تنشيط cGAS الورمي بواسطة TET2 يحفز تنشيط STING البطاني لتنظيم إعادة تشكيل الأوعية الدموية والمناعة المضادة للأورام في سرطان الكبد TET2-mediated tumor cGAS triggers endothelial STING activation to regulate vasculature remodeling and anti-tumor immunity in liver cancer
يُعد تحفيز تطبيع الأوعية الدموية للورم إجراءً حيويًا لتعزيز فعالية العلاج المناعي. يُعتبر مسار cGAS-STING ضروريًا للمناعة المضادة للأورام، لكن دوره في أوعية الورم غير واضح. هنا، باستخدام نماذج سريرية أولية لسرطان الكبد في ذكور الفئران الناقصة لـ Cgas/Sting، نبلغ أن التداخل بين cGAS في الورم وSTING في العائل يُوسط تطبيع الأوعية الدموية واستجابة المناعة المضادة للورم. من الناحية الميكانيكية، يقوم TET2 بتنظيم إشارات IL-2/STAT5A بشكل إبجيني لزيادة تعبير cGAS في الورم وإنتاج cGAMP. بعد ذلك، يُنقل cGAMP عبر قنوات LRRC8C لتنشيط STING في الخلايا البطانية، مما يعزز تجنيد وهجرة اللمفاويات عبر البطانة. تكشف الدراسات الحية في ذكور الفئران أيضًا أن إعطاء فيتامين C، وهو عامل واعد مضاد للسرطان، يحفز نشاط TET2، ويُحدث تطبيعًا في أوعية الورم، ويعزز فعالية علاج مضاد PD-L1 بمفرده أو بالاشتراك مع IL-2. توضح نتائجنا تواصلاً بين خلايا الورم والخلايا البطانية الوعائية في البيئة المناعية للورم، مما يوفر استراتيجيات لتعزيز فعالية العلاج المناعي التكاملي لسرطان الكبد.
سرطان الكبد، وخاصة سرطان الخلايا الكبدية (HCC)، هو واحد من أكثر الأورام الخبيثة شيوعًا وثالث الأسباب الرئيسية للوفاة المرتبطة بالسرطان على مستوى العالم.حاليًا، حققت العلاجات المناعية القائمة على حجب نقاط التفتيش المناعية، وخاصة الأجسام المضادة التي تستهدف مسار موت الخلية المبرمج-1 (PD-1)/ليجاند موت الخلية المبرمج 1 (PD-L1)، نجاحًا ملحوظًا في علاج مختلف الأورام الخبيثة بما في ذلك الحالات المتقدمة. ومع ذلك، العلاج الأحادي بمثبطات PD-1/PD-L1 يُحدث استجابات دائمة في جزء فقط من مرضى السرطان وبعض المستجيبين ينتكسون بعد فترة من الاستجابةالأوعية الدموية غير الطبيعية للأورام ونقص الأكسجة الناتج وبيئة الورم الدقيقة المثبطة للمناعة هي عوامل رئيسية تعيق فعالية العلاج المناعي، خاصة في سرطان الكبد الذي يتميز بتكوّن أوعية دموية كثيف.يمكن للعلاج المضاد لتكوين الأوعية الدموية أن يطبع الأوعية الدموية الشاذة في الورم لتحسين تسلل خلايا الجهاز المناعي الفعالة داخل الورم، وليس فقط ذلك
كما يعكس البيئة الدقيقة المثبطة للمناعة التي يسببها المحفزات الوعائية الجديدة، وخاصة عامل نمو بطانة الأوعية الدموية (VEGF)، مما يوفر مبررًا للجمع بين العلاجات المضادة لتكوين الأوعية الدموية وعرقلة نقاط التفتيش المناعية.مؤخرًا، أظهرت تركيبة العلاج المضاد لتكوين الأوعية الدموية وحجب نقاط التفتيش المناعية فعالية في علاج سرطان الكبد المتقدم، مما يجعلها العلاج الأولي.ومع ذلك، فإن ‘نافذة الوقت’ لتطبيع الأوعية الدموية في العلاج المضاد لتكوين الأوعية الحالي ضيقة وعابرة، مما يحد من تأثيره المعزز على العلاج المناعي. لذلك، هناك حاجة ماسة إلى استراتيجيات فعالة لتحفيز تطبيع الأوعية الدموية في الأورام بشكل مستقر وتعزيز فعالية العلاج المناعي.
كمستشعر للحمض النووي في السيتوسول، يقوم إنزيم سينثاز GMP-AMP الدوري (cGAS) بتحفيز الاستجابات المناعية الفطرية من خلال إنتاج الرسول الثاني GMP-AMP الدوري (cGAMP)، الذي يرتبط وينشط محفز جينات الإنترفيرون (STING).يقوم STING المنشط بتجنيد كيناز ربط TANK 1 (TBK1) ثم يفسفر عامل تنظيم الإنترفيرون 3 (IRF3) يتبعه تحفيز النوع الأول من الإنترفيرون (IFN) والكيموكينات مثل CCL5 و CXCL10يلعب مسار cGAS-STING المنشط دورًا حيويًا في المناعة المضادة للأورام من خلال تحفيز خلايا Tومع ذلك، لا يزال تنظيم وآلية مسار cGAS-STING في مناعة السرطان غير مفهومة بشكل كامل. بالإضافة إلى تنشيط cGAS-STING الداخلي في خلايا المناعة أو خلايا الورم، يسمح مسار cGAS-STING بالتواصل المتبادل بين خلايا الورم والخلايا المحيطة غير الورمية (أو خلايا المضيف) لتنظيم المناعة المضادة للورم.. يتم نقل cGAMP أو DNA الذي تفرزه الأورام إلى الخلايا العارضة للمستضدات (APCs)، مثل الخلايا التغصنية (DC) والبلعميات، ثم يقوم بتنشيط مسار إشارة IFN من النوع الأول المستحث بواسطة STING، مما يؤدي في النهاية إلى تحفيز الاستجابة المناعية المضادة للأورام التي تتوسطها خلايا CD8الخلايا أو خلايا القاتل الطبيعيعلى الرغم من اعتبار خلايا DC النوع الخلوي الرئيسي الذي يستجيب لـ cGAMP المستمد من الورم، فإن العديد من الخلايا الأخرى في البيئة الدقيقة للورم التي تعبر عن STING بشكل كبير، وخاصة خلايا البطانة الوعائية، قد تكتشف أيضًا cGAMP المستمد من الورم. ومع ذلك، لا يزال من غير المعروف إلى حد كبير ما إذا كان وكيفية وساطة مسار cGAS-STING للتفاعل بين خلايا الورم وخلايا البطانة، بالإضافة إلى دور مسار cGAS-STING في الأوعية الدموية للورم.
تم بذل جهود مكثفة لتطوير منشطات cGAS-STING وقد أظهرت عدة منشطات STING وعدًا كبيرًا في علاج السرطان المناعي في النماذج قبل السريرية.لسوء الحظ، فإن منشطات STING الحالية، التي معظمها نظائر صناعية لـ cGAMP، تُعطى بشكل رئيسي داخل الورم بسبب ضعف التوافر البيولوجي، مما يحد من تطبيقها السريري وفعاليتها العلاجية النهائية.لذلك، تُبذل جهود كبيرة لتطوير منشطات cGAS-STING أكثر فعالية وانتقائية لتعزيز العلاج المناعي للسرطان.
هنا، نصف التفاعل المتبادل بين cGAS في خلايا الورم وSTING في الخلايا البطانية، والذي يعزز الهجرة عبر البطانة للخلايا اللمفاوية، وتطبيع الأوعية الدموية، والمناعة المضادة للورم. استنادًا إلى التنظيم اللاجيني لتعبير cGAS في الورم بواسطة إنزيم تين إليفن ترانسلوكيشن-2 (TET2) ميثيلسيتوسين ديوكسجيناز بالتعاون مع إشارة STAT5A، نقوم بتحفيز نشاط TET2 باستخدام فيتامين C (VC) لتحفيز تفعيل مسار cGAS-cGAMP- STING البطاني في الورم، مما يؤدي إلى تطبيع الأوعية الدموية للورم، وبالتالي تعزيز الفعالية العلاجية لحجب نقاط التفتيش المناعية في الجسم الحي.
النتائج
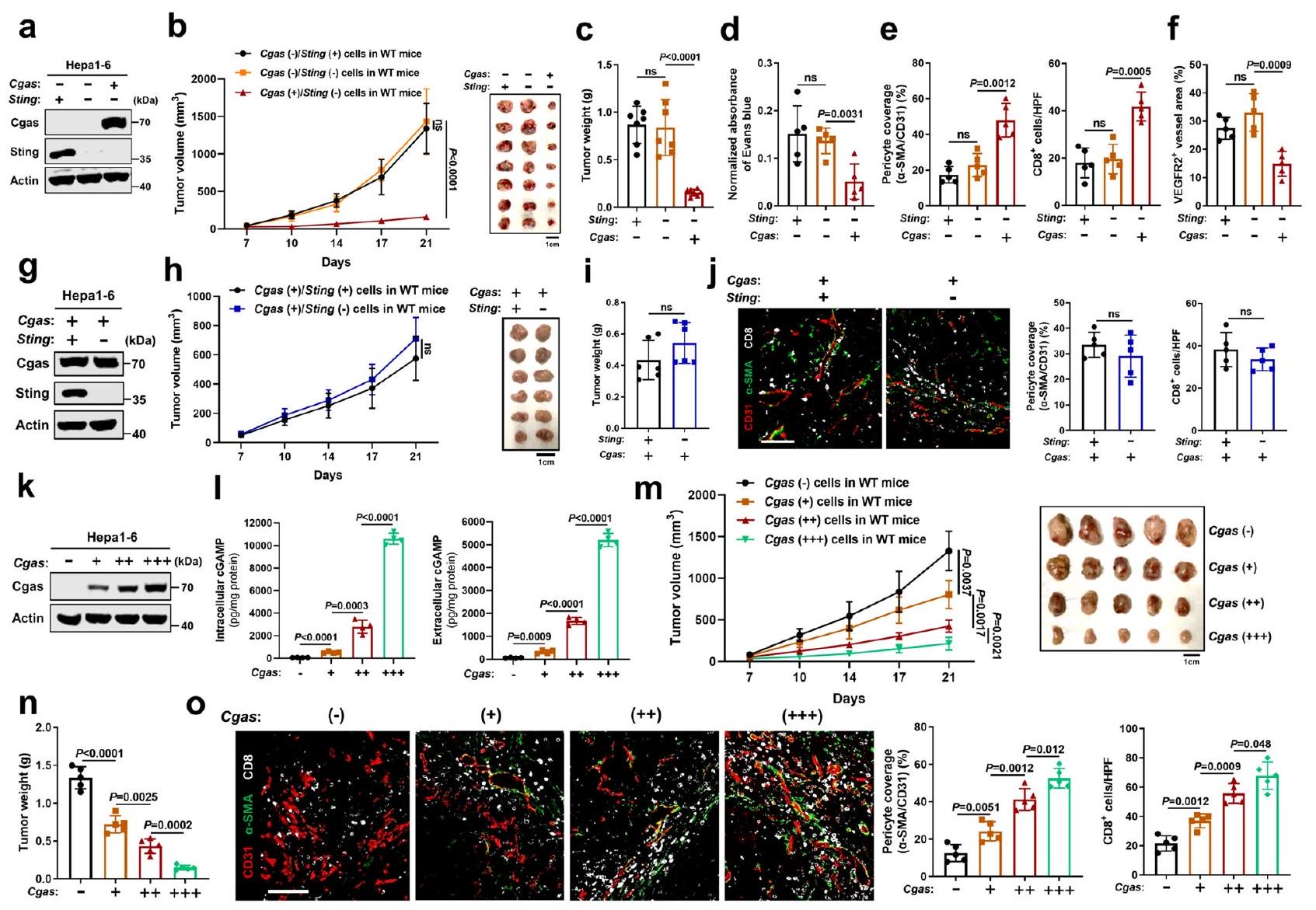
ينظم إنزيم cGAS في الورم تطبيع الأوعية الدموية والاستجابة المناعية المضادة للورم بطريقة داخلية مستقلة عن STING. أولاً، بحثنا دور cGAS وSTING الداخليين في خلايا السرطان في تطبيع الأوعية الدموية للورم والمناعة المضادة للورم. تم التعبير قسرًا عن cGAS في خط خلايا سرطان الكبد الفأري (Hepal-6) الذي يحتوي على مستويات منخفضة من cGAS الذاتية ويعبر عن STING بشكل سليم، بعد استنفاد STING باستخدام تقنية CRISPR-Cas9 أو عدم استنفاده، لإنشاء ثلاث خلايا من نفس الأصل مع cGAS.لدغة، Cgas لدغة، وCgas لدغةالتعبير (الشكل 1أ، الشكل التكميلي 1أ). ثم، قمنا بتحدي الفئران البرية النوع (WT) بهذه الخلايا الثلاث على التوالي. ومن المثير للاهتمام، لم يؤدِّ حذف Sting في خلايا Hepal-6 (Cgas (-)/Sting (-)) إلى تغيير في نمو الورم مقارنة بالخلايا الأصلية (Cgas (-)/Sting (+)) (الشكل 1ب، ج). بالإضافة إلى ذلك، لم يُلاحظ فرق واضح فيتغطية الخلايا المحيطة للأوعية الدمويةالأوعية الدموية للأورام، والتي تُعد واحدة من علامات تطبيع أوعية الأورام، نفاذية الأوعية الدموية، أو تسلل خلايا T داخل الورم (الشكل 1د، هـ، الشكل التكميلي 2أ-ج)، مما يشير إلى أن تعبير STING الجوهري في الورم غير مطلوب لتطبيع الأوعية الدموية والمناعة المضادة للورم في ظل خلفية منخفضة من cGAS في الورم. على النقيض من ذلك، في ظل نقص Sting، أدى فرط التعبير عن Cgas في خلايا Hepal-6 (Cgas (+)/Sting (-)) إلى إبطاء نمو الورم بشكل كبير (الشكل 1ب، ج) وتكوين الأوعية الدموية، وزيادة تغطية الخلايا الداعمة لأوعية الورم، مصحوبًا بتقليل نفاذية الأوعية الدموية وارتفاع تسلل خلايا T داخل الورم (الشكل 1د، هـ، الشكل التكميلي 2أ، ب). علاوة على ذلك، كانت أوعية الورم عمومًا أكثر نضجًا في أورام فرط التعبير عن Cgas، كما يتضح من أن غالبية الأوعية تعبر عن مستويات منخفضة من مستقبل عامل نمو بطانة الأوعية الدموية 2 (VEGFR2)، وهو علامة جزيئية يعبر عنها بشكل عالي في الأوعية النامية وغير الناضجة.(الشكل 1f، الشكل التكميلي 2c). على النقيض من ذلك، لم يظهر نقص Sting الداخلي في خلايا السرطان تأثيرًا كبيرًا على نضج الأوعية الدموية للورم. لاستبعاد دور STING الداخلي للورم في ظل زيادة تعبير cGAS الداخلي للورم، قمنا بحذف Sting في خلايا ذات تعبير مفرط لـ Cgas (الشكل 1g) ووجدنا أن نقص Sting (Cgas (+)/Sting (-)) لم يغير من نمو الورم مقارنة بالخلايا الأصلية (Cgas (+)/Sting (+)) (الشكل 1h، i). علاوة على ذلك، لم يُلاحظ فرق واضح فيتغطية الخلايا المحيطة للأوعية الدمويةالأوعية الدموية للورم أو تسلل خلايا T داخل الورم (الشكل 1j). معًا، تشير هذه النتائج إلى أن cGAS في الورم يتحكم في تطبيع الأوعية الدموية والاستجابة المناعية المضادة للورم بطريقة داخلية مستقلة عن STING.
لتقليد محاكاة أكثر وفاءً لمستويات مسار cGAS/STING الداخلي التي لوحظت في سرطان الكبد البشري (HCC)، تم اختيار سلسلة من النسائل المتميزة ذات مستويات تعبير مختلفة لـ Cgas (الشكل 1k). كما هو موضح في الشكل 11، لاحظنا نطاقًا أوسع من تركيزات cGAMP داخل الخلايا وخارجها، تتراوح من بضع مئات من بيكوغرام لكل ملغ من البروتين إلى عشرات الآلاف.البروتين. في المختبر، كانت معدلات تكاثر الخلايا متشابهة بعد زيادة التعبير عن Cgas (الشكل التكميلي 1ب). من المهم أننا وجدنا أنه مع زيادة تعبير Cgas في خلايا السرطان، لوحظت أعباء أورام أصغر (الشكل 1م، ن)، في حين تم الكشف عن تغطية أكبر للخلايا المحيطة بالأوعية الدموية الورمية وتسلل خلايا T داخل الورم (الشكل 1و)، مما يشير إلى أن cGAS الورمي يوسّط كبح الورم، وتطبيع الأوعية الدموية، والاستجابة المناعية المضادة للورم بطريقة تعتمد على مستوى تعبير cGAS. للتحقق أكثر من هذه الظاهرة في سرطان الكبد البشري، أنشأنا خلايا سرطان كبد بشري (Huh7) تعبر عن cGAS بشكل مستقر، والتي تفتقر إلى التعبير الذاتي عن cGAS وSTING، ولم يؤثر التعبير القسري لـ cGAS على تكاثر خلايا Huh7 في الثقافة (الشكل التكميلي 1ج، د). ومن الجدير بالذكر أن الأورام المشتقة من خلايا cGAS في الفئران العارية أظهرت نموًا متأخرًا (الشكل التكميلي 3أ، ب) وزيادة في تغطية الخلايا المحيطة بالأوعية، مصحوبة بانخفاض نقص الأكسجة داخل الورم وتسلل أكبر لخلايا NK مقارنة بالأورام المشتقة من خلايا Ctrl (الشكل التكميلي 3ج، د). بالإضافة إلى ذلك، جينات خاصة بالفئران بما في ذلك الجينات التابعة لتنشيط STING IFNوتم تنظيم جينات ISGs، وجينات تثبيت الأوعية الدموية، وجزيئات الالتصاق المرتبطة بتفاعل البطانة اللمفاوية بشكل مرتفع في الأورام التي تحتوي على cGAS (الشكل التكميلي 3e)، مما يشير إلى تنشيط STING في المضيف وتطبيع الأوعية الدموية. في المقابل، كان لزيادة تعبير STING في خلايا cGAS (cGAS (+)/STING (+)) تأثير ضئيل على نمو الورم وتطبيع الأوعية الدموية مقارنة بالخلايا cGAS (+)/STING (-) (الأشكال التكميلية 3f-i).
إنزيم cGAS في الورم ومستقبل STING في المضيف يتوسطان تطبيع الأوعية الدموية والاستجابة المناعية المضادة للورم بطريقة تعتمد على التداخل المتبادل
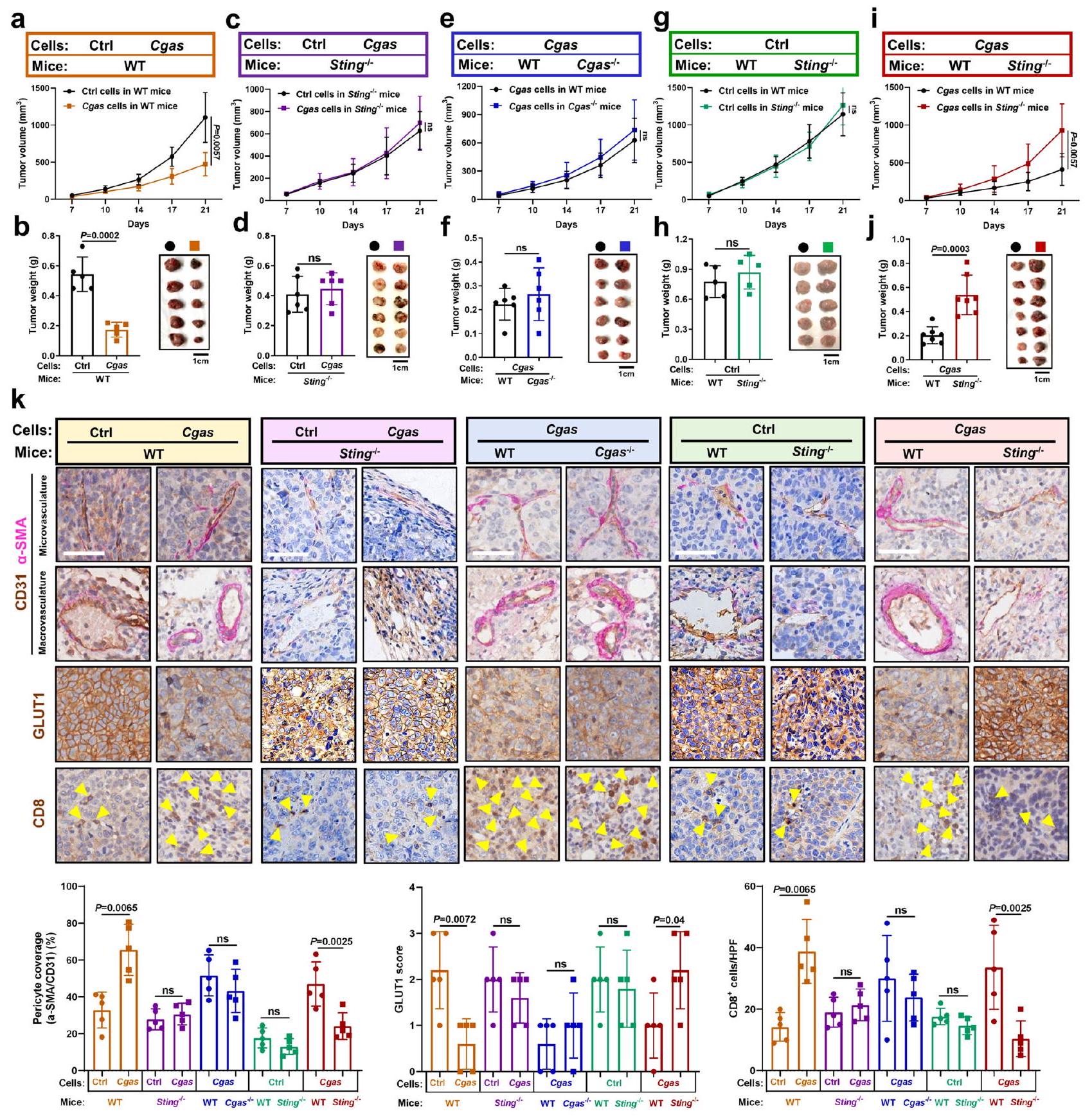
قمنا بتقييم أدوار STING المضيف في تطبيع الأوعية الدموية التي يسببها cGAS في الورم والاستجابة المناعية المضادة للورم. تم زرع خلايا ذات cGAS فعّال وخلايا تحكم في فئران ناقصة STING (Sting-1-) أو في فئران طبيعية (WT) على التوالي. وجدنا أن الفئران الطبيعية (WT)
الشكل 1 | ينظم cGAS الورمي تطبيع الأوعية الدموية والاستجابة المناعية المضادة للأورام بطريقة داخلية مستقلة عن STING. أ مستويات بروتين Cgas وSting في خلايا Hepal-6 مع حذف Sting بعد زيادة تعبير Cgas. منحنيات نمو الورم (ب) وأعباء الورم (ج) في الفئران البرية التي تم حقنها تحت الجلد بالخلايا المشار إليها لمدة 3 أسابيع (الفئران لكل مجموعة). د الامتصاصية المعادلة للون إيفانز الأزرق في الأورام المشار إليها (). التحديد الكمي للخلايا المحيطة بالأوعية () تغطية أوعية الورم (CD31 ) و الخلايا في الأورام المشار إليها (). HPF، مجال قوة عالية.الكمية لمستقبل VEGFR2الأوعية الدموية للأورام) في الأورام المشار إليها (). مستويات بروتين Cgas وSting في خلايا Hepal-6 مع زيادة تعبير Cgas بعد حذف Sting. منحنيات نمو الورم (h) وأعباء الورم (i) في الفئران البرية (WT) التي تم حقنها تحت الجلد بالخلايا المشار إليها لمدة 3 أسابيع (فئران لكل مجموعة).صور مناعية فلورية تمثيلية والكمية لـ الخلايا المحيطة بالأوعية الدموية (pericyte)) تغطية أوعية الورم () و الخلايا في الأورام المشار إليها (). HPF، مجال قوة عالية. أشرطة المقياس،مستويات بروتين Cgas في خلايا Hepa1-6 مع زيادة تعبير Cgas. 1 مستويات cGAMP داخل الخلايا وخارجها من خلايا Hepa1-6 مع زيادة تعبير Cgas (). منحنيات نمو الورم (m) وأعباء الورم (n) في الفئران البرية (WT) التي تم حقنها تحت الجلد بالخلايا المشار إليها لمدة 3 أسابيع (الفئران لكل مجموعة). صور مناعية فلورية تمثيلية وكمية للخلايا المحيطة بالأوعية () تغطية أوعية الورم () و الخلايا في الأورام المشار إليها (). HPF، مجال قوة عالية. أشرطة المقياس،يتم حساب القيم باستخدام تحليل التباين ثنائي الاتجاه (b, h, m)، تحليل التباين أحادي الاتجاه (c-f, l, n, o) واختبار الطالب غير المرتبط ذو الذيليناختبار (). غير مهم إحصائياً. ممثل عنتجارب مستقلة (). يتم توفير بيانات المصدر كمصدر ملف البيانات. التي تم زرعها بخلايا ذات كفاءة Cgas طورت أورامًا أصغر بشكل ملحوظ مقارنة بالفئران البرية (WT) التي تم حقنها بالخلايا الأصلية (الشكل 2أ، ب)، في حين أن الفئران Sting-/- التي تم زرعها بخلايا ذات كفاءة Cgas طورت أورامًا مماثلة لتلك الناتجة عن خلايا التحكم الأصلية (الشكل 2ج، د)، مما يشير إلى أن تثبيط الورم الناجم عن cGAS في الورم يعتمد على STING في المضيف. على عكس الأورام الناتجة عن خلايا التحكم، لوحظ زيادة ملحوظة في تغطية الخلايا المحيطة لأوعية الورم في الأورام ذات كفاءة Cgas في الفئران البرية (الشكل 2ك). علاوة على ذلك، لوحظ انخفاض كبير في نقص الأكسجة داخل الورم، كما هو موضح بواسطة مستوى مؤشر نقص الأكسجة ناقل الجلوكوز 1 (GLUT1)، وزيادة كبيرة في تم الكشف عن تسلل الخلايا في الأورام التي تحتوي على Cgas مقارنة بالأورام الضابطة في الفئران البرية (الشكل 2ك). بالإضافة إلى ذلك، بعد تنشيط STING، تم الكشف عن IFNوجينات المحفزة بواسطة الإنترفيرون (ISGs)، وجينات تثبيت الأوعية الدموية، وجزيئات الالتصاق المرتبطة بتفاعل البطانة اللمفاوية تم تنظيمها صعودًا في الأورام التي تحتوي على Cgas مقارنة بالأورام الضابطة في الفئران البرية (WT) (الشكل التكميلي 4أ). ومن المهم أن نلاحظ عدم وجود فروق واضحة في أعباء الأورام، وتطبيع الأوعية الدموية،تم العثور على تسلل الخلايا في الفئران التي تفتقر إلى STING (Sting-/) المزروعة بخلايا ذات كفاءة في Cgas مقابل خلايا التحكم (الشكل 2ج، د، ك، الشكل التكميلي 4ب). تشير هذه النتائج مجتمعة إلى أن cGAS الوسيط في الورم كبح الورم، تطبيع الأوعية الدموية، والاستجابة المناعية المضادة للورم تعتمد على STING المضيف.
بعد ذلك تحدينا Cgasأو فئران WT مع خلايا ذات كفاءة Cgas لمعالجة ما إذا كانت هذه الظاهرة التي يسببها cGAS في الورم تحددها cGAS في العائل. ومن المثير للاهتمام، لم يتم الكشف عن فرق كبير في نمو الورم بينو فئران WT (الشكل 2e، f). علاوة على ذلك، أظهرت الفئران التي تفتقر إلى Cgas أو فئران WT التي تم تحديها بخلايا تمتلك Cgas تغطية مماثلة للأوعية الدموية الورمية بواسطة الخلايا المحيطة بالأوعية، ونقص الأكسجة داخل الورم،تسلل الخلايا التائية، وتعبير جينات تطبيع الأوعية الدموية (الشكل 2ك، الشكل التكميلي 4ج). تشير هذه النتائج إلى أن cGAS المضيف غير مطلوب لتطبيع الأوعية الدموية الذي يتم بواسطة cGAS الورمي والاستجابة المناعية المضادة للورم.
لاختبار ما إذا كان STING المضيف يعتمد على cGAS الورمي لتنظيم تطبيع الأوعية الدموية والاستجابة المناعية المضادة للأورام، قمنا بتحدي فئران Sting-/- وفئران WT بخلايا ذات cGAS كفء أو خلايا تحكم، على التوالي. طورت خلايا التحكم أعباء أورام مماثلة في فئران Sting-/- مقابل فئران WT (الشكل 2g، h)، في حين أظهرت الخلايا ذات cGAS الكفء نموًا أسرع في فئران Sting-/- مقابل فئران WT (الشكل 2i، j). على عكس الأورام المشتقة من خلايا التحكم، لوحظ انخفاض ملحوظ في
الشكل 2 | إنزيم cGAS في الورم ومستقبل STING في المضيف يتوسطان تطبيع الأوعية الدموية والاستجابة المناعية المضادة للورم بطريقة تعتمد على بعضهما البعض. منحنيات نمو الورم (أ) وأعباء الورم (ب) في الفئران البرية (WT) التي تم حقنها تحت الجلد بخلايا Hepal-6-Cgas أو خلايا التحكم لمدة 3 أسابيع (الفئران لكل مجموعة). منحنيات نمو الورم (ج) وأعباء الورم (د) في فئران Sting-/- التي تم حقنها تحت الجلد بخلايا Hepal-6-Cgas أو Ctrl لمدة 3 أسابيع (الفئران لكل مجموعة). منحنيات نمو الورم (هـ) وأعباء الورم (و) فيأو فئران WT التي تم حقنها تحت الجلد بخلايا Hepa1-6-Cgas لمدة 3 أسابيع (فئران لكل مجموعة).منحنيات نمو الورم (g) وأعباء الورم (h) في الفئران Sting-/- أو WT التي تم حقنها تحت الجلد بخلايا Hepal-6-Ctrl لمدة 3 أسابيع ( الفئران لكل مجموعة). منحنيات نمو الورم (i) وأعباء الورم (j) في Stingأو الفئران البرية التي تم حقنها تحت الجلد بخلايا Hepal-6-Cgas لمدة 3 أسابيع (فئران لكل مجموعة).صور مناعية نسيجية تمثيلية وقياس كمية للخلايا المحيطة بالأوعية () تغطية السفن ()، GLUT1 منطقة نقص الأكسجة، وCD8الخلايا التائية في الأورام المشار إليها (). تمثل الأسهم الصفراء الخلايا. أشرطة المقياس، تم حساب القيم باستخدام تحليل التباين ثنائي الاتجاه (a, c, e, g, i) واختبار الطالب غير المرتبط ذو الطرفيناختبار (). غير مهم. يتم توفير بيانات المصدر كملف بيانات مصدر. تغطية الخلايا المحيطة للأوعية الدموية في الورم وتم ملاحظة تسلل الخلايا، ولكن تم تسجيل زيادة كبيرة في نقص الأكسجة داخل الورم في الأورام التي تحتوي على Cgas من Sting.الفئران مقابل الفئران البرية (الشكل 2ك). بالإضافة إلى ذلك، لا توجد فروق واضحة في تفعيل IFN أسفل STINGوتمت ملاحظة جينات ISGs، وجينات تثبيت الأوعية الدموية، وجزيئات الالتصاق المرتبطة بتفاعل الخلايا البطانية مع اللمفاويات في أورام التحكم من الفئران Sting-/- مقارنة بالفئران البرية WT، ولكن هذه تم تقليل تنظيم الجينات في الأورام التي تحتوي على Cgas في الفئران التي تفتقر إلى Sting مقارنة بالفئران البرية (الشكل التكميلي 4د، هـ). توضح هذه النتائج أن كبت الورم الذي يتم بوساطة STING في المضيف، وتطبيع الأوعية الدموية، والاستجابة المناعية المضادة للورم تعتمد على cGAS في الورم. مجتمعة، تشير هذه النتائج إلى أن cGAS في الورم وSTING في المضيف يتوسطان تطبيع الأوعية الدموية والاستجابة المناعية المضادة للورم بطريقة تعتمد على بعضهما البعض.
ينتج cGAS الورمي cGAMP لتنشيط STING البطاني وتعزيز حركة اللمفاويات
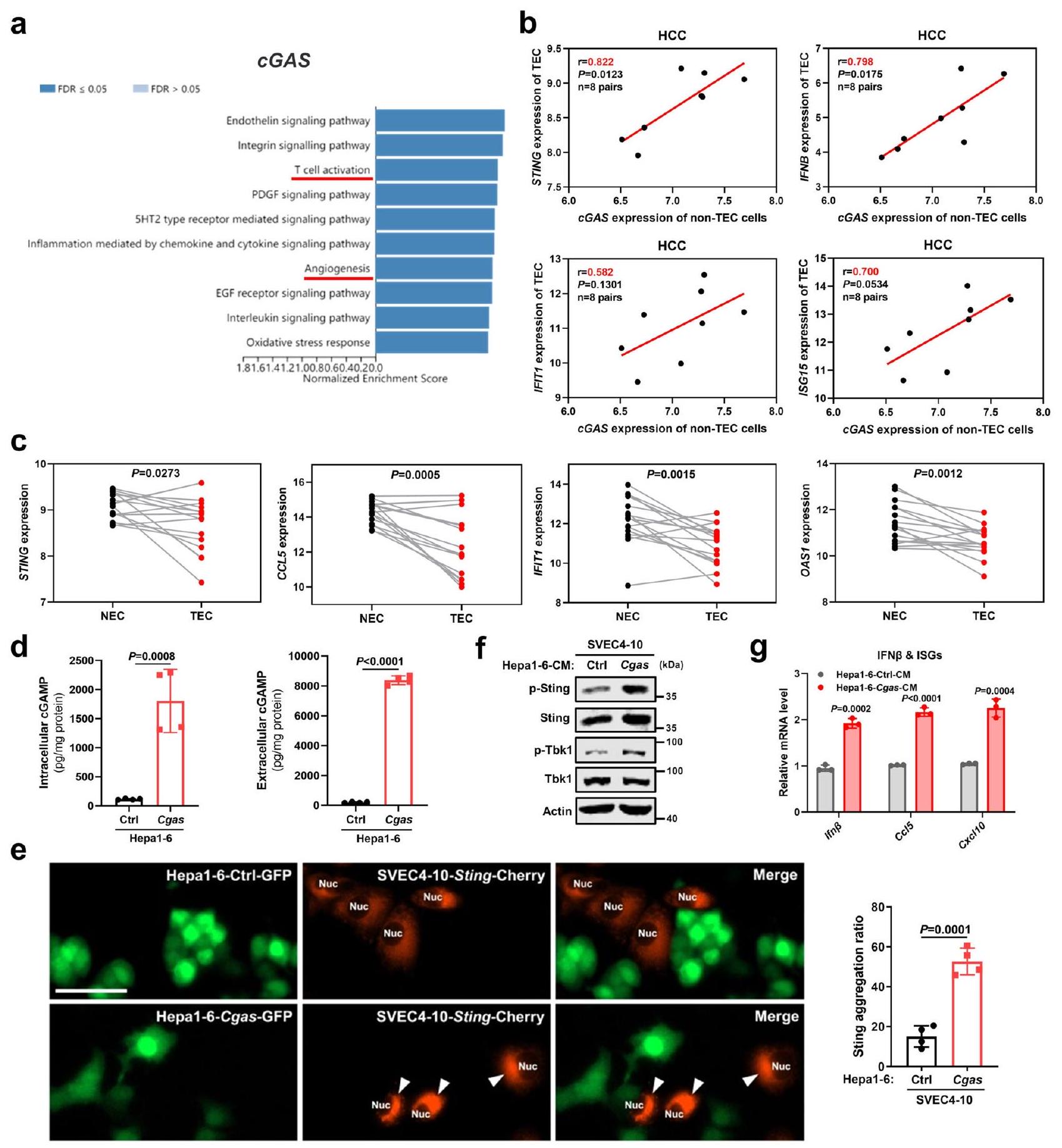
لاستكشاف المزيد من التفاعل المتبادل بين cGAS-STING بين خلايا الورم وخلايا المضيف، قمنا أولاً بتقييم تعبير cGAS وSTING في بيئة الورم الدقيقة. في أطلس البروتين البشري (HPA)، كان تعبير cGAS إيجابيًا في خلايا سرطان الكبد لمعظم المرضى، لكن تم الكشف عن تعبير STING الإيجابي في خلايا السرطان في مريض واحد فقط من بين 12 مريضًا (الشكل التكميلي 5أ). وبالمثل،أظهر 103 مرضى سرطان الكبد من مستشفانا (EHBH) تعبيرًا إيجابيًا عن cGAS في خلايا السرطان، في حين أن أقل منأظهر عدد من المرضى تعبيرًا إيجابيًا لبروتين STING في خلايا السرطان (الشكل التكميلي 5ب). بالمقابل، تم تحديد تعبير مميز لبروتين STING في خلايا البطانية مقارنة بالخلايا المضيفة الأخرى في بيئة الورم الكبدي البشري من خلال تجمعات نوع الخلية المفردة.(الشكل التوضيحي التكميلي 5ج). علاوة على ذلك، تعبير STING فيتم التحقق من أوعية الورم في مقاطع متسلسلة من عينات سرطان الكبد من نفس المريض (الشكل التكميلي 5د). لذلك، تشير هذه النتائج إلى أن خلايا سرطان الكبد تعبر عن cGAS، لكنها بالكاد تعبر عن STING، في حين تظهر الخلايا البطانية تعبيرًا واضحًا لـ STING وهي الهدف الأهم لـ cGAMP، لأنها أكثر وفرة من الخلايا المضيفة الأخرى في بيئة الورم الدقيقة. بالإضافة إلى ذلك، على الرغم من أن الخلايا التائية تساهم بشكل مهم في تطبيع أوعية الورم، كان cGAS الورمي كافياً لكبح تكوين الأوعية الدموية الورمية وتحفيز تطبيع الأوعية الدموية في الفئران المناعية المعوقة (الشكل التكميلي 4a-e)، مما يستبعد الدور الأساسي لـ STING الداخلي للخلايا التائية في إعادة تشكيل الأوعية الدموية الورمية التي يسببها cGAS الورمي. معاً، تدعم هذه البيانات التفاعل التفضيلي بين cGAS الورمي وSTING البطاني في السرطان الحي، الذي يتميز بارتفاع تكوين الأوعية الدموية.
كشف تحليل إثراء مجموعة الجينات (GSEA) (تحليل مسار بانثر) أن cGAS الورمي مرتبط بشكل كبير بكل من تنشيط الخلايا التائية وتكوين الأوعية الدموية في سرطان الكبد من TCGA (الشكل 3أ)، مما يشير إلى الدور التنظيمي لـ cGAS الورمي في إعادة تشكيل الأوعية الدموية. قمنا بمزيد من التقييم للعلاقة بين cGAS الورمي وتنشيط STING البطاني في سرطان الكبد البشري. CD31تم فرز الخلايا البطانية المرتبطة بالأورام (TECs) والخلايا غير المرتبطة بالـ TEC (خاصة خلايا النسيج الورمي) من أنسجة سرطان الكبد لنفس المريض. ومن المهم أن تعبير cGAS في الخلايا غير المرتبطة بالـ TEC كان مرتبطًا إيجابيًا بتعبير STING وجينات الاستجابة للإنترفيرون (ISGs) في خلايا TEC المقابلة (الشكل 3ب). علاوة على ذلك، مقارنةً بخلايا TEC المطابقة، كان CD31…عبّرت الخلايا البطانية الطبيعية (NECs) عن زيادة في STING وجينات الاستجابة للإنترفيرون (ISGs) (الشكل 3ج)، مما يؤكد دور STING البطاني في تطبيع الأوعية الدموية. بعد ذلك، تحققنا من تنشيط STING البطاني بواسطة cGAS في خلايا سرطان الكبد من خلال تجارب مخبرية. بعد التأكد من أن زيادة تعبير Cgas عززت إفراز cGAMP في خلايا سرطان الكبد عن طريق قياس مستويات cGAMP في الوسط داخل الخلايا وخارجها (الشكل 3د)، اختبرنا بعد ذلك ما إذا كان cGAMP المنتج بواسطة خلايا سرطان الكبد يمكن أن ينشط STING في الخلايا البطانية المحيطة، باستخدام تكوين تجمعات STING حول النواة كمؤشر على تنشيط STING بواسطة cGAMP.تم تأكيد تكوين تجمعات Sting عند تحفيزها بـ cGAMP لأول مرة في الخلايا البطانية التي تعبر عن بلازميد StingCherry بعد استنفاد Sting الداخلي (الشكل التكميلي 6a، b). ثم، تم تعايش الخلايا البطانية المعبرة عن Sting-Cherry مع خلايا Cgas الموسومة بـ GFP وخلايا Ctrl الموسومة بـ GFP على التوالي. وجدنا أن تجمعات Sting كانت أكثر وضوحًا بكثير في الخلايا البطانية المتعايشة مع خلايا Cgas مقارنة بخلايا Ctrl (الشكل 3e). بالإضافة إلى ذلك، مقارنةً بالوسط المشروط (CM) المشتق من خلايا Ctrl، أدى تحفيز الخلايا البطانية بواسطة الوسط المشروط المشتق من خلايا Cgas (تم تأكيد وجود كمية أكبر بكثير من cGAMP في الوسط من خلايا Cgas مقارنة بخلايا Ctrl في الشكل 3d) إلى تنشيط Sting، كما يتضح من زيادة فسفرة Sting وTbk1، بالإضافة إلى زيادة Ifn.وتعبيرات جينات الاستجابة للإنترفيرون (الشكل 3f، g).
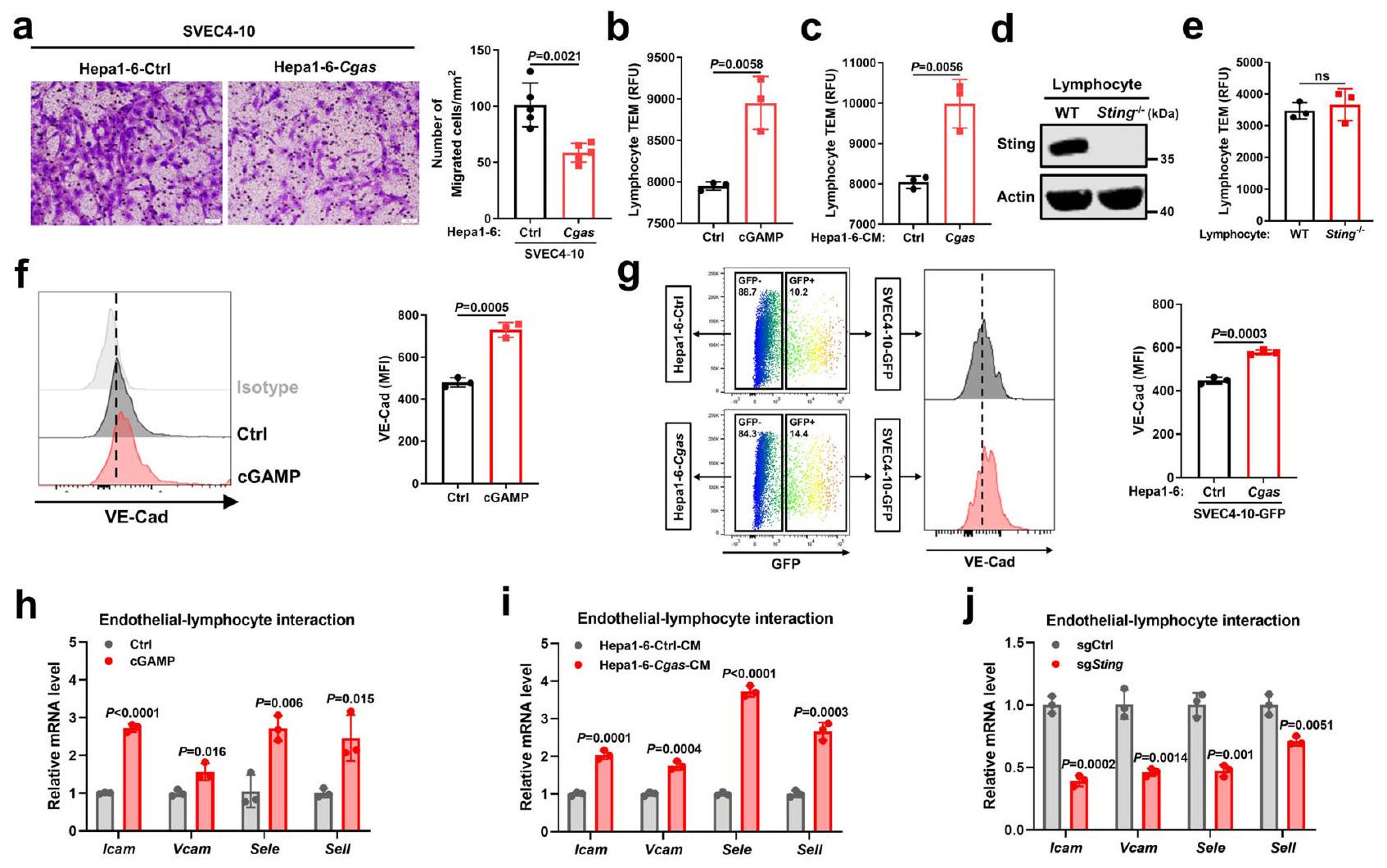
بعد ذلك، بحثنا دور تنشيط STING في الخلايا البطانية بواسطة خلايا سرطان الكبد في وظائف الخلايا البطانية. أدى التنشيط المباشر لمسار Sting بواسطة cGAMP (تم تأكيد تنشيط مسار STING المحفز بواسطة cGAMP في الشكل التكميلي 6ج، د) إلى تثبيط الخلايا البطانية تكاثرها وقدرتها على تكوين هياكل أنبوبية في اختبار ماتريجل (الشكل التكميلي 6e، g). وبالمثل، أدى علاج الخلايا البطانية الوسطية المستمدة من خلايا Cgas إلى كبح جزئي لتكاثر الخلايا البطانية وتكوين الأنابيب (الشكل التكميلي 6f، h). بالإضافة إلى ذلك، تم قمع الخصائص الهجومية للخلايا البطانية بعد التعايش مع خلايا Cgas، كما هو موضح في اختبار كيميائية الأورام (الشكل 4a). نظرًا لأن تطبيع الأوعية الدموية للأورام يحسن تسلل الخلايا اللمفاوية إلى الأورام، قمنا بتقييم ما إذا كانت الخلايا البطانية المنشّطة بواسطة STING تدعم تسلل المزيد من الخلايا اللمفاوية. من خلال استخدام اختبار هجرة الخلايا اللمفاوية عبر البطانة المعدل كما وُصف سابقًا، وجدنا أن المزيد من اللمفاويات عبرت حاجز الخلايا البطانية المعالجة بـ cGAMP مقارنة بحاجز التحكم (الشكل 4ب) أو حاجز الخلايا البطانية المتعايش مع خلايا Cgas مقارنة بخلايا التحكم (الشكل 4ج). ومع ذلك، لم تظهر اللمفاويات الناقصة لـ Sting واللمفاويات البرية أي اختلاف في الهجرة عبر الخلايا البطانية (الشكل 4د، هـ). لذلك، تعزز خلايا السرطان التي تعبر عن cGAS بشكل عالي هجرة اللمفاويات عبر الخلايا البطانية من خلال تنشيط STING في الخلايا البطانية، وليس من خلال مسار STING في اللمفاويات، عبر إفراز cGAMP.
من الناحية الميكانيكية، أظهرت خلايا البطانية زيادة في تعبير الكادهرين البطاني الوعائي (VE-Cad) بعد علاجها بـ cGAMP (الشكل 4f) أو في الثقافة المختلطة مع خلايا Cgas مقارنة بخلايا Ctrl (الشكل 4g)، مما يشير إلى أن تنشيط STING في خلايا البطانية الناتج عن خلايا Cgas يحافظ على استقرار وصلات خلايا البطانية، والتي تعد ضرورية لحركة الكريات البيضاء.. بالإضافة إلى ذلك، بعد تنشيط Sting بواسطة cGAMP أو تحفيز CM المستمد من خلايا Cgas، أظهرت الخلايا البطانية زيادة في تعبير جزيئات الالتصاق المشاركة في التفاعل بين الخلايا البطانية واللمفاويات بما في ذلك Icam وVcam وE-selectin (Sele) وL-selectin (Sell) (الشكل 4h، i). في المقابل، أدى نقص Sting إلى إضعاف تعبير هذه الجزيئات الالتصاقية (الشكل 4j). علاوة على ذلك، فإن حجب IFNمسار الإشارة بواسطة مثبط تنشيط STAT1 فلودارابين (فلورا) عكس تعبير Ccl5 المحفز بواسطة cGAMP، ولكن ليس تعبير Icam (الشكل التكميلي 6i)، وIFNالعلاج زاد من تعبير Ccl 5، لكنه لم يؤثر بشكل واضح على تعبير Icam في الخلايا البطانية (الشكل التكميلي 6j)، مما يشير إلى أن تنظيم جزيئات الالتصاق المرتبطة بتفاعل الخلايا البطانية مع اللمفاويات يتم بشكل رئيسي عبر مسار STING، بدلاً من جيناته المستهدفة IFN.وISGs. تشير هذه البيانات مجتمعة إلى أن cGAS في الورم ينتج cGAMP لتنشيط مسار STING في الخلايا البطانية، مما يثبط تكوين الأوعية الدموية ويعزز حركة اللمفاويات من خلال الحفاظ على استقرار وصلات الخلايا البطانية وزيادة التعبير عن جزيئات الالتصاق المرتبطة بتفاعل الخلايا البطانية مع اللمفاويات.
نقل cGAMP المشتق من الورم عبر قنوات LRRC8C لتنشيط STING في الخلايا البطانية
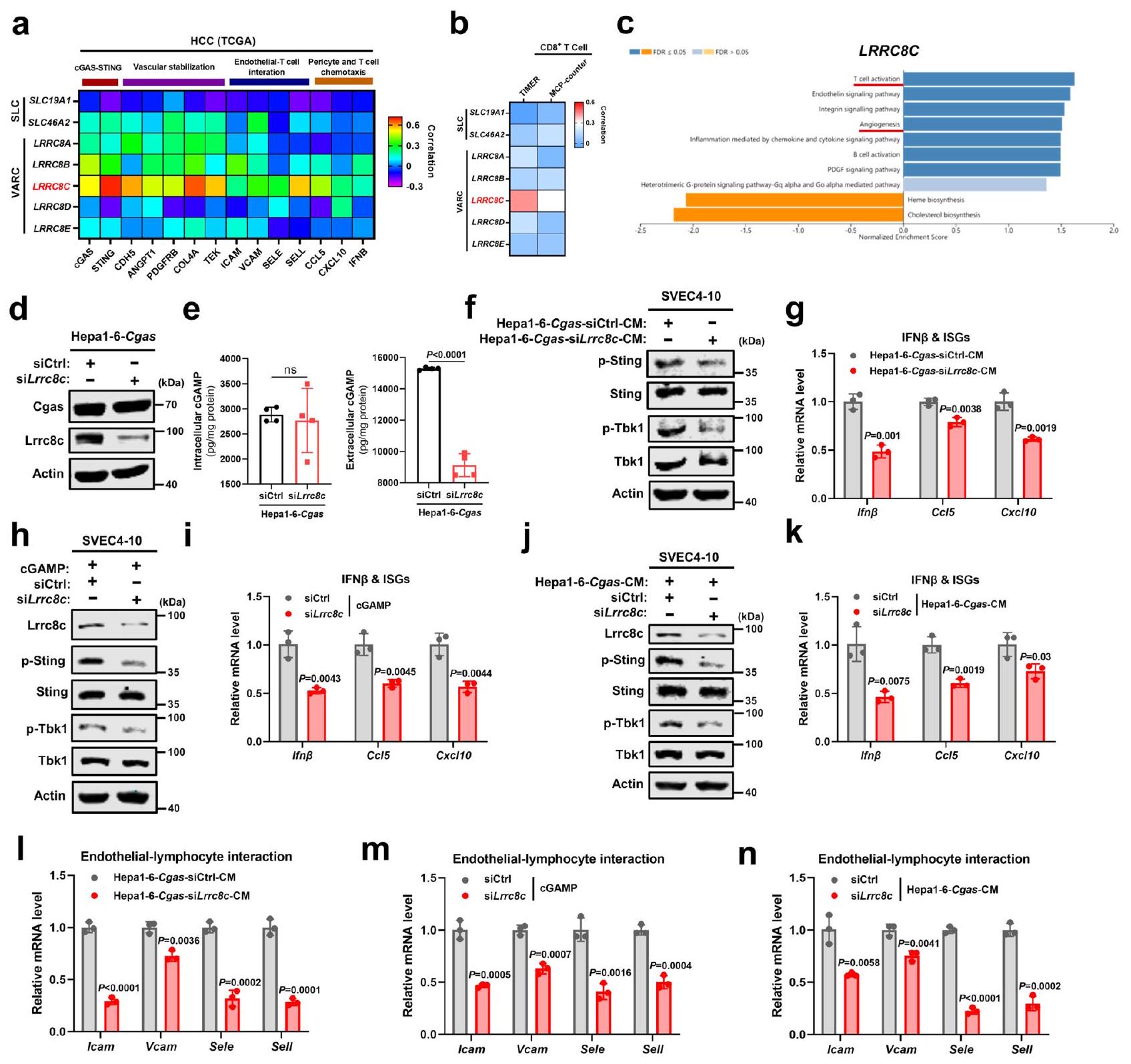
نحقق بعد ذلك في كيفية نقل cGAMP الذي تنتجه cGAS في خلايا سرطان الكبد إلى الخلايا البطانية. بالإضافة إلى الوصلات الفجوية، التي تربط الخلايا المجاورة مباشرةً، أظهرت دراسات حديثة أن cGAMP ينتقل بين الخلايا عبر ناقلات المذاب (SLCs) بما في ذلك SLC19A1 وSLC46A2، وقنوات الأنيون المنظمة بالحجم (VRACs)، التي تتكون من هتيرومرات LRRC8نظرًا لأن الوسط الخلوي المستمد من خلايا Cgas كان كافيًا لتنشيط STING البطاني (الشكل 3e-g)، تم استبعاد الوصلات الفجوية. ومن المثير للاهتمام، من بين ناقلات SLCs وVRACs المذكورة أعلاه، كان تعبير LRRC8C في الورم هو الأكثر ارتباطًا إيجابيًا مع جينات cGAS-STING والجينات المرتبطة بتطبيع الأوعية الدموية بما في ذلك استقرار الأوعية، التفاعل بين البطاني والخلايا اللمفاوية، الخلايا المحيطة بالأوعية، وكيمياء الخلايا التائية في قاعدة بيانات TCGA (الشكل 5a). بالإضافة إلى ذلك، تم العثور على أقوى ارتباط إيجابي بين تعبير LRRC8C في الورم وتسلل الخلايا التائية (الشكل 5ب). من بين LRRC8A-E، تنبأت التعبيرات العالية لـ LRRC8C في الورم بتوقع أفضل لمرضى سرطان الكبد (الشكل التكميلي 7أ-هـ). وبالمثل مع cGAS في الورم، أظهرت تحليل GSEA (تحليل مسار بانثر) أن LRRC8C في الورم مرتبط بشكل كبير بكل من تنشيط الخلايا التائية وتكوين الأوعية الدموية في سرطان الكبد من مجموعة بيانات TCGA (الشكل 5ج). لذلك، تشير هذه النتائج من خلال التحليل المعلوماتي الحيوي إلى أن LRRC8C قد يكون وسيطًا في…
الشكل 3 | ينتج cGAS الورمي cGAMP لتنشيط STING في الخلايا البطانية. تحليل GSEA (تحليل مسار Panther) لـ cGAS في سرطان الكبد من TCGA (). ب تحليل الارتباط بين تعبير cGAS في خلايا البطانية غير المرتبطة بالأورام (non-TEC) مع تعبيرات STING وISGs في خلايا TEC المقابلة من أنسجة سرطان الكبد لنفس المريض عبر الفرز المغناطيسي المنشط (MACS) في مجموعة بيانات GSE51401 (أزواج). ج تعبيرات STING وISGs في خلايا TECs مقابل خلايا البطانة الطبيعية (NECs) في مجموعة بيانات GSE51401 (أزواج). د مستويات cGAMP داخل الخلايا وخارجها من خلايا Hepa1-6-Cgas أو خلايا Ctrl (). صور مناعية فلورية تمثيلية وقياس لتجمعات Sting في SVEC4-10 الخلايا التي تعبر بشكل مفرط عن Sting-Cherry والمزروعة مع خلايا Hepal-6-Cgas أو Ctrl الموسومة بـ GFP (). تمثل الأسهم البيضاء تجمعات ستينغ في خلايا SVEC4-10. أشرطة المقياس،. ف مستويات البروتين لمؤشرات في مسار Sting في خلايا SVEC4-10 بعد التعرض لـ CM من خلايا Hepal-6-Cgas أو Ctrl. تمثيلي لـتجارب مستقلة.مستويات mRNA لـ Ifnو ISGs في خلايا SVEC4-10 بعد التعرض لـ CM من خلايا Hepal-6-Cgas أو Ctrl ().يتم حساب القيم باستخدام معامل ارتباط بيرسون ذو الذيلين (ب)، واختبار الطالب المزدوج ذو الذيلين للعينات المزدوجةاختبار (c) واختبار الطالب غير المزدوجاختبار (). يتم توفير بيانات المصدر كملف بيانات مصدر. انتقال cGAMP بين خلايا سرطان الكبد وخلايا البطانة الوعائية.
لاختبار دور LRRC8C في نقل cGAMP بشكل أعمق، تم تثبيط Lrrc8c في خلايا Cgas (الشكل 5د). وُجد أن مستوى cGAMP داخل الخلية لم يتأثر بشكل واضح، في حين أن انخفض مستوى cGAMP خارج الخلية بشكل ملحوظ (الشكل 5e)، مما يشير إلى أن LRRC8 يتوسط إفراز cGAMP في خلايا سرطان الكبد. علاوة على ذلك، تم إضعاف تنشيط Sting في الخلايا البطانية بواسطة الوسط المستخلص من خلايا Cgas بشكل كبير بعد تثبيط Lrrc8c، كما هو موضح بانخفاض فسفرة Sting وTbk1، بالإضافة إلى Ifn.و ISGs
الشكل 4 | تنشيط STING البطاني الوعائي الناتج عن cGAS في الورم يعزز حركة الخلايا اللمفاوية. أ الصور التمثيلية والقياسات الكمية لخلايا SVEC4-10 التي هاجرت نحو خلايا Hepa1-6-Cgas أو خلايا التحكم في تجارب Transwell (). ب الهجرة عبر البطانة (TEM) للخلايا اللمفاوية الطحالية للفأر عبر حاجز خلايا SVEC410 مع أو بدون المعالجة المسبقة بـ cGAMP ( ). تصوير المجهر الإلكتروني النافذ لخلايا اللمفاويات الطحالية للفأر عبر حاجز خلايا SVEC4-10 مع المعالجة المسبقة بمادة CM من خلايا Hepal-6-Cgas أو خلايا Ctrl ().مستويات بروتين Sting في الخلايا اللمفاوية الطحالية من Stingأو فئران WT. ممثل عنتجارب مستقلة. المجهر الإلكتروني النافذ لـ تحليل التدفق الخلوي وشدة الفلورة المتوسطة (MFI) لـ VECad السطحي في خلايا SVEC4-10 بعد علاج cGAMP (f) ()، مزروعة مع خلايا Hepal-6Cgas أو خلايا Ctrlمستويات mRNA للجينات المشار إليها في خلايا SVEC4-10 بعد علاج cGAMP ( ). مستويات mRNA للجينات المشار إليها في خلايا SVEC4-10 المتعايشة مع خلايا Hepal-6-Cgas أو خلايا Ctrl ().مستويات mRNA للجينات المشار إليها في خلايا SVEC4-10 مع تعطيل Sting (sgSting) أو خلايا التحكم (sgCtrl) ().يتم حساب القيم باستخدام اختبار الطالب غير المزدوجاختبار (a-c، e-j). ns، غير معنوي. تم توفير بيانات المصدر كملف بيانات مصدر. اللمفاويات المستمدة من فئران Sting-/- أو WT عبر حاجز خلايا SVEC4-10 ().
التعبيرات (الشكل 5f، g)، مما يشير معًا إلى أن cGAMP الذي تنتجه خلايا سرطان الكبد يُفرز خارج الخلية عبر قنوات LRRC8C، وبالتالي يُفعّل STING في الخلايا البطانية. نظرًا لأن قنوات LRRC8 ثبت أنها ناقل ثنائي الاتجاه لـ cGAMP، قمنا بعد ذلك بتحديد ما إذا كان cGAMP الذي تفرزه خلايا السرطان يدخل خلايا البطانة أيضًا عبر LRRC8C. عندما تم حجب تعبير Lrrc8c في خلايا البطانة، فشل كل من cGAMP (الشكل 5h، i) أو الوسط المستخلص من خلايا Cgas (الشكل 5j، k) في تنشيط مسار Sting في خلايا البطانة، كما يتضح من انخفاض فسفرة Sting وTbk1، بالإضافة إلى انخفاض Ifnوتعبيرات ISGs، مما يشير إلى أن خلايا البطانية تستخدم قنوات LRRC8C لاستيراد cGAMP، مماثلة للتقارير السابقة. علاوة على ذلك، أدى تثبيط Lrrc8c في خلايا Cgas أو الخلايا البطانية إلى كبح كبير في تعبير جزيئات الالتصاق المرتبطة بتفاعل الخلايا البطانية مع اللمفاويات الناتج عن cGAMP أو الوسط المستمد من خلايا Cgas في الخلايا البطانية (الشكل 51-ن). تشير هذه البيانات مجتمعة إلى أن قنوات LRRC8C تتوسط نقل cGAMP من خلايا سرطان الكبد إلى الخلايا البطانية الوعائية، مما يؤدي إلى تنشيط مسار STING في الخلايا البطانية.
TET2 يتعاون مع إشارة IL-2/STAT5A لتعزيز تعبير cGAS في الورم وإفراز cGAMP
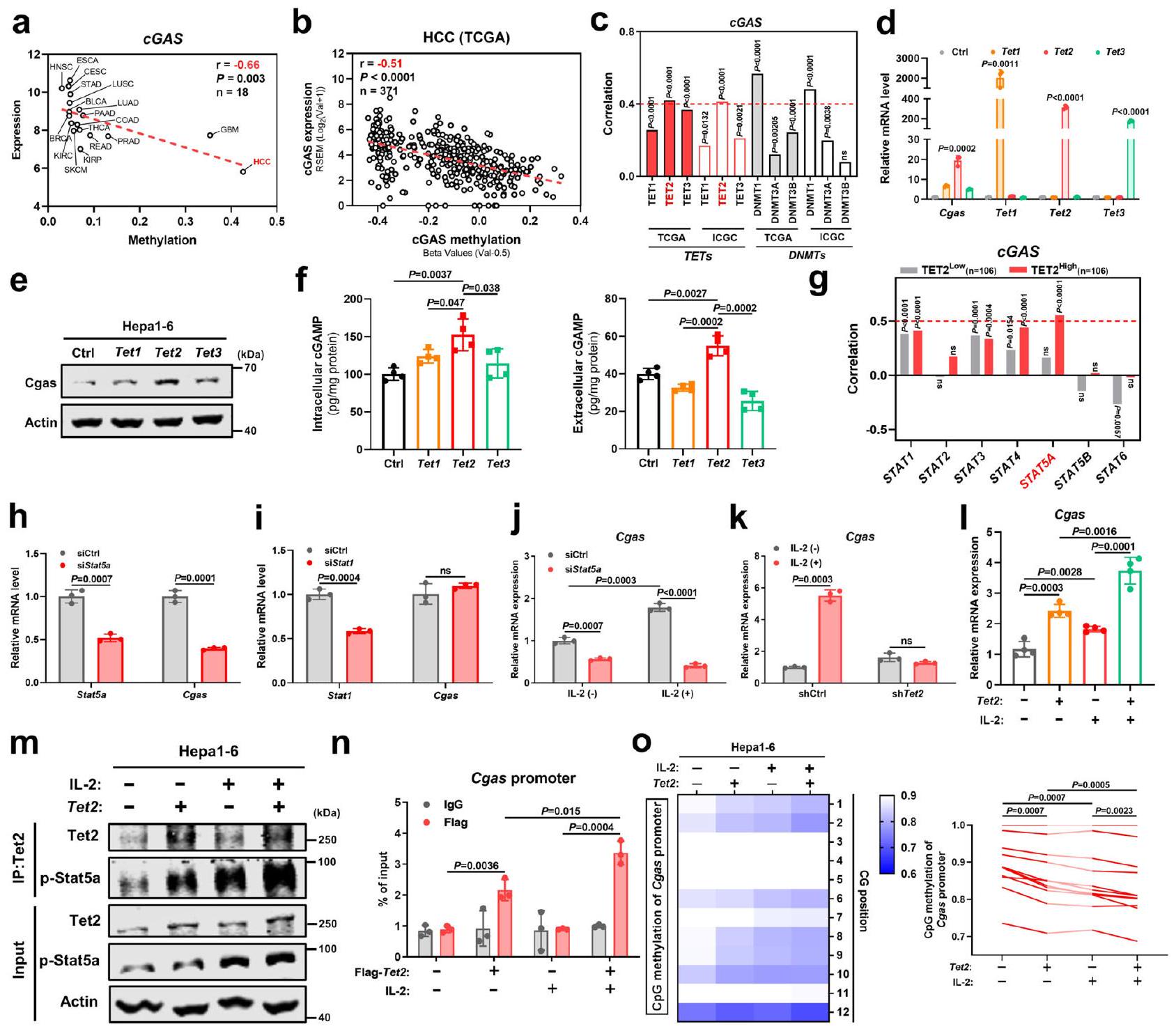
على الرغم من أن cGAS كان معبرًا عنه إيجابيًا في سرطان الكبد الكبدي (HCC)، إلا أن تعبير cGAS على مستوى النسخ كان الأدنى بين جميع أنواع السرطان، مصحوبًا بأعلى نسبة مثيلة لمروج جين cGAS.(الشكل 6أ). في الواقع، كانت مستويات تعبير cGAS مرتبطة سلبًا بمستويات مثيلتها () في سرطان الكبد من مجموعة بيانات TCGA (الشكل 6ب). إزالة مثيلة الحمض النووي يتم التحكم فيها بشكل رئيسي بواسطة إنزيمات TET ثنائية الأكسجين، والتي قادرون على تحويل 5-ميثيل السيتوزين (5mC) إلى 5-هيدروكسي ميثيل السيتوزين (5 hmCمن المثير للاهتمام، من بين أعضاء عائلة TET، كان تعبير مثبط الورم TET2 هو الأكثر ارتباطًا إيجابيًا بتعبير cGAS ولم يُلاحظ أي ارتباط سلبي بين تعبير cGAS في الورم وإنزيمات مثيلة الحمض النووي (DNMTs) في سرطان الكبد من مجموعات بيانات TCGA وICGC (الشكل 6ج). للتحقيق بشكل أعمق فيما إذا كان TET2 ينظم تعبير cGAS، قمنا بزيادة التعبير عن Tet1 وTet2 وTet3 في خلايا سرطان الكبد على التوالي. ومن الجدير بالذكر أن زيادة التعبير عن Tet2 زادت بشكل ملحوظ من تعبير Cgas مقارنة بـ Tet1 وTet3، على المستويين النصي والبروتيني (الشكل 6د، هـ). علاوة على ذلك، عززت زيادة التعبير عن Tet2 أيضًا نشاط إنزيم Cgas، كما تدعمه المستويات المرتفعة من cGAMP داخل وخارج الخلايا (الشكل 6و). مجتمعة، تشير هذه النتائج إلى أن TET2 هو الوسيط الرئيسي لتعبير cGAS في سرطان الكبد.
لأن عائلة TET غالبًا ما تتفاعل مع إشارات أخرى، وخاصة مسار JAK/STAT، لتنسيق التعبير الجيني المستهدف بشكل تآزري“، تساءلنا عما إذا كان TET2 يتعاون مع إشارات STAT لتنظيم تعبير cGAS. ومن المدهش أنه من بين أعضاء عائلة STAT (STAT1-6)، أظهر STAT5A أعلى ارتباط إيجابي مع cGAS في مجموعة التعبير العالي لـ TET2 وليس في مجموعة التعبير المنخفض لـ TET2 من مجموعات بيانات HCC ICGC (الشكل 6g). في المختبر، أدى تعطيل Stat5a، وليس Stat1، الذي أظهر ارتباطًا إيجابيًا مع cGAS بغض النظر عن تعبير TET2 (الشكل 6g)، إلى تراجع تعبير Cgas في خلايا سرطان الكبد (الشكل 6h، i). وعلى العكس، أدى تنشيط Stat5a عند تحفيز IL-2 إلى زيادة تعبير Cgas، وتم عكس هذا الارتفاع في Cgas الناتج عن IL-2 بعد تثبيط Stat5a (الشكل 6j)، مما يشير إلى أنَّ…
الشكل 5 | نقل cGAMP المشتق من الورم عبر قناة LRRC8C لتنشيط STING في الخلايا البطانية. أ. معامل ارتباط بيرسون لتعبيرات SLC (SLC19A1/SLC46A2) وVARC (LRRC8A-E) مع الجينات المتعلقة بـ cGAS-STING، استقرار الأوعية الدموية، تفاعل الخلايا البطانية مع الخلايا التائية، وكيمياء جذب الخلايا المحيطة/الخلايا التائية في سرطان الكبد الكبدي من مجموعة بيانات TCGA (). ب ارتباط بيرسون بين تعبيرات SLC (SLC19A1/SLC46A2) وVARC (LRRC8A-E) معتسلل الخلايا التائية في سرطان الكبدة الخلوي من مجموعة بيانات TCGA من خلال TIMER وMCP-counter (). ج تحليل GSEA (تحليل مسار بانثر) لـ LRRC8C في سرطان الكبد من TCGA ().مستويات البروتين لـ Cgas و Lrrc8c في خلايا Hepal-6-Cgas مع تثبيط Lrrc8c بواسطة siRNA (Hepal-6-Cgas-siLrrc8c) أو خلايا التحكم (Hepa1-6-Cgas-siCtrl). هـ مستويات cGAMP داخل الخلايا وخارجها من خلايا Hepal-6-Cgas-siLrrc8c أو Cgas-siCtrl.).مستويات البروتين للعلامات في مسار Sting في خلايا SVEC4-10 بعد التعرض لـ CM من خلايا Hepal-6-Cgas-siLrrc8c أو Cgas-siCtrl.مستويات mRNA لـ Ifnو ISGs في خلايا SVEC4-10 بعد التعرض لـ CM من خلايا Hepa1-6-Cgas-siLrrc8c أو Cgas-siCtrl ().مستويات البروتين في
Lrrc8c والعلامات في مسار Sting في خلايا SVEC4-10-siLrrc8c أو siCtrl بعد علاج cGAMP. مستويات mRNA لـ Ifnو ISGs في خلايا SVEC4-10-siLrrc8c أو siCtrl بعد علاج cGAMP ().مستويات البروتين لـ Lrrc8c والعلامات في مسار Sting في خلايا SVEC4-10-siLrrc8c أو siCtrl بعد التعرض لـ CM من خلايا Hepal-6Cgas.مستويات mRNA لـ Ifnو ISGs في خلايا SVEC4-10-siLrrc8c أو siCtrl بعد التعرض لـ CM من خلايا Hepal-6-Cgas ( ). 1 مستويات mRNA للجينات المشار إليها في خلايا SVEC4-10 بعد التعرض لـ CM من خلايا Hepal-6-Cgas-siLrrc8c أو Cgas-siCtrl ().مستويات mRNA للجينات المشار إليها في خلايا SVEC4-10-siLrrc8c أو siCtrl بعد علاج cGAMP ().مستويات mRNA للجينات المشار إليها في خلايا SVEC4-10-siLrrc8c أو siCtrl بعد التعرض لـ CM من خلايا Hepal-6-Cgas ().يتم حساب القيم باستخدام اختبار الطالب غير المرتبط ذو الطرفيناختبار (). غير مهم إحصائياً. ممثل لـتجارب مستقلة (). يتم توفير بيانات المصدر كملف بيانات مصدر. الدور التنظيمي لإشارة IL-2/STAT5A في تعبير cGAS. ومع ذلك، فشل علاج IL-2 في تحفيز تعبير Cgas بعد نقص Tet2 (الشكل 6k). على النقيض من ذلك، أدى الإفراط في التعبير عن Tet2 مع تحفيز IL-2 إلى زيادة أكبر في تعبير Cgas مقارنة بكل منهما على حدة (الشكل 61)، مما يشير إلى أن TET2 يتآزر مع إشارة STAT5A لتعزيز تعبير cGAS في الورم. من الناحية الآلية، تفاعل Tet2 مع Stat5a المنشط (p-Stat5a) وكان هذا التفاعل أقوى عند
تحفيز IL-2 (الشكل 6m). علاوة على ذلك، ارتبط Tet2 بمُحفز Cgas وتم تعزيز هذا الارتباط بشكل كبير في وجود IL-2 (الشكل 6n). للتحقق بشكل أكبر من دور Tet2 في إزالة مثيلة Cgas، كان الإفراط في التعبير عن Tet2 كافياً لتقليل المثيلة داخل مُحفز Cgas وتم ملاحظة تقليل أكبر في مثيلة مُحفز Cgas عند الجمع مع تحفيز IL-2 (الشكل 60). على الرغم من أن IL-2 معروف جيدًا بدفع استجابات الخلايا التائية من خلال الارتباط بمستقبله
الشكل 6 | يعمل TET2 بتآزر مع إشارة IL-2/STAT5A لتعزيز تعبير cGAS في الأورام وإفراز cGAMP. أ العلاقة بين التعبير الوسيط والميثلة لـ cGAS في 18 نوعًا من الأورام من مجموعات بيانات TCGA.الارتباط بين تعبير cGAS والميثلة في سرطان الكبد الكبدي (HCC) من مجموعة بيانات TCGA (). ج الارتباط بين تعبيرات cGAS وTETs أو DNMTs في سرطان الكبد الكبدي من TCGA () و ICGC () مجموعات البيانات.مستويات mRNA لـ Cgas و Tet1-3 في خلايا Hepal-6 مع زيادة تعبير Tet1-3 بواسطة البلازميدات (). مستوى بروتين Cgas في خلايا Hepal-6 مع زيادة التعبير عن Tet1-3 بواسطة البلازميدات. ف مستويات cGAMP داخل الخلايا وخارجها من خلايا Hepal-6 مع زيادة التعبير عن Tet1-3 بواسطة البلازميدات. ج الارتباط بين تعبيرات STAT1-6 و cGAS في مجموعات التعبير المنخفض والعالي لـ TET2 من مجموعة بيانات HCC-ICGC. تم تقسيم مجموعات التعبير المنخفض والعالي نسبةً إلى قيم التعبير الوسيط. ح مستويات mRNA لكل من Stat5a و Cgas في خلايا Hepa1-6 مع تثبيط Stat5a بواسطة siRNA (siStat5a) ( ). مستويات mRNA لكل من Stat1 وCgas في خلايا Hepal-6 مع تثبيط Stat1 بواسطة siRNA (siStat1) ().مستوى mRNA لـ Cgas في خلايا Hepa1-6-siStat5a أو siCtrl عند تحفيز IL-2 ().مستوى mRNA لجين Cgas في خلايا Hepal-6 مع تثبيط Tet2 بواسطة shRNA (تت2 ). مستوى mRNA لـ I Cgas في خلايا Hepal-6 مع زيادة تعبير Tet2 عند تحفيز IL-2 (). تحليل Co-IP لـ Tet2 مع p-Stat5a في خلايا Hepal-6 مع زيادة تعبير Tet2 عند تحفيز IL-2. n تحليل ChIP-qPCR لنشاط ارتباط Tet2 بمروج Cgas في خلايا Hepal-6 مع زيادة تعبير FlagTet2 عند تحفيز IL-2 (). تحليل التسلسل الناري وقياس حالة مثيلة المحفز لجين Cgas في خلايا Hepal-6 مع زيادة تعبير Tet2 عند تحفيز IL-2 ().يتم حساب القيم باستخدام معامل ارتباط بيرسون ذو الذيلين ()، تحليل التباين الأحادي الاتجاه () واختبار الطالب غير المرتبط ذو الطرفيناختبار (). غير مهم إحصائياً. ممثل عنتجارب مستقلة (). يتم توفير بيانات المصدر كملف بيانات مصدر. المستقبلات، التي تتكون من ثلاث وحدات فرعية بما في ذلك IL-2R(IL2RA)، مستقبل IL-2(IL2RB)، و IL-2R(IL2RG)، وجدنا أن كل من IL2RB و IL2RG معبر عنهما في العديد من خطوط خلايا سرطان الكبد، خاصة IL2RG، في قاعدة بيانات CCLE (الشكل التكميلي 8أ، ب) وتم تأكيد نتائج مماثلة في عينات سرطان الكبد من مجموعة بيانات TCGA (الشكل التكميلي 8ج). نظرًا لأن IL2RB و IL2RG كافيان لتشكيل مستقبل وظيفي لنقل الإشارات من IL-، يمكن لخلايا سرطان الكبد أيضًا الاستجابة لـ IL-2 الذي تفرزه خلايا T مما يؤدي إلى تنشيط STAT5A. تشير هذه النتائج معًا إلى أن STAT5A المنشط بواسطة IL-2 يجند ارتباط TET2 بموقع cGAS لتعزيز إزالة مثيلة cGAS ونسخه، مما يؤدي إلى زيادة تعبير cGAS في سرطان الكبد.
تحفيز TET2 وتسرب dsDNA بواسطة VC ينشطان مسار cGAS-cGAMP-STING البطاني الوعائي في الورم ويعززان حركة اللمفاويات
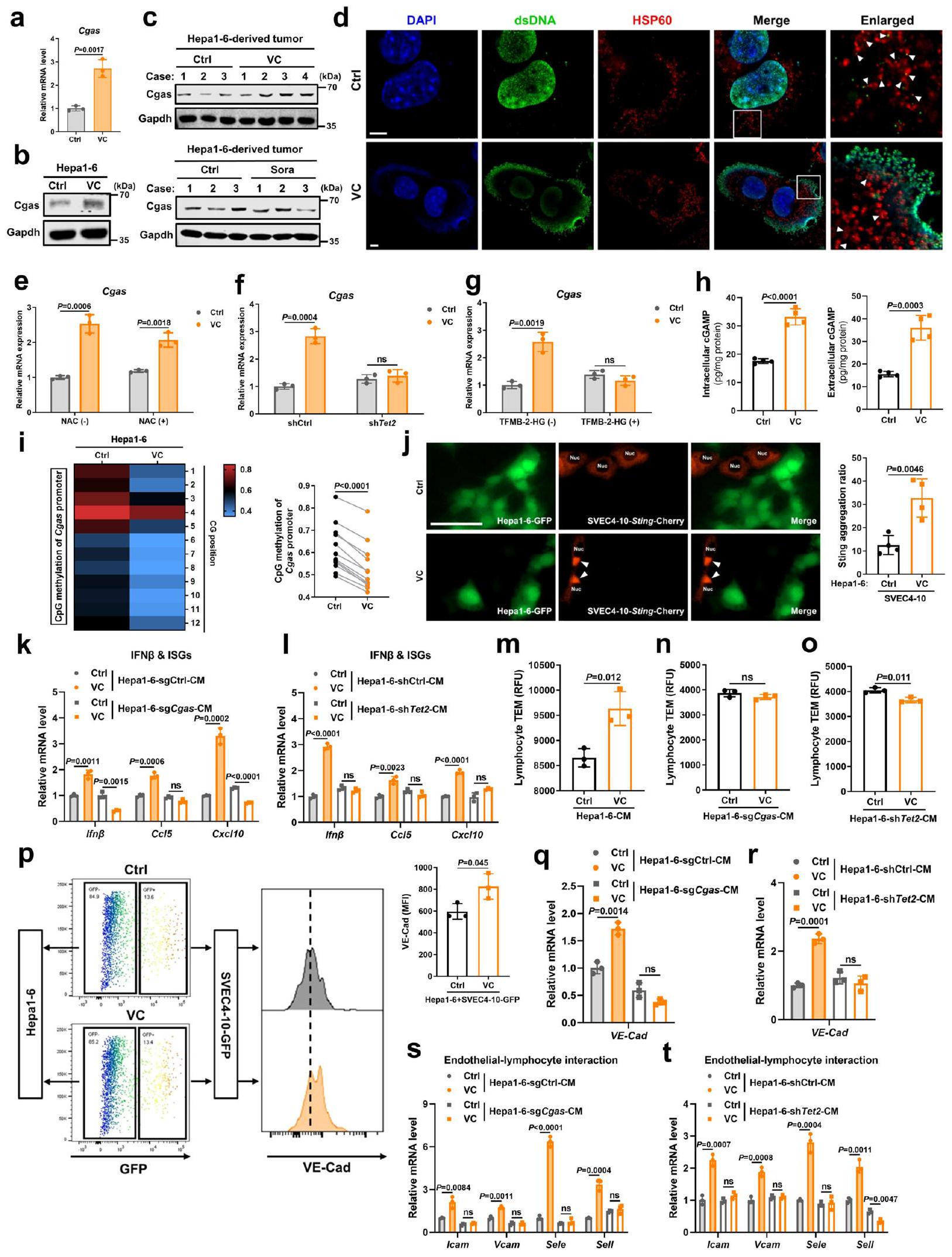
يقل تعبير TET2 بشكل كبير في سرطان الكبدة الخلوي (HCC)، لكنه نادراً ما يتحور.لذلك، دفعنا فقدان نشاط TET2 غير الناتج عن الطفرات في سرطان الكبد إلى استكشاف إمكانية إعادة تنشيط TET2 كاستراتيجية لتحفيز تعبير cGAS بشكل فعال. يعمل VC (حمض الأسكوربيك أو الأسكوربات) كعامل مساعد، ويعزز نشاط TETs، وقد تم اقتراحه كعامل واعد مضاد للسرطان.من المثير أن علاج VC أدى إلى تحفيز تعبير cGAS بشكل كبير على مستوى النسخ والبروتين في خلايا سرطان الكبد في المختبر (الشكل 7أ، ب). وبما يتفق مع
في النتائج المختبرية، تم زيادة تعبير cGAS في أورام
النشاط عبر الإجهاد التأكسدي الناتج عن بيروكسيد الهيدروجينوجدنا أن علاج VC يسبب تلفًا في الحمض النووي (الشكل التوضيحي التكميلي 9أ، ب) ويتسبب في تسرب الحمض النووي النووي والحمض النووي الميتوكوندري (mtDNA) إلى السيتوبلازم (الشكل 7د)، وهو ما يُعرف بأنه ينشط cGAS مباشرةً.. ومع ذلك، فإن تثبيط تلف الحمض النووي الناتج عن VC، كما هو مؤشر عليه بفوسفرة الهيستون 2AX ()، بواسطة مزيل جذور الأكسجين التفاعلية N-أسيتيل-إل-سيستئين
الشكل 7 | تحفيز TET2 وتسرب dsDNA بواسطة VC ينشطان مسار cGAS-cGAMP- STING البطاني الوعائي في الورم ويعززان حركة اللمفاويات. أ، ب مستويات mRNA لـ Cgas (أ) والبروتين (ب) في خلايا Hepal-6 المعالجة بـ VC (). ج مستوى بروتين Cgas في الورم المشتق من خلايا Hepal-6 بعد علاج VC أو Sora لمدة أسبوعين. د التلوين المناعي الفلوري المشترك لـ dsDNA وHSP60 وDAPI في خلايا Hepa16 المعالجة بـ VC. أشرطة المقياس،. مستوى mRNA لـ Cgas في خلايا Hepal-6 المعالجة بـ VC بعد المعالجة المسبقة بـ NAC ().مستوى mRNA لجين Cgas في خلايا Hepal-6 المعالجة بـ VC والخلايا shTet2 و shCtrl. مستوى mRNA لـ g Cgas في خلايا Hepal-6 المعالجة بـ VC بعد المعالجة المسبقة بـ TFMB-2HG (). ه مستويات cGAMP داخل الخلايا وخارجها من خلايا Hepal-6 المعالجة بـ VC (). تحليل التسلسل الناري وقياس حالة مثيلة المحفز لجين Cgas في خلايا Hepa1-6 المعالجة بـ VC (). تلوين المناعة المناعية الكمية لتجمعات Sting في خلايا SVEC4-10 مع زيادة تعبير Sting-Cherry المزرعة مع خلايا Hepa1-6 الموسومة بـ GFP بعد علاج VC (). تمثل الأسهم البيضاء تجمعات Sting في SVEC4-10 الخلايا. أشرطة المقياس، مستويات mRNA لـ Ifnو ISGs في خلايا SVEC4-10 بعد التعرض لـ CM من خلايا Hepal-6 sgCgas المعالجة بـ VC مقابل خلايا sgCtrl (k) أو خلايا Hepal-6 shTet2 مقابل خلايا shCtrl (l) (). م-و تصوير المجهر الإلكتروني النافذ لخلايا اللمف في الطحال للفأر عبر حاجز خلايا SVEC4-10 مع المعالجة المسبقة بمستخلص الخلايا من خلايا Hepal-6 (م)، خلايا Hepa1-6 sgCgas (ن)، أو خلايا Hepa1-6 shTet2 (ع) المعالجة بـ VC أو غير المعالجة ().تحليل التدفق الخلوي ومتوسط شدة الفلورة (MFI) لـ VE-Cad السطحي في خلايا SVEC4-10-GFP المتعايشة مع خلايا Hepal-6 بعد علاج VC ().، مستويات mRNA لـ VECad () والجينات المحددة () في خلايا SVEC4-10 بعد التعرض إلى CM من خلايا Hepa1-6 sgCgas المعالجة بـ VC مقابل خلايا sgCtrl () أو خلايا Hepal-6 shTet2 مقابل خلايا shCtrl () ().يتم حساب القيم باستخدام اختبار الطالب غير المرتبط ذو الطرفيناختبار (a، e-h، j، k-t) واختبار الطالب المزدوجاختبار (i). ns، غير معنوي. ممثل عنتجارب مستقلة (). يتم توفير بيانات المصدر كملف بيانات مصدر. (ن-أس-سي)، فشل في عكس التعبير المرتفع لـ Cgas بعد تحفيز VC (الشكل التكميلي 9أ والشكل 7هـ). تشير هذه النتائج مجتمعة إلى أنه على الرغم من أن VC الدوائي ينشط cGAS عبر تسرب dsDNA الناتج عن ROS، فإن التعبير عن cGAS الذي يسببه VC مستقل عن الإجهاد التأكسدي. كما حددنا ما إذا كان VC يعزز تعبير cGAS من خلال إزالة مثيلة الحمض النووي المعتمدة على TET2. أدى حجب Tet2 إما عن طريق تثبيط Tet2 أو مثبط إنزيم Tet (2-هيدروكسيغلوتارات القابل لاختراق الخلايا (2-HG)) إلى انخفاض كبير في تعبير Cgas الناتج عن VC في خلايا سرطان الكبد (الشكل 7و، ز). وبما يتوافق مع زيادة تعبير Tet2 (الشكل 60)، قلل علاج VC بشكل واضح من مستوى مثيلة محفز Cgas (الشكل 7ط). مجتمعة، تشير هذه النتائج إلى أن VC الدوائي لا ينشط فقط cGAS الورمي من خلال تسرب dsDNA في السيتوبلازم، بل يزيد أيضًا من تعبير cGAS الورمي عبر التعديلات اللاجينية بواسطة إزالة مثيلة الحمض النووي المعتمدة على Tet2.
لتقييم ما إذا كان cGAS الورمي المحفز بواسطة VC ينشط STING في الخلايا البطانية ويعزز حركة الخلايا اللمفاوية، تم التأكد أولاً من زيادة واضحة في إنتاج وإفراز cGAMP في خلايا سرطان الكبد بعد علاج VC (الشكل 7ه). أدى علاج VC إلى تكوين تجمعات STING حول النواة بشكل أكثر وضوحًا، والتي تُعد مؤشراً على تنشيط STING بواسطة cGAMP.“، في خلايا البطانة المعبر عنها بـ Sting-Cherry عند التعايش المشترك مع خلايا Hepa16 الموسومة بـ GFP (الشكل 7j)، في حين أن علاج خلايا البطانة بـ VC وحدها لم يحفز تكوين المزيد من تجمعات Sting ولا رفع تعبير Cgas (الشكل التكميلي 10a-c)، مما يشير إلى أن VC ينشط Sting في خلايا البطانة بشكل غير مباشر من خلال الخلايا السرطانية. وبالمثل، فإن الوسط المستمد من خلايا Hepal-6 المعالجة بـ VC نشط بشكل كبير Sting في خلايا البطانة، كما يتضح من زيادة فسفرة Sting وTbk1، بالإضافة إلى زيادة Ifnوتعبيرات ISGs (الشكل التكميلي 9ج، د)، في حين أن علاج الخلايا البطانية بـ VC وحده لم يُظهر فرقًا واضحًا في تنشيط Sting البطاني (الشكل التكميلي 10د، هـ). والأهم من ذلك، عندما تم حجب تعبير Cgas أو Tet2 (الشكل التكميلي 9هـ)، فشلت CM المشتقة من هذه الخلايا بعد تحفيزها بـ VC في تنشيط STING البطاني (الشكل التكميلي 9و، ز والشكل 7ك، ل). علاوة على ذلك، حددنا أن المزيد من الخلايا اللمفاوية عبرت الحواجز الخلوية البطانية المحفزة بواسطة CM من خلايا Hepa1-6 المعالجة بـ VC مقارنة بالخلايا المعالجة بـ PBS (الشكل 7م). بعد التعرض لـ CM من خلايا Hepa1-6 المعالجة بـ VC والتي تعاني من نقص في Cgas أو Tet2، لم تعزز الحواجز الخلوية البطانية هجرة الخلايا اللمفاوية عبر البطانية (الشكل 7ن، و). لذلك، تشير هذه النتائج إلى أن TET2 المحفز بواسطة VC يرفع من تعبير cGAS في الورم لتنشيط مسار STING في الخلايا البطانية ويعزز حركة الخلايا اللمفاوية.
من الناحية الميكانيكية، بعد التعرض لـ VC، أظهرت الخلايا البطانية في الثقافة المختلطة مع خلايا Hepal-6 زيادة في تعبير VE-Cad، وهو علامة على استقرار وصلات الخلايا البطانية، في حين أن علاج الخلايا البطانية وحدها بـ VC لم يكن له تأثير ملحوظ على تعبير VE-Cad الخاص بها (الشكل التكميلي 10f). وبالمثل، أدى الوسط المستمد من خلايا Hepa1-6 المعالجة بـ VC إلى زيادة تعبير VE-Cad وجزيئات الالتصاق المرتبطة بتفاعل الخلايا البطانية مع الخلايا اللمفاوية في الخلايا البطانية مقارنة بالوسط المستمد من خلايا Hepa1-6 المعالجة بـ PBS (الشكل 7p و)
(الشكل التكميلي 9h، i)، في حين لم يُلاحظ أي تغيير في الجينات المرتبطة بتفاعل الخلايا البطانية مع اللمفاويات في الخلايا البطانية بعد العلاج المباشر بـ VC (الشكل التكميلي 10g). على النقيض من ذلك، أدى تعطيل إما Cgas أو Tet2 إلى إلغاء زيادة تعبير VE-Cad وجزيئات الالتصاق المرتبطة بتفاعل الخلايا البطانية مع اللمفاويات في الخلايا البطانية التي يحفزها الوسط المستمد من خلايا Hepal-6 المعالجة بـ VC (الشكل 7q-t). معًا، تشير هذه البيانات إلى أن VC يحفز تنشيط TET2 لزيادة تعبير cGAS في الورم، مما يؤدي إلى إنتاج cGAMP لتنشيط STING في الخلايا البطانية وتعزيز هجرة اللمفاويات عبر البطانة من خلال الحفاظ على استقرار وصلات الخلايا البطانية وزيادة تعبير جزيئات الالتصاق المرتبطة بتفاعل الخلايا البطانية مع اللمفاويات.
محور الورم TET2-p-STAT5A-cGAS-LRRC8C-بطانة الأوعية الدموية STING يرتبط بتطبيع الأوعية الدموية وتسلل المناعة في سرطان الكبد البشري
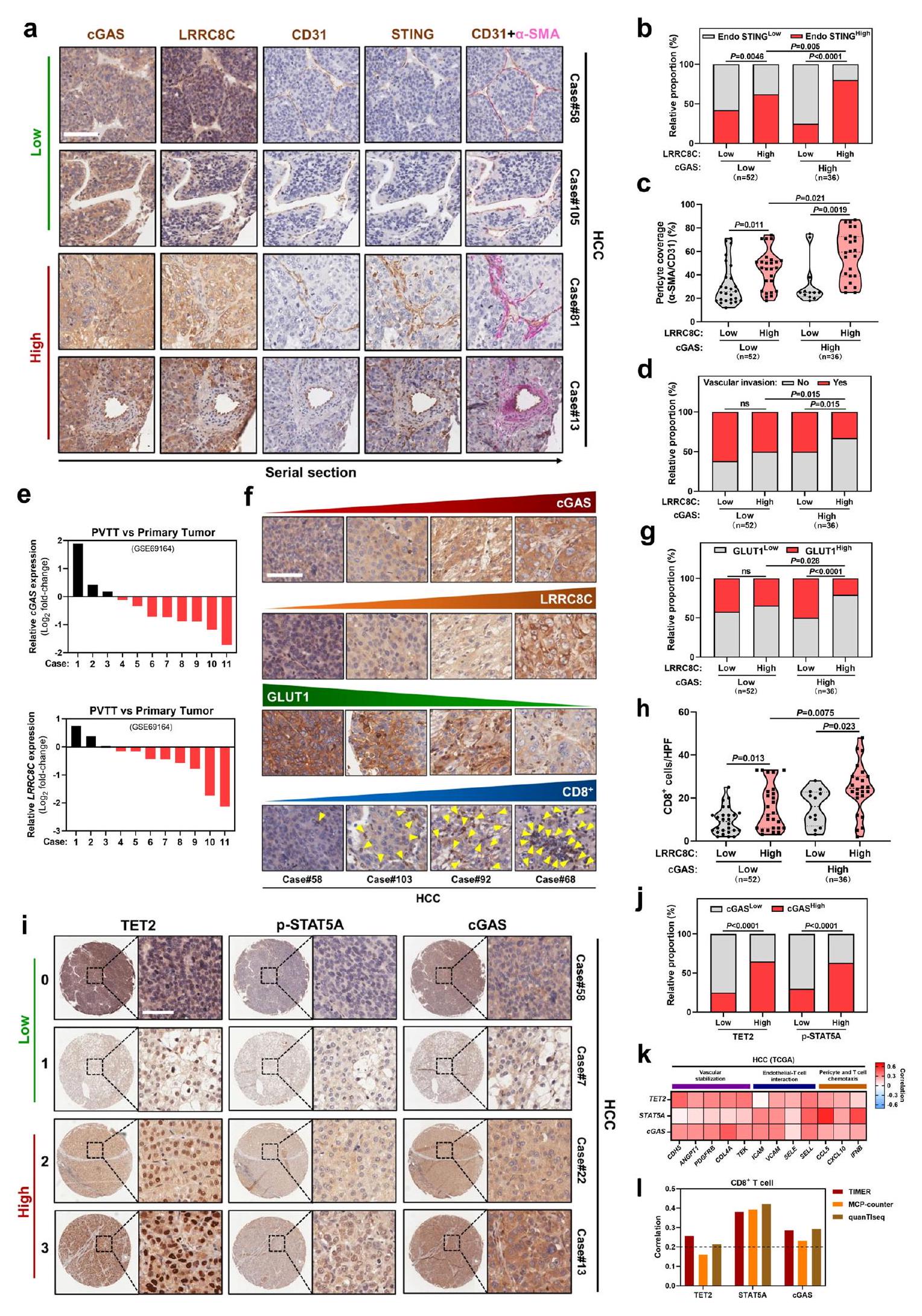
بعد ذلك، قمنا بدراسة مستويات تعبير البروتين لمحور الورم TET2-p-STAT5A-cGAS-LRRC8C-البطانة STING في خزعات الأورام من مرضى سرطان الكبد. في مقاطع متسلسلة من عينات سرطان الكبد من نفس المريض، كان التعبير العالي لـ LRRC8C في الورم مرتبطًا بالتعبير العالي لـ STING في CD31الخلايا البطانية الوعائية في مجموعة تعبير cGAS الورمي العالي أو المنخفض (الشكل 8أ، ب). من المهم، عندما كان تعبير LRRC8C الورمي مرتفعًا، كان تعبير STING البطاني بشكل ملحوظ أعلى في مجموعة تعبير cGAS الورمي العالي مقارنة بمجموعة تعبير cGAS الورمي المنخفض (الشكل 8أ، ب)، مما يشير إلى أن تعبير cGAS الورمي يرتبط بتنشيط STING البطاني اعتمادًا على LRRC8C.
بالمقارنة مع الأورام التي تعبر عن مستويات منخفضة من cGAS أو LRRC8C،تغطية الخلايا المحيطة للأوعية الدمويةالأوعية الدموية للأورام، والتي تُعد واحدة من علامات تطبيع أوعية الأورام، زاد بشكل كبير في الأورام التي تعبر عن كل من cGAS وLRRC8C بمستويات عالية (الشكل 8أ، ج). كان تعبير LRRC8C في الورم مرتبطًا سلبًا بالغزو الوعائي عندما كان تعبير cGAS عاليًا، في حين لم يظهر أي ارتباط واضح في مجموعة التعبير المنخفض لـ cGAS (الشكل 8أ، د). وبشكل متسق، تم تقليل تعبير cGAS وLRRC8C في ثمانية من 11 حالة من تجلط الورم في الوريد البابي (PVTT) مقارنة بالأورام الأولية المقابلة (الشكل 8هـ). علاوة على ذلك، كان التعبير العالي لـ LRRC8C في الورم مرتبطًا بتعبير منخفض لمؤشر نقص الأكسجة GLUT1 في مجموعة التعبير العالي لـ cGAS، ولكن ليس عندما كان تعبير cGAS منخفضًا في أنسجة سرطان الكبد (الشكل 8و، ز). لتقييم المزيد من التسلل المناعي، كان تعبير LRRC8C في الورم مرتبطًا إيجابيًا مع داخل الورم تسلل الخلايا، خاصة عندما كان تعبير cGAS في الورم مرتفعًا (الشكل 8f، h). معًا، تشير هذه النتائج إلى أن محور cGAS-LRRC8C-ستينغ البطاني في الورم مرتبط بتطبيع الأوعية الدموية وتسلل المناعة في سرطان الكبد البشري.
قمنا بمزيد من التحقق من تنظيم cGAS الورمي بواسطة إشارات TET2 وSTAT5A في عينات سرطان الكبد البشري ووجدنا أن التعبيرات العالية لـ TET2 الورمي وp-STAT5A كانت مرتبطة بتعبير عالي لـ cGAS الورمي (الشكل 8i، j). في قاعدة بيانات TCGA، كان تعبير TET2 الورمي وSTAT5A وcGAS مرتبطًا إيجابيًا مع الأوعية الدموية الجينات المرتبطة بالتطبيع بما في ذلك استقرار الأوعية الدموية، التفاعل بين الخلايا البطانية واللمفاوية، الخلايا المحيطة بالأوعية الكيميائية وتحرك الخلايا التائية (الشكل 8ك). بالإضافة إلى ذلك، وُجد ارتباط إيجابي بين تعبير TET2 وSTAT5A وcGAS في الورم وبين داخل الورمتسلل الخلايا التائية (الشكل 81). علاوة على ذلك، مرضى سرطان الكبد الكبدي (HCC) الذين لديهم مستويات عالية من TET2 في الورم، كان تعبير p-STAT5A وcGAS وLRRC8C وSTING البطاني مرتبطًا بنتائج سريرية أفضل (الشكل التكميلي 11a-e). مجتمعة، تُظهر هذه البيانات أن محور الورم TET2-p-STAT5A-cGAS-LRRC8C-STING البطاني مرتبط بتطبيع الأوعية الدموية وتسلل المناعة في سرطان الكبد البشري.
الشكل 8 | محور الورم TET2-p-STAT5A-cGAS-LRRC8C-الإندوثيلي STING يرتبط بتطبيع الأوعية الدموية وتسلل المناعة في سرطان الكبد البشري. صور مناعية نسيجية تمثيلية (أ) وتحليل الارتباط بين تعبيرات cGAS وLRRC8C في الورم مع تعبير STING في الخلايا البطانية (ب)، الخلايا المحيطة بالأوعية () تغطية الأوعية الدموية (CD31) (ج)، وغزو الأوعية الدموية (د) في مقاطع متسلسلة من عينات سرطان الكبد الخلوي من نفس المريض (). بناءً على شدة التلوين (تعبير البروتين)، تم تقسيم المرضى إلى مجموعتين: مجموعة التعبير المنخفض (درجة التلوين 0-1) ومجموعة التعبير العالي (درجة التلوين 2-3). أشرطة المقياس،. هـ و مستويات mRNA بين PVTT وعينات HCC الأولية المقابلة (أزواج) (GEO: GSE69164). صور مناعية نسيجية تمثيلية (و) وتحليل الارتباط بين تعبيرات cGAS الورمية وLRRC8C مع تعبير GLUT1 (ز) أوتسلل الخلايا التائية (h) في عينات سرطان الكبد البشري ( ). الخط المنقط عبر مخططات الكمان يمثل الربيعات والخط الكامل يوضح الوسيط. أشرطة المقياس،. صور مناعية نسيجية تمثيلية (i) وتحليل الارتباط (j) لتعبيرات TET2 وp-STAT5A في الورم مع تعبير cGAS في عينات سرطان الكبد البشري (). أشرطة المقياس،. معامل ارتباط بيرسون لتعبيرات TET2 وSTAT5A وcGAS مع الجينات المرتبطة بتثبيت الأوعية الدموية، وتفاعل الخلايا البطانية مع الخلايا التائية، والكيموتاكيس للخلايا المحيطة/الخلايا التائية في سرطان الكبدة من مجموعة بيانات TCGA (). معامل ارتباط بيرسون لـ TET2 وSTAT5A وتعبيرات معتسلل الخلايا في سرطان الكبدة الخلوي من مجموعة بيانات TCGA () من خلال TIMER وMCP-counter وquanTIseq.يتم حساب القيم باستخدام اختبار كاي تربيع (b, d, g, j) وتحليل التباين الأحادي الاتجاه (c, h). ns، غير معنوي. يتم توفير بيانات المصدر كملف بيانات مصدر.
علاج VC يحفز تطبيع الأوعية الدموية ويعزز فعالية علاج مضاد PD-L1 أو العلاج المركب لمضاد PD-L1 مع IL-2
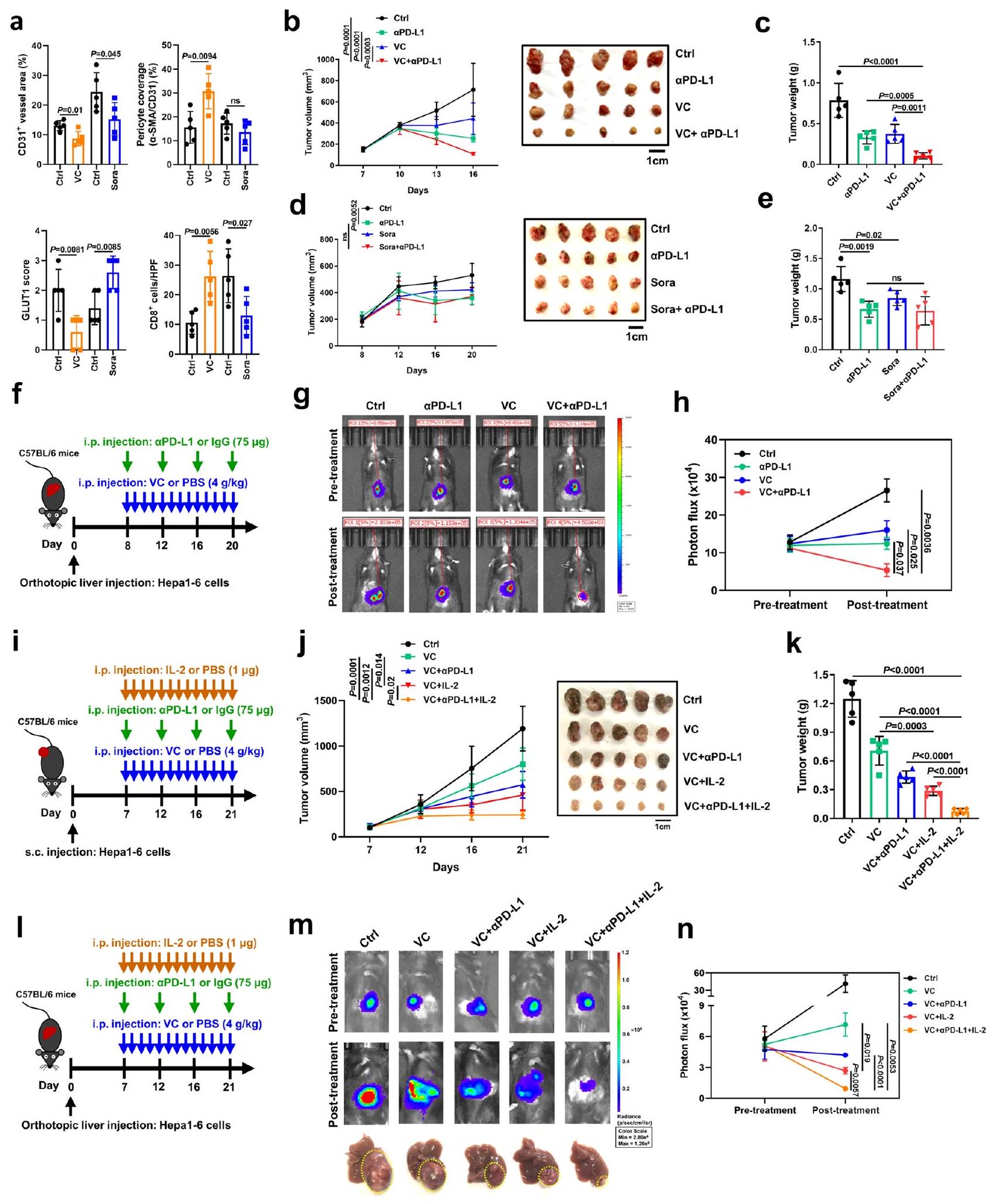
بعد ذلك، قمنا بالتحقيق في تأثير علاج فيتامين سي بجرعة عالية على الأوعية الدموية للأورام والتسلل المناعي في الجسم الحي. تم حقن فيتامين سي داخل الصفاقرأس المال المغامر (ما يعادلعن طريق الوريد)، جرعة دوائية تستخدم على نطاق واسع في نماذج الفئران وتظهر تأثيرات مثبطة ضد نمو أورام مختلفة بما في ذلك سرطان الكبد، تم إعطاؤه لفئران ذات جهاز مناعي سليم تحمل أورام Hepal-6 تحت الجلد (الشكل التكميلي 12أ، ب). وبشكل مشابه للتأثير الناتج عن تنشيط cGAS في الورم وSTING في العائل، أدى علاج VC إلى تقليل فيالأوعية الدموية الورمية ونقص الأكسجة داخل الورم، كما يتضح من انخفاض تعبير GLUT1، ولكن هناك زيادة واضحة فيتغطية الخلايا المحيطة بالأوعية لـالأوعية الدموية الورمية وداخل الورمتسلل خلايا T (الشكل 9أ، الشكل التكميلي 12هـ). تماشيًا مع النتائج المختبرية، بعد تنشيط STING يتم إنتاج IFNوتم تنظيم جينات ISGs، وجينات تثبيت الأوعية الدموية، وجزيئات الالتصاق المرتبطة بتفاعل البطانة اللمفاوية للأوعية الدموية بشكل مرتفع في الأورام المعالجة بـ VC مقارنة بالأورام المعالجة بـ PBS (الشكل التكميلي 12f). وعلى النقيض من ذلك، على الرغم من أن علاج السورا، وهو الدواء الخط الأول لسرطان الكبد، قد أخر نمو الورم وقلل من كثافة أوعية الورم في الفئران، إلا أنه فشل في تطبيع أوعية الورم، كما يتضح من عدم وجود فرق في تغطية الخلايا المحيطة بأوعية الورم وزيادة نقص الأكسجة داخل الورم، مما أدى إلى انخفاض…تسلل الخلايا (الشكل 9أ، الشكل التكميلي 12ج-هـ). يتماشى هذا مع الملاحظات السابقة التي أشارت إلى أن سورا يعزز القمع المناعي الناتج عن نقص الأكسجة.لذلك، تشير هذه النتائج إلى أن علاج VC، وليس Sora، يحفز تطبيع الأوعية الدموية للورم ويحسن تسلل الجهاز المناعي في سرطان الكبد.
قمنا بتقييم مجموعة بيانات حديثة لسرطان الكبد الأولي (HCC) لمرضى خضعوا لعلاج مضاد لـ PD-1/PD-L1. ووجد أن استفاد المرضى الذين يعانون من تعبيرات عالية لجينات الاستجابة للإنترفيرون (ISGs) من علاج مضادات PD-1/PD-L1، في حين استجاب فقط 8-16% من المرضى الذين يعانون من تعبيرات منخفضة لجينات الاستجابة للإنترفيرون للعلاج بمضادات PD-1/PD-L1 (الشكل التكميلي 13أ)، مما يشير إلى أن تنشيط مسار cGAS-STING قد يحسن فعالية حجب نقاط التفتيش المناعية في سرطان الكبد. لاستبعاد أن تكون هذه الملاحظات ناتجة عن تأثير حجم الورم على فعالية علاج مضاد PD-L1، قمنا بضبط أعداد الخلايا التي تعبر عن Cgas واخترنا أورامًا ذات أحجام مشابهة لأورام مجموعة التحكم لعلاج مضاد PD-L1. كما هو متوقع، مقارنة بمجموعة التحكم، أدى علاج مضاد PD-L1 إلى أورام أصغر بكثير (الشكل التكميلي 13ب، ج)، تزامن ذلك مع زيادة في داخل الورمتسلل خلايا T السامة للخلايا في مجموعة فرط التعبير عن Cgas بأحجام مماثلة لأورام Ctrl قبل العلاج (الشكل التكميلي 13د). لذلك، تؤكد هذه النتائج الفعالية المعززة لعلاج مضاد PD-L1 بواسطة تعبير cGAS في سرطان الكبد بغض النظر عن حجم الورم.
نظرًا لأن فيتامين سي يحفز تنشيط cGAS في الأورام وتنشيط STING في الخلايا البطانية، كما هو موضح في الشكل 6، قمنا باختبار ما إذا كان علاج فيتامين سي يجعل سرطان الكبد أكثر حساسية للعلاج المناعي. في نماذج الأورام المزروعة تحت الجلد، لوحظ تراجع ملحوظ في الأورام لدى الفئران التي عولجت بمزيج من فيتامين سي ومضاد PD-L1 مقارنة بالعلاج الفردي بفيتامين سي أو مضاد PD-L1 (الشكل 9ب، ج)، مما يشير إلى أن فيتامين سي يعزز الاستجابات المضادة للأورام التي يسببها مثبط نقطة التفتيش PD-L1. على النقيض من ذلك، وبما يتوافق مع دوره في تثبيط المناعة، فإن سورا لم يُظهر العلاج أي نشاط مضاد للأورام إضافي عند دمجه مع جسم مضاد مضاد لـ PD-L1 (الشكل 9د، هـ)، مماثل للنتائج السابقةمن الجدير بالذكر أن العلاج المركب باستخدام VC ومضاد PD-L1 أظهر تحقيقًا ملحوظًا لكبت كامل للورم بعد حوالي 28 يومًا من فترة علاج استمرت 21 يومًا في الفئران (الشكل التكميلي 15أ). لذلك، تشير هذه النتائج إلى أن الجمع بين VC والعلاج بمضاد PD-L1 يحقق كبتًا كاملاً للورم بعد فترة علاج طويلة. وبما أن نماذج الأورام المزروعة تحت الجلد قد لا تمثل بشكل كامل البيئة المرضية الدقيقة، فقد استخدمنا أيضًا نماذج سرطان الكبد الأورثوتوبية، والتي تعتبر ممثلة أكثر موثوقية لسرطان الكبد البشري.. تسلل أقل لخلايا المناعة بما في ذلك تم العثور على خلايا T، وخلايا NK، والبلعميات في النماذج تحت الجلدية مقابل النماذج الأورثوتوبية (الشكل التكميلي 14a-d)، مما يشير إلى أن الفعالية العلاجية لمضاد PD-L1 قد تكون أقل في النماذج الأورثوتوبية. ومع ذلك، وبما يتماشى مع النتائج في نماذج الزرع تحت الجلد، أدى العلاج المشترك لـ VC ومضاد PD-L1 إلى أعباء أورام أصغر بشكل ملحوظ مقارنة بمجموعة التحكم بـ IgG المفردة، أو مجموعة VC، أو مجموعة مضاد PD-L1 في نماذج الأورام الأورثوتوبية (الشكل 9f-h).
استنادًا إلى التآزر بين TET2 وإشارة IL-2/STAT5A (الشكل 6)، قمنا بمزيد من الفحص لمعرفة ما إذا كان VC يعزز فعالية العلاج المركب بمضاد PD-L1 وIL-2، والذي يُعتبر مرشحًا واعدًا محتملًا لعلاج السرطان.من المثير للاهتمام، بالإضافة إلى التأثير المضاد للأورام المشترك لـ VC وanti-PD-L1، عزز علاج VC فعالية العلاج المركب لـ anti-PD-L1 مع IL-2 في كل من نماذج الأورام المزروعة تحت الجلد (الشكل 9i-k) ونماذج سرطان الكبد الأورثوتوبية (الشكل 91-n). ومن اللافت أننا لاحظنا أن هذا التأثير المعزز كان متقاربًا بين نماذج الأورام الأورثوتوبية وتحت الجلد. مجتمعة، يحفز علاج VC تطبيع الأوعية الدموية ويعزز فعالية علاج anti-PD-L1 أو العلاج المركب لـ anti-PD-L1 مع IL-2.
محور الورم TET2-STAT5A-cGAS-المضيف STING يتوسط تطبيع الأوعية الدموية الناتج عن VC والفعالية العلاجية لـ VC المدمج مع مضاد PD-L1
لتقييم ما إذا كان هذا التأثير المضاد للأورام التآزري يتم عبرتم علاج الفئران الحاملة للأورام بمركب VC وأجسام مضادة مضادة لـ PD-L1 في وجود إما IgG Ctrl أو أجسام مضادة مضادة لـ CD8 لاستنزافالخلايا (الشكل التوضيحي التكميلي 15ب). كما هو متوقع، تزامن التراجع المعزز للأورام بواسطة العلاج المشترك لـ VC و مضاد PD-L1 مع زيادة تسلل داخل الورمخلايا T السامة للخلايا في مجموعات IgG Ctrl (الشكل التكميلي 15ج-هـ). ومع ذلك، بعد استنفاد CD8، الذي تم تأكيده بواسطة قياس التدفق الخلوي (الشكل التكميلي 15و)، لم يكن هناك تحسن في الفائدة المضادة للأورام عند استخدام VC مع مضاد PD-L1 مقارنة بالعلاج الفردي بـ VC أو مضاد PD-L1 (الشكل التكميلي 15ج-هـ)، مما يشير إلى أن فعالية الجمع بين علاج VC ومضاد PD-L1 تعتمد علىاستجابة مناعية مضادة للأورام ناتجة عن الخلايا.
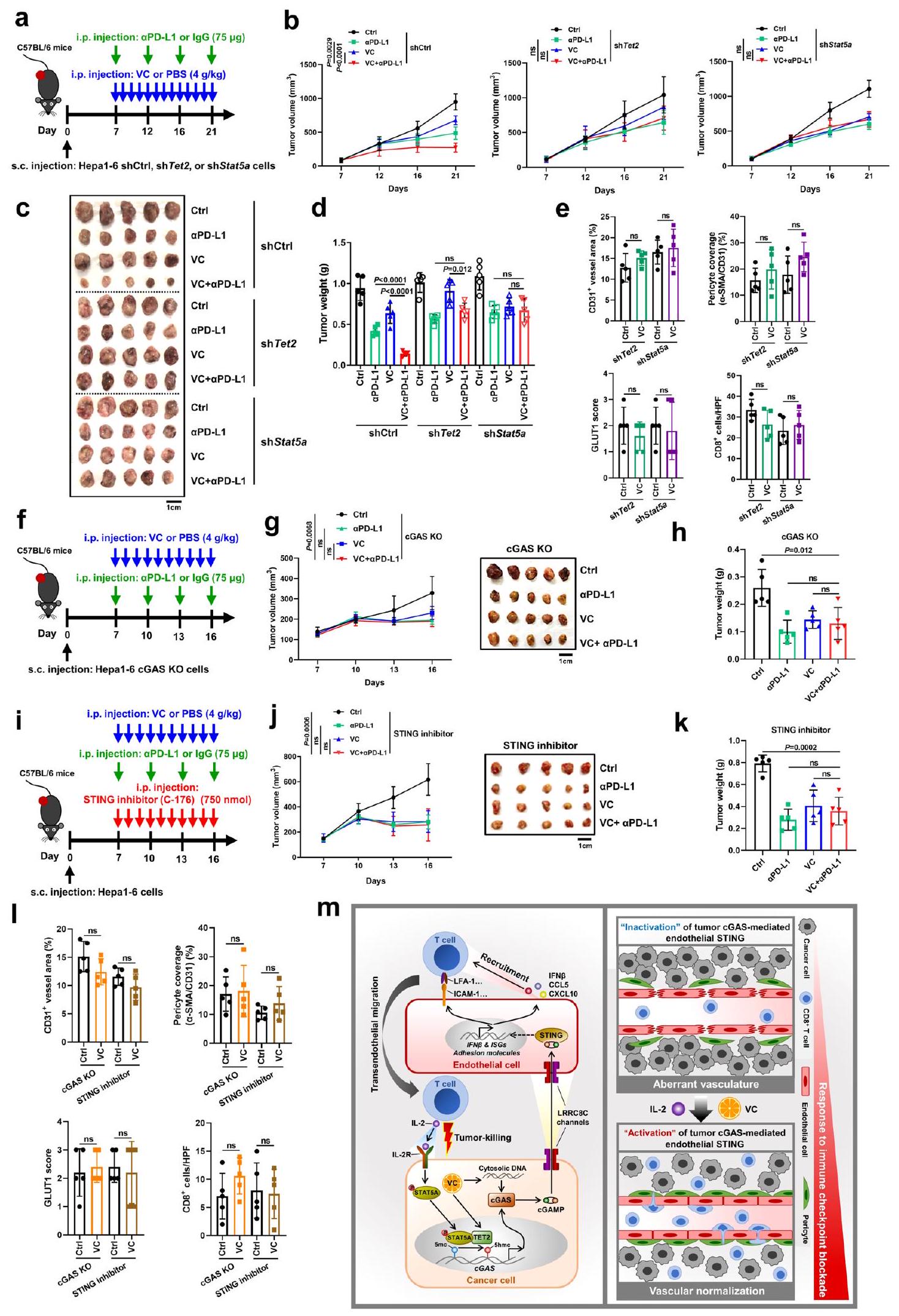
من المهم أن نلاحظ أنه في الأورام المشتقة من خلايا ناقصة Tet2 أو Stat5a، لم يُظهر الجمع بين VC و مضاد PD-L1 تغييرات واضحة في أعباء الأورام مقارنة بالعلاج الفردي بـ VC أو مضاد PD-L1 (الشكل 10 أ-د). علاوة على ذلك، لم يُظهر علاج VC تأثيرًا كبيرًا على تغطية الأوعية الدموية في الورم بواسطة الخلايا المحيطة بالأوعية، ومستويات نقص الأكسجة داخل الورم، و تسلل الخلايا في هذه الأورام مع نقص Tet2 أو Stat5a (الشكل 10e، الشكل التكميلي 12g). علاوة على ذلك، أدى تثبيط Tet2 أو Stat5a في الأورام إلى إلغاء الزيادة التي تسببها معالجة VC في IFN.وجينات ISGs، وجينات تثبيت الأوعية الدموية، وجزيئات الالتصاق المرتبطة بتفاعل البطانة اللمفاوية (الشكل التكميلي 12ه). معًا، توفر هذه النتائج دليلاً قاطعًا على الدور الحاسم لـ Tet2 و Stat5a في الورم في التأثيرات المعيارية للأوعية الدموية التي يسببها VC، وكفاءة الجمع بين علاج VC وعلاج مضاد PD-L1 في سرطان الكبد.
علاوة على ذلك، قمنا بإزالة cGAS في الورم واستخدمنا مثبط STING C-176 لمنع تنشيط STING في المضيف. نظرًا للمستوى المنخفض الأولي لتعبير Cgas في خلايا Hepal-6 الأصلية (الشكل التكميلي 1أ)، أظهر حذف Cgas (KO) في خلايا Hepal-6 زيادة طفيفة في نمو الورم والأعباء، والتي لم تصل إلى دلالة إحصائية (الشكل التكميلي 15ج، ح). ومع ذلك، أدى حذف Cgas في خلايا Hepal-6 إلى إلغاء كامل لزيادة تعبير Cgas التي يسببها علاج VC، وفي الأورام المشتقة من خلايا cGAS KO، لم يكن هناك تغييرات واضحة في الورم عند الجمع بين VC وanti-PD-L1.
الشكل 9 | علاج VC يحفز تطبيع الأوعية الدموية ويعزز فعالية علاج مضاد PD-L1 أو العلاج المركب بمضاد PD-L1 مع IL-2.
تحديد كمي لـ CD31كثافة الأوعية، الخلايا المحيطة بالأوعية () تغطية السفن (), منطقة نقص الأكسجين، والخلايا في الأورام المعالجة بـ VC أو Sora (). منحنيات نمو الورم () وأعباء الأورام () لفئران C57BL/6 التي تم حقنها تحت الجلد بخلايا Hepal-6 مع العلاج بـ PD-L1 و VC سواء بمفردهما أو معًا ((الفئران لكل مجموعة). منحنيات نمو الورم (د) وأعباء الورم (هـ) لفئران C57BL/6 التي تم حقنها تحت الجلد بخلايا Hepa1-6 مع علاج بـ PD-L1 وسورا إما بمفردهما أو معًا ((الفئران لكل مجموعة). مخطط يوضح الإجراء التجريبي (و)، صور ممثلة للبيولومينيسنس المعتمدة على اللوسيفيراز (ز) والكمية (ح) لفئران C57BL/6 المحقونة في الموقع مع خلايا Hepal-6 مع العلاج المسبق واللاحق لـ PD-L1 و VC سواء بمفردهما أو معًا ((الفئران لكل مجموعة). مخطط يوضح الإجراء التجريبي (i)، منحنيات نمو الورم (j)، وأعباء الورم (k) لفئران C57BL/6 التي تم حقنها تحت الجلد بخلايا Hepal-6 مع علاج بـ VC فقط أو مع العلاج المشترك معPD-L1، IL-2، أوPD-L1 + IL-2 (الفئران لكل مجموعة). مخطط يوضح الإجراء التجريبي (I)، صور ممثلة للتألق الحيوي المعتمد على اللوسيفيراز () والكمية () من فئران C57BL/6 المحقونة في الموقع بخلايا Hepal-6 مع العلاج المسبق واللاحق بـ VC وحده أو بالاشتراك معL1، IL-2، أوPD-L1 + IL-2 (فئران لكل مجموعة).يتم حساب القيم باستخدام اختبار الطالب غير المرتبط ذو الطرفيناختبار (أ)، تحليل التباين ثنائي الاتجاه (ب، د، ي) وتحليل التباين أحادي الاتجاه (ج، هـ، ح، ك، ن). غير معنوي. تم توفير بيانات المصدر كملف بيانات مصدر. الأعباء مقارنة بالعلاج باستخدام VC واحد أو مضاد PD-L1 (الشكل 10f-h). وبالمثل، أدى تثبيط STING في المضيف بواسطة علاج C-176 إلى إلغاء التأثير التآزري المضاد للورم لكل من VC وحجب PD-L1 في الجسم الحي (الشكل 10i-k). علاوة على ذلك، لم يكن لعلاج VC تأثير كبير على تغطية الأوعية الدموية للورم بواسطة الخلايا المحيطة بالأوعية، ونقص الأكسجة داخل الورم، وتسلل الخلايا بعد نقص cGAS في الورم وتثبيط STING في المضيف (الشكل 10I، الشكل التكميلي 12i). بالإضافة إلى ذلك، ألغى حذف cGAS في الورم أو تثبيط STING في المضيف الزيادة في التعبير عن IFN التي يسببها VC.وجينات ISGs، وجينات تثبيت الأوعية الدموية، وجزيئات الالتصاق المرتبطة بتفاعل البطانة اللمفاوية في الأورام (الشكل التكميلي 12j). معًا، تؤكد هذه النتائج الدور الحيوي لـ cGAS في الورم وتنشيط STING في المضيف في تأثيرات تطبيع الأوعية الدموية التي يسببها VC والكفاءة التوافقية للعلاج المشترك بين VC وanti-PD-L1 في سرطان الكبد. بشكل عام، توضح نتائجنا أن محور TET2-STAT5A-cGAS في الورم وSTING في المضيف يتوسط تطبيع الأوعية الدموية الناتج عن VC والفعالية العلاجية لـ VC عند دمجه مع anti-PD-L1.
مناقشة
مسار cGAS-STING ضروري للمناعة المضادة للأورام. بالإضافة إلى التأثير المناعي المضاد للأورام الذي تم دراسته جيدًا لمسار cGAS-STING في الخلايا المناعية، لقد تبين مؤخرًا أن تنشيط مسار cGAS-STING الداخلي في خلايا السرطان يحدد أيضًا قابليتها للمناعة ويجعل الأورام نشطة مناعياً. ومع ذلك، لا يزال التداخل في مسار cGAS-STING بين خلايا الورم وخلايا المضيف في بيئة الورم غير واضح إلى حد كبير. من جهة، يوضح هذا العمل أن التعبير الداخلي لخلايا السرطان عن cGAS، وليس STING، يحدد تطبيع الأوعية الدموية للورم والاستجابة المناعية المضادة للورم بطريقة تعتمد على STING في المضيف. من جهة أخرى، فإن cGAS في المضيف غير ضروري، لكن cGAS في الورم ضروري لتطبيع الأوعية الدموية المناعي المضاد للورم الذي يتوسطه STING في المضيف. معًا، نحدد الاعتماد المتبادل بين cGAS في الورم وSTING في المضيف في تنظيم إعادة تشكيل الأوعية الدموية والمناعة المضادة للورم في سرطان الكبد. من الناحية الميكانيكية، ينتج cGAS في الورم جزيء cGAMP، الذي ينتقل عبر قنوات LRRC8C لتنشيط STING في الخلايا البطانية، مما يعزز تجنيد وهجرة الخلايا اللمفاوية عبر البطانة. بدوره، تفرز الخلايا اللمفاوية المتسللة داخل الورم IL-2 لتنشيط إشارة STAT5A في الورم، والتي تتعاون مع TET2 لزيادة التعبير الجيني لـ cGAS في الورم بطريقة إبجينية، مما يشير إلى حلقة تغذية راجعة إيجابية. وبناءً عليه، قد يؤدي تحفيز TET2 في الورم بواسطة VC لتسريع هذه الحلقة الإيجابية إلى تحفيز تطبيع الأوعية الدموية للورم وتعزيز فعالية العلاج المناعي.
حاليًا، تم الإبلاغ عن أن تنشيط STING داخل الورم باستخدام منشطات STING يعمل على تطبيع الأوعية الدموية للورم. ومع ذلك، لا يزال من غير المحدد ما إذا كان STING الورمي أو STING المضيف يلعب الدور السائد. باستخدام فئران ناقصة STING، نحدد أن STING المضيف، وليس STING الجوهري في خلايا السرطان، هو الذي يحدد تطبيع الأوعية الدموية والاستجابة المناعية المضادة للأورام في سرطان الكبد عالي التكوّن الوعائي. يمكن تفسير هذه الظاهرة بعدة أسباب. أولاً، بالإضافة إلى تجنيد وتنشيط خلايا T والخلايا المحيطة بالأوعية الدموية بواسطة جينات الهدف في مسار STING IFNومثل ISGs مثل CCL5 وCXCL10، يلعب مسار STING في الخلايا البطانية دورًا حيويًا في تنظيم حركة الخلايا التائية ووظيفة الخلايا البطانية. على سبيل المثال، تم إظهار أن عامل النسخ النشط downstream لـ STING وهو IRF3 يرتبط إلى محفز ICAM-1 وتحفيز تعبير ICAM-1، وهو أحد جزيئات الالتصاق الرئيسية لهجرة الخلايا التائية عبر البطانة، في الخلايا البطانية. وبالمثل، نجد أن حجب IFNمسار الإشارة بواسطة مثبط تنشيط STAT1 يفشل في عكس تعبير ICAM-1 المحفز بواسطة cGAMP وIFNالعلاج له تأثير ضئيل على تعبير ICAM-1 في الخلايا البطانية، مما يشير إلى أن تنظيم تعبير ICAM-1 البطاني يتم بشكل رئيسي عبر مسار STING، بدلاً من جيناته المستهدفة IFNو ISGs. بالإضافة إلى ذلك، كشفت الدراسات الحديثة أنه عندما يتم تنشيط مسار STING في الخلايا البطانية، يقوم الكيناز التالي TBK1 بتثبيط تكاثر الخلايا البطانية عبر قمع مسار YAP.لذلك، على الرغم من أن تنشيط STING في خلايا السرطان قد يعزز أيضًا IFNوإنتاج جينات الاستجابة للإنترفيرون (ISGs) لتجنيد خلايا T، فإنه لا يغير تعبير جزيئات الالتصاق في الخلايا البطانية أو الهجرة عبر البطانة لخلايا T. علاوة على ذلك، بما أن الخلايا البطانية قد ثبت أنها المصدر الرئيسي للإنترفيرونات من النوع الأول في الأورام النامية، والإنترفيرون المشتق من الخلايا البطانيةيبدأالمناعة المضادة للأورام المعتمدة على الخلايا، مساهمة الإنترفيرون التي تنتجها خلايا السرطان لتحفيز خلايا T قد تكون ضئيلة. بشكل عام، يعتبر STING في الخلايا البطانية أفضل من STING في الورم في المناعة المضادة للورم، حيث أن تفعيل STING في الخلايا البطانية لا يعزز فقط تجنيد وتنشيط خلايا T، بل يعزز أيضًا تطبيع الأوعية الدموية للورم من أجلتجوال الخلايا.
يتم تنشيط STING بشكل كلاسيكي بواسطة cGAMP المنتج من cGAS ومن المعروف أن cGAMP ينتقل بين الخلايا.لذلك، قد يتم التحكم في تنشيط STING في المضيف وتأثيره على تطبيع الأوعية الدموية بواسطة cGAS في الورم أو cGAS في المضيف. ومن المدهش أننا نجد أن تطبيع الأوعية الدموية الذي يتم بوساطة STING في المضيف والاستجابة المناعية المضادة للورم تعتمد على cGAS في الورم، وليس على cGAS في المضيف. ومن المقبول على نطاق واسع أن خلايا السرطان ليست فقط المكون الرئيسي للورم، بل غالبًا ما تكون غنية بشكل مستمر بالحمض النووي مزدوج الشريط في السيتوبلازم، والذي يزداد أكثر عند العلاجات التي تسبب تلف الحمض النووي مثل العلاج الكيميائي لتنشيط cGAS مباشرة لإنتاج cGAMP.لذلك، تعتبر خلايا السرطان المصدر الرئيسي لـ cGAMP في بيئة الورم الدقيقة، خاصة عندما يتم تنظيم تعبيرات cGAS لديها بواسطة TET2 في هذه الدراسة. علاوة على ذلك، نُظهر أن معظم خلايا سرطان الكبد تعبر عن cGAS، لكنها بالكاد تعبر عن STING، في عينات سرطان الكبدة الخلوي، وهو ما يتوافق مع تقارير سابقة أظهرت فقدان STING الداخلي في خلايا السرطان في الميلانوما وسرطان القولون والمستقيم.. على النقيض من ذلك، تظهر الخلايا المضيفة، وخاصة خلايا البطانية، في بيئة الورم الدقيقة تعبيرًا مميزًا لبروتين STING. نظرًا لأن cGAMP معروف بانتقاله بين الخلايا، cGAMP الذي ينتجه cGAS في خلايا السرطان، والتي غالبًا ما تفتقر إلى STING الداخلي، يكون عرضة للطرح في البيئة الدقيقة للورم، مما يؤدي إلى تنشيط STING في العائل. قد تفسر هذه الأسباب لماذا يكون cGAS في الورم ضروريًا لتنشيط STING في العائل وبالتالي لتطبيع الأوعية الدموية الذي يتم بوساطة STING في العائل والاستجابة المناعية المضادة للورم، في حين أن cGAS في خلايا العائل غير ضروري. يعمل إنزيم cGAS كمستشعر أساسي للحمض النووي، حيث يكتشف الحمض النووي مزدوج الشريط في السيتوبلازم وينشط الاستجابة المناعية المضادة للأورام.. ومع ذلك، لا يزال آلية تنظيم تعبير ونشاط cGAS غير مفهومة بالكامل. يرتبط فرط مثيلة محفز جين cGAS بانخفاض تنظيمه في سرطان الكبد، لكن آلية التنظيم المعتمدة على المثيلة غير واضحة. العمل الحالي يحدد ميثيل السيتوزين TET2 إنزيم الديوكسجيناز كإنزيم مسؤول عن إزالة مثيلة cGAS ونكشف أن STAT5A المنشط يجند ارتباط TET2 بموقع cGAS لتعزيز إزالة المثيلة ونسخ cGAS، مما يؤدي إلى زيادة تعبير cGAS في خلايا سرطان الكبد. كما هو متوقع، نثبت أيضًا أن علاج VC كمحفز لإنزيمات TETs يزيد من تعبير cGAS عبر إزالة المثيلة المعتمدة على TET2 في خلايا سرطان الكبد.
قد يكون تعبير cGAS الذي يتم بوساطة TET2 خاصًا بخلايا السرطان، حيث لا يؤثر التعرض لـ VC بشكل واضح على تعبير cGAS في الخلايا البطانية. بالإضافة إلى إزالة الميثيل التي يتم بوساطتها بواسطة TET2، هناك حاجة إلى مزيد من التحقيق لتوضيح ما إذا كانت عائلة DNMT تلعب دورًا رئيسيًا في التأثير على حالة مثيلة الحمض النووي لمروج cGAS في نموذجنا.
الشكل 10 | محور الورم TET2-STAT5A-cGAS-المضيف STING يتوسط التماثل الوعائي الناتج عن VC والفعالية العلاجية لـ VC المدمج مع مضاد PD- L1. مخطط يوضح الإجراء التجريبي (أ)، منحنيات نمو الورم (ب)، صور الأورام (ج)، وأعباء الأورم (د) لفئران C57BL/6 التي تم حقنها تحت الجلد بخلايا Hepa1-6-shTet2، خلايا shStat5a، أو خلايا shCtrl مع العلاج بـ PD-L1 و VC إما بمفردهما أو معًا (). التكميم لـكثافة الأوعية، الخلايا المحيطة بالأوعية () تغطية السفن ()، منطقة نقص الأكسجة، وتسلل الخلايا في أورام shTet2 و shStat5a بعد علاج VC (). مخطط يوضح الإجراء التجريبي (و)، منحنيات نمو الورم (ز)، وأعباء الورم (ح) لفئران C57BL/6 التي تم حقنها تحت الجلد بخلايا Hepa1-6-sgCgas (حذف cGAS) مع العلاج بـPD-L1 و VC سواء بمفردهما أو معًا. مخطط يوضح الإجراء التجريبي (i)، منحنيات نمو الورم (j)، وأعباء الورم (k) لفئران C57BL/6 التي تم حقنها تحت الجلد بخلايا Hepal-6 مع علاج PD-L1 و VC إما بمفردهما أو معًا بعد علاج مثبط STING C-176 (). 1 التكميم لـكثافة الأوعية، الخلايا المحيطة بالأوعية ( ) تغطية السفن ()، GLUT1 منطقة نقص الأكسجين، وتسلل الخلايا في الأورام المعالجة بـ cGAS KO ومثبط STING بعد علاج VC (). رسم تخطيطي يوضح تحفيز cGAS في الورم بواسطة TET2 الذي يؤدي إلى تنشيط STING في الخلايا البطانية لتنظيم إعادة تشكيل الأوعية الدموية والمناعة المضادة للورم. اليسار: ينتج cGAS في الورم جزيء cGAMP، الذي ينتقل عبر قنوات LRRC8C لتنشيط STING في الخلايا البطانية، مما يعزز تجنيد وهجرة الخلايا اللمفاوية عبر البطانة. بدوره، تفرز الخلايا اللمفاوية المتسللة داخل الورم IL2 لتنشيط إشارة STAT5A في الورم، والتي تتعاون مع TET2 لزيادة التعبير الجيني لـ cGAS في الورم بطريقة إبجينية، مما يشير إلى حلقة تغذية راجعة إيجابية. اليمين: تحفيز TET2 في الورم بواسطة VC لتسريع هذه الحلقة الإيجابية قد يؤدي بفعالية إلى تطبيع الأوعية الدموية في الورم وتعزيز فعالية العلاج المناعي.يتم حساب القيم باستخدام تحليل التباين ثنائي الاتجاه (b، g، j)، تحليل التباين أحادي الاتجاه ()، واختبار الطالب غير المرتبط ذو الطرفيناختبار (). غير مهم. تم توفير بيانات المصدر كملف بيانات مصدر.
تشير الأدلة الناشئة إلى أن إنزيمات TET ميثيلسايتوسين ديوكسجيناز تنظم مناعة الأورام.لقد أثبتنا أنالأيض يحفز الإنترفيرونتعبير نقطة تفتيش PD-L1 المحفز بواسطة TET1 ويعزز التهرب المناعي للورم. بالإضافة إلى تحفيز PD-L1، أُثبت أن TET2 يرفع من تنظيم الكيموكينات ويزيد من الخلايا اللمفاوية المخترقة للأورام. لقد ثبت أن TETs الورمية تنظم بشكل مباشر التفاعل بين خلايا الورم والخلايا المناعية عبر نقاط التفتيش المناعية والكيموكينات حتى الآن. ومع ذلك، فإن الأوعية الدموية غير الطبيعية والغير وظيفية بشكل كبير في الأورام تشكل حاجزًا حاسمًا أمام تسلل الخلايا المناعية إلى نسيج الورم، مما يحد من الاستجابات المناعية المضادة للورم.لذلك، لا يزال دور إنزيمات TET في الأوعية الدموية للأورام بحاجة إلى التحليل. تُظهر الدراسة الحالية أن TET2 يرفع من تعبير cGAS في الورم لإنتاج cGAMP، الذي ينشط بدوره مسار STING في الخلايا البطانية، مما يؤدي إلى تطبيع الأوعية الدموية للورم وتعزيز التغلغل داخل الورم.تسلل الخلايا التائية. تكشف نتائجنا عن دور مثبط الورم TET2 في المناعة المضادة للأورام من خلال تطبيع الأوعية الدموية الوسيط بواسطة cGAS-STING.
لقد ظهر فيتامين سي الدوائي مؤخرًا كعامل واعد مضاد للسرطان بسمية منخفضة وتكلفة مالية منخفضة وفقًا لمختلف التجارب قبل السريرية والسريرية.من المعروف أن فيتامين سي يمارس نشاطًا مضادًا للأورام من خلال الإجهاد التأكسدي الناتج عن بيروكسيد الهيدروجين لقتل الخلايا السرطانية بشكل تفضيلي عند استخدامه بجرعات عالية أو من خلال التحكم في نشاط إنزيمات TET لاستعادة نمط مثيلة الحمض النووي غير الطبيعي الذي يُلاحظ في العديد من الأورام.حتى الآن، أظهرت سلسلة من التجارب السريرية من المرحلة الأولى والثانية سلامة وفعالية فيتامين سي بجرعات عالية عن طريق الوريد كعلاج وحيد أو بالاشتراك مع الإشعاع والعلاجات الكيميائية التقليدية لمختلف أنواع السرطان.لقد أظهرنا سابقًا أن فيتامين سي الدوائي يقضي بشكل تفضيلي على خلايا جذور سرطان الكبد، وأن استخدام فيتامين سي عن طريق الوريد مرتبط بتحسن بقاء مرضى سرطان الكبد.. على الرغم من أن فيتامين سي بجرعات عالية قد تم الإبلاغ عنه حالياً لتعزيز العلاج المناعي، ومع ذلك، لم تُدرس التأثيرات المناعية الدوائية لفيتامين سي بشكل كامل. في هذا العمل، نقدم دليلاً على أن تحفيز نشاط TET2 بواسطة فيتامين سي، إلى جانب تسرب الحمض النووي المزدوج الشريطة الناتج عن فيتامين سي، ينشط مسار cGAS-cGAMP- STING البطاني الوعائي في الورم ويعزز حركة اللمفاويات. هناك آليات مختلفة تحفز تصدير شظايا الحمض النووي التالفة المشتقة من النواة و/أو الميتوكوندريا، مثل تكوين نواة دقيقة غير طبيعية، تدهور الميتوكوندريا وانخفاض جهد الغشاء، والالتهام الذاتي. هناك حاجة إلى دراسات إضافية لتوضيح الآليات التي من خلالها يحفز علاج VC تصدير شظايا الحمض النووي التالفة المشتقة من النواة و/أو الميتوكوندريا، وللتأكد من خصوصية هذا التعديل تجاه الخلايا السرطانية. ومن المهم أننا نكشف أيضًا أن علاج VC يحفز تطبيع الأوعية الدموية للورم ويعزز فعالية علاج مضاد PD-L1 أو العلاج المركب مضاد PD-L1 مع IL-2 في الجسم الحي، مما يوفر استراتيجيات واعدة لتعزيز العلاج المناعي المركب لسرطان الكبد.
باختصار، توضح نتائجنا وجود تواصل متبادل بين الخلايا الورمية وخلايا البطانة الوعائية في البيئة الدقيقة للورم عبر cGAS و
يعد STING ضروريًا لتطبيع الأوعية الدموية للأورام والمناعة المضادة للأورام (الشكل 10m). استنادًا إلى التنظيم اللاجيني لـ cGAS في الورم بواسطة TET2 بالتعاون مع إشارة STAT5A، يؤدي تحفيز نشاط TET2 بواسطة VC إلى تنشيط مسار cGAS-cGAMP-STING البطاني الوعائي في الورم ويحفز تطبيع الأوعية الدموية للورم، مما يعزز فعالية العلاج المناعي. لا يكشف هذا العمل فقط عن آلية التنظيم اللاجيني لـ cGAS في الورم، بل يكشف أيضًا عن الأدوار المناعية التنظيمية لـ VC وTET2 في سرطان الكبد، مما يوفر استراتيجية لتحفيز تطبيع الأوعية الدموية للورم وأساسًا علميًا لإجراء تجارب سريرية مستقبلية تجمع بين VC والعلاج المناعي.
الطرق
الموافقة على الدراسة
تمت الموافقة على جميع بروتوكولات الحيوانات المستخدمة في هذه الدراسة من قبل لجنة الجامعة لاستخدام ورعاية الحيوانات في الجامعة الطبية العسكرية الثانية. تم الحصول على أنسجة الأورام من مرضى سرطان الكبد الكبدي (HCC) من مستشفى جراحة الكبد والقنوات الصفراوية الشرقية (EHBH)، شنغهاي، الصين، بنسبة ذكور إلى إناث تبلغ، متوافقًا مع الفوارق الجنسية في سرطان الخلايا الكبدية، الذي يتميز بغلبة الذكور بشكل كبيرتم الحصول على موافقة المريض قبل بدء الدراسة. جميع الإجراءات التي أُجريت في الدراسة تمت الموافقة عليها من قبل اللجنة الأخلاقية للجامعة الطبية العسكرية الثانية ووفقًا لإعلان هلسنكي.
زراعة الخلايا
تم شراء خط خلايا سرطان الكبد الفأري Hepa1-6 (SCSP-512) وخطوط خلايا سرطان الكبد البشري Huh7 (SCSP-526) من بنك خلايا مجموعة الثقافة النوعية للأكاديمية الصينية للعلوم (CBTCCCAS، شنغهاي، الصين). تم شراء خط الخلايا البطانية الفأري SVEC4-10 (CRL-2181) من مجموعة الثقافة النوعية الأمريكية (ATCC؛ ماناساس، فيرجينيا، الولايات المتحدة الأمريكية). تم التحقق من صحة خطوط الخلايا بواسطة تحليل البصمة الجينية STR وتم التأكد من خلوها من الميكوبلازما. تم زراعة جميع الخلايا في وسط DMEM (Gibco) مع في و .
تم شراء الأجسام المضادة المضادة لـ NKp46 للفأر (ab233558، 1:200)، وHSP60 للبشر/الفأر (ab46798، 1:200) من شركة Abcam. تم شراء الجسم المضاد المضاد لـ VEGFR2 للبشر/الفأر (sc-6251، 1:1000) من Santa Cruz. تم شراء الأجسام المضادة المضادة لـ VE-Cad (CD144) للفأر (138011، 1:100)، وCD45 للفأر (103112/103108، 1:100)، وCD3 للفأر (100214، 1:100)، وCD8 للفأر (100706، 1:100)، وNK1.1 للفأر (108714، 1:100)، وCD11b للفأر (101224، 1:100)، وF4/80 للفأر (123116، 1:100) من Biolegend. تم شراء الأجسام المضادة InVivoMab المضادة لـ PD-L1 للفأر (BE0101)، وCD8 للفأر (BE0004)، ونوع النظير IgG2a (BE0089) من BioXCell.
تم شراء IL-2 للفأر (212-12) من Peprotech. تم شراء IL-2 البشري المؤتلف (BT-002-AFL) من R&D systems. تم شراء L-ascorbate (VC) (A4034)، NAC (A9165)، كولاجيناز IV (C5138)، و DNase I (D5025) من Sigma. تم شراء فلودارابين (S1491) من Selleck. تم شراء 2’3′-cGAMP (HY-100564)، TFMB-(R)-2-HG (HY129079)، مثبط STING C-176 (HY-112906)، وسورافينيب (Sora) (HY10201) من MCE. تم شراء مؤشر الميتوكس سوكس الأحمر للأكسيد الفائق الميتوكوندري (M36008) من Invitrogen.
الفئران
فئران معطلة جين Cgas (Cgas)) (رقم المخزون: 026554) وفئران النوك أوت لـ Sting (Sting “) (رقم المخزون: 025805)، كلاهما على خلفية C57BL/6، تم شراؤهما من مختبر جاكسون. تم استخدام فئران C57BL/6 كفئران نوع بري (WT) في هذه الدراسة وتم الحصول عليها من موارد حيوانات المختبر، الأكاديمية الصينية للعلوم (شنغهاي، الصين). الفئران الذكور بعمر 5-6 أسابيع المستخدمة في التجارب تم الحصول عليها من شركة GemPharmatech المحدودة (جيانغسو، الصين). تم تربية الفئران تحت دورة ضوء/ظلام 12/12، ودرجات حرارة تبلغ مع الرطوبة.
نماذج حيوانية
لإنشاء نماذج أورام تحت الجلد،تم زرع خلايا سرطان الكبد الفأرية (Hepa1-6) أو خلايا سرطان الكبد البشرية (Huh7) تحت الجلد في أفخاذ الفئران الذكور من نوع C57BL/6 أو الفئران العارية في عمر 5-6 أسابيع، على التوالي. تم مراقبة نمو الورم عن طريق قياس حجم الورم (الطول)العرض) مرتين في الأسبوع بعد الزرع. لم يتجاوز الحجم الأقصى المسموح به للورم 1.5 سم في أكبر قطر. في بعض الحالات، تم تجاوز هذا الحد في آخر يوم من القياس وتم إعدام الفئران على الفور. بعد 3-4 أسابيع من تأسيس الورم، تم التضحية بجميع الفئران لجمع الأورام تحت الجلد لقياس الوزن وإجراء تحليلات نسيجية إضافية. لإنشاء نماذج أورام أورثوتوبية، تم إجراء جراحة بطنية لزرع خلايا Hepal-6 التي تحمل جين مراسل اللوسيفيراز في الفص الأيسر من الكبد بعد تخدير الفئران.
للتصوير بالضوء الحيوي، تم حقن الفئران داخل الصفاق بدواء دي-لوسيفيرين. بعد 15-20 دقيقة من الحقن، خضعت الفئران للتصوير باستخدام جهاز IVIS Lumina III (PerkinElmer) وتم قياس الضوء الحيوي باستخدام برنامج Living Image. تم تحديد حجم الورم بناءً على التدفق الكلي (الفوتونات في الثانية).
العلاجات داخل الجسم الحي واستنفادخلايا
قبل بدء العلاجات، تم توزيع الفئران عشوائيًا على مجموعات مختلفة ذات أحجام أورام متوسطة متشابهة. للعلاجات داخل الجسم الحي، VC (وزن الجسم؛ يوميًا؛ حقن داخل الصفاق) (سيغما) أو محلول فوسفات البفر (PBS) كعينة تحكم، سورا (وزن الجسم؛ يوميًا؛ عن طريق الفم) (MCE)؛ أجسام مضادة لـ PD-L1 (لكل فأر؛ مرتين في الأسبوع؛ حقن داخل الصفاق) (BioXCell) أو تحكم النوع النظيري IgG، rlL-2 ((لكل فأر؛ يوميًا؛ حقن داخل الصفاق) (أنظمة البحث والتطوير) أو PBS كعينة تحكم، وC-176 (750 نانومول لكل فأر؛ يوميًا؛ حقن داخل الصفاق) (MCE) أو المذيب تم حقنها لمدة أسبوعين بدءًا من اليوم السابع بعد إنشاء نماذج سرطان الكبد في الفئران. لاستنزافالخلايا في الجسم الحي، تم حقن الفئران داخل الصفاق بـ من جسم مضاد مضاد لـ CD8 (BioXCell) أو تحكم النظير IgG (BioXCell) قبل 3 أيام ويوم واحد من زرع الورم ومرتين أسبوعيًا بعد ذلك لضمان استنفاد مستمر لـ مجموعة الخلايا الفرعية خلال الفترة التجريبية.
اختبار النفاذية الوعائية في الجسم الحي
تم حقن الفئران الحاملة للأورام عن طريق الوريد بـ 0.5 ملغ من صبغة إيفانز بلو (سيغما) في محلول PBS. بعد ساعة من الدوران، تم ضخ الفئران عبر القلب بمحلول PBS يحتوي على 2 م م EDTA لإزالة صبغة إيفانز بلو داخل الأوعية الدموية. تم جمع الأورام، وتقطيعها إلى قطع، وحضانتها في 1 مل من الفورماميد لمدة 24 ساعة عندلاستخلاص اللون الأزرق إيفانز من الورم. بعد الطرد المركزي، تم جمع السائل الفوقي وتخفيفه إلى تركيز نهائي يعادل وزن الورم نفسه لكل مل من الفورماميد. ثم تم قياس الامتصاصية عند 655 نانومتر و750 نانومتر باستخدام مقياس الطيف الضوئي. لإزالة تأثير صبغة الهيم المتبقية في الدم، تم استخدام الصيغة التالية: الامتصاصية المصححة (A) A655 نانومتر – ( A750 نانومتر + 0.03) (7).
siRNA، shRNA، تعطيل CRISPR/Cas9، والتعبير الزائد
تم إجراء استخدام siRNAs لتثبيط جينات Lrrc8c و Stat1 و Stat5a في الفئران بواسطة شركة Biotend (شنغهاي، الصين). تم نقل siRNAs إلى الخلايا باستخدام Lipofectamine 2000 (Invitrogen) وفقًا لتعليمات الشركة المصنعة. تم شراء نواقل التعبير عن shRNA المعتمدة على الفيروسات اللاجنسية والتي تستهدف جين Tet2 في الفئران (تسلسل الهدف: 5′-GCTCTAAAT-GATGTAGCTTTG-3′) و Stat5a (تسلسل الهدف: 5′-GACGTGA-GATTCAAGTCTAAC-3′) من شركتي Genomeditech (شنغهاي، الصين) و Genechem (شنغهاي، الصين) على التوالي. تم شراء ناقل CRISPR/Cas9 المعتمد على الفيروسات اللاجنسية لعمل حذف (KO) لجين Sting في الفئران (تسلسل الهدف: 5′-CAGCCTGATGATCCTTTGGG-3′) وجين Cgas (تسلسل الهدف: …-CGGCGGGCAGCTCCGGATCC-3′) تم شراؤه من شركة أوبيو تكنولوجي (شنغهاي، الصين). تم تصنيع بلازميد اللنتيفيروس المعبر عن جين Cgas الفأري بواسطة شركة بيوتند (شنغهاي، الصين). بلازميدات اللنتيفيروس المعبرة عن الجين البشري، وتم شراء Sting-Cherry الفأري من Genechem (شنغهاي، الصين). تم نقل الخلايا (بمعدل تلاصق 30%) باستخدام تخفيفات مثالية من الفيروسات اللامتناهية المختلطة مع بوليبرين. ثم، تم معالجة الخلايا المنقولة بالبيروميسين لاختيار الخلايا المنقولة المستقرة. تم تأكيد تعطيل الجين المطلوب بواسطة تحليل التلطيخ المناعي للبروتينات المستهدفة.
الترسيب الغربي والترسيب المناعي المشترك (co-IP)
تم تحضير مستخلصات الخلايا الكاملة باستخدام محلول تحلل الخلايا (بيوتايم للتكنولوجيا الحيوية) وغليها في محلول تحميل عينات SDS. ثم، تم استخدام محتوى بروتين متساوٍ ( من البروتين لكل مسار) تم تحميله إلى تم إجراء الرحلان الكهربائي باستخدام PAGE ونُقلت إلى أغشية PVDF. تم حجب الأغشية في 5% حليب في محلول TBST لمدة ساعة واحدة في درجة حرارة الغرفة، ثم تم حضنها طوال الليل مع الأجسام المضادة الأولية المناسبة عند. بعد الحضانة مع الأجسام المضادة الثانوية المرتبطة بالفلوريسئين لمدة ساعة واحدة في درجة حرارة الغرفة، تم تصوير اللطخات المناعية باستخدام ماسح ضوئي فلوري من نوع Odyssey (Li-Cor، لينكولن، نبراسكا، الولايات المتحدة الأمريكية). بالنسبة للتسريب المناعي المشترك (co-IP)، مستخلصات الخلايا ( من البروتين) تم خلطها مع مضاد Tet2 (GeneTex) طوال الليل عندثم تم سحبه باستخدام خرزة مغناطيسية للبروتين G (إنفيتروجين) عندلمدة ساعتين. ثم تم غسل حبات IP بثلاث مرات بمحلول التحلل، وتسخينها في محلول تحميل عينات SDS عندلمدة 5 دقائق لتحليل اللطخة المناعية.
تفاعل البوليميراز المتسلسل الكمي في الوقت الحقيقي (qRT-PCR)
تم استخراج الحمض النووي الريبي الكلي من الخلايا باستخدام كاشف TRIzol (Invitrogen) وتم نسخه عكسيًا إلى الحمض النووي التكميلي باستخدام بادئات عشوائية باستخدام إنزيم النسخ العكسي Superscript III (Invitrogen). تم استخدام الحمض النووي التكميلي بعد ذلك كقالب لـ رد فعل. تم التنفيذ باستخدام خليط SYBR Green PCR Master Mix (شركة أبلايد بيوسيستمز) على نظام كشف التسلسل ABI PRISM 7300HT (شركة أبلايد بيوسيستمز).– تم استخدام الأكتين كعنصر تحكم للتطبيع. تم سرد البوادئ في الجدول التكميلي 1.
ترسيب المناعة للنواة (ChIP)
تم إجراء تفاعل ترسيب الكروماتين المناعي (ChIP) باستخدام مجموعة EpiQuik لترسيب الكروماتين المناعي (ChIP) من شركة Epigentek وفقًا لتعليمات الشركة المصنعة. التعليمات. باختصار، تم تثبيت الخلايا وربط الكروماتين واستخراجه وتجزئته. تم إجراء التفاعل المناعي باستخدام جسم مضاد مضاد للعلم (Abcam). تم إجراء تفاعل البوليميراز المتسلسل الكمي (qRT-PCR) على الحمض النووي المناعي باستخدام خليط SYBR Green PCR Master Mix (Applied Biosystems) على نظام الكشف عن التسلسل ABI PRISM 7300 HT (Applied Biosystems). تم تطبيع قيم الحمض النووي المناعي إلى إشارات الإدخال. تم سرد بادئات محفز Cgas في الجدول التكميلي 1.
تحليل التدفق الخلوي
تم تقطيع أورام الفئران ميكانيكيًا واحتُضنت في كولاجيناز IV (، سيغما) و DNase I (، سيغما) لمدة 30 دقيقة عند مع الهز. تم ترشيح الخلايا المفككة من خلالمصفاة الخلايا (BD). ثم، تم حضانة المعلقات الخلوية المفردة الناتجة مع حاجز Fc وتلوينها بالأجسام المضادة السطحية المحددة لمدة 20 دقيقة عندتم تحليل الخلايا الملونة فورًا باستخدام جهاز قياس التدفق LSRFortessa (بي دي بيوسسينس) وتم تحليلها باستخدام برنامج FlowJo.
قياس cGAMP
تم قياس مستويات cGAMP في مستخلصات الخلايا ووسط الزرع باستخدام مجموعة ELISA لـ 2’3′-cGAMP (Cayman) وفقًا لبروتوكول الشركة المصنعة. لتحضير العينات، تم تحلل الخلايا في M-PERكاشف استخراج البروتينات من الثدييات (ثيرموفيشر ساينتيفيك).
اختبار هجرة اللمفاويات عبر البطانة الداخلية للأوعية الدموية
تم تقييم هجرة اللمفاويات عبر حاجز بطاني في المختبر باستخدام CytoSelectمجموعة اختبار عبور الكريات البيضاء (Cell Biolabs) وفقًا لتعليمات الشركة المصنعة. تم عزل الخلايا اللمفاوية المستخدمة في الدراسة من طحال الفئران البرية (WT) أو الفئران المعدلة جينيًا (Sting-/-) باستخدام وسط فصل الخلايا اللمفاوية للفئران (Dakewe Biotechnology) وفقًا لبروتوكول الشركة المصنعة، ثم تم تمييزها بـ LeukoTracker.تم تعريض طبقات الخلايا البطانية للعلاج المشار إليه لمدة 24 ساعة قبل وضع جهاز تتبع الكريات البيضاء.الخلايا اللمفاوية الموسومة على طبقة الخلايا البطانية الأحادية. تم حساب الوفرة النسبية للخلايا اللمفاوية التي هاجرت عبر الخلايا البطانية من خلال قياس الفلورة في العينات عند.
تلوين التألق المناعي
تم تثبيت مقاطع أورام الفئران المجمدة أو الخلايا باستخدام 4% بارافورمالدهيد لمدة 20 دقيقة في درجة حرارة الغرفة وتمت تغلغلها بمحلول 1% ترايتون إكس-100 (سولاربيو) لمدة 15 دقيقة. بعد ذلك، تم استخدام 5% بي إس إيه في محلول فوسفات البفر (PBS) معتم استخدام التريتون لحجب مواقع الارتباط غير النوعي في درجة حرارة الغرفة لمدة ساعة واحدة. تم تلطيخ مقاطع الورم بالأجسام المضادة الأولية المحددة ضد CD31 و-SMA، وتم تلطيخ الخلايا بأجسام مضادة أولية ضد dsDNA وHSP60 طوال الليل عندتبع ذلك استخدام الأجسام المضادة الثانوية المفلورة في درجة حرارة الغرفة لمدة ساعة واحدة. تم استخدام DAPI (شركة لايف تكنولوجيز) لصبغ النواة. تم عرض جميع الشرائح وتوثيقها تصويريًا باستخدام مجهر كونفوكال STELLARIS 5 (شركة ليكا للأنظمة الميكروسكوبية).
اختبار تكاثر وهجرة الخلايا البطانية
تم زرع خلايا SVEC4-10 بكثافة 3000-5000 خلية لكل بئر في لوحة ذات 96 بئراً. بعد المعالجة المحددة لمدة 24 ساعة، تم قياس حيوية الخلايا باستخدام اختبار عد الخلايا Kit-8 (CCK-8) (Vazyme) وفقًا لتعليمات الشركة المصنعة. لاختبار الهجرة،تم زرع خلايا SVEC4-10 المحرومة من المصل طوال الليل على غشاء PET (حجم المسام) للإدخال. تم تعليق الإدخالات على لوحة دعم ذات 24 بئراً، والتي كانت تحتوي على طبقة أحادية متماسكة بنسبة 70-80% من خلايا Hepa1-6-Ctrl أو Hepa1-6-Cgas المزروعة مع وسط نمو كامل. بعد 24 ساعة من الحضانة، تم تثبيت الإدخالات بواسطة 4% بارافورمالدهيد وصبغها بمحلول البنفسجي البلوري. تم تصوير الحقول التمثيلية وتم قياس عدد الخلايا المهاجرة باستخدام برنامج ImageJ (المعهد الوطني للصحة).
اختبار تكوين الأنابيب
تم تعريض خلايا SVEC4-10 للعلاج المشار إليه لمدة 24 ساعة، ثم تم زرعها على أطباق بآبار 24 مغطاة بماتريجل (كورنينج). خلايا/بئر) لمدة 6 ساعات عند قبل التقاط الصور. تم تطبيق وحدة تحليل تكوين الأوعية الدموية في برنامج ImageJ (المعهد الوطني للصحة) لقياس عدد الشبكات والطول الكلي للأنابيب.
التلوين المناعي النسيجي (IHC)
تم إجراء تلوين المناعة المناعية الكيميائية على مقاطع نسيجية ممثلة من كتل نسيجية مثبتة بالفورمالين ومضمّنة في البارافين من سرطان الكبد البشري (HCC) وزرعات الأورام في الفئران باستخدام الأجسام المضادة المشار إليها. بناءً على طريقة درجة التفاعل المناعي، تم تقييم شدة تلوين مصفوفات الأنسجة لسرطان الكبد البشري (تعبير البروتين) على النحو التالي: 0 (تلوين سلبي)، 1 (تلوين ضعيف)، 2 (تلوين متوسط)، و3 (تلوين قوي). ثم تم تقسيم المرضى إلى مجموعتين: مجموعة التعبير المنخفض (تلوين سلبي أو ضعيف) ومجموعة التعبير العالي (تلوين متوسط أو قوي). بالنسبة لـ CD31/-تلوين مزدوج IHC لـ SMA، تم تلوين CD31 أولاً باستخدام 3،3-ديامينوبنزيدين (DAB) (صبغة بنية)-تم اختيار SMA ثانيًا للتلوين باستخدام سينانين 3 (الكروموجين الأحمر). تم تقدير تغطية الخلايا المحيطة للأوعية الدموية في الورم عن طريق قياس الأوعية التي تلوّنت إيجابيًا لكل من CD31 وSMA (علامة الخلايا المحيطة بالأوعية) بين جميع الأوعية الدموية الإيجابية لـ CD31. تم عرض النتائج كنسبة مئوية من أوعية الدم الورمية التي تحتوي على-تغطية الخلايا المحيطة الإيجابية لـ SMA.
تسلسل البيروسيكوينس للميثلة الحمض النووي
تم قياس مستويات مثيلة Cgas باستخدام التسلسل النيوكليوتيدي الفسفوري لفحص حالة المثيلة في مواقع CpG في محفز Cgas. تم توفير خدمة التسلسل والتحليل المعلوماتي الحيوي من قبل شركة شنغهاي للتكنولوجيا الحيوية (شنغهاي، الصين). باختصار، تم استخراج الحمض النووي الجينومي من الخلايا ثم خضعت عينات الحمض النووي لتحويل البيسلفيت باستخدام مجموعة EZ DNA Methylation Kit (Zymo Research) وفقًا لتعليمات الشركة المصنعة. تم استخدام الحمض النووي المعالج بالبيسلفيت كقالب لتضخيم الجزء المستهدف من محفز Cgas باستخدام زوج البادئات المحدد. تم تسلسل منتجات تفاعل البوليميراز المتسلسل (PCR) بواسطة التسلسل النيوكليوتيدي الفسفوري باستخدام جهاز PyroMark Q96 (QIAGEN).
تحليل قاعدة البيانات عبر الإنترنت
تم الحصول على تعبيرات بروتين cGAS (C6orf150، MB21D1) وSTING (TMEM173) في أنسجة سرطان الكبد التي تم تقييمها بواسطة الكيمياء المناعية من أطلس البروتين البشري (HPA،)http://www.proteinatlas.org) تم الحصول على الترابطات بين تعبيرات الجينات المختلفة في سرطان الكبدة الخلوي من أطلس جينوم السرطان (TCGA،)https://portal.gdc.cancer.gov) والمجموعة الدولية لجينوم السرطان (ICGC، https://dcc. icgc.org) قواعد البيانات. تم إجراء تحليل تسلل الخلايا وفقًا لتعبيرات جينية مختلفة في سرطان الكبدة الخلوي (HCC) من قاعدة بيانات TCGA باستخدام مورد تقدير المناعة الورمية (TIMER).تم الحصول على بيانات مثيلة الحمض النووي من TCGA من قاعدة بيانات التفاعل البصري لمثيلة الحمض النووي (DNMIVD)تم إجراء تحليل إثراء مجموعة الجينات (GSEA) وتحليل مسار بانثر في مجموعات بيانات HCC TCGA على منصة LinkedOmics.تم الحصول على تعبيرات مستقبلات IL-2 في خطوط خلايا سرطان الكبد البشري من موسوعة خطوط خلايا السرطان (CCLE،)https://sites.broadinstitute.org/ccle). تم التحقق من اختلافات البقاء على قيد الحياة على مستوى التعبير الجيني في HCC TCGA من خلال قاعدة بيانات عبر الإنترنت Kaplan-Meier Plotter (KM-Plotter) .
الإحصائيات
تم إجراء التحليل الإحصائي باستخدام برنامج GraphPad Prism 8 (GraphPad Software، سان دييغو، كاليفورنيا). تُعرض البيانات كمتوسطالخطأ المعياري (الانحراف المعياري) ما لم يُذكر خلاف ذلك. تم تكرار التجارب ثلاث مرات على الأقل مع نتائج مماثلة. الاختبارات الإحصائية المحددة المطبقة مذكورة في تسميات الأشكال الخاصة بها.قيمةتم اعتباره ذا دلالة إحصائية.
ملخص التقرير
مزيد من المعلومات حول تصميم البحث متاحة في ملخص تقرير مجموعة نيتشر المرتبط بهذا المقال.
توفر البيانات
مجموعة البيانات المتاحة للجمهور المذكورة في هذا البحث مأخوذة من قواعد بيانات GEO (GSE51401، GSE69164، GSE146409، GSE140901)، أطلس جينوم السرطان (TCGA،https://portal.gdc.cancer.gov)، الاتحاد الدولي لجينوم السرطان (ICGC، https://dcc.icgc.org)، أطلس البروتين البشري (HPA، http://www.proteinatlas.org)، وموسوعة خطوط خلايا السرطان (CCLE، https://sites.broadinstitute.org/ccle). البيانات المتبقية متاحة داخل المقالة، المعلومات التكميلية أو ملف بيانات المصدر. يتم توفير بيانات المصدر مع هذه الورقة.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209-249 (2021).
Cheng, A. L., Hsu, C., Chan, S. L., Choo, S. P. & Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 72, 307-319 (2020).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707-723 (2017).
Huang, Y. et al. Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 18, 195-203 (2018).
Zheng, X. et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J. Clin. Investig. 128, 2104-2115 (2018).
Martin, J. D., Seano, G. & Jain, R. K. Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu. Rev. Physiol. 81, 505-534 (2019).
Tian, L. et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544, 250-254 (2017).
Khan, K. A. & Kerbel, R. S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 15, 310-324 (2018).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786-791 (2013).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788-792 (2009).
Kwon, J. & Bakhoum, S. F. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 10, 26-39 (2020).
Guey, B. & Ablasser, A. Emerging dimensions of cellular cGASSTING signaling. Curr. Opin. Immunol. 74, 164-171 (2022).
Corrales, L. & Gajewski, T. F. Molecular pathways: targeting the stimulator of interferon genes (STING) in the immunotherapy of cancer. Clin. Cancer Res. 21, 4774-4779 (2015).
Amouzegar, A., Chelvanambi, M., Filderman, J. N., Storkus, W. J. & Luke, J. J. STING agonists as cancer therapeutics. Cancers 13, 2695 (2021).
Park, J. S. et al. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell 30, 953-967 (2016).
Heidenreich, R., Kappel, A. & Breier, G. Tumor endothelium-specific transgene expression directed by vascular endothelial growth factor receptor-2 (Flk-1) promoter/enhancer sequences. Cancer Res. 60, 6142-6147 (2000).
Massalha, H. et al. A single cell atlas of the human liver tumor microenvironment. Mol. Syst. Biol. 16, e9682 (2020).
Saitoh, T. et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl Acad. Sci. USA 106, 20842-20846 (2009).
Ding, Z., Xiong, K. & Issekutz, T. B. Regulation of chemokine-induced transendothelial migration of T lymphocytes by endothelial activation: differential effects on naive and memory T cells. J. Leukoc. Biol. 67, 825-833 (2000).
Vestweber, D., Wessel, F. & Nottebaum, A. F. Similarities and differences in the regulation of leukocyte extravasation and vascular permeability. Semin. Immunopathol. 36, 177-192 (2014).
Chen, Q. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493-498 (2016).
Ritchie, C., Cordova, A. F., Hess, G. T., Bassik, M. C. & Li, L. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol. Cell 75, 372-381 (2019).
Cordova, A. F., Ritchie, C., Böhnert, V. & Li, L. Human SLC46A2 is the dominant cGAMP importer in extracellular cGAMP-sensing macrophages and monocytes. ACS Cent. Sci. 7, 1073-1088 (2021).
Lahey, L. J. et al. LRRC8A:C/E heteromeric channels are ubiquitous transporters of cGAMP. Mol. Cell 80, 578-591 (2020).
Zhou, C. et al. Transfer of cGAMP into Bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral immunity. Immunity 52, 767-781 (2020).
An, X. et al. An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol. Ther. Nucleic Acids 14, 80-89 (2019).
Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597-610 (2005).
Yang, R. et al. Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory cell differentiation and maintain immune homeostasis. Immunity 43, 251-263 (2015).
Malek, T. R. The biology of interleukin-2. Annu. Rev. Immunol. 26, 453-479 (2008).
Liu, J. et al. Global DNA 5-hydroxymethylcytosine and 5-formylcytosine contents are decreased in the early stage of hepatocellular carcinoma. Hepatology 69, 196-208 (2019).
Cimmino, L. et al. Restoration of TET2 function blocks aberrant selfrenewal and leukemia progression. Cell 170, 1079-1095.e20 (2017).
Agathocleous, M. et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476-481 (2017).
Wilhelm, S. et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5, 835-844 (2006).
Ngo, B., Riper, Van, Cantley, J. M., Yun, L. C. & Targeting, J. cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 19, 271-282 (2019).
Lv, H. et al. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis. Oncol 2, 1 (2018).
Chen, Y. et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 61, 1591-1602 (2015).
Chang, C. J. et al. Targeting tumor-infiltrating Ly6G+ myeloid cells improves sorafenib efficacy in mouse orthotopic hepatocellular carcinoma. Int. J. Cancer 142, 1878-1889 (2018).
Hsu, C. et al. Exploring markers of exhausted CD8 T cells to predict response to immune checkpoint inhibitor therapy for hepatocellular carcinoma. Liver Cancer 10, 346-359 (2021).
Newell, P., Villanueva, A., Friedman, S. L., Koike, K. & Llovet, J. M. Experimental models of hepatocellular carcinoma. J. Hepatol. 48, 858-879 (2008).
West, E. E. et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Investig. 123, 2604-2615 (2013).
Hashimoto, M. et al. PD-1 combination therapy with IL-2 modifies CD8+ T cell exhaustion program. Nature 610, 173-181 (2022).
Falahat, R. et al. STING signaling in melanoma cells shapes antigenicity and can promote antitumor T-cell activity. Cancer Immunol. Res. 7, 1837-1848 (2019).
Mahadevan, N. R. et al. Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov. 11, 1952-1969 (2021).
Yang, H. et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig. 129, 4350-4364 (2019).
Chelvanambi, M., Fecek, R. J., Taylor, J. L. & Storkus, W. J. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J. Immunother. Cancer 9, e001906 (2021).
Mao, Y. et al. STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in dietinduced obesity. Arterioscler. Thromb. Vasc. Biol. 37, 920-929 (2017).
Huang, L. S. et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52, 475-486.e5 (2020).
Corrales, L. et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11, 1018-1030 (2015).
Demaria, O. et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl Acad. Sci. USA 112, 15408-15413 (2015).
Shen, Y. J. et al. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep. 11, 460-473 (2015).
Konno, H. et al. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37, 2037-2051 (2018).
Xia, T., Konno, H., Ahn, J. & Barber, G. N. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14, 282-297 (2016).
Lv, H. et al. NAD+ metabolism maintains inducible PD-L1 expression to drive tumor immune evasion. Cell Metab. 33, 110-127.e5 (2021).
Xu, Y. P. et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 129, 4316-4331 (2019).
Slaney, C. Y., Kershaw, M. H. & Darcy, P. K. Trafficking of T cells into tumors. Cancer Res. 74, 7168-7174 (2014).
Nauman, G., Gray, J. C., Parkinson, R., Levine, M. & Paller, C. J. Systematic review of intravenous ascorbate in cancer clinical trials. Antioxidants 7, 89 (2018).
Luchtel, R. A. et al. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl Acad. Sci. USA 117, 1666-1677 (2020).
Magri, A. et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 12, eaay8707 (2020).
Lan, Y. Y., Londoño, D., Bouley, R., Rooney, M. S. & Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 9, 180-192 (2014).
Gehrke, N. et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STINGdependent immune sensing. Immunity 39, 482-495 (2013).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509-W514 (2020).
Ding, W. et al. DNMIVD: DNA methylation interactive visualization database. Nucleic Acids Res. 48, D856-D862 (2020).
Vasaikar, S. V., Straub, P., Wang, J. & Zhang, B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46, D956-D963 (2018).
Menyhárt, O., Nagy, Á. & Győrffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 5, 181006 (2018).
الشكر والتقدير
نشكر الدكتور ووهان شياو والدكتور غوليانغ شو (الأكاديمية الصينية للعلوم) على بلازميدات Tet، ودان كاو، شان هوا تانغ، لينا قوه، لو تشن، مينغشوانغ شو، شين نيان غي، وكي يانغ على مساعدتهم الفنية. تم دعم هذا العمل بمنح من المؤسسة الوطنية للعلوم الطبيعية في الصين (82273176، 81902894، 81972779، 81903036، 81830054، 91859205، و81988101)، والمشروع الوطني الرئيسي الصيني (2018ZX10723204-006-003)، ومشروع لجنة التعليم البلدية في شنغهاي (201901070007E00065)، وبرنامج قائد البحث الأكاديمي في شنغهاي (23XD1404800).
مساهمات المؤلف
صمم W.Y. و H.L. و H.W. الدراسة. قام H.L. و Q.Z. و C.C. و W.X. و F.X. و G.L. بإجراء التجارب وتحليل البيانات. قدم G.J و B.Y. و X.S. و Y.M. و L.W. و Z.W. و X.C. الدعم الفني. كتب H.L. و W.Y. المخطوطة. نظم H.W. و W.Y. الدراسة وأشرفا عليها.
يجب توجيه المراسلات وطلبات المواد إلى هونغيانغ وانغ أو وين يانغ.
معلومات مراجعة الأقران تشكر مجلة نيتشر كوميونيكيشنز والتر ستوركوس والمراجع(ين) الآخر(ين) المجهول(ين) على مساهمتهم في مراجعة الأقران لهذا العمل. ملف مراجعة الأقران متاح.
مختبر التعاون الدولي في نقل الإشارات، مستشفى جراحة الكبد والقنوات الصفراوية الشرقي، الجامعة الطبية البحرية (الجامعة الطبية العسكرية الثانية)، شنغهاي 200438، الصين.المركز الوطني لسرطان الكبد، جامعة الطب البحري (الجامعة الطبية العسكرية الثانية)، شنغهاي 201805، الصين.مركز أبحاث السرطان، المستشفى الأول التابع لجامعة العلوم والتكنولوجيا الصينية، قسم علوم الحياة والطب، جامعة العلوم والتكنولوجيا الصينية، هيفي، أنهوي 230027، الصين.قسم جراحة الكبد والقنوات الصفراوية، كلية الطب الإكلينيكي، جامعة يانغتشو، يانغتشو، جيانغسو 225000، الصين.مستشفى جامعة تشنغتشو، تشنغتشو، خنان 450000، الصين.المختبر الرئيسي لبيولوجيا أورام الكبد والمرارة في شنغهاي، شنغهاي 200438، الصين.المختبر الرئيسي لتنظيم الإشارات والعلاج المستهدف لسرطان الكبد، وزارة التعليم، شنغهاي 200438، الصين.ساهم هؤلاء المؤلفون بالتساوي: هونغوي لو، تشياني زونغ، سيان تشن. البريد الإلكتروني:hywangk@vip.sina.com; woodeasy66@hotmail.com
TET2-mediated tumor cGAS triggers endothelial STING activation to regulate vasculature remodeling and anti-tumor immunity in liver cancer
Received: 3 April 2023
Accepted: 17 November 2023
Published online: 04 January 2024
(4) Check for updates
Hongwei Lv , Qianni Zong , Cian Chen , Guishuai Lv , Wei Xiang , Fuxue Xing , Guoqing Jiang , Bing Yan , Xiaoyan Sun , Yue Ma , Liang Wang , Zixin Wu , Xiuliang Cui , Hongyang Wang & Wen Yang
Induction of tumor vascular normalization is a crucial measure to enhance immunotherapy efficacy. cGAS-STING pathway is vital for anti-tumor immunity, but its role in tumor vasculature is unclear. Herein, using preclinical liver cancer models in Cgas/Sting-deficient male mice, we report that the interdependence between tumor cGAS and host STING mediates vascular normalization and anti-tumor immune response. Mechanistically, TET2 mediated IL-2/STAT5A signaling epigenetically upregulates tumor cGAS expression and produces cGAMP. Subsequently, cGAMP is transported via LRRC8C channels to activate STING in endothelial cells, enhancing recruitment and transendothelial migration of lymphocytes. In vivo studies in male mice also reveal that administration of vitamin C, a promising anti-cancer agent, stimulates TET2 activity, induces tumor vascular normalization and enhances the efficacy of anti-PD-L1 therapy alone or in combination with IL-2. Our findings elucidate a crosstalk between tumor and vascular endothelial cells in the tumor immune microenvironment, providing strategies to enhance the efficacy of combinational immunotherapy for liver cancer.
Liver cancer, primarily hepatocellular carcinoma (HCC), is one of the most frequent malignancies and the third leading causes of cancerrelated death globally . Currently, immune checkpoint blockade-based immunotherapies, particularly antibodies targeting programmed cell death-1 (PD-1)/programmed cell death ligand 1 (PD-L1) pathway have achieved remarkable success in the treatment of various malignancies including advanced . Nevertheless, anti-PD-1/PD-L1 monotherapy
induces durable responses in only a subset of cancer patients and some responders relapse after a period of response . Abnormal tumor blood vessels and the resulting hypoxia and immunosuppressive tumor microenvironment are key factors hindering immunotherapy efficacy, especially in liver cancer, which is highly angiogenic . Antiangiogenic therapy can not only normalize aberrant tumor blood vessels to improve intratumoral immune effector cell infiltration, but
also reverse the immunosuppressive microenvironment induced by angiogenic inducers, especially vascular endothelial growth factor (VEGF), providing rational for the combination of anti-angiogenic therapies with immune checkpoint blockade . Recently, combination of anti-angiogenic therapy and immune checkpoint blockade have demonstrated efficacy in treating advanced HCC, making it the firstline therapy . However, the vascular normalization ‘time window’ of current anti-angiogenic therapy is narrow and transient, limiting its enhancing effect on immunotherapy. Therefore, effective strategies to stably induce tumor vascular normalization and boost immunotherapy efficacy are urgently needed.
As a cytosolic DNA sensor, cyclic GMP-AMP synthase (cGAS) triggers innate immune responses through production of the second messenger cyclic GMP-AMP (cGAMP), which binds and activates the stimulator of interferon genes (STING) . The activated STING recruits TANK binding kinase 1 (TBK1) and then phosphorylates interferon (IFN) regulatory factor 3 (IRF3) followed by induction of type I IFN and chemokines such as CCL5 and CXCL10 . Activated cGAS-STING pathway plays a vital role in anti-tumor immunity via T cell priming . Yet, the regulation and mechanism of cGAS-STING pathway in cancer immunity remain to be fully understood. In addition to immune cell or tumor cell-intrinsic cGAS-STING activation, cGAS-STING pathway allows the cross-talk between tumor cells and surrounding non-tumor cells (or host cells) to regulate anti-tumor immunity . The tumorsecreted cGAMP or DNA is transported to antigen presenting cells (APCs), such as dendritic cell (DC) and macrophage, and then activates the STING-induced type I IFN signaling pathway, finally triggering the anti-tumor immune response mediated by CD8 cells or NK cells . Although DC is considered as the major cell type responding to tumorderived cGAMP, many other cells in the tumor microenvironment with substantial STING expression, especially vascular endothelial cells, may also detect tumor-derived cGAMP. However, whether and how cGAS-STING pathway mediates the interaction between tumor cells and endothelial cells, as well as the role of cGAS-STING pathway in tumor vasculature remain largely unknown.
Intensive efforts have been invested to develop cGAS-STING agonists and several STING agonists have shown great promise in cancer immunotherapy in pre-clinical models . Unfortunately, current STING agonists, most of which are synthetic analogs of cGAMP, are mainly delivered intratumorally due to poor bioavailability, limiting their clinical application and final therapeutic efficacy . Therefore, major efforts are ongoing to develop more potent and selective cGAS-STING agonists to boost cancer immunotherapy.
Here, we describe the cross-talk between cGAS in tumor cells and STING in endothelial cells, which enhances transendothelial migration of lymphocytes, vascular normalization, and anti-tumor immunity. Based on the epigenetic regulation of tumor cGAS expression by teneleven translocation-2 (TET2) methylcytosine dioxygenase synergized with STAT5A signaling, we further stimulate TET2 activity using vitamin C (VC) to trigger tumor cGAS-cGAMP-endothelial STING pathway activation and induce tumor vascular normalization, thereby potentiating the therapeutic efficacy of immune checkpoint blockade in vivo.
Results
Tumor cGAS regulates vascular normalization and anti-tumor immune response in an intrinsic STING-independent manner We first investigated the role of cancer cell-intrinsic cGAS and STING in tumor vascular normalization and anti-tumor immunity. The murine liver cancer cell line (Hepal-6) with low endogenous Cgas and intact Sting expression was forcedly expressed with Cgas following depletion of its Sting via CRISPER-Cas9 or not to establish three cells of same origin with Cgas Sting , Cgas Sting , and Cgas Sting expression (Fig. 1a, Supplementary Fig. 1a). Then, we challenged wildtype (WT) mice with these three cells respectively. Interestingly,
knockout of Sting in Hepal-6 cells (Cgas (-)/Sting (-)) did not alter tumor growth compared to parental cells (Cgas (-)/Sting (+)) (Fig. 1b, c). Additionally, no obvious difference was observed in pericyte coverage of tumor vessels, which is one of the hallmarks of tumor vessel normalization , vascular permeability, or intratumoral T cell infiltration (Fig. 1d, e, Supplementary Fig. 2a-c), indicating that tumorintrinsic STING expression is not required for vascular normalization and anti-tumor immunity under low tumor cGAS background. In contrast, on the background of Sting deficiency, Cgas overexpression in Hepal-6 cells (Cgas (+)/Sting (-)) dramatically retarded tumor growth (Fig. 1b, c) and angiogenesis, increased pericyte coverage of tumor vessels, accompanied by mitigated vascular permeability and elevated intratumoral T cell infiltration (Fig. 1d, e, Supplementary Fig. 2a, b). Moreover, tumor vessels were generally more mature in Cgas overexpression tumors, as evidenced by the majority of the vessels expressing low levels of VEGF receptor 2 (VEGFR2), a molecular marker highly expressed by growing and immature vessels (Fig. 1f, Supplementary Fig. 2c). In contrast, cancer cell-intrinsic Sting depletion exhibited no substantial effect on tumor blood vessel maturation. To further exclude the role of tumor-intrinsic STING under tumorintrinsic cGAS overexpression background, we knocked out Sting in Cgas overexpression cells (Fig. 1g) and found that Sting deficiency (Cgas (+)/Sting (-)) did not alter tumor growth compared to parental cells (Cgas (+)/Sting (+)) (Fig. 1h, i). Moreover, no obvious difference was observed in pericyte coverage of tumor vessels or intratumoral T cell infiltration (Fig. 1j). Together, these results suggest that tumor cGAS controls vascular normalization and anti-tumor immune response in an intrinsic STING-independent manner.
To mimic a more faithful emulation of the endogenous cGAS/ STING pathway levels observed in human HCC, a series of distinct clones with differential Cgas expression levels were selected (Fig. 1k). As shown in Fig. 11, we observed a broader range of intracellular and extracellular cGAMP concentrations, spanning from a few hundred pg/ mg protein to tens of thousands protein. In vitro, the cell proliferation rates were similar after Cgas overexpression (Supplementary Fig. 1b). Importantly, we found that with higher Cgas expression in cancer cells, smaller tumor burdens were observed (Fig. 1m, n), while more pericyte coverage of tumor vessels and intratumoral T cell infiltration were detected (Fig. 1o), suggesting that tumor cGAS mediates tumor repression, vascular normalization, and anti-tumor immune response in a cGAS expression level-dependent manner. To further verify this phenomenon in human liver cancer, we established cGAS stably expressed human liver cancer cells (Huh7), which lack intrinsic cGAS and STING expressions, and forced cGAS expression did not alter Huh7 cells proliferation in culture (Supplementary Fig. 1c, d). Of note, cGAS cells-derived tumors in nude mice exhibited delayed growth (Supplementary Fig. 3a, b) and increased pericyte coverage of vessels, accompanied by reduced intratumoral hypoxia and more NK cells infiltration as compared to Ctrl cells-derived tumors (Supplementary Fig. 3c, d). Additionally, mouse-specific genes including downstream of STING activation IFN and ISGs, vascular stabilizing genes, endothelial-lymphocyte interaction-associated adhesion molecules were up-regulated in cGAS-proficient tumors (Supplementary Fig. 3e), indicating host STING activation and vascular normalization. In contrast, STING overexpression in cGAS cells (cGAS (+)/STING (+)) had little effect on tumor growth and vascular normalization compared with cGAS (+)/STING (-) cells (Supplementary Fig. 3f-i).
Tumor cGAS and host STING mediates vascular normalization and anti-tumor immune response in an interdependence manner
We further assessed the roles of host STING in tumor cGAS-mediated vascular normalization and anti-tumor immune response. Cgasproficient cells and ctrl cells were implanted in Sting-deficient (Sting-1-) mice or WT mice, respectively. We found that WT mice
Fig. 1 | Tumor cGAS regulates vascular normalization and anti-tumor immune response in an intrinsic STING-independent manner. a Cgas and Sting protein levels in Hepal-6 cells with Sting knockout following Cgas overexpression. Tumor growth curves (b) and tumor burdens (c) in WT mice injected subcutaneously with indicated cells for 3 weeks ( mice per group). d Normalized absorbance of Evans blue in indicated tumors ( ). e Quantification for pericyte ( ) coverage of tumor vessels (CD31 ) and cells in indicated tumors ( ). HPF, high power field. Quantification for VEGFR2 tumor vessels ( ) in indicated tumors ( ). g Cgas and Sting protein levels in Hepal-6 cells with Cgas overexpression following Sting knockout. Tumor growth curves (h) and tumor burdens (i) in WT mice injected subcutaneously with indicated cells for 3 weeks ( mice per group). Representative immunofluorescence images and quantification for
pericyte ( ) coverage of tumor vessels ( ) and cells in indicated tumors ( ). HPF, high power field. Scale bars, Cgas protein levels in Hepa1-6 cells with Cgas overexpression. 1 Intercellular and extracellular cGAMP levels from Hepal-6 cells with Cgas overexpression ( ). Tumor growth curves (m) and tumor burdens (n) in WT mice injected subcutaneously with indicated cells for 3 weeks ( mice per group). o Representative immunofluorescence images and quantification for pericyte ( ) coverage of tumor vessels ( ) and cells in indicated tumors ( ). HPF, high power field. Scale bars, values are calculated using two-way ANOVA (b, h, m), one-way ANOVA (c-f, l, n, o) and two-tailed unpaired Student’s test ( ). ns not significant. Representative of independent experiments ( ). Source data are provided as a Source
Data file.
implanted with Cgas-proficient cells developed significantly smaller tumors than WT mice injected with parental cells (Fig. 2a, b), while Sting-/- mice implanted with Cgas-proficient cells developed comparable tumors than parental ctrl cells (Fig. 2c, d), suggesting that tumor cGAS-caused tumor repression depends on host STING. In contrast to Ctrl cells-derived tumors, a marked increase in pericyte coverage of tumor vessels was observed in Cgas-proficient tumors in WT mice (Fig. 2k). Furthermore, a significant reduction in intratumoral hypoxia, as shown by a hypoxia marker glucose transporter 1 (GLUT1) level , and a substantial increase in cells infiltration were detected in Cgas-proficient tumors versus Ctrl tumors in WT mice (Fig. 2k). Additionally, downstream of STING activation IFN and IFN-stimulated genes (ISGs), vascular stabilizing genes, and endothelial-lymphocyte interaction-associated adhesion molecules were up-regulated in Cgas-proficient tumors versus Ctrl tumors in WT mice (Supplementary Fig. 4a). Importantly, no obvious differences in tumor burdens, vascular normalization, cells infiltration were found in STING-deficient (Sting-/) mice implanted with Cgas-proficient cells versus Ctrl cells (Fig. 2c, d, k, Supplementary Fig. 4b). Together, these results indicate that tumor cGAS-mediated
tumor repression, vascular normalization, and anti-tumor immune response rely on host STING.
We next challenged Cgas or WT mice with Cgas-proficient cells to address whether these tumor cGAS-mediated phenomenon are determined by host cGAS. Intriguingly, no significant difference in tumor growth was detected between and WT mice (Fig. 2e, f). Moreover, Cgas-deficient mice or WT mice challenged with Cgasproficient cells showed comparable tumor vessel coverage by pericytes, intratumoral hypoxia, T cell infiltration, and vascular normalizing genes expressions (Fig. 2k, Supplementary Fig. 4c). These findings suggest that host cGAS is not required for tumor cGASmediated mediated vascular normalization and anti-tumor immune response.
To test whether host STING relies on tumor cGAS to regulate vascular normalization and anti-tumor immune response, we challenged Sting-/- mice and WT mice with Cgas-proficient cells or ctrl cells, respectively. Ctrl cells developed similar tumor burdens in Sting-/- mice versus WT mice (Fig. 2g, h), whereas Cgas-proficient cells exhibited faster growth in Sting-/- mice versus WT mice (Fig. 2i, j). In contrast to Ctrl cells-derived tumors, a marked reduction in
Fig. 2 | Tumor cGAS and host STING mediates vascular normalization and antitumor immune response in an interdependence manner. Tumor growth curves (a) and tumor burdens (b) in WT mice injected subcutaneously with Hepal-6-Cgas or Ctrl cells for 3 weeks ( mice per group). Tumor growth curves (c) and tumor burdens (d) in Sting-/- mice injected subcutaneously with Hepal-6-Cgas or Ctrl cells for 3 weeks ( mice per group). Tumor growth curves (e) and tumor burdens (f) in or WT mice injected subcutaneously with Hepa1-6-Cgas cells for 3 weeks ( mice per group). Tumor growth curves (g) and tumor burdens (h) in Sting-/- or WT mice injected subcutaneously with Hepal-6-Ctrl cells for 3 weeks
( mice per group). Tumor growth curves (i) and tumor burdens (j) in Sting or WT mice injected subcutaneously with Hepal-6-Cgas cells for 3 weeks ( mice per group). Representative immunohistochemical images and quantification for pericyte ( ) coverage of vessels ( ), GLUT1 hypoxic area, and CD8 T cells in indicated tumors ( ). The yellow arrows represent cells. Scale bars, values are calculated using two-way ANOVA (a, c, e, g, i) and twotailed unpaired Student’s test ( ). ns, not significant. Source data are provided as a Source Data file.
pericyte coverage of tumor vessels and cells infiltration, but a significant increase in intratumoral hypoxia were observed in Cgasproficient tumors from Sting mice versus WT mice (Fig. 2k). Additionally, no obvious differences in downstream of STING activation IFN and ISGs, vascular stabilizing genes, and endotheliallymphocyte interaction-associated adhesion molecules were observed in ctrl tumors from Sting-/- mice versus WT mice, but these
genes were down-regulated in Cgas-proficient tumors from Sting-/mice versus WT mice (Supplementary Fig. 4d, e). These findings demonstrate that host STING-mediated tumor repression, vascular normalization, and anti-tumor immune response depend on tumor cGAS. Collectively, these findings indicate that tumor cGAS and host STING mediates vascular normalization and anti-tumor immune response in an interdependence manner.
Tumor cGAS produces cGAMP to activate endothelial STING and promote lymphocyte trafficking
To further explore the crosstalk of cGAS-STING between tumor cells and host cells, we first assessed cGAS and STING expression in tumor microenvironment. In Human Protein Atlas (HPA), cGAS was positive expressed in liver cancer cells from most patients, but positive STING expression in cancer cells was only detected in one of 12 patients (Supplementary Fig. 5a). Similarly, of the 103 HCC patients from our hospital (EHBH) showed positive expression of cGAS in cancer cells, while only less than of the patients displayed positive STING expression in cancer cells (Supplementary Fig. 5b). In contrast, most distinct STING expression was identified in endothelial cells compared with other host cells in human liver tumor microenvironment by single cell type clusters (Supplementary Fig. 5c). Furthermore, STING expression in tumor vessels was verified in serial sections of HCC samples from the same patient (Supplementary Fig. 5d). Therefore, these results suggest that liver cancer cells express cGAS, but hardly express STING, whereas endothelial cells show most distinct STING expression and they are most important target of cGAMP, as they are more abundant than other host cells in tumor microenvironment. Additionally, although T cell is an important contributor to tumor vessel normalization , tumor cGAS was sufficient to repress tumor angiogenesis and induce vascular normalization in immunodeficient mice (Supplementary Fig. 4a-e), excluding the essential role of T cellintrinsic STING in tumor cGAS-mediated tumor vasculature remodeling. Together, these data support the preferential crosstalk between tumor cGAS and endothelial STING in live caner, which is highly angiogenic.
Gene set enrichment analysis (GSEA) (Panther pathway analysis) revealed that tumor cGAS was highly interrelated with both T cell activation and angiogenesis in liver cancer from TCGA (Fig. 3a), indicating the regulatory role of tumor cGAS in vasculature remodeling. We further assessed the correlation between tumor cGAS and endothelial STING activation in human liver cancer. CD31 tumor-associated endothelial cells (TECs) and the remaining non-TEC cells (mainly tumor parenchymal cells) were sorted from liver cancer tissues of the same patient. Importantly, cGAS expression in non-TEC cells was positively correlated with STING and ISGs expressions in paired TECs (Fig. 3b). Moreover, compared to the matched TECs, CD31 normal endothelial cells (NECs) expressed increased STING and ISGs (Fig. 3c), confirming the role of endothelial STING in vascular normalization. Next, we verified the activation of endothelial STING by liver cancer cell cGAS through in vitro experiments. After confirming that Cgas overexpression boosted cGAMP secretion in liver cancer cells by detecting the levels of cGAMP in intracellular and extracellular media (Fig. 3d), we then tested whether cGAMP produced by liver cancer cells could activate STING in surrounding endothelial cells, using the formation of perinuclear STING aggregates as readout for STING activation by cGAMP . The formation of Sting aggregates upon cGAMP stimulation was first confirmed in endothelial cells expressing StingCherry plasmid following endogenous Sting depletion (Supplementary Fig. 6a, b). Then, Sting-Cherry-expressed endothelial cells were cocultured with GFP-labeled Cgas and GFP-labeled Ctrl cells, respectively. We found that Sting aggregates were much more prominent in endothelial cells co-cultured with Cgas cells versus Ctrl cells (Fig. 3e). Additionally, compared with Ctrl cells-derived conditioned medium (CM), stimulation of endothelial cells by Cgas cell-derived CM (much more cGAMP in CM form Cgas cells versus Ctrl cells were confirmed in Fig. 3d) led to Sting activation, as shown by elevated phosphorylation of Sting and Tbk1, as well as increased Ifn and ISGs expressions (Fig. 3f, g).
We next investigated the role of endothelial STING activation by liver cancer cells in endothelial cells functionalities. Direct activation of Sting pathway by cGAMP (cGAMP-induced STING pathway activation was confirmed in Supplementary Fig. 6c, d) inhibited endothelial cell
proliferation and their tube-like structure formation ability in the Matrigel assay (Supplementary Fig. 6e, g). Similarly, Cgas cell-derived CM treatment also partially repressed endothelial cell proliferation and tube formation (Supplementary Fig. 6f, h). Additionally, the migratory characteristics of endothelial cells were suppressed after coculture with Cgas cells, as shown in the tumor-chemotaxis assay (Fig. 4a). Since tumor vascular normalization improves lymphocyte infiltration into tumors , we evaluated whether STING-activated endothelial cells supported more lymphocytes infiltration. By utilizing a modified lymphocytes transendothelial migration assay as described previously , we found that more lymphocytes transmigrated through the cGAMP-treated endothelial cell barrier versus Ctrl barrier (Fig. 4b) or endothelial cell barrier co-cultured with Cgas cells versus Ctrl cells (Fig. 4c). However, Sting-deficient lymphocytes and WT lymphocytes did not display any difference in transendothelial migration (Fig. 4d, e). Therefore, high cGAS expressed cancer cells enhances lymphocytes transendothelial migration by activation of endothelial STING, rather than lymphocytes STING pathway, through cGAMP secretion.
Mechanistically, endothelial cells exhibited increased vascular endothelial (VE)-cadherin (VE-Cad) expression following cGAMP treatment (Fig. 4f) or mixed culture with Cgas cells versus Ctrl cells (Fig. 4g), suggesting that Cgas cell-induced endothelial STING activation maintains the stability of endothelial cell junctions, which are crucial for leukocyte trafficking . Additionally, after Sting activation by cGAMP or Cgas cell-derived CM stimulation, endothelial cells displayed increased expression of adhesion molecules involved in endothelial-lymphocyte interaction including Icam, Vcam, E-selectin (Sele), and L-selectin (Sell) (Fig. 4h, i). In contrast, Sting deficiency impaired these adhesion molecules expressions (Fig. 4j). Furthermore, blocking the IFN signaling pathway by STAT1 activation inhibitor fludarabine (Flura) reversed cGAMP-induced Ccl5 expression, but not Icam expression (Supplementary Fig. 6i), and IFN treatment increased Ccl 5 expression, but has no obvious effect on Icam expression in endothelial cells (Supplementary Fig. 6j), suggesting the regulation of endothelial-lymphocyte interaction-associated adhesion molecules mainly by STING pathway, rather than its target genes IFN and ISGs. Collectively, these data indicate that tumor cGAS produces cGAMP to activate STING pathway in endothelial cells, further suppressing angiogenesis and promoting lymphocyte trafficking via maintenance of endothelial cell junction stability and upregulation of endothelial-lymphocyte interaction-associated adhesion molecules.
Tumor-derived cGAMP transport via LRRC8C channels to activate STING in endothelial cells
We next investigate how cGAMP produced by cGAS in liver cancer cells is transported to endothelial cells. In addition to the gap junction, which directly connects adjacent cells , recent studies have reported that cGAMP is transmitted between cells through solute carriers (SLCs) including SLC19A1 and SLC46A2, and volume-regulated anion channels (VRACs), formed by LRRC8 heteromers . Since Cgas cellsderived CM was sufficient to activate endothelial STING (Fig. 3e-g), gap junctions were excluded. Interestingly, among above reported SLCs and VRACs, tumor LRRC8C expression was most positively correlated with cGAS-STING and vascular normalization-associated genes including vascular stabilization, endothelial-lymphocyte interaction, pericytes and T cell chemotaxis in TCGA database (Fig. 5a). Additionally, the strongest positive correlation was found between tumor LRRC8C expression and T cell infiltration (Fig. 5b). Among LRRC8A-E, high tumor LRRC8C expression predicted better prognosis in liver cancer patients (Supplementary Fig. 7a-e). Similar to tumor cGAS, GSEA (Panther pathway analysis) showed that tumor LRRC8C was highly interrelated with both T cell activation and angiogenesis in liver cancer from TCGA dataset (Fig. 5c). Therefore, these results by bioinformatic analysis imply that LRRC8C may mediate the
Fig. 3 | Tumor cGAS produces cGAMP to activate STING in endothelial cells.
a GSEA (Panther pathway analysis) of cGAS in liver cancer from TCGA ( ).
b Correlation analysis of cGAS expression in non-tumor-associated endothelial cell (non-TEC) cells with STING and ISGs expressions in paired TECs from liver cancer tissues of the same patient via magnetic-activated cell sorting (MACS) in the GSE51401 dataset ( pairs). c STING and ISGs expressions of TECs versus normal endothelial cells (NECs) in the GSE51401 dataset ( pairs). d Intercellular and extracellular cGAMP levels from Hepa1-6-Cgas or Ctrl cells ( ). e Representative immunofluorescence images and quantification for Sting aggregates in SVEC4-10
cells with Sting-Cherry overexpression co-cultured with GFP-labeled Hepal-6-Cgas or Ctrl cells ( ). The white arrows represent Sting aggregates in SVEC4-10 cells. Scale bars, . f Protein levels of markers in the Sting pathway in SVEC4-10 cells after exposure to CM from Hepal-6-Cgas or Ctrl cells. Representative of independent experiments. mRNA levels of Ifn and ISGs in SVEC4-10 cells after exposure to CM from Hepal-6-Cgas or Ctrl cells ( ). values are calculated using two-tailed Pearson correlation coefficient (b), two-tailed paired Student’s test (c) and unpaired Student’s test ( ). Source data are provided as a Source Data file.
transmission of cGAMP between liver cancer cells and vascular endothelial cells.
To further test the role of LRRC8C in cGAMP transportion, Lrrc8c was knocked down in Cgas cells (Fig. 5d). It was found that the intracellular level of cGAMP was not obvious affected, while the
extracellular level of cGAMP was significantly reduced (Fig. 5e), indicating that LRRC8 mediates the secretion of cGAMP in liver cancer cells. Furthermore, endothelial Sting activation by Cgas cells-derived CM was dramatically weakened after Lrrc8c knockdown, as shown by decreased phosphorylation of Sting and Tbk1, as well as Ifn and ISGs
Fig. 4 | Tumor cGAS-induced endothelial STING activation promotes lymphocyte trafficking. a Representative images and quantifications of SVEC4-10 cells that migrated towards Hepa1-6-Cgas or Ctrl cells in Transwell assays ( ). b Transendothelial migration (TEM) of mouse splenic lymphocytes through SVEC410 cell barrier with cGAMP pre-treatment or not ( ). c TEM of mouse splenic lymphocytes through SVEC4-10 cell barrier with pre-treatment of CM from Hepal-6-Cgas or Ctrl cells ( ). Protein levels of Sting in splenic lymphocytes from Sting or WT mice. Representative of independent experiments. e TEM of Flow cytometric analysis and mean fluorescence intensity (MFI) of surface VECad in SVEC4-10 cells after cGAMP treatment (f) ( ), co-cultured with Hepal-6Cgas or Ctrl cells mRNA levels of indicated genes in SVEC4-10 cells after cGAMP treatment ( ). i mRNA levels of indicated genes in SVEC4-10 cells cocultured with Hepal-6-Cgas or Ctrl cells ( ). mRNA levels of indicated genes in SVEC4-10 cells with Sting knocked out (sgSting) or Ctrl cells (sgCtrl) ( ). values are calculated using unpaired Student’s test (a-c, e-j). ns, not significant. Source data are provided as a Source Data file. Sting-/- or WT mice-derived lymphocytes through SVEC4-10 cell barrier ( ).
expressions (Fig. 5f, g), together suggesting that cGAMP produced by liver cancer cells is secreted out of the cell through LRRC8C channels, thereby activating STING of endothelial cells. Since LRRC8 channels have been proved to be a bidirectional cGAMP transporter , we next determined whether cGAMP secreted by cancer cells enters endothelial cells also through LRRC8C. When Lrrc8c expression was blocked in endothelial cells, either cGAMP (Fig. 5h, i) or Cgas cells-derived CM (Fig. 5j, k) failed to activate endothelial Sting pathway, as evidenced by reduced phosphorylation of Sting and Tbk1, as well as decreased Ifn and ISGs expressions, indicating that endothelial cells use LRRC8C channels to import cGAMP, similar to previous reports . Furthermore, knockdown of Lrrc8c in Cgas cells or endothelial cells also significantly repressed cGAMP or Cgas cells-derived CM induced endotheliallymphocyte interaction-associated adhesion molecules expressions in endothelial cells (Fig. 51-n). Together, these data indicate that LRRC8C channels mediate cGAMP transmission from liver cancer cells to vascular endothelial cells, thereby activating endothelial STING pathway.
TET2 synergizes with IL-2/STAT5A signaling to promote tumor cGAS expression and cGAMP secretion
Although cGAS was positive expressed in HCC, its cGAS expression at transcription level was the lowest among pan-cancer, accompanied by the highest methylation of cGAS gene promoter (Fig. 6a). Indeed, cGAS expression levels were negatively correlated with their methylation levels ( ) in HCC from TCGA dataset (Fig. 6b). DNA demethylation is mainly controlled by TET dioxygenases, which
are capable of converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine ( 5 hmC. Interestingly, among TET family members, tumor suppressor TET2 expression was most positively correlated with cGAS expression and no negative correlation between tumor cGAS expression and DNA methyltransferases (DNMTs) was observed in HCC from TCGA and ICGC datasets (Fig. 6c). To further investigate whether TET2 regulates cGAS expression, we overexpressed Tet1, Tet2, and Tet3 in liver cancer cells, respectively. Notably, Tet2 overexpression significantly increased Cgas expression in comparison to Tet1 and Tet3, at both the transcript and protein levels (Fig. 6d, e). Furthermore, Tet2 overexpression also enhanced Cgas enzyme activity, as supported by the elevated intracellular and extracellular levels of cGAMP (Fig. 6f). Together, these findings suggest that TET2 is a major mediator of cGAS expression in liver cancer.
Because TET family often interacts with other signaling, especially JAK/STAT pathway, to synergistically mediate target gene expression , we questioned whether TET2 cooperated with STAT signaling to regulate cGAS expression. Surprisingly, among STAT family members (STAT1-6), STAT5A showed a most positive correlation with cGAS in the high TET2 expression group rather than in low TET2 expression group from HCC ICGC datasets (Fig. 6g). In vitro, disruption of Stat5a, but not Stat1, which exhibited a positive correlation with cGAS regardless of TET2 expression (Fig. 6g), impaired Cgas expression in liver cancer cells (Fig. 6h, i). Conversely, activation of Stat5a upon IL-2 stimulation upregulated Cgas and this IL-2-induced Cgas was reversed after Stat5a knockdown (Fig. 6j), indicating the
Fig. 5 | Tumor-derived cGAMP transport via LRRC8C channel to activate STING in endothelial cells. a Pearson’s correlation of SLC (SLC19A1/SLC46A2) and VARC (LRRC8A-E) expressions with genes related to cGAS-STING, vascular stabilization, endothelial-T cell interaction, and pericyte/T cell chemotaxis in HCC from TCGA dataset ( ). b Pearson’s correlation of SLC (SLC19A1/SLC46A2) and VARC (LRRC8A-E) expressions with T cell infiltration in HCC from TCGA dataset through TIMER and MCP-counter ( ). c GSEA (Panther pathway analysis) of LRRC8C in liver cancer from TCGA ( ). Protein levels of Cgas and Lrrc8c in Hepal-6-Cgas cells with Lrrc8c knocked down by siRNA (Hepal-6-Cgas-siLrrc8c) or Ctrl cells (Hepa1-6-Cgas-siCtrl). e Intercellular and extracellular cGAMP levels from Hepal-6-Cgas-siLrrc8c or Cgas-siCtrl cells ( ). Protein levels of markers in the Sting pathway in SVEC4-10 cells after exposure to CM from Hepal-6-Cgas-siLrrc8c or Cgas-siCtrl cells. mRNA levels of Ifn and ISGs in SVEC4-10 cells after exposure to CM from Hepa1-6-Cgas-siLrrc8c or Cgas-siCtrl cells ( ). Protein levels of
Lrrc8c and markers in the Sting pathway in SVEC4-10-siLrrc8c or siCtrl cells after cGAMP treatment. i mRNA levels of Ifn and ISGs in SVEC4-10-siLrrc8c or siCtrl cells after cGAMP treatment ( ). Protein levels of Lrrc8c and markers in the Sting pathway in SVEC4-10-siLrrc8c or siCtrl cells after exposure to CM from Hepal-6Cgas cells. mRNA levels of Ifn and ISGs in SVEC4-10-siLrrc8c or siCtrl cells after exposure to CM from Hepal-6-Cgas cells ( ). 1 mRNA levels of indicated genes in SVEC4-10 cells after exposure to CM from Hepal-6-Cgas-siLrrc8c or Cgas-siCtrl cells ( ). mRNA levels of indicated genes in SVEC4-10-siLrrc8c or siCtrl cells after cGAMP treatment ( ). mRNA levels of indicated genes in SVEC4-10-siLrrc8c or siCtrl cells after exposure to CM from Hepal-6-Cgas cells ( ). values are calculated using two-tailed unpaired Student’s test ( ). ns, not significant. Representative of independent experiments ( ). Source data are provided as a Source Data file.
regulatory role of IL-2/STAT5A signaling in cGAS expression. However, IL-2 treatment failed to induce Cgas expression after Tet2 deficiency (Fig. 6k). In contrast, Tet2 overexpression in combination with IL-2 stimulation led to more increase in Cgas expression than either alone (Fig. 61), suggesting that TET2 synergizes with STAT5A signaling to promote tumor cGAS expression. Mechanistically, Tet2 interacted with activated Stat5a (p-Stat5a) and this interaction was stronger upon
IL-2 stimulation (Fig. 6m). Furthermore, Tet2 bound to the Cgas promoter and this binding was significantly enhanced in the presence of IL-2 (Fig. 6n). To further verify the role of Tet2 in Cgas demethylation, Tet2 overexpression was sufficient to attenuate methylation within the promoter of Cgas and more reduction in Cgas promoter methylation was observed when combined with IL-2 stimulation (Fig. 60). Although IL-2 is well known for driving T cell responses by binding to its
Fig. 6 | TET2 synergizes with IL-2/STAT5A signaling to promote tumor cGAS expression and cGAMP secretion. a Correlation between the median expression and methylation of cGAS in 18 types of tumors from TCGA datasets. Correlation between cGAS expression and methylation in HCC from TCGA dataset ( ). c Correlation between cGAS and TETs or DNMTs expressions in HCC from TCGA ( ) and ICGC ( ) datasets. Cgas and Tet1-3 mRNA levels in Hepal-6 cells with respective Tet1-3 overexpression by plasmids ( ). e Cgas protein level in Hepal-6 cells with respective Tet1-3 overexpression by plasmids. f Intercellular and extracellular cGAMP levels from Hepal-6 cells with respective Tet1-3 overexpression by plasmids . g Correlation between STAT1-6 and cGAS expressions in low and high TET2 expression groups from HCC-ICGC dataset. The low and high expression groups were divided relative to the median expression values. h Stat5a and Cgas mRNA levels in Hepa1-6 cells with Stat5a knocked down by siRNA
(siStat5a) ( ). i Stat1 and Cgas mRNA levels in Hepal-6 cells with Stat1 knocked down by siRNA (siStat1) ( ). Cgas mRNA level in Hepa1-6-siStat5a or siCtrl cells upon IL-2 stimulation ( ). Cgas mRNA level in Hepal-6 cells with Tet2 knocked down by shRNA ( Tet2 ) ( ). I Cgas mRNA level in Hepal-6 cells with Tet2 overexpression upon IL-2 stimulation ( ). m Co-IP analysis of Tet2 with p-Stat5a in Hepal-6 cells with Tet2 overexpression upon IL-2 stimulation. n ChIP-qPCR analysis of Tet2 binding activity to the promoter of Cgas in Hepal-6 cells with FlagTet2 overexpression upon IL-2 stimulation ( ). o Pyrosequencing analysis and quantification of the promoter methylation status of Cgas in Hepal-6 cells with Tet2 overexpression upon IL-2 stimulation ( ). values are calculated using twotailed Pearson correlation coefficient ( ), one-way ANOVA ( ) and twotailed unpaired Student’s test ( ). ns not significant. Representative of independent experiments ( ). Source data are provided as a Source Data file.
receptors, which consists of three subunits including IL-2R (IL2RA), IL-2R (IL2RB), and IL-2R (IL2RG) , we found that both IL2RB and IL2RG were expressed in numerous liver cancer cell lines, especially IL2RG, in CCLE database (Supplementary Fig. 8a, b) and similar results were confirmed in liver cancer samples from TCGA dataset (Supplementary Fig. 8c). Since IL2RB and IL2RG are sufficient to form functional receptor for transmitting signals from IL- , liver cancer cells could also respond to IL-2 secreted by T cells and led to STAT5A activation. Together, these results indicate that IL-2-activated STAT5A recruits TET2 binding to the cGAS locus to promote cGAS demethylation and transcription, leading to cGAS upregulation in liver cancer.
VC-induced TET2 and dsDNA leakage activate tumor cGAS-cGAMP-endothelial STING pathway and promote lymphocyte trafficking
TET2 expression is significantly decreased in HCC, but rarely mutated . Therefore, non-mutational loss of TET2 activity in HCC prompted us to explore the possibility of reactivating TET2 as a strategy to efficiently stimulate cGAS expression. Acting as a cofactor, VC (ascorbic acid or ascorbate) promotes TETs activity and has been proposed as a promising anti-cancer agent . Excitingly, VC treatment dramatically induced cGAS expression at both the transcript and protein levels in liver cancer cells in vitro (Fig. 7a, b). Consistent with
in vitro results, cGAS expression was upregulated in the tumors of
activity via hydrogen peroxide-induced oxidative stress . We found that VC treatment induced DNA damage (Supplementary Fig. 9a, b) and cause cytoplasmic leakage of nuclear DNA and mitochondrial DNA (mtDNA) (Fig. 7d), which is known to directly activate cGAS . However, inhibition of VC-induced DNA damage, as marker by phosphorylation of histone 2AX ( ), by ROS scavenger N-acetyl-L-cysteine
Fig. 7 | VC-induced TET2 and dsDNA leakage activate tumor cGAS-cGAMPendothelial STING pathway and promote lymphocyte trafficking. a, b Cgas mRNA (a) and protein (b) levels in VC-treated Hepal-6 cells ( ). c Cgas protein level in Hepal-6 cells-derived tumor after VC or Sora treatment for 2 weeks. d Immunofluorescence costaining of dsDNA, HSP60, and DAPI in VC-treated Hepa16 cells. Scale bars, . e Cgas mRNA level in VC-treated Hepal-6 cells following NAC pretreatment ( ). Cgas mRNA level in VC-treated Hepal-6 shTet2 cells and shCtrl cells . g Cgas mRNA level in VC-treated Hepal-6 cells following TFMB-2HG pretreatment ( ). h Intercellular and extracellular cGAMP levels from VCtreated Hepal-6 cells ( ). i Pyrosequencing analysis and quantification of the promoter methylation status of Cgas in VC-treated Hepa1-6 cells ( ). j Immunofluorescence staining and quantification for Sting aggregates in SVEC4-10 cells with Sting-Cherry overexpression co-cultured with GFP-labeled Hepa1-6 cells after VC treatment ( ). The white arrows represent Sting aggregates in SVEC4-10
cells. Scale bars, mRNA levels of Ifn and ISGs in SVEC4-10 cells after exposure to CM from VC-treated Hepal-6 sgCgas cells versus sgCtrl cells (k) or Hepal-6 shTet2 cells versus shCtrl cells (l) ( ). m-o TEM of mouse splenic lymphocytes through SVEC4-10 cell barrier with pretreatment of CM from Hepal-6 cells (m), Hepa1-6 sgCgas cells (n), or Hepa1-6 shTet2 cells (o) treated with VC or not ( ). Flow cytometric analysis and MFI of surface VE-Cad in SVEC4-10-GFP cells co-cultured with Hepal-6 cells after VC treatment ( ). , mRNA levels of VECad ( ) and indicated genes ( ) in SVEC4-10 cells after exposure to CM from VCtreated Hepa1-6 sgCgas cells versus sgCtrl cells ( ) or Hepal-6 shTet2 cells versus shCtrl cells ( ) ( ). values are calculated using two-tailed unpaired Student’s test (a, e-h, j, k-t) and paired Student’s test (i). ns, not significant. Representative of independent experiments ( ). Source data are provided as a Source Data file.
(NAC), failed to reverse the elevated expression of Cgas after VC stimulation (Supplementary Fig. 9a and Fig. 7e). Together, these findings indicate that despite pharmacological VC activates cGAS via ROSinduced dsDNA leakage, VC-mediated cGAS expression is independent of oxidative stress. We further determined whether VC promotes cGAS expression through TET2-dependent DNA demethylation. Tet2 blockade by either Tet2 knockdown or Tet enzyme inhibitor (cellpermeable 2-hydroxyglutarate (2-HG)) significantly decreased VCinduced Cgas expression in liver cancer cells (Fig. 7f, g). Consistent with Tet2 overexpression (Fig. 60), VC treatment obviously reduced the methylation level of Cgas promoter (Fig. 7i). Collectively, these findings imply that pharmacological VC not only activates tumor cGAS through cytoplasmic dsDNA leakage, but also increases tumor cGAS expression via epigenetic modifications by Tet2-dependent DNA demethylation.
To next assessed whether VC-induced tumoral cGAS activates endothelial STING and enhances lymphocyte trafficking, an obvious increase in cGAMP production and secretion in liver cancer cells after VC treatment was firstly confirmed (Fig. 7h). VC treatment resulted in much more marked formation of perinuclear Sting aggregates, which are readout for STING activation by cGAMP , in Sting-Cherryexpressed endothelial cells when co-cultured with GFP-labeled Hepa16 cells (Fig. 7j), whereas VC treatment on endothelial cells alone neither induced more Sting aggregates nor upregulated Cgas expression (Supplementary Fig. 10a-c), implying that VC indirectly activates endothelial Sting through cancer cells. Similarly, CM derived from VCtreated Hepal-6 cells significantly activated endothelial Sting, as evidenced by elevated phosphorylation of Sting and Tbk1, as well as increased Ifn and ISGs expressions (Supplementary Fig. 9c, d), whereas VC treatment on endothelial cells alone led to no obvious difference in endothelial Sting activation (Supplementary Fig. 10d, e). Importantly, when the Cgas or Tet2 expression was blocked (Supplementary Fig. 9e), these cells-derived CM after VC stimulation failed to activate endothelial STING (Supplementary Fig. 9f, g and Fig. 7k, l). Furthermore, we identified that more lymphocytes transmigrated through endothelial cell barriers stimulated by CM from VC-treated Hepal-6 cells versus PBS-treated cells (Fig. 7m). After exposure to CM from VC-treated Hepa1-6 cells with Cgas or Tet2 deficiency, endothelial cell barriers did not promote transendothelial migration of lymphocytes (Fig. 7n, o). Therefore, these results suggest that VC-induced TET2 upregulates tumor cGAS to activate STING pathway in endothelial cells and promote lymphocyte trafficking.
Mechanistically, after exposure to VC, endothelial cells in mixed culture with Hepal-6 cells displayed increased VE-Cad expression, a marker of endothelial cell junction stability, whereas VC treatment on endothelial cells alone had no notable effect on their VE-Cad expression (Supplementary Fig. 10f). Similarly, CM derived from VC-treated Hepa1-6 cells upregulated VE-Cad and endothelial-lymphocyte inter-action-associated adhesion molecules in endothelial cells compared with CM derived from PBS-treated Hepa1-6 cells (Fig. 7p and
Supplementary Fig. 9h, i), whereas no change in endotheliallymphocyte interaction-associated genes were observed in endothelial cells after direct VC treatment (Supplementary Fig. 10g). In contrast, disruption of either Cgas or Tet2 abrogated increased VE-Cad and endothelial-lymphocyte interaction-associated adhesion molecules expressions in endothelial cells induced by CM derived from VCtreated Hepal-6 cells (Fig. 7q-t). Together, these data indicate that VC induces TET2 activation to upregulates tumor cGAS, further producing cGAMP to activate endothelial STING and enhance transendothelial migration of lymphocytes via maintenance of endothelial cell junction stability and upregulation of endothelial-lymphocyte interactionassociated adhesion molecules.
Tumor TET2-p-STAT5A-cGAS-LRRC8C-endothelial STING axis correlates with vascular normalization and immune infiltration in human liver cancer
Next, we investigated the protein expression levels of tumor TET2-p-STAT5A-cGAS-LRRC8C-endothelial STING axis in tumor biopsies from liver cancer patients. In serial sections of HCC samples from the same patient, high tumor LRRC8C expression was associated with high STING expression in CD31 vascular endothelial cells in either high or low tumor cGAS expression group (Fig. 8a, b). Importantly, when tumor LRRC8C expression was high, endothelial STING expression was significantly higher in high tumor cGAS expression group versus low tumor cGAS expression group (Fig. 8a, b), indicating that tumor cGAS expression correlates with endothelial STING activation depending on LRRC8C.
Compared with tumors with either low cGAS or LRRC8C expression, pericyte coverage of tumor vessels, which is one of the hallmarks of tumor vessel normalization , was dramatically increased in tumors with both high cGAS and LRRC8C expressions (Fig. 8a, c). Tumor LRRC8C expression negatively correlated with vascular invasion when cGAS was highly expressed, whereas no obvious correlation was shown in low cGAS expression group (Fig. 8a, d). Consistently, cGAS and LRRC8C expressions were downregulated in eight of 11 portal vein tumor thrombosis (PVTT) in comparison to corresponding primary tumors (Fig. 8e). Furthermore, high tumor LRRC8C expression was associated with low hypoxia marker GLUT1 expression in the high cGAS expression group, but not when cGAS was low expressed in HCC tissues (Fig. 8f, g). To further assess immune infiltration, tumor LRRC8C expression positively correlated with intratumoral cell infiltration, especially when tumor cGAS expression was high (Fig. 8f, h). Together, these results suggest that tumor cGAS-LRRC8C-endothelial STING axis was associated with vascular normalization and immune infiltration in human liver cancer.
We further verified the regulation of tumor cGAS by TET2 and STAT5A signaling in human liver cancer samples and found that high tumor TET2 and p-STAT5A expressions were associated with high tumor cGAS expression (Fig. 8i, j). In TCGA database, tumor TET2, STAT5A, and cGAS expressions positively correlated with vascular
normalization-associated genes including vascular stabilization, endothelial-lymphocyte interaction, pericytes and T cell chemotaxis (Fig. 8k). Additionally, a positive correlation was found between tumor TET2, STAT5A, cGAS expression and intratumoral T cell infiltration (Fig. 81). Furthermore, HCC patients with high tumor TET2,
p-STAT5A, cGAS, LRRC8C, and endothelial STING expression had better clinical outcome (Supplementary Fig. 11a-e). Collectively, these data show that tumor TET2-p-STAT5A-cGAS-LRRC8C-endothelial STING axis is linked to vascular normalization and immune infiltration in human liver cancer.
Fig. 8 | Tumor TET2-p-STAT5A-cGAS-LRRC8C-endothelial STING axis correlates with vascular normalization and immune infiltration in human liver cancer.
Representative immunohistochemical images (a) and correlation analysis of tumor cGAS and LRRC8C expressions with endothelial STING expression (b), pericyte ( ) coverage of vessels (CD31 ) (c), and vascular invasion (d) in serial sections of HCC samples from the same patient ( ). Based on the intensity of staining (protein expression), the patients were subdivided into two groups: low (staining score 0-1) and high (staining score 2-3) expression group. Scale bars, . e and mRNA levels between PVTT and the corresponding primary HCC samples ( pairs) (GEO: GSE69164). Representative immunohistochemical images (f) and correlation analysis of tumor cGAS and LRRC8C expressions with GLUT1 expression (g) or T cell infiltration (h) in human liver cancer samples
( ). The dashed line across the violin plots represents the quartiles and the full line depicts the median. Scale bars, . Representative immunohistochemical images (i) and correlation analysis (j) of tumor TET2 and p-STAT5A expressions with cGAS expression in human liver cancer samples ( ). Scale bars, . k Pearson’s correlation of TET2, STAT5A, and cGAS expressions with genes related to vascular stabilization, endothelial-T cell interaction, and pericyte/T cell chemotaxis in HCC from TCGA dataset ( ). I Pearson’s correlation of TET2, STAT5A, and expressions with cell infiltration in HCC from TCGA dataset ( ) through TIMER, MCP-counter, and quanTIseq. values are calculated using Chi-square test (b, d, g, j) and one-way ANOVA (c, h). ns, not significant. Source data are provided as a Source Data file.
VC treatment induces vascular normalization and boosts efficacy of anti-PD-L1 therapy or anti-PD-L1 combination therapy with IL-2
We next investigated the impact of high-dose VC treatment on tumor vasculatures and immune infiltration in vivo. Intraperitoneal injection of VC (equivalent to intravenously), a pharmacological dose widely used in mouse models and showing inhibitory effects against various tumors growth including liver cancer , was administered to immunocompetent mice subcutaneously bearing Hepal-6 tumors (Supplementary Fig. 12a, b). Similar to the effect caused by tumor cGAS and host STING activation, VC treatment led to a reduction in tumor vessels and intratumoral hypoxia, as evidenced by decreased GLUT1 expression, but an obvious increase in pericyte coverage of tumor vessels and intratumoral T cells infiltration (Fig. 9a, Supplementary Fig. 12e). In accordance with in vitro findings, downstream of STING activation IFN and ISGs, vascular stabilizing genes, endothelial-lymphocyte interaction-associated adhesion molecules were up-regulated in VC-treated tumors compared with PBS-treated tumors (Supplementary Fig. 12f). In contrast, although HCC first-line drug Sora treatment also delayed tumor growth and reduced tumor vessel density in mice, it failed to normalize tumor vessels, as shown by no difference in pericyte coverage of tumor vessels and enhanced intratumoral hypoxia, and thereby led to decreased cells infiltration (Fig. 9a, Supplementary Fig. 12c-e). This is in line with previous observations that Sora promoted hypoxiamediated immunosuppression . Therefore, these results suggest that VC treatment, but not Sora, induces normalization of the tumor vasculature and improves immune infiltration in liver cancer.
We evaluated a recent HCC dataset with patients who underwent anti-PD-1/PD-L1 therapy and found that patients with high ISGs expressions benefitted from anti-PD-1/PD-L1 treatment, whereas only 8-16% patients with low ISGs expressions responded to anti-PD-1/ PD-L1 therapy (Supplementary Fig. 13a), imply that cGAS-STING activation may improve the efficacy of immune checkpoint blockade in liver cancer. To exclude that these observations resulted from an impact of tumor size on efficacy of anti-PD-L1 therapy, we adjusted cell numbers of Cgas overexpression cells and chose tumors with similar sizes to Ctrl tumors for anti-PD-L1 therapy. As expected, compared with Ctrl group, anti-PD-L1 treatment resulted in much smaller tumors (Supplementary Fig. 13b, c), coincided with more intratumoral cytotoxic T cells infiltration in Cgas overexpression group with similar sizes to Ctrl tumors before therapy (Supplementary Fig. 13d). Therefore, these findings confirm the enhanced efficacy of anti-PD-L1 therapy by cGAS expression in liver cancer regardless of tumor size.
Since VC is a stimulator of tumor cGAS and endothelial STING activation, as shown in Fig. 6, we tested whether VC treatment sensitizes liver cancer to immunotherapy. In subcutaneously implanted tumor models, striking tumor regressions were observed in mice treated with the combination of VC and anti-PD-L1 in comparison to the single VC or anti-PD-L1 treatment (Fig. 9b, c), suggesting that VC augments PD-L1 checkpoint inhibitor-induced anti-tumor responses. In contrast, consistent with its role in immunosuppression, Sora
treatment displayed no additional anti-tumor activity when combined with anti-PD-L1 antibody (Fig. 9d, e), similar to previous findings . Of note, the combination therapy of VC and anti-PD-L1 exhibited a notable achievement of complete tumor repression approximately 28 days following a treatment period of 21 days in mice (Supplementary Fig. 15a). Therefore, these results indicate that VC combined with anti-PD-L1 treatment achieves complete tumor repression after a long period of therapy. Because subcutaneously implanted tumor models may not fully represent the pathologic microenvironment, we further employed orthotopic liver cancer models, a more reliable representative of human liver cancer . Less infiltration of immune cells including T cells, NK cells, and macrophages were found in subcutaneous versus orthotopic models (Supplementary Fig. 14a-d), indicating that the therapeutic efficacy of anti-PD-L1 may be diminished in orthotopic models. Nevertheless, in line with the results in subcutaneous xenograft models, combined treatment of VC and anti-PD-L1 led to significant smaller tumor burdens than the single IgG Ctrl, VC or anti-PD-L1-treated group in orthotopic tumor models (Fig. 9f-h).
Based on the synergy between TET2 to IL-2/STAT5A signaling (Fig. 6), we further examined whether VC enhanced the efficacy of anti-PD-L1 and IL-2 combination therapy, which is a potentially promising candidate for cancer therapy . Excitingly, in addition to the combined anti-tumor effect of VC and anti-PD-L1, VC treatment further boosted efficacy of anti-PD-L1 combination therapy with IL-2 in both subcutaneously implanted tumor models (Fig. 9i-k) and orthotopic liver cancer models (Fig. 91-n). Remarkably, we observed that this enhanced effect was comparable between orthotopic and subcutaneous tumor models. Collectively, VC treatment induces vascular normalization and boosts efficacy of anti-PD-L1 therapy or anti-PD-L1 combination therapy with IL-2.
Tumor TET2-STAT5A-cGAS-host STING axis mediates VCinduced vascular normalization and therapeutic efficacy of VC combined with anti-PD-L1
To assess whether this synergistic anti-tumor effect is mediated through T cell population, tumor-bearing mice were treated with VC and anti-PD-L1 antibody in the presence of either IgG Ctrl or antiCD 8 antibody to deplete cells (Supplementary Fig. 15b). As expected, the enhanced tumor regressions by combined therapy of VC and anti-PD-L1 coincided with increased infiltration of intratumoral cytotoxic T cells in IgG Ctrl groups (Supplementary Fig. 15c-e). However, after CD8 depletion, confirmed by flow cytometry (Supplementary Fig. 15f), the improved anti-tumor benefit of VC combined with anti-PD-L1 was absent in comparison to single VC or anti-PD-L1 treatment (Supplementary Fig. 15c-e), suggesting the combinational efficiency of VC and anti-PD-L1 therapy depending on cellinduced anti-tumor immune response.
Importantly, in Tet2 or Stat5a deficiency cells-derived tumors, the combination of VC and anti-PD-L1 had no obvious changes in tumor burdens as compared to single VC or anti-PD-L1 therapy (Fig. 10a-d). Moreover, VC therapy did not exert a significant impact on tumor vessel coverage by pericytes, intratumoral hypoxia levels, and
cell infiltration in these tumors with Tet2 or Stat5a deficiency (Fig. 10e, Supplementary Fig. 12g). Furthermore, the knockdown of Tet2 or Stat5a in tumors negated the upregulation induced by VC treatment of IFN and ISGs, vascular stabilizing genes, endothelial-lymphocyte interaction-associated adhesion molecules (Supplementary Fig. 12h). Together, these findings provide conclusive evidence for the crucial involvement of tumor Tet2 and Stat5a in VC-mediated vascular normalizing effects and the combinational efficiency of VC and anti-PD-L1 therapy in liver cancer.
Furthermore, we depleted tumor cGAS and employed STING inhibitor C-176 to block host STING activation. Given the initial low expression level of Cgas in native Hepal-6 cells (Supplementary Fig. 1a), Cgas knockout (KO) in Hepal-6 cells exhibited a marginal augmentation of tumor growth and burdens, which did not reach statistical significance (Supplementary Fig. 15g, h). Nevertheless, Cgas KO in Hepal-6 cells completely abrogated Cgas upregulation induced by VC treatment and in tumors derived from cGAS KO cells, the combination of VC and anti-PD-L1 had no obvious changes in tumor
Fig. 9 | VC treatment induces vascular normalization and boosts efficacy of anti-PD-L1 therapy or anti-PD-L1 combination therapy with IL-2.
a Quantification for CD31 vessels density, pericyte ( ) coverage of vessels ( ), hypoxic area, and cells in VC-treated or Sora-treated tumors ( ). Tumor growth curves ( ) and tumor burdens ( ) of C57BL/6 mice injected subcutaneously with Hepal-6 cells with treatment of PD-L1 and VC either alone or in combination ( mice per group). Tumor growth curves (d) and and tumor burdens (e) of C57BL/6 mice injected subcutaneously with Hepa1-6 cells with treatment of PD-L1 and Sora either alone or in combination ( mice per group). Scheme representing the experimental procedure (f), representative luciferasebased bioluminescence images (g) and quantification (h) of C57BL/6 mice injected
in situ with Hepal-6 cells with pre- and post-treatment of PD-L1 and VC either alone or in combination ( mice per group). Scheme representing the experimental procedure (i), tumor growth curves (j), and tumor burdens (k) of C57BL/6 mice injected subcutaneously with Hepal-6 cells with treatment of VC alone or combined with PD-L1, IL-2, or PD-L1 + IL-2 ( mice per group). Scheme representing the experimental procedure (I), representative luciferase-based bioluminescence images ( ) and quantification ( ) of C57BL/6 mice injected in situ with Hepal-6 cells with pre- and post-treatment of VC alone or combined with L1, IL-2, or PD-L1 + IL-2 ( mice per group). values are calculated using twotailed unpaired Student’s test (a), two-way ANOVA (b, d, j) and one-way ANOVA (c, e, h, k, n). ns not significant. Source data are provided as a Source Data file.
burdens as compared to single VC or anti-PD-L1 therapy (Fig. 10f-h). Similarly, host STING inhibition by C-176 treatment also abrogated the synergistic anti-tumor effect of VC and PD-L1 blockade in vivo (Fig. 10i-k). Moreover, VC therapy had no substantial effect on tumor vessel coverage by pericytes, intratumoral hypoxia, and cell infiltration after tumor cGAS deficiency and host STING inhibition (Fig. 10I, Supplementary Fig. 12i). Additionally, tumoral cGAS KO or host STING inhibition nullified the VC-induced upregulation of IFN and ISGs, vascular stabilizing genes, endothelial-lymphocyte interac-tion-associated adhesion molecules in tumors (Supplementary Fig. 12j). Together, these results confirm the vital role of tumor cGAS and host STING activation in VC-mediated vascular normalizing effects and the combinational efficiency of VC and anti-PD-L1 therapy in liver cancer. Overall, our findings demonstrate that tumor TET2-STAT5A-cGAS-host STING axis mediates VC-induced vascular normalization and therapeutic efficacy of VC combined with anti-PD-L1.
Discussion
cGAS-STING pathway is vital for anti-tumor immunity . In addition to the well-studied anti-tumor immune effect of the cGAS-STING pathway in immune cells , cancer cell-intrinsic cGAS-STING pathway activation has also been recently shown to define their immunogenicity and make tumors hot . However, the crosstalk of cGAS-STING pathway between tumor cells and host cells in tumor microenvironment remains largely unclear. On one side, this work demonstrates that cancer cell-intrinsic expression of cGAS, but not STING, determines tumor vascular normalization and anti-tumor immune response in a host STING-dependent manner. On the other side, host cGAS is dispensable but tumor cGAS is necessary for host STING-mediated vascular normalization and anti-tumor immunity. Together, we identify the interdependence between tumor cGAS and host STING in the regulation of vasculature remodeling and anti-tumor immunity in liver cancer. Mechanistically, tumor cGAS produces cGAMP, which further transports via LRRC8C channels to activate STING in endothelial cells, enhancing recruitment and transendothelial migration of lymphocytes. In turn, intratumoral infiltrating lymphocytes secrete IL-2 to activate tumor STAT5A signaling, which further synergizes with TET2 to epigenetically upregulate tumor cGAS, indicating a positive feedback loop. Accordingly, stimulating tumor TET2 by VC to accelerate this positive feedback loop may effectively induce tumor vascular normalization and boost immunotherapy efficacy.
Currently, intratumoral STING activation with STING agonists has been reported to normalize tumor vasculature . However, whether tumor STING or host STING plays a dominant role remains to be defined. Using Sting-deficient mice, we identify that host STING, rather than cancer cell-intrinsic STING, determines vascular normalization and anti-tumor immune response in highly angiogenic liver cancer. This phenomenon may be explained by several reasons. First, in addition to the recruitment and activation of T cells and pericytes by STING pathway target genes IFN and ISGs such as CCL5 and CXCL10, endothelial STING pathway plays a crucial role in regulating T cells trafficking and endothelial cell function. For instance, the activated STING downstream transcriptional factor IRF3 has been shown to bind
to the promoter of ICAM-1 and induce ICAM-1 expression, one of the key adhesion molecules for transendothelial migration of T cells, in endothelial cells . Similarly, we find that blocking the IFN signaling pathway by STAT1 activation inhibitor fails to reverse cGAMP-induced ICAM-1 expression and IFN treatment has little effect on ICAM-1 expression in endothelial cells, suggesting the regulation of endothelial ICAM-1 expression mainly by STING pathway, rather than its target genes IFN and ISGs. Additionally, recent studies have revealed that when endothelial STING pathway is activated, the downstream kinase TBK1 inhibits endothelial cell proliferation via suppressing YAP pathway . Therefore, although STING activation in cancer cells may also enhance IFN and ISGs production for T cell recruitment, it does not alter the expression of adhesion molecules in endothelial cells or the transendothelial migration of T cells. Furthermore, since endothelial cells have been demonstrated to be the principal source of type I IFNs in growing tumors and endothelial cell-derived IFN initiates cell-mediated anti-tumor immunity , the contribution of IFN produced by cancer cells to T cell priming may be minimal. Collectively, endothelial STING is superior to tumor STING in antitumor immunity, as STING activation in endothelial cells not only enhances T cells recruitment and activation, but also promotes tumor vascular normalization for cells trafficking.
STING is classically activated by cGAS-produced cGAMP and cGAMP is known to be transmitted between cells . Therefore, host STING activation and its vascular normalizing effect may be controlled by tumor cGAS or host cGAS. Surprisingly, we find that host STINGmediated vascular normalization and anti-tumor immune response depends on tumor cGAS, but not host cGAS. It is well accepted that cancer cells are not only the major component of tumor, but also are often constitutively rich in cytoplasmic dsDNA, which further increases upon DNA damaging therapies such as chemotherapy to directly activate cGAS to produce cGAMP . Therefore, cancer cells are the dominant source of cGAMP in the tumor microenvironment, especially when their cGAS expressions are upregulated by TET2 in this study. Furthermore, we show that most liver cancer cells express cGAS, but hardly express STING, in HCC samples, consistent with previous reports showing loss of cancer cell-intrinsic STING in melanoma and colorectal cancer . In contrast, host cells, especially endothelial cells, in tumor microenvironment exhibit distinct STING expression. Since cGAMP is known to be transmitted between cells , cGAMP produced by cGAS in cancer cells, which are frequently lack of intrinsic STING, is prone to be secreted to tumor microenvironment, leading to host STING activation. These reasons may explain why tumor cGAS is essential for host STING activation and thereby host STING-mediated vascular normalization and anti-tumor immune response, whereas cGAS of host cells is dispensable.
cGAS functions as an essential DNA sensor, which senses the cytoplasmic dsDNA and activates the anti-tumor immune response . However, the regulatory mechanism of cGAS expression and activity remained to be fully understood. Hypermethylation of the cGAS gene promoter is associated with its downregulation in liver cancer , but the methylation-dependent regulatory mechanism is unclear. The present work identifies TET2 methylcytosine
dioxygenase as a responsible enzyme for cGAS demethylation and reveal that activated STAT5A recruits TET2 binding to the cGAS locus to promote cGAS demethylation and transcription, resulting in cGAS upregulation in liver cancer cells. Expectedly, we further demonstrate that as a TETs enzyme activator, VC treatment increases cGAS expression via TET2-dependent demethylation in liver cancer cells.
TET2-mediated cGAS expression may be cancer cell-specific, as VC exposure has no obvious effect on cGAS expression in endothelial cells. In addition to TET2-mediated demethylation, further investigation is needed to clarify whether the DNMT family plays a predominant role in influencing the DNA methylation status of the cGAS promoter in our model.
Fig. 10 | Tumor TET2-STAT5A-cGAS-host STING axis mediates VC-induced vascular normalization and therapeutic efficacy of VC combined with anti-PD-
L1. Scheme representing the experimental procedure (a), tumor growth curves (b), tumor images (c), and tumor burdens (d) of C57BL/6 mice injected subcutaneously with Hepa1-6-shTet2 cells, shStat5a cells, or shCtrl cells with treatment of PD-L1 and VC either alone or in combination ( ). e Quantification for vessels density, pericyte ( ) coverage of vessels ( ), hypoxic area, and cells infiltration in shTet2 and shStat5a tumors after VC treatment ( ). Scheme representing the experimental procedure (f), tumor growth curves (g), and tumor burdens (h) of C57BL/6 mice injected subcutaneously with Hepa1-6-sgCgas cells (cGAS KO) with treatment of PD-L1 and VC either alone or in combination . Scheme representing the experimental procedure (i), tumor growth curves (j), and tumor burdens (k) of C57BL/6 mice injected subcutaneously with Hepal-6 cells with treatment of PD-L1 and VC either alone or in combination after STING inhibitor C-176 treatment ( ). 1 Quantification for vessels density, pericyte
( ) coverage of vessels ( ), GLUT1 hypoxic area, and cells infiltration in cGAS KO and STING inhibitor-treated tumors after VC treatment ( ). m Schematic illustration of TET2-mediated tumor cGAS triggering endothelial STING activation to regulate vasculature remodeling and anti-tumor immunity. Left: tumor cGAS produces cGAMP, which further transports via LRRC8C channels to activate STING in endothelial cells, enhancing recruitment and transendothelial migration of lymphocytes. In turn, intratumoral infiltrating lymphocytes secrete IL2 to activate tumor STAT5A signaling, which further synergizes with TET2 to epigenetically upregulate tumor cGAS, indicating a positive feedback loop. Right: stimulating tumor TET2 by VC to accelerate this positive feedback loop may effectively induce tumor vascular normalization and boost immunotherapy efficacy. values are calculated using two-way ANOVA (b, g, j), one-way ANOVA ( ), and two-tailed unpaired Student’s test ( ). ns, not significant. Source data are provided as a Source Data file.
Emerging evidences show that TET methylcytosine dioxygenases regulate tumor immunity . We have demonstrated that metabolism drives IFN -induced PD-L1 checkpoint expression via TET1 and promotes tumor immune evasion . In addition to PD-L1 induction, TET2 has been shown to upregulate chemokines and increase tumorinfiltrating lymphocytes . Collectively, tumoral TETs have been shown to directly regulate interaction between tumor cells and immune cells via immune checkpoint and chemokines so far. However, the highly abnormal and dysfunctional vasculature of tumors is a crucial barrier of immune cell infiltration into tumor parenchyma, thus limiting antitumor immune responses . Therefore, the role of TETs in tumor vasculature remains to be dissected. The present study shows that TET2 upregulates tumor cGAS expression to produce cGAMP, which further activates STING pathway in endothelial cells, leading to tumor vascular normalization and enhanced intratumoral T cell infiltration. Our findings uncover the role of tumor suppressor TET2 in anti-tumor immunity via cGAS-STING-mediated vascular normalization.
Pharmacologic VC has recently emerged as a promising anticancer agent with low toxicity and financial cost according to different pre-clinical and clinical trials . VC is known to exert anti-tumor activity through hydrogen peroxide-induced oxidative stress to preferentially kill cancer cells when used at high-doses or through control of TET enzymes activity to restore the abnormal DNA methylation pattern observed in many tumors . To date, a series of phase I/II clinical trials have demonstrated the safety and efficacy of intravenous high-dose VC as a monotherapy or in combination with radiation and conventional chemotherapies for various cancers . We have previously shown that pharmacologic VC preferentially eradicates liver cancer stem cells and intravenous VC use is linked to improved survival of HCC patients . Although high-dose VC has currently been reported to enhance immunotherapy , yet the immunomodulatory effects of pharmacologic VC have been incompletely studied. In this work, we provide evidence that stimulating TET2 activity by VC combined with VC-induced dsDNA leakage activate tumor cGAS-cGAMP-endothelial STING pathway and promote lymphocyte trafficking. There are various mechanisms that trigger the export of damaged DNA fragments derived from the nucleus and/or mitochondria, such as formation of abnormal micronucleus, mitochondrial degeneration and membrane potential reduction, autophagy . Further studies are needed to clarify the mechanisms underlying how VC treatment triggers the export of damaged DNA fragments derived from the nucleus and/or mitochondria, and to ascertain the specificity of this modulation towards tumor cells. Importantly, we further reveal that VC treatment induces tumor vascular normalization and boosts efficacy of anti-PD-L1 therapy or anti-PD-L1 combination therapy with IL-2 in vivo, provides promising strategies to enhance the combinational immunotherapy for liver cancer.
In summary, our findings elucidate a crosstalk between tumor and vascular endothelial cells in the tumor microenvironment via cGAS and
STING essential for tumor vascular normalization and anti-tumor immunity (Fig. 10m). Based on the epigenetic regulation of tumor cGAS by TET2 synergized with STAT5A signaling, stimulating TET2 activity by VC triggers tumor cGAS-cGAMP-endothelial STING pathway activation and induces tumor vascular normalization, thereby potentiating immunotherapy efficacy. This work not only uncovers the epigenetic regulatory mechanism of tumor cGAS, but also reveals the immunomodulatory roles of VC and TET2 in liver cancer, providing a strategy to induce tumor vascular normalization and a scientific rationale for further clinical trials combining VC with immunotherapy.
Methods
Study approval
All animal protocols used in this study were approved by The University Committee on Use and Care of Animals of Second Military Medical University. Tumor tissues from patients with HCC were obtained from the Eastern Hepatobiliary Surgery Hospital (EHBH), Shanghai, China, with male to female ratio of , conforming to the sex disparities in HCC, which has a strong male predominance . Patient consent was obtained prior to the commencement of the study. All procedures performed in the study were approved by the Ethical Committee of the Second Military Medical University and in accordance with the Declaration of Helsinki.
Cell culture
The murine liver cancer cell line Hepa1-6 (SCSP-512) and human HCC cell lines Huh7 (SCSP-526) were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (CBTCCCAS, Shanghai, China). The murine endothelial cell line SVEC4-10 (CRL-2181) was purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). Cell lines were authenticated by STR profiling and verified to be mycoplasma negative. All cells were grown in DMEM (Gibco) with at and .
Antibodies and reagents
Anti-human cGAS (79978, 1:1000), anti-mouse cGAS (31659, 1:1000), anti-human/mouse STING (13647, 1:1000), anti-mouse p-STING (Ser365) (72971, 1:1000), anti-human/mouse TBK1 (38066, 1:1000), anti-human/mouse p-TBK1(Ser172) (5483, 1:1000), anti-human/mouse -SMA (19245, 1:100) antibodies, and anti-human/mouse (9718, 1:1000) were purchased from Cell Signaling Technology. Anti-human/ mouse LRRC8C (21601-1-AP, 1:1000), anti-human/mouse Flag (20543-1AP, 1:1000), anti-human/mouse GAPDH (60004-1-Ig, 1:10,000), and anti-human/mouse -Actin (66009-1-Ig, 1:10000) were purchased from Proteintech. Anti-human/mouse TET2 (GTX124205, 1:1000) and anti-human/mouse p-STAT5A (GTX13593, 1:1000) were purchased from GeneTex. Anti-human/mouse CD31 (ab182981, 1:100), antihuman/mouse GLUT1 (ab115730, 1:500), anti-human CD8 (ab93278, 1:100), anti-mouse CD8 (ab209775, 1:100), anti-dsDNA (ab27156,
1:500), anti-mouse NKp46 (ab233558, 1:200), and anti-human/mouse HSP60 (ab46798, 1:200) were purchased from Abcam. Anti-human/ mouse VEGFR2 (sc-6251, 1:1000) was purchased from Santa Cruz. Antimouse VE-Cad (CD144) (138011, 1:100), anti-mouse CD45 (103112/ 103108, 1:100), anti-mouse CD3 (100214, 1:100), anti-mouse CD8 (100706, 1:100), anti-mouse NK1.1 (108714, 1:100), anti-mouse CD11b (101224, 1:100), and anti-mouse F4/80 (123116, 1:100) antibodies were purchased from Biolegend. InVivoMab anti-mouse PD-L1 (BE0101), anti-mouse CD8 (BE0004), and IgG2a isotype (BE0089) antibodies were purchased from BioXCell.
Mouse IL-2 (212-12) was purchased from Peprotech. Recombinant human IL-2 (BT-002-AFL) was purchased from R&D systems. L-ascorbate (VC) (A4034), NAC (A9165), collagenase IV (C5138), and DNase I (D5025) were purchased from Sigma. Fludarabine (S1491) was purchased from Selleck. 2’3′-cGAMP (HY-100564), TFMB-(R)-2-HG (HY129079), STING inhibitor C-176 (HY-112906), and Sorafenib (Sora) (HY10201) were purchased from MCE. MitoSOX Red mitochondrial superoxide indicator (M36008) was purchased from Invitrogen.
Mice
Cgas knockout mice ( Cgas ) (stock number: 026554) and Sting knockout mice (Sting ) (stock number: 025805), both on C57BL/6 background, were purchased from Jackson Laboratory. C57BL/6 mice were used as wild-type (WT) mice in this study and obtained from Laboratory Animal Resources, Chinese Academy of Sciences (Shanghai, China). Male mice at 5-6 weeks of age used in the experiments were obtained from the GemPharmatech Co., Ltd (Jiangsu, China). Mice were housed under 12 light/12 dark cycle, temperatures of with humidity.
Animal models
To establish subcutaneous tumor models, murine liver cancer cells (Hepa1-6) or human liver cancer cells (Huh7) were implanted subcutaneously into the left thighs of male C57BL/6 mice or Nude mice at 5-6 weeks of age, respectively. Tumor growth was monitored by measuring the tumor size (length width ) twice per week after implantation. The permitted maximal tumor size did not exceed 1.5 cm at the largest diameter. In some cases, this limit has been exceeded the last day of measurement and the mice were immediately euthanized. At 3-4 weeks following tumor establishment, all the mice were sacrificed to harvest subcutaneous tumors for weight measurement and further tissue analyses. To establish orthotopic tumor models, laparotomy was administered to implant Hepal-6 cells carrying a luciferase reporter gene into the left lobe of the liver after the mice were anaesthetized.
For bioluminescence imaging, mice were intraperitoneally injected with d-luciferin. After 15-20 min of injection, mice were subjected to imaging using IVIS Lumina III (PerkinElmer) and bioluminescence was quantified using Living Image software. Tumor volume was determined on the basis of the total flux (photons per second).
In vivo treatments and depletion of cells
Prior to treatments initiation, mice were randomly assigned to different groups with similar average tumor volumes. For in vivo treatments, VC ( body weight; daily; i.p.) (Sigma) or PBS control, Sora ( body weight; daily; i.g.) (MCE); PD-L1 antibodies ( per mouse; twice a week; i.p.) (BioXCell) or IgG isotype control, rlL-2 ( per mouse; daily; i.p.) (R&D systems) or PBS control, and C-176 ( 750 nmol per mouse; daily; i.p.) (MCE) or solvent were injected for 2 weeks beginning on day 7 after the establishment of mice liver cancer models. To deplete cells in vivo, mice were intraperitoneally injected with of anti-CD8 antibody (BioXCell) or IgG isotype control (BioXCell) 3 days and 1 day before tumor implantation and twice weekly thereafter to ensure sustained depletion of cell subset during the experimental period.
In vivo vascular permeability assay
Tumor-bearing mice were injected intravenously with 0.5 mg of Evans blue dye (Sigma) in PBS. After 1 h of circulation, the mice were perfused through the heart with PBS containing 2 mM EDTA to remove intravascular Evans blue dye. The tumors were collected, cut into pieces, and incubated in 1 ml formamide for 24 h at to extract the Evans blue from the tumor. After centrifugation, the supernatant was collected and diluted to a final concentration of same tumor weight per ml formamide. Then, the absorbances at 655 nm and 750 nm were measured with a spectrophotometer. To eliminate the effect of residual haem pigment in the blood, the following formula was used: corrected absorbance (A) A655 nm – ( A750 nm + 0.03) (7).
siRNA, shRNA, CRISPR/Cas9 knockout, and overexpression
siRNAs used to knock down murine Lrrc8c, Stat1, and Stat5a were conducted by Biotend (Shanghai, China). siRNAs were transfected into cells using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. The Lentivirus-mediated shRNA expressing vectors targeting murine Tet2 (target sequence: 5′-GCTCTAAAT-GATGTAGCTTTG-3′) and Stat5a (target sequence: 5′-GACGTGA-GATTCAAGTCTAAC-3′) were purchased from Genomeditech (Shanghai, China) and Genechem (Shanghai, China), respectively. Lentivirus-mediated CRISPR/Cas9 knockout (KO) vector targeting murine Sting (target sequence: 5′-CAGCCTGATGATCCTTTGGG-3′) and Cgas (target sequence: -CGGCGGGCAGCTCCGGATCC-3′) was purchased from Obio Technology (Shanghai, China). The lentiviral plasmid expressing murine Cgas was conducted by Biotend (Shanghai, China). The lentiviral plasmids expressing human , and murine Sting-Cherry were purchased from Genechem (Shanghai, China). Cells (30% confluency) were transfected with optimal dilutions of lentivirus mixed with polybrene. Then, the transfected cells were treated with puromycin to select for stable transfected cells. Desired gene disruption was confirmed by immunoblot analysis of target proteins.
Western blotting and co-immunoprecipitation (co-IP)
Whole cell lysates were prepared with cell lysis buffer (Beyotime Biotechnology) and boiled in SDS sample loading buffer. Then, an equal protein content ( of protein per lane) was loaded to PAGE electrophoresis and transferred onto PVDF membranes. The membranes were blocked in 5% milk in TBST for 1 h at room temperature and subsequently incubated overnight with the appropriate primary antibodies at . After incubation with fluorescein-conjugated secondary antibody for 1 h at room temperature, the immunoblots were visualized using an Odyssey fluorescence scanner (Li-Cor, Lincoln, NE, USA). For co-IP, cells lysates ( of protein) were mixed with anti-Tet2 (GeneTex) overnight at and then pulled down with protein G magnetic bead (Invitrogen) at for 2 h . IP beads were then washed with lysis buffer three times, and heated in SDS sample loading buffer at for 5 min for immunoblot analysis.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) and reversely transcribed into cDNA with random primers using Superscript III reverse transcriptase (Invitrogen). The cDNA was subsequently used as the template for the reaction. was performed using SYBR Green PCR Master Mix (Applied Biosystems) on ABI PRISM 7300HT Sequence Detection System (Applied Biosystems). -Actin was used as a control for normalization. The primers were listed in Supplementary Table 1.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed by using EpiQuik Chromatin Immunoprecipitation (ChIP) Kit (Epigentek) according to the manufacturer’s
instructions. In brief, cells were crosslinked and chromatin was extracted and sheared. The samples were immunoprecipitated with anti-Flag antibody (Abcam). qRT-PCR was performed on the immunoprecipitated DNA using SYBR Green PCR Master Mix (Applied Biosystems) on ABI PRISM 7300 HT Sequence Detection System (Applied Biosystems). The values for immunoprecipitated DNA were normalized to input signals. The primers of Cgas promoter were listed in Supplementary Table 1.
Flow cytometric analysis
Mice tumors were mechanically minced and incubated in collagenase IV ( , Sigma) and DNase I ( , Sigma) for 30 min at with shaking. The dissociated cells were filtered through a cell strainer (BD). Then, the resulting single-cell suspensions were incubated with Fc block and stained with the indicated surface antibodies for 20 min at . The stained cells were analyzed immediately by a LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software.
cGAMP measurement
cGAMP levels in cell lysates and culture medium were measured using a 2’3′-cGAMP ELISA Kit (Cayman) according to manufacturer’s protocol. For sample preparation, cells were lysed in M-PER Mammalian Protein Extraction Reagent (ThermoFisher Scientific).
Lymphocytes transendothelial migration assay
Lymphocytes migration across an endothelial barrier in vitro was assessed using CytoSelect Leukocyte Transmigration Assay kit (Cell Biolabs) according to the manufacturers’ instruction. Lymphocytes used in the study were isolated from the spleens of WT or Sting-/- mice with Mouse Lymphocyte Separation Medium (Dakewe Biotechnology) following manufacturer’s protocol, and then labeled with LeukoTracker . The endothelial monolayers were exposed to indicated treatment for 24 h before placing the LeukoTracker labeled lymphocytes on the endothelial monolayer. The relative abundance of transendothelial migrated lymphocytes was calculated by measuring the fluorescence of the samples at .
Immunofluorescence staining
Frozen mouse tumor sections or cells were fixed with 4% paraformaldehyde for 20 min at room temperature and were permeabilized with 1% Triton X-100 (Solarbio) solution for 15 min. Next, 5% BSA in PBS with triton was used to block non-specific binding sites at room temperature for 1 h . Tumor sections were stained with indicated primary antibodies against CD31 and -SMA, and cells were stained with primary antibodies against dsDNA and HSP60 overnight at followed by fluorescent secondary antibodies at room temperature for 1 h . DAPI (Life Technologies) was used for nuclear staining. All the slides were visualized and photo-documented using a STELLARIS 5 confocal microscope (Leica Microsystems).
Endothelial cell proliferation and migration assay
SVEC4-10 cells were seeded at a density of 3000-5000 cells per well in a 96-well plate. After indicated treatment for 24 h , cell viability was measured using Cell Counting Kit-8 (CCK-8) assay (Vazyme) following manufacturer’s instruction. For migration assay, overnight serum-starved SVEC4-10 cells were seeded on the PET membrane ( pore size) of an insert. The inserts were hanged on a 24 -well support plate, which had a 70-80% confluent monolayer of cultured Hepa1-6-Ctrl or Hepa1-6-Cgas cells with complete growth media. After 24 h of incubation, the inserts were fixed with 4% paraformaldehyde and stained with crystal violet solution. Representative fields were photographed and the migrated cells were quantified by ImageJ software (NIH).
Tube formation assay
SVEC4-10 cells were exposed to indicated treatment for 24 h , and subsequently plated on Matrigel (Corning) coated 24 -well plates ( cells/well) for 6 h at before taking pictures. Angiogenesis analyzer module of ImageJ software (NIH) was applied to quantify the number of meshes and total tube length.
Immunohistochemical (IHC) staining
IHC staining was performed on representative tissue sections from formalin-fixed and paraffin-embedded tissue blocks from human HCC and mice tumor xenografts using the indicated antibodies. Based on the immunoreactive score method, the intensity of human HCC tissue microarrays staining (protein expression) was scored as 0 (negative staining), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). Then, the patients were subdivided into two groups: low expression group (negative or weak staining) and high expression group (moderate or strong staining). For CD31/ -SMA dual IHC staining, CD31 was stained first with 3,3-diaminobenzidine (DAB) (brown chromogen) and -SMA was selected second to stain with Cynanine3 (red chromogen). Pericyte coverage of tumor vessels was estimated by measuring the vessels which stained positively for both CD31 and SMA (pericytes marker) among all CD31-positive vessels. The results were shown as the percentage of tumor blood vessels with -SMApositive pericyte coverage.
DNA methylation pyrosequencing
Cgas methylation levels were measured using pyrosequencing to examine the methylation status at CpG sites in the Cgas promoter. The sequencing service and bioinformatics analysis were provided by Shanghai Biotechnology Corporation (Shanghai, China). Briefly, genomic DNA was extracted from cells and then the DNA samples were subjected to bisulfite conversion using the EZ DNA Methylation Kit (Zymo Research) according to the manufacturer’s instructions. Bisulfite-treated DNA was used as template to amplify the target fragment of Cgas promoter with the specific primer pair. PCR products were sequenced by pyrosequencing using PyroMark Q96 (QIAGEN).
Online database analysis
cGAS (C6orf150, MB21D1) and STING (TMEM173) protein expressions in liver cancer tissues assessed by immunohistochemistry were obtained from the Human Protein Atlas (HPA, http://www.proteinatlas.org) . The correlations between different genes expressions in HCC were obtained from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov) and the International Cancer Genome Consortium (ICGC, https://dcc. icgc.org) databases. cell infiltration analysis according to different genes expressions in HCC from TCGA database was conducted using Tumor Immune Estimation Resource (TIMER) . The DNA methylation data from TCGA were derived from DNA methylation interactive visualization database (DNMIVD) . Gene Set Enrichment Analysis (GSEA) and Panther Pathway Analysis in HCC TCGA datasets was performed in the LinkedOmics platform . The expressions of IL-2 receptors in human liver cancer cell lines were obtained from the Cancer Cell Line Encyclopedia (CCLE, https://sites.broadinstitute.org/ccle). Survival differences were validated at the gene expression level in HCC TCGA by an online database Kaplan-Meier Plotter (KM-Plotter) .
Statistics
Statistical analysis was performed using GraphPad Prism 8 software (GraphPad Software, San Diego, CA). Data are presented as mean standard error (SD) unless otherwise stated. Experiments were repeated at least three times with similar results. The specific statistical tests applied are given in the respective figure legends. value was considered to be statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Publicly available datasets reported in this paper are from the GEO databases (GSE51401, GSE69164, GSE146409, GSE140901), The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov), the International Cancer Genome Consortium (ICGC, https://dcc.icgc.org), the Human Protein Atlas (HPA, http://www.proteinatlas.org), and the Cancer Cell Line Encyclopedia (CCLE, https://sites.broadinstitute.org/ccle). The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209-249 (2021).
Cheng, A. L., Hsu, C., Chan, S. L., Choo, S. P. & Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 72, 307-319 (2020).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707-723 (2017).
Huang, Y. et al. Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 18, 195-203 (2018).
Zheng, X. et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. J. Clin. Investig. 128, 2104-2115 (2018).
Martin, J. D., Seano, G. & Jain, R. K. Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu. Rev. Physiol. 81, 505-534 (2019).
Tian, L. et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544, 250-254 (2017).
Khan, K. A. & Kerbel, R. S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 15, 310-324 (2018).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786-791 (2013).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788-792 (2009).
Kwon, J. & Bakhoum, S. F. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 10, 26-39 (2020).
Guey, B. & Ablasser, A. Emerging dimensions of cellular cGASSTING signaling. Curr. Opin. Immunol. 74, 164-171 (2022).
Corrales, L. & Gajewski, T. F. Molecular pathways: targeting the stimulator of interferon genes (STING) in the immunotherapy of cancer. Clin. Cancer Res. 21, 4774-4779 (2015).
Amouzegar, A., Chelvanambi, M., Filderman, J. N., Storkus, W. J. & Luke, J. J. STING agonists as cancer therapeutics. Cancers 13, 2695 (2021).
Park, J. S. et al. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell 30, 953-967 (2016).
Heidenreich, R., Kappel, A. & Breier, G. Tumor endothelium-specific transgene expression directed by vascular endothelial growth factor receptor-2 (Flk-1) promoter/enhancer sequences. Cancer Res. 60, 6142-6147 (2000).
Massalha, H. et al. A single cell atlas of the human liver tumor microenvironment. Mol. Syst. Biol. 16, e9682 (2020).
Saitoh, T. et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl Acad. Sci. USA 106, 20842-20846 (2009).
Ding, Z., Xiong, K. & Issekutz, T. B. Regulation of chemokine-induced transendothelial migration of T lymphocytes by endothelial activation: differential effects on naive and memory T cells. J. Leukoc. Biol. 67, 825-833 (2000).
Vestweber, D., Wessel, F. & Nottebaum, A. F. Similarities and differences in the regulation of leukocyte extravasation and vascular permeability. Semin. Immunopathol. 36, 177-192 (2014).
Chen, Q. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493-498 (2016).
Ritchie, C., Cordova, A. F., Hess, G. T., Bassik, M. C. & Li, L. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol. Cell 75, 372-381 (2019).
Cordova, A. F., Ritchie, C., Böhnert, V. & Li, L. Human SLC46A2 is the dominant cGAMP importer in extracellular cGAMP-sensing macrophages and monocytes. ACS Cent. Sci. 7, 1073-1088 (2021).
Lahey, L. J. et al. LRRC8A:C/E heteromeric channels are ubiquitous transporters of cGAMP. Mol. Cell 80, 578-591 (2020).
Zhou, C. et al. Transfer of cGAMP into Bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral immunity. Immunity 52, 767-781 (2020).
An, X. et al. An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol. Ther. Nucleic Acids 14, 80-89 (2019).
Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597-610 (2005).
Yang, R. et al. Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory cell differentiation and maintain immune homeostasis. Immunity 43, 251-263 (2015).
Malek, T. R. The biology of interleukin-2. Annu. Rev. Immunol. 26, 453-479 (2008).
Liu, J. et al. Global DNA 5-hydroxymethylcytosine and 5-formylcytosine contents are decreased in the early stage of hepatocellular carcinoma. Hepatology 69, 196-208 (2019).
Cimmino, L. et al. Restoration of TET2 function blocks aberrant selfrenewal and leukemia progression. Cell 170, 1079-1095.e20 (2017).
Agathocleous, M. et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476-481 (2017).
Wilhelm, S. et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5, 835-844 (2006).
Ngo, B., Riper, Van, Cantley, J. M., Yun, L. C. & Targeting, J. cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 19, 271-282 (2019).
Lv, H. et al. Vitamin C preferentially kills cancer stem cells in hepatocellular carcinoma via SVCT-2. NPJ Precis. Oncol 2, 1 (2018).
Chen, Y. et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 61, 1591-1602 (2015).
Chang, C. J. et al. Targeting tumor-infiltrating Ly6G+ myeloid cells improves sorafenib efficacy in mouse orthotopic hepatocellular carcinoma. Int. J. Cancer 142, 1878-1889 (2018).
Hsu, C. et al. Exploring markers of exhausted CD8 T cells to predict response to immune checkpoint inhibitor therapy for hepatocellular carcinoma. Liver Cancer 10, 346-359 (2021).
Newell, P., Villanueva, A., Friedman, S. L., Koike, K. & Llovet, J. M. Experimental models of hepatocellular carcinoma. J. Hepatol. 48, 858-879 (2008).
West, E. E. et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Investig. 123, 2604-2615 (2013).
Hashimoto, M. et al. PD-1 combination therapy with IL-2 modifies CD8+ T cell exhaustion program. Nature 610, 173-181 (2022).
Falahat, R. et al. STING signaling in melanoma cells shapes antigenicity and can promote antitumor T-cell activity. Cancer Immunol. Res. 7, 1837-1848 (2019).
Mahadevan, N. R. et al. Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov. 11, 1952-1969 (2021).
Yang, H. et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig. 129, 4350-4364 (2019).
Chelvanambi, M., Fecek, R. J., Taylor, J. L. & Storkus, W. J. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J. Immunother. Cancer 9, e001906 (2021).
Mao, Y. et al. STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in dietinduced obesity. Arterioscler. Thromb. Vasc. Biol. 37, 920-929 (2017).
Huang, L. S. et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52, 475-486.e5 (2020).
Corrales, L. et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11, 1018-1030 (2015).
Demaria, O. et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl Acad. Sci. USA 112, 15408-15413 (2015).
Shen, Y. J. et al. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep. 11, 460-473 (2015).
Konno, H. et al. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37, 2037-2051 (2018).
Xia, T., Konno, H., Ahn, J. & Barber, G. N. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14, 282-297 (2016).
Lv, H. et al. NAD+ metabolism maintains inducible PD-L1 expression to drive tumor immune evasion. Cell Metab. 33, 110-127.e5 (2021).
Xu, Y. P. et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 129, 4316-4331 (2019).
Slaney, C. Y., Kershaw, M. H. & Darcy, P. K. Trafficking of T cells into tumors. Cancer Res. 74, 7168-7174 (2014).
Nauman, G., Gray, J. C., Parkinson, R., Levine, M. & Paller, C. J. Systematic review of intravenous ascorbate in cancer clinical trials. Antioxidants 7, 89 (2018).
Luchtel, R. A. et al. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl Acad. Sci. USA 117, 1666-1677 (2020).
Magri, A. et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 12, eaay8707 (2020).
Lan, Y. Y., Londoño, D., Bouley, R., Rooney, M. S. & Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 9, 180-192 (2014).
Gehrke, N. et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STINGdependent immune sensing. Immunity 39, 482-495 (2013).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509-W514 (2020).
Ding, W. et al. DNMIVD: DNA methylation interactive visualization database. Nucleic Acids Res. 48, D856-D862 (2020).
Vasaikar, S. V., Straub, P., Wang, J. & Zhang, B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46, D956-D963 (2018).
Menyhárt, O., Nagy, Á. & Győrffy, B. Determining consistent prognostic biomarkers of overall survival and vascular invasion in hepatocellular carcinoma. R. Soc. Open Sci. 5, 181006 (2018).
Acknowledgements
We thank Dr. Wuhan Xiao and Dr. Guoliang Xu (Chinese Academy of Sciences) for Tet plasmids, and Dan Cao, Shanhua Tang, Linna Guo, Lu Chen, Mingshuang Xu, Shennian Ge, and Qi Yang for their technical assistance. This work was supported by grants from the National Natural Science Foundation of China (82273176, 81902894, 81972779, 81903036, 81830054, 91859205, and 81988101), Chinese National Key Project (2018ZX10723204-006-003), Shanghai Municipal Commission of Education Project (201901070007E00065), and Program of Shanghai Academic Research Leader (23XD1404800).
Author contributions
W.Y., H.L., and H.W. designed the study. H.L., Q.Z., C.C., W.X., F.X., and G.L. performed the experiments and data analysis. G.J, B.Y., X.S., Y.M., L.W., Z.W., and X.C. provided technical support. H.L. and W.Y. wrote the manuscript. H.W. and W.Y. organized and supervised the study.
Correspondence and requests for materials should be addressed to Hongyang Wang or Wen Yang.
Peer review information Nature Communications thanks Walter Storkus and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
International Co-operation Laboratory on Signal Transduction, Eastern Hepatobiliary Surgery Hospital, Naval Medical University (Second Military Medical University), Shanghai 200438, China. National Center for Liver Cancer, Naval Medical University (Second Military Medical University), Shanghai 201805, China. Cancer Research Center, First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui 230027, China. Department of Hepatobiliary Surgery, Clinical Medical College, Yangzhou University, Yangzhou, Jiangsu 225000, China. Hospital of Zhengzhou University, Zhengzhou, Henan 450000, China. Shanghai Key Laboratory of Hepato-biliary Tumor Biology, Shanghai 200438, China. Key Laboratory of Signaling Regulation and Targeting Therapy of Liver Cancer, Ministry of Education, Shanghai 200438, China. These authors contributed equally: Hongwei Lv, Qianni Zong, Cian Chen. e-mail: hywangk@vip.sina.com; woodeasy66@hotmail.com