DOI: https://doi.org/10.1038/s41929-024-01153-0
PMID: https://pubmed.ncbi.nlm.nih.gov/38947227
تاريخ النشر: 2024-04-29
تفعيل الروابط C–H الأليفاتية بالضوء لتمكين الديكاربوفنكشنالية الانتقائية للإنانتيومر في الألكينات المحفزة بالنيكل
تم القبول: 25 مارس 2024
نُشر على الإنترنت: 29 أبريل 2024
(أ) التحقق من التحديثات
الملخص
تطوير استراتيجيات جديدة لبناء جزيئات كيرالية معقدة بسرعة من المواد الأولية المتاحة بسهولة هو سعي طويل الأمد في مجتمع الكيمياء. تمثل التفاعلات المزدوجة للالكينات التي تتوسطها الجذور منصة ممتازة نحو هذا الهدف. ومع ذلك، تظل النسخ غير المتماثلة تحديًا كبيرًا، والأهم من ذلك، أن الأمثلة التي تتضمن هيدروكربونات بسيطة كشركاء في التفاعل نادرة. هنا نبلغ عن تفاعل مزدوج غير متماثل للالكينات من ثلاثة مكونات يستفيد من التنشيط المباشر لـ
تفعيل الوظائف
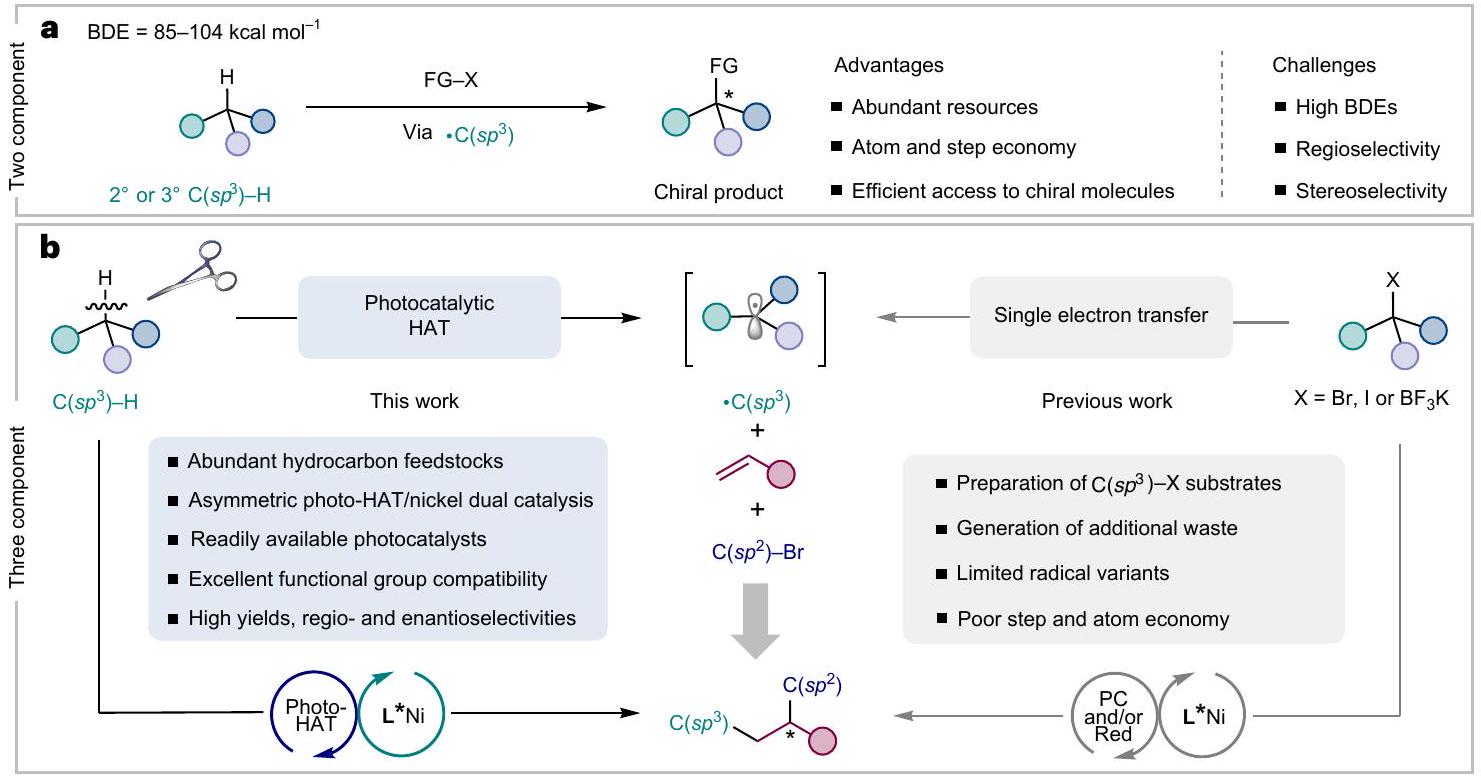
التفعيل من خلال عملية HAT التآزرية والمعادن الانتقالية
تحفيز. ب، الاستراتيجيات السابقة وتصميمنا للتفاعل غير المتماثل ثلاثي المكونات لتفاعل الديكاربوفنكشنالization الجذري للألكينات. BDE، طاقة تفكك الرابطة؛ FG، مجموعة وظيفية؛ PC، محفز ضوئي؛ Red، مختزل.
المذيب و/أو الشركاء المشاركون في التفاعل يؤثرون على كفاءة واختيارية العملية الكلية.
النتائج
تحسين ظروف التفاعل
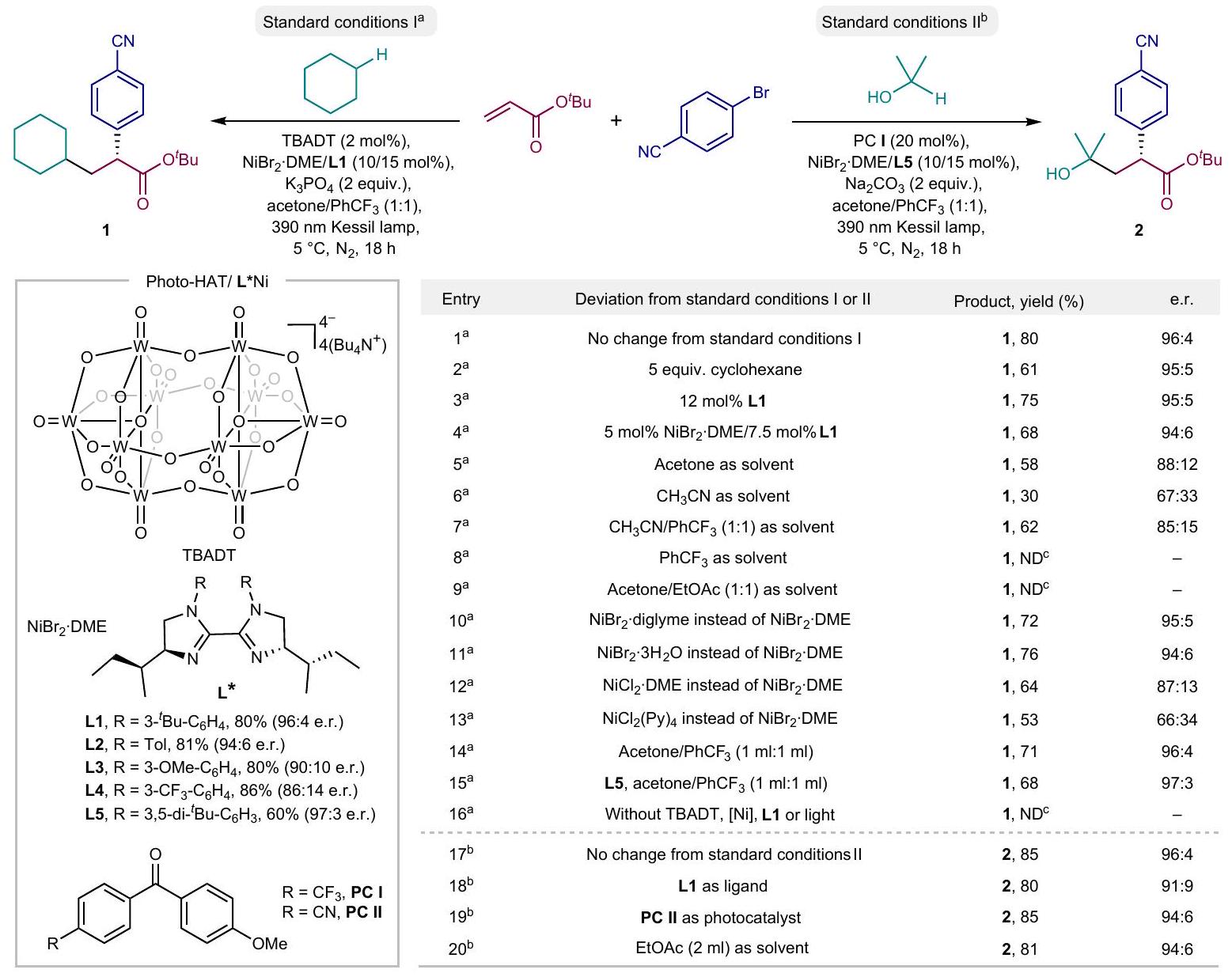
محفزات النيكل مثل
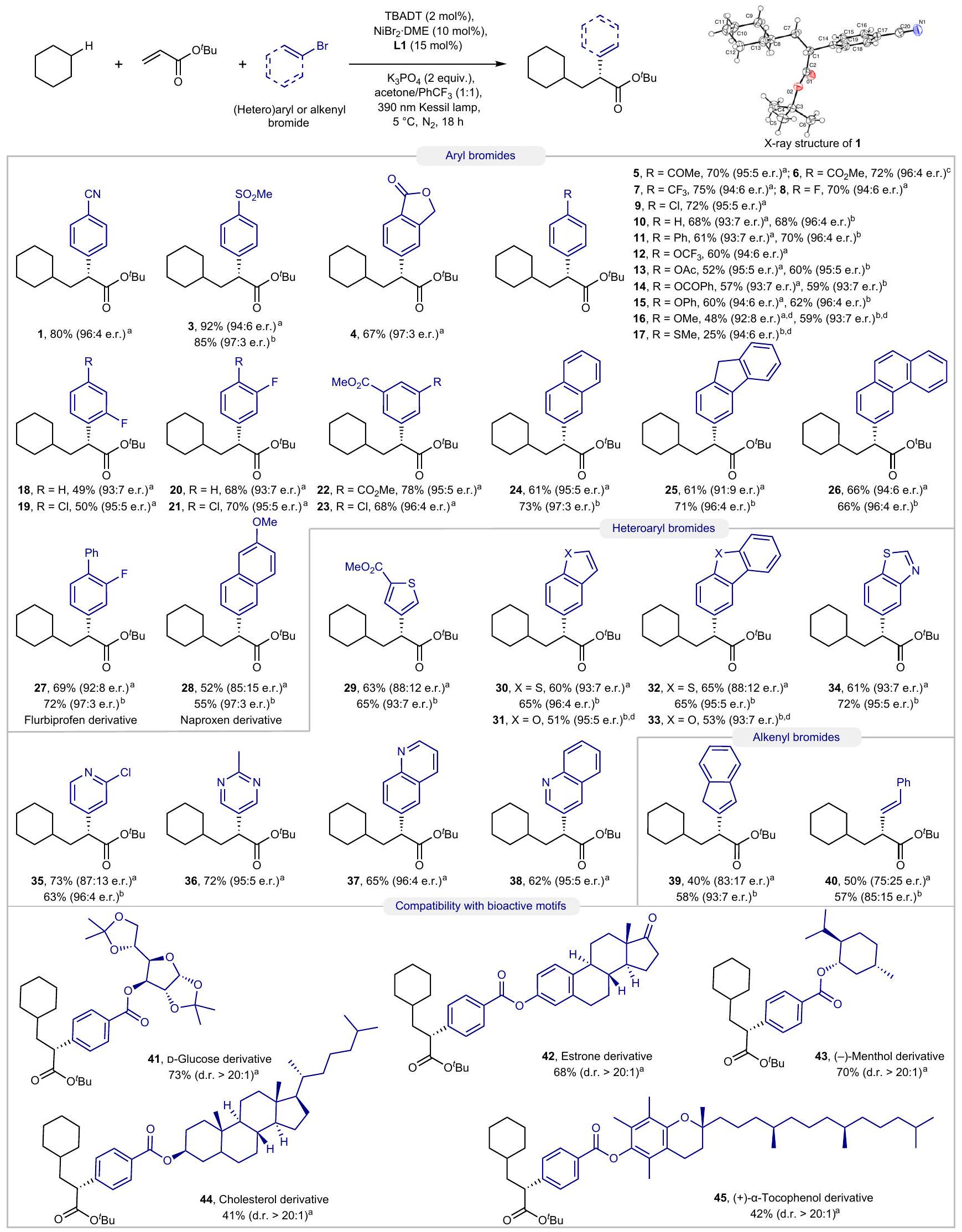
نطاق الركيزة
مقارنةً بالمواد المحايدة إلكترونياً (10) وتلك الغنية بالإلكترونات. لم تكن هاليدات الأريل التي تحمل مجموعات مانحة قوية للإلكترونات، مثل 4-بروموانيسول و4-بروموثيوانيسول، قابلة للتفاعل تحت ظروف التفاعل القياسية. من خلال زيادة شدة الضوء في التفاعل، يمكن تحقيق تكوين المنتجات الناتجة عن الديكاربوفنكشنالization بنجاح في هذه الحالات.
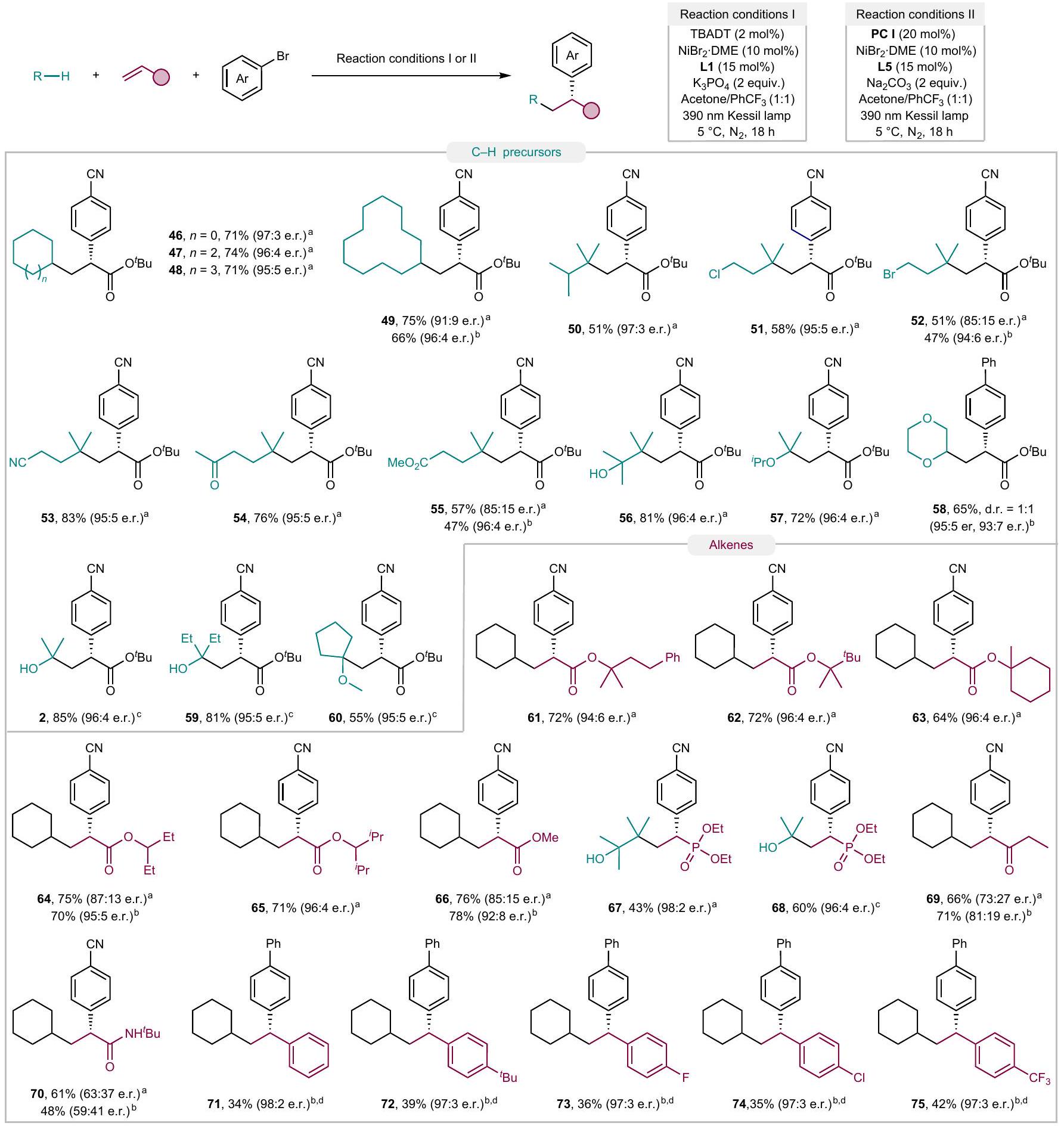
(
دراسات الفولتموجرام لـ TBADT و
المستقبلات، على الرغم من أن المنتجات (69 و70) تم الحصول عليها بانتقائية متوسطة. قدمت الأكريلاميدات الثلاثية كمية ضئيلة من المنتجات المرغوبة من التفاعل الثنائي الكربوني (الجدول التكميلي 9). كانت الفينيل أرينات، وهي فئة من الأوليفينات المنشطة، غير متوافقة في التقارير السابقة.
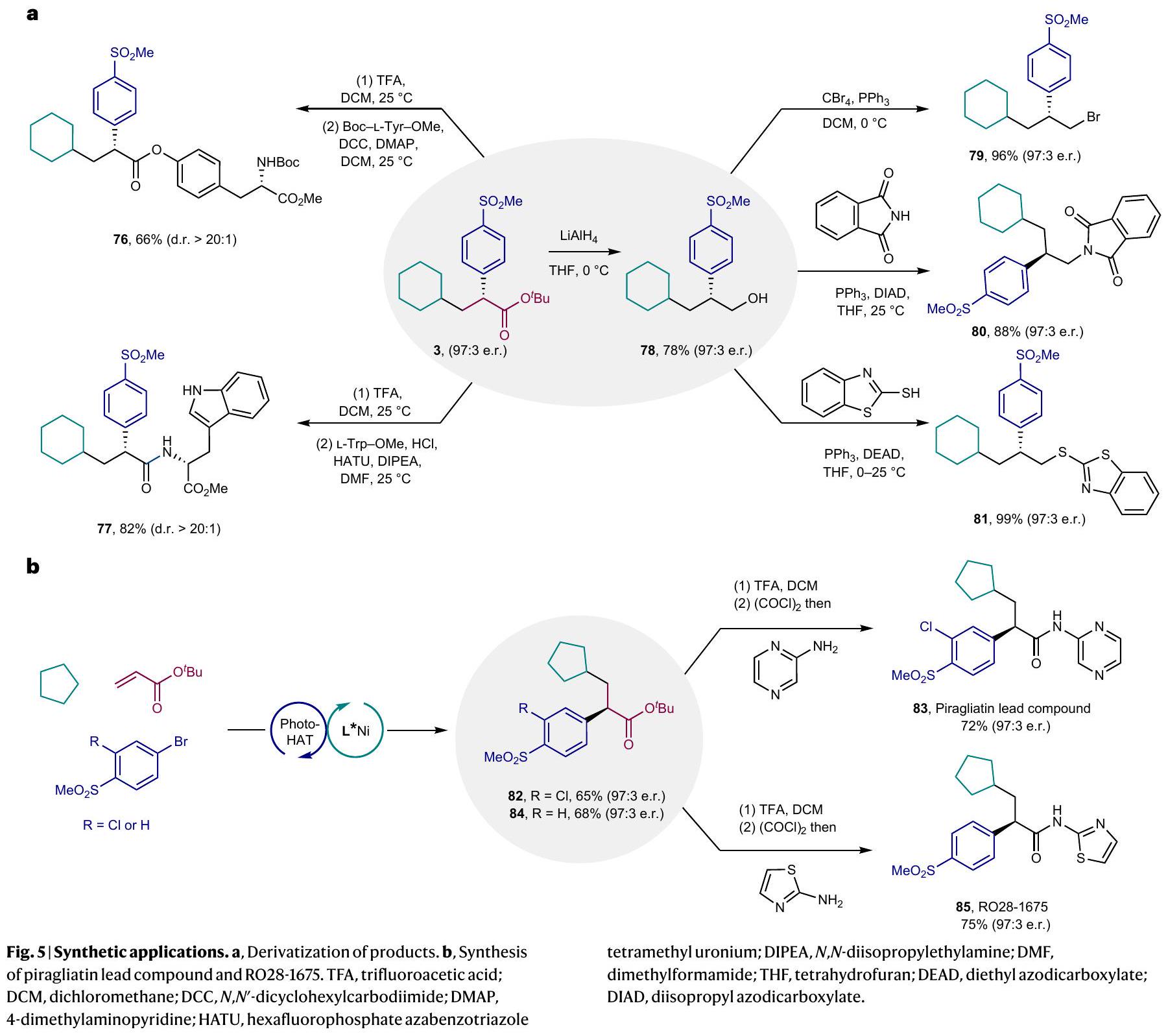
التطبيقات الاصطناعية
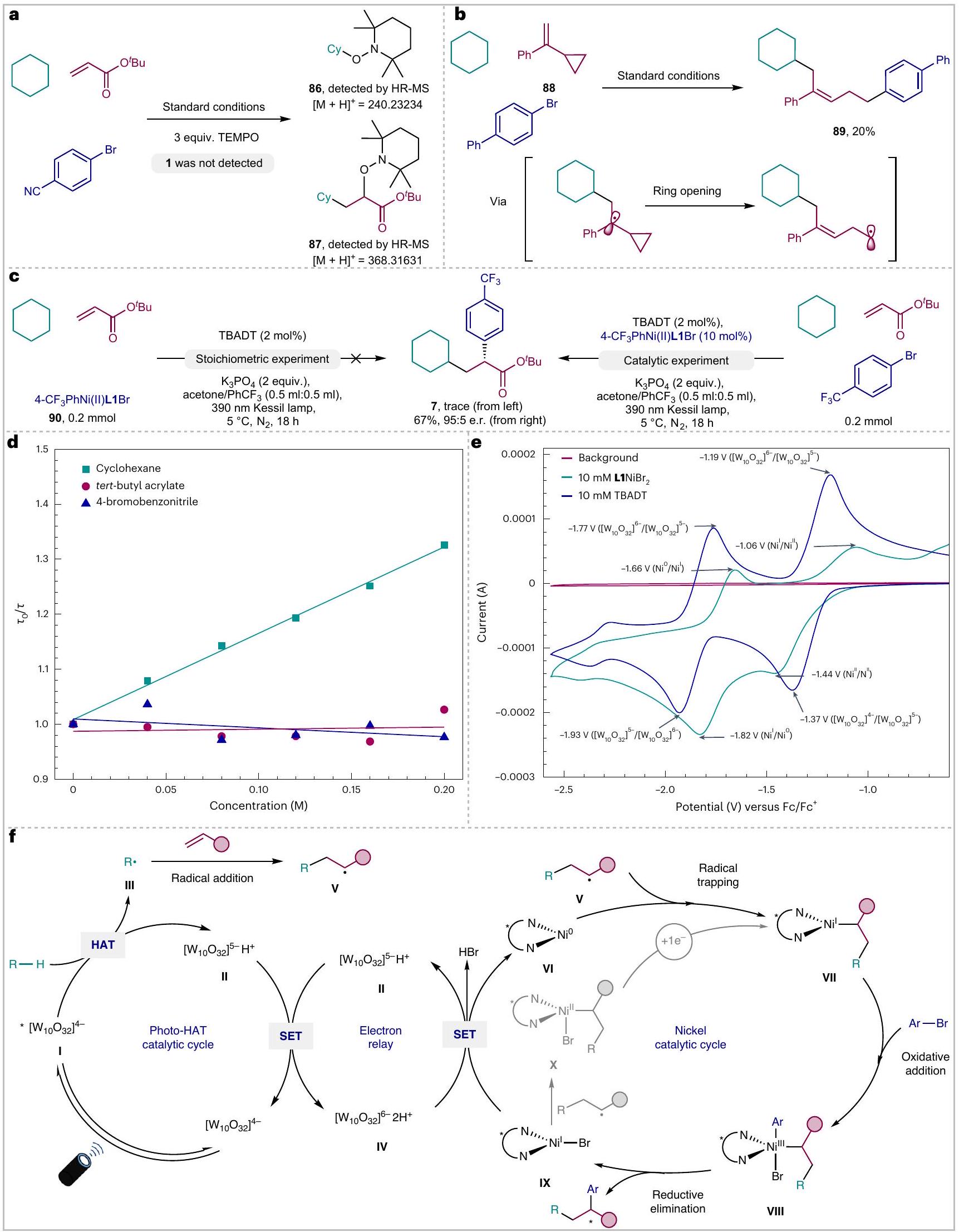
الدراسات الميكانيكية
المتوسط في الدورة الحفازة الرئيسية. بالإضافة إلى ذلك، تم إجراء تجارب تفريغ الليزر لدراسة إخماد الحالة المثارة لـ TBADT (TBADT*) في وجود تركيزات متزايدة من السيكلوهكسان، أكريلات التيرت-بيوتيل و4-بروموبنزونيتريل، على التوالي. لوحظ تدهور واضح لـ TBADT* في وجود السيكلوهكسان متبعًا سلوك ستيرن-فولمر الخطي (ثابت معدل ثنائي الجزيئات
الاستنتاجات
طرق
الطريقة أ
الطريقة ب
توفر البيانات
References
- Labinger, J. A. & Bercaw, J. E. Understanding and exploiting C-H bond activation. Nature 417, 507-514 (2002).
- Zhang, C., Li, Z. L., Gu, Q. S. & Liu, X. Y. Catalytic enantioselective
functionalization involving radical intermediates. Nat. Commun. 12, 475 (2021). - Saint-Denis, T. G. et al. Enantioselective
bond activation by chiral transition metal catalysts. Science 359, eaao4798 (2018). - Olden, D. L., Suh, S. E. & Stahl, S. S. Radical C(sp³)-H functionalization and cross-coupling reactions. Nat. Rev. Chem. 6, 405-427 (2022).
- Capaldo, L., Ravelli, D. & Fagnoni, M. Direct photocatalyzed hydrogen atom transfer (HAT) for aliphatic C-H bonds elaboration. Chem. Rev. 122, 1875-1924 (2022).
- Cao, H., Tang, X., Tang, H., Yuan, Y. & Wu, J. Photoinduced intermolecular hydrogen atom transfer reactions in organic synthesis. Chem. Catal. 1, 523-598 (2021).
- Capaldo, L. & Ravelli, D. Hydrogen atom transfer (HAT): a versatile strategy for substrate activation in photocatalyzed organic synthesis. Eur. J. Org. Chem. 2017, 2056-2071 (2017).
- Ravelli, D., Fagnoni, M., Fukuyama, T., Nishikawa, T. & Ryu, I. Site-selective
functionalization by decatungstate anion photocatalysis: synergistic control by polar and steric effects expands the reaction scope. ACS Catal. 8, 701-713 (2017). - Tzirakis, M. D., Lykakis, I. N. & Orfanopoulos, M. Decatungstate as an efficient photocatalyst in organic chemistry. Chem. Soc. Rev. 38, 2609-2621 (2009).
- West, J. G., Huang, D. & Sorensen, E. J. Acceptorless dehydrogenation of small molecules through cooperative base metal catalysis. Nat. Commun. 6, 10093 (2015).
- Cao, H. et al. Photoinduced site-selective alkenylation of alkanes and aldehydes with aryl alkenes. Nat. Commun. 11, 1956 (2020).
- Perry, I. B. et al. Direct arylation of strong aliphatic C-H bonds. Nature 560, 70-75 (2018).
- Shen, Y., Gu, Y. & Martin, R.
arylation and alkylation enabled by the synergy of triplet excited ketones and nickel catalysts. J. Am. Chem. Soc. 140, 12200-12209 (2018). - Sarver, P. J. et al. The merger of decatungstate and copper catalysis to enable aliphatic
-H trifluoromethylation. Nat. Chem. 12, 459-467 (2020). - Murphy, J. J., Bastida, D., Paria, S., Fagnoni, M. & Melchiorre, P. Asymmetric catalytic formation of quaternary carbons by iminium ion trapping of radicals. Nature 532, 218-222 (2016).
- Laudadio, G. et al.
functionalizations of light hydrocarbons using decatungstate photocatalysis in flow. Science 369, 92-96 (2020). - Laudadio, G. et al. Selective
aerobic oxidation enabled by decatungstate photocatalysis in flow. Angew. Chem. Int. Ed. 57, 4078-4082 (2018). - Schultz, D. M. et al. Oxyfunctionalization of the remote
bonds of aliphatic amines by decatungstate photocatalysis. Angew. Chem. Int. Ed. 56, 15274-15278 (2017). - Ryu, I. et al. Efficient
and conversion via decatungstate-photoinduced alkylation of diisopropyl azodicarboxylate. Org. Lett. 15, 2554-2557 (2013). - Wan, T. et al. Decatungstate-mediated
heteroarylation via radical-polar crossover in batch and flow. Angew. Chem. Int. Ed. 60, 17893-17897 (2021). - Halperin, S. D., Fan, H., Chang, S., Martin, R. E. & Britton, R. A convenient photocatalytic fluorination of unactivated
bonds. Angew. Chem. Int. Ed. 53, 4690-4693 (2014). - Xia, J. B., Zhu, C. & Chen, C. Visible light-promoted metal-free C-H activation: diarylketone-catalyzed selective benzylic mono- and difluorination. J. Am. Chem. Soc. 135, 17494-17500 (2013).
- Sarver, P. J., Bissonnette, N. B. & MacMillan, D. W. C. Decatungstate-catalyzed
sulfinylation: rapid access to diverse organosulfur functionality. J. Am. Chem. Soc. 143, 9737-9743 (2021). - Xu, S., Chen, H., Zhou, Z. & Kong, W. Three-component alkene difunctionalization by direct and selective activation of aliphatic C-H bonds. Angew. Chem. Int. Ed. 60, 7405-7411 (2021).
- Campbell, M. W., Yuan, M., Polites, V. C., Gutierrez, O. & Molander, G. A. Photochemical C-H activation enables nickel-catalyzed olefin dicarbofunctionalization. J. Am. Chem. Soc. 143, 3901-3910 (2021).
- Lu, F. D., He, G. F., Lu, L. Q. & Xiao, W. J. Metallaphotoredox catalysis for multicomponent coupling reactions. Green Chem. 23, 5379-5393 (2021).
- Wickham, L. M. & Giri, R. Transition metal (Ni, Cu, Pd)-catalyzed alkene dicarbofunctionalization reactions. Acc. Chem. Res. 54, 3415-3437 (2021).
- Derosa, J., Apolinar, O., Kang, T., Tran, V. T. & Engle, K. M. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci. 11, 4287-4296 (2020).
- Qi, X. & Diao, T. Nickel-catalyzed dicarbofunctionalization of alkenes. ACS Catal. 10, 8542-8556 (2020).
- Badir, S. O. & Molander, G. A. Developments in photoredox/ nickel dual-catalyzed 1,2-difunctionalizations. Chem 6, 1327-1339 (2020).
- Zhu, C., Yue, H., Chu, L. & Rueping, M. Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity. Chem. Sci. 11, 4051-4064 (2020).
- Campbell, M. W., Compton, J. S., Kelly, C. B. & Molander, G. A. Three-component olefin dicarbofunctionalization enabled by nickel/photoredox dual catalysis. J. Am. Chem. Soc. 141, 20069-20078 (2019).
- Garcia-Dominguez, A., Mondal, R. & Nevado, C. Dual photoredox/ nickel-catalyzed three-component carbofunctionalization of alkenes. Angew. Chem. Int. Ed. 58, 12286-12290 (2019).
- Guo, L., Tu, H. Y., Zhu, S. & Chu, L. Selective, intermolecular alkylarylation of alkenes via photoredox/nickel dual catalysis. Org. Lett. 21, 4771-4776 (2019).
- Mega, R. S., Duong, V. K., Noble, A. & Aggarwal, V. K. Decarboxylative conjunctive cross-coupling of vinyl boronic esters using metallaphotoredox catalysis. Angew. Chem. Int. Ed. 59, 4375-4379 (2020).
- Zheng, S. et al. Selective 1,2-aryl-aminoalkylation of alkenes enabled by metallaphotoredox catalysis. Angew. Chem. Int. Ed. 59, 17910-17916 (2020).
- Li, Z. L., Fang, G. C., Gu, Q. S. & Liu, X. Y. Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 49, 32-48 (2020).
- Fu, L., Zhou, S., Wan, X., Chen, P. & Liu, G. Enantioselective trifluoromethylalkynylation of alkenes via copper-catalyzed radical relay. J. Am. Chem. Soc. 140, 10965-10969 (2018).
- Lin, J. S. et al. Cu/chiral phosphoric acid-catalyzed asymmetric three-component radical-initiated 1,2-dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 141, 1074-1083 (2019).
- Wang, F. et al. Enantioselective copper-catalyzed intermolecular cyanotrifluoromethylation of alkenes via radical process. J. Am. Chem. Soc. 138, 15547-15550 (2016).
- Wang, P. Z. et al. Asymmetric three-component olefin dicarbofunctionalization enabled by photoredox and copper dual catalysis. Nat. Commun. 12, 1815 (2021).
- Wu, L., Wang, F., Chen, P. & Liu, G. Enantioselective construction of quaternary all-carbon centers via copper-catalyzed arylation of tertiary carbon-centered radicals. J. Am. Chem. Soc. 141, 1887-1892 (2019).
- Wu, L. et al. Asymmetric Cu-catalyzed intermolecular trifluoromethylarylation of styrenes: enantioselective arylation of benzylic radicals. J. Am. Chem. Soc. 139, 2904-2907 (2017).
- Zhu, S., Zhao, X., Li, H. & Chu, L. Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel. Chem. Soc. Rev. 50, 10836-10856 (2021).
- Chierchia, M., Xu, P., Lovinger, G. J. & Morken, J. P. Enantioselective radical addition/cross-coupling of organozinc reagents, alkyl iodides, and alkenyl boron reagents. Angew. Chem. Int. Ed. 58, 14245-14249 (2019).
- Tu, H. Y. et al. Enantioselective three-component fluoroalkylarylation of unactivated olefins through nickel-catalyzed cross-electrophile coupling. J. Am. Chem. Soc. 142, 9604-9611 (2020).
- Wei, X., Shu, W., Garcia-Dominguez, A., Merino, E. & Nevado, C. Asymmetric Ni-catalyzed radical relayed reductive coupling. J. Am. Chem. Soc. 142, 13515-13522 (2020).
- Wang, F., Pan, S., Zhu, S. & Chu, L. Selective three-component reductive alkylalkenylation of unbiased alkenes via carbonyl-directed nickel catalysis. ACS Catal. 12, 9779-9789 (2022).
- Qian, P. et al. Catalytic enantioselective reductive domino alkyl arylation of acrylates via nickel/photoredox catalysis. Nat. Commun. 12, 6613 (2021).
- Guo, L. et al. General method for enantioselective threecomponent carboarylation of alkenes enabled by visible-light dual photoredox/nickel catalysis. J. Am. Chem. Soc. 142, 2039020399 (2020).
- Li, X. et al. Three-component enantioselective alkenylation of organophosphonates via nickel metallaphotoredox catalysis. Chem 9, 154-169 (2023).
- Du, X. Y., Cheng-Sánchez, I. & Nevado, C. Dual nickel/ photoredox-catalyzed asymmetric carbosulfonylation of alkenes. J. Am. Chem. Soc. 145, 12532-12540 (2023).
- Cheng, X., Lu, H. & Lu, Z. Enantioselective benzylic C-H arylation via photoredox and nickel dual catalysis. Nat. Commun. 10, 3549 (2019).
- Rand, A. W. et al. Dual catalytic platform for enabling
arylation and alkylation of benzamides. ACS Catal. 10, 4671-4676 (2020). - Shu, X., Huan, L., Huang, Q. & Huo, H. Direct enantioselective
acylation for the synthesis of -amino ketones. J. Am. Chem. Soc. 142, 19058-19064 (2020). - Cheng, X. et al. Stereo- and enantioselective benzylic C-H alkenylation via photoredox/nickel dual catalysis. ACS Catal. 11, 11059-11065 (2021).
- Xu, J., Li, Z., Xu, Y., Shu, X. & Huo, H. Stereodivergent synthesis of both
– and -alkenes by photoinduced, Ni-catalyzed enantioselective alkenylation. ACS Catal. 11, 13567-13574 (2021). - Shu, X., Zhong, D., Lin, Y., Qin, X. & Huo, H. Modular access to chiral
-(hetero)aryl amines via Ni/photoredox-catalyzed enantioselective cross-coupling. J. Am. Chem. Soc. 144, 8797-8806 (2022). - Shu, X., Zhong, D., Huang, Q., Huan, L. & Huo, H. Site- and enantioselective cross-coupling of saturated N -heterocycles with carboxylic acids by cooperative Ni/photoredox catalysis. Nat. Commun. 14, 125 (2023).
-
. et al. Enantioselective functionalization of oxacycles via photo-HAT/nickel dual catalysis. J. Am. Chem. Soc. 145, 5231-5241 (2023). - Zhang, W. et al. Enantioselective cyanation of benzylic C-H bonds via copper-catalyzed radical relay. Science 353, 1014-1018 (2016).
- Li, J. et al. Site-specific allylic C-H bond functionalization with a copper-bound N-centred radical. Nature 574, 516-521 (2019).
- Li, Y., Lei, M. & Gong, L. Photocatalytic regio- and stereoselective
functionalization of benzylic and allylic hydrocarbons as well as unactivated alkanes. Nat. Catal. 2, 1016-1026 (2019). - Xu, P., Fan, W., Chen, P. & Liu, G. Enantioselective radical trifluoromethylation of benzylic C-H bonds via cooperative photoredox and copper catalysis. J. Am. Chem. Soc. 144, 13468-13474 (2022).
- Waele, V. D., Poizat, O., Fagnoni, M., Bagno, A. & Ravelli, D. Unraveling the key features of the reactive state of decatungstate anion in hydrogen atom transfer (HAT) photocatalysis. ACS Catal. 6, 7174-7182 (2016).
- Bhutani, P. et al. FDA approved drugs from 2015-June 2020: a perspective. J. Med. Chem. 64, 2339-2381 (2021).
- Lamberth, C. & Dinges, J. (eds) Bioactive Carboxylic Compound Classes: Pharmaceuticals and Agrochemicals (Wiley-VCH, 2016).
- Horsman, G. P. & Zechel, D. L. Phosphonate biochemistry. Chem. Rev. 117, 5704-5783 (2017).
- Ameen, D. & Snape, T. J. Chiral 1,1-diaryl compounds as important pharmacophores. MedChemComm 4, 893-907 (2013).
شكر وتقدير
مساهمات المؤلفين
تمويل
المصالح المتنافسة
معلومات إضافية
(ج) المؤلف(ون) 2024
- ¹قسم الكيمياء، جامعة زيورخ، زيورخ، سويسرا.
معهد الدراسات المتقدمة، جامعة ووهان، ووهان، الصين. قسم الكيمياء، جامعة بنسلفانيا، فيلادلفيا، بنسلفانيا، الولايات المتحدة الأمريكية. البريد الإلكتروني: cristina.nevado@chem.uzh.ch
DOI: https://doi.org/10.1038/s41929-024-01153-0
PMID: https://pubmed.ncbi.nlm.nih.gov/38947227
Publication Date: 2024-04-29
Nickel-catalysed enantioselective alkene dicarbofunctionalization enabled by photochemical aliphatic C-Hbond activation
Accepted: 25 March 2024
Published online: 29 April 2024
(a) Check for updates
Abstract
The development of novel strategies to rapidly construct complex chiral molecules from readily available feedstocks is a long-term pursuit in the chemistry community. Radical-mediated alkene difunctionalizations represent an excellent platform towards this goal. However, asymmetric versions remain highly challenging, and more importantly, examples featuring simple hydrocarbons as reaction partners are elusive. Here we report an asymmetric three-component alkene dicarbofunctionalization capitalizing on the direct activation of
functionalizations
functionalization through a synergistic HAT process and transition metal
catalysis. b, Previous strategies and our design for asymmetric three-component radical dicarbofunctionalization of alkenes. BDE, bond dissociation energy;FG, functional group; PC, photocatalyst; Red, reductant.
solvent and/or the participating reaction partners compromise the efficiency and selectivity of the overall process.
Results
Optimization of the reaction conditions
nickel catalysts such as
Substrate scope
compared to electron-neutral (10) and electron-rich ones. Aryl halides bearing strong electron-donating groups, such as 4-bromoanisole and 4-bromothioanisole, were not amenable to the standard reaction conditions. By increasing the light intensity in the reaction, the formation of the corresponding dicarbofunctionalization adducts could be successfully accomplished in these cases (
(
voltammogram studies of TBADT and
acceptors, although the products ( 69 and 70) were obtained with moderate enantioselectivity. Tertiary acrylamides furnished a trace amount of the desired dicarbofunctionalization adducts (Supplementary Table 9). Vinylarenes, a class of activated olefins that was not compatible in previous reports
Synthetic applications
Mechanistic studies
intermediate in the main catalytic cycle. In addition, laser flash photolysis experiments were performed to study the quenching of the excited state of TBADT (TBADT*) in the presence of increasing concentrations of cyclohexane, tert-butyl acrylate and 4-bromobenzonitrile, respectively. A clear decay of TBADT* was observed in the presence of cyclohexane following a linear Stern-Volmer behaviour (bimolecular rate constant
Conclusions
Methods
Method A
Method B
Data availability
References
- Labinger, J. A. & Bercaw, J. E. Understanding and exploiting C-H bond activation. Nature 417, 507-514 (2002).
- Zhang, C., Li, Z. L., Gu, Q. S. & Liu, X. Y. Catalytic enantioselective
functionalization involving radical intermediates. Nat. Commun. 12, 475 (2021). - Saint-Denis, T. G. et al. Enantioselective
bond activation by chiral transition metal catalysts. Science 359, eaao4798 (2018). - Olden, D. L., Suh, S. E. & Stahl, S. S. Radical C(sp³)-H functionalization and cross-coupling reactions. Nat. Rev. Chem. 6, 405-427 (2022).
- Capaldo, L., Ravelli, D. & Fagnoni, M. Direct photocatalyzed hydrogen atom transfer (HAT) for aliphatic C-H bonds elaboration. Chem. Rev. 122, 1875-1924 (2022).
- Cao, H., Tang, X., Tang, H., Yuan, Y. & Wu, J. Photoinduced intermolecular hydrogen atom transfer reactions in organic synthesis. Chem. Catal. 1, 523-598 (2021).
- Capaldo, L. & Ravelli, D. Hydrogen atom transfer (HAT): a versatile strategy for substrate activation in photocatalyzed organic synthesis. Eur. J. Org. Chem. 2017, 2056-2071 (2017).
- Ravelli, D., Fagnoni, M., Fukuyama, T., Nishikawa, T. & Ryu, I. Site-selective
functionalization by decatungstate anion photocatalysis: synergistic control by polar and steric effects expands the reaction scope. ACS Catal. 8, 701-713 (2017). - Tzirakis, M. D., Lykakis, I. N. & Orfanopoulos, M. Decatungstate as an efficient photocatalyst in organic chemistry. Chem. Soc. Rev. 38, 2609-2621 (2009).
- West, J. G., Huang, D. & Sorensen, E. J. Acceptorless dehydrogenation of small molecules through cooperative base metal catalysis. Nat. Commun. 6, 10093 (2015).
- Cao, H. et al. Photoinduced site-selective alkenylation of alkanes and aldehydes with aryl alkenes. Nat. Commun. 11, 1956 (2020).
- Perry, I. B. et al. Direct arylation of strong aliphatic C-H bonds. Nature 560, 70-75 (2018).
- Shen, Y., Gu, Y. & Martin, R.
arylation and alkylation enabled by the synergy of triplet excited ketones and nickel catalysts. J. Am. Chem. Soc. 140, 12200-12209 (2018). - Sarver, P. J. et al. The merger of decatungstate and copper catalysis to enable aliphatic
-H trifluoromethylation. Nat. Chem. 12, 459-467 (2020). - Murphy, J. J., Bastida, D., Paria, S., Fagnoni, M. & Melchiorre, P. Asymmetric catalytic formation of quaternary carbons by iminium ion trapping of radicals. Nature 532, 218-222 (2016).
- Laudadio, G. et al.
functionalizations of light hydrocarbons using decatungstate photocatalysis in flow. Science 369, 92-96 (2020). - Laudadio, G. et al. Selective
aerobic oxidation enabled by decatungstate photocatalysis in flow. Angew. Chem. Int. Ed. 57, 4078-4082 (2018). - Schultz, D. M. et al. Oxyfunctionalization of the remote
bonds of aliphatic amines by decatungstate photocatalysis. Angew. Chem. Int. Ed. 56, 15274-15278 (2017). - Ryu, I. et al. Efficient
and conversion via decatungstate-photoinduced alkylation of diisopropyl azodicarboxylate. Org. Lett. 15, 2554-2557 (2013). - Wan, T. et al. Decatungstate-mediated
heteroarylation via radical-polar crossover in batch and flow. Angew. Chem. Int. Ed. 60, 17893-17897 (2021). - Halperin, S. D., Fan, H., Chang, S., Martin, R. E. & Britton, R. A convenient photocatalytic fluorination of unactivated
bonds. Angew. Chem. Int. Ed. 53, 4690-4693 (2014). - Xia, J. B., Zhu, C. & Chen, C. Visible light-promoted metal-free C-H activation: diarylketone-catalyzed selective benzylic mono- and difluorination. J. Am. Chem. Soc. 135, 17494-17500 (2013).
- Sarver, P. J., Bissonnette, N. B. & MacMillan, D. W. C. Decatungstate-catalyzed
sulfinylation: rapid access to diverse organosulfur functionality. J. Am. Chem. Soc. 143, 9737-9743 (2021). - Xu, S., Chen, H., Zhou, Z. & Kong, W. Three-component alkene difunctionalization by direct and selective activation of aliphatic C-H bonds. Angew. Chem. Int. Ed. 60, 7405-7411 (2021).
- Campbell, M. W., Yuan, M., Polites, V. C., Gutierrez, O. & Molander, G. A. Photochemical C-H activation enables nickel-catalyzed olefin dicarbofunctionalization. J. Am. Chem. Soc. 143, 3901-3910 (2021).
- Lu, F. D., He, G. F., Lu, L. Q. & Xiao, W. J. Metallaphotoredox catalysis for multicomponent coupling reactions. Green Chem. 23, 5379-5393 (2021).
- Wickham, L. M. & Giri, R. Transition metal (Ni, Cu, Pd)-catalyzed alkene dicarbofunctionalization reactions. Acc. Chem. Res. 54, 3415-3437 (2021).
- Derosa, J., Apolinar, O., Kang, T., Tran, V. T. & Engle, K. M. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem. Sci. 11, 4287-4296 (2020).
- Qi, X. & Diao, T. Nickel-catalyzed dicarbofunctionalization of alkenes. ACS Catal. 10, 8542-8556 (2020).
- Badir, S. O. & Molander, G. A. Developments in photoredox/ nickel dual-catalyzed 1,2-difunctionalizations. Chem 6, 1327-1339 (2020).
- Zhu, C., Yue, H., Chu, L. & Rueping, M. Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity. Chem. Sci. 11, 4051-4064 (2020).
- Campbell, M. W., Compton, J. S., Kelly, C. B. & Molander, G. A. Three-component olefin dicarbofunctionalization enabled by nickel/photoredox dual catalysis. J. Am. Chem. Soc. 141, 20069-20078 (2019).
- Garcia-Dominguez, A., Mondal, R. & Nevado, C. Dual photoredox/ nickel-catalyzed three-component carbofunctionalization of alkenes. Angew. Chem. Int. Ed. 58, 12286-12290 (2019).
- Guo, L., Tu, H. Y., Zhu, S. & Chu, L. Selective, intermolecular alkylarylation of alkenes via photoredox/nickel dual catalysis. Org. Lett. 21, 4771-4776 (2019).
- Mega, R. S., Duong, V. K., Noble, A. & Aggarwal, V. K. Decarboxylative conjunctive cross-coupling of vinyl boronic esters using metallaphotoredox catalysis. Angew. Chem. Int. Ed. 59, 4375-4379 (2020).
- Zheng, S. et al. Selective 1,2-aryl-aminoalkylation of alkenes enabled by metallaphotoredox catalysis. Angew. Chem. Int. Ed. 59, 17910-17916 (2020).
- Li, Z. L., Fang, G. C., Gu, Q. S. & Liu, X. Y. Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 49, 32-48 (2020).
- Fu, L., Zhou, S., Wan, X., Chen, P. & Liu, G. Enantioselective trifluoromethylalkynylation of alkenes via copper-catalyzed radical relay. J. Am. Chem. Soc. 140, 10965-10969 (2018).
- Lin, J. S. et al. Cu/chiral phosphoric acid-catalyzed asymmetric three-component radical-initiated 1,2-dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 141, 1074-1083 (2019).
- Wang, F. et al. Enantioselective copper-catalyzed intermolecular cyanotrifluoromethylation of alkenes via radical process. J. Am. Chem. Soc. 138, 15547-15550 (2016).
- Wang, P. Z. et al. Asymmetric three-component olefin dicarbofunctionalization enabled by photoredox and copper dual catalysis. Nat. Commun. 12, 1815 (2021).
- Wu, L., Wang, F., Chen, P. & Liu, G. Enantioselective construction of quaternary all-carbon centers via copper-catalyzed arylation of tertiary carbon-centered radicals. J. Am. Chem. Soc. 141, 1887-1892 (2019).
- Wu, L. et al. Asymmetric Cu-catalyzed intermolecular trifluoromethylarylation of styrenes: enantioselective arylation of benzylic radicals. J. Am. Chem. Soc. 139, 2904-2907 (2017).
- Zhu, S., Zhao, X., Li, H. & Chu, L. Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel. Chem. Soc. Rev. 50, 10836-10856 (2021).
- Chierchia, M., Xu, P., Lovinger, G. J. & Morken, J. P. Enantioselective radical addition/cross-coupling of organozinc reagents, alkyl iodides, and alkenyl boron reagents. Angew. Chem. Int. Ed. 58, 14245-14249 (2019).
- Tu, H. Y. et al. Enantioselective three-component fluoroalkylarylation of unactivated olefins through nickel-catalyzed cross-electrophile coupling. J. Am. Chem. Soc. 142, 9604-9611 (2020).
- Wei, X., Shu, W., Garcia-Dominguez, A., Merino, E. & Nevado, C. Asymmetric Ni-catalyzed radical relayed reductive coupling. J. Am. Chem. Soc. 142, 13515-13522 (2020).
- Wang, F., Pan, S., Zhu, S. & Chu, L. Selective three-component reductive alkylalkenylation of unbiased alkenes via carbonyl-directed nickel catalysis. ACS Catal. 12, 9779-9789 (2022).
- Qian, P. et al. Catalytic enantioselective reductive domino alkyl arylation of acrylates via nickel/photoredox catalysis. Nat. Commun. 12, 6613 (2021).
- Guo, L. et al. General method for enantioselective threecomponent carboarylation of alkenes enabled by visible-light dual photoredox/nickel catalysis. J. Am. Chem. Soc. 142, 2039020399 (2020).
- Li, X. et al. Three-component enantioselective alkenylation of organophosphonates via nickel metallaphotoredox catalysis. Chem 9, 154-169 (2023).
- Du, X. Y., Cheng-Sánchez, I. & Nevado, C. Dual nickel/ photoredox-catalyzed asymmetric carbosulfonylation of alkenes. J. Am. Chem. Soc. 145, 12532-12540 (2023).
- Cheng, X., Lu, H. & Lu, Z. Enantioselective benzylic C-H arylation via photoredox and nickel dual catalysis. Nat. Commun. 10, 3549 (2019).
- Rand, A. W. et al. Dual catalytic platform for enabling
arylation and alkylation of benzamides. ACS Catal. 10, 4671-4676 (2020). - Shu, X., Huan, L., Huang, Q. & Huo, H. Direct enantioselective
acylation for the synthesis of -amino ketones. J. Am. Chem. Soc. 142, 19058-19064 (2020). - Cheng, X. et al. Stereo- and enantioselective benzylic C-H alkenylation via photoredox/nickel dual catalysis. ACS Catal. 11, 11059-11065 (2021).
- Xu, J., Li, Z., Xu, Y., Shu, X. & Huo, H. Stereodivergent synthesis of both
– and -alkenes by photoinduced, Ni-catalyzed enantioselective alkenylation. ACS Catal. 11, 13567-13574 (2021). - Shu, X., Zhong, D., Lin, Y., Qin, X. & Huo, H. Modular access to chiral
-(hetero)aryl amines via Ni/photoredox-catalyzed enantioselective cross-coupling. J. Am. Chem. Soc. 144, 8797-8806 (2022). - Shu, X., Zhong, D., Huang, Q., Huan, L. & Huo, H. Site- and enantioselective cross-coupling of saturated N -heterocycles with carboxylic acids by cooperative Ni/photoredox catalysis. Nat. Commun. 14, 125 (2023).
-
. et al. Enantioselective functionalization of oxacycles via photo-HAT/nickel dual catalysis. J. Am. Chem. Soc. 145, 5231-5241 (2023). - Zhang, W. et al. Enantioselective cyanation of benzylic C-H bonds via copper-catalyzed radical relay. Science 353, 1014-1018 (2016).
- Li, J. et al. Site-specific allylic C-H bond functionalization with a copper-bound N-centred radical. Nature 574, 516-521 (2019).
- Li, Y., Lei, M. & Gong, L. Photocatalytic regio- and stereoselective
functionalization of benzylic and allylic hydrocarbons as well as unactivated alkanes. Nat. Catal. 2, 1016-1026 (2019). - Xu, P., Fan, W., Chen, P. & Liu, G. Enantioselective radical trifluoromethylation of benzylic C-H bonds via cooperative photoredox and copper catalysis. J. Am. Chem. Soc. 144, 13468-13474 (2022).
- Waele, V. D., Poizat, O., Fagnoni, M., Bagno, A. & Ravelli, D. Unraveling the key features of the reactive state of decatungstate anion in hydrogen atom transfer (HAT) photocatalysis. ACS Catal. 6, 7174-7182 (2016).
- Bhutani, P. et al. FDA approved drugs from 2015-June 2020: a perspective. J. Med. Chem. 64, 2339-2381 (2021).
- Lamberth, C. & Dinges, J. (eds) Bioactive Carboxylic Compound Classes: Pharmaceuticals and Agrochemicals (Wiley-VCH, 2016).
- Horsman, G. P. & Zechel, D. L. Phosphonate biochemistry. Chem. Rev. 117, 5704-5783 (2017).
- Ameen, D. & Snape, T. J. Chiral 1,1-diaryl compounds as important pharmacophores. MedChemComm 4, 893-907 (2013).
Acknowledgements
Author contributions
Funding
Competing interests
Additional information
(c) The Author(s) 2024
- ¹Department of Chemistry, University of Zurich, Zurich, Switzerland.
The Institute for Advanced Studies, Wuhan University, Wuhan, China. Department of Chemistry, University of Pennsylvania, Philadelphia, PA, USA. e-mail: cristina.nevado@chem.uzh.ch