DOI: https://doi.org/10.1038/s41467-024-47624-7
PMID: https://pubmed.ncbi.nlm.nih.gov/38615063
تاريخ النشر: 2024-04-13
تعزيز إنتاج H2O2 الضوئي المساعد باستخدام المحفزات المساعدة من الذهب من خلال تعديل الهيكل الإلكتروني
تم القبول: 8 أبريل 2024
نُشر على الإنترنت: 13 أبريل 2024
(أ) التحقق من التحديثات
الملخص
المحفزات المساعدة القائمة على الذهب هي فئة واعدة من المواد ذات التطبيقات المحتملة في التحفيز الضوئي
يجب أن يكون هناك طاقة ربط مثالية بين المواقع النشطة والمواد الممتصة. تشير الأبحاث الإضافية إلى أن التوزيع الإلكتروني للمواقع النشطة يحدد بشكل أساسي تفاعلها مع المواد الممتصة، مما يؤثر على أدائها في الامتصاص/الإزالة.
تقليل انتقائي لاحتلال المدارات المضادة للرابطة
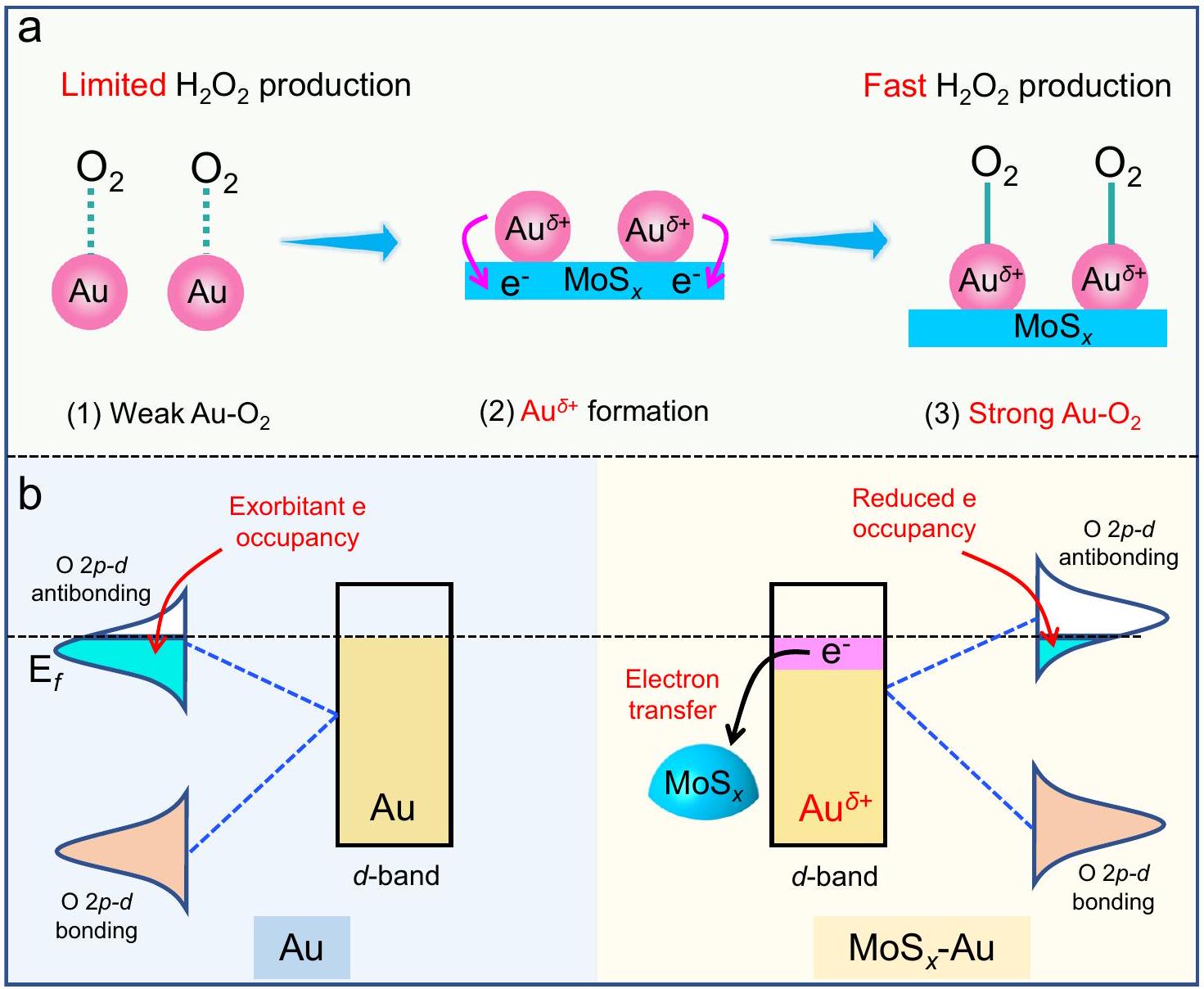
قوي
نهج لتعزيز
النتائج والمناقشة
تركيب وتوصيف
المحفز الضوئي. يمكن دعم النتيجة أعلاه بشكل أكبر بواسطة حيود الأشعة السينية (XRD) وطيف رامان. بالمقارنة مع
أداء التحفيز الضوئي
تركيز (
آلية التحفيز الضوئي لـ
فرق
المحفز المساعد. تشكيل ما سبق من نقص الإلكترونات
مخططات مدارات 5d في الذهب (Au) وموكسيد الموليبدينوم (MoS)
أقوى
تحسين كبير في الانتقائية والنشاط للضوء المحفز
في
طرق
تحضير
تحضير
تحضير
توصيف
تحفيز ضوئي
محفز ضوئي مُعد مسبقًا و100 مل من محلول الإيثانول
القياسات الضوئية الكهروكيميائية
متوسط زمن التحلل (
حساب العائد الكمي الظاهر (AQY)
اختبارات الامتصاص العابر فائق السرعة (TA)
مذبذب الياقوت (Coherent Vitara،
التفاصيل الحاسوبية
توفر البيانات
References
- Zhao, Y. et al. Mechanistic analysis of multiple processes controlling solar-driven
synthesis using engineered polymeric carbon nitride. Nat. Commun. 12, 3701 (2021). - Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374-384 (2021).
- Liu, T. et al. Overall photosynthesis of
by an inorganic semiconductor. Nat. Commun. 13, 1034 (2022). - Zhang, Y. et al.
generation from and on a near-infrared absorbing porphyrin supramolecular photocatalyst. Nat. Energy 8, 361-371 (2023). - Zhang, X. et al. S-scheme heterojunction of core-shell biphase (1T
nanorod arrays for enhanced photoelectrocatalytic production of hydrogen peroxide. Chem. Eng. J. 429, 131312 (2022). - He, Q. et al. Multiscale structural engineering of carbon nitride for enhanced photocatalytic
production. Nano. Res. 16, 4524-4530 (2021). - Yang, Y. et al. Molecular engineering of polymeric carbon nitride for highly efficient photocatalytic oxytetracycline degradation and
production. Appl. Catal. B Environ. 272, 118970 (2020). - Hou, H., Zeng, X. & Zhang, X. Production of hydrogen peroxide by photocatalytic processes. Angew. Chem. Int. Ed. 59, 17356-17376 (2020).
- Wang, L. et al. Inorganic metal-oxide photocatalyst for
production. Small 18, 2104561 (2021). - Zhang, J. et al. The critical role of furfural alcohol in photocatalytic
production on TiO2. Appl. Catal. B Environ. 269, 118770 (2020). - Song, S. et al. Structure effect of graphene on the photocatalytic performance of plasmonic
-rGO for photocatalytic elimination of pollutants. Appl. Catal. B Environ. 181, 71-78 (2016). - Lin, Z. et al. Apparent potential difference boosting directional electron transfer for full solar spectrum-irradiated catalytic
evolution. Adv. Funct. Mater. 30, 1908797 (2019). - Li, K., Zhang, S., Li, Y., Fan, J. & Lv, K. MXenes as noble-metalalternative co-catalysts in photocatalysis. Chin. J. Catal. 42, 3-14 (2021).
- Chu, C. et al. Electronic tuning of metal nanoparticles for highly efficient photocatalytic hydrogen peroxide production. ACS Catal 9, 626-631 (2018).
- Zhang,
. et al. PDA -Scheme heterojunction with enhanced photocatalytic production performance and its mechanism. Adv. Sustain. Syst. 7, 2200113 (2023). - Zhang, Y., Liang, C., Feng, H. & Liu, W. Nickel single atoms anchored on ultrathin carbon nitride for selective hydrogen peroxide generation with enhanced photocatalytic activity. Chem. Eng. J. 446, 137379 (2022).
- Teng, Z. et al. Photoexcited single metal atom catalysts for heterogeneous photocatalytic
production: Pragmatic guidelines for predicting charge separation. Appl. Catal. B Environ. 282, 119589 (2021). - Choi, C. et al. Hydrogen peroxide synthesis via enhanced twoelectron oxygen reduction pathway on carbon-coated Pt surface. J. Phys. Chem. C 118, 30063-30070 (2014).
- Yang, Y. et al. Bifunctional
-scheme photocatalyst with enhanced production and furoic acid synthesis mechanism. Appl. Catal. B Environ. 333, 122780 (2023). - Koper, M. & Bouwman, E. Electrochemical hydrogen production: bridging homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 49, 3723-3725 (2010).
- Gao, D., Long, H., Wang, X., Yu, J. & Yu, H. Tailoring antibondingorbital occupancy state of selenium in Se-enriched ReSe
cocatalyst for exceptional evolution of photocatalyst. Adv. Funct. Mater. 33, 2209994 (2023). - Wang, K. et al. Optimizing oxygen and intermediate
adsorption of Cu-Pd alloy cocatalyst for boosting photocatalytic production of . Adv. Sustain. Syst. 6, 2200144 (2022). -
. et al. Palladium-copper nanodot as novel -evolution cocatalyst: optimizing interfacial hydrogen desorption for highly efficient photocatalytic activity. Chin. J. Catal. 43, 215-225 (2022). - Shi, H. et al. Mass-transfer control for selective deposition of welldispersed AuPd cocatalysts to boost photocatalytic
production of . Chem. Eng. J. 443, 136429 (2022). - Fuku, K. et al. Photocatalytic
production from under visible light irradiation over phosphate ion-coated Pd nanoparticlessupported . Appl. Catal. B Environ. 272, 119003 (2020). - Teranishi, M., Naya, S. & Tada, H. In situ liquid phase synthesis of hydrogen peroxide from molecular oxygen using gold nanoparticle-loaded titanium(IV) dioxide photocatalyst. J. Am. Chem. Soc. 132, 7850-7851 (2010).
- Hirakawa, H. et al. Au nanoparticles supported on
: effective inorganic photocatalysts for production from water and under visible light. ACS Catal 6, 4976-4982 (2016). - Kobayashi, H., Teranishi, M., Negishi, R., Naya, S. & Tada, H. Reaction mechanism of the multiple-electron oxygen reduction reaction on the surfaces of gold and platinum nanoparticles loaded on titanium(IV) oxide. J. Phys. Chem. Lett. 7, 5002-5007 (2016).
- Wang, Y. et al. Efficient production of
on under visible light and the influencing factors. Appl. Catal. B Environ. 284, 119691 (2021). - Zuo, G. et al. Finely dispersed Au nanoparticles on graphitic carbon nitride as highly active photocatalyst for hydrogen peroxide production. Catal. Commun. 123, 69-72 (2019).
- Kim, K. et al. Solid-phase photocatalysts: physical vapor deposition of Au nanoislands on porous
films for millimolar production within a few minutes. ACS Catal 9, 9206-9211 (2019). - An, R. et al. Decoration of Au NPs on hollow structured BiOBr with surface oxygen vacancies for enhanced visible light photocatalytic
evolution. J. Solid State Chem. 306, 122722 (2022). - Liu, Z., Gong, X., Kohanoff, J., Sanchez, C. & Hu, P. Catalytic role of metal oxides in gold-based catalysts: a first principles study of CO oxidation on
supported Au. Phys. Rev. Lett. 91, 266102 (2003). - Li, L. et al. Tailoring selectivity of electrochemical hydrogen peroxide generation by tunable pyrrolic-nitrogen-carbon. Adv. Energy Mater. 10, 2000789 (2020).
- Tsukamoto, D. et al. Photocatalytic
production from ethanol/ system using loaded with Au-Ag bimetallic alloy nanoparticles. ACS Catal 2, 599-603 (2012). - Wang, K. et al.
Microparticles decorated with coreshell nanostructures for photocatalytic production. ACS Appl. Nano Mater. 4, 13158-13166 (2021). - Men, Y. et al. Oxygen-inserted top-surface layers of Ni for boosting alkaline hydrogen oxidation electrocatalysis. J. Am. Chem. Soc. 144, 12661-12672 (2022).
- Yadav, V. et al. Modulating the structure and hydrogen evolution reactivity of metal chalcogenide complexes through ligand exchange onto colloidal Au nanoparticles. ACS Catal 10, 13305-13313 (2020).
- Gao, D. et al. Reversing free-electron transfer of
cocatalyst for optimizing antibonding-orbital occupancy enables high photocatalytic evolution. Angew. Chem. Int. Ed. 62, e202304559 (2023). - Gao, D. et al. Optimizing atomic hydrogen desorption of sulfur-rich
cocatalyst for boosting photocatalytic evolution. Adv. Mater. 34, 2108475 (2022). - Castelló, L. et al. Nano-gold decorated ZnO : an alternative photocatalyst promising for
degradation. Chem. Eng. Sci. 267, 118377 (2023). - Inagaki, M., Motobayashi, K. & Ikeda, K. In situ surface-enhanced electronic and vibrational Raman scattering spectroscopy at metal/ molecule interfaces. Nanoscale 12, 22988-22994 (2020).
- Liu, Q. et al. Unraveling the roles of hot electrons and cocatalyst toward broad spectrum photocatalytic
generation of nanotube. Sol. RRL 5, 2000504 (2021). - Zhong, W. et al. Novel core-shell Ag@AgSe nanoparticle co-catalyst: in situ surface selenization for efficient photocatalytic
production of . Chin. J. Catal. 43, 1074-1083 (2022). - Tao, J. et al. A novel CoP@AAH cocatalyst leads to excellent stability and enhanced photocatalytic
evolution of CdS by structurally separating the photogenerated carriers. Appl. Catal. B Environ. 32O, 122004 (2023). - Yue, X., Cheng, L., Fan, J. & Xiang, Q. 2D/2D BiVO4
S-scheme heterojunction for photocatalytic reduction: Insights into structure regulation and Fermi level modulation. Appl. Catal. B Environ. 304, 120979 (2022). - Zhong, W., Zhao, B., Wang, X., Wang, P. & Yu, H. Synchronously enhancing water adsorption and strengthening
bonds in Se-rich cocatalyst for efficient alkaline photocatalytic production. ACS Catal 13, 749-756 (2023). - Yang, C., Wan, S., Zhu, B., Yu, J. & Cao, S. Calcination-regulated microstructures of donor-acceptor polymers towards enhanced and stable photocatalytic
production in pure water. Angew. Chem. Int. Ed. 134, e202208438 (2022). - Li, K., Blatov, V. & Wang, J. Discovery of electrides in electron-rich Non-electride materials via energy modification of interstitial electrons. Adv. Funct. Mater. 32, 2112198 (2022).
- Zhang, X. et al. Optimizing the Pd sites in pure metallic aerogels for efficient electrocatalytic
production. Adv. Mater. 35, 2211512 (2023). - Zhong, W. et al. Charging d-orbital electron of
cocatalyst enables splendid alkaline photocatalytic evolution. Adv. Funct. Mater. 33, 2302325 (2023). - Wang, J. et al. Cooperative coupling of photocatalytic production of
and oxidation of organic pollutants over gadolinium ion doped nanocomposite. Chin. Chem. Lett. 34, 108157 (2023). - Li, F. et al. Understanding the unique S-scheme charge migration in triazine/heptazine crystalline carbon nitride homojunction. Nat. Commun. 14, 3901 (2023).
- Zhao, M. et al. A novel S-scheme 3D
heterostructure for improved hydrogen production under visible light irradiation. Chin. J. Catal. 43, 2615-2624 (2022). - Chen, R., Fan, F., Dittrich, T. & Li, C. Imaging photogenerated charge carriers on surfaces and interfaces of photocatalysts with surface photovoltage microscopy. Chem. Soc. Rev. 47, 8238-8262 (2018).
- Chen, R. et al. Charge separation via asymmetric illumination in photocatalytic
particles. Nat. Energy 3, 655-663 (2018). - Gu, M. et al. Efficient sacrificial-agent-free solar
production over all-inorganic S-scheme composites. Appl. Catal. B Environ. 324, 122227 (2023). - He, B. et al. Cooperative coupling of
production and organic synthesis over a floatable polystyrene-sphere-supported S-Scheme photocatalyst. Adv. Mater. 34, 2203225 (2022). - Cheng, C. et al. Verifying the charge-transfer mechanism in SScheme heterojunctions using femtosecond transient absorption spectroscopy. Angew. Chem. Int. Ed. 135, e202218688 (2023).
- Wang, L. et al. Dynamics of photogenerated charge carriers in inorganic/organic S-Scheme heterojunctions. J. Phys. Chem. Lett. 13, 4695-4700 (2022).
شكر وتقدير
مساهمات المؤلفين
حسابات المواد. قام X.Z. و D.G. بإجراء الاختبار الضوئي التحفيزي. ساهم X.Z. و B.C. و J.Y. و H.Y. في تحليل البيانات. أشرف J.Y. و H.Y. على المشروع. كتب X.Z. و H.Y. المخطوطة. قام B.C. و J.Y. و H.Y. بمراجعة المخطوطة. ناقش جميع المؤلفين النتائج وعلقوا على المخطوطة.
المصالح المتنافسة
معلومات إضافية
المواد التكميلية متاحة على
https://doi.org/10.1038/s41467-024-47624-7.
http://www.nature.com/reprints
(ج) المؤلف(ون) 2024
مختبر الوقود الشمسي، كلية علوم المواد والكيمياء، جامعة علوم الأرض الصينية، 68 شارع جينتشينغ، ووهان، جمهورية الصين الشعبية. المختبر الوطني الرئيسي لمواد السيليكات للهندسة المعمارية، جامعة ووهان للتكنولوجيا، 122 طريق لوشي، ووهان، جمهورية الصين الشعبية. المختبر الوطني الرئيسي للتكنولوجيا المتقدمة لتخليق المواد ومعالجتها، جامعة ووهان للتكنولوجيا، ووهان، جمهورية الصين الشعبية. البريد الإلكتروني: yuhuogen@cug.edu.cn
DOI: https://doi.org/10.1038/s41467-024-47624-7
PMID: https://pubmed.ncbi.nlm.nih.gov/38615063
Publication Date: 2024-04-13
Enhancing photocatalytic
Accepted: 8 April 2024
Published online: 13 April 2024
(A) Check for updates
Abstract
Gold-based co-catalysts are a promising class of materials with potential applications in photocatalytic
the interaction between active sites and adsorbates must have an optimal binding energy. Further research indicates that the electron configuration of active sites fundamentally determines their interaction with adsorbates, influencing their adsorption/desorption performances
selectively decreasing the antibonding-orbital occupancy of
strong
approach to enhance
Results and discussion
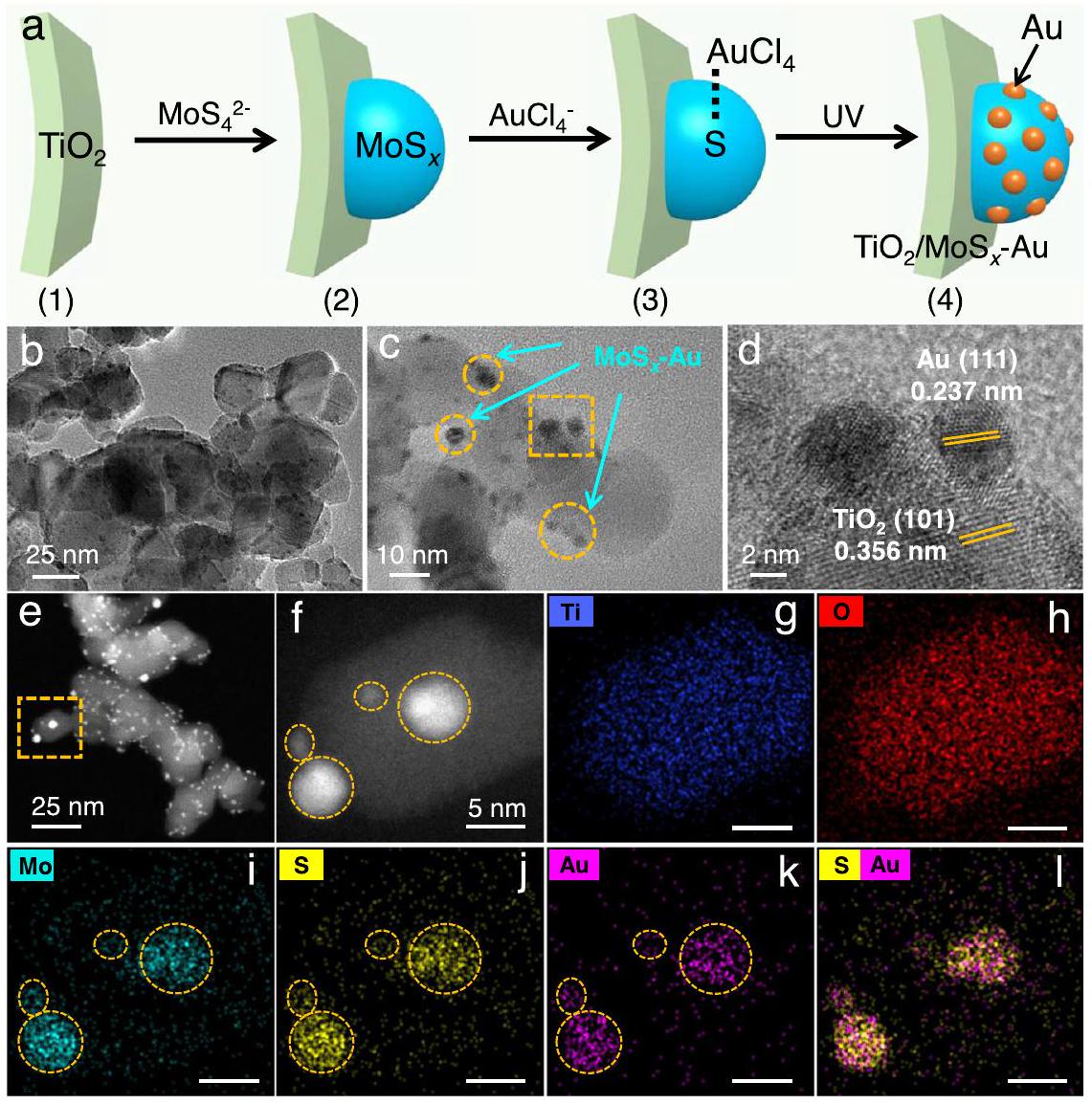
Synthesis and characterization of
photocatalyst. The above result can further be supported by X-ray diffraction (XRD) and Raman spectra. Compared with the
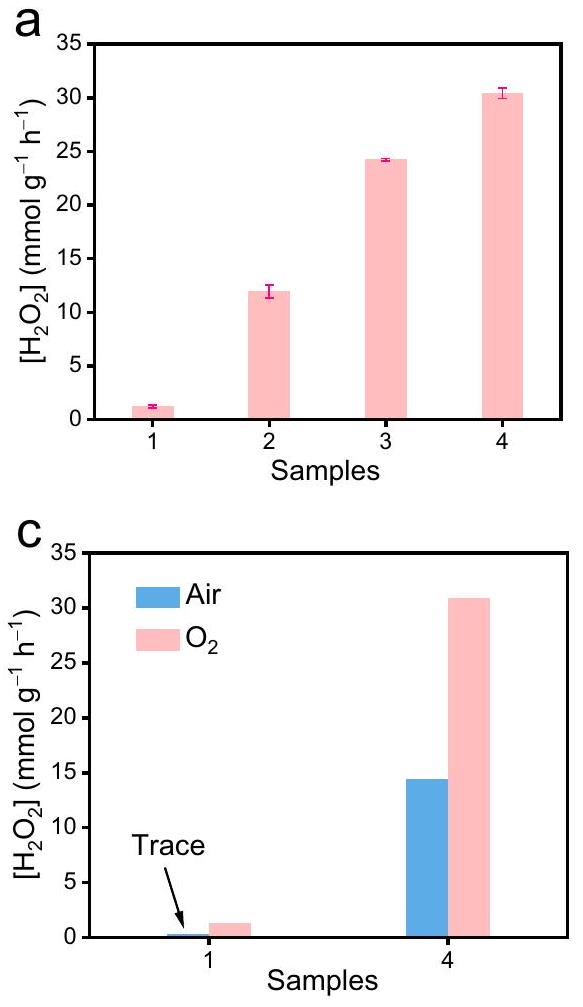
Photocatalytic performance
concentration (
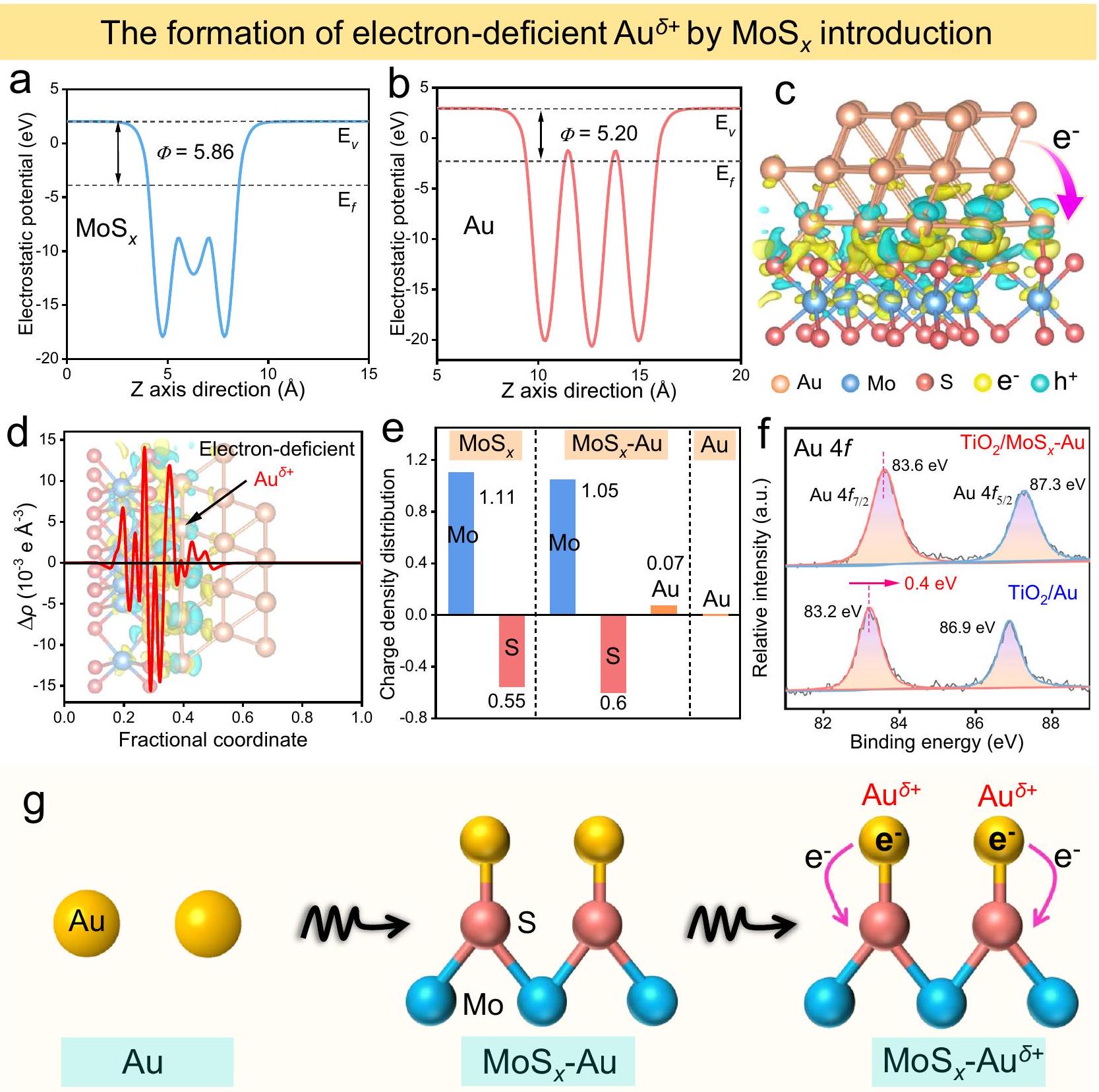
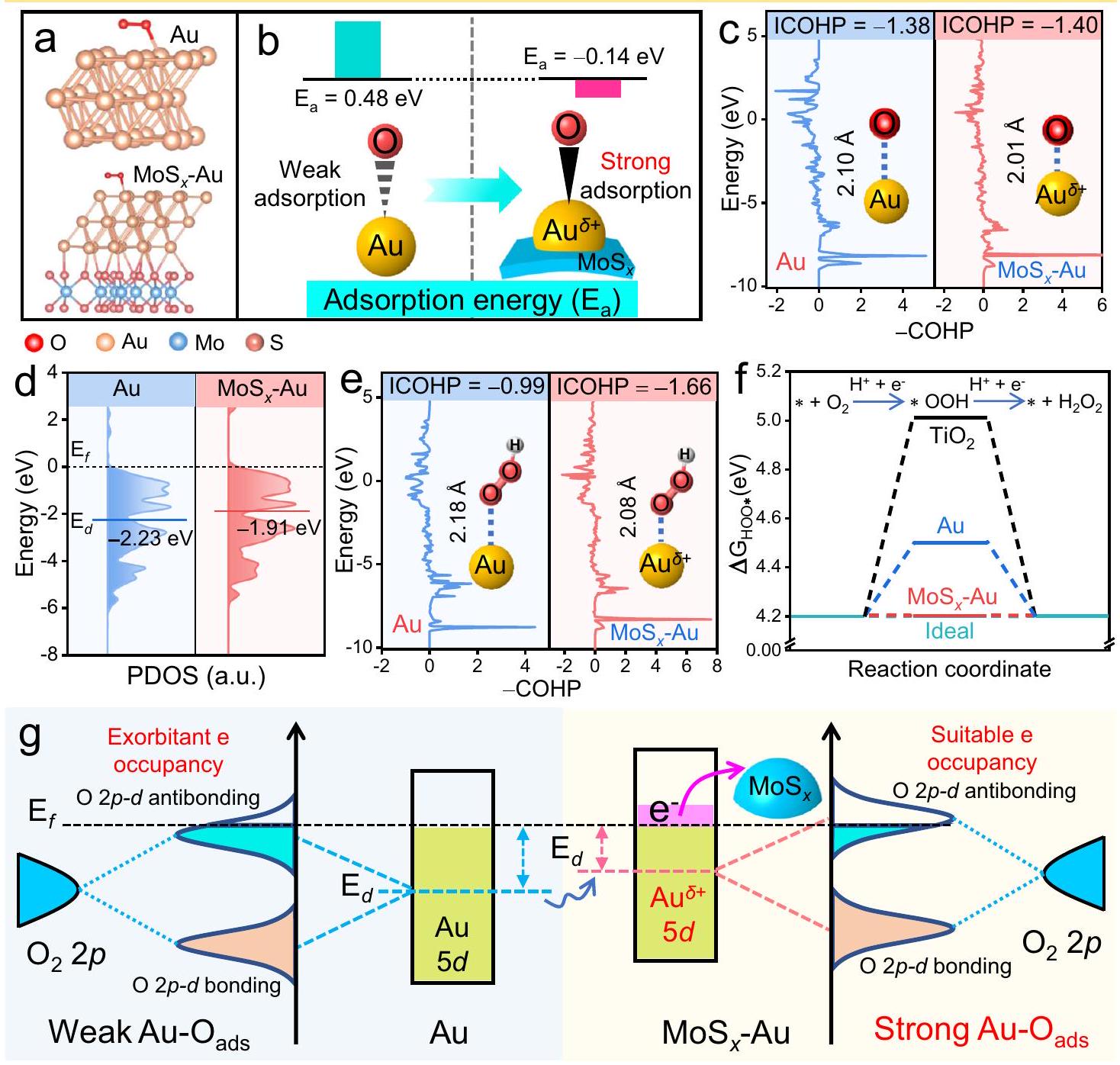
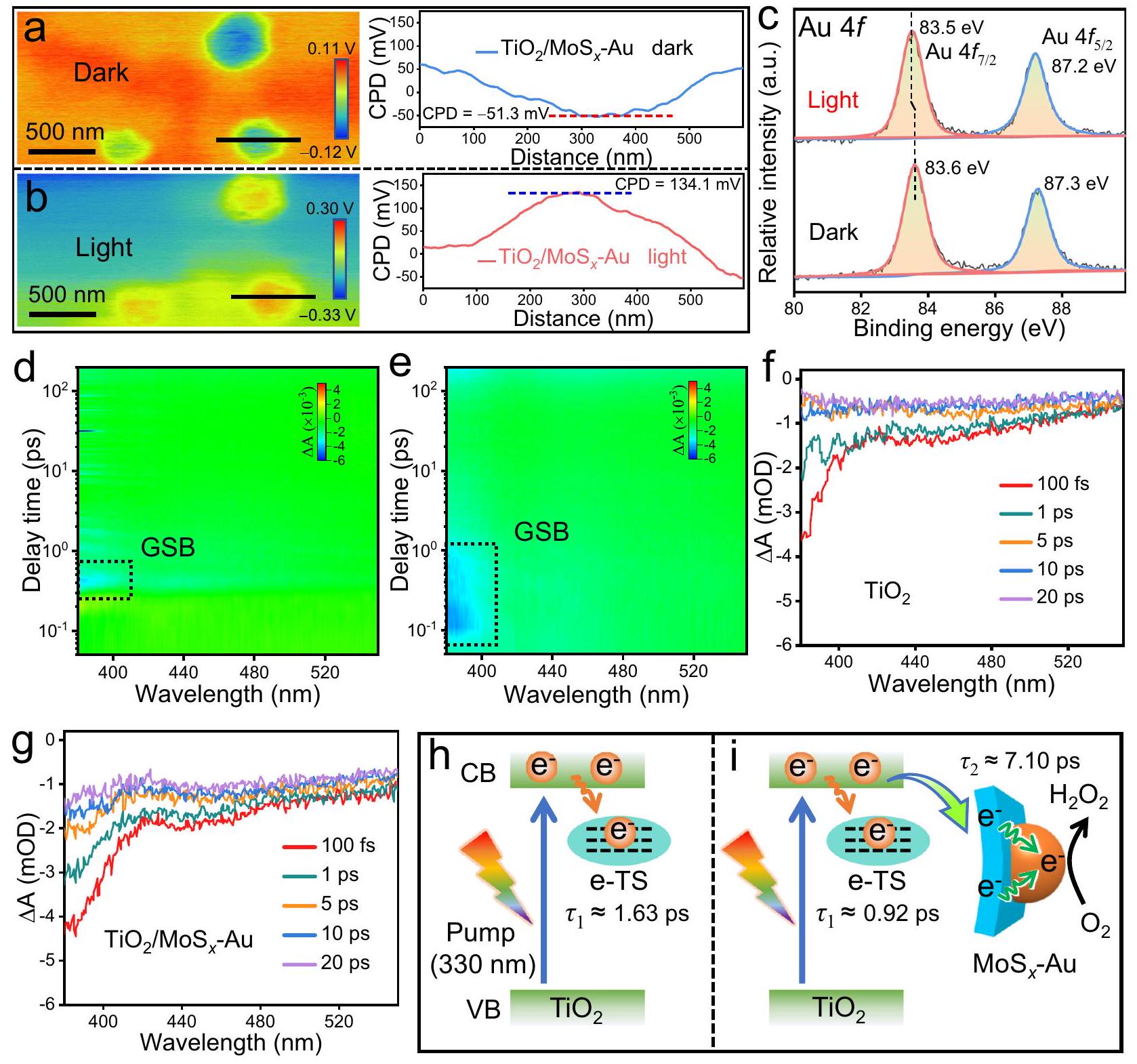
Photocatalytic mechanism of
difference
cocatalyst. The formation of above electron-deficient
diagrams of Au 5d orbitals in Au and MoS
stronger
greatly improving the selectivity and activity of photocatalytic
in the
Methods
Preparation of
Preparation of
Preparation of
Characterization
Photocatalytic
as-prepared photocatalyst and 100 mL of ethanol solution (
Photoelectrochemical measurements
Average decay time (
Apparent quantum yield (AQY) calculation
Ultrafast transient absorption (TA) tests
sapphire oscillator (Coherent Vitara,
Computational details
Data availability
References
- Zhao, Y. et al. Mechanistic analysis of multiple processes controlling solar-driven
synthesis using engineered polymeric carbon nitride. Nat. Commun. 12, 3701 (2021). - Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374-384 (2021).
- Liu, T. et al. Overall photosynthesis of
by an inorganic semiconductor. Nat. Commun. 13, 1034 (2022). - Zhang, Y. et al.
generation from and on a near-infrared absorbing porphyrin supramolecular photocatalyst. Nat. Energy 8, 361-371 (2023). - Zhang, X. et al. S-scheme heterojunction of core-shell biphase (1T
nanorod arrays for enhanced photoelectrocatalytic production of hydrogen peroxide. Chem. Eng. J. 429, 131312 (2022). - He, Q. et al. Multiscale structural engineering of carbon nitride for enhanced photocatalytic
production. Nano. Res. 16, 4524-4530 (2021). - Yang, Y. et al. Molecular engineering of polymeric carbon nitride for highly efficient photocatalytic oxytetracycline degradation and
production. Appl. Catal. B Environ. 272, 118970 (2020). - Hou, H., Zeng, X. & Zhang, X. Production of hydrogen peroxide by photocatalytic processes. Angew. Chem. Int. Ed. 59, 17356-17376 (2020).
- Wang, L. et al. Inorganic metal-oxide photocatalyst for
production. Small 18, 2104561 (2021). - Zhang, J. et al. The critical role of furfural alcohol in photocatalytic
production on TiO2. Appl. Catal. B Environ. 269, 118770 (2020). - Song, S. et al. Structure effect of graphene on the photocatalytic performance of plasmonic
-rGO for photocatalytic elimination of pollutants. Appl. Catal. B Environ. 181, 71-78 (2016). - Lin, Z. et al. Apparent potential difference boosting directional electron transfer for full solar spectrum-irradiated catalytic
evolution. Adv. Funct. Mater. 30, 1908797 (2019). - Li, K., Zhang, S., Li, Y., Fan, J. & Lv, K. MXenes as noble-metalalternative co-catalysts in photocatalysis. Chin. J. Catal. 42, 3-14 (2021).
- Chu, C. et al. Electronic tuning of metal nanoparticles for highly efficient photocatalytic hydrogen peroxide production. ACS Catal 9, 626-631 (2018).
- Zhang,
. et al. PDA -Scheme heterojunction with enhanced photocatalytic production performance and its mechanism. Adv. Sustain. Syst. 7, 2200113 (2023). - Zhang, Y., Liang, C., Feng, H. & Liu, W. Nickel single atoms anchored on ultrathin carbon nitride for selective hydrogen peroxide generation with enhanced photocatalytic activity. Chem. Eng. J. 446, 137379 (2022).
- Teng, Z. et al. Photoexcited single metal atom catalysts for heterogeneous photocatalytic
production: Pragmatic guidelines for predicting charge separation. Appl. Catal. B Environ. 282, 119589 (2021). - Choi, C. et al. Hydrogen peroxide synthesis via enhanced twoelectron oxygen reduction pathway on carbon-coated Pt surface. J. Phys. Chem. C 118, 30063-30070 (2014).
- Yang, Y. et al. Bifunctional
-scheme photocatalyst with enhanced production and furoic acid synthesis mechanism. Appl. Catal. B Environ. 333, 122780 (2023). - Koper, M. & Bouwman, E. Electrochemical hydrogen production: bridging homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 49, 3723-3725 (2010).
- Gao, D., Long, H., Wang, X., Yu, J. & Yu, H. Tailoring antibondingorbital occupancy state of selenium in Se-enriched ReSe
cocatalyst for exceptional evolution of photocatalyst. Adv. Funct. Mater. 33, 2209994 (2023). - Wang, K. et al. Optimizing oxygen and intermediate
adsorption of Cu-Pd alloy cocatalyst for boosting photocatalytic production of . Adv. Sustain. Syst. 6, 2200144 (2022). -
. et al. Palladium-copper nanodot as novel -evolution cocatalyst: optimizing interfacial hydrogen desorption for highly efficient photocatalytic activity. Chin. J. Catal. 43, 215-225 (2022). - Shi, H. et al. Mass-transfer control for selective deposition of welldispersed AuPd cocatalysts to boost photocatalytic
production of . Chem. Eng. J. 443, 136429 (2022). - Fuku, K. et al. Photocatalytic
production from under visible light irradiation over phosphate ion-coated Pd nanoparticlessupported . Appl. Catal. B Environ. 272, 119003 (2020). - Teranishi, M., Naya, S. & Tada, H. In situ liquid phase synthesis of hydrogen peroxide from molecular oxygen using gold nanoparticle-loaded titanium(IV) dioxide photocatalyst. J. Am. Chem. Soc. 132, 7850-7851 (2010).
- Hirakawa, H. et al. Au nanoparticles supported on
: effective inorganic photocatalysts for production from water and under visible light. ACS Catal 6, 4976-4982 (2016). - Kobayashi, H., Teranishi, M., Negishi, R., Naya, S. & Tada, H. Reaction mechanism of the multiple-electron oxygen reduction reaction on the surfaces of gold and platinum nanoparticles loaded on titanium(IV) oxide. J. Phys. Chem. Lett. 7, 5002-5007 (2016).
- Wang, Y. et al. Efficient production of
on under visible light and the influencing factors. Appl. Catal. B Environ. 284, 119691 (2021). - Zuo, G. et al. Finely dispersed Au nanoparticles on graphitic carbon nitride as highly active photocatalyst for hydrogen peroxide production. Catal. Commun. 123, 69-72 (2019).
- Kim, K. et al. Solid-phase photocatalysts: physical vapor deposition of Au nanoislands on porous
films for millimolar production within a few minutes. ACS Catal 9, 9206-9211 (2019). - An, R. et al. Decoration of Au NPs on hollow structured BiOBr with surface oxygen vacancies for enhanced visible light photocatalytic
evolution. J. Solid State Chem. 306, 122722 (2022). - Liu, Z., Gong, X., Kohanoff, J., Sanchez, C. & Hu, P. Catalytic role of metal oxides in gold-based catalysts: a first principles study of CO oxidation on
supported Au. Phys. Rev. Lett. 91, 266102 (2003). - Li, L. et al. Tailoring selectivity of electrochemical hydrogen peroxide generation by tunable pyrrolic-nitrogen-carbon. Adv. Energy Mater. 10, 2000789 (2020).
- Tsukamoto, D. et al. Photocatalytic
production from ethanol/ system using loaded with Au-Ag bimetallic alloy nanoparticles. ACS Catal 2, 599-603 (2012). - Wang, K. et al.
Microparticles decorated with coreshell nanostructures for photocatalytic production. ACS Appl. Nano Mater. 4, 13158-13166 (2021). - Men, Y. et al. Oxygen-inserted top-surface layers of Ni for boosting alkaline hydrogen oxidation electrocatalysis. J. Am. Chem. Soc. 144, 12661-12672 (2022).
- Yadav, V. et al. Modulating the structure and hydrogen evolution reactivity of metal chalcogenide complexes through ligand exchange onto colloidal Au nanoparticles. ACS Catal 10, 13305-13313 (2020).
- Gao, D. et al. Reversing free-electron transfer of
cocatalyst for optimizing antibonding-orbital occupancy enables high photocatalytic evolution. Angew. Chem. Int. Ed. 62, e202304559 (2023). - Gao, D. et al. Optimizing atomic hydrogen desorption of sulfur-rich
cocatalyst for boosting photocatalytic evolution. Adv. Mater. 34, 2108475 (2022). - Castelló, L. et al. Nano-gold decorated ZnO : an alternative photocatalyst promising for
degradation. Chem. Eng. Sci. 267, 118377 (2023). - Inagaki, M., Motobayashi, K. & Ikeda, K. In situ surface-enhanced electronic and vibrational Raman scattering spectroscopy at metal/ molecule interfaces. Nanoscale 12, 22988-22994 (2020).
- Liu, Q. et al. Unraveling the roles of hot electrons and cocatalyst toward broad spectrum photocatalytic
generation of nanotube. Sol. RRL 5, 2000504 (2021). - Zhong, W. et al. Novel core-shell Ag@AgSe nanoparticle co-catalyst: in situ surface selenization for efficient photocatalytic
production of . Chin. J. Catal. 43, 1074-1083 (2022). - Tao, J. et al. A novel CoP@AAH cocatalyst leads to excellent stability and enhanced photocatalytic
evolution of CdS by structurally separating the photogenerated carriers. Appl. Catal. B Environ. 32O, 122004 (2023). - Yue, X., Cheng, L., Fan, J. & Xiang, Q. 2D/2D BiVO4
S-scheme heterojunction for photocatalytic reduction: Insights into structure regulation and Fermi level modulation. Appl. Catal. B Environ. 304, 120979 (2022). - Zhong, W., Zhao, B., Wang, X., Wang, P. & Yu, H. Synchronously enhancing water adsorption and strengthening
bonds in Se-rich cocatalyst for efficient alkaline photocatalytic production. ACS Catal 13, 749-756 (2023). - Yang, C., Wan, S., Zhu, B., Yu, J. & Cao, S. Calcination-regulated microstructures of donor-acceptor polymers towards enhanced and stable photocatalytic
production in pure water. Angew. Chem. Int. Ed. 134, e202208438 (2022). - Li, K., Blatov, V. & Wang, J. Discovery of electrides in electron-rich Non-electride materials via energy modification of interstitial electrons. Adv. Funct. Mater. 32, 2112198 (2022).
- Zhang, X. et al. Optimizing the Pd sites in pure metallic aerogels for efficient electrocatalytic
production. Adv. Mater. 35, 2211512 (2023). - Zhong, W. et al. Charging d-orbital electron of
cocatalyst enables splendid alkaline photocatalytic evolution. Adv. Funct. Mater. 33, 2302325 (2023). - Wang, J. et al. Cooperative coupling of photocatalytic production of
and oxidation of organic pollutants over gadolinium ion doped nanocomposite. Chin. Chem. Lett. 34, 108157 (2023). - Li, F. et al. Understanding the unique S-scheme charge migration in triazine/heptazine crystalline carbon nitride homojunction. Nat. Commun. 14, 3901 (2023).
- Zhao, M. et al. A novel S-scheme 3D
heterostructure for improved hydrogen production under visible light irradiation. Chin. J. Catal. 43, 2615-2624 (2022). - Chen, R., Fan, F., Dittrich, T. & Li, C. Imaging photogenerated charge carriers on surfaces and interfaces of photocatalysts with surface photovoltage microscopy. Chem. Soc. Rev. 47, 8238-8262 (2018).
- Chen, R. et al. Charge separation via asymmetric illumination in photocatalytic
particles. Nat. Energy 3, 655-663 (2018). - Gu, M. et al. Efficient sacrificial-agent-free solar
production over all-inorganic S-scheme composites. Appl. Catal. B Environ. 324, 122227 (2023). - He, B. et al. Cooperative coupling of
production and organic synthesis over a floatable polystyrene-sphere-supported S-Scheme photocatalyst. Adv. Mater. 34, 2203225 (2022). - Cheng, C. et al. Verifying the charge-transfer mechanism in SScheme heterojunctions using femtosecond transient absorption spectroscopy. Angew. Chem. Int. Ed. 135, e202218688 (2023).
- Wang, L. et al. Dynamics of photogenerated charge carriers in inorganic/organic S-Scheme heterojunctions. J. Phys. Chem. Lett. 13, 4695-4700 (2022).
Acknowledgements
Author contributions
calculations of the materials. X.Z. and D.G. carried out the photocatalytic test. X.Z., B.C., J.Y., and H.Y. contributed to data analysis. J.Y. and H.Y. supervised the project. X.Z. and H.Y. wrote the manuscript. B.C., J.Y., and H.Y. revised and reviewed the manuscript. All authors discussed the results and commented on the manuscript.
Competing interests
Additional information
supplementary material available at
https://doi.org/10.1038/s41467-024-47624-7.
http://www.nature.com/reprints
(c) The Author(s) 2024
Laboratory of Solar Fuel, Faculty of Materials Science and Chemistry, China University of Geosciences, 68 Jincheng Street, Wuhan, P. R. China. State Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, 122 Luoshi Road, Wuhan, P. R. China. State Key Laboratory of Advanced Technology for Material Synthesis and Processing, Wuhan University of Technology, Wuhan, P. R. China. e-mail: yuhuogen@cug.edu.cn