمقارنة (الشكل 4e). من الملحوظ وجود نطاق امتصاص جديد عنديلاحظ ذلك مع انخفاض الجهد المطبق. حيث أن قمم الاهتزاز تحت الحمراء لنوع *OOH عادة ما تظهر في منطقةنطاق الامتصاص عنديمكن تعيينه إلى. فقط نطاق الامتصاص لنوع *O الذي لوحظ لـ CR-Co/ClNC خلال عملية ORR يشير بوضوح إلى الانقسام السريع لـرابطة في *متوسط في التطور الديناميكيالمواقع. لذلك، يتم تحسين قوة الامتزاز لمفتاح *OOH بشكل فعال للتطور بسرعة إلى أنواع *O لـ CR-Co/ClNC بسبب الديناميكيةتطور الإلكترونفي حالة التفاعل المبكر، مما يعزز بشكل فعال حركيات تفاعل الأربعة إلكترونات ويزيد من نشاط واختيارية اختزال الأكسجين.
فوق كل شيء، يتم عرض التطور الديناميكي للمواقع النشطة تحت ظروف العمل بشكل تخطيطي في الشكل 4f. أولاً، يتم إطلاق مراكز الكوبالت ديناميكيًا من ركيزة لتشكيل مركب منسق مخفضالهيكل النشط في حالة التفاعل المبكر (عند ظروف 1.0 فولت)، كاشفًا عن انخفاض-ملء الإلكترونات في النطاق. من المRemarkably، هذه التنسيق المخفضةتعمل الوحدات بشكل واضح على تحسين امتصاص الوسائط المحتوية على الأكسجين في الموقع وتعزز انقسام الـرابطة لسرعة تفاعلات كيميائية سريعة. أخيرًا، الـ *تظل البنية النشطة محتفظة بها مع انخفاض الجهد بشكل مستمر إلى 0.75 فولت، مما يحقق عملية اختزال الأكسجين عالية الكفاءة. علاوة على ذلك، تعود الهياكل الإلكترونية والتنسيقية المتطورة ديناميكيًا لمواقع الكوبالت إلى حالتها الأصلية بعد التفاعل وفقًا لنتائج XAFS (الشكل التكميلي 33)، مما يشير إلى أن تطور البنية لموقع الكوبالت النشط هو عملية ديناميكية قابلة للعكس. تشير هذه النتائج إلى أن التعديل في الموقعيمكن أن يؤدي ملء الإلكترونات في نطاق Co من خلال إدخال Cl تسريع كبير في حركية تفاعل الأكسدة الاختزالية، مما يمنح محفز CR-Co/CINC إمكانيات ممتازة للتطبيقات الصناعية.
أداء بطارية الزنك-هواء (ZAB)
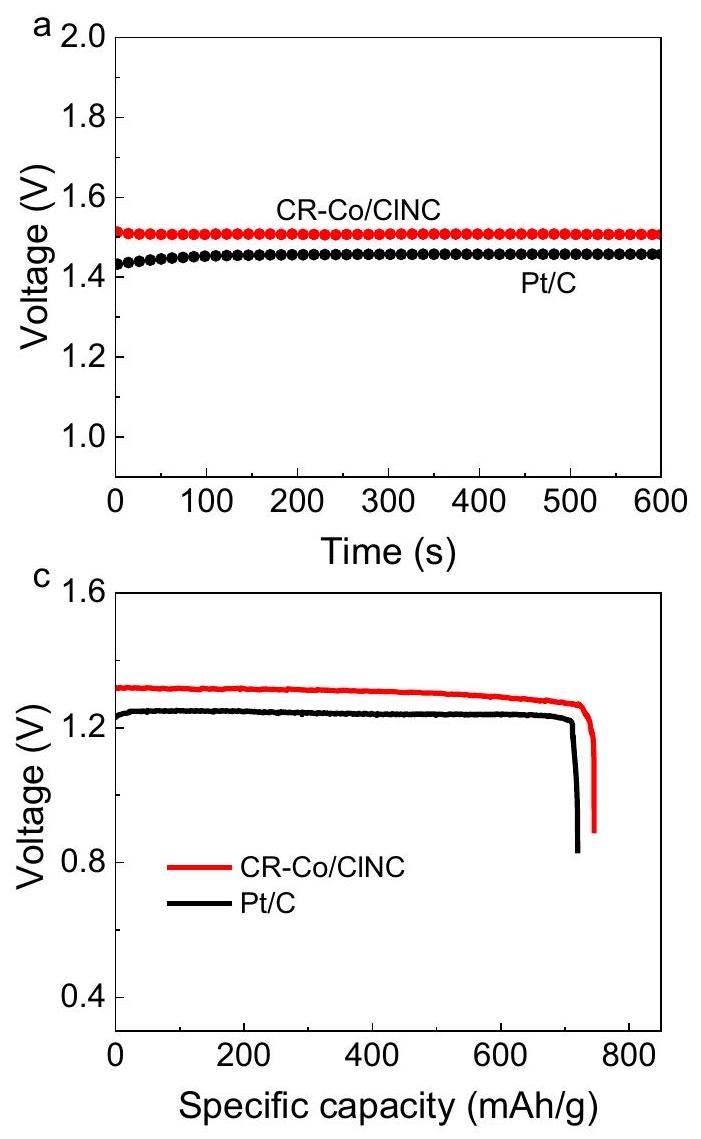
في ضوء الأداء المتفوق لـ ORR، تم استخدام CR-Co/CINC المُعد كعامل حفاز للقطب السالب في بطاريات الزنك-هواء الأولية المائية (ZABs) للتحقيق في قابليته للاستخدام (الشكل التوضيحي 34). تم تجميع نفس الجهاز باستخدام Pt/C التجارية للمقارنة. تظهر خلية الزنك-هواء المدمجة مع CR-Co/ClNC جهد مفتوح دائري مثير للإعجاب يبلغ 1.50 فولت (الشكل 5a)، وهو أعلى من ذلك الخاص بخلية الزنك-هواء المعتمدة على Pt/C (1.45 فولت)، مما يشير إلى جهد خرج أعلى للبطارية عند استخدام CR-Co/ClNC كعامل حفاز للقطب السالب. من منحنيات استقطاب التفريغ ومخططات كثافة الطاقة لبطارية الزنك-هواء (الشكل 5b)، يمكن ملاحظة أن ZAB المعتمد على CR-Co/ClNC يقدم هضبة جهد تفريغ أعلى مع كثافة طاقة قصوى قريبة منالذي يتفوق علىنظير (كثافة الطاقة لبطارية الزنك-هواء القائمة على CR-Co/CINC تتجاوز معظم SACs المبلغ عنها (الجدول التكميلي 4). ومن الجدير بالذكر أن بطارية الزنك-هواء مع CR-Co/CINC تقدم سعة محددة تبلغ 745 مللي أمبير ساعة.عند كثافة تيار التفريغ في الشكل 5c، يتفوق على أداء زAB القائم على Pt/C ( ). كانت ملاحظات التفريغ الجلفاني ثابتة عند كثافات تيار تتراوح من 5 إلى موضحة في الشكل 5d. من الواضح أن بطارية الزنك-هواء المعتمدة على CR-Co/CINC تحافظ على فولتية تفريغ أعلى من-المعتمد ZAB ضمن نطاقات الكثافة الحالية ثم العودة بشكل دوري إلى. بعد تفريغ عالي المعدل في ، يعود جهد التفريغ لبطارية الزنك الهوائية المعتمدة على CR-Co/CINC إلى المرحلة الابتدائية ( )، مما يدل على قدرة المعدل الجيدة والقابلية للعكس لبطارية الزنك-هواء القائمة على CR-Co/CINC، والتي يمكن أن تُعزى إلى النشاط الفعال لتفاعل اختزال الأكسجين واستقرار جيد لمحفز CR-Co/CINC. ومن الملحوظ أنه لم يتم تسجيل أي تدهور ملحوظ بعد 3 دورات لبطارية الزنك-هواء مع المحفز الكهربائي CR-Co/CINC على مدى 30 ساعة في (الشكل التوضيحي 35). بالإضافة إلى ذلك، تظهر قياسات التفريغ – الشحن الجلفاني أن هناك تدهورًا ضئيلًا في جهد التفريغ – الشحن، مما يعكس المتانة الواعدة لـ CR-Co/CINC في بطاريات الزنك-هواء (الشكل التوضيحي 36). تشير هذه النتائج إلى أن محفز CR-Co/CINC يحمل إمكانيات جيدة للتطبيق في بطاريات الزنك-هواء.
باختصار، لقد قمنا بإنشاء نوع من المحفز CR-Co/CINC الموزع ذريًا والمنظم بالتنسيق مع كسر التناظرالمكونات عبر استراتيجية التحلل الحراري السهلة ذات الخطوتين، مع تجنب إدخال الشوائب بنجاح وتحقيق اقتران الكلور المحوري علىجزء. على المستوى الذري، لقد أظهرنا أن إدخال ذرات الكلور المحورية في الموقع النشط يمكن أن يكسر بشكل فعال توزيع الإلكترونات المتماثل للكوبر.أجزاء، تحقيق انخفاض-ملء الإلكترونات في النطاق من أجل التنظيم في الموقع لقوة الامتزاز للمواد الماصة للأكسجين حول مواقع الكوبالت. وقد كشفت الدراسات الآلية التي تستخدم تقنيات SRIR وXAFS في الموقع عن التطور الديناميكي للتنسيق المخفض.الهيكل يمكن أن يحسن-شغل الإلكترونات في مواقع الكوبالت في الموقع في حالة التفاعل المبكرة، وهو ما يكون مفيدًا جدًا لتفكيك الـ (*أنواع OOH) إلى *O وسائط نحو نشاط انتقائي ممتاز في التحفيز الكهربائي لتفاعل اختزال الأكسجين (ORR). يوفر المحفز CR-Co/CINC المحدد جيدًا انتقائية ملحوظة لأربعة إلكترونات وكثافة تيار حركي عالية تتجاوز تلك الخاصة بـ Pt/C. علاوة على ذلك، فإن البطارية القابلة لإعادة الشحن (ZAB) مع محفز CR-Co/CINC تظهر كفاءة عالية وثبات قوي عند كثافة تيار عالية. يبرز هذا العمل أهمية تعديل الهيكل الإلكتروني ويعمق الفهم للتطور الديناميكي لمحفزات الذرات الفردية في تعزيز أداء ORR.
طرق
تركيب ZIF-8
في إجراء نموذجي، تم إذابة 2-ميثيل إيميدازول (14.575 جرام) في الميثانول (250 مل). بعد ذلك، تم إضافة ميثانول (250 مل) يحتوي علىتمت إضافته إلى الحل أعلاه تحت
الشكل 5 | أداء بطارية الزنك-هواء (ZAB). أ مخططات جهد الدائرة المفتوحة لبطاريات الزنك-هواء المجمعة مع CR-Co/CINC و Pt/C. ب منحنيات استقطاب التفريغ ومخططات كثافة الطاقة لبطاريات الزنك-هواء المعتمدة على CR-Co/ClNC و Pt/C. ج محدد
سعة تطبيع الكتلة عند كثافة تيار . منحنيات التفريغ الجلفاني الاستاتيكي تحت كثافات تيار مختلفة.
تحريك مستمر لمدة 120 دقيقة في درجة حرارة الغرفة. بعد ذلك، تم الاحتفاظ بالتعليق الحليبي الأبيض أعلاه في درجة حرارة الغرفة لمدة 12 ساعة دون تحريك. ثم تم طرد العينة الناتجة في جهاز الطرد المركزي عند، تم غسله بالميثانول ثلاث مرات وتجفيفه تحت الفراغ لمدة 12 ساعة في.
تركيب أوراق نانوية من NC
تم خلط ZIF-8 (2.5 جرام) الذي تم الحصول عليه كما هو موضح أعلاه مع 10 جرام من KCl وطحنهما جيدًا، تلاه إذابة في 200 مل من الماء. بعد الموجات فوق الصوتية لمدة ساعتين، تمت إزالة المحلول المائي في العينة عن طريق الطرد المركزي المباشر وتجفيفه عندبين عشية وضحاها. تم طحن المسحوق الناتج بالكامل وتسخينه إلى ) في جو من الأرجون لمدة 3 ساعات. بعد ذلك، تم طحن العينة المستخرجة بالكامل، وتحريكها مع لمدة ساعتين، تم غسلها بـ HCl ثلاث مرات، ثم تم غسلها بالماء والإيثانول مرتين لكل منهما، وأخيرًا تم تجفيفها طوال الليل تحت الفراغ في .
تركيب محفز CR-Co/CINC
تم أولاً توزيع الـ NC (100 ملغ) في محلول إيثانول بتركيز 20 مل و subjected إلى الموجات فوق الصوتية لمدة 10 دقائق. بعد التوزيع المتجانس، تم إضافة 20 ملغ من كلوريد الكوبالت سداسي الماء إلى المحلول أعلاه وتم تعريضه للموجات فوق الصوتية لمدة 10 دقائق. ثم، تم إغلاق المحلول وتحريكه مغناطيسياً لمدة 12 ساعة تحت التبخر الدوار عندتم نقل العينة الناتجة إلى فرن تجفيف مفرغ عندعندما كانت المحلول قد تبخر تقريبًا. تم طحن المسحوق الناتج بالكامل وتسخينه إلىفي جو من الأرجون لمدة 5 ساعات. بعد ذلك، تم غسل المسحوق ثلاث مرات بمحلول مختلط من الماء والإيثانول، وتم الطرد المركزي بعد الموجات فوق الصوتية وتجفيفه تحت الفراغ عند.
تركيبمحفز
التم إعداد العينة وفقًا لنفس إجراء CRلكن تم تلدينه عند.
توصيف الشكل والبنية
تم الحصول على قياسات حيود الأشعة السينية بالمسحوق (XRD) باستخدام جهاز حيود الأشعة السينية Philips X’ Pert Pro Super.الإشعاع. تم تسجيل طيف الإلكترون الضوئي بالأشعة السينية (XPS) على جهاز ESCALAB MKII معكمصدر للإثارة. تم إجراء تحليلات المجهر الإلكتروني الماسح (SEM) على مجهر إلكتروني ماسح (Gemini SEM 500). تم إجراء المجهر الإلكتروني الناقل (TEM) والمجهر الإلكتروني الناقل الماسح مع التحليل الطيفي للطاقة المشتتة (STEM-EDS) باستخدام مجهر (JEM-2100F، عند 200 كيلوفولت)، وتم إجراء المجهر الإلكتروني الناقل الماسح بتصحيح التشوهات (AC-HAADF-TEM، JEM-ARM200F) عند 200 كيلوفولت مع مصحح تشوهات كروية للنبضة.
التوصيف الكهروكيميائي
تم تحضير حبر المحفز عن طريق استخدام الموجات فوق الصوتية للمزيج الذي يتكون من المحفز (5 ملغ) محلول نافيون ( )، والمذيب ( ماء/إيثانول ) لعدة ساعات حتى تم تشكيل معلق متجانس. بعد ذلك، تم أخذ كمية معينة من حبر المحفز تم طلاء وتجفيف على سطح القطب الكربوني الزجاجي المصقول (3 مم) في درجة حرارة الغرفة، وكتلة المحفز المودعة على القطب هيتم إجراء جميع القياسات الكهروكيميائية على جهاز العمل الكهروكيميائي CHI760E (CH Instruments، الصين) باستخدام نظام ثلاثي الأقطاب قياسي يتضمن قضيب كربوني،إلكترود (كلوريد البوتاسيوم المشبع)، و إلكترود كربون زجاجي مغطى بمحفز يعمل كإلكترود مضاد، وإلكترود مرجعي، وإلكترود عمل، على التوالي. تم تسجيل منحنيات الاستقطاب لتفاعل اختزال الأكسجين عند درجة حرارة الغرفة فيمحلول مائي مشبع من KOH بتركيز 0.1 م عند سرعات دوران مختلفة ). لاختبار الكرونوأمبريومترية في الوقت الحالي، تم نفخها في محلول 0.1 م من هيدروكسيد البوتاسيوم لمدة 30 دقيقة قبل التجربة وتدفقتم الحفاظ على ذلك فوق الإلكتروليت خلال الاختبار لضمان تشبع الأكسجين. كانت عملية الاختبار ثابتة عند 0.7 فولت. في هذا العمل، تم معايرة الجهد النهائي إلى قطب هيدروجين قابل للعكس (RHE)، ما لم يُذكر خلاف ذلك.
تم حساب عدد الإلكترونات المنقولة (n) لتفاعل اختزال الأكسجين وفقًا لمعادلة كوتيكي-ليفش أدناه:
أين هو كثافة التيار المقاسة و و هي حركيات نقل الشحنة وكثافات التيار المحدودة بالانتشار، على التوالي.يمثل السرعة الزاوية للإلكترود الدوار ( ) ، هو عدد نقل الإلكترونات في اختزال الأكسجين، هو ثابت فاراداي ( )، و هو تركيز الكتلة للأكسجين. يشير إلى معامل انتشار الأكسجين، و هي اللزوجة الحركية للإلكتروليت. و في محلول إلكتروليتي 0.1 م من هيدروكسيد البوتاسيوم كانت و ، على التوالي.
تم إجراء قياسات RRDE لحساب إنتاج بيروكسيد الهيدروجين. ) ورقم نقل الإلكترون المقابل ( ) كما يلي:
أين و هي تيارات القرص والحلقة، على التوالي. كفاءة جمع تيار حلقة البلاتينتم تحديده على أنه 0.37.
حساب النشاط الكتلي وتردد الدوران
يمكن تحديد النشاط الكتلي من خلال اعتبار مواقع المعادن كمواقع نشطة. في البداية، تم حساب محتوى المعدن في محفز CR-Co/CINC من كتلة تحميل الكوبالت على القطب. بعد ذلك، تم حساب النشاط الكتلي ( ) تم حسابها وفقًا للمعادلة التالية: المساحة الهندسية للقطب الكهربائي ( ) من وكتلة تحميل المعدن من المحفز الكهربائي ( ) تم أخذها بعين الاعتبار.
وبالمثل، من خلال افتراض أن كل ذرة كوبالت في المحفز تعتبر موقعًا نشطًا فرديًا، يمكن بعد ذلك حساب عدد المواقع النشطة في محفز CR-Co/CINC بناءً على كتلة تحميل الكوبالت على القطب. يمكن اشتقاق تردد التحويل (TOF) من المعادلة التالية:
أين هو كثافة التيار الحركي ( ) ، هو عدد الإلكترونات لكل كولوم ) ، هو كتلة تحميل المعدن على القطب، هو ثابت أفوجادرو ( )، و هو الكتلة المولية لـ.
تجميع واختبار كهربائي كيميائي لبطاريات الزنك-هواء (ZAB)
تم تقييم السائل ZAB باستخدام جهاز مصنوع يدويًا بشكل دوري تحت ظروف جوية محيطة. كان المحلول الكهربائي مكونًا من 6 م كOH مع 0.2 م أسيتات الزنك، وتدفق منتم الحفاظ على ذلك في الإلكتروليت خلال الاختبار لضمانالتشبع. تم استخدام المحفزات المطلية على ورق الكربون كتركيب إلكترود الغشاء (MEA) للقطب السالب (تم التحكم في منطقة طلاء المحفزات عند )، ولوح زنك بمساحة فعالة قدرها 1 سم عمل كأنود. تم تسجيل منحنيات الاستقطاب بواسطة LSV في درجة حرارة الغرفة على جهاز العمل الكهربائي CHI 760E. تم تطبيع كل من كثافة التيار وكثافة الطاقة على المساحة السطحية الفعالة للقطب الكهربائي الهوائي. تم حساب السعة النوعية وفقًا للمعادلات التالية:
أين هو التيار المطبق (أمبير)، هو وقت التقديم (ث)، و يمثل وزن الزنك المستهلك (غ).
قياسات XAFS في الموقع
تم إجراء قياسات XAFS في الموقع في محطة 1W1B في منشأة الإشعاع السنكروتروني في بكين (BSRF)، الصين. الحد الأقصى للتيار هو 250 مللي أمبير من حلقة التخزين في BSRF. استخدمت المحطة الخطية مقياس الطيف الثنائي البلورات Si (111)، وتمت عملية ضبط إضافية.لإزالة التوافقيات العليا عند تنفيذ الـ Co-قياسات XAFS عند حافة. خلال قياسات XAFS في الموقع، تم اختيار أقمشة الكربون المطلية بمحفز الكوبالت كقطب عمل في محلول قلوي من خلال خلية مصنوعة في المنزل عبر نظام ثلاثي الأقطاب. على وجه التحديد، تم توزيع عينة 5 ملغ بالتساوي في 1 مل من المحلول (ماء/إيثانول ) يحتوي على نافيوين. ثم، تم ترسيب حبر المحفز كالكاثود العامل على ورق الكربون. )، مع تثبيت فيلم الكابتون على ظهر ورق الكربون لضمان أن المزيد من مواقع الفتح يمكن أن تشارك في تفاعل اختزال الأكسجين. الجهود التمثيلية ( ) تم اختيارها للحصول على تطور الهيكل الذري والإلكتروني لمواقع الكوبالت خلال عملية اختزال الأكسجين. عند جمع بيانات قياس XAFS، استخدمنا عينة معيارية من رقائق الكوبالت لمعايرة موضع حافة الامتصاص ( )، وجمعنا جميع بيانات XAFS خلال فترة زمنية محددة. استخدمنا كاشف Lytle (وضع الفلورية) لتسجيل طيف XAFS أثناء التفاعلات الكهروكيميائية.
قياسات SRIR في الموقع
قمنا بإجراء قياسات SRIR في الموقع في خط الشعاع BL01B من مختبر الإشعاع السنكروتروني الوطني (NSRL، الصين). من أجل الحصول على إشارات تحت الحمراء أفضل، تم إجراء اختبارات كيميائية كهربائية في خلية مصنوعة يدويًا ذات لوحة علوية. وبشكل محدد، مع الأخذ في الاعتبار امتصاص الاهتزاز لجزيئات الماء، استخدمنا نافذة بلورية من ZnSe وضغطنا الكاثود المحفز بالقرب من النافذة لتقليل فقدان الضوء تحت الأحمر. تم استخدام اختبارات SRIR في وضع الانعكاس بدقة طيفية منتمت قياسات ORR النظامية بعد قياس طيف الخلفية لقطب التحفيز عند جهد دائرة مفتوحة، وكان جهد ORR يتراوح بين 1.00 و 0.65 فولت.
توفر البيانات
البيانات التي تدعم نتائج هذه الدراسة متاحة ضمن المقال وفي ملفات المعلومات التكميلية. جميع البيانات متاحة من المؤلفين عند الطلب.
References
Luo, F. et al. P-block single-metal-site tin/nitrogen-doped carbon fuel cell cathode catalyst for oxygen reduction reaction. Nat. Mater. 19, 1215-1223 (2020).
Gao, R. et al. Pt/Fe2O3 with Pt-Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy 6, 614-623 (2021).
Li, Y. & Dai, H. Recent advances in zinc-air batteries. Chem. Soc. Rev. 43, 5257-5275 (2014).
Xie, X. et al. Performance enhancement and degradation mechanism identification of a single-atom catalyst for proton exchange membrane fuel cells. Nat. Catal. 3, 1044-1054 (2020).
Zaman, S. et al. Oxygen reduction electrocatalysts toward practical fuel cells: progress and perspectives. Angew. Chem. 133, 17976-17996 (2021).
Zhao, Y. et al. S and O Cocoordinated Mo single sites in hierarchically porous tubes from sulfur-enamine copolymerization for oxygen reduction and evolution. J. Am. Chem. Soc. 144, 20571-20581 (2022).
Fu, J. et al. Electrically rechargeable zinc-air batteries: progress, challenges, and perspectives. Adv. Mater. 29, 1604685 (2017).
Lai, W. H. et al. General п-electron-assisted strategy for Ir, Pt, Ru, Pd, Fe, Ni single-atom electrocatalysts with bifunctional active sites for highly efficient water splitting. Angew. Chem. Int. Ed. 58, 11868-11873 (2019).
Kodama, K., Nagai, T., Kuwaki, A., Jinnouchi, R. & Morimoto, Y. Challenges in applying highly active Pt-based nanostructured catalysts for oxygen reduction reactions to fuel cell vehicles. Nat. Nanotechnol. 16, 140-147 (2021).
Yuan, Y. et al. Zirconium nitride catalysts surpass platinum for oxygen reduction. Nat. Mater. 19, 282-286 (2020).
Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Z. Design of electrocatalysts for oxygen-and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 44, 2060-2086 (2015).
Lian, Y. et al. Unpaired 3d electrons on atomically dispersed cobalt centres in coordination polymers regulate both oxygen reduction reaction (ORR) activity and selectivity for use in zinc-air batteries. Angew. Chem. Int. Ed. 59, 286-294 (2020).
Shang, H. et al. Engineering unsymmetrically coordinated Cu-S1N3 single atom sites with enhanced oxygen reduction activity. Nat. Commun. 11, 3049 (2020).
Zhou, W. et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 13, 6414 (2022).
Zhang, H. et al. Symbiotic synergy enabling moderate oxo-hydroxy adsorption capacity for high-selectivity oxygen reduction. Nano Energy 101, 107587 (2022).
Liu, M. et al. Synergetic Dual-Ion Centers Boosting Metal Organic Framework Alloy Catalysts toward Efficient Two Electron Oxygen Reduction. Small 18, 2202248 (2022).
Qu, Y. et al. Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms. Nat. Catal. 1, 781-786 (2018).
An, Q. et al. Engineering Unsymmetrically Coordinated Fe Sites via Heteroatom Pairs Synergetic Contribution for Efficient Oxygen Reduction. Small 19, 2304303 (2023).
Su, H. et al. Hetero-N-coordinated Co single sites with high turnover frequency for efficient electrocatalytic oxygen evolution in an acidic medium. ACS Energy Lett. 4, 1816-1822 (2019).
Mun, Y. et al. Versatile strategy for tuning ORR activity of a single FeN4 site by controlling electron-withdrawing/donating properties of a carbon plane. J. Am. Chem. Soc. 141, 6254-6262 (2019).
Zheng, X. et al. Enriched d-band holes enabling fast oxygen evolution kinetics on atomic-layered defect-rich lithium cobalt oxide nanosheets. Adv. Func. Mater. 32, 2200663 (2022).
Zu, L. et al. Self-assembly of Ir-based nanosheets with ordered interlayer space for enhanced electrocatalytic water oxidation. J. Am. Chem. Soc. 144, 2208-2217 (2022).
Grimaud, A. et al. Activation of surface oxygen sites on an iridiumbased model catalyst for the oxygen evolution reaction. Nat. Energy 2, 1-10 (2016).
Jia, C. et al. Sulfur-Dopant-Promoted Electroreduction of over Coordinatively Unsaturated Ni-N2 Moieties. Angew. Chem. Int. Ed. 60, 23342-23348 (2021).
Liu, M. et al. Self-nanocavity-confined halogen anions boosting the high selectivity of the two-electron oxygen reduction pathway over Ni-based MOFs. J. Phys. Chem. Lett. 12, 8706-8712 (2021).
Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
Zhou, W. et al. Identification of the evolving dynamics of coordination-unsaturated iron atomic active sites under reaction conditions. ACS Energy Lett. 6, 3359-3366 (2021).
Su, H., Soldatov, M. A., Roldugin, V. & Liu, Q. Platinum single-atom catalyst with self-adjustable valence state for large-current-density acidic water oxidation. eScience 2, 102-109 (2022).
Hai, X. et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 17, 174-181 (2022).
Zhang, Z. et al. Ultraconformal chemo-mechanical stable cathode interface for high-performance all-solid-state batteries at wide temperatures. Energy Environ. Sci. 16, 4453-4463 (2023).
Sun, T. et al. Single-atomic cobalt sites embedded in hierarchically ordered porous nitrogen-doped carbon as a superior bifunctional electrocatalyst. Proc. Natl Acad. Sci. 115, 12692-12697 (2018).
Gong, C., Yang, C., Zhou, W., Su, H. & Liu, Q. Modulation of Electronic States in Bimetallic-doped Nitrogen-Carbon Based Nanoparticles for Enhanced Oxygen Reduction Kinetics. Chinese J. Chem. Phys. https://doi.org/10.1063/1674-0068/ cjcp2304033 (2023).
Liang, Y. et al. Covalent hybrid of spinel manganese-cobalt oxide and graphene as advanced oxygen reduction electrocatalysts. J. Am. Chem. Soc. 134, 3517-3523 (2012).
Yi, J.-D. et al. Atomically dispersed iron-nitrogen active sites within porphyrinic triazine-based frameworks for oxygen reduction reaction in both alkaline and acidic media. ACS Energy Lett. 3, 883-889 (2018).
He, C. et al. Molecular evidence for metallic cobalt boosting CO2 electroreduction on pyridinic nitrogen. Angew. Chem. 132, 4944-4949 (2020).
Edgington, J., Schweitzer, N., Alayoglu, S. & Seitz, L. C. Constant change: exploring dynamic oxygen evolution reaction catalysis and material transformations in strontium zinc iridate perovskite in acid. J. Am. Chem. Soc. 143, 9961-9971 (2021).
Cheng, W., Su, H. & Liu, Q. Tracking the Oxygen Dynamics of Solid-Liquid Electrochemical Interfaces by Correlative In Situ Synchrotron Spectroscopies. Acc. Chem. Res. 55, 1949-1959 (2022).
Su, H. et al. Dynamic evolution of solid-liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 142, 12306-12313 (2020).
Cheng, W. et al. Lattice-strained metal-organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 4, 115-122 (2019).
Yang, C. et al. Dynamically-evolved surface heterojunction in iridium nanocrystals boosting acidic oxygen evolution and overall water splitting. J. Energy Chem. 78, 374-380 (2023).
Zandi, O. & Hamann, T. W. Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy. Nat. Chem. 8, 778-783 (2016).
Rao, R. R. et al. Operando identification of site-dependent water oxidation activity on ruthenium dioxide single-crystal surfaces. Nat. Catal. 3, 516-525 (2020).
Sivasankar, N., Weare, W. W. & Frei, H. Direct observation of a hydroperoxide surface intermediate upon visible light-driven water oxidation at an Ir oxide nanocluster catalyst by rapid-scan FT-IR spectroscopy. J. Am. Chem. Soc. 133, 12976-12979 (2011).
شكر وتقدير
تم دعم هذا العمل من قبل البرنامج الوطني الرئيسي للبحث والتطوير في الصين (2022YFA1502903 (Q.L.))، ومؤسسة العلوم الطبيعية الوطنية في الصين (12205300 (H.S.)، 22241202 (Q.L.)، و12135012 (H.S.))، ومؤسسة العلوم الطبيعية في مقاطعة آنهوي (2208085JO1 (Q.L.) و2208085QA28 (H.S.))، وبرنامج البحث والتطوير الرئيسي لمركز علوم هيفي، الأكاديمية الصينية للعلوم (2022HSC-KPRD003 (W.Z.)). تم تنفيذ هذا العمل جزئيًا في مركز الأدوات للعلوم الفيزيائية، جامعة العلوم والتكنولوجيا في الصين.
مساهمات المؤلفين
قام Q.L. وH.S. وJ.P. بتصور المشروع. قام M.L. وJ.Z. بتنفيذ التجارب. قام Q.L. وH.S. وM.L. وY.J. وW.Z. وC.Y. وS.B. بتحليل البيانات التجريبية. تم كتابة المخطوطة بواسطة M.L. وH.S. وQ.L. مع مساهمات من جميع المؤلفين.
مختبر الإشعاع السنكروتروني الوطني، جامعة العلوم والتكنولوجيا في الصين، هيفي 230029 آنهوي، الصين.المختبر الوطني الرئيسي لعلوم المعادن المسحوقة، جامعة جنوب الوسط، تشانغشا 410083 هونان، الصين.المختبر الرئيسي لمواد تحويل الطاقة الضوئية في كلية مقاطعة هونان، كلية الكيمياء والهندسة الكيميائية، جامعة هونان العادية، تشانغشا 410081 هونان، الصين.ساهم هؤلاء المؤلفون بالتساوي: ميهوان ليو، جينغ زانغ.البريد الإلكتروني:suhui@ustc.edu.cn;jun.pan@csu.edu.cn;qhliu@ustc.edu.cn
Single-atom catalysts, especially those with metal- moieties, hold great promise for facilitating the oxygen reduction reaction. However, the symmetrical distribution of electrons within the metal- moiety results in unsatisfactory adsorption strength of intermediates, thereby limiting their performance improvements. Herein, we present atomically coordinationregulated Co single-atom catalysts that comprise a symmetry-broken moiety, which serves to break the symmetrical electron distribution. In situ characterizations reveal the dynamic evolution of the symmetry-broken Cl moiety into a coordination-reduced structure, effectively optimizing the electron filling of Co sites toward a reduced -band electron occupancy ( ) under reaction conditions for a fast four-electron oxygen reduction reaction process. As a result, the coordination-regulated Co single-atom catalysts deliver a large half-potential of 0.93 V and a mass activity of . Importantly, a Zn-air battery using the coordinationregulated Co single-atom catalysts as the cathode also exhibits a large power density and excellent stability.
The electrochemical oxygen reduction reaction (ORR) serves as a pivotal component in emerging energy technologies such as fuel cells and metal-air batteries, presenting a promising and environmentally friendly avenue for modern sustainable energy conversion and storage . Unfortunately, the ORR is a multi-step process involving multi-electron and proton coupling that suffers from sluggish kinetics and intricate mechanisms . Although platinum-based materials have historically demonstrated high catalytic efficiency in facilitating the ORR, the high cost, scarcity, and poor stability of platinum have hindered their widespread commercial application . Consequently, materials aspiring to function as effective ORR electrocatalysts must possess sufficient reactivity for cleaving the bond, an optimized geometric construction to facilitate efficient diffusion and mass transfer, and robust reactive sites for long-term durability . Among
these, single-atom catalysts (SACs), especially those housing isolated single-metal sites anchored in a nitrogen-doped carbon matrix ( -C), have garnered significant attention for ORR owing to their tunable electronic properties, distinctive geometric configurations, maximal atomic utilization efficiency, and low . Consequently, the pursuit of developing exemplary -based ORR electrocatalysts with high selectivity, sufficient reactivity, and industrial applicability, is increasingly receiving serious attention, yet remains a formidable challenge. materials, especially those featuring symmetrical planar configurations with four nitrogen-coordinated metal sites ( moieties), have been extensively investigated and have demonstrated favorable catalytic performance . Nevertheless, their ORR activity and four-electron ( ) selectivity still fall short when compared to the
benchmark Pt/C. This deficiency is attributed to the unfavorable adsorption capacity for oxygen-related intermediates, resulting from the symmetric distribution of electrons within the planar moieties . Noticeably, the activity and selectivity of SACs are closely related to the electron occupancy of orbitals, as they engage in bonding with oxygen-related species . Regrettably, for the transition metals, the more -band electrons in the symmetrical moieties can lead to reduced orbital overlap with electrons, necessitating higher energy for the formation and evolution of *OOH species . To address this challenge, the modulation of the coordination environment of metal centers using elements with varying atomic radii and electronegativity is considered a promising strategy to break the planar symmetry of electron density . Furthermore, the potential-dependent surface rearrangement of electrocatalysts has been predicted in theoretical calculations and observed in experiments, which can in situ optimize the electron distribution of the active sites to accelerate the adsorption and dissociation of reactive species . Fallaciously, the stable symmetrical moiety impedes the self-optimization of active centers during the reaction, posing an obstacle for improving their ORR activity. Thus, breaking the symmetric electron distribution of single-atom metal centers is impressive for actuating structural self-reconstruction, in situ optimizing the adsorption strength of oxygen species, and subsequently enhancing the four-electron ORR properties.
Herein, we have successfully fabricated coordination-regulated Co sites (CR-Co) confined in N-doped carbon nanosheets through a facile two-step pyrolysis strategy. At the atomic level, both local electric quadrupole transition (1s to transition) and local electric dipole transition (1s to transition) are achieved by introducing Cl bonding to form the symmetry-broken moiety in the N -doped carbon matrix (CR-Co/ClNC), thereby evidencing the breakage of symmetric electron distribution. In situ X-ray absorption fine structure (XAFS) spectroscopy reveals that the moiety dynamically releases two N coordination to form an unsaturated structure at the early reaction state. Especially, this structural self-rearrangement can in situ optimize the electron filling of Co sites toward a low -band electron occupancy ( ) during the reaction. This, in turn, promotes the transformation of the bond from absorbed oxygencontaining species into and further evolution into the ( ) structure. This transformation has been corroborated by in situ synchrotron radiation infrared spectroscopy (SRIR) results, and it subsequently enhances the four-electron ORR properties. The welldesigned CR-Co/CINC catalyst exhibits significantly improved ORR selectivity (98%) with a half-wave potential of 0.93 V and a high mass activity of at 0.85 V . This represents an approximate 47fold increase compared to that of commercial . As a proof of concept, the Zn-air battery assembled with the CR-Co/CINC catalyst delivers a superior power density of compared to Pt/C ( ), which suggests excellent potential for practical application in metal-air batteries.
Results and discussion
Morphology and structure characterizations of electrocatalysts
The atomically dispersed cobalt electrocatalyst in which coordinationregulated Co sites were confined within N-doped carbon nanosheets was prepared via a facile two-step pyrolysis strategy. Initially, ZIF-8 was blended with KCl powder and subjected to pyrolysis under a flowing Ar atmosphere to yield nitrogen-doped carbon (NC) nanosheets with accessible active sites. Subsequently, to regulate the Co coordination field, the coordination of metal sites in the Co salt containing Cl was introduced onto the surface of the NC nanosheet by pyrolysis at different temperatures, in which was selected to obtain CR-Co/ CINC. In this process, the Co atoms were simultaneously trapped and anchored in NC due to the strong coupling effect with lone-pair electrons of N species. Meanwhile, the sample was prepared as a
reference by elevating the pyrolysis temperature to to exclude the Cl component .
Transmission electron microscopy (TEM) images demonstrate that the obtained CR-Co/CINC and Co/NC catalysts possess a sheet-like structure (Fig. 1a and Supplementary Figs. 1 and 2) without obvious nanoparticles. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, Fig. 1b) of CR-Co/CINC confirms the presence of dense and isolated speckled bright dots marked by red circles, signifying atomically dispersed Co atoms anchored on the NC substrate. This can be further proven by the powder X-ray diffraction (XRD) results in Supplementary Fig. 3, which show the absence of diffraction peaks corresponding to Co species. To further clarify the morphological structure of Co sites, elemental mapping images reveal the homogeneous distribution of , and Cl in the entire CINC architecture (Fig. 1c) and homogeneously dispersed C, N, and Co in the sample (Fig. 1d). The physisorption isotherms reveal a slightly greater specific surface area of CR-Co/CINC and similar pore architecture in comparison with (Supplementary Fig. 4). Significantly, the Co loadings for CR-Co/CINC and Co/NC were and , respectively, as determined from inductively coupled plasma-optical emission spectrometry (ICP-OES) analysis. These results confirm the uniform dispersion of Co atoms on the surface sites of the NC substrate and the introduction of Cl in .
Additionally, in the high-resolution X-ray photoelectron spectroscopy (XPS) spectrum (Supplementary Fig. 5) of CR-Co/CINC, the characteristic peak at 197.7 eV corresponds to the species, while peaks at and are attributed to the bond . The content of Cl in is determined to be 2.15 by XPS. The N 1s spectra (Supplementary Fig. 6) also show a peak at 399.4 eV , which can be assigned to the bond. Collectively, the appearance of and bonds in the XPS spectra of CR-Co/ CINC indicates the coordination of Co atoms with both Cl and N atoms. More importantly, extended X-ray absorption fine structure (EXAFS) spectra were obtained to further identify the coordination environment of cobalt (Fig. 1e). To exclude the high-frequency noise in the high region and ensure sufficient independent free points during EXAFS fitting, a range of was selected for all Co samples during the fast Fourier transform (Supplementary Fig. 7). In contrast to the Co foil and CoOOH references, the prominent peak located at can be assigned to the bond, owing to the presence of N and Cl elements, with no scattering peak appeared in both and , validating an atomic dispersion of Co in the NC support . Following the introduction of Cl into , the Co peak exhibits a positive shift of compared to the sample, suggesting the formation of a coordination structure due to the larger atomic radius of Cl . Furthermore, to elucidate the local coordination structure of Co sites, the fitted curves of the weighted Co -edge EXAFS spectra in Fig. 1e and Supplementary Fig. 8, along with the corresponding data of and listed in Supplementary Table 1, show that the coordination number (CN) of the bond for is 4.1 . For of 3.9 and 1.0 for the bond and an axial coordination depicted in Fig. 1e are obtained, respectively. To highlight the local structure of the Co metal site and axial Cl coordination, the local structure model is shown in Fig. 1e. The above results imply that the proposed local coordination environment of CR-Co/ClNC is composed of a symmetry-broken Cl-Co moiety, suggesting the successful regulation of the coordination field of the Co center by introducing a highly electronegative Cl element.
To gain atomic-level insights into the local electronics of Co, soft X-ray absorption near-edge structure spectroscopy (XANES) for N was performed, as shown in Fig. 2a. The peak at is attributed to the excitation of hybrid orbital of , while the peak at arises from transitions . Upon the introduction of Co sites in the NC substrate, new peaks at 399.4 eV were
Fig. 1 | Morphology and structure characterizations. a TEM and b HAADF-STEM images of the CR-Co/CINC catalyst. TEM-EDS mapping images for CR-Co/CINC and Co/NC. e FT-EXAFS spectra of the -edge for the catalyst and
reference samples, and the corresponding fitting curves for and NC . The inset shows , and C atoms indicated by cyan, purple, dark blue, and gray.
Fig. 2 | Chemical state and atomic local structure. a -edge XANES spectra of and NC. b N XPS spectra of CR-Co/ClNC and Co/NC. c Co edge XANES spectra for CR-Co/CINC and reference samples. The inset shows the
fitted pre-edge region. The fitted average formal -band electron counts. The error bars are the standard deviations of three replicate calculation. e Co XPS spectra of and .
observed for both CR-Co/CINC and , which can be assigned to the excitation of . For , the peak displays a slight positive shift of in comparison with , suggesting a reduced electron transfer of after the introduction of Cl elements . Furthermore, the high-resolution XPS N 1s spectra of both catalysts can be deconvoluted into pyridinic , pyrrolic N , and graphitic N species (Fig. 2 b. The corresponding ratios of the
deconvoluted N species are summarized in Supplementary Fig. 9. It reveals the presence of N types in similar proportions, thus excluding the influence of the N type in the substrate on ORR performance. Meanwhile, the high-energy shift of binding energy ( ) for Co -N in CR-Co/CINC proves that the reduced interaction between Co and N is attributed to the introduced Cl atoms, which matches well with the -edge XANES results .
Fig. 3 | Electrochemical oxygen reduction performance. a Polarization curves for CR-Co/ClNC and references under -saturated , scan rate of The contrast between CR-Co/CINC and references for and . The error bars are the standard deviations of three replicate calculation. Tafel slopes and Mass activity (MA) and turnover frequency (TOF) for CR-Co/CINC and
reference samples. The error bars error bars are the standard deviations of three replicate calculation. e Electron transfer number (top) and yield (bottom) vs potential of CR-Co/CINC and Pt/C. f Current-time chronoamperometric responses of and .
To obtain in-depth insight into the local electronic structure changes of Co sites after introducing Cl at the atomic scale, XANES spectra of the Co -edge were adopted (Fig. 2c). The XANES spectrum of CR-Co/ClNC display different peak shapes and intensities, indicating a different coordination environment and local electronic structure as compared to . The enlarged pre-edge feature (peak A) of CR-Co/ CINC (inset, magnified image in Fig. 2c) suggests the presence of both local electric quadrupole transition ( to transition) and local electric dipole transition (1s to transition) in the catalyst, clearly confirming a noncentrosymmetric electron distribution of Co sites in . The maximum value of the first derivative of CINC exceeds that of CoOOH , revealing a higher Co oxidation state in CR-Co/CINC related to CoOOH (Supplementary Fig. 10). Additionally, the high-energy shift of the absorption edge and the enhanced intensity of the white-line peak in CR-Co/CINC demonstrate an increased valence state of Co following the introduction of . The valence state can be obtained by linear fitting of the absorption edge position (Supplementary Fig. 11). Figure 2d illustrates the absorption edge positions of the catalysts as a function of the formal -band electron count obtained from the standard. Notably, exhibits a distinct lower number of -band electrons (5.80) compared with (5.96), suggesting a pronounced hybridization of states, effectively breaking the symmetric electron distribution of Co-N moieties. The Co peak in CR-Co/CINC shifts to a higher binding energy than that of (Fig. 2e), indicating a higher valence state of Co following Cl regulation, which is consistent with the previous XANES results. Consequently, these findings demonstrate that the symmetry-broken moieties in CR-Co/ClNC effectively break the symmetric electron distribution of Co sites in a typical moiety, leading to a reduced number of -band electrons, and ultimately optimizing the performance of the active sites.
Electrochemical oxygen reduction performance
To assess the ORR performances, the as-prepared samples and commercial Pt/C were subjected to evaluation using a rotating disk electrode (RDE) in -saturated 0.1 M KOH electrolyte. The comparison of linear scan voltammogram (LSV) curves for CR-Co/CINC, Co/NC, NC,
and catalysts are illustrated in Fig. 3a. The exhibits an optimal ORR activity with a half-wave potential ( ) of 0.93 V vs a reversible hydrogen electrode (RHE, the potentials mentioned below are all relative to RHE) (Fig. 3b), obviously outperforming those of commercial and NC. A higher onset potential of 1.008 V is required for CR-Co/CINC to achieve 5% of the diffusion-limited current (Supplementary Fig. 12), slightly surpassing that of Pt/C ( 0.994 V ). Furthermore, the mass transfer limiting current density for CR-Co/ CINC reaches , exceeding that observed for the commercial Pt/C catalyst ( ). This higher limiting current density seems to be a consequence of the fast desorption rates of product on the CR-Co/CINC electrode. In addition, CR-Co/CINC possesses a superior kinetic current density ( ) up to at 0.85 V , fold that of . In contrast, displays inferior ORR activity with reduced and a more negative ( 0.79 V ), demonstrating the optimal intrinsic ORR activity of CR-Co/ CINC attributed to suitable modulation of the coordination field of Co sites with Cl elements. Meanwhile, the ORR performance achieved by CR-Co/CINC is superior to those of most recently reported SACs (Supplementary Table 2). The faster ORR kinetics for CR-Co/CINC are further confirmed by its smallest Tafel slope ( ) compared to , and NC ( ) (Fig. 3c). Additionally, as shown in Fig. 3d, CR-Co/ClNC delivers a high turnover frequency (TOF) of and a large mass activity (MA) of at 0.85 V , almost 15 and 47 times higher than those of commercial and . To eliminate the influence of test errors, including the loading of catalyst, the uniformity of electrode film, the flatness of the surface of glassy carbon electrode, and the penetration of electrolyte on the electrode surface, CR-Co/ CINC and were tested three times in the repetitive process to obtain the error bars of ECSA (Supplementary Figs. 13-15). Moreover, similar electrochemically active surface area (ECSA) for CR-Co/CINC ( with an error bar of ) and with an error bar of ) indicates that the source of enhanced activity of is the structure formed by axial Cl coordination, which optimizes the electronic structure to achieve increased intrinsic activity of the active site, rather than the
Fig. 4 | In situ XAFS and SRIR characterizations. a The curve-fitting analysis of EXAFS spectra and XANES spectra of Co -edge recorded at different applied potentials during the ORR process for CR-Co/CINC. Inset, magnified white-line peak region. c The fitted average formal -band electron counts of Co at CR-Co/ClNC
under ex situ, 1.00 V , and 0.90 V conditions based on the absorption edge of Co K edge XANES spectra. The error bars are the standard deviations of three replicate calculation. In situ SRIR measurements under various potentials for (d) CR-Co/CINC and (e) . ORR schematics of .
increase in the number of active sites (Supplementary Figs. 16-18). These results demonstrate that the introduction of axial Cl atoms in the symmetry-broken moieties with low -band electron filling can significantly enhance the ORR activity.
Apart from the ORR catalytic activity, catalytic selectivity is an important index to evaluate the electrocatalytic oxygen reduction performance. As illustrated in Fig. 3e, the electron transfer number of CR-Co/CINC was calculated to be according to the rotating ring disk electrode (RRDE) measurements, confirming the preference of the CR-Co/CINC catalyst for the pathway. Moreover, the ORR pathway is further proven by the Koutechy-Levich (K-L) equation applied in the diffusion-controlled region at various rotation speeds (Supplementary Figs. 19 and 20). Clearly, the peroxide yield for CR-Co/CINC in the same potential range is below 3%, lower than those of the other samples (Supplementary Fig. 21), indicating the highest four-electron selectivity toward the ORR pathway for CR-Co/CINC. Additionally, the stability and durability of the CR-Co/CINC catalyst were measured via chronoamperometry (CA) and accelerated durability testing (ADT). The CR-Co/CINC exhibits reliable stability with better methanol resistance and lower current density attenuation (<8%) after 120 h of CA testing at 0.7 V , and the CR-Co/CINC catalyst only experiences a 20 mV loss in after the ADT, suggesting its long-term durability for the ORR (Fig. 3f and Supplementary Figs. 22 and 23). The morphology and electronic characterizations, such as HAADF-STEM and XAFS of CR-Co/CINC (Supplementary Figs. 24 and 25) after long-term electrolysis, were performed without observation of Co species and nanoparticles. These results indicate the superior structural robustness and excellent stability of CR-Co/CINC.
In situ characterizations of the evolution of active sites
To deeply elucidate the underlying mechanisms responsible for the exceptional four-electron selectivity of CR-Co/CINC during the ORR process, in situ XAFS measurements of the Co -edge were conducted using a homemade in situ cell (Supplementary Fig. 26a) .Firstly, the evolution of the local coordination environment of the Co sites under applied voltages was clarified by in situ EXAFS results (Supplementary Fig. 26). It is evident that a dominant peak at showed a damping in peak intensity and a positive shift of for CR-Co/ CINC as the potential changed from ex situ (immersed in solution without applied voltage) to 1.00 V conditions. The reduced peak intensity in EXAFS clearly signifies the potential-driven reduction in the coordination of Co single sites and no adsorption by oxygencontaining species at the early reaction state. The peak displays a discernible increase in intensity and a slight negative shift in location when applying voltages of 0.90 and 0.75 V , implying the adsorption of oxygen-containing species during the ORR process. Quantitatively, the EXAFS fitting results in Fig. 4a, Supplementary Figs. 27-30, and Supplementary Table 3 exhibit a coordination number of four bonds and one axial bond under ex situ states, resembling the EXAFS result measured under air conditions. Interestingly, when applying a potential of 1.00 V , the coordination number of the bonds is evidently reduced to two, indicating that the potential-driven structural evolution of Co single sites truly occurs under ORR conditions by releasing Co centers from the N-C substrate to form a Cl-Co- active site. It is of high interest that this coordination-unsaturated structure favors the surface adsorption of oxygen molecules. As the potential continually decreases to 0.90 V , the coordination combined with an additional coordination is retained for the Co
sites, which might be driven from the adsorption of key reactive oxygen-containing intermediates.
The dynamic evolution of the local coordination structure often accompanies the optimization of the electronic structure of the active site under working conditions. XANES analysis is employed to clarify the changes in the electronic structure. In situ XANES spectra of the Co -edge for CR-Co/CINC at different applied voltages are depicted in Fig. 4b. Compared with the ex situ conditions, the absorption edge shows a slight positive-energy shift, and the white-line peak displays a modest increase in intensity at an applied voltage of 1.0 V . To quantify the change in -band electron filling, the absorption edge shifts are correlated with the -band electron counts of Co using as a standard (Fig. 4c and Supplementary Fig. 31). A lower Co 3d electron filling count in CR-Co/CINC (5.28) is observed at 1.0 V . The dynamically evolved experiences a rapid decrease of 0.52 electrons at the early reaction state (corresponding to a dynamic electron evolution of ), implying that there are more -band vacant orbitals for coupling with orbitals to regulate the adsorption of oxygen-containing species. Notably, the Co electron filling count in CR-Co/CINC depopulates much more rapidly and violently with the applied potential than that of (Supplementary Fig. 32), which is beneficial for optimizing the adsorption kinetics of intermediates. These results reveal that a fast in situ modulation of -band electrons occurs at the early reaction state for CR-Co/CINC, as the symmetrybroken moiety rapidly evolves into the coordinationreduced .
To investigate the adsorption properties of key intermediate species over the Co sites with dynamic electron evolution, the surface-sensitive in situ synchrotron SRIR technique was adopted by a dedicated cell . As shown in Fig. 4d, when the applied potential is less than 1.00 V for CR-Co/CINC, the new absorption band at shows a potential-dependent behavior. This can be attributed to the accumulation of crucial intermediates *O over the moieties under ORR conditions because the stretching vibration of the oxygen species (*O) is usually in the range of . To further verify the effect of the coordinated Cl bond, in situ SRIR signals of were also acquired under the same typical potentials for comparison (Fig. 4e). Noticeably, a new absorption band at is observed as the applied voltage decreases. Since the infrared vibration peaks of the *OOH species usually appear in the region of , the absorption band at can be assigned to . Only the absorption band of *O species observed for CR-Co/ClNC during the ORR process clearly implies the rapid cleavage of the bond in the * intermediate on the dynamically evolved sites. Therefore, the adsorption strength of the key *OOH is effectively optimized to rapidly evolve into *O species for CR-Co/ClNC owing to the dynamic electron evolution ( ) at the early reaction state, effectively promoting the four-electron reaction kinetics and enhancing ORR activity and selectivity.
Above all, the dynamic evolution of active sites under working conditions is schematically shown in Fig. 4f. Firstly, the Co centers are dynamically released from the substrate to form a coordinationreduced active structure at the early reaction state (at 1.0 V conditions), revealing a lower -band electron filling. Remarkably, these coordination-reduced moieties obviously optimize the adsorption of oxygen-containing intermediates in situ and promote the cleavage of the bond for fast reaction kinetics. Finally, the *O active structure is retained as the potential steadily decreases to 0.75 V , thereby realizing a highly efficient ORR process. Moreover, the dynamically evolved electron and coordination structures of the Co sites return to their original state after the reaction according to the XAFS results (Supplementary Fig. 33), suggesting that the structural evolution of the Co active site is a dynamic reversible process. These results suggest that the in situ modulated -band electron filling of Co sites through the introduction of Cl can yield
significantly accelerated ORR kinetics, which endows the CR-Co/CINC catalyst with excellent potential for industrial applications.
Zn-air battery (ZAB) performance
In light of the superior ORR performance, the as-prepared CR-Co/CINC was served as a cathode catalyst in aqueous primary zinc-air batteries (ZABs) to investigate its practicability (Supplementary Fig. 34). The same device was assembled using commercial Pt/C for comparison. The CR-Co/ClNC-incorporated Zn-air cell exhibits an impressive opencircuit voltage of 1.50 V (Fig. 5a), which is higher than that of the Pt/Cbased Zn-air cell ( 1.45 V ), indicating a higher output voltage for the battery when using CR-Co/ClNC as the cathode catalyst. From the discharge polarization curves and power density plots for the Zn-air battery (Fig. 5b), it can be seen that the CR-Co/ClNC-based ZAB presents a higher discharging voltage plateau with a maximum power density close to , which outperforms the counterpart ( ). The power density of CR-Co/CINC based ZAB surpasses most of the reported SACs (Supplementary Table 4). Notably, the ZAB with CR-Co/CINC delivers a specific capacity of 745 mAh at a discharging current density of in Fig. 5c, outperforming that of Pt/C-based ZAB ( ). The galvanostatic discharge observations at current densities ranging from 5 to are shown in Fig. 5d. The CR-Co/CINC-based ZAB obviously maintains higher discharge voltages than -based ZAB under the current density ranges and then periodically return to . Following a high-rate discharge at , the discharge potential for the CR-Co/CINC-based ZAB recovers to the starting stage ( ), indicating the good rate capability and reversibility of the CR-Co/CINC-based ZAB, which can be attributed to the efficient ORR activity and good stability of the CR-Co/CINC catalyst. Noticeably, no discernible degradation was recorded after 3 cycles for ZAB with the CR-Co/CINC electrocatalyst over a duration of 30 h at (Supplementary Fig. 35). In addition, the galvanostatic discharge -charge measurements show negligible deterioration of the discharge -charge voltage, reflecting the promising durability of the CR-Co/CINC in Zn-air batteries (Supplementary Fig. 36). These results suggest that the CR-Co/CINC catalyst holds good potential for application in ZABs.
In summary, we have constructed a kind of atomically dispersed and coordination-regulated CR-Co/CINC catalyst with symmetrybroken moieties via a facile two-step pyrolysis strategy, successfully avoiding the introduction of impurities and realizing the coupling of axial Cl over the moiety. At the atomic scale, we have demonstrated that the introduction of axial Cl atoms in the active site could effectively break the symmetric electron distribution of Co moieties, achieving low -band electron filling for in situ regulation of the adsorption strength of oxygen adsorbates around the Co sites. Mechanistic studies employing in situ SRIR and XAFS spectroscopies have revealed the dynamic evolution of the coordinationreduced structure could optimize the -band electron occupancy of Co sites in situ at the early reaction state, which is quite beneficial for the cleavage of the (*OOH species) into *O intermediates toward excellent electrocatalytic ORR activity and selectivity. The well-defined CR-Co/CINC catalyst delivers an appreciable fourelectron selectivity and a high kinetic current density exceeding those of Pt/C. Furthermore, the ZAB with the CR-Co/CINC catalyst presents high efficiency and robust stability at a high current density. This work highlights the importance of modulating the electronic structure and deepens the understanding of the dynamic evolution of single-atom catalysts in promoting ORR performance.
Methods
Synthesis of ZIF-8
In a typical procedure, 2-methylimidazole ( 14.575 g ) was dissolved in methanol ( 250 mL ). Subsequently, methanol ( 250 mL ) containing was added to the above solution under
Fig. 5 | Zn-air battery (ZAB) performance. a Open-circuit voltage plots of Zn-air batteries assembled with CR-Co/CINC and Pt/C. b Discharge polarization curves and power density plots of CR-Co/ClNC and Pt/C-based Zn-air batteries. c Specific
capacity by mass normalization at a current density of . d Galvanostatic discharge curves under different current densities.
continuous stirring for 120 min at room temperature. After that, the above milky white dispersion was kept at room temperature for 12 h without stirring. The resultant sample was then centrifuged at , washed with methanol three times and further dried under vacuum for 12 h at .
Synthesis of NC nanosheets
The ZIF-8 ( 2.5 g ) obtained as described above and 10 g KCl were mixed together and ground thoroughly, followed by dissolution in 200 mL of water. After ultrasonication for 2 h , the aqueous solution in the sample was removed by direct centrifugation and dried at overnight. The obtained powder was fully ground and heated to ) in an Ar atmosphere for 3 h . Subsequently, the obtained sample was fully ground, stirred with for 2 h , washed with HCl three times, then washed with water and ethanol twice each, and finally dried overnight under vacuum at .
Synthesis of the CR-Co/CINC catalyst
The NC ( 100 mg ) was first dispersed into a 20 mL ethanol solution and subjected to ultrasonication for 10 min . After uniform dispersion, 20 mg cobalt chloride hexahydrate was added to the above solution and ultrasonicated for 10 min . Then, the solution was sealed and magnetically stirred for 12 h under rotary evaporation at . The resulting sample was transferred to a vacuum drying oven at when the solution was almost volatilized. The obtained powder was fully ground and heated to in an Ar atmosphere for 5 h . Thereafter, the powder was washed three times with a mixed solution of water and ethanol, centrifuged after ultrasonication and dried under vacuum at .
Synthesis of catalyst
The sample was prepared following the same procedure of CR but annealed at .
Morphology and structure characterization
Powder X-ray diffraction (XRD) measurements were acquired on a Philips X’ Pert Pro Super X-ray diffractometer with radiation. X-ray photoelectron spectroscopy (XPS) was recorded on an ESCALAB MKII with as the excitation source. Scanning electron microscopy (SEM) analyses were carried out on a scanning electron microscope (Gemini SEM 500). Transmission electron microscopy (TEM) and scanning transmission electron microscopyenergy dispersive spectroscopy (STEM-EDS) were conducted using a microscope (JEM-2100F, at 200 kV ), and aberration corrector highangle annular dark-field transmission electron microscopy (AC-HAADF-TEM, JEM-ARM200F) was performed at 200 kV with a probe spherical aberration corrector.
Electrochemical characterization
Catalyst ink was prepared by sonicating the mixture comprising the catalyst ( 5 mg ), Nafion solution ( ), and solvent ( , water/ethanol ) for several hours until a homogeneous suspension was formed. Afterward, a certain amount of the catalyst ink was coated and dried on the surface of the polished glassy carbon electrode ( 3 mm ) at room temperature, and the mass of catalyst deposited on the electrode is . All electrochemical measurements were performed on a CHI760E electrochemical workstation (CH Instruments, China) with a standard three-electrode system using a carbon rod, an (saturated KCl ) electrode, and
a glassy carbon electrode coated with catalyst serving as the counter, reference, and working electrodes, respectively. Polarization curves for the ORR were recorded at room temperature in saturated 0.1 M KOH aqueous solution at various rotation rates ( ). For the current-time chronoamperometric test, was bubbled into 0.1 M KOH electrolyte for 30 min prior to the experiment and a flow of was maintained over the electrolyte during the test to ensure oxygen saturation. The test process was constant at 0.7 V . In this work, the final potential was calibrated into a reversible hydrogen electrode (RHE), unless otherwise noted.
The transfer electron number ( n ) of the ORR was calculated according to the Koutecky-Levich equation below:
where is the measured current density and and are the chargetransfer kinetics and the diffusion-limited current densities, respectively. stands for the angular velocity of the rotating electrode ( ), is the electron-transfer number in ORR, is the Faraday constant ( ), and is the bulk concentration of oxygen. indicates the diffusion coefficient of oxygen, and is the kinematic viscosity of the electrolyte. and in 0.1 M KOH electrolyte were and , respectively.
RRDE measurements were carried out to calculate the yield of hydrogen peroxide ( ) and the corresponding electron transfer number ( ) as follows:
where and are the disk and ring currents, respectively. The Pt ring current collection efficiency was determined to be 0.37 .
Calculation for mass activity and turnover frequency
The mass activity can be determined by considering the metal sites as the active sites. Initially, the metal content of the CR-Co/CINC catalyst was calculated from the Co loading mass on the electrode. Afterwards, the mass activity ( ) was calculated according to the following equation: the geometric electrode area ( ) of and the metal loading mass of the electrocatalyst ( ) were taken into account.
Similarly, by assuming each Co atom in the catalyst as an active single-site, the number of active sites in the CR-Co/CINC catalyst could then be calculated based on the Co loading mass on the electrode. The turnover frequency (TOF) can be derived from the following equation:
where is the kinetic current density ( ), is the electron number per Coulomb ( ), is the metal loading mass on the electrode, is Avogadro’s constant ( ), and is the molar mass of .
Assembly and electrochemical testing of Zn-air batteries (ZAB)
The liquid ZAB was evaluated using a cyclically home-made instrument under ambient atmospheric conditions. The electrolyte was composed of 6 M KOH with 0.2 M zinc acetate, and a flow of was maintained into the electrolyte during the test to ensure saturation. The catalysts coated on the carbon paper were used as the membrane electrode assembly (MEA) of the cathode (catalysts coating area was controlled at ), and a zinc plate with an effective area of 1 cm served as the anode. The polarization curves were recorded by LSV at room temperature on a CHI 760E electrochemical workstation. Both the current density and power density were normalized to the effective surface area of the air electrode. The specific capacity was calculated according to the following equations:
where is the applied current (A), is the serving time (s), and stands for the weight of zinc consumed (g).
In situ XAFS measurements
The in situ XAFS measurements were performed at the 1W1B station in the Beijing Synchrotron Radiation Facility (BSRF), China. The maximum current is 250 mA of the storage ring of BSRF. The line station used a Si (111) double-crystal monochromator, and further detuning of to remove higher harmonics when performing the Co -edge XAFS measurements. During In situ XAFS measurements, the Co catalyst-coated carbon cloths was selected as working electrode in an alkaline solution by a homemade cell through a three-electrode system. Specifically, the 5 mg sample was evenly dispersed in 1 mL solution (water/ethanol ) containing Nafion. Then, the catalyst ink was deposited as the working electrode on the carbon paper ( ), with the Capton film fixed on the back of the carbon paper to ensure that more opening sites can participate in ORR. The representative potentials ( ) were selected to obtain the evolution of atomic and electronic structure of the Co sites during the ORR. When collecting XAFS measurement data, we used a Co foil standard sample to calibrate the position of the absorption edge ( ), and collected all XAFS data over a beam time period. We used a Lytle detector (fluorescence mode) to record XAFS spectra during electrochemical reactions.
In situ SRIR measurements
We conducted in situ SRIR measurements at the beamline BL01B of the National Synchrotron Radiation Laboratory (NSRL, China). In order to gain better infrared signals, electrochemical tests were performed in a homemade top-plate cell. Specifically, considering the vibration absorption of water molecules, we used a ZnSe crystal window and pressed the catalyst electrode close to the window to reduce the loss of infrared light. The SRIR tests were employed in reflection mode with a spectral resolution of . Systemic ORR measurements were made after the background spectrum of the catalyst electrode was measured at an open-circuit voltage, and the ORR potential ranged from 1.00 to 0.65 V .
Data availability
The data supporting the findings of this study are available within the article and in Supplementary information files. All data are available from the authors upon request.
References
Luo, F. et al. P-block single-metal-site tin/nitrogen-doped carbon fuel cell cathode catalyst for oxygen reduction reaction. Nat. Mater. 19, 1215-1223 (2020).
Gao, R. et al. Pt/Fe2O3 with Pt-Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy 6, 614-623 (2021).
Li, Y. & Dai, H. Recent advances in zinc-air batteries. Chem. Soc. Rev. 43, 5257-5275 (2014).
Xie, X. et al. Performance enhancement and degradation mechanism identification of a single-atom catalyst for proton exchange membrane fuel cells. Nat. Catal. 3, 1044-1054 (2020).
Zaman, S. et al. Oxygen reduction electrocatalysts toward practical fuel cells: progress and perspectives. Angew. Chem. 133, 17976-17996 (2021).
Zhao, Y. et al. S and O Cocoordinated Mo single sites in hierarchically porous tubes from sulfur-enamine copolymerization for oxygen reduction and evolution. J. Am. Chem. Soc. 144, 20571-20581 (2022).
Fu, J. et al. Electrically rechargeable zinc-air batteries: progress, challenges, and perspectives. Adv. Mater. 29, 1604685 (2017).
Lai, W. H. et al. General п-electron-assisted strategy for Ir, Pt, Ru, Pd, Fe, Ni single-atom electrocatalysts with bifunctional active sites for highly efficient water splitting. Angew. Chem. Int. Ed. 58, 11868-11873 (2019).
Kodama, K., Nagai, T., Kuwaki, A., Jinnouchi, R. & Morimoto, Y. Challenges in applying highly active Pt-based nanostructured catalysts for oxygen reduction reactions to fuel cell vehicles. Nat. Nanotechnol. 16, 140-147 (2021).
Yuan, Y. et al. Zirconium nitride catalysts surpass platinum for oxygen reduction. Nat. Mater. 19, 282-286 (2020).
Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Z. Design of electrocatalysts for oxygen-and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 44, 2060-2086 (2015).
Lian, Y. et al. Unpaired 3d electrons on atomically dispersed cobalt centres in coordination polymers regulate both oxygen reduction reaction (ORR) activity and selectivity for use in zinc-air batteries. Angew. Chem. Int. Ed. 59, 286-294 (2020).
Shang, H. et al. Engineering unsymmetrically coordinated Cu-S1N3 single atom sites with enhanced oxygen reduction activity. Nat. Commun. 11, 3049 (2020).
Zhou, W. et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 13, 6414 (2022).
Zhang, H. et al. Symbiotic synergy enabling moderate oxo-hydroxy adsorption capacity for high-selectivity oxygen reduction. Nano Energy 101, 107587 (2022).
Liu, M. et al. Synergetic Dual-Ion Centers Boosting Metal Organic Framework Alloy Catalysts toward Efficient Two Electron Oxygen Reduction. Small 18, 2202248 (2022).
Qu, Y. et al. Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms. Nat. Catal. 1, 781-786 (2018).
An, Q. et al. Engineering Unsymmetrically Coordinated Fe Sites via Heteroatom Pairs Synergetic Contribution for Efficient Oxygen Reduction. Small 19, 2304303 (2023).
Su, H. et al. Hetero-N-coordinated Co single sites with high turnover frequency for efficient electrocatalytic oxygen evolution in an acidic medium. ACS Energy Lett. 4, 1816-1822 (2019).
Mun, Y. et al. Versatile strategy for tuning ORR activity of a single FeN4 site by controlling electron-withdrawing/donating properties of a carbon plane. J. Am. Chem. Soc. 141, 6254-6262 (2019).
Zheng, X. et al. Enriched d-band holes enabling fast oxygen evolution kinetics on atomic-layered defect-rich lithium cobalt oxide nanosheets. Adv. Func. Mater. 32, 2200663 (2022).
Zu, L. et al. Self-assembly of Ir-based nanosheets with ordered interlayer space for enhanced electrocatalytic water oxidation. J. Am. Chem. Soc. 144, 2208-2217 (2022).
Grimaud, A. et al. Activation of surface oxygen sites on an iridiumbased model catalyst for the oxygen evolution reaction. Nat. Energy 2, 1-10 (2016).
Jia, C. et al. Sulfur-Dopant-Promoted Electroreduction of over Coordinatively Unsaturated Ni-N2 Moieties. Angew. Chem. Int. Ed. 60, 23342-23348 (2021).
Liu, M. et al. Self-nanocavity-confined halogen anions boosting the high selectivity of the two-electron oxygen reduction pathway over Ni-based MOFs. J. Phys. Chem. Lett. 12, 8706-8712 (2021).
Su, H. et al. In-situ spectroscopic observation of dynamic-coupling oxygen on atomically dispersed iridium electrocatalyst for acidic water oxidation. Nat. Commun. 12, 6118 (2021).
Zhou, W. et al. Identification of the evolving dynamics of coordination-unsaturated iron atomic active sites under reaction conditions. ACS Energy Lett. 6, 3359-3366 (2021).
Su, H., Soldatov, M. A., Roldugin, V. & Liu, Q. Platinum single-atom catalyst with self-adjustable valence state for large-current-density acidic water oxidation. eScience 2, 102-109 (2022).
Hai, X. et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 17, 174-181 (2022).
Zhang, Z. et al. Ultraconformal chemo-mechanical stable cathode interface for high-performance all-solid-state batteries at wide temperatures. Energy Environ. Sci. 16, 4453-4463 (2023).
Sun, T. et al. Single-atomic cobalt sites embedded in hierarchically ordered porous nitrogen-doped carbon as a superior bifunctional electrocatalyst. Proc. Natl Acad. Sci. 115, 12692-12697 (2018).
Gong, C., Yang, C., Zhou, W., Su, H. & Liu, Q. Modulation of Electronic States in Bimetallic-doped Nitrogen-Carbon Based Nanoparticles for Enhanced Oxygen Reduction Kinetics. Chinese J. Chem. Phys. https://doi.org/10.1063/1674-0068/ cjcp2304033 (2023).
Liang, Y. et al. Covalent hybrid of spinel manganese-cobalt oxide and graphene as advanced oxygen reduction electrocatalysts. J. Am. Chem. Soc. 134, 3517-3523 (2012).
Yi, J.-D. et al. Atomically dispersed iron-nitrogen active sites within porphyrinic triazine-based frameworks for oxygen reduction reaction in both alkaline and acidic media. ACS Energy Lett. 3, 883-889 (2018).
He, C. et al. Molecular evidence for metallic cobalt boosting CO2 electroreduction on pyridinic nitrogen. Angew. Chem. 132, 4944-4949 (2020).
Edgington, J., Schweitzer, N., Alayoglu, S. & Seitz, L. C. Constant change: exploring dynamic oxygen evolution reaction catalysis and material transformations in strontium zinc iridate perovskite in acid. J. Am. Chem. Soc. 143, 9961-9971 (2021).
Cheng, W., Su, H. & Liu, Q. Tracking the Oxygen Dynamics of Solid-Liquid Electrochemical Interfaces by Correlative In Situ Synchrotron Spectroscopies. Acc. Chem. Res. 55, 1949-1959 (2022).
Su, H. et al. Dynamic evolution of solid-liquid electrochemical interfaces over single-atom active sites. J. Am. Chem. Soc. 142, 12306-12313 (2020).
Cheng, W. et al. Lattice-strained metal-organic-framework arrays for bifunctional oxygen electrocatalysis. Nat. Energy 4, 115-122 (2019).
Yang, C. et al. Dynamically-evolved surface heterojunction in iridium nanocrystals boosting acidic oxygen evolution and overall water splitting. J. Energy Chem. 78, 374-380 (2023).
Zandi, O. & Hamann, T. W. Determination of photoelectrochemical water oxidation intermediates on haematite electrode surfaces using operando infrared spectroscopy. Nat. Chem. 8, 778-783 (2016).
Rao, R. R. et al. Operando identification of site-dependent water oxidation activity on ruthenium dioxide single-crystal surfaces. Nat. Catal. 3, 516-525 (2020).
Sivasankar, N., Weare, W. W. & Frei, H. Direct observation of a hydroperoxide surface intermediate upon visible light-driven water oxidation at an Ir oxide nanocluster catalyst by rapid-scan FT-IR spectroscopy. J. Am. Chem. Soc. 133, 12976-12979 (2011).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1502903 (Q.L.)), the National Natural Science Foundation of China (12205300 (H.S.), 22241202 (Q.L.), and 12135012 (H.S.)), the Natural Science Foundation of Anhui Province (2208085JO1 (Q.L.) and 2208085QA28 (H.S.)), and the Key Program of Research and Development of Hefei Science Center, CAS (2022HSC-KPRD003 (W.Z.)). This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China.
Author contributions
Q.L., H.S. and J.P. conceived the project. M.L. and J.Z. carried out the experiments. Q.L., H.S., M.L., Y.J., W.Z., C.Y., and S.B. analyzed the experimental data. The manuscript was written by M.L., H.S. and Q.L. with contributions from all authors.
Correspondence and requests for materials should be addressed to Hui Su, Jun Pan or Qinghua Liu.
Peer review information Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available
National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei 230029 Anhui, China. State Key Laboratory for Powder Metallurgy, Central South University, Changsha 410083 Hunan, China. Key Laboratory of Light Energy Conversion Materials of Hunan Province College, College of Chemistry and Chemical Engineering, Hunan Normal University, Changsha 410081 Hunan, China. These authors contributed equally: Meihuan Liu, Jing Zhang. e-mail: suhui@ustc.edu.cn; jun.pan@csu.edu.cn; qhliu@ustc.edu.cn