تفاعل الدعم المعدني الإلكتروني يعدل الهياكل الإلكترونية للنحاس من أجل الاختزال الكهربائي لثاني أكسيد الكربون إلى المنتجات المرغوبة Electronic metal-support interaction modulates Cu electronic structures for CO2 electroreduction to desired products
تفاعل الدعم المعدني الإلكتروني يعدل الهياكل الإلكترونية للنحاس من أجل الاختزال الكهربائي لثاني أكسيد الكربون إلى المنتجات المرغوبة
تاريخ الاستلام: 23 أغسطس 2024
تم القبول: 18 فبراير 2025
نُشر على الإنترنت: 25 فبراير 2025 تحقق من التحديثات
يونغ زانغفي في في تشينشيني يانغ ييران قوه شينغhua تشانغهونغ دونغويهوى وانغفنج لوزُنْمِينغ لوهوي ليوهوي ليو®، ياو شياوياهوى تشينغ®
في هذا العمل، تم دعم المحفزات أحادية الذرة من النحاس (SACs) بواسطة أكاسيد المعادن، و تُستخدم كنماذج نظرية لاستكشاف العلاقات بين الهياكل الإلكترونية والأداء. بالنسبة لهذه المحفزات، فإن التفاعل الإلكتروني بين المعدن والدعم (EMSI) الناتج عن نقل الشحنة بين مواقع النحاس والدعائم يعدل بشكل دقيق التركيب الإلكتروني للنحاس لتشكيل أعلى مستوى مشغول مختلف. أعلى مستوى مشغولمداريعزز قوة امتصاص CO ويضعفروابط من خلالالتبرع بالإلكترونات. هذا يقلل من حاجز الطاقة لـالترابط، مما يعزز تشكيل متعدد الكربون على. الأعلى مشغولاًمداريُسرعالتفعيل، ويقلل من طاقة التفاعل لتكوين. هذا مفرط النشاط، بدوره، يعزز تفاعل تطور الهيدروجين المتنافس (HER)، مما يعيق الإنتاج عالي الانتقائية لـعلى. مع أعلى إشغالترويج مداريالتفعيل وحالته الإلكترونية الموضعية تعيق اقتران C-C. النشاط المائي المعتدل لـيسهل * الهدرجة العميقة لثاني أكسيد الكربون دون تنشيط مفرط لتفاعل تطور الهيدروجين. ومن ثم،يعرض أعلىكفاءة فارادايفي.
تفاعل اختزال ثاني أكسيد الكربون المدفوع بالطاقة المتجددة ) هي طريقة واعدة لتحقيق دورة الكربون وإنتاج نظيف للمواد الكيميائية . تتقلص الجزيئات بشكل عميق على سطح المحفز القائم على النحاس إلى هيدروكربونات ومركبات تحتوي على الأكسجين مثل الميثانالإيثيلينالإيثانول (EtOH) ، إلخ ، من خلال خطوات متعددة مرتبطة بالإلكترونات والبروتونات. على الرغم من أن هذه المواد الكيميائية عالية القيمة والوقود عالي الكثافة الطاقية لها أسواق واسعة، إلا أن التنفيذ على نطاق صناعي لـلا يزال أمامه طريق طويل. من ناحية، في الوسائط المائية، الماءتعمل الجزيئات كمصدر للبروتونات للتفاعلات الكهروكيميائية، مما يؤدي إلى صراع بينوتفاعل تطور الهيدروجين (HER)خصائص اقتران البروتون والإلكترون يتطلب تفعيلًا فعالًاونقل البروتون بسلاسة لتجنب التنشيط المفرط لـوإلا فإن HER ستتفوق بشكل تنافسيمن ناحية أخرى، غالبًا ما تتواجد الهدرجة العميقة لثاني أكسيد الكربون الممتص (*CO) وترابط C-C معًا وتتنافسان مع بعضهما البعض، مما يؤدي إلى انخفاض انتقائية المنتج.إن الامتزاز والتغطية للوسطاء الرئيسيين *CO على سطح المحفز أمران حاسمان في التحكم في الانتقائيةالمنتجات. ومن ثم، فإن التصميم العقلاني والتخليق القابل للتحكم في المحفزات استنادًا إلى الفهم العميق لآلية التفاعل وعلاقة البنية بالنشاط أمر بالغ الأهمية للتنظيم الدقيق للمسارات التنافسية من أجل.
تعديل الهيكل الإلكتروني للعامل الحفاز وتوضيح تأثيره على النشاط الحفاز هو نهج فعال لدراسة
علاقات الهيكل-النشاط. لتعديل الهيكل الإلكتروني للنحاس، تركزت اهتمامات البحث على السبائكهندسة التلاعبتعديل تفاعل الدعم المعدني (MSI)، وما إلى ذلك. في التحفيز غير المتجانس، يؤثر تفاعل المعادن والدعم (MSI) بشكل كبير على الأداء التحفيزي حيث أنه يعدل الهياكل الإلكترونية والهندسية للمعادن بالإضافة إلى بيئات التنسيق. تم اقتراح تفاعل المعادن والدعم الإلكتروني (EMSI) من قبل كامبل، والذي يتجاوز MSI ويقدم تفسيرًا أكثر تفصيلًا للخصائص المحسنة للمحفزات المدعومة مقارنةً بـ MSI.ترتبط EMSI بإعادة الهجين المداري ونقل الشحنة عبر واجهة المعدن والدعم، مما يؤدي إلى تكوين روابط كيميائية جديدة وإعادة ترتيب مستويات الطاقة الجزيئية.من خلال التحكم الدقيق في هيكل نطاق d للمعادن من خلال EMSI، يمكن تعطيل علاقة قياس الامتصاص لتنظيم امتصاص الوسائط الرئيسية. هذا أمر حاسم للتحكم الدقيق فيمسار لأن البروتونات تشكل وسائط رئيسية (مثل *COOH وصعب على سطح الامتزاز الضعيف، بينما سيؤدي تعزيز قوة طاقة الامتزاز إلى تفضيل تفاعل تقليل الهيدروجين وفقًا لعلاقة قياس الامتزاز.ومع ذلك، فإن القيود المفروضة من تأثيرات الكتلة الجوهرية تجعل من الصعب تحديد المعلومات المدارية لجزيئات المعادن أو الكتل بدقة، مما يشكل تحديات في تقديم إرشادات التصميم أو تفسيرات تحسين الأداء للمواد الحفازة المدعومة..
يوفر نظام EMSI القائم على المحفزات ذات الذرة الواحدة (SACs) جسرًا للدراسات النظرية للهيكل الإلكتروني وتصميم المحفزات غير المتجانسة، لأن هيكلها الإلكتروني يمكن تمييزه بسهولة من خلال التجارب والحسابات النظرية.EMSI القوي لا ي stabilizes فقط المعادن ذات الذرة الواحدة بسبب تكوين الروابط بين المعدن والدعم المفضل حرارياً، بل يؤدي أيضاً إلى إعادة توزيع الشحنات الناتجة عن نقل الإلكترونات، مما يؤثر على توزيع مستويات الطاقة للأوربيتال 3d للذرات الفردية.. على سبيل المثال، أفاد ما وآخرون أن بيئة التنسيق لـيرفع مستوى الطاقة لـمداري مقارنةً بـ، مما يعزز امتصاص COOH ويعزز *إزالة CO. لذلك، مستوى الطاقة من قد يتم التلاعب بالحالة من خلال اضطرابات هيكلية منسقة لذرة النحاس.. على الرغم من أن SACs قد تم استخدامها على نطاق واسع لـالعلاقات العامة بين الهياكل الإلكترونية وسلوكيات التحفيز لـ SAC لا تزال غير واضحة. لذلك، هناك حاجة ملحة لفهم شامل على المستوى الذري لعلاقة الهيكل بالنشاط لـ Cu SAC لتوجيه تنظيم الهيدروجين العميق لـ * CO أو الاقتران.
هنا، تدعم Cu SACs بـ-كوساك، ، و تم بناؤها بتقنية ترسيب الطبقات الذرية (ALD) والارتباطات بين خصائص الهياكل الإلكترونية و يتم تبرير الأداء من خلال توصيف مفصل وحسابات نظرية الكثافة الوظيفية (DFT). إن تغيير الدعامات يعدل بشكل دقيق التركيب الإلكتروني لمواقع النحاس لتشكيل ثلاثة مواقع نشطة فردية (SACs) بأعلى مدارات مشغولة مختلفة تمامًا.مداري لـمداري لـ، و مداري لـ” ). الـمنيميل إلى التفاعل معمدار مضاد للرابطة لجزيء CO، الذي يعزز امتصاص CO علىوإضعافروابط من خلالالتبرع بالإلكترونات. في الوقت نفسه، يزيد عدم توطين إلكترونات النحاس من طاقات الامتصاص المجاورة لـ CO على – . هذه تعزز الربط بين الكربون – الكربون على – ، مما أدى إلى أدنى نسبة من كفاءة فاراداي (FE) إلى. الـمنيعرض تزاوجًا قويًا معرابطة ومدارات مضادة للرابطة لـ، مما يعزز الامتزاز لـويعززالانفصال عن طريق إضعاف روابط O-H. لذلك،يعرض أعلىمن 4.14. ومع ذلك، فإن هذا تم تنشيطه بشكل مفرط، بدوره، يعزز المنافسة في HER، مما يعيق الإنتاج عالي الانتقائية لـعلى – . الـيُعزز المداري بشكل فعالتفعيل وحالة الإلكترون النحاسي الموضعية تعيق اقتران C -C. هذا يجعلالعرض أمن 3.16. في هذه الأثناء، النشاط المائي المعتدل لـيسهلالهيدروجين العميق دون تنشيط HER بشكل مفرط، مما يؤدي إلى أعلىFE منفي.
النتائج
التحليل النظري للهيكل الإلكتروني
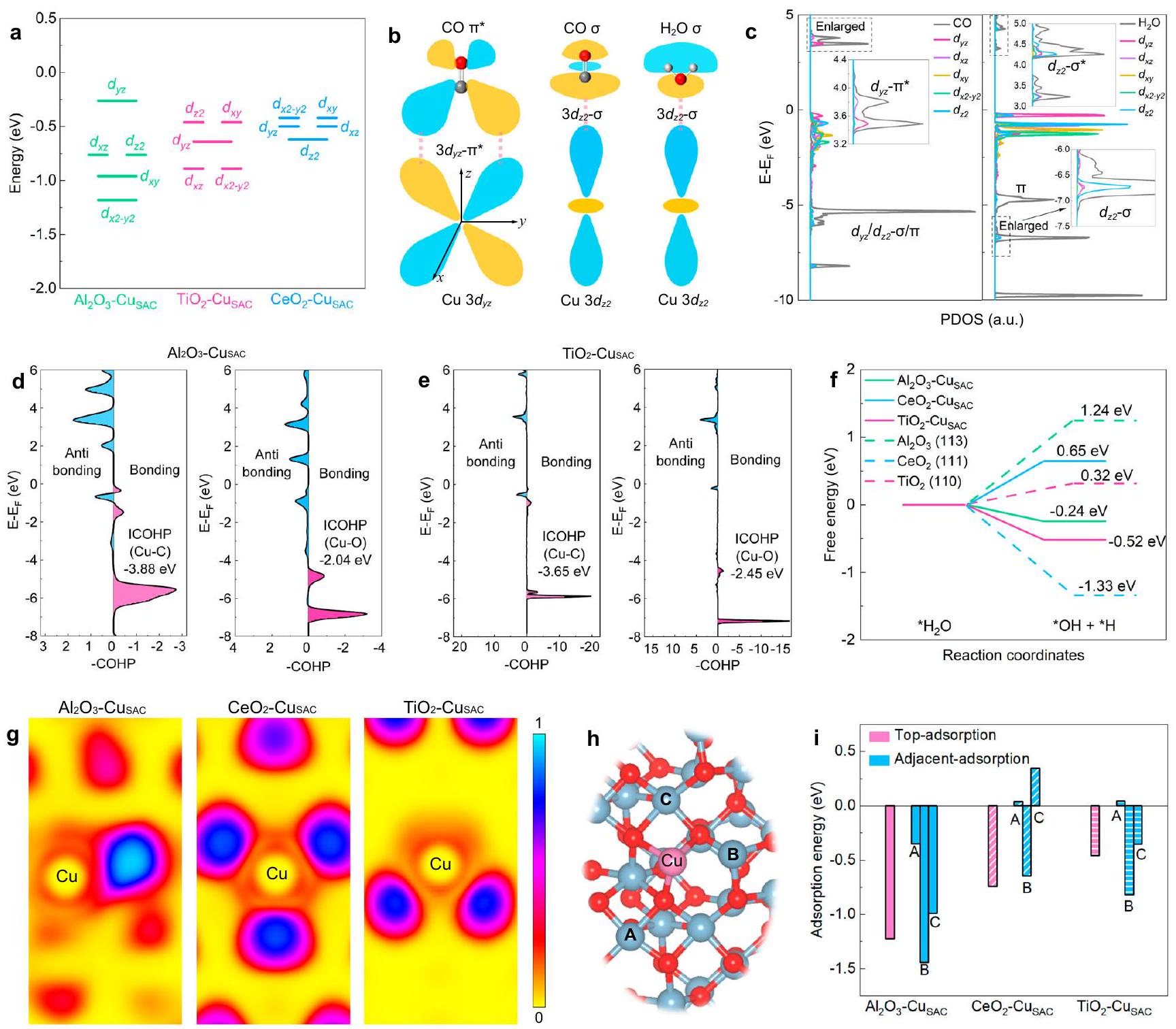
لاستكشاف علاقة الهيكل بالنشاط لهيكل الإلكترون الفردي للنحاس في الهيدروجين العميق الانتقائي لثاني أكسيد الكربون أو الاقتران، تم إجراء تحليل نظري استنادًا إلى حسابات DFT + U. من خلال تحميل ذرات النحاس الفردية على، و يدعم تعديل الهيكل الإلكتروني لذرة النحاس المفردة (الشكل التكميلي 1 والبيانات التكمالية 1)، المسمى بـ، و كما هو موضح في الشكل التوضيحي 2، الكثافة الجزئية للحالات (PDOS) لـمدارات، و تُحسب الأسطح، ومستويات الطاقة المعنية، ، و تم رسمها بناءً على مركز نطاق d الخاص بها (الشكل 1أ). بدلاً من تشكيل نطاق d واسع، فإن كل حالة 3d محلية بشكل كبير في الفضاء مع نافذة طاقة ضيقة. على، الـ هو أعلى مدار مشغول؛ على ، الـ هو أعلى مدار مشغول؛ بينما على ، الـهو أعلى مدار مشغول. من الواضح أن تغيير الدعم يمكن أن يؤثر بشكل كبير على أعلى مدار مشغول لذرة النحاس الفردية بسبب إعادة تهجين المدار ونقل الشحنة عبر واجهة المعدن والدعم (الشكل التكميلي 3)، و كل ذرة نحاس مفردة تقدم ثلاثة مدارات مشغولة أعلى مختلفة تمامًا. غالبًا ما تلعب المدارات d المشغولة الأعلى دورًا مهمًا في تنظيم أنماط الربط للوسطاء لأن الإلكترونات المملوءة في المدار الأقرب إلى مستوى فيرمي. ) أكثر نشاطًا من دوال الموجة المدارية، يمكن ملاحظة أنمداري (تحديد فيتماثل المحور) والتوافق الطاقي معمدار CO، مما يسهل تشكيل الروابط (الشكل 1ب). تناظر المدارات والتوافق الطاقي معمدار CO بالإضافة إلىمدار، مما يساعد في تشكيل الروابط (الشكل 1ب والشكل التكميلي 4). تناظر المدارات والتوافق الطاقي معمداراتتعزيز تشكيلسندات.
الشكل 1c والأشكال التكميلية 5 و 6 تظهر الكثافة النسبية لحالة الإلكترون (PDOS) لثاني أكسيد الكربون (CO) والمدارات الجزيئية الحدودية بالإضافة إلى الكثافة الإلكترونية لحالة الطاقة الجزئية لـ CO و الامتزاز على ، و الأسطح. يثبت الـ PDOS أن التهجين القوي بين ذرة CO وذرة Cu يساهم بشكل رئيسي من و مداري، والتهجين بينويساهم النحاس بشكل رئيسي منمدار. التهجين لـمداري معيساهم بشكل أساسي فيمداري، بينما المساهمة فييأتي أساسًا منمداري. يتماشى هذا مع تحليل دوال الموجات المدارية. على، الـمدار النحاس يتداخل معمدار CO، بينما يحدث تزاوج أقوى بين أعلى مستوى مشغولمداري ومدار CO أكثر من و مركز نطاق dعلىأقرب إلىأكثر من ذلك على و ، مما يجعل المزيد من الإلكترونات النشطة فيالتبرع بسهولة مرة أخرى إلىمن CO كتشكل لـسندات. علاوة على ذلك، بسبب خاصية عدم الترابط لـمداري، الالرابطة ستضعف الروابط في *CO، التي يمكن أن تسهل التفاعلات اللاحقة لـ *CO (الهيدروجين أو الاقتران) . بالمقابل، أعلى مشغولمداريعرض تزاوجًا قويًا معمدار CO، بينما التهجين بين و أقل من ذلك لـ – (الشكل التوضيحي 6a). بسبب المدارات الرابطة والمدارات المضادة للرابطة لـ هو و التزاوج بين و علىأقوى بشكل ملحوظ من ذلك على و (الشكل 1 ج والشكل التكميلية 6 ب، د)، مما يؤدي إلى قدرة معززة على يمتص. بالإضافة إلى ذلك، التفاعل بين المدار والمضاد للرابطةيضعف المداري الروابط، مما يعزز تفكك . على الأعلى المشغولالمدار يميل إلى الاقتران مع الأدنىمدار (الشكل التوضيحي 7)، ضعف
الشكل 1 | تحليل الهيكل الإلكتروني. أمخططات لـ، و ، على التوالي. ب أوضاع ارتباط CO والتفاعل مع موقع النحاس. الـ من – CuSAC مع CO الممتص و، بالإضافة إلى PDOS لـ CO والمدارات بعد الامتصاص (الإطار هو عرض مكبر للخط المتقطع في ج). منحنيات COHP لـالسندات والسندات في د و رسم بياني للطاقة الحرة لتفكك. الجني، و رسم تخطيطي لامتصاص CO المتجاورسطح. طاقة امتصاص CO لـ، و تم توفير بيانات المصدر ذات الصلة كملف بيانات المصدر. الروابط وتفضيل الهدرجة على ذرة الأكسجين ذات التنسيق المنخفض لتشكيل الوسيط الحاسم *COOH، مما يعزز تفعيل لتشكيل * .
تتبع طاقات امتصاص CO لذرات النحاس المفردة على هذه الدعائم الثلاثة الترتيب، متسق مع ترتيب مستويات الطاقة لـمداري (الشكل التوضيحي 8 والشكل 1أ).تكون طاقات الامتزاز مرتبطة ارتباطًا وثيقًا بموقع الـمستوى الطاقة المداري، موضحًا الترتيبتم استخدام توزيع هاملتون للسكان المداري البلوري (COHP) لتحليلقوة الرابطة في و الـ قوة الرابطة في (الشكل 1د، هـ والشكل التكميلي 9) . على ، يظهر ذرة النحاس الفردية أقوىترابط مع COHP مدمج (ICOHP) بقيمة -3.88 eV، يليه و . أسفل الـهناك عدد أقل من الإلكترونات المملوءة في المدارات المضادة للرابطة لـ مقارنةً بـ على، مشيرًا أن هو أكثر ملاءمة للتواصل مع CO. على النقيض، يعرض أعلى قيمة لـ ICOHP تبلغ -2.45 eV، مما يشير إلى أنالمدار هو مفضل لـالسندات. علاوة على ذلك،يعرض الأدنىطاقة التفكك -0.52 إلكترون فولت، تليها و (الشكل 1f). هذا يوفر بروتونات مفعلة كافية لعملية الهيدروجين العميق لـ *CO لتكوين. ومع ذلك، فإن التنشيط المفرط لـيمكن أن يعزز HER التنافسي، وطاقة التكوين لـعلىيصل إلى 0.18 إلكترون فولت، مما قد يثبط بشكل كبير النشاط (الشكل التوضيحي 10). على النقيض من (111) السطح بعيدًا عن مواقع الذرات الفردية للنحاس، طاقة التفكك لـهو 1.33 إلكترون فولت، والذي يمكن أن يوفر أيضًا بروتونات كافية لعملية هدرجة CO الم adsorb على مواقع الذرات المفردة من النحاس.
بعد التنسيق بين النحاس والأكسجين، يتجمع مقدار كبير من الشحنة حول ذرة الأكسجين، بينما يحمل النحاس الشحنة الموجبة بسبب الكهربية السالبة الأعلى للأكسجين مقارنة بالنحاس. تظهر تحليل شحنة بادر أن شحنة بادر للنحاس هيفي، “في، و في (تكميلي
الشكل 11). تشير وظيفة تحديد موضع الإلكترون (ELF) المستمدة من حسابات DFT إلى أنيجذب المزيد من الإلكترونات من النحاس، مما يؤدي إلى عدم تحديد موضع الإلكترونات حول ذرة النحاس المفردة (الشكل 1g)تم إجراء مزيد من الحسابات لطاقة الامتصاص العليا وطاقة الامتصاص المجاورة لثاني أكسيد الكربون على ذرة النحاس المفردة، وتظهر أوضاع الامتصاص المقابلة في الشكل 1h والشكل التكميلي 12. على، هناك انخفاض طفيف فقط في طاقات الامتصاص المجاورة لـ CO (من -0.35 إلى -1.44 eV) مقارنةً بطاقة الامتصاص العلوية (الشكل 1i). وهذا يشير إلى أن الحالة الإلكترونية غير المحلية للنحاس على – يمكن أن يمكّن المزيد من جزيئات CO من الامتصاص حول ذرة النحاس مناقتران. على ، فإن الكهربية السالبة للنحاس (Cu) أعلى من تلك الموجودة في السيريوم (Ce)، مما يمكّن النحاس من جذب الإلكترونات من السيريوم، مما يقلل بشكل كبير من انتشار الإلكترونات حول ذرة النحاس المفردة (الشكل 1g). ونتيجة لذلك، فإن طاقات الامتزاز المجاورة لـإلىعلى ذرة النحاس، ينخفض بشكل كبير مقارنةً بطاقة الامتصاص العلوية، مما يجعل *CO أكثر احتمالاً للخضوع للهيدروجين العميق في شكل امتصاص علوي فردي بدلاً من اقتران C-C.
التركيب والتوصيف
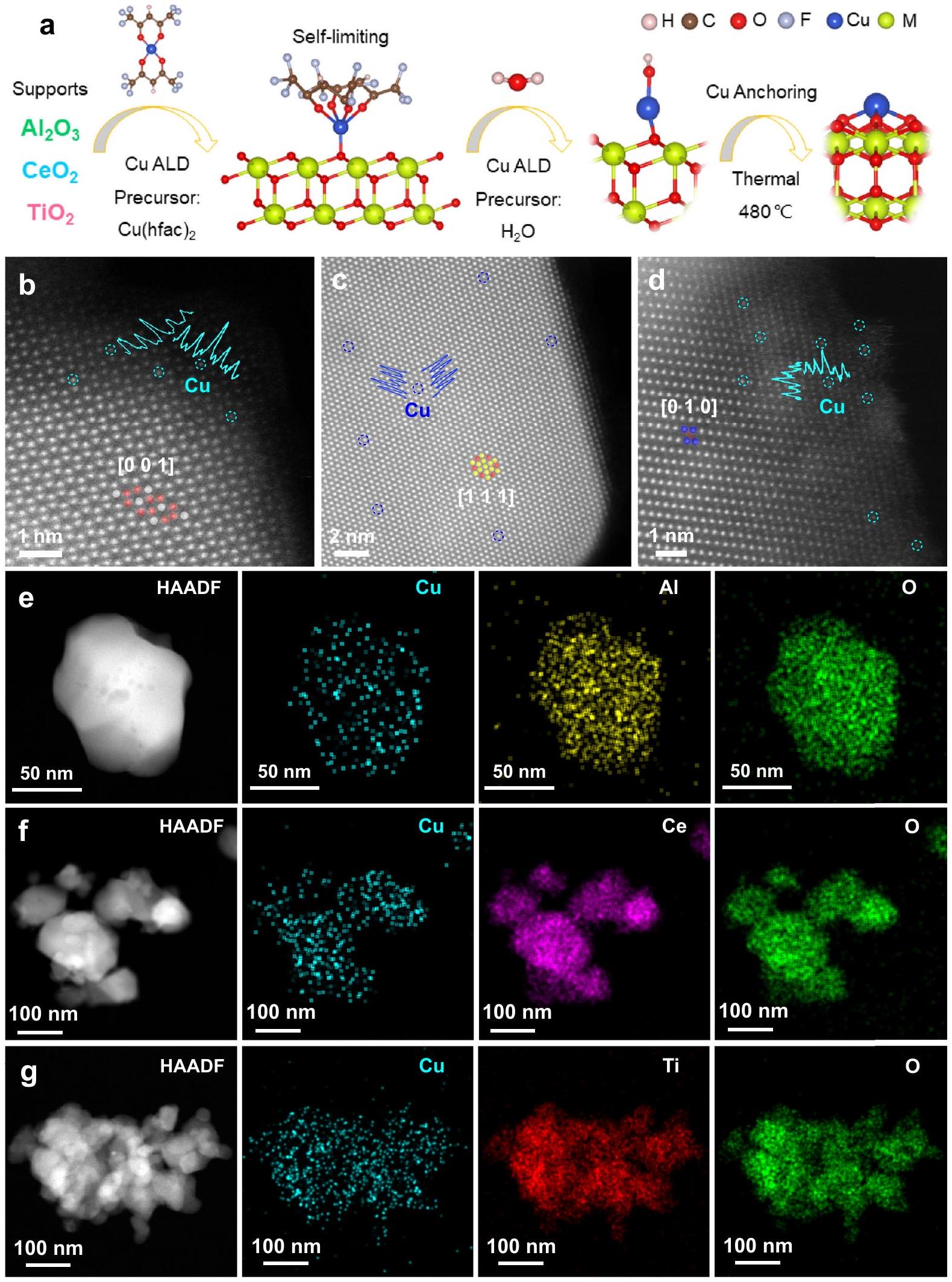
للتحقق من نتائج حسابات DFT، تم ترسيب ذرات النحاس علىروتيل، والفلوريتيدعم استخدام تقنية ALD. تشير صور المجهر الإلكتروني الماسح (SEM) والمجهر الإلكتروني الناقل (TEM) إلى أن أشكال هذه الدعائم الثلاث تتكون من جزيئات نانوية غير منتظمة (NPs) بأحجام جزيئات أكبر من 50 نانومتر، مما يتجنب تأثيرات الحجم للدعائم (الأشكال التكميلية 13 و 14).تؤكد أنماط حيود الأشعة السينية (XRD) وصور المجهر الإلكتروني عالي الدقة (HRTEM) أن الدعامات هي مراحل نقية من، و (الأشكال التكميلية 14 و 15). الاستفادة من تقنية ALD التي تعتمد على تفاعلات سطحية ذاتية التقييد على المستوى الجزيئي بشكل متسلسليمكن لذرة النحاس أن ترتبط بالدعائم في شكل ذرة واحدة (الشكل 2أ). تشير قياسات مطياف الانبعاث الضوئي البلازمي المقترن بالحث (ICP-OES) إلى أن كميات تحميل النحاس لـ، و هم، و ، على التوالي (الجدول التكميلي 1). تظهر صور TEM الماسح عالي الزاوية ذات الحقل المظلم عالي الدقة المصححة للانحراف أن ذرات النحاس موزعة ذريًا في، و الدعائم (الشكل 2ب-د)، دون وجود أي تجمعات مرئية عند كل من التكبير المنخفض والعالي (الشكل التكميلي 16). تؤكد أنماط حيود الأشعة السينية (XRD) وصور المجهر الإلكتروني الماسح (SEM) للعينات بعد الترسيب الكيميائي للبخار (ALD) أنه لا توجد جزيئات نانوية من النحاس (Cu NPs) مرئية على الدعائم (الأشكال التكميلية 17 و18). تشير صور التصوير المجهري الإلكتروني عالي الكثافة (HAADF-STEM) وتحليل الطيف بالأشعة السينية المشتتة للطاقة (EDS) إلى أن النحاس (Cu) موزع بالتساوي في جميع أنحاء، و يدعم (الشكل 2e، f). من خلال التحكم في عملية ALD، تم أيضًا إعداد محفزات غير متجانسة محملة بجزيئات النحاس النانوية لتكون بمثابة توسيع لمحفزات EMSI (الشكل التكميلي 19). المحفزات المحملة بجزيئات النحاس النانوية على و تسمىجزيئات النحاس النانوية-جزيئات النحاس النانوية، على التوالي. كميات تحميل النحاس لـ-جزيئات النحاس النانوية-جزيئات النحاس النانوية و ، على التوالي (الجدول التكميلي 2). توضح توزيع حجم الجسيمات الإحصائي لجزيئات النحاس النانوية بناءً على صور TEM أن متوسط قطر جزيئات النحاس النانوية المحملة على و هم و ، على التوالي (الأشكال التكميلية 20 و21). صور HRTEM لـ-جزيئات النحاس النانوية-CuNPs كلاهما يكشف عن واجهات بين جزيئات النحاس النانوية والدعامات، حيث تكون جزيئات النحاس النانوية مثبتة جزئيًا وبقوة على سطح الدعامة (الأشكال التكميلية 20e و21b).
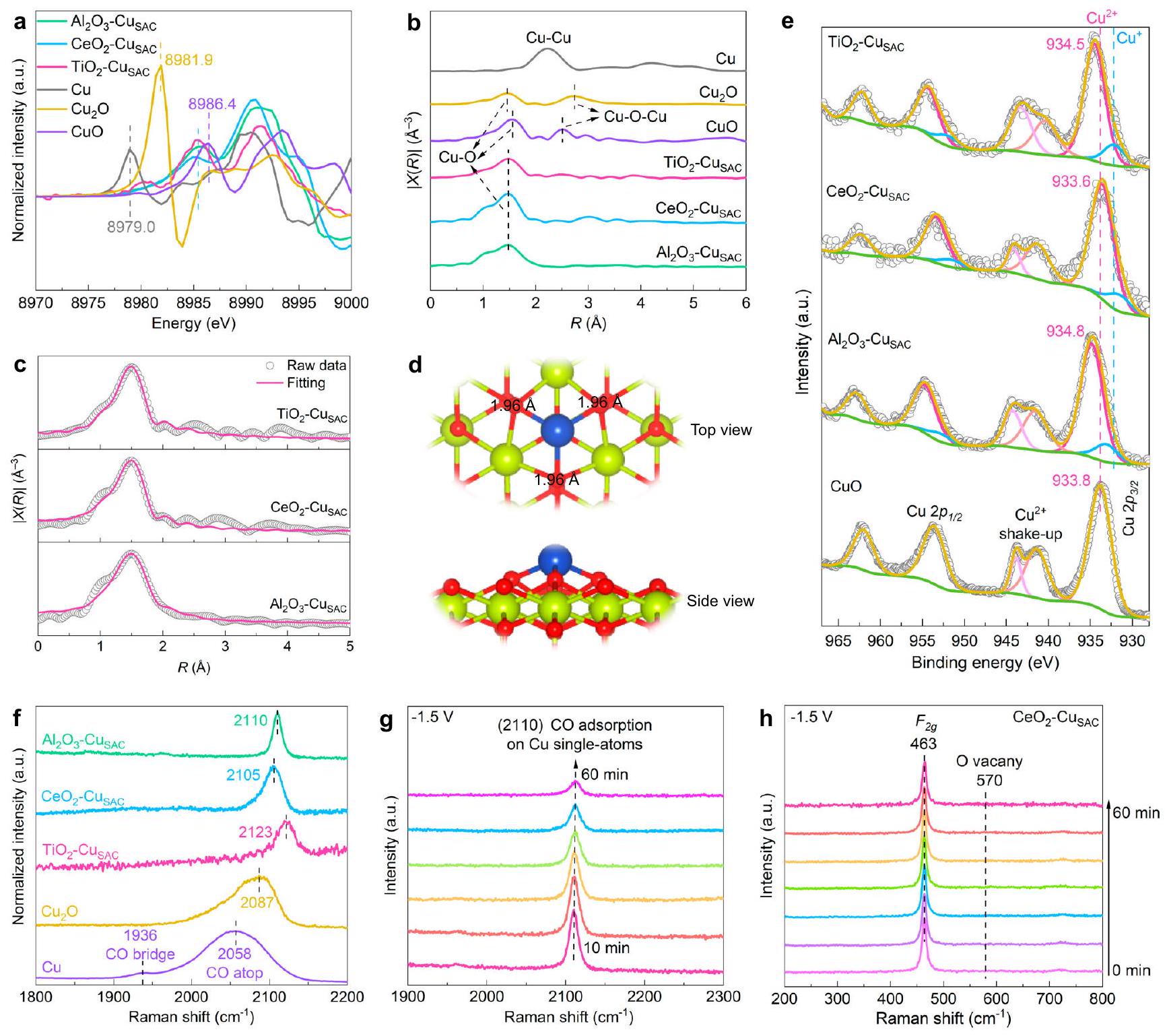
للحصول على المعلومات الهيكلية لـتم إجراء طيف الامتصاص بالأشعة السينية القائم على الأشعة السينية للأكسيد المعدني، وطيف الامتصاص بالأشعة السينية القائم على السنكروترون، وطيف الإلكترون الضوئي بالأشعة السينية (XPS). في طيف هيكل الامتصاص القريب من الحافة بالأشعة السينية (XANES)، كانت حواف الامتصاص لـ، و الانتقال إلى طاقة أعلى من تلك الخاصة بورق النحاس، مما يشير إلى أن ذرات النحاس في حالة مؤكسدة (الشكل التوضيحي 22)أول قمم، و تشبه وتقترب منفي البداية مشتق طيف XANES (الشكل 3أ)، مما يشير إلى حالة أكسدة أنواع النحاس، و قريبون منقمة واحدة ظاهرة فقط فييمكن الكشف عن القشرة التنسيقية الأولى لتشتت النحاس-الأكسجين في طيف الامتصاص بالأشعة السينية الممتد (EXAFS) لـ، و (الشكل 3ب). هذا يؤكد أن ذرات النحاس موزعة على المستوى الذري لأنه لا يوجد أو تلاحظ الروابط المعدنية. بيانات تركيب EXAFS لـ، و موضحة في الشكل 3c، الشكل التوضيحي 23، والجدول التوضيحي 3. المتوسطأرقام التنسيق لـ، ، و هي 3.37 و 3.67 و 3.41، على التوالي. بالاقتران مع نتائج الحسابات النظرية ( يمتلك أقل طاقة حرة؛ الشكل التوضيحي الإضافي 1)، تكشف هذه البيانات أن الهيكل المحلي لـيتكون SAC من أكاسيد المعادن حيث يمكن لذرة النحاس المعزولة أن تتنسق مع حوالي ثلاث ذرات أكسجين.وحدات) في هيكل الهرم الثلاثي بين ذرات النحاس والأكسجين (الشكل 3د)تركيبتشير طيفيات XPS إلى أن حالة الأكسدة السائدة على أسطح، و هو ، مع نسب من ، و ، على التوالي. (الشكل 3e). بالمقارنة مع CuO، فإنقمم XPS لـ و الانتقال إلى طاقة ربط أعلى، بينماوالقمم XPS تنتقل إلى طاقة ربط أقل من تلك الخاصة بالنقي و (الشكل التوضيحي 24). وهذا يعني أن الإلكترون ينتقل من النحاس إلى الجوار و الدعائم، التي تنتج عن التفاعل القوي بين النحاس والدعائم المعدنية-الأكسيد. على النقيض، فإن النحاسيتم تحويل قمة XPS إلى طاقة ارتباط أقل على سطح، مما يشير إلى أن النحاس يجذب الإلكترونات من يدعم، ربما بسبب الكهربية السالبة الأعلى لذرة النحاس مقارنة بذرة السيريوم. هذه النتائج لانتقال الإلكترون بين النحاس والدعائم تتماشى مع حسابات نظرية الوظيفة الكثيفة.
باستخدام CO كجزيء استكشافي، تم دراسة التشتت الذري لذرات النحاس واستقرار ذرات النحاس الفردية خلاليمكن تمييز العملية بواسطة مطيافية رامان في الموقع (الشكل 3f). في محلول مشبع بـ 0.1 م من أول أكسيد الكربونتمت ملاحظة قمم رامان المنسوبة إلى الامتصاص العلوي الخطي لـ CO (اهتزاز تمدد C-O) عند 2087 ولـو Cu ، على التواليتمت ملاحظة الامتزاز المربوط لثاني أكسيد الكربون (أي، ثاني أكسيد الكربون الممتز على ذرتين متجاورتين من النحاس) أيضًا على سطح النحاس. بالمقابل، لم يتم الكشف عن الامتزاز المربوط لثاني أكسيد الكربون على سطح النحاس SAC. تظهر طيف رامان المجمعة على الدعامات أنه لا يوجد ذروة في النطاق منإلى، مما يشير إلى عدم وجود امتصاص لثاني أكسيد الكربون على الدعامات (الشكل التكميلي 25). لذلك، فإن قمم رامان الموجودة عند 2110 و2105 ويمكن أن يُعزى إلى CO المتواجد على مواقع النحاس الفردية، و تكشف حسابات DFT المستندة إلى نموذج بلاي هولدر عن اختلافات في تردداهتزازات التمدد بين مواقع النحاس الفردية ومواقع تجمعات النحاس (الشكل التكميلي 26)، مما يوفر الأساس للقياسات الطيفية لتقييم استقرار ذرات النحاس الفردية. يرتبط استقرار ذرات النحاس الفردية بشكل جيد مع الألفة بين مواقع النحاس والدعامات، وبشكل خاص، تؤدي التفاعلات القوية بين النحاس والأكسجين إلى استقرار عالٍ.لتقييم استقرار ذرات النحاس الفردية خلال عملية الاختزال الكهربائي، تم تطبيق جهد ثابت قدره -1.5 فولت (جميع الجهود في هذا العمل هي مقابل القطب الهيدروجيني القابل للعكس، RHE).القطب الكهربائي لمدة 60 دقيقة. بعد التحليل الكهربائي عند -1.5 فولت لمدة 60 دقيقة، لم تُلاحظ أي قمم رامان المقابلة لامتصاص جسر CO على مواقع النحاس المعدنية (الشكل 3g)، مما يشير إلى أن النحاس يبقى مثبتًا في الدعامات في شكل ذرات مفردة خلال عملية التحليل الكهربائي. يمكن أن يُعزى الانخفاض في شدة قمم رامان لـ CO إلى تراكم الفقاعات خلال التحليل الكهربائي طويل الأمد. وبالمثل، يتم الحفاظ على استقرار الدعامات خلال التحليل الكهربائي بسبب ارتفاع إمكانيات الاختزال.، و . على وجه التحديد، بعد فترة طويلة منالتحليل الكهربائيلا تظهر قمم رامان الناتجة عن عيوب الأكسجين في، والأول من الدرجةذروة رامان المميزة لـيبقى حاضرًا طوال الوقتعملية (الشكل 3 ح)في الموقع
الشكل 2 | التخليق والتوصيف الهيكلي. أ رسم تخطيطي لعملية التخليق لدعم Cu SAC مع أكسيد المعدن. صور HAADF-STEM عالية الدقة لـ، و نماذج ذرية: أل، رمادي؛ س، الأصفر؛ التيتانيوم، الأزرق؛ والأكسجين، الأحمر. صور رسم خرائط العناصر باستخدام HAADF-STEM وEDS لـ e، و .
الشكل 3 | توصيف مواقع النحاس الفردية. أ المشتقات الأولى لطيف XANES.المقابل-طيف تحويل فورييه الموزون لـ EXAFS. ج تركيب لـ-بيانات EXAFS الموزونة لـ، و في نطاق 1.0-2.5 Å. د المقترحتكوين SAC أكسيد المعدن. النماذج الذرية: المعدن، أصفر، النحاس، أزرق؛ والأكسجين، أحمر. طيف XPS. طيف رامان لامتصاص CO علىأكسيد المعدن، و طيف رامان في الموقع لامتصاص CO علىلإجراء التحليل الكهربائي على المدى الطويل عند -1.5 فولت دونتصحيح. طيف رامان في الموقع لـلإجراء التحليل الكهربائي على المدى الطويل عند -1.5 فولت دونتصحيح. تم توفير بيانات المصدر ذات الصلة كملف بيانات المصدر.
طيف رامان لـ و كما تؤكد استقرارها خلال التحليل الكهربائي على المدى الطويل (الشكل التوضيحي الإضافي 27).
أداء
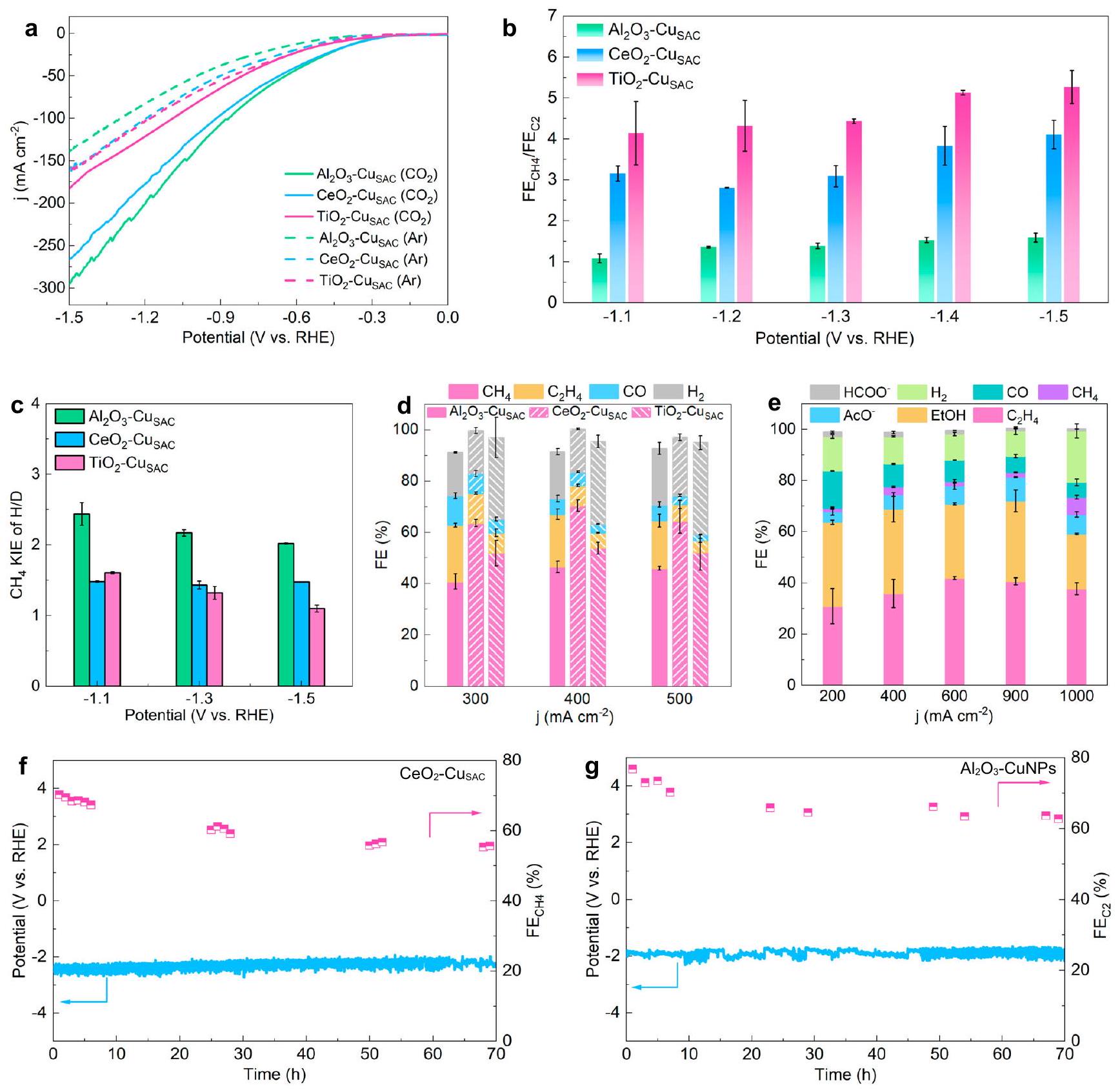
تجارب كيميائية كهربائية على، و تمت في خلية تدفق مع التحليل الكهربائي عند جهد ثابت. كانت المنتجات الغازية الرئيسية التي لوحظت خلالهم، CO ، و ، مع منتجات سائلة طفيفة (الشكل التوضيحي التكميلي 28). قبل تقييم أداء، و تمت مراعاة شاملة لاستبعاد التأثيرات المحتملة لامتصاص الهيدروكسيد السطحي، ومقاومة نقل الشحنة للدعم، وكميات تحميل النحاس علىأداء المحفزات من خلال سلسلة من القياسات الكهروكيميائية، BET، وSEM (الأشكال التكميلية 29-31 والملاحظة التكميلية 1). تشير منحنيات الفولتامترية المسحية الخطية (LSV) إلى أن كثافات التيار لـتزداد بسرعة أكبر مع الإمكانية، تليها و (الشكل 4أ). في هذه الأثناء، كثافات التيار من العينات التي تم قياسها بـالغاز عند جميع الجهود المطبقة أعلى من تلك المسجلة مع غاز الأرجون، مما يشير إلى حدوث. أصغر فرق في قيم كثافة التيار بين القياسات لـفيوتمثل الغلافين الجويين للأرجونتظهر أعلى نشاط HER وقدرة الانفصال، تليها و يتم عرض توزيع المنتج من -1.1 فولت إلى -1.5 فولت في الشكل التوضيحي 28. عند -1.1 فولت، تكون نسبةإلىلـ، و هو ، و4.14 على التوالي، وتزداد كلما أصبح الجهد أكثر سلبية (الشكل 4ب). كما تم التنبؤ به من خلال التركيب الإلكتروني،يعرض أعلى درجة من الانتقائيةبين هذه المحفزات الثلاثة، بينما و إظهار انتقائية تجاهتحليل لـأثر النظائر الحركية (KIE)، يُعرف بأنه نسبةمعدل الجيل في و الالكتروليتات القائمة على -، تم إجراءها لتقييم تأثير الهياكل الإلكترونية على عملية نقل البروتون لـ (الشكل 4c والشكل التكميلي 32). قيم KIE لـأعلى من
شكل. | أداء. منحنيات LSV لـ، و معو Ar كغازات إمداد. بمن، و عند الجهود من -1.1 فولت إلى -1.5 فولت. c KIE لـ H/D لـ إلىالتحويل عند إمكانيات مختلفة. د القيم المقابلة عند كثافات تيار عالية. هـ القيم الخاصة بـ-جزيئات النحاس النانوية عند كثافات تيار مختلفة. على المدى الطويل عدم الاستقرار و -CuNPs عند كثافة تيار ثابتة منلاتم تطبيق تصحيح لحساب الجهد المطبق. تمثل أشرطة الخطأ الانحرافات المعيارية من ثلاثة قياسات مستقلة على الأقل. تم توفير البيانات المصدر ذات الصلة كملف بيانات مصدر. و عند جميع الإمكانيات وقريبًا من 2، مما يشير إلى أن خطوته المحددة للسرعة تتضمن انفصال. لديه أدنى قيمة KIE، مما يشير إلى أنتسارع مداري بشكل كبير عملية التفعيل لـبينماتظهر معدلات انفصال معتدلة من. بالنظر إلى طاقة امتصاص ثاني أكسيد الكربون المعتدلة والحالة الإلكترونية الموضعية لـ، يظهر الأعلىمن، خاصة عند كثافة تيار عالية من (الشكل 4د). و لديها منخفضة نسبيًاقيم من و على التوالي. – يظهر أداءً ممتازًا من حيثكثافة التيار الجزئي و FE مقارنةً بالعوامل الحفازة الممتازة المبلغ عنها سابقًا (الشكل التكميلي 33 والجدول التكميلي 4).
تم إجراء مزيد من الاختبارات علىأداء-جزيئات النحاس النانوية-CuNPs للتحقق من علاقات الهيكل-النشاط بناءً على EMSI لتوجيه تصميم المحفزات المدعومة. بسبب تعزيزالاقتران بواسطة الهيكل الإلكتروني للنحاس في، فإنه يظهر مستوى أعلىعند توسيع ذرات النحاس الفردية إلى جزيئات النحاس النانوية، يمكن أن تؤدي المزيد من المواقع لامتصاص CO أو CHO والتزاوج على سطح النحاس إلى زيادة الانتقائية لـالمنتج (الشكل التوضيحي التكميلي 34). كانت النسب المئوية المختلفة لـالمنتجات، بما في ذلكأسيتات، و الإيثانول، فوق – CuNPs ضمن نطاق كثافة التيار من 200 إلىموضحة في الشكل 4e. بشكل محدد، تحت شرط التحليل الكهربائي بتيار ثابت، تم تعزيزانتقائيةيتم ملاحظة -CuNPs، محققًا أعلىمنفي. على النقيض، فإن انتقائية
الشكل 5 | حسابات DFT و ATR-IRAS في الموقع. أ مخطط الطاقة الحرة لـلتشكيل. ب مخطط الطاقة الحرة لاقتران *CO-*CHO. طيف ATR-IRAS في الموقع لـ فوق و e تحليل الطيف تحت الأشعة تحت الحمراء باستخدام تقنية ATR-IRAS لتمدد O-H في الموقع اهتزاز لـ، و . جزء من الحريةفي واجهة القطب الكهربائي – الإلكتروليت. c-i لاتم تطبيق تصحيح لحساب الجهد المطبق. تم توفير بيانات المصدر ذات الصلة كملف بيانات المصدر. غير مفعل – لا تزيد CuNPs بشكل ملحوظ مع زيادة المواقع لتزاوج C – C، مع من و منفي (الشكل التوضيحي 35). كما تكشف منحنيات LSV أن -CuNPs يظهر حجمًا أكبركثافة الاستجابة الحالية من CuNPs (الشكل التوضيحي 36). المتانة على المدى الطويل لـ و -تم تقييم CuNPs من خلال الاستمرار فيفي، مع الاستبدال الفوري للإلكتروليت وتنظيف GDE لتجنب ترسب الملح (الشكل 4f، g). الجهد المطبق لـيحافظ على الاستقرار لمدة 70 ساعة، معالبقاء باستمرار فوق-CuNPs تظهر أيضًا استقرارًا لمدة 70 ساعة مع مستقر فوق. أخيرًا، تم إجراء قياسات SEM وXRD وXPS وTEM على و المحفزات بعد اختبارات الاستقرار، جنبًا إلى جنب مع قياس ICP-MS على الإلكتروليت (الأشكال التكميلية 37، 38 والجدول التكميلية 5). تؤكد هذه النتائج مجتمعة استقرار المحفزات خلال فترة طويلة.اختبارات (ملاحظة إضافية. 2).
حسابات DFT ورؤى طيفية في
بعد ذلك، تم إجراء حسابات DFT بشكل إضافي لكشف تأثير الهياكل الإلكترونية على الانتقائية لـ المنتجات. أولاً، الطاقة المطلوبة لتحويلتم حساب * CO لهذه النماذج الثلاثة، تليها طاقة التكوين لـمن خلال الهيدروجين العميق لـ *CO (الشكل 5 أ والجداول التكميلية 6، 7). متسق مع الاستنتاجات من تحليل الهيكل الإلكتروني، فإن أعلى مستوى مشغولالمدار يقلل من طاقة التفاعل لـتنشيط لتكوين * COOH على، وهو ما يقل بشكل كبير عن ذلك على و الأسطح. يتم اختزال الوسط *COOH إلىنوع من خلال التفاعل مع بروتون وإطلاقالجزيء. بعد ذلك، في فرع *هدرجة أول أكسيد الكربون إلى *CHO أو *COH، يجب أن يكون *CHO هو النوع الرئيسي المتكون من هدرجة *CO على هذه الأسطح لأن الطاقة التفاعلية المطلوبة لتكوين *CHO أقل من تلك المطلوبة لتكوين *COH (الشكل التكميلي 39). الطاقة التفاعلية لهدرجة *CO على هو الأدنى بمقدار 0.02 إلكترون فولت، يليه من 0.61 إلكترون فولت ومن 0.66 إلكترون فولت. في المسار لتكوين *CHOتفاعل * CHOH مع بروتون وإطلاقلتشكيلهو الخطوة المحددة للسرعة لـRR-toطاقة التفاعل لـ * CHOH إلى * CH على و تصل إلى 1.31 إلكترون فولت و0.90 إلكترون فولت، على التوالي. بالمقابل، يتم تقليل طاقة التفاعل لـ *CH إلى 0.46 إلكترون فولت بسبب النشاط بروتونات على. بعد ذلك، الحواجز الطاقية لـتم حساب الاقتران على هذه الأسطح الثلاثة، وهو أمر حاسم لتكوينالمنتجات وتتنافس مع *CO الهيدروجين العميق. على وجه التحديد، فإن حاجز الطاقة لاقتران *CO-*CHO هو الأدنى حيث يبلغ 0.37 إلكترون فولت على، تليها من 0.61 إلكترون فولت و – من 0.80 إلكترون فولت (الشكل 5ب والشكل التكميلي 40). من الجدير بالذكر أنه على جميع الأسطح الثلاثة، فإن طاقة التفاعل لتكوين ثنائي *CO أعلى من تلك الخاصة بالاقتران غير المتماثل لـ *CO-*CHO (الشكل التكميلي 41)، مما يشير إلى آلية اقتران على أسطح Cu SAC حيث يتم هدرجة *CO إلى *CHO ويتزاوج مع وسائط CO المحيطة. تشير حسابات DFT أيضًا إلى أنه عندما تكون المواقع النشطة على يدعم التحول من ذرة النحاس المفردة إلى تجمع النحاس، حيث تمنع المواقع المتتالية للنحاس بشكل قوي التوليد الديناميكي الحراري لـمع الحفاظ على انتقائية عالية تجاهالمنتجات (الشكل التوضيحي التكميلي 42).
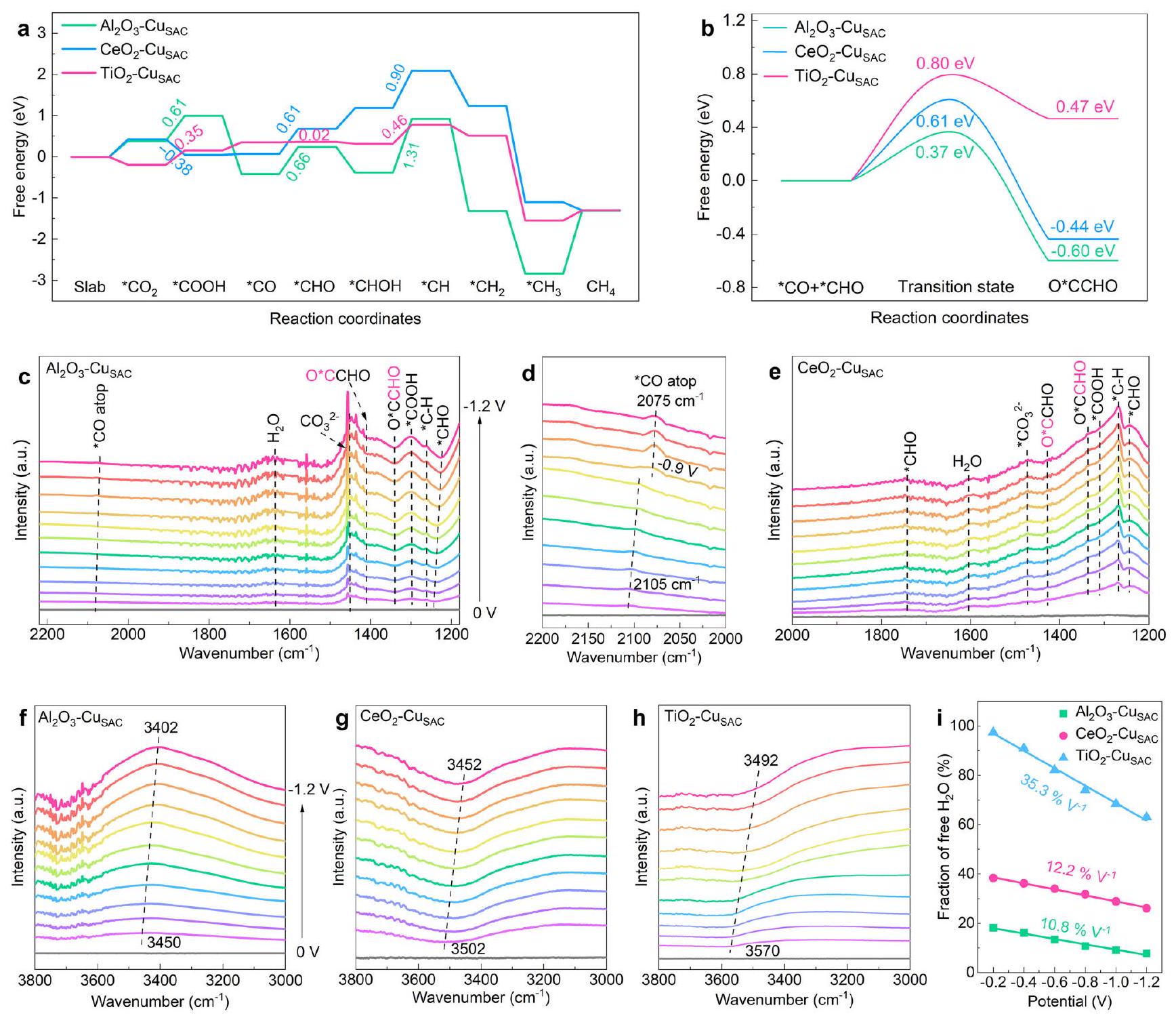
تم إجراء مطيافية الامتصاص بالأشعة تحت الحمراء بتقنية الانعكاس الكلي المخفف في الموقع (ATR-IRAS) عند إمكانيات مختلفة للكشف عن طرق امتصاص الوسطاء في مسارات التفاعل المختلفة. كانت الأطياف تحت الحمراء (IR) عند و تُخصص للوسائط المقابلة بناءً على الدراسات المبلغ عنها بشكل مستقل (الجدول التكميلي 8). نطاق الأشعة تحت الحمراء عنديمكن أن يُعزى إلىتشوه * COOH، وهو وسيط حرج فيمسار تحويل CO إلى * (الشكل 5c، e). نطاقات الأشعة تحت الحمراء عند 1242 ويمكن أن يُعزى إلىاهتزاز التمدد لـوتشويه *C-H في المسار لتحويل *CO إلى الهيدروجين العميق لإنتاج. على فرقة الأشعة تحت الحمراء ( المنسوبة إلى يمكن أيضًا اكتشاف اهتزاز الشد لـ * CHO. نطاقات الأشعة تحت الحمراء عند 1340 ويمكن تعيينه إلىالوسط الذي يتكون بعد الاقتران غير المتماثل لـ * CO و *. جزء CHO من في، وجزء CO من في. نطاق الأشعة تحت الحمراء في علىيمكن أن يُعزى ذلك إلى الامتصاص العلوي لـ CO (الشكل 5d)، بينما لم يتم الكشف عن أي وسيط *CO على (الشكل التوضيحي 43)، ربما بسبب انخفاض تركيز *CO أو الاستهلاك السريع من خلال الهدرجةالنطاق الواسع للأشعة تحت الحمراء في نطاقيمكن أن يُعزى إلىانحناء H للمواد الممتصة، مع الذروة فيالسطح يبدو أكبر بشكل ملحوظ من ذلك علىالسطح، متسق مع حسابات DFT. لتجنب تأثير النطاق الواسع للأشعة تحت الحمراء منوتحقق مما إذا كانت الأنواع الممتصة علىتحتوي السطح على الهيدروجين، وتم تكرار ATR-IRAS في الموقع باستخدامبدلاً منمشبعفي. الـتختفي الفرقة وتتحول إلى انزياح أحمر إلى، وهو متسق مع اهتزاز الانحناء في المستوى (D-O-D) لـ (الشكل التوضيحي 44) تم تحويل نطاق الكربونات الممتصة إلى اللون الأزرق في النظام المديوتري إلى، وهو ما يتماشى مع الأعمال المبلغ عنها مؤخرًا. من المحتمل أن يكون ذلك بسبب تغيير الديوتريوم في توازن الكربونات والبيكربونات وعملية امتصاص الكربونات غير المتناظراهتزاز التمدد لـيمكن اكتشافه عند. الشهرة، نطاقات الأشعة تحت الحمراء حول (*COOH)، و انتقل بوضوح إلى، 1257 (*C-D)، 1285 (COOD)، و 1317 (OCCDOفي، مما يشير إلى علاقتهم بالهيدروجين. على الـعلى السطح، تخضع نطاقات الأشعة تحت الحمراء لـ *CHO و *CO لكلاهما لانزياح نحو الأحمر مع زيادة الجهد، وهو ما يسببه تأثير ستارك (الشكل 5c، d). هذا يشير إلى أن *CO و *CHO يتم توليدهما في الموقع، وتحدث تفاعلات اقتران بينهما.علاوة على ذلك، شدةأعلى بكثير من ذلك لـ *، مما يشير إلى أن أكثر ملاءمة لـتفاعلات الاقتران. على النقيض، فيالسطح، شدة *C-H أعلى بكثير من تلك الخاصة بـ، مما يشير إلى أن * CO على السطح أكثر ملاءمة للهدرجة، مما يؤدي إلى توليد كمية كبيرة من.
دولةالجزيئات بالقرب من واجهات القطب الكهربائي – الإلكتروليت، و تم تحليلها بعد ذلك. النطاق العريض في 3000-3800يمثل وضع اهتزاز الشد لرابطة O-H (الشكل 5f-h)، ويمكن أن يكون النطاق العريض مقسمة إلى ثلاثة قمم بالقرب من 3610 و 3450 ومن خلال التناسب الغاوسي وفقًا لبيئات الروابط الهيدروجينية المميزة للماء (الشكل التكميلي 45). إنهم ينتمون إلى الماء بدون روابط هيدروجينية (حر ) ، مع روابط هيدروجينية ضعيفة (مثل السائل ) ، وروابط هيدروجينية قوية (مثل الجليد )، على التوالي. بشكل عام، فإن زيادة درجة الروابط الهيدروجينية تخفض من طاقة امتداد O-H، وبالتالي طاقات التفكك لـزاد بالترتيب: مجانيمثل السائلمثل الجليد. على النسبة الأولية للحر هو أعلى بكثير منمن و من. هذا يشير إلى أن السطح مُغَنى بالحر، مما يوفر بروتونات نشطة أكثر لعملية الهيدروجين العميق لثاني أكسيد الكربون وتفاعل HER. مع ازدياد السلبية في الجهد، تتزايد نسبة البروتونات الحرةعلى أسطح الأقطاب الثلاثة يتناقص تدريجياً، مما يشير إلى استهلاك الحرالمشاركة في التفاعلات السطحية (الشكل 5i). معدل استهلاك الحرةعلى السطح هو الأسرع بسبب القدرة الهيدروليتية القوية لـ. ومع ذلك، فإن زيادة الهيدروجين النشط تعزز تفاعل تقليل الهيدروجين المنافس، مما يزيد من من. على النقيض، فإن الندرة المنخفضة لـ وقدرة التحلل المائي الضعيفة لـ – يجعل * CO أكثر عرضة للتزاوج بدلاً من الهدرجة العميقة.موجودة بين و ، يوازن * الهدرجة العميقة للهيدروجين وHER بشكل جيد، مما يؤدي إلى بقدر ما.
نقاش
باختصار،، و تم بناؤها بنجاح بواسطة تقنية ALD، واستنادًا إلى هذه المحفزات، فإن العلاقات بين خصائص الهياكل الإلكترونية و يتم اقتراح الأداء من خلال توصيف مفصل وحسابات DFT. EMSI بين مواقع النحاس وترتيب الدعمالمدار المداري كأعلى مدار مشغول، مما يعزز قوة الامتزاز لـ CO ويضعف روابط C -O من خلالالتبرع بالإلكترونات. هذا يقلل من حاجز الطاقة لربط الكربون-كربون، مما يعزز تكوينالمنتجات على. EMSI بين مواقع النحاس وترتيب الدعمالمدار المداري كأعلى مدار مشغول، مما يعزز الامتصاص لـويعززالانفصال، مما يقلل من حاجز الطاقة لتشكيل. ومع ذلك، فإن هذا النشاط المفرط، بدوره، يعزز المنافسة في HER، مما يعيق الإنتاج عالي الانتقائية لـعلى. EMSI بين مواقع النحاس وترتيب الدعمالمدار المداري كأعلى مدار مشغول، مما يعززالتفعيل والبروتنة.يوازن بشكل فعال بين قوة امتصاص CO وتنشيط، مما أدى إلى أعلى منبين الثلاثة SACs في. يمكن أن توفر هذه العلاقة بين التركيب والنشاط المستندة إلى EMSI إلهامًا لتصميم المحفزات المدعومة. تحت هذا التوجيه، يُتوقع أن يتم تحميل جزيئات النحاس النانوية علىيعرض أعلىانتقائية أكثر من تلك على، و منفيتحقق. لا يوفر هذا العمل محفزًا فعالًا للتطبيقات الصناعية المحتملة فحسب، بل يمنح أيضًا فهمًا عميقًا لـآليات قد تلهم التصميم العقلاني لمحفزات أخرى للتحكم في.
طرق
تحضير، و
النقيفلوريتوروتيلتم شراءها من شركة شنغهاي ياوي سبائك المواد المحدودة. لوضع ذرات النحاس المفردة على، و الدعائم، تم استخدام طريقة ترسيب الطبقة الذرية (ALD). أولاً، تم توزيع 0.1 جرام من الدعائم بشكل متساوٍ في قرص، ثم تم وضع القرص داخل غرفة الفراغ، حيث حدثت عملية ALD. تم الحفاظ على درجة حرارة غرفة الفراغ عندخلال عملية ALD. ثنائي (هيكسا فلوروأستيل أسيتوناتو) النحاس(II) (Cu(hfac)تم استخدام شركة نانجينغ آي مو يوان لمعدات العلوم المحدودة كمواد سابقة للنحاس، وتم استخدامه كعامل مختزل.تم الاحتفاظ بـ و الـ ناقل تم الاحتفاظ بالغاز في. كانت دورة ALD تتبع تسلسلًا دقيقًا: تقديم من خلال صمام الملف اللولبي لمدة 50 مللي ثانية وتفريغ الغرفة بـ لمدة 12 ثانية. في هذه النقطة، كانت استجابة الضغط في غرفة الفراغ 0.5 باسكال. بعد ذلك، تم حقنه لمدة 100 مللي ثانية وجولة أخرى منتم تنفيذ التنظيف لمدة 30 ثانية. تم تكرار هذه الدورة من الترسيب 150 مرة. بعد انتهاء دورات ALD، تم تسخين غرفة الفراغ على الفور إلىمعوتم الاحتفاظ بـلمدة 3 ساعات لتثبيت ذرات النحاس الفردية بشكل قوي على الدعائم.
إعداد – جزيئات النحاس النانوية (CuNPs) و – كان CuNPs مشابهًا عمومًا للطريقة المستخدمة في SAC. الاختلاف كان: إدخالمن خلال صمام الملف اللولبي لمدة 3 ثوانٍ وتفريغ الحجرة بـ لمدة 12 ثانية. في هذه المرحلة، كانت استجابة الضغط في غرفة الفراغ 10 باسكال. بعد ذلك، تم حقنه لمدة 200 مللي ثانية وجولة أخرى منتم تنفيذ التنظيف لمدة 15 ثانية. تم تكرار دورة الترسيب هذه 100 مرة.
الكيمياء الكهربائيةالقياسات
تم تحويل المحفزات المستخرجة إلى حبر إلكترودي لـالقياسات. تم تفريق 2 ملغ من المحفز بالموجات فوق الصوتية في محلول يحتوي على من الإيزوبروبانول ( شركة ميرير المحدودة،من الماء المنزوع الأيونات، ومن نافيون (شركة ألفا أيسر المحدودة) لتشكيل حبر الإلكترود.اختبار، إسقاطمن حبر الإلكترود على الإلكترود النافذ للغاز (GDE،المنطقة النشطة) ثم تم تسخين GDE عند لمدة 0.5 ساعة لإزالة الإيزوبروبانول المتبقي. تم اختبار النشاط والانتقائية للمحفزات الناتجة في خلية تدفق تتكون من كاثود GDE (YLS-30T، سينورو إنرجي مول)، وغشاء تبادل أنيون (Fumasep FAB-PK-130، مول سينورو للطاقةإلكترود، و الـ شبكةسمك) الأنود. توفير 20 سنتيمتر مكعب قياسي في الدقيقةمنإلى جانب الكاثود وتيار الأنوليت الذي تدفق عبر الأنود بمعدلحل لـتم استخدامه كإلكتروليت كاثودي.تم تقييم أداء المحفزات التي تم الحصول عليها من خلال تطبيق تيارات مختلفة باستخدام مضخم تيار في نظام الثلاثة أقطاب في محطة العمل الكهروكيميائية (CHI660E، تشينهوا) مع مضخم تيار. تم إجراء اختبار منحنيات LSV عند إمكانيات تتراوح من 0 فولت إلى -1.5 فولت مقابل RHE بمعدل مسحالإمكانات مقابلتم تحويل أقطاب المرجع إلى مقياس مرجع RHE باستخدام المعادلة التالية دونتعويض:
تم تحديد كمية منتجات الغاز بواسطة كروماتوغرافيا الغاز (GC، FULI INSTRUMENTS GC9790 Plus) المزودة بكاشف الموصلية الحرارية (TCD) وكاشف التأين اللهبي (FID). تم معايرة جهاز الكروماتوغرافيا الغازية بواسطة خمسة غازات معيارية. و في ) قبل الاستخدام. تم حساب FEs للمنتجات السائلة باستخدام الكمية الإجمالية للمنتجات المجمعة عند الأنودات والكاثودات. تم الكشف عن المنتج السائل بواسطة السائل طيف الرنين المغناطيسي النووي (NMR) (بروكير، AVANCE III 400 ميغاهرتز NanoBAY)، ومن الإلكتروليت المؤين،منشركة ميرير المحدودة. من DMSO (، تم خلط شركة MERYER المحدودة بشكل موحد في أنبوب NMR. FE من الـتم حساب المنتجات من:
حيث F هو ثابت فاراداي هو الناتج الكلي (بالمول)، هو عدد الإلكترونات المنقولة لكل منتج، و هو التكامل الزمني الحالي لمقدار الشحنة المكتسبة.
توصيفات
تم الحصول على أنماط حيود الأشعة السينية للبودرة على جهاز حيود البودرة (Rigaku Smart Lab 3 kW) باستخدام-مصدر الأشعة. تم الحصول على صور SEM للعينات باستخدام مجهر SEM JSM-7800F. تم الحصول على صور TEM باستخدام مجهر Talos 200X G2 للميكروسكوب الناقل عند 200 كيلوفولت. تم إجراء خصائص الميكروسكوب الإلكتروني الناقل ذو المجال المظلم الحلقي العالي الزاوية (HAADF-STEM) على جهاز FEI.تم تجهيز G2 60-300 بمصححات انحراف مزدوج، وتم تشغيله عند 200 كيلو فولت. تم الحصول على طيف امتصاص الأشعة السينية عند حافة النحاس K في خطوط الأشعة BL14W في منشأة الإشعاع المتزامن في شنغهاي (SSRF) (شنغهاي، الصين)، باستخدام مقياس الطيف البلوري Si (111) الذي يعمل في وضع النقل. تم معالجة وتحليل الأطياف بواسطة برامج Athena وArtemis. تم إجراء XPS على جهاز Thermo Scientific ESCALAB 250Xi باستخدام-مصدر الأشعة. تم معايرة طاقة الربط للطيف المجمّع بواسطة طاقة الربط C 1s البالغة 284.8 إلكترون فولت. تم تحديد بيانات مطيافية الكتلة المتصلة بالتحليل الطيفي (ICP-MS) باستخدام نظام Aglient 7850 (Ms). تم الحصول على منحنيات امتصاص النيتروجين عند 77 كلفن على جهاز Micromeritics ASAP 2460.
قياسات طيف رامان في الموقع
تم تسجيل طيف رامان في الموقع على جهاز مطياف رامان بالليزر (LabRAM HR Evolution) باستخدام ليزر بقدرة 785 نانومتر.الشدة). تم إجراء قياسات طيف رامان في الموقع في خلية رامان (شركة تيانجين غاوس يونيون للتكنولوجيا المحدودة) مع نافذة بصرية من الكوارتز،إلكترود مرجعي وإلكترود مضاد من البلاتين. تم تسجيل كل طيف رامان لثلاثة تراكمات مع وقت اكتساب قدره 20 ثانية. تم ملء خلية رامان بمحلول مشبع بـ 0.1 م من ثاني أكسيد الكربون.الإلكتروليت، وتم الحفاظ على معدل تدفق CO عند 2 SCCM خلال قياسات امتصاص CO. التطور الهيكلي للدعائم على، و خلالتم إجراء العملية فيمشبعالمحلول الكهربائي، ومعدل تدفق المحلول الكهربائي تم تحديده على 2 SCCM لإزالة الفقاعات.
قياسات ATR-IRAS في الموقع
تم إجراء ATR-IRAS في الموقع على جهاز طيفي Nicolet iS50 مزود بكاشف HgCdTe (MCT) وملحق VeeMax III (تقنيات PIKE). تم إجراء القياس في خلية كهربائية فردية مزودة بـ Pt وكأقطاب مضادة ومرجعية. تم ملء الخلية بـ-مشبعالكهرباء الساكنة. منشور سيليكون بزاوية ثابتة ( ) المغلفة بمحفزات مدفونة في قاع الخلية عملت كالكاثود العامل. تم استخدام الكرونوأمبروغرافيا لـ اختبار وكان مصحوبًا بجمع الطيف (32 مسحًا،تم طرح جميع الأطياف من الخلفية.
حسابات DFT
تم إجراء جميع حسابات DFT باستخدام حزمة المحاكاة الأولية فيينا (VASP)تُعبر تفاعلات الأيونات والإلكترونات بواسطة طريقة الموجة المعززة بالمشروعات (PAW) وتُعبر تبادل الإلكترونات والتداخل بواسطة تقريب التدرج العام (GGA) مع دالة تبادل-تداخل بيردو-بورك-إرنزرهوف (PBE).. تم توسيع حالات التكافؤ لكوهين-شام في مجموعة أساس الموجات المسطحة مع طاقة قطع تبلغ 450 إلكترون فولت وتم إجراء تكاملات منطقة بريلويين باستخدام ( شبكة مونكهورست-باك أثناء التحسين. تم تثبيت الطبقات الذرية الثلاث السفلية وكان الفضاء الفراغيلتجنب التفاعلات مع الصور الدورية لها في جميع الحسابات. يتقارب الهيكل حتى تصبح جميع القوى على الذرات الحرة أقل منتم تعيين دورات المجال الذاتي المتسق الإلكتروني عندتم إجراء الحساب باستخدام تقريب التدرج العام مع DFT +U ولـلـلـتم استخدام طريقة DFT +U لحساب الهياكل النهائية والطاقة دون اعتبار للإنتروبيا. تمثل الجهد الكيميائي لزوج البروتون-إلكترون (تمت معادلته بـ الجهد الكيميائي لجزيء الهيدروجين عند جهد القطب الهيدروجيني القياسي (SHE) تم تطبيق طريقة الصورة المتسلقة المدفوعة بواسطة الشريط المرن (CI-NEB) لتحديد الهياكل الانتقالية وتم بناء ملف سطح الطاقة الكامنة (PES) وفقًا لذلك.تم تطبيق حسابات التردد للتحقق من الوسائط الممتصة وحالات الانتقال (مع تردد تخيلي واحد فقط). يمكن العثور على عملية حساب DFT التفصيلية والوصف في الأشكال التكميلية 46 و47 والملاحظة التكميلية 3.
توفر البيانات
تم توفير بيانات المصدر مع هذه الورقة وهي متاحة من المؤلفين المقابلين عند الطلب. يتم توفير بيانات المصدر كملف بيانات مصدر. تم توفير بيانات المصدر مع هذه الورقة.
References
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610-7672 (2019).
Bushuyev, O. S. et al. What should we make with and how can we make it? Joule 2, 825-832 (2018).
Wang, Z. et al. Advanced catalyst design and reactor configuration upgrade in electrochemical carbon dioxide conversion. Adv. Mater. 35, e2303052 (2023).
Jouny, M., Luc, W. & Jiao, F. General techno-economic analysis of electrolysis systems. Ind. Eng. Chem. Res. 57, 2165-2177 (2018).
Ge, W. et al. Dynamically formed surfactant assembly at the electrified electrode-electrolyte interface boosting CO2 electroreduction. J. Am. Chem. Soc. 144, 6613-6622 (2022).
Ni, W. et al. Molecular engineering of cation solvation structure for highly selective carbon dioxide electroreduction. Angew. Chem. Int. Ed. 62, e202303233 (2023).
Liu, J. et al. Switching between products and in electrolysis by tuning the composition and structure of rare-earth/ copper catalysts. J. Am. Chem. Soc. 145, 23037-23047 (2023).
Lin, Y. et al. Tunable electroreduction to ethanol and ethylene with controllable interfacial wettability. Nat. Commun. 14, 3575 (2023).
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732-745 (2019).
Zheng, M. et al. Electrocatalytic -to- with ampere-level current on heteroatom-engineered copper via tuning *CO intermediate coverage. J. Am. Chem. Soc. 144, 14936-14944 (2022).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Li, Y. C. et al. Binding site diversity promotes electroreduction to ethanol. J. Am. Chem. Soc. 141, 8584-8591 (2019).
Zhong, M. et al. Accelerated discovery of electrocatalysts using active machine learning. Nature 581, 178-183 (2020).
Sun, Y. et al. Boosting electroreduction to via unconventional hybridization: high-order and O 2 p interaction in for stabilizing . ACS Nano 17, 13974-13984 (2023).
Zhang, L. et al. Oxophilicity-controlled electroreduction to alcohols over Lewis acid metal-doped catalysts. J. Am. Chem. Soc. 145, 21945-21954 (2023).
Lee, C. W. et al. Metal-oxide interfaces for selective electrochemical C-C coupling reactions. ACS Energy Lett 4, 2241-2248 (2019).
Belgamwar, R. et al. Defects tune the strong metal-support interactions in copper supported on defected titanium dioxide catalysts for reduction. J. Am. Chem. Soc. 145, 8634-8646 (2023).
Bruix, A. et al. A new type of strong metal-support interaction and the production of through the transformation of water on Pt/ (111) and Pt/CeOx/TiO2(110) catalysts. J. Am. Chem. Soc. 134, 8968-8974 (2012).
Campbell, C. T. Catalyst-support interactions: electronic perturbations. Nat. Chem. 4, 597-598 (2012).
Yang, J., Li, W., Wang, D. & Li, Y. Electronic metal-support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 32, e2003300 (2020).
Shi, Y. et al. Electronic metal-support interaction modulates singleatom platinum catalysis for hydrogen evolution reaction. Nat. Commun. 12, 3021 (2021).
Nørskov, J. K., Bligaard, T., Rossmeisl, J. & Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37-46 (2009).
Liang, X., Fu, N., Yao, S., Li, Z. & Li, Y. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 144, 18155-18174 (2022).
Liu, D., He, Q., Ding, S. & Song, L. Structural regulation and support coupling effect of single-atom catalysts for heterogeneous catalysis. Adv Energy Mater 10, 202001482 (2020).
Lee, B.-H. et al. Electronic interaction between transition metal single-atoms and anatase boosts photoreduction with . Energy Environ. Sci. 15, 601-609 (2022).
Huang, J. R. et al. Single-product faradaic efficiency for electrocatalytic of to CO at current density larger than in neutral aqueous solution by a single-atom nanozyme. Angew. Chem. Int. Ed. 61, e202210985 (2022).
Wu, Q. J. et al. Atomically precise copper nanoclusters for highly efficient electroreduction of towards hydrocarbons via breaking the coordination symmetry of Cu site. Angew. Chem. Int. Ed. 62, e202306822 (2023).
Wang, J. et al. Atomically dispersed metal-nitrogen-carbon catalysts with d-orbital electronic configuration-dependent selectivity for electrochemical CO2-to-CO reduction. ACS Catal 13, 2374-2385 (2023).
Zhu, W. et al. Activating *CO by strengthening -backbonding to enhance two-carbon products formation toward electroreduction on sites. Adv. Funct. Mater. 34, 202402537 (2024).
He, C., Lee, C. H., Meng, L., Chen, H. T. & Li, Z. Selective orbital coupling: an adsorption mechanism in single-atom catalysis. J. Am. Chem. Soc. 146, 12395-12400 (2024).
Wang, Q. et al. Atomically dispersed s-block magnesium sites for electroreduction of to CO. Angew. Chem. Int. Ed. 60, 25241-25245 (2021).
Ding, J. et al. Atomic high-spin cobalt(II) center for highly selective electrochemical CO reduction to . Nat. Commun. 14, 6550 (2023).
Wang, J. et al. Direct electrochemical synthesis of acetamide from and on a single-atom alloy catalyst. ACS Appl. Mater. Interfaces 15, 53436-53445 (2023).
Ouyang, Y. et al. Selectivity of electrochemical CO2 reduction toward ethanol and ethylene: the key role of surface-active hydrogen. ACS Catal. 13, 15448-15456 (2023).
Yang, J. et al. The electronic metal-support interaction directing the design of single atomic site catalysts: achieving high efficiency towards hydrogen evolution. Angew. Chem. Int. Ed. 60, 19085-19091 (2021).
Zhou, Y. et al. Dopant-induced electron localization drives reduction to hydrocarbons. Nat. Chem. 10, 974-980 (2018).
Parastaev, A. et al. Boosting hydrogenation via size-dependent metal-support interactions in cobalt/ceria-based catalysts. Nat. Catal. 3, 526-533 (2020).
Li, J. et al. Highly active and stable metal single-atom catalysts achieved by strong electronic metal-support interactions. J. Am. Chem. Soc. 141, 14515-14519 (2019).
Gao, J. et al. Solar reduction of carbon dioxide on copper-tin electrocatalysts with energy conversion efficiency near 20%. Nat. Commun. 13, 5898 (2022).
Jiang, Y. et al. Pushing the performance limit of catalyst in electroreduction: a cluster model study for loading single atoms. ACS Nano 17, 2620-2628 (2023).
Zhao, H. et al. The role of species in single-atom catalyst for hydrogenation. Nat. Catal. 5, 818-831 (2022).
Chen, S. et al. Lewis acid site-promoted single-atomic Cu catalyzes electrochemical methanation. Nano. Lett. 21, 7325-7331 (2021).
Zhang, L. et al. Elucidating the structure-stability relationship of Cu single-atom catalysts using operando surface-enhanced infrared absorption spectroscopy. Nat. Commun. 14, 8311 (2023).
Gunathunge, C. M., Li, J., Li, X., Hong, J. J. & Waegele, M. M. Revealing the predominant surface facets of rough Cu electrodes under electrochemical conditions. ACS Catal. 10, 6908-6923 (2020).
Yang, T. et al. Catalytic structure design by AI generating with spectroscopic descriptors. J. Am. Chem. Soc. 145, 26817-26823 (2023).
Wang, F. et al. Active site dependent reaction mechanism over Ru/ catalyst toward methanation. J. Am. Chem. Soc. 138, 6298-6305 (2016).
Ilie, A. G. et al. Principal component analysis of Raman spectra for nanoparticle characterization. Appl. Surf. Sci. 417, 93-103 (2017).
Zhou, X. et al. Stabilizing ions by solid solutions to promote electroreduction to methane. J. Am. Chem. Soc. 144, 2079-2084 (2022).
Zhang, X. D. et al. Asymmetric low-frequency pulsed strategy enables ultralong reduction stability and controllable product selectivity. J. Am. Chem. Soc. 145, 2195-2206 (2023).
Firet, N. J. & Smith, W. A. Probing the reaction mechanism of electroreduction over Ag films via operando infrared spectroscopy. ACS Catal. 7, 606-612 (2016).
Shao, F. et al. In situ spectroelectrochemical probing of CO redox landscape on copper single-crystal surfaces. Proc. Natl Acad. Sci. USA 119, e2118166119 (2022).
Shan, W. et al. In situ surface-enhanced Raman spectroscopic evidence on the origin of selectivity in electrocatalytic reduction. ACS Nano 14, 11363-11372 (2020).
Yan, X. et al. Synergy of interface and cu nanoparticles dual catalytic regions in electrolysis of CO to acetic acid. Angew. Chem. Int. Ed. 62, e202301507 (2023).
Kim, Y. et al. Time-resolved observation of C-C coupling intermediates on Cu electrodes for selective electrochemical CO2 reduction. Energy Environ. Sci. 13, 4301-4311 (2020).
Delmo, E. P. et al. In situ infrared spectroscopic evidence of enhanced electrochemical CO2 reduction and C-C coupling on oxide-derived copper. J. Am. Chem. Soc. 146, 1935-1945 (2024).
Tao, Z., Pearce, A. J., Mayer, J. M. & Wang, H. Bridge sites of Au surfaces are active for electrocatalytic reduction. J. Am. Chem. Soc. 144, 8641-8648 (2022).
Chernyshova, I. V., Somasundaran, P. & Ponnurangam, S. On the origin of the elusive first intermediate of electroreduction. Proc. Natl Acad. Sci. USA 115, E9261-E9270 (2018).
Liang, Y. et al. Stabilizing copper sites in coordination polymers toward efficient electrochemical C-C coupling. Nat. Commun. 14, 474 (2023).
Wang, Y. et al. Strong hydrogen-bonded interfacial water inhibiting hydrogen evolution kinetics to promote electrochemical CO2 reduction to . ACS Catal. 14, 3457-3465 (2024).
Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81-85 (2021).
Kressea, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15-50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311-1315 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J Chem. Phys. 113, 9901-9904 (2000).
شكر وتقدير
تم دعم هذا العمل من خلال المنح التالية: المؤسسة الوطنية للعلوم الطبيعية في الصين (رقم 52071183، 51871122). يود المؤلفون أن يشكروا مييو لي من مختبر شينغيا.www.shiyanjia.com) لاختبارات XANES و EXAFS.
مساهمات المؤلفين
قام Y.C. و H.L. (جامعة نانكاي) و H.L. (جامعة تيانجين) بالإشراف على هذا المشروع. صمم Y.Z. وأجرى معظم التجارب وحلل البيانات التجريبية. قام F.C. بإجراء قياسات SEM وتحليل النتائج. أجرى Y.Z. و X.Y. و Y.G. الاختبارات الحفزية. قام X.Z. و Z.L. بإجراء قياسات رامان. أجرى Y.Z. و H.L. (جامعة تيانجين) قياسات ATR-IRAS. قام W.W. و F.L. و D.H. و Y.X. و Y.Z. بإجراء الحسابات النظرية. أجرى Y.Z. و Y.C. و F.L. قياسات TEM وحللوا النتائج. قام Y.X. و Y.Z. بإجراء قياسات BET و XAFS. شارك جميع المؤلفين في مناقشة البحث.
ملاحظة الناشر: تظل شركة سبرينجر ناتشر محايدة فيما يتعلق بالمطالبات القضائية في الخرائط المنشورة والانتماءات المؤسسية.
الوصول المفتوح هذه المقالة مرخصة بموجب رخصة المشاع الإبداعي النسبية-غير التجارية-بدون اشتقاقات 4.0 الدولية، التي تسمح بأي استخدام غير تجاري، ومشاركة، وتوزيع، وإعادة إنتاج في أي وسيلة أو صيغة، طالما أنك تعطي الائتمان المناسب للمؤلفين الأصليين والمصدر، وتوفر رابطًا لرخصة المشاع الإبداعي، وتوضح إذا قمت بتعديل المادة المرخصة. ليس لديك إذن بموجب هذه الرخصة لمشاركة المواد المعدلة المشتقة من هذه المقالة أو أجزاء منها. الصور أو المواد الأخرى من طرف ثالث في هذه المقالة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة، ما لم يُشار إلى خلاف ذلك في سطر الائتمان للمادة. إذا لم تكن المادة مشمولة في رخصة المشاع الإبداعي الخاصة بالمقالة وكان استخدامك المقصود غير مسموح به بموجب اللوائح القانونية أو يتجاوز الاستخدام المسموح به، فستحتاج إلى الحصول على إذن مباشرة من صاحب حقوق الطبع والنشر. لعرض نسخة من هذه الرخصة، قم بزيارة http:// creativecommons.org/licenses/by-nc-nd/4.0/. (ج) المؤلف(ون) 2025
¹قسم العلوم والهندسة الإلكترونية، جامعة نانكاي، تيانجين، الصين.مدرسة علوم المواد والهندسة، جامعة خبي للتكنولوجيا، تيانجين، الصين.معهد مواد الطاقة الجديدة، جامعة تيانجين، تيانجين، الصين.كلية الكيمياء وهندسة المواد، جامعة ونتشو، ونتشو، الصين. البريد الإلكتروني:hui_liu@tju.edu.cn; liuhui@nankai.edu.cn; xiaoyao@wzu.edu.cn; chengyahui@nankai.edu.cn
Electronic metal-support interaction modulates Cu electronic structures for electroreduction to desired products
Received: 23 August 2024
Accepted: 18 February 2025
Published online: 25 February 2025
Check for updates
Yong Zhang , Feifei Chen , Xinyi Yang , Yiran Guo , Xinghua Zhang , Hong Dong , Weihua Wang , Feng Lu , Zunming Lu , Hui Liu , Hui Liu ®, Yao Xiao Yahui Cheng ®
In this work, the Cu single-atom catalysts (SACs) supported by metal-oxides , and are used as theoretical models to explore the correlations between electronic structures and performances. For these catalysts, the electronic metal-support interaction (EMSI) induced by charge transfer between Cu sites and supports subtly modulates the Cu electronic structure to form different highest occupied-orbital. The highest occupied orbital of enhances the adsorption strength of CO and weakens bonds through electron back-donation. This reduces the energy barrier for coupling, thereby promoting multicarbon formation on . The highest occupied orbital of accelerates the activation, and lowers the reaction energy for forming . This over activated , in turn, intensifies competing hydrogen evolution reaction (HER), which hinders the high-selectivity production of on . with highest occupied orbital promotes activation and its localized electronic state inhibits C-C coupling. The moderate water activity of facilitates * CO deep hydrogenation without excessively activating HER. Hence, exhibits the highest Faradaic efficiency of at .
Renewable energy-driven electrochemical carbon dioxide reduction reaction ( ) is a promising method for achieving carbon cycle and clean production of chemicals . molecules are deeply reduced on the Cu-based catalyst surface into hydrocarbons and oxygen-containing compounds such as methane ( ), ethylene , ethanol (EtOH), etc., through multiple electron-proton coupled steps of . Although these high value chemicals and high energy density fuels have broad markets, industrial-scale implementation of still has a long way to . On the one hand, in aqueous media, water molecules serve as the proton source for electrochemical reactions, leading to a conflict between and hydrogen evolution reaction (HER) . The proton-electron coupling properties of require effective activation of and smooth proton transfer to avoid excessive activation of , otherwise HER would competitively overwhelm . On the other hand, deep hydrogenation of adsorbed CO (* CO ) and C -C coupling often coexist and compete with each other, resulting in low product selectivity . The adsorption and coverage of key *CO intermediate on the catalyst surface are crucial in controlling the selectivity of products . Hence, the rational design and controllable synthesis of catalysts based on the deep understanding of reaction mechanism and structure-activity relationship is crucial for precise regulation of the competitive pathways for .
Modulating the electronic structure of a catalyst and elucidating its influence over catalytic activity is an effective approach for studying
the structure-activity relationships. To modulate Cu electronic structure, research interest has concentrated on alloying , doping engineering , metal-support interaction (MSI) modulation , and so on. In heterogeneous catalysis, MSI significantly affects the catalytic performance as it modulates the electronic and geometric structures of metal as well as coordination environments. The electronic metalsupport interaction (EMSI) was further proposed by Campbell, which goes beyond MSI and provides a much more detailed explanation of the enhanced properties of supported catalysts than MSI . EMSI is associated with the orbital rehybridization and charge transfer across the metal-support interface, leading to the formation of new chemical bonds and the realignment of molecular energy levels . By precisely controlling the d-band structure of metal through EMSI, the adsorption scaling relation can be disrupted to regulate the adsorption of key intermediates. This is crucial for the precise control of pathway because protonating to form key intermediates (such as *COOH and ) is difficult on weakly adsorption surface, while overstrengthening of the adsorption energy would favor HER according to the adsorption scaling relation . However, limited by the intrinsic bulk effects, accurately identifying the orbital information of metal particles or clusters still poses challenges in providing design guidance or performance enhancement explanations for supported catalysts .
The EMSI based on single-atom catalysts (SACs) provides a bridge for theoretical electronic structure studies and the design of heterogenous catalysts because their electronic structure can be easily characterized through experiments and theoretical calculations . Strong EMSI not only stabilizes the single-atom metals due to the thermodynamically favorable metal-support bond formation, but also leads to the charge redistribution induced via electron transfer, affecting the energy level distribution of the 3 d orbitals of single atoms . For example, Ma et al. reported that the coordination environment of raises the energy level of orbital compared to that of , thereby enhancing COOH adsorption and promoting *CO desorption . Therefore, the energy level of state may be manipulated by coordination structural perturbations of the Cu atom . Although SACs have been widely used for , general relationships between electronic structures and the catalytic behaviors of SAC remain unclear. Hence, a comprehensive atomic-level insights into the structure-activity relationship of Cu SAC is urgently needed for guiding the regulation of * CO deep hydrogenation or coupling.
Herein, the Cu SACs supported by -CusAC , , and are constructed by atomic layer deposition (ALD) technique and the correlations between the characteristics of electronic structures and the performance are rationalized through detailed characterization and density functional theory (DFT) calculations. The switching of supports subtly modulates the electronic structure of Cu sites to form three SACs with completely different highest occupied orbitals ( orbital for orbital for , and orbital for ). The of tends to interact with the anti-bonding orbital of CO , which enhance CO adsorption on and weaken the bonds through electron back-donation. Meanwhile, the Cu electron delocalization increases the CO adjacent-adsorption energies on – . These enhance the C – C coupling on – , resulting in a lowest ratio of Faradaic efficiency (FE) to . The of exhibits strong hybridization with the bonding and anti-bonding orbitals of , which enhances the adsorption of and promotes dissociation by weakening O-H bonds. Therefore, exhibits a highest of 4.14. However, this over activated , in turn, intensifies competing HER, which hinders the high-selectivity production of on – . The orbital effectively promotes activation and its localized Cu electronic state inhibits C -C coupling. This makes exhibit a of 3.16. Meanwhile, the moderate water activity of facilitates deep hydrogenation
without excessively activating HER, resulting in the highest FE of at .
Results
Theoretical analysis of electronic structure
To explore the structure-activity relationship of Cu single-atom electronic structure in selective *CO deep hydrogenation or coupling, theoretical analysis based on DFT + U calculations was conducted. By loading Cu single atoms on , and supports to modulate the electronic structure of Cu single-atom (Supplementary Fig. 1 and Supplementary data 1 ), named as , and . As shown in Supplementary Fig. 2, the partial density of states (PDOS) of orbitals of , and surfaces are calculated, and the respective energy levels of , , and are plotted based on their d-band center (Fig. 1a) . Instead of forming a broad d-band, each 3d state is spatially highly localized with a narrow energy window. On , the is the highest occupied-orbital; on , the is the highest occupied-orbital; while on , the is the highest occupied-orbital. Clearly, changing the support can significantly affect the highest occupied-orbital of the Cu single-atom due to the orbital rehybridization and charge transfer across the metal-support interface (Supplementary Fig. 3) , and each present Cu single-atoms with three completely different highest occupied-orbitals. The highest occupied d-orbitals often play an important role in regulating the binding modes of intermediates because the electrons filled on the orbital closer to the Fermi level ( ) are more active . From the orbital wave functions, it can be observed that the orbital (localize in axis) symmetry and energy-match with the orbital of CO , facilitating the formation of bonds (Fig. 1b). The orbital symmetry and energy-match with the orbital of CO as well as orbital of , thereby aiding in the formation of bonds (Fig. 1b and Supplementary Fig. 4). The orbital symmetry and energy-match with the orbitals of , promoting the formation of bonds.
Figure 1c and Supplementary Figs. 5, 6 show the PDOS of CO and molecular frontier orbitals as well as the PDOS of CO and adsorption on , and surfaces. The PDOS proves that the strong hybridization between CO and Cu atom mainly contributes from the and orbital, and the hybridization between and Cu mainly contributes from the orbital. The hybridization of the orbital with the is primarily contributed by the orbital, while the contribution to the is mainly from the orbital. This aligns with the analysis of the orbital wave functions. On , the orbital of Cu hybridize with orbital of CO, while stronger hybridization occurs between the highest occupied orbital and orbital of CO than that of and . The d-band center of on are closer to the than that on and , making more active electrons in easily back-donate to the of CO as formation of bonds . Moreover, due to antibonding feature of the orbital, the bonding will weaken bonds in *CO, which can facilitate the subsequent reactions of *CO (hydrogenation or coupling) . In contrast, the highest occupied orbital of exhibits strong hybridization with the orbital of CO , while the hybridization between and is lower than that of – (Supplementary Fig. 6a). Due to the bonding and antibonding orbitals of is and , the hybridization between and on are remarkably stronger than that on and (Fig. 1c and Supplementary Fig. 6b, d), leading to an enhanced ability for to adsorb . In addition, the interaction between the orbital and the anti-bonding orbital weakens the bonds, thereby promoting the dissociation of . On , the highest occupied orbital tends to couple with the lowest orbital of (Supplementary Fig. 7), weakening
Fig. 1 | Electronic structure analysis. a The diagrams of , and , respectively. b The binding modes of CO and interacting with Cu site. The of – CuSAC with adsorbed CO and , as well as PDOS of CO and orbitals after adsorbed (The inset is an enlarged view of the dashed line in c). The COHP curves of bonds and bonds in
d and . f Free energy diagram for the dissociation of . The ELF of , and . h Schematic diagram of CO adjacent-adsorption on surface. i The CO adsorption energy of , and . Relevant source data are provided as a Source Data file. bonds and favoring hydrogenation on the low-coordinate O atom to form the crucial *COOH intermediate, thereby promoting the activation of to form * .
The CO adsorption energies of Cu single-atom on these three supports follows the order , consistent with the energy level arrangement of the orbital (Supplementary Fig. 8 and Fig. 1a). The adsorption energies are strongly related to the position of the orbital energy level, showing the order . The crystal orbital Hamilton population (COHP) was further used to analyze the bond strength in and the bond strength in (Fig. 1d, e and Supplementary Fig. 9) . On , the Cu single-atom exhibits the strongest bonding with an integrated COHP (ICOHP) of -3.88 eV , followed by and . Below the , there are fewer filling electrons in the anti-bonding orbitals for compared to that of on , indicating
that is more favorable for bonding with CO . In contrast, exhibits the highest ICOHP of -2.45 eV , suggesting that the orbital is favor for bonds. Moreover, exhibits the lowest dissociation energy of -0.52 eV , succeeded by and (Fig. 1f). This provides sufficient activated protons for *CO deep hydrogenation to form . However, excessive activation of could enhance competitive HER, and the formation energy of on is as low as 0.18 eV , which may significantly suppress activity (Supplementary Fig. 10). On the pure (111) surface far away from Cu single-atom sites, the dissociation energy of is 1.33 eV , which can also provide sufficient protons for CO adsorbed on Cu single-atom sites hydrogenation.
After coordination between Cu and O , a large amount of charge accumulates around the O atom, while Cu carries the positive charge due to the higher electronegativity of O than that of Cu . Bader charge analysis shows that the Bader charge of Cu is in , in , and in (Supplementary
Fig. 11). The electron localization function (ELF) obtained from DFT calculations indicates that attracts more electrons from Cu , leading to electron delocalization around the Cu single atom (Fig. 1g) . Further calculations are performed for the top-adsorption energy and adjacent-adsorption energies of CO on the Cu single-atom, and the corresponding adsorption modes are shown in Fig. 1h and Supplementary Fig. 12. On , there is only a slight decrease in the CO adjacent-adsorption energies ( -0.35 to -1.44 eV ) compared to the top-adsorption energy (Fig. 1i). This indicates that the delocalized electronic state of Cu on – enables more CO molecules to adsorb around the Cu atom for coupling . On , the electronegativity of Cu is higher than that of Ce , enabling Cu to attract electrons from Ce, significantly reducing the electron delocalization around the Cu single-atom (Fig. 1g). As a result, the adjacentadsorption energies of to on the Cu atom significantly decreases compared to top-adsorption energy, making *CO more likely to undergo deep hydrogenation in the form of individual top-adsorption rather than C-C coupling.
Synthesis and characterization
To verify the results of DFT calculations, Cu atoms were deposited on , rutile , and fluorite supports using ALD technique. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images indicate that the morphologies of these three supports consist of irregular nanoparticles (NPs) with particle sizes larger than 50 nm , thus avoiding size effects of supports (Supplementary Figs. 13 and 14. X-ray diffraction (XRD) patterns and highresolution TEM (HRTEM) images confirm that the supports are pure phases of , and (Supplementary Figs. 14 and 15). Benefiting from the ALD technique relying on sequential molecular-level self-limiting surface reactions , the Cu atom can anchor on supports in the form of a single atom (Fig. 2a). The Inductively coupled plasma optical emission spectrometer (ICP-OES) measurements indicate that the Cu loading amounts for , and are , and , respectively (Supplementary Table 1). Aberration-corrected high-resolution high-angle annular dark-field scanning TEM (HAADF-STEM) images show that Cu atoms are atomically dispersed in , and supports (Fig. 2b-d), without the presence of any visible clusters at both low and high magnifications (Supplementary Fig. 16). XRD patterns and SEM images of the samples after ALD further confirm that no observable Cu NPs are deposited on supports (Supplementary Figs. 17 and 18). HAADF-STEM and energy dispersive X-ray spectroscopy (EDS) elementmapping images indicate that Cu is evenly distributed throughout the , and supports (Fig. 2e, f). By controlling the ALD process, Cu NPs loaded heterogenous catalysts were also prepared to serve as an expansion of EMSI catalysts (Supplementary Fig. 19). The catalysts loaded with Cu NPs on and are named CuNPs and -CuNPs, respectively. The Cu loading amounts for -CuNPs and -CuNPs are and , respectively (Supplementary Table 2). The statistical particle size distribution of Cu NPs based on TEM images displays that the average diameter of Cu NPs loaded onto and are and , respectively (Supplementary Figs. 20 and 21). HRTEM images of -CuNPs and -CuNPs both reveal interfaces between Cu NPs and the supports, with the Cu NPs partly and firmly socketed onto the support surface (Supplementary Figs. 20e and 21b).
To acquire the structural information of metal-oxides SAC, synchrotron-based X-ray adsorption spectroscopy and X-ray photoelectron spectroscopy (XPS) were performed. In the X-ray absorption near-edge structure (XANES) spectra, the absorption edges for , and shift to higher energy than that of Cu -foil, indicating that the Cu atoms are in an oxidized state (Supplementary Fig. 22) . The first peaks of , and are similar and close to that of in the first
derivative of XANES spectra (Fig. 3a), indicating that the valence of Cu species in , and are close to . Only one apparent peak at corresponding to the first coordination shell of Cu-O scattering can be detected in the extended X-ray absorption fine structure (EXAFS) spectra of , and (Fig. 3b). This confirms that Cu atoms are atomically dispersed because no or metallic bonds are observed. The EXAFS fitting data for , and are shown in Fig. 3c, Supplementary Fig. 23, and Supplementary Table 3. The average coordination numbers of , , and are 3.37, 3.67, and 3.41, respectively. Combined with the results of theoretical calculations ( has the lowest free energy; Supplementary Fig. 1), these data reveal that the local structure of metal-oxides SAC comprised one isolated Cu atom may coordinate with about three oxygen atoms ( units) in the triangular pyramid structure between the Cu and O atoms (Fig. 3d) . The fitting of XPS spectra indicate that the predominant oxidation state on the surfaces of , and is , with proportions of , and , respectively. (Fig. 3e). Compared to CuO , the XPS peaks of and shift to higher binding energy, while and Al XPS peaks shift to lower binding energy than that of pure and (Supplementary Fig. 24). It means that the electron transfers from Cu to the adjacent and supports, which results from the strong EMSI between Cu and metal-oxide supports. In contrast, the Cu XPS peak shifts to lower binding energy on the surface of , indicating that Cu attracts electrons from supports, possibly due to the higher electronegativity of Cu compared to Ce atom. These results of electron transfer between Cu and the supports are consistent with DFT calculations.
Using CO as a probe molecule, the atomic dispersion of Cu atoms and the stability of Cu single-atoms during the process can be characterized by in-situ Raman spectroscopy (Fig. 3f) . In the 0.1 M CO-saturated solution, Raman peaks attributed to linear top adsorption (atop) of CO (C-O stretching vibration) are observed at 2087 and for and Cu , respectively . Bridged adsorption of CO (i.e., CO adsorbed on two adjacent Cu atoms) is also observed on the Cu surface. In contrast, bridged adsorption of CO is not detected on the Cu SAC surface. Raman spectra collected on the supports show that there is no peak in the range from to , suggesting that there is no CO adsorption on the supports (Supplementary Fig. 25). Therefore, the Raman peaks located at 2110, 2105 , and can be attributed to CO atop on the Cu single-sites of , and . DFT calculations based on the Blyholder model reveal differences in the frequency of stretching vibrations between Cu single sites and Cu cluster sites (Supplementary Fig. 26), providing the basis for spectroscopic measurements to assess the stability of Cu single atoms. The stability of Cu single-atoms correlates well with the affinity between the Cu sites and supports, specifically, strong interactions between Cu and O lead to high stability . To evaluate the stability of Cu single-atoms during the electroreduction process, a constant potential of -1.5 V (all potentials in this work are vs. the reversible hydrogen electrode, RHE) was applied to electrode for 60 min . After electrolysis at -1.5 V for 60 min , no corresponding Raman peaks are observed for CO bridge-adsorption on metal Cu sites (Fig. 3g), indicating that Cu remains anchored in supports in the form of single-atoms during the electrolysis process. The reduce in intensity of CO atop Raman peaks could be attributed to the accumulation of bubbles during long-term electrolysis. Similarly, the stability of the supports during electrolysis is maintained due to the high reduction potentials of , and . Specifically, after prolonged electrolysis, does not show Raman peaks caused by oxygen defects of , and the first-order Raman peak characteristic of remains present during the whole process (Fig. 3 h. In-situ
Fig. 2 | Synthesis and structural characterization. a Schematic of the synthesis process for Cu SAC support with metal-oxide. High-resolution HAADF-STEM images of , and . Atomic models: Al , gray; Ce ,
yellow; Ti , bule; and O , red. HAADF-STEM and EDS element-mapping images of e , and .
Fig. 3 | Characterization of Cu single-sites. a The first derivatives of XANES spectra. The corresponding -weighted Fourier transform spectra of EXAFS. c Fitting of -weighted EXAFS data of , and in the region of 1.0-2.5 Å. d The proposed configuration of metal-oxide SAC. Atomic models: Metal, yellow, Cu , bule; and O , red. e The XPS spectra.
f The Raman spectra of CO adsorption on metal-oxide , and Insitu Raman spectra of CO adsorption on for long-term electrolysis at -1.5 V without correction. h In-situ Raman spectra of for long-term electrolysis at -1.5 V without correction. Relevant source data are provided as a Source Data file.
Raman spectra of and also confirm their stability during long-term electrolysis (Supplementary Fig. 27) .
performance
Electrochemical experiments on , and were conducted in a flow cell with constant potential electrolysis. The main gas products observed during are , CO , and , with minor liquid products (Supplementary Fig. 28). Prior to assessing the performance of , and , thorough consideration was given to exclude the potential impacts of surface hydroxide adsorption, charge transfer resistance of the support, and Cu loading amounts on the performance of catalysts through a series of electrochemical, BET, and SEM measurements (Supplementary Figs. 29-31 and Supplementary Note 1). The linear sweep voltammetry (LSV) curves indicate that the current densities of increase the fastest with potential, followed by and (Fig. 4a). Meanwhile, the current densities
of the samples measured with gas at all applied potentials are higher than those recorded with Ar gas, indicating the occurrence of . The smallest difference in current density values between measurements of in and Ar atmospheres indicates that exhibits the highest HER activity and dissociation ability, followed by and . The product distribution from -1.1 V to -1.5 V is displayed in Supplementary Fig. 28. At -1.1 V , the ratio of to for , and is , and 4.14 respectively, and it increases as the potential becomes more negative (Fig. 4b). As predicted by the electronic structure, exhibits the highest selectivity of among these three catalysts, while and show selectivity towards . An analysis of the kinetic isotopic effect (KIE), defined as the ratio of generation rate in and -based electrolytes, was performed to assess the impact of electronic structures on the proton transfer process for (Fig. 4c and Supplementary Fig. 32). The KIE values of are higher than
Fig. | performance. a The LSV curves of , and with and Ar as supply gases. b The of , and at the potentials form -1.1 V to -1.5 V . c KIE of H/D for -to- conversion at different potentials. d The corresponding FEs at high current densities. e FEs of -CuNPs for at different current densities. Long-term
stability of and -CuNPs at a constant current density of No correction was applied to calculate the applied potential. The error bars represent standard deviations from at least three independent measurements. Relevant source data are provided as a Source Data file. and at all potentials and close to 2, indicating that its rate-determining step involves dissociation . has the lowest KIE value, indicating that the orbital significantly accelerates the activation process of , while shows moderate dissociation rates of . Considering the moderate CO adsorption energy and the localized electronic state of , demonstrates the highest of , especially at high current density of (Fig. 4d). and have relatively low values of and , respectively. – exhibits excellent performance in terms of the FE and partial current density compared to previously reported excellent catalysts (Supplementary Fig. 33 and Supplementary Table 4) .
Further testing was conducted on the performance of -CuNPs and -CuNPs to validate the structure-activity relationships based on EMSI for guiding the design of supported catalysts. Due to the promotion of coupling by the Cu electronic structure in , it exhibits a higher . When extending Cu singleatoms to Cu nanoparticles, more sites for CO or CHO adsorption and coupling on the Cu surface can lead to the increased selectivity of product (Supplementary Fig. 34). The FEs of various products, including , acetate , and EtOH , over the – CuNPs within a current density range of 200 to , are depicted in Fig. 4e. Specifically, under the condition of constant current electrolysis, the enhanced selectivity of -CuNPs is observed, achieving the highest of at . In contrast, the selectivity of
Fig. 5 | DFT calculations and in-situ ATR-IRAS. a Free energy diagram for to form . b Free energy diagram for *CO-*CHO coupling. In-situ ATR-IRAS spectra of over and e . In-situ ATR-IRAS of O-H stretching
vibration for , and . i Fraction of free in the electrode-electrolyte interface. c-i No correction was applied to calculate the applied potential. Relevant source data are provided as a Source Data file.
on – CuNPs do not significancy increase as the sites for C – C coupling increased, with the of and of at (Supplementary Fig. 35). The LSV curves also reveal that -CuNPs displays a larger response current density than CuNPs (Supplementary Fig. 36). The long-term durability of and -CuNPs were evaluated by continuous at , with timely replacement of the electrolyte and clean the GDE to avoid salt deposit (Fig. 4f, g). The applied potential of maintains stability for 70 h , with the consistently remaining above -CuNPs also shows stability for 70 h with a stable above . Finally, SEM, XRD, XPS, and TEM measurements were conducted on and catalysts after stability tests, along with ICP-MS measurement on the electrolyte (Supplementary Figs. 37, 38 and Supplementary Table 5). These results collectively confirm the stability of the catalysts during long-term tests (Supplementary Note. 2).
DFT calculations and spectroscopic insights into
Afterward, the DFT calculations were further carried out to reveal the effect of the electronic structures on the selectivity of
products. First, the energy required for converting to * CO was calculated for these three models, followed by the formation energy of through deep hydrogenation of *CO (Fig. 5a and Supplementary Tables 6, 7). Consistent with the conclusions from electronic structure analysis, the highest occupied orbital reduces the reaction energy for activation to form * COOH on , which is significantly lower than that on and surfaces. The *COOH intermediate is reduced to the species by reacting with a proton and releasing a molecule. Subsequently, in the branch of *CO hydrogenation to * CHO or * COH , the * CHO should be the major species formed from *CO hydrogenation on these surfaces because the reaction energy required for *CHO formation is lower than that for *COH (Supplementary Fig. 39). The reaction energy for * CO hydrogenation on is the lowest with 0.02 eV , followed by of 0.61 eV and of 0.66 eV . In the pathway for *CHO to form , the reaction of * CHOH with a proton and release of to form is the rate-limiting step for RR-to. The reaction energies for * CHOH to * CH on and are as high as 1.31 eV and 0.90 eV , respectively. In contrast, the reaction energy for *CH is reduced to 0.46 eV due to the active
protons on . Subsequently, the energy barriers for coupling on these three surfaces were calculated, which is crucial for the formation of products and competes with *CO deep hydrogenation. Specifically, the energy barrier for *CO-*CHO coupling is the lowest with 0.37 eV on , followed by of 0.61 eV and – of 0.80 eV (Fig. 5b and Supplementary Fig. 40). It is noteworthy that on all three surfaces, the reaction energy for *CO dimerization is higher than that for asymmetric coupling of *CO-*CHO (Supplementary Fig. 41), suggesting a coupling mechanism on Cu SAC surfaces where *CO is hydrogenated to *CHO and couples with surrounding CO intermediates. DFT calculations further indicate that when the active sites on the support switch from Cu single-atom to Cu cluster, the consecutive Cu sites strongly inhibit the thermodynamic generation of while maintaining high selectivity towards products (Supplementary Fig. 42).
The in-situ attenuated total reflectance infrared absorption spectroscopy (ATR-IRAS) was further carried out at different potentials to reveal the intermediate absorption manners in different reaction pathways. The infrared (IR) bands at and are assigned to corresponding intermediates based on independently reported studies (Supplementary Table 8). The IR band at can be attributed to deformation of * COOH , a critical intermediate in the to * CO conversion pathway (Fig. 5c, e) . The IR bands at 1242 and can be attributed to stretching vibration of and *C-H deformation in the pathway for *CO deep hydrogenation to produce . On , IR band ( ) attributed to the stretching vibration of * CHO can also be detected . The IR bands at 1340 and can be assigned to the intermediate formed after asymmetric coupling of * CO and * . The CHO part of is at , and the CO part of is at . The IR band at on can be attributed to top-adsorption of CO (Fig. 5d), while no *CO intermediate is detected on (Supplementary Fig. 43), possibly due to low *CO concentration or rapid consumption through hydrogenation . The broad IR band in the range of can be attributed to H bend of adsorbed , with the peak on surface appearing noticeably larger than that on surface, consistent with DFT calculations. To avoid the influence of the broad IR band from and verify whether the adsorbed species on surface contain hydrogen, in-situ ATR-IRAS were repeated using instead of -saturated in . The band disappears and is redshifted to , which is consistent with the in-plane bending vibration (D-O-D) of (Supplementary Fig. 44) . The adsorbed carbonate band is blueshifted in the deuterated system to , which is consistent with recent reported work . This is likely due to the deuteration altering the carbonate-bicarbonate equilibrium and the carbonate adsorption process . The asymmetric stretching vibration of can be detected at . Notability, the IR bands around (*COOH), and obviously shifted to , 1257 (*C-D), 1285 (COOD), and 1317 (OCCDO) in , suggesting their relation with hydrogen . On the surface, the IR bands of *CHO and *CO both undergo a redshift with increasing potential, which is caused by the Stark effect (Fig. 5c, d) . This indicates that *CO and *CHO are in-situ generated, and coupling reactions occur between them . Moreover, the intensity of is significantly higher than that of * , indicating that the is more favorable for coupling reactions. In contrast, on the surface, the intensity of *C-H is significancy higher than that of , suggesting that * CO on the surface is more conducive to hydrogenation, leading to the generation of a large amount of .
The state of molecules near the electrode-electrolyte interfaces of , and were then analyzed. The broad band in 3000-3800 represents the stretching vibration mode of the O-H bond (Fig. 5f-h), and the broad band can be
divided into three peaks near 3610,3450 , and through Gaussian fitting according to the distinct hydrogen bonding environments of water (Supplementary Fig. 45) . They belong to water without hydrogen-bonds (free ), with weak hydrogen-bonds (liquid-like ), and strong hydrogen-bonds (ice-like ), respectively. In general, an increased degree of hydrogen-bonds lowers the energy of the O-H stretch, and thus the dissociation energies of increased in the order: free liquid-like ice-like . On , the initial proportion of free is , much higher than of and of . This suggests that the surface is enriched with free , providing more active protons for *CO deep hydrogenation and the HER reaction. As the potential becomes more negative, the proportion of free on the three electrode surfaces gradually decreases, indicating the consumption of free involved in surface reactions (Fig. 5i). The consumption rate of free on the surface is the fastest ( ) due to the strong hydrolytic ability of . However, an excess of active hydrogen enhances competing HER, increasing the of . In contrast, the low abundance of free and weak hydrolytic ability of – make * CO more prone to coupling rather than deep hydrogenation. , positioned in between and , balances * CO deep hydrogenation and the HER well, resulting in a as high as .
Discussion
In summary, , and are successfully constructed by ALD technique, and based on these catalysts, the correlations between the characteristics of electronic structures and the performance are proposed through detailed characterization and DFT calculations. The EMSI between Cu sites and support ranks the orbital as the highest occupied orbital, which enhances the adsorption strength of CO and weakens C -O bonds through electron back-donation. This reduces the energy barrier for C-C coupling, thereby promoting the formation of products on . The EMSI between Cu sites and support ranks the orbital as the highest occupied orbital, which enhances the adsorption of and promotes dissociation, thus lowering the energy barrier for forming . However, this over activated , in turn, intensifies competing HER, which hinders the high-selectivity production of on . The EMSI between Cu sites and support ranks the orbital as the highest occupied orbital, which promotes activation and protonation. effectively balances the CO adsorption strength with the activation of , resulting in the highest of among the three SACs at . This structure-activity relationship based on EMSI can provide inspiration for the design of supported catalysts. Under this guidance, it is predicted that Cu nanoparticles loaded on exhibit higher selectivity than those on , and a of at is achieved. This work not only provides an efficient catalyst for potential industrial application, but also gives an in-depth understanding of the mechanisms that may inspire the rational design of other catalysts for the controlled .
Methods
Preparation of , and
The pure , fluorite , and rutile were purchased from Shanghai Yaoyi Alloy Material Co., Ltd. To deposit Cu single-atoms on , and supports, the atomic layer deposition (ALD) method was used. Firstly, 0.1 g of supports was evenly dispersed in a crucible, and then the crucible was placed inside the vacuum chamber, where the ALD took place. The vacuum chamber temperature was maintained at during the ALD process. Bis(hexa-fluoroacetylacetonato) copper(II) (Cu(hfac) , Nanjing Ai Mou Yuan Scientific Equipment Co. Ltd.) was used as Cu precursors, and was used as reducing agent. was kept at and the carrier
gas was kept at . The ALD cycle followed a precise sequence: introducing through a solenoid valve for 50 ms and purging the chamber with for 12 s . At this point, the pressure response in the vacuum chamber was 0.5 Pa . Thereafter, was injected for 100 ms and another round of cleaning was executed for 30 s . This deposition cycle was repeated 150 times. After ending the ALD cycles, the vacuum chamber was immediately heated to with and kept at for 3 h to firmly anchor Cu single atoms on supports.
Preparation of – CuNPs and – CuNPs
The preparation of – CuNPs and – CuNPs was generally similar to the method used for SAC. The difference was: introducing through a solenoid valve for 3 s and purging the chamber with for 12 s . At this point, the pressure response in the vacuum chamber was 10 Pa . Thereafter, was injected for 200 ms and another round of cleaning was executed for 15 s . This deposition cycle was repeated 100 times.
Electrochemical measurements
The obtained catalysts were made into electrode ink for measurements. 2 mg of catalyst was ultrasonically dispersed into a solution containing of isopropanol ( , MERYER Co. Ltd.), of deionized water, and of Nafion ( , Alfa Aesar Co. Ltd.) to form electrode ink. For test, drop of electrode ink onto the gas diffusion electrode (GDE, active area) and then GDE were heated at for 0.5 h to remove residual isopropanol. The activity and selectivity of the obtained catalysts were tested in a flow cell comprising the GDE cathode (YLS-30T, Sinoro Energy Mall), anionexchange membrane (Fumasep FAB-PK-130, , Sinoro Energy Mall), electrode , and the mesh thickness) anode. Supply 20 standard cubic centimeter per minutes of to the cathode side and anolyte stream that flowed through the anode at a rate of . A solution of was used as cathode electrolyte. The performance for the obtained catalysts was evaluated by applying different currents with a current amplifier in the threeelectrode system at the electrochemical workstation (CHI660E, Chenhua) with a current amplifier. The LSV curves test was conducted at potentials from 0 V to -1.5 V vs. RHE with the scan rate of . Potentials vs. reference electrode were converted to RHE reference scale using the following equation without compensation:
The gas products were quantified by gas chromatography (GC, FULI INSTRUMENTS GC9790 Plus) equipped with thermal conductivity detector (TCD) and flame ionization detector (FID) detectors. The GC was calibrated by five standard gases ( and in ) before use. The FEs of liquid products were calculated using the total amount of the products collected at anodes and cathodes. The liquid product was detected by liquid nuclear magnetic resonance (NMR) spectroscopy (Bruker, AVANCE III 400 MHz NanoBAY), and of electrolyzed electrolyte, of , MERYER Co. Ltd.) and of DMSO ( , MERYER Co. Ltd.) were mixed uniformly in the NMR tube. FE of the products were computed from:
where F is the Faraday constant is the total product (in mole), is the number of transferred electrons for each product, and is the current time integral the amount of charge obtained.
Characterizations
The powder XRD patterns were obtained on a powder diffractometer (Rigaku Smart Lab 3 kW ) using -ray source. The SEM images of the samples were obtained using a JSM-7800F SEM. The TEM images were obtained with a Talos 200X G2 transmission scanning microscopy at 200 kV . High-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM) characterizations were carried out on a FEI G2 60-300 equipped with double aberration correctors, which was operated at 200 kV . The X-ray absorption spectroscopy at the Cu K-edge was obtained at the BL14W beamlines at the Shanghai Synchrotron Radiation Facility (SSRF) (Shanghai, China), using a Si (111) crystal monochromator operated in transmission mode. The spectra were processed and analyzed by the software codes Athena and Artemis. XPS was performed on a Thermo Scientific ESCALAB 250Xi instrument using -ray source. The binding energy of the collected spectra was calibrated by the C 1s binding energy of 284.8 eV . The inductively coupled plasma mass spectrometry (ICP-MS) datas were determined using an Aglient 7850 (Ms) system. Nitrogen sorption isotherms at 77 K were obtained on Micromeritics ASAP 2460.
In-situ Raman spectroscopy measurements
In-situ Raman spectra was recorded on laser Raman spectrometer (LabRAM HR Evolution) with 785 nm laser ( intensity). The in-situ Raman spectroscopy measurements were performed in the Raman cell (Tianjin Gaoss Union Technology Co. Ltd.) with a quartz optical window, an reference electrode and a Pt counter electrode. Each Raman spectra was recorded for three accumulations with an acquisition time of 20 s . The Raman cell was filled with 0.1 M CO -saturated electrolyte, and the CO flow rate was maintained at 2 sccm during the CO adsorption measurements. The structural evolution of the supports on , and during the process was conducted in -saturated electrolyte, and the flow rate of electrolyte was sat as 2 sccm to remove bubbles.
In-situ ATR-IRAS measurements
In situ ATR-IRAS was performed on a Nicolet iS50 spectrometer equipped with an HgCdTe (MCT) detector and a VeeMax III (PIKE Technologies) accessory. The measurement was conducted in an electrochemical single-cell furnished with Pt and as counter and reference electrodes. The cell was filled with -saturated electrolyte. A fixed-angle Si prism ( ) coated with catalysts embed into the bottom of the cell served as the working electrode. Chronoamperometry was used for test and was accompanied by the spectrum collection ( 32 scans, resolution). All spectra were subtracted with the background.
DFT calculations
All DFT calculations were performed using Vienna ab initio simulation package (VASP) . The ion-electron interactions are represented by the projector augmented wave (PAW) method and the electron exchangecorrelation by the generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional . The Kohn-Sham valence states were expanded in a plane-wave basis set with a cut-off energy of 450 eV and Brillouin-zone integrations were performed using a ( ) Monkhorst-Pack mesh during the optimization. The bottom three atomic layers were fixed and the vacuum space was to avoid interactions with their periodic images in all calculations. The structure converges until all the forces on the free atoms were less than . The electronic self-consistent field cycles were set at . The calculation was performed using generalized gradient approximation with DFT +U and for for for . The DFT +U method was employed to calculate final structures and energies without entropy. The chemical potential of the proton-electron pair ( ) was equated with the
chemical potential of a hydrogen molecule at the standard hydrogen electrode (SHE) potential reference . The climbing-image-nudged-elastic-band (CI-NEB) method was applied to locate transition structures and the profile of the potential-energy surface (PES) was constructed accordingly . Frequency calculations were applied to verify the adsorbed intermediates and the transition states (with only one imaginary frequency). The detailed DFT calculation process and descriptions can be found in Supplementary Figs. 46, 47 and Supplementary Note 3.
Data availability
Source data are provided with this paper and are available from the corresponding authors upon request. Source data are provided as a Source Data file. Source data are provided with this paper.
References
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610-7672 (2019).
Bushuyev, O. S. et al. What should we make with and how can we make it? Joule 2, 825-832 (2018).
Wang, Z. et al. Advanced catalyst design and reactor configuration upgrade in electrochemical carbon dioxide conversion. Adv. Mater. 35, e2303052 (2023).
Jouny, M., Luc, W. & Jiao, F. General techno-economic analysis of electrolysis systems. Ind. Eng. Chem. Res. 57, 2165-2177 (2018).
Ge, W. et al. Dynamically formed surfactant assembly at the electrified electrode-electrolyte interface boosting CO2 electroreduction. J. Am. Chem. Soc. 144, 6613-6622 (2022).
Ni, W. et al. Molecular engineering of cation solvation structure for highly selective carbon dioxide electroreduction. Angew. Chem. Int. Ed. 62, e202303233 (2023).
Liu, J. et al. Switching between products and in electrolysis by tuning the composition and structure of rare-earth/ copper catalysts. J. Am. Chem. Soc. 145, 23037-23047 (2023).
Lin, Y. et al. Tunable electroreduction to ethanol and ethylene with controllable interfacial wettability. Nat. Commun. 14, 3575 (2023).
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732-745 (2019).
Zheng, M. et al. Electrocatalytic -to- with ampere-level current on heteroatom-engineered copper via tuning *CO intermediate coverage. J. Am. Chem. Soc. 144, 14936-14944 (2022).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Li, Y. C. et al. Binding site diversity promotes electroreduction to ethanol. J. Am. Chem. Soc. 141, 8584-8591 (2019).
Zhong, M. et al. Accelerated discovery of electrocatalysts using active machine learning. Nature 581, 178-183 (2020).
Sun, Y. et al. Boosting electroreduction to via unconventional hybridization: high-order and O 2 p interaction in for stabilizing . ACS Nano 17, 13974-13984 (2023).
Zhang, L. et al. Oxophilicity-controlled electroreduction to alcohols over Lewis acid metal-doped catalysts. J. Am. Chem. Soc. 145, 21945-21954 (2023).
Lee, C. W. et al. Metal-oxide interfaces for selective electrochemical C-C coupling reactions. ACS Energy Lett 4, 2241-2248 (2019).
Belgamwar, R. et al. Defects tune the strong metal-support interactions in copper supported on defected titanium dioxide catalysts for reduction. J. Am. Chem. Soc. 145, 8634-8646 (2023).
Bruix, A. et al. A new type of strong metal-support interaction and the production of through the transformation of water on Pt/ (111) and Pt/CeOx/TiO2(110) catalysts. J. Am. Chem. Soc. 134, 8968-8974 (2012).
Campbell, C. T. Catalyst-support interactions: electronic perturbations. Nat. Chem. 4, 597-598 (2012).
Yang, J., Li, W., Wang, D. & Li, Y. Electronic metal-support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 32, e2003300 (2020).
Shi, Y. et al. Electronic metal-support interaction modulates singleatom platinum catalysis for hydrogen evolution reaction. Nat. Commun. 12, 3021 (2021).
Nørskov, J. K., Bligaard, T., Rossmeisl, J. & Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37-46 (2009).
Liang, X., Fu, N., Yao, S., Li, Z. & Li, Y. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 144, 18155-18174 (2022).
Liu, D., He, Q., Ding, S. & Song, L. Structural regulation and support coupling effect of single-atom catalysts for heterogeneous catalysis. Adv Energy Mater 10, 202001482 (2020).
Lee, B.-H. et al. Electronic interaction between transition metal single-atoms and anatase boosts photoreduction with . Energy Environ. Sci. 15, 601-609 (2022).
Huang, J. R. et al. Single-product faradaic efficiency for electrocatalytic of to CO at current density larger than in neutral aqueous solution by a single-atom nanozyme. Angew. Chem. Int. Ed. 61, e202210985 (2022).
Wu, Q. J. et al. Atomically precise copper nanoclusters for highly efficient electroreduction of towards hydrocarbons via breaking the coordination symmetry of Cu site. Angew. Chem. Int. Ed. 62, e202306822 (2023).
Wang, J. et al. Atomically dispersed metal-nitrogen-carbon catalysts with d-orbital electronic configuration-dependent selectivity for electrochemical CO2-to-CO reduction. ACS Catal 13, 2374-2385 (2023).
Zhu, W. et al. Activating *CO by strengthening -backbonding to enhance two-carbon products formation toward electroreduction on sites. Adv. Funct. Mater. 34, 202402537 (2024).
He, C., Lee, C. H., Meng, L., Chen, H. T. & Li, Z. Selective orbital coupling: an adsorption mechanism in single-atom catalysis. J. Am. Chem. Soc. 146, 12395-12400 (2024).
Wang, Q. et al. Atomically dispersed s-block magnesium sites for electroreduction of to CO. Angew. Chem. Int. Ed. 60, 25241-25245 (2021).
Ding, J. et al. Atomic high-spin cobalt(II) center for highly selective electrochemical CO reduction to . Nat. Commun. 14, 6550 (2023).
Wang, J. et al. Direct electrochemical synthesis of acetamide from and on a single-atom alloy catalyst. ACS Appl. Mater. Interfaces 15, 53436-53445 (2023).
Ouyang, Y. et al. Selectivity of electrochemical CO2 reduction toward ethanol and ethylene: the key role of surface-active hydrogen. ACS Catal. 13, 15448-15456 (2023).
Yang, J. et al. The electronic metal-support interaction directing the design of single atomic site catalysts: achieving high efficiency towards hydrogen evolution. Angew. Chem. Int. Ed. 60, 19085-19091 (2021).
Zhou, Y. et al. Dopant-induced electron localization drives reduction to hydrocarbons. Nat. Chem. 10, 974-980 (2018).
Parastaev, A. et al. Boosting hydrogenation via size-dependent metal-support interactions in cobalt/ceria-based catalysts. Nat. Catal. 3, 526-533 (2020).
Li, J. et al. Highly active and stable metal single-atom catalysts achieved by strong electronic metal-support interactions. J. Am. Chem. Soc. 141, 14515-14519 (2019).
Gao, J. et al. Solar reduction of carbon dioxide on copper-tin electrocatalysts with energy conversion efficiency near 20%. Nat. Commun. 13, 5898 (2022).
Jiang, Y. et al. Pushing the performance limit of catalyst in electroreduction: a cluster model study for loading single atoms. ACS Nano 17, 2620-2628 (2023).
Zhao, H. et al. The role of species in single-atom catalyst for hydrogenation. Nat. Catal. 5, 818-831 (2022).
Chen, S. et al. Lewis acid site-promoted single-atomic Cu catalyzes electrochemical methanation. Nano. Lett. 21, 7325-7331 (2021).
Zhang, L. et al. Elucidating the structure-stability relationship of Cu single-atom catalysts using operando surface-enhanced infrared absorption spectroscopy. Nat. Commun. 14, 8311 (2023).
Gunathunge, C. M., Li, J., Li, X., Hong, J. J. & Waegele, M. M. Revealing the predominant surface facets of rough Cu electrodes under electrochemical conditions. ACS Catal. 10, 6908-6923 (2020).
Yang, T. et al. Catalytic structure design by AI generating with spectroscopic descriptors. J. Am. Chem. Soc. 145, 26817-26823 (2023).
Wang, F. et al. Active site dependent reaction mechanism over Ru/ catalyst toward methanation. J. Am. Chem. Soc. 138, 6298-6305 (2016).
Ilie, A. G. et al. Principal component analysis of Raman spectra for nanoparticle characterization. Appl. Surf. Sci. 417, 93-103 (2017).
Zhou, X. et al. Stabilizing ions by solid solutions to promote electroreduction to methane. J. Am. Chem. Soc. 144, 2079-2084 (2022).
Zhang, X. D. et al. Asymmetric low-frequency pulsed strategy enables ultralong reduction stability and controllable product selectivity. J. Am. Chem. Soc. 145, 2195-2206 (2023).
Firet, N. J. & Smith, W. A. Probing the reaction mechanism of electroreduction over Ag films via operando infrared spectroscopy. ACS Catal. 7, 606-612 (2016).
Shao, F. et al. In situ spectroelectrochemical probing of CO redox landscape on copper single-crystal surfaces. Proc. Natl Acad. Sci. USA 119, e2118166119 (2022).
Shan, W. et al. In situ surface-enhanced Raman spectroscopic evidence on the origin of selectivity in electrocatalytic reduction. ACS Nano 14, 11363-11372 (2020).
Yan, X. et al. Synergy of interface and cu nanoparticles dual catalytic regions in electrolysis of CO to acetic acid. Angew. Chem. Int. Ed. 62, e202301507 (2023).
Kim, Y. et al. Time-resolved observation of C-C coupling intermediates on Cu electrodes for selective electrochemical CO2 reduction. Energy Environ. Sci. 13, 4301-4311 (2020).
Delmo, E. P. et al. In situ infrared spectroscopic evidence of enhanced electrochemical CO2 reduction and C-C coupling on oxide-derived copper. J. Am. Chem. Soc. 146, 1935-1945 (2024).
Tao, Z., Pearce, A. J., Mayer, J. M. & Wang, H. Bridge sites of Au surfaces are active for electrocatalytic reduction. J. Am. Chem. Soc. 144, 8641-8648 (2022).
Chernyshova, I. V., Somasundaran, P. & Ponnurangam, S. On the origin of the elusive first intermediate of electroreduction. Proc. Natl Acad. Sci. USA 115, E9261-E9270 (2018).
Liang, Y. et al. Stabilizing copper sites in coordination polymers toward efficient electrochemical C-C coupling. Nat. Commun. 14, 474 (2023).
Wang, Y. et al. Strong hydrogen-bonded interfacial water inhibiting hydrogen evolution kinetics to promote electrochemical CO2 reduction to . ACS Catal. 14, 3457-3465 (2024).
Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81-85 (2021).
Kressea, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15-50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758-1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311-1315 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J Chem. Phys. 113, 9901-9904 (2000).
Acknowledgements
This work was supported by the following grants: National Natural Science Foundation of China (No. 52071183, 51871122). The authors would like to thank Meiyu Li from Shiyanjia Lab (www.shiyanjia.com) for tests of the XANES and EXAFS.
Author contributions
Y.C., H.L., (Nankai University), and H.L. (Tianjin University) supervised this project. Y.Z. designed and performed most of the experiments and analyzed the experimental data. F. C. performed the SEM measurements and analyzed the results. Y. Z., X.Y., and Y. G. conducted the catalytic tests. X.Z. and Z.L. performed the Raman measurements. Y. Z. and H. L. (Tianjin University) performed the ATR-IRAS measurements. W.W., F.L., D.H., Y.X., and Y. Z. carried out the theoretical calculations. Y.Z., Y.C., and F.L. performed the TEM measurements and analyzed the results. Y.X. and Y.Z. performed the BET and XAFS measurements. All authors participated in the discussion of the research.
Correspondence and requests for materials should be addressed to Hui Liu, Hui Liu, Yao Xiao or Yahui Cheng.
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http:// creativecommons.org/licenses/by-nc-nd/4.0/.
(c) The Author(s) 2025
¹Department of Electronic Science and Engineering, Nankai University, Tianjin, China. School of Material Science and Engineering, Hebei University of Technology, Tianjin, China. Institute of New-Energy Materials, Tianjin University, Tianjin, China. College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, China. e-mail: hui_liu@tju.edu.cn; liuhui@nankai.edu.cn; xiaoyao@wzu.edu.cn; chengyahui@nankai.edu.cn